Vitamins
The term vitamin is derived from the words vital and amine, because vitamins are required for life and were originally thought to be amines. Although not all vitamins are amines, they are organic compounds required by humans in small amounts from the diet. An organic compound is considered a vitamin if a lack of that compound in the diet results in overt symptoms of deficiency.
The information from the Linus Pauling Institute's Micronutrient Information Center on vitamins and minerals is now available in a book titled, An Evidence-based Approach to Vitamins and Minerals: Health Benefits and Intake Recommendations. The book can be purchased from the Linus Pauling Institute or Thieme Medical Publishers.
Select a vitamin from the list for more information.
Biotin
Contents
Summary
- Water-soluble biotin is an essential cofactor to enzymes in intermediary metabolism and a key regulator of gene expression. (More information)
- Both parenteral nutrition devoid of biotin and prolonged consumption of raw egg white have been associated with symptoms of frank biotin deficiency, including hair loss, dermatitis, skin rash, ataxia, seizures, and other neurologic dysfunctions. (More information)
- Biotinidase deficiency is a rare hereditary disorder that impairs biotin absorption and recycling, resulting in secondary biotin deficiency. (More information)
- The recommended adequate intake (AI) of biotin is set at 30 micrograms (μg)/day in adults. Biotin requirements are likely increased during pregnancy and breast-feeding. (More information)
- Animal studies have shown that biotin sufficiency is essential for normal fetal development. Yet, it is not known if marginal biotin deficiency during pregnancy increases the risk for congenital anomalies in humans. (More information)
- Biotin is used in the treatment of biotin-thiamin responsive basal ganglia disease, an inherited disorder of thiamin transport. (More information)
- Recent randomized controlled trials have not found high-dose biotin supplementation to be beneficial in the treatment of multiple sclerosis. Yet, results of animal studies and meta-analyses of human clinical trials are promising. (More information)
- Definitive evidence that establishes whether biotin supplementation improves glucose and lipid homeostasis in individuals with type 2 diabetes mellitus is currently lacking, but suggestive observations have been published. (More information)
- Biotin cannot be synthesized by mammalian cells and must be obtained from exogenous sources. Biotin is widely found in food, and good dietary sources include egg yolk, liver, whole-grain cereal, and some vegetables. (More information)
- Long-term anticonvulsant (anti-seizure) therapy may increase the dietary requirement for biotin because anticonvulsants can interfere with the intestinal absorption and renal re-absorption of biotin and likely also increase degradation of biotin to inactive metabolites. (More information)
- Use of high-dose biotin supplements can result in aberrant results of laboratory tests that utilize the very high affinity biotin-streptavidin (avidin) interaction. (More information)
Biotin is a water-soluble vitamin that is generally classified as a B-complex vitamin. After its initial discovery in 1927, 40 years of additional research was required to unequivocally establish biotin as a vitamin (1). Biotin is required by all organisms but can be synthesized by some strains of bacteria, yeast, mold, algae, and some plant species (2).
Function
Biotinylation
Biotin functions as a covalently bound cofactor required for the biological activity of the five known mammalian biotin-dependent carboxylases (see below). Such non-protein cofactors are termed “prosthetic groups” and are common in water-soluble vitamins. The covalent attachment of biotin to the apocarboxylase (i.e., the carboxylase protein without the biotin prosthetic group and is catalytically inactive) is catalyzed by the enzyme, holocarboxylase synthetase (HCS). The term “biotinylation” refers to the covalent addition of biotin to any molecule, including the apocarboxylases and histones. HCS catalyzes the post-translational biotinylation of the epsilon amino group of a lysine residue at the active site of each apocarboxylase, converting the inactive apocarboxylase into a fully active holocarboxylase (Figure 1a). Particular lysine residues within the N-terminal tail of specific histone that help package DNA in eukaryotic nuclei can also be biotinylated (3). Biotinidase is the enzyme that catalyzes the release of biotin from biotinylated histones and from the peptide products of holocarboxylase breakdown (Figure 1b).


Enzyme cofactor
Five mammalian carboxylases catalyze essential metabolic reactions:
• Both acetyl-Coenzyme A (CoA) carboxylase 1 (ACC1) and acetyl-CoA carboxylase 2 (ACC2) catalyze the conversion of acetyl-CoA to malonyl-CoA using bicarbonate and ATP; however, the two enzymes have different roles in metabolism and different intracellular locations. ACC1 is located in the cytosol, and the malonyl CoA generated by ACC1 is a rate-limiting substrate for the synthesis of fatty acids (Figure 2). ACC1 is found in all tissues and is particularly active in lipogenic tissues (i.e., liver, white adipose tissue, and mammary gland), heart, and pancreatic islets. ACC2 is located on the outer mitochondrial membrane, and the malonyl CoA generated via ACC2 inhibits CPT1, an enzyme that regulates malonyl-CoA entry into the inner mitochondria, thereby regulating fatty acid oxidation (Figure 3). ACC2 is especially abundant in skeletal muscle and heart (4).

• Pyruvate carboxylase is a critical enzyme in gluconeogenesis (the formation of glucose from sources other than carbohydrates, such as pyruvate, lactate, glycerol, and the glucogenic amino acids). Pyruvate carboxylase catalyzes the ATP-dependent incorporation of bicarbonate into pyruvate, producing oxaloacetate; hence, pyruvate carboxylase is anaplerotic for the citric acid cycle (Figure 3). Oxaloacetate can then be converted to phosphoenolpyruvate and eventually to glucose.

• Methylcrotonyl-CoA carboxylase catalyzes an essential step in the catabolism of leucine, an essential branched-chain amino acid. This enzyme catalyzes the production of 3-methylglutaconyl-CoA from methylcrotonyl-CoA (Figure 4a).
• Propionyl-CoA carboxylase produces D-malonylmalonyl-CoA from propionyl-CoA, a by-product in the β-oxidation of fatty acids with an odd number of carbon atoms (Figure 4a). The conversion of propionyl-CoA to D-malonylmalonyl-CoA is also required in the catabolic pathways of two branched-chain amino acids (isoleucine and valine) and the side chain of cholesterol (Figure 4a) and of the amino acids methionine and threonine (Figure 4b).


Regulation of chromatin structure and gene expression
In eukaryotic nuclei, DNA is packaged into compact structures to form nucleosomes — fundamental units of chromatin. Each nucleosome is composed of 147 base pairs of DNA wrapped around eight histones (paired histones: H2A, H2B, H3, and H4). The H1 linker histone is located at the outer surface of each nucleosome and serves as an anchor to fix the DNA around the histone core. The compact packaging of chromatin must be relaxed for DNA replication and transcription. Chemical modifications of DNA and histones affect the folding of chromatin, increasing or reducing DNA accessibility to factors involved in replication and transcription. DNA methylation and a number of chemical modifications within the N-terminal tail of core histones modify their electric charge and structure, thereby changing chromatin conformation and transcriptional activity of genes.
The modifications of histone tails ("marks"), including acetylation, methylation, phosphorylation, ubiquitination, SUMOylation, ADP-ribosylation, carbonylation, deimination, hydroxylation, and biotinylation, have various regulatory functions. Several sites of biotinylation have been identified in histones H2A, H3, and H4 (5). Amongst them, histone H4 biotinylation at lysine (K) 12 (noted H4K12bio) appears to be enriched in heterochromatin, a tightly condensed chromatin associated with repeat regions in (peri)centromeres and telomeres. H4 biotinylation appears to be enriched in transposable elements known as long terminal repeats (3). These biotinylation marks also co-localize with well-known gene repression marks like methylated lysine 9 in histone H3 (H3K9me) in transcriptionally competent chromatin (6). For example, H4K12bio can be found at the promoter of the gene SLC5A6 that codes for the transporter mediating biotin uptake into cells, the human sodium-dependent multivitamin transporter (hSMVT). When biotin is abundant, HCS can biotinylate histones H4 in the SLC5A6 promoter, which down regulates hSMVT synthesis and reduces biotin uptake. Conversely, in biotin-deficient cells, biotinylation marks in the SLC5A6 promoter are removed increasing gene expression and enabling the synthesis of hSMVT and uptake of biotin (7).
Deficiency
Although clinically overt biotin deficiency is very rare, the human requirement for dietary biotin has been demonstrated in three different situations: prolonged intravenous feeding (parenteral) without biotin supplementation, infants fed an elemental formula devoid of biotin, and consumption of raw egg white for a prolonged period (many weeks to years) (8). Raw egg white contains avidin; this antimicrobial protein binds biotin with an affinity and specificity that is almost unique as a reversible binding. Because native avidin is resistant to mammalian and microbial digestion, avidin prevents biotin absorption. Cooking egg white denatures avidin, rendering it susceptible to digestion and therefore unable to block the absorption of dietary biotin (5).
Signs and symptoms of biotin deficiency
Signs of overt biotin deficiency include hair loss (alopecia) and a scaly red rash around the eyes, nose, mouth, and genital area. Neurologic symptoms in adults have included depression, lethargy, hallucinations, numbness and tingling of the extremities, ataxia, and seizures. The characteristic facial rash, together with unusual facial fat distribution, has been termed the "biotin deficient facies" by some investigators (1). Individuals with hereditary disorders of biotin metabolism (see Inborn metabolic disorders) that result in functional biotin deficiency often have similar physical findings, impaired immune system function, and increased susceptibility to bacterial and fungal infections (9, 10).
Risk factors for biotin deficiency
Aside from prolonged consumption of raw egg white or total intravenous nutritional support lacking biotin, other conditions may increase the risk of biotin depletion. Smoking has been associated with increased biotin catabolism (11). The rapidly dividing cells of the developing fetus require biotin for synthesis of essential carboxylases and for histone biotinylation; hence, the maternal biotin requirement is likely increased during pregnancy. Research suggests that a substantial number of women develop marginal or subclinical biotin deficiency during normal pregnancy (see also Disease Prevention) (8, 12, 13). Moreover, certain types of liver disease may decrease biotinidase activity and theoretically increase the requirement for biotin. For example, a study of 62 children with chronic liver disease and 27 healthy controls found serum biotinidase activity to be abnormally low in those with severely impaired liver function due to cirrhosis (14). However, this study did not provide evidence of biotin deficiency. Additionally, anticonvulsant medications used to prevent seizures in individuals with epilepsy increase the risk of biotin depletion (for more information on biotin and anticonvulsants, see Drug interactions).
Inborn metabolic disorders
Biotinidase deficiency
Biotinidase deficiency is an autosomal recessive inherited disorder that is often detected upon newborn screening for metabolic disorders, although late-onset forms have been recently described (15-17). Biotinidase deficiency leads to secondary biotin deficiency in several ways. Intestinal absorption is decreased because deficient biotinidase impairs release of biotin from dietary protein (18). Further, recycling of intracellular biotin bound to carboxylases and histones is also impaired, and urinary loss of biocytin (N-biotinyl-lysine) and biotin is increased (see Figure 1 above) (5). Biotinidase deficiency responds to supplemental biotin. Oral supplementation with as much as 5 to 20 milligrams (mg) of biotin daily is sometimes required (19, 20), although smaller doses may be sufficient, especially later in childhood (reviewed in 20, 21). Prognosis is characteristically good when biotin therapy is introduced in infancy or early childhood and reliably continued for life (10).
Holocarboxylase synthetase deficiency
Holocarboxylase synthetase deficiency results in decreased formation of all holocarboxylases at physiological blood biotin concentrations; thus, high-dose biotin supplementation (10-80 mg of biotin daily) is required (10). Holocarboxylase synthetase deficiency responds to supplementation with pharmacologic doses of biotin in some cases but not others. The prognosis of holocarboxylase synthetase is usually, but not always, good if biotin therapy is introduced early (even antenatally) and continued for life (10, 22).
Biotin transport deficiency
There has been one case report of a child with biotin transport deficiency who responded to high-dose biotin supplementation (23). Of note, the presence of a defective human sodium-dependent multivitamin transporter (hSMVT) was ruled out as a cause of biotin transport deficiency.
Markers of biotin status
Four measures of marginal biotin deficiency have been validated as indicators of biotin status: and (1) reduced levels of holo-methylcrotonyl-CoA carboxylase and holo-propionyl-CoA carboxylase in lymphocytes, the most reliable indicators of biotin status (24); (2) reduced propionyl-CoA carboxylase activity in peripheral blood lymphocytes (5); (3) high urinary excretion of an organic acid, 3-hydroxyisovaleric acid, and its derivative, 3-hydroxyisovaleryl carnitine, both of which reflect decreased activity of biotin-dependent methylcrotonyl-CoA carboxylase; (4) reduced urinary excretion of biotin and some of its catabolites. These markers have been only validated in men and nonpregnant women, and they may not accurately reflect biotin status in pregnant or breast-feeding women (12, 25).
The Adequate Intake (AI)
Sufficient scientific evidence is lacking to estimate the dietary requirement for biotin; thus, no Recommended Dietary Allowance (RDA) for biotin has been established. Instead, the Food and Nutrition Board of the National Academy of Medicine set recommendations for an Adequate Intake (AI; Table 1). The AI for adults (30 μg/day) was extrapolated from the AI for infants exclusively fed human milk and probably overestimates the dietary requirement for biotin for most adults. Dietary intakes of generally healthy adults have been estimated to be 40 to 60 micrograms (μg) of biotin daily (1). The requirement for biotin in pregnancy may be increased (26).
Disease Prevention
Congenital anomalies
Current research indicates that at least one-third of women develop marginal biotin deficiency during pregnancy (8). Small observational studies in pregnant women have reported an abnormally high urinary excretion of 3-hydroxyisovaleric acid in both early and late pregnancy, suggesting decreased activity of biotin-dependent methylcrotonyl-CoA carboxylase (27, 28). In a randomized, single-blinded intervention study in 26 pregnant women, supplementation with 300 μg/day of biotin for two weeks limited the excretion of 3-hydroxyisovaleric acid compared to placebo, confirming that increased 3-hydroxyisovaleric acid excretion indeed reflected marginal biotin deficiency in pregnancy (29). A small cross-sectional study in 22 pregnant women reported an incidence of low lymphocyte propionyl-CoA carboxylase activity greater than 80% (13). Although these levels of biotin deficiency are not associated with overt signs of deficiency in pregnant women, such observations are sources of concern because subclinical biotin deficiency has been shown to cause cleft palate and limb hypoplasia in several animal species (reviewed in 13). In addition, biotin depletion has been found to suppress the expression of biotin-dependent carboxylases, remove biotin marks from histones, and decrease the proliferation in human embryonic palatal mesenchymal cells in culture (30). Impaired carboxylase activity may result in alterations in lipid metabolism, which have been linked to cleft palate and skeletal abnormalities in animals. Further, biotin deficiency leading to reduced histone biotinylation at specific genomic loci may increase genomic instability and result in chromosome anomalies and fetal malformations (13).
Analogous to pregnant women who are advised to consume supplemental folic acid prior to and during pregnancy to prevent neural tube defects (see Folate), it would also be prudent to ensure adequate biotin intake throughout pregnancy. The current AI for pregnant women is 30 μg/day of biotin, and no toxicity has ever been reported at this level of intake (see Safety).
Disease Treatment
Biotin-thiamin-responsive basal ganglia disease
Biotin-thiamin-responsive basal ganglia disease (also called biotin-responsive basal ganglia disease, thiamin transporter-2 deficiency, and thiamin metabolism dysfunction syndrome-2) is caused by an autosomal recessive mutation in the SLC19A3 gene that codes for thiamin transporter-2 (THTR-2). The disease usually presents around 3 to 10 years of age (31), but an early infantile form of the disease exists with onset as early as one month of age (32). Clinical features include subacute encephalopathy (confusion, drowsiness, altered level of consciousness), ataxia, and seizures.
A retrospective study of 18 affected individuals from the same family or the same tribe in Saudi Arabia showed that biotin monotherapy (5-10 mg/kg/day) efficiently abolished the clinical manifestations of the disease, although one-third of the patients suffered from recurrent acute crises. Often associated with poor outcomes, acute crises were not observed after thiamin supplementation started (300-400 mg/day) and during a five-year follow-up period, early diagnosis and immediate treatment with biotin and thiamin led to positive outcomes (33). Although the specific mechanism for therapeutic effects of biotin in biotin-thiamin-responsive basal ganglia disease remains unknown, lifelong high-dose supplementation with a combination of biotin and thiamin is the recommended treatment (31). Early diagnosis and treatment is important to ensure a better prognosis (32, 34).
Multiple sclerosis
Multiple sclerosis (MS) is an autoimmune disease characterized by progressive damage to the myelin sheath surrounding nerve fibers (axons) and neuronal loss in the brain and spinal cord of affected individuals in anatomic locations that vary widely among affected individuals producing variable signs and symptoms. The progression of neurologic disabilities in MS patients is often assessed by the Expanded Disability Status Scale (EDSS) with scores from 1 to 10, from minimal signs of motor dysfunction (score of 1) to death by MS (score of 10). ATP deficiency due to mitochondrial dysfunction and increased oxidative stress may be partly responsible for the progressive degeneration of neurons in MS (35). Given its role in energy production by intermediary metabolism and fatty acid oxidation and in fatty acid synthesis (required for myelin formation) (see Function), high-dose biotin supplementation it has been hypothesized that to exert beneficial effects that would limit or reverse MS-associated functional impairments (35).
The mechanism of action of high-dose biotin has been investigated in a genetic mouse model of chronic axon injury caused by oxidative damage and bioenergetic failure. High-dose biotin restored redox homeostasis, mitochondria biogenesis, and ATP levels, and reversed axonal death and locomotor impairment. Dysregulation of the transcriptional program for lipid synthesis and degradation in the spinal cord was also normalized, possibly as the result of hyperactivation of a nutrient/energy/redox sensor that controls protein synthesis restoring lipid homeostasis.
A nonrandomized, uncontrolled pilot study in 23 patients with progressive MS found high doses of biotin (100-600 mg/day) to be associated with sustained clinical improvements in five (out of five) patients with progressive visual loss and 16 (out of 18) patients with partial paralysis of the limbs after a mean three months following treatment onset (36). Additionally, a multicenter, randomized, placebo-controlled trial in 154 subjects with progressive MS reported that 13 out of 103 patients supplemented with high-dose, pharmaceutical-grade biotin (300 mg/day) for 12 months achieved MS-related disability reversal — assessed by improved EDSS or 25-foot walk time (37). In comparison, none of the 51 patients randomized to the placebo group showed significant clinical improvements (37). However, when this regimen of high-dose biotin supplementation was examined in a larger, international cohort of patients with progressive MS (326 patients receiving biotin and 316 patients receiving placebo), no benefits on EDSS or walk time were seen after 12 months (38). Moreover, a randomized, double-blind, placebo-controlled trial in 93 MS patients with chronic visual loss found that 300 mg/day of pharmaceutical-grade biotin for six months did not improve visual acuity, but an interesting trend favoring the biotin group was observed in the subgroup of patients with progressive optic neuritis (39). Moreover, a meta-analysis of three randomized controlled trials (2 on disability; 3 on adverse effects), involving 889 individuals diagnosed with MS (the preponderance of participants [830] had progressive MS while only 59 had remitting relapsing MS) was conducted (40). Pooling results of two trials found no benefit of high-dose biotin on MS-related disability, but there was significant heterogeneity between the trials. When the subgroup progressive MS was analyzed separately, a moderate certainty of evidence suggested a potential benefit in favor of high-dose biotin for the 25-foot minute walk time (40). On balance, studies remain inconclusive but promising.
Diabetes mellitus
Overt biotin deficiency has been shown to impair glucose utilization in mice (41) and cause fatal hypoglycemia in chickens. Overt biotin deficiency likely also causes abnormalities in glucose regulation in humans (see Function). One early human study reported lower serum biotin concentrations in 43 patients with type 2 diabetes mellitus compared to 64 control subjects without the disease; an inverse relationship between fasting blood glucose and biotin concentrations was observed as well (42). In a small, randomized, placebo-controlled intervention study in 28 patients with type 2 diabetes, daily supplementation with 9 milligrams (mg) of biotin for one month resulted in a 45% decrease in mean fasting blood glucose concentrations (42). Yet, another small study in 10 patients with type 2 diabetes and 7 controls without diabetes found no effect of biotin supplementation (15 mg/day) for 28 days on fasting blood glucose concentrations in either group (43). A more recent double-blind, placebo-controlled study by the same research group showed that the same biotin regimen lowered plasma triglyceride concentrations in patients with hypertriglyceridemia — independent of whether they had type 2 diabetes (44). In this study, biotin administration did not affect blood glucose concentrations in either patient group. Additionally, a few studies have shown that co-supplementation with biotin and chromium picolinate may be a beneficial adjunct therapy in patients with type 2 diabetes (45-48). For information on chromium supplementation as a monotherapy for type 2 diabetes, see the article on Chromium.
Potential mechanisms for the glucose and lipid effects have been suggested. As a cofactor of carboxylases required for fatty acid synthesis, biotin may increase the utilization of glucose for fat synthesis. Also, biotin stimulates glucokinase, a liver enzyme that increases synthesis of glycogen, the storage form of glucose. Biotin also triggers the secretion of insulin in the pancreas of rats and improves glucose homeostasis (50). Yet, reduced activity of ACC1 and ACC2 would be expected to reduce fatty acid synthesis and increase fatty acid oxidation, respectively. Hence, whether pharmacologic doses of biotin benefits the management of hyperglycemia in patients with impaired glucose tolerance remains unclear. Moreover, whether supplemental biotin lowers the risk of cardiovascular complications in patients with diabetes by reducing serum triglycerides and LDL-cholesterol remains to be proven (44-46).
Brittle fingernails (onychorrhexis)
The finding that biotin supplements were effective in treating hoof abnormalities in hoofed animals suggested that biotin might also be helpful in strengthening brittle fingernails in humans (50-52). Three uncontrolled trials examining the effects of biotin supplementation (2.5 mg/day for several months) in women with brittle fingernails have been published (53-55). In two of the trials, subjective evidence of clinical improvement was reported in 67%-91% of the participants available for follow-up at the end of the treatment period (53, 54). One trial that used scanning electron microscopy to assess fingernail brittleness reported less fingernail splitting and a 25% increase in the thickness of the nail plate in patients supplemented with biotin for 6 to 15 months (55). Biotin supplementation (5 mg/day) was also found to be effective in controlling unruly hair and splitting nails in two toddlers with inherited uncombable hair syndrome (56). Although preliminary evidence suggests that supplemental biotin may help strengthen fragile nails (reviewed in 57), larger placebo-controlled trials are needed to assess the efficacy of high-dose biotin supplementation for the treatment of brittle fingernails.
Hair loss (alopecia)
Biotin administration has been associated with alopecia reversal in children treated with the anticonvulsant valproic acid (see Drug interactions), as well as with hair regrowth or normal hair growth in some children with inborn errors of biotin metabolism or other genetic disorders (i.e., uncombable hair syndrome) (reviewed in 58). Yet, while hair loss is a symptom of severe biotin deficiency (see Deficiency), there are no published scientific studies that support the claim that high-dose biotin supplements are effective in preventing or treating hair loss in men or women (59, 60). Randomized, placebo-controlled trials in healthy individuals would be needed to evaluate this claim.
Sources
Food sources
Biotin is found in many foods, either as the free (i.e., unbound) form that is directly taken up by enterocytes or as biotin bound to dietary proteins. Egg yolk, liver, and yeast are rich sources of biotin. Estimates of average daily intakes of biotin from small studies ranged between 40 and 60 micrograms (μg) per day in adults (1). However, US national nutritional surveys have not yet been able to estimate biotin intake due to the scarcity and unreliability of data regarding biotin content of food. Food composition tables for biotin are incomplete such that dietary intakes cannot be reliably estimated in humans. A study by Staggs et al. (61) employed a high-performance liquid chromatography method rather than bioassays (62) and reported relatively different biotin content for some selected foods. Table 2 lists some food sources of biotin, along with their content in μg.
| Food | Serving | Biotin (μg) |
|---|---|---|
| Yeast | 1 packet (7 grams) | 1.4-14 |
| Bread, whole-wheat | 1 slice | 0.02-6 |
| Egg, cooked | 1 large | 13-25 |
| Cheese, cheddar | 1 ounce | 0.4-2 |
| Liver, cooked | 3 ounces* | 27-35 |
| Pork, cooked | 3 ounces* | 2-4 |
| Salmon, cooked | 3 ounces* | 4-5 |
| Avocado | 1 whole | 2-6 |
| Raspberries | 1 cup | 0.2-2 |
| Cauliflower, raw | 1 cup | 0.2-4 |
| *A three-ounce serving of meat is about the size of a deck of cards. | ||
Bacterial synthesis
A majority of bacteria that normally colonize the small and large intestine (colon) synthesize biotin (63). Whether the biotin is released and absorbed by humans in meaningful amounts remains unknown. The uptake of free biotin into intestinal cells via the human sodium-dependent multivitamin transporter (hSMVT) has been identified in cultured cells derived from the lining of the small intestine and colon (64), suggesting that humans may be able to absorb biotin produced by enteric bacteria — a phenomenon documented in swine.
Supplements
Biotin is available as a single-nutrient supplement in various doses (many containing 5,000 μg [5 mg] of biotin) and is often included in B-complex and multivitamin-mineral supplements. Several multivitamin supplements contain 30 μg of biotin, although the amount varies by product (65).
Safety
Toxicity
Biotin is not known to be toxic. In people without disorders of biotin metabolism, doses of up to 5 mg/day (5,000 μg) for two years were not associated with adverse effects (66). Oral biotin supplementation has been well tolerated in doses up to 200 mg/day (nearly 7,000 times the AI) in people with hereditary disorders of biotin metabolism (1). Daily supplementation with a highly concentrated formulation of biotin (100-600 mg) for several months was also found to be well tolerated in individuals with progressive multiple sclerosis (36, 67). However, there is one case report of life-threatening eosinophilic pleuropericardial effusion in an elderly woman who took a combination of 10 mg/day of biotin and 300 mg/day of pantothenic acid (vitamin B5) for two months (68). Because reports of adverse events were lacking when the Dietary Reference Intakes (DRIs) for biotin were established in 1998, the Food and Nutrition Board did not establish a tolerable upper intake level (UL) for biotin (1).
Nutrient interactions
Large doses of pantothenic acid (vitamin B5) have the potential to compete with biotin for intestinal and cellular uptake by the human sodium-dependent multivitamin transporter (hSMVT) (69, 70). Biotin also shares the hSMVT with lipoic acid (71). Pharmacologic (very high) doses of lipoic acid have been found to decrease the activity of biotin-dependent carboxylases in rats, but such an effect has not been demonstrated in humans (72).
Drug interactions
Individuals on long-term anticonvulsant (anti-seizure) therapy reportedly have reduced blood biotin concentrations, as well as an increased urinary excretion of organic acids (e.g., 3-hydroxyisovaleric acid) that indicate decreased carboxylase activity (see Markers of biotin status) (5). Potential mechanisms of biotin depletion by the anticonvulsants, primidone (Mysoline), phenytoin (Dilantin, Phenytek), and carbamazepine (Carbatrol, Epitol, Equetro, Tegretol), include inhibition of biotin intestinal absorption and renal reabsorption, as well as increased biotin catabolism (73). Use of the anticonvulsant valproic acid in children has resulted in hair loss reversed by biotin supplementation (74-77). Long-term treatment with antibacterial sulfonamide (sulfa) drugs or other antibiotics may decrease bacterial synthesis of biotin. Yet, given that the extent to which bacterial synthesis contributes to biotin intake in humans is not known, effects of antimicrobial drugs on biotin nutritional status remain uncertain (73).
Interference with lab assays
In 2017, the US Food and Drug Administration (FDA) released a safety communication regarding high-dose biotin supplementation and its potential interference with streptavidin (avidin)-biotin immunoassays, including assays of thyroid hormones, reproductive hormones, and the cardiac protein, troponin. Such interference can cause aberrant results — either falsely high or falsely low depending on the method of the particular assay — and potential misdiagnosis of disease (78-81). An updated FDA communication in 2019 focused on biotin interference in certain lab assays of troponin that have not addressed the concern of falsely low blood concentrations (82). Troponin in blood is a marker of cardiac damage and often used in the clinical diagnosis of a myocardial infarction (heart attack). When blood work is planned, patients should routinely inform their health care provider concerning supplementation of biotin at doses substantially greater than those in usual diets or in routine daily multivitamins.
Linus Pauling Institute Recommendation
Little is known regarding the amount of dietary biotin required to promote optimal health or prevent chronic disease. The Linus Pauling Institute supports the recommendation made by the National Academy of Medicine, which is 30 micrograms (μg) of biotin per day for adults. A varied diet should provide enough biotin for most people. However, following the Linus Pauling Institute recommendation to take a daily multivitamin-mineral supplement will generally provide an intake of at least 30 μg/day of biotin.
Older adults (>50 years)
Presently, there is no indication that older adults have an increased requirement for biotin. If dietary biotin intake is not sufficient, a daily multivitamin-mineral supplement will generally provide an intake of at least 30 μg of biotin per day.
Authors and Reviewers
Originally written in 2000 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in June 2004 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in August 2008 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in July 2015 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in July 2022 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in March 2023 by:
Donald Mock, M.D., Ph.D.
Professor Emeritus
Departments of Biochemistry and Molecular Biology and Pediatrics
University of Arkansas for Medical Sciences
Copyright 2000-2024 Linus Pauling Institute
References
1. Food and Nutrition Board, Institute of Medicine. Biotin. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B-6, Vitamin B-12, Pantothenic Acid, Biotin, and Choline. Washington, D.C.: National Academy Press; 1998:374-389. (National Academy Press)
2. Mock DM. Biotin. Handbook of vitamins. 4th ed. Boca Raton, FL: CRC Press; 2007:361-383.
3. Zempleni J, Teixeira DC, Kuroishi T, Cordonier EL, Baier S. Biotin requirements for DNA damage prevention. Mutat Res. 2012;733(1-2):58-60. (PubMed)
4. Saggerson D. Malonyl-CoA, a key signaling molecule in mammalian cells. Annu Rev Nutr. 2008;28:253-272. (PubMed)
5. Zempleni J, Wijeratne SSK, Kuroishi T. Biotin. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed: John Wiley & Sons, Inc.; 2012:359-374.
6. Zempleni J, Li Y, Xue J, Cordonier EL. The role of holocarboxylase synthetase in genome stability is mediated partly by epigenomic synergies between methylation and biotinylation events. Epigenetics. 2011;6(7):892-894. (PubMed)
7. Zempleni J, Gralla M, Camporeale G, Hassan YI. Sodium-dependent multivitamin transporter gene is regulated at the chromatin level by histone biotinylation in human Jurkat lymphoblastoma cells. J Nutr. 2009;139(1):163-166. (PubMed)
8. Mock DM. Biotin. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed: Lippincott Williams & Wilkins; 2014:390-398.
9. Baumgartner ER, Suormala T. Inherited defects of biotin metabolism. Biofactors. 1999;10(2-3):287-290. (PubMed)
10. Elrefai S, Wolf B. Disorders of biotin metabolism. In: Rosenberg RN, Pascual JM, eds. Rosenberg's Molecular and Genetic basis of Neurological and Psychiatric Disease. 5th ed. United States of America: Elsevier; 2015:531-539.
11. Sealey WM, Teague AM, Stratton SL, Mock DM. Smoking accelerates biotin catabolism in women. Am J Clin Nutr. 2004;80(4):932-935. (PubMed)
12. Perry CA, West AA, Gayle A, et al. Pregnancy and lactation alter biomarkers of biotin metabolism in women consuming a controlled diet. J Nutr. 2014;144(12):1977-1984. (PubMed)
13. Mock DM. Marginal biotin deficiency is common in normal human pregnancy and is highly teratogenic in mice. J Nutr. 2009;139(1):154-157. (PubMed)
14. Pabuccuoglu A, Aydogdu S, Bas M. Serum biotinidase activity in children with chronic liver disease and its clinical significance. J Pediatr Gastroenterol Nutr. 2002;34(1):59-62. (PubMed)
15. Yang Y, Yang JY, Chen XJ. Biotinidase deficiency characterized by skin and hair findings. Clin Dermatol. 2020;38(4):477-483. (PubMed)
16. Mohite K, Nair KV, Sapare A, et al. Late onset subacute profound biotinidase deficiency caused by a novel homozygous variant c.466-3T>G in the BTD gene. Indian J Pediatr. 2022;89(6):594-596. (PubMed)
17. Radelfahr F, Riedhammer KM, Keidel LF, et al. Biotinidase deficiency: A treatable cause of hereditary spastic paraparesis. Neurol Genet. 2020;6(6):e525. (PubMed)
18. Zempleni J, Hassan YI, Wijeratne SS. Biotin and biotinidase deficiency. Expert Rev Endocrinol Metab. 2008;3(6):715-724. (PubMed)
19. Saleem H, Simpson B. Biotinidase deficiency. StatPearls. Treasure Island (FL); 2022. (PubMed)
20. Canda E, Kalkan Ucar S, Coker M. Biotinidase deficiency: prevalence, impact and management strategies. Pediatric Health Med Ther. 2020;11:127-133. (PubMed)
21. Wolf B. Biotinidase deficiency: "if you have to have an inherited metabolic disease, this is the one to have". Genet Med. 2012;14(6):565-575. (PubMed)
22. Bandaralage SP, Farnaghi S, Dulhunty JM, Kothari A. Antenatal and postnatal radiologic diagnosis of holocarboxylase synthetase deficiency: a systematic review. Pediatr Radiol. 2016;46(3):357-364. (PubMed)
23. Mardach R, Zempleni J, Wolf B, et al. Biotin dependency due to a defect in biotin transport. J Clin Invest. 2002;109(12):1617-1623. (PubMed)
24. Eng WK, Giraud D, Schlegel VL, Wang D, Lee BH, Zempleni J. Identification and assessment of markers of biotin status in healthy adults. Br J Nutr. 2013;110(2):321-329. (PubMed)
25. Bogusiewicz A, Boysen G, Mock DM. In HepG2 cells, coexisting carnitine deficiency masks important indicators of marginal biotin deficiency. J Nutr. 2015;145(1):32-40. (PubMed)
26. Mock DM. Adequate intake of biotin in pregnancy: why bother? J Nutr. 2014;144(12):1885-1886. (PubMed)
27. Mock DM, Stadler DD. Conflicting indicators of biotin status from a cross-sectional study of normal pregnancy. J Am Coll Nutr. 1997;16(3):252-257. (PubMed)
28. Mock DM, Stadler DD, Stratton SL, Mock NI. Biotin status assessed longitudinally in pregnant women. J Nutr. 1997;127(5):710-716. (PubMed)
29. Mock DM, Quirk JG, Mock NI. Marginal biotin deficiency during normal pregnancy. Am J Clin Nutr. 2002;75(2):295-299. (PubMed)
30. Takechi R, Taniguchi A, Ebara S, Fukui T, Watanabe T. Biotin deficiency affects the proliferation of human embryonic palatal mesenchymal cells in culture. J Nutr. 2008;138(4):680-684. (PubMed)
31. Tabarki B, Al-Hashem A, Alfadhel M. Biotin-thiamine-responsive basal ganglia disease. In: Adam MP, Mirzaa GM, Pagon RA, et al., eds. GeneReviews((R)). Seattle (WA); 1993-2022. (PubMed)
32. Kilic B, Topcu Y, Dursun S, et al. Single gene, two diseases, and multiple clinical presentations: Biotin-thiamine-responsive basal ganglia disease. Brain Dev. 2020;42(8):572-580. (PubMed)
33. Alfadhel M, Almuntashri M, Jadah RH, et al. Biotin-responsive basal ganglia disease should be renamed biotin-thiamine-responsive basal ganglia disease: a retrospective review of the clinical, radiological and molecular findings of 18 new cases. Orphanet J Rare Dis. 2013;8:83. (PubMed)
34. Algahtani H, Ghamdi S, Shirah B, Alharbi B, Algahtani R, Bazaid A. Biotin-thiamine-responsive basal ganglia disease: catastrophic consequences of delay in diagnosis and treatment. Neurol Res. 2017;39(2):117-125. (PubMed)
35. Sedel F, Bernard D, Mock DM, Tourbah A. Targeting demyelination and virtual hypoxia with high-dose biotin as a treatment for progressive multiple sclerosis. Neuropharmacology. 2016;110(Pt B):644-653. (PubMed)
36. Sedel F, Papeix C, Bellanger A, et al. High doses of biotin in chronic progressive multiple sclerosis: a pilot study. Mult Scler Relat Disord. 2015;4(2):159-169. (PubMed)
37. Tourbah A, Lebrun-Frenay C, Edan G, et al. MD1003 (high-dose biotin) for the treatment of progressive multiple sclerosis: A randomised, double-blind, placebo-controlled study. Mult Scler. 2016;22(13):1719-1731. (PubMed)
38. Cree BAC, Cutter G, Wolinsky JS, et al. Safety and efficacy of MD1003 (high-dose biotin) in patients with progressive multiple sclerosis (SPI2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2020;19(12):988-997. (PubMed)
39. Tourbah A, Gout O, Vighetto A, et al. MD1003 (high-dose pharmaceutical-grade biotin) for the treatment of chronic visual loss related to optic neuritis in multiple sclerosis: a randomized, double-blind, placebo-controlled study. CNS Drugs. 2018;32(7):661-672. (PubMed)
40. Espiritu AI, Remalante-Rayco PPM. High-dose biotin for multiple sclerosis: A systematic review and meta-analyses of randomized controlled trials. Mult Scler Relat Disord. 2021;55:103159. (PubMed)
41. Larrieta E, Vega-Monroy ML, Vital P, et al. Effects of biotin deficiency on pancreatic islet morphology, insulin sensitivity and glucose homeostasis. J Nutr Biochem. 2012;23(4):392-399. (PubMed)
42. Maebashi M, Makino Y, Furukawa Y, Ohinata K, Kimura S, Sato T. Therapeutic evaluation of the effect of biotin on hyperglycemia in pateints with non-insulin dependent diabetes mellitus. J Clin Biochem Nutr.1993;14:211-218.
43. Baez-Saldana A, Zendejas-Ruiz I, Revilla-Monsalve C, et al. Effects of biotin on pyruvate carboxylase, acetyl-CoA carboxylase, propionyl-CoA carboxylase, and markers for glucose and lipid homeostasis in type 2 diabetic patients and nondiabetic subjects. Am J Clin Nutr. 2004;79(2):238-243. (PubMed)
44. Revilla-Monsalve C, Zendejas-Ruiz I, Islas-Andrade S, et al. Biotin supplementation reduces plasma triacylglycerol and VLDL in type 2 diabetic patients and in nondiabetic subjects with hypertriglyceridemia. Biomed Pharmacother. 2006;60(4):182-185. (PubMed)
45. Geohas J, Daly A, Juturu V, Finch M, Komorowski JR. Chromium picolinate and biotin combination reduces atherogenic index of plasma in patients with type 2 diabetes mellitus: a placebo-controlled, double-blinded, randomized clinical trial. Am J Med Sci. 2007;333(3):145-153. (PubMed)
46. Albarracin C, Fuqua B, Geohas J, Juturu V, Finch MR, Komorowski JR. Combination of chromium and biotin improves coronary risk factors in hypercholesterolemic type 2 diabetes mellitus: a placebo-controlled, double-blind randomized clinical trial. J Cardiometab Syndr. 2007;2(2):91-97. (PubMed)
47. Singer GM, Geohas J. The effect of chromium picolinate and biotin supplementation on glycemic control in poorly controlled patients with type 2 diabetes mellitus: a placebo-controlled, double-blinded, randomized trial. Diabetes Technol Ther. 2006;8(6):636-643. (PubMed)
48. Albarracin CA, Fuqua BC, Evans JL, Goldfine ID. Chromium picolinate and biotin combination improves glucose metabolism in treated, uncontrolled overweight to obese patients with type 2 diabetes. Diabetes Metab Res Rev. 2008;24(1):41-51. (PubMed)
49. Lazo de la Vega-Monroy ML, Larrieta E, German MS, Baez-Saldana A, Fernandez-Mejia C. Effects of biotin supplementation in the diet on insulin secretion, islet gene expression, glucose homeostasis and beta-cell proportion. J Nutr Biochem. 2013;24(1):169-177. (PubMed)
50. Randhawa SS, Dua K, Randhawa CS, Randhawa SS, Munshi SK. Effect of biotin supplementation on hoof health and ceramide composition in dairy cattle. Vet Res Commun. 2008;32(8):599-608. (PubMed)
51. Reilly JD, Cottrell DF, Martin RJ, Cuddeford DJ. Effect of supplementary dietary biotin on hoof growth and hoof growth rate in ponies: a controlled trial. Equine Vet J Suppl.1998(26):51-57. (PubMed)
52. Zenker W, Josseck H, Geyer H. Histological and physical assessment of poor hoof horn quality in Lipizzaner horses and a therapeutic trial with biotin and a placebo. Equine Vet J.1995;27(3):183-191. (PubMed)
53. Romero-Navarro G, Cabrera-Valladares G, German MS, et al. Biotin regulation of pancreatic glucokinase and insulin in primary cultured rat islets and in biotin-deficient rats. Endocrinology.1999;140(10):4595-4600. (PubMed)
54. Floersheim GL. [Treatment of brittle fingernails with biotin]. Z Hautkr.1989;64(1):41-48. (PubMed)
55. Hochman LG, Scher RK, Meyerson MS. Brittle nails: response to daily biotin supplementation. Cutis.1993;51(4):303-305. (PubMed)
56. Boccaletti V, Zendri E, Giordano G, Gnetti L, De Panfilis G. Familial uncombable hair syndrome: ultrastructural hair sudy and response to biotin. Pediatr Dermatol. 2007;24(3):E14-16. (PubMed)
57. Lipner SR, Scher RK. Biotin for the treatment of nail disease: what is the evidence? J Dermatolog Treat. 2018;29(4):411-414. (PubMed)
58. Walth CB, Wessman LL, Wipf A, Carina A, Hordinsky MK, Farah RS. Response to: "Rethinking biotin therapy for hair, nail, and skin disorders". J Am Acad Dermatol. 2018;79(6):e121-e124. (PubMed)
59. Famenini S, Goh C. Evidence for supplemental treatments in androgenetic alopecia. J Drugs Dermatol. 2014;13(7):809-812. (PubMed)
60. Patel DP, Swink SM, Castelo-Soccio L. A review of the use of biotin for hair loss. Skin Appendage Disord. 2017;3(3):166-169. (PubMed)
61. Staggs CG, Sealey WM, McCabe BJ, Teague AM, Mock DM. Determination of the biotin content of select foods using accurate and sensitive HPLC/avidin binding. J Food Compost Anal. 2004;17(6):767-776. (PubMed)
62. Briggs DR, Wahlqvist ML. Food facts: the complete no-fads-plain-facts guide to healthy eating. Victoria, Australia: Penguin Books; 1988.
63. Magnusdottir S, Ravcheev D, de Crecy-Lagard V, Thiele I. Systematic genome assessment of B-vitamin biosynthesis suggests co-operation among gut microbes. Front Genet. 2015;6:148. (PubMed)
64. Said HM. Cell and molecular aspects of human intestinal biotin absorption. J Nutr. 2009;139(1):158-162. (PubMed)
65. US Department of Health and Human Services, National Institutes of Health, Office of Dietary Supplements. Dietary Supplement Label Database (DSLD). [Internet]. [Accessed 7/5/2022]. Available at: https://dsld.od.nih.gov/.
66. Koutsikos D, Agroyannis B, Tzanatos-Exarchou H. Biotin for diabetic peripheral neuropathy. Biomed Pharmacother.1990;44(10):511-514. (PubMed)
67. Tourbah A LFC, Edan G, Clanet M, Papeix C, Vukusic S, et al. Effect of MD1003 (high doses of biotin) in progressive multiple sclerosis: results of a pivotal phase III randomized double blind placebo controlled study. Paper presented at: American Association of Neurological Surgeons (AANS) Annual Scientific Meeting 2015; Washington, D.C.
68. Debourdeau PM, Djezzar S, Estival JL, Zammit CM, Richard RC, Castot AC. Life-threatening eosinophilic pleuropericardial effusion related to vitamins B5 and H. Ann Pharmacother. 2001;35(4):424-426. (PubMed)
69. Chirapu SR, Rotter CJ, Miller EL, Varma MV, Dow RL, Finn MG. High specificity in response of the sodium-dependent multivitamin transporter to derivatives of pantothenic acid. Curr Top Med Chem. 2013;13(7):837-842. (PubMed)
70. Said HM, Ortiz A, McCloud E, Dyer D, Moyer MP, Rubin S. Biotin uptake by human colonic epithelial NCM460 cells: a carrier-mediated process shared with pantothenic acid. Am J Physiol.1998;275(5 Pt 1):C1365-1371. (PubMed)
71. Prasad PD, Wang H, Kekuda R, et al. Cloning and functional expression of a cDNA encoding a mammalian sodium-dependent vitamin transporter mediating the uptake of pantothenate, biotin, and lipoate. J Biol Chem.1998;273(13):7501-7506. (PubMed)
72. Zempleni J, Trusty TA, Mock DM. Lipoic acid reduces the activities of biotin-dependent carboxylases in rat liver. J Nutr.1997;127(9):1776-1781. (PubMed)
73. Natural-Medicines. Biotin/Drug interactions. www.naturaldatabase.com/. 2014.
74. Castro-Gago M, Gomez-Lado C, Eiris-Punal J, Diaz-Mayo I, Castineiras-Ramos DE. Serum biotinidase activity in children treated with valproic acid and carbamazepine. J Child Neurol. 2010;25(1):32-35. (PubMed)
75. Castro-Gago M, Perez-Gay L, Gomez-Lado C, Castineiras-Ramos DE, Otero-Martinez S, Rodriguez-Segade S. The influence of valproic acid and carbamazepine treatment on serum biotin and zinc levels and on biotinidase activity. J Child Neurol. 2011;26(12):1522-1524. (PubMed)
76. Schulpis KH, Karikas GA, Tjamouranis J, Regoutas S, Tsakiris S. Low serum biotinidase activity in children with valproic acid monotherapy. Epilepsia. 2001;42(10):1359-1362. (PubMed)
77. Yilmaz Y, Tasdemir HA, Paksu MS. The influence of valproic acid treatment on hair and serum zinc levels and serum biotinidase activity. Eur J Paediatr Neurol. 2009;13(5):439-443. (PubMed)
78. Mock DM. Biotin: from nutrition to therapeutics. J Nutr. 2017;147(8):1487-1492. (PubMed)
79. Li J, Wagar EA, Meng QH. Comprehensive assessment of biotin interference in immunoassays. Clin Chim Acta. 2018;487:293-298. (PubMed)
80. Gifford JL, de Koning L, Sadrzadeh SMH. Strategies for mitigating risk posed by biotin interference on clinical immunoassays. Clin Biochem. 2019;65:61-63. (PubMed)
81. Bowen R, Benavides R, Colon-Franco JM, et al. Best practices in mitigating the risk of biotin interference with laboratory testing. Clin Biochem. 2019;74:1-11. (PubMed)
82. US Food and Drug Administration. Biotin interference with troponin lab tests — assays subject to biotin interference. Available at: https://www.fda.gov/medical-devices/in-vitro-diagnostics/biotin-interference-troponin-lab-tests-assays-subject-biotin-interference. Accessed 7/5/2022.
Folate
Contents
Summary
- Folate is a generic term referring to both natural folates in food and folic acid, the synthetic form used in supplements and fortified food. Folate is critical in the metabolism of nucleic acid precursors and several amino acids, as well as in methylation reactions. (More information)
- Severe deficiency in either folate or vitamin B12 can lead to megaloblastic anemia, which causes fatigue, weakness, and shortness of breath. Improper treatment of vitamin B12-dependent megaloblastic anemia with high-dose supplemental folic acid can potentially delay the diagnosis of vitamin B12 deficiency and thus leave the individual at risk of developing irreversible brain and nervous system damage. (More information)
- Folate status is influenced by the presence of genetic variations in folate metabolism enzymes, particularly polymorphisms of the 5,10-methylenetetrahydrofolate reductase (MTHFR) gene. (More information)
- Inadequate folate status during early pregnancy increases the risk of congenital anomalies. The introduction of mandatory folic acid fortification of refined grain products in the US in 1998 has reduced the prevalence of neural tube defects (NTDs) in newborns. Yet, folate status is considered inadequate in a majority of women of childbearing age worldwide. Moreover, genetic factors might modify the risk of NTDs by increasing the susceptibility to folate deficiency during pregnancy. Several studies have investigated the role of folic acid supplementation in the prevention of congenital anomalies other than NTDs. (More information)
- Folate deficiency and elevated concentrations of homocysteine in the blood are associated with increased risk of cardiovascular disease. Although folic acid supplementation has been proven effective to control circulating homocysteine concentrations, homocysteine lowering has not affected the incidence of cardiovascular disease in supplementation trials. Yet, supplementation with folic acid and other B vitamins does appear to lower the risk of stroke. (More information)
- Low folate status has been linked to increased cancer risk. However, intervention trials with high doses of folic acid have not generally shown any benefit on cancer incidence. There is some concern that high doses of supplemental folic acid might increase the risk of certain cancers, but further study is needed. (More information)
- Prospective cohort studies have reported an inverse association between folate status and colorectal cancer risk, especially among men. However, the relationship between folate status and cancer risk is complex and requires further research. (More information)
- Folate is essential for brain development and function. Low folate status and/or high homocysteine concentrations are associated with cognitive dysfunction in aging (from mild impairments to dementia). However, whether supplemental folic acid confers long-term benefits in maintaining cognitive health is not yet known. (More information)
-
Several autosomal recessive disorders affecting folate transport and metabolism can be treated with high doses of folinic acid, a folate derivative. (More information)
Folate is a water-soluble B-vitamin, which is also known as vitamin B9 or folacin. Naturally occurring folates exist in many chemical forms; folates are found in food, as well as in metabolically active forms in the human body. Folic acid is the major synthetic form found in fortified foods and vitamin supplements. Other synthetic forms include folinic acid (Figure 1) and levomefolic acid. Folic acid has no biological activity unless converted into folates (1). In the following discussion, forms found in food or the body are referred to as "folates," while the form found in supplements or fortified food is referred to as "folic acid."

Function
One-carbon metabolism
The only function of folate coenzymes in the body appears to be in mediating the transfer of one-carbon units (2). Folate coenzymes act as acceptors and donors of one-carbon units in a variety of reactions critical to the metabolism of nucleic acids and amino acids (Figure 2) (3).

Nucleic acid metabolism
Folate coenzymes play a vital role in DNA metabolism through two different pathways: (1) The synthesis of DNA from its precursors (thymidine and purines) is dependent on folate coenzymes. (2) A folate coenzyme is required for the synthesis of methionine from homocysteine, and methionine is required for the synthesis of S-adenosylmethionine (SAM). SAM is a methyl group (one-carbon unit) donor used in most biological methylation reactions, including the methylation of a number of sites within DNA, RNA, proteins, and phospholipids. The methylation of DNA plays a role in controlling gene expression and is critical during cell differentiation. Aberrations in DNA methylation have been linked to the development of cancer (see Cancer).
Amino acid metabolism
Folate coenzymes are required for the metabolism of several important amino acids, namely methionine, cysteine, serine, glycine, and histidine. The synthesis of methionine from homocysteine is catalyzed by methionine synthase, an enzyme that requires not only folate (as 5-methyltetrahydrofolate) but also vitamin B12. Thus, folate (and/or vitamin B12) deficiency can result in decreased synthesis of methionine and an accumulation of homocysteine. Elevated blood concentrations of homocysteine have been considered for many years to be a risk factor for some chronic diseases, including cardiovascular disease and dementia (see Disease Prevention).
Nutrient interactions
Vitamin B12 and vitamin B6
The metabolism of homocysteine, an intermediate in the metabolism of sulfur-containing amino acids, provides an example of the interrelationships among nutrients necessary for optimal physiological function and health. Healthy individuals utilize two different pathways to metabolize homocysteine (Figure 3). One pathway (methionine synthase) synthesizes methionine from homocysteine and is dependent on both folate and vitamin B12 as cofactors. The other pathway converts homocysteine to another amino acid, cysteine, and requires two vitamin B6-dependent enzymes. Thus, the concentration of homocysteine in the blood is regulated by three B-vitamins: folate, vitamin B12, and vitamin B6 (4). In some individuals, riboflavin (vitamin B2) is also involved in the regulation of homocysteine concentrations (see the article on Riboflavin).

Riboflavin
Although less well recognized, folate has an important metabolic interaction with riboflavin. Riboflavin is a precursor of flavin adenine dinucleotide (FAD), a coenzyme required for the activity of the folate-metabolizing enzyme, 5,10-methylenetetrahydrofolate reductase (MTHFR). FAD-dependent MTHFR in turn catalyzes the reaction that generates 5-methyltetrahydrofolate (see Figure 2). This active form of folate is required to form methionine from homocysteine. Along with other B-vitamins, higher riboflavin intakes have been associated with decreased plasma homocysteine concentrations (5). The effects of riboflavin on folate metabolism appear to be greatest in individuals homozygous for the common c.677C>T polymorphism (i.e., TT genotype) in the MTHFR gene (see Genetic variation in folate requirements) (6). These individuals (up to 32% of the population) typically present with low folate status, along with elevated homocysteine concentrations, particularly when folate and/or riboflavin intake is suboptimal. The elevated homocysteine concentration in these individuals, however, is highly responsive to lowering with riboflavin supplementation. In addition, riboflavin supplementation has been shown to reduce blood pressure in hypertensive individuals homozygous for the MTHFR c.677C>T polymorphism (7, 8), confirming the importance of the riboflavin-MTHFR interaction (9, 10).
Bioavailability
Dietary folates exist predominantly in the polyglutamyl form (containing several glutamate residues), whereas folic acid — the synthetic vitamin form — is a monoglutamate, containing just one glutamate moiety. In addition, natural folates are reduced molecules, whereas folic acid is fully oxidized. These chemical differences have major implications for the bioavailability of the vitamin such that folic acid is considerably more bioavailable than naturally occurring food folates at equivalent intake levels.
The intestinal absorption of dietary folates is a two-step process that involves the hydrolysis of folate polyglutamates to the corresponding monoglutamyl derivatives, followed by their transport into intestinal cells (11). There, folic acid is converted into a naturally occurring folate, namely 5-methyltetrahydrofolate, which is the major circulating form of folate in the human body (see Figure 1).
The bioavailability of naturally occurring folates is inherently limited and variable. There is much variability in the ease with which folates are released from different food matrices, and the polyglutamyl "tail" is removed (de-conjugation) before uptake by intestinal cells. Also, other dietary constituents can contribute to instability of labile folates during the processes of digestion. As a result, naturally occurring folates show incomplete bioavailability compared with folic acid. The bioavailability of folic acid, in contrast, is assumed to be 100% when ingested as a supplement, while folic acid in fortified food is estimated to have about 85% the bioavailability of supplemental folic acid.
Of note, folate recommendations in the US and certain other countries are now expressed as Dietary Folate Equivalents (DFEs), a calculation that was devised to take into account the greater bioavailability of folic acid compared to naturally occurring dietary folates (see The Recommended Dietary Allowance).
Transport
Folate and its coenzymes require transporters to cross cell membranes. Folate transporters include the reduced folate carrier (RFC), the proton-coupled folate transporter (PCFT), and the folate receptor proteins, FRα and FRβ. Folate homeostasis is supported by the ubiquitous distribution of folate transporters, although abundance and importance vary among tissues (12). PCFT plays a major role in folate intestinal transport since mutations affecting the gene encoding PCFT cause hereditary folate malabsorption. Defective PCFT also leads to impaired folate transport into the brain (see Disease Treatment). FRα and RFC are also critical for folate transport across the blood-brain barrier when extracellular folate is either low or high, respectively. Folate is essential for the proper development of the embryo and the fetus. The placenta is known to concentrate folate to the fetal circulation, leading to higher folate concentrations in the fetus compared to those found in the pregnant woman. All three types of receptors have been associated with folate transport across the placenta during pregnancy (13).
Deficiency
Causes
Although folate deficiency is uncommon in the United States, it is estimated to be more prevalent worldwide (11). Most often caused by a dietary insufficiency, folate deficiency can also occur in a number of other situations. Chronic, heavy alcohol consumption is associated with diminished absorption of folate (in addition to low dietary intake), which can lead to folate deficiency (14). Smoking is also associated with low folate status. In one study, folate concentrations in blood were about 15% lower in smokers compared to nonsmokers (15). Additionally, impaired folate transport to the fetus has been described in pregnant women who either smoked or abused alcohol during their pregnancy (16, 17).
Pregnancy is a time when the folate requirement is greatly increased to sustain the demand for rapid cell replication and growth of fetal, placental, and maternal tissue. Conditions such as cancer or inflammation can also result in increased rates of cell division and metabolism, causing an increase in the body's demand for folate (18). Moreover, folate deficiency can result from some malabsorptive conditions, including inflammatory bowel diseases (Crohn’s disease and ulcerative colitis) and celiac disease (11, 19). Several medications can contribute to folate deficiency (see Drug interactions). Finally, a number of genetic diseases affecting folate absorption, transport, or metabolism can cause folate deficiency or impede its metabolic functions (see Disease Treatment).
Symptoms
Clinical folate deficiency leads to megaloblastic anemia, which is reversible with folic acid treatment. Rapidly dividing cells like those derived from bone marrow are most vulnerable to the effects of folate deficiency since DNA synthesis and cell division are dependent on folate coenzymes. When folate supply to the rapidly dividing cells of the bone marrow is inadequate, blood cell division is reduced, resulting in fewer but larger red blood cells. This type of anemia is called megaloblastic or macrocytic anemia, referring to the enlarged, immature red blood cells. Neutrophils, a type of white blood cell, become hypersegmented, a change that can be found by examining a blood sample microscopically. Because normal red blood cells have a lifetime in the circulation of approximately four months, it can take months for folate-deficient individuals to develop the characteristic megaloblastic anemia. Progression of such an anemia leads to a decreased oxygen carrying capacity of the blood and may ultimately result in symptoms of fatigue, weakness, and shortness of breath (1). It is important to point out that megaloblastic anemia resulting from folate deficiency is identical to the megaloblastic anemia resulting from vitamin B12 deficiency, and further clinical testing is required to diagnose the true cause of megaloblastic anemia (see Toxicity).
Individuals in the early stages of folate deficiency may not show obvious symptoms, but blood concentrations of homocysteine may increase (see Disease Prevention). Yet, the concentration of circulating homocysteine is not a specific indicator of folate status, as elevated homocysteine can be the result of vitamin B12 and other B-vitamin deficiencies, lifestyle factors, and renal insufficiency. Subclinical deficiency is typically detected by measurement of folate concentrations in serum/plasma or in red blood cells.
The Recommended Dietary Allowance (RDA)
Determination of the RDA
Traditionally, the dietary folate requirement was defined as the amount needed to prevent a deficiency severe enough to cause symptoms like anemia. The most recent RDA (1998; Table 1) was based primarily on the adequacy of red blood cell folate concentrations at different levels of folate intake, as judged by the absence of abnormal hematological indicators. Red cell folate has been shown to correlate with liver folate stores and is used as an indicator of long-term folate status. Plasma or serum folate reflects recent folate intake and is not a reliable biomarker for folate status, especially when used as a one-time assessment (11). Maintenance of normal blood homocysteine concentrations, an indicator of one-carbon metabolism, was considered only as an ancillary indicator of adequate folate intake.
Because pregnancy is associated with a significant increase in cell division and other metabolic processes that require folate coenzymes, the RDA for pregnant women is considerably higher than for women who are not pregnant (3). However, the prevention of neural tube defects (NTDs) was not considered when setting the RDA for pregnant women. Rather, reducing the risk of NTDs was considered in a separate recommendation for women capable of becoming pregnant (see Disease Prevention), because the crucial events in the development of the neural tube occur before many women are aware that they are pregnant (20).
Dietary Folate Equivalents (DFEs)
When the Food and Nutrition Board of the US Institute of Medicine (now the National Academy of Medicine) last revised the dietary recommendations for folate, they introduced a new unit, the Dietary Folate Equivalent (DFE) (1). Use of the DFE reflects the higher bioavailability of synthetic folic acid found in supplements and fortified food compared to that of naturally occurring food folates (20).
- 1 microgram (μg) of food folate provides 1 μg of DFEs
- 1 μg of folic acid taken with meals or as fortified food provides 1.7 μg of DFEs
- 1 μg of folic acid (supplement) taken on an empty stomach provides 2 μg of DFEs
For example, a serving of food containing 60 μg of folate would provide 60 μg of DFEs, while a serving of pasta fortified with 60 μg of folic acid would provide 1.7 x 60 = 102 μg of DFEs due to the higher bioavailability of folic acid. A folic acid supplement of 400 μg taken on an empty stomach would provide 800 μg of DFEs. It should be noted that DFEs were determined in studies with adults and whether folic acid in infant formula is more bioavailable than folates in mother's milk has not been studied. Use of DFEs to determine a folate requirement for the infant would not be desirable.
| Life Stage | Age | Males (μg/day) | Females (μg/day) |
|---|---|---|---|
| Infants | 0-6 months | 65 (AI) | 65 (AI) |
| Infants | 7-12 months | 80 (AI) | 80 (AI) |
| Children | 1-3 years | 150 | 150 |
| Children | 4-8 years | 200 | 200 |
| Children | 9-13 years | 300 | 300 |
| Adolescents | 14-18 years | 400 | 400 |
| Adults | 19 years and older | 400 | 400 |
| Pregnancy | all ages | - | 600 |
| Breast-feeding | all ages | - | 500 |
Genetic variation in folate requirements
A common polymorphism or variation in the sequence of the gene for the enzyme, 5, 10-methylenetetrahydrofolate reductase (MTHFR), known as the MTHFR c.677C>T polymorphism, results in a thermolabile enzyme (21). The substitution of a cytosine (C) by a thymine (T) at nucleotide 677 in the exon 4 of MTHFR gene leads to an alanine-to-valine transition in the catalytic domain of the enzyme. Depending on the population, 20% to 53% of individuals may have inherited one T copy (677C/T genotype), and 3% to 32% of individuals may have inherited two T copies (677T/T genotype) for the MTHFR gene (22). MTHFR catalyzes the reduction of 5,10-methylenetetrahydrofolate (5,10-methylene THF) into 5-methyl tetrahydrofolate (5-MeTHF). The latter is the folate coenzyme required to form methionine from homocysteine (see Figure 2). MTHFR activity is greatly diminished in heterozygous 677C/T (-30%) and homozygous 677T/T (-65%) individuals compared to those with the 677C/C genotype (23). Homozygosity for the mutation (677T/T) is linked to lower concentrations of folate in red blood cells and higher blood concentrations of homocysteine (24, 25). Improving folate nutritional status in elderly women with the T allele reduced plasma homocysteine concentration (26). An important unanswered question about folate is whether the present RDA is enough to compensate for the reduced MTHFR enzyme activity in individuals with at least one T allele, or whether those individuals have a higher folate requirement than the RDA (27).
Disease Prevention
Adverse pregnancy outcomes
Neural tube defects
Fetal growth and development are characterized by widespread cell division. Adequate folate is critical for DNA and RNA synthesis. Neural tube defects (NTDs) arise from failure of embryonic neural tube closure between the 21st and 28th days after conception, a time when many women may not even realize they are pregnant (28). NTDs include various malformations, such as lesions of the brain (e.g., anencephaly, encephalocele) or lesions of the spine (spina bifida), which are devastating and life-threatening (29). The worldwide prevalence of NTDs is estimated to be 2 per 1,000 births, which translates to 214,000-322,000 cases annually (30).
Results of randomized trials have demonstrated 60% to 100% reductions in NTD cases when women consumed folic acid supplements in addition to a varied diet during the periconceptional period (about one month before and at least one month after conception) (31, 32). To decrease the incidence of NTDs in the US (about 1 NTD case per 1,000 pregnancies; 1), the Food and Drug Administration implemented legislation in 1998 requiring the fortification of all enriched grain products with 1.4 mg of folic acid per kg of grain (see Sources). The required level of folic acid fortification in the US was initially estimated to provide 100 μg of additional folic acid in the average person's diet, though it probably provides even more due to overuse of folic acid by food manufacturers (27, 33). As a result of this fortification mandate, a 30% decrease in the prevalence of NTDs was noted compared to the pre-fortification period; the post-fortification prevalence of NTDs was found to be 0.69 cases per 1,000 live births and fetal deaths (34). Recent estimates indicate an NTD prevalence less than 0.5 cases per 1,000 live births and that fortification strategies in the US prevent 1,326 NTDs each year (35). To date, 58 nations worldwide have established mandatory programs of folic acid fortification of staple grains (30, 36), and an estimated 22% of NTDs have been prevented through such programs (36). Many countries in Europe, Asia, and Africa, however, have not mandated folic acid fortification of staple foods (37).
The US Public Health Service recommends that all persons capable of becoming pregnant consume 400 μg of folic acid daily to prevent NTDs. Those with a previously affected pregnancy were also advised to receive 4,000 μg (4 mg) of folic acid daily in order to reduce NTD recurrence (38). These recommendations were made to all persons of childbearing age because adequate folate must be available very early in pregnancy, and because many pregnancies in the US are unplanned (39). Despite the effectiveness of folic acid supplementation in improving folate status, it appears that globally only 30% of women who become pregnant correctly follow the recommendation, and there is some concern that young women from minority ethnic groups and lower socioeconomic backgrounds are the least likely to follow the recommendation (40-43).
Also, a genetic component in NTD etiology is evidenced by the increased risk in women with a family history of an NTD and also by variations in risk among ethnicities (44). Moreover, NTD occurrence can be attributed to specific folate-gene interactions. The MTHFR c.677C>T polymorphism and other genetic variations can increase the folate requirement and susceptibility for an NTD-affected pregnancy. Prior to the fortification era, a case-control study showed that both red blood cell and serum folate concentrations were significantly lower in pregnant women with the T/T and C/T variants compared to the wild-type C/C genotype (24), suggesting inadequate folate metabolism with specific maternal genotypes. A meta-analysis of 34 case-control studies, including 3,018 case mothers and 8,746 control mothers, showed a positive association between the maternal MTHFR c.677C>T polymorphism and NTDs (45). Another MTHFR variant, an A-to-C change at position 1298, has also been associated with reduced MTHFR activity and increased NTD risk in some (46, 47), but not all (48), populations. Individuals heterozygous for both of these MTHFR variants (677C/T + 1298A/C) exhibit lower plasma folate and higher homocysteine concentrations than individuals with 677C/T + 1298A/A (49). Combined genotypes with homozygosity G/G for the reduced folate carrier transporter (RFC-1) polymorphism (c.80A>G) could further contribute to NTD occurrence (50). The degree of NTD risk was also assessed with additional MTHFR polymorphisms (c.116C>T, c.1793G>A) (51), as well as with mutations affecting other enzymes of the one-carbon metabolism, including methionine synthase (MTR c.2756A>G) (52), methionine synthase reductase (MTRR c.66A>G) (53), and methylenetetrahydrofolate dehydrogenase (MTHFD1 c.1958G>A) (54, 55). While maternal genotype can impact pregnancy outcome, it appears that gene-gene interactions between mother and fetus influence it further. The risk of NTD was increased by certain genetic combinations, including maternal (MTHFR c.677C>T)-fetal (MTHFR c.677C>T) and maternal (MTRR c.66A>G)-fetal (MTHFR c.677C>T) interactions (52, 53, 56). Finally, vitamin B12 status has been associated with NTD risk modification in the presence of specific polymorphisms in one-carbon metabolism (57).
Cardiovascular malformations
Congenital anomalies of the heart are a major cause of infant mortality but also cause deaths in adulthood (58). Using data from the European Registration of Congenital Anomalies and Twins (EUROCAT) database, a case-control study, involving 596 cases and 2,359 controls, found that consumption of at least 400 μg/day of folic acid during the periconceptual period (one month before conception through eight weeks' post-conception, covering the period of embryonic heart development) was associated with an 18% reduced risk of congenital heart defects (59). Meta-analyses of 20 to 25 case-control and family-based studies observed positive associations between maternal, fetal, or paternal MTHFR c.677C>T variant and incidence of congenital heart defects (60, 61). Additional studies are needed to elucidate the effects of gene-nutrient interactions on the risk of congenital heart defects; however, the currently available research indicates that adequate folate intake may play an important role.
Orofacial clefts
Maternal folate status during pregnancy may influence the risk of congenital anomalies called orofacial clefts, namely cleft lip with or without cleft palate (CL/P) (62). Most orofacial clefts are non-syndromic clefts, meaning no other congenital malformations are present (63). In a recent systematic review and meta-analysis of 39 observational studies, use of folic acid-containing supplements preconceptionally or during pregnancy was associated with a 42% lower risk of CL/P (95% CI, 0.49-0.70) (64). While some studies have suggested that polymorphisms in the cystathionine β-synthase (CBS) gene (c.699C>T) or MTHFR gene (c.677C>T) might be protective of orofacial clefts at low folate intakes (65, 66), this pooled analysis did not find an association between MTHFR polymorphisms and orofacial clefts (64). An earlier meta-analysis of observational studies evaluating mandatory programs of folic acid fortification globally found that folic acid fortification was only associated with a reduced risk of non-syndromic CL/P (5 studies) and not with orofacial clefts in total (24 studies), CL/P (16 studies), or cleft palate only (16 studies) (67). It is important to note that the observational studies to date are highly heterogeneous, which presents challenges when pooling study results in a meta-analysis.
Other adverse pregnancy outcomes
Low birth weight has been associated with increased risk of mortality during the first year of life and may also influence health outcomes during adulthood (68). A systematic review and meta-analysis of eight randomized controlled trials found a positive association between folic acid supplementation and birth weight; no association with length of gestation was observed (69). Additionally, a prospective cohort study of 306 pregnant adolescents associated low folate intakes and maternal folate status during the third trimester of pregnancy with higher incidence of small for gestational age births (birth weight <10th percentile) (70). Moreover, the maternal c.677C>T MTHFR genotype and increased homocysteine concentrations, considered an indicator of functional folate deficiency, have been linked to lower birth weights (71).
Elevated blood homocysteine concentrations have also been associated with increased incidence of miscarriage and other pregnancy complications, including preeclampsia and placental abruption (72). A large retrospective study showed that high plasma homocysteine in Norwegian women was strongly related to adverse outcomes and complications, including preeclampsia, premature delivery, and very low birth weight, in previous pregnancies (73). A meta-analysis of 51 prospective cohort studies linked the c.677C>T MTHFR variant with increased risk of preeclampsia in Caucasian and East Asian populations, reinforcing the notion that folate metabolism may play a role in the condition (74). However, it is not known whether maternal folic acid supplementation during pregnancy reduces the risk of preeclampsia, although results of a meta-analysis of 12 studies (11 cohort studies and 1 randomized controlled trial) suggested a lower risk (RR, 0.69; 95% CI 0.58-0.83) (75). A large international, multicenter, double-blind, placebo-controlled trial, the Folic Acid Clinical Trial (FACT), was initiated to evaluate whether high-dose folic acid supplementation throughout pregnancy could prevent preeclampsia and other adverse outcomes (e.g., maternal death, placental abruption, preterm delivery) (76). In this trial in 2,301 pregnant women at high risk of preeclampsia, 4.0-5.1 mg/day of supplemental folic acid from 8 to 16 weeks’ gestation until delivery did not lower risk of preeclampsia or any other adverse outcome when compared to placebo (subjects in the placebo group could take up to 1.1 mg/day of folic acid) (77).
Overall, findings of observational studies on the association of maternal folic acid supplementation and autism spectrum disorders in the offspring have been inconsistent (reviewed in 78, 79). Additionally, systematic reviews of observational studies have found no evidence of an association between folate exposure during pregnancy and childhood asthma or allergies (80, 81).
Adequate folate intake throughout pregnancy is known to protect against megaloblastic anemia (82). Thus, it seems reasonable to maintain folic acid supplementation throughout pregnancy, even after closure of the neural tube, in order to decrease the risk of other problems during pregnancy.
Cardiovascular disease
Homocysteine and cardiovascular disease
The results of more than 80 observational studies indicate that even moderately elevated concentrations of homocysteine in the blood increase the risk of cardiovascular disease (CVD) (4). Possible predispositions to vascular accidents have also been linked to genetic deficiencies in homocysteine metabolism in certain populations (83). The mechanism by which homocysteine may increase the risk of vascular disease has been the subject of a great deal of research, but it may involve adverse effects of homocysteine on blood clotting, arterial vasodilation, and thickening of arterial walls (84). Although increased homocysteine concentrations in the blood have been consistently associated with increased risk of CVD, it is unclear whether lowering circulating homocysteine will reduce CVD risk (see Folate and homocysteine). Elevated homocysteine may instead be a biomarker of cardiovascular disease rather than a causative factor (85).
Research had initially predicted that a prolonged decrease in serum homocysteine level of 3 micromoles/liter would lower the risk of CVD by up to 25% and be a reasonable treatment goal for individuals at high risk (86, 87). However, the analysis of clinical trials of B-vitamin supplementation has shown that lowering homocysteine concentrations did not prevent the occurrence of a second cardiovascular event in patients with existing CVD (88-90). Consequently, the American Heart Association recommends screening for elevated total homocysteine concentrations only in "high risk" individuals, for example, in those with personal or family history of premature cardiovascular disease, malnutrition or malabsorption syndromes, hypothyroidism, kidney failure, lupus, or for individuals taking certain medications (nicotinic acid, theophylline, bile acid-binding resins, methotrexate, and L-dopa.
Folate and homocysteine
Folate-rich diets have been associated with decreased risk of CVD, including coronary artery disease, myocardial infarction (heart attack), and stroke. A study that followed 1,980 Finnish men for 10 years found that those who consumed the most dietary folate had a 55% lower risk of an acute coronary event when compared to those who consumed the least dietary folate (91). Of the three B vitamins that regulate homocysteine concentrations, folic acid has been shown to have the greatest effect in lowering basal concentrations of homocysteine in the blood when there is no coexisting deficiency of vitamin B12 or vitamin B6 (see Nutrient interactions) (92). Increasing folate intake through folate-rich food or supplements has been found to reduce homocysteine concentrations (93). Moreover, blood homocysteine concentrations have declined since the FDA mandated folic acid fortification of the grain supply in the US (27). A meta-analysis of 25 randomized controlled trials, including almost 3,000 subjects, found that folic acid supplementation with 800 μg/day or more could achieve a maximal 25% reduction in plasma homocysteine concentrations. In this meta-analysis, daily doses of 200 μg and 400 μg of folic acid were associated with a 13% and 20% reduction in plasma homocysteine, respectively (94). A supplement regimen of 400 μg of folic acid, 2 mg of vitamin B6, and 6 μg of vitamin B12 has been advocated by the American Heart Association if an initial trial of a folate-rich diet (see Sources) is not successful in adequately lowering homocysteine concentrations (95).
Several polymorphisms in folate/one-carbon metabolism modify homocysteine concentrations in blood (96). In particular, the effect of the c.677C>T MTHFR variant has been examined in relation to folic acid fortification policies worldwide. The analysis of randomized trials, including 59,995 subjects without a history of CVD, revealed that the difference in homocysteine concentrations between T/T and C/C genotypes was greater in low-folate regions compared to regions with food fortification policy (3.12 vs. 0.13 micromoles/liter) (97). Although folic acid supplementation effectively decreases homocysteine concentrations, it is not yet clear whether it also decreases risk for CVD. A meta-analysis of 19 randomized clinical trials, including 47,921 subjects with preexisting cardiovascular or renal disease, found that homocysteine lowering through folic acid and other B-vitamin supplementation failed to reduce the incidence of CVD despite significant reductions in plasma homocysteine concentrations (90). Other meta-analyses have confirmed the lack of causality between the lowering of homocysteine and the risk of CVD (96-98). Consequently, the American Heart Association removed its recommendation for using folic acid to prevent cardiovascular disease in high-risk women (99). However, more recent evidence suggests that homocysteine-lowering therapy with B vitamins may help prevent stroke in particular. A meta-analysis of three randomized controlled trials in patients with existing vascular disease found a benefit of B-vitamin supplementation on prevention of recurrent stroke (HR, 0.71; 95% CI, 0.58-0.88) (100). Other recent meta-analyses of randomized controlled trials have found that folic acid supplementation decreases the risk of stroke or cerebrovascular events in patients with existing CVD (101-103), as well as in those without CVD (104). A recent systematic review and meta-analysis of 21 randomized controlled trials — mostly in patients with existing CVD — found that folic acid supplementation slightly lowers systolic (1.10 mm Hg) and diastolic (-0.24 mm Hg) blood pressure (105).
Moreover, some studies have investigated the effect of folic acid supplementation on the development of atherosclerosis, a known risk factor for vascular accidents. The measurement of the carotid intima-media thickness (CIMT) is a surrogate endpoint for early atherosclerosis and a predictor for cardiovascular events (106). The meta-analysis of 10 randomized trials testing the effect of folic acid supplementation showed a significant reduction in CIMT in subjects with chronic kidney diseases and in those at risk for CVD, but not in healthy participants (107). Endothelial dysfunction is a common feature in atherosclerosis and vascular disease. High doses of folic acid (400-10,000 μg/day) have been associated with improvements in vascular health in both healthy and CVD subjects (108). A meta-analysis of 21 randomized, placebo-controlled trials found that folic acid supplementation improves endothelial function by increasing flow-mediated vasodilation (109).
Although some trials have failed to demonstrate any cardiovascular protection from folic acid supplementation, low folate intake is a known risk factor for vascular disease, and more research is needed to explore the role of folate in maintaining vascular health (110).
Cancer
Cancer is thought to arise from DNA damage in excess of ongoing DNA repair and/or the inappropriate expression of critical genes. Because of the important roles played by folate in DNA and RNA synthesis and methylation, it is possible that inadequate folate intake contributes to genome instability and chromosome breakage that often characterize cancer development. In particular, DNA replication and repair are critical for genome maintenance, and the shortage in nucleotides caused by folate deficiency might lead to genome instability and DNA mutations. A decrease in 5,10-methylene THF can compromise the conversion of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP) by the enzyme thymidylate synthase (TYMS), causing uracil accumulation and thymine depletion. This could then lead to uracil misincorporation into DNA during replication or repair, and cause DNA damage, including point mutations and strand breaks (111). Since 5,10-methylene THF is also the MTHFR enzyme substrate, it is plausible that a reduction of MTHFR activity with the c.677C>T polymorphism may increase the use of 5,10-methylene THF for thymidylate synthesis and prevent DNA damage. However, this hypothesis might only be valid in a situation of folate deficiency (112). Conversely, it was argued that folic acid supplementation could fuel DNA synthesis, therefore promoting tumor growth. This is supported by the observation that TYMS can function like a tumor promoter (oncogene), while a reduction in TYMS activity is linked to a lower risk of cancer (113, 114). Additionally, antifolate molecules that block the thymidylate synthesis pathway are successfully used in cancer therapy (115). Folate also controls the homocysteine/methionine cycle and the pool of S-adenosylmethionine (SAM), the methyl donor for methylation reactions. Thus, folate deficiency may impair DNA and protein methylation and alter the expression of genes involved in DNA repair, cell proliferation, and cell death. Global DNA hypomethylation, a typical hallmark of cancer, causes genome instability and chromosome breaks (reviewed in 116).
The consumption of at least five servings of fruit and vegetables daily has been consistently associated with a decreased incidence of cancer (117, 118). Fruit and vegetables are excellent sources of folate, which may play a role in their anti-carcinogenic effect. Observational studies have found diminished folate status to be associated with site-specific cancers. While food fortification is mandatory in the US (since 1998; see Sources), concerns about the impact of high folic acid intakes on health have delayed the practice in several other countries (119). However, meta-analyses of folic acid intervention trials (supplemental doses ranging from 500 to 5,000 μg/day for at least one year) did not show any specific benefit or harm regarding total and site-specific cancer incidence (120, 121). In 2015, an expert panel convened by the US National Toxicology Program and the US Office of Dietary Supplements concluded that folic acid supplementation does not reduce cancer incidence in folate replete individuals and that further research is needed to determine whether supplemental folic acid might promote cancer growth (122).
Colorectal cancer
A pooled analysis of 13 prospective cohort studies, which followed a total of 725,134 individuals for a 7 to 20-year period, revealed a modest, inverse association between dietary and total (from food and supplements) folate intake and colon cancer risk. Specifically, a 2% decrease in colon cancer risk was estimated for every 100 μg/day increase in total folate intake (123). A large US prospective study, which followed 525,488 subjects, ages 50 to 71 years between 1995 and 2006, correlated higher intakes of dietary folate, supplemental folic acid, and total folate (diet and supplements combined) with decreased colorectal cancer (CRC) risk (124). However, when stratified by gender, there was no association between dietary folate intake and CRC risk in women (124, 125). A lack of association between CRC risk and dietary, supplemental, and total folate intakes was also reported in another prospective study that followed more than 90,000 US postmenopausal women during an 11-year period encompassing pre- and post-fortification periods (126). Yet, in the long-term follow up (followed for up to 24 years) of 86,320 women participating in the prospective Nurses’ Health Study, higher total intakes of folate at baseline, as well as higher supplemental folic acid intakes, were associated with lower risks of CRC (127).
Finally, a meta-analysis of 18 case-control studies found a slight reduction in CRC risk with folate from food (128). However, it is important to note that the case-control studies were highly heterogeneous, and that the authors stated that dietary fiber, vitamins, and alcohol intake could have confounded their results. Moreover, the lower limit of the highest quantile of folate intake was highly variable, ranging from 270 to 1,367 μg/day (128).
While most observational research shows a protective association of folate against colorectal cancer development, it has been suggested that high doses of supplemental folic acid may actually accelerate tumor growth in cancer patients (129). Whereas higher folate status within the normal dietary range is widely considered to be protective against cancer, some investigators remain concerned that exposure to excessively high folic acid intakes may increase the growth of pre-existing neoplasms (129). Several clinical trials addressed the effect of folic acid supplementation in patients with a history of colorectal adenoma, with trials finding a risk reduction or no effect of supplemental folic acid (130-133). A meta-analysis of three large randomized controlled trials in high-risk subjects did not demonstrate any increase in colorectal adenoma recurrence in subjects supplemented with 500 or 1,000 μg/day of folic acid for 24 to 42 months when compared with placebo treatment (134). The B-PROOF study was a randomized, double-blind, placebo-controlled trial examining whether supplemental folic acid and vitamin B12 affected risk of osteoporotic fracture in older adults (≥65 years) with hyperhomocysteinemia (135). In a secondary analysis of 2,524 older adults participating in B-PROOF, co-supplementation with folic acid (400 μg/day) and vitamin B12 (500 μg/day) for 2 to 3 years significantly increased risk of colorectal cancer (HR 1.77; 95% CI, 1.08-2.90) (136).
As suggested above, the MTHFR 677T/T genotype might prevent uracil misincorporation and protect DNA integrity and stability under low-folate conditions. A meta-analysis of 62 case-control and two cohort studies revealed that while the T/T variant reduces CRC risk by 12% compared to both C/T and C/C genotypes, the risk was decreased by 30% with high (348-1,583 μg/day) versus low total folate intakes (264-450 μg/day), irrespective of the genotype (137). A common polymorphism (c.2756A>G) in the MTR gene, which codes for methionine synthase, was also examined in relation with the risk of colorectal adenoma and cancer. Methionine synthase catalyzes the simultaneous conversion of homocysteine and 5-methylene THF into methionine and TFH, respectively. Meta-analyses of case-control studies have found no association between MTR variants and colorectal cancer risk (138, 139).
Alcohol consumption interferes with the absorption and metabolism of folate (18). One case-control and five prospective cohort studies have reported either reduction in CRC risk among nondrinkers compared to drinkers or a lack of association (128). In a large prospective study that followed more than 28,000 male health professionals for 22 years, intake of more than two alcoholic drinks (>30 grams of alcohol) per day augmented CRC risk by 42% during the pre-fortification period. CRC risk was not increased during the post-fortification period, suggesting that it is the combination of high alcohol and low folate intake that might increase CRC risk. Yet, another prospective study that followed more than 69,000 female nurses for 28 years did not report a significant increase in CRC risk with alcohol intake before and after the mandatory folic acid fortification (140). In some studies, individuals who are homozygous for the c.677C>T MTHFR polymorphism (T/T) have been found to be at decreased risk for colon cancer when folate intake is adequate. However, when folate intake is low and/or alcohol intake is high, individuals with the (T/T) genotype have been found to be at increased risk of colorectal cancer (141, 142).
Breast cancer
Several prospective cohort and case-control studies investigating whether folate intake is associated with breast cancer have reported mixed results (143). A meta-analysis of 39 prospective cohort and case-control studies found dietary folate intake to be inversely associated with risk of breast cancer, but data stratification revealed the association was evident in premenopausal women and not in postmenopausal women (144). Another meta-analysis of only prospective cohort studies (n=23) found a similar protective association in premenopausal women (145).
Moderate alcohol intake has been associated with increased risk of breast cancer in women (146). Results from prospective studies have also suggested that increased folate intake may reduce the risk of breast cancer in women who regularly consume alcohol (145, 147-150). Thus, high folate intake might be associated with a risk reduction only in women whose breast cancer risk is raised by alcohol consumption. A very large prospective study in more than 88,000 nurses reported that folic acid intake was not associated with breast cancer in women who consumed less than one alcoholic drink per day. However, in women consuming at least one alcoholic drink per day, folic acid intake of at least 600 μg daily resulted in about half the risk of breast cancer compared with women who consumed less than 300 μg of folic acid daily (149). Nevertheless, how alcohol consumption increases breast cancer risk is still subject to discussion (151).
Finally, meta-analyses evaluating the influence of polymorphisms in one-carbon metabolism on cancer risk found that specific variants in the genes encoding thymidylate synthase (152, 153), methionine synthase (154), methionine synthase reductase (154), and MTHFR (154) increase the risk of breast cancer in certain ethnic populations.
Childhood cancers
The incidence of Wilms' tumors (kidney cancer) and certain types of brain cancers (neuroblastoma, ganglioneuroblastoma, and ependymoma) in children has decreased since the mandatory fortification of the US grain supply in 1998 (155). However, incidence rates were unchanged between the pre- and post-fortification periods for leukemia – a predominant childhood malignancy. Overall, the findings of observational studies of maternal folic acid supplementation during pregnancy and childhood cancers have been mixed. A recent meta-analysis of case-control studies found a protective association of maternal folic acid supplementation during pregnancy with childhood acute lymphoblastic leukemia (ALL; OR, 0.75; 95% CI 0.66-0.86; 11 studies) but not with acute myeloid leukemia (5 studies) or childhood brain cancers (6 studies) (156).
Additionally, several meta-analyses have found little to no protective association of MTHFR polymorphisms; however, the most recent meta-analysis of 66 case-control studies found a reduction in the risk of ALL with the c.677C>T variant, driven mainly by a protective association in Caucasians and Asians (157).
Alzheimer's disease and cognitive impairment
Alzheimer’s disease (AD) is the most common form of dementia, affecting more than 5 million people 65 years or older in the US (158). β-amyloid plaque deposition, Tau protein-forming tangles, and increased cell death in the brain of AD patients have been associated with cognitive decline and memory loss. One study associated increased consumption of fruit and vegetables, which are abundant sources of folate, with a reduced risk of developing dementia and AD in women (159). In the Baltimore Longitudinal Study of Aging that followed 579 nondemented older adults for an average of 9.3 years, daily folate intakes of at least 400 μg at baseline were associated with a 55% lower risk of developing Alzheimer’s disease compared to lower intakes (160). Through its role in nucleic acid synthesis and methyl donor provision for methylation reactions, folate is critical for normal brain development and function, not only throughout pregnancy and postnatally but also later in life (161). In one cross-sectional study of elderly women, AD patients had significantly higher homocysteine and lower red blood cell folate concentrations compared to healthy individuals. However, there was no difference in the level of serum folate between groups, suggesting that long-term folate status, rather than recent folate intake, may be associated with the risk of AD (162).
Several investigators have described associations between increased homocysteine concentrations and cognitive impairment in the elderly (163), but prospective cohort studies examining dietary or total daily folate intake and cognition have reported mixed results (164-169). Higher homocysteine concentrations were found in individuals suffering from dementia, including AD and vascular dementia, compared to healthy subjects (170, 171).
Over the past few decades, hyperhomocysteinemia in older adults has become recognized as a modifiable risk factor for cognitive decline, dementia, and Alzheimer’s disease (172-174). A Cochrane review of 14 placebo-controlled trials found that B-vitamin supplementation (folic acid, vitamin B6, vitamin B12) in older adults who were primarily "cognitively healthy" (but many had cardiovascular disease or other conditions) had no significant benefits on global cognitive function (175). Moreover, a recent meta-analysis of 14 randomized, placebo-controlled trials in older adults found that any benefits of B-vitamin supplementation were limited to slowing cognitive decline (176). Stratification of the data revealed that cognitive benefits were seen only in those without dementia at baseline and in trials of more than one year in duration (176). However, not every trial included in these meta-analyses supplemented with folic acid.
Some trials have examined the effects of B-vitamin supplementation in older adults experiencing cognitive impairment, with a few finding cognitive benefit. A two-year randomized, placebo-controlled trial in 168 elderly subjects with mild cognitive impairment described the benefits of a daily regimen of 800 μg of folic acid, 500 μg of vitamin B12, and 20 mg of vitamin B6 (177, 178). Atrophy of specific brain regions affected by AD was observed in individuals of both groups, and this atrophy correlated with cognitive decline; however, the B-vitamin treatment group experienced a smaller loss of gray matter compared to the placebo group (0.5% vs. 3.7%). A greater benefit was seen in subjects with higher baseline homocysteine concentrations, suggesting the importance of lowering circulating homocysteine in prevention of cognitive decline and dementia. Three randomized controlled trials conducted in Chinese adults ages 65 years or older found that supplementation with 400 μg/day of folic acid for 6 to 24 months improved some measures of cognitive function compared to a no-treatment control/conventional treatment (179-181), suggesting that supplemental folic acid in populations without mandated folic acid fortification may confer cognitive benefit. Large double-blind, placebo-controlled trials are needed to understand whether folic acid supplementation late in life might help decrease age-related cognitive impairments or the risk of Alzheimer’s disease and other dementias. Yet, it is prudent to ensure adequate intakes of folate and other B vitamins throughout life for brain — and overall — health.
Disease Treatment
Metabolic diseases
Folinic acid (see Figure 1; also known as leucovorin), a tetrahydrofolic acid derivative, is used in the clinical management of rare inborn errors that affect folate transport or metabolism (reviewed in 182). Such conditions are of autosomal recessive inheritance, meaning only individuals receiving two copies of the mutated gene (one from each parent) develop the disease. Folinic acid can be administered orally, intravenously, or intramuscularly.
Hereditary folate malabsorption
Hereditary folate malabsorption is caused by mutations in the SLC46A1 gene coding for the folate transporter PCFT and typically affects gastrointestinal folate absorption and folate transport into the brain (183). Patients present with low to undetectable concentrations of folate in serum and cerebrospinal fluid; anemia (often macrocytic), thrombocytopenia, and/or pancytopenia (low number of all blood cells); impaired immune responses that increase susceptibility to infections; and a general failure to thrive (184, 185). Neurologic symptoms, including developmental delays, cognitive disorders, and seizures, have also been observed (185, 186). Clinical improvements have been recorded following provision of folinic acid parenterally (187, 188) as well as intramuscularly (188, 189).
Cerebral Folate Deficiency (CFD) syndrome
CFD is characterized by low levels of folate coenzymes in cerebrospinal fluid despite often normal concentrations of folate in blood. Folate transport across the blood-brain barrier is compromised in CFD and has been linked either to the presence of autoantibodies blocking the folate receptor-alpha (FRα) or to mutations in the FOLR1 gene encoding FRα (190-193). Neurological abnormalities, along with visual and hearing impairments, have been described in children with CFD; autism spectrum disorder (ASD) is present in some cases. CFD has also been described in adults presenting with neurological symptoms (194).
Folinic acid can enter the brain and normalize the level of folate coenzymes and has been shown to normalize folate concentrations and improve various social interactions in CFD, including mood, behavior, and verbal communication in children with ASD (190, 195, 196).
Dihydrofolate reductase (DHFR) deficiency
DHFR is the NADPH-dependent enzyme that catalyzes the reduction of dihydrofolic acid to tetrahydrofolic acid. DHFR is also required to convert folic acid to DHF. DHFR deficiency is characterized by megaloblastic anemia and cerebral folate deficiency causing intractable seizures and mental deficits. Although folinic acid treatment can alleviate the symptoms of DHFR deficiency, early diagnosis is essential to prevent irreversible brain damage and improve clinical outcomes (197, 198).
Sources
Food sources
Green leafy vegetables (foliage) are rich sources of folate and provide the basis for its name. Citrus fruit juices, legumes, and fortified foods are also excellent sources of folate (1); the folate content of fortified cereal varies greatly. A number of folate-rich foods are listed in Table 2, along with their folate content in micrograms (μg). For more information on the nutrient content of specific foods, search USDA's FoodData Central.
| Food | Serving | Folate (μg DFEs) |
|---|---|---|
| Lentils (cooked, boiled) | ½ cup | 179 |
| Garbanzo beans (chickpeas, boiled) | ½ cup | 141 |
| Asparagus (boiled) | ½ cup (~6 spears) | 134 |
| Spinach (boiled) | ½ cup | 132 |
| Lima beans (boiled) | ½ cup | 78 |
| Orange juice | 6 fl. oz. | 56 |
| Bread (enriched) | 1 slice | 50* |
| Spaghetti (enriched, cooked) | 1 cup | 180* |
| White rice (enriched, cooked) | 1 cup | 153-180* |
| *To help prevent neural tube defects, the US FDA required the addition of 1.4 milligrams (mg) of folic acid per kilogram (kg) of grain to be added to refined grain products, which were already enriched with niacin, thiamin, riboflavin, and iron, as of January 1, 1998. The addition of nutrients to food in order to prevent a nutritional deficiency or restore nutrients lost in processing is known as fortification. The FDA initially estimated that this level of fortification would increase dietary intake by an average of 100 μg folic acid/day (28). However, further evaluations based on observational studies suggested increases twice that predicted by the FDA (33). On April 14, 2016, the FDA approved the voluntary fortification of corn masa flour up to 1.5 mg of folic acid per kg corn flour (199). | ||
Supplements
The principal form of supplemental folate is folic acid. It is available as a single-ingredient supplement and also included in combination products, such as B-complex vitamins and multivitamins. Doses of 1 mg or greater require a prescription (200). 5-Methyltetrahydrofolate is also available as a supplement (201).
Additionally, folinic acid, a tetrahydrofolic acid derivative, is used to manage certain metabolic diseases (see Disease Treatment). Further, the US FDA has approved the supplementation of folate in oral contraceptives. The addition of levomefolate calcium (the calcium salt of MeTHF; 451 μg/tablet) to oral contraceptives is intended to raise folate status in women of childbearing age (202). According to a US national survey, only 24% of non-pregnant women aged 15-44 years are meeting the current recommendation of 400 μg/day of folic acid (203).
Safety
Toxicity
No adverse effects have been associated with the consumption of excess folate from food. Concerns regarding safety are limited to synthetic folic acid intake. Deficiency of vitamin B12, though often undiagnosed, may affect a significant number of people, especially older adults (see the separate article on Vitamin B12). One symptom of vitamin B12 deficiency is megaloblastic anemia, which is indistinguishable from that associated with folate deficiency (see Deficiency). There is concern that large doses of folic acid given to an individual with an undiagnosed vitamin B12 deficiency could correct megaloblastic anemia without correcting the underlying vitamin B12 deficiency, leaving the individual at risk of developing irreversible neurologic damage. Such cases of neurologic progression in vitamin B12 deficiency have been mostly seen in case studies at folic acid doses of 5,000 μg (5 mg) and above. In order to be very sure of preventing irreversible neurologic damage in vitamin B12-deficient individuals, the Food and Nutrition Board of the US National Academy of Medicine advises that all adults limit their intake of folic acid (supplements and fortification) to 1,000 μg (1 mg) daily (Table 3). The Board also noted that vitamin B12 deficiency is very rare in women in their childbearing years, making the consumption of folic acid at or above 1,000 μg/day unlikely to cause problems (1); however, there are limited data on the effects of large doses (78).
The saturation of DHFR metabolic capacity by oral doses of folic acid has been associated with the appearance of unmetabolized folic acid in blood (204). Exposure to folic acid in the food supply is also associated with unmetabolized folic acid in the blood. A US national cross-sectional survey, NHANES 2007-2008, detected unmetabolized folic acid in 96% of serum samples of Americans (≥1 year of age) (205). In an ancillary study of the Folic Acid Clinical Trial, maternal supplementation with 4.0-5.1 mg/day of folic acid during pregnancy (starting at 8 to 16 weeks’ gestation) led to higher maternal blood concentrations of unmetabolized folic acid and 5-methylTHF at 24 to 26 weeks’ gestation compared to dosages of 1.1 mg/day or less (206). However, no differences were seen in other markers of one-carbon metabolism, including serum concentrations of THF, 5-formylTHF, 5,10-methenylTHF, vitamin B12, and pyridoxal 5’-phosphate (206). A randomized controlled trial in 126 pregnant women found that continuing folic acid supplementation of 400 μg/day beyond the first trimester of pregnancy (i.e., gestational weeks 14 throughout the second and third trimesters) did not increase blood concentrations of unmetabolized folic acid at 36 weeks’ gestation compared to placebo despite increases in plasma total folate, plasma 5-MTHF, and red blood cell folate (207). Maternal folic acid supplementation throughout pregnancy also did not increase unmetabolized folic acid detected in neonatal cord blood samples compared to placebo (207).
Evidence of adverse health effects of unmetabolized folic acid is quite limited. Hematologic abnormalities and poorer cognition have been associated with the presence of unmetabolized folic acid in vitamin B12-deficient older adults (≥60 years) (208, 209). A small study conducted in postmenopausal women also raised concerns about the effect of exposure to unmetabolized folic acid on immune function (210). Recent observational studies have found no link between circulating maternal unmetabolized folate during pregnancy and allergic disease in infancy (211) or autistic traits or language impairment in early childhood (212). In a small, randomized, open-label trial in 38 women of reproductive age receiving 30 weeks of daily multivitamin supplements, daily supplementation with either 1.1 mg or 5 mg of folic acid resulted in the transient appearance of unmetabolized folic acid in blood over the first 12 weeks of supplementation (213). However, unmetabolized folic acid concentrations returned to baseline levels at the end of the study, suggesting that adaptive mechanisms eventually converted folic acid to reduced forms of folate. Nonetheless, the use of supplemental 5-methyltetrahydrofolate may provide an alternative to prevent any potential negative effects of unconverted folic acid in older adults.
Drug interactions
When nonsteroidal anti-inflammatory drugs (NSAIDs), such as aspirin or ibuprofen, are taken in very large therapeutic dosages (i.e., to treat severe arthritis), they may interfere with folate metabolism. In contrast, routine use of NSAIDs has not been found to adversely affect folate status. The anticonvulsant, phenytoin, has been shown to inhibit the intestinal absorption of folate, and several studies have associated decreased folate status with long-term use of the anticonvulsants, phenytoin, phenobarbital, and primidone (214). However, few studies controlled for differences in dietary folate intake between anticonvulsant users and nonusers. Also, taking folic acid at the same time as the cholesterol-lowering agents, cholestyramine and colestipol, may decrease the absorption of folic acid (200). Methotrexate is a folic acid antagonist used to treat a number of diseases, including cancer, rheumatoid arthritis, and psoriasis. Some of the side effects of methotrexate are similar to those of severe folate deficiency, and supplementation with folic or folinic acid is used to reduce antifolate toxicity. Other antifolate molecules currently used in cancer therapy include aminopterin, pemetrexed, pralatrexate, and raltitrexed (11, 115). Further, a number of other medications have been shown to have antifolate activity, including trimethoprim (an antibiotic), pyrimethamine (an antimalarial), triamterene (a blood pressure medication), and sulfasalazine (a treatment for ulcerative colitis). Early studies of oral contraceptives (birth control pills) containing high doses of estrogen indicated adverse effects on folate status; however, this finding has not been supported in more recent studies that used low-dose oral contraceptives and controlled for dietary folate (215).
Linus Pauling Institute Recommendation
The available scientific evidence shows that adequate folate intake prevents neural tube defects and other poor outcomes of pregnancy; is helpful in lowering the risk of some forms of cancer, especially in genetically susceptible individuals; and may lower the risk of stroke. The Linus Pauling Institute recommends that adults take a daily multivitamin/mineral supplement, which typically contains 400 μg of folic acid, the Daily Value (DV). Even with a larger than average intake of folic acid from fortified food, it is unlikely that an individual's daily folic acid intake would regularly exceed the tolerable upper intake level of 1,000 μg/day established by the Institute of Medicine (see Safety).
Older adults (>50 years)
The recommendation for 400 μg/day of supplemental folic acid as part of a daily multivitamin/mineral supplement, in addition to a folate-rich diet, is especially important for older adults because blood homocysteine concentrations tend to increase with age (see Disease Prevention).
Authors and Reviewers
Originally written in 2000 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2002 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in September 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in June 2014 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in October 2023 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in December 2023 by:
Martha S. Field, Ph.D.
Division of Nutritional Sciences
Cornell University
Copyright 2000-2024 Linus Pauling Institute
References
1. Food and Nutrition Board, Institute of Medicine. Folate. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B-6, Folate, Vitamin B-12, Pantothenic Acid, Biotin, and Choline. Washington D.C.: National Academy Press; 1998:196-305. (National Academy Press)
2. Choi SW, Mason JB. Folate and carcinogenesis: an integrated scheme. J Nutr. 2000;130(2):129-132. (PubMed)
3. Bailey LB, Gregory JF, 3rd. Folate metabolism and requirements. J Nutr. 1999;129(4):779-782. (PubMed)
4. Gerhard GT, Duell PB. Homocysteine and atherosclerosis. Curr Opin Lipidol. 1999;10(5):417-428. (PubMed)
5. Jacques PF, Bostom AG, Wilson PW, Rich S, Rosenberg IH, Selhub J. Determinants of plasma total homocysteine concentration in the Framingham Offspring cohort. Am J Clin Nutr. 2001;73(3):613-621. (PubMed)
6. Jacques PF, Kalmbach R, Bagley PJ, et al. The relationship between riboflavin and plasma total homocysteine in the Framingham Offspring cohort is influenced by folate status and the C677T transition in the methylenetetrahydrofolate reductase gene. J Nutr. 2002;132(2):283-288. (PubMed)
7. Wilson CP, McNulty H, Ward M, et al. Blood pressure in treated hypertensive individuals with the MTHFR 677TT genotype is responsive to intervention with riboflavin: findings of a targeted randomized trial. Hypertension. 2013;61(6):1302-1308. (PubMed)
8. Wilson CP, Ward M, McNulty H, et al. Riboflavin offers a targeted strategy for managing hypertension in patients with the MTHFR 677TT genotype: a 4-y follow-up. Am J Clin Nutr. 2012;95(3):766-772. (PubMed)
9. McNulty H, Dowey le RC, Strain JJ, et al. Riboflavin lowers homocysteine in individuals homozygous for the MTHFR 677C->T polymorphism. Circulation. 2006;113(1):74-80. (PubMed)
10. McNulty H, Ward M, Hoey L, Hughes CF, Pentieva K. Addressing optimal folate and related B-vitamin status through the lifecycle: health impacts and challenges. Proc Nutr Soc. 2019;78(3):449-462. (PubMed)
11. Shulpekova Y, Nechaev V, Kardasheva S, et al. The concept of folic acid in health and disease. Molecules. 2021;26(12):3731. (PubMed)
12. Desmoulin SK, Hou Z, Gangjee A, Matherly LH. The human proton-coupled folate transporter: Biology and therapeutic applications to cancer. Cancer Biol Ther. 2012;13(14):1355-1373. (PubMed)
13. Solanky N, Requena Jimenez A, D'Souza SW, Sibley CP, Glazier JD. Expression of folate transporters in human placenta and implications for homocysteine metabolism. Placenta. 2010;31(2):134-143. (PubMed)
14. Halsted CH, Villanueva JA, Devlin AM, Chandler CJ. Metabolic interactions of alcohol and folate. J Nutr. 2002;132(8 Suppl):2367S-2372S. (PubMed)
15. Pfeiffer CM, Sternberg MR, Schleicher RL, Rybak ME. Dietary supplement use and smoking are important correlates of biomarkers of water-soluble vitamin status after adjusting for sociodemographic and lifestyle variables in a representative sample of U.S. adults. J Nutr. 2013;143(6):957S-965S. (PubMed)
16. Stark KD, Pawlosky RJ, Sokol RJ, Hannigan JH, Salem N, Jr. Maternal smoking is associated with decreased 5-methyltetrahydrofolate in cord plasma. Am J Clin Nutr. 2007;85(3):796-802. (PubMed)
17. Hutson JR, Stade B, Lehotay DC, Collier CP, Kapur BM. Folic acid transport to the human fetus is decreased in pregnancies with chronic alcohol exposure. PLoS One. 2012;7(5):e38057. (PubMed)
18. Herbert V. Folic acid. In: Shils M, Olson, J.A., Shike M., Ross A.C., ed. Modern Nutrition in Health and Disease. Baltimore: Williams & Wilkins; 1999:433-446.
19. Stabler SP. Clinical folate deficiency. In: Bailey LB, ed. Folate in Health and Disease. 2nd ed. Boca Raton, FL: CRC press, Taylor & Francis Group; 2010:409-428.
20. Bailey LB. Dietary reference intakes for folate: the debut of dietary folate equivalents. Nutr Rev. 1998;56(10):294-299. (PubMed)
21. Bailey LB, Gregory JF, 3rd. Polymorphisms of methylenetetrahydrofolate reductase and other enzymes: metabolic significance, risks and impact on folate requirement. J Nutr. 1999;129(5):919-922. (PubMed)
22. Wilcken B, Bamforth F, Li Z, et al. Geographical and ethnic variation of the 677C>T allele of 5,10 methylenetetrahydrofolate reductase (MTHFR): findings from over 7000 newborns from 16 areas world wide. J Med Genet. 2003;40(8):619-625. (PubMed)
23. Guenther BD, Sheppard CA, Tran P, Rozen R, Matthews RG, Ludwig ML. The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomocysteinemia. Nat Struct Biol. 1999;6(4):359-365. (PubMed)
24. Molloy AM, Daly S, Mills JL, et al. Thermolabile variant of 5,10-methylenetetrahydrofolate reductase associated with low red-cell folates: implications for folate intake recommendations. Lancet. 1997;349(9065):1591-1593. (PubMed)
25. Rozen R. Genetic predisposition to hyperhomocysteinemia: deficiency of methylenetetrahydrofolate reductase (MTHFR). Thromb Haemost. 1997;78(1):523-526. (PubMed)
26. Kauwell GP, Wilsky CE, Cerda JJ, et al. Methylenetetrahydrofolate reductase mutation (677C-->T) negatively influences plasma homocysteine response to marginal folate intake in elderly women. Metabolism. 2000;49(11):1440-1443. (PubMed)
27. Shane B. Folic acid, vitamin B-12, and vitamin B-6. In: Stipanuk M, ed. Biochemical and Physiological Aspects of Human Nutrition. Philadelphia: W.B. Saunders Co.; 2000:483-518.
28. Eskes TK. Open or closed? A world of difference: a history of homocysteine research. Nutr Rev. 1998;56(8):236-244. (PubMed)
29. Czeizel AE, Dudas I, Vereczkey A, Banhidy F. Folate deficiency and folic acid supplementation: the prevention of neural-tube defects and congenital heart defects. Nutrients. 2013;5(11):4760-4775. (PubMed)
30. Kancherla V. Neural tube defects: a review of global prevalence, causes, and primary prevention. Childs Nerv Syst. 2023;39(7):1703-1710. (PubMed)
31. MRC Vitamin Study Research Group. Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet. 1991;338(8760):131-137. (PubMed)
32. Czeizel AE, Dudas I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med. 1992;327(26):1832-1835. (PubMed)
33. Quinlivan EP, Gregory JF, 3rd. Effect of food fortification on folic acid intake in the United States. Am J Clin Nutr. 2003;77(1):221-225. (PubMed)
34. National Birth Defects Prevention Network. Neural Tube Defect Ascertainment Project. Available at: NTD fact sheet 01-10 for website.pdf. Accessed 12/16/14.
35. Williams J, Mai CT, Mulinare J, et al. Updated estimates of neural tube defects prevented by mandatory folic Acid fortification - United States, 1995-2011. MMWR Morb Mortal Wkly Rep. 2015;64(1):1-5. (PubMed)
36. Kancherla V, Wagh K, Priyadarshini P, Pachon H, Oakley GP, Jr. A global update on the status of prevention of folic acid-preventable spina bifida and anencephaly in year 2020: 30-Year anniversary of gaining knowledge about folic acid's prevention potential for neural tube defects. Birth Defects Res. 2022;114(20):1392-1403. (PubMed)
37. Kancherla V, Botto LD, Rowe LA, et al. Preventing birth defects, saving lives, and promoting health equity: an urgent call to action for universal mandatory food fortification with folic acid. Lancet Glob Health. 2022;10(7):e1053-e1057. (PubMed)
38. Talaulikar VS, Arulkumaran S. Folic acid in obstetric practice: a review. Obstet Gynecol Surv. 2011;66(4):240-247. (PubMed)
39. American College of Obstetricians and Gynecologists (ACOG). Neural tube defects. Washington, DC. 2003. http://www.guideline.gov/content.aspx?id=3994. Accessed 12/19/14.
40. McNulty B, Pentieva K, Marshall B, et al. Women's compliance with current folic acid recommendations and achievement of optimal vitamin status for preventing neural tube defects. Hum Reprod. 2011;26(6):1530-1536. (PubMed)
41. Nilsen RM, Vollset SE, Gjessing HK, et al. Patterns and predictors of folic acid supplement use among pregnant women: the Norwegian Mother and Child Cohort Study. Am J Clin Nutr. 2006;84(5):1134-1141. (PubMed)
42. Ray JG, Singh G, Burrows RF. Evidence for suboptimal use of periconceptional folic acid supplements globally. BJOG. 2004;111(5):399-408. (PubMed)
43. Rogers LM, Cordero AM, Pfeiffer CM, et al. Global folate status in women of reproductive age: a systematic review with emphasis on methodological issues. Ann N Y Acad Sci. 2018;1431(1):35-57. (PubMed)
44. Copp AJ, Stanier P, Greene ND. Neural tube defects: recent advances, unsolved questions, and controversies. Lancet Neurol. 2013;12(8):799-810. (PubMed)
45. Yang Y, Chen J, Wang B, Ding C, Liu H. Association between MTHFR C677T polymorphism and neural tube defect risks: A comprehensive evaluation in three groups of NTD patients, mothers, and fathers. Birth Defects Res A Clin Mol Teratol. 2015;103(6):488-500. (PubMed)
46. De Marco P, Calevo MG, Moroni A, et al. Study of MTHFR and MS polymorphisms as risk factors for NTD in the Italian population. J Hum Genet. 2002;47(6):319-324. (PubMed)
47. Boduroglu K, Alanay Y, Alikasifoglu M, Aktas D, Tuncbilek E. Analysis of MTHFR 1298A>C in addition to MTHFR 677C>T polymorphism as a risk factor for neural tube defects in the Turkish population. Turk J Pediatr. 2005;47(4):327-333. (PubMed)
48. Wang Y, Liu Y, Ji W, et al. Variants in MTHFR gene and neural tube defects susceptibility in China. Metab Brain Dis. 2015;30(4):1017-1026. (PubMed)
49. van der Put NM, Gabreels F, Stevens EM, et al. A second common mutation in the methylenetetrahydrofolate reductase gene: an additional risk factor for neural-tube defects? Am J Hum Genet. 1998;62(5):1044-1051. (PubMed)
50. De Marco P, Calevo MG, Moroni A, et al. Reduced folate carrier polymorphism (80A-->G) and neural tube defects. Eur J Hum Genet. 2003;11(3):245-252. (PubMed)
51. O'Leary VB, Mills JL, Parle-McDermott A, et al. Screening for new MTHFR polymorphisms and NTD risk. Am J Med Genet A. 2005;138A(2):99-106. (PubMed)
52. Christensen B, Arbour L, Tran P, et al. Genetic polymorphisms in methylenetetrahydrofolate reductase and methionine synthase, folate levels in red blood cells, and risk of neural tube defects. Am J Med Genet. 1999;84(2):151-157. (PubMed)
53. Relton CL, Wilding CS, Pearce MS, et al. Gene-gene interaction in folate-related genes and risk of neural tube defects in a UK population. J Med Genet. 2004;41(4):256-260. (PubMed)
54. Brody LC, Conley M, Cox C, et al. A polymorphism, R653Q, in the trifunctional enzyme methylenetetrahydrofolate dehydrogenase/methenyltetrahydrofolate cyclohydrolase/formyltetrahydrofolate synthetase is a maternal genetic risk factor for neural tube defects: report of the Birth Defects Research Group. Am J Hum Genet. 2002;71(5):1207-1215. (PubMed)
55. Meng J, Han L, Zhuang B. Association between MTHFD1 polymorphisms and neural tube defect susceptibility. J Neurol Sci. 2015;348(1-2):188-194. (PubMed)
56. van der Put NM, van den Heuvel LP, Steegers-Theunissen RP, et al. Decreased methylene tetrahydrofolate reductase activity due to the 677C-->T mutation in families with spina bifida offspring. J Mol Med (Berl). 1996;74(11):691-694. (PubMed)
57. Wilson A, Platt R, Wu Q, et al. A common variant in methionine synthase reductase combined with low cobalamin (vitamin B12) increases risk for spina bifida. Mol Genet Metab. 1999;67(4):317-323. (PubMed)
58. Gilboa SM, Salemi JL, Nembhard WN, Fixler DE, Correa A. Mortality resulting from congenital heart disease among children and adults in the United States, 1999 to 2006. Circulation. 2010;122(22):2254-2263. (PubMed)
59. van Beynum IM, Kapusta L, Bakker MK, den Heijer M, Blom HJ, de Walle HE. Protective effect of periconceptional folic acid supplements on the risk of congenital heart defects: a registry-based case-control study in the northern Netherlands. Eur Heart J. 2010;31(4):464-471. (PubMed)
60. Yin M, Dong L, Zheng J, Zhang H, Liu J, Xu Z. Meta analysis of the association between MTHFR C677T polymorphism and the risk of congenital heart defects. Ann Hum Genet. 2012;76(1):9-16. (PubMed)
61. Wang W, Wang Y, Gong F, Zhu W, Fu S. MTHFR C677T polymorphism and risk of congenital heart defects: evidence from 29 case-control and TDT studies. PLoS One. 2013;8(3):e58041. (PubMed)
62. Badovinac RL, Werler MM, Williams PL, Kelsey KT, Hayes C. Folic acid-containing supplement consumption during pregnancy and risk for oral clefts: a meta-analysis. Birth Defects Res A Clin Mol Teratol. 2007;79(1):8-15. (PubMed)
63. Watkins SE, Meyer RE, Strauss RP, Aylsworth AS. Classification, epidemiology, and genetics of orofacial clefts. Clin Plast Surg. 2014;41(2):149-163. (PubMed)
64. Zhou Y, Sinnathamby V, Yu Y, et al. Folate intake, markers of folate status and oral clefts: An updated set of systematic reviews and meta-analyses. Birth Defects Res. 2020;112(19):1699-1719. (PubMed)
65. Boyles AL, Wilcox AJ, Taylor JA, et al. Folate and one-carbon metabolism gene polymorphisms and their associations with oral facial clefts. Am J Med Genet A. 2008;146A(4):440-449. (PubMed)
66. Boyles AL, Wilcox AJ, Taylor JA, et al. Oral facial clefts and gene polymorphisms in metabolism of folate/one-carbon and vitamin A: a pathway-wide association study. Genet Epidemiol. 2009;33(3):247-255. (PubMed)
67. Millacura N, Pardo R, Cifuentes L, Suazo J. Effects of folic acid fortification on orofacial clefts prevalence: a meta-analysis. Public Health Nutr. 2017;20(12):2260-2268. (PubMed)
68. Wilcox AJ. On the importance--and the unimportance--of birthweight. Int J Epidemiol. 2001;30(6):1233-1241. (PubMed)
69. Fekete K, Berti C, Trovato M, et al. Effect of folate intake on health outcomes in pregnancy: a systematic review and meta-analysis on birth weight, placental weight and length of gestation. Nutr J. 2012;11:75. (PubMed)
70. Baker PN, Wheeler SJ, Sanders TA, et al. A prospective study of micronutrient status in adolescent pregnancy. Am J Clin Nutr. 2009;89(4):1114-1124. (PubMed)
71. Lee HA, Park EA, Cho SJ, et al. Mendelian randomization analysis of the effect of maternal homocysteine during pregnancy, as represented by maternal MTHFR C677T genotype, on birth weight. J Epidemiol. 2013;23(5):371-375. (PubMed)
72. Scholl TO, Johnson WG. Folic acid: influence on the outcome of pregnancy. Am J Clin Nutr. 2000;71(5 Suppl):1295S-1303S. (PubMed)
73. Vollset SE, Refsum H, Irgens LM, et al. Plasma total homocysteine, pregnancy complications, and adverse pregnancy outcomes: the Hordaland Homocysteine study. Am J Clin Nutr. 2000;71(4):962-968. (PubMed)
74. Wang XM, Wu HY, Qiu XJ. Methylenetetrahydrofolate reductase (MTHFR) gene C677T polymorphism and risk of preeclampsia: an updated meta-analysis based on 51 studies. Arch Med Res. 2013;44(3):159-168. (PubMed)
75. Liu C, Liu C, Wang Q, Zhang Z. Supplementation of folic acid in pregnancy and the risk of preeclampsia and gestational hypertension: a meta-analysis. Arch Gynecol Obstet. 2018;298(4):697-704. (PubMed)
76. Wen SW, Champagne J, Rennicks White R, et al. Effect of folic acid supplementation in pregnancy on preeclampsia: the folic acid clinical trial study. J Pregnancy. 2013;2013:294312. (PubMed)
77. Wen SW, White RR, Rybak N, et al. Effect of high dose folic acid supplementation in pregnancy on pre-eclampsia (FACT): double blind, phase III, randomised controlled, international, multicentre trial. BMJ. 2018;362:k3478. (PubMed)
78. Field MS, Stover PJ. Safety of folic acid. Ann N Y Acad Sci. 2018;1414(1):59-71. (PubMed)
79. Hoxha B, Hoxha M, Domi E, et al. Folic acid and autism: a systematic review of the current state of knowledge. Cells. 2021;10(8):1976. (PubMed)
80. Crider KS, Cordero AM, Qi YP, Mulinare J, Dowling NF, Berry RJ. Prenatal folic acid and risk of asthma in children: a systematic review and meta-analysis. Am J Clin Nutr. 2013;98(5):1272-1281. (PubMed)
81. Brown SB, Reeves KW, Bertone-Johnson ER. Maternal folate exposure in pregnancy and childhood asthma and allergy: a systematic review. Nutr Rev. 2014;72(1):55-64. (PubMed)
82. Lassi ZS, Salam RA, Haider BA, Bhutta ZA. Folic acid supplementation during pregnancy for maternal health and pregnancy outcomes. Cochrane Database Syst Rev. 2013;3:CD006896. (PubMed)
83. Ding R, Lin S, Chen D. The association of cystathionine beta synthase (CBS) T833C polymorphism and the risk of stroke: a meta-analysis. J Neurol Sci. 2012;312(1-2):26-30. (PubMed)
84. Seshadri N, Robinson K. Homocysteine, B vitamins, and coronary artery disease. Med Clin North Am. 2000;84(1):215-237, x. (PubMed)
85. Smith AD, Refsum H. Homocysteine - from disease biomarker to disease prevention. J Intern Med. 2021;290(4):826-854. (PubMed)
86. Wald DS, Law M, Morris JK. Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis. BMJ. 2002;325(7374):1202. (PubMed)
87. Homocysteine Studies Collaboration. Homocysteine and risk of ischemic heart disease and stroke: a meta-analysis. JAMA. 2002;288(16):2015-2022. (PubMed)
88. Clarke R, Halsey J, Lewington S, et al. Effects of lowering homocysteine levels with B vitamins on cardiovascular disease, cancer, and cause-specific mortality: Meta-analysis of 8 randomized trials involving 37 485 individuals. Arch Intern Med. 2010;170(18):1622-1631. (PubMed)
89. Clarke R, Halsey J, Bennett D, Lewington S. Homocysteine and vascular disease: review of published results of the homocysteine-lowering trials. J Inherit Metab Dis. 2011;34(1):83-91. (PubMed)
90. Huang T, Chen Y, Yang B, Yang J, Wahlqvist ML, Li D. Meta-analysis of B vitamin supplementation on plasma homocysteine, cardiovascular and all-cause mortality. Clin Nutr. 2012;31(4):448-454. (PubMed)
91. Voutilainen S, Rissanen TH, Virtanen J, Lakka TA, Salonen JT. Low dietary folate intake is associated with an excess incidence of acute coronary events: The Kuopio Ischemic Heart Disease Risk Factor Study. Circulation. 2001;103(22):2674-2680. (PubMed)
92. Brattstrom L. Vitamins as homocysteine-lowering agents. J Nutr. 1996;126(4 Suppl):1276S-1280S. (PubMed)
93. Rader JI. Folic acid fortification, folate status and plasma homocysteine. J Nutr. 2002;132(8 Suppl):2466S-2470S. (PubMed)
94. Homocysteine Lowering Trialists Collaboration. Dose-dependent effects of folic acid on blood concentrations of homocysteine: a meta-analysis of the randomized trials. Am J Clin Nutr. 2005;82(4):806-812. (PubMed)
95. Malinow MR, Bostom AG, Krauss RM. Homocyst(e)ine, diet, and cardiovascular diseases: a statement for healthcare professionals from the Nutrition Committee, American Heart Association. Circulation. 1999;99(1):178-182. (PubMed)
96. van Meurs JB, Pare G, Schwartz SM, et al. Common genetic loci influencing plasma homocysteine concentrations and their effect on risk of coronary artery disease. Am J Clin Nutr. 2013;98(3):668-676. (PubMed)
97. Holmes MV, Newcombe P, Hubacek JA, et al. Effect modification by population dietary folate on the association between MTHFR genotype, homocysteine, and stroke risk: a meta-analysis of genetic studies and randomised trials. Lancet. 2011;378(9791):584-594. (PubMed)
98. Clarke R, Bennett DA, Parish S, et al. Homocysteine and coronary heart disease: meta-analysis of MTHFR case-control studies, avoiding publication bias. PLoS Med. 2012;9(2):e1001177. (PubMed)
99. Mosca L, Benjamin EJ, Berra K, et al. Effectiveness-based guidelines for the prevention of cardiovascular disease in women--2011 update: a guideline from the American Heart Association. J Am Coll Cardiol. 2011;57(12):1404-1423. (PubMed)
100. Park J-H, Saposnik G, Ovbiagele B, Markovic D, Towfighi A. Effect of B-vitamins on stroke risk among individuals with vascular disease who are not on antiplatelets: A meta-analysis. Int J Stroke.11(2):206-211. (PubMed)
101. Tian T, Yang KQ, Cui JG, Zhou LL, Zhou XL. Folic acid supplementation for stroke prevention in patients with cardiovascular disease. Am J Med Sci. 2017;354(4):379-387. (PubMed)
102. Wang WW, Wang XS, Zhang ZR, He JC, Xie CL. A meta-analysis of folic acid in combination with anti-hypertension drugs in patients with hypertension and hyperhomocysteinemia. Front Pharmacol. 2017;8:585. (PubMed)
103. Wang Y, Jin Y, Wang Y, et al. The effect of folic acid in patients with cardiovascular disease: A systematic review and meta-analysis. Medicine (Baltimore). 2019;98(37):e17095. (PubMed)
104. Li Y, Huang T, Zheng Y, Muka T, Troup J, Hu FB. Folic acid supplementation and the risk of cardiovascular diseases: a meta-analysis of randomized controlled trials. J Am Heart Assoc. 2016;5(8):e003768. (PubMed)
105. Asbaghi O, Salehpour S, Rezaei Kelishadi M, et al. Folic acid supplementation and blood pressure: a GRADE-assessed systematic review and dose-response meta-analysis of 41,633 participants. Crit Rev Food Sci Nutr. 2023;63(13):1846-1861. (PubMed)
106. Lorenz MW, Markus HS, Bots ML, Rosvall M, Sitzer M. Prediction of clinical cardiovascular events with carotid intima-media thickness: a systematic review and meta-analysis. Circulation. 2007;115(4):459-467. (PubMed)
107. Qin X, Xu M, Zhang Y, et al. Effect of folic acid supplementation on the progression of carotid intima-media thickness: a meta-analysis of randomized controlled trials. Atherosclerosis. 2012;222(2):307-313. (PubMed)
108. de Bree A, van Mierlo LA, Draijer R. Folic acid improves vascular reactivity in humans: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2007;86(3):610-617. (PubMed)
109. Zamani M, Rezaiian F, Saadati S, et al. The effects of folic acid supplementation on endothelial function in adults: a systematic review and dose-response meta-analysis of randomized controlled trials. Nutr J. 2023;22(1):12. (PubMed)
110. McNeil CJ, Beattie JH, Gordon MJ, Pirie LP, Duthie SJ. Nutritional B vitamin deficiency disrupts lipid metabolism causing accumulation of proatherogenic lipoproteins in the aorta adventitia of ApoE null mice. Mol Nutr Food Res. 2012;56(7):1122-1130. (PubMed)
111. Blount BC, Mack MM, Wehr CM, et al. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. Proc Natl Acad Sci U S A. 1997;94(7):3290-3295. (PubMed)
112. Narayanan S, McConnell J, Little J, et al. Associations between two common variants C677T and A1298C in the methylenetetrahydrofolate reductase gene and measures of folate metabolism and DNA stability (strand breaks, misincorporated uracil, and DNA methylation status) in human lymphocytes in vivo. Cancer Epidemiol Biomarkers Prev. 2004;13(9):1436-1443. (PubMed)
113. Rahman L, Voeller D, Rahman M, et al. Thymidylate synthase as an oncogene: a novel role for an essential DNA synthesis enzyme. Cancer Cell. 2004;5(4):341-351. (PubMed)
114. Hubner RA, Liu JF, Sellick GS, Logan RF, Houlston RS, Muir KR. Thymidylate synthase polymorphisms, folate and B-vitamin intake, and risk of colorectal adenoma. Br J Cancer. 2007;97(10):1449-1456. (PubMed)
115. Desmoulin SK, Wang L, Polin L, et al. Functional loss of the reduced folate carrier enhances the antitumor activities of novel antifolates with selective uptake by the proton-coupled folate transporter. Mol Pharmacol. 2012;82(4):591-600. (PubMed)
116. Crider KS, Yang TP, Berry RJ, Bailey LB. Folate and DNA methylation: a review of molecular mechanisms and the evidence for folate's role. Adv Nutr. 2012;3(1):21-38. (PubMed)
117. Butrum RR, Clifford CK, Lanza E. NCI dietary guidelines: rationale. Am J Clin Nutr. 1988;48(3 Suppl):888-895. (PubMed)
118. World Cancer Research Fund/American Institute for Cancer Research. Diet, Nutrition, Physical Activity, and Cancer: a Global Perspective 2018. Available at: https://www.wcrf.org/wp-content/uploads/2021/02/Summary-of-Third-Expert-Report-2018.pdf. Accessed 12/21/2023.
119. Crider KS, Bailey LB, Berry RJ. Folic acid food fortification-its history, effect, concerns, and future directions. Nutrients. 2011;3(3):370-384. (PubMed)
120. Qin X, Cui Y, Shen L, et al. Folic acid supplementation and cancer risk: A meta-analysis of randomized controlled trials. Int J Cancer. 2013;133(5):1033-1041. (PubMed)
121. Vollset SE, Clarke R, Lewington S, et al. Effects of folic acid supplementation on overall and site-specific cancer incidence during the randomised trials: meta-analyses of data on 50,000 individuals. Lancet. 2013;381(9871):1029-1036. (PubMed)
122. National Toxicology Program. Identifying research needs for assessing safe use of high intakes of folic acid. NTP Monograph. Research Triangle Park, NC: National Toxicology Program, 2015.
123. Kim DH, Smith-Warner SA, Spiegelman D, et al. Pooled analyses of 13 prospective cohort studies on folate intake and colon cancer. Cancer Causes Control. 2010;21(11):1919-1930. (PubMed)
124. Gibson TM, Weinstein SJ, Pfeiffer RM, et al. Pre- and postfortification intake of folate and risk of colorectal cancer in a large prospective cohort study in the United States. Am J Clin Nutr. 2011;94(4):1053-1062. (PubMed)
125. Stevens VL, McCullough ML, Sun J, Jacobs EJ, Campbell PT, Gapstur SM. High levels of folate from supplements and fortification are not associated with increased risk of colorectal cancer. Gastroenterology. 2011;141(1):98-105, 105 e101. (PubMed)
126. Zschabitz S, Cheng TY, Neuhouser ML, et al. B vitamin intakes and incidence of colorectal cancer: results from the Women's Health Initiative Observational Study cohort. Am J Clin Nutr. 2013;97(2):332-343. (PubMed)
127. Wang F, Wu K, Li Y, et al. Association of folate intake and colorectal cancer risk in the postfortification era in US women. Am J Clin Nutr. 2021;114(1):49-58. (PubMed)
128. Kennedy DA, Stern SJ, Moretti M, et al. Folate intake and the risk of colorectal cancer: a systematic review and meta-analysis. Cancer Epidemiol. 2011;35(1):2-10. (PubMed)
129. Kim YI. Folate: a magic bullet or a double edged sword for colorectal cancer prevention? Gut. 2006;55(10):1387-1389. (PubMed)
130. Paspatis GA, Kalafatis E, Oros L, Xourgias V, Koutsioumpa P, Karamanolis DG. Folate status and adenomatous colonic polyps. A colonoscopically controlled study. Dis Colon Rectum. 1995;38(1):64-67; discussion 67-68. (PubMed)
131. Jaszewski R, Misra S, Tobi M, et al. Folic acid supplementation inhibits recurrence of colorectal adenomas: a randomized chemoprevention trial. World J Gastroenterol. 2008;14(28):4492-4498. (PubMed)
132. Wu K, Platz EA, Willett WC, et al. A randomized trial on folic acid supplementation and risk of recurrent colorectal adenoma. Am J Clin Nutr. 2009;90(6):1623-1631. (PubMed)
133. Logan RF, Grainge MJ, Shepherd VC, Armitage NC, Muir KR, ukCAP Trial Group. Aspirin and folic acid for the prevention of recurrent colorectal adenomas. Gastroenterology. 2008;134(1):29-38. (PubMed)
134. Figueiredo JC, Mott LA, Giovannucci E, et al. Folic acid and prevention of colorectal adenomas: a combined analysis of randomized clinical trials. Int J Cancer. 2011;129(1):192-203. (PubMed)
135. van Wijngaarden JP, Dhonukshe-Rutten RA, van Schoor NM, et al. Rationale and design of the B-PROOF study, a randomized controlled trial on the effect of supplemental intake of vitamin B12 and folic acid on fracture incidence. BMC Geriatr. 2011;11:80. (PubMed)
136. Oliai Araghi S, Kiefte-de Jong JC, van Dijk SC, et al. Folic acid and vitamin B12 supplementation and the risk of cancer: long-term follow-up of the B Vitamins for the Prevention of Osteoporotic Fractures (B-PROOF) trial. Cancer Epidemiol Biomarkers Prev. 2019;28(2):275-282. (PubMed)
137. Kennedy DA, Stern SJ, Matok I, et al. Folate intake, MTHFR polymorphisms, and the risk of colorectal cancer: a systematic review and meta-analysis. J Cancer Epidemiol. 2012;2012:952508. (PubMed)
138. Ding W, Zhou DL, Jiang X, Lu LS. Methionine synthase A2756G polymorphism and risk of colorectal adenoma and cancer: evidence based on 27 studies. PLoS One. 2013;8(4):e60508. (PubMed)
139. Haerian MS, Haerian BS, Molanaei S, et al. MTRR rs1801394 and its interaction with MTHFR rs1801133 in colorectal cancer: a case-control study and meta-analysis. Pharmacogenomics. 2017;18(11):1075-1084. (PubMed)
140. Nan H, Lee JE, Rimm EB, Fuchs CS, Giovannucci EL, Cho E. Prospective study of alcohol consumption and the risk of colorectal cancer before and after folic acid fortification in the United States. Ann Epidemiol. 2013;23(9):558-563. (PubMed)
141. Slattery ML, Potter JD, Samowitz W, Schaffer D, Leppert M. Methylenetetrahydrofolate reductase, diet, and risk of colon cancer. Cancer Epidemiol Biomarkers Prev. 1999;8(6):513-518. (PubMed)
142. Ma J, Stampfer MJ, Giovannucci E, et al. Methylenetetrahydrofolate reductase polymorphism, dietary interactions, and risk of colorectal cancer. Cancer Res. 1997;57(6):1098-1102. (PubMed)
143. Larsson SC, Giovannucci E, Wolk A. Folate and risk of breast cancer: a meta-analysis. J Natl Cancer Inst. 2007;99(1):64-76. (PubMed)
144. Ren X, Xu P, Zhang D, et al. Association of folate intake and plasma folate level with the risk of breast cancer: a dose-response meta-analysis of observational studies. Aging (Albany NY). 2020;12(21):21355-21375. (PubMed)
145. Zeng J, Wang K, Ye F, et al. Folate intake and the risk of breast cancer: an up-to-date meta-analysis of prospective studies. Eur J Clin Nutr. 2019;73(12):1657-1660. (PubMed)
146. Brooks PJ, Zakhari S. Moderate alcohol consumption and breast cancer in women: from epidemiology to mechanisms and interventions. Alcohol Clin Exp Res. 2013;37(1):23-30. (PubMed)
147. Rohan TE, Jain MG, Howe GR, Miller AB. Dietary folate consumption and breast cancer risk. J Natl Cancer Inst. 2000;92(3):266-269. (PubMed)
148. Sellers TA, Kushi LH, Cerhan JR, et al. Dietary folate intake, alcohol, and risk of breast cancer in a prospective study of postmenopausal women. Epidemiology. 2001;12(4):420-428. (PubMed)
149. Zhang S, Hunter DJ, Hankinson SE, et al. A prospective study of folate intake and the risk of breast cancer. JAMA. 1999;281(17):1632-1637. (PubMed)
150. Fagherazzi G, Vilier A, Boutron-Ruault MC, Mesrine S, Clavel-Chapelon F. Alcohol consumption and breast cancer risk subtypes in the E3N-EPIC cohort. Eur J Cancer Prev. 2015;24(3):209-214. (PubMed)
151. Freudenheim JL. Alcohol's effects on breast cancer in women. Alcohol Res. 2020;40(2):11. (PubMed)
152. Wang J, Wang B, Bi J, Di J. The association between two polymorphisms in the TYMS gene and breast cancer risk: a meta-analysis. Breast Cancer Res Treat. 2011;128(1):203-209. (PubMed)
153. Weiner AS, Boyarskikh UA, Voronina EN, et al. Polymorphisms in the folate-metabolizing genes MTR, MTRR, and CBS and breast cancer risk. Cancer Epidemiol. 2012;36(2):e95-e100. (PubMed)
154. Mo W, Ding Y, Zheng Y, Zou D, Ding X. Associations between folate metabolism enzyme polymorphisms and breast cancer: A meta-analysis. Breast J. 2020;26(3):484-487. (PubMed)
155. Linabery AM, Johnson KJ, Ross JA. Childhood cancer incidence trends in association with US folic acid fortification (1986-2008). Pediatrics. 2012;129(6):1125-1133. (PubMed)
156. Wan Ismail WR, Abdul Rahman R, Rahman NAA, Atil A, Nawi AM. The protective effect of maternal folic acid supplementation on childhood cancer: a systematic review and meta-analysis of case-control studies. J Prev Med Public Health. 2019;52(4):205-213. (PubMed)
157. Frikha R. Assessment of the relationship between methylenetetrahydrofolate reductase polymorphism and acute lymphoblastic leukemia: Evidence from an updated meta-analysis. J Oncol Pharm Pract. 2020;26(7):1598-1610. (PubMed)
158. Matthews KA, Xu W, Gaglioti AH, et al. Racial and ethnic estimates of Alzheimer's disease and related dementias in the United States (2015-2060) in adults aged ≥65 years. Alzheimers Dement. 2019;15(1):17-24. (PubMed)
159. Hughes TF, Andel R, Small BJ, et al. Midlife fruit and vegetable consumption and risk of dementia in later life in Swedish twins. Am J Geriatr Psychiatry. 2010;18(5):413-420. (PubMed)
160. Corrada MM, Kawas CH, Hallfrisch J, Muller D, Brookmeyer R. Reduced risk of Alzheimer's disease with high folate intake: the Baltimore Longitudinal Study of Aging. Alzheimers Dement. 2005;1(1):11-18. (PubMed)
161. Weir DG, Scott JM. Brain function in the elderly: role of vitamin B12 and folate. Br Med Bull. 1999;55(3):669-682. (PubMed)
162. Faux NG, Ellis KA, Porter L, et al. Homocysteine, vitamin B12, and folic acid levels in Alzheimer's disease, mild cognitive impairment, and healthy elderly: baseline characteristics in subjects of the Australian Imaging Biomarker Lifestyle study. J Alzheimers Dis. 2011;27(4):909-922. (PubMed)
163. Van Dam F, Van Gool WA. Hyperhomocysteinemia and Alzheimer's disease: A systematic review. Arch Gerontol Geriatr. 2009;48(3):425-430. (PubMed)
164. Morris MC, Evans DA, Bienias JL, et al. Dietary folate and vitamin B12 intake and cognitive decline among community-dwelling older persons. Arch Neurol. 2005;62(4):641-645. (PubMed)
165. Morris MC, Evans DA, Schneider JA, Tangney CC, Bienias JL, Aggarwal NT. Dietary folate and vitamins B-12 and B-6 not associated with incident Alzheimer's disease. J Alzheimers Dis. 2006;9(4):435-443. (PubMed)
166. Qin B, Xun P, Jacobs DR, Jr., et al. Intake of niacin, folate, vitamin B-6, and vitamin B-12 through young adulthood and cognitive function in midlife: the Coronary Artery Risk Development in Young Adults (CARDIA) study. Am J Clin Nutr. 2017;106(4):1032-1040. (PubMed)
167. Agnew-Blais JC, Wassertheil-Smoller S, Kang JH, et al. Folate, vitamin B-6, and vitamin B-12 intake and mild cognitive impairment and probable dementia in the Women's Health Initiative Memory Study. J Acad Nutr Diet. 2015;115(2):231-241. (PubMed)
168. Sheng LT, Jiang YW, Pan XF, et al. Association between dietary intakes of B vitamins in midlife and cognitive impairment in late-life: the Singapore Chinese Health Study. J Gerontol A Biol Sci Med Sci. 2020;75(6):1222-1227. (PubMed)
169. Zanin Palchetti C, Gomes Goncalves N, Vidal Ferreira N, et al. Dietary folate intake and its association with longitudinal changes in cognition function. Clin Nutr ESPEN. 2023;55:332-339. (PubMed)
170. Wald DS, Kasturiratne A, Simmonds M. Serum homocysteine and dementia: meta-analysis of eight cohort studies including 8669 participants. Alzheimers Dement. 2011;7(4):412-417. (PubMed)
171. Ho RC, Cheung MW, Fu E, et al. Is high homocysteine level a risk factor for cognitive decline in elderly? A systematic review, meta-analysis, and meta-regression. Am J Geriatr Psychiatry. 2011;19(7):607-617. (PubMed)
172. McCaddon A, Miller JW. Assessing the association between homocysteine and cognition: reflections on Bradford Hill, meta-analyses, and causality. Nutr Rev. 2015;73(10):723-735. (PubMed)
173. Smith AD, Refsum H. Homocysteine, B vitamins, and cognitive impairment. Annu Rev Nutr. 2016;36:211-239. (PubMed)
174. Smith AD, Refsum H, Bottiglieri T, et al. Homocysteine and dementia: an International Consensus Statement. J Alzheimers Dis. 2018;62(2):561-570. (PubMed)
175. Rutjes AW, Denton DA, Di Nisio M, et al. Vitamin and mineral supplementation for maintaining cognitive function in cognitively healthy people in mid and late life. Cochrane Database Syst Rev. 2018;12(12):CD011906. (PubMed)
176. Wang Z, Zhu W, Xing Y, Jia J, Tang Y. B vitamins and prevention of cognitive decline and incident dementia: a systematic review and meta-analysis. Nutr Rev. 2022;80(4):931-949. (PubMed)
177. Smith AD, Smith SM, de Jager CA, et al. Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: a randomized controlled trial. PLoS One. 2010;5(9):e12244. (PubMed)
178. Douaud G, Refsum H, de Jager CA, et al. Preventing Alzheimer's disease-related gray matter atrophy by B-vitamin treatment. Proc Natl Acad Sci U S A. 2013;110(23):9523-9528. (PubMed)
179. Ma F, Wu T, Zhao J, et al. Folic acid supplementation improves cognitive function by reducing the levels of peripheral inflammatory cytokines in elderly Chinese subjects with MCI. Sci Rep. 2016;6:37486. (PubMed)
180. Ma F, Wu T, Zhao J, et al. Effects of 6-month folic acid supplementation on cognitive function and blood biomarkers in mild cognitive impairment: a randomized controlled trial in China. J Gerontol A Biol Sci Med Sci. 2016;71(10):1376-1383. (PubMed)
181. Ma F, Li Q, Zhou X, et al. Effects of folic acid supplementation on cognitive function and Abeta-related biomarkers in mild cognitive impairment: a randomized controlled trial. Eur J Nutr. 2019;58(1):345-356. (PubMed)
182. Watkins D, Rosenblatt DS. Update and new concepts in vitamin responsive disorders of folate transport and metabolism. J Inherit Metab Dis. 2012;35(4):665-670. (PubMed)
183. Zhao R, Min SH, Qiu A, et al. The spectrum of mutations in the PCFT gene, coding for an intestinal folate transporter, that are the basis for hereditary folate malabsorption. Blood. 2007;110(4):1147-1152. (PubMed)
184. Borzutzky A, Crompton B, Bergmann AK, et al. Reversible severe combined immunodeficiency phenotype secondary to a mutation of the proton-coupled folate transporter. Clin Immunol. 2009;133(3):287-294. (PubMed)
185. Goldman ID. Hereditary folate malabsorption. In: Adam MP, Mirzaa GM, Pagon RA, et al., eds. GeneReviews((R)). Seattle (WA); 1993. (PubMed)
186. Sofer Y, Harel L, Sharkia M, Amir J, Schoenfeld T, Straussberg R. Neurological manifestations of folate transport defect: case report and review of the literature. J Child Neurol. 2007;22(6):783-786. (PubMed)
187. Diop-Bove N, Kronn D, Goldman ID. Hereditary folate malabsorption. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, eds. GeneReviews™ [Internet]. Seattle, WA: University of Washington, Seattle; 2008. (PubMed)
188. Lubout CMA, Goorden SMI, van den Hurk K, et al. Successful treatment of hereditary folate malabsorption with intramuscular folinic acid. Pediatr Neurol. 2020;102:62-66. (PubMed)
189. Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: 229050: 7/9/2016: . World Wide Web URL: https://www.omim.org/entry/229050.
190. Frye RE, Sequeira JM, Quadros EV, James SJ, Rossignol DA. Cerebral folate receptor autoantibodies in autism spectrum disorder. Mol Psychiatry. 2013;18(3):369-381. (PubMed)
191. Grapp M, Just IA, Linnankivi T, et al. Molecular characterization of folate receptor 1 mutations delineates cerebral folate transport deficiency. Brain. 2012;135(Pt 7):2022-2031. (PubMed)
192. Kanmaz S, Simsek E, Yilmaz S, Durmaz A, Serin HM, Gokben S. Cerebral folate transporter deficiency: a potentially treatable neurometabolic disorder. Acta Neurol Belg. 2023;123(1):121-127. (PubMed)
193. Ramaekers VT, Quadros EV. Cerebral folate deficiency syndrome: early diagnosis, intervention and treatment strategies. Nutrients. 2022;14(15):3096. (PubMed)
194. Masingue M, Benoist JF, Roze E, et al. Cerebral folate deficiency in adults: A heterogeneous potentially treatable condition. J Neurol Sci. 2019;396:112-118. (PubMed)
195. Ramaekers VT, Blau N, Sequeira JM, Nassogne MC, Quadros EV. Folate receptor autoimmunity and cerebral folate deficiency in low-functioning autism with neurological deficits. Neuropediatrics. 2007;38(6):276-281. (PubMed)
196. Ramaekers VT, Hausler M, Opladen T, Heimann G, Blau N. Psychomotor retardation, spastic paraplegia, cerebellar ataxia and dyskinesia associated with low 5-methyltetrahydrofolate in cerebrospinal fluid: a novel neurometabolic condition responding to folinic acid substitution. Neuropediatrics. 2002;33(6):301-308. (PubMed)
197. Banka S, Blom HJ, Walter J, et al. Identification and characterization of an inborn error of metabolism caused by dihydrofolate reductase deficiency. Am J Hum Genet. 2011;88(2):216-225. (PubMed)
198. Cario H, Smith DE, Blom H, et al. Dihydrofolate reductase deficiency due to a homozygous DHFR mutation causes megaloblastic anemia and cerebral folate deficiency leading to severe neurologic disease. Am J Hum Genet. 2011;88(2):226-231. (PubMed)
199. US Food & Drug Admininstration. FDA News Release. FDA approves folic acid fortification of corn masa flour. April 14, 2016. Available at: https://www.fda.gov/news-events/press-announcements/fda-approves-folic-acid-fortification-corn-masa-flour. Accessed 7/17/2023.
200. Folate. In: Hendler SS, Rorvik, D.R., ed. PDR for Nutritional Supplements. 2nd ed. Montvale: Physicians' Desk Reference Inc.; 2008.
201. US Department of Health and Human Services, National Institutes of Health, Office of Dietary Supplements. Dietary Supplement Label Database (DSLD). [Internet]. [cited 7/17/2023]. Available from: https://dsld.nlm.nih.gov/dsld/.
202. Wiesinger H, Eydeler U, Richard F, et al. Bioequivalence evaluation of a folate-supplemented oral contraceptive containing ethinylestradiol/drospirenone/levomefolate calcium versus ethinylestradiol/drospirenone and levomefolate calcium alone. Clin Drug Investig. 2012;32(10):673-684. (PubMed)
203. Tinker SC, Cogswell ME, Devine O, Berry RJ. Folic acid intake among U.S. women aged 15-44 years, National Health and Nutrition Examination Survey, 2003-2006. Am J Prev Med. 2010;38(5):534-542. (PubMed)
204. Kelly P, McPartlin J, Goggins M, Weir DG, Scott JM. Unmetabolized folic acid in serum: acute studies in subjects consuming fortified food and supplements. Am J Clin Nutr. 1997;65(6):1790-1795. (PubMed)
205. Pfeiffer CM, Sternberg MR, Fazili Z, et al. Unmetabolized folic acid is detected in nearly all serum samples from US children, adolescents, and adults. J Nutr. 2015;145(3):520-531. (PubMed)
206. Murphy MSQ, Muldoon KA, Sheyholislami H, et al. Impact of high-dose folic acid supplementation in pregnancy on biomarkers of folate status and 1-carbon metabolism: An ancillary study of the Folic Acid Clinical Trial (FACT). Am J Clin Nutr. 2021;113(5):1361-1371. (PubMed)
207. Pentieva K, Selhub J, Paul L, et al. Evidence from a randomized trial that exposure to supplemental folic acid at recommended levels during pregnancy does not lead to increased unmetabolized folic acid concentrations in maternal or cord blood. J Nutr. 2016;146(3):494-500. (PubMed)
208. Morris MS, Jacques PF, Rosenberg IH, Selhub J. Folate and vitamin B-12 status in relation to anemia, macrocytosis, and cognitive impairment in older Americans in the age of folic acid fortification. Am J Clin Nutr. 2007;85(1):193-200. (PubMed)
209. Morris MS, Jacques PF, Rosenberg IH, Selhub J. Circulating unmetabolized folic acid and 5-methyltetrahydrofolate in relation to anemia, macrocytosis, and cognitive test performance in American seniors. Am J Clin Nutr. 2010;91(6):1733-1744. (PubMed)
210. Troen AM, Mitchell B, Sorensen B, et al. Unmetabolized folic acid in plasma is associated with reduced natural killer cell cytotoxicity among postmenopausal women. J Nutr. 2006;136(1):189-194. (PubMed)
211. Best KP, Green TJ, Sulistyoningrum DC, et al. Maternal late-pregnancy serum unmetabolized folic acid concentrations are not associated with infant allergic disease: a prospective cohort study. J Nutr. 2021;151(6):1553-1560. (PubMed)
212. Husebye ESN, Wendel AWK, Gilhus NE, Riedel B, Bjork MH. Plasma unmetabolized folic acid in pregnancy and risk of autistic traits and language impairment in antiseizure medication-exposed children of women with epilepsy. Am J Clin Nutr. 2022;115(5):1432-1440. (PubMed)
213. Tam C, O'Connor D, Koren G. Circulating unmetabolized folic acid: relationship to folate status and effect of supplementation. Obstet Gynecol Int. 2012;2012:485179. (PubMed)
214. Apeland T, Mansoor MA, Strandjord RE. Antiepileptic drugs as independent predictors of plasma total homocysteine levels. Epilepsy Res. 2001;47(1-2):27-35. (PubMed)
215. Wilson SM, Bivins BN, Russell KA, Bailey LB. Oral contraceptive use: impact on folate, vitamin B(6), and vitamin B(1)(2) status. Nutr Rev. 2011;69(10):572-583. (PubMed)
Niacin
Contents
Summary
- Dietary precursors of nicotinamide adenine dinucleotide (NAD), including nicotinic acid, nicotinamide, and nicotinamide riboside, are collectively referred to as niacin or vitamin B3. The essential amino acid tryptophan can also be converted into NAD via the kynurenine pathway. (More information)
- NAD can be phosphorylated (NADP) and reduced (NADH and NADPH). NAD functions in oxidation-reduction (redox) reactions and non-redox reactions. (More information)
- Pellagra is the disease of severe niacin deficiency. It is characterized by symptoms affecting the skin, the digestive system, and the nervous system; pellagra can lead to death if left untreated. (More information)
- Causes of niacin deficiency include inadequate oral intake, poor bioavailability from unlimed grains, defective tryptophan absorption, metabolic disorders, and the long-term use of chemotherapeutic treatments. (More information)
- Dietary intake requirements for niacin are based on the urinary excretion of niacin metabolites. (More information)
- NAD is the sole substrate for PARP enzymes and sirtuins involved in DNA repair activities; thus, NAD is critical for genome stability. Several studies, mostly using in vitro and animal models, suggest a possible role for niacin in cancer prevention. In a recent phase III trial, a daily pharmacologic dose of nicotinamide was found to reduce the rate of premalignant skin lesions and nonmelanoma cancers in high-risk subjects. (More information)
- Despite promising initial results, nicotinamide administration has failed to prevent or delay the onset of type 1 diabetes mellitus in high-risk relatives of type 1 diabetic patients. Future research might explore the use of nicotinamide in combined therapy and evaluate activators of NAD-dependent enzymes. (More information)
- At pharmacologic doses, nicotinic acid improved lipid profiles of patients with a history of vascular disease yet failed to reduce recurrent cardiovascular events or mortality. (More information)
- Elevated tryptophan breakdown in the kynurenine pathway and niacin deficiency have been reported in HIV-positive people. However, at present, a better understanding of the role of kynurenine pathway and other NAD biosynthetic pathways during HIV infection is needed to establish whether this population could benefit from niacin supplementation. (More information)
- Most adverse effects of niacin (nicotinic acid and nicotinamide) have been reported with pharmacological doses of nicotinic acid. The tolerable upper intake level (UL) for niacin is based on preventing skin flushing, nicotinic acid’s most prominent side effect. The co-administration of laropiprant — a prostaglandin D2 receptor-1 antagonist — helps reduce nicotinic acid-induced skin flushing. (More information)
Niacin or vitamin B3 is a water-soluble vitamin used by the body to form the nicotinamide coenzyme, NAD+. The term ‘niacin’ is often used to refer to nicotinic acid (pyridine-3-carboxylic acid) only, although other vitamers with a pyridine ring, including nicotinamide (pyridine-3-carboxamide) and nicotinamide riboside, also contribute to NAD+ formation (1). None of the vitamers are related to the nicotine found in tobacco, although their names are similar. Likewise, nicotine — but not nicotinic acid — is an agonist of the nicotinic receptors that respond to the neurotransmitter, acetylcholine.
Metabolism
Essential to all forms of life, the nicotinamide coenzyme NAD+ is synthesized in the body from four precursors that are provided in the diet: nicotinic acid, nicotinamide, nicotinamide riboside, and tryptophan (Figure 1).
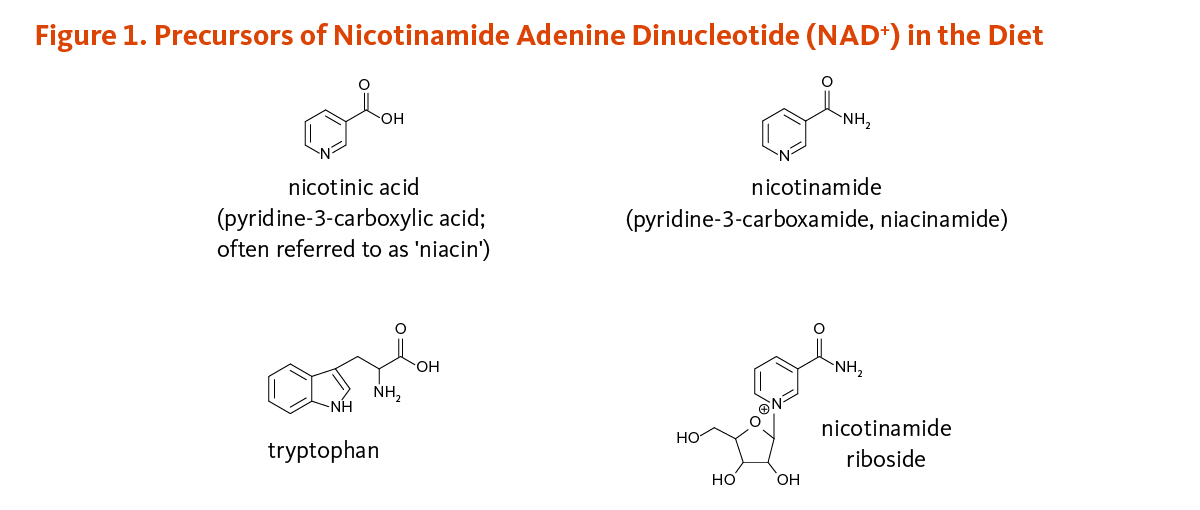
Figure 2 illustrates the separate biosynthetic pathways that lead to NAD+ production from the various dietary precursors. NAD+ is synthesized from nicotinamide and nicotinamide riboside via two enzymatic reactions, while the pathway that yields NAD+ from nicotinic acid - known as the Preiss-Handler pathway — includes three steps. The kynurenine pathway is the longest NAD+ biosynthetic pathway: the catabolism of tryptophan through kynurenine produces quinolinic acid, which is then converted to nicotinic acid mononucleotide, an intermediate in NAD+ metabolism. NAD+ is then synthesized from nicotinic acid mononucleotide in the Preiss-Handler pathway (2).
All pathways generate intermediary mononucleotides — either nicotinic acid mononucleotide or nicotinamide mononucleotide. Specific enzymes, known as phosphoribosyltransferases, catalyze the addition of a phosphoribose moiety onto nicotinic acid or quinolinic acid to produce nicotinic acid mononucleotide or onto nicotinamide to generate nicotinamide mononucleotide. Nicotinamide mononucleotide is also generated by the phosphorylation of nicotinamide riboside, catalyzed by nicotinamide riboside kinases (NRKs). Further, adenylyltransferases catalyze the adenylation of these mononucleotides to form either nicotinic acid adenine dinucleotide or NAD+. Nicotinic acid adenine dinucleotide is then converted to NAD+ by glutamine-dependent NAD+ synthetase (NADSYN), which uses glutamine as an amide group donor (Figure 2) (2). Of note, nicotinic acid adenine dinucleotide has been reported to form following the administration of high-dose nicotinamide riboside, suggesting that a potential deamidation could occur to convert NAD+ to nicotinic acid adenine dinucleotide when the pool of NAD+ is high (1).
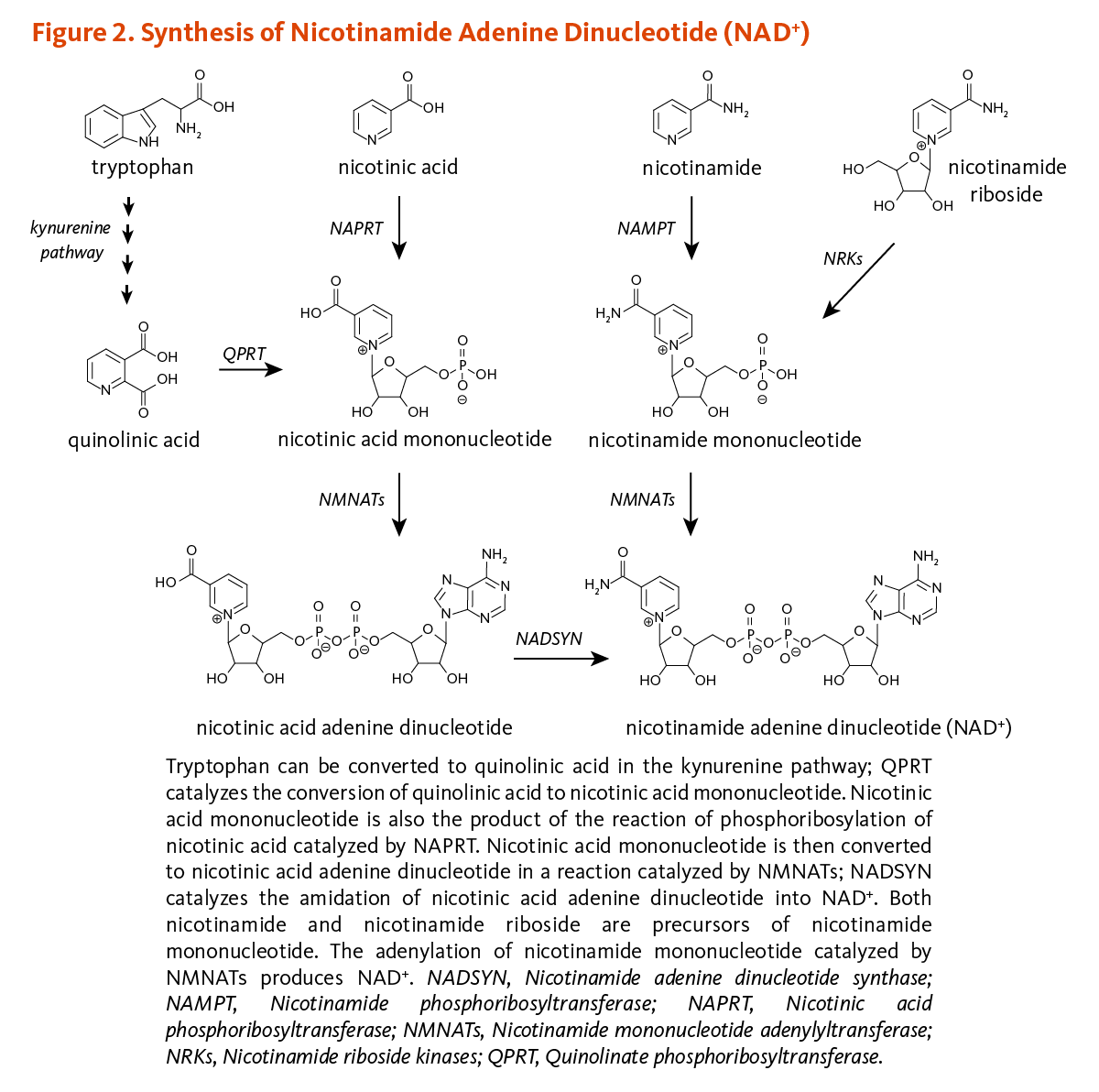
NAD kinase catalyzes the phosphorylation of NAD into NADP using adenosyl triphosphate (ATP) as the phosphoryl donor (3). The oxidation-reduction (redox) properties of the dinucleotide are not affected by the phosphorylation such that the redox pairs NAD+/NADH and NADP+/NADPH show similar redox potentials (4). Oxidation and reduction of the C-4 position of the nicotinamide moiety of NADand its phosphorylated form are essential for electron-transfer reactions supporting vital metabolic and bioenergetic functions in all cells (see Function). Thus, NAD and NADP are recycled back and forth between oxidized (NAD+ and NADP+) and reduced forms (NADH and NADPH), as shown in Figure 3.
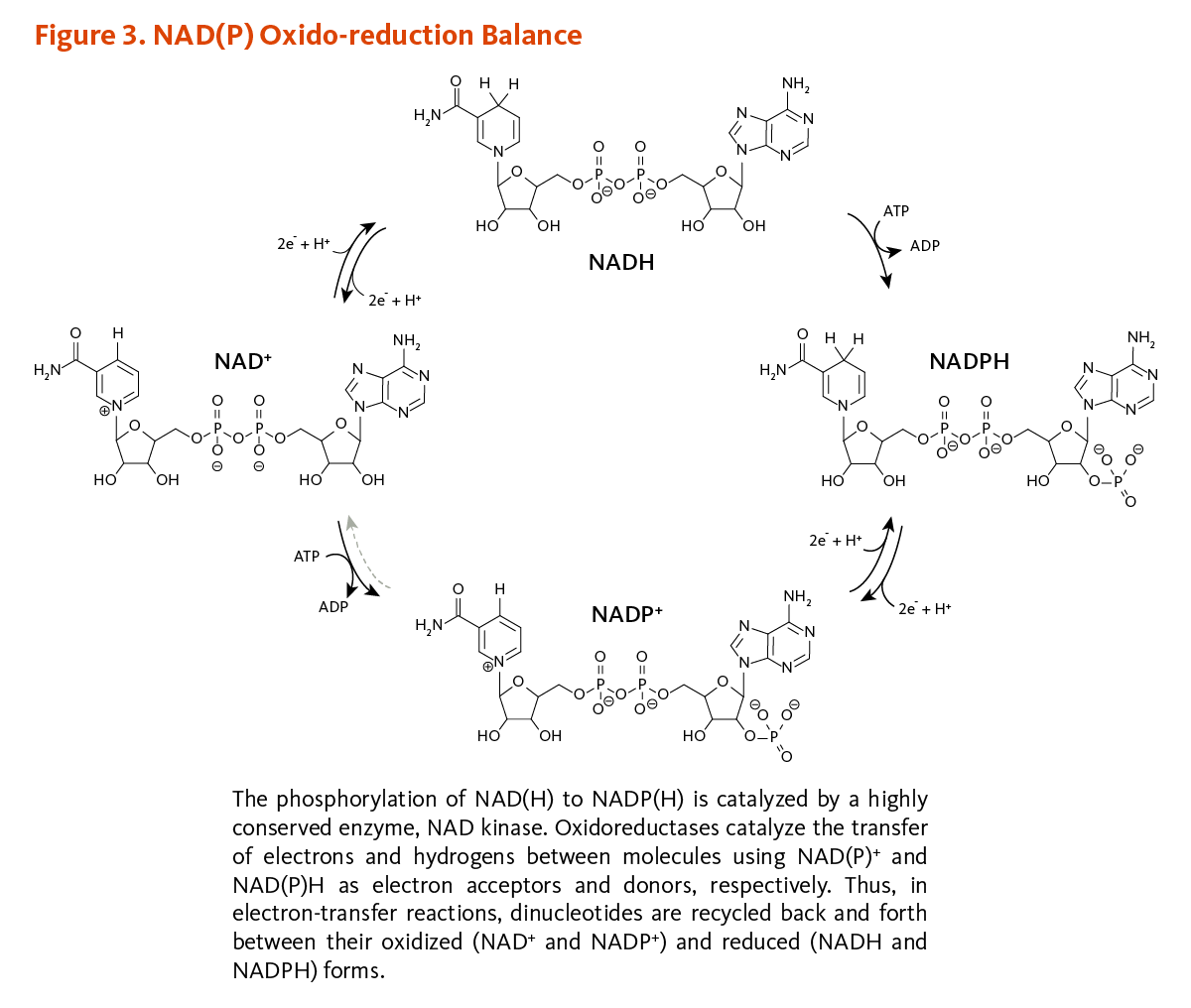
Function
NAD as a coenzyme in electron-transfer reactions
Living organisms derive most of their energy from redox reactions, which are processes involving the transfer of electrons. Over 400 enzymes require the niacin coenzymes, NAD and NADP, mainly to accept or donate electrons for redox reactions (5). NAD and NADP appear to support distinct functions (Figure 4). NAD functions most often in energy-producing reactions involving the degradation (catabolism) of carbohydrates, fats, proteins, and alcohol. NADP generally serves in biosynthetic (anabolic) reactions, such as in the synthesis of fatty acids, steroids (e.g., cholesterol, bile acids, and steroid hormones), and building blocks of other macromolecules (4). NADP is also essential for the regeneration of components of detoxification and antioxidant systems (4). To support these functions, the cell maintains NAD in a largely oxidized state (NAD+) to serve as oxidizing agent for catabolic reactions, while NADP is kept largely in a reduced state (NADPH) to readily donate electrons for reductive cellular processes (4, 6).
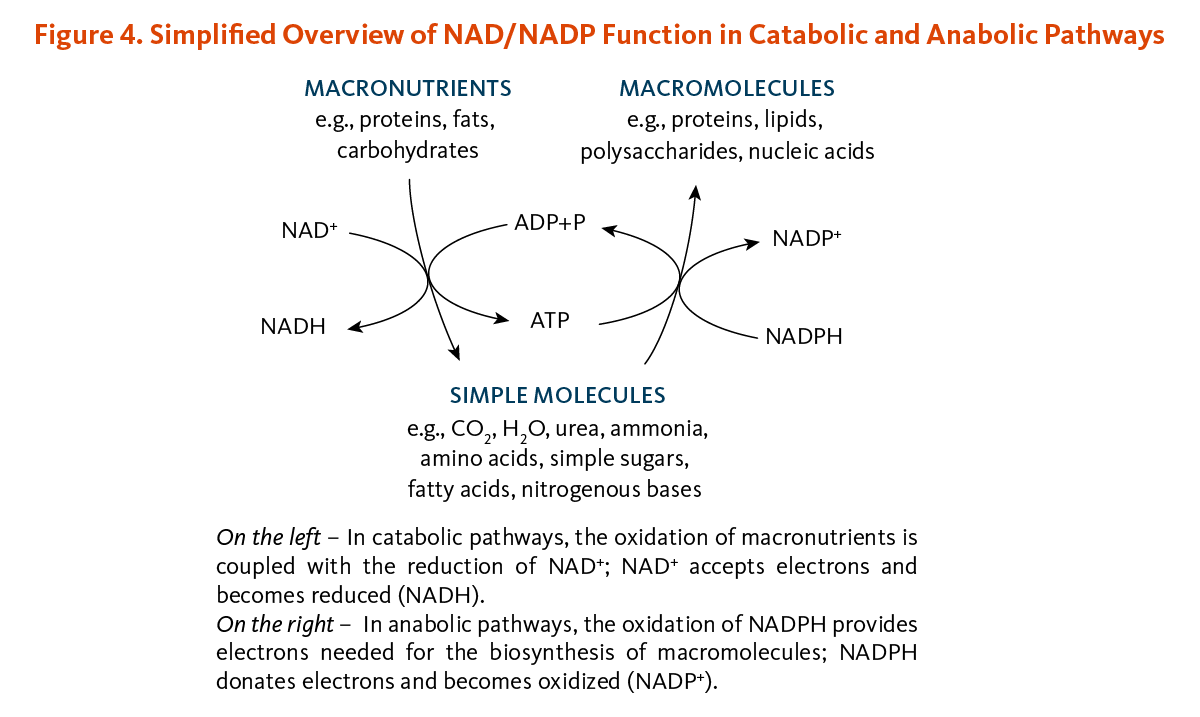
NAD as a substrate for NAD-consuming enzymes
The niacin coenzyme, NAD, is the substrate (reactant) for at least four classes of enzymes. Two classes of enzymes with mono adenosine diphosphate (ADP)-ribosyltransferase and/or poly (ADP-ribose) polymerase activities catalyze ADP-ribosyl transfer reactions. Silent information regulator-2 (Sir2)-like proteins (sirtuins) catalyze the removal of acetyl groups from acetylated proteins, utilizing ADP-ribose from NAD as an acceptor for acetyl groups. Finally, ADP-ribosylcyclases are involved in the regulation of intracellular calcium signaling.
ADP-ribosylation
Enzymes with ADP-ribosyltransferase activities were formerly divided between mono ADP-ribosyltransferases (ARTs) and poly (ADP-ribose) polymerases (PARPs). ARTs were first discovered in certain pathogenic bacteria — like those causing cholera or diphtheria — where they mediate the actions of toxins. These enzymes transfer an ADP-ribose residue moiety from NAD to a specific amino acid of a target protein, with the creation of an ADP-ribosylated protein and the release of nicotinamide.
Because most PARPs have been found to exhibit only mono ADP-ribosyltransferase activities, a new nomenclature was proposed for enzymes catalyzing ADP-ribosylation: A family of mono ADP-ribosyltransferases with homology to bacterial diphteria toxins was named ARTD, while enzymes with either mono or poly ADP-ribosyltransferase activities and related to C2 and C3 clostridial toxins were included in the ARTC family (7, 8).
- ARTCs are extracellular enzymes that catalyze the mono ADP-ribosylation of membrane or secreted proteins involved in innate immunity and cell communication (2).
- ARTDs are intracellular enzymes with either mono or poly ADP-ribosyltransferase activities. At least 18 ARTDs have been identified. All ARTDs possess a diphtheria toxin-like catalytic domain that binds NAD+. Only ARTDs 1, 2, 5, and 6 catalyze poly (ADP-ribose) transfers; the others have mono ADP-ribosyltransferase activities. ARTDs were shown to be involved in DNA repair and stress responses, cell signaling, transcription regulation, apoptosis, cell differentiation, maintenance of genomic integrity, and antiviral defense (reviewed in 8).
NAD-dependent deacetylation
Seven sirtuins (SIRT 1-7) have been identified in humans. Sirtuins are a class of NAD-dependent deacetylase enzymes that remove acetyl groups from the acetylated lysine residues of target proteins. During the deacetylation process, the acetyl group is transferred onto the ADP-ribose moiety cleaved off NAD, producing O-acetyl-ADP-ribose. Nicotinamide can exert feedback inhibition to the deacetylation reaction (9). Like ADP-ribosylation, acetylation is a post-translational modification that affects the function of target proteins. The initial interest in sirtuins followed the discovery that their activation could mimic caloric restriction, which has been shown to increase lifespan in lower organisms. Such a role in mammals is controversial, although sirtuins are energy-sensing regulators involved in signaling pathways that could play important roles in delaying the onset of age-related diseases (e.g., cardiovascular disease, cancer, dementia, arthritis). To date, the spectrum of their biological functions includes gene silencing, DNA damage repair, cell cycle regulation, and cell differentiation (10).
Calcium mobilization
In humans, CD38 and CD157 belong to a family of NAD+ glycohydrolases/ADP-ribosylcyclases. These enzymes catalyze the formation of key regulators of calcium signaling, namely (linear) ADP-ribose, cyclic ADP-ribose, and nicotinic acid adenine dinucleotide phosphate (Figure 5). Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate works within cells to provoke the release of calcium ions from internal storage sites (i.e., endoplasmic reticulum, lysosomes, mitochondria), whereas ADP-ribose stimulates extracellular calcium entry through cell membrane TRPM2 cation channels (2). Another TRPM2 agonist, 2’-deoxy-ADP-ribose, was recently identified in vitro. CD38 was found to catalyze the synthesis of 2’-deoxy-ADP-ribose from nicotinamide mononucleotide and 2’-deoxy-ATP (11). O-acetyl-ADP-ribose generated by the activity of sirtuins also controls calcium entry through TRPM2 channels (6). Intracellular calcium-mediated signal transduction is regulated by transient calcium entry into the cell or release of calcium from intracellular stores. Calcium signaling is critically involved in processes like neurotransmission, insulin release from pancreatic β-cells, muscle cell contraction, and T-lymphocyte activation (6).
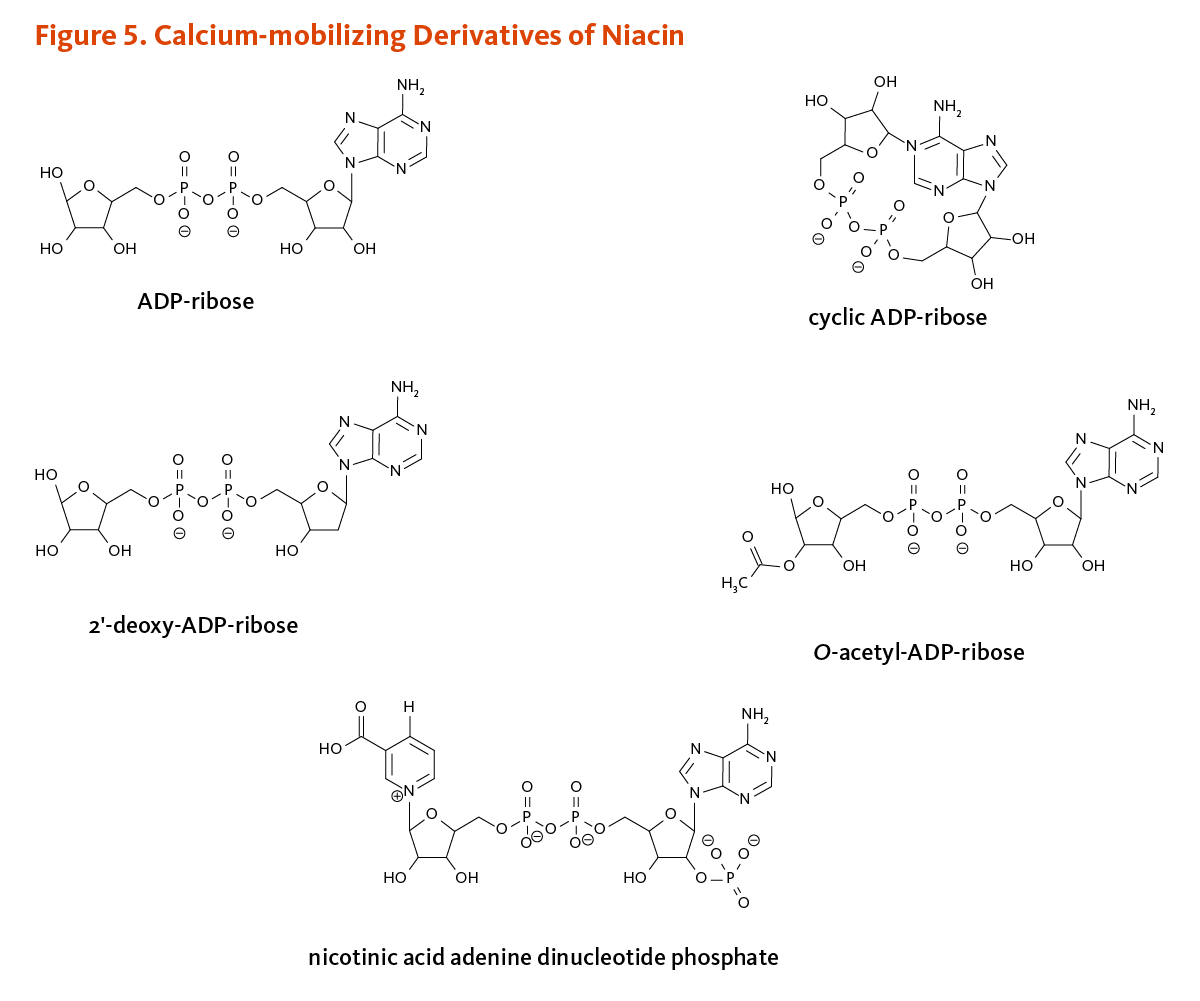
NAD as a ligand
NAD+ has been identified as an endogenous agonist of purinergic membrane receptors of the P2Y subclass. In particular, NAD was found to bind to P2Y1 receptor and act as an inhibitory neurotransmitter at neuromuscular junctions in visceral smooth muscles (12). Extracellular NAD+ was also found to behave like a proinflammatory cytokine, triggering the activation of isolated granulocytes. NAD+ binding to the P2Y11 receptor at the granulocyte surface activated a signaling cascade involving cyclic ADP-ribose and the rise of intracellular calcium, eventually stimulating superoxide generation and chemotaxis (13). Similar observations were made with lipopolysaccharide-activated monocytes (14). Extracellular NAADP+ and ADP-ribose might also bind to P2Y receptors and trigger intracellular NAADP+- and ADP-ribose-dependent calcium mobilization (see Calcium mobilization) (15, 16).
Lipid-lowering effects with pharmacologic doses of nicotinic acid
For over half a century, pharmacologic doses of nicotinic acid, but not nicotinamide, have been known to reduce serum cholesterol (see Disease Treatment) (17). However, the exact mechanisms underlying the lipid-lowering effect of nicotinic acid remain speculative. Two G-protein-coupled membrane receptors, GPR109A and GPR109B, bind nicotinic acid with high and low affinity, respectively. These nicotinic acid receptors are primarily expressed in adipose tissue and immune cells (but not lymphocytes). They are also found in retinal pigmented and colonic epithelial cells, keratinocytes, breast cells, microglia, and possibly at low levels in the liver (18). Thus, lipid-modifying effects of nicotinic acid are likely to be mediated by receptor-independent mechanisms in major tissues of lipid metabolism like liver and skeletal muscle. Early in vitro data suggested that nicotinic acid could impair very-low-density lipoprotein (VLDL) secretion by inhibiting triglyceride synthesis and triggering ApoB lipoprotein degradation in hepatocytes (19). In another study, nicotinic acid affected the hepatic uptake of ApoAI lipoprotein, thereby reducing high-density lipoprotein (HDL) removal from the circulation (reviewed in 20). In adipocytes, the binding of nicotinic acid to GPR109A was found to initiate a signal transduction cascade resulting in reductions in free fatty acid production via the inhibition of hormone-sensitive lipase involved in triglyceride lipolysis (21). Nonetheless, recent observations have suggested that the lipid-lowering effect of nicotinic acid was not due to its anti-lipolytic activity (22). Trials showed that synthetic agonists of GPR109A acutely lowered free fatty acids yet failed to affect serum lipids (22). Aside from its impact on HDL and other plasma lipids, nicotinic acid has exhibited anti-atherosclerotic activities in cultured monocytes, macrophages, or vascular endothelial cells, by modulating inflammation and oxidative stress and regulating cell adhesion, migration, and differentiation (reviewed in 18).
Deficiency
Pellagra
Causes
The late stage of severe niacin deficiency is known as pellagra. Early records of pellagra followed the widespread cultivation of corn in Europe in the 1700s (23). The disease is generally associated with poorer social classes whose chief dietary staple consisted of cereal like corn or sorghum. Pellagra was also common in the southern United States during the early 1900s where income was low and corn products were a major dietary staple (24). Interestingly, pellagra was not known in Mexico, where corn was also an important dietary staple and much of the population was also poor. In fact, if corn contains appreciable amounts of niacin, it is present in a bound form that is not nutritionally available to humans. The traditional preparation of corn tortillas in Mexico involves soaking the corn in a lime (calcium oxide) solution, prior to cooking. Heating the corn in an alkaline solution results in the release of bound niacin, increasing its bioavailability (25). Pellagra epidemics were also unknown to Native Americans who consumed immature corn that contains predominantly unbound (bioavailable) niacin (24).
Niacin deficiency or pellagra may result from inadequate dietary intake of NAD precursors, including tryptophan. Niacin deficiency — often associated with malnutrition — is observed in the homeless population, in individuals suffering from anorexia nervosa or obesity, and in consumers of diets high in maize and poor in animal protein (26-29). Deficiencies of other B vitamins and some trace minerals may aggravate niacin deficiency (30, 31). Malabsorptive disorders that can lead to pellagra include Crohn’s disease and megaduodenum (32, 33). Patients with Hartnup’s disease, a hereditary disorder resulting in defective tryptophan absorption, have developed pellagra (see Niacin-responsive genetic disorders). Carcinoid syndrome, a condition of increased secretion of serotonin and other catecholamines by carcinoid tumors, may also result in pellagra due to increased utilization of dietary tryptophan for serotonin rather than niacin synthesis. Further, prolonged treatment with the anti-tuberculosis drug isoniazid has resulted in niacin deficiency (34). Other pharmaceutical agents, including the immunosuppressive drugs azathioprine (Imuran) and 6-mercaptopurine, the anti-cancer drug 5-fluorouracil (5-FU, Adrucil), and levodopa/carbidopa (Sinemet; two drugs given to people with Parkinson’s disease), are known to increase the reliance on dietary niacin by interfering with the tryptophan-kynurenine-niacin pathway (35). Finally, other populations at risk for niacin deficiency include dialysis patients, cancer patients (36, 37), individuals suffering from chronic alcoholism (38), and people with HIV (see HIV/AIDS below). Further, chronic alcohol intake can lead to severe niacin deficiency through reducing dietary niacin intake and interfering with the tryptophan-to-NAD conversion (30).
Symptoms of deficiency
The most common symptoms of niacin deficiency involve the skin, the digestive system, and the nervous system. The symptoms of pellagra are commonly referred to as the three "Ds": sun-sensitive dermatitis, diarrhea, and dementia. A fourth "D," death, occurs if pellagra is left untreated (5). In the skin, a thick, scaly, darkly pigmented rash develops symmetrically in areas exposed to sunlight. In fact, the word "pellagra" comes from "pelle agra," the Italian phrase for rough skin. Symptoms related to the digestive system include inflammation of the mouth and tongue ("bright red tongue"), vomiting, constipation, abdominal pain, and ultimately, diarrhea. Gastrointestinal disorders and diarrhea contribute to the ongoing malnourishment of the patients. Neurologic symptoms include headache, apathy, fatigue, depression, disorientation, and memory loss and are more consistent with delirium than with the historically described dementia (38). Disease presentations vary in appearance since the classic triad rarely presents in its entirety. The absence of dermatitis, for example, is known as pellagra sine pellagra.
Treatment
To treat pellagra, the World Health Organization (WHO) recommends administering nicotinamide to avoid the flushing commonly caused by nicotinic acid (see Safety). Treatment guidelines suggest using 300 mg/day of oral nicotinamide in divided doses, or 100 mg/day administered parenterally in divided doses, for three to four weeks (37, 39). Because patients with pellagra often display additional vitamin deficiencies, administration of a vitamin B-complex preparation is advised (39).
The Recommended Dietary Allowance (RDA)
Niacin equivalent (NE)
The term "niacin equivalent" (NE) is used to describe the contribution to dietary intake of all the forms of niacin that are available to the body. In healthy individuals, less than 2% of dietary tryptophan is converted to NAD in the kynurenine pathway (40). The synthesis of NAD from tryptophan is fairly inefficient and depends on enzymes requiring vitamin B6 and riboflavin, as well as a heme (iron)-containing enzyme. Nonetheless, tryptophan is essential as a precursor for NAD+. Inherited defects in tryptophan transport and metabolism result in severe clinical disorders attributed to NAD+ depletion (see Niacin-responsive genetic disorders). On average, 60 milligrams (mg) of tryptophan are considered to correspond to 1 mg of niacin or 1 mg of NE.
Daily recommended intakes
The recommended dietary allowance (RDA) for niacin is based on the prevention of deficiency. Pellagra can be prevented by about 11 mg NE/day, but 12 mg to 16 mg NE/day has been found to normalize the urinary excretion of niacin metabolites (breakdown products) in healthy young adults. Because pellagra represents severe deficiency, the Food and Nutrition Board (FNB) of the US Institute of Medicine chose to use the excretion of niacin metabolites as an indicator of niacin nutritional status rather than symptoms of pellagra (41). However, it has been argued that cellular NAD and NADP content may be more relevant indicators of niacin nutritional status (24).
| Life Stage | Age | Males (mg NE*/day) | Females (mg NE/day) |
|---|---|---|---|
| Infants | 0-6 months | 2 (AI) | 2 (AI) |
| Infants | 7-12 months | 4 (AI) | 4 (AI) |
| Children | 1-3 years | 6 | 6 |
| Children | 4-8 years | 8 | 8 |
| Children | 9-13 years | 12 | 12 |
| Adolescents | 14-18 years | 16 | 14 |
| Adults | 19 years and older | 16 | 14 |
| Pregnancy | all ages | - | 18 |
| Breast-feeding | all ages | - | 17 |
| *NE, niacin equivalent: 1 mg NE = 60 mg of tryptophan = 1 mg niacin | |||
Disease Prevention
Cancer
Studies of cultured cells (in vitro) provide evidence that NAD content influences mechanisms that maintain genomic stability. Loss of genomic stability, characterized by a high rate of damage to DNA and chromosomes, is a hallmark of cancer (42). The current understanding is that the pool of NAD is decreased during niacin deficiency and that it affects the activity of NAD-consuming enzymes rather than redox and metabolic functions (43). Among NAD-dependent reactions, poly ADP-ribosylations catalyzed by PARP enzymes (ARTDs) are critical for the cellular response to DNA injury. After DNA damage, PARPs are activated; the subsequent poly ADP-ribosylations of a number of signaling and structural molecules by PARPs were shown to facilitate DNA repair at DNA strand breaks (44). Cellular depletion of NAD has been found to decrease levels of the tumor suppressor protein p53, a target for poly ADP-ribosylation, in human breast, skin, and lung cells (45). The expression of p53 was also altered by niacin deficiency in rat bone marrow cells (46). Impairment of DNA repair caused by niacin deficiency could lead to genomic instability and drive tumor development in rat models (47, 48). Both PARPs and sirtuins have been recently involved in the maintenance of heterochromatin, a chromosomal domain associated with genome stability, as well as in transcriptional gene silencing, telomere integrity, and chromosome segregation during cell division (49, 50). Neither the cellular NAD content nor the dietary intake of NAD precursors necessary for optimizing protective responses following DNA damage has been determined, but both are likely to be higher than that required for the prevention of pellagra.
Bone marrow
Cancer patients often suffer from bone marrow suppression following chemotherapy, given that bone marrow is one of the most proliferative tissues in the body and thus a primary target for chemotherapeutic agents. Niacin deficiency was found to decrease bone marrow NAD and poly-ADP-ribose levels and increase the risk of chemically induced leukemia in rats (51). Conversely, a pharmacologic dose of either nicotinic acid or nicotinamide was able to increase NAD and poly ADP-ribose in bone marrow and decrease the development of leukemia in rats (52). It has been suggested that niacin deficiency often observed in cancer patients could sensitize bone marrow tissue to the suppressive effect of chemotherapy. However, little is known regarding cellular NAD levels and the prevention of DNA damage or cancer in humans. One study in two healthy individuals involved elevating NAD levels in blood lymphocytes by supplementation with 100 mg/day of nicotinic acid for eight weeks. Compared to non-supplemented individuals, the supplemented individuals had reduced DNA strand breaks in lymphocytes exposed to free radicals in a test tube assay (53). However, nicotinic acid supplementation of up to 100 mg/day for 14 weeks in 21 healthy smokers failed to provide any evidence of a decrease in cigarette smoke-induced genetic damage in blood lymphocytes compared to placebo (54). More recently, the frequency of chromosome translocation was used to evaluate DNA damage in peripheral blood lymphocytes of 82 pilots chronically exposed to ionizing radiation, a known human carcinogen. In this observational study, the rate of chromosome aberrations was significantly lower in subjects with higher (28.4 mg/day) compared to lower (20.5 mg/day) dietary niacin intake (55). Higher availability of NAD+ in X-irradiated peripheral blood lymphocytes was found to favor DNA repair by enhancing survival, particularly through SIRT-mediated p53 deacetylation (56).
Upper digestive tract
Generally, relationships between dietary factors and cancer are established first in epidemiological studies and followed up by basic cancer research at the cellular level. In the case of niacin, research on biochemical and cellular aspects of DNA repair has stimulated an interest in the relationship between niacin intake and cancer risk in human populations (57). A large case-control study found increased consumption of niacin, along with antioxidant nutrients, to be associated with decreased incidence of oral (mouth), pharyngeal (throat), and esophageal cancers in northern Italy and Switzerland. An increase in daily niacin intake of 6.2 mg was associated with about a 40% decrease in cases of cancers of the mouth and throat, while a 5.2 mg increase in daily niacin intake was associated with a similar decrease in cases of esophageal cancer (58, 59).
Skin
Niacin deficiency can lead to severe sunlight sensitivity in exposed skin. Given the implication of NAD-dependent enzymes in DNA repair, there has been some interest in the effect of niacin on skin health. In vitro and animal experiments have helped gather information, but human data on niacin/NAD status and skin cancer are very limited. One study reported that niacin supplementation decreased the risk of ultraviolet light (UV)-induced skin cancers in mice, despite the fact that mice convert tryptophan to NAD more efficiently than rats and humans and thus do not get severely deficient (60). Hyper-proliferation and impaired differentiation of skin cells can alter the integrity of the skin barrier and increase the occurrence of pre-malignant and malignant skin conditions. A protective effect of niacin was suggested by topical application of myristyl nicotinate, a niacin derivative, which successfully increased the expression of epidermal differentiation markers in subjects with photodamaged skin (61). The activation of the nicotinic acid receptors, GPR109A and GPR109B, by pharmacologic doses of niacin could be involved in improving skin barrier function. Conversely, differentiation defects in skin cancer cells were linked to the abnormal cellular localization of defective nicotinic acid receptors (62). Nicotinamide restriction with subsequent depletion of cellular NAD was shown to increase oxidative stress-induced DNA damage in a precancerous skin cell model, implying a protective role of NAD-dependent pathways in cancer (63). Altered NAD availability also affects sirtuin expression and activity in UV-exposed human skin cells. Along with PARPs, NAD-consuming sirtuins could play an important role in the cellular response to photodamage and skin homeostasis (64).
A pooled analysis of two large US prospective cohort studies that followed 41,808 men and 72,308 women for up to 26 years suggested that higher versus lower intake of niacin (from diet and supplements) might be protective against squamous-cell carcinoma but not against basal-cell carcinoma and melanoma (65). A phase III, randomized, double-blind, placebo-controlled trial in 386 subjects with a history of nonmelanoma skin cancer recently examined the effect of daily nicotinamide supplementation (1 g) for 12 months on skin cancer recurrence at three-month intervals over an 18-month period (66). Nicotinamide effectively reduced the rate of premalignant actinic keratose (-11%), squamous-cell carcinoma (-30%), and basal-cell carcinoma (-20%) compared to placebo after 12 months, yet this protection was not sustained during the six-month post-supplementation period (66). Larger trials are needed to assess whether nicotinamide could reduce the risk of melanomas, which are not as common as other skin cancer but are more deadly (67).
Type 1 diabetes mellitus
Type 1 diabetes mellitus in children is caused by the autoimmune destruction of insulin-secreting β-cells in the pancreas. Prior to the onset of symptomatic diabetes, specific antibodies, including islet cell autoantibodies (ICA), can be detected in the blood of high-risk individuals (68). In an experimental animal model of diabetes, high levels of nicotinamide are administered to protect β-cells from damage caused by streptozotocin (69).
Yet, pharmacologic doses of nicotinamide (up to 3 g/day) have not been found to be effective in delaying or preventing the onset of type 1 diabetes in at-risk subjects. An analysis of 10 trials, of which five were placebo-controlled, found evidence of improved β-cell function after one year of treatment with nicotinamide, but the analysis failed to find any clinical evidence of improved glycemic control (70). A large, multicenter randomized controlled trial of nicotinamide in ICA-positive siblings (ages, 3-12 years) of type 1 diabetic patients also failed to find a difference in the incidence of type 1 diabetes after three years (70). A randomized, double-blind, placebo-controlled multicenter trial of nicotinamide (maximum of 3 g/day) was conducted in 552 ICA-positive relatives of patients with type 1 diabetes. The proportion of relatives who developed type 1 diabetes within five years was comparable whether they were treated with nicotinamide or placebo (71). Nicotinamide could reduce inflammation-related parameters in these high-risk subjects yet was ineffective to prevent disease onset (72). More recently, case reports of the combined use of nicotinamide (25 mg/kg/day) and acetyl-L-carnitine (50 mg/kg/day) in children at risk for type 1 diabetes showed promising results, warranting further investigation (73).
Disease Treatment
Niacin supplements at pharmacologic doses (i.e., doses much larger than those needed to prevent deficiency) have been used in an attempt to treat a range of conditions, some of which are discussed below.
Niacin-responsive genetic disorders
Congenital NAD deficiency-related disorders can result from mutations in genes involved in the uptake and transport of the various dietary NAD+ precursors or in the distinct metabolic pathways leading to NAD+ production (see Metabolism). Some of these disorders might respond to niacin supplementation. For example, defective transport of tryptophan into cells results in Hartnup disease, which features signs of severe niacin deficiency (74). Hartnup disease is due to mutations in the SLCA19 gene, which codes for a sodium-dependent neutral amino acid transporter expressed primarily in the kidneys and intestine. Disease management involves supplementation with nicotinic acid or nicotinamide (75). Recessive mutations in genes coding for enzymes of the kynurenine pathway — namely kynureninase and 3-hydroxyanthranilic-acid 3,4-dioxygenase — lead to combined vertebral, anal, cardiac, tracheo-esophageal, renal, and limb (VACTERL) congenital malformations (76). Depletion of NAD+, rather than accumulation of intermediate metabolites in the kynurenine pathway, was found to be responsible for these malformations. Niacin supplementation throughout pregnancy ensured adequate levels of NAD+ and prevented congenital anomalies in mice with kynurenine pathway mutations (76). In humans, the dose of NAD+ precursors necessary to avert NAD deficiency-induced congenital VACTERL malformations has yet to be defined (77).
Nicotinamide may also rescue NAD+ depletion secondary to an ultra-rare inborn error of glutamine metabolism (78). Glutamine is required for the conversion of nicotinic acid adenine dinucleotide to NAD+ catalyzed by NAD+ synthetase (Figure 2). Thus, inherited glutamine synthetase deficiency specifically affects the synthesis of NAD+ from the NAD+ precursors, tryptophan and nicotinic acid. If the combined deficiencies of glutamine and NAD+ are responsible for the severe clinical phenotype of subjects with inherited glutamine synthetase deficiency, it is likely that supplementation with both glutamine and nicotinamide would provide some relief (78).
Finally, many inborn errors of metabolism result from genetic mutations decreasing cofactor binding affinity and, subsequently, enzyme efficiency (79). In many cases, the administration of high doses of the vitamins serving as precursors of cofactors can restore enzymatic activity — at least partially — and lessen signs of the genetic diseases (79). Given the large number of enzymes requiring NAD, it is speculated that many of the conditions due to defective enzymes might be rescued by niacin supplementation (5).
Cardiovascular disease
Cardiovascular risk factors
Nicotinic acid is a well-known lipid-lowering agent: Nicotinic acid therapy markedly increases high-density lipoprotein (HDL) cholesterol concentrations, decreases serum lipoprotein(a) concentrations, and shifts small, dense low-density lipoprotein (LDL) particles to large, buoyant LDL particles. All of these changes in the blood lipid profile are considered cardioprotective. Low concentrations of HDL-cholesterol are one major risk factor for coronary heart disease (CHD), and an increase in HDL concentrations is associated with a reduction of that risk (80). Because of the adverse side effects associated with high doses of nicotinic acid (see Safety), nicotinic acid has most often been used in combination with other lipid-lowering medications at slightly lower doses (17). In particular, low-dose nicotinic acid is often co-administered with 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors (statins), the cornerstone of treatment of hyperlipidemia, a major risk factor for CHD. A placebo-controlled study in 39 patients taking statins (cerivastatin, atorvastatin, or simvastatin) found that a very low dose of nicotinic acid (100 mg/day) increased HDL-cholesterol by only 2.1 mg/dL, and the combination had no effect on LDL-cholesterol, total cholesterol, or triglyceride concentrations (81).
The Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2 study — a double-blind, placebo-controlled trial — investigated the incremental effect of adding nicotinic acid (1 g/day) to statin therapy in 167 patients with known CHD and low HDL concentrations on carotid intima-media thickness (CIMT) (82), a surrogate endpoint for the development of atherosclerosis. The addition of extended-release nicotinic acid to simvastatin prevented the increase in CIMT compared to simvastatin monotherapy. A post-hoc analysis of data from ARBITER2 showed that the blockade of atherosclerotic progression was related to the increase in HDL concentrations in patients with normal glycemic status. However, in the presence of additional risk factors, such as impaired fasting glucose or diabetes mellitus, the increase in HDL concentrations was not predictive of CIMT reduction and atherosclerotic retardation (83). A comparative efficacy trial (ARBITER6) also showed a significant reduction of baseline CIMT with extended-release nicotinic acid (2 g/day for 14 months), as opposed to ezetimibe (a cholesterol-lowering drug), in patients taking statins (84).
Additional studies have examined the impact of nicotinic acid on endothelium-dependent brachial flow-mediated dilation (FMD) in patients at risk of CHD or with established CHD. The measurement of FMD is often used as a surrogate marker of endothelial function; FMD values are inversely correlated with the risk of future cardiovascular events (85). A meta-analysis of seven randomized controlled trials, including 441 participants, showed a significant, 2% increase with nicotinic acid (1-2 g/day) administered for 12 weeks to one year (86).
Cardiovascular events
Several randomized, placebo-controlled, multicenter trials have investigated the efficacy and safety of nicotinic acid therapy, alone or in combination with other lipid-lowering agents, on outcomes of cardiovascular disease (CVD). Specifically, the Coronary Drug Project (CDP) followed over 8,000 men with a previous myocardial infarction for six years (87). Compared to the placebo group, patients who took 3 g of immediate-release nicotinic acid daily experienced an average 10% reduction in total blood cholesterol, a 26% decrease in triglycerides, a 27% reduction in recurrent nonfatal myocardial infarction, and a 26% reduction in cerebrovascular events (stroke and transient ischemic attacks). In addition, nine-year follow-up post-trial revealed a 10% reduction in total deaths with nicotinic acid treatment.
The HDL-Atherosclerosis Treatment Study (HATS), a three-year randomized controlled trial in 160 patients with documented CHD and low HDL concentrations, found that a combination of simvastatin and nicotinic acid (2-3 g/day) increased HDL concentrations, inhibited the progression of coronary artery stenosis (narrowing), and decreased the frequency of cardiovascular events, including myocardial infarction and stroke, compared to placebo (88). A subgroup analysis of the HATS patients with metabolic syndrome showed a reduction in rate of primary clinical events even though glucose and insulin metabolism were moderately impaired by nicotinic acid (89). Moreover, a review of nicotinic acid safety and tolerability among the HATS subjects showed glycemic control in diabetic patients returned to pretreatment values following eight months of disease management with medication and diet (90). Similarly, the cardiovascular benefit of long-term nicotinic acid therapy outweighed the modest increase in risk of newly onset type 2 diabetes in patients from the CDP study (91).
In contrast, the AIM-HIGH (Atherothrombosis intervention in metabolic syndrome with low HDL/high triglycerides: impact on global health outcomes) trial, which examined the incremental effect of extended-release nicotinic acid (1.5-2 g/day) on 3,414 patients who had established CVD and atherogenic dyslipidemia and were treated with simvastatin (+/- ezetimibe), provided disappointing results. Indeed, in these patients who had achieved target concentrations of LDL-cholesterol (<70 mg/dL) before randomization, the HDL-raising effect of nicotinic acid treatment failed to reduce the number of cardiovascular events after a mean follow-up of three years (92, 93). While some limitations, like a greater use of simvastatin and ezetimibe in the control group, may have confounded the results, it was also suggested that low HDL-cholesterol might be a marker of risk rather than a causal risk factor for predicting CVD (93). In addition, a post-hoc analysis of 505 participants with stage 3 chronic kidney disease found an increase in all-cause mortality in those randomized to nicotinic acid compared to those in the placebo group (94).
Although nicotinic acid failed to reduce the number of cardiovascular events in simvastatin-treated patients with low LDL-cholesterol, these results cannot be extrapolated to patients with higher LDL-cholesterol at baseline. A much larger multicenter, randomized, double-blind, placebo-controlled trial — the HPS2-THRIVE (Heart protection study 2: treatment of HDL to reduce the incidence of vascular events) trial — in 25,673 participants with vascular disease examined the incremental effect of extended-release nicotinic acid (2 g/day) and laropiprant (a prostaglandin D2 receptor-1 antagonist; 40 mg/day) on the incidence of vascular events. Compared to placebo, nicotinic acid/laropiprant reduced LDL-cholesterol by an average of 10 mg/dL, decreased triglycerides by 33 mg/dL, and increased HDL-cholesterol by 6 mg/dL after a median 3.9-year follow-up period. Nonetheless, nicotinic acid/laropiprant showed no effect on the incidence of major vascular events and death of any cause (95).
A recent meta-analysis of 23 randomized controlled trials — including the CDP, AIM-HIGH, and HPS2-THRIVE trials — in 39,195 subjects with a history of vascular disease compared the effect of nicotinic acid alone or as an add-on to other lipid-lowering agents. No cardiovascular benefits were associated with nicotinic acid therapy: the number of fatal and non-fatal myocardial infarctions and strokes was not decreased with nicotinic acid supplementation (median dose of 2 g/day for a median period of 11.5 months) (96).
Despite the lack of evidence for a role of nicotinic acid in CVD prevention (96, 97), the use of nicotinic acid therapy has rapidly increased over the years in the US (98).
Friedreich’s ataxia
Friedreich’s ataxia, a common form of inherited ataxia, is an early onset recessive disorder with clinical features that includes progressive ataxia, scoliosis, dysarthria, cardiomyopathy, and diabetes mellitus (99). Most affected subjects carry homozygous guanine-adenine-adenine (GAA) repeat expansions in the first intron of the gene FXN coding for the protein frataxin. These abnormal and unstable GAA repeats trigger gene silencing through heterochromatin formation, leading to significantly reduced frataxin expression (100). Frataxin is a mitochondrial protein needed for the making of iron-sulfur clusters (ISC). ISC-containing subunits are especially important for the mitochondrial respiratory chain and for the synthesis of heme-containing proteins (99).
Predominantly localized in the nucleus, SIRT1 is a NAD+-dependent deacetylase that promotes gene silencing through heterochromatin formation. Nicotinamide has been shown to antagonize heterochromatization of the FXN locus and upregulate frataxin expression in lymphoblastoid cells derived from Friedreich’s ataxia-affected patients, possibly through inhibiting SIRT1 activity (100). In an open-label, dose escalating pilot trial in 10 adult patients with Friedreich’s ataxia, single and repeated doses of nicotinamide (2-8 g) for up to eight weeks were found to be well tolerated (101). Repeated daily doses of 3.5 to 6 g of nicotinamide led to significant increases in frataxin concentration in peripheral white blood cells (101). Yet, no neurological improvements were reported, suggesting that the duration of the treatment was too short and/or the nervous system of the participants was unresponsive to increases in frataxin (102). To our knowledge, there is currently no ongoing trial designed to further investigate the effect of nicotinamide in Friedreich’s ataxia-affected patients.
HIV/AIDS
The first step in the kynurenine pathway is catalyzed by the extrahepatic enzyme, indoleamine 2,3-dioxygenase (IDO), which is responsible for the oxidative cleavage of tryptophan. The chronic stimulation of tryptophan oxidation, mediated by an increased activity of IDO and/or inadequate dietary niacin intakes, is observed with the infection of human immunodeficiency virus (HIV), the virus that causes acquired immunodeficiency syndrome (AIDS). Interferon-gamma (IFN-γ) is a cytokine produced by cells of the immune system in response to infection. Through stimulating the enzyme IDO, IFN-γ increases the breakdown of tryptophan, thus supporting the finding that the average tryptophan concentration in blood is significantly lower in HIV patients compared to uninfected subjects (40). An increased degradation of tryptophan via the kynurenine pathway appears to coexist with intracellular niacin/NAD deficiency in HIV infection (103). An explanatory model for these paradoxical observations incriminates the oxidative stress induced by multiple nutrient deficiencies in HIV patients (103). In particular, the activation of PARP enzymes (ARTDs) by oxidative damage to DNA could be responsible for inducing niacin/NAD depletion (see Function). The breakdown of tryptophan would then be a compensatory response to inadequate niacin/NAD levels.
However, metabolites derived from the oxidation of tryptophan in the kynurenine pathway regulate specific T-lymphocyte subgroups. As mentioned above, circulating IFN-γ, but also viral and bacterial products, can activate IDO during HIV infection. The overstimulation of the tryptophan pathway has been involved in the loss of normal T-lymphocyte function, which characterizes HIV infection (104, 105). The increased IDO activity has been linked to the altered immune response that contributes to the persistence of HIV (104). Antiretroviral therapy (ART) only partially restores normal IDO activity, without normalizing it, yet induces viral suppression and CD4 T-cell recovery (106). In a monkey model for HIV infection, a partial and transient blockade of IDO with IDO inhibitor 1-methyl tryptophan proved ineffective to reduce the viral load in plasma and intestinal tissues beyond the level achieved by ART (107). At present, a better understanding of the role of kynurenine pathway and other NAD biosynthetic pathways during HIV infection is needed before the relevance and clinical implications of niacin supplementation in HIV treatment could be considered.
Nonetheless, pharmacologic doses of nicotinic acid have been shown to be well tolerated in HIV patients with hyperlipidemia (108). Abnormal lipid profiles observed in patients have been attributed to the HIV infection and to the highly active antiretroviral treatment (HAART) (109). Moreover, insulin resistance has been detected together with dyslipidemia in ART-treated patients (110). Cardiovascular disease (CVD) is the second most frequent cause of deaths in the HIV population, and the rate of CVD is predicted to increase further as patients are living longer due to successful antiretroviral therapies. As for the general population, statin-based therapy appears to benefit HIV patients in terms of atherogenic protection and CVD risk reduction, although contraindications exist due to drug interactions with ART. Other first-line treatments include lipid-lowering fibrates, which are preferred to nicotinic acid due to the increased risk of glucose intolerance and insulin resistance (111). Nevertheless, an unblinded, controlled pilot study showed that extended-release nicotinic acid (0.5-1.5 g/day for 12 weeks) could effectively improve endothelial function of the brachial artery in ART-treated HIV subjects with low HDL-cholesterol and no history of CVD (112). In addition, a combined treatment of fibrates, extended-release nicotinic acid (0.5-2 g/day), and lifestyle changes (low-fat diet and exercise) for 24 weeks was effective in normalizing lipid parameters in a cohort of 191 ART-treated patients. Increased risk of liver dysfunction was detected in subjects receiving both fibrates and niacin, but insulin sensitivity was not affected by nicotinic acid treatment given alone or when combined with fibrates (113). Another 24-week, open-label, uncontrolled trial in 99 ART-treated patients found that randomization to extended-release nicotinic acid (0.5-2 g/day) or fenofibrates increased blood HDL-cholesterol but did not reduce inflammatory markers or improve endothelial function when compared to baseline (114).
Schizophrenia
Schizophrenia is a neurologic disorder with unclear etiology that is diagnosed purely from its clinical presentation. Because neurologic disorders associated with pellagra resemble acute schizophrenia, niacin-based therapy for the condition was investigated during the 1950s-70s (reviewed in 115). The adjunctive use of nutrients like niacin to correct deficiencies associated with neurologic symptoms is called orthomolecular psychiatry (116). Such an approach has not been included in psychiatric practice; practitioners have instead relied solely on antipsychotic drugs to eliminate the clinical symptoms of schizophrenia. Nevertheless, recent scientific advances and new hypotheses on the benefit of nutrient supplementation in the treatment of psychiatric disorders have suggested the re-assessment of orthomolecular medicine by the medical community (117, 118).
Skin flushing is one major side effect of the therapeutic use of nicotinic acid and the primary reason for non-adherence to treatment (see Toxicity). Flushing is caused by the activation of phospholipase A2, an enzyme that stimulates the production of a specific lipid from the prostanoid family called prostaglandin D2. Prostaglandin D2, synthesized by antigen-presenting cells of the skin and mucosa (i.e., the Langerhans cells), can induce the dilation of blood vessels and trigger a flushing response. Interestingly, patients with schizophrenia tend not to flush following treatment with nicotinic acid. This blunted skin flushing response suggests abnormal prostanoid signaling in schizophrenic patients (119, 120). An association has been found between the altered niacin sensitivity and greater functional impairment in schizophrenic subjects (121), which supports other findings suggesting that altered lipid metabolism could critically impair brain development and contribute to the disease (122). Interestingly, blunted skin flushing responses are more prevalent in first-degree relatives of people with schizophrenia than in the general population, suggesting that reduced niacin sensitivity is a heritable trait within affected families (123).
Sources
Food sources
Good sources of niacin include yeast, meat, poultry, red fish (e.g., tuna, salmon), cereal (especially fortified cereal), legumes, and seeds. Milk, green leafy vegetables, coffee, and tea also provide some niacin (124). In plants, especially mature cereal grains like corn and wheat, niacin may be bound to sugar molecules in the form of glycosides, which significantly decrease its bioavailability (25).
In the United States, the average dietary intake of niacin is about 30 mg/day for young adult men and 20 mg/day for young adult women. In a sample of adults over the age of 60, men and women were found to have an average dietary intake of 21 mg/day and 17 mg/day, respectively (41). Some foods with substantial amounts of niacin are listed in Table 2, along with their niacin content in milligrams (mg). Food composition tables generally list niacin content without including niacin equivalents (NE) from tryptophan or any adjustment for niacin bioavailability. For more information on the nutrient content of specific foods, search USDA's FoodData Central database; data included in Table 2 are from this database (125).
Supplements
Niacin supplements are available as nicotinamide or nicotinic acid. Nicotinamide is the form of niacin typically used in nutritional supplements and in food fortification. Nicotinic acid is available over the counter and with a prescription as a cholesterol-lowering agent (126). Nicotinic acid for anti-hyperlipidemic use is available in three formulations: immediate-release (crystalline) nicotinic acid (absorption time, 1-2 hrs), extended-release nicotinic acid (absorption time, 8-12 hrs), and sustained-release nicotinic acid (absorption time, >12 hrs) (127). At the pharmacologic dose required for cholesterol-lowering effects, the use of nicotinic acid should be approached as if it were a drug (see Safety). Individuals should only undertake cholesterol-lowering therapy with nicotinic acid under the supervision of a qualified health care provider in order to minimize potentially adverse effects and maximize therapeutic benefits.
Safety
Toxicity
Nicotinic acid
Common side effects of nicotinic acid include flushing, pruritus (severe itching of the skin), skin rashes, and gastrointestinal disturbances, such as nausea and vomiting (97). Transient episodes of low blood pressure (hypotension) and headache have also been reported. Hepatotoxicity (liver cell damage), including elevated liver enzymes and jaundice, has been observed at intakes as low as 750 mg/day of nicotinic acid (128). Although hepatitis has been observed with extended-release nicotinic acid at dosages as little as 500 mg/day for two months, almost all reports of severe hepatitis have been associated with doses of 3 to 9 g/day used to treat high cholesterol for months or years (41). It is unclear whether immediate-release (crystalline) nicotinic acid is less toxic to the liver than extended-release forms (41). Yet, immediate-release nicotinic acid is often used at higher doses than extended-release forms, and severe liver toxicity has occurred in individuals who substituted extended-release nicotinic acid for immediate-release nicotinic acid at equivalent doses (126). Large doses of nicotinic acid have been observed to impair glucose tolerance, likely because of a decrease in insulin sensitivity. Impaired glucose tolerance in susceptible (pre-diabetic) individuals could result in elevated blood glucose concentrations and clinical type 2 diabetes mellitus. An analysis of the HPS2-THRIVE trial (see Cardiovascular disease), using data from 17,374 participants without type 2 diabetes at baseline, found a significantly higher proportion of newly diagnosed cases among those randomized to nicotinic acid/laropiprant than to placebo (5.7% versus 4.3%) over a 3.9 year-period (95). Likewise, randomization to nicotinic acid/laropiprant significantly increased the risk of serious disturbances in diabetes control (leading to hospitalization) compared to placebo among 8,299 participants with diabetes at baseline (95). Elevated blood concentrations of uric acid, occasionally resulting in attacks of gout in susceptible individuals, have also been observed with high-dose nicotinic acid therapy (126). Niacin at doses of 1.5 to 5 g/day has resulted in a few case reports of blurred vision and other eye problems, which have generally been reversible upon discontinuation (41). People with abnormal liver function or a history of liver disease, diabetes, active peptic ulcer disease, gout, cardiac arrhythmias, inflammatory bowel disease, migraine headaches, or alcoholism may be more susceptible to the adverse effects of excess niacin intake than the general population (41).
Nicotinamide
Nicotinamide is generally better tolerated than nicotinic acid; it does not generally cause flushing (126). However, nausea, vomiting, and signs of liver toxicity (elevated liver enzymes, jaundice) have been observed at very high doses (≥10 g/day) (126).
Nicotinamide riboside
A study in 12 healthy subjects found that nicotinamide riboside at three single doses (100 mg, 300 mg, and 1,000 mg) safely increased blood NAD+. Two of the participants self-reported skin flushing after taking the 300-mg dose, and two others reported feeling hot following intake of 1,000 mg of nicotinamide riboside (1). In a recent randomized, placebo-controlled trial in 120 healthy adults (ages, 60-80 years), daily supplementation with nicotinamide riboside (250 mg or 500 mg) and pterostilbene (a SIRT activator; 50 mg or 100 mg) for eight weeks showed a favorable side effect profile, with no evidence of higher incidence of adverse effects compared to placebo (129). Most recently, a randomized, placebo-controlled trial in 40 obese men (ages, 40-70 years) found that daily supplementation with nicotinamide riboside (2,000 mg/day divided into two daily dosages) for 12 weeks was associated with reports of only minor side effects, including excessive sweating, pruritus, and mild gastrointestinal symptoms like bloating (139).
The tolerable upper intake level (UL)
Flushing of the skin primarily on the face, arms, and chest is a common side effect of nicotinic acid and may occur initially at doses as low as 30 mg/day. Although flushing from nicotinamide is rare, the Food and Nutrition Board set the tolerable upper intake level (UL) for niacin (nicotinic acid and nicotinamide) at 35 mg/day in adults to avoid the adverse effect of flushing (41). Analysis of data from the US National Health and Nutrition Examination Survey (NHANES) 2003-2006 found that 15.8% of children and adolescents (ages 2-18 years) and 8.5% of adults (≥19 years) had total usual niacin intakes exceeding the UL (130). The UL applies to the general population and is not meant to apply to individuals who are being treated with a nutrient under medical supervision (e.g., high-dose nicotinic acid for elevated blood cholesterol concentrations).
Drug interactions
The occurrence of rhabdomyolysis is increased in patients treated with statins (HMG-CoA reductase inhibitors). Rhabdomyolysis is a relatively uncommon condition in which muscle cells are broken down, releasing enzymes and electrolytes into the blood, and sometimes resulting in kidney failure (131). Co-administration of nicotinic acid with a statin seems to enhance the risk of rhabdomyolosis (132). A new drug, laropiprant, blocks prostanoid receptors and reduces nicotinic acid-induced flushing (133). A randomized, placebo-controlled trial was designed to identify possible adverse effects of the niacin/laropiprant combination in over 25,000 simvastatin-treated subjects (134). When added to the statin therapy, niacin/laropiprant increased the risk of myopathy and rhabdomyolosis, particularly in Asian subjects. It is possible that the niacin/laropiprant combination further reduces the poor tolerability to statin treatment observed in certain populations (135).
In the three-year, randomized controlled HATS study, concurrent therapy with antioxidants (1,000 mg/day of vitamin C, 800 IU/day of RRR-α-tocopherol, 100 µg/day of selenium, and 25 mg/day of β-carotene) diminished the protective effects of the simvastatin-nicotinic acid combination (136). Although the mechanism for these effects is not known, the benefit of concurrent antioxidant therapy in patients on lipid-lowering agents has been questioned (137).
Adverse effects of large doses of nicotinic acid may be exacerbated by the concomitant use of certain medications. The risk of myopathy may be further increased in those taking nicotinic acid and bile acid sequestrants (e.g., cholestyramine, colestipol) or the anti-lipidemic drug, gemfibrozil (Lopid), and the risk of hepatotoxicity observed with nicotinic acid might be enhanced by drugs like paracetamol, amiodarone (Cordarone), or carbamazepine (Tegretol) (35). In addition, large doses of nicotinic acid may reduce uric acid excretion, thereby opposing the action of uricosuric agents like probenecid (Probalan) (35).
Several other medications may interact with niacin therapy or with absorption and metabolism of the vitamin (126). Estrogen and estrogen-containing oral contraceptives increase the efficiency of niacin synthesis from tryptophan, resulting in a decreased dietary requirement for niacin (138). Long-term administration of chemotherapy agents has been reported to cause symptoms of pellagra; therefore, niacin supplementation may be needed (see Pellagra causes).
Linus Pauling Institute Recommendation
The optimum intake of niacin for health promotion and chronic disease prevention is not yet known. The RDA (16 mg NE/day for men and 14 mg NE/day for women) is easily obtainable by consuming a varied diet and should prevent deficiency in most people. Following the Linus Pauling Institute recommendation to take a daily multivitamin/mineral supplement, containing 100% of the Daily Value (DV) for niacin, will provide at least 20 mg of niacin daily.
Authors and Reviewers
Originally written in 2000 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in August 2002 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in June 2007 by:
Victoria J. Drake, Ph.D
Linus Pauling Institute
Oregon State University
Updated in July 2013 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in December 2017 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in March 2018 by:
Mirella Meyer-Ficca, Ph.D.
Research Assistant Professor
Utah State University
The 2017 update of this article was supported by a grant from ChromaDex, Inc.
Last updated 8/10/18 Copyright 2000-2024 Linus Pauling Institute
References
1. Trammell SA, Schmidt MS, Weidemann BJ, et al. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun. 2016;7:12948. (PubMed)
2. Nikiforov A, Kulikova V, Ziegler M. The human NAD metabolome: functions, metabolism and compartmentalization. Crit Rev Biochem Mol Biol. 2015;50(4):284-297. (PubMed)
3. Kawai S, Murata K. Structure and function of NAD kinase and NADP phosphatase: key enzymes that regulate the intracellular balance of NAD(H) and NADP(H). Biosci Biotechnol Biochem. 2008;72(4):919-930. (PubMed)
4. Agledal L, Niere M, Ziegler M. The phosphate makes a difference: cellular functions of NADP. Redox Rep. 2010;15(1):2-10. (PubMed)
5. Penberthy WT, Kirkland JB. Niacin. In: Erdman JW, MacDonald I, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Ames: International Life Sciences Institute; 2012:293-306.
6. Kirkland JB. Niacin. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore: Lippincott Williams & Wilkins; 2014:331-340.
7. Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci. 2010;35(4):208-219. (PubMed)
8. Liu C, Yu X. ADP-ribosyltransferases and poly ADP-ribosylation. Curr Protein Pept Sci. 2015;16(6):491-501. (PubMed)
9. Hwang ES, Song SB. Nicotinamide is an inhibitor of SIRT1 in vitro, but can be a stimulator in cells. Cell Mol Life Sci. 2017;74(18):3347-3362. (PubMed)
10. Morris BJ. Seven sirtuins for seven deadly diseases of aging. Free Radic Biol Med. 2013;56:133-171. (PubMed)
11. Fliegert R, Bauche A, Wolf Perez AM, et al. 2'-Deoxyadenosine 5'-diphosphoribose is an endogenous TRPM2 superagonist. Nat Chem Biol. 2017;13(9):1036-1044. (PubMed)
12. Mutafova-Yambolieva VN, Hwang SJ, Hao X, et al. Beta-nicotinamide adenine dinucleotide is an inhibitory neurotransmitter in visceral smooth muscle. Proc Natl Acad Sci U S A. 2007;104(41):16359-16364. (PubMed)
13. Moreschi I, Bruzzone S, Nicholas RA, et al. Extracellular NAD+ is an agonist of the human P2Y11 purinergic receptor in human granulocytes. J Biol Chem. 2006;281(42):31419-31429. (PubMed)
14. Klein C, Grahnert A, Abdelrahman A, Muller CE, Hauschildt S. Extracellular NAD(+) induces a rise in [Ca(2+)](i) in activated human monocytes via engagement of P2Y(1) and P2Y(11) receptors. Cell Calcium. 2009;46(4):263-272. (PubMed)
15. Moreschi I, Bruzzone S, Bodrato N, et al. NAADP+ is an agonist of the human P2Y11 purinergic receptor. Cell Calcium. 2008;43(4):344-355. (PubMed)
16. Huang C, Hu J, Subedi KP, et al. Extracellular adenosine diphosphate ribose mobilizes intracellular Ca2+ via purinergic-dependent Ca2+ pathways in rat pulmonary artery smooth muscle cells. Cell Physiol Biochem. 2015;37(5):2043-2059. (PubMed)
17. Knopp RH. Drug treatment of lipid disorders. N Engl J Med. 1999;341(7):498-511. (PubMed)
18. Graff EC, Fang H, Wanders D, Judd RL. Anti-inflammatory effects of the hydroxycarboxylic acid receptor 2. Metabolism. 2016;65(2):102-113. (PubMed)
19. Jin FY, Kamanna VS, Kashyap ML. Niacin accelerates intracellular ApoB degradation by inhibiting triacylglycerol synthesis in human hepatoblastoma (HepG2) cells. Arterioscler Thromb Vasc Biol. 1999;19(4):1051-1059. (PubMed)
20. Kamanna VS, Ganji SH, Kashyap ML. Recent advances in niacin and lipid metabolism. Curr Opin Lipidol. 2013;24(3):239-245. (PubMed)
21. Carlson LA. Studies on the effect of nicotinic acid on catecholamine stimulated lipolysis in adipose tissue in vitro. Acta Med Scand. 1963;173:719-722. (PubMed)
22. Lauring B, Taggart AK, Tata JR, et al. Niacin lipid efficacy is independent of both the niacin receptor GPR109A and free fatty acid suppression. Sci Transl Med. 2012;4(148):148ra115. (PubMed)
23. Brody T. Nutritional Biochemistry. 2nd ed. San Diego: Academic Press; 1999.
24. Kirkland JB. Niacin. In: Zempleni J, Suttie JW, Gregory III JF, Stover PJ, eds. Handbook of Vitamins. 5th ed. Boca Raton: CRC Press; 2013:149-190.
25. Gregory JF, 3rd. Nutritional properties and significance of vitamin glycosides. Annu Rev Nutr. 1998;18:277-296. (PubMed)
26. Dawson B, Favaloro EJ, Taylor J, Aggarwal A. Unrecognized pellagra masquerading as odynophagia. Intern Med J. 2006;36(7):472-474. (PubMed)
27. Jagielska G, Tomaszewicz-Libudzic EC, Brzozowska A. Pellagra: a rare complication of anorexia nervosa. Eur Child Adolesc Psychiatry. 2007;16(7):417-420. (PubMed)
28. Kertesz SG. Pellagra in 2 homeless men. Mayo Clin Proc. 2001;76(3):315-318. (PubMed)
29. Prakash R, Gandotra S, Singh LK, Das B, Lakra A. Rapid resolution of delusional parasitosis in pellagra with niacin augmentation therapy. Gen Hosp Psychiatry. 2008;30(6):581-584. (PubMed)
30. Badawy AA. Pellagra and alcoholism: a biochemical perspective. Alcohol Alcohol. 2014;49(3):238-250. (PubMed)
31. Majewski M, Kozlowska A, Thoene M, Lepiarczyk E, Grzegorzewski WJ. Overview of the role of vitamins and minerals on the kynurenine pathway in health and disease. J Physiol Pharmacol. 2016;67(1):3-19. (PubMed)
32. Rosmaninho A, Sanches M, Fernandes IC, et al. Letter: Pellagra as the initial presentation of Crohn disease. Dermatol Online J. 2012;18(4):12. (PubMed)
33. Zaraa I, Belghith I, El Euch D, et al. A case of pellagra associated with megaduodenum in a young woman. Nutr Clin Pract. 2013;28(2):218-222. (PubMed)
34. Bilgili SG, Karadag AS, Calka O, Altun F. Isoniazid-induced pellagra. Cutan Ocul Toxicol. 2011;30(4):317-319. (PubMed)
35. Natural Medicines. Professional Monograph - Niacin/Interactions with drugs. Available at: https://naturalmedicines.therapeuticresearch.com/. Accessed 8/2/17.
36. Dreizen S, McCredie KB, Keating MJ, Andersson BS. Nutritional deficiencies in patients receiving cancer chemotherapy. Postgrad Med. 1990;87(1):163-167, 170. (PubMed)
37. Nogueira A, Duarte AF, Magina S, Azevedo F. Pellagra associated with esophageal carcinoma and alcoholism. Dermatol Online J. 2009;15(5):8. (PubMed)
38. Oldham MA, Ivkovic A. Pellagrous encephalopathy presenting as alcohol withdrawal delirium: a case series and literature review. Addict Sci Clin Pract. 2012;7(1):12. (PubMed)
39. World Health Organization, United Nations High Commissions for Refugees. Pellagra and its prevention and control in major emergencies. World Health Organization. 2000. Available at: http://www.who.int/nutrition/publications/emergencies/WHO_NHD_00.10/en/. Accessed 6/20/13.
40. Murray MF. Tryptophan depletion and HIV infection: a metabolic link to pathogenesis. Lancet Infect Dis. 2003;3(10):644-652. (PubMed)
41. Food and Nutrition Board, Institute of Medicine. Niacin. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B-6, Vitamin B-12, Pantothenic Acid, Biotin, and Choline. Washington, D.C.: The National Academies Press; 1998:123-149. (The National Academies Press)
42. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11(3):220-228. (PubMed)
43. Kirkland JB. Niacin requirements for genomic stability. Mutat Res. 2012;733(1-2):14-20. (PubMed)
44. Burkle A. Poly(ADP-ribose). The most elaborate metabolite of NAD+. FEBS J. 2005;272(18):4576-4589. (PubMed)
45. Jacobson EL, Shieh WM, Huang AC. Mapping the role of NAD metabolism in prevention and treatment of carcinogenesis. Mol Cell Biochem. 1999;193(1-2):69-74. (PubMed)
46. Spronck JC, Nickerson JL, Kirkland JB. Niacin deficiency alters p53 expression and impairs etoposide-induced cell cycle arrest and apoptosis in rat bone marrow cells. Nutr Cancer. 2007;57(1):88-99. (PubMed)
47. Spronck JC, Kirkland JB. Niacin deficiency increases spontaneous and etoposide-induced chromosomal instability in rat bone marrow cells in vivo. Mutat Res. 2002;508(1-2):83-97. (PubMed)
48. Kostecki LM, Thomas M, Linford G, et al. Niacin deficiency delays DNA excision repair and increases spontaneous and nitrosourea-induced chromosomal instability in rat bone marrow. Mutat Res. 2007;625(1-2):50-61. (PubMed)
49. Dantzer F, Santoro R. The expanding role of PARPs in the establishment and maintenance of heterochromatin. FEBS J. 2013;280(15):3508-3518. (PubMed)
50. El Ramy R, Magroun N, Messadecq N, et al. Functional interplay between Parp-1 and SirT1 in genome integrity and chromatin-based processes. Cell Mol Life Sci. 2009;66(19):3219-3234. (PubMed)
51. Boyonoski AC, Spronck JC, Gallacher LM, et al. Niacin deficiency decreases bone marrow poly(ADP-ribose) and the latency of ethylnitrosourea-induced carcinogenesis in rats. J Nutr. 2002;132(1):108-114. (PubMed)
52. Boyonoski AC, Spronck JC, Jacobs RM, Shah GM, Poirier GG, Kirkland JB. Pharmacological intakes of niacin increase bone marrow poly(ADP-ribose) and the latency of ethylnitrosourea-induced carcinogenesis in rats. J Nutr. 2002;132(1):115-120. (PubMed)
53. Weitberg AB. Effect of nicotinic acid supplementation in vivo on oxygen radical-induced genetic damage in human lymphocytes. Mutat Res. 1989;216(4):197-201. (PubMed)
54. Hageman GJ, Stierum RH, van Herwijnen MH, van der Veer MS, Kleinjans JC. Nicotinic acid supplementation: effects on niacin status, cytogenetic damage, and poly(ADP-ribosylation) in lymphocytes of smokers. Nutr Cancer. 1998;32(2):113-120. (PubMed)
55. Yong LC, Petersen MR. High dietary niacin intake is associated with decreased chromosome translocation frequency in airline pilots. Br J Nutr. 2011;105(4):496-505. (PubMed)
56. Weidele K, Beneke S, Burkle A. The NAD+ precursor nicotinic acid improves genomic integrity in human peripheral blood mononuclear cells after X-irradiation. DNA Repair (Amst). 2017;52:12-23. (PubMed)
57. Jacobson EL. Niacin deficiency and cancer in women. J Am Coll Nutr. 1993;12(4):412-416. (PubMed)
58. Negri E, Franceschi S, Bosetti C, et al. Selected micronutrients and oral and pharyngeal cancer. Int J Cancer. 2000;86(1):122-127. (PubMed)
59. Franceschi S, Bidoli E, Negri E, et al. Role of macronutrients, vitamins and minerals in the aetiology of squamous-cell carcinoma of the oesophagus. Int J Cancer. 2000;86(5):626-631. (PubMed)
60. Gensler HL, Williams T, Huang AC, Jacobson EL. Oral niacin prevents photocarcinogenesis and photoimmunosuppression in mice. Nutr Cancer. 1999;34(1):36-41. (PubMed)
61. Jacobson EL, Kim H, Kim M, et al. A topical lipophilic niacin derivative increases NAD, epidermal differentiation and barrier function in photodamaged skin. Exp Dermatol. 2007;16(6):490-499. (PubMed)
62. Bermudez Y, Benavente CA, Meyer RG, Coyle WR, Jacobson MK, Jacobson EL. Nicotinic acid receptor abnormalities in human skin cancer: implications for a role in epidermal differentiation. PLoS One. 2011;6(5):e20487. (PubMed)
63. Benavente CA, Jacobson EL. Niacin restriction upregulates NADPH oxidase and reactive oxygen species (ROS) in human keratinocytes. Free Radic Biol Med. 2008;44(4):527-537. (PubMed)
64. Benavente CA, Schnell SA, Jacobson EL. Effects of niacin restriction on sirtuin and PARP responses to photodamage in human skin. PLoS One. 2012;7(7):e42276. (PubMed)
65. Park SM, Li T, Wu S, et al. Niacin intake and risk of skin cancer in US women and men. Int J Cancer. 2017;140(9):2023-2031. (PubMed)
66. Chen AC, Martin AJ, Choy B, et al. A phase 3 randomized trial of nicotinamide for skin-cancer chemoprevention. N Engl J Med. 2015;373(17):1618-1626. (PubMed)
67. Minocha R, Damian DL, Halliday GM. Melanoma and nonmelanoma skin cancer chemoprevention: A role for nicotinamide? Photodermatol Photoimmunol Photomed. 2018;34(1):5-12. (PubMed)
68. Orban T, Sosenko JM, Cuthbertson D, et al. Pancreatic islet autoantibodies as predictors of type 1 diabetes in the Diabetes Prevention Trial-Type 1. Diabetes Care. 2009;32(12):2269-2274. (PubMed)
69. Szkudelski T. Streptozotocin-nicotinamide-induced diabetes in the rat. Characteristics of the experimental model. Exp Biol Med (Maywood). 2012;237(5):481-490. (PubMed)
70. Lampeter EF, Klinghammer A, Scherbaum WA, et al. The Deutsche Nicotinamide Intervention Study: an attempt to prevent type 1 diabetes. DENIS Group. Diabetes. 1998;47(6):980-984. (PubMed)
71. Gale EA, Bingley PJ, Emmett CL, Collier T, European Nicotinamide Diabetes Intervention Trial Group. European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet. 2004;363(9413):925-931. (PubMed)
72. Hedman M, Ludvigsson J, Faresjo MK. Nicotinamide reduces high secretion of IFN-gamma in high-risk relatives even though it does not prevent type 1 diabetes. J Interferon Cytokine Res. 2006;26(4):207-213. (PubMed)
73. Fernandez IC, Del Carmen Camberos M, Passicot GA, Martucci LC, Cresto JC. Children at risk of diabetes type 1. Treatment with acetyl-L-carnitine plus nicotinamide - Case reports. J Pediatr Endocrinol Metab. 2013;26(3-4):347-355. (PubMed)
74. Patel AB, Prabhu AS. Hartnup disease. Indian J Dermatol. 2008;53(1):31-32. (PubMed)
75. Oakley A, Wallace J. Hartnup disease presenting in an adult. Clin Exp Dermatol. 1994;19(5):407-408. (PubMed)
76. Shi H, Enriquez A, Rapadas M, et al. NAD deficiency, congenital malformations, and niacin supplementation. N Engl J Med. 2017;377(6):544-552. (PubMed)
77. Vander Heiden MG. Metabolism and congenital malformations - NAD's effects on development. N Engl J Med. 2017;377(6):509-511. (PubMed)
78. Hu L, Ibrahim K, Stucki M, et al. Secondary NAD+ deficiency in the inherited defect of glutamine synthetase. J Inherit Metab Dis. 2015;38(6):1075-1083. (PubMed)
79. Ames BN, Elson-Schwab I, Silver EA. High-dose vitamin therapy stimulates variant enzymes with decreased coenzyme binding affinity (increased K(m)): relevance to genetic disease and polymorphisms. Am J Clin Nutr. 2002;75(4):616-658. (PubMed)
80. Bays HE, Shah A, Lin J, Sisk CM, Dong Q, Maccubbin D. Consistency of extended-release niacin/laropiprant effects on Lp(a), ApoB, non-HDL-C, Apo A1, and ApoB/ApoA1 ratio across patient subgroups. Am J Cardiovasc Drugs. 2012;12(3):197-206. (PubMed)
81. Wink J, Giacoppe G, King J. Effect of very-low-dose niacin on high-density lipoprotein in patients undergoing long-term statin therapy. Am Heart J. 2002;143(3):514-518. (PubMed)
82. Taylor AJ, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation. 2004;110(23):3512-3517. (PubMed)
83. Taylor AJ, Zhu D, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Relationship between glycemic status and progression of carotid intima-media thickness during treatment with combined statin and extended-release niacin in ARBITER 2. Vasc Health Risk Manag. 2007;3(1):159-164. (PubMed)
84. Villines TC, Stanek EJ, Devine PJ, et al. The ARBITER 6-HALTS Trial (Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol 6-HDL and LDL Treatment Strategies in Atherosclerosis): final results and the impact of medication adherence, dose, and treatment duration. J Am Coll Cardiol. 2010;55(24):2721-2726. (PubMed)
85. Ras RT, Streppel MT, Draijer R, Zock PL. Flow-mediated dilation and cardiovascular risk prediction: a systematic review with meta-analysis. Int J Cardiol. 2013;168(1):344-351. (PubMed)
86. Sahebkar A. Effect of niacin on endothelial function: a systematic review and meta-analysis of randomized controlled trials. Vasc Med. 2014;19(1):54-66. (PubMed)
87. Canner PL, Berge KG, Wenger NK, et al. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol. 1986;8(6):1245-1255. (PubMed)
88. Brown BG, Zhao XQ, Chait A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345(22):1583-1592. (PubMed)
89. Vittone F, Chait A, Morse JS, Fish B, Brown BG, Zhao XQ. Niacin plus simvastatin reduces coronary stenosis progression among patients with metabolic syndrome despite a modest increase in insulin resistance: a subgroup analysis of the HDL-Atherosclerosis Treatment Study (HATS). J Clin Lipidol. 2007;1(3):203-210. (PubMed)
90. Zhao XQ, Morse JS, Dowdy AA, et al. Safety and tolerability of simvastatin plus niacin in patients with coronary artery disease and low high-density lipoprotein cholesterol (The HDL Atherosclerosis Treatment Study). Am J Cardiol. 2004;93(3):307-312. (PubMed)
91. Sazonov V, Maccubbin D, Sisk CM, Canner PL. Effects of niacin on the incidence of new onset diabetes and cardiovascular events in patients with normoglycaemia and impaired fasting glucose. Int J Clin Pract. 2013;67(4):297-302. (PubMed)
92. Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255-2267. (PubMed)
93. Michos ED, Sibley CT, Baer JT, Blaha MJ, Blumenthal RS. Niacin and statin combination therapy for atherosclerosis regression and prevention of cardiovascular disease events: reconciling the AIM-HIGH (Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides: Impact on Global Health Outcomes) trial with previous surrogate endpoint trials. J Am Coll Cardiol. 2012;59(23):2058-2064. (PubMed)
94. Kalil RS, Wang JH, de Boer IH, et al. Effect of extended-release niacin on cardiovascular events and kidney function in chronic kidney disease: a post hoc analysis of the AIM-HIGH trial. Kidney Int. 2015;87(6):1250-1257. (PubMed)
95. Landray MJ, Haynes R, Hopewell JC, et al. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371(3):203-212. (PubMed)
96. Schandelmaier S, Briel M, Saccilotto R, et al. Niacin for primary and secondary prevention of cardiovascular events. Cochrane Database Syst Rev. 2017;6:Cd009744. (PubMed)
97. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 Pt B):2889-2934. (PubMed)
98. Jackevicius CA, Tu JV, Ko DT, de Leon N, Krumholz HM. Use of niacin in the United States and Canada. JAMA Intern Med. 2013;173(14):1379-1381. (PubMed)
99. Burk K. Friedreich ataxia: current status and future prospects. Cerebellum Ataxias. 2017;4:4. (PubMed)
100. Chan PK, Torres R, Yandim C, et al. Heterochromatinization induced by GAA-repeat hyperexpansion in Friedreich's ataxia can be reduced upon HDAC inhibition by vitamin B3. Hum Mol Genet. 2013;22(13):2662-2675. (PubMed)
101. Libri V, Yandim C, Athanasopoulos S, et al. Epigenetic and neurological effects and safety of high-dose nicotinamide in patients with Friedreich's ataxia: an exploratory, open-label, dose-escalation study. Lancet. 2014;384(9942):504-513. (PubMed)
102. Lynch DR, Fischbeck KH. Nicotinamide in Friedreich's ataxia: useful or not? Lancet. 2014;384(9942):474-475. (PubMed)
103. Taylor EW. The oxidative stress-induced niacin sink (OSINS) model for HIV pathogenesis. Toxicology. 2010;278(1):124-130. (PubMed)
104. Favre D, Mold J, Hunt PW, et al. Tryptophan catabolism by indoleamine 2,3-dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci Transl Med. 2010;2(32):32ra36. (PubMed)
105. Jenabian MA, Patel M, Kema I, et al. Distinct tryptophan catabolism and Th17/Treg balance in HIV progressors and elite controllers. PLoS One. 2013;8(10):e78146. (PubMed)
106. Chen J, Shao J, Cai R, et al. Anti-retroviral therapy decreases but does not normalize indoleamine 2,3-dioxygenase activity in HIV-infected patients. PLoS One. 2014;9(7):e100446. (PubMed)
107. Dunham RM, Gordon SN, Vaccari M, et al. Preclinical evaluation of HIV eradication strategies in the simian immunodeficiency virus-infected rhesus macaque: a pilot study testing inhibition of indoleamine 2,3-dioxygenase. AIDS Res Hum Retroviruses. 2013;29(2):207-214. (PubMed)
108. Souza SA, Chow DC, Walsh EJ, Ford S, 3rd, Shikuma C. Pilot study on the safety and tolerability of extended release niacin for HIV-infected patients with hypertriglyceridemia. Hawaii Med J. 2010;69(5):122-125. (PubMed)
109. Dube MP, Lipshultz SE, Fichtenbaum CJ, et al. Effects of HIV infection and antiretroviral therapy on the heart and vasculature. Circulation. 2008;118(2):e36-40. (PubMed)
110. Carr A, Samaras K, Burton S, et al. A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS. 1998;12(7):F51-58. (PubMed)
111. Giannarelli C, Klein RS, Badimon JJ. Cardiovascular implications of HIV-induced dyslipidemia. Atherosclerosis. 2011;219(2):384-389. (PubMed)
112. Chow DC, Stein JH, Seto TB, et al. Short-term effects of extended-release niacin on endothelial function in HIV-infected patients on stable antiretroviral therapy. AIDS. 2010;24(7):1019-1023. (PubMed)
113. Balasubramanyam A, Coraza I, Smith EO, et al. Combination of niacin and fenofibrate with lifestyle changes improves dyslipidemia and hypoadiponectinemia in HIV patients on antiretroviral therapy: results of "heart positive," a randomized, controlled trial. J Clin Endocrinol Metab. 2011;96(7):2236-2247. (PubMed)
114. Dube MP, Komarow L, Fichtenbaum CJ, et al. Extended-release niacin versus fenofibrate in HIV-infected participants with low high-density lipoprotein cholesterol: effects on endothelial function, lipoproteins, and inflammation. Clin Infect Dis. 2015;61(5):840-849. (PubMed)
115. Hoffer LJ. Vitamin therapy in schizophrenia. Isr J Psychiatry Relat Sci. 2008;45(1):3-10. (PubMed)
116. Pauling L. Orthomolecular psychiatry. Varying the concentrations of substances normally present in the human body may control mental disease. Science. 1968;160(3825):265-271. (PubMed)
117. Seybolt SE. Is it time to reassess alpha lipoic acid and niacinamide therapy in schizophrenia? Med Hypotheses. 2010;75(6):572-575. (PubMed)
118. Zell M, Grundmann O. An orthomolecular approach to the prevention and treatment of psychiatric disorders. Adv Mind Body Med. 2012;26(2):14-28. (PubMed)
119. Yao JK, Dougherty GG, Jr., Gautier CH, et al. Prevalence and specificity of the abnormal niacin response: a potential endophenotype marker in schizophrenia. Schizophr Bull. 2016;42(2):369-376. (PubMed)
120. Sun L, Yang X, Jiang J, et al. Identification of the niacin-blunted subgroup of schizophrenia patients from mood disorders and healthy individuals in Chinese population. Schizophr Bull. 2017; doi: 10.1093/schbul/sbx150. [Epub ahead of print]. (PubMed)
121. Messamore E. Niacin subsensitivity is associated with functional impairment in schizophrenia. Schizophr Res. 2012;137(1-3):180-184. (PubMed)
122. Horrobin DF. The membrane phospholipid hypothesis as a biochemical basis for the neurodevelopmental concept of schizophrenia. Schizophr Res. 1998;30(3):193-208. (PubMed)
123. Messamore E. The niacin response biomarker as a schizophrenia endophenotype: A status update. Prostaglandins Leukot Essent Fatty Acids. 2017; pii: S0952-3278(16)30249-6. doi: 10.1016/j.plefa.2017.06.014. [Epub ahead of print]. (PubMed)
124. Jacob R, Swenseid M. Niacin. In: Ziegler E, Filer L, eds. Present Knowledge in Nutrition. 7th ed. Washington D.C.: ILSI Press; 1996:185-190.
125. US Department of Agriculture. USDA National Nutrient Database for Standard Reference, Release 25. 2012. https://ndb.nal.usda.gov/ndb/. Accessed 7/30/17.
126. Hendler SS, Rorvik DR. PDR for Nutritional Supplements. 2nd ed. Montvale: Thomson Reuters; 2008.
127. Minto C, Vecchio MG, Lamprecht M, Gregori D. Definition of a tolerable upper intake level of niacin: a systematic review and meta-analysis of the dose-dependent effects of nicotinamide and nicotinic acid supplementation. Nutr Rev. 2017;75(6):471-490. (PubMed)
128. MacKay D, Hathcock J, Guarneri E. Niacin: chemical forms, bioavailability, and health effects. Nutr Rev. 2012;70(6):357-366. (PubMed)
129. Dellinger RW, Santos SR, Morris M, et al. Repeat dose NRPT (nicotinamide riboside and pterostilbene) increases NAD(+) levels in humans safely and sustainably: a randomized, double-blind, placebo-controlled study. NPJ Aging Mech Dis. 2017;3:17. (PubMed)
130. Fulgoni VL, 3rd, Keast DR, Bailey RL, Dwyer J. Foods, fortificants, and supplements: Where do Americans get their nutrients? J Nutr. 2011;141(10):1847-1854. (PubMed)
131. Kar S, Chockalingam A. Statin-associated rhabdomyolysis with acute renal failure complicated by intradialytic NSTEMI: a review of lipid management considerations. Am J Ther. 2013;20(1):57-60. (PubMed)
132. Cziraky MJ, Willey VJ, McKenney JM, et al. Risk of hospitalized rhabdomyolysis associated with lipid-lowering drugs in a real-world clinical setting. J Clin Lipidol. 2013;7(2):102-108. (PubMed)
133. Maccubbin DL, Chen F, Anderson JW, et al. Effectiveness and safety of laropiprant on niacin-induced flushing. Am J Cardiol. 2012;110(6):817-822. (PubMed)
134. Hps Thrive Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J. 2013;34(17):1279-1291. (PubMed)
135. Lewey J, Shrank WH, Bowry AD, Kilabuk E, Brennan TA, Choudhry NK. Gender and racial disparities in adherence to statin therapy: a meta-analysis. Am Heart J. 2013;165(5):665-678, 678 e661. (PubMed)
136. Cheung MC, Zhao XQ, Chait A, Albers JJ, Brown BG. Antioxidant supplements block the response of HDL to simvastatin-niacin therapy in patients with coronary artery disease and low HDL. Arterioscler Thromb Vasc Biol. 2001;21(8):1320-1326. (PubMed)
137. Brown BG, Cheung MC, Lee AC, Zhao XQ, Chait A. Antioxidant vitamins and lipid therapy: end of a long romance? Arterioscler Thromb Vasc Biol. 2002;22(10):1535-1546. (PubMed)
138. Rios-Avila L, Coats B, Chi YY, et al. Metabolite profile analysis reveals association of vitamin B-6 with metabolites related to one-carbon metabolism and tryptophan catabolism but not with biomarkers of inflammation in oral contraceptive users and reveals the effects of oral contraceptives on these processes. J Nutr. 2015;145(1):87-95. (PubMed)
139. Dollerup OL, Christensen B,Svart M, et al. A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: safety, insulin-sensitivity, and lipid-mobilizing effects. Am J Clin Nutr. 2018;108:343-353. (PubMed)
Pantothenic Acid
Contents
Summary
- Pantothenic acid — also known as vitamin B5 — is a water-soluble vitamin that is a precursor in the synthesis of coenzyme A. Coenzyme A is essential to many biochemical reactions that sustain life. Also, the phosphopantetheinyl moiety of coenzyme A is required for the biological activity of several proteins, including the acyl-carrier protein involved in fatty acid synthesis. (More information)
- Pantothenic acid is essential to all forms of life. It is ubiquitously found in foods of plant and animal origin, and dietary deficiency is very rare. (More information)
- The Food and Nutrition Board of the National Academy of Medicine set an adequate intake (AI) of 5 milligrams (mg)/day for adults based on the estimated daily average intake of pantothenic acid. (More information)
- Evidence from limited intervention studies suggests that pantothenic acid and/or pantothenol (alcohol analog) might improve the healing process of skin wounds. Yet, additional larger studies are warranted. (More information)
- Treatment with high-dose pantethine — a pantothenic acid derivative — has been shown to lower serum cholesterol and lipid concentrations. Although pantethine therapy appears to be well tolerated, medical supervision is indispensable. (More information)
- Foods rich in pantothenic acid include animal organs (liver and kidney), fish, shellfish, milk products, eggs, avocados, legumes, mushrooms, and sweet potatoes. (More information)
- Little or no toxicity has been associated with dietary and supplemental pantothenic acid such that no tolerable upper intake level (UL) has been set. (More information)
Pantothenic acid, also known as vitamin B5, is essential to all forms of life (1). Pantothenic acid is found throughout all branches of life in the form of coenzyme A, a vital coenzyme in numerous chemical reactions (2, 3).
Function
Synthesis of pantothenic acid cofactors
Coenzyme A
Pantothenic acid is a precursor in the biosynthesis of coenzyme A (CoA) (Figure 1), an essential coenzyme in a variety of biochemical reactions that sustain life (see Coenzyme A). Pantothenic acid kinase 2 (PANK2) catalyzes the initial step of phosphorylation of pantothenic acid to 4’-phosphopantothenic acid. Coenzyme A and its derivatives inhibit the synthesis of 4’-phosphopantothenic acid, but the inhibition can be reversed by carnitine, required for the transport of fatty acids into the mitochondria (4). The subsequent reactions in this biosynthetic pathway include the synthesis of the intermediate 4’-phosphopantetheine, as well as the recycling of coenzyme A to 4’-phosphopantetheine (Figure 1).

4’-Phosphopantetheine
The 4’-phosphopantetheinyl moiety of coenzyme A can be transferred to enzymes in which 4’-phosphopantetheine is an essential cofactor for their biological activities (see 4’-Phosphopantetheinylation).
Cofactor and co-substrate function
Coenzyme A
Coenzyme A reacts with acyl groups, giving rise to thioester derivatives, such as acetyl-CoA, succinyl-CoA, malonyl-CoA, and 3-hydroxy-3-methylglutaryl (HMG)-CoA. Coenzyme A and its acyl derivatives are required for reactions that generate energy from the degradation of dietary fat, carbohydrates, and proteins. In addition, coenzyme A in the form of acetyl-CoA and succinyl-CoA is involved in the citric acid cycle; in the synthesis of essential fats, cholesterol, steroid hormones, vitamins A and D, the neurotransmitter acetylcholine; and in the fatty acid β-oxidation pathway. Coenzyme A derivatives are also required for the synthesis of the hormone, melatonin, and for a component of hemoglobin called heme. Further, metabolism of a number of drugs and toxins by the liver requires coenzyme A (5).
Coenzyme A was named for its role in acetylation reactions. Most acetylated proteins in the body have been modified by the addition of an acetate group that was donated by the coenzyme A thioester derivative, acetyl-CoA. Protein acetylation alters the overall charge of proteins, modifying their three-dimensional structure and, potentially, their function. For example, acetylation is a mechanism that regulates the activity of peptide hormones, including those produced by the pituitary gland (6). Also, protein acetylation, like other posttranslational modifications, has been shown to regulate the subcellular localization, the function, and the half-life of many signaling molecules, transcription factors, and enzymes. Notably, the acetylation of histones plays a role in the regulation of gene expression by facilitating transcription (i.e., mRNA synthesis), while deacetylated histones are usually associated with chromatin compaction and gene silencing. The acetylation of histones was found to result in structural changes of the chromatin, which affect both DNA-protein and protein-protein interactions. Crosstalk between acetylation marks and other posttranscriptional modifications of the histones also facilitate the recruitment of transcriptional regulators to the promoter of genes that are subsequently transcribed (reviewed in 7).
Finally, a number of signaling molecules are modified by the attachment of long-chain fatty acids donated by coenzyme A. These modifications are known as protein acylation and have central roles in cell-signaling pathways (5).
4’-Phosphopantetheinylation
Specific multi-enzyme complexes, which need to carry out several reactions in an orderly manner, may require the covalent attachment of a 4’-phosphopantetheine arm to a “carrier” domain (or protein). This carrier domain holds substrates or reaction intermediates during the progression through the various enzymatic reactions. In mammals, the transfer of the 4’-phosphopantetheinyl moiety from coenzyme A to a conserved serine residue of a specific carrier domain is catalyzed by one unique phosphopantetheinyl transferase (8). The 4’-phosphopantetheinylation is necessary for the conversion of apo-enzymes into fully active holo-enzymes (see below).
Acyl-carrier protein
Lipids are fat molecules essential for normal physiological function and, among other types, include sphingolipids (essential components of the myelin sheath that enhances nerve transmission), phospholipids (important structural components of cell membranes), and fatty acids. Fatty acid synthase (FAS) is a multi-enzyme complex that catalyzes the synthesis of fatty acids. Within the FAS complex, the acyl-carrier protein (ACP) requires pantothenic acid in the form of 4'-phosphopantetheine for its activity as a carrier protein (4). A group, such as the 4’-phosphopantetheinyl moiety for ACP, is called a prosthetic group; the prosthetic group is not composed of amino acids and is a tightly bound cofactor required for the biological activity of some proteins (Figure 2). Acetyl-CoA, malonyl-CoA, and ACP are all required for the synthesis of fatty acids in the cytosol. During fatty acid synthesis, the acyl groups of acetyl-CoA and malonyl-CoA are transferred to the sulfhydryl group (-SH) of the 4’-phosphopantetheinyl moiety of ACP. The prosthetic group is used as a flexible arm to transfer the growing fatty acid chain to each of the enzymatic centers of the type I FAS complex. In the mitochondria, 4'-phosphopantetheine also serves as a prosthetic group for an ACP homolog present in mitochondrial type II FAS complex (9).

10-Formyltetrahydrofolate dehydrogenase
The enzyme 10-formyltetrahydrofolate dehydrogenase (FDH) catalyzes the conversion of 10-formyltetrahydrofolate to tetrahydrofolate, an essential cofactor in the metabolism of nucleic acids and amino acids (Figure 3). Similar to ACP, FDH requires a 4’-phosphopantetheine prosthetic group for its biological activity. The prosthetic group acts as a swinging arm to couple the activities of the two catalytic domains of FDH (10, 11). A homolog of FDH in mitochondria also requires 4’-phosphopantetheinylation to be biologically active (12).

α-Aminoadipate semialdehyde synthase
4’- Phosphopantetheinylation is required for the biological activity of the apo-enzyme a-aminoadipate semialdehyde synthase (AASS). AASS catalyzes the initial reactions in the mitochondrial pathway for the degradation of lysine — an essential amino acid for humans. AASS is made of two catalytic domains. The lysine-ketoglutarate reductase domain first catalyzes the conversion of lysine to saccharopine. Saccharopine is further converted to a-aminoadipate semialdehyde in a reaction catalyzed by the saccharopine dehydrogenase domain (Figure 4).

Deficiency
Naturally occurring pantothenic acid deficiency in humans is very rare and has been observed only in cases of severe malnutrition. World War II prisoners in the Philippines, Burma, and Japan experienced numbness and painful burning and tingling in their feet; these symptoms were relieved specifically by pantothenic acid supplementation (5). Pantothenic acid deficiency in humans has been induced experimentally by co-administering a pantothenic acid kinase inhibitor (ω-methylpantothenate; Figure 1) and a pantothenic acid-deficient diet. Participants in this experiment complained of headache, fatigue, insomnia, intestinal disturbances, and numbness and tingling of their hands and feet (13). In another study, participants fed only a pantothenic acid-free diet did not develop clinical signs of deficiency, although some appeared listless and complained of fatigue (14).
Calcium homopantothenate (or hopantenate) is a pantothenic acid antagonist with cholinergic effects (i.e., similar to those of the neurotransmitter, acetylcholine). This compound is used in Japan to enhance mental function, especially in Alzheimer’s disease. A rare side effect was the development of hepatic encephalopathy, a condition of abnormal brain function resulting from the failure of the liver to eliminate toxins. The encephalopathy was reversed by pantothenic acid supplementation, suggesting that it was due to homopantothenate-induced pantothenic acid deficiency (15). Of note, autosomal recessive mutations in the human gene PANK2, which codes for pantothenic acid kinase 2 (Figure 1), result in impaired synthesis of 4'-phosphopantetheine and coenzyme A (see Function). This rare disorder, called pantothenate kinase-associated neurodegeneration (PKAN), is associated with iron accumulation in the brain and characterized by visual and intellectual impairments, dystonia, speech abnormalities, behavioral difficulties, and personality disorders (16-18). Two forms of the disorder have been described: classic PKAN with symptom onset early in life (before age 10 years) and atypical PKAN with symptom onset after age 10, sometimes in early adulthood (19).
Yet, because pantothenic acid is widely distributed in nature and deficiency is extremely rare in humans, most information regarding the consequences of deficiency has been gathered from experimental research in animals (reviewed in 4). Pantothenic acid-deficient rats developed damage to the adrenal glands, while pantothenic acid-deficient monkeys developed anemia due to decreased synthesis of heme, a component of hemoglobin. Dogs with pantothenic acid deficiency developed low blood glucose, rapid breathing and heart rates, and convulsions. Chickens developed skin irritation, feather abnormalities, and spinal nerve damage associated with the degeneration of the myelin sheath. Pantothenic acid-deficient mice showed decreased exercise tolerance and diminished storage of glucose (in the form of glycogen) in muscle and liver. Mice also developed skin irritation and graying of the fur that was reversed with pantothenic acid administration.
The diversity of symptoms emphasizes the numerous functions of pantothenic acid in its coenzyme forms.
Moreover, some recent case-control studies have documented cerebral pantothenic acid deficiency in patients with Alzheimer’s disease (20) and in those with Huntington’s disease (21).
The Adequate Intake (AI)
Because there was little information on the requirements of pantothenic acid in humans, the Food and Nutrition Board of the Institute of Medicine (now the National Academy of Medicine) set an adequate intake (AI) based on observed dietary intakes in healthy population groups (Table 1) (22).
Disease Treatment
Wound healing
The addition of calcium D-pantothenate and/or pantothenol (Figure 5) to the medium of cultured skin fibroblasts given an artificial wound was found to increase cell proliferation and migration, thus accelerating wound healing in vitro (23, 24). Likewise, in vitro deficiency in pantothenic acid induced the expression of differentiation markers in proliferating skin fibroblasts and inhibited proliferation in human keratinocytes (25). The application of ointments containing either calcium D-pantothenate or pantothenol& — also known as D-panthenol or dexpanthenol — to the skin has been shown to accelerate the closure of skin wounds and increase the strength of scar tissue in animals (4).
The effects of topical dexpanthenol on wound healing in humans are unclear. In a placebo-controlled study that included 12 healthy volunteers, the application of dexpanthenol-containing ointment (every 12 hours for 1 to 6 days) in a model of skin wound healing was associated with an enhanced expression of markers of proliferation, inflammation, and tissue repair (26). However, the study failed to report whether these changes in response to dexpanthenol treatment improved the wound-repair process compared to placebo (26). In a randomized, double-blind, placebo-controlled study, the use of dexpanthenol pastilles (300 mg/day for up to 14 days post surgery) was found to accelerate mucosal healing after tonsillectomy in children (27).
Few studies have explored the effects of oral supplementation with pantothenic acid. Early randomized controlled trials in patients undergoing surgery for tattoo removal found that daily co-supplementation with 1 gram or 3 grams of vitamin C and 200 mg or 900 mg of pantothenic acid for 21 days did not significantly improve the wound-healing process (28, 29).
High cholesterol
Early studies suggested that pharmacological doses of pantethine, a pantothenic acid derivative, might have a cholesterol-lowering effect (30, 31). Pantethine is made of two molecules of pantetheine joined by a disulfide bond (chemical bond between two molecules of sulfur) (Figure 5). Pantethine is structurally related to coenzyme A and is found in the prosthetic group that is required for the biological function of acyl-carrier protein, formyltetrahydrofolate dehydrogenase, and α-aminoadipate semialdehyde synthase (see Function)
In a 16-week, randomized, double-blind, placebo-controlled study, daily pantethine supplementation (600 mg/day for 8 weeks, followed by 900 mg/day for another 8 weeks) significantly improved the profile of lipid parameters in 120 individuals at low-to-moderate risk of cardiovascular disease. After adjusting to baseline, pantethine was found to be significantly more effective than placebo in lowering the concentrations of low-density lipoprotein (LDL) cholesterol and apolipoprotein B (apoB), as well as reducing the ratio of triglycerides to high-density lipoprotein-cholesterol (TG:HDL-C) (32). In a randomized controlled trial of 24 individuals at low-to-moderate cardiovascular risk, pantethine supplementation (600 mg/day of pantethine for 8 weeks, followed by 900 mg for 8 weeks) decreased concentrations of total cholesterol, LDL cholesterol, and non-HDL cholesterol at 16 weeks compared to placebo (33). Although it appears to be well tolerated and potentially beneficial in improving cholesterol metabolism, pantethine is not a vitamin, and the decision to use pharmacological doses of pantethine to treat elevated blood cholesterol or triglycerides should only be made in collaboration with a qualified healthcare provider who provides appropriate follow up.

Graying of hair
Mice that are deficient in pantothenic acid developed skin irritation and graying of the fur, which is reversed by pantothenic acid administration (34). In humans, evidence that taking pantothenic acid as supplements or using shampoos containing pantothenic acid can prevent or restore hair color is lacking.
Hair loss
A case report of nine patients (35) and two small studies (36, 37) suggested some benefit of intramuscular injections of dexpanthenol in the treatment of alopecia or pattern hair loss; however, placebo-controlled trials would be needed to determine whether the drug has any effects on hair loss.
Sources
Food sources
Pantothenic acid is available in a variety of foods, usually as a component of coenzyme A (CoA) and 4’-phosphopantetheine (see Figure 1). Upon ingestion, dietary coenzyme A and phosphopantetheine are hydrolyzed to pantothenic acid prior to intestinal absorption (4). Animal liver and kidney, fish, shellfish, pork, chicken, egg yolk, milk, yogurt, legumes, mushrooms, avocados, broccoli, and sweet potatoes are good sources of pantothenic acid. Whole grains are also good sources of pantothenic acid; processing and refining grains result in significant losses, up to 55% for wheat and up to 88% for maize (38). Freezing and canning of foods result in similar losses (22). Food preparation and cooking methods don’t substantially affect the vitamin due to its stability, but like other water-soluble vitamins, pantothenic acid can leach into cooking liquids (38).
Large nutritional surveys in the United States have failed to estimate pantothenic acid intake, mainly because of the scarcity of data on the pantothenic acid content of food (22). Smaller studies estimated average daily intakes of pantothenic acid to be between 4 and 7 mg/day in adults. Table 2 lists some rich sources of pantothenic acid, along with their content in milligrams (mg). For more information on the nutrient content of foods, search USDA's FoodData Central.
Intestinal bacteria
The bacteria that normally colonize the colon (large intestine) are capable of synthesizing pantothenic acid. A specialized transporter for the uptake of biotin and pantothenic acid was identified in cultured cells derived from the lining of the colon, suggesting that humans may be able to absorb pantothenic acid and biotin produced by intestinal bacteria (39). However, the extent to which bacterial synthesis contributes to pantothenic acid intake in humans is not known (38, 40).
Supplements
Pantothenol and pantothenate
Supplements commonly contain pantothenol (panthenol), a stable alcohol analog of pantothenic acid, which can be rapidly converted to pantothenic acid by humans. Calcium D-pantothenate, the calcium salt of pantothenic acid, is also available as a supplement (41).
Pantethine
Pantethine is used as a cholesterol-lowering agent in Japan and is available in the US as a dietary supplement (42).
Safety
Toxicity
Pantothenic acid is not known to be toxic in humans. The only adverse effect noted was diarrhea resulting from very high intakes of 10 to 20 g/day of calcium D-pantothenate (43). However, there is one case report of life-threatening eosinophilic pleuropericardial effusion in an elderly woman who took a combination of 10 mg/day of biotin and 300 mg/day of pantothenic acid for two months (44). Due to the lack of reports of adverse effects when the Dietary Reference Intakes (DRI) for pantothenic acid were established in 1998, the Food and Nutrition Board of the Institute of Medicine (now the National Academy of Medicine) did not establish a tolerable upper intake level (UL) for pantothenic acid (22). Pantethine is generally well tolerated in doses up to 1,200 mg/day. However, gastrointestinal side effects, such as nausea and heartburn, have been reported (42). Also, topical formulations containing up to 5% of dexpanthenol (D-panthenol) have been safely used for up to one month. Yet, several cases of skin irritation, allergic contact dermatitis, and eczema have been reported with the use of dexpanthenol-containing ointments (45-49).
Nutrient interactions
Large doses of pantothenic acid have the potential to compete with biotin for intestinal and cellular uptake by the human sodium-dependent multivitamin transporter (hSMVT) (39, 50).
Drug interactions
Oral contraceptives (birth control pills) containing estrogen and progestin may increase the requirement for pantothenic acid (43). Use of pantethine in combination with cholesterol-lowering drugs called statins (HMG-CoA reductase inhibitors) or with nicotinic acid (see the article on Niacin) may produce additive effects on blood lipids (42).
Linus Pauling Institute Recommendation
More data are needed to define the amount of dietary pantothenic acid required to promote optimal health or prevent chronic disease. The Linus Pauling Institute supports the recommendation by the Food and Nutrition Board of 5 mg/day of pantothenic acid for adults. A varied diet should provide enough pantothenic acid for most people. Following the Linus Pauling Institute recommendation to take a daily multivitamin/mineral supplement that contains 100% of the Daily Value (DV) for pantothenic acid will ensure an intake of at least 5 mg/day.
Older adults (>50 years)
There is currently little evidence that older adults differ in their intake of or their requirement for pantothenic acid. Most multivitamin/mineral supplements provide at least 5 mg/day of pantothenic acid.
Authors and Reviewers
Originally written in 2000 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in August 2002 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in May 2004 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2008 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2015 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2023 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in July 2015 by:
Robert B. Rucker, Ph.D.
Distinguished Professor Emeritus
Department of Nutrition and School of Medicine
University of California, Davis
Copyright 2000-2024 Linus Pauling Institute
References
1. Trumbo PR. Pantothenic acid. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed: Lippincott Williams & Wilkins; 2014:351-357.
2. Martinez DL, Tsuchiya Y, Gout I. Coenzyme A biosynthetic machinery in mammalian cells. Biochem Soc Trans. 2014;42(4):1112-1117. (PubMed)
3. Said HM. Water-soluble vitamins. World Rev Nutr Diet. 2015;111:30-37. (PubMed)
4. Miller JW, Rucker RB. Pantothenic acid. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed: John Wiley & Sons, Inc.; 2012:375-390.
5. Bauerly K, Rucker RB. Pantothenic acid. In: Zempleni J, Rucker RB, McCormick DB, Suttie JW, eds. Handbook of vitamins. 4th ed. Boca Raton, Fl: CRC Press; 2007:289-314.
6. Takahashi A, Mizusawa K. Posttranslational modifications of proopiomelanocortin in vertebrates and their biological significance. Front Endocrinol (Lausanne). 2013;4:143. (PubMed)
7. Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014;15(8):536-550. (PubMed)
8. Beld J, Sonnenschein EC, Vickery CR, Noel JP, Burkart MD. The phosphopantetheinyl transferases: catalysis of a post-translational modification crucial for life. Nat Prod Rep. 2014;31(1):61-108. (PubMed)
9. Bunkoczi G, Pasta S, Joshi A, et al. Mechanism and substrate recognition of human holo ACP synthase. Chem Biol. 2007;14(11):1243-1253. (PubMed)
10. Donato H, Krupenko NI, Tsybovsky Y, Krupenko SA. 10-formyltetrahydrofolate dehydrogenase requires a 4'-phosphopantetheine prosthetic group for catalysis. J Biol Chem. 2007;282(47):34159-34166. (PubMed)
11. Strickland KC, Hoeferlin LA, Oleinik NV, Krupenko NI, Krupenko SA. Acyl carrier protein-specific 4'-phosphopantetheinyl transferase activates 10-formyltetrahydrofolate dehydrogenase. J Biol Chem. 2010;285(3):1627-1633. (PubMed)
12. Strickland KC, Krupenko NI, Dubard ME, Hu CJ, Tsybovsky Y, Krupenko SA. Enzymatic properties of ALDH1L2, a mitochondrial 10-formyltetrahydrofolate dehydrogenase. Chem Biol Interact. 2011;191(1-3):129-136. (PubMed)
13. Hodges RE, Ohlson MA, Bean WB. Pantothenic acid deficiency in man. J Clin Invest. 1958;37(11):1642-1657. (PubMed)
14. Fry PC, Fox HM, Tao HG. Metabolic response to a pantothenic acid deficient diet in humans. J Nutr Sci Vitaminol (Tokyo). 1976;22(4):339-346. (PubMed)
15. Bender DA. Optimum nutrition: thiamin, biotin and pantothenate. Proc Nutr Soc. 1999;58(2):427-433. (PubMed)
16. Kurian MA, Hayflick SJ. Pantothenate kinase-associated neurodegeneration (PKAN) and PLA2G6-associated neurodegeneration (PLAN): review of two major neurodegeneration with brain iron accumulation (NBIA) phenotypes. Int Rev Neurobiol. 2013;110:49-71. (PubMed)
17. Hayflick SJ. Defective pantothenate metabolism and neurodegeneration. Biochem Soc Trans. 2014;42(4):1063-1068. (PubMed)
18. Chang X, Zhang J, Jiang Y, Wang J, Wu Y. Natural history and genotype-phenotype correlation of pantothenate kinase-associated neurodegeneration. CNS Neurosci Ther. 2020;26(7):754-761. (PubMed)
19. Gregory A, Hayflick SJ. Pantothenate kinase-associated neurodegeneration. In: Adam MP, Mirzaa GM, Pagon RA, et al., eds. GeneReviews((R)). Seattle (WA); 1993. (PubMed)
20. Xu J, Patassini S, Begley P, et al. Cerebral deficiency of vitamin B5 (d-pantothenic acid; pantothenate) as a potentially-reversible cause of neurodegeneration and dementia in sporadic Alzheimer's disease. Biochem Biophys Res Commun. 2020;527(3):676-681. (PubMed)
21. Patassini S, Begley P, Xu J, et al. Cerebral vitamin B5 (D-pantothenic acid) deficiency as a potential cause of metabolic perturbation and neurodegeneration in Huntington's disease. Metabolites. 2019;9(6):113. (PubMed)
22. Food and Nutrition Board, Institute of Medicine. Pantothenic acid. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B-6, Vitamin B-12, Pantothenic Acid, Biotin, and Choline. Washington, D.C.: National Academy Press; 1998:357-373. (National Academy Press)
23. Weimann BI, Hermann D. Studies on wound healing: effects of calcium D-pantothenate on the migration, proliferation and protein synthesis of human dermal fibroblasts in culture. Int J Vitam Nutr Res. 1999;69(2):113-119. (PubMed)
24. Wiederholt T, Heise R, Skazik C, et al. Calcium pantothenate modulates gene expression in proliferating human dermal fibroblasts. Exp Dermatol. 2009;18(11):969-978. (PubMed)
25. Kobayashi D, Kusama M, Onda M, Nakahata N. The effect of pantothenic acid deficiency on keratinocyte proliferation and the synthesis of keratinocyte growth factor and collagen in fibroblasts. J Pharmacol Sci. 2011;115(2):230-234. (PubMed)
26. Heise R, Skazik C, Marquardt Y, et al. Dexpanthenol modulates gene expression in skin wound healing in vivo. Skin Pharmacol Physiol. 2012;25(5):241-248. (PubMed)
27. Celebi S, Tepe C, Yelken K, Celik O. Efficacy of dexpanthenol for pediatric post-tonsillectomy pain and wound healing. Ann Otol Rhinol Laryngol. 2013;122(7):464-467. (PubMed)
28. Vaxman F, Olender S, Lambert A, et al. Effect of pantothenic acid and ascorbic acid supplementation on human skin wound healing process. A double-blind, prospective and randomized trial. Eur Surg Res. 1995;27(3):158-166. (PubMed)
29. Vaxman F, Olender S, Lambert A, Nisand G, Grenier JF. Can the wound healing process be improved by vitamin supplementation? Experimental study on humans. Eur Surg Res. 1996;28(4):306-314. (PubMed)
30. Coronel F, Tornero F, Torrente J, et al. Treatment of hyperlipemia in diabetic patients on dialysis with a physiological substance. Am J Nephrol. 1991;11(1):32-36. (PubMed)
31. Gaddi A, Descovich GC, Noseda G, et al. Controlled evaluation of pantethine, a natural hypolipidemic compound, in patients with different forms of hyperlipoproteinemia. Atherosclerosis. 1984;50(1):73-83. (PubMed)
32. Rumberger JA, Napolitano J, Azumano I, Kamiya T, Evans M. Pantethine, a derivative of vitamin B(5) used as a nutritional supplement, favorably alters low-density lipoprotein cholesterol metabolism in low- to moderate-cardiovascular risk North American subjects: a triple-blinded placebo and diet-controlled investigation. Nutr Res. 2011;31(8):608-615. (PubMed)
33. Evans M, Rumberger JA, Azumano I, Napolitano JJ, Citrolo D, Kamiya T. Pantethine, a derivative of vitamin B5, favorably alters total, LDL and non-HDL cholesterol in low to moderate cardiovascular risk subjects eligible for statin therapy: a triple-blinded placebo and diet-controlled investigation. Vasc Health Risk Manag. 2014;10:89-100. (PubMed)
34. Kuo YM, Hayflick SJ, Gitschier J. Deprivation of pantothenic acid elicits a movement disorder and azoospermia in a mouse model of pantothenate kinase-associated neurodegeneration. J Inherit Metab Dis. 2007;30(3):310-317. (PubMed)
35. Kutlu O. Dexpanthenol may be a novel treatment for male androgenetic alopecia: Analysis of nine cases. Dermatol Ther. 2020;33(3):e13381. (PubMed)
36. Samadi A, Ketabi Y, Firooz R, Firooz A. Efficacy of intramuscular injections of biotin and dexpanthenol in the treatment of diffuse hair loss: A randomized, double-blind controlled study comparing two brands. Dermatol Ther. 2022;35(9):e15695. (PubMed)
37. Kutlu O, Metin A. Systemic dexpanthenol as a novel treatment for female pattern hair loss. J Cosmet Dermatol. 2021;20(4):1325-1330. (PubMed)
38. Hrubsa M, Siatka T, Nejmanova I, et al. Biological properties of vitamins of the B-complex, part 1: vitamins B(1), B(2), B(3), and B(5). Nutrients. 2022;14(3):484. (PubMed)
39. Said HM, Ortiz A, McCloud E, Dyer D, Moyer MP, Rubin S. Biotin uptake by human colonic epithelial NCM460 cells: a carrier-mediated process shared with pantothenic acid. Am J Physiol. 1998;275(5 Pt 1):C1365-1371. (PubMed)
40. Said HM. Intestinal absorption of water-soluble vitamins in health and disease. Biochem J. 2011;437(3):357-372. (PubMed)
41. US Department of Health and Human Services, National Institutes of Health, Office of Dietary Supplements. Dietary Supplement Label Database (DSLD). [Internet]. [cited 5/1/23)]. Available from: https://dsld.od.nih.gov/.
42. Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. 2nd ed: Thomson Reuters; 2008.
43. Flodin N. Pharmacology of micronutrients. New York: Alan R. Liss, Inc.; 1988.
44. Debourdeau PM, Djezzar S, Estival JL, Zammit CM, Richard RC, Castot AC. Life-threatening eosinophilic pleuropericardial effusion related to vitamins B5 and H. Ann Pharmacother. 2001;35(4):424-426. (PubMed)
45. Herbst RA, Uter W, Pirker C, Geier J, Frosch PJ. Allergic and non-allergic periorbital dermatitis: patch test results of the Information Network of the Departments of Dermatology during a 5-year period. Contact Dermatitis. 2004;51(1):13-19. (PubMed)
46. Schmuth M, Wimmer MA, Hofer S, et al. Topical corticosteroid therapy for acute radiation dermatitis: a prospective, randomized, double-blind study. Br J Dermatol. 2002;146(6):983-991. (PubMed)
47. Blanchard G, Kerre S, Walker A, et al. Allergic contact dermatitis from pantolactone and dexpanthenol in wound healing creams. Contact Dermatitis. 2022;87(5):468-471. (PubMed)
48. Fernandes RA, Santiago L, Gouveia M, Goncalo M. Allergic contact dermatitis caused by dexpanthenol-Probably a frequent allergen. Contact Dermatitis. 2018;79(5):276-280. (PubMed)
49. Miroux-Catarino A, Silva L, Amaro C, Viana I. Allergic contact dermatitis caused dexpanthenol-But is that all? Contact Dermatitis. 2019;81(5):391-392. (PubMed)
50. Chirapu SR, Rotter CJ, Miller EL, Varma MV, Dow RL, Finn MG. High specificity in response of the sodium-dependent multivitamin transporter to derivatives of pantothenic acid. Curr Top Med Chem. 2013;13(7):837-842. (PubMed)
Riboflavin
Contents
Summary
- Riboflavin is the precursor of the coenzymes flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN). FAD and FMN act as electron carriers in a number of oxidation-reduction (redox) reactions involved in energy production, cellular antioxidant function, and in numerous metabolic pathways. (More information)
- Riboflavin (as FAD or FMN) is required for the metabolism of iron and vitamin B6, and in the synthesis of niacin from tryptophan. It also plays an essential role in folate and related one-carbon metabolism, where FAD is required as a cofactor for methylenetetrahydrofolate reductase (MTHFR), a key folate-metabolizing enzyme. (More information)
- Clinical signs of riboflavin deficiency include skin disorders, angular stomatitis, cheilosis, glossitis, and corneal vascularization. Riboflavin deficiency occurs commonly in low- and middle-income countries. (More information)
- Subclinical deficiency (low status) of riboflavin without clinical signs may be widespread, including in high-income countries, but it usually goes undetected because riboflavin biomarkers are very rarely measured in human studies. (More information)
- Low riboflavin status is associated with preeclampsia in pregnant women, a condition that may progress to eclampsia and cause severe bleeding and death (More information). It is also linked with cataracts in older individuals. (More information)
- Deficient/low status of riboflavin interferes with the metabolism of folate, particularly in individuals with the variant 677TT genotype in MTHFR. There is some evidence that these individuals exhibit a higher risk of cancer. (More information)
- Case reports have shown that patients with autosomal recessive disorders of riboflavin metabolism may benefit from riboflavin supplementation. (More information)
- The MTHFR C677T polymorphism is associated with an increased risk of hypertension. Intervention studies have shown that supplementation with riboflavin (the MTHFR cofactor) effectively lowers blood pressure, specifically in individuals with the variant MTHFR 677TT genotype. (More information)
- Riboflavin supplementation has been evaluated as a prophylactic agent for migraine headache, but the results are conflicting. (More information)
-
Riboflavin supplementation has been studied as a potential adjunct therapy in cancer, certain eye disorders, and multiple sclerosis. (More information)
Riboflavin is a water-soluble B vitamin, also known as vitamin B2. Riboflavin is primarily found as an integral component of the coenzymes flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN) (1). Coenzymes derived from riboflavin are termed flavocoenzymes, and enzymes that use a flavocoenzyme are called flavoproteins (2).
Function
Oxidation-reduction (redox) reactions
Living organisms derive most of their energy from redox reactions, which are processes that involve the transfer of electrons. Flavocoenzymes participate in redox reactions in numerous metabolic pathways (3). They are critical for the metabolism of carbohydrates, lipids, and proteins. FAD is part of the electron transport (respiratory) chain, which is central to energy production. In conjunction with cytochrome P-450, flavocoenzymes also participate in the metabolism of drugs and toxins (4).
Antioxidant functions
Glutathione reductase is a FAD-dependent enzyme that participates in the redox cycle of glutathione. The glutathione redox cycle plays a major role in protecting organisms from reactive oxygen species, such as hydroperoxides. Glutathione reductase requires FAD to regenerate two molecules of reduced glutathione from oxidized glutathione. Riboflavin deficiency has been associated with increased oxidative stress (4) Measurement of glutathione reductase activity in red blood cells is commonly used to assess riboflavin nutritional status (5).
Glutathione peroxidases (GPx), selenium-containing enzymes, require two molecules of reduced glutathione to break down hydroperoxides. GPx are involved in the glutathione oxidation-reduction (redox) cycle (Figure 1).

Xanthine oxidase, another FAD-dependent enzyme, catalyzes the oxidation of hypoxanthine and xanthine to uric acid. Uric acid is one of the most effective water-soluble antioxidants in the blood. Riboflavin deficiency can result in decreased xanthine oxidase activity, reducing blood uric acid levels (6).
Nutrient interactions
Riboflavin (as FAD or FMN) is required for the synthesis of niacin from tryptophan, and in the metabolism of vitamin B6 and iron. It is also essential for folate and related one-carbon metabolism, where FAD is required as a cofactor for methylenetetrahydrofolate reductase (MTHFR), a key folate-metabolizing enzyme.
B-complex vitamins
Flavoproteins are involved in the metabolism of several other vitamins: vitamin B6, niacin, vitamin B12, and folate. Therefore, low and deficient riboflavin status can affect several enzyme systems. The conversion of vitamin B6 to its active coenzyme form in tissues, pyridoxal 5'-phosphate (PLP), requires the FMN-dependent enzyme, pyridoxine 5'-phosphate oxidase (PPO) (7). Human studies have provided evidence of the metabolic dependency of vitamin B6 on riboflavin status in older (8-10) and younger (10) adults. The synthesis of the niacin-containing coenzymes, NAD and NADP, from the amino acid tryptophan, requires the FAD-dependent enzyme, kynurenine 3-monooxygenase. Severe riboflavin deficiency can thus decrease the conversion of tryptophan to NAD and NADP, increasing the risk of niacin deficiency (3).
Methylenetetrahydrofolate reductase (MTHFR) is an FAD-dependent enzyme that plays a key role in one-carbon metabolism by catalyzing the reduction of 5,10 methyleneTHF to 5 methylTHF. Once formed, 5 methylTHF is used by methionine synthase for the vitamin B12-dependent conversion of homocysteine to methionine and the formation of THF (Figure 2). Both FMN and FAD are coenzymes for the enzyme methionine synthase reductase, which is responsible for the regeneration of methylcobalamin, the biologically active form of vitamin B12 acting as a coenzyme for methionine synthase (11). Along with other B vitamins (folate, vitamin B12, and vitamin B6), higher dietary riboflavin intakes have been associated with lower plasma concentrations of homocysteine (12). In individuals homozygous for the C677T polymorphism in the MTHFR gene, low riboflavin status is associated with elevated plasma homocysteine, and in turn linked with a higher risk of cardiovascular disease and other chronic diseases (13, 14). Furthermore, supplementation with riboflavin results in marked lowering of homocysteine concentrations specifically in individuals with the variant MTHFR 677TT genotype (15). Such results illustrate that chronic disease risk may be influenced by complex interactions between genetic and dietary factors.

Iron
Riboflavin deficiency alters iron metabolism. Although the mechanism is not clear, research in animals suggests that riboflavin deficiency may impair iron absorption, increase intestinal loss of iron, and/or impair iron utilization for the synthesis of hemoglobin (16). In humans, low dietary intake of riboflavin has been associated with an increased risk for anemia (17), and improving riboflavin nutritional status has been found to increase circulating hemoglobin levels (18). Correction of riboflavin deficiency in individuals who are both riboflavin and iron deficient improves the response of iron-deficiency anemia to iron therapy (19). Anemia during pregnancy, a worldwide public health problem, is responsible for considerable perinatal morbidity and mortality (20, 21). The management of maternal anemia typically involves supplementation with iron alone or iron in combination with folic acid (22). It is possible that the inclusion of riboflavin could enhance the effects of iron-folic acid supplementation in treating maternal anemia, but the evidence is limited. There are, however, randomized, double-blind intervention trials conducted in pregnant women with anemia in Southeast Asia showing that a combination of folic acid, iron, vitamin A, and riboflavin improved hemoglobin levels and decreased anemia prevalence compared to iron-folic acid supplementation alone (23, 24).
Deficiency
Ariboflavinosis is the medical name for clinical riboflavin deficiency, which occurs commonly in low- and middle-income countries. Riboflavin deficiency is rarely found in isolation; it typically occurs in combination with deficiencies of other water-soluble vitamins. Clinical signs of riboflavin deficiency include sore throat, redness and swelling of the lining of the mouth and throat, cracks or sores on the outsides of the lips (cheliosis) and at the corners of the mouth (angular stomatitis), inflammation and redness of the tongue (magenta tongue), and a moist, scaly skin inflammation (seborrheic dermatitis). Other signs may involve the formation of blood vessels in the clear covering of the eye (vascularization of the cornea) and decreased red blood cell count in which the existing red blood cells contain normal levels of hemoglobin and are of normal size (normochromic normocytic anemia) (1, 3). Subclinical deficiency (low status) of riboflavin without clinical signs may be widespread, including in high-income countries, but usually goes undetected because riboflavin biomarkers are very rarely measured in human studies. Low or deficient riboflavin status may result in decreased conversion of vitamin B6 to its active coenzyme form (PLP) and decreased conversion of tryptophan to niacin (see Nutrient interactions).
Preeclampsia is defined as the presence of elevated blood pressure, protein in the urine, and edema (significant swelling) during pregnancy. About 5% of women with preeclampsia progress to eclampsia, a significant cause of maternal and fetal death. Eclampsia is characterized by seizures, in addition to high blood pressure and increased risk of hemorrhage (severe bleeding) (25). A study in 154 pregnant women at increased risk of preeclampsia found that those who were riboflavin deficient were 4.7 times more likely to develop preeclampsia than those who had adequate riboflavin nutritional status (26). The cause of preeclampsia-eclampsia is not known. Decreased intracellular levels of flavocoenzymes could cause mitochondrial dysfunction, increase oxidative stress, and interfere with nitric oxide release and thus blood vessel dilation – all of these changes have been associated with preeclampsia (26).
A 2015 meta-analysis of 54 case-control studies found that the MTHFR C677T polymorphism was associated with an increased risk of preeclampsia, especially in Caucasian and Asian populations (27). The reduction in the flavoprotein MTHFR activity observed in individuals with the variant MTHFR 677TT genotype leads to an increase in plasma homocysteine (14); higher homocysteine concentrations have been associated with preeclampsia (28). One small randomized controlled trial in 450 pregnant women in West Africa, without specified MTHFR genotype but at high risk for preeclampsia, found that supplementation with 15 mg of riboflavin daily was not effective in preventing the condition (29), but the study was likely underpowered to detect a significant effect. Further studies are needed to assess the potential benefit of riboflavin supplementation in reducing perinatal complications generally and specifically in preeclamptic women with the MTHFR 677TT genotype.
Risk factors for riboflavin deficiency
Alcoholics are at an increased risk of riboflavin deficiency, likely due to decreased dietary intake, decreased absorption, and/or impaired utilization of riboflavin. Interestingly, the elevated blood homocysteine concentrations associated with riboflavin deficiency rapidly decline during alcohol withdrawal (30). Additionally, people with anorexia rarely consume adequate dietary riboflavin, and those who are lactose intolerant are unlikely to meet requirements due to the avoidance of dairy products, the major dietary sources of riboflavin. The conversion of riboflavin into the active cofactor forms FAD and FMN is impaired in hypothyroidism and adrenal insufficiency (3, 4). Further, people who are very active physically (athletes, laborers) may have slightly increased riboflavin requirements. However, riboflavin supplementation has not generally been found to increase exercise tolerance or performance (31) unless the individuals are riboflavin deficient (32).
The Recommended Dietary Allowance (RDA)
The RDA for riboflavin, revised in 1998, is based on the prevention of deficiency (Table 1). Clinical signs of deficiency in humans appear at intakes of less than 0.5 to 0.6 milligrams (mg)/day, and urinary excretion of riboflavin is seen at intake levels of approximately 1 mg/day (1).
| Life Stage | Age | Males (mg/day) | Females (mg/day) |
|---|---|---|---|
| Infants | 0-6 months | 0.3 (AI) | 0.3 (AI) |
| Infants | 7-12 months | 0.4 (AI) | 0.4 (AI) |
| Children | 1-3 years | 0.5 | 0.5 |
| Children | 4-8 years | 0.6 | 0.6 |
| Children | 9-13 years | 0.9 | 0.9 |
| Adolescents | 14-18 years | 1.3 | 1.0 |
| Adults | 19 years and older | 1.3 | 1.1 |
| Pregnancy | all ages | - | 1.4 |
| Breast-feeding | all ages | - | 1.6 |
Disease Prevention
Cataracts
Age-related cataracts are the leading cause of visual disability in the US and other developed countries. Research has focused on the role of nutritional antioxidants because of evidence that light-induced oxidative damage of lens proteins may lead to the development of age-related cataracts. A case-control study found significantly decreased risk of age-related cataracts (33% to 51%) in men and women in the highest quintile of dietary riboflavin intake (median of 1.6 to 2.2 mg/day) compared to those in the lowest quintile (median of 0.08 mg/day in both men and women) (33). Another case-control study reported that individuals in the highest quintile of riboflavin status, as measured by red blood cell glutathione reductase activity, had approximately one-half the occurrence of age-related cataract as those in the lowest quintile of riboflavin status, though the results were not statistically significant (34). A cross-sectional study of 2,900 Australian men and women, 49 years of age and older, found that those in the highest quintile of riboflavin intake were 50% less likely to have cataracts than those in the lowest quintile (35). A prospective cohort study of more than 50,000 women did not observe a difference between rates of cataract extraction between women in the highest quintile of riboflavin intake (median of 1.5 mg/day) and women in the lowest quintile (median of 1.2 mg/day) (36). However, the range between the highest and lowest quintiles was small, and median intake levels for both quintiles were above the RDA for riboflavin. A study in 408 women found that higher dietary intakes of riboflavin were inversely associated with a five-year change in lens opacification (37). A randomized controlled trial using a fractional factorial design showed that compared with placebo, the combined supplementation with riboflavin (3 mg/day) and niacin (40 mg/day) for five to six years reduced the prevalence of nuclear cataract but increased the progression of posterior subcapsular cataracts in population affected by multiple nutrient deficiency living in rural China (38). Of note is that the results of this trial are somewhat conflicting, and the study design does not allow the effects of riboflavin and niacin to be differentiated. In summary, there is some evidence predominantly from observational studies, that suggests higher riboflavin status might be beneficial; however, more evidence from well-designed, randomized controlled trials is needed to confirm a role for riboflavin in the prevention of cataracts.
Cancer
The flavoprotein, methylenetetrahydrofolate reductase (MTHFR), plays a pivotal role in folate-mediated one-carbon metabolism. MTHFR converts 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate, the cofactor form necessary for the re-methylation of homocysteine to methionine (see Figure 2 above). The conversion of homocysteine to methionine is of importance for homocysteine detoxification and for the production of S-adenosylmethionine (SAM), the methyl donor for the methylation of DNA and histones. Folate deficiency and elevated homocysteine concentrations may increase cancer risk (see the article on Folate). Aberrant methylation changes are also known to alter the structure and function of DNA and histones during cancer development (39). Since MTHFR controls the detoxification of homocysteine and the supply of methyl groups for SAM synthesis, a reduction in its activity can affect homocysteine metabolism and disturb cellular methylation processes. The substitution of a cytosine by a thymine in position 677 (c.677C>T) in the MTHFR gene is a polymorphism that affects the binding of FAD and leads to an increased propensity for MTHFR to lose its flavin coenzyme (40). Subjects homozygous for this mutation (MTHFR 677TT genotype) exhibit reduced MTHFR activity and increased risk for a wide variety of cancers (41-43), but the evidence of an association between this polymorphism and cancer is inconsistent, with some reports suggesting a reduction in colorectal cancer risk with the T allele (44).
As mentioned above (see B-complex vitamins), riboflavin intake is a determinant of homocysteine concentration. This suggests that riboflavin status can influence MTHFR activity and the metabolism of folate, thereby affecting cancer risk (43). In a randomized, double-blind, placebo-controlled study, 93 subjects with colorectal polyps and 86 healthy subjects were given a placebo, folic acid (400 or 1,200 μg/day), or folic acid (400 μg/day) plus riboflavin (5 mg/day) for 45 days. These interventions significantly improved folate and riboflavin status in vitamin-supplemented individuals compared to those taking the placebo. Interestingly, riboflavin enhanced the effect of 400 μg folic acid on circulating 5-methyltetrahydrofolate (5-MTHF) specifically in the polyp patients with the C677T genetic variant (45). This suggests that riboflavin may improve the response to folic acid supplementation in individuals with a reduced MTHFR activity. Additionally, a prospective cohort study of 88,045 postmenopausal women found total (dietary plus supplemental) intake of riboflavin to be inversely correlated with colorectal cancer risk when comparing the highest (>3.97 mg) and lowest (<1.80 mg) quartiles of daily intake (46); intake in the reference group was well above the RDA for riboflavin of 1.1 mg/day. The subjects in this study were not prescreened to identify those with the variant MTHFR 677TT genotype, and the association between this polymorphism and colorectal cancer remains unclear, with some reports suggesting a reduction in cancer risk with the T allele (44). Two meta-analyses have found inverse associations between riboflavin intake and risk of colorectal cancer (47, 48). The most recent of these was a dose-response meta-analysis that pooled results from five prospective cohort studies, nine case-control studies, and two studies reporting blood concentrations of riboflavin. This analysis found that higher intakes of riboflavin were associated with a significantly lower risk of colorectal cancer (RR=0.87; 95% CI, 0.81-0.93); inverse associations were observed for both dietary riboflavin intake and total daily intake from the diet and supplements (48).
Associations between riboflavin intake and cancer risk have also been evaluated in other types of cancer. A seven-year intervention study evaluated the use of riboflavin-fortified salt in 22,093 individuals at high risk for esophageal cancer in China. Riboflavin status and esophageal pathology (percent normal, dysplastic, and cancerous tissues) improved in the intervention group compared to the control group, but the lower incidence of esophageal cancer found in the intervention group was not statistically significant (49). Additionally, a 25-year follow up of an intervention trial in patients at high risk for gastric cancer found that dietary supplementation with riboflavin (3.2 mg/day) and niacin (40 mg/day) for five years decreased the risk of mortality from esophageal cancer by 8% but had no effect on mortality from gastric cancer (50). In the Melbourne Collaborative Cohort Study, which followed 41,514 men and women over a 15-year period, weak inverse associations were found between riboflavin intake and lung cancer (51) and breast cancer (52); no association of riboflavin intake with prostate cancer was observed in this cohort (53). A 2017 meta-analysis of 10 observational studies found an overall inverse association of riboflavin intake and breast cancer incidence and reported a 6% lower risk with each 1 mg/day increment of riboflavin intake (54). Further, studies to date have not found riboflavin intake or measures of riboflavin status to be associated with renal cell carcinoma, as reviewed in a recent meta-analysis (55).
Disease Treatment
Migraine headaches
Some evidence indicates that impaired mitochondrial oxygen metabolism in the brain may play a role in the pathology of migraine headaches. Since riboflavin is the precursor of the two flavocoenzymes (FAD and FMN) required by the flavoproteins of the mitochondrial electron transport chain, supplemental riboflavin has been investigated as a treatment for migraine. A randomized controlled trial examined the effect of very high dose riboflavin (400 mg/day) for three months on migraine prevention in 54 men and women with a history of recurrent migraine headaches (56). Riboflavin compared to placebo reduced attack frequency and the number of headache days, though the beneficial effect was most pronounced during the third month of treatment (56). Another study by the same investigators found that treatment with either a β-blocker drug or high-dose riboflavin (400 mg/day) for four months resulted in clinical improvement, but each therapy appeared to act on a distinct pathological mechanism: β-blockers on abnormal cortical information processing and riboflavin on decreased brain mitochondrial energy reserve (57). A small study in 23 patients reported a reduction in median migraine attack frequency after supplementation with 400 mg of riboflavin daily for three months (58). A single-blinded, randomized, parallel group trial in 85 patients with migraine headaches (ages 15-55 years), high-dose riboflavin supplementation (400 mg/day) for 12 weeks decreased migraine frequency, duration, and severity compared to baseline and was as effective as sodium valproate (500 mg/day) (59), a medication with established efficacy in migraine preventative therapy (60). Riboflavin elicited significantly fewer adverse effects compared to the drug (59). Thus, although the available trials have been small and short term, most studies to date suggest that high-dose riboflavin supplementation might be a useful adjunct therapy in adults with migraine headaches.
A few randomized controlled trials have investigated the effect of riboflavin supplementation on the frequency and severity of headache attacks in children with migraines. An initial study evaluated riboflavin at 200 mg/day for 12 weeks in 48 children of ages 5 to 15 years old (61). A second study was a cross-over trial with half of the 42 children, ages 6 to 13, receiving 50 mg/day riboflavin for 16 weeks then placebo (100 mg/day carotene) for 16 weeks (with a four-week washout period in between each), while the other half were first given the placebo then riboflavin (62). Neither study showed differences in the frequency, duration, or intensity of migraines between treatments. However, a more recent trial found a benefit of intervention with higher dose riboflavin: children with migraine treated with 400 mg/day of riboflavin for 12 weeks (n=30) had reductions in migraine frequency and duration, but not intensity, compared to placebo (n=30), yet no benefit was seen in children taking 200 mg/day for 12 weeks in this study (63). Additionally, a randomized controlled trial in 98 adolescents, ages 12 to 19 years, found that 400 mg/day of riboflavin for three months decreased both headache frequency and duration and improved migraine-related disability compared to placebo (64). Retrospective studies of children and adolescents suffering from migraine have also suggested some benefit associated with supplemental riboflavin (65-67). Thus, studies to date are somewhat conflicting, and more research is needed to understand whether riboflavin supplementation might have utility in the treatment of childhood migraine and the most effective dose required for any beneficial effects.
Metabolic disorders
Increasing evidence from case reports indicates that patients with autosomal recessive disorders caused by defective FAD-dependent enzymes could benefit from riboflavin supplementation.
Multiple acyl-CoA dehydrogenase deficiency (MADD)
MADD, also known as type II glutaric aciduria (or acidemia), is a fatty acid metabolism disorder characterized by the accumulation of short-, medium-, and long-chain acyl-carnitines in various tissues. MADD is classified into three separate types based on age of onset and clinical symptoms: type I MADD is evident in the neonatal period and is characterized by the presence of congenital anomalies; type II MADD is present in the neonatal period but lacks congenital defects; and type III is characterized by late onset, from infancy through adulthood (68), and even as late as the seventh decade of life (69). Clinical symptoms of type I and II MADD present shortly after birth and include hypoglycemia, hyperammonemia, metabolic acidosis, hepatomegaly, and respiratory distress (68, 70); these forms of MADD are often fatal in infancy, even if treated. Type III MADD usually presents later in life and includes milder symptoms, varying from periodic vomiting, rhabdomyolysis, muscle pain and weakness, and exercise intolerance (68, 70). Peripheral neuropathy has also recently been reported as a symptom of adult-onset MADD (71).
MADD is caused by autosomal recessive mutations in genes that impair the activity of enzymes involved in the transfer of electrons from acyl-coenzyme A (acyl-CoA) to coenzyme Q10/ubiquinone inside the mitochondria (Figure 3). ETFA, ETFB, and ETFDH code for the two subunits of the electron transfer flavoprotein (ETF-A and -B) and for ETF dehydrogenase/ubiquinone oxidoreductase (ETFDH/ETFQO), respectively. Deficiencies in these enzymes (ETF or ETFDH) lead to a decrease in oxidized FAD, which becomes unavailable for FAD-dependent dehydrogenation reactions, including the first step in β-oxidation – a major fatty acid catabolic process that takes place in the mitochondria. A defect in fatty acid β-oxidation causes lipid accumulation in skeletal muscles, leading to lipid storage myopathy characterized by muscle pain and weakness and exercise intolerance.
Together with a low-fat, high-carbohydrate diet, riboflavin supplementation has led to significant clinical improvements in patients with ETFDH mutations. The specific type of the mutation in ETF/ETFDH contributes to age of onset, severity, and responsiveness to riboflavin treatment (70, 72). Additionally, the report of a 20-year-old man with riboflavin-responsive MADD failed to find mutations in ETF and ETFDH genes, suggesting that other sites of mutation should not be excluded (73). Finally, secondary deficiencies in the respiratory chain are observed in MADD and appear to respond favorably to riboflavin supplementation (72, 74).

Acyl-CoA dehydrogenase 9 deficiency (ACAD9)
Acyl-CoA dehydrogenase family member 9 (ACAD9) is an FAD-dependent enzyme with important roles in both the electron transport chain and β-oxidation of fatty acids in the mitochondria. Recessive mutations in the ACAD9 gene coding for ACAD9 have been found in patients with mitochondrial complex I deficiency, a respiratory chain disorder (75). Complex I carries electrons from NADH to coenzyme Q10 in the electron transport chain. Defective oxidative phosphorylation (ATP synthesis by the respiratory chain) due to complex I deficiency has been linked to a broad variety of clinical manifestations, from neonatal death to late-onset neurodegenerative diseases. The clinical symptoms of complex I deficiency due to ACAD9 mutations typically include muscle weakness, exercise intolerance, lactic acidosis, and hypertrophic cardiomyopathy (76). However, symptoms can be of varying severity, likely due to the remaining functional activity of ACAD9. For example, affected patients have been reported to exhibit a spectrum of cardiac deficits, including isolated, mild ventricular hypertrophy to severe hypertrophic cardiomyopathy (77).
Riboflavin supplementation (100-300 mg/day) has been shown to increase complex I activity in patients with childhood-onset clinical forms of ACAD9 deficiency. Improvements in muscle strength and exercise tolerance have also been associated with riboflavin supplementation (78-80). A review of cases of ACAD9 deficiency presenting in infancy (i.e., cases with severe symptoms) found riboflavin treatment to be associated with improved survival: 7 of 22 patients treated with riboflavin succumbed to the illness compared to 16 out of 17 untreated patients (76).
Defective riboflavin transport-associated disorders
SLC52A1, SLC52A2, and SLC52A3 genes code for the human riboflavin transporters RFVT1, RFVT2, and RFVT3, respectively. Mutations in these genes lead to riboflavin transporter deficiency, a rare neurodegenerative condition with variable age of onset, from infancy to early stages of adulthood. Autosomal recessive mutations in SLC52A2 or SLC52A3 respectively cause disorders known as riboflavin transporter deficiency type 2 (RFVT2 deficiency) and riboflavin transporter deficiency type 3 (RFVT3 deficiency). These genetic disorders were formerly called Brown-Vialetto-Van Laere syndrome and Fazio-Londe syndrome (81). Riboflavin transporter deficiency caused by mutation of SLC52A1 is exceedingly rare and has been reported in only three cases (reviewed in 82).
Clinical features of riboflavin transporter deficiency can include muscle weakness in the arms and legs, sensory ataxia, bulbar palsy with hypotonia and facial weakness, sensorineural deafness, and respiratory insufficiency (83, 84). High-dose, oral supplementation with riboflavin improves many of these symptoms in the majority of affected patients; such treatment should be given at the time of suspected riboflavin transporter deficiency for a better prognosis (84). A 2016 review of the published literature found that oral supplementation with riboflavin – at doses ranging from 7 to 60 mg/kg/day – led to improved symptoms in 71% of the patients (n=39) and to no deaths (83). In contrast, all of the untreated patients (n=31) had a progression of the disease and a mortality rate of at least 48% (83).
Riboflavin-responsive trimethylaminuria
Primary trimethylaminuria is caused by defective oxidation of trimethylamine by a liver flavoprotein called flavin containing mono-oxygenase 3 (FMO3). Individuals with FMO3 deficiency have increased levels of trimethylamine in urine, sweat, and breath (85). This socially distressing condition is known as "fish odor syndrome" due to the fishy odor and volatile nature of trimethylamine. FMO3 gene mutations are usually associated with mild or intermittent trimethylaminuria; the condition is sometimes limited to peri-menstrual periods in female subjects or to the consumption of trimethylamine-rich food. The clinical management of the condition includes dietary restriction of trimethylamine and its precursors, such as foods rich in choline and seafood, as well as cruciferous vegetables that contain both trimethylamine precursors and FMO3 antagonists (86). The use of riboflavin supplements was reported in a 17-year-old female patient affected by pyridoxine non-responsive homocystinuria (87). The disease was initially treated with betaine (a choline derivative), which caused body odor secondary to FMO3 deficiency. Riboflavin supplementation (200 mg/day) reduced trimethylamine excretion and the betaine treatment-related body odor. Similar effects were seen with riboflavin supplementation in two pediatric patients (88). The data suggest that riboflavin might help maximize residual FMO3 enzyme activity in patients with primary trimethylaminuria. Moreover, a recent case report in a 35-year-old male with HIV described supplemental riboflavin as an effective treatment for secondary trimethylaminuria caused by antiretroviral therapy (89).
Hypertension
Hypertension in adulthood is recognized as the leading risk factor contributing to mortality worldwide primarily from cardiovascular disease, while hypertension in pregnancy leads to serious adverse fetal and maternal outcomes. A number of risk factors are recognized to contribute to the development of hypertension. In recent years, evidence has emerged from genetic and clinical studies pointing to the role of one-carbon metabolism in blood pressure (90). The common MTHFR C677T polymorphism, affecting 1 in 10 adults globally, is associated with higher blood pressure, although this is much less well recognized compared with the phenotype of elevated homocysteine concentrations that was established at the time of discovery of this polymorphism and its link with cardiovascular disease (91). Meta-analyses show that this polymorphism is associated with an increased risk of hypertension by up to 87% and of heart disease and stroke by up to 40% (92). The MTHFR C677T polymorphism is also associated with a significantly higher risk of hypertension in pregnancy (93) and with preeclampsia (27).
Since FAD is required as a cofactor for the MTHFR enzyme and the MTHFR C677T polymorphism results in decreased MTHFR activity, studies have investigated whether affected individuals may benefit from riboflavin supplementation. In an initial randomized controlled trial in 77 healthy young adults stratified by MTHFR genotype, riboflavin supplementation at dietary levels (1.6 mg/day for 12 weeks) resulted in marked lowering of homocysteine concentrations in the MTHFR 677TT genotype group, but not in the 677CC or 677CT genotype groups who exhibited normal plasma homocysteine at baseline (15). Three randomized controlled trials subsequently investigated the effect of riboflavin on blood pressure in patients with hypertension with or without overt cardiovascular disease (91, 94, 95). The results of these trials showed that supplementation with low-dose riboflavin (1.6 mg/day for 16 weeks) resulted in significant lowering of blood pressure and reduction in incidence of hypertension specifically in those patients with the variant MTHFR 677TT genotype. Riboflavin intervention reduced mean systolic/diastolic blood pressure in those with the TT genotype from 144/87 to 131/80 mm Hg, with no response observed in those without the genetic variant (i.e., the CT or CC genotypes) (89). Notably, the 13 mm Hg decrease in systolic blood pressure occurred even though over 80% of the patients were taking one or more antihypertensive drugs at recruitment, and the addition of supplemental riboflavin was shown to greatly enhance the achievement of goal blood pressure with routine antihypertensive drugs (89, 91). Furthermore, the magnitude of blood pressure response achieved with riboflavin in these trials compares very favorably with typical decreases from other interventions, such as dietary salt reductions of 3 g/day (3.6/1.9 mm Hg) and 6 g/day (7.1/3.9 mm Hg). The trial findings therefore suggest that the excess risk of hypertension linked to this genetic polymorphism can be overcome by low-dose riboflavin supplementation. Also, analysis of plasma samples from individuals participating in these trials showed lower concentrations of S-adenosylmethionine (SAM), an important methyl group donor for methylation reactions, in those with the MTHFR 677TT genotype versus the CC genotype (96). However, riboflavin supplementation (1.6 mg/day) for 12 weeks was shown to increase plasma concentrations of SAM and another one-carbon metabolite, cystathionine (96), and thus may have potential in correcting the altered one-carbon metabolism arising with the variant TT genotype.
Thus, studies to date indicate that riboflavin supplementation may have benefits in lowering blood pressure and reducing hypertension in individuals (and sub-populations) affected by the common MTHFR C677T polymorphism. However, the mechanisms explaining the blood pressure phenotype and its responsiveness to riboflavin remain unclear. Future studies examining the effects of riboflavin supplementation on one-carbon metabolism may help to elucidate the biological mechanisms involved. Interestingly, a recent randomized controlled trial found that riboflavin supplementation in those with the variant MTHFR 677TT genotype resulted in altered DNA methylation of certain genes known to be involved in blood pressure regulation (97).
Cancer
Anticancer agents often display various side effects that may force patients to limit the dose or to discontinue the treatment. The antioxidant effect of co-administering riboflavin (10 mg/day), niacin (50 mg/day), and coenzyme Q10 (100 mg/day) was evaluated in 78 postmenopausal patients with breast cancer treated with tamoxifen for 90 days. This supplementation effectively prevented the oxidative stress associated with tamoxifen treatment (98).
Riboflavin can also act as a photosensitizer, and this property may have value in photodynamic therapy of cancer. A mouse model was used to assess the effect of riboflavin in combination with cisplatin, one of the most effective anticancer agents. Under light exposure, riboflavin administration reduced cisplatin-induced DNA damage in the liver and kidneys (99). These results are promising, but human studies are needed to examine whether riboflavin is an effective adjunct to chemotherapy.
Corneal disorders
Corneal ectasia is an eye condition characterized by irregularities of the cornea that affect vision. Corneal cross-linking – a fairly new procedure used by professionals to limit the progression of corneal damage –involves the use of topical riboflavin in conjunction with ultraviolet-A irradiation. Riboflavin functions as a photosensitizer in the reaction. Cross-linking modifies the properties of the cornea and strengthens its architecture (100, 101).
Multiple sclerosis
Multiple sclerosis (MS) is an autoimmune disease of unknown etiology that is characterized by the progressive destruction of myelin and nerve fibers in the central nervous system, causing neurological symptoms in affected individuals (102). Riboflavin appears to have a role in the formation of myelin (103), and oxidative stress has been implicated in the pathogenesis of MS; thus, riboflavin may be helpful in treatment of the disease. A strong inverse association between dietary riboflavin intake and risk for MS was initially observed in a case-control study (104). In a mouse model of MS (i.e., experimental autoimmune encephalomyelitis), riboflavin supplementation improved clinical measures of the disease (105). However, a randomized, double-blind, placebo-controlled pilot study in 29 patients with MS found that supplementation with 10 mg/day of riboflavin for six months had no effect on MS-related disability, assessed by the Expanded Disability Status Scale (106). Large-scale randomized, placebo-controlled trials are needed to determine whether riboflavin supplementation has a beneficial effect in the treatment of MS.
Sources
Food sources
Most plant- and animal-derived foods contain at least small quantities of riboflavin. In the US, wheat flour and bread have been enriched with riboflavin (as well as thiamin, niacin, and iron) since 1943. Data from a US national survey indicate that the average dietary intake of riboflavin is 2.5 mg/day for men and 1.8 mg/day for women (107); these intakes are well above the RDA values of 1.3 mg/day for men and 1.1 mg/day for women. Surveys of adults of ages 70 years or older showed similar intakes: 2.2 mg/day for older men and 1.8 mg day for older women (107).
Riboflavin is heat-stable, but it is easily destroyed upon exposure to light. For instance, up to 50% of the riboflavin in milk contained in a clear glass bottle can be destroyed after two hours of exposure to bright sunlight (6). Nationally representative surveys from the US, Ireland, and the UK showed that milk and other dairy products were the main dietary contributors to riboflavin intake, followed by meat and ready-to-eat breakfast cereals (108-110). Some foods with substantial amounts of riboflavin are listed in Table 2, along with their riboflavin content in milligrams (mg). For more information on the nutrient content of food, search USDA's FoodData Central.
The bioavailability of riboflavin from food is reported to be very high, nearly 95% (108). Limited data exist for the relative bioavailability of riboflavin from different food sources, however a cross-over study in healthy women using stable isotopes and kinetic modeling did not find significant differences in riboflavin absorption from milk and spinach (111).
Supplements
The most common forms of riboflavin available in supplements are riboflavin and riboflavin 5'-monophosphate. Riboflavin is commonly found in multivitamin and vitamin B-complex preparations (112).
Safety
Toxicity
No toxic or adverse effects of high riboflavin intake in humans are known. Studies in cell culture indicate that excess riboflavin may increase the risk of DNA strand breaks in the presence of chromium (VI), a known carcinogen (113). This may be of concern to workers exposed to chrome, yet no data in humans are available. High-dose riboflavin therapy has been found to intensify urine color to a bright yellow (flavinuria), but this is a harmless side effect. The Food and Nutrition Board did not establish a tolerable upper intake level (UL) when the RDA was revised in 1998 (1).
Drug interactions
Several early reports indicated that women taking high-dose oral contraceptives had diminished riboflavin biomarker status. However, when investigators controlled for dietary riboflavin intake, no differences between users of oral contraceptives and non-users were found (1). Phenothiazine derivatives like the anti-psychotic medication, chlorpromazine (Thorazine), and tricyclic antidepressants inhibit the conversion of riboflavin to FAD and FMN, as do the anti-malarial medication, quinacrine, and the cancer chemotherapy agent, adriamycin (4). Long-term use of the anticonvulsant, phenobarbitol, may increase destruction of riboflavin by liver enzymes, increasing the risk of deficiency (3). Additionally, chronic alcohol consumption has been associated with riboflavin deficiency. In rats chronically fed alcohol, the inhibition of riboflavin transporters caused impairment in intestinal absorption and renal re-uptake of the vitamin (114).
Linus Pauling Institute Recommendation
The RDA for riboflavin (1.3 mg/day for men and 1.1 mg/day for women), which should prevent deficiency in most individuals, is easily met by eating a varied diet. Consuming a varied diet should supply 1.5 mg to 2 mg of riboflavin a day. Following the Linus Pauling Institute recommendation to take a multivitamin/mineral supplement containing 100% of the Daily Values (DV) will ensure an intake of at least 1.3 mg/day of riboflavin.
Older adults (>50 years)
Some experts in nutrition and aging feel that the RDA (1.3 mg/day for men and 1.1 mg/day for women) leaves little margin for error in people over 50 years of age (115, 116). A study of independently living people between 65 and 90 years of age found that almost 25% consumed less than the recommended riboflavin intake, and 10% had biochemical evidence of deficiency (117). Epidemiological studies of cataract prevalence indicate that riboflavin intakes of 1.6 to 2.2 mg/day may reduce the risk of developing age-related cataracts. Additionally, older people suffering from acute ischemic stroke were found to be deficient for riboflavin (118), and riboflavin deficiency has been linked to a higher risk of fracture in postmenopausal women with the MTHFR 677T variant (119). Individuals whose diets may not supply adequate riboflavin, especially those over 50 years of age, should consider taking a multivitamin/mineral supplement, which generally provides at least 1.3 mg/day of riboflavin.
Authors and Reviewers
Originally written in 2000 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in September 2002 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in June 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in July 2013 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in August 2021 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in July 2022 by:
Kristina Pentieva, MD, Ph.D. and Helene McNulty, RD, Ph.D.
Nutrition Innovation Centre for Food and Health (NICHE)
Ulster University, Coleraine, Northern Ireland
Copyright 2000-2024 Linus Pauling Institute
References
1. Food and Nutrition Board, Institute of Medicine. Riboflavin. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B-6, Vitamin B-12, Pantothenic Acid, Biotin, and Choline. Washington D.C.: National Academy Press; 1998:87-122. (National Academy Press)
2. Brody T. Nutritional Biochemistry. 2nd ed. San Diego: Academic Press; 1999.
3. McCormick D. Riboflavin. In: Shils M, Olson J, Shike M, Ross A, eds. Nutrition in Health and Disease. 9th ed. Baltimore: Williams & Wilkins; 1999:391-399.
4. Powers HJ. Current knowledge concerning optimum nutritional status of riboflavin, niacin and pyridoxine. Proc Nutr Soc. 1999;58(2):435-440. (PubMed)
5. Rivlin R. Riboflavin. In: Ziegler E, Filer L, eds. Present Knowledge in Nutrition. 7th ed. Washington D.C.: ILSI Press; 1996:167-173.
6. Bohles H. Antioxidative vitamins in prematurely and maturely born infants. Int J Vitam Nutr Res. 1997;67(5):321-328. (PubMed)
7. McCormick DB. Two interconnected B vitamins: riboflavin and pyridoxine. Physiol Rev. 1989;69(4):1170-1198. (PubMed)
8. Madigan SM, Tracey F, McNulty H, et al. Riboflavin and vitamin B-6 intakes and status and biochemical response to riboflavin supplementation in free-living elderly people. Am J Clin Nutr. 1998;68(2):389-395. (PubMed)
9. Lowik MR, van den Berg H, Kistemaker C, Brants HA, Brussaard JH. Interrelationships between riboflavin and vitamin B6 among elderly people (Dutch Nutrition Surveillance System). Int J Vitam Nutr Res. 1994;64(3):198-203. (PubMed)
10. Jungert A, McNulty H, Hoey L, et al. Riboflavin is an important determinant of vitamin B-6 status in healthy adults. J Nutr. 2020;150(10):2699-2706. (PubMed)
11. Wolthers KR, Scrutton NS. Cobalamin uptake and reactivation occurs through specific protein interactions in the methionine synthase-methionine synthase reductase complex. FEBS J. 2009;276(7):1942-1951. (PubMed)
12. Jacques PF, Bostom AG, Wilson PW, Rich S, Rosenberg IH, Selhub J. Determinants of plasma total homocysteine concentration in the Framingham Offspring cohort. Am J Clin Nutr. 2001;73(3):613-621. (PubMed)
13. Jacques PF, Kalmbach R, Bagley PJ, et al. The relationship between riboflavin and plasma total homocysteine in the Framingham Offspring cohort is influenced by folate status and the C677T transition in the methylenetetrahydrofolate reductase gene. J Nutr. 2002;132(2):283-288. (PubMed)
14. McNulty H, McKinley MC, Wilson B, et al. Impaired functioning of thermolabile methylenetetrahydrofolate reductase is dependent on riboflavin status: implications for riboflavin requirements. Am J Clin Nutr. 2002;76(2):436-441. (PubMed)
15. McNulty H, Dowey le RC, Strain JJ, et al. Riboflavin lowers homocysteine in individuals homozygous for the MTHFR 677C->T polymorphism. Circulation. 2006;113(1):74-80. (PubMed)
16. Powers HJ, Weaver LT, Austin S, Beresford JK. A proposed intestinal mechanism for the effect of riboflavin deficiency on iron loss in the rat. Br J Nutr. 1993;69(2):553-561. (PubMed)
17. Shi Z, Zhen S, Wittert GA, Yuan B, Zuo H, Taylor AW. Inadequate riboflavin intake and anemia risk in a Chinese population: five-year follow up of the Jiangsu Nutrition Study. PLoS One. 2014;9(2):e88862. (PubMed)
18. Powers HJ, Hill MH, Mushtaq S, Dainty JR, Majsak-Newman G, Williams EA. Correcting a marginal riboflavin deficiency improves hematologic status in young women in the United Kingdom (RIBOFEM). Am J Clin Nutr. 2011;93(6):1274-1284. (PubMed)
19. Powers HJ. Riboflavin-iron interactions with particular emphasis on the gastrointestinal tract. Proc Nutr Soc. 1995;54(2):509-517. (PubMed)
20. Kalaivani K. Prevalence & consequences of anaemia in pregnancy. Indian J Med Res. 2009;130(5):627-633. (PubMed)
21. Worldwide prevalence of anaemia 1993-2005: WHO global database on anaemia. de Benoist B, McLean E, Egli I, Cogswell M, eds. 2008; World Health Organization Press. Available at: http://www.who.int/nutrition/publications/micronutrients/anaemia_iron_deficiency/9789241596657/en/index.html. Accessed 7/22/13.
22. Pena-Rosas JP, Viteri FE. Effects of routine oral iron supplementation with or without folic acid for women during pregnancy. Cochrane Database Syst Rev. 2006(3):CD004736. (PubMed)
23. Suprapto B, Widardo, Suhanantyo. Effect of low-dosage vitamin A and riboflavin on iron-folate supplementation in anaemic pregnant women. Asia Pac J Clin Nutr. 2002;11(4):263-267. (PubMed)
24. Ma AG, Schouten EG, Zhang FZ, et al. Retinol and riboflavin supplementation decreases the prevalence of anemia in Chinese pregnant women taking iron and folic Acid supplements. J Nutr. 2008;138(10):1946-1950. (PubMed)
25. Crombleholme W. Obstetrics. In: Tierney L, McPhee S, Papadakis M, eds. Current Medical Treatment and Diagnosis. Stamford: Appleton and Lange; 1998:731-734.
26. Wacker J, Fruhauf J, Schulz M, Chiwora FM, Volz J, Becker K. Riboflavin deficiency and preeclampsia. Obstet Gynecol. 2000;96(1):38-44. (PubMed)
27. Wu X, Yang K, Tang X, et al. Folate metabolism gene polymorphisms MTHFR C677T and A1298C and risk for preeclampsia: a meta-analysis. J Assist Reprod Genet. 2015;32(5):797-805. (PubMed)
28. Braekke K, Ueland PM, Harsem NK, Karlsen A, Blomhoff R, Staff AC. Homocysteine, cysteine, and related metabolites in maternal and fetal plasma in preeclampsia. Pediatr Res. 2007;62(3):319-324. (PubMed)
29. Neugebauer J, Zanre Y, Wacker J. Riboflavin supplementation and preeclampsia. Int J Gynaecol Obstet. 2006;93(2):136-137. (PubMed)
30. Heese P, Linnebank M, Semmler A, et al. Alterations of homocysteine serum levels during alcohol withdrawal are influenced by folate and riboflavin: results from the German Investigation on Neurobiology in Alcoholism (GINA). Alcohol Alcohol. 2012;47(5):497-500. (PubMed)
31. Soares MJ, Satyanarayana K, Bamji MS, Jacob CM, Ramana YV, Rao SS. The effect of exercise on the riboflavin status of adult men. Br J Nutr. 1993;69(2):541-551. (PubMed)
32. Suboticanec K, Stavljenic A, Schalch W, Buzina R. Effects of pyridoxine and riboflavin supplementation on physical fitness in young adolescents. Int J Vitam Nutr Res. 1990;60(1):81-88. (PubMed)
33. Mares-Perlman JA, Brady WE, Klein BE, et al. Diet and nuclear lens opacities. Am J Epidemiol. 1995;141(4):322-334. (PubMed)
34. Leske MC, Wu SY, Hyman L, et al. Biochemical factors in the lens opacities. Case-control study. The Lens Opacities Case-Control Study Group. Arch Ophthalmol. 1995;113(9):1113-1119. (PubMed)
35. Cumming RG, Mitchell P, Smith W. Diet and cataract: the Blue Mountains Eye Study. Ophthalmology. 2000;107(3):450-456. (PubMed)
36. Hankinson SE, Stampfer MJ, Seddon JM, et al. Nutrient intake and cataract extraction in women: a prospective study. BMJ. 1992;305(6849):335-339. (PubMed)
37. Jacques PF, Taylor A, Moeller S, et al. Long-term nutrient intake and 5-year change in nuclear lens opacities. Arch Ophthalmol. 2005;123(4):517-526. (PubMed)
38. Sperduto RD, Hu TS, Milton RC, et al. The Linxian cataract studies. Two nutrition intervention trials. Arch Ophthalmol. 1993;111(9):1246-1253. (PubMed)
39. McGlynn AP, Wasson GR, O'Reilly SL, et al. Low colonocyte folate is associated with uracil misincorporation and global DNA hypomethylation in human colorectum. J Nutr. 2013;143(1):27-33. (PubMed)
40. Guenther BD, Sheppard CA, Tran P, Rozen R, Matthews RG, Ludwig ML. The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomocysteinemia. Nat Struct Biol. 1999;6(4):359-365. (PubMed)
41. Yin G, Ming H, Zheng X, Xuan Y, Liang J, Jin X. Methylenetetrahydrofolate reductase C677T gene polymorphism and colorectal cancer risk: A case-control study. Oncol Lett. 2012;4(2):365-369. (PubMed)
42. Gao S, Ding LH, Wang JW, Li CB, Wang ZY. Diet folate, DNA methylation and polymorphisms in methylenetetrahydrofolate reductase in association with the susceptibility to gastric cancer. Asian Pac J Cancer Prev. 2013;14(1):299-302. (PubMed)
43. Wen YY, Yang SJ, Zhang JX, Chen XY. Methylenetetrahydrofolate reductase genetic polymorphisms and esophageal squamous cell carcinoma susceptibility: a meta-analysis of case-control studies. Asian Pac J Cancer Prev. 2013;14(1):21-25. (PubMed)
44. Kennedy DA, Stern SJ, Matok I, et al. Folate intake, MTHFR polymorphisms, and the risk of colorectal cancer: a systematic review and meta-analysis. J Cancer Epidemiol. 2012;2012:952508. (PubMed)
45. Powers HJ, Hill MH, Welfare M, et al. Responses of biomarkers of folate and riboflavin status to folate and riboflavin supplementation in healthy and colorectal polyp patients (the FAB2 Study). Cancer Epidemiol Biomarkers Prev. 2007;16(10):2128-2135. (PubMed)
46. Zschabitz S, Cheng TY, Neuhouser ML, et al. B vitamin intakes and incidence of colorectal cancer: results from the Women's Health Initiative Observational Study cohort. Am J Clin Nutr. 2013;97(2):332-343. (PubMed)
47. Liu Y, Yu QY, Zhu ZL, Tang PY, Li K. Vitamin B2 intake and the risk of colorectal cancer: a meta-analysis of observational studies. Asian Pac J Cancer Prev. 2015;16(3):909-913. (PubMed)
48. Ben S, Du M, Ma G, et al. Vitamin B2 intake reduces the risk for colorectal cancer: a dose-response analysis. Eur J Nutr. 2019;58(4):1591-1602. (PubMed)
49. He Y, Ye L, Shan B, Song G, Meng F, Wang S. Effect of riboflavin-fortified salt nutrition intervention on esophageal squamous cell carcinoma in a high incidence area, China. Asian Pac J Cancer Prev. 2009;10(4):619-622. (PubMed)
50. Wang SM, Taylor PR, Fan JH, et al. Effects of nutrition intervention on total and cancer mortality: 25-year post-trial follow-up of the 5.25-year Linxian Nutrition Intervention Trial. J Natl Cancer Inst. 2018;110(11):1229-1238. (PubMed)
51. Bassett JK, Hodge AM, English DR, et al. Dietary intake of B vitamins and methionine and risk of lung cancer. Eur J Clin Nutr. 2012;66(2):182-187. (PubMed)
52. Bassett JK, Baglietto L, Hodge AM, et al. Dietary intake of B vitamins and methionine and breast cancer risk. Cancer Causes Control. 2013;24(8):1555-1563. (PubMed)
53. Bassett JK, Severi G, Hodge AM, et al. Dietary intake of B vitamins and methionine and prostate cancer incidence and mortality. Cancer Causes Control. 2012;23(6):855-863. (PubMed)
54. Yu L, Tan Y, Zhu L. Dietary vitamin B2 intake and breast cancer risk: a systematic review and meta-analysis. Arch Gynecol Obstet. 2017;295(3):721-729. (PubMed)
55. Clasen JL, Heath AK, Scelo G, Muller DC. Components of one-carbon metabolism and renal cell carcinoma: a systematic review and meta-analysis. Eur J Nutr. 2020;59(8):3801-3813. (PubMed)
56. Schoenen J, Jacquy J, Lenaerts M. Effectiveness of high-dose riboflavin in migraine prophylaxis. A randomized controlled trial. Neurology. 1998;50(2):466-470. (PubMed)
57. Sandor PS, Afra J, Ambrosini A, Schoenen J. Prophylactic treatment of migraine with beta-blockers and riboflavin: differential effects on the intensity dependence of auditory evoked cortical potentials. Headache. 2000;40(1):30-35. (PubMed)
58. Boehnke C, Reuter U, Flach U, Schuh-Hofer S, Einhaupl KM, Arnold G. High-dose riboflavin treatment is efficacious in migraine prophylaxis: an open study in a tertiary care centre. Eur J Neurol. 2004;11(7):475-477. (PubMed)
59. Rahimdel A, Zeinali A, Yazdian-Anari P, Hajizadeh R, Arefnia E. Effectiveness of vitamin B2 versus sodium valproate in migraine prophylaxis: a randomized clinical trial. Electron Physician. 2015;7(6):1344-1348. (PubMed)
60. Silberstein SD, Holland S, Freitag F, et al. Evidence-based guideline update: pharmacologic treatment for episodic migraine prevention in adults: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Neurology. 2012;78(17):1337-1345. (PubMed)
61. MacLennan SC, Wade FM, Forrest KM, Ratanayake PD, Fagan E, Antony J. High-dose riboflavin for migraine prophylaxis in children: a double-blind, randomized, placebo-controlled trial. J Child Neurol. 2008;23(11):1300-1304. (PubMed)
62. Bruijn J, Duivenvoorden H, Passchier J, Locher H, Dijkstra N, Arts WF. Medium-dose riboflavin as a prophylactic agent in children with migraine: a preliminary placebo-controlled, randomised, double-blind, cross-over trial. Cephalalgia. 2010;30(12):1426-1434. (PubMed)
63. Talebian A, Soltani B, Banafshe HR, Moosavi GA, Talebian M, Soltani S. Prophylactic effect of riboflavin on pediatric migraine: a randomized, double-blind, placebo-controlled trial. Electron Physician. 2018;10(2):6279-6285. (PubMed)
64. Athaillah A, Y. D, Saing JH, Saing B, Hakimi H, Lelo A. Riboflavin as migraine prophylaxis in adolescents. Paediatr Indones. 2012;52(3):132-137.
65. Condo M, Posar A, Arbizzani A, Parmeggiani A. Riboflavin prophylaxis in pediatric and adolescent migraine. J Headache Pain. 2009;10(5):361-365. (PubMed)
66. Das R, Qubty W. Retrospective observational study on riboflavin prophylaxis in child and adolescent migraine. Pediatr Neurol. 2021;114:5-8. (PubMed)
67. Yamanaka G, Suzuki S, Takeshita M, et al. Effectiveness of low-dose riboflavin as a prophylactic agent in pediatric migraine. Brain Dev. 2020;42(7):523-528. (PubMed)
68. Prasun P. Multiple acyl-CoA dehydrogenase deficiency. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle (WA); 1993. (PubMed)
69. Macchione F, Salviati L, Bordugo A, et al. Multiple acyl-COA dehydrogenase deficiency in elderly carriers. J Neurol. 2020;267(5):1414-1419. (PubMed)
70. Yildiz Y, Talim B, Haliloglu G, et al. Determinants of riboflavin responsiveness in multiple acyl-CoA dehydrogenase deficiency. Pediatr Neurol. 2019;99:69-75. (PubMed)
71. Huang K, Duan HQ, Li QX, Luo YB, Yang H. Investigation of adult-onset multiple acyl-CoA dehydrogenase deficiency associated with peripheral neuropathy. Neuropathology. 2020;40(6):531-539. (PubMed)
72. Olsen RK, Olpin SE, Andresen BS, et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain. 2007;130(Pt 8):2045-2054. (PubMed)
73. Cotelli MS, Vielmi V, Rimoldi M, et al. Riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency with unknown genetic defect. Neurol Sci. 2012;33(6):1383-1387. (PubMed)
74. Liang WC, Ohkuma A, Hayashi YK, et al. ETFDH mutations, CoQ10 levels, and respiratory chain activities in patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Neuromuscul Disord. 2009;19(3):212-216. (PubMed)
75. Haack TB, Danhauser K, Haberberger B, et al. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat Genet. 2010;42(12):1131-1134. (PubMed)
76. Repp BM, Mastantuono E, Alston CL, et al. Clinical, biochemical and genetic spectrum of 70 patients with ACAD9 deficiency: is riboflavin supplementation effective? Orphanet J Rare Dis. 2018;13(1):120. (PubMed)
77. Dewulf JP, Barrea C, Vincent MF, et al. Evidence of a wide spectrum of cardiac involvement due to ACAD9 mutations: Report on nine patients. Mol Genet Metab. 2016;118(3):185-189. (PubMed)
78. Scholte HR, Busch HF, Bakker HD, Bogaard JM, Luyt-Houwen IE, Kuyt LP. Riboflavin-responsive complex I deficiency. Biochim Biophys Acta. 1995;1271(1):75-83. (PubMed)
79. Gerards M, van den Bosch BJ, Danhauser K, et al. Riboflavin-responsive oxidative phosphorylation complex I deficiency caused by defective ACAD9: new function for an old gene. Brain. 2011;134(Pt 1):210-219. (PubMed)
80. Garone C, Donati MA, Sacchini M, et al. Mitochondrial encephalomyopathy due to a novel mutation in ACAD9. JAMA Neurol. 2013:1-3. (PubMed)
81. Bosch AM, Abeling NG, Ijlst L, et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: a new inborn error of metabolism with potential treatment. J Inherit Metab Dis. 2011;34(1):159-164. (PubMed)
82. Mereis M, Wanders RJA, Schoonen M, Dercksen M, Smuts I, van der Westhuizen FH. Disorders of flavin adenine dinucleotide metabolism: MADD and related deficiencies. Int J Biochem Cell Biol. 2021;132:105899. (PubMed)
83. Jaeger B, Bosch AM. Clinical presentation and outcome of riboflavin transporter deficiency: mini review after five years of experience. J Inherit Metab Dis. 2016;39(4):559-564. (PubMed)
84. O'Callaghan B, Bosch AM, Houlden H. An update on the genetics, clinical presentation, and pathomechanisms of human riboflavin transporter deficiency. J Inherit Metab Dis. 2019;42(4):598-607. (PubMed)
85. Mackay RJ, McEntyre CJ, Henderson C, Lever M, George PM. Trimethylaminuria: causes and diagnosis of a socially distressing condition. Clin Biochem Rev. 2011;32(1):33-43.
86. Phillips IR, Shephard EA. Trimethylaminuria. 2007 Oct 8 [Updated 2011 Apr 19]. In: Pagon RA, Adam MP, Bird TD, et al., editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 1993-2013. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1103/.
87. Manning NJ, Allen EK, Kirk RJ, Sharrard MJ, Smith EJ. Riboflavin-responsive trimethylaminuria in a patient with homocystinuria on betaine therapy. JIMD Rep. 2012;5:71-75. (PubMed)
88. Bouchemal N, Ouss L, Brassier A, et al. Diagnosis and phenotypic assessment of trimethylaminuria, and its treatment with riboflavin: (1)H NMR spectroscopy and genetic testing. Orphanet J Rare Dis. 2019;14(1):222. (PubMed)
89. Scimone C, Alibrandi S, Donato L, et al. Antiretroviral treatment leading to secondary trimethylaminuria: Genetic associations and successful management with riboflavin. J Clin Pharm Ther. 2021;46(2):304-309. (PubMed)
90. McNulty H, Strain JJ, Hughes CF, Ward M. Riboflavin, MTHFR genotype and blood pressure: A personalized approach to prevention and treatment of hypertension. Mol Aspects Med. 2017;53:2-9. (PubMed)
91. Frosst P, Blom HJ, Milos R, et al. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet. 1995;10(1):111-113. (PubMed)
92. McNulty H, Strain JJ, Hughes CF, Pentieva K, Ward M. Evidence of a role for one-carbon metabolism in blood pressure: can B vitamin intervention address the genetic risk of hypertension owing to a common folate polymorphism? Curr Dev Nutr. 2020;4(1):nzz102. (PubMed)
93. Yang B, Fan S, Zhi X, et al. Associations of MTHFR gene polymorphisms with hypertension and hypertension in pregnancy: a meta-analysis from 114 studies with 15411 cases and 21970 controls. PLoS One. 2014;9(2):e87497. (PubMed)
94. Horigan G, McNulty H, Ward M, Strain JJ, Purvis J, Scott JM. Riboflavin lowers blood pressure in cardiovascular disease patients homozygous for the 677C-->T polymorphism in MTHFR. J Hypertens. 2010;28(3):478-486. (PubMed)
95. Wilson CP, Ward M, McNulty H, et al. Riboflavin offers a targeted strategy for managing hypertension in patients with the MTHFR 677TT genotype: a 4-y follow-up. Am J Clin Nutr. 2012;95(3):766-772. (PubMed)
96. Rooney M, Bottiglieri T, Wasek-Patterson B, et al. Impact of the MTHFR C677T polymorphism on one-carbon metabolites: Evidence from a randomised trial of riboflavin supplementation. Biochimie. 2020;173:91-99. (PubMed)
97. Amenyah SD, Ward M, McMahon A, et al. DNA methylation of hypertension-related genes and effect of riboflavin supplementation in adults stratified by genotype for the MTHFR C677T polymorphism. Int J Cardiol. 2021;322:233-239. (PubMed)
98. Yuvaraj S, Premkumar VG, Vijayasarathy K, Gangadaran SG, Sachdanandam P. Augmented antioxidant status in Tamoxifen treated postmenopausal women with breast cancer on co-administration with Coenzyme Q10, Niacin and Riboflavin. Cancer Chemother Pharmacol. 2008;61(6):933-941. (PubMed)
99. Hassan I, Chibber S, Khan AA, Naseem I. Riboflavin ameliorates cisplatin induced toxicities under photoillumination. PLoS One. 2012;7(5):e36273. (PubMed)
100. Raiskup F, Spoerl E. Corneal crosslinking with riboflavin and ultraviolet A. I. Principles. Ocul Surf. 2013;11(2):65-74. (PubMed)
101. Beckman KA, Gupta PK, Farid M, et al. Corneal crosslinking: Current protocols and clinical approach. J Cataract Refract Surg. 2019;45(11):1670-1679. (PubMed)
102. Definition of MS. National Multiple Sclerosis Society. Available at: https://www.nationalmssociety.org/What-is-MS/Definition-of-MS. Accessed 8/25/21.
103. Parks NE, Jackson-Tarlton CS, Vacchi L, Merdad R, Johnston BC. Dietary interventions for multiple sclerosis-related outcomes. Cochrane Database Syst Rev. 2020;5:CD004192. (PubMed)
104. Ghadirian P, Jain M, Ducic S, Shatenstein B, Morisset R. Nutritional factors in the aetiology of multiple sclerosis: a case-control study in Montreal, Canada. Int J Epidemiol. 1998;27(5):845-852. (PubMed)
105. Naghashpour M, Amani R, Sarkaki A, et al. Brain-derived neurotrophic and immunologic factors: beneficial effects of riboflavin on motor disability in murine model of multiple sclerosis. Iran J Basic Med Sci. 2016;19(4):439-448. (PubMed)
106. Naghashpour M, Majdinasab N, Shakerinejad G, et al. Riboflavin supplementation to patients with multiple sclerosis does not improve disability status nor is riboflavin supplementation correlated to homocysteine. Int J Vitam Nutr Res. 2013;83(5):281-290. (PubMed)
107. US Department of Agriculture, Agricultural Research Service. 2020. Nutrient Intakes from Food and Beverages: Mean Amounts Consumed per Individual, by Gender and Age, What We Eat in America, NHANES 2017-2018.
108. Food and Nutrition Board, Institute of Medicine. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, D.C.: National Academy Press; 2000. (National Academy Press)
109. National Adult Nutrition Survey (NANS, 2008-2010). Summary Report, 2011. Accessed March 2022. Available at: www.iuna.net/surveyreports.
110. Bates B, Cox L, Nicholson S, Page P, Prentice A, Steer T, Swan G. National Diet and Nutrition Survey Results from Years 5 and 6 (combined) of the Rolling Programme (2012/2013 – 2013/2014). A survey carried out on behalf of the Department of Health and the Food Standards Agency, 2016. Accessed March 2022. Available at: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/551352/NDNS_Y5_6_UK_Main_Text.pdf
111. Dainty JR, Bullock NR, Hart DJ, et al. Quantification of the bioavailability of riboflavin from foods by use of stable-isotope labels and kinetic modeling. Am J Clin Nutr. 2007;85(6):1557-1564. (PubMed)
112. Hendler S, Rorvik D, eds. PDR for Nutritional Supplements. Montvale: Medical Economics Company, Inc.; 2001.
113. Sugiyama M. Role of physiological antioxidants in chromium(VI)-induced cellular injury. Free Radic Biol Med. 1992;12(5):397-407. (PubMed)
114. Subramanian VS, Subramanya SB, Ghosal A, Said HM. Chronic alcohol feeding inhibits physiological and molecular parameters of intestinal and renal riboflavin transport. Am J Physiol Cell Physiol. 2013;305(5):C539-46. (PubMed)
115. Russell RM, Suter PM. Vitamin requirements of elderly people: an update. Am J Clin Nutr. 1993;58(1):4-14. (PubMed)
116. Blumberg J. Nutritional needs of seniors. J Am Coll Nutr. 1997;16(6):517-523. (PubMed)
117. Lopez-Sobaler AM, Ortega RM, Quintas ME, et al. The influence of vitamin b2 intake on the activation coefficient of erythrocyte glutation reductase in the elderly. J Nutr Health Aging. 2002;6(1):60-62. (PubMed)
118. Gariballa S, Ullegaddi R. Riboflavin status in acute ischaemic stroke. Eur J Clin Nutr. 2007;61(10):1237-1240. (PubMed)
119. Yazdanpanah N, Uitterlinden AG, Zillikens MC, et al. Low dietary riboflavin but not folate predicts increased fracture risk in postmenopausal women homozygous for the MTHFR 677 T allele. J Bone Miner Res. 2008;23(1):86-94. (PubMed)
Thiamin
Contents
Summary
- Thiamin pyrophosphate (TPP), the active form of thiamin, is involved in several enzyme functions associated with the metabolism of carbohydrates, branched-chain amino acids, and fatty acids. (More information)
- Severe thiamin deficiency leads to beriberi, a disease that affects multiple organ systems, including the central and peripheral nervous systems. (More information)
- Wernicke’s encephalopathy refers to an acute neurologic disorder secondary to thiamin deficiency. The Wernicke-Korsakoff syndrome results in persistent alterations in memory formation, along with the encephalopathy-related symptoms. (More information)
- Thiamin deficiency can result from poor dietary intake, inadequate provision in parenteral nutrition, reduced gastrointestinal absorption, increased metabolic requirements, or excessive loss of thiamin. Chronic alcohol consumption is the primary cause of thiamin deficiency in industrialized countries. (More information)
- Alteration in glucose metabolism has been associated with decreased plasma thiamin concentrations in patients with diabetes. Correction of thiamin deficiency may reduce the risk of vascular complications in these patients. (More information)
- Alzheimer’s disease has been associated with altered glucose metabolism and thiamin deficiency. Although some promising results have been observed in animal models, it is not known whether supplementation with thiamin or benfotiamine (a synthetic precursor of thiamin) might slow the cognitive decline in patients with Alzheimer’s disease. (More information)
- A recent study found decreased levels of thiamin in the brain of patients with Huntington’s disease. Clinical trials are needed to evaluate whether vitamin supplementation might be a potential therapy. (More information)
- Diuretic-induced thiamin excretion may increase the risk of thiamin deficiency and disease severity in subjects with congestive heart failure. Further studies are needed to assess the inclusion of thiamin supplementation in the management of this disease. (More information)
- Intravenous thiamin has been studied as potential treatment for sepsis, either as a monotherapy or in combination with other agents like vitamin C and corticosteroids. Large-scale clinical trials are needed to determine its efficacy. (More information)
Thiamin (also spelled thiamine) is a water-soluble B vitamin, also known as vitamin B1 or aneurine (1). Isolated and characterized in the 1930s, thiamin was one of the first organic compounds to be recognized as a vitamin (2). Thiamin occurs in the human body as free thiamin and as various phosphorylated forms: thiamin monophosphate (TMP), thiamin triphosphate, adenosine thiamin triphosphate, and thiamin pyrophosphate (TPP), which is also known as thiamin diphosphate.
Function
Coenzyme function
The synthesis of TPP from free thiamin requires magnesium, adenosine triphosphate (ATP), and the enzyme, thiamin pyrophosphokinase. In humans, TPP is required as a coenzyme in the metabolism of carbohydrates and branched-chain amino acids. Forms of thiamin are also needed for ribose synthesis and for α-oxidation of 3-methyl-branched fatty acids.
Pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, 2-oxoadipate dehydrogenase, and branched-chain α-ketoacid dehydrogenase (BCKDH) each comprise a different enzyme complex found within cellular organelles called mitochondria. They catalyze the decarboxylation of pyruvate, α-ketoglutarate, 2-oxoadipate, and branched-chain amino acids (BCAA) to form acetyl-coenzyme A (CoA), succinyl-CoA, glutaryl-CoA, and derivatives of BCAA, respectively (Figure 1). All products play critical roles in the production of energy from food through their connection to the citric acid (Krebs) cycle (2). BCAA, including leucine, isoleucine, and valine, are eventually degraded into acetyl-CoA and succinyl-CoA to fuel the citric acid cycle. The catabolism of the three BCAAs also contributes to the production of cholesterol and donates nitrogen for the synthesis of the neurotransmitters, glutamate and g-aminobutyric acid (GABA) (3). In addition to the thiamin coenzyme (TPP), each dehydrogenase complex requires a niacin-containing coenzyme (NAD), a riboflavin-containing coenzyme (FAD), and lipoic acid.
Transketolase catalyzes critical reactions in another metabolic pathway occurring in the cytosol, known as the pentose phosphate pathway. One of the most important intermediates of this pathway is ribose-5-phosphate, a phosphorylated 5-carbon sugar required for the synthesis of the high-energy ribonucleotides, such as ATP and guanosine triphosphate (GTP). Nucleotides are the building blocks of nucleic acids, DNA, and RNA. The pentose phosphate pathway also supplies various anabolic pathways, including fatty acid synthesis, with the niacin-containing coenzyme NADPH (1, 4). Because transketolase decreases early in thiamin deficiency and, unlike most thiamin-dependent enzymes, is present in red blood cells, measurement of its activity in red blood cells has been used to assess thiamin nutritional status (2, 5, 6).
2-Hydroxyacyl-CoA lyase is a TPP-dependent enzyme in peroxisomes that catalyzes the catabolism of 3-methyl-branched fatty acids through the process of α-oxidation, the oxidative removal of a single carbon atom from fatty acids like phytanic acid (7).

Deficiency
Beriberi, the disease resulting from severe thiamin deficiency, was described in Chinese literature as early as 2600 B.C. Thiamin deficiency affects the cardiovascular, muscular, gastrointestinal, and central and peripheral nervous systems (2). Beriberi has been subdivided into dry, wet, cerebral, or gastrointestinal, depending on the systems affected by severe thiamin deficiency (1, 8).
Dry beriberi
The main feature of dry (paralytic or nervous) beriberi is peripheral neuropathy. Early in the course of the neuropathy, "burning feet syndrome" may occur. Other symptoms include abnormal (exaggerated) reflexes, as well as diminished sensation and weakness in the legs and arms. Muscle pain and tenderness and difficulty rising from a squatting position have also been observed (9).
Wet beriberi
In addition to neurologic symptoms, wet (cardiac) beriberi is characterized by cardiovascular manifestations of thiamin deficiency, which include rapid heart rate, enlargement of the heart, severe swelling (edema), difficulty breathing, and ultimately, congestive heart failure. The Japanese literature describes the acute fulminant form of wet beriberi as “shoshin” (10).
Cerebral beriberi
Cerebral beriberi may lead to Wernicke's encephalopathy and Korsakoff's psychosis, especially in people who abuse alcohol. The diagnosis of Wernicke's encephalopathy is based on a "triad" of signs, which include abnormal eye movements, stance and gait ataxia, and cognitive impairment. Due in part to an overlap of symptoms with alcoholic delirium, Wernicke’s encephalopathy is thought to be underdiagnosed (11). If left untreated, the irreversible neurologic damage can cause additional clinical manifestations known as Korsakoff’s psychosis. This syndrome – also called Korsakoff’s dementia, Korsakoff's amnesia, or amnestic confabulatory syndrome – involves a confused, apathetic state and a profound memory disorder, with severe amnesia and loss of recent and working memory.
Thiamin deficiency affecting the central nervous system is referred to as Wernicke's disease when the amnesic state is not present and Wernicke-Korsakoff syndrome (WKS) when the amnesic symptoms are present along with the eye-movement and gait disorders. Rarer neurologic manifestations can include seizures (12). Most WKS sufferers are alcoholics, although it has been observed in other disorders of gross malnutrition, including stomach cancer and AIDS. Administration of intravenous thiamin to WKS patients generally results in prompt improvement of the eye symptoms, but improvements in motor coordination and memory may be less, depending on how long the symptoms have been present. Evidence of increased immune cell activation and increased free radical production in the areas of the brain that are selectively damaged suggests that oxidative stress plays an important role in the neurologic pathology of thiamin deficiency (13).
Gastrointestinal beriberi
TPP is critical for metabolic reactions that utilize glucose in glycolysis and the citric acid cycle (see Figure 1). A decrease in the activity of thiamin-dependent enzymes limits the conversion of pyruvate to acetyl-CoA and the utilization of the citric acid cycle, leading to accumulation of pyruvate and lactate. Lactic acidosis, a condition resulting from the accumulation of lactate, is often associated with nausea, vomiting, and severe abdominal pain in a syndrome described as gastrointestinal beriberi (8).
Causes of thiamin deficiency
Thiamin deficiency may result from inadequate thiamin intake, increased requirement for thiamin, excessive loss of thiamin from the body, consumption of anti-thiamin factors in food, or a combination of these factors.
Inadequate intake
Inadequate consumption of thiamin is the main cause of thiamin deficiency in developing countries (2). Thiamin deficiency is common in low-income populations whose diets are high in carbohydrate and low in thiamin (e.g., milled or polished rice). Breast-fed infants whose mothers are thiamin deficient are vulnerable to developing infantile beriberi. Alcoholism, which is associated with low intake of thiamin among other nutrients, is the primary cause of thiamin deficiency in industrialized countries. Some of the non-alcoholic conditions associated with WKS include anorexia nervosa, bariatric surgery (weight-loss surgery), gastrointestinal malignancies, and malabsorption syndromes (14-17). Obese individuals may also be at heightened risk of thiamin deficiency (18, 19). Moreover, cases of Wernicke’s encephalopathy have been linked with hyperemesis gravidarum (severe nausea and vomiting during pregnancy) (20, 21), and with parenteral nutrition lacking vitamin supplementation (22, 23).
Increased requirement
Conditions resulting in an increased requirement for thiamin include strenuous physical exertion, fever, pregnancy, breast-feeding, and adolescent growth. Such conditions place individuals with marginal thiamin intake at risk for developing symptomatic thiamin deficiency.
Malaria patients in Southeast Asia were found to be thiamin deficient more frequently than non-infected individuals (24, 25). Malarial infection leads to a large increase in the metabolic demand for glucose. Because thiamin is required for enzymes involved in glucose metabolism, the stresses induced by malarial infection could exacerbate thiamin deficiency in predisposed individuals. HIV-infected individuals, whether or not they had developed AIDS, were also found to be at increased risk for thiamin deficiency (26). Further, chronic alcohol abuse impairs intestinal absorption and utilization of thiamin (1); thus, alcoholics have increased requirements for thiamin. Thiamin deficiency is also observed as a complication of the refeeding syndrome: the introduction of carbohydrates in severely starved individuals leads to an increased demand for thiamin in glycolysis and the citric acid cycle that precipitates thiamin deficiency (27).
Excessive loss
Excessive loss of thiamin may precipitate thiamin deficiency. By increasing urinary flow, diuretics may prevent reabsorption of thiamin by the kidneys and increase its excretion in the urine (28, 29). The risk of thiamin deficiency is increased in diuretic-treated patients with marginal thiamin intake (30) and in individuals receiving long-term, diuretic therapy (31). Individuals with kidney failure requiring hemodialysis lose thiamin at an increased rate and are at risk for thiamin deficiency (32). Alcoholics who maintain a high fluid intake and high urine flow rate may also experience increased loss of thiamin, exacerbating the effects of low thiamin intake (33).
Anti-thiamin factors (ATF)
The presence of anti-thiamin factors (ATF) in foods contributes to the risk of thiamin deficiency. Certain plants contain ATF, which react with thiamin to form an oxidized, inactive product. Consuming very large amounts of tea or coffee (including decaffeinated), as well as chewing tea leaves and betel nuts, might lower thiamin status due to the presence of ATF (34, 35). ATF include mycotoxins (molds) and thiaminases that break down thiamin in food. Individuals who habitually eat certain raw fresh-water fish, raw shellfish, or ferns are at higher risk of thiamin deficiency because these foods contain thiaminase that normally is inactivated by heat in cooking (1, 6). In Nigeria, an acute, neurologic syndrome (seasonal ataxia) has been associated with thiamin deficiency precipitated by a thiaminase in African silkworms, a traditional, high-protein food for some Nigerians (36).
The Recommended Dietary Allowance (RDA)
The RDA for thiamin, revised in 1998 by the Food and Nutrition Board of the Institute of Medicine, was based on the prevention of deficiency in generally healthy individuals (37; Table 1).
| Life Stage | Age | Males (mg/day) | Females (mg/day) |
|---|---|---|---|
| Infants | 0-6 months | 0.2 (AI) | 0.2 (AI) |
| Infants | 7-12 months | 0.3 (AI) | 0.3 (AI) |
| Children | 1-3 years | 0.5 | 0.5 |
| Children | 4-8 years | 0.6 | 0.6 |
| Children | 9-13 years | 0.9 | 0.9 |
| Adolescents | 14-18 years | 1.2 | 1.0 |
| Adults | 19 years and older | 1.2 | 1.1 |
| Pregnancy | all ages | - | 1.4 |
| Breast-feeding | all ages | - | 1.4 |
Disease Prevention
Cataracts
A cross-sectional study of 2,900 Australian men and women, 49 years of age and older, found that those in the highest quintile of thiamin intake were 40% less likely to have nuclear cataracts than those in the lowest quintile (38). In addition, a study in 408 US women found that higher dietary intakes of thiamin were inversely associated with five-year change in lens opacification (39). However, these cross-sectional associations have yet to be elucidated by studies of causation.
Diabetes mellitus and vascular complications
Patients with diabetes mellitus have been reported to have low plasma concentrations and high renal clearance of thiamin (40, 41), suggesting that individuals with type 1 or type 2 diabetes are at increased risk for thiamin deficiency. Two thiamin transporters, thiamin transporter-1 (THTR-1) and THTR-2, are involved in thiamin uptake by enterocytes in the small intestine and re-uptake in the proximal tubules of the kidneys. One study suggested that hyperglycemia in patients with diabetes could affect thiamin re-uptake by decreasing the expression of thiamin transporters in the kidneys (42). Conversely, thiamin deficiency appears to impair the normal endocrine function of the pancreas and exacerbate hyperglycemia. Early studies showed that insulin synthesis and secretion were altered in the endocrine pancreatic cells of thiamin-deficient rats (43, 44). In humans, thiamin deficiency caused by recessive mutations in the gene encoding THTR-1 leads to diabetes mellitus in the thiamin-responsive megaloblastic anemia syndrome (see Metabolic diseases below).
In a randomized, double-blind pilot study, high-dose thiamin supplements (300 mg/day) were given for six weeks to hyperglycemic individuals (either glucose intolerant or newly diagnosed with type 2 diabetes). Thiamin supplementation prevented any further increase in fasting glucose and insulin concentrations compared with placebo treatment but did not reduce the hyperglycemia (45). However, one study suggested that thiamin supplementation might improve fasting glucose concentrations in early stages of type 2 diabetes (i.e., pre-diabetes) (46).
Disease Treatment
Alzheimer's disease
Some older adults are at increased risk for developing subclinical thiamin deficiency secondary to poor dietary intake, reduced gastrointestinal absorption, and multiple medical conditions (47, 48). Since thiamin deficiency can result in a form of dementia (Wernicke-Korsakoff syndrome), its relationship to Alzheimer's disease (AD) and other forms of dementia have been investigated. AD is characterized by a decline in cognitive function in elderly people, accompanied by pathologic features that include β-amyloid plaque deposition and neurofibrillary tangles formed by hyperphosphorylated Tau protein (49).
Using positron emission tomography (PET) scanning, reduced glucose metabolism has been observed in brains of AD patients (50). A large, multicenter PET study using a radiolabeled glucose analog, 18F-Fluoro-deoxyglucose (FDG), correlated a reduction in FDG uptake (a surrogate marker for glucose metabolism) with the extent of cognitive impairment in AD patients. This study, which included 822 subjects over 55 years of age that were cognitively normal (n=229), displayed mild cognitive impairment (n=405), or had mild AD (n=188), demonstrated that brain glucose utilization could predict the progression from mild cognitive impairment to AD (51). A nine-year longitudinal study associated the presence of diabetes mellitus in older people (above 55 years old) with an increased risk for developing AD (52). Emerging evidence links type 2 diabetes and AD, conditions that may involve insulin resistance in the brain (reviewed in 53).
A reduction in thiamin-dependent processes in the brain appears to be related to the altered glucose metabolism in patients with AD (54-56). Case-control studies have found blood levels of thiamin, TPP, and TMP to be lower in those with dementia of Alzheimer's type (DAT) compared to control subjects (57, 58). Moreover, several investigators have found evidence of decreased activity of TPP-dependent enzymes, α-ketoglutarate dehydrogenase and transketolase, in the brains of patients who died of AD (59). The finding of decreased brain levels of TPP in the presence of normal levels of free thiamin and TMP suggests altered TPP synthesis rather than poor thiamin bioavailability. However, it is not clear whether the activities of TPP-metabolizing enzymes (including thiamin pyrophosphokinase) are altered in AD patients (60, 61). Chronic administration of the thiamin derivative benfotiamine alleviated cognitive alterations and decreased the number of β-amyloid plaques in a mouse model of AD without increasing TMP and TPP levels in the brain. This suggested that the beneficial effects of benfotiamine in the brain were likely mediated by the stimulation of TPP-independent pathways (62). Chronic benfotiamine administration was also shown to decrease the number of neurofibrillary tangles in certain brain regions and improve survival in a mouse model (63). In a rat model of neurodegeneration, long-term oral benfotiamine supplementation increased thiamin pyrophosphate concentrations and led to improvements in insulin signaling and cognitive deficits (64).
Thiamin deficiency has been linked to increased β-amyloid production in cultured neuronal cells and to plaque formation in animal models (65, 66). These pathological hallmarks of AD could be reversed by thiamin supplementation, suggesting that thiamin could be protective in AD. Other disorders, including mitochondrial dysfunction and chronic oxidative stress, have been linked to both thiamin deficiency and AD pathogenesis and progression (13, 55, 67). Presently, there is only slight and inconsistent evidence that thiamin supplements are of benefit in AD. A double-blind, placebo-controlled study of 15 patients (10 completed the study) reported no beneficial effect of 3 grams/day of thiamin on cognitive decline over a 12-month period (68). A preliminary report from another study claimed a mild benefit of 3 to 8 grams of thiamin per day in DAT, but no additional data from that study are available (69). A mild beneficial effect in patients with AD was reported after 12 weeks of treatment with 100 mg/day of a thiamin derivative (thiamin tetrahydrofurfuryl disulfide), but this study was not placebo-controlled (70). A 2001 systematic review of randomized, double-blind, placebo-controlled trials of thiamin in patients with DAT found no evidence that thiamin was a useful treatment for the symptoms of Alzheimer's disease (71). More recently, a small uncontrolled study in five patients with mild-to-moderate AD reported cognitive improvement, measured by the Mini-Mental Status Examination, following supplementation with 300 mg/day of benfotiamine for 18 months (72). In a placebo-controlled study of 70 β-amyloid positive patients with either amnestic mild cognitive impairment (a precursor to AD) or mild AD, those receiving 600 mg/day of benfotiamine for 12 months experienced less cognitive decline compared to placebo, but the differences did not reach statistical significance (p=0.125; 73). Large-scale, randomized controlled trials are needed to determine whether supplemental thiamin or benfotiamine might help slow progression of cognitive decline in those with Alzheimer’s disease.
Huntington’s disease
Huntington’s disease is an inherited neurodegenerative disorder characterized by selective degeneration of nerve cells known as striatal spiny neurons. Symptoms, such as movement disorders and impaired cognitive function, typically develop in the fourth decade of life and progressively deteriorate over time. A recent study found decreased levels of the thiamin transporter-2 (THTR-2) protein in the striatum and frontal cortex of patients with Huntington’s disease compared to age-and sex-matched healthy controls (74). Compared to control subjects, this study also found lower concentrations of TPP in the striatum and lower concentrations of TMP in the cerebrospinal fluid of patients with Huntington’s disease (74). Mutations in the SLC19A3 gene that encodes THTR-2 causes biotin-thiamin-responsive basal ganglia disease, which is treated with high-dose co-supplementation with biotin and thiamin for life (see Biotin-thiamin-responsive basal ganglia disease below). In a mouse model of Huntington’s disease, high-dose supplementation with both of these vitamins improved neuropathological and motor deficits but had no effect on lifespan (p=0.15) (74). A phase II, open-label clinical trial evaluating the effect of combined thiamin-biotin supplementation, at moderate (600 mg/day of thiamin and 150 mg/day of biotin) and high (1,200 mg/day of thiamin and 300 mg/day of biotin) dosages, in Huntington’s disease is currently underway (75).
Congestive heart failure
Severe thiamin deficiency (wet beriberi) can lead to impaired cardiac function and ultimately congestive heart failure (CHF). Although cardiac manifestations of beriberi are rarely encountered in industrialized countries, CHF due to other causes is common, especially in the elderly. Loop diuretics used in the treatment of CHF, notably furosemide, increase thiamin excretion, potentially leading to thiamin deficiency (76, 77). Patients with CHF might also have altered thiamin metabolism, including reduced absorption of thiamin in the small intestine (78). A 2015 meta-analysis of nine observational studies found a 2.5 times higher risk of thiamin deficiency in patients with heart failure compared to control subjects (78). As in the general population, older CHF patients were found to be at higher risk of thiamin deficiency than younger ones (79).
An important measure of cardiac function in CHF is the left ventricular ejection fraction (LVEF), which can be assessed by echocardiography. One study in 25 patients found that furosemide use at doses of 80 mg/day or greater was associated with a 98% prevalence of thiamin deficiency (31). In a randomized, double-blind study of 30 CHF patients, all of whom had been taking furosemide (80 mg/day) for at least three months, intravenous (IV) thiamin therapy (200 mg/day) for seven days resulted in an improved LVEF compared to IV placebo (80). When all 30 of the CHF patients in that study subsequently received six weeks of oral thiamin therapy (200 mg/day), the average LVEF improved by 22%. This finding may be relevant because improvements in LVEF have been associated with improved survival in CHF patients (81). However, clinical trials of oral thiamin supplementation in heart failure patients have not found any benefit. In a randomized, double-blind, placebo-controlled trial in 52 patients with systolic heart failure, 300 mg/day of supplemental thiamin for one month did not improve LVEF compared to placebo (82). A randomized, double-blind, placebo-controlled trial in 64 patients with heart failure reported that 200 mg/day or supplemental thiamin for six months did not improve LVEF (83).
Although little evidence supports the routine use of supplemental thiamin in CHF patients, trials specifically in CHF patients with marginal thiamin status have not been done, and some suggest that it may be prudent to screen patients on long-term diuretic therapy for thiamin deficiency and treat accordingly (84).
Type 2 diabetes mellitus and vascular complications
Chronic hyperglycemia in individuals with diabetes mellitus contributes to the pathogenesis of microvascular diseases. Diabetes-related vascular damage can affect the heart (cardiomyopathy), kidneys (nephropathy), retina (retinopathy), and peripheral nervous system (neuropathy). In subjects with diabetes, hyperglycemia alters the function of bone marrow-derived endothelial progenitor cells (EPC) that are critical for the growth of blood vessels (85). Interestingly, a higher daily intake of thiamin from the diet was correlated with more circulating EPC and with better vascular endothelial health in 88 individuals with type 2 diabetes (86). An inverse association has also been found between plasma concentrations of thiamin and the presence of soluble vascular adhesion molecule-1 (sVCAM-1), a marker of vascular dysfunction, in patients with diabetes (40, 87). Early markers of diabetic nephropathy include the presence of serum albumin in the urine, known as microalbuminuria. Administration of thiamin or benfotiamine (a thiamin derivative) prevented the development of renal complications in chemically-induced diabetic rats (88). A randomized, double-blind, placebo-controlled study conducted in 40 patients with type 2 diabetes with microalbuminuria found that high-dose thiamin supplementation (300 mg/day) decreased excretion of urinary albumin over a three-month period (87). Since thiamin treatment has shown promising results in cultured cells and animal models (89-91), the effects of thiamin and its derivatives on vascular complications should be examined in patients with diabetes.
Cancer
Thiamin deficiency and Wernicke-Korsakoff syndrome have been observed in some cancer patients with rapidly growing tumors (92, 93). Research in cell culture and animal models indicates that rapidly dividing cancer cells have a high requirement for thiamin (94). All rapidly dividing cells require nucleic acids at an increased rate, and some cancer cells appear to rely heavily on the TPP-dependent enzyme, transketolase, to provide the ribose-5-phosphate necessary for nucleic acid synthesis. One study found that the levels of THTR-1, transketolase, and TPP mitochondrial transporters were increased in samples of human breast cancer tissue compared to normal tissue, suggesting an adaptation in thiamin homeostasis in support of cancer metabolism (95). Other studies have found that the gene encoding THTR-2 is downregulated in certain cancers (96). Moreover, use of the chemotherapeutic drug, 5-fluorouracil, inhibits phosphorylation of thiamin to thiamin pyrophosphate and may thus lead to thiamin deficiency (97, 98).
Thiamin supplementation in cancer patients is common to prevent thiamin deficiency, but Boros et al. caution that too much thiamin may actually fuel the growth of some malignant tumors (99), suggesting that thiamin supplementation be reserved for those cancer patients who are actually deficient in thiamin. Presently, there is no evidence available from studies in humans to support or refute this theory. However, it would be prudent for individuals with cancer who are considering thiamin supplementation to discuss it with the clinician managing their cancer therapy. Intravenous, high-dose thiamin has been suggested as a treatment for cancer patients with confirmed Wernicke-Korsakoff (93).
Sepsis
Sepsis is a life-threatening critical illness caused by a dysregulated host response to an infection. The widespread inflammation can lead to tissue and organ damage and to death (100). Because thiamin deficiency is common among septic patients (101), several studies have investigated the treatment effect of intravenous thiamin – alone or in combination with other agents like vitamin C and hydrocortisone.
Observational studies examining the association of intravenous thiamin as a monotherapy have mainly looked at its association with lactic acidosis, which commonly occurs in both thiamin deficiency and sepsis, and with mortality. One retrospective study in 123 septic patients and 246 matched controls found that intravenous thiamin administration within 24 hours of hospital admission was linked to improvements in both lactate clearance and 28-day mortality (102). In a small retrospective study of 53 alcohol-use disorder patients presenting with septic shock, lower mortality was observed in the 34 patients who received intravenous thiamin compared to the 19 patients who did not (103).
A few randomized controlled trials have evaluated the effect of intravenous thiamin in the treatment of sepsis. A randomized, double-blind, placebo-controlled trial in 88 patients with sepsis and elevated blood concentrations of lactate reported that intravenous thiamin (200 mg twice daily for seven days or until discharge from the hospital) did not decrease lactate concentrations at 24 hours post initiation of treatment – the primary endpoint of the trial (104). No differences between the treatment and placebo groups were found for the secondary endpoints, which included survival (104). In a subsequent analysis of data from this trial, the septic patients that were given parenteral thiamin (n=31) had lower creatinine concentrations throughout the treatment and were less likely to need renal replacement therapy compared to placebo (n=39; 105).
A 2020 meta-analysis of four studies – one observational and three randomized controlled trials – found no benefit of intravenous thiamin for improving lactate concentrations, length of hospital stay in intensive care, or overall survival (106). Large-scale clinical trials are needed to determine whether parenteral administration of thiamin is beneficial in the treatment of sepsis. Administering thiamin in combination with vitamin C and corticosteroids may be more efficacious to treat sepsis (107); some clinical trials of such treatments are currently underway (see clinicaltrials.gov/).
Metabolic diseases
Thiamin supplementation is included in the clinical management of genetic diseases that affect the metabolism of carbohydrates and branched-chain amino acids (BCAAs).
Thiamin-responsive pyruvate dehydrogenase complex (PDHC) deficiency
Mutations in PDHC prevent the efficient oxidation of carbohydrates in affected individuals. PDHC deficiency is commonly characterized by lactic acidosis, neurologic and neuromuscular degeneration, and death during childhood. The patients who respond to thiamin treatment (from a few mg/day to doses above 1,000 mg/day) exhibit PDHC deficiency due to the decreased affinity of PDHC for TPP (108, 109). Although thiamin supplementation can reduce lactate accumulation and improve the clinical features in thiamin-responsive patients, it does not constitute a cure (110).
Maple syrup urine disease
Inborn errors of BCAA metabolism lead to thiamin-responsive branched-chain ketoaciduria, also known as maple syrup urine disease. Alterations in the BCAA catabolic pathway result in neurologic dysfunction caused by the accumulation of BCAAs and their derivatives, branched-chain ketoacids (BCKA). The therapeutic approach includes a synthetic diet with reduced BCAA content, and thiamin (10-1,000 mg/day) is supplemented to patients with mutations in the E2 subunit of the BCKDH complex (111). In thiamin-responsive individuals, the supplementation has been proven effective to correct the phenotype without recourse to the BCAA restriction diet.
Thiamin-responsive megaloblastic anemia
Mutations in the SLC19A2 gene that encodes THTR-1 impairs intestinal thiamin uptake and causes thiamin deficiency, leading to thiamin-responsive megaloblastic anemia (112). This syndrome, which is also called thiamin metabolism dysfunction syndrome-1, is characterized by megaloblastic anemia, diabetes mellitus, and deafness. A review of 30 cases reported additional neurologic, visual, and cardiac impairments (113). High-dose oral supplementation with thiamin (up to 300 mg/day) helps to maintain health and correct hyperglycemia in prepubescent children. A recent study in 32 individuals with found no additional benefit of oral doses above 150 mg/day (114). After puberty, a decline in pancreatic function results in the requirement of insulin together with thiamin to control the hyperglycemia. One study also reported that the treatment of a four-month-old girl with 100 mg/day of thiamin did not prevent hearing loss at 20 months of age (115). Early diagnosis of the syndrome and early treatment with thiamin is important for a better prognosis (114).
Biotin-thiamin-responsive basal ganglia disease
Biotin-thiamin-responsive basal ganglia disease (also called biotin-responsive basal ganglia disease, thiamin transporter-2 deficiency, and thiamin metabolism dysfunction syndrome-2) is caused by an autosomal recessive mutation in the SLC19A3 gene that codes for THTR-2. The disease usually presents around 3 to 10 years of age (116), but an early infantile form of the disease exists with onset as early as one month of age (117). Clinical features include subacute encephalopathy (confusion, drowsiness, altered level of consciousness), ataxia, and seizures.
A retrospective study of 18 affected individuals from the same family or the same tribe in Saudi Arabia showed that biotin monotherapy (5-10 mg/kg/day) efficiently abolished the clinical manifestations of the disease, although one-third of the patients suffered from recurrent acute crises. Often associated with poor outcomes, acute crises were not observed for a five-year follow-up period following thiamin supplementation (300-400 mg/day) – early diagnosis and immediate treatment with biotin and thiamin led to positive outcomes (118). Recent studies have found supplemental thiamin to be important in treating the condition. In an open-label study of 20 pediatric patients with the disease, supplemental thiamin alone was as effective as combined biotin-thiamin supplementation when given for 30 months (119). Lifelong high-dose supplementation with a combination of biotin and thiamin is generally the treatment for biotin-thiamin-responsive basal ganglia disease (116). Early diagnosis and treatment is important to ensure a better prognosis (117, 120).
Other thiamin metabolism dysfunction syndromes
Supplemental thiamin has limited utility in treating other inborn errors of thiamin metabolism. Mutations in the SLC25A19 gene that codes for the mitochondrial TPP transporter can result in either thiamin metabolism dysfunction syndrome-3 (THMD3) or thiamin metabolism dysfunction syndrome-4 (THMD4). Of these rare syndromes, THMD3 (also called Amish-type microcephaly or Amish lethal microcephaly) has the more severe phenotype, resulting in a congenital microcephaly, elevated concentrations of α-ketoglutarate in urine, and usually death in infancy (121). THMD4 is characterized by episodic encephalopathy and weakness, which often presents following a viral infection or febrile illness in childhood. Some patients affected with THMD4 may respond to high-dose thiamin supplementation (122).
Mutations in the TPK1 gene result in thiamin pyrophosphokinase 1 deficiency and thiamin metabolism dysfunction syndrome-5 (THMD5), which usually manifests in early childhood. While the clinical presentation of THMD5 varies, affected individuals often experience episodic ataxia, dystonia, and lactic acidosis (123). Only a few cases of THMD5 have been reported to date; two of these patients experienced limited improvement of symptoms upon supplementation with thiamin, in conjunction with adherence to a high-fat diet (124).
Sources
Humans obtain thiamin from dietary sources and from the normal microflora of the colon, although the contribution of the latter towards the body’s requirement for thiamin is not clear (125).
Food sources
A varied diet should provide most individuals with adequate thiamin to prevent deficiency. In the US the average dietary thiamin intake for young adult men is about 2 mg/day and 1.2 mg/day for young adult women. A survey of people over the age of 60 found an average dietary thiamin intake of 1.4 mg/day for men and 1.1 mg/day for women (37). However, institutionalization and poverty both increase the likelihood of inadequate thiamin intake in the elderly (126). Whole-grain cereals, legumes (e.g., beans and lentils), nuts, lean pork, and yeast are rich sources of thiamin (1). Because most of the thiamin is lost during the production of white flour and polished (milled) rice, white rice and foods made from white flour (e.g., bread and pasta) are fortified with thiamin in many Western countries. A number of thiamin-rich foods are listed in the table below, along with their thiamin content in milligrams (mg). For more information on the nutrient content of foods, search USDA's FoodData Central.
Supplements
Thiamin is available in dietary supplements and in fortified foods, most commonly as thiamin hydrochloride or thiamin mononitrate (127). Multivitamin supplements typically contain at least 1.2 mg of thiamin, the Daily Value (DV) for adults and children 4 years and older (128).
Benfotiamine is a synthetic, lipid-soluble precursor of thiamin that is available as a dietary supplement. It has higher bioavailability compared to thiamin (129).
Safety
Toxicity
The Food and Nutrition Board did not set a tolerable upper intake level (UL) for thiamin because there are no well-established toxic effects from consumption of excess thiamin in food or through long-term, oral supplementation (up to 200 mg/day). A small number of life-threatening anaphylactic reactions have been observed with large intravenous doses of thiamin (37).
Drug interactions
Reduced blood concentrations of thiamin have been reported in individuals with seizure disorders (epilepsy) taking the anticonvulsant medication, phenytoin, for long periods of time (130). 5-Fluorouracil, a drug used in cancer therapy, inhibits the phosphorylation of thiamin to TPP (131). Diuretics, especially furosemide, may increase the risk of thiamin deficiency in individuals with marginal thiamin intake due to increased urinary excretion of thiamin (29). Moreover, chronic alcohol abuse is associated with thiamin deficiency due to low dietary intake, impaired absorption and utilization, and increased excretion of the vitamin (1). Chronic alcohol feeding to rats showed a decrease in the active absorption of thiamin linked to the inhibition of thiamin membrane transporter THTR-1 in the intestinal epithelium (132). Alcohol consumption in rats also decreases the levels of THTR-1 and THTR-2 in renal epithelial cells, thus limiting thiamin re-uptake by the kidneys (133).
Linus Pauling Institute Recommendation
The Linus Pauling Institute supports the recommendation by the Food and Nutrition Board of 1.2 mg/day of thiamin for men and 1.1 mg/day for women. A varied diet should provide enough thiamin for most people. Following the Linus Pauling Institute recommendation to take a daily multivitamin/mineral supplement, containing 100% of the Daily Values (DV), will ensure an intake of at least 1.5 mg/day of thiamin.
Older adults (>50 years)
Presently, there is no evidence that the requirement for thiamin is increased in older adults, but some studies have found inadequate dietary intake and thiamin insufficiency to be more common in elderly populations (126). Thus, it would be prudent for older adults to take a multivitamin/mineral supplement, which will generally provide at least 1.5 mg/day of thiamin.
Authors and Reviewers
Originally written in 2000 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in September 2002 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in June 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in June 2013 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in July 2021 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in October 2021 by:
Lucien Bettendorff, Ph.D.
Research Director, F.R.S.-FNRS
University of Liège, Belgium
Copyright 2000-2024 Linus Pauling Institute
References
1. Tanphaichitr V. Thiamin. In: Shils M, ed. Modern Nutrition in Health and Disease. 9th ed. Baltimore: Williams & Wilkins; 1999:381-389.
2. Rindi G. Thiamin. In: Ziegler E, Filer L, eds. Present Knowledge in Nutrition. Washington D.C.: ILSI Press; 1996:160-166.
3. Hutson SM, Sweatt AJ, Lanoue KF. Branched-chain [corrected] amino acid metabolism: implications for establishing safe intakes. J Nutr. 2005;135(6 Suppl):1557S-1564S. (PubMed)
4. Brody T. Nutritional Biochemistry. 2nd ed. San Diego: Academic Press; 1999.
5. Whitfield KC, Bourassa MW, Adamolekun B, et al. Thiamine deficiency disorders: diagnosis, prevalence, and a roadmap for global control programs. Ann N Y Acad Sci. 2018;1430(1):3-43. (PubMed)
6. Bettendorff L. Thiamin. In: Erdman Jr. JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Ames: Wiley-Blackwell; 2012:261-279.
7. Foulon V, Antonenkov VD, Croes K, et al. Purification, molecular cloning, and expression of 2-hydroxyphytanoyl-CoA lyase, a peroxisomal thiamine pyrophosphate-dependent enzyme that catalyzes the carbon-carbon bond cleavage during alpha-oxidation of 3-methyl-branched fatty acids. Proc Natl Acad Sci U S A. 1999;96(18):10039-10044. (PubMed)
8. Donnino M. Gastrointestinal beriberi: a previously unrecognized syndrome. Ann Intern Med. 2004;141(11):898-899. (PubMed)
9. McDowell L. Thiamin. Vitamins in Animal and Human Nutrition. 2nd ed. Ames: Iowa State University Press; 2000:265-310.
10. Yamasaki H, Tada H, Kawano S, Aonuma K. Reversible pulmonary hypertension, lactic acidosis, and rapidly evolving multiple organ failure as manifestations of shoshin beriberi. Circ J. 2010;74(9):1983-1985. (PubMed)
11. Chandrakumar A, Bhardwaj A, t Jong GW. Review of thiamine deficiency disorders: Wernicke encephalopathy and Korsakoff psychosis. J Basic Clin Physiol Pharmacol. 2018;30(2):153-162. (PubMed)
12. Doss A, Mahad D, Romanowski CA. Wernicke encephalopathy: unusual findings in nonalcoholic patients. J Comput Assist Tomogr. 2003;27(2):235-240. (PubMed)
13. Hazell AS, Faim S, Wertheimer G, Silva VR, Marques CS. The impact of oxidative stress in thiamine deficiency: a multifactorial targeting issue. Neurochem Int. 2013;62(5):796-802. (PubMed)
14. Saad L, Silva LF, Banzato CE, Dantas CR, Garcia C, Jr. Anorexia nervosa and Wernicke-Korsakoff syndrome: a case report. J Med Case Rep. 2010;4:217. (PubMed)
15. Becker DA, Balcer LJ, Galetta SL. The neurological complications of nutritional deficiency following bariatric surgery. J Obes. 2012;2012:608534. (PubMed)
16. Jung ES, Kwon O, Lee SH, et al. Wernicke's encephalopathy in advanced gastric cancer. Cancer Res Treat. 2010;42(2):77-81. (PubMed)
17. Greenspon J, Perrone EE, Alaish SM. Shoshin beriberi mimicking central line sepsis in a child with short bowel syndrome. World J Pediatr. 2010;6(4):366-368. (PubMed)
18. Nath A, Tran T, Shope TR, Koch TR. Prevalence of clinical thiamine deficiency in individuals with medically complicated obesity. Nutr Res. 2017;37:29-36. (PubMed)
19. Polegato BF, Pereira AG, Azevedo PS, et al. Role of thiamin in health and disease. Nutr Clin Pract. 2019;34(4):558-564. (PubMed)
20. Oudman E, Wijnia JW, Oey M, van Dam M, Painter RC, Postma A. Wernicke's encephalopathy in hyperemesis gravidarum: A systematic review. Eur J Obstet Gynecol Reprod Biol. 2019;236:84-93. (PubMed)
21. Meggs WJ, Lee SK, Parker-Cote JN. Wernicke encephalopathy associated with hyperemesis gravidarum. Am J Emerg Med. 2020;38(3):690 e693-690 e695. (PubMed)
22. Sequeira Lopes da Silva JT, Almaraz Velarde R, Olgado Ferrero F, et al. Wernicke's encephalopathy induced by total parental nutrition. Nutr Hosp. 2010;25(6):1034-1036. (PubMed)
23. Francini-Pesenti F, Brocadello F, Manara R, Santelli L, Laroni A, Caregaro L. Wernicke's syndrome during parenteral feeding: not an unusual complication. Nutrition. 2009;25(2):142-146. (PubMed)
24. Krishna S, Taylor AM, Supanaranond W, et al. Thiamine deficiency and malaria in adults from southeast Asia. Lancet. 1999;353(9152):546-549. (PubMed)
25. Mayxay M, Taylor AM, Khanthavong M, et al. Thiamin deficiency and uncomplicated falciparum malaria in Laos. Trop Med Int Health. 2007;12(3):363-369. (PubMed)
26. Muri RM, Von Overbeck J, Furrer J, Ballmer PE. Thiamin deficiency in HIV-positive patients: evaluation by erythrocyte transketolase activity and thiamin pyrophosphate effect. Clin Nutr. 1999;18(6):375-378. (PubMed)
27. Stanga Z, Brunner A, Leuenberger M, et al. Nutrition in clinical practice-the refeeding syndrome: illustrative cases and guidelines for prevention and treatment. Eur J Clin Nutr. 2008;62(6):687-694. (PubMed)
28. Suter PM, Haller J, Hany A, Vetter W. Diuretic use: a risk for subclinical thiamine deficiency in elderly patients. J Nutr Health Aging. 2000;4(2):69-71. (PubMed)
29. Rieck J, Halkin H, Almog S, et al. Urinary loss of thiamine is increased by low doses of furosemide in healthy volunteers. J Lab Clin Med. 1999;134(3):238-243. (PubMed)
30. Sica DA. Loop diuretic therapy, thiamine balance, and heart failure. Congest Heart Fail. 2007;13(4):244-247. (PubMed)
31. Zenuk C, Healey J, Donnelly J, Vaillancourt R, Almalki Y, Smith S. Thiamine deficiency in congestive heart failure patients receiving long term furosemide therapy. Can J Clin Pharmacol. 2003;10(4):184-188. (PubMed)
32. Hung SC, Hung SH, Tarng DC, Yang WC, Chen TW, Huang TP. Thiamine deficiency and unexplained encephalopathy in hemodialysis and peritoneal dialysis patients. Am J Kidney Dis. 2001;38(5):941-947. (PubMed)
33. Wilcox CS. Do diuretics cause thiamine deficiency? J Lab Clin Med. 1999;134(3):192-193. (PubMed)
34. Vimokesant SL, Hilker DM, Nakornchai S, Rungruangsak K, Dhanamitta S. Effects of betel nut and fermented fish on the thiamin status of northeastern Thais. Am J Clin Nutr. 1975;28(12):1458-1463. (PubMed)
35. Ventura A, Mafe MC, Bourguet M, Tornero C. Wernicke's encephalopathy secondary to hyperthyroidism and ingestion of thiaminase-rich products. Neurologia. 2013;28(4):257-259. (PubMed)
36. Nishimune T, Watanabe Y, Okazaki H, Akai H. Thiamin is decomposed due to Anaphe spp. entomophagy in seasonal ataxia patients in Nigeria. J Nutr. 2000;130(6):1625-1628. (PubMed)
37. Food and Nutrition Board, Institute of Medicine. Thiamin. Thiamin, Riboflavin, Niacin, Vitamin B-6, Vitamin B-12, Pantothenic Acid, Biotin, and Choline. Washington D.C.: National Academy Press; 1998:58-86.
38. Cumming RG, Mitchell P, Smith W. Diet and cataract: the Blue Mountains Eye Study. Ophthalmology. 2000;107(3):450-456. (PubMed)
39. Jacques PF, Taylor A, Moeller S, et al. Long-term nutrient intake and 5-year change in nuclear lens opacities. Arch Ophthalmol. 2005;123(4):517-526. (PubMed)
40. Thornalley PJ, Babaei-Jadidi R, Al Ali H, et al. High prevalence of low plasma thiamine concentration in diabetes linked to a marker of vascular disease. Diabetologia. 2007;50(10):2164-2170. (PubMed)
41. Rosner EA, Strezlecki KD, Clark JA, Lieh-Lai M. Low thiamine levels in children with type 1 diabetes and diabetic ketoacidosis: a pilot study. Pediatr Crit Care Med. 2015;16(2):114-118. (PubMed)
42. Larkin JR, Zhang F, Godfrey L, et al. Glucose-induced down regulation of thiamine transporters in the kidney proximal tubular epithelium produces thiamine insufficiency in diabetes. PLoS One. 2012;7(12):e53175. (PubMed)
43. Rathanaswami P, Sundaresan R. Effects of thiamine deficiency on the biosynthesis of insulin in rats. Biochem Int. 1991;24(6):1057-1062. (PubMed)
44. Rathanaswami P, Pourany A, Sundaresan R. Effects of thiamine deficiency on the secretion of insulin and the metabolism of glucose in isolated rat pancreatic islets. Biochem Int. 1991;25(3):577-583. (PubMed)
45. Alaei Shahmiri F, Soares MJ, Zhao Y, Sherriff J. High-dose thiamine supplementation improves glucose tolerance in hyperglycemic individuals: a randomized, double-blind cross-over trial. Eur J Nutr. 2013; 52(7):1821-4. (PubMed)
46. Gonzalez-Ortiz M, Martinez-Abundis E, Robles-Cervantes JA, Ramirez-Ramirez V, Ramos-Zavala MG. Effect of thiamine administration on metabolic profile, cytokines and inflammatory markers in drug-naive patients with type 2 diabetes. Eur J Nutr. 2011;50(2):145-149. (PubMed)
47. Lee DC, Chu J, Satz W, Silbergleit R. Low plasma thiamine levels in elder patients admitted through the emergency department. Acad Emerg Med. 2000;7(10):1156-1159. (PubMed)
48. Ito Y, Yamanaka K, Susaki H, Igata A. A cross-investigation between thiamin deficiency and the physical condition of elderly people who require nursing care. J Nutr Sci Vitaminol (Tokyo). 2012;58(3):210-216. (PubMed)
49. Prvulovic D, Hampel H. Amyloid beta (Abeta) and phospho-tau (p-tau) as diagnostic biomarkers in Alzheimer's disease. Clin Chem Lab Med. 2011;49(3):367-374. (PubMed)
50. Kish SJ. Brain energy metabolizing enzymes in Alzheimer's disease: alpha-ketoglutarate dehydrogenase complex and cytochrome oxidase. Ann N Y Acad Sci. 1997;826:218-228. (PubMed)
51. Langbaum JB, Chen K, Lee W, et al. Categorical and correlational analyses of baseline fluorodeoxyglucose positron emission tomography images from the Alzheimer's Disease Neuroimaging Initiative (ADNI). Neuroimage. 2009;45(4):1107-1116. (PubMed)
52. Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 2004;61(5):661-666. (PubMed)
53. Arnold SE, Arvanitakis Z, Macauley-Rambach SL, et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol. 2018;14(3):168-181. (PubMed)
54. Gibson GE, Hirsch JA, Cirio RT, Jordan BD, Fonzetti P, Elder J. Abnormal thiamine-dependent processes in Alzheimer's Disease. Lessons from diabetes. Mol Cell Neurosci. 2013;55:17-25. (PubMed)
55. Butterfield DA, Halliwell B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci. 2019;20(3):148-160. (PubMed)
56. Gibson GE, Hirsch JA, Fonzetti P, Jordan BD, Cirio RT, Elder J. Vitamin B1 (thiamine) and dementia. Ann N Y Acad Sci. 2016;1367(1):21-30. (PubMed)
57. Glaso M, Nordbo G, Diep L, Bohmer T. Reduced concentrations of several vitamins in normal weight patients with late-onset dementia of the Alzheimer type without vascular disease. J Nutr Health Aging. 2004;8(5):407-413. (PubMed)
58. Pan X, Sang S, Fei G, et al. Enhanced activities of blood thiamine diphosphatase and monophosphatase in Alzheimer's disease. PLoS One. 2017;12(1):e0167273. (PubMed)
59. Bender DA. Optimum nutrition: thiamin, biotin and pantothenate. Proc Nutr Soc. 1999;58(2):427-433. (PubMed)
60. Mastrogiacoma F, Bettendorff L, Grisar T, Kish SJ. Brain thiamine, its phosphate esters, and its metabolizing enzymes in Alzheimer's disease. Ann Neurol. 1996;39(5):585-591. (PubMed)
61. Heroux M, Raghavendra Rao VL, Lavoie J, Richardson JS, Butterworth RF. Alterations of thiamine phosphorylation and of thiamine-dependent enzymes in Alzheimer's disease. Metab Brain Dis. 1996;11(1):81-88. (PubMed)
62. Pan X, Gong N, Zhao J, et al. Powerful beneficial effects of benfotiamine on cognitive impairment and beta-amyloid deposition in amyloid precursor protein/presenilin-1 transgenic mice. Brain. 2010;133(Pt 5):1342-1351. (PubMed)
63. Tapias V, Jainuddin S, Ahuja M, et al. Benfotiamine treatment activates the Nrf2/ARE pathway and is neuroprotective in a transgenic mouse model of tauopathy. Hum Mol Genet. 2018;27(16):2874-2892. (PubMed)
64. Moraes RCM, Singulani MP, Goncalves AC, Portari GV, Torrao ADS. Oral benfotiamine reverts cognitive deficit and increase thiamine diphosphate levels in the brain of a rat model of neurodegeneration. Exp Gerontol. 2020;141:111097. (PubMed)
65. Karuppagounder SS, Xu H, Shi Q, et al. Thiamine deficiency induces oxidative stress and exacerbates the plaque pathology in Alzheimer's mouse model. Neurobiol Aging. 2009;30(10):1587-1600. (PubMed)
66. Zhang Q, Yang G, Li W, et al. Thiamine deficiency increases beta-secretase activity and accumulation of beta-amyloid peptides. Neurobiol Aging. 2011;32(1):42-53. (PubMed)
67. Dumont M, Beal MF. Neuroprotective strategies involving ROS in Alzheimer disease. Free Radic Biol Med. 2011;51(5):1014-1026. (PubMed)
68. Nolan KA, Black RS, Sheu KF, Langberg J, Blass JP. A trial of thiamine in Alzheimer's disease. Arch Neurol. 1991;48(1):81-83. (PubMed)
69. Meador K, Loring D, Nichols M, et al. Preliminary findings of high-dose thiamine in dementia of Alzheimer's type. J Geriatr Psychiatry Neurol. 1993;6(4):222-229. (PubMed)
70. Mimori Y, Katsuoka H, Nakamura S. Thiamine therapy in Alzheimer's disease. Metab Brain Dis. 1996;11(1):89-94. (PubMed)
71. Rodriguez-Martin JL, Qizilbash N, Lopez-Arrieta JM. Thiamine for Alzheimer's disease (Cochrane Review). Cochrane Database Syst Rev. 2001;2:CD001498. (PubMed)
72. Pan X, Chen Z, Fei G, et al. Long-term cognitive improvement after benfotiamine administration in patients with Alzheimer's disease. Neurosci Bull. 2016;32(6):591-596. (PubMed)
73. Gibson GE, Luchsinger JA, Cirio R, et al. Benfotiamine and cognitive decline in Alzheimer's disease: results of a randomized placebo-controlled phase IIa clinical trial. J Alzheimers Dis. 2020;78(3):989-1010. (PubMed)
74. Pico S, Parras A, Santos-Galindo M, et al. CPEB alteration and aberrant transcriptome-polyadenylation lead to a treatable SLC19A3 deficiency in Huntington's disease. Sci Transl Med. 2021;13(613):eabe7104. (PubMed)
75. US National Library of Medicine. ClinicalTrials.gov. Trial of the combined use of thiamine and biotin in patients With Huntington's disease (HUNTIAM). Available at: https://clinicaltrials.gov/ct2/show/NCT04478734.
76. Hanninen SA, Darling PB, Sole MJ, Barr A, Keith ME. The prevalence of thiamin deficiency in hospitalized patients with congestive heart failure. J Am Coll Cardiol. 2006;47(2):354-361. (PubMed)
77. Katta N, Balla S, Alpert MA. Does long-term furosemide therapy cause thiamine deficiency in patients with heart failure? A focused review. Am J Med. 2016;129(7):753 e757-753 e711. (PubMed)
78. Jain A, Mehta R, Al-Ani M, Hill JA, Winchester DE. Determining the role of thiamine deficiency in systolic heart failure: a meta-analysis and systematic review. J Card Fail. 2015;21(12):1000-1007. (PubMed)
79. Wilkinson TJ, Hanger HC, George PM, Sainsbury R. Is thiamine deficiency in elderly people related to age or co-morbidity? Age Ageing. 2000;29(2):111-116. (PubMed)
80. Shimon I, Almog S, Vered Z, et al. Improved left ventricular function after thiamine supplementation in patients with congestive heart failure receiving long-term furosemide therapy. Am J Med. 1995;98(5):485-490. (PubMed)
81. Leslie D, Gheorghiade M. Is there a role for thiamine supplementation in the management of heart failure? Am Heart J. 1996;131(6):1248-1250. (PubMed)
82. Thiamine supplementation in patients with chronic heart failure receiving optimum medical treatment. J Cardiol Curr Res. 2017;9(2):00316.
83. Keith M, Quach S, Ahmed M, et al. Thiamin supplementation does not improve left ventricular ejection fraction in ambulatory heart failure patients: a randomized controlled trial. Am J Clin Nutr. 2019;110(6):1287-1295. (PubMed)
84. Goel A, Kattoor AJ, Mehta JL. Thiamin therapy for chronic heart failure: is there any future for this vitamin? Am J Clin Nutr. 2019;110(6):1270-1271. (PubMed)
85. Tepper OM, Galiano RD, Capla JM, et al. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106(22):2781-2786. (PubMed)
86. Wong CY, Qiuwaxi J, Chen H, et al. Daily intake of thiamine correlates with the circulating level of endothelial progenitor cells and the endothelial function in patients with type II diabetes. Mol Nutr Food Res. 2008;52(12):1421-1427. (PubMed)
87. Rabbani N, Alam SS, Riaz S, et al. High-dose thiamine therapy for patients with type 2 diabetes and microalbuminuria: a randomised, double-blind placebo-controlled pilot study. Diabetologia. 2009;52(2):208-212. (PubMed)
88. Babaei-Jadidi R, Karachalias N, Ahmed N, Battah S, Thornalley PJ. Prevention of incipient diabetic nephropathy by high-dose thiamine and benfotiamine. Diabetes. 2003;52(8):2110-2120. (PubMed)
89. Hammes HP, Du X, Edelstein D, et al. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat Med. 2003;9(3):294-299. (PubMed)
90. Varkonyi T, Kempler P. Diabetic neuropathy: new strategies for treatment. Diabetes Obes Metab. 2008;10(2):99-108. (PubMed)
91. Kohda Y, Shirakawa H, Yamane K, et al. Prevention of incipient diabetic cardiomyopathy by high-dose thiamine. J Toxicol Sci. 2008;33(4):459-472. (PubMed)
92. Seligmann H, Levi R, Konijn AM, Prokocimer M. Thiamine deficiency in patients with B-chronic lymphocytic leukaemia: a pilot study. Postgrad Med J. 2001;77(911):582-585. (PubMed)
93. Isenberg-Grzeda E, Alici Y, Hatzoglou V, Nelson C, Breitbart W. Nonalcoholic thiamine-related encephalopathy (Wernicke-Korsakoff syndrome) among inpatients with cancer: a series of 18 cases. Psychosomatics. 2016;57(1):71-81. (PubMed)
94. Comin-Anduix B, Boren J, Martinez S, et al. The effect of thiamine supplementation on tumour proliferation. A metabolic control analysis study. Eur J Biochem. 2001;268(15):4177-4182. (PubMed)
95. Zastre JA, Hanberry BS, Sweet RL, et al. Up-regulation of vitamin B1 homeostasis genes in breast cancer. J Nutr Biochem. 2013;24(9):1616-24. (PubMed)
96. Lu'o'ng KV, Nguyen LT. The role of thiamine in cancer: possible genetic and cellular signaling mechanisms. Cancer Genomics Proteomics. 2013;10(4):169-185. (PubMed)
97. Aksoy M, Basu TK, Brient J, Dickerson JW. Thiamin status of patients treated with drug combinations containing 5-fluorouracil. Eur J Cancer. 1980;16(8):1041-1045. (PubMed)
98. Basu TK. Vitamins - cytotoxic drug interaction. Int J Vitam Nutr Res Suppl. 1983;24:225-233. (PubMed)
99. Boros LG, Brandes JL, Lee WN, et al. Thiamine supplementation to cancer patients: a double edged sword. Anticancer Res. 1998;18(1B):595-602. (PubMed)
100. Centers for Disease Control and Prevention. What is sepsis? January 27, 2021. Available at: https://www.cdc.gov/sepsis/what-is-sepsis.html. Accessed 7/21/21.
101. Moskowitz A, Donnino MW. Thiamine (vitamin B1) in septic shock: a targeted therapy. J Thorac Dis. 2020;12(Suppl 1):S78-S83. (PubMed)
102. Woolum JA, Abner EL, Kelly A, Thompson Bastin ML, Morris PE, Flannery AH. Effect of thiamine administration on lactate clearance and mortality in patients with septic shock. Crit Care Med. 2018;46(11):1747-1752. (PubMed)
103. Holmberg MJ, Moskowitz A, Patel PV, et al. Thiamine in septic shock patients with alcohol use disorders: An observational pilot study. J Crit Care. 2018;43:61-64. (PubMed)
104. Donnino MW, Andersen LW, Chase M, et al. Randomized, double-blind, placebo-controlled trial of thiamine as a metabolic resuscitator in septic shock: a pilot study. Crit Care Med. 2016;44(2):360-367. (PubMed)
105. Moskowitz A, Andersen LW, Cocchi MN, Karlsson M, Patel PV, Donnino MW. Thiamine as a renal protective agent in septic shock. A secondary analysis of a randomized, double-blind, placebo-controlled trial. Ann Am Thorac Soc. 2017;14(5):737-741. (PubMed)
106. Qian X, Zhang Z, Li F, Wu L. Intravenous thiamine for septic shock: A meta-analysis of randomized controlled trials. Am J Emerg Med. 2020;38(12):2718-2722. (PubMed)
107. Marik PE, Khangoora V, Rivera R, Hooper MH, Catravas J. Hydrocortisone, vitamin C, and thiamine for the treatment of severe sepsis and septic shock: A retrospective before-after study. Chest. 2017;151(6):1229-1238. (PubMed)
108. Naito E, Ito M, Yokota I, Saijo T, Ogawa Y, Kuroda Y. Diagnosis and molecular analysis of three male patients with thiamine-responsive pyruvate dehydrogenase complex deficiency. J Neurol Sci. 2002;201(1-2):33-37. (PubMed)
109. Patel KP, O'Brien TW, Subramony SH, Shuster J, Stacpoole PW. The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol Genet Metab. 2012;106(3):385-394. (PubMed)
110. Lee EH, Ahn MS, Hwang JS, Ryu KH, Kim SJ, Kim SH. A Korean female patient with thiamine-responsive pyruvate dehydrogenase complex deficiency due to a novel point mutation (Y161C)in the PDHA1 gene. J Korean Med Sci. 2006;21(5):800-804. (PubMed)
111. Chuang DT, Chuang JL, Wynn RM. Lessons from genetic disorders of branched-chain amino acid metabolism. J Nutr. 2006;136(1 Suppl):243S-249S. (PubMed)
112. Labay V, Raz T, Baron D, et al. Mutations in SLC19A2 cause thiamine-responsive megaloblastic anaemia associated with diabetes mellitus and deafness. Nat Genet. 1999;22(3):300-304. (PubMed)
113. Shaw-Smith C, Flanagan SE, Patch AM, et al. Recessive SLC19A2 mutations are a cause of neonatal diabetes mellitus in thiamine-responsive megaloblastic anaemia. Pediatr Diabetes. 2012;13(4):314-321. (PubMed)
114. Habeb AM, Flanagan SE, Zulali MA, et al. Pharmacogenomics in diabetes: outcomes of thiamine therapy in TRMA syndrome. Diabetologia. 2018;61(5):1027-1036. (PubMed)
115. Akin L, Kurtoglu S, Kendirci M, Akin MA, Karakukcu M. Does early treatment prevent deafness in thiamine-responsive megaloblastic anaemia syndrome? J Clin Res Pediatr Endocrinol. 2011;3(1):36-39. (PubMed)
116. Tabarki B, Al-Hashem A, Alfadhel M. Biotin-thiamine-responsive basal ganglia disease. In: Adam MP, Ardinger HH, Pagon RA, eds. GeneReviews [Internet]. Seattle: University of Washington, Seattle; 1993-2021. (PubMed)
117. Kilic B, Topcu Y, Dursun S, et al. Single gene, two diseases, and multiple clinical presentations: Biotin-thiamine-responsive basal ganglia disease. Brain Dev. 2020;42(8):572-580. (PubMed)
118. Alfadhel M, Almuntashri M, Jadah RH, et al. Biotin-responsive basal ganglia disease should be renamed biotin-thiamine-responsive basal ganglia disease: a retrospective review of the clinical, radiological and molecular findings of 18 new cases. Orphanet J Rare Dis. 2013;8:83. (PubMed)
119. Tabarki B, Alfadhel M, AlShahwan S, Hundallah K, AlShafi S, AlHashem A. Treatment of biotin-responsive basal ganglia disease: Open comparative study between the combination of biotin plus thiamine versus thiamine alone. Eur J Paediatr Neurol. 2015;19(5):547-552. (PubMed)
120. Algahtani H, Ghamdi S, Shirah B, Alharbi B, Algahtani R, Bazaid A. Biotin-thiamine-responsive basal ganglia disease: catastrophic consequences of delay in diagnosis and treatment. Neurol Res. 2017;39(2):117-125. (PubMed)
121. Biesecker LG. Amish Lethal Microcephaly. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle (WA); 1993. (PubMed)
122. Chen Y, Fang B, Hu X, et al. Identification and functional analysis of novel SLC25A19 variants causing thiamine metabolism dysfunction syndrome 4. Orphanet J Rare Dis. 2021;16(1):403. (PubMed)
123. Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: 614458: 6/8/17. Available at: https://omim.org/entry/614458.
124. Mayr JA, Freisinger P, Schlachter K, et al. Thiamine pyrophosphokinase deficiency in encephalopathic children with defects in the pyruvate oxidation pathway. Am J Hum Genet. 2011;89(6):806-812. (PubMed)
125. LeBlanc JG, Milani C, de Giori GS, Sesma F, van Sinderen D, Ventura M. Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr Opin Biotechnol. 2013;24(2):160-168. (PubMed)
126. Russell RM, Suter PM. Vitamin requirements of elderly people: an update. Am J Clin Nutr. 1993;58(1):4-14. (PubMed)
127. US National Institutes of Health, Office of Dietary Supplements. Dietary Supplement Label Database, Version 7.0.12. August 2020. Available at: https://dsld.od.nih.gov/dsld/index.jsp.
128. US Department of Health and Human Services, Food and Drug Administration, 21 CFR Part 101 [Docket No. FDA-20120N-1210] RIN 0910-AF22. Food Labeling: Revision of the Nutrition and Supplement Fact Labels. Final rule. Federal Register. Vol. 81, No. 103. Available at: https://www.federalregister.gov/documents/2016/05/27/2016-11867/food-labeling-revision-of-the-nutrition-and-supplement-facts-labels.
129. Volvert ML, Seyen S, Piette M, et al. Benfotiamine, a synthetic S-acyl thiamine derivative, has different mechanisms of action and a different pharmacological profile than lipid-soluble thiamine disulfide derivatives. BMC Pharmacol. 2008;8:10. (PubMed)
130. Flodin N. Pharmacology of micronutrients. New York: Alan R. Liss, Inc.; 1988.
131. Schumann K. Interactions between drugs and vitamins at advanced age. Int J Vitam Nutr Res. 1999;69(3):173-178. (PubMed)
132. Subramanya SB, Subramanian VS, Said HM. Chronic alcohol consumption and intestinal thiamin absorption: effects on physiological and molecular parameters of the uptake process. Am J Physiol Gastrointest Liver Physiol. 2010;299(1):G23-31. (PubMed)
133. Subramanian VS, Subramanya SB, Tsukamoto H, Said HM. Effect of chronic alcohol feeding on physiological and molecular parameters of renal thiamin transport. Am J Physiol Renal Physiol. 2010;299(1):F28-34. (PubMed)
Vitamin A
Contents
Summary
- Vitamin A is a generic term that refers to fat-soluble compounds found as preformed vitamin A (retinol) in animal products and as provitamin A carotenoids in fruit and vegetables. The three active forms of vitamin A in the body are retinol, retinal, and retinoic acid. (More information)
- Vitamin A is involved in regulating the growth and specialization (differentiation) of virtually all cells in the human body. Vitamin A has important roles in embryonic development, organ formation during fetal development, normal immune functions, and eye development and vision. (More information)
- Vitamin A deficiency is a major cause of preventable blindness in the world. It is most prevalent among children and women of childbearing age. Vitamin A deficiency is associated with an increased susceptibility to infections, as well as to thyroid and skin disorders. (More information)
- The recommended dietary allowance (RDA) is 700 micrograms of retinol activity equivalents (μg RAE)/day for women and 900 μg RAE/day for men. (More information)
- Vitamin A prophylaxis appears to significantly reduce childhood mortality (especially from diarrheal illnesses) in regions at high risk of vitamin A deficiency. Further, high-dose vitamin A supplementation is widely recommended for children over six months of age when they are infected with measles while malnourished, immunodeficient, or are at risk of measles complications. (More information)
- Retinoic acid and analogs are used at pharmacological doses in the treatment of acute promyelocytic leukemia and various skin diseases. (More information)
- Animal food sources rich in preformed vitamin A include dairy products, fortified cereal, liver, and fish oils. Rich sources of provitamin A carotenoids include orange and green vegetables, such as sweet potato and spinach. (More information)
-
Overconsumption of preformed vitamin A can be highly toxic and is especially contraindicated prior to and during pregnancy as it can result in severe birth defects. The tolerable upper intake level (UL) for vitamin A in adults is set at 3,000 μg RAE/day. The UL does not apply to vitamin A derived from carotenoids. (More information)
Vitamin A is a generic term that encompasses a number of related compounds (Figure 1). Retinol and retinyl esters are often referred to as preformed vitamin A. Retinol can be converted by the body to retinal, which can be in turn be oxidized to retinoic acid, the form of vitamin A known to regulate gene transcription. Retinol, retinal, retinoic acid, and related compounds are known as retinoids. β-Carotene and other food carotenoids that can be converted by the body into retinol are referred to as provitamin A carotenoids (see the article on Carotenoids). Hundreds of different carotenoids are synthesized by plants, but only about 10% of them are capable of being converted to retinol (1). The following discussion will focus mainly on preformed vitamin A compounds and retinoic acid.

Function
Vitamin A compounds are essential fat-soluble molecules predominantly stored in the liver in the form of retinyl esters (e.g., retinyl palmitate). When appropriate, retinyl esters are hydrolyzed to generate all-trans-retinol, which binds to retinol binding protein (RBP) before being released in the bloodstream. The all-trans-retinol/RBP complex circulates bound to the protein, transthyretin, which delivers all-trans-retinol to peripheral tissues (reviewed in 2). Vitamin A as retinyl esters in chylomicrons was also found to have an appreciable role in delivering vitamin A to extrahepatic tissues, especially in early life (3, 4).
Visual system and eyesight
Located at the back of the eye, the retina contains two main types of light-sensitive receptor cells — known as rod and cone photoreceptor cells. Photons (particles of light) that pass through the lens are sensed by the photoreceptor cells of the retina and converted to nerve impulses (electric signals) for interpretation by the brain. All-trans-retinol is transported to the retina via the circulation and accumulates in retinal pigment epithelial cells (Figure 2) (5). Here, all-trans-retinol is esterified to form a retinyl ester, which can be stored. When needed, retinyl esters are broken apart (hydrolyzed) and isomerized to form 11-cis-retinol, which can be oxidized to form 11-cis-retinal. 11-cis-retinal can be shuttled across the interphotoreceptor space to the rod photoreceptor cell that is specialized for vision in low-light conditions and for detection of motion. In rod cells, 11-cis-retinal binds to a protein called opsin to form the visual pigment rhodopsin (also known as visual purple). Absorption of a photon of light catalyzes the isomerization of 11-cis-retinal to all-trans-retinal that is released from the opsin molecule. This photoisomerization triggers a cascade of events, leading to the generation of a nerve impulse conveyed by the optic nerve to the brain’s visual cortex. All-trans-retinal is converted to all-trans-retinol and transported across the interstitial space to the retinal pigment epithelial cells, thereby completing the visual cycle.
A similar cycle occurs in cone cells that contain red, green, or blue opsin proteins required for the absorption of photons from the visible light spectrum (2). Vitamin A is also essential for mammalian eye development (6). Thus, because vitamin A is required for the normal functioning of the retina, dim-light vision, and color vision, inadequate retinol and retinal available to the retina result in impaired dark adaptation. In the severest cases of vitamin A deficiency, thinning and ulceration of the cornea leads to blindness (see Deficiency).

Regulation of gene expression
Regulatory capacity of retinoic acid
In cells, all-trans-retinol can be either stored (in the form of retinyl ester) or oxidized to all-trans-retinal by alcohol dehydrogenases. In turn, retinaldehyde dehydrogenases can catalyze the conversion of all-trans-retinal into two biologically active isomers of retinoic acid (RA): all-trans-RA and 9-cis-RA. RA isomers act as hormones to affect gene expression and thereby influence numerous physiological processes. All-trans-RA and 9-cis-RA are transported to the nucleus of the cell bound to cellular retinoic acid-binding proteins (CRABP). Within the nucleus, RA isomers bind to specific nuclear receptor proteins that are ligand-dependent transcription factors. In vitro studies have indicated that both all-trans-RA and 9-cis-RA can bind to retinoic acid receptors (RARα, RARβ, and RARγ) and that 9-cis-RA can bind to retinoid X receptors (RXR) (7). RAR and RXR subtypes form either complexes of two of the same protein (RAR/RAR and RXR/RXR homodimers) or complexes of two different proteins (RAR/RXR heterodimers). RAR/RXR heterodimers can bind to a regulatory DNA sequence called retinoic acid response element (RARE) located within the promoter of retinoid-responsive genes. The transcriptional activity of RAR/RXR heterodimers appears to be mainly driven by the binding of all-trans-RA to RAR.
The activation of RAR by RA binding triggers the recruitment of transcriptional coregulators to target promoters, thereby inhibiting or allowing the transcription of genes (8). RXR also forms heterodimers with several other nuclear receptors, including thyroid hormone receptor, vitamin D receptor, steroid receptors, and peroxisome proliferator-activated receptor (PPAR) (9). In this way, vitamin A may interact with thyroid hormone, vitamin D, steroids (e.g., estrogen), or PPAR ligands signaling pathways and influence the transcription of a broad range of genes.
There is also evidence that RA/RAR can affect gene expression in a RARE-independent manner. For example, it was reported that RAR could interfere with TGFβ/Smad signaling pathway through direct interaction of RAR with the heterodimeric transcription factor, Smad3/Smad4. In the absence of RA, RAR was found to act as a coactivator of Smad3/Smad4-mediated transcription, while RAR agonists repressed the transcriptional activity of Smad3/Smad4 (10). In retinoblastoma cells, RAR was also involved in RA-induced activation of signaling cascades mediated by tyrosine kinases known as phosphoinositide 3-kinase (PI3K) and leading to cell differentiation (11, 12). RA also appeared to induce neuronal differentiation by activating ERK1/2 MAP kinase signaling pathway that phosphorylated transcription factor, CREB (cyclic AMP response element binding protein). Phosphorylated CREB can subsequently bind to the CREB response element in the promoter of genes involved in cell differentiation (13). Also, independently of RAR, RA was found to inhibit ERK1/2 phosphorylation/activation and subsequent AP1-mediated expression of interleukin-6 in synovial cells (14). Hence, RA can influence the expression of genes whose promoters do not contain RARE.
By regulating the expression of over 500 retinoid-responsive genes (including several genes involved in vitamin A metabolism itself), retinoic acid isomers play major roles in cellular proliferation and differentiation (i.e., cell commitment to highly specialized functions).
Regulatory capacity of retinol
In the eye and tissues like white adipose and muscle, retinol plasma membrane receptor/transporter STRA6 accepts retinol from extracellular RBP and unloads it to intracellular retinol-binding protein (CRBP). STRA6 also cooperates with lecithin:retinol acyltransferase (LRAT), an enzyme that catalyzes retinol esterification and storage, to maintain an inward concentration gradient of retinol (15). Interestingly, retinol uptake by STRA6 was found to trigger the activation of a signaling cascade mediated by tyrosine kinases known as Janus kinases (JAK) and associated transcription factors (STAT). JAK/STAT signaling pathway regulates the expression of a wide range of cytokines, hormones, and growth factors (16). Animal studies have reported that an increased expression of genes, such as SOCS3 by the JAK/STAT pathway, could result in the inhibition of insulin signaling. Hence, obese mice lacking LRAT or STRA6 appear to be protected from retinol/STRA6-induced insulin resistance (17, 18).
Regulatory capacity of retinal
Apart from its role as a ligand for opsin in the visual cascade (see Visual system and eyesight), retinal has been specifically implicated in the regulation of genes important for lipid metabolism. In humans, two types of adipose tissue have been distinguished based on their respective functions: white adipose tissue (WAT) stores fatty acids as triglycerides, and brown adipose tissue (BAT) oxidizes fatty acids to generate heat (thermogenesis). In the mitochondrial respiratory chain of brown adipose cells, the processes of electron transport and ATP production are uncoupled (dissociated) to permit the rapid production of heat from fatty acid oxidation (19).
Retinaldehyde dehydrogenase 1 (RALDH1), which converts retinal to retinoic acid, is highly expressed in WAT but not in BAT. The suppression of RALDH1 expression in WAT can induce a thermogenic phenotype resembling that of BAT (20). During adipocyte differentiation, the stimulation of cells with all-trans retinal has been found to activate the UCP1 gene required for thermogenesis while inhibiting genes promoting adipogenesis, such as PPARγ (20). Retinal also appeared to regulate lipid metabolism and adiposity in bone marrow by inhibiting PPARγ/RXR heterodimer-mediated gene expression (21). In addition, retinal was found to inhibit gluconeogenic gene expression and glucose production in the liver of mice deficient in RALDH1 (22).
Immunity
Vitamin A was initially coined "the anti-infective vitamin" because of its importance in the normal functioning of the immune system (23). The skin and mucosal cells, lining the airways, digestive tract, and urinary tract function as a barrier and form the body's first line of defense against infection. Retinoic acid (RA) is produced by antigen-presenting cells (APCs), including macrophages and dendritic cells, found in these mucosal interfaces and associated lymph nodes. RA appears to act on dendritic cells themselves to regulate their differentiation, migration, and antigen-presenting capacity. In addition, the production of RA by APCs is required for the differentiation of naïve CD4 T-lymphocytes into induced regulatory T-lymphocytes (Tregs). Critical to the maintenance of mucosal integrity, the differentiation of Tregs is driven by all-trans-RA through RARα-mediated regulation of gene expression (see Regulation of gene expression). Also, during inflammation, all-trans-RA/RARα signaling pathway promotes the conversion of naïve CD4 T-lymphocytes into effector T-lymphocytes — type 1 helper T-cells (Th1) — (rather than into Tregs) and induces the production of proinflammatory cytokines by effector T-lymphocytes in response to infection.
A recent meta-analysis of randomized, placebo-controlled trials found that vitamin A supplementation decreased serum concentrations of TNF-α (5 studies) and IL-6 (9 studies) but increased serum C-reactive protein (CRP) concentrations (9 studies) (24). There is also substantial evidence to suggest that RA may help prevent the development of autoimmunity (reviewed in 25).
Prenatal and postnatal development
Both vitamin A excess and deficiency are known to cause birth defects. Retinoid signaling begins soon after the early phase of embryonic development known as gastrulation. During fetal development, RA is critical for the development of organs, including the heart, eyes, ears, lungs, as well as other limbs and visceral organs. Vitamin A has been implicated in fetal lung maturation (2). Vitamin A status is lower in preterm newborns than in full-term infants (26). There is some evidence to suggest that vitamin A supplementation may help reduce the incidence of chronic lung disease and mortality in preterm newborns (see Disease Prevention). Retinoid signaling is also involved in the expression of many proteins of the extracellular matrix (ECM; material surrounding cells), including collagen, laminin, and proteoglycans (27). Vitamin A deficiency may then result in alterations of the ECM composition, thus disrupting organ morphology and function (reviewed in 27).
Red blood cell production (erythropoiesis)
Red blood cells (erythrocytes), like all blood cells, are derived from pluripotent stem cells in the bone marrow. Studies involving in vitro culture systems have suggested a role for retinoids in stem cell commitment and differentiation to the red blood cell lineage. Retinoids might also regulate apoptosis (programmed cell death) of red blood cell precursors (erythropoietic progenitor cells) (28). However, whether retinoids regulate erythropoiesis in vivo has not been established. Yet, vitamin A supplementation in vitamin A deficient-individuals has been shown to increase hemoglobin concentrations. Additionally, vitamin A appears to facilitate the mobilization of iron from storage sites to the developing red blood cell for incorporation into hemoglobin, the oxygen carrier in red blood cells (28, 29).
Nutrient interactions
Zinc
Zinc deficiency is thought to interfere with vitamin A metabolism in several ways (30): (1) zinc deficiency results in decreased synthesis of retinol-binding protein (RBP), which transports retinol through the circulation to peripheral tissues and protects the organism against potential toxicity of retinol; (2) zinc deficiency results in decreased activity of the enzyme that releases retinol from its storage form, retinyl palmitate, in the liver; and (3) zinc is required for the enzyme that converts retinol into retinal (31). The health consequences of zinc deficiency on vitamin A nutritional status in humans are yet to be defined (30).
Iron
Vitamin A deficiency often coexists with iron deficiency and may exacerbate iron deficiency anemia by altering iron metabolism (28). Vitamin A supplementation has beneficial effects on iron deficiency anemia and improves iron nutritional status among children and pregnant women (28, 29, 32). The combination of supplemental vitamin A and iron seems to reduce anemia more effectively than either supplemental iron or vitamin A alone (33). Moreover, studies in rats have shown that vitamin A deficiency interferes with erythropoiesis (34) and iron deficiency also alters plasma and liver levels of vitamin A (35, 36).
Deficiency
Vitamin A deficiency usually results from inadequate intakes of vitamin A from animal products (as preformed vitamin A) and fruit and vegetables (as provitamin A carotenoids). In developing countries, vitamin A deficiency and associated disorders predominantly affect children and women of reproductive age. Other individuals at risk of vitamin A deficiency are those with poor absorption of lipids due to impaired pancreatic or biliary secretion and those with inflammatory bowel diseases, such as Crohn’s disease and celiac disease (2).
Subclinical vitamin A deficiency is often defined by serum retinol concentrations lower than 0.70 μmol/L (20 μg/dL). In severe vitamin A deficiency, vitamin A body stores are depleted and serum retinol concentrations fall below 0.35 μmol/L (10 μg/dL). Other biomarkers have been calibrated to assess vitamin A nutritional status (reviewed in 37). Of note, the World Health Organization considers vitamin A deficiency in a population to be a moderate public health problem when the prevalence of low serum retinol (<0.70 μmol/L) among children ages 6-71 months is at least 10% but less than 20% and a severe public health problem when the prevalence is 20% or greater (38).
Vitamin A deficiency-related disorders
Disease of the eye and blindness
With an estimated 250,000 to 500,000 children becoming blind annually, vitamin A deficiency constitutes the leading preventable cause of blindness in low- and middle-income nations (39). The earliest symptom of vitamin A deficiency is impaired dark adaptation known as night blindness or nyctalopia. The next clinical stage is the occurrence of abnormal changes in the conjunctiva (corner of the eye), manifested by the presence of Bitot's spots. Severe or prolonged vitamin A deficiency eventually results in a condition called xerophthalmia (Greek for dry eye), characterized by changes in the cells of the cornea (clear covering of the eye) that ultimately result in corneal ulcers, scarring, and blindness (40). Immediate administration of 200,000 international units (IU; 60 mg RAE) of vitamin A for two consecutive days is required to prevent blinding xerophthalmia (40).
There is an estimated 19.1 million pregnant women worldwide (especially in Sub-Saharan Africa, Southeast Asia, and Central America) with vitamin A deficiency and over half of them are affected by night blindness (41). The prevalence of vitamin A deficiency and night blindness is especially high during the third trimester of pregnancy due to accelerated fetal growth. Prenatal vitamin A supplementation lowers the risk of maternal night blindness (42). Approximately 190 million preschool-age children have low serum retinol concentrations (<0.70 μmol/L), with 5.2 million suffering from night blindness. Moreover, half of the children affected by severe vitamin A deficiency-induced blinding xerophthalmia are estimated to die within a year of becoming blind (41). The World Health Organization (WHO) and the United Nations Children’s Fund (UNICEF) promote vitamin A supplementation as a public health intervention to reduce child mortality in areas and populations where vitamin A deficiency is prevalent (see the section on Childhood morbidity and mortality) (43).
Susceptibility to infectious diseases
Infectious diseases have been associated with depletion of vitamin A hepatic reserves (already limited in vitamin A-deficient subjects), reduced serum retinol concentrations, and increased loss of vitamin A in the urine (41). Infection with the measles virus was found to precipitate conjunctival and corneal damage, leading to blindness in children with poor vitamin A status (44). Conversely, vitamin A deficiency can be considered a nutritionally acquired immunodeficiency disease (45). Even children who are only mildly deficient in vitamin A have a higher incidence of respiratory complications and diarrhea, as well as a higher rate of mortality from measles infection compared to children consuming sufficient vitamin A (46). Because vitamin A supplementation may decrease both the severity and incidence of measles complications in developing countries (see Disease Prevention), WHO recommends that children aged at least one year receive 200,000 IU of vitamin A (60 mg RAE) for two consecutive days in addition to standard treatment when they are infected with measles virus and live in areas of vitamin A deficiency (47).
A prospective cohort study, conducted in 2,774 Colombian children (ages, 5-12 years old) followed for a median 128 days, also reported an inverse relationship between plasma retinol concentrations and rates of diarrhea with vomiting and cough with fever, the latter being a strong predictor of influenza infection (flu) (48). A review of five randomized, placebo-controlled studies that included 7,528 HIV-positive pregnant or breast-feeding women found no substantial benefit of vitamin A supplementation in reducing the mother-to-child transmission of HIV (49). One early observational study found that HIV-infected women who were vitamin A deficient were three to four times more likely to transmit HIV to their infants (50). Yet, no trial to date has provided any information on potential adverse effects of vitamin A supplementation on mother-to-child HIV transmission (51).
Thyroid dysfunction
In North and West Africa, vitamin A deficiency and iodine deficiency induced-goiter can coexist in up to 50% of children. The response to iodine prophylaxis in iodine-deficient populations appears to depend on various nutritional factors, including vitamin A status (52, 53). Vitamin A deficiency in animal models was found to interfere with the pituitary-thyroid axis by (1) increasing the synthesis and secretion of thyroid-stimulating hormone (TSH) by the pituitary gland, (2) increasing the size of the thyroid gland, (3) reducing iodine uptake by the thyroid gland and impairing the synthesis and iodination of thyroglobulin, and (4) increasing circulating concentrations of thyroid hormones (reviewed in 54, 55). A cross-sectional study of 138 children with concurrent vitamin A and iodine deficiencies found that the severity of vitamin A deficiency was associated with higher risk of goiter and higher concentrations of circulating TSH and thyroid hormones (53). These children received iodine-enriched salt with either vitamin A (200,000 IU [60 mg RAE] at baseline and 5 months) or placebo in a randomized, double-blind, 10-month trial. This vitamin A supplementation significantly decreased TSH concentration and thyroid volume compared to placebo (53). In another trial, supplementation of vitamin A to iodine-deficient children had no additional effect to iodine on thyroid status compared to placebo, but vitamin A supplementation alone (without iodine) reduced the volume of the thyroid gland, as well as TSH and thyroglobulin concentrations (56).
Other disorders
Phrynoderma or follicular hyperkeratosis is a skin condition characterized by an excessive production of keratin in hair follicles. The lesions first appear on the extremities, shoulders, and buttocks and may spread over the entire body in the severest cases (57). While vitamin A deficiency may contribute to the occurrence of phrynoderma, the condition has been strongly associated with multiple nutritional deficiencies and is considered a sign of general malnutrition. A rare case of esophagitis (inflammation of the esophagus) has been attributed to hyperkeratosis secondary to vitamin A deficiency (58).
Also, vitamin A deficiency affects iron mobilization, impairs hemoglobin synthesis, and precipitates iron deficiency anemia that is only alleviated with supplementation of both vitamin A and iron (see Nutrient interactions) (28).
The Recommended Dietary Allowance (RDA)
Retinol Activity Equivalents (RAE)
Vitamin A can be obtained from food as preformed vitamin A in animal products or as provitamin A carotenoids in fruit and vegetables (see Food sources). Yet, while preformed vitamin A is effectively absorbed, stored, and hydrolyzed to form retinol, provitamin A carotenoids like β-carotene are less easily digested and absorbed, and must be converted to retinol and other retinoids by the body after uptake into the small intestine. The efficiency of conversion of provitamin A carotenes into retinol is highly variable, depending on factors such as food matrix, food preparation, and one’s digestive and absorptive capacities (59).
The most recent international standard of measure for vitamin A is retinol activity equivalents (RAE), which represent vitamin A activity as retinol. It has been determined that 2 micrograms (μg) of β-carotene in oil provided as a supplement could be converted by the body to 1 μg of retinol giving it an RAE ratio of 2:1. However, 12 μg of β-carotene from food are required to provide the body with 1 μg of retinol, giving dietary β-carotene an RAE ratio of 12:1. Other provitamin A carotenoids in food are less easily absorbed than β-carotene, resulting in RAE ratios of 24:1. RAE ratios are shown in Table 1 (60).
The RDA for vitamin A was revised by the Food and Nutrition Board of the US National Academy of Medicine in 2001. The RDA is based on the Estimated Average Requirement (EAR), which is defined as the biological requirement for 50% of the population. The RDA is the recommended intake needed by nearly all of the population to ensure adequate hepatic stores of vitamin A in the body (20 μg/g for four months if the person consumes a vitamin A-deficient diet) to support normal reproductive function, immune function, gene expression, and vision (for details of calculations, see 60). Table 2 lists the RDA values in micrograms (μg) of Retinol Activity Equivalents (RAE) per day.
| Life Stage | Age | Males (μg/day) | Females (μg/day) |
|---|---|---|---|
| Infants | 0-6 months | 400 (AI) | 400 (AI) |
| Infants | 7-12 months | 500 (AI) | 500 (AI) |
| Children | 1-3 years | 300 | 300 |
| Children | 4-8 years | 400 | 400 |
| Children | 9-13 years | 600 | 600 |
| Adolescents | 14-18 years | 900 | 700 |
| Adults | 19 years and older | 900 | 700 |
| Pregnancy | 18 years and younger | - | 750 |
| Pregnancy | 19 years and older | - | 770 |
| Breast-feeding | 18 years and younger | - | 1,200 |
| Breast-feeding | 19 years and older | - | 1,300 |
Disease Prevention
Bronchopulmonary dysplasia in preterm infants
Preterm infants are born with inadequate body stores of vitamin A, placing them at risk of developing diseases of the eye and the respiratory and gastrointestinal tracts. About one-third of preterm infants born between 22 and 28 weeks of gestation develop bronchopulmonary dysplasia (BPD), a chronic lung disease that can be fatal or result in life-long morbidities in survivors. A few randomized controlled trials have investigated the effect of postnatal vitamin A administration on the incidence of BPD and the risk of mortality in very low birth weight infants (VLBW; ≤1,500 g) requiring respiratory support (61-63). In the largest, multicenter, randomized, blinded, placebo-controlled trial that enrolled 807 extremely low birth weight (ELBW; ≤1,000 g) preterm newborns, the intramuscular administration of 5,000 IU (1,500 μg RAE) of vitamin A three times a week for four weeks significantly, though modestly, reduced the risk of BPD or death at 36 weeks’ postmenstrual age (gestational age plus chronological age) (62). While vitamin A supplementation was included in some neonatal programs after this trial (64), a national shortage in vitamin A supply that has affected US neonatal intensive care units since 2010 has led to a significant reduction in the use of vitamin A supplementation in premature newborns (401-1,000 g at birth) with respiratory failure (65, 66). However, a retrospective analysis of US nationwide data from 6,210 preterm infants born between 2010 and 2012 found that a reduction in vitamin A prophylaxis from 27.2% to 2.1% during the same period had no significant impact on the incidence of BPD or death before hospital discharge (66). A Cochrane review that pooled six randomized controlled trials in VLBW (≤1,500 g) or premature (less than 32 weeks' gestation) infants found vitamin A administration (5 trials of intramuscular administration and one trial or oral administration) only slightly reduced the risk of chronic lung disease or death at 28 days post birth (RR, 0.93; 95% CI, 0.88-0.99) (67).
In a retrospective study, the nonrandomized use of vitamin A supplementation with inhaled nitric oxide (iNO) was found to result in a lower incidence of BPD (but not mortality) compared to iNO therapy alone in preterm newborns with a birth weight of 750-999 g (68). Neurodevelopment index scores at one year of age were also improved in the vitamin A group of newborns weighing 500-749 g at birth. Yet, caution is advised with the interpretation of the results, especially because the trial was not designed to assess the effect of vitamin A. In Germany, a large, multicenter, randomized study — the NeoVitaA trial — is underway to explore the effect of high-dose oral vitamin A (5,000 IU/kg/day) for 28 days on the incidence of BPD and mortality at 36 weeks' postmenstrual age (69, 70).
While high doses of vitamin A during early pregnancy can cause birth defects (see Safety), vitamin A supplementation during late pregnancy may improve maternal and fetal vitamin A status (71). However, it is not known whether vitamin A supplementation during pregnancy might reduce BPD incidence in infants.
Childhood morbidity and mortality
A 2022 Cochrane review and meta-analysis of randomized controlled trials evaluating the preventive effect of vitamin A on childhood mortality indicated that high-dose vitamin A supplementation in children ages 6 to 59 months reduced all-cause mortality by 12% (19 studies) and diarrhea-specific mortality by 12% (9 studies) (72). However, vitamin A administration in this age group had no preventive effect on mortality from respiratory disease (9 studies), measles (6 studies), or meningitis (3 studies). However, in this pooled analysis, vitamin A supplementation reduced the incidence of diarrhea by 15% (15 studies), measles by 50% (6 studies), Bitot’s spots by 58% (5 studies) and night blindness by 68% (2 studies) but had no effect on the incidence of respiratory disease (72).
Current WHO policy recommends vitamin A supplementation at routine vaccination contacts in children after six months of age living in regions at high risk of vitamin A deficiency (73). Supplementation with high doses of vitamin A — 100,000 IU (30 mg RAE) for infants 6 to 11 months of age and 200,000 IU (60 mg RAE) for children 12 to 59 months of age — is thought to provide adequate protection for up to six months (74). See the WHO website for the current guidelines. Because beneficial effects are lacking, WHO does not recommend vitamin A supplementation in the neonatal period (in the 28 days following birth) (75) or in children under 6 months of age (reviewed in 76) to prevent infant morbidity and mortality.
There is some concern that vitamin A supplementation may interfere with vaccine effectiveness in young children. The timing of vitamin A interventions needs to be further examined in relation to the timing of vaccinations in order to maximize their benefits. Future studies should also examine whether the effects of vitamin A supplementation are modified by sex (75), as observed in at least one randomized controlled trial (77).
Complications from measles infection
An earlier meta-analysis of seven randomized controlled trials examining specifically the role of vitamin A supplementation in 2,069 children with measles found no overall reduction on the risk of mortality (78). Yet, the pooled analysis of four studies that reported the age distribution of participants found an 83% lower risk of mortality with two doses of 200,000 IU (60 mg RAE) of vitamin A in children younger than two years. In addition, the pooled analysis of three studies indicated a 67% reduction in the risk of pneumonia-led mortality (78). Similar to WHO and UNICEF guidelines, the American Academy of Pediatrics recommends vitamin A supplementation for children over six months of age when they are infected with measles while malnourished, immunodeficient, or are at risk of measles complications or vitamin A deficiency disorders (79). Although measles infection has been associated with vitamin A deficiency and blindness, there is currently no evidence to suggest that vitamin A supplementation reduces the risk of blindness in children infected with measles (80).
Cancer
Studies in cell culture and animal models have documented the capacity for natural and synthetic retinoids to reduce carcinogenesis significantly in skin, breast, liver, colon, prostate, and other sites. However, the results of human studies examining the relationship between the consumption of preformed vitamin A and cancer do not currently suggest that consuming vitamin A at intakes greater than the RDA benefit in the prevention of cancer (2).
Lung cancer
The results of the β-Carotene And Retinol Efficacy Trial (CARET) suggested that high-dose supplementation of preformed vitamin A and β-carotene should be avoided in people at high risk for lung cancer (81). In the CARET study, about 9,000 people (smokers and people with asbestos exposure) were assigned a daily regimen of 25,000 IU (7,500 μg RAE) of retinyl palmitate and 30 mg of β-carotene, while a similar number of people were assigned a placebo. After four years of follow-up, the incidence of lung cancer was 28% higher in the supplemented group compared to the placebo group; however, the incidence was not different six years after the intervention ended (82). A possible explanation for an increase in lung cancer is that the oxidative environment of the lung, created by smoke or asbestos exposure, could give rise to unusual carotenoid cleavage products, which might promote carcinogenesis (83). Interestingly, a case-control study that included 749 lung cancer cases and 679 controls from the CARET trial found a significant association between lung cancer risk reduction and high vitamin D intakes (≥400 IU/day) in individuals who received the active CARET supplements or in those with vitamin A intakes equal to or greater than 1,500 μg RAE/day (84). Further, a meta-analysis of four randomized controlled trials, including a total of 202,924 participants at low risk of lung cancer, indicated that supplementation with retinol and/or β-carotene had no significant effect on lung cancer incidence (85). While an inverse association has been observed between blood retinol concentration and lung cancer incidence in a dose-response meta-analysis of eight prospective cohort studies (86), randomized controlled trials have not shown that preformed vitamin A (e.g., retinol) lowers the risk of lung cancer.
Disease Treatment
Retinoids may be used at pharmacological doses to treat several conditions, including, acute promyelocytic leukemia, retinitis pigmentosa, and various skin diseases. It is important to note that treatment with high doses of natural or synthetic retinoids overrides the body's own control mechanisms; therefore, retinoid therapies are associated with potential side effects and toxicities. Additionally, all of the retinoid compounds have been found to cause fetal deformations. Thus, women who have a chance of becoming pregnant should avoid treatment with these medications. Retinoids tend to be very long acting: side effects and birth defects have been reported to occur months after discontinuing retinoid therapy (2). The retinoids discussed below are prescription drugs and should not be used without medical supervision.
Acute promyelocytic leukemia
Normal differentiation of myeloid stem cells in the bone marrow gives rise to platelets, red blood cells, and white blood cells (also called leukocytes) that are important for the immune response. Altered differentiation of myeloid cells can result in the proliferation of immature white blood cells, giving rise to leukemia. Reciprocal chromosome translocations involving the promyelocytic leukemia (PML) gene and the gene coding for retinoic acid receptor α (RARα) lead to a specific type of acute myeloid leukemia called acute promyelocytic leukemia (APL). The fusion protein PML/RARα represses transcription by binding to RARE in the promoter of retinoid-responsive genes involved in hematopoietic cell differentiation. Gene repression by PML/RARα is achieved by the recruitment of several chromatin modifiers, including histone deacetylases (HDACs) and DNA methyltransferases (DNMTs). Contrary to RARα wild-type receptor, PML/RARα appears to be insensitive to physiological concentrations of retinoic acid (RA) such that only treatments with high doses of all-trans-RA can restore normal differentiation and lead to significant improvements and complete remission in some APL patients (87). Combination therapy with arsenic trioxide (or chemotherapy) improves long-term remission (reviewed in 88).
More information on APL treatment can be found on the Leukemia & Lymphoma Society website.
Diseases of the skin
Both natural and synthetic retinoids have been used as pharmacologic agents to treat disorders of the skin. Acitretin is a synthetic retinoid that has been FDA-approved for the treatment for psoriasis (89, 90). Topical tretinoin (all-trans-retinoic acid) and oral isotretinoin (13-cis-retinoic acid) have been used successfully to treat mild-to-severe acne vulgaris (91, 92). Retinoids exhibit anti-inflammatory properties and regulate the proliferation and differentiation of skin epithelial cells, as well as the production of sebum. Use of pharmacological doses of retinoids (especially oral isotretinoin) by pregnant women causes birth defects and is therefore contraindicated prior to and during pregnancy (see Safety in pregnancy).
For more information on the use of retinoids in the management of acne, see the article on Vitamin A and Skin Health.
Retinitis pigmentosa
Retinitis pigmentosa (RP) affects approximately 1.5 million people worldwide and is a leading cause of inherited blindness. RP describes a broad spectrum of genetic disorders that result in the progressive loss of photoreceptor cells (rods and cones) in the retina of the eye (93). While at least 45 loci have been associated with RP, mutations in the rhodopsin gene (RHO), the usherin gene (USH2A), and the RP GTPase regulator gene (RPGR) account for about 30% of all RP cases (94).
Early symptoms of RP include impaired dark adaptation and night blindness, followed by the progressive loss of peripheral and central vision over time (94). The results of a randomized controlled trial in 601 patients with common forms of RP indicated that supplementation with 4,500 μg RAE/day of retinyl palmitate significantly slowed the loss of retinal function over a period of four to six years (95). In contrast, supplementation with 400 IU/day of vitamin E (dl-α-tocopherol) modestly but significantly increased the loss of retinal function, suggesting that patients with common forms of RP may benefit from long-term vitamin A supplementation but should avoid high-dose vitamin E supplementation. Up to 12 years of follow-up in these patients did not reveal any signs of liver toxicity as a result of excess vitamin A intake (96). Because neither children nor adults affected by less common forms of RP were included in the trial, no formal recommendation about vitamins A and E could be made (94). Evidence of a beneficial effect of vitamin A supplementation on the progression of RP is lacking, as concluded in a 2020 Cochrane review (97). Nevertheless, use of high-dose vitamin A supplementation in RP would require close medical supervision and must be discontinued if there is a possibility of pregnancy (see Safety).
Sources
Food sources
Free retinol is not generally found in food. Retinyl esters (including retinyl palmitate) are the storage form of retinol in animals and thus the main precursors of retinol in food from animals. Plants contain carotenoids, some of which are precursors for vitamin A (e.g., α-carotene, β-carotene, and β-cryptoxanthin). Yellow- and orange-colored vegetables contain significant quantities of carotenoids. Green vegetables also contain carotenoids, though yellow-to-red pigments are masked by the green pigment of chlorophyll (1). Additionally, certain foods, including milk, margarines, and breakfast cereals, may be fortified with retinyl esters or β-carotene (2).
Table 3 lists a number of good food sources of vitamin A, including fruit and vegetables, along with their vitamin A content. The retinol activity is indicated in micrograms of retinol activity equivalents (μg RAE). For information on this unit of measurement, see the section on RAE. In addition, use the USDA’s FoodData Central database to check foods for their content of carotenoids without vitamin A activity, such as lycopene, lutein, and zeaxanthin.
Vitamin A international units (IUs)
In the past, vitamin A was listed on food and supplement labels in international units instead of μg RAE ('μg' on labels). In contrast to RAE, the number of IUs of vitamin A does not reflect the bioavailability of vitamin A from different food sources. Conversion rates between IUs and μg RAE are as follows:
- 1 IU of retinol is equivalent to 0.3 μg RAE
- 1 IU of supplemental β-carotene is equivalent to 0.3 μg RAE
- 1 IU of dietary β-carotene is equivalent to 0.05 μg RAE
- 1 IU of α-carotene or β-cryptoxanthin to 0.025 μg RAE
Table 3 lists some foods rich in vitamin A; the amounts of preformed vitamin A (retinol) and the RAE, which account for bioavailability, are both listed.
| Food | Serving | Preformed Vitamin A (Retinol), μg | Vitamin A, μg RAE |
|---|---|---|---|
| Beef liver, cooked | 1 slice (68 g) | 6,410* | 6,420* |
| Cod liver oil | 1 teaspoon | 1,350 | 1,350 |
| Fortified breakfast cereal (oats) | 1 serving (1 oz) | 230 | 766 |
| Egg | 1 large | 97 | 98 |
| Butter | 1 tablespoon | 95 | 97 |
| Whole milk | 1 cup (8 fl oz) | 110 | 112 |
| 2% fat milk (vitamin A added) | 1 cup (8 fl oz) | 134 | 134 |
| Nonfat milk (vitamin A added) | 1 cup (8 fl oz) | 157 | 157 |
| Sweet potato (canned) | ½ cup | 0 | 555 |
| Sweet potato (baked) | ½ cup | 0 | 960 |
| Pumpkin (canned) | ½ cup | 0 | 955 |
| Carrot (raw) | ½ cup | 0 | 534 |
| Cantaloupe | ½ medium melon | 0 | 467 |
| Mango | 1 fruit | 0 | 181 |
| Spinach (cooked) | ½ cup | 0 | 472 |
| Broccoli (cooked) | ½ cup | 0 | 60 |
| Kale (cooked) | ½ cup | 0 | 86 |
| Collards (cooked) | ½ cup | 0 | 361 |
| Squash, butternut (cooked) | ½ cup | 0 | 570 |
| *Above the tolerable upper intake level (UL) of 3,000 μg RAE/day. | |||
Supplements
The principal forms of preformed vitamin A in supplements are retinyl palmitate and retinyl acetate. β-Carotene is also a common source of vitamin A in supplements, and many supplements provide a combination of retinol and β-carotene (98). If a percentage of the total vitamin A content of a supplement comes from β-carotene, this information may be included in the Supplement Facts label under vitamin A. While many multivitamins available in the US contain the daily value of 900 μg RAE of vitamin A, some may provide up to 1,500 μg RAE from preformed vitamin A, which is substantially more than the current RDA for vitamin A.
Safety
Toxicity
The condition caused by vitamin A toxicity is called hypervitaminosis A. It is caused by overconsumption of preformed vitamin A, not carotenoids. Preformed vitamin A is rapidly absorbed and slowly cleared from the body. Therefore, toxicity from preformed vitamin A may result acutely from high-dose exposure over a short period of time or chronically from a much lower intake (2). Acute vitamin A toxicity is relatively rare, and symptoms include nausea, headache, fatigue, loss of appetite, dizziness, dry skin, desquamation, and cerebral edema. Signs of chronic toxicity include dry itchy skin, desquamation, anorexia, weight loss, headache, cerebral edema, enlarged liver, enlarged spleen, anemia, and bone and joint pain. Also, symptoms of vitamin A toxicity in infants include bulging fontanels. Severe cases of hypervitaminosis A may result in liver damage, hemorrhage, and coma. Generally, signs of toxicity are associated with long-term consumption of vitamin A in excess of 10 times the RDA (8,000-10,000 μg RAE/day). However, more research is necessary to determine if subclinical vitamin A toxicity is a concern in certain populations (99). There is evidence that some populations may be more susceptible to toxicity at lower doses, including the elderly, chronic alcohol users, and some people with a genetic predisposition to high cholesterol (100). In January 2001, the Food and Nutrition Board of the US National Academy of Medicine set the tolerable upper level (UL) of vitamin A intake for adults at 3,000 μg RAE/day of preformed vitamin A (60).
Safety in pregnancy
Although normal fetal development requires sufficient vitamin A intake, consumption of excess preformed vitamin A (such as retinol) during early pregnancy is known to cause birth defects, mainly of the cardiovascular and central nervous systems (101). No increase in the risk of vitamin A-associated birth defects has been observed at doses of preformed vitamin A from supplements below 3,000 μg RAE/day (60). Of note, in 2011, the World Health Organization (WHO) recommended vitamin A supplementation (up to 3,000 μg RAE/day or 7,500 μg RAE/week) during pregnancy in areas with high prevalence of vitamin A deficiency for the prevention of blindness (102). In industrialized countries, pregnant or potentially pregnant women should monitor their intake of vitamin A from fortified food and food naturally high in preformed vitamin A (e.g., liver) and avoid taking daily multivitamin supplements that contain more than 1,500 μg RAE of vitamin A. There is no evidence that consumption of vitamin A from β-carotene might increase the risk of birth defects.
The synthetic derivative of retinol, isotretinoin, is known to cause serious birth defects and should not be taken during pregnancy or if there is a possibility of becoming pregnant (91). Tretinoin (all-trans-retinoic acid), another retinol derivative, is prescribed as a topical preparation that is applied to the skin. Although percutaneous absorption of topical tretinoin is minimal, its use during pregnancy is not recommended (103).
Do high intakes of vitamin A increase the risk of osteoporosis?
Results from some prospective studies have suggested that long-term intakes of preformed vitamin A in excess of 1,500 μg RAE/day are associated with reduced bone mineral density (BMD) and increased risk of osteoporotic fracture in older adults (104-106). However, other investigators failed to observe such detrimental effects on BMD and/or fracture risk (107-110). A meta-analysis of four prospective studies, including nearly 183,000 participants over 40 years of age, found that highest versus lowest quintiles of retinol (preformed vitamin A) intake significantly increased the risk of hip fracture (111). A 2017 meta-analysis found similar results, and additionally, did not find higher retinol intakes to be associated with an increased risk of total fracture (112). Only excess intakes of retinol, not β-carotene, have been associated with adverse effects on bone health. The earlier meta-analysis indicated a U-shaped relationship between circulating retinol and risk of hip fracture, suggesting that both elevated and reduced retinol concentrations in the blood were associated with an increased risk of hip fracture (111). It is important to note that the available data on retinol intake and bone fracture come from observational studies, not randomized controlled trials.
To date, limited experimental data have suggested that vitamin A (as all-trans-retinoic acid) may affect the development of bone-remodeling cells and stimulate bone matrix degradation (resorption) (reviewed in 113). Vitamin A may also interfere with the ability of vitamin D to maintain calcium balance (114). In the large Women’s Health Initiative prospective study, the highest versus lowest quintile of retinol intake (≥1,426 μg/day vs. <474 μg/day) was found to be significantly associated with increased risk of fracture only in women with the lowest vitamin D intakes (≤440 IU/day) (115).
It is advisable for older individuals to consume multivitamin supplements that contain no more than 750 μg of preformed vitamin A (usually labeled vitamin A acetate or vitamin A palmitate) and no more than 750 μg RAE of additional vitamin A as β-carotene.
Drug interactions
Chronic alcohol consumption results in depletion of liver stores of vitamin A and may contribute to alcohol-induced liver damage (cirrhosis) (116). However, the liver toxicity of preformed vitamin A (retinol) is enhanced by chronic alcohol consumption, thus narrowing the therapeutic window for vitamin A supplementation in alcoholics (116). Oral contraceptives that contain estrogen and progestin increase retinol binding protein (RBP) synthesis by the liver, increasing the export of all-trans-retinol/RBP complex to the circulation. Whether this increases the dietary requirement of vitamin A is not known. Also, the use of cholesterol-lowering medications (like cholestyramine and colestipol), as well as orlistat, mineral oil, and the fat substitute, olestra, which interfere with fat absorption, may affect the absorption of fat-soluble vitamins, including vitamin A (98). Further, intake of large doses of vitamin A may decrease the absorption of vitamin K. Retinoids or retinoid analogs, including acitretin, all-trans-retinoic acid, bexarotene, etretinate, and isotretinoin, should not be used in combination with single-nutrient vitamin A supplements, because they may increase the risk of vitamin A toxicity (98).
Linus Pauling Institute Recommendation
The RDA for vitamin A (700 μg RAE/day for women and 900 μg RAE/day for men) is sufficient to support normal gene expression, immune function, and vision. However, following the Linus Pauling Institute’s recommendation to take a multivitamin/mineral supplement daily could supply as much as 1,500 μg RAE/day of vitamin A as retinol, the amount that has been associated with adverse effects on bone health in older adults. For this reason, we recommend taking a multivitamin/mineral supplement that provides no more than 750 μg RAE of preformed vitamin A (usually labeled vitamin A acetate or vitamin A palmitate) and no more than 750 μg RAE of additional vitamin A as β-carotene. High potency vitamin A supplements should not be used without medical supervision due to the risk of toxicity.
Older adults (>50 years)
Presently, there is little evidence that the requirement for vitamin A in older adults differs from that of younger adults. Additionally, vitamin A toxicity may occur at lower doses in older adults than in younger adults. Further, data from observational studies suggested an inverse association between intakes of preformed vitamin A in excess of 1,500 μg RAE day and risk of hip fracture in older people (see Safety). Yet, following the Linus Pauling Institute’s recommendation to take a multivitamin/mineral supplement daily could supply as much as 1,500 μg RAE/day of retinol, the amount that has been associated with adverse effects on bone health in older adults in some studies. For this reason, the Linus Pauling Institute recommends taking a multivitamin/mineral supplement that provides no more than 750 μg RAE of preformed vitamin A (usually labeled vitamin A acetate or vitamin A palmitate) and no more than 750 μg RAE of additional vitamin A as β-carotene. As for all age groups, high potency vitamin A supplements should not be used without medical supervision due to the risk of toxicity.
Authors and Reviewers
Originally written in 2000 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in December 2003 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in November 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in January 2015:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in January 2024 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in February 2024 by:
A. Catharine Ross, Ph.D.
Professor of Nutrition, Texas A&M University
Professor of Nutrition Emeritus, The Pennsylvania State University
Copyright 2000-2024 Linus Pauling Institute
References
1. Groff JL. Advanced Nutrition and Human Metabolism. 2nd ed. St. Paul: West Publishing; 1995.
2. Ross AC. Vitamin A. In: Ross A, Caballero B, Cousins R, Tucker K, Ziegler T, eds. Modern Nutrition in Health and Disease. 11th ed: Lippincott Williams & Wilkins; 2014:260-277.
3. Tan L, Green MH, Ross AC. Vitamin A kinetics in neonatal rats vs. adult rats: comparisons from model-based compartmental analysis. J Nutr. 2015; 145(3):403-10. (PubMed)
4. Tan L, Wray AE, Green MH, Ross AC. Compartmental modeling of whole-body vitamin A kinetics in unsupplemented and vitamin A-retinoic acid-supplemented neonatal rats. J Lipid Res. 2014;55(8):1738-1749. (PubMed)
5. Zhong M, Kawaguchi R, Ter-Stepanian M, Kassai M, Sun H. Vitamin A transport and the transmembrane pore in the cell-surface receptor for plasma retinol binding protein. PLoS One. 2013;8(11):e73838. (PubMed)
6. See AW, Clagett-Dame M. The temporal requirement for vitamin A in the developing eye: mechanism of action in optic fissure closure and new roles for the vitamin in regulating cell proliferation and adhesion in the embryonic retina. Dev Biol. 2009;325(1):94-105. (PubMed)
7. Theodosiou M, Laudet V, Schubert M. From carrot to clinic: an overview of the retinoic acid signaling pathway. Cell Mol Life Sci. 2010;67(9):1423-1445. (PubMed)
8. Lefebvre P, Martin PJ, Flajollet S, Dedieu S, Billaut X, Lefebvre B. Transcriptional activities of retinoic acid receptors. Vitam Horm. 2005;70:199-264. (PubMed)
9. Amann PM, Eichmuller SB, Schmidt J, Bazhin AV. Regulation of gene expression by retinoids. Curr Med Chem. 2011;18(9):1405-1412. (PubMed)
10. Pendaries V, Verrecchia F, Michel S, Mauviel A. Retinoic acid receptors interfere with the TGF-beta/Smad signaling pathway in a ligand-specific manner. Oncogene. 2003;22(50):8212-8220. (PubMed)
11. Masia S, Alvarez S, de Lera AR, Barettino D. Rapid, nongenomic actions of retinoic acid on phosphatidylinositol-3-kinase signaling pathway mediated by the retinoic acid receptor. Mol Endocrinol. 2007;21(10):2391-2402. (PubMed)
12. Qiao J, Paul P, Lee S, et al. PI3K/AKT and ERK regulate retinoic acid-induced neuroblastoma cellular differentiation. Biochem Biophys Res Commun. 2012;424(3):421-426. (PubMed)
13. Canon E, Cosgaya JM, Scsucova S, Aranda A. Rapid effects of retinoic acid on CREB and ERK phosphorylation in neuronal cells. Mol Biol Cell. 2004;15(12):5583-5592. (PubMed)
14. Kirchmeyer M, Koufany M, Sebillaud S, Netter P, Jouzeau JY, Bianchi A. All-trans retinoic acid suppresses interleukin-6 expression in interleukin-1-stimulated synovial fibroblasts by inhibition of ERK1/2 pathway independently of RAR activation. Arthritis Res Ther. 2008;10(6):R141. (PubMed)
15. Amengual J, Golczak M, Palczewski K, von Lintig J. Lecithin:retinol acyltransferase is critical for cellular uptake of vitamin A from serum retinol-binding protein. J Biol Chem. 2012;287(29):24216-24227. (PubMed)
16. Noy N. Signaling by retinol and its serum binding protein. Prostaglandins Leukot Essent Fatty Acids. 2015; 93:3-7. (PubMed)
17. Berry DC, Jacobs H, Marwarha G, et al. The STRA6 receptor is essential for retinol-binding protein-induced insulin resistance but not for maintaining vitamin A homeostasis in tissues other than the eye. J Biol Chem. 2013;288(34):24528-24539. (PubMed)
18. Marwarha G, Berry DC, Croniger CM, Noy N. The retinol esterifying enzyme LRAT supports cell signaling by retinol-binding protein and its receptor STRA6. FASEB J. 2014;28(1):26-34. (PubMed)
19. Farmer SR. Molecular determinants of brown adipocyte formation and function. Genes Dev. 2008;22(10):1269-1275. (PubMed)
20. Kiefer FW, Vernochet C, O'Brien P, et al. Retinaldehyde dehydrogenase 1 regulates a thermogenic program in white adipose tissue. Nat Med. 2012;18(6):918-925. (PubMed)
21. Nallamshetty S, Le PT, Wang H, et al. Retinaldehyde dehydrogenase 1 deficiency inhibits PPARgamma-mediated bone loss and marrow adiposity. Bone. 2014;67:281-291. (PubMed)
22. Kiefer FW, Orasanu G, Nallamshetty S, et al. Retinaldehyde dehydrogenase 1 coordinates hepatic gluconeogenesis and lipid metabolism. Endocrinology. 2012;153(7):3089-3099. (PubMed)
23. Green HN, Mellanby E. Vitamin A as an anti-infective agent. Br Med J. 1928;2(3537):691-696. (PubMed)
24. Gholizadeh M, Basafa Roodi P, Abaj F, et al. Influence of vitamin A supplementation on inflammatory biomarkers in adults: a systematic review and meta-analysis of randomized clinical trials. Sci Rep. 2022;12(1):21384. (PubMed)
25. Raverdeau M, Mills KH. Modulation of T cell and innate immune responses by retinoic acid. J Immunol. 2014;192(7):2953-2958. (PubMed)
26. Spears K, Cheney C, Zerzan J. Low plasma retinol concentrations increase the risk of developing bronchopulmonary dysplasia and long-term respiratory disability in very-low-birth-weight infants. Am J Clin Nutr. 2004;80(6):1589-1594. (PubMed)
27. Barber T, Esteban-Pretel G, Marin MP, Timoneda J. Vitamin A deficiency and alterations in the extracellular matrix. Nutrients. 2014;6(11):4984-5017. (PubMed)
28. Semba RD, Bloem MW. The anemia of vitamin A deficiency: epidemiology and pathogenesis. Eur J Clin Nutr. 2002;56(4):271-281. (PubMed)
29. Allen LH. Iron supplements: scientific issues concerning efficacy and implications for research and programs. J Nutr. 2002;132(4 Suppl):813S-819S. (PubMed)
30. Christian P, West KP, Jr. Interactions between zinc and vitamin A: an update. Am J Clin Nutr. 1998;68(2 Suppl):435S-441S. (PubMed)
31. Auld DS, Bergman T. Medium- and short-chain dehydrogenase/reductase gene and protein families : The role of zinc for alcohol dehydrogenase structure and function. Cell Mol Life Sci. 2008;65(24):3961-3970. (PubMed)
32. da Cunha MSB, Campos Hankins NA, Arruda SF. Effect of vitamin A supplementation on iron status in humans: A systematic review and meta-analysis. Crit Rev Food Sci Nutr. 2019;59(11):1767-1781. (PubMed)
33. Suharno D, West CE, Muhilal, Karyadi D, Hautvast JG. Supplementation with vitamin A and iron for nutritional anaemia in pregnant women in West Java, Indonesia. Lancet. 1993;342(8883):1325-1328. (PubMed)
34. da Cunha MS, Siqueira EM, Trindade LS, Arruda SF. Vitamin A deficiency modulates iron metabolism via ineffective erythropoiesis. J Nutr Biochem. 2014;25(10):1035-1044. (PubMed)
35. Jang JT, Green JB, Beard JL, Green MH. Kinetic analysis shows that iron deficiency decreases liver vitamin A mobilization in rats. J Nutr. 2000;130(5):1291-1296. (PubMed)
36. Rosales FJ, Jang JT, Pinero DJ, Erikson KM, Beard JL, Ross AC. Iron deficiency in young rats alters the distribution of vitamin A between plasma and liver and between hepatic retinol and retinyl esters. J Nutr. 1999;129(6):1223-1228. (PubMed)
37. Tanumihardjo SA. Biological evidence to define a vitamin A deficiency cutoff using total liver vitamin A reserves. Exp Biol Med (Maywood). 2021;246(9):1045-1053. (PubMed)
38. World Health Organization. Serum retinol concentrations for determining the prevalence of vitamin A deficiency in populations. Vitamin and Mineral Nutrition Information System. Geneva, 2011. https://www.who.int/publications/i/item/WHO-NMH-NHD-MNM-11.3. Accessed 1/25/2024.
39. Underwood BA, Arthur P. The contribution of vitamin A to public health. Faseb J. 1996;10(9):1040-1048. (PubMed)
40. Solomons NW. Vitamin A. In: Erdman JJ, Macdonald I, Zeisel S, eds. Present Knowledge in Nutrition. 10th ed: John Wiley & Sons, Ltd.; 2012:149-184.
41. Sherwin JC, Reacher MH, Dean WH, Ngondi J. Epidemiology of vitamin A deficiency and xerophthalmia in at-risk populations. Trans R Soc Trop Med Hyg. 2012;106(4):205-214. (PubMed)
42. McCauley ME, van den Broek N, Dou L, Othman M. Vitamin A supplementation during pregnancy for maternal and newborn outcomes. Cochrane Database Syst Rev. 2015;2015(10):CD008666. (PubMed)
43. World Health Organization. Guideline: Vitamin A supplementation in infants and children 6–59 months of age. Geneva, 2011. Available at: https://www.who.int/publications/i/item/9789241501767. Accessed 1/19/24.
44. Gilbert C, Awan H. Blindness in children. BMJ. 2003;327(7418):760-761. (PubMed)
45. Semba RD. Vitamin A and human immunodeficiency virus infection. Proc Nutr Soc. 1997;56(1B):459-469. (PubMed)
46. Field CJ, Johnson IR, Schley PD. Nutrients and their role in host resistance to infection. J Leukoc Biol. 2002;71(1):16-32. (PubMed)
47. World Health Organization, UNICEF, IVACG Task Force. Vitamin A supplements: a guide to their use in the treatment and prevention of vitamin A deficiency and xerophthalmia. Geneva: World Health Organization; 1997.
48. Thornton KA, Mora-Plazas M, Marin C, Villamor E. Vitamin A deficiency is associated with gastrointestinal and respiratory morbidity in school-age children. J Nutr. 2014;144(4):496-503. (PubMed)
49. Wiysonge CS, Shey M, Kongnyuy EJ, Sterne JA, Brocklehurst P. Vitamin A supplementation for reducing the risk of mother-to-child transmission of HIV infection. Cochrane Database Syst Rev. 2011(1):CD003648. (PubMed)
50. Semba RD, Miotti PG, Chiphangwi JD, et al. Maternal vitamin A deficiency and mother-to-child transmission of HIV-1. Lancet. 1994;343(8913):1593-1597. (PubMed)
51. Wiysonge CS, Ndze VN, Kongnyuy EJ, Shey MS. Vitamin A supplements for reducing mother-to-child HIV transmission. Cochrane Database Syst Rev. 2017;9(9):CD003648. (PubMed)
52. Zimmermann MB, Adou P, Torresani T, Zeder C, Hurrell RF. Effect of oral iodized oil on thyroid size and thyroid hormone metabolism in children with concurrent selenium and iodine deficiency. Eur J Clin Nutr. 2000;54(3):209-213. (PubMed)
53. Zimmermann MB, Wegmuller R, Zeder C, Chaouki N, Torresani T. The effects of vitamin A deficiency and vitamin A supplementation on thyroid function in goitrous children. J Clin Endocrinol Metab. 2004;89(11):5441-5447. (PubMed)
54. Zimmermann MB. Interactions of vitamin A and iodine deficiencies: effects on the pituitary-thyroid axis. Int J Vitam Nutr Res. 2007;77(3):236-240. (PubMed)
55. Capriello S, Stramazzo I, Bagaglini MF, Brusca N, Virili C, Centanni M. The relationship between thyroid disorders and vitamin A.: A narrative minireview. Front Endocrinol (Lausanne). 2022;13:968215. (PubMed)
56. Zimmermann MB, Jooste PL, Mabapa NS, et al. Vitamin A supplementation in iodine-deficient African children decreases thyrotropin stimulation of the thyroid and reduces the goiter rate. Am J Clin Nutr. 2007;86(4):1040-1044. (PubMed)
57. Maronn M, Allen DM, Esterly NB. Phrynoderma: a manifestation of vitamin A deficiency?... The rest of the story. Pediatr Dermatol. 2005;22(1):60-63. (PubMed)
58. Herring W, Nowicki MJ, Jones JK. An uncommon cause of esophagitis. Answer to the clinical challenges and images in GI question: image 1: esophageal hyperkeratosis secondary to vitamin A deficiency. Gastroenterology. 2010;139(2):e6-7. (PubMed)
59. Weber D, Grune T. The contribution of beta-carotene to vitamin A supply of humans. Mol Nutr Food Res. 2012;56(2):251-258. (PubMed)
60. Food and Nutrition Board, Institute of Medicine. Vitamin A. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Washington D.C.: National Academy Press; 2001:65-126. (National Academy Press)
61. Ravishankar C, Nafday S, Green RS, et al. A trial of vitamin A therapy to facilitate ductal closure in premature infants. J Pediatr. 2003;143(5):644-648. (PubMed)
62. Tyson JE, Wright LL, Oh W, et al. Vitamin A supplementation for extremely-low-birth-weight infants. National Institute of Child Health and Human Development Neonatal Research Network. N Engl J Med. 1999;340(25):1962-1968. (PubMed)
63. Wardle SP, Hughes A, Chen S, Shaw NJ. Randomised controlled trial of oral vitamin A supplementation in preterm infants to prevent chronic lung disease. Arch Dis Child Fetal Neonatal Ed. 2001;84(1):F9-F13. (PubMed)
64. Ambalavanan N, Kennedy K, Tyson J, Carlo WA. Survey of vitamin A supplementation for extremely-low-birth-weight infants: is clinical practice consistent with the evidence? J Pediatr. 2004;145(3):304-307. (PubMed)
65. Laughon MM. Vitamin A shortage and risk of bronchopulmonary dysplasia. JAMA Pediatr. 2014;168(11):995-996. (PubMed)
66. Tolia VN, Murthy K, McKinley PS, Bennett MM, Clark RH. The effect of the national shortage of vitamin a on death or chronic lung disease in extremely low-birth-weight infants. JAMA Pediatr. 2014;168(11):1039-1044. (PubMed)
67. Darlow BA, Graham PJ, Rojas-Reyes MX. Vitamin A supplementation to prevent mortality and short- and long-term morbidity in very low birth weight infants. Cochrane Database Syst Rev. 2016;2016(8):CD000501. (PubMed)
68. Gadhia MM, Cutter GR, Abman SH, Kinsella JP. Effects of early inhaled nitric oxide therapy and vitamin A supplementation on the risk for bronchopulmonary dysplasia in premature newborns with respiratory failure. J Pediatr. 2014;164(4):744-748. (PubMed)
69. Meyer S, Gortner L, NeoVita ATI. Early postnatal additional high-dose oral vitamin A supplementation versus placebo for 28 days for preventing bronchopulmonary dysplasia or death in extremely low birth weight infants. Neonatology. 2014;105(3):182-188. (PubMed)
70. Meyer S, Gortner L, NeoVita ATi. Up-date on the NeoVitaA Trial: Obstacles, challenges, perspectives, and local experiences. Wien Med Wochenschr. 2017;167(11-12):264-270. (PubMed)
71. Babu TA, Sharmila V. Vitamin A supplementation in late pregnancy can decrease the incidence of bronchopulmonary dysplasia in newborns. J Matern Fetal Neonatal Med. 2010;23(12):1468-1469. (PubMed)
72. Imdad A, Mayo-Wilson E, Haykal MR, et al. Vitamin A supplementation for preventing morbidity and mortality in children from six months to five years of age. Cochrane Database Syst Rev. 2022;3(3):CD008524. (PubMed)
73. World Health Organization. Essential Programme on Immunization. Vitamin A supplementation. https://www.who.int/teams/immunization-vaccines-and-biologicals/essential-programme-on-immunization/integration/linking-with-other-health-interventions/vitamin-a. Accessed 1/25/24.
74. World Health Organization. Guideline - Vitamin A supplementation for infants and children 6-59 months of age - Guideline. Geneva 2011. Available at: https://www.who.int/publications/i/item/9789241501767. Accessed 2/23/2024.
75. World Health Organization. Guideline - Neonatal vitamin A supplementation Geneva 2011. Available at: https://iris.who.int/bitstream/handle/10665/44626/9789241501798_eng.pdf?sequence=1. Accessed 2/23/2024.
76. Benn CS, Bale C, Sommerfelt H, Friis H, Aaby P. Hypothesis: Vitamin A supplementation and childhood mortality: amplification of the non-specific effects of vaccines? Int J Epidemiol. 2003;32(5):822-828. (PubMed)
77. Fisker AB, Bale C, Rodrigues A, et al. High-dose vitamin A with vaccination after 6 months of age: a randomized trial. Pediatrics. 2014;134(3):e739-748. (PubMed)
78. Huiming Y, Chaomin W, Meng M. Vitamin A for treating measles in children. Cochrane Database Syst Rev. 2005(4):CD001479. (PubMed)
79. American Academy of Pediatrics Committee on Infectious Diseases: Vitamin A treatment of measles. Pediatrics. 1993;91(5):1014-1015. (PubMed)
80. Bello S, Meremikwu MM, Ejemot-Nwadiaro RI, Oduwole O. Routine vitamin A supplementation for the prevention of blindness due to measles infection in children. Cochrane Database Syst Rev. 2016;2016(8):CD007719. (PubMed)
81. Omenn GS, Goodman GE, Thornquist MD, et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N Engl J Med. 1996;334(18):1150-1155. (PubMed)
82. Goodman GE, Thornquist MD, Balmes J, et al. The Beta-Carotene and Retinol Efficacy Trial: incidence of lung cancer and cardiovascular disease mortality during 6-year follow-up after stopping beta-carotene and retinol supplements. J Natl Cancer Inst. 2004;96(23):1743-1750. (PubMed)
83. Palozza P, Simone R, Mele MC. Interplay of carotenoids with cigarette smoking: implications in lung cancer. Curr Med Chem. 2008;15(9):844-854. (PubMed)
84. Cheng TY, Goodman GE, Thornquist MD, et al. Estimated intake of vitamin D and its interaction with vitamin A on lung cancer risk among smokers. Int J Cancer. 2014;135(9):2135-2145. (PubMed)
85. Cortes-Jofre M, Rueda JR, Corsini-Munoz G, Fonseca-Cortes C, Caraballoso M, Bonfill Cosp X. Drugs for preventing lung cancer in healthy people. Cochrane Database Syst Rev. 2012;10:CD002141. (PubMed)
86. Abar L, Vieira AR, Aune D, et al. Blood concentrations of carotenoids and retinol and lung cancer risk: an update of the WCRF-AICR systematic review of published prospective studies. Cancer Med. 2016;5(8):2069-2083. (PubMed)
87. Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369(2):111-121. (PubMed)
88. Yilmaz M, Kantarjian H, Ravandi F. Acute promyelocytic leukemia current treatment algorithms. Blood Cancer J. 2021;11(6):123. (PubMed)
89. Booij MT, Van De Kerkhof PC. Acitretin revisited in the era of biologics. J Dermatolog Treat. 2011;22(2):86-89. (PubMed)
90. Armstrong AW, Read C. Pathophysiology, clinical presentation, and treatment of psoriasis: a review. JAMA. 2020;323(19):1945-1960. (PubMed)
91. Orfanos CE, Zouboulis CC. Oral retinoids in the treatment of seborrhoea and acne. Dermatology. 1998;196(1):140-147. (PubMed)
92. Thielitz A, Gollnick H. Topical retinoids in acne vulgaris: update on efficacy and safety. Am J Clin Dermatol. 2008;9(6):369-381. (PubMed)
93. Vishwanathan R, Johnson EJ. Eye disease. In: Erdman JJ, Macdonald I, Zeisel S, eds. Present Knowledge in Nutrition. 10th ed: John Wiley & Sons, Ltd; 2012:939-981.
94. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795-1809. (PubMed)
95. Berson EL, Rosner B, Sandberg MA, et al. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(6):761-772. (PubMed)
96. Sibulesky L, Hayes KC, Pronczuk A, Weigel-DiFranco C, Rosner B, Berson EL. Safety of <7500 RE (<25000 IU) vitamin A daily in adults with retinitis pigmentosa. Am J Clin Nutr. 1999;69(4):656-663. (PubMed)
97. Schwartz SG, Wang X, Chavis P, Kuriyan AE, Abariga SA. Vitamin A and fish oils for preventing the progression of retinitis pigmentosa. Cochrane Database Syst Rev. 2020;6(6):CD008428. (PubMed)
98. Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. 2nd ed: Thomson Reuters; 2008.
99. Penniston KL, Tanumihardjo SA. The acute and chronic toxic effects of vitamin A. Am J Clin Nutr. 2006;83(2):191-201. (PubMed)
100. Russell RM. The vitamin A spectrum: from deficiency to toxicity. Am J Clin Nutr. 2000;71(4):878-884. (PubMed)
101. Bastos Maia S, Rolland Souza AS, Costa Caminha MF, et al. Vitamin A and pregnancy: a narrative review. Nutrients. 2019;11(3):681. (PubMed)
102. World Health Organization Organization. Guideline - Vitamin A supplementation in pregnant women. Geneva 2011.
103. Bozzo P, Chua-Gocheco A, Einarson A. Safety of skin care products during pregnancy. Can Fam Physician. 2011;57(6):665-667. (PubMed)
104. Michaelsson K, Lithell H, Vessby B, Melhus H. Serum retinol levels and the risk of fracture. N Engl J Med. 2003;348(4):287-294. (PubMed)
105. Promislow JH, Goodman-Gruen D, Slymen DJ, Barrett-Connor E. Retinol intake and bone mineral density in the elderly: the Rancho Bernardo Study. J Bone Miner Res. 2002;17(8):1349-1358. (PubMed)
106. Feskanich D, Singh V, Willett WC, Colditz GA. Vitamin A intake and hip fractures among postmenopausal women. JAMA. 2002;287(1):47-54. (PubMed)
107. Rejnmark L, Vestergaard P, Charles P, et al. No effect of vitamin A intake on bone mineral density and fracture risk in perimenopausal women. Osteoporos Int. 2004;15(11):872-880. (PubMed)
108. Sowers MF, Wallace RB. Retinol, supplemental vitamin A and bone status. J Clin Epidemiol. 1990;43(7):693-699. (PubMed)
109. Ballew C, Galuska D, Gillespie C. High serum retinyl esters are not associated with reduced bone mineral density in the Third National Health And Nutrition Examination Survey, 1988-1994. J Bone Miner Res. 2001;16(12):2306-2312. (PubMed)
110. de Jonge EA, Kiefte-de Jong JC, Campos-Obando N, et al. Dietary vitamin A intake and bone health in the elderly: the Rotterdam Study. Eur J Clin Nutr. 2015;69(12):1360-1368. (PubMed)
111. Wu AM, Huang CQ, Lin ZK, et al. The relationship between vitamin A and risk of fracture: meta-analysis of prospective studies. J Bone Miner Res. 2014;29(9):2032-2039. (PubMed)
112. Zhang X, Zhang R, Moore JB, et al. The effect of vitamin A on fracture risk: a meta-analysis of cohort studies. Int J Environ Res Public Health. 2017;14(9):1043. (PubMed)
113. Conaway HH, Henning P, Lerner UH. Vitamin A metabolism, action, and role in skeletal homeostasis. Endocr Rev. 2013;34(6):766-797. (PubMed)
114. Johansson S, Melhus H. Vitamin A antagonizes calcium response to vitamin D in man. J Bone Miner Res. 2001;16(10):1899-1905. (PubMed)
115. Caire-Juvera G, Ritenbaugh C, Wactawski-Wende J, Snetselaar LG, Chen Z. Vitamin A and retinol intakes and the risk of fractures among participants of the Women's Health Initiative Observational Study. Am J Clin Nutr. 2009;89(1):323-330. (PubMed)
116. Lieber CS. Relationships between nutrition, alcohol use, and liver disease. Alcohol Res Health. 2003;27(3):220-231. (PubMed)
Vitamin B6
Contents
Summary
- Vitamin B6 and its derivative pyridoxal 5'-phosphate (PLP) are essential to over 100 enzymes mostly involved in protein metabolism. (More information)
- High levels of circulating homocysteine are associated with an increased risk of cardiovascular disease. Randomized controlled trials have demonstrated that supplementation with B vitamins, including vitamin B6, could effectively reduce homocysteine levels. However, homocysteine lowering by B vitamins has failed to lower the risk of adverse cardiovascular outcomes in high-risk individuals. (More information)
- Growing evidence from experimental and clinical studies suggests that systemic inflammation underlying most chronic diseases may impair vitamin B6 metabolism. (More information)
- Although supplementation with vitamin B6 and other B vitamins has not been associated with improved cognitive performance or delayed cognitive deterioration in the elderly, recent studies suggest that vitamin B6 might help reduce the risk of late-life depression. (More information)
- Pharmacologic doses of vitamin B6 are used to treat seizures in rare inborn errors of vitamin B6 metabolism. Also, randomized controlled trials support the use of vitamin B6 to treat morning sickness in pregnant women and suggest a possible benefit in the management of premenstrual syndrome and carpal tunnel syndrome. (More information)
- Vitamin B6 is found in a variety of foods, including fish, poultry, nuts, legumes, potatoes, and bananas. (More information)
-
Several medications, including anti-tuberculosis drugs, anti-parkinsonians, nonsteroidal anti-inflammatory drugs, and oral contraceptives, may interfere with vitamin B6 metabolism. (More information)
Vitamin B6 is a water-soluble vitamin that was first isolated in the 1930s. The term vitamin B6 refers to six common forms, namely pyridoxal, pyridoxine (pyridoxol), pyridoxamine, and their phosphorylated forms. The phosphate ester derivative pyridoxal 5'-phosphate (PLP) is the bioactive coenzyme form involved in over 4% of all enzymatic reactions (Figure 1) (1-3).

Function
Vitamin B6 must be obtained from the diet because humans cannot synthesize it. PLP plays a vital role in the function of over 100 enzymes that catalyze essential chemical reactions in the human body (4). PLP-dependent enzymes have been classified into five structural classes known as Fold Type I-V (5):
- Fold Type I - aspartate aminotransferase family
- Fold Type II - tryptophan synthase family
- Fold Type III - alanine racemase family
- Fold Type IV - D-amino acid aminotransferase family
- Fold Type V - glycogen phosphorylase family
The many biochemical reactions catalyzed by PLP-dependent enzymes are involved in essential biological processes, such as hemoglobin and amino acid biosynthesis, as well as fatty acid metabolism. Of note, PLP also functions as a coenzyme for glycogen phosphorylase, an enzyme that catalyzes the release of glucose from stored glycogen. Much of the PLP in the human body is found in muscle bound to glycogen phosphorylase. PLP is also a coenzyme for reactions that generate glucose from amino acids, a process known as gluconeogenesis (6).
Nervous system function
In the brain, the PLP-dependent enzyme aromatic L-amino acid decarboxylase catalyzes the synthesis of two major neurotransmitters: serotonin from the amino acid tryptophan and dopamine from L-3,4-dihydroxyphenylalanine (L-Dopa). Other neurotransmitters, including glycine, D-serine, glutamate, histamine, and γ-aminobutyric acid (GABA), are also synthesized in reactions catalyzed by PLP-dependent enzymes (7).
Hemoglobin synthesis and function
PLP functions as a coenzyme of 5-aminolevulinic acid synthase, which is involved in the synthesis of heme, an iron-containing component of hemoglobin. Hemoglobin is found in red blood cells and is critical to their ability to transport oxygen throughout the body. Both pyridoxal and PLP are able to bind to the hemoglobin molecule and affect its ability to pick up and release oxygen. However, the impact of this on normal oxygen delivery to tissues is not known (6, 8). Vitamin B6 deficiency may impair hemoglobin synthesis and lead to microcytic anemia (3).
Tryptophan metabolism
Deficiency in another B vitamin, niacin, is easily prevented by adequate dietary intakes. The dietary requirement for niacin and the niacin coenzyme, nicotinamide adenine dinucleotide (NAD), can be also met, though to a fairly limited extent, by the catabolism of the essential amino acid tryptophan in the tryptophan-kynurenine pathway (Figure 2). Key reactions in this pathway are PLP-dependent; in particular, PLP is the cofactor for the enzyme kynureninase, which catalyzes the conversion of 3-hydroxykynurenine to 3-hydroxyanthranilic acid. A reduction in PLP availability appears to primarily affect kynureninase activity, limiting NAD production and leading to higher concentrations of kynurenine, 3-hydroxykynurenine, and xanthurenic acid in blood and urine (Figure 2) (9). Thus, while dietary vitamin B6 restriction impairs NAD synthesis from tryptophan, adequate PLP levels help maintain NAD formation from tryptophan (10). The effect of vitamin B6 inadequacy on immune activation and inflammation may be partly related to the role of PLP in the tryptophan-kynurenine metabolism (see Disease Prevention).
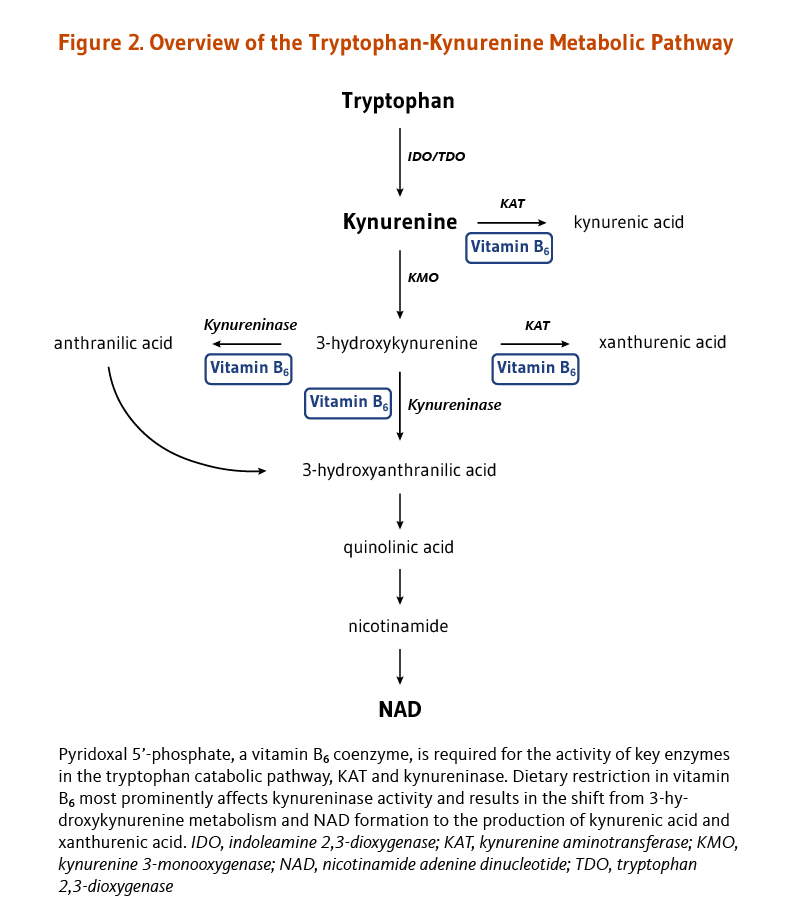
Hormone function
Steroid hormones, such as estrogen and testosterone, exert their effects in the body by binding to steroid hormone receptors in the nucleus of target cells. The nuclear receptors themselves bind to specific regulatory sequences in DNA and alter the transcription of target genes. Experimental studies have uncovered a mechanism by which PLP may affect the activity of steroid receptors and decrease their effects on gene expression. It was found that PLP could interact with RIP140/NRIP1, a repressor of nuclear receptors known for its role in reproductive biology (11). Yet, additional research is needed to confirm that this interaction can enhance RIP140/NRIP1 repressive activity on steroid receptor-mediated gene expression. If the activity of steroid receptors for estrogen, progesterone, testosterone, or other steroid hormones can be inhibited by PLP, it is possible that vitamin B6 status may influence one's risk of developing diseases driven by steroid hormones, such as breast and prostate cancers (6).
Nucleic acid synthesis
The synthesis of nucleic acids from precursors thymidine and purines is dependent on folate coenzymes. The de novo thymidylate (dTMP) biosynthesis pathway involves three enzymes: dihydrofolate reductase (DHFR), serine hydroxymethyltransferase (SHMT), and thymidylate synthase (TYMS) (Figure 3). PLP serves as a coenzyme for SHMT, which catalyzes the simultaneous conversions of serine to glycine and tetrahydrofolate (THF) to 5,10-methylene THF. The latter molecule is the one-carbon donor for the generation of dTMP from deoxyuridine monophosphate (dUMP) by TYMS.
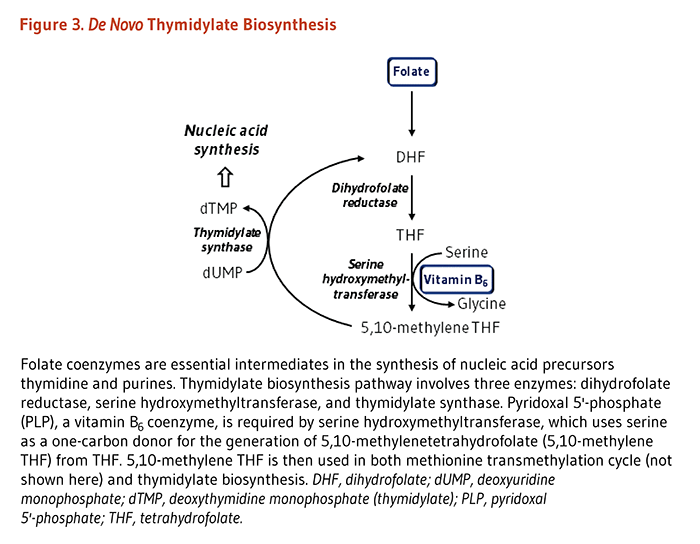
Deficiency
Severe deficiency of vitamin B6 is uncommon. Alcoholics are thought to be most at risk of vitamin B6 deficiency due to low dietary intakes and impaired metabolism of the vitamin. In the early 1950s, seizures were observed in infants as a result of severe vitamin B6 deficiency caused by an error in the manufacture of infant formula (7). Abnormal electroencephalogram (EEG) patterns have also been reported in vitamin B6-deficient adults. Other neurologic symptoms observed in severe vitamin B6 deficiency include irritability, depression, and confusion; additional symptoms include inflammation of the tongue, sores or ulcers of the mouth, and ulcers of the skin at the corners of the mouth (12).
The Recommended Dietary Allowance (RDA)
Since vitamin B6 is involved in many aspects of metabolism, especially in amino acid metabolic pathways, an individual's protein intake is likely to influence the requirement for vitamin B6 (13). Unlike previous recommendations issued by the Food and Nutrition Board (FNB) of the Institute of Medicine, the most recent RDA for vitamin B6 was not expressed in terms of protein intake, although the relationship was considered in setting the RDA (14). The current RDA was revised by the FNB in 1998 and is presented in Table 1.
| Life Stage | Age | Males (mg/day) | Females (mg/day) |
|---|---|---|---|
| Infants | 0-6 months | 0.1 (AI) | 0.1 (AI) |
| Infants | 7-12 months | 0.3 (AI) | 0.3 (AI) |
| Children | 1-3 years | 0.5 | 0.5 |
| Children | 4-8 years | 0.6 | 0.6 |
| Children | 9-13 years | 1.0 | 1.0 |
| Adolescents | 14-18 years | 1.3 | 1.2 |
| Adults | 19-50 years | 1.3 | 1.3 |
| Adults | 51 years and older | 1.7 | 1.5 |
| Pregnancy | all ages | - | 1.9 |
| Breast-feeding | all ages | - | 2.0 |
Disease Prevention
Immune dysfunction
Several enzymatic reactions in the tryptophan-kynurenine pathway are dependent on vitamin B6 coenzyme, pyridoxal 5'-phosphate (PLP) (see Figure 2 above) (see Tryptophan metabolism). This pathway is known to be activated during pro-inflammatory immune responses and plays a critical role in immune tolerance of the fetus during pregnancy (15). Key intermediates in the tryptophan-kynurenine pathway are involved in the regulation of immune responses. Several tryptophan derivatives have been found to induce the death (apoptosis) or block the proliferation of certain types of immune cells, such as lymphocytes (in particular T-helper 1). They can also inhibit the production of pro-inflammatory cytokines (reviewed in 15). There is evidence to suggest that adequate vitamin B6 intake is important for optimal immune system function, especially in older individuals (16, 17). Yet, chronic inflammation that triggers tryptophan degradation and underlies many diseases (e.g., cardiovascular disease and cancers) may precipitate the loss of PLP and increase vitamin B6 requirements. Additional research is needed to evaluate whether vitamin B6 intakes higher than the current RDA could prevent and/or reverse immune system impairments (see also Vitamin B6 and inflammation).
Cardiovascular disease
The use of multivitamin supplements (including vitamin B6) has been associated with a 24% lower risk of incidental coronary artery disease (CAD) in a large prospective study of 80,082 women from the US Nurses' Health Study cohort (18). Using food frequency questionnaires, the authors observed that women in the highest quintile of vitamin B6 intakes from both food and supplements (median, 4.6 mg/day) had a 34% lower risk of CAD compared to those in the lowest quintile (median, 1.1 mg/day). CAD is characterized by the abnormal stenosis (narrowing) of coronary arteries (generally due to atherosclerosis), which can result in a potentially fatal myocardial infarction (heart attack). More recently, a prospective study that followed a Japanese cohort of over 40,000 middle-aged individuals for 11.5 years reported a 48% lower risk of myocardial infarction in those in the highest (mean, 1.6 mg/day) versus lowest quintile (mean, 1.3 mg/day) of vitamin B6 intakes in non-supplement users (19).
Early observational studies have also demonstrated an association between suboptimal pyridoxal 5'-phosphate (PLP) plasma level, elevated homocysteine blood level, and increased risk of cardiovascular disease (20-22). More recent research has confirmed that low plasma PLP status is a risk factor for CAD. In a case-control study, which included 184 participants with CAD and 516 healthy controls, low plasma PLP levels (<30 nanomoles/liter) were associated with a near doubling of CAD risk when compared to higher PLP levels (≥30 nanomoles/liter) (23). In a nested case-control study based on the Nurses' Health Study cohort and including 144 cases of myocardial infarction (of which 21 were fatal), women in the highest quartile of blood PLP levels (≥70 nanomoles/liter) had a 79% lower risk of myocardial infarction compared to those in the lowest quartile (<27.9 nanomoles/liter) (24).
Vitamin B6 and homocysteine
Even moderately elevated levels of homocysteine in the blood have been associated with increased risk for cardiovascular disease (CVD), including cardiac insufficiency (heart failure), CAD, myocardial infarction, and cerebrovascular attack (stroke) (25). During protein digestion, amino acids, including methionine, are released. Methionine is an essential amino acid and precursor of S-adenosylmethionine (SAM), the universal methyl donor for most methylation reactions, including the methylation of DNA, RNA, proteins, and phospholipids (Figure 4). Homocysteine is an intermediate in the metabolism of methionine. Healthy individuals utilize two different pathways to regenerate methionine from homocysteine in the methionine remethylation cycle (Figure 5). One pathway relies on the vitamin B12-dependent methionine synthase and the methyl donor, 5-methyl tetrahydrofolate (a folate derivative), to convert homocysteine back to methionine. The other reaction is catalyzed by betaine homocysteine methyltransferase, which uses betaine as a source of methyl groups for the formation of methionine from homocysteine. Moreover, two PLP-dependent enzymes are required to convert homocysteine to the amino acid cysteine in homocysteine transsulfuration pathway: cystathionine β synthase and cystathionine γ lyase (Figure 5). Thus, the amount of homocysteine in the blood may be influenced by nutritional status of at least three B vitamins, namely folate, vitamin B12, and vitamin B6.
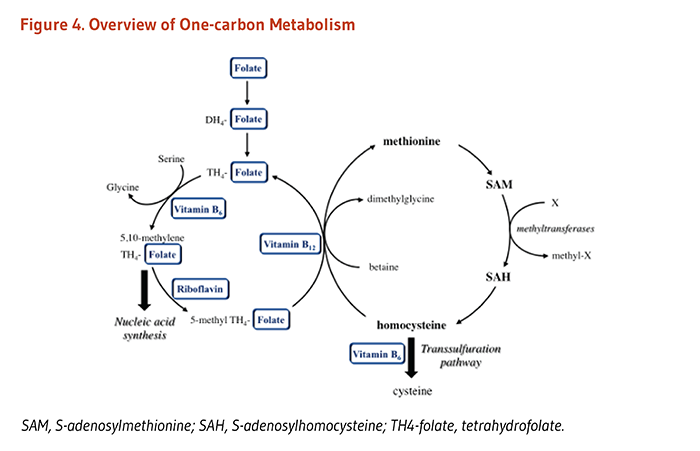
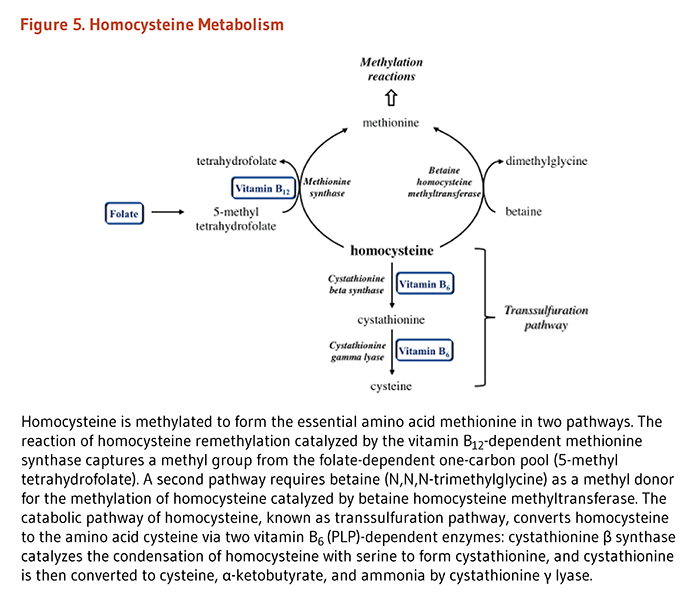
Deficiencies in one or all of these B vitamins may affect both remethylation and transsulfuration processes and result in abnormally elevated homocysteine levels. An early study found that vitamin B6 supplementation could lower blood homocysteine levels after an oral dose of methionine was given (i.e., a methionine load test) (26), but vitamin B6 supplementation might not be effective in decreasing fasting levels of homocysteine. In a recent study conducted in nine healthy young volunteers, the rise of homocysteine during the postprandial period (after a meal) was found to be greater with marginal vitamin B6 deficiency (mean plasma PLP level of 19 nanomoles/liter) as compared to vitamin B6 sufficiency (mean PLP level of 49 nanomoles/liter) (27). The authors reported an increased rate of cystathionine synthesis with vitamin B6 restriction, suggesting that homocysteine catabolism in the transsulfuration may be maintained or enhanced in response to a marginal reduction in PLP availability. Yet, the flux ratio between methionine cycle and transsulfuration pathway appeared to favor homocysteine clearance by remethylation rather than transsulfuration in six out of nine participants (27).
Numerous randomized controlled trials, many in subjects with existing hyperhomocysteinemia and vascular dysfunction, have demonstrated that supplementation with folic acid, alone or combined with vitamin B6 and vitamin B12, could effectively reduce fasting plasma homocysteine concentrations. In 19 intervention studies recently included in a meta-analysis, reductions in homocysteine level in the blood following B-vitamin supplementation ranged between 7.6% and 51.7% compared to baseline levels (28). In contrast, studies supplementing individuals with only vitamin B6 have usually failed to show an effect on fasting levels of homocysteine (29, 30). Of the three supplemental B vitamins, folic acid appears to be the main determinant in the regulation of fasting homocysteine levels when there is no coexisting deficiency of vitamin B12 or vitamin B6 (31). Yet, the effect of homocysteine lowering on CVD risk reduction is debated. A recent meta-analysis of nine randomized controlled trials reported a 10% reduction in stroke events with supplemental B vitamins, with greater benefits for high-risk subjects (e.g., those with kidney disease) (32). However, most systematic reviews and meta-analyses of B-vitamin intervention studies to date have indicated a lack of causality between the decrease of fasting homocysteine levels and the prevention of cardiovascular events (28, 33-35). Moreover, B-vitamin supplementation trials in high-risk subjects have not resulted in significant changes in carotid intima-media thickness (CIMT) and flow-mediated dilation (FMD) of the brachial artery, two markers of vascular health used to assess atherosclerotic progression (36). Finally, in the Western Norway B Vitamin Intervention Trial (WENBIT), a randomized, double-blind, placebo-controlled trial in 87 subjects with suspected CAD, vitamin B6 supplementation (40 mg/day of pyridoxine) for a median of 10 months had no effect on coronary stenosis progression, assessed by quantitative angiography (37).
It has been suggested that antiplatelet therapy used in the primary prevention of CVD might interfere with the effect of homocysteine lowering by B vitamins on CVD risk (38). In this context, a post-hoc subgroup analysis of the multicenter, randomized, double blind, placebo-controlled VITATOPS trial (39) proposed that the small benefit of homocysteine-lowering by B vitamins might be cancelled in patients treated with antiplatelet drugs (40). Yet, the benefit of B-vitamin supplementation in primary prevention (i.e., in non-antiplatelet users) remains to be established.
Vitamin B6 and inflammation
A growing body of evidence currently suggests that low vitamin B6 status may increase the risk of cardiovascular disease through mechanisms independent of homocysteine lowering (41-43). Markers of immune activation and inflammation have been associated with hyperhomocysteinemia (homocysteine levels >15 micromoles/liter) in individuals with coronary artery disease (CAD) (44). In fact, inflammation is involved in the early steps of atherosclerosis in which lipids deposit in plaques (known as atheromas) within arterial walls and increase the risk of CAD (45). In a case-control study that included 267 patients with CAD and 475 healthy controls, plasma PLP concentrations were inversely correlated with the levels of two markers of systemic inflammation, C-reactive protein (CRP) and fibrinogen (46). Yet, the study suggested that suboptimal PLP levels (<36.3 nanomoles/liter) might contribute to an increased risk of CAD independently of inflammation since the risk was unchanged after adjustment for inflammation markers (unadjusted odds ratio (OR): 1.71 vs. multivariate adjusted OR: 1.73). Furthermore, the analysis of inflammation markers in 2,686 participants of the US National Health and Nutrition Examination Survey (NHANES) 2003-2004 indicated that serum CRP concentrations were inversely related to total vitamin B6 intakes (from both food and supplements). Specifically, the risk of having serum CRP levels greater than 10 mg/L (corresponding to high inflammatory activity) was 57% higher in individuals with vitamin B6 intakes lower than 2 mg/day compared to those with intakes equal to and above 5 mg/day (41). In addition, the prevalence of inadequate vitamin B6 status (plasma PLP levels <20 nanomoles/liter) with intakes lower than 5 mg/day was systematically greater in individuals with high versus low serum CRP concentrations (>10 mg/L vs. ≤3 mg/L), suggesting that inflammation might impair vitamin B6 metabolism. These observations were confirmed in the study of another cohort (Framingham Offspring Study) in which vitamin B6 status was linked to an overall inflammatory score based on the levels of 13 inflammation markers (including CRP, fibrinogen, tumor necrosis factor-α, and interleukin-6) (42). Specifically, plasma PLP levels were 24% lower in individuals in the highest versus lowest tertile of inflammatory score. Moreover, the inverse correlation between PLP levels and inflammatory scores remained significant regardless of vitamin B6 intakes, questioning again the nature of this relationship. Interestingly, a recent analysis of data collected in the WENBIT study demonstrated that systemic inflammation was associated with an increased degradation of pyridoxal (PL) to 4-pyridoxic acid (PA), supporting the use of the ratio PA:(PL+PLP) as a marker of both vitamin B6 status and systemic inflammation (47). Finally, while inflammation may contribute to lower vitamin B6 status, current evidence fails to support a role for vitamin B6 in the control of inflammation in patients with cardiovascular disease (48, 49).
Cognitive decline and Alzheimer's disease
A few observational studies have linked cognitive decline and Alzheimer's disease (AD) in the elderly with inadequate status of folate, vitamin B12, and vitamin B6 (50). Yet, the relationship between B vitamins and cognitive health in aging is complicated by both the high prevalence of hyperhomocysteinemia and signs of systemic inflammation in elderly people (51). On the one hand, since inflammation may impair vitamin B6 metabolism, low serum PLP levels may well be caused by processes related to aging rather than by malnutrition. On the other hand, high serum homocysteine may possibly be a risk factor for cognitive decline in the elderly, although the matter remains under debate. Specifically, the meta-analysis of 19 randomized, placebo-controlled trials of B-vitamin supplementation failed to report any difference in several measures of cognitive function between treatment and placebo groups, despite the treatment effectively lowering homocysteine levels (52). In a recent randomized, double-blind, placebo-controlled study of 2,695 stroke survivors with or without cognitive impairments, daily supplementation with 2 mg of folic acid, 0.5 mg of vitamin B12, and 25 mg of vitamin B6 for 3.4 years resulted in significant reductions in mean homocysteine levels (by 28% and 43% in cognitively unimpaired and impaired patients, respectively) compared to placebo. Yet, the B-vitamin intervention had no effect on either the incidence of newly diagnosed cognitive impairments or on measures of cognitive performance when compared to placebo (53). In contrast, another recent placebo-controlled trial found that a daily B-vitamin regimen that led to significant homocysteine lowering in high-risk elderly individuals could limit the progressive atrophy of gray matter brain regions associated with the AD process (54). Yet, the authors attributed the changes in homocysteine levels primarily to vitamin B12. Because of mixed findings, it is presently unclear whether supplementation with B vitamins might blunt cognitive decline in the elderly. Evidence is needed to determine whether marginal B-vitamin deficiencies, which are relatively common in the elderly, even contribute to age-associated declines in cognitive function, or whether both result from processes associated with aging and/or disease.
Depression
Late-life depression is a common disorder sometimes occurring after acute illnesses, such as hip fracture or stroke (55, 56). Coexistence of symptoms of depression and low vitamin B6 status (plasma PLP level ≤20 nanomoles/liter) has been reported in a few cross-sectional studies (57, 58). In a prospective study of 3,503 free-living people aged 65 and older from the Chicago Health and Aging Project, total vitamin B6 intakes (but not dietary intakes alone) were inversely correlated with the incidence of depressive symptoms during a mean follow-up period of 7.2 years (59). In a randomized, double-blind, placebo-controlled trial in 563 individuals who suffered from a recent stroke, daily supplementation of 2 mg of folic acid, 0.5 mg of vitamin B12, and 25 mg of vitamin B6 halved the risk of developing a major depressive episode during a mean follow-up period of 7.1 years (60). This reduction in risk was associated with a 25% lower level of plasma homocysteine in supplemented patients compared to controls. Additional evidence is needed to evaluate whether B vitamins could be included in the routine management of older people at high risk for depression.
Cancer
Chronic inflammation that underlies most cancers may enhance vitamin B6 degradation (see Vitamin B6 and inflammation). In addition, because PLP is required for the methionine cycle, homocysteine catabolism, and thymidylate synthesis, low vitamin B6 status might affect these pathways and potentially increase the risk for chronic conditions. The systematic review of nine prospective studies found either inverse or positive associations between vitamin B6 intakes and colorectal cancer (CRC) risk (61). Inconsistent evidence regarding the link between vitamin B6 intakes and breast cancer was also recently reported in a meta-analysis (62). Yet, a prospective study that followed nearly 500,000 older adults for nine years observed that the risk of esophageal and stomach cancers was lower in participants in the highest versus lowest quintile of total vitamin B6 intakes (median values, 2.7 mg/day vs. 1.4 mg/day) (63). Additionally, a meta-analysis of four nested case-control studies reported a 48% reduction in CRC risk in the highest versus lowest quartile of blood PLP level (61). Another meta-analysis of five nested case-control studies found higher versus lower serum PLP levels to be associated with a 29% lower risk of breast cancer in postmenopausal, but not premenopausal, women (62).
Very few randomized, placebo-controlled trials investigating the nature of the association between B vitamins and cancer risk have focused on vitamin B6. Two earlier studies conducted in subjects with coronary artery disease failed to observe any benefit of supplemental vitamin B6 (40 mg/day) on CRC risk and mortality (reviewed in 64). A recent randomized, double-blind, placebo-controlled study conducted in 1,470 women with high cardiovascular risk showed that daily supplementation with 2.5 mg of folic acid, 1 mg of vitamin B12, and 50 mg of vitamin B6 for a mean treatment period of 7.3 years had no effect on the risk of developing colorectal adenoma when compared to placebo (65).
Kidney stones
A large prospective study examined the relationship between vitamin B6 intake and the occurrence of symptomatic kidney stones in women. A group of more than 85,000 women without a prior history of kidney stones were followed over 14 years, and those who consumed 40 mg or more of vitamin B6 daily had only two-thirds the risk of developing kidney stones compared with those who consumed 3 mg or less (66). However, in a group of more than 45,000 men followed for 14 years, no association was found between vitamin B6 intake and the occurrence of kidney stones (67). Limited experimental data have suggested that supplementation with high doses of pyridoxamine may help decrease the formation of calcium oxalate kidney stones and reduce urinary oxalate levels, an important determinant of calcium oxalate kidney stone formation (68, 69). Presently, the relationship between vitamin B6 intake and the risk of developing kidney stones requires further study before any recommendations can be made.
Disease Treatment
Vitamin B6 supplements at pharmacologic doses (i.e., doses much larger than those needed to prevent deficiency) have been used in an attempt to treat a wide variety of conditions, some of which are discussed below.
Metabolic diseases
A few rare inborn metabolic disorders, including pyridoxine-dependent epilepsy (PDE) and pyridoxamine 5'-phosphate oxidase (PNPO) deficiency, are the cause of early-onset epileptic encephalopathies that are found to be responsive to pharmacologic doses of vitamin B6. In individuals affected by PDE and PNPO deficiency, PLP bioavailability is limited, and treatment with pyridoxine and/or PLP have been used to alleviate or abolish epileptic seizures characterizing these conditions (70, 71). Pyridoxine therapy, along with dietary protein restriction, is also used in the management of vitamin B6 responsive homocystinuria caused by the deficiency of the PLP-dependent enzyme, cystathionine β synthase (72).
Morning sickness
Nausea and vomiting in pregnancy (NVP), often referred to as morning sickness, can affect up to 85% of women during early pregnancy and usually lasts between 12 and 16 weeks (73). Vitamin B6 has been used since the 1940s to treat nausea during pregnancy. Vitamin B6 was originally included in the medication Bendectin, which was prescribed for NVP treatment and later withdrawn from the market due to unproven concerns that it increased the risk for birth defects. Vitamin B6 itself is considered safe during pregnancy and has been used in pregnant women without any evidence of fetal harm (74). The results of two double-blind, placebo-controlled trials, including 401 pregnant women that used 25 mg of pyridoxine every eight hours for three days (75) or 10 mg of pyridoxine every eight hours for five days (76), suggested that vitamin B6 may be beneficial in reducing nausea. A recent systematic review of randomized controlled trials on NVP symptoms during early pregnancy found supplemental vitamin B6 to be somewhat effective (77). It should be noted that NVP usually resolves without any treatment, making it difficult to perform well-controlled trials. More recently, NVP symptoms were evaluated using Pregnancy Unique Quantification of Emesis (PUQE) scores in a randomized, double-blind, placebo-controlled study conducted in 256 pregnant women (7-14 weeks' gestation) suffering from NVP (78). Supplementation with pyridoxine and the drug doxylamine significantly improved NVP symptoms, as assessed by lower PUQE scores compared to placebo. Moreover, more women supplemented with pyridoxine-doxylamine (48.9%) than placebo-treated (32.8%) asked to continue their treatment at the end of the 15-day trial. The American and Canadian Colleges of Obstetrics and Gynecology have recommended the use of vitamin B6 (pyridoxine hydrochloride, 10 mg) and doxylamine succinate (10 mg) as first-line therapy for NVP (73).
Premenstrual syndrome
Premenstrual syndrome (PMS) refers to a cluster of symptoms, including but not limited to fatigue, irritability, moodiness/depression, fluid retention, and breast tenderness, that begin sometime after ovulation (mid-cycle) and subside with the onset of menstruation (the monthly period). A systematic review and meta-analysis of nine randomized, placebo-controlled trials suggested that supplemental vitamin B6, up to 100 mg/day, may be of value to treat PMS, including mood symptoms; however, only limited conclusions could be drawn because most of the studies were of poor quality (79). Another more recent review of 13 randomized controlled studies also emphasized the need for conclusive evidence before recommendations can be made (80).
Depression
The importance of PLP-dependent enzymes in the synthesis of several neurotransmitters (see Nervous system function) has led researchers to consider whether vitamin B6 deficiency may contribute to the onset of depressive symptoms (see Disease Prevention). There is limited evidence suggesting that supplemental vitamin B6 may have therapeutic efficacy in the management of depression. In a randomized, placebo-controlled trial conducted in 225 elderly patients hospitalized for acute illness, a six-month intervention with daily multivitamin/mineral supplements improved nutritional B-vitamin status and decreased the number and severity of depressive symptoms when compared to placebo (81). In addition, while supplement intake effectively reduced plasma homocysteine levels compared to placebo, the effect of supplementation on depressive symptoms at the end of the trial was greater in treated subjects in the lowest versus highest quartile of homocysteine levels (≤10 micromoles/liter vs. ≥16.1 micromoles/liter) (82). Yet, the etiology of late-onset depression is unclear and evidence is currently lacking to suggest whether supplemental B vitamins (including vitamin B6) could relieve depressive symptoms.
Carpal tunnel syndrome
Carpal tunnel syndrome (CTS) causes numbness, pain, and weakness of the hand and fingers due to compression of the median nerve at the wrist. It may result from repetitive stress injury of the wrist or from soft-tissue swelling, which sometimes occurs with pregnancy or hypothyroidism. Early studies by the same investigator suggested that supplementation with 100-200 mg/day of vitamin B6 for several months might improve CTS symptoms in individuals with low vitamin B6 status (83, 84). In addition, a cross-sectional study in 137 men not taking vitamin supplements found that decreased blood levels of PLP were associated with increased pain, tingling, and nocturnal awakening—all symptoms of CTS (85). However, studies using electrophysiological measurements of median nerve conduction have largely failed to find an association between vitamin B6 deficiency and CTS (86). While a few studies have noted some symptomatic relief with vitamin B6 supplementation, double-blind, placebo-controlled trials have not generally found vitamin B6 to be effective in treating CTS (86). Yet, despite its equivocal effectiveness, vitamin B6 supplementation is sometimes used in complementary therapy in an attempt to avoid hand surgery. Patients taking high doses of vitamin B6 should be advised by a physician and monitored for vitamin B6-related toxicity symptoms (see Toxicity) (87).
Sources
Food sources
The analysis of data collected in the US NHANES 2003-2004 has indicated that vitamin B6 intakes from food only averaged about 1.9 mg/day (88). Yet, despite values well above the current RDA, total vitamin B6 intakes (combining food and supplements) below 2 mg/day appear to be associated with relatively high proportions of low vitamin B6 status in all age groups (see Supplements). Many plant foods contain a unique form of vitamin B6 called pyridoxine glucoside; this form of vitamin B6 appears to be only about half as bioavailable as vitamin B6 from other food sources or supplements (7). Vitamin B6 in a mixed diet has been found to be approximately 75% bioavailable (14). In most cases, including foods in the diet that are rich in vitamin B6 should supply enough to meet the current RDA. However, those who follow a very restricted vegetarian diet might need to increase their vitamin B6 intake by eating foods fortified with vitamin B6 or by taking a supplement. Some foods that are relatively rich in vitamin B6 and their vitamin B6 content in milligrams (mg) are listed in Table 2. For more information on the nutrient content of specific foods, search USDA's FoodData Central.
Supplements
Vitamin B6 is available as pyridoxine hydrochloride in multivitamin, vitamin B-complex, and vitamin B6 supplements (89). In NHANES 2003-2004, low vitamin B6 status (plasma PLP level <20 nanomoles/liter) was reported in 24% of non users of supplements and 11% of supplement users. Moreover, total vitamin B6 intakes (from food and supplements) lower than 2 mg/day were associated with high proportions of low plasma PLP levels: 16% in men aged 13-54 years, 24% in menstruating women, and 26% in individuals aged 65 years and older. Finally, the prevalence of low PLP levels was found to be greater in individuals consuming less than 2 mg/day of vitamin B6 compared to higher intakes. For example, only 14% of men and women aged 65 and older had low PLP values with total vitamin B6 intakes of 2-2.9 mg/day compared to 26% in those consuming less than 2 mg/day of vitamin B6 (88).
Safety
Toxicity
Because adverse effects have only been documented from vitamin B6 supplements and never from food sources, safety concerning only the supplemental form of vitamin B6 (pyridoxine) is discussed. Although vitamin B6 is a water-soluble vitamin and is excreted in the urine, long-term supplementation with very high doses of pyridoxine may result in painful neurological symptoms known as sensory neuropathy. Symptoms include pain and numbness of the extremities and in severe cases, difficulty walking. Sensory neuropathy typically develops at doses of pyridoxine in excess of 1,000 mg per day. However, there have been a few case reports of individuals who developed sensory neuropathies at doses of less than 500 mg daily over a period of months. Yet, none of the studies in which an objective neurological examination was performed reported evidence of sensory nerve damage at intakes below 200 mg pyridoxine daily (90). To prevent sensory neuropathy in virtually all individuals, the Food and Nutrition Board of the Institute of Medicine set the tolerable upper intake level (UL) for pyridoxine at 100 mg/day for adults (Table 3) (14). Because placebo-controlled studies have generally failed to show therapeutic benefits of high doses of pyridoxine, there is little reason to exceed the UL of 100 mg/day.
Drug interactions
Certain medications interfere with the metabolism of vitamin B6; therefore, some individuals may be vulnerable to a vitamin B6 deficiency if supplemental vitamin B6 is not taken. In the NHANES 2003-2004 analysis, significantly more current and past users of oral contraceptives (OCs) among menstruating women had low plasma PLP levels compared to women who have never used OCs, suggesting that the estrogen content of OCs may interfere with vitamin B6 metabolism (see Side effects of oral contraceptives) (88). Anti-tuberculosis medications (e.g., isoniazid and cycloserine), the metal chelator penicillamine, and anti-parkinsonian drugs like L-Dopa can all form complexes with vitamin B6 and limit its bioavailability, thus creating a functional deficiency. PLP bioavailability may also be reduced by methylxanthines, such as theophylline used to treat certain respiratory conditions (7). The long-term use of nonsteroidal anti-inflammatory drugs (NSAIDs; e.g. celecoxib and naproxen) may also impair vitamin B6 metabolism (91). Conversely, high doses of vitamin B6 have been found to decrease the efficacy of two anticonvulsants, phenobarbital and phenytoin, and of L-Dopa (6, 90).
Side effects of oral contraceptives
Because vitamin B6 is required for the metabolism of the amino acid tryptophan, the tryptophan load test (an assay of tryptophan metabolites after an oral dose of tryptophan) has been used as a functional assessment of vitamin B6 status. Abnormal tryptophan load tests in women taking high-dose oral contraceptives (OCs) in the 1960s and 1970s suggested that these women were vitamin B6 deficient, which led to the prescription of high doses of vitamin B6 (100-150 mg/day) to women taking OCs. However, most other indices of vitamin B6 status were normal in women on high-dose OCs, and the estrogen content of OCs appeared to be more likely responsible for the abnormality in tryptophan metabolism (88). Yet, more recently, the use of lower dose formulations has also been associated with vitamin B6 inadequacy (88, 92). Although it is not known whether OCs actually impair vitamin B6 metabolism or merely affect the tissue distribution of PLP, the use of OCs may place women at higher risk of vitamin B6 deficiency when they discontinue OCs and become pregnant (93). Whether OC users are at higher risk of cardiovascular disease despite normal homocysteine levels also needs to be determined. Finally, although high doses of vitamin B6 (pyridoxine) have demonstrated no benefit in preventing the risk of side effects from OCs (94), the use of vitamin B6 supplements may be warranted in current and past OC users.
Linus Pauling Institute Recommendation
The Linus Pauling Institute supports the RDA for vitamin B6. LPI recommends that all adults take a daily multivitamin/mineral supplement, which usually contains at least 2 mg of vitamin B6. This amount is slightly above the RDA but still 50 times lower than the tolerable upper intake level (UL) set by the Food and Nutrition Board (see Safety).
Older adults (>50 years)
Early metabolic studies have indicated that the requirement for vitamin B6 in older adults is approximately 2 mg daily (95). Yet, the analysis of the US population survey (NHANES) 2003-2004 showed that adequate vitamin B6 status and low homocysteine levels were associated with total vitamin B6 intakes equal to and above 3 mg/day in people aged 65 years and older (88). The Linus Pauling Institute recommends that older adults take a multivitamin/mineral supplement, which provides at least 2.0 mg of vitamin B6 daily.
Authors and Reviewers
Originally written in 2000 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in February 2002 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in November 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in May 2014 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in June 2014 by:
Jesse F. Gregory, Ph.D.
Professor, Food Science and Human Nutrition
University of Florida
The 2014 update of this article was underwritten, in part, by a grant from Bayer Consumer Care AG, Basel, Switzerland.
Copyright 2000-2024 Linus Pauling Institute
References
1. Dakshinamurti S, Dakshinamurti K. Vitamin B6. In: Zempleni J, Rucker RB, McCormick DB, Suttie JW, eds. Handbook of Vitamins. 4th ed. New York: CRC Press (Taylor & Fracis Group); 2007:315-359.
2. Galluzzi L, Vacchelli E, Michels J, et al. Effects of vitamin B6 metabolism on oncogenesis, tumor progression and therapeutic responses. Oncogene. 2013;32(42):4995-5004. (PubMed)
3. McCormick DB. Vitamin B6. In: Bowman BA, Russell RM, eds. Present Knowledge in Nutrition. Vol. I. Washington, D.C.: International Life Sciences Institute; 2006:269-277.
4. Da Silva VR, Russell KA, Gregory JF 3rd. Vitamin B6. In: Erdman JW Jr., Macdonald IA, Zeisel SH. Present Knowldege in Nutrition. 10th ed: Wiley-Blackwell; 2012:307-320.
5. Eliot AC, Kirsch JF. Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu Rev Biochem. 2004;73:383-415. (PubMed)
6. Leklem JE. Vitamin B-6. In: Shils M, Olson JA, Shike M, Ross AC, eds. Modern Nutrition in Health and Disease. 9th ed. Baltimore: Williams & Wilkins; 1999:413-422.
7. Clayton PT. B6-responsive disorders: a model of vitamin dependency. J Inherit Metab Dis. 2006;29(2-3):317-326. (PubMed)
8. Schnackerz KD, Benesch RE, Kwong S, Benesch R, Helmreich EJ. Specific receptor sites for pyridoxal 5'-phosphate and pyridoxal 5'-deoxymethylenephosphonate at the α and β NH2-terminal regions of hemoglobin. J Biol Chem. 1983;258(2):872-875. (PubMed)
9. Rios-Avila L, Nijhout HF, Reed MC, Sitren HS, Gregory JF, 3rd. A mathematical model of tryptophan metabolism via the kynurenine pathway provides insights into the effects of vitamin B-6 deficiency, tryptophan loading, and induction of tryptophan 2,3-dioxygenase on tryptophan metabolites. J Nutr. 2013;143(9):1509-1519. (PubMed)
10. Oxenkrug G. Insulin resistance and dysregulation of tryptophan-kynurenine and kynurenine-nicotinamide adenine dinucleotide metabolic pathways. Mol Neurobiol. 2013;48(2):294-301. (PubMed)
11. Huq MD, Tsai NP, Lin YP, Higgins L, Wei LN. Vitamin B6 conjugation to nuclear corepressor RIP140 and its role in gene regulation. Nat Chem Biol. 2007;3(3):161-165. (PubMed)
12. Leklem JE. Vitamin B6. In: Machlin L, ed. Handbook of Vitamins. New York: Marcel Decker Inc; 1991:341-378.
13. Hansen CM, Shultz TD, Kwak HK, Memon HS, Leklem JE. Assessment of vitamin B-6 status in young women consuming a controlled diet containing four levels of vitamin B-6 provides an estimated average requirement and recommended dietary allowance. J Nutr. 2001;131(6):1777-1786. (PubMed)
14. Food and Nutrition Board, Institute of Medicine. Vitamin B6. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B6, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington D.C.: National Academies Press; 1998:150-195. (National Academies Press)
15. Paul L, Ueland PM, Selhub J. Mechanistic perspective on the relationship between pyridoxal 5'-phosphate and inflammation. Nutr Rev. 2013;71(4):239-244. (PubMed)
16. Meydani SN, Ribaya-Mercado JD, Russell RM, Sahyoun N, Morrow FD, Gershoff SN. Vitamin B-6 deficiency impairs interleukin 2 production and lymphocyte proliferation in elderly adults. Am J Clin Nutr. 1991;53(5):1275-1280. (PubMed)
17. Talbott MC, Miller LT, Kerkvliet NI. Pyridoxine supplementation: effect on lymphocyte responses in elderly persons. Am J Clin Nutr. 1987;46(4):659-664. (PubMed)
18. Rimm EB, Willett WC, Hu FB, et al. Folate and vitamin B6 from diet and supplements in relation to risk of coronary heart disease among women. JAMA. 1998;279(5):359-364. (PubMed)
19. Ishihara J, Iso H, Inoue M, et al. Intake of folate, vitamin B6 and vitamin B12 and the risk of CHD: the Japan Public Health Center-Based Prospective Study Cohort I. J Am Coll Nutr. 2008;27(1):127-136. (PubMed)
20. Folsom AR, Nieto FJ, McGovern PG, et al. Prospective study of coronary heart disease incidence in relation to fasting total homocysteine, related genetic polymorphisms, and B vitamins: the Atherosclerosis Risk in Communities (ARIC) study. Circulation. 1998;98(3):204-210. (PubMed)
21. Robinson K, Arheart K, Refsum H, et al. Low circulating folate and vitamin B6 concentrations: risk factors for stroke, peripheral vascular disease, and coronary artery disease. European COMAC Group.Circulation. 1998;97(5):437-443. (PubMed)
22. Robinson K, Mayer EL, Miller DP, et al. Hyperhomocysteinemia and low pyridoxal phosphate. Common and independent reversible risk factors for coronary artery disease. Circulation. 1995;92(10):2825-2830. (PubMed)
23. Lin PT, Cheng CH, Liaw YP, Lee BJ, Lee TW, Huang YC. Low pyridoxal 5'-phosphate is associated with increased risk of coronary artery disease. Nutrition. 2006;22(11-12):1146-1151. (PubMed)
24. Page JH, Ma J, Chiuve SE, et al. Plasma vitamin B(6) and risk of myocardial infarction in women. Circulation. 2009;120(8):649-655. (PubMed)
25. Gerhard GT, Duell PB. Homocysteine and atherosclerosis. Curr Opin Lipidol. 1999;10(5):417-428. (PubMed)
26. Ubbink JB, Vermaak WJ, van der Merwe A, Becker PJ, Delport R, Potgieter HC. Vitamin requirements for the treatment of hyperhomocysteinemia in humans. J Nutr. 1994;124(10):1927-1933. (PubMed)
27. Lamers Y, Coats B, Ralat M, Quinlivan EP, Stacpoole PW, Gregory JF, 3rd. Moderate vitamin B-6 restriction does not alter postprandial methionine cycle rates of remethylation, transmethylation, and total transsulfuration but increases the fractional synthesis rate of cystathionine in healthy young men and women. J Nutr. 2011;141(5):835-842. (PubMed)
28. Huang T, Chen Y, Yang B, Yang J, Wahlqvist ML, Li D. Meta-analysis of B vitamin supplementation on plasma homocysteine, cardiovascular and all-cause mortality. Clin Nutr. 2012;31(4):448-454. (PubMed)
29. Bosy-Westphal A, Holzapfel A, Czech N, Muller MJ. Plasma folate but not vitamin B(12) or homocysteine concentrations are reduced after short-term vitamin B(6) supplementation. Ann Nutr Metab. 2001;45(6):255-258. (PubMed)
30. Lee BJ, Huang MC, Chung LJ, et al. Folic acid and vitamin B12 are more effective than vitamin B6 in lowering fasting plasma homocysteine concentration in patients with coronary artery disease. Eur J Clin Nutr. 2004;58(3):481-487. (PubMed)
31. Bostom AG, Carpenter MA, Kusek JW, et al. Homocysteine-lowering and cardiovascular disease outcomes in kidney transplant recipients: primary results from the Folic Acid for Vascular Outcome Reduction in Transplantation trial. Circulation. 2011;123(16):1763-1770. (PubMed)
32. Qin X, Huo Y, Xie D, Hou F, Xu X, Wang X. Homocysteine-lowering therapy with folic acid is effective in cardiovascular disease prevention in patients with kidney disease: a meta-analysis of randomized controlled trials. Clin Nutr. 2013;32(5):722-727. (PubMed)
33. Clarke R, Halsey J, Bennett D, Lewington S. Homocysteine and vascular disease: review of published results of the homocysteine-lowering trials. J Inherit Metab Dis. 2011;34(1):83-91. (PubMed)
34. Marti-Carvajal AJ, Sola I, Lathyris D, Karakitsiou DE, Simancas-Racines D. Homocysteine-lowering interventions for preventing cardiovascular events. Cochrane Database Syst Rev. 2013;1:CD006612. (PubMed)
35. Zhang C, Chi FL, Xie TH, Zhou YH. Effect of B-vitamin supplementation on stroke: a meta-analysis of randomized controlled trials. PLoS One. 2013;8(11):e81577. (PubMed)
36. Potter K, Hankey GJ, Green DJ, Eikelboom J, Jamrozik K, Arnolda LF. The effect of long-term homocysteine-lowering on carotid intima-media thickness and flow-mediated vasodilation in stroke patients: a randomized controlled trial and meta-analysis. BMC Cardiovasc Disord. 2008;8:24. (PubMed)
37. Loland KH, Bleie O, Blix AJ, et al. Effect of homocysteine-lowering B vitamin treatment on angiographic progression of coronary artery disease: a Western Norway B Vitamin Intervention Trial (WENBIT) substudy. Am J Cardiol. 2010;105(11):1577-1584. (PubMed)
38. Wang X, Qin X, Demirtas H, et al. Efficacy of folic acid supplementation in stroke prevention: a meta-analysis. Lancet. 2007;369(9576):1876-1882. (PubMed)
39. Vitatops Trial Study Group. B vitamins in patients with recent transient ischaemic attack or stroke in the VITAmins TO Prevent Stroke (VITATOPS) trial: a randomised, double-blind, parallel, placebo-controlled trial. Lancet Neurol. 2010;9(9):855-865. (PubMed)
40. Hankey GJ, Eikelboom JW, Yi Q, et al. Antiplatelet therapy and the effects of B vitamins in patients with previous stroke or transient ischaemic attack: a post-hoc subanalysis of VITATOPS, a randomised, placebo-controlled trial. Lancet Neurol. 2012;11(6):512-520. (PubMed)
41. Morris MS, Sakakeeny L, Jacques PF, Picciano MF, Selhub J. Vitamin B-6 intake is inversely related to, and the requirement is affected by, inflammation status. J Nutr. 2010;140(1):103-110. (PubMed)
42. Sakakeeny L, Roubenoff R, Obin M, et al. Plasma pyridoxal-5-phosphate is inversely associated with systemic markers of inflammation in a population of US adults. J Nutr. 2012;142(7):1280-1285. (PubMed)
43. Shen J, Lai CQ, Mattei J, Ordovas JM, Tucker KL. Association of vitamin B-6 status with inflammation, oxidative stress, and chronic inflammatory conditions: the Boston Puerto Rican Health Study. Am J Clin Nutr. 2010;91(2):337-342. (PubMed)
44. Schroecksnadel K, Grammer TB, Boehm BO, Marz W, Fuchs D. Total homocysteine in patients with angiographic coronary artery disease correlates with inflammation markers. Thromb Haemost. 2010;103(5):926-935. (PubMed)
45. Hartman J, Frishman WH. Inflammation and atherosclerosis: a review of the role of interleukin-6 in the development of atherosclerosis and the potential for targeted drug therapy. Cardiol Rev. 2014;22(3):147-151. (PubMed)
46. Friso S, Girelli D, Martinelli N, et al. Low plasma vitamin B-6 concentrations and modulation of coronary artery disease risk. Am J Clin Nutr. 2004;79(6):992-998. (PubMed)
47. Ulvik A, Midttun O, Pedersen ER, Eussen SJ, Nygard O, Ueland PM. Evidence for increased catabolism of vitamin B-6 during systemic inflammation. Am J Clin Nutr. 2014;100(1):250-255. [Epub ahead of print] (PubMed)
48. Bleie O, Semb AG, Grundt H, et al. Homocysteine-lowering therapy does not affect inflammatory markers of atherosclerosis in patients with stable coronary artery disease. J Intern Med. 2007;262(2):244-253. (PubMed)
49. Potter K, Lenzo N, Eikelboom JW, Arnolda LF, Beer C, Hankey GJ. Effect of long-term homocysteine reduction with B vitamins on arterial wall inflammation assessed by fluorodeoxyglucose positron emission tomography: a randomised double-blind, placebo-controlled trial. Cerebrovasc Dis. 2009;27(3):259-265. (PubMed)
50. Selhub J, Bagley LC, Miller J, Rosenberg IH. B vitamins, homocysteine, and neurocognitive function in the elderly. Am J Clin Nutr. 2000;71(2):614S-620S. (PubMed)
51. Pawelec G, Goldeck D, Derhovanessian E. Inflammation, ageing and chronic disease. Curr Opin Immunol. 2014;29C:23-28. (PubMed)
52. Ford AH, Almeida OP. Effect of homocysteine lowering treatment on cognitive function: a systematic review and meta-analysis of randomized controlled trials. J Alzheimers Dis. 2012;29(1):133-149. (PubMed)
53. Hankey GJ, Ford AH, Yi Q, et al. Effect of B vitamins and lowering homocysteine on cognitive impairment in patients with previous stroke or transient ischemic attack: a prespecified secondary analysis of a randomized, placebo-controlled trial and meta-analysis. Stroke. 2013;44(8):2232-2239. (PubMed)
54. Douaud G, Refsum H, de Jager CA, et al. Preventing Alzheimer's disease-related gray matter atrophy by B-vitamin treatment. Proc Natl Acad Sci U S A. 2013;110(23):9523-9528. (PubMed)
55. Hackett ML, Yapa C, Parag V, Anderson CS. Frequency of depression after stroke: a systematic review of observational studies. Stroke. 2005;36(6):1330-1340. (PubMed)
56. Lenze EJ, Munin MC, Skidmore ER, et al. Onset of depression in elderly persons after hip fracture: implications for prevention and early intervention of late-life depression. J Am Geriatr Soc. 2007;55(1):81-86. (PubMed)
57. Merete C, Falcon LM, Tucker KL. Vitamin B6 is associated with depressive symptomatology in Massachusetts elders. J Am Coll Nutr. 2008;27(3):421-427. (PubMed)
58. Pan WH, Chang YP, Yeh WT, et al. Co-occurrence of anemia, marginal vitamin B6, and folate status and depressive symptoms in older adults. J Geriatr Psychiatry Neurol. 2012;25(3):170-178. (PubMed)
59. Skarupski KA, Tangney C, Li H, Ouyang B, Evans DA, Morris MC. Longitudinal association of vitamin B-6, folate, and vitamin B-12 with depressive symptoms among older adults over time. Am J Clin Nutr. 2010;92(2):330-335. (PubMed)
60. Almeida OP, Marsh K, Alfonso H, Flicker L, Davis TM, Hankey GJ. B-vitamins reduce the long-term risk of depression after stroke: The VITATOPS-DEP trial. Ann Neurol. 2010;68(4):503-510. (PubMed)
61. Larsson SC, Orsini N, Wolk A. Vitamin B6 and risk of colorectal cancer: a meta-analysis of prospective studies. JAMA. 2010;303(11):1077-1083. (PubMed)
62. Wu W, Kang S, Zhang D. Association of vitamin B6, vitamin B12 and methionine with risk of breast cancer: a dose-response meta-analysis. Br J Cancer. 2013;109(7):1926-1944. (PubMed)
63. Xiao Q, Freedman ND, Ren J, Hollenbeck AR, Abnet CC, Park Y. Intakes of folate, methionine, vitamin B6, and vitamin B12 with risk of esophageal and gastric cancer in a large cohort study. Br J Cancer. 2014;110(5):1328-1333. (PubMed)
64. Zhang XH, Ma J, Smith-Warner SA, Lee JE, Giovannucci E. Vitamin B6 and colorectal cancer: current evidence and future directions. World J Gastroenterol. 2013;19(7):1005-1010. (PubMed)
65. Song Y, Manson JE, Lee IM, et al. Effect of combined folic acid, vitamin B(6), and vitamin B(12) on colorectal adenoma. J Natl Cancer Inst. 2012;104(20):1562-1575. (PubMed)
66. Curhan GC, Willett WC, Speizer FE, Stampfer MJ. Intake of vitamins B6 and C and the risk of kidney stones in women. J Am Soc Nephrol. 1999;10(4):840-845. (PubMed)
67. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol. 2004;15(12):3225-3232. (PubMed)
68. Chetyrkin SV, Kim D, Belmont JM, Scheinman JI, Hudson BG, Voziyan PA. Pyridoxamine lowers kidney crystals in experimental hyperoxaluria: a potential therapy for primary hyperoxaluria. Kidney Int. 2005;67(1):53-60. (PubMed)
69. Scheinman JI, Voziyan PA, Belmont JM, Chetyrkin SV, Kim D, Hudson BG. Pyridoxamine lowers oxalate excretion and kidney crystals in experimental hyperoxaluria: a potential therapy for primary hyperoxaluria. Urol Res. 2005;33(5):368-371. (PubMed)
70. Pearl PL, Gospe SM, Jr. Pyridoxine or pyridoxal-5'-phosphate for neonatal epilepsy: The distinction just got murkier. Neurology. 2014;82(16):1392-1394. (PubMed)
71. Stockler S, Plecko B, Gospe SM, Jr., et al. Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up. Mol Genet Metab. 2011;104(1-2):48-60. (PubMed)
72. Picker JD, Levy HL. Homocystinuria caused by cystathionine β-synthase deficiency. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews®. Seattle, Washington: University of Washington, Seattle 1993-2014. (PubMed)
73. Maltepe C, Koren G. The management of nausea and vomiting of pregnancy and hyperemesis gravidarum--a 2013 update. J Popul Ther Clin Pharmacol. 2013;20(2):e184-192. (PubMed)
74. Magee LA, Mazzotta P, Koren G. Evidence-based view of safety and effectiveness of pharmacologic therapy for nausea and vomiting of pregnancy (NVP). Am J Obstet Gynecol. 2002;186(5 Suppl Understanding):S256-261. (PubMed)
75. Sahakian V, Rouse D, Sipes S, Rose N, Niebyl J. Vitamin B6 is effective therapy for nausea and vomiting of pregnancy: a randomized, double-blind placebo-controlled study. Obstet Gynecol. 1991;78(1):33-36. (PubMed)
76. Vutyavanich T, Wongtra-ngan S, Ruangsri R. Pyridoxine for nausea and vomiting of pregnancy: a randomized, double-blind, placebo-controlled trial. Am J Obstet Gynecol. 1995;173(3 Pt 1):881-884. (PubMed)
77. Matthews A, Haas DM, O'Mathuna DP, Dowswell T, Doyle M. Interventions for nausea and vomiting in early pregnancy. Cochrane Database Syst Rev. 2014;3:CD007575. (PubMed)
78. Koren G, Clark S, Hankins GD, et al. Effectiveness of delayed-release doxylamine and pyridoxine for nausea and vomiting of pregnancy: a randomized placebo controlled trial. Am J Obstet Gynecol. 2010;203(6):571 e571-577. (PubMed)
79. Wyatt KM, Dimmock PW, Jones PW, Shaughn O'Brien PM. Efficacy of vitamin B-6 in the treatment of premenstrual syndrome: systematic review. BMJ. 1999;318(7195):1375-1381. (PubMed)
80. Whelan AM, Jurgens TM, Naylor H. Herbs, vitamins and minerals in the treatment of premenstrual syndrome: a systematic review. Can J Clin Pharmacol. 2009;16(3):e407-429. (PubMed)
81. Gariballa S, Forster S. Effects of dietary supplements on depressive symptoms in older patients: a randomised double-blind placebo-controlled trial. Clin Nutr. 2007;26(5):545-551. (PubMed)
82. Gariballa S. Testing homocysteine-induced neurotransmitter deficiency, and depression of mood hypothesis in clinical practice. Age Ageing. 2011;40(6):702-705. (PubMed)
83. Ellis J, Folkers K, Watanabe T, et al. Clinical results of a cross-over treatment with pyridoxine and placebo of the carpal tunnel syndrome. Am J Clin Nutr. 1979;32(10):2040-2046. (PubMed)
84. Ellis JM, Kishi T, Azuma J, Folkers K. Vitamin B6 deficiency in patients with a clinical syndrome including the carpal tunnel defect. Biochemical and clinical response to therapy with pyridoxine. Res Commun Chem Pathol Pharmacol. 1976;13(4):743-757. (PubMed)
85. Keniston RC, Nathan PA, Leklem JE, Lockwood RS. Vitamin B6, vitamin C, and carpal tunnel syndrome. A cross-sectional study of 441 adults. J Occup Environ Med. 1997;39(10):949-959. (PubMed)
86. Aufiero E, Stitik TP, Foye PM, Chen B. Pyridoxine hydrochloride treatment of carpal tunnel syndrome: a review. Nutr Rev. 2004;62(3):96-104. (PubMed)
87. Ryan-Harshman M, Aldoori W. Carpal tunnel syndrome and vitamin B6. Can Fam Physician. 2007;53(7):1161-1162. (PubMed)
88. Morris MS, Picciano MF, Jacques PF, Selhub J. Plasma pyridoxal 5'-phosphate in the US population: the National Health and Nutrition Examination Survey, 2003-2004. Am J Clin Nutr. 2008;87(5):1446-1454. (PubMed)
89. Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. Montvale: Medical Economics Company, Inc; 2001.
90. Bender DA. Non-nutritional uses of vitamin B6. Br J Nutr. 1999;81(1):7-20 (PubMed)
91. Chang HY, Tang FY, Chen DY, et al. Clinical use of cyclooxygenase inhibitors impairs vitamin B-6 metabolism. Am J Clin Nutr. 2013;98(6):1440-1449. (PubMed)
92. Lussana F, Zighetti ML, Bucciarelli P, Cugno M, Cattaneo M. Blood levels of homocysteine, folate, vitamin B6 and B12 in women using oral contraceptives compared to non-users. Thromb Res. 2003;112(1-2):37-41. (PubMed)
93. Wilson SM, Bivins BN, Russell KA, Bailey LB. Oral contraceptive use: impact on folate, vitamin B(6), and vitamin B(1)(2) status. Nutr Rev. 2011;69(10):572-583. (PubMed)
94. Villegas-Salas E, Ponce de Leon R, Juarez-Perez MA, Grubb GS. Effect of vitamin B6 on the side effects of a low-dose combined oral contraceptive. Contraception. 1997;55(4):245-248. (PubMed)
95. Ribaya-Mercado JD, Russell RM, Sahyoun N, Morrow FD, Gershoff SN. Vitamin B-6 requirements of elderly men and women. J Nutr. 1991;121(7):1062-1074. (PubMed)
Vitamin B12
Contents
Summary
- Vitamin B12 or cobalamin plays essential roles in folate metabolism and in the synthesis of the citric acid cycle intermediate, succinyl-CoA. (More information)
- Vitamin B12 deficiency is commonly associated with chronic stomach inflammation, which may contribute to an autoimmune vitamin B12 malabsorption syndrome called pernicious anemia and to a food-bound vitamin B12 malabsorption syndrome. Impairment of vitamin B12 absorption can cause megaloblastic anemia and neurologic disorders in deficient subjects. (More information)
- Normal function of the digestive system required for food-bound vitamin B12 absorption is commonly impaired in individuals over 60 years of age, placing them at risk for vitamin B12 deficiency. (More information)
- Vitamin B12 and folate are important for homocysteine metabolism. Elevated blood homocysteine concentration is a risk factor for cardiovascular disease. However, most intervention trials of B-vitamin supplementation have found homocysteine lowering does not reduce risk of heart attack. B-vitamin supplementation may slightly reduce risk of stroke, but these benefits may not be seen in patients with chronic kidney disease. (More information)
- The preservation of DNA integrity is dependent on folate and vitamin B12 availability. Poor vitamin B12 status has been linked to increased risk of breast cancer in some, but not all, observational studies. Additional studies are needed to clarify the relationship of vitamin B12 and breast cancer. (More information)
- Low maternal vitamin B12 status has been associated with an increased risk of neural tube defects (NTD), but it is not known whether vitamin B12 supplementation could help reduce the risk of NTD. (More information)
- Vitamin B12 is essential for the preservation of the myelin sheath around neurons and for the synthesis of neurotransmitters. While hyperhomocysteinemia may increase the risk of cognitive impairment and age-related neurodegenerative disease (e.g., Alzheimer’s disease), it is not clear whether vitamin B12 deficiency contributes to the risk of dementia in the elderly. Although B-vitamin supplementation lowers blood homocysteine levels in older subjects, the long-term benefit is not yet known. (More information)
- Both depression and osteoporosis have been linked to diminished vitamin B12 status and high homocysteine levels. (More information)
- Products of animal origin constitute the primary source of vitamin B12. Older individuals and vegans are advised to use vitamin B12 fortified foods and supplements to meet their needs. (More information)
- The long-term use of certain medications, such as inhibitors of stomach acid secretion and the anti-diabetes drug, metformin, can adversely affect vitamin B12 absorption. (More information)
Vitamin B12 has the largest and most complex chemical structure of all the vitamins. It is unique among vitamins in that it contains a metal ion, cobalt. For this reason cobalamin is the term used to refer to compounds having vitamin B12 activity. Methylcobalamin and 5-deoxyadenosylcobalamin are the forms of vitamin B12 used in the human body (1). The form of cobalamin used in most nutritional supplements and fortified foods, cyanocobalamin, is readily converted to 5-deoxyadenosylcobalamin and methylcobalamin in the body. In mammals, cobalamin is a cofactor for only two enzymes, methionine synthase and L-methylmalonyl-coenzyme A mutase (2).
Function
Cofactor for methionine synthase
Methylcobalamin is required for the function of the folate-dependent enzyme, methionine synthase. This enzyme is required for the synthesis of the amino acid, methionine, from homocysteine. Methionine in turn is required for the synthesis of S-adenosylmethionine, a methyl group donor used in many biological methylation reactions, including the methylation of a number of sites within DNA, RNA, and proteins (Figure 1) (3). Aberrant methylation of DNA and proteins, which causes alterations in chromatin structure and gene expression, are a common feature of cancer cells. Inadequate function of methionine synthase can lead to an accumulation of homocysteine, which has been associated with increased risk of cardiovascular disease and age-related neurodegenerative disease (e.g., Alzheimer’s disease).

Cofactor for L-methylmalonyl-coenzyme A mutase
5-Deoxyadenosylcobalamin is required by the enzyme that catalyzes the conversion of L-methylmalonyl-coenzyme A to succinyl-coenzyme A (succinyl-CoA), which then enters the citric acid cycle (Figure 2). Succinyl-CoA plays an important role in the production of energy from lipids and proteins and is also required for the synthesis of hemoglobin, the oxygen-carrying pigment in red blood cells (3).

Deficiency
In healthy adults, overt vitamin B12 deficiency is uncommon, mainly because total body stores can exceed 2,500 μg, daily turnover is slow, and dietary intake of only 2.4 μg/day is sufficient to maintain adequate vitamin B12 status (see RDA) (4). Among elderly individuals, vitamin B12 deficiency is more common, primarily because of impaired intestinal absorption that can result in marginal to severe vitamin B12 deficiency in this population. In certain regions of the world, marginal or subclinical vitamin B12 deficiency may be fairly common in other age groups as well, including children, premenopausal women, and pregnant women, primarily due to low intake of animal-source foods (reviewed in 5).
Causes of vitamin B12 deficiency
Absorption of vitamin B12 from food requires normal function of the stomach, pancreas, and small intestine. Stomach acid and enzymes free vitamin B12 from food, allowing it to bind to R-protein (formerly known as transcobalamin I and now called haptocorrin), found in saliva and gastric fluids. In the alkaline environment of the small intestine, R-proteins are degraded by pancreatic enzymes, freeing vitamin B12 to bind to intrinsic factor (IF), a protein secreted by specialized cells in the stomach (parietal cells). Receptors on the surface of the terminal ileum (the distal portion of the small intestine) take up the IF-B12 complex only in the presence of calcium, which is supplied by the pancreas (6). Vitamin B12 can also be absorbed by passive diffusion, but this process is very inefficient — only about 1% to 2% of the oral vitamin B12 dose is absorbed passively (2). The prevalent causes of vitamin B12 deficiency are (1) an autoimmune condition known as pernicious anemia, and (2) a disorder called food-bound vitamin B12 malabsorption. Both conditions have been associated with a chronic inflammatory disease of the stomach known as atrophic gastritis.
Atrophic gastritis
Atrophic gastritis is thought to affect 10%-30% of people over 60 years of age (7). The condition is frequently associated with the presence of autoantibodies directed towards stomach cells (see Pernicious anemia) and/or infection by the bacteria, Helicobacter pylori (H. pylori) (8). H. pylori infection induces chronic inflammation of the stomach, which may progress to peptic ulcer disease, atrophic gastritis, and/or gastric cancer in some individuals. Diminished gastric function in individuals with atrophic gastritis can result in bacterial overgrowth in the small intestine and cause food-bound vitamin B12 malabsorption. Vitamin B12 levels in serum, plasma, and gastric fluids are significantly decreased in individuals with H. pylori infection, and eradication of the bacteria has been shown to significantly improve vitamin B12 serum concentrations (9).
Pernicious anemia
Pernicious anemia has been estimated to affect approximately 2% to 3% of individuals over 65 years of age (5). Although anemia is often a symptom, the condition is actually the end stage of an autoimmune inflammation of the stomach known as autoimmune atrophic gastritis, resulting in destruction of stomach cells by one's own antibodies (autoantibodies). Progressive destruction of the cells that line the stomach causes decreased secretion of acid and enzymes required to release food-bound vitamin B12 (5). Antibodies to intrinsic factor (IF) bind to IF and prevent formation of the IF-B12 complex, further inhibiting vitamin B12 absorption. Absorption of vitamin B12 is inhibited from both dietary sources and from the bile (enterohepatic circulation) and thus depletion of vitamin B12 body stores can occur relatively quickly. About 20% of the relatives of pernicious anemia patients also have the condition, suggesting a genetic predisposition. It is also thought that H. pylori infection could be involved in initiating the autoimmune response in a subset of individuals (10). Further, co-occurrence of autoimmune atrophic gastritis with other autoimmune conditions, especially autoimmune thyroiditis and type 1 diabetes mellitus, has been reported (11, 12).
Treatment of pernicious anemia generally requires intramuscular injections of vitamin B12 to bypass intestinal absorption. High-dose oral supplementation may be another treatment option, because consuming 1,000 μg (1 mg) per day of vitamin B12 orally should result in the absorption of about 10 μg/day (1% of dose) by passive diffusion. However, the effect of oral vitamin B12 varies among patients (13), and the available randomized controlled trials comparing the two treatment regimens are considered to be of low quality (14). Large-scale, well-designed trials with proper randomization and blinding are needed to determine whether high-dose oral supplementation is as effective as intramuscular injection for increasing vitamin B12 status and alleviating clinical symptoms of vitamin B12 deficiency.
Food-bound vitamin B12 malabsorption
Food-bound vitamin B12 malabsorption is defined as an impaired ability to absorb food- or protein-bound vitamin B12; individuals with this condition can fully absorb the free form (15). While the condition is the major cause of poor vitamin B12 status in the elderly population, it is usually associated with atrophic gastritis, a chronic inflammation of the lining of the stomach that ultimately results in the loss of glands in the stomach (atrophy) and decreased stomach acid production (see Atrophic gastritis). Because stomach acid is required for the release of vitamin B12 from the proteins in food, vitamin B12 absorption is diminished. Decreased stomach acid production also provides an environment conducive to the overgrowth of anaerobic bacteria in the stomach, which further interferes with vitamin B12 absorption (3). Because vitamin B12 in supplements is not bound to protein, and because intrinsic factor (IF) is still available, the absorption of supplemental vitamin B12 is not reduced as it is in pernicious anemia. Thus, individuals with food-bound vitamin B12 malabsorption do not have an increased requirement for vitamin B12; they simply need it in the crystalline form found in fortified foods and dietary supplements.
Other causes of vitamin B12 deficiency
Other causes of vitamin B12 deficiency include surgical resection of the stomach or portions of the small intestine where receptors for the IF-B12 complex are located. Conditions affecting the small intestine, such as malabsorption syndromes (celiac disease and tropical sprue), may also result in vitamin B12 deficiency. Because the pancreas provides critical enzymes, as well as calcium required for vitamin B12 absorption, pancreatic insufficiency may contribute to vitamin B12 deficiency. Since vitamin B12 is found predominantly in foods of animal origin, strict vegetarian and vegan diets can result in vitamin B12 deficiency (2, 16). Moreover, alcoholics may experience reduced intestinal absorption of vitamin B12 (2), and individuals with acquired immunodeficiency syndrome (AIDS) appear to be at increased risk of deficiency, possibly related to a failure of the IF-B12 receptor to take up the IF-B12 complex (3). Further, long-term use of acid-reducing drugs and the anti-diabetes drug metformin and repeated exposure to nitrous oxide have been implicated in vitamin B12 deficiency (see Drug interactions).
Inherited disorders of vitamin B12 absorption
Rare cases of inborn errors of vitamin B12 metabolism have been reported in the literature (reviewed in 6). Imerslund-Gräsbeck syndrome is an inherited vitamin B12 malabsorption syndrome that causes megaloblastic anemia and neurologic disorders of variable severity in affected subjects. Similar clinical symptoms are found in individuals with hereditary IF deficiency (also called congenital pernicious anemia) in whom the lack of IF results in the defective absorption of vitamin B12. Additionally, mutations affecting vitamin B12 transport and intracellular metabolism have been identified (17).
Symptoms of vitamin B12 deficiency
Vitamin B12 deficiency results in impairment of the activities of vitamin B12-requiring enzymes. Impaired activity of methionine synthase results in elevated blood concentrations of homocysteine, while impaired activity of L-methylmalonyl-CoA mutase results in increased concentrations of a metabolite of methylmalonyl-CoA called methylmalonic acid (MMA) in blood and urine. While individuals with mild vitamin B12 deficiency may not experience symptoms, blood homocysteine and/or MMA may be elevated (18). While elevated MMA blood concentration is considered the specific indicator of deficiency (19), other clinical assays are often used as biomarkers of vitamin B12 status — sometimes in combination. These include blood concentrations of total cobalamin, holo-transcobalamin (vitamin B12 bound to transcobalamin, one of the two carrier proteins in blood; called 'active vitamin B12'), and homocysteine (5). Yet, there is no ‘gold standard’ blood test, and the manifestation of clinical symptoms is important in the diagnosis of vitamin B12 deficiency (13).
Megaloblastic anemia
Diminished activity of methionine synthase in vitamin B12 deficiency inhibits the regeneration of tetrahydrofolate (THF) and traps folate in a form that is not usable by the body (Figure 3), resulting in symptoms of folate deficiency even in the presence of adequate folate. Thus, in both folate and vitamin B12 deficiencies, folate is unavailable to participate in DNA synthesis. This impairment of DNA synthesis affects the rapidly dividing cells of the bone marrow earlier than other cells, resulting in the production of large, immature, hemoglobin-poor red blood cells (macrocytosis), as well as effects on white blood cells, including abnormal (hypersegmented) neutrophils and reduced overall cell counts (pancytopenia). The resulting anemia is known as megaloblastic anemia and is the symptom for which the disease, pernicious anemia, was named (3). Supplementation with folic acid will provide enough usable folate to restore normal red blood cell formation. However, if vitamin B12 deficiency is the cause, it will persist despite the resolution of the anemia. Thus, megaloblastic anemia should not be treated with folic acid until the underlying cause has been determined (20).

Neurologic symptoms
The neurologic symptoms of vitamin B12 deficiency are myriad and include numbness and tingling of the hands and, more commonly, the feet (peripheral neuropathy); difficulty walking (gait ataxia); memory loss and other cognitive impairments; disorientation; alterations in mood, including depression and anxiety; and dementia that can resemble Alzheimer’s disease (13). Although the progression of neurologic complications is generally gradual, such symptoms may not be reversed with treatment of vitamin B12 deficiency, especially if they have been present for a long time. Neurologic complications are not always associated with megaloblastic anemia and are the only clinical symptom of vitamin B12 deficiency in about 25% of cases (21). Although vitamin B12 deficiency is known to damage the myelin sheath covering cranial, spinal, and peripheral nerves, the biochemical processes leading to neurologic damage in vitamin B12 deficiency are not yet fully understood (22).
Gastrointestinal symptoms
Tongue soreness, appetite loss, and constipation have also been associated with vitamin B12 deficiency. The origins of these symptoms are unclear, but they may be related to the stomach inflammation underlying some cases of vitamin B12 deficiency and to the progressive destruction of the lining of the stomach (21).
The Recommended Dietary Allowance (RDA)
The RDA for vitamin B12 was last revised by the Food and Nutrition Board (FNB) of the US Institute of Medicine (now the National Academy of Medicine) in 1998 (Table 1). Because of the increased risk of food-bound vitamin B12 malabsorption in older adults, the FNB recommended that adults over 50 years of age get most of the RDA from fortified food or vitamin B12-containing supplements (21).
| Life Stage | Age | Males (μg/day) | Females (μg/day) |
|---|---|---|---|
| Infants | 0-6 months | 0.4 (AI) | 0.4 (AI) |
| Infants | 7-12 months | 0.5 (AI) | 0.5 (AI) |
| Children | 1-3 years | 0.9 | 0.9 |
| Children | 4-8 years | 1.2 | 1.2 |
| Children | 9-13 years | 1.8 | 1.8 |
| Adolescents | 14-18 years | 2.4 | 2.4 |
| Adults | 19-50 years | 2.4 | 2.4 |
| Adults | 51 years and older | 2.4* | 2.4* |
| Pregnancy | all ages | - | 2.6 |
| Breast-feeding | all ages | - | 2.8 |
| *Vitamin B12 intake should be from supplements or fortified foods due to the age-related increase in food-bound malabsorption. | |||
Disease Prevention
Cardiovascular disease
As mentioned above, chronic atrophic gastritis and infection by H. pylori can cause deficiency in vitamin B12 secondary to malabsorption disorders (see Causes of vitamin B12 deficiency). Some studies, but not all, have found H. pylori infection or chronic atrophic gastritis to be associated with adverse cardiovascular events, including myocardial infarction and stroke (reviewed in 23-25).
Homocysteine and cardiovascular disease
Observational studies indicate that even moderately elevated blood concentrations of homocysteine raise the risk of cardiovascular disease (CVD), although randomized controlled trials of homocysteine-lowering therapy have generally not translated to reductions in adverse cardiovascular outcomes (26, 27). The mechanism by which homocysteine might increase CVD risk remains the subject of a great deal of research (28). The amount of homocysteine in the blood is regulated by at least four vitamins: folate, vitamin B6, riboflavin, and vitamin B12 (see Figure 1). An early analysis of the results of 12 randomized controlled trials showed that folic acid supplementation (0.5-5 mg/day) had the greatest lowering effect on blood homocysteine concentrations (25% decrease); co-supplementation with folic acid and vitamin B12 (500 μg/day) provided an additional 7% reduction (32% decrease) in blood homocysteine concentrations (29). The results of a sequential supplementation trial in 53 men and women indicated that after optimization of folate status with folic acid supplements, vitamin B12 became the major determinant of plasma homocysteine levels (30). In addition, in a population of older adults (age >60 years) exposed to folic acid fortification, the main determinants of plasma homocysteine were renal function and vitamin B12 (31). It is thought that the elevation of homocysteine levels might be partly due to vitamin B12 deficiency in individuals over 60 years of age. Two studies found blood methylmalonic acid (MMA) levels to be elevated in more than 60% of elderly individuals with elevated homocysteine levels. In the absence of impaired kidney function, an elevated MMA level in conjunction with elevated homocysteine suggests either a vitamin B12 deficiency or a combined vitamin B12 and folate deficiency (32). Thus, it appears important to evaluate vitamin B12 status, as well as kidney function, in older individuals with elevated homocysteine levels prior to initiating homocysteine-lowering therapy. For more information regarding homocysteine and CVD, see the article on folate.
Intervention studies
Although increased intake of folic acid and vitamin B12 is effective in decreasing blood concentrations of homocysteine, whether these B vitamins lower risk of CVD remains controversial. Several randomized, placebo-controlled trials have been conducted to determine whether homocysteine-lowering through folic acid, vitamin B12, and vitamin B6 supplementation reduces the incidence of CVD. A 2017 meta-analysis of data from 15 trials, including more than 71,000 participants at risk of or with existing CVD, showed that B-vitamin supplementation had no significant effect on risk of myocardial infarction (heart attack) or all-cause mortality but reduced risk of stroke by 10% (RR, 0.90; 95% CI, 0.82-0.99) (33). This analysis excluded trials in those with end-stage renal disease, who are at high risk of cardiovascular events and mortality. Two recent meta-analyses that pooled data from patients with end-stage renal disease found no benefit of B-vitamin supplementation on stroke outcomes (34, 35). Some have raised concern over potential toxicity of high-dose supplemental cyanocobalamin in individuals with impaired kidney function (34, 36); alternate supplement formulations of vitamin B12, such as methylcobalamin and hydroxycobalamin, have been suggested for this patient population.
A meta-analysis of 12 clinical trials measuring flow-mediated vasodilation (FMD; a surrogate marker of vascular health) in response to homocysteine reduction revealed that B-vitamin supplementation was accompanied by improved FMD in short-term (<8 weeks) but not in long-term studies conducted in subjects with preexisting vascular diseases (37). Some of the studies included in these meta-analyses did not use vitamin B12, and folate administration on its own has shown a protective role on vascular function and stroke risk (38, 39). Also, the high prevalence of malabsorption disorders and vitamin B12 deficiency in elderly individuals might warrant the use of higher doses of vitamin B12 than those used in these trials (40).
Cancer
Folate is required for synthesis of DNA, and there is evidence that decreased availability of folate results in strands of DNA that are more susceptible to damage. Deficiency of vitamin B12 traps folate in a form that is unusable by the body for DNA synthesis. Both vitamin B12 and folate deficiencies result in a diminished capacity for methylation reactions (see Figure 3). Thus, vitamin B12 deficiency may lead to an elevated rate of DNA damage and altered methylation of DNA, both of which are important risk factors for cancer. A series of studies in young adults and older men indicated that increased levels of homocysteine and decreased levels of vitamin B12 in the blood were associated with a biomarker of chromosome breakage in white blood cells (reviewed in 41). In a double-blind, placebo-controlled study, the same biomarker of chromosome breakage was minimized in young adults who were supplemented with 700 μg of folic acid and 7 μg of vitamin B12 daily in cereal for two months (42).
Breast cancer
While results from two early case-control studies suggested higher intakes of vitamin B12 might help protect against breast cancer (43, 44), nested case-control studies and prospective cohort studies in different populations have found no associations between breast cancer incidence and dietary or total intake of vitamin B12 (dietary plus supplemental) (45-51) or blood concentrations of vitamin B12 (52-54). However, a nested case-control study within the Women’s Health Study (participants ≥45 years at baseline) reported a positive association between the highest quintile of total vitamin B12 intake (>10.5 μg/day) and breast cancer compared to the lowest quintile (≤4.3 μg/day; RR, 1.44; 95% CI, 1.02-2.04), but this study did not find an association between plasma concentrations of vitamin B12 and breast cancer (55). A more recent nested case-control study within the Nurses’ Health Study II reported a positive association between plasma concentrations of vitamin B12 and breast cancer risk; participants in this study were mostly premenopausal women (56). While additional studies are needed to determine whether elevated vitamin B12 status may be related to cancer development, including evaluating whether it might be a potential marker of cancer, it is important for observational studies to adequately control for potential confounders, such as age, menopausal status, alcohol use, and intake of the B-vitamin folate.
Neural tube defects
Neural tube defects (NTD) may result in anencephaly or spina bifida, which are often fatal congenital malformations of the central nervous system. The defects arise from failure of the embryonic neural tube to close, which occurs between the 21st and 28th days after conception, a time when most women are unaware of their pregnancy (57). Randomized controlled trials have demonstrated 60 to 100% reductions in NTD cases when women consumed folic acid supplements in addition to a varied diet during the month before and the month after conception. The homocysteine-lowering effect of folic acid may play a critical role in reducing the risk of NTD (58, 59). Homocysteine accumulates in the blood when there is inadequate folate and/or vitamin B12 for effective functioning of the methionine synthase enzyme (see Function). However, disruption in folate metabolism may be the underlying cause of folic acid-responsive NTD, and elevated homocysteine may simply be coincidental.
While the etiology of NTD is multifactorial, some studies have found that blood concentrations of homocysteine or holo-transcobalamin to be lower in NTD-affected infants and mothers compared to non-affected controls (reviewed in 59). Polymorphisms in various genes involved in vitamin B12 metabolism, including the genes encoding the intrinsic factor-cobalamin receptor (60) and the holo-transcobalamin receptor (61), have been associated with increased NTD risk in some studies. However, it is not known whether vitamin B12 supplementation, in addition to folic acid supplementation, may be beneficial in the prevention of NTD (62) — randomized controlled trials are needed to address this question.
Cognitive decline, dementia, and Alzheimer's disease
The occurrence of vitamin B12 deficiency prevails in the elderly population and has been frequently associated with Alzheimer's disease (reviewed in 63, 64). One study found lower vitamin B12 levels in the cerebrospinal fluid of patients with Alzheimer's disease than in patients with other types of dementia, though blood levels of vitamin B12 did not differ (65). The reason for the association of low vitamin B12 status with Alzheimer's disease is not clear. Vitamin B12 deficiency, like folate deficiency, may lead to decreased synthesis of methionine and S-adenosylmethionine (SAM), thereby adversely affecting methylation reactions. Methylation reactions are essential for the metabolism of components of the myelin sheath of nerve cells, as well as for synthesis and metabolism of neurotransmitters (22). Other metabolic implications of vitamin B12 deficiency include the accumulation of homocysteine and methylmalonic acid, which might contribute to the neuropathologic features of dementia (63). Indeed, elevated homocysteine is now recognized as a strong risk factor for cognitive impairment and dementia, including Alzheimer’s disease (66-68).
Observational studies
A large number of cross-sectional and prospective cohort studies have associated elevated blood homocysteine concentrations with measures of poor cognitive scores and increased risk of dementia, including Alzheimer's disease (reviewed in 67, 69). A case-control study of 164 patients with dementia of Alzheimer's type included 76 cases in which the diagnosis of Alzheimer's disease was confirmed by examination of brain cells after death. Compared to 108 control subjects without evidence of dementia, subjects with dementia of Alzheimer's type and confirmed Alzheimer's disease had higher blood levels of homocysteine and lower blood levels of folate and vitamin B12. Measures of general nutritional status indicated that the association of increased homocysteine levels and diminished vitamin B12 status with Alzheimer's disease was not due to dementia-related malnutrition (70). In a sample of 1,092 men and women without dementia followed for an average of 10 years, those with higher plasma homocysteine levels at baseline had a significantly higher risk of developing Alzheimer's disease and other types of dementia. Specifically, those with plasma homocysteine levels greater than 14 μmol/L had nearly double the risk of developing Alzheimer's disease (71). A study in 650 elderly men and women reported that the risk of elevated plasma homocysteine levels was significantly higher in those with lower cognitive function scores (72). A prospective study in 816 elderly men and women reported that those with hyperhomocysteinemia (homocysteine levels >15 μmol/L) had a significantly higher risk of developing Alzheimer's disease or dementia. Although raised homocysteine levels might be partly due to a poor vitamin B12 status, the latter was not related to risk of dementia or Alzheimer's disease in this study (73). Several meta-analyses of observational studies have found elevated blood concentrations of homocysteine to be linked with an increased risk of dementia (74) or Alzheimer’s disease (68, 75-78).
A systematic review of 35 prospective cohort studies assessing the association between vitamin B12 status and cognitive deterioration in older individuals with or without dementia at baseline did not support a relationship between serum vitamin B12 concentrations and cognitive decline, dementia, or Alzheimer’s disease (79). More recent meta-analyses of prospective cohort studies in older adults have not found a relationship between blood cobalamin concentration or dietary vitamin B12 intake and cognitive decline or dementia (80, 81). Nevertheless, studies utilizing more specific biomarkers of vitamin B12 status, including measures of holo-transcobalamin and methylmalonic acid, have shown more consistent results and a trend towards associations between poor vitamin B12 status and faster cognitive decline and increased risk of Alzheimer’s disease (82-86). Interestingly, two longitudinal studies in older adults (>60 years) found lower blood concentrations of holo-transcobalamin (and cobalamin) at baseline were associated with increased brain volume loss over a period of five (87) and six years (88).
Intervention studies
High-dose B-vitamin supplementation has been proven effective for treating hyperhomocysteinemia in elderly individuals with or without cognitive impairment. However, homocysteine-lowering trials have produced equivocal results regarding the prevention of cognitive deterioration in this population. A recent systematic review and meta-analysis of 21 randomized, placebo-controlled trials examining the effect of B-vitamin supplementation found the supplementation decreased homocysteine concentrations and improved measures global cognitive function but had no effect on individual measures of executive function, information processing speed, or episodic memory (89). This analysis pooled results from trials of participants with mild cognitive impairment (6 trials) and trials of participants without cognitive impairment (15 trials). A separate meta-analysis that pooled results of trials in older adults with mild cognitive impairment found B-vitamin supplementation (6 to 24 months, depending on the trial) had no benefits on various cognitive parameters, including executive function, information processing speed, and episodic memory (90). The trials of this meta-analysis employed different dosages of B vitamins, with vitamin B12 included in four of five trials.
A two-year, randomized, placebo-controlled study in older adults (>70 years) with mild cognitive impairment reported that a daily regimen of 800 μg of folic acid, 500 μg of vitamin B12, and 20 mg of vitamin B6 significantly reduced the rate of brain atrophy compared to placebo treatment (0.5% vs. 3.7%) (91, 92). Compared to placebo, B-vitamin supplementation in these older subjects also improved a measure of executive function, a secondary outcome of the trial (93). Interestingly, greater benefits were seen in those with high compared to low homocysteine concentrations at baseline, suggesting the importance of lowering homocysteine levels in prevention of brain atrophy and cognitive decline (91-93). The authors attributed the changes in homocysteine levels primarily to vitamin B12 (92). Yet, very few clinical trials have looked at the effect of vitamin B12, solely, on cognitive endpoints. A randomized, double-blind, placebo-controlled trial in 191 older adults (≥75 years) with moderate vitamin B12 deficiency (serum cobalamin concentrations of 107-210 pmol/L) without anemia provided participants with 1 mg/day of cyanocobalamin for one year (94). Compared to baseline, the vitamin B12 supplementation increased serum cobalamin and holo-transcobalamin levels and decreased blood homocysteine concentrations, but had no effect on measures of cognitive function, including assessments of memory, processing speed, and executive function (94). It is important to note that the participants in this trial were not experiencing clinical symptoms of vitamin B12 deficiency.
While it is known that B-vitamin supplementation effectively treats hyperhomocysteinemia, there is a need for well-designed, large trials to evaluate the effect of B-vitamin supplementation on long-term outcomes, such as age-related cognitive decline and the incidence of Alzheimer’s disease. The authors of one trial (95) included in the above-mentioned meta-analysis (89) noted that little cognitive decline was observed in the placebo group over their two-year trial, which may have limited their power to detect an effect of the B-vitamin intervention (95).
Depression
A few observational studies have found higher dietary intakes of vitamin B12 to be associated with lower risk of developing depression, especially among women. One study found as many as 30% of patients hospitalized for depression were deficient in vitamin B12 (96). A cross-sectional study of 700 community-living, physically disabled women over the age of 65 found that vitamin B12-deficient women were twice as likely to be severely depressed as non-deficient women (97). A population-based study in 3,884 elderly men and women with depressive disorders found that those with vitamin B12 deficiency were almost 70% more likely to experience depression than those with normal vitamin B12 status (98). A recent meta-analysis of 12 observational studies found that higher dietary intake of vitamin B12 was associated with a 23% lower risk of depression compared to lower intakes, but stratifying the data revealed a significant protective association in females, not in males (99). Similarly, a meta-analysis of nine observational studies in older adults found low serum vitamin B12 concentration to be associated with increased risk for depression, with significant association only in women (100). More recently, a longitudinal study among 3,849 community-living older adults in Ireland found that low vitamin B12 serum concentrations (<185 pmol/L) at baseline were associated with a 51% increased risk of developing symptoms of depression over a four-year period compared to those with normal concentrations (>258-601 pmol/L) (101).
The reasons for a link between vitamin B12 deficiency and depression are not clear but may involve a shortage in S-adenosylmethionine (SAM). SAM is a methyl group donor for numerous methylation reactions in the brain, including those involved in the metabolism of neurotransmitters whose deficiency has been related to depression (102). Severe vitamin B12 deficiency in a mouse model showed dramatic alterations in the level of DNA methylation in the brain, which might lead to neurologic impairments (103). This hypothesis is supported by several studies that have shown supplementation with SAM improves depressive symptoms (104-107).
Increased blood concentration of homocysteine is another nonspecific biomarker of vitamin B12 deficiency that has been linked to depressive symptoms in the elderly (108). However, few studies have examined the relationship of vitamin B12 status, homocysteine concentrations, and the development of depression over time. In a randomized, placebo-controlled intervention study with more than 900 older participants experiencing psychological distress, daily supplementation with folic acid (400 μg) and vitamin B12 (100 μg) for two years did not reduce the occurrence of symptoms of depression despite significantly improving blood folate, vitamin B12, and homocysteine levels compared to placebo (109). Additionally, a randomized, double-blind, placebo-controlled trial in 2,588 healthy older adults with mild hyperhomocysteinemia found that daily supplementation with 500 μg of vitamin B12 and 400 μg of folic acid for two years reduced blood homocysteine concentrations but had no benefit on depressive symptoms in those with or without depressive symptoms at baseline (110). In a long-term randomized, double-blind, placebo-controlled study among sufferers of cerebrovascular accidents at high risk of depression, daily supplementation with 2 mg of folic acid, 25 mg of vitamin B6, and 500 μg vitamin B12 significantly lowered the risk of major depressive episodes during a seven-year follow-up period compared to placebo (111). A randomized, double-blind, placebo-controlled trial in 153 older adults taking the antidepressant citalopram found daily supplementation with 500 μg vitamin B12, 2 mg of folic acid, and 25 mg of vitamin B6 for one year did not reduce the severity of depressive symptoms but increased the response to drug treatment and helped to prevent symptom relapse (112), suggesting B-vitamin supplementation might provide some benefit as an adjunct therapy.
Although it cannot yet be determined whether vitamin B12 deficiency plays a causal role in depression, it may be beneficial to screen for vitamin B12 deficiency in older individuals as part of a medical evaluation for depression.
Osteoporosis
High blood concentrations of homocysteine may affect bone remodeling by increasing bone resorption (breakdown), decreasing bone formation, and reducing bone blood flow. Another proposed mechanism involves the binding of homocysteine to the collagenous matrix of bone, which may modify collagen properties and reduce bone strength (reviewed in 113). Alterations of bone biomechanical properties can contribute to osteoporosis and increase the risk of fractures in the elderly. Since vitamin B12 is a determinant of homocysteine metabolism, it was suggested that the risk of osteoporotic fractures in older subjects might be enhanced by vitamin B12 deficiency. A meta-analysis of four observational studies, following a total of 7,475 older individuals for 3 to 16 years, found a weak association between an elevation in vitamin B12 of 50 pmol/L in blood and a reduction in fracture risk (114). Moreover, a US national study of 2,806 older women (≥50 years) found increased plasma concentration of methylmalonic acid — the metabolic indicator of vitamin B12 deficiency — to be associated with increased risk of lumbar spine osteoporosis (115).
Randomized controlled trials evaluating the role of vitamin B12 in bone health have used vitamin B12 in combination with other B-vitamins. A randomized controlled trial in 167 older adults (≥50 years) found two-year supplementation with vitamin B12 (10 μg/day), along with folic acid (200 μg/day), vitamin B6 (10 mg/day), riboflavin (5 mg/day), and vitamin D (10 μg/day), had no effect on bone mineral density (total hip, femoral neck, or lumbar spine) compared to the control group only receiving vitamin D (10 μg/day ) (116). However, in a subanalysis of participants with low vitamin B12 status at baseline (n= 101), the B-vitamin supplementation slowed the decline in bone mineral density (total hip and femoral neck but not lumbar spine) (116). A randomized, placebo-controlled trial in 559 elderly individuals with low serum levels of folate and vitamin B12 and at increased risk of fracture evaluated the combined supplementation of very high doses of folic acid (5 mg/day) and vitamin B12 (1.5 mg/day). The two-year study found that the supplementation improved B-vitamin status, decreased homocysteine concentrations, and reduced risk of total fractures compared to placebo (117). However, a multicenter study in 5,485 subjects with cardiovascular disease or diabetes mellitus showed that daily supplementation with folic acid (2.5 mg), vitamin B12 (1 mg), and vitamin B6 (50 mg) lowered homocysteine concentrations but had no effect on fracture risk compared to placebo (118). Another small, randomized, double-blind trial in 93 individuals with low vitamin D status found no additional benefit of B-vitamin supplementation (50 mg/day of vitamin B6, 0.5 mg/day of folic acid, and 0.5 mg/day of vitamin B12) on markers of bone health over a one-year period beyond that associated with vitamin D and calcium supplementation. Yet, the short duration of the study did not permit a conclusion on whether the lowering of homocysteine through B-vitamin supplementation could have long-term benefits on bone strength and fracture risk (119). In a randomized, double-blind, placebo-controlled trial in 2,919 participants (≥65 years) with elevated blood concentrations of homocysteine, supplementation with vitamin B12 (500 μg/day) and folic acid (400 μg/day) for two years did not decrease the risk of osteoporotic fracture despite reductions in homocysteine (120, 121). However, stratification of the data revealed a protective effect against osteoporotic fracture in participants older than 80 years (120). Further, a secondary analysis of a trial in women at high risk for or with existing cardiovascular disease reported that high-dose supplemental B-vitamins (1 mg/day of vitamin B12, 50 mg/day of vitamin B6, and 2.5 mg/day of folic acid) for 7.3 years had no effect on risk of non-vertebral fractures (122). In general, the available data from intervention trials do not support the use of supplemental B-vitamins to prevent osteoporotic fractures.
Sources
Food sources
Only certain bacteria and archaea can synthesize vitamin B12 (123, 124). Vitamin B12 is present in animal products, such as meat, poultry, fish (including shellfish), and to a lesser extent, dairy products and eggs (1). Strict vegetarians who eat no animal products (vegans) need supplemental vitamin B12 to meet their requirements. A few plant-source foods, such as certain fermented beans and vegetables and edible algae and mushrooms, may contain bioactive vitamin B12 (125). Together with B-vitamin fortified food (e.g., cereal, nutritional yeast) and supplements, these foods might contribute, though modestly, to prevent vitamin B12 deficiency in individuals consuming vegetarian diets. Individuals over the age of 50 years should augment their dietary intake with vitamin B12 from supplements or fortified foods because of the increased likelihood of food-bound vitamin B12 malabsorption with advanced age.
Most people do not have a problem obtaining the RDA of 2.4 μg/day of vitamin B12 from food. According to a US national survey, the average dietary intake of vitamin B12 is 5.9 μg/day for adult men and 3.8 μg/day for adult women. Men and women 60 years or older have average dietary intakes of 5.6 μg/day and 3.7 μg/day, respectively (126). However, consumption of any type of vegetarian diet dramatically increases the prevalence of vitamin B12 deficiency in individuals across all age groups (127, 128). Some foods with substantial amounts of vitamin B12 are listed in Table 2, along with their vitamin B12 content in micrograms (μg). For more information on the nutrient content of specific foods, search USDA's FoodData Central.
Supplements
Vitamin B12 is available over-the-counter as a single-nutrient supplement and also as a component of multivitamin and vitamin B-complex supplements (129). Cyanocobalamin is the principal form used in oral supplements in the United States, while hydroxycobalamin is used primarily in Europe. Other forms, including methylcobalamin and adenosylcobalamin, are available as well (130). Sublingual vitamin B12 (placed under the tongue until dissolved) is available over-the-counter. Some studies suggest that sublingual cyanocobalamin (131-133) and sublingual methylcobalamin (134, 135) are effective at increasing vitamin B12 status. Further, cyanocobalamin and hydroxocobalamin are available by prescription in an injectable form to treat pernicious anemia, and cyanocobalamin is available as a prescription nasal spray.
Safety
Toxicity
No toxic or adverse effects have been associated with large intakes of vitamin B12 from food or supplements in healthy people. Doses as high as 2 mg (2,000 μg) daily by mouth or 1 mg monthly by intramuscular (IM) injection have been used to treat pernicious anemia without significant side effects (136). When high doses of vitamin B12 are given orally, only a small percentage can be absorbed, which may explain the low toxicity (4). Because of the low toxicity of vitamin B12, no tolerable upper intake level (UL) has been set by the US Food and Nutrition Board (21). Some have raised concern over potential toxicity of high-dose cyanocobalamin supplements in those with impaired kidney function (renal disease) (34, 36); alternate supplement formulations of vitamin B12, such as methylcobalamin and hydroxycobalamin, have been suggested for this patient population.
Drug interactions
A number of drugs reduce the absorption of vitamin B12. Proton-pump inhibitors (e.g., omeprazole, esomeprazole, and lansoprazole), used to treat Zollinger-Ellison syndrome and gastroesophageal reflux disease (GERD), markedly decrease stomach acid secretion required for the release of vitamin B12 from food but not from supplements. Long-term use of proton-pump inhibitors has been found to decrease blood vitamin B12 concentrations. However, vitamin B12 deficiency does not generally develop until after at least two-to-three years of continuous therapy (137-139). A recent systematic review and meta-analysis of 25 observational studies found proton-pump inhibitor use was associated with a small increased risk of vitamin B12 deficiency; however, the studies included in the pooled analysis were too heterogeneous to inform an association (140). Another class of gastric acid inhibitors known as histamine2 (H2)-receptor antagonists (e.g., cimetidine, famotidine, and ranitidine), often used to treat peptic ulcer disease, has also been found to decrease the absorption of vitamin B12 from food. It is not clear whether the long-term use of H2-receptor antagonists could cause overt vitamin B12 deficiency (141, 142). Individuals taking drugs that inhibit gastric acid secretion should consider taking vitamin B12 in the form of a supplement because gastric acid is not required for its absorption.
Other drugs found to inhibit vitamin B12 absorption from food include cholestyramine (a bile acid-binding resin used in the treatment of high cholesterol), chloramphenicol and neomycin (antibiotics), and colchicine (medicine for gout treatment). Metformin, a medication for individuals with type 2 diabetes mellitus, was found to decrease vitamin B12 absorption possibly by tying up free calcium required for absorption of the IF-B12 complex (143). However, the clinical significance of this is unclear (144). It is not known whether calcium supplementation can reverse vitamin B12 malabsorption; therefore, calcium supplementation is not currently prescribed for the prevention or treatment of metformin-induced vitamin B12 deficiency (145). Nevertheless, use of metformin may decrease vitamin B12 status: a meta-analysis of four short-term intervention trials (up to 4 months’ duration) in patients with type 2 diabetes found that metformin use decreased serum cobalamin concentrations by 57 pmol/L, which could have clinical implications if such patients have suboptimal vitamin B12 status (146). Previous reports that megadoses of vitamin C destroy vitamin B12 have not been supported (147) and may have been an artifact of the assay used to measure vitamin B12 status (21).
Nitrous oxide, a commonly used anesthetic, oxidizes and inactivates vitamin B12, thus inhibiting both of the vitamin B12-dependent enzymes, and can produce many of the clinical features of vitamin B12 deficiency, such as megaloblastic anemia and neuropathy. This is of particular risk in chronic recreational users of nitrous oxide. Since nitrous oxide is commonly used for surgery in the elderly, some experts feel vitamin B12 deficiency should be ruled out prior to its use or prophylactic vitamin B12 supplementation implemented before and after exposure to the gas (7, 18).
Large doses of folic acid given to an individual with an undiagnosed vitamin B12 deficiency can correct megaloblastic anemia without correcting the underlying vitamin B12 deficiency, leaving the individual at risk of developing irreversible neurologic damage (21). For this reason, the Food and Nutrition Board of the National Academy of Medicine advises that all adults limit their intake of folic acid (supplements and fortification) to 1,000 μg (1 mg) daily. There is no upper level set for reduced forms of folate (i.e., forms other than folic acid) found in foods.
Linus Pauling Institute Recommendation
A varied diet should provide enough vitamin B12 to prevent deficiency in most individuals 50 years of age and younger. Strict vegetarians and women planning to become pregnant should take a multivitamin supplement daily or eat fortified cereal, which would ensure a daily intake of 6 to 30 μg of vitamin B12 in a form that is easily absorbed. Higher doses of vitamin B12 supplements are recommended for patients taking medications that interfere with its absorption (see Drug interactions).
Older adults (>50 years)
Because vitamin B12 malabsorption and vitamin B12 deficiency are more common in older adults, the Linus Pauling Institute recommends that adults older than 50 years take 100 to 400 μg/day of supplemental vitamin B12.
Authors and Reviewers
Originally written in 2000 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in March 2003 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in August 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in January 2014 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in October 2023 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in November 2023 by:
Joshua W. Miller, Ph.D.
Professor and Chair, Department of Nutritional Sciences
Rutgers, The State University of New Jersey
Copyright 2000-2024 Linus Pauling Institute
References
1. Brody T. Nutritional Biochemistry. 2nd ed. San Diego: Academic Press; 1999.
2. Carmel R. Cobalamin (Vitamin B-12). In: Shils M, Shike M, Ross A, Caballero B, RJ C, eds. Modern Nutrition in Health and Disease. Philadelphia: Lippincott, Williams & Wilkins; 2006:482-497.
3. Shane B. Folic acid, vitamin B-12, and vitamin B-6. In: Stipanuk M, ed. Biochemical and Physiological Aspects of Human Nutrition. Philadelphia: W.B. Saunders Co.; 2000:483-518.
4. Carmel R. How I treat cobalamin (vitamin B12) deficiency. Blood. 2008;112(6):2214-2221. (PubMed)
5. Green R, Allen LH, Bjorke-Monsen AL, et al. Vitamin B12 deficiency. Nat Rev Dis Primers. 2017;3:17040. (PubMed)
6. Kozyraki R, Cases O. Vitamin B12 absorption: mammalian physiology and acquired and inherited disorders. Biochimie. 2013;95(5):1002-1007. (PubMed)
7. Baik HW, Russell RM. Vitamin B12 deficiency in the elderly. Annu Rev Nutr. 1999;19:357-377. (PubMed)
8. Neumann WL, Coss E, Rugge M, Genta RM. Autoimmune atrophic gastritis--pathogenesis, pathology and management. Nat Rev Gastroenterol Hepatol. 2013;10(9):529-541. (PubMed)
9. Lahner E, Persechino S, Annibale B. Micronutrients (Other than iron) and Helicobacter pylori infection: a systematic review. Helicobacter. 2012;17(1):1-15. (PubMed)
10. Banka S, Ryan K, Thomson W, Newman WG. Pernicious anemia - genetic insights. Autoimmun Rev. 2011;10(8):455-459. (PubMed)
11. Lam-Tse WK, Batstra MR, Koeleman BP, et al. The association between autoimmune thyroiditis, autoimmune gastritis and type 1 diabetes. Pediatr Endocrinol Rev. 2003;1(1):22-37. (PubMed)
12. Checchi S, Montanaro A, Ciuoli C, et al. Prevalence of parietal cell antibodies in a large cohort of patients with autoimmune thyroiditis. Thyroid. 2010;20(12):1385-1389. (PubMed)
13. Wolffenbuttel BH, Owen PJ, Ward M, Green R. Vitamin B(12). BMJ. 2023;383:e071725. (PubMed)
14. Wang H, Li L, Qin LL, Song Y, Vidal-Alaball J, Liu TH. Oral vitamin B12 versus intramuscular vitamin B12 for vitamin B12 deficiency. Cochrane Database Syst Rev. 2018;3:CD004655. (PubMed)
15. Ho C, Kauwell GP, Bailey LB. Practitioners' guide to meeting the vitamin B-12 recommended dietary allowance for people aged 51 years and older. J Am Diet Assoc. 1999;99(6):725-727. (PubMed)
16. Desmond MA, Sobiecki JG, Jaworski M, et al. Growth, body composition, and cardiovascular and nutritional risk of 5- to 10-y-old children consuming vegetarian, vegan, or omnivore diets. Am J Clin Nutr. 2021;113(6):1565-1577. (PubMed)
17. Watkins D, Rosenblatt DS. Lessons in biology from patients with inborn errors of vitamin B12 metabolism. Biochimie. 2013;95(5):1019-1022. (PubMed)
18. Weir DG, Scott JM. Vitamin B-12 "Cobalamin". In: Shils M, ed. Modern Nutrition in Health and Disease. 9th ed. Baltimore: Williams & Wilkins; 1999:447-458.
19. Wolffenbuttel BHR, Wouters H, de Jong WHA, Huls G, van der Klauw MM. Association of vitamin B12, methylmalonic acid, and functional parameters. Neth J Med. 2020;78(1):10-24. (PubMed)
20. Herbert V. Vitamin B-12. In: Ziegler E, Filer L, eds. Present Knowledge in Nutrition. 7th ed. Washington D.C.: ILSI Press; 1996:191-205.
21. Food and Nutrition Board, Institute of Medicine. Vitamin B-12. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B-6, Vitamin B-12, Pantothenic Acid, Biotin, and Choline. Washington D.C.: National Academy Press; 1998:306-356. (National Academy Press)
22. Scalabrino G. The multi-faceted basis of vitamin B12 (cobalamin) neurotrophism in adult central nervous system: Lessons learned from its deficiency. Prog Neurobiol. 2009;88(3):203-220. (PubMed)
23. Liu J, Wang F, Shi S. Helicobacter pylori infection increase the risk of myocardial infarction: a meta-analysis of 26 studies involving more than 20,000 participants. Helicobacter. 2015;20(3):176-183. (PubMed)
24. Sun J, Rangan P, Bhat SS, Liu L. A meta-analysis of the association between Helicobacter pylori infection and risk of coronary heart disease from published prospective studies. Helicobacter. 2016;21(1):11-23. (PubMed)
25. Wang B, Yu M, Zhang R, Chen S, Xi Y, Duan G. A meta-analysis of the association between Helicobacter pylori infection and risk of atherosclerotic cardiovascular disease. Helicobacter. 2020;25(6):e12761. (PubMed)
26. Chrysant SG, Chrysant GS. The current status of homocysteine as a risk factor for cardiovascular disease: a mini review. Expert Rev Cardiovasc Ther. 2018;16(8):559-565. (PubMed)
27. Yuan S, Mason AM, Carter P, Burgess S, Larsson SC. Homocysteine, B vitamins, and cardiovascular disease: a Mendelian randomization study. BMC Med. 2021;19(1):97. (PubMed)
28. Cacciapuoti F. Hyper-homocysteinemia: a novel risk factor or a powerful marker for cardiovascular diseases? Pathogenetic and therapeutical uncertainties. J Thromb Thrombolysis. 2011;32(1):82-88. (PubMed)
29. Lowering blood homocysteine with folic acid based supplements: meta-analysis of randomised trials. Homocysteine Lowering Trialists' Collaboration. BMJ. 1998;316(7135):894-898. (PubMed)
30. Quinlivan EP, McPartlin J, McNulty H, et al. Importance of both folic acid and vitamin B12 in reduction of risk of vascular disease. Lancet. 2002;359(9302):227-228. (PubMed)
31. Green R, Miller JW. Vitamin B12 deficiency is the dominant nutritional cause of hyperhomocysteinemia in a folic acid-fortified population. Clin Chem Lab Med. 2005;43(10):1048-1051. (PubMed)
32. Stabler SP, Lindenbaum J, Allen RH. Vitamin B-12 deficiency in the elderly: current dilemmas. Am J Clin Nutr. 1997;66(4):741-749. (PubMed)
33. Marti-Carvajal AJ, Sola I, Lathyris D, Dayer M. Homocysteine-lowering interventions for preventing cardiovascular events. Cochrane Database Syst Rev. 2017;8:CD006612. (PubMed)
34. Spence JD, Yi Q, Hankey GJ. B vitamins in stroke prevention: time to reconsider. Lancet Neurol. 2017;16(9):750-760. (PubMed)
35. Hankey GJ. B vitamins for stroke prevention. Stroke Vasc Neurol. 2018;3(2):51-58. (PubMed)
36. Capelli I, Cianciolo G, Gasperoni L, et al. Folic acid and vitamin B12 administration in CKD, why not? Nutrients. 2019;11(2):383. (PubMed)
37. Potter K, Hankey GJ, Green DJ, Eikelboom J, Jamrozik K, Arnolda LF. The effect of long-term homocysteine-lowering on carotid intima-media thickness and flow-mediated vasodilation in stroke patients: a randomized controlled trial and meta-analysis. BMC Cardiovasc Disord. 2008;8:24. (PubMed)
38. Qin X, Xu M, Zhang Y, et al. Effect of folic acid supplementation on the progression of carotid intima-media thickness: a meta-analysis of randomized controlled trials. Atherosclerosis. 2012;222(2):307-313. (PubMed)
39. Zhao M, Wu G, Li Y, et al. Meta-analysis of folic acid efficacy trials in stroke prevention: Insight into effect modifiers. Neurology. 2017;88(19):1830-1838. (PubMed)
40. Spence JD. B vitamin therapy for homocysteine: renal function and vitamin B12 determine cardiovascular outcomes. Clin Chem Lab Med. 2013;51(3):633-637. (PubMed)
41. Fenech M. Folate (vitamin B9) and vitamin B12 and their function in the maintenance of nuclear and mitochondrial genome integrity. Mutat Res. 2012;733(1-2):21-33. (PubMed)
42. Fenech M. Micronucleus frequency in human lymphocytes is related to plasma vitamin B12 and homocysteine. Mutat Res. 1999;428(1-2):299-304. (PubMed)
43. Wu K, Helzlsouer KJ, Comstock GW, Hoffman SC, Nadeau MR, Selhub J. A prospective study on folate, B12, and pyridoxal 5'-phosphate (B6) and breast cancer. Cancer Epidemiol Biomarkers Prev. 1999;8(3):209-217. (PubMed)
44. Lajous M, Lazcano-Ponce E, Hernandez-Avila M, Willett W, Romieu I. Folate, vitamin B(6), and vitamin B(12) intake and the risk of breast cancer among Mexican women. Cancer Epidemiol Biomarkers Prev. 2006;15(3):443-448. (PubMed)
45. Sellers TA, Kushi LH, Cerhan JR, et al. Dietary folate intake, alcohol, and risk of breast cancer in a prospective study of postmenopausal women. Epidemiology. 2001;12(4):420-428. (PubMed)
46. Maruti SS, Ulrich CM, White E. Folate and one-carbon metabolism nutrients from supplements and diet in relation to breast cancer risk. Am J Clin Nutr. 2009;89(2):624-633. (PubMed)
47. Shrubsole MJ, Shu XO, Li HL, et al. Dietary B vitamin and methionine intakes and breast cancer risk among Chinese women. Am J Epidemiol. 2011;173(10):1171-1182. (PubMed)
48. Stevens VL, McCullough ML, Sun J, Gapstur SM. Folate and other one-carbon metabolism-related nutrients and risk of postmenopausal breast cancer in the Cancer Prevention Study II Nutrition Cohort. Am J Clin Nutr. 2010;91(6):1708-1715. (PubMed)
49. Bassett JK, Baglietto L, Hodge AM, et al. Dietary intake of B vitamins and methionine and breast cancer risk. Cancer Causes Control. 2013;24(8):1555-1563. (PubMed)
50. de Batlle J, Ferrari P, Chajes V, et al. Dietary folate intake and breast cancer risk: European prospective investigation into cancer and nutrition. J Natl Cancer Inst. 2015;107(1):367. (PubMed)
51. Egnell M, Fassier P, Lecuyer L, et al. B-vitamin intake from diet and supplements and breast cancer risk in middle-aged women: results from the prospective NutriNet-Sante cohort. Nutrients. 2017;9(5):488. (PubMed)
52. Houghton SC, Eliassen AH, Zhang SM, et al. Plasma B-vitamin and one-carbon metabolites and risk of breast cancer before and after folic acid fortification in the United States. Int J Cancer. 2019;144(8):1929-1940. (PubMed)
53. Agnoli C, Grioni S, Krogh V, et al. Plasma riboflavin and vitamin B-6, but not homocysteine, folate, or vitamin B-12, are inversely associated with breast cancer risk in the European Prospective Investigation into Cancer and Nutrition-Varese cohort. J Nutr. 2016;146(6):1227-1234. (PubMed)
54. Essen A, Santaolalla A, Garmo H, et al. Baseline serum folate, vitamin B12 and the risk of prostate and breast cancer using data from the Swedish AMORIS cohort. Cancer Causes Control. 2019;30(6):603-615. (PubMed)
55. Lin J, Lee IM, Cook NR, et al. Plasma folate, vitamin B-6, vitamin B-12, and risk of breast cancer in women. Am J Clin Nutr. 2008;87(3):734-743. (PubMed)
56. Houghton SC, Eliassen AH, Zhang SM, et al. Plasma B-vitamins and one-carbon metabolites and the risk of breast cancer in younger women. Breast Cancer Res Treat. 2019;176(1):191-203. (PubMed)
57. Eskes TK. Open or closed? A world of difference: a history of homocysteine research. Nutr Rev. 1998;56(8):236-244. (PubMed)
58. Mills JL, Scott JM, Kirke PN, et al. Homocysteine and neural tube defects. J Nutr. 1996;126(3):756S-760S. (PubMed)
59. Wahbeh F, Manyama M. The role of vitamin B12 and genetic risk factors in the etiology of neural tube defects: A systematic review. Int J Dev Neurosci. 2021;81(5):386-406. (PubMed)
60. Franke B, Vermeulen SH, Steegers-Theunissen RP, et al. An association study of 45 folate-related genes in spina bifida: Involvement of cubilin (CUBN) and tRNA aspartic acid methyltransferase 1 (TRDMT1). Birth Defects Res A Clin Mol Teratol. 2009;85(3):216-226. (PubMed)
61. Pangilinan F, Mitchell A, VanderMeer J, et al. Transcobalamin II receptor polymorphisms are associated with increased risk for neural tube defects. J Med Genet. 2010;47(10):677-685. (PubMed)
62. Molloy AM. Should vitamin B12 status be considered in assessing risk of neural tube defects? Ann N Y Acad Sci. 2018;1414(1):109-125. (PubMed)
63. McCaddon A. Vitamin B12 in neurology and ageing; clinical and genetic aspects. Biochimie. 2013;95(5):1066-1076. (PubMed)
64. Lauer AA, Grimm HS, Apel B, et al. Mechanistic link between vitamin B12 and Alzheimer's disease. Biomolecules. 2022;12(1):129. (PubMed)
65. Nourhashemi F, Gillette-Guyonnet S, Andrieu S, et al. Alzheimer disease: protective factors. Am J Clin Nutr. 2000;71(2):643S-649S. (PubMed)
66. Smith AD, Refsum H. Homocysteine, B vitamins, and cognitive impairment. Annu Rev Nutr. 2016;36:211-239. (PubMed)
67. Smith AD, Refsum H, Bottiglieri T, et al. Homocysteine and dementia: an international Consensus Statement. J Alzheimers Dis. 2018;62(2):561-570. (PubMed)
68. Zuin M, Cervellati C, Brombo G, Trentini A, Roncon L, Zuliani G. Elevated blood homocysteine and risk of Alzheimer's dementia: an updated systematic review and meta-analysis based on prospective studies. J Prev Alzheimers Dis. 2021;8(3):329-334. (PubMed)
69. Smith AD. The worldwide challenge of the dementias: a role for B vitamins and homocysteine? Food Nutr Bull. 2008;29(2 Suppl):S143-172. (PubMed)
70. Clarke R, Smith AD, Jobst KA, Refsum H, Sutton L, Ueland PM. Folate, vitamin B12, and serum total homocysteine levels in confirmed Alzheimer disease. Arch Neurol. 1998;55(11):1449-1455. (PubMed)
71. Seshadri S, Beiser A, Selhub J, et al. Plasma homocysteine as a risk factor for dementia and Alzheimer's disease. N Engl J Med. 2002;346(7):476-483. (PubMed)
72. Ravaglia G, Forti P, Maioli F, et al. Homocysteine and cognitive function in healthy elderly community dwellers in Italy. Am J Clin Nutr. 2003;77(3):668-673. (PubMed)
73. Ravaglia G, Forti P, Maioli F, et al. Homocysteine and folate as risk factors for dementia and Alzheimer disease. Am J Clin Nutr. 2005;82(3):636-643. (PubMed)
74. Wald DS, Kasturiratne A, Simmonds M. Serum homocysteine and dementia: meta-analysis of eight cohort studies including 8669 participants. Alzheimers Dement. 2011;7(4):412-417. (PubMed)
75. Shen L, Ji HF. Associations between homocysteine, folic acid, vitamin B12 and Alzheimer's disease: insights from meta-analyses. J Alzheimers Dis. 2015;46(3):777-790. (PubMed)
76. Van Dam F, Van Gool WA. Hyperhomocysteinemia and Alzheimer's disease: A systematic review. Arch Gerontol Geriatr. 2009;48(3):425-430. (PubMed)
77. Beydoun MA, Beydoun HA, Gamaldo AA, Teel A, Zonderman AB, Wang Y. Epidemiologic studies of modifiable factors associated with cognition and dementia: systematic review and meta-analysis. BMC Public Health. 2014;14:643. (PubMed)
78. Zhou F, Chen S. Hyperhomocysteinemia and risk of incident cognitive outcomes: An updated dose-response meta-analysis of prospective cohort studies. Ageing Res Rev. 2019;51:55-66. (PubMed)
79. O'Leary F, Allman-Farinelli M, Samman S. Vitamin B(1)(2) status, cognitive decline and dementia: a systematic review of prospective cohort studies. Br J Nutr. 2012;108(11):1948-1961. (PubMed)
80. Zhang C, Luo J, Yuan C, Ding D. Vitamin B12, B6, or folate and cognitive function in community-dwelling older adults: a systematic review and meta-analysis. J Alzheimers Dis. 2020;77(2):781-794. (PubMed)
81. Zhou J, Sun Y, Ji M, Li X, Wang Z. Association of vitamin B status with risk of dementia in cohort studies: a systematic review and meta-analysis. J Am Med Dir Assoc. 2022;23(11):1826 e1821-1826 e1835. (PubMed)
82. Clarke R, Birks J, Nexo E, et al. Low vitamin B-12 status and risk of cognitive decline in older adults. Am J Clin Nutr. 2007;86(5):1384-1391. (PubMed)
83. Tangney CC, Tang Y, Evans DA, Morris MC. Biochemical indicators of vitamin B12 and folate insufficiency and cognitive decline. Neurology. 2009;72(4):361-367. (PubMed)
84. Kivipelto M, Annerbo S, Hultdin J, et al. Homocysteine and holo-transcobalamin and the risk of dementia and Alzheimers disease: a prospective study. Eur J Neurol. 2009;16(7):808-813. (PubMed)
85. Hooshmand B, Solomon A, Kareholt I, et al. Homocysteine and holotranscobalamin and the risk of Alzheimer disease: a longitudinal study. Neurology. 2010;75(16):1408-1414. (PubMed)
86. Hooshmand B, Solomon A, Kareholt I, et al. Associations between serum homocysteine, holotranscobalamin, folate and cognition in the elderly: a longitudinal study. J Intern Med. 2012;271(2):204-212. (PubMed)
87. Vogiatzoglou A, Refsum H, Johnston C, et al. Vitamin B12 status and rate of brain volume loss in community-dwelling elderly. Neurology. 2008;71(11):826-832. (PubMed)
88. Hooshmand B, Mangialasche F, Kalpouzos G, et al. Association of vitamin B12, folate, and sulfur amino acids with brain magnetic resonance imaging measures in older adults: a longitudinal population-based study. JAMA Psychiatry. 2016;73(6):606-613. (PubMed)
89. Li S, Guo Y, Men J, Fu H, Xu T. The preventive efficacy of vitamin B supplements on the cognitive decline of elderly adults: a systematic review and meta-analysis. BMC Geriatr. 2021;21(1):367. (PubMed)
90. McCleery J, Abraham RP, Denton DA, et al. Vitamin and mineral supplementation for preventing dementia or delaying cognitive decline in people with mild cognitive impairment. Cochrane Database Syst Rev. 2018;11:CD011905. (PubMed)
91. Smith AD, Smith SM, de Jager CA, et al. Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: a randomized controlled trial. PLoS One. 2010;5(9):e12244. (PubMed)
92. Douaud G, Refsum H, de Jager CA, et al. Preventing Alzheimer's disease-related gray matter atrophy by B-vitamin treatment. Proc Natl Acad Sci U S A. 2013;110(23):9523-9528. (PubMed)
93. de Jager CA, Oulhaj A, Jacoby R, Refsum H, Smith AD. Cognitive and clinical outcomes of homocysteine-lowering B-vitamin treatment in mild cognitive impairment: a randomized controlled trial. Int J Geriatr Psychiatry. 2012;27(6):592-600. (PubMed)
94. Dangour AD, Allen E, Clarke R, et al. Effects of vitamin B-12 supplementation on neurologic and cognitive function in older people: a randomized controlled trial. Am J Clin Nutr. 2015;102(3):639-647. (PubMed)
95. Kwok T, Wu Y, Lee J, et al. A randomized placebo-controlled trial of using B vitamins to prevent cognitive decline in older mild cognitive impairment patients. Clin Nutr. 2020;39(8):2399-2405. (PubMed)
96. Hutto BR. Folate and cobalamin in psychiatric illness. Compr Psychiatry. 1997;38(6):305-314. (PubMed)
97. Penninx BW, Guralnik JM, Ferrucci L, Fried LP, Allen RH, Stabler SP. Vitamin B(12) deficiency and depression in physically disabled older women: epidemiologic evidence from the Women's Health and Aging Study. Am J Psychiatry. 2000;157(5):715-721. (PubMed)
98. Tiemeier H, van Tuijl HR, Hofman A, Meijer J, Kiliaan AJ, Breteler MM. Vitamin B12, folate, and homocysteine in depression: the Rotterdam Study. Am J Psychiatry. 2002;159(12):2099-2101. (PubMed)
99. Wu Y, Zhang L, Li S, Zhang D. Associations of dietary vitamin B1, vitamin B2, vitamin B6, and vitamin B12 with the risk of depression: a systematic review and meta-analysis. Nutr Rev. 2022;80(3):351-366. (PubMed)
100. Petridou ET, Kousoulis AA, Michelakos T, et al. Folate and B12 serum levels in association with depression in the aged: a systematic review and meta-analysis. Aging Ment Health. 2016;20(9):965-973. (PubMed)
101. Laird E, O'Halloran AM, Molloy AM, et al. Low vitamin B12 but not folate is associated with incident depressive symptoms in community-dwelling older adults: a 4 year longitudinal study. Br J Nutr. 2021:1-22. (PubMed)
102. Mischoulon D, Fava M. Role of S-adenosyl-L-methionine in the treatment of depression: a review of the evidence. Am J Clin Nutr. 2002;76(5):1158S-1161S. (PubMed)
103. Fernandez-Roig S, Lai SC, Murphy MM, Fernandez-Ballart J, Quadros EV. Vitamin B12 deficiency in the brain leads to DNA hypomethylation in the TCblR/CD320 knockout mouse. Nutr Metab (Lond). 2012;9:41. (PubMed)
104. Bressa GM. S-adenosyl-l-methionine (SAMe) as antidepressant: meta-analysis of clinical studies. Acta Neurol Scand Suppl. 1994;154:7-14. (PubMed)
105. Bell KM, Plon L, Bunney WE, Jr., Potkin SG. S-adenosylmethionine treatment of depression: a controlled clinical trial. Am J Psychiatry. 1988;145(9):1110-1114. (PubMed)
106. Delle Chiaie R, Pancheri P, Scapicchio P. Efficacy and tolerability of oral and intramuscular S-adenosyl-L-methionine 1,4-butanedisulfonate (SAMe) in the treatment of major depression: comparison with imipramine in 2 multicenter studies. Am J Clin Nutr. 2002;76(5):1172S-1176S. (PubMed)
107. Williams AL, Girard C, Jui D, Sabina A, Katz DL. S-adenosylmethionine (SAMe) as treatment for depression: a systematic review. Clin Invest Med. 2005;28(3):132-139. (PubMed)
108. Almeida OP, McCaul K, Hankey GJ, Norman P, Jamrozik K, Flicker L. Homocysteine and depression in later life. Arch Gen Psychiatry. 2008;65(11):1286-1294. (PubMed)
109. Walker JG, Mackinnon AJ, Batterham P, et al. Mental health literacy, folic acid and vitamin B12, and physical activity for the prevention of depression in older adults: randomised controlled trial. Br J Psychiatry. 2010;197(1):45-54. (PubMed)
110. de Koning EJ, van der Zwaluw NL, van Wijngaarden JP, et al. Effects of two-year vitamin B12 and folic acid supplementation on depressive symptoms and quality of life in older adults with elevated homocysteine concentrations: additional results from the B-PROOF study, an RCT. Nutrients. 2016;8(11):748. (PubMed)
111. Almeida OP, Marsh K, Alfonso H, Flicker L, Davis TM, Hankey GJ. B-vitamins reduce the long-term risk of depression after stroke: The VITATOPS-DEP trial. Ann Neurol. 2010;68(4):503-510. (PubMed)
112. Almeida OP, Ford AH, Hirani V, et al. B vitamins to enhance treatment response to antidepressants in middle-aged and older adults: results from the B-VITAGE randomised, double-blind, placebo-controlled trial. Br J Psychiatry. 2014;205(6):450-457. (PubMed)
113. Vacek TP, Kalani A, Voor MJ, Tyagi SC, Tyagi N. The role of homocysteine in bone remodeling. Clin Chem Lab Med. 2013;51(3):579-590. (PubMed)
114. van Wijngaarden JP, Doets EL, Szczecinska A, et al. Vitamin B12, folate, homocysteine, and bone health in adults and elderly people: a systematic review with meta-analyses. J Nutr Metab. 2013;2013:486186. (PubMed)
115. Bailey RL, Looker AC, Lu Z, et al. B-vitamin status and bone mineral density and risk of lumbar osteoporosis in older females in the United States. Am J Clin Nutr. 2015;102(3):687-694. (PubMed)
116. Clements M, Heffernan M, Ward M, et al. A 2-year randomized controlled trial with low-dose B-vitamin supplementation shows benefits on bone mineral density in adults with lower B12 status. J Bone Miner Res. 2022;37(12):2443-2455. (PubMed)
117. Sato Y, Honda Y, Iwamoto J, Kanoko T, Satoh K. Effect of folate and mecobalamin on hip fractures in patients with stroke: a randomized controlled trial. JAMA. 2005;293(9):1082-1088. (PubMed)
118. Sawka AM, Ray JG, Yi Q, Josse RG, Lonn E. Randomized clinical trial of homocysteine level lowering therapy and fractures. Arch Intern Med. 2007;167(19):2136-2139. (PubMed)
119. Herrmann W, Kirsch SH, Kruse V, et al. One year B and D vitamins supplementation improves metabolic bone markers. Clin Chem Lab Med. 2013;51(3):639-647. (PubMed)
120. van Wijngaarden JP, Swart KM, Enneman AW, et al. Effect of daily vitamin B-12 and folic acid supplementation on fracture incidence in elderly individuals with an elevated plasma homocysteine concentration: B-PROOF, a randomized controlled trial. Am J Clin Nutr. 2014;100(6):1578-1586. (PubMed)
121. Oliai Araghi S, Kiefte-de Jong JC, van Dijk SC, et al. Long-term effects of folic acid and vitamin-B12 supplementation on fracture risk and cardiovascular disease: Extended follow-up of the B-PROOF trial. Clin Nutr. 2021;40(3):1199-1206. (PubMed)
122. Stone KL, Lui LY, Christen WG, et al. Effect of combination folic acid, vitamin B6 , and vitamin B12 supplementation on fracture risk in women: a randomized, controlled trial. J Bone Miner Res. 2017;32(12):2331-2338. (PubMed)
123. Fang H, Kang J, Zhang D. Microbial production of vitamin B12: a review and future perspectives. Microb Cell Fact. 2017;16(1):15. (PubMed)
124. Watanabe F, Bito T. Vitamin B12 sources and microbial interaction. Exp Biol Med (Maywood). 2018;243(2):148-158. (PubMed)
125. Watanabe F, Yabuta Y, Tanioka Y, Bito T. Biologically active vitamin B12 compounds in foods for preventing deficiency among vegetarians and elderly subjects. J Agric Food Chem. 2013;61(28):6769-6775. (PubMed)
126. US Department of Agriculture, Agricultural Research Service. 2020. Total Nutrient Intakes: Percent Reporting and Mean Amounts of Selected Vitamins and Minerals from Food and Beverages and Dietary Supplements, by Gender and Age, What We Eat in America, NHANES 2017-2018. Available: www.ars.usda.gov/nea/bhnrc/fsrg.
127. Pawlak R, Parrott SJ, Raj S, Cullum-Dugan D, Lucus D. How prevalent is vitamin B(12) deficiency among vegetarians? Nutr Rev. 2013;71(2):110-117. (PubMed)
128. Pawlak R, Lester SE, Babatunde T. The prevalence of cobalamin deficiency among vegetarians assessed by serum vitamin B12: a review of literature. Eur J Clin Nutr. 2014;68(5):541-548. (PubMed)
129. Hendler S, Rorvik D, eds. PDR for Nutritional Supplements. Montvale: Medical Economics Company, Inc.; 2001.
130. US Department of Health and Human Services, National Institutes of Health, Office of Dietary Supplements. Dietary Supplement Label Database (DSLD). [Internet]. [cited 2/7/22]. Available from: https://dsld.od.nih.gov/.
131. Del Bo C, Riso P, Gardana C, Brusamolino A, Battezzati A, Ciappellano S. Effect of two different sublingual dosages of vitamin B12 on cobalamin nutritional status in vegans and vegetarians with a marginal deficiency: A randomized controlled trial. Clin Nutr. 2019;38(2):575-583. (PubMed)
132. Sharabi A, Cohen E, Sulkes J, Garty M. Replacement therapy for vitamin B12 deficiency: comparison between the sublingual and oral route. Br J Clin Pharmacol. 2003;56(6):635-638. (PubMed)
133. Bensky MJ, Ayalon-Dangur I, Ayalon-Dangur R, et al. Comparison of sublingual vs. intramuscular administration of vitamin B12 for the treatment of patients with vitamin B12 deficiency. Drug Deliv Transl Res. 2019;9(3):625-630. (PubMed)
134. Orhan Kilic B, Kilic S, Sahin Eroglu E, Gul E, Belen Apak FB. Sublingual methylcobalamin treatment is as effective as intramuscular and peroral cyanocobalamin in children age 0-3 years. Hematology. 2021;26(1):1013-1017. (PubMed)
135. Varkal MA, Karabocuoglu M. Efficiency of the sublingual route in treating B12 deficiency in infants. Int J Vitam Nutr Res. 2021;(3):226-232. (PubMed)
136. Kuzminski AM, Del Giacco EJ, Allen RH, Stabler SP, Lindenbaum J. Effective treatment of cobalamin deficiency with oral cobalamin. Blood. 1998;92(4):1191-1198. (PubMed)
137. Dharmarajan TS, Kanagala MR, Murakonda P, Lebelt AS, Norkus EP. Do acid-lowering agents affect vitamin B12 status in older adults? J Am Med Dir Assoc. 2008;9(3):162-167. (PubMed)
138. Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6(4):443-451. (PubMed)
139. Lam JR, Schneider JL, Zhao W, Corley DA. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency. JAMA. 2013;310(22):2435-2442. (PubMed)
140. Choudhury A, Jena A, Jearth V, et al. Vitamin B12 deficiency and use of proton pump inhibitors: a systematic review and meta-analysis. Expert Rev Gastroenterol Hepatol. 2023;17(5):479-487. (PubMed)
141. Valuck RJ, Ruscin JM. A case-control study on adverse effects: H2 blocker or proton pump inhibitor use and risk of vitamin B12 deficiency in older adults. J Clin Epidemiol. 2004;57(4):422-428. (PubMed)
142. Termanini B, Gibril F, Sutliff VE, Yu F, Venzon DJ, Jensen RT. Effect of long-term gastric acid suppressive therapy on serum vitamin B12 levels in patients with Zollinger-Ellison syndrome. Am J Med. 1998;104(5):422-430. (PubMed)
143. Bauman WA, Shaw S, Jayatilleke E, Spungen AM, Herbert V. Increased intake of calcium reverses vitamin B12 malabsorption induced by metformin. Diabetes Care. 2000;23(9):1227-1231. (PubMed)
144. Obeid R. Metformin causing vitamin B12 deficiency: a guilty verdict without sufficient evidence. Diabetes Care. 2014;37(2):e22-23. (PubMed)
145. Mazokopakis EE, Starakis IK. Recommendations for diagnosis and management of metformin-induced vitamin B12 (Cbl) deficiency. Diabetes Res Clin Pract. 2012;97(3):359-367. (PubMed)
146. Chapman LE, Darling AL, Brown JE. Association between metformin and vitamin B12 deficiency in patients with type 2 diabetes: A systematic review and meta-analysis. Diabetes Metab. 2016;42(5):316-327. (PubMed)
147. Simon JA, Hudes ES. Relation of serum ascorbic acid to serum vitamin B12, serum ferritin, and kidney stones in US adults. Arch Intern Med. 1999;159(6):619-624. (PubMed)
Vitamin C
Contents
Summary
- Vitamin C, also known as L-ascorbic acid, is a water-soluble vitamin. Unlike most mammals and other animals, humans do not have the ability to synthesize vitamin C and must obtain it from the diet. (More information)
- Vitamin C is an essential cofactor in numerous enzymatic reactions, e.g., in the biosynthesis of collagen, carnitine, and neuropeptides, and in the regulation of gene expression. It is also a potent antioxidant. (More information)
- Prospective cohort studies indicate that higher vitamin C status, assessed by measuring circulating vitamin C, is associated with lower risks of hypertension, coronary heart disease, and stroke. (More information)
- There is some evidence to suggest that vitamin C may be a useful adjunct to conventional medical practice to reduce myocardial injury and arrhythmia following a cardiac procedure or surgery in patients with cardiovascular disease. (More information)
- There are insufficient data to suggest a link between vitamin C status and the risk of developing a given type of cancer. Most observational studies examining vitamin C intake in relation to cancer incidence have found no association. Randomized controlled trials have reported no effect of vitamin C supplementation on cancer risk. (More information)
- Current evidence of the efficacy of intravenous vitamin C in cancer patients is limited to observational studies, uncontrolled interventions, and case reports. There is a need for large, longer-duration phase II clinical trials that test the efficacy of intravenous vitamin C in cancer progression and overall survival. (More information)
- Overall, regular use of vitamin C supplements shortens the duration of the common cold but does not reduce the risk of becoming ill. Taking supplements once cold symptoms have already begun has no proven benefits. (More information)
- Vitamin C supplements are available in many forms, but there is little scientific evidence that any one form is better absorbed or more effective than another. (More information)
- There is no scientific evidence that large amounts of vitamin C (up to 10 grams [g]/day in adults) exert any adverse or toxic effects. An upper intake level of 2 g/day is recommended in order to prevent some adults from experiencing diarrhea and gastrointestinal disturbances. (More information)
- Supplemental vitamin C increases urinary oxalate concentrations, but whether an increase in urinary oxalate elevates the risk for kidney stones is not yet known. Those predisposed for kidney stone formation may consider avoiding high-dose (≥1 g/day) vitamin C supplementation. (More information)
Function
Vitamin C (L-ascorbic acid) is a potent reducing agent, meaning that it readily donates electrons to recipient molecules (Figure 1). Related to this oxidation-reduction (redox) potential, two major functions of vitamin C are as an antioxidant and as an enzyme cofactor (1).
Vitamin C is the primary water-soluble, non-enzymatic antioxidant in plasma and tissues. Even in small amounts, vitamin C can protect indispensable molecules in the body, such as proteins, lipids (fats), carbohydrates, and nucleic acids (DNA and RNA), from damage by free radicals and reactive oxygen species (ROS) that are generated during normal metabolism, by active immune cells, and through exposure to toxins and pollutants (e.g., certain chemotherapy drugs and cigarette smoke). Vitamin C also participates in redox recycling of other important antioxidants; for example, vitamin C is known to regenerate vitamin E from its oxidized form (see the article on Vitamin E).
The role of vitamin C as a cofactor is also related to its redox potential. By maintaining enzyme-bound metals in their reduced forms, vitamin C assists mixed-function oxidases in the synthesis of several critical biomolecules (1). These enzymes are either monooxygenases or dioxygenases (see Table 1). Symptoms of vitamin C deficiency, such as poor wound healing and lethargy, likely result from the impairment of these vitamin C-dependent enzymatic reactions leading to the insufficient synthesis of collagen, carnitine, and catecholamines (see Deficiency). Moreover, several dioxygenases involved in the regulation of gene expression and the maintenance of genome integrity require vitamin C as a cofactor. Indeed, research has recently uncovered the crucial role played by enzymes, such as the TET dioxygenases and Jumonji domain-containing histone demethylases, in the fate of cells and tissues (see Table 1). These enzymes contribute to the epigenetic regulation of gene expression by catalyzing reactions involved in the demethylation of DNA and histones.
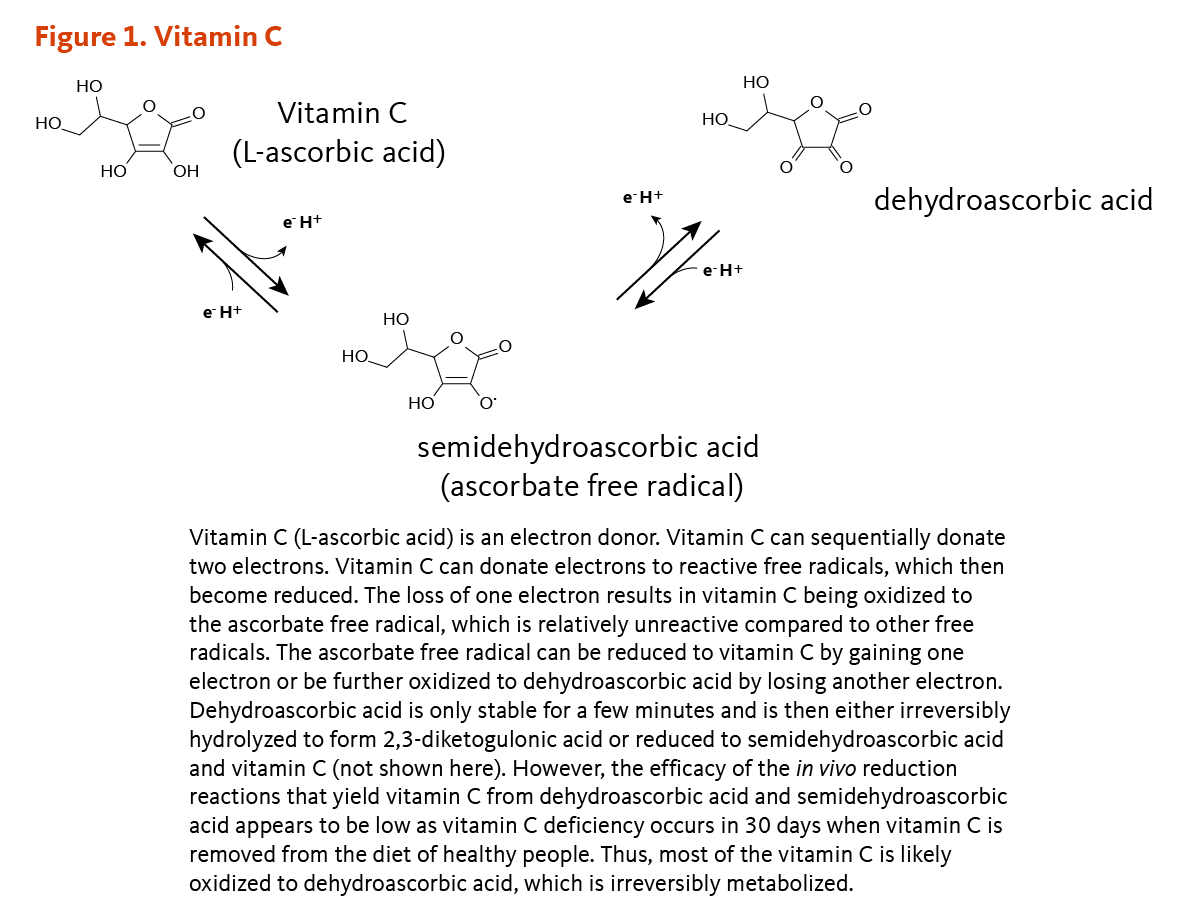
| Enzymes* | Functions |
|---|---|
| Monooxygenases | |
| Dopamine β-monooxygenase | Norepinephrine (Noradrenaline) biosynthesis |
| Peptidyl-glycine α-amidating monooxygenase | Amidation of peptide hormones |
| Dioxygenases | |
| 3 Prolyl 4-hydroxylase isoenzymes | Collagen hydroxylation |
| 3 Prolyl 3-hydroxylase isoenzymes | Collagen hydroxylation |
| 3 Lysyl hydroxylase isoenzymes | Collagen hydroxylation |
| 4 Hypoxia-inducible factor (HIF) isoenzymes | HIF hydroxylation |
| Trimethyllysine hydroxylase | Carnitine biosynthesis |
| γ-Butyrobetaine hydroxylase | Carnitine biosynthesis |
| 4-Hydroxyphenylpyruvate dioxygenase | Tyrosine metabolism |
| Ten-eleven translocation (TET) family of dioxygenases | Demethylation of DNA |
| Jumonji domain-containing histone demethylases | Demethylation of histones |
| *Monooxygenases catalyze the hydroxylation of one substrate, whereas dioxygenases catalyze a reaction that couples the hydroxylation of a specific substrate with the conversion (decarboxylation) of α-ketoglutarate into succinate. | |
The capacity of vitamin C to influence the methylation status of DNA and histones in mammalian cells supports a role for the vitamin in health and disease beyond what was previously understood, in particular by safeguarding genome integrity (3, 4).
Role in immunity
Vitamin C affects several components of the human immune system in vitro; for example, vitamin C has been shown to stimulate both the production (5-9) and function (10, 11) of leukocytes (white blood cells), especially neutrophils, lymphocytes, and phagocytes. Specific measures of functions stimulated by vitamin C include cellular motility (10), chemotaxis (10, 11), and phagocytosis (11). Neutrophils, mononuclear phagocytes, and lymphocytes accumulate vitamin C to high concentrations, which can protect these cell types from oxidative damage (12-14). In response to invading microorganisms, phagocytic leukocytes release non-specific toxins, such as superoxide radicals, hypochlorous acid ("bleach"), and peroxynitrite; these reactive oxygen species kill pathogens and, in the process, can damage the leukocytes themselves (15). Vitamin C, through its antioxidant functions, has been shown to protect leukocytes from self-inflicted oxidative damage (14). Phagocytic leukocytes also produce and release cytokines, including interferons, which have antiviral activity (16). Vitamin C has been shown to increase interferon production in vitro (17). Additional studies have reported that vitamin C enhances the chemotactic and microbial killing capacities of neutrophils and stimulates the proliferation and differentiation of B- and T-lymphocytes (reviewed in 18).
It is widely thought by the general public that vitamin C boosts immune function, yet human studies published to date are conflicting. Disparate results are likely due to study design issues, often linked to a lack of understanding of vitamin C pharmacokinetics and requirements (19, 20).
Finally, vitamin C increases the bioavailability of iron from foods by enhancing intestinal absorption of non-heme iron (see the article on Iron) (21).
Bioavailability
Depletion-repletion pharmacokinetic experiments demonstrated that plasma vitamin C concentration is tightly controlled by three primary mechanisms: intestinal absorption, tissue transport, and renal reabsorption (22). In response to increasing oral doses of vitamin C, plasma vitamin C concentration rises steeply at intakes between 30 and 100 mg/day. Plasma concentrations of ascorbate reach steady-state at concentrations between 60 and 80 micromoles/L (μmol/L). This is typically observed at doses between 200 to 400 mg/day in healthy young adults, with some degree of individual variation (23, 24).
One hundred percent absorption efficiency is observed when ingesting vitamin C at doses up to 200 mg at a time. Higher doses (>500 mg) result in fractionally less vitamin C being absorbed as the dose increases. Once plasma vitamin C concentrations reach saturation, additional vitamin C is largely excreted in the urine. Notably, intravenous administration of vitamin C bypasses absorptive control in the intestine such that very high concentrations of vitamin C can be achieved in the plasma; within a few hours, renal excretion restores vitamin C to baseline plasma concentrations (see Cancer Treatment) (25).
While plasma vitamin C concentration reflects recent dietary intake, leukocyte (white blood cell) vitamin C is thought to be an indicator of body stores. However, leukocyte vitamin C concentration does not accurately reflect vitamin C in several tissues and may specifically underestimate vitamin C uptake into skeletal muscle (26). Yet, plasma concentrations of vitamin C ≥50 μmol/L are sufficient to saturate muscle tissue vitamin C.
There is also some limited evidence suggesting that individuals who carry certain polymorphisms in genes involved in vitamin C transport and detoxification mechanisms may have lower plasma vitamin C concentrations even with high vitamin C intakes (see also Vascular complications of diabetes mellitus) (reviewed in 27).
Due to the pharmacokinetics and tight regulation of plasma vitamin C, supplementation with vitamin C will have variable effects in vitamin C-replete (plasma concentrations near saturation) versus sub-optimal (plasma concentrations <50 μmol/L), marginally deficient (plasma concentrations <23 μmol/L), or severely deficient (plasma concentrations <11 μmol/L) individuals (28). Scientific studies investigating vitamin C efficacy to prevent or treat disease need to assess baseline vitamin C status before embarking on an intervention or statistical analysis (22, 29-31).
For a more detailed discussion on the bioavailability of different forms of vitamin C, see the article, The Bioavailability of Different Forms of Vitamin C.
Deficiency
Severe vitamin C deficiency has been known for many centuries as the potentially fatal disease, scurvy. By the late 1700s, the British navy was aware that scurvy could be cured by eating oranges or lemons, even though vitamin C would not be isolated until the early 1930s. Symptoms of scurvy include subcutaneous bleeding, poor wound closure, and bruising easily, hair and tooth loss, and joint pain and swelling. Such symptoms appear to be related to the weakening of blood vessels, connective tissue, and bone, which all contain collagen. Early symptoms of scurvy like fatigue may result from diminished levels of carnitine, which is needed to derive energy from fat, or from decreased synthesis of the catecholamine norepinephrine (see Function). Scurvy is rare in developed countries because it can be prevented by as little as 10 mg of vitamin C daily (32). However, cases have occurred in children and the elderly on very restricted diets (33, 34).
The Recommended Dietary Allowance (RDA)
The recommended dietary allowance (RDA) for vitamin C is based on the amount of vitamin C intake necessary to maintain neutrophil vitamin C concentration with minimal urinary excretion of vitamin C and is proposed to provide sufficient antioxidant protection (Table 2) (35). The recommended intake for smokers is 35 mg/day higher than for nonsmokers, because smokers are under increased oxidative stress from the toxins in cigarette smoke and generally have lower blood concentrations of vitamin C (36).
| Life Stage | Age | Males (mg/day) | Females (mg/day) |
|---|---|---|---|
| Infants | 0-6 months | 40 (AI) | 40 (AI) |
| Infants | 7-12 months | 50 (AI) | 50 (AI) |
| Children | 1-3 years | 15 | 15 |
| Children | 4-8 years | 25 | 25 |
| Children | 9-13 years | 45 | 45 |
| Adolescents | 14-18 years | 75 | 65 |
| Adults | 19 years and older | 90 | 75 |
| Smokers | 19 years and older | 125 | 110 |
| Pregnancy | 18 years and younger | - | 80 |
| Pregnancy | 19 years and older | - | 85 |
| Breast-feeding | 18 years and younger | - | 115 |
| Breast-feeding | 19 years and older | - | 120 |
Disease Prevention
The amount of vitamin C required to help prevent chronic disease is higher than the amount required for prevention of scurvy. Information regarding vitamin C and the prevention of chronic disease is based on both observational prospective cohort studies and randomized controlled trials (29, 37). Prospective cohort studies can examine the incidence of a specific disease in relation to vitamin C intake or body status in a cohort of participants who are followed over time. In contrast, trials are intervention studies that can establish a causal relationship between an exposure and an outcome, e.g., by evaluating the effect of vitamin C supplementation on the incidence of chronic disease in participants randomly assigned to receive either vitamin C or placebo for a given length of time.
Cardiovascular disease
Endothelial dysfunction
Endothelial dysfunction is considered to be an early step in the development of atherosclerosis. Alterations in the structure and function of the vascular endothelium that lines the inner surface of all blood vessels are associated with the loss of normal nitric oxide-mediated endothelium-dependent vasodilation. Endothelial dysfunction results in widespread vasoconstriction and coagulation abnormalities. The measurement of brachial artery flow-mediated dilation (FMD) is often used as a functional marker of endothelial function; FMD values are inversely correlated with the risk of future cardiovascular events (38). A 2014 meta-analysis of 44 randomized controlled trials in subjects with or without chronic diseases summarized the effect of supplemental vitamin C on endothelial function by measuring FMD (19 studies), assessing forearm blood flow (20 studies), or by pulse wave analysis (5 trials) (39). Short-term supplementation with vitamin C was found to reduce endothelial dysfunction in subjects with heart failure, atherosclerosis, or diabetes mellitus, but it had no effect in those with hypertension. Vitamin C also limited endothelial dysfunction that was experimentally induced in healthy volunteers (39). Improved endothelial function was observed with daily vitamin C doses above 500 mg (39).
Hypertension
Hypertension is a major risk factor for cardiovascular disease, including coronary heart disease, stroke, and atrial fibrillation. An analysis that combined data from three, large, independent prospective cohorts, (1) Nurses’ Health Study 1 (NHS1; 88,540 women, median age 49 years); (2) Nurses’ Health Study 2 (NHS2; 97,315 women, median age 36 years); and (3) Health Professionals Follow-up Study (HPFS; 37,375 men, median age 52 years), found no association between the level of vitamin C intake and risk of developing hypertension (40). On the other hand, when plasma vitamin C concentration was measured, cross-sectional studies have consistently indicated an inverse relationship between plasma vitamin C concentration and blood pressure in both men and women (41-43). A 15-year follow-up of about 2,500 participants in the Coronary Artery Risk Development in Young Adults (CARDIA) study found that higher plasma vitamin C and a higher diet quality score were independently associated with a reduced risk of developing hypertension (44). Interestingly, there was no relationship between diet score and risk of hypertension in those with the lowest plasma vitamin C, and plasma vitamin C was positively associated with risk of hypertension in those with low diet scores (44).
A meta-analysis of 29 small randomized controlled trials of short durations (median duration, 8 weeks) in 1,407 participants (10 to 120 subjects per trial; including both normotensive and hypertensive subjects) found that daily supplementation with 60 to 4,000 mg of vitamin C (median dose, 500 mg) reduced systolic blood pressure by 3.84 mm Hg and diastolic blood pressure by 1.48 mm Hg (45). Good quality long-term trials are needed to examine whether the anti-hypertensive effect of vitamin C is sustained over time and eventually results in a reduced risk of cardiovascular events.
It is important for individuals with significantly elevated blood pressure not to rely on vitamin C supplementation alone to reduce their hypertension. They should instead seek or continue treatment with anti-hypertensive medication and make dietary and lifestyle changes in consultation with their health care provider.
Cardiovascular disease risk
Coronary heart disease (CHD) is characterized by the buildup of plaque inside the arteries that supply blood to the heart (atherosclerosis). Over years of buildup and accumulated damage to the coronary arteries, CHD may culminate in a myocardial infarction or heart attack. Many prospective cohort studies have examined the relationship between vitamin C intake from diet and supplements and CHD risk, the results of which have been pooled and analyzed in two separate analyses (46, 47). In 2004, a pooled analysis of nine prospective cohort studies found that supplemental vitamin C intake (≥400 mg/day for a mean of 10 years), but not dietary vitamin C intake, was inversely associated with CHD risk (46). Conversely, a 2008 meta-analysis of 14 cohort studies concluded that dietary, but not supplemental, vitamin C intake was inversely related to CHD risk (47). The most recent large prospective cohort study found an inverse association between dietary vitamin C intake and CHD mortality in Japanese women, but not in men (48). In spite of the variable association depending on source, these analyses indicate an overall inverse association between higher vitamin C intakes and CHD risk.
Limitations inherent to dietary assessment methodology, such as recall bias, measurement error, and residual confounding, may account for some of the inconsistent associations between vitamin C intake and CHD risk. In order to overcome such limitations, some prospective studies measured plasma or serum concentrations of vitamin C as a more reliable index of vitamin C intake and biomarker of body vitamin C status.
The European Investigation into Cancer and Nutrition (EPIC)-Norfolk prospective cohort study investigated the relationship between vitamin C status and incident heart failure in healthy adults (9,187 men and 11,112 women, aged 58.1+/-9.2 years) (49). After a mean follow-up of 12.8 years, plasma vitamin C was inversely associated with incident cases of heart failure. Specifically, plasma vitamin C ranged from approximately 23 to 70 μmol/L in men and 33 to 82 μmol/L in women; across this range, every 20 μmol/L increase in plasma vitamin C was associated with a 9% reduction in risk of heart failure. Although a primary source of dietary vitamin C, consumption of fruit and vegetables — assessed by food frequency questionnaire — was not found to be associated with a lower risk of congestive heart failure (49). This highlights the fact that limitations associated with dietary assessment methods such as food frequency questionnaires may be overcome by using biomarkers of nutrient intake (50, 51).
A 2017 review of eight published randomized controlled trials found inconsistent results from seven trials reporting on the effect of vitamin C supplementation on serum cholesterol and triglycerides, established risk factors for cardiovascular disease (52). Only one large trial in more than 14,000 older men participating in the Physicians’ Health Study II (PHS II) reported on cardiovascular outcomes. PHS II found that vitamin C supplementation (500 mg/day) for an average of eight years had no significant effect on major cardiovascular events, total myocardial infarction, or cardiovascular mortality (53). Notably, this study had several limitations (54), including no measurement of vitamin C status and the recruitment of a well-nourished study population.
There is a need for better quality studies to examine the effect of vitamin C on cardiovascular endpoints in participants with elevated risk of cardiovascular disease.
Stroke
A cerebrovascular event, or stroke, can be classified as hemorrhagic or ischemic. Hemorrhagic stroke occurs when a weakened blood vessel ruptures and bleeds into the surrounding brain tissue. Ischemic stroke occurs when an obstruction within a blood vessel blocks blood flow to the brain. Most (~80%) cerebrovascular events in high-income countries are ischemic in nature and associated with atherosclerosis as an underlying condition (55, 56).
With respect to vitamin C and cerebrovascular disease, a prospective cohort study that followed more than 2,000 residents of a rural Japanese community for 20 years found that the risk of stroke in those with the highest serum concentrations of vitamin C was 29% lower than in those with the lowest serum concentrations of vitamin C (57). Similarly, the EPIC-Norfolk study, a 10-year prospective cohort study in 20,649 adults, found that individuals with plasma vitamin C concentrations in the top quartile (25%) had a 42% lower risk of stroke compared to those in the lowest quartile (≥66 μmol/L vs. <41 μmol/L) (58). In both the Japanese (57) and EPIC-Norfolk (58) populations, blood vitamin C concentrations were highly correlated with fruit and vegetable intake. Therefore, as in many studies of vitamin C intake and chronic disease risk, it is difficult to separate the effects of vitamin C from the effects of other components of fruit and vegetables. For example, potassium — found at high levels in bananas, potatoes, and other fruit and vegetables — is known to be important in blood pressure regulation, and elevated blood pressure is a major risk factor for stroke (see the article on Potassium). A 2013 meta-analysis of 17 prospective cohort studies reported a 19% lower risk of stroke with the highest versus lowest dietary vitamin C intakes and a 38% lower risk with the highest versus lowest circulating vitamin C concentrations (59).
A randomized, double-blind, placebo-controlled trial in more than 14,000 older men participating in the Physicians’ Health Study II (PHS II) found that vitamin C supplementation (500 mg/day) for an average of eight years had no significant effect on the incidence of or mortality from any type of stroke (53). Other trials also failed to show any evidence of an effect of vitamin C on the risk of stroke. A meta-analysis of 10 trials that examined antioxidant vitamins, of which five included vitamin C, found no association between any antioxidant vitamin (vitamin C, vitamin E, or β-carotene), administered alone or in combination, and risk of stroke (60).
Cancer
Overall, observational prospective cohort studies have reported no or modest inverse associations between vitamin C intake and the risk of developing a given type of cancer (37, 61-63). Additional detail is provided below for those cancer subtypes with substantial scientific information obtained from prospective cohort studies. Randomized, double-blind, placebo-controlled trials that have tested the effect of vitamin C supplementation (alone or in combination with other antioxidant nutrients) on cancer incidence or mortality have shown no effect (64).
Breast cancer
Two large prospective studies found dietary vitamin C intake to be inversely associated with breast cancer incidence in certain subgroups. In the Nurses' Health Study, premenopausal women with a family history of breast cancer who consumed an average of 205 mg/day of vitamin C from food had a 63% lower risk of breast cancer than those who consumed an average of 70 mg/day (65). In the Swedish Mammography Cohort, overweight women who consumed an average of 110 mg/day of vitamin C had a 39% lower risk of breast cancer compared to overweight women who consumed an average of 31 mg/day (66). More recent prospective cohort studies have reported no association between dietary and/or supplemental vitamin C intake and breast cancer (67-69).
Stomach cancer
A number of observational studies have found increased dietary vitamin C intake to be associated with decreased risk of gastric (stomach) cancer, and laboratory experiments indicate that vitamin C inhibits the formation of carcinogenic N-nitroso compounds in the stomach (70-72). A nested case-control study in the EPIC study found a 45% lower risk of gastric cancer incidence in individuals in the highest (≥51 μmol/L) versus lowest (<29 μmol/L) quartile of plasma vitamin C concentration; no association was observed between dietary vitamin C intake and gastric cancer (73).
Infection with the bacteria, Helicobacter pylori (H. pylori), is known to increase the risk of stomach cancer and is associated with lower vitamin C content of stomach secretions (74, 75). Although two intervention studies failed to show a reduction in stomach cancer incidence with vitamin C supplementation (35), some research suggests that vitamin C supplementation may be a useful addition to standard H. pylori eradication therapy in reducing the risk of gastric cancer (76). Because vitamin C can inactivate urease (an enzyme that facilitates H. pylori survival and colonization of the gastric mucosa at low pH) in vitro, vitamin C may be most effective as a prophylactic agent in those without achlorhydria (77, 78).
Colon cancer
By pooling data from 13 prospective cohort studies comprising 676,141 participants, it was determined that dietary intake of vitamin C was not associated with colon cancer, while total intake of vitamin C (i.e., from food and supplements) was associated with a 19% reduced risk of colon cancer (79). Each of the cohort studies used self-administered food frequency questionnaires at baseline to assess vitamin C intake. Although the analysis adjusted for several lifestyle and known risk factors, the authors noted that other healthy behaviors and/or folate intake may have confounded the association.
Non-Hodgkin lymphoma
A population-based, prospective study, the Iowa Women’s Health Study, collected baseline data on diet and supplement use in 35,159 women (aged 55-69 years) and evaluated the risk of developing non-Hodgkin lymphoma (NHL) over 19 years of follow-up (80). Overall, an inverse association between fruit and vegetable intake and risk of NHL was observed. Additionally, dietary, but not supplemental, intake of vitamin C and other antioxidant nutrients (carotenoids, proanthocyanidins, and manganese) was inversely associated with NHL risk. Another large, multi-center, prospective study — the Women’s Health Initiative — that followed 154,363 postmenopausal women for 11 years found that dietary and supplemental vitamin C intake at baseline was inversely associated with diffuse B-cell lymphoma, a subtype of NHL (81).
Other site-specific cancer types
The Physicians’ Health Study II was a randomized, placebo-controlled trial that examined the effect of vitamin E (400 IU/day), vitamin C (500 mg/day), and a multivitamin supplement on the risk of cancer in 14,641 middle-aged male physicians over 10.3 years (7.6 years of active treatment plus 2.8 years post-treatment follow-up) (82). Supplementation with vitamin C had no effect on the overall risk of cancer or on the risk of prostate, bladder, or pancreatic cancer; there was a marginal reduction in colorectal cancer incidence with vitamin C compared to placebo (82).
Type 2 diabetes mellitus
In the National Institutes of Health (NIH)-American Association of Retired Persons (AARP) Diet and Health study that included 232,007 participants, the use of vitamin C supplements for at least seven times a week was associated with a 9% lower risk of developing type 2 diabetes mellitus compared to non-supplement use (83). In a cohort of 21,831 adults followed for 12 years in the EPIC-Norfolk study, high plasma vitamin C was found to be strongly associated with a reduced risk of diabetes (84). Additionally, several cross-sectional studies reported inverse associations between circulating vitamin C concentrations and markers of insulin resistance or glucose intolerance, such as glycated hemoglobin (HbA1c) concentration (50, 85, 86). Yet, short-term randomized controlled studies have found no effect of vitamin C supplementation on fasting glucose, fasting insulin, and HbA1c concentrations in healthy individuals (87). It is not known whether supplemental vitamin C could improve markers of glycemic control in subjects at risk of diabetes.
Adverse pregnancy outcomes
A 2015 meta-analysis of 29 randomized controlled trials found that administration of vitamin C during pregnancy, alone or in combination with a few other supplements, failed to reduce the risks of stillbirth, perinatal death, intrauterine growth restriction, preterm birth, premature rupture of membranes, and preeclampsia (88). Nonetheless, vitamin C supplementation led to a 36% lower risk of placental abruption and to a significant increase in gestational age at birth (88). Another meta-analysis of 40 randomized controlled trials in 276,820 women found no effect of vitamin C, alone or combined with vitamin E or multivitamins, when supplemented during pregnancy (starting prior to 20 weeks’ gestation), on the risks of overall fetal loss, miscarriage, stillbirth, and congenital malformation (89).
Cigarette smoking during pregnancy causes intrauterine growth restriction and preterm birth, among other pregnancy complications (90, 91), and is the primary cause of childhood respiratory illness (92). For some still unclear reasons, smoking has been associated with a lower risk of preeclampsia during pregnancy (93). A secondary analysis of a multicenter, randomized, double-blind, placebo-controlled trial in nearly 10,000 pregnant women found no reduction in the risk of preeclampsia with supplemental vitamin C (1,000 mg/day) and vitamin E (400 IU/day), regardless of women’s smoking status during pregnancy. However, antioxidant supplementation resulted in reduced risks of placental abruption and preterm birth in women who smoked during pregnancy but not in non-smokers (94). Another pilot multicenter trial found better lung function during the first week of life and lower risk of wheezing through one year of age in infants whose smoking mothers were randomized to receive vitamin C (500 mg/day) rather than a placebo during pregnancy (95). The Vitamin C to Decrease the Effects of Smoking in Pregnancy on Infant Lung Function [VCSIP] study is an ongoing trial designed to confirm these preliminary observations using more accurate measurements of pulmonary function in a larger sample of women randomized to receive supplemental vitamin C or placebo (96).
Alzheimer's disease
In the US, Alzheimer’s disease (AD) is the most common form of dementia, affecting 5.5 million individuals 65 years and over (97). Oxidative stress, neuroinflammation, β-amyloid plaque deposition, Tau protein-forming tangles, and neuronal cell death in the brain of subjects affected by AD have been associated with cognitive decline and memory loss. Lower vitamin C concentrations in the cerebrospinal fluid (CSF) and brain extracellular matrix of a mouse model of AD were found to increase oxidative stress and accelerate amyloid deposition and disease progression (98). In another AD mouse model that was lacking the ability to synthesize vitamin C, supplementation with a high versus low dose of vitamin C reduced amyloid deposition in the cortex and hippocampus and limited blood-brain barrier impairments and mitochondrial dysfunction (99).
The majority of large, population-based studies examining the relationship of vitamin C intake or supplementation with AD incidence have reported null results (100). In contrast, observational studies reported lower plasma vitamin C concentrations in AD patients compared to cognitively healthy subjects (101) and found better cognitive function or lower risk of cognitive impairment with higher plasma vitamin C (100).
Few studies have measured vitamin C concentration in the CSF, which more closely reflects the vitamin C status of the brain. Vitamin C is concentrated in the brain through a combination of active transport into brain tissue and retention via the blood-brain barrier (100). Although CSF vitamin C is maintained at concentrations several-fold higher than plasma vitamin C, the precise function of vitamin C in cognitive function and AD etiology is not yet fully understood (102). In a small, longitudinal biomarker study in 32 individuals with probable AD, a higher CSF-to-plasma vitamin C ratio at baseline was associated with a slower rate of cognitive decline at one year of follow-up (103). Impaired blood-brain barrier integrity may affect the brain’s ability to retain vitamin C and thus to maintain a high CSF-to-plasma vitamin C ratio. The significance of the CSF-to-plasma vitamin C ratio in AD progression requires further study.
The effect of vitamin C supplementation, in combination with other antioxidants, on CSF biomarkers and cognitive function has been examined in only a few trials involving AD patients. In a small (n=23), open-label trial, combined supplementation with vitamin C (1,000 mg/day) and vitamin E (400 IU/day) to AD patients taking a cholinesterase inhibitor significantly increased antioxidant levels and decreased lipoprotein oxidation in CSF after one year, but had no effect on the clinical course of AD compared to controls (104). A similar finding was obtained in a double-blind, randomized controlled trial in which combined supplementation with vitamin C (500 mg/day), vitamin E (800 IU/day), and α-lipoic acid (900 mg/day) for 16 weeks reduced lipoprotein oxidation in CSF but elicited no clinical benefit in individuals with mild-to-moderate AD (n=78) (105). In this latter trial, a greater decline in the Mini Mental State Examination (MMSE) score was observed in the supplemented group, however, the significance of this observation remains unclear. A third placebo-controlled trial in mildly cognitively impaired older adults (ages, 60-75 years) found that one-year supplementation with vitamin C (400 mg/day) and vitamin E (300 mg/day) improved antioxidant blood capacity but had no effect on MMSE scores (106).
At this time, avoidance of vitamin C deficiency or insufficiency, rather than supplementation in replete individuals, seems prudent for the promotion of healthy brain aging (101).
Cataracts
The lens of the eye focuses light, producing a clear, sharp image on the retina, a layer of tissue on the inside back wall of the eyeball. Age-related changes to the lens (thickening, loss of flexibility) and oxidative damage contribute to the formation of cataract, i.e., cloudiness or opacity in the lens that interferes with the clear focusing of images on the retina.
In humans, vitamin C concentration is about 15 to 20 times higher in the aqueous humor — fluid that fills the anterior and posterior chambers of the eye — than in plasma, suggesting that the vitamin may be playing an important role in the eye (107). Decreased vitamin C concentrations in the lens of the eye have been associated with increased severity of cataracts (108). A meta-analysis of observational studies found that a reduced risk of age-related cataract with higher dietary vitamin C intakes in case-control studies and with higher circulating vitamin C concentrations in cross-sectional studies. However, no such associations were found in pooled analyses of prospective cohort studies (109). In fact, two prospective cohort studies in Swedish men (110) and women (111) reported that high-dose single nutrient supplements of vitamin C were associated with an increased risk of cataract, especially in those on corticosteroid therapy.
A 2012 review of nine randomized controlled trials found no substantial effect of β-carotene, vitamin C, and vitamin E, administered individually or in combination over 2.1 to 12 years, on the risk of cataracts or cataract surgery (112). Although trials do not currently support the use of high-dose supplementation with vitamin C in cataract prevention, there is a consistent inverse association observed between high daily intake of fruit and/or vegetables (>5 servings/day) and risk of cataract (113).
Gout
Gout, a condition that afflicts more than 4% of US adults (114), is characterized by abnormally high blood concentrations of uric acid (urate) (115). Urate crystals may form in joints, resulting in inflammation and pain, as well as in the kidneys and urinary tract, resulting in kidney stones. The tendency to exhibit elevated blood uric acid concentrations and develop gout is often inherited; however, dietary and lifestyle modification may be helpful in both the prevention and treatment of gout (116). In an observational study that included 1,387 men, higher intakes of vitamin C were associated with lower serum concentrations of uric acid (117). In a cross-sectional study conducted in 4,576 African Americans, the odds of having hyperuricemia was associated with dietary intakes high in fructose, low in vitamin C, or with high fructose-to-vitamin C ratios (118). A prospective study that followed a cohort of 46,994 men for 20 years found that total daily vitamin C intake was inversely associated with incidence of gout, with higher intakes being associated with greater risk reductions (119). The results of this study also indicated that supplemental vitamin C may be helpful in the prevention of gout (119).
A 2011 meta-analysis of 13 randomized controlled trials in healthy individuals with elevated serum uric acid revealed that vitamin C supplementation (a median dose of 500 mg/day for a median duration of 30 days) modestly reduced serum uric acid concentrations by 0.35 mg/dL compared to placebo (120). Such a reduction falls within the range of assay variability and is unlikely to be clinically significant (121). An eight-week, open-label, controlled trial randomized 40 subjects with gout to receive either allopurinol (standard-of-care), vitamin C, or both treatments (122). The effect of vitamin C, alone or with allopurinol, decreasing serum uric acid was modest and much less than that of allopurinol alone. The trial did not examine the effect of vitamin C on other outcomes associated with gout (122).
Although observational studies suggested that supplemental vitamin C may be helpful to prevent incident and recurrent gout, this has not been demonstrated by intervention studies undertaken thus far. In addition, there is currently little evidence to support a role for vitamin C in the management of patients with gout (123).
Mortality
Two large prospective cohort studies assessed the relationship between dietary and supplemental vitamin C intakes and mortality. In the Vitamins and Lifestyle Study, 77,719 men and women (ages 50-76 years) were questioned at baseline on their use of dietary supplements during the previous 10 years (124). After five years of follow-up, vitamin C supplement use was associated with a small decreased risk of total mortality, although no association was found with cardiovascular disease- or cancer-specific mortality. In the second prospective cohort study, the Diet, Cancer and Health Study, 55,543 Danish adults (ages 50-64 years) were questioned at baseline about their lifestyle, diet, and supplement use during the previous 12 months (125). No association between dietary or supplemental intake of vitamin C and mortality was found after approximately 14 years of follow-up. In contrast, a 2014 meta-analysis of 10 prospective cohort studies in 17,696 women with breast cancer found a lower risk of total and breast cancer-specific mortality with higher supplemental and dietary vitamin C intakes (126). A 2012 meta-analysis of 29 trials found no effect of oral vitamin C, given alone or in combination with other antioxidants, on all-cause mortality (127).
In parallel to these dietary assessment studies, a strong inverse association between plasma vitamin C and mortality from all-causes, cardiovascular disease, and ischemic heart disease (and cancer in men only) was observed in the EPIC-Norfolk multicenter, prospective cohort study (128). After approximately four years of follow-up in 19,496 men and women (ages 45-79 years), a dose-response relationship was observed such that each 20 μmol/L increase in plasma vitamin C was associated with an estimated 20% risk reduction in all-cause mortality. Similarly, higher serum vitamin C concentrations were associated with decreased risks of cancer-specific and all-cause mortality in 16,008 adults from the US National Health and Nutrition Examination Survey (NHANES) III (1994-1998) (129).
Disease Treatment
Cardiovascular disease
Complications of cardiac procedures and surgeries
Periprocedural myocardial injury: Coronary angioplasty (also called percutaneous transluminal coronary angioplasty) is a nonsurgical procedure for treating obstructive coronary heart disease (CHD), including unstable angina pectoris, acute myocardial infarction, and multivessel CHD. Angioplasty involves temporarily inserting and inflating a tiny balloon into the clogged artery to help restore the blood flow to the heart. Periprocedural myocardial injury that occurs in up to one-third of patients undergoing otherwise uncomplicated angioplasty increases the risk of morbidity and mortality at follow-up.
One randomized, placebo-controlled trial has examined the effect of intravenous vitamin C administered to patients with stable angina undergoing elective coronary angioplasty (130). Administration of a 1-gram (g) vitamin C infusion one hour prior to the angioplasty reduced the concentrations of oxidative stress markers and improved microcirculatory perfusion compared to placebo (130). Another trial randomized 532 patients to receive a 3-g vitamin C infusion or a placebo (saline solution) within six hours prior to coronary angioplasty (131). Vitamin C treatment substantially reduced the incidence of periprocedural myocardial injury, as assessed by a reduction in the concentrations of two markers of myocardial injury, namely creatine kinase and troponin-I (131). A recent randomized controlled trial assessed the effect of vitamin C and vitamin E administration on reperfusion damage in patients who experienced acute myocardial infarction and underwent coronary angioplasty (see below) (132).
Myocardial reperfusion injury: Reperfusion injury refers to tissue damage occurring at the time of blood flow restoration (reperfusion) following transient ischemia. The heart muscle may become oxygen-deprived (ischemic) as the result of myocardial infarction or with aortic clamping during coronary artery bypass graft (CABG) surgery. Increased generation of reactive oxygen species (ROS) when the heart muscle's oxygen supply is restored might be an important contributor to myocardial damage occurring at reperfusion (133). Myocardial reperfusion injury leads to complications, such as reperfusion arrhythmias (see Atrial fibrillation) and myocardial stunning.
Vitamin C is depleted during and following cardiac surgery (134) and this might be due to the direct quenching of ROS, the regeneration of other antioxidants, and/or a massive synthesis of catecholamines (dopamine, epinephrine, norepinephrine) (135). Two randomized controlled trials conducted in the 1990s reported a reduction in reperfusion-induced oxidative stress and myocardial injury with intravenous (136) or oral (137) vitamin C administration prior to CABG surgery (reviewed in 135). A more recent randomized, double-blind, placebo-controlled trial has been designed to examine the effect of vitamin C and vitamin E administration on ischemia-reperfusion damage in 99 patients with acute myocardial infarction undergoing coronary angioplasty (132). Vitamin C infusion (sodium ascorbate: 3.20 mmol/min for 1 hour then 0.96 mmol/min for 2 hours) prior to reperfusion followed by oral supplementation with vitamin C (1 g/day) and vitamin E (400 IU/day) for 84 days effectively prevented a reduction in antioxidant capacity at reperfusion and for the next six to eight hours. The protocol also limited microvascular dysfunction (i.e., improved microcirculatory perfusion) and improved left ventricular ejection fraction at discharge (on day 84) (138, 139). However, no difference in the infarct size between antioxidant vitamin treatment and placebo was seen (138).
Atrial fibrillation: Atrial fibrillation is the most common type of cardiac arrhythmia. It is also a common post-cardiac surgery complication, leading to an increased risk of cardiovascular morbidity (e.g., heart failure, stroke) and mortality. Three meta-analyses of prospective cohort studies and randomized controlled trials have reported an overall reduction in the risk of post-operative atrial fibrillation following administration of primarily oral vitamin C (140-142). In most trials, participants received 2 g of vitamin C prior to undergoing CABG or valve replacement surgery and 1 to 2 g/day for five days post-surgery. Although only a minority of trials delivered vitamin C intravenously, this administration route appeared to be more effective at reducing the risk of atrial fibrillation — presumably due to higher plasma concentrations achieved (140). Of note, a subgroup analysis in one of the meta-analyses showed a reduction of post-operative atrial fibrillation with vitamin C in non US-based trials (10 trials) but no effect of vitamin C in US-based trials (5 trials) (140).
Cerebral ischemia-reperfusion injury
A small randomized controlled trial performed in 60 ischemic stroke patients showed that intravenous vitamin C administration (500 mg/day for 10 days, initiated day 1 post-stroke) had no effect on serum markers of oxidative stress or neurological outcomes compared to placebo (143).
Vascular complications of diabetes mellitus
Cardiovascular disease (CVD) is the leading cause of death in individuals with diabetes mellitus. The role of increased oxidative stress in the occurrence of vascular complications in subjects with diabetes has led to hypothesis that higher intakes of antioxidant nutrients could help lower the risk of CVD in diabetic subjects (144). A 2018 meta-analysis of randomized controlled trials investigating the effect of antioxidant vitamin supplementation in patients with type 2 diabetes found that most improvement in markers of oxidative stress and blood glucose control could be attributed to vitamin E (145). Another meta-analysis of trials found no effect of vitamins E and C, alone or in combination, on measures of β-cell function and insulin resistance (146). Yet, most studies were small and of short duration and thus did not assess the consequence of long-term use of antioxidant vitamins on the risk of vascular complications in diabetic patients. One 12-month randomized placebo-controlled trial in 456 participants with type 2 diabetes treated with metformin examined the effect of vitamin C (500 mg/day) or acetylsalicylic acid (aspirin; 100 mg/day) on risk factors for diabetes-related complications such as CVD (147). Both vitamin C and aspirin reduced fasting blood glucose and HbA1c concentrations and improved blood lipid profile in metformin-treated patients. Compared to placebo, both treatments were found to be more likely to limit risk factors contributing to diabetes-related complications, as well as to lower the risk of future cardiovascular events over a 10-year period (estimated using the Framingham risk score) (147).
Of note, it is possible that genetic differences among diabetic patients influence the effect of vitamin C supplementation on cardiovascular risk. In particular, a specific allele of the haptoglobin gene (Hp), namely Hp2, appears to be associated with an increased risk of diabetic vascular complications. Carriers of two copies of the Hp2 allele (Hp2-2) express a Hp protein that has a lower capacity to bind and remove pro-oxidant, free hemoglobin (Hb) from plasma, compared to Hp proteins coded by the Hp1-1 and Hp1-2 genotypes. When the results of the Women’s Antioxidant Vitamin Estrogen (WAVE) trial were reanalyzed based on Hp genotype, antioxidant therapy (1,000 mg/day of vitamin C + 800 IU/day of vitamin E) was associated with improvement of coronary atherosclerosis in diabetic women with Hp1-1 genotype but worsening of coronary atherosclerosis in those carrying the Hp2-2 genotype (148). Results from another study by the same investigators suggested that vitamin C could not prevent the oxidation of high-density lipoprotein (HDL)-cholesterol by glycated Hb-Hp2-2 complexes in vitro nor restore impaired HDL function in diabetic mice carrying the Hp2-2 genotype (149).
Sepsis
Sepsis and septic shock — defined as persistent sepsis-induced low blood pressure — are associated with elevated mortality rates in critically ill patients (150, 151). Because systemic inflammatory responses involve excessive oxidative stress, it has been suggested that providing antioxidant nutrients like vitamin C may improve the outcome of critically ill patients in intensive care units. In addition, hypovitaminosis C is common in critically ill patients, especially in those with septic shock, and persists despite enteral/parenteral nutritional therapy providing recommended amounts of vitamin C (152). Vitamin C requirements are likely to be increased in this population due to the hypermetabolic response driven by the systemic inflammatory reaction (152, 153). Intravenous administration of 50 mg or 200 mg of vitamin C per kg per day for 96 hours to patients with sepsis admitted in intensive care unit was found to correct vitamin C deficiency. Vitamin C also prevented the rise of Sequential Organ Failure Assessment (SOFA) and Acute Physiologic Assessment and Chronic Health Evaluation (APACHE) II scores — used to assess severity of illness and risk of mortality — observed in placebo-treated patients (154). Vitamin C infusion also lowered the concentration of markers of inflammation and endothelial injury in patients compared to placebo (154). In another randomized, double-blind, controlled trial in 28 critically ill patients with septic shock, infusion of 25 mg of vitamin C per kg every six hours for 72 hours significantly limited the requirement to vasopressor norepinephrine — decreasing both the dose and duration of treatment — and dramatically improved the 28-day survival rate (155). Similar results have been reported in septic patients given intravenous vitamin C (1.5 g/6 h), hydrocortisone (50 mg/6 h), and thiamin (200 mg/12 h) until hospital discharge. Compared to standard-of-care, this intervention cocktail more than halved the mean duration of vasopressor use (18.3 h versus 54.9 h) and reduced the odds of mortality by nearly 90% (156). Although intravenous vitamin C administration appears to be safe and well tolerated, there is a non-negligible risk of oxalate nephropathy (a rare cause of kidney failure) in these critically ill patients (157).
See also the section on Sepsis in the Symposium on Vitamin C — Part of the LPI’s 9th International Conference on Diet and Optimum Health.
Cancer
Route of administration
Studies in the 1970s and 1980s conducted by Linus Pauling, Ewan Cameron, and colleagues suggested that large doses of vitamin C (10 g/day infused intravenously for 10 days followed by at least 10 g/day orally indefinitely) were helpful in increasing the survival time and improving the quality of life of terminal cancer patients (158). Controversy surrounding the efficacy of vitamin C in cancer treatment ensued, leading to the recognition that the route of vitamin C administration is critical (22, 159). Compared to orally administered vitamin C, intravenous vitamin C can result in 30 to 70-fold higher plasma vitamin C concentrations (25). Higher plasma concentrations achieved via intravenous vitamin C administration are comparable to those that are toxic to cancer cells in culture. The anticancer mechanism of intravenous vitamin C action is under investigation. It may involve the production of high levels of hydrogen peroxide, selectively toxic to cancer cells (22, 160-162), or the deactivation of hypoxia inducible factor, a prosurvival transcription factor that protects cancer cells from various forms of stress (159, 163, 164). Vitamin C likely also plays a role in the maintenance of genome integrity and in the protection against cellular transformation through regulating DNA and histone demethylating enzymes (see Function) (165).
Safety
Current evidence from controlled clinical trials indicates that intravenous vitamin C is generally safe and well tolerated in cancer patients. Of note, because intravenous administration of 80 g of vitamin C precipitated hemolytic anemia in two subjects with glucose-6-phosphate dehydrogenase deficiency, patients due to receive high-dose vitamin C infusion are systematically screened for this genetic disorder (166). Four phase I clinical trials in patients with advanced cancer found that intravenous administration of vitamin C at doses up to 1.5 g/kg of body weight (equivalent to about 100 g/day for an average weight [70 kg] person) and 70 to 80 g/m2 was well tolerated and safe in pre-screened patients (167-170). A few observational studies in cancer patients undergoing chemotherapy and/or radiotherapy reported that complementary intravenous vitamin C treatment was associated with a reduction in treatment-associated side effects and an improved quality of life (171). A phase I study in nine patients with metastatic pancreatic cancer showed that millimolar concentrations of plasma vitamin C could be reached safely when administered in conjunction with the cancer chemotherapy drugs, gemcitabine and erlotinib (168).
Sensitivity to vitamin C
Retrospective in vitro colony formation assays revealed that patient leukemic cells displayed variable sensitivity to vitamin C treatment: leukemic cells from seven out of the nine patients who experienced a significant clinical benefit were sensitive to vitamin C in vitro (i.e., "responders"); the leukemic cells from the remaining six patients were not sensitive to vitamin C (i.e., "non-responders"). Thus, in vitro vitamin C sensitivity assays may provide predictive value for the clinical response to intravenous vitamin C treatment. The mechanisms underlying differential sensitivity to vitamin C are under investigation. In vitro experiments performed using 11 different cancer cell lines demonstrated that sensitivity to vitamin C correlated with the expression of catalase, an enzyme involved in the decomposition of hydrogen peroxide (172). Approximately one-half of the cell lines tested were resistant to vitamin C cytotoxicity, a response associated with high levels of catalase activity.
Sensitivity to vitamin C may also be determined by the expression of sodium-dependent vitamin C transporter-2 (SVCT-2), which transports vitamin C into cells (173). Higher SVCT-2 levels were associated with enhanced sensitivity to vitamin C in nine different breast cancer cell lines. Moreover, SVCT-2 was significantly expressed in 20 breast cancer tissue samples, but weakly expressed in normal tissues. Finally, mutations in genes coding for vitamin C-dependent TET demethylases, mutations that are common in cancer cells, may also contribute to resistance to vitamin C treatment (165).
Efficacy
Current evidence of the efficacy of intravenous vitamin C in cancer patients is limited to observational studies, uncontrolled interventions, and case reports (174, 175). There is a need for larger, longer-duration phase II clinical trials that test the efficacy of intravenous vitamin C in disease progression and overall survival (176).
See also the section on Cancer in the Symposium on Vitamin C — Part of the LPI’s 9th International Conference on Diet and Optimum Health.
Common cold
The work of Linus Pauling stimulated public interest in the use of doses greater than 1 g/day of vitamin C to prevent the common cold (177). In the past 40 years, numerous placebo-controlled trials have examined the effect of vitamin C supplementation on the prevention and treatment of colds. A 2013 meta-analysis of 53 placebo-controlled trials evaluated the effect of vitamin C supplementation on the incidence, duration, or severity of the common cold when taken as a continuous daily supplement (43 trials) or as therapy upon onset of cold symptoms (10 trials) (178). Regarding the incidence of colds, a difference was observed between two groups of participants. Regular supplementation with vitamin C (0.25 to 2 g/day) did not reduce the incidence of colds in the general population (23 trials); however, in participants undergoing heavy physical stress (e.g., marathon runners, skiers, or soldiers in subarctic conditions), vitamin C supplementation halved the incidence of colds (5 trials). A benefit of regular vitamin C supplementation was also seen in the duration of colds, with a greater benefit in children than in adults: The pooled effect of vitamin C supplementation was a 14% reduction in cold duration in children and an 8% reduction in adults. Finally, no significant effect of vitamin C supplementation (1-8 g/day) was observed in therapeutic trials in which vitamin C was administered after cold symptoms occurred.
In addition, a 2013 systematic review by the same investigators identified only two small randomized, double-blind, placebo-controlled trials that examined the effect of vitamin C on the incidence of respiratory infection-induced asthma (179). One trial found that vitamin C supplementation (1 g/day) for 14 weeks reduced the risk of asthma attacks precipitated by respiratory infection. The other trial randomized subjects diagnosed with infection-related asthma to receive 5 g/day of vitamin C or a placebo for one week; a lower proportion of participants was found to present with bronchial hypersensitivity to histamine — which characterizes chronic asthma — in the vitamin C group compared to the control group (reviewed in 179). These observations need to be confirmed in larger, well-designed trials.
Asthma
A 2013 systematic review identified 11 randomized controlled studies that evaluated the effect of vitamin C on asthma (eight trials) or exercise-induced bronchoconstriction (three trials) (180). Exercise-induced bronchoconstriction is a transient narrowing of the airways that occurs after exercise and is indicated by a ≥10% decline in Forced Expiratory Volume in 1 second (FEV1). In the three trials that included a total of 40 participants with exercise-induced bronchoconstriction, vitamin C administration before exercise (a 0.5-g dose on two subsequent days in one trial, a single dose of 2 g in the second trial, and 1.5 g daily for two weeks in the third trial) significantly reduced the exercise-induced decline in FEV1. Among the five out of eight trials in asthmatic subjects that reported on FEV1 outcomes, none found a difference between vitamin C supplementation and placebo (180).
Lead toxicity
Although the use of lead paint and leaded gasoline has been discontinued in the US, lead toxicity continues to be a significant health problem, especially in children living in urban areas. Abnormal growth and development have been observed in infants of women exposed to lead during pregnancy, while children who are chronically exposed to lead are more likely to develop learning disabilities, behavioral problems, and to have a low IQ. In adults, lead toxicity may result in kidney damage, high blood pressure, and anemia.
Several cross-sectional studies have reported an inverse association between vitamin C status and blood lead concentration. For instance, in a study of 747 older men, blood lead concentration was significantly higher in those who reported total dietary vitamin C intakes averaging less than 109 mg/day compared to those with higher vitamin C intakes (181). A much larger study of 19,578 people, including 4,214 children from 6 to 16 years of age, found higher serum vitamin C concentrations to be associated with significantly lower blood lead concentrations (182). A US national survey of more than 10,000 adults found that blood lead concentrations were inversely related to serum vitamin C concentrations (183).
Cigarette smoking or second-hand exposure to cigarette smoke contributes to increased blood lead concentration and a state of chronic low-level lead exposure. An intervention trial in 75 adult male smokers found that supplementation with 1,000 mg/day of vitamin C resulted in significantly lower blood lead concentration over a four-week treatment period compared to placebo (184). A lower dose of 200 mg/day did not significantly affect blood lead concentration, although serum vitamin C concentrations were not different from those in the group who took 1,000 mg/day.
The mechanism(s) by which vitamin C reduces blood lead concentration is not known, yet it has been proposed that vitamin C could inhibit intestinal absorption (184) or enhance urinary excretion of lead (185).
Sources
Unlike plants and most animals, humans have lost the ability to synthesize vitamin C endogenously and therefore have an essential dietary requirement for this vitamin (see The Recommended Dietary Allowance). Results from 7,277 participants in the US National Health and Nutrition Examination Survey (NHANES) 2003-2004 indicated that an estimated 7.1% of individuals ages ≥6 years were deficient in vitamin C — based on serum vitamin C concentrations <11.4 μmol/L (36). The national study identified smokers and those of lower socioeconomic status to both be at higher risk for vitamin C deficiency (36).
Food sources
As shown in Table 3, different fruit and vegetables vary in their vitamin C content, but five servings (2½ cup-equivalents) of a variety of fruit and vegetables should average out to about 150 to 200 mg of vitamin C, especially if vitamin C-rich fruits are consumed. If you wish to check foods for their vitamin C content, search USDA's FoodData Central.
Supplements
Vitamin C (L-ascorbic acid) is available in many forms, but there is little scientific evidence that any one form is better absorbed or more effective than another. Most experimental and clinical research uses ascorbic acid or its sodium salt, called sodium ascorbate. Natural and synthetic L-ascorbic acid are chemically identical and there are no known differences regarding biological activities or bioavailability (186).
Mineral ascorbates
Mineral salts of vitamin C are considered less acidic than vitamin C and therefore are considered "buffered." Some people find them less irritating to the gastrointestinal tract than ascorbic acid. Sodium ascorbate and calcium ascorbate are the most common forms, although a number of other mineral ascorbates are available. Sodium ascorbate provides 111 mg of sodium (889 mg of ascorbic acid) per 1,000 mg of sodium ascorbate, and calcium ascorbate generally provides 90 to 110 mg of calcium (890-910 mg of ascorbic acid) per 1,000 mg of calcium ascorbate.
Vitamin C with flavonoids
Flavonoids are a class of water-soluble plant pigments that are often found in vitamin C-rich fruit and vegetables, especially citrus fruit and berries (see the article on Flavonoids). There is little evidence that the flavonoids in most commercial preparations increase the bioavailability or efficacy of vitamin C (187). Some, yet not all, studies in animal models such as vitamin C-deficient guinea pigs or genetically scorbutic rats found an increased uptake of vitamin C in peripheral circulation and specific organs in the presence of flavonoids. However, studies conducted in humans found no differences in bioavailability of vitamin C from flavonoid-rich whole fruit or fruit juice and synthetic vitamin C (reviewed in 186).
Vitamin C and metabolites
One supplement, Ester-C®, contains mainly calcium ascorbate and includes small amounts of the vitamin C metabolites, dehydroascorbic acid (oxidized ascorbic acid), calcium threonate, and trace amounts of xylonate and lyxonate. Although these metabolites are purported to increase the bioavailability of vitamin C, the only published study in humans addressing this issue found no difference between Ester-C® and commercially available vitamin C tablets with respect to the absorption and urinary excretion of vitamin C (187). Ester-C® should not be confused with ascorbyl palmitate, which is also marketed as "vitamin C ester" (see below).
Ascorbyl palmitate
Ascorbyl palmitate is a vitamin C ester (i.e., ascorbic acid linked to a fatty acid). In this case, vitamin C is esterified to the saturated fatty acid, palmitic acid, resulting in a fat-soluble form of vitamin C. Ascorbyl palmitate has been added to a number of skin creams due to interest in its antioxidant properties, as well as its importance in collagen synthesis (see the separate article, Vitamin C and Skin Health) (188). Although ascorbyl palmitate is also available as an oral supplement, most of it is likely hydrolyzed to ascorbic acid and palmitic acid in the digestive tract before it is absorbed (189). Ascorbyl palmitate is marketed as "vitamin C ester," which should not be confused with Ester-C® (see above).
Other formulations of vitamin C
One small placebo-controlled, cross-over trial in 11 men showed that the oral administration of 4 g of vitamin C resulted in a greater vitamin C concentration in plasma over a four-hour period when vitamin C was encapsulated in liposomes compared to unencapsulated vitamin C (190). Although liposomal encapsulation could increase vitamin C bioavailability, plasma vitamin C concentrations were much lower than those achieved with intravenous vitamin C administration (190).
For a more detailed review of scientific research on the bioavailability of different forms of vitamin C, see The Bioavailability of Different Forms of Vitamin C.
Safety
Toxicity
A number of possible adverse health effects of very large doses of vitamin C have been identified, mainly based on in vitro experiments or isolated case reports, and include genetic mutations, birth defects, cancer, atherosclerosis, kidney stones, "rebound scurvy," increased oxidative stress, excess iron absorption, vitamin B12 deficiency, and erosion of dental enamel. However, none of these alleged adverse health effects have been confirmed in subsequent studies, and there is no reliable scientific evidence that doses of vitamin C up to 10 g/day in adults are toxic or detrimental to health. The concern of kidney stone formation with vitamin C supplementation is discussed below.
With the latest RDA published in 2000, a tolerable upper intake level (UL) for vitamin C was set for the first time (Table 4). A UL of 2 g (2,000 mg) daily was recommended in order to prevent generally healthy adults from experiencing diarrhea and gastrointestinal disturbances (35). Such symptoms are not generally serious, especially if they resolve with temporary discontinuation of vitamin C supplementation.
Kidney stones
Because oxalate is a metabolite of vitamin C, there is some concern that high vitamin C intake could increase the risk of calcium oxalate kidney stones. Some (24, 191, 192), but not all (193-195), studies have reported that supplemental vitamin C increases urinary oxalate concentrations. Whether any increase in oxalate levels would translate to an elevation in risk for kidney stones has been examined in several epidemiological studies. Two large prospective cohort studies, one following 45,251 men for six years and the other following 85,557 women for 14 years, reported that consumption of ≥1,500 mg of vitamin C daily did not increase the risk of kidney stone formation compared to those consuming <250 mg daily (196, 197). On the other hand, two other large prospective studies reported that a high intake of vitamin C was associated with an increased risk of kidney stone formation in men (198, 199). Specifically, the Health Professionals Follow-Up Study collected data on dietary and supplemental vitamin C intake every four years in 45,619 male health professionals (ages 40-75 years) (198). After 14 years of follow-up, it was found that men who consumed ≥1,000 mg/day of vitamin C had a 41% higher risk of kidney stones compared to men consuming <90 mg of vitamin C daily. In the Cohort of Swedish Men study, self-reported use of single-nutrient vitamin C supplements (taken seven or more times per week) at baseline was associated with a two-fold higher risk of incident kidney stones among 48,840 men (ages 45-79 years) followed for 11 years (199). Despite conflicting results, it may be prudent for individuals predisposed to oxalate kidney stone formation to avoid high-dose vitamin C supplementation.
Drug interactions
Overall, evidence suggesting specific drugs can lower blood vitamin C concentrations in humans is limited. Dihydropyridine calcium channel blockers (e.g., nicardipine, nifedipine) can inhibit vitamin C uptake by intestinal cells in vitro. However, a reduction in blood vitamin C concentrations with these drugs has not been reported in humans (200). Aspirin can impair vitamin C status if taken frequently (201).
Conversely, there are case reports suggesting that supplemental vitamin C may lower blood concentrations of some medications, such as fluphenazine (the antipsychotic drug, Prolixin) and indinavir (the antiretroviral drug, Crixivan) (200). There is some evidence, though controversial, that vitamin C interacts with anticoagulant medications like warfarin (Coumadin). Large doses of vitamin C may block the action of warfarin and thus lower its effectiveness. Individuals on anticoagulants should limit their vitamin C intake to <1 g/day and have their prothrombin time monitored by the clinician following their anticoagulant therapy (200). In addition, vitamin C may bind aluminum in the gut and increase the absorption of aluminum-containing compounds (e.g., aluminum-containing antacids, aluminum-containing phosphate binders). People with impaired kidney function may be at risk for aluminum toxicity when supplemental vitamin C is taken at the same time as these compounds (200, 201). Finally, supplemental vitamin C may increase blood estrogen concentrations in women using oral contraceptives or hormone replacement therapy (200).
The potential effect of antioxidants during chemotherapy is not well understood, yet only likely to be an issue if a specific chemotherapeutic agent acts through an oxidative mechanism, which is uncommon (171). It is not clear whether vitamin C given parenterally could diminish or increase the efficacy of chemotherapy drugs — in particular, akylating agents (e.g., cyclophosphamide, busulfan), antitumor antibiotics (e.g., doxorubicin, bleomycin), and arsenic trioxide. Patients are advised to discuss with their oncologist before using vitamin C supplements (200, 201).
Because high doses of vitamin C have also been found to interfere with the interpretation of certain laboratory tests (e.g., serum bilirubin, serum creatinine, and the stool guaiac assay for occult blood), it is important to inform one's health care provider of any recent supplement use.
Antioxidant supplements and HMG-CoA reductase inhibitors (statins)
A three-year randomized controlled trial in 160 patients with documented coronary heart disease and low blood HDL concentrations found that a combination of simvastatin (Zocor) and niacin increased HDL concentration, inhibited the progression of coronary artery stenosis (narrowing), and decreased the frequency of cardiovascular events, such as myocardial infarction and stroke (202). Surprisingly, when an antioxidant combination (1,000 mg vitamin C, 800 IU vitamin E, 100 µg selenium, and 25 mg β-carotene daily) was taken with the simvastatin-niacin combination, the protective effects were diminished. Since the antioxidants were taken together in this trial, the individual contribution of vitamin C cannot be determined. In contrast, a much larger trial in more than 20,000 men and women with coronary heart disease or diabetes mellitus found that simvastatin and an antioxidant combination (600 mg vitamin E, 250 mg vitamin C, and 20 mg β-carotene daily) did not diminish the cardioprotective effects of simvastatin therapy over a five-year period (203). These contradictory findings indicate that further research is needed on potential interactions between antioxidant supplements and cholesterol-lowering drugs, such as HMG-CoA reductase inhibitors (statins).
Does vitamin C promote oxidative damage under physiological conditions?
Vitamin C is known to function as a highly effective antioxidant in living organisms. However, in test tube experiments, vitamin C can interact with some free metal ions and lead to the generation of potentially damaging free radicals. Although free metal ions are not generally found under physiological conditions, the idea that high doses of vitamin C might be able to promote oxidative damage in vivo has received a great deal of attention. Widespread publicity has been given to a few studies suggesting a pro-oxidant effect of vitamin C (204, 205), but these studies turned out to be either flawed or of no physiological relevance. A comprehensive review of the literature found no credible scientific evidence that supplemental vitamin C promotes oxidative damage under physiological conditions or in humans (206).
Linus Pauling Institute Recommendation
Combined evidence from metabolic, pharmacokinetic, and observational studies, and from randomized controlled trials supports consuming sufficient vitamin C to achieve plasma concentrations of at least 60 μmol/L. While most generally healthy young adults can achieve these plasma concentrations with daily vitamin C intake of at least 200 mg/day, some individuals may have a lower vitamin C absorptive capacity than what is currently documented. Thus, the Linus Pauling Institute recommends a vitamin C intake of 400 mg daily for adults to ensure replete tissue concentrations (29) — an amount substantially higher than the RDA yet with minimal risk of side effects.
This recommendation can be met through food if the diet includes at least several servings of vitamin C-rich fruit and vegetables (e.g., citrus fruit, kiwifruit, peppers; see Food sources) as part of the daily recommended fruit and vegetable intake (see article on Fruit and Vegetables). Most multivitamin supplements provide at least 60 mg of vitamin C.
Older adults (>50 years)
Whether older adults have higher requirements for vitamin C is not yet known with certainty, yet some older populations have been found to have vitamin C intakes considerably below the RDA of 75 and 90 mg/day for women and men, respectively (207). A vitamin C intake of at least 400 mg daily may be particularly important for older adults who are at higher risk for age-related chronic diseases. Pharmacokinetic studies in older adults have not yet been conducted, but there is some evidence suggesting that the efficiency of one of the molecular mechanisms for the cellular uptake of vitamin C declines with age (208). Because maximizing blood concentrations of vitamin C may be important in protecting against oxidative damage to cells and biological molecules, a vitamin C intake of at least 400 mg daily might benefit older adults who are at higher risk for chronic diseases caused, in part, by oxidative damage, such as heart disease, stroke, certain cancers, and cataract.
For more information on the difference between Dr Linus Pauling's recommendation and the Linus Pauling Institute's recommendation for vitamin C intake, select the highlighted text.
Authors and Reviewers
Originally written in 2000 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in November 2002 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in September 2003 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in December 2004 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in January 2006 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in September 2009 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in November 2013 by:
Giana Angelo, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in July 2018 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in December 2018 by:
Anitra C. Carr, Ph.D.
Research Associate Professor
Department of Pathology & Biomedical Science
University of Otago
Christchurch, New Zealand
Reviewed in December 2018 by:
Alexander J. Michels, Ph.D.
Research Associate
Linus Pauling Institute
Oregon State University
Copyright 2000-2024 Linus Pauling Institute
References:
1. Levine M, Padayatty SJ. Vitamin C. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease, 11th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2014:416-426.
2. Englard S, Seifter S. The biochemical functions of ascorbic acid. Annu Rev Nutr. 1986;6:365-406. (PubMed)
3 Camarena V, Wang G. The epigenetic role of vitamin C in health and disease. Cell Mol Life Sci. 2016;73(8):1645-1658. (PubMed)
4 Young JI, Zuchner S, Wang G. Regulation of the epigenome by vitamin C. Annu Rev Nutr. 2015;35:545-564. (PubMed)
5 Jariwalla RJ, Harakeh S. Antiviral and immunomodulatory activities of ascorbic acid. In: Harris JR, ed. Subcellular Biochemistry. Vol. 25. Ascorbic Acid: Biochemistry and Biomedical Cell Biology. New York: Plenum Press; 1996:215-231.
6 Kennes B, Dumont I, Brohee D, Hubert C, Neve P. Effect of vitamin C supplements on cell-mediated immunity in old people. Gerontology. 1983;29(5):305-310. (PubMed)
7 Panush RS, Delafuente JC, Katz P, Johnson J. Modulation of certain immunologic responses by vitamin C. III. Potentiation of in vitro and in vivo lymphocyte responses. Int J Vitam Nutr Res Suppl. 1982;23:35-47. (PubMed)
8 Prinz W, Bortz R, Bregin B, Hersch M. The effect of ascorbic acid supplementation on some parameters of the human immunological defence system. Int J Vitam Nutr Res. 1977;47(3):248-257. (PubMed)
9 Vallance S. Relationships between ascorbic acid and serum proteins of the immune system. Br Med J. 1977;2(6084):437-438. (PubMed)
10. Anderson R, Oosthuizen R, Maritz R, Theron A, Van Rensburg AJ. The effects of increasing weekly doses of ascorbate on certain cellular and humoral immune functions in normal volunteers. Am J Clin Nutr. 1980;33(1):71-76. (PubMed)
11. Levy R, Shriker O, Porath A, Riesenberg K, Schlaeffer F. Vitamin C for the treatment of recurrent furunculosis in patients with impaired neutrophil functions. J Infect Dis. 1996;173(6):1502-1505. (PubMed)
12. Bergsten P, Amitai G, Kehrl J, Dhariwal KR, Klein HG, Levine M. Millimolar concentrations of ascorbic acid in purified human mononuclear leukocytes. Depletion and reaccumulation. J Biol Chem. 1990;265(5):2584-2587. (PubMed)
13. Evans RM, Currie L, Campbell A. The distribution of ascorbic acid between various cellular components of blood, in normal individuals, and its relation to the plasma concentration. Br J Nutr. 1982;47(3):473-482. (PubMed)
14. Jariwalla RJ, Harakeh S. Mechanisms underlying the action of vitamin C in viral and immunodeficiency disease. In: Packer L, Fuchs J, eds. Vitamin C in Health and Disease. New York: Macel Dekker, Inc.; 1997:309-322.
15. Alberts B, Bray D, Lewis J, Raff M. Differentiated cells and the maintenance of tissues. Molecular Biology of the Cell. 3rd ed. New York: Garland Publishing, Inc.; 1994:1139-1193.
16. Pauling L. The immune system. How to Live Longer and Feel Better. 20th Anniversary ed. Corvallis: Oregon State University Press; 2006:105-111.
17. Dahl H, Degre M. The effect of ascorbic acid on production of human interferon and the antiviral activity in vitro. Acta Pathol Microbiol Scand B. 1976;84B(5):280-284. (PubMed)
18. Carr AC, Maggini S. Vitamin C and immune function. Nutrients. 2017;9(11). (PubMed)
19. Lykkesfeldt J, Poulsen HE. Is vitamin C supplementation beneficial? Lessons learned from randomised controlled trials. Br J Nutr. 2010;103(9):1251-1259. (PubMed)
20. Michels AJ, Frei B. Myths, artifacts, and fatal flaws: identifying limitations and opportunities in vitamin C research. Nutrients. 2013;5(12):5161-5192. (PubMed)
21. Johnston CS. Vitamin C. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Ames, Iowa: Wiley-Blackwell; 2012:248-260.
22. Levine M, Padayatty SJ, Espey MG. Vitamin C: a concentration-function approach yields pharmacology and therapeutic discoveries. Adv Nutr. 2011;2(2):78-88. (PubMed)
23. Levine M, Wang Y, Padayatty SJ, Morrow J. A new recommended dietary allowance of vitamin C for healthy young women. Proc Natl Acad Sci U S A. 2001;98(17):9842-9846. (PubMed)
24. Levine M, Conry-Cantilena C, Wang Y, et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci U S A. 1996;93(8):3704-3709. (PubMed)
25. Padayatty SJ, Sun H, Wang Y, et al. Vitamin C pharmacokinetics: implications for oral and intravenous use. Ann Intern Med. 2004;140(7):533-537. (PubMed)
26. Carr AC, Bozonet SM, Pullar JM, Simcock JW, Vissers MC. Human skeletal muscle ascorbate is highly responsive to changes in vitamin C intake and plasma concentrations. Am J Clin Nutr. 2013;97(4):800-807. (PubMed)
27. Michels AJ, Hagen TM, Frei B. Human genetic variation influences vitamin C homeostasis by altering vitamin C transport and antioxidant enzyme function. Annu Rev Nutr. 2013;33:45-70. (PubMed)
28. Carr AC, Pullar JM, Bozonet SM, Vissers MC. Marginal ascorbate status (hypovitaminosis C) results in an attenuated response to vitamin C supplementation. Nutrients. 2016;8(6). (PubMed)
29. Frei B, Birlouez-Aragon I, Lykkesfeldt J. Authors' perspective: What is the optimum intake of vitamin C in humans? Crit Rev Food Sci Nutr. 2012;52(9):815-829. (PubMed)
30. Levine M, Rumsey SC, Daruwala R, Park JB, Wang Y. Criteria and recommendations for vitamin C intake. JAMA. 1999;281(15):1415-1423. (PubMed)
31. Lykkesfeldt J, Christen S, Wallock LM, Chang HH, Jacob RA, Ames BN. Ascorbate is depleted by smoking and repleted by moderate supplementation: a study in male smokers and nonsmokers with matched dietary antioxidant intakes. Am J Clin Nutr. 2000;71(2):530-536. (PubMed)
32. Sauberlich HE. A history of scurvy and vitamin C. In: Packer L, Fuchs J, eds. Vitamin C in health and disease. New York: Marcel Decker, Inc.; 1997:1-24.
33. Stephen R, Utecht T. Scurvy identified in the emergency department: a case report. J Emerg Med. 2001;21(3):235-237. (PubMed)
34. Weinstein M, Babyn P, Zlotkin S. An orange a day keeps the doctor away: scurvy in the year 2000. Pediatrics. 2001;108(3):E55. (PubMed)
35. Food and Nutrition Board, Institute of Medicine. Vitamin C. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids. Washington, D.C.: National Academy Press; 2000:95-185. (National Academy Press)
36. Schleicher RL, Carroll MD, Ford ES, Lacher DA. Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003-2004 National Health and Nutrition Examination Survey (NHANES). Am J Clin Nutr. 2009;90(5):1252-1263. (PubMed)
37. Carr AC, Frei B. Toward a new recommended dietary allowance for vitamin C based on antioxidant and health effects in humans. Am J Clin Nutr. 1999;69(6):1086-1107. (PubMed)
38. Matsuzawa Y, Kwon TG, Lennon RJ, Lerman LO, Lerman A. Prognostic value of flow-mediated vasodilation in brachial artery and fingertip artery for cardiovascular events: a systematic review and meta-analysis. J Am Heart Assoc. 2015;4(11). (PubMed)
39. Ashor AW, Lara J, Mathers JC, Siervo M. Effect of vitamin C on endothelial function in health and disease: a systematic review and meta-analysis of randomised controlled trials. Atherosclerosis. 2014;235(1):9-20. (PubMed)
40. Forman JP, Choi H, Curhan GC. Fructose and vitamin C intake do not influence risk for developing hypertension. J Am Soc Nephrol. 2009;20(4):863-871. (PubMed)
41. Block G, Jensen CD, Norkus EP, Hudes M, Crawford PB. Vitamin C in plasma is inversely related to blood pressure and change in blood pressure during the previous year in young Black and White women. Nutr J. 2008;7:35. (PubMed)
42. Moran JP, Cohen L, Greene JM, et al. Plasma ascorbic acid concentrations relate inversely to blood pressure in human subjects. Am J Clin Nutr. 1993;57(2):213-217. (PubMed)
43. Myint PK, Luben RN, Wareham NJ, Khaw KT. Association between plasma vitamin C concentrations and blood pressure in the European prospective investigation into cancer-Norfolk population-based study. Hypertension. 2011;58(3):372-379. (PubMed)
44. Buijsse B, Jacobs DR, Jr., Steffen LM, Kromhout D, Gross MD. Plasma ascorbic acid, a priori diet quality score, and incident hypertension: a prospective cohort study. PLoS One. 2015;10(12):e0144920. (PubMed)
45. Juraschek SP, Guallar E, Appel LJ, Miller ER, 3rd. Effects of vitamin C supplementation on blood pressure: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2012;95(5):1079-1088. (PubMed)
46 Knekt P, Ritz J, Pereira MA, et al. Antioxidant vitamins and coronary heart disease risk: a pooled analysis of 9 cohorts. Am J Clin Nutr. 2004;80(6):1508-1520. (PubMed)
47. Ye Z, Song H. Antioxidant vitamins intake and the risk of coronary heart disease: meta-analysis of cohort studies. Eur J Cardiovasc Prev Rehabil. 2008;15(1):26-34. (PubMed)
48 Kubota Y, Iso H, Date C, et al. Dietary intakes of antioxidant vitamins and mortality from cardiovascular disease: the Japan Collaborative Cohort Study (JACC) study. Stroke. 2011;42(6):1665-1672. (PubMed)
49. Pfister R, Sharp SJ, Luben R, Wareham NJ, Khaw KT. Plasma vitamin C predicts incident heart failure in men and women in European Prospective Investigation into Cancer and Nutrition-Norfolk prospective study. Am Heart J. 2011;162(2):246-253. (PubMed)
50. Carter P, Gray LJ, Troughton J, Khunti K, Davies MJ. Fruit and vegetable intake and incidence of type 2 diabetes mellitus: systematic review and meta-analysis. BMJ. 2010;341:c4229. (PubMed)
51. Dehghan M, Akhtar-Danesh N, McMillan CR, Thabane L. Is plasma vitamin C an appropriate biomarker of vitamin C intake? A systematic review and meta-analysis. Nutr J. 2007;6:41. (PubMed)
52. Al-Khudairy L, Flowers N, Wheelhouse R, et al. Vitamin C supplementation for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2017;3:Cd011114. (PubMed)
53. Sesso HD, Buring JE, Christen WG, et al. Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians' Health Study II randomized controlled trial. JAMA. 2008;300(18):2123-2133. (PubMed)
54. Roberts LJ, 2nd, Traber MG, Frei B. Vitamins E and C in the prevention of cardiovascular disease and cancer in men. Free Radic Biol Med. 2009;46(11):1558. (PubMed)
55. Hankey GJ. The global and regional burden of stroke. Lancet Glob Health. 2013;1(5):e239-240. (PubMed)
56. Tsivgoulis G, Safouris A, Kim DE, Alexandrov AV. Recent Advances in Primary and Secondary Prevention of Atherosclerotic Stroke. J Stroke. 2018;20(2):145-166. (PubMed)
57. Yokoyama T, Date C, Kokubo Y, Yoshiike N, Matsumura Y, Tanaka H. Serum vitamin C concentration was inversely associated with subsequent 20-year incidence of stroke in a Japanese rural community. The Shibata study. Stroke. 2000;31(10):2287-2294. (PubMed)
58. Myint PK, Luben RN, Welch AA, Bingham SA, Wareham NJ, Khaw KT. Plasma vitamin C concentrations predict risk of incident stroke over 10 y in 20 649 participants of the European Prospective Investigation into Cancer Norfolk prospective population study. Am J Clin Nutr. 2008;87(1):64-69. (PubMed)
59. Chen GC, Lu DB, Pang Z, Liu QF. Vitamin C intake, circulating vitamin C and risk of stroke: a meta-analysis of prospective studies. J Am Heart Assoc. 2013;2(6):e000329. (PubMed)
60. Ye Y, Li J, Yuan Z. Effect of antioxidant vitamin supplementation on cardiovascular outcomes: a meta-analysis of randomized controlled trials. PLoS One. 2013;8(2):e56803. (PubMed)
61. Bertoia M, Albanes D, Mayne ST, Mannisto S, Virtamo J, Wright ME. No association between fruit, vegetables, antioxidant nutrients and risk of renal cell carcinoma. Int J Cancer. 2010;126(6):1504-1512. (PubMed)
62. Heinen MM, Verhage BA, Goldbohm RA, van den Brandt PA. Intake of vegetables, fruits, carotenoids and vitamins C and E and pancreatic cancer risk in The Netherlands Cohort Study. Int J Cancer. 2012;130(1):147-158. (PubMed)
63. Roswall N, Olsen A, Christensen J, Dragsted LO, Overvad K, Tjonneland A. Micronutrient intake and risk of urothelial carcinoma in a prospective Danish cohort. Eur Urol. 2009;56(5):764-770. (PubMed)
64. Goodman M, Bostick RM, Kucuk O, Jones DP. Clinical trials of antioxidants as cancer prevention agents: past, present, and future. Free Radic Biol Med. 2011;51(5):1068-1084. (PubMed)
65. Zhang S, Hunter DJ, Forman MR, et al. Dietary carotenoids and vitamins A, C, and E and risk of breast cancer. J Natl Cancer Inst. 1999;91(6):547-556. (PubMed)
66. Michels KB, Holmberg L, Bergkvist L, Ljung H, Bruce A, Wolk A. Dietary antioxidant vitamins, retinol, and breast cancer incidence in a cohort of Swedish women. Int J Cancer. 2001;91(4):563-567. (PubMed)
67. Hutchinson J, Lentjes MA, Greenwood DC, et al. Vitamin C intake from diary recordings and risk of breast cancer in the UK Dietary Cohort Consortium. Eur J Clin Nutr. 2012;66(5):561-568. (PubMed)
68 Nagel G, Linseisen J, van Gils CH, et al. Dietary beta-carotene, vitamin C and E intake and breast cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC). Breast Cancer Res Treat. 2010;119(3):753-765. (PubMed)
69. Roswall N, Olsen A, Christensen J, Dragsted LO, Overvad K, Tjonneland A. Micronutrient intake and breast cancer characteristics among postmenopausal women. Eur J Cancer Prev. 2010;19(5):360-365. (PubMed)
70. Liu C, Russell RM. Nutrition and gastric cancer risk: an update. Nutr Rev. 2008;66(5):237-249. (PubMed)
71. Mirvish SS, Wallcave L, Eagen M, Shubik P. Ascorbate-nitrite reaction: possible means of blocking the formation of carcinogenic N-nitroso compounds. Science. 1972;177(4043):65-68. (PubMed)
72. Tsugane S, Sasazuki S. Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer. 2007;10(2):75-83. (PubMed)
73 Jenab M, Riboli E, Ferrari P, et al. Plasma and dietary vitamin C levels and risk of gastric cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC-EURGAST). Carcinogenesis. 2006;27(11):2250-2257. (PubMed)
74. Banerjee S, Hawksby C, Miller S, Dahill S, Beattie AD, McColl KE. Effect of Helicobacter pylori and its eradication on gastric juice ascorbic acid. Gut. 1994;35(3):317-322. (PubMed)
75. Zhang ZW, Patchett SE, Perrett D, Katelaris PH, Domizio P, Farthing MJ. The relation between gastric vitamin C concentrations, mucosal histology, and CagA seropositivity in the human stomach. Gut. 1998;43(3):322-326. (PubMed)
76. Chuang CH, Sheu BS, Kao AW, et al. Adjuvant effect of vitamin C on omeprazole-amoxicillin-clarithromycin triple therapy for Helicobacter pylori eradication. Hepatogastroenterology. 2007;54(73):320-324. (PubMed)
77. Krajewska B, Brindell M. Urease activity and L-ascorbic acid. J Enzyme Inhib Med Chem. 2011;26(3):309-318. (PubMed)
78. Pal J, Sanal MG, Gopal GJ. Vitamin-C as anti-Helicobacter pylori agent: More prophylactic than curative- Critical review. Indian J Pharmacol. 2011;43(6):624-627. (PubMed)
79 Park Y, Spiegelman D, Hunter DJ, et al. Intakes of vitamins A, C, and E and use of multiple vitamin supplements and risk of colon cancer: a pooled analysis of prospective cohort studies. Cancer Causes Control. 2010;21(11):1745-1757. (PubMed)
80. Thompson CA, Cerhan JR. Fruit and vegetable intake and survival from non-Hodgkin lymphoma: does an apple a day keep the doctor away? Leuk Lymphoma. 2010;51(6):963-964. (PubMed)
81. Kabat GC, Kim MY, Wactawski-Wende J, Shikany JM, Vitolins MZ, Rohan TE. Intake of antioxidant nutrients and risk of non-Hodgkin's Lymphoma in the Women's Health Initiative. Nutr Cancer. 2012;64(2):245-254. (PubMed)
82. Wang L, Sesso HD, Glynn RJ, et al. Vitamin E and C supplementation and risk of cancer in men: posttrial follow-up in the Physicians' Health Study II randomized trial. Am J Clin Nutr. 2014;100(3):915-923. (PubMed)
83. Song Y, Xu Q, Park Y, Hollenbeck A, Schatzkin A, Chen H. Multivitamins, individual vitamin and mineral supplements, and risk of diabetes among older U.S. adults. Diabetes Care. 2011;34(1):108-114. (PubMed)
84. Harding AH, Wareham NJ, Bingham SA, et al. Plasma vitamin C level, fruit and vegetable consumption, and the risk of new-onset type 2 diabetes mellitus: the European prospective investigation of cancer--Norfolk prospective study. Arch Intern Med. 2008;168(14):1493-1499. (PubMed)
85. Donin AS, Dent JE, Nightingale CM, et al. Fruit, vegetable and vitamin C intakes and plasma vitamin C: cross-sectional associations with insulin resistance and glycaemia in 9-10 year-old children. Diabet Med. 2016;33(3):307-315. (PubMed)
86. Kositsawat J, Freeman VL. Vitamin C and A1c relationship in the National Health and Nutrition Examination Survey (NHANES) 2003-2006. J Am Coll Nutr. 2011;30(6):477-483. (PubMed)
87. Ashor AW, Werner AD, Lara J, Willis ND, Mathers JC, Siervo M. Effects of vitamin C supplementation on glycaemic control: a systematic review and meta-analysis of randomised controlled trials. Eur J Clin Nutr. 2017;71(12):1371-1380. (PubMed)
88. Rumbold A, Ota E, Nagata C, Shahrook S, Crowther CA. Vitamin C supplementation in pregnancy. Cochrane Database Syst Rev. 2015(9):Cd004072. (PubMed)
89. Balogun OO, da Silva Lopes K, Ota E, et al. Vitamin supplementation for preventing miscarriage. Cochrane Database Syst Rev. 2016(5):Cd004073. (PubMed)
90. Hammoud AO, Bujold E, Sorokin Y, Schild C, Krapp M, Baumann P. Smoking in pregnancy revisited: findings from a large population-based study. Am J Obstet Gynecol. 2005;192(6):1856-1862; discussion 1862-1853. (PubMed)
91. Kitsantas P, Christopher KE. Smoking and respiratory conditions in pregnancy: associations with adverse pregnancy outcomes. South Med J. 2013;106(5):310-315. (PubMed)
92. Milner AD, Rao H, Greenough A. The effects of antenatal smoking on lung function and respiratory symptoms in infants and children. Early Hum Dev. 2007;83(11):707-711. (PubMed)
93. Conde-Agudelo A, Althabe F, Belizan JM, Kafury-Goeta AC. Cigarette smoking during pregnancy and risk of preeclampsia: a systematic review. Am J Obstet Gynecol. 1999;181(4):1026-1035. (PubMed)
94. Abramovici A, Gandley RE, Clifton RG, et al. Prenatal vitamin C and E supplementation in smokers is associated with reduced placental abruption and preterm birth: a secondary analysis. Bjog. 2015;122(13):1740-1747. (PubMed)
95. McEvoy CT, Schilling D, Clay N, et al. Vitamin C supplementation for pregnant smoking women and pulmonary function in their newborn infants: a randomized clinical trial. JAMA. 2014;311(20):2074-2082. (PubMed)
96. McEvoy CT, Milner KF, Scherman AJ, et al. Vitamin C to Decrease the Effects of Smoking in Pregnancy on Infant Lung Function (VCSIP): Rationale, design, and methods of a randomized, controlled trial of vitamin C supplementation in pregnancy for the primary prevention of effects of in utero tobacco smoke exposure on infant lung function and respiratory health. Contemp Clin Trials. 2017;58:66-77. (PubMed)
97. Alzheimer's Association. 2018 Alzheimer's Disease Facts and Figures. Available at: https://www.alz.org/alzheimers-dementia/facts-figures. Accessed 6/22/18.
98. Dixit S, Bernardo A, Walker JM, et al. Vitamin C deficiency in the brain impairs cognition, increases amyloid accumulation and deposition, and oxidative stress in APP/PSEN1 and normally aging mice. ACS Chem Neurosci. 2015;6(4):570-581. (PubMed)
99. Kook SY, Lee KM, Kim Y, et al. High-dose of vitamin C supplementation reduces amyloid plaque burden and ameliorates pathological changes in the brain of 5XFAD mice. Cell Death Dis. 2014;5:e1083. (PubMed)
100. Bowman GL. Ascorbic acid, cognitive function, and Alzheimer's disease: a current review and future direction. Biofactors. 2012;38(2):114-122. (PubMed)
101. Harrison J, Rentz DM, McLaughlin T, et al. Cognition in MCI and Alzheimer's disease: baseline data from a longitudinal study of the NTB. Clin Neuropsychol. 2014;28(2):252-268. (PubMed)
102. Hansen SN, Tveden-Nyborg P, Lykkesfeldt J. Does vitamin C deficiency affect cognitive development and function? Nutrients. 2014;6(9):3818-3846. (PubMed)
103. Bowman GL, Dodge H, Frei B, et al. Ascorbic acid and rates of cognitive decline in Alzheimer's disease. J Alzheimers Dis. 2009;16(1):93-98. (PubMed)
104. Arlt S, Muller-Thomsen T, Beisiegel U, Kontush A. Effect of one-year vitamin C- and E-supplementation on cerebrospinal fluid oxidation parameters and clinical course in Alzheimer's disease. Neurochem Res. 2012;37(12):2706-2714. (PubMed)
105. Galasko DR, Peskind E, Clark CM, et al. Antioxidants for Alzheimer disease: a randomized clinical trial with cerebrospinal fluid biomarker measures. Arch Neurol. 2012;69(7):836-841. (PubMed)
106. Naeini AM, Elmadfa I, Djazayery A, et al. The effect of antioxidant vitamins E and C on cognitive performance of the elderly with mild cognitive impairment in Isfahan, Iran: a double-blind, randomized, placebo-controlled trial. Eur J Nutr. 2014;53(5):1255-1262. (PubMed)
107. Reiss GR, Werness PG, Zollman PE, Brubaker RF. Ascorbic acid levels in the aqueous humor of nocturnal and diurnal mammals. Arch Ophthalmol. 1986;104(5):753-755. (PubMed)
108. Tessier F, Moreaux V, Birlouez-Aragon I, Junes P, Mondon H. Decrease in vitamin C concentration in human lenses during cataract progression. Int J Vitam Nutr Res. 1998;68(5):309-315. (PubMed)
109. Wei L, Liang G, Cai C, Lv J. Association of vitamin C with the risk of age-related cataract: a meta-analysis. Acta Ophthalmol. 2016;94(3):e170-176. (PubMed)
110. Zheng Selin J, Rautiainen S, Lindblad BE, Morgenstern R, Wolk A. High-dose supplements of vitamins C and E, low-dose multivitamins, and the risk of age-related cataract: a population-based prospective cohort study of men. Am J Epidemiol. 2013;177(6):548-555. (PubMed)
111. Rautiainen S, Lindblad BE, Morgenstern R, Wolk A. Vitamin C supplements and the risk of age-related cataract: a population-based prospective cohort study in women. Am J Clin Nutr. 2010;91(2):487-493. (PubMed)
112. Mathew MC, Ervin AM, Tao J, Davis RM. Antioxidant vitamin supplementation for preventing and slowing the progression of age-related cataract. Cochrane Database Syst Rev. 2012(6):Cd004567. (PubMed)
113. Pastor-Valero M. Fruit and vegetable intake and vitamins C and E are associated with a reduced prevalence of cataract in a Spanish Mediterranean population. BMC Ophthalmol. 2013;13:52. (PubMed)
114. Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63(10):3136-3141. (PubMed)
115. Saag KG, Choi H. Epidemiology, risk factors, and lifestyle modifications for gout. Arthritis Res Ther. 2006;8 Suppl 1:S2. (PubMed)
116. Choi HK, Curhan G. Gout: epidemiology and lifestyle choices. Curr Opin Rheumatol. 2005;17(3):341-345. (PubMed)
117. Gao X, Curhan G, Forman JP, Ascherio A, Choi HK. Vitamin C intake and serum uric acid concentration in men. J Rheumatol. 2008;35(9):1853-1858. (PubMed)
118. Zheng Z, Harman JL, Coresh J, et al. The dietary fructose: vitamin C intake ratio is associated with hyperuricemia in African-American adults. J Nutr. 2018;148(3):419-426. (PubMed)
119. Choi HK, Gao X, Curhan G. Vitamin C intake and the risk of gout in men: a prospective study. Arch Intern Med. 2009;169(5):502-507. (PubMed)
120. Juraschek SP, Miller ER, 3rd, Gelber AC. Effect of oral vitamin C supplementation on serum uric acid: a meta-analysis of randomized controlled trials. Arthritis Care Res (Hoboken). 2011;63(9):1295-1306. (PubMed)
121. Stamp LK, Zhu X, Dalbeth N, Jordan S, Edwards NL, Taylor W. Serum urate as a soluble biomarker in chronic gout-evidence that serum urate fulfills the OMERACT validation criteria for soluble biomarkers. Semin Arthritis Rheum. 2011;40(6):483-500. (PubMed)
122. Stamp LK, O'Donnell JL, Frampton C, Drake JM, Zhang M, Chapman PT. Clinically insignificant effect of supplemental vitamin C on serum urate in patients with gout: a pilot randomized controlled trial. Arthritis Rheum. 2013;65(6):1636-1642. (PubMed)
123. Andres M, Sivera F, Falzon L, Buchbinder R, Carmona L. Dietary supplements for chronic gout. Cochrane Database Syst Rev. 2014(10):Cd010156. (PubMed)
124. Pocobelli G, Peters U, Kristal AR, White E. Use of supplements of multivitamins, vitamin C, and vitamin E in relation to mortality. Am J Epidemiol. 2009;170(4):472-483. (PubMed)
125. Roswall N, Olsen A, Christensen J, et al. Micronutrient intake in relation to all-cause mortality in a prospective Danish cohort. Food Nutr Res. 2012;56. (PubMed)
126. Harris HR, Orsini N, Wolk A. Vitamin C and survival among women with breast cancer: a meta-analysis. Eur J Cancer. 2014;50(7):1223-1231. (PubMed)
127. Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst Rev. 2012(3):Cd007176. (PubMed)
128. Khaw KT, Bingham S, Welch A, et al. Relation between plasma ascorbic acid and mortality in men and women in EPIC-Norfolk prospective study: a prospective population study. European Prospective Investigation into Cancer and Nutrition. Lancet. 2001;357(9257):657-663. (PubMed)
129. Goyal A, Terry MB, Siegel AB. Serum antioxidant nutrients, vitamin A, and mortality in U.S. Adults. Cancer Epidemiol Biomarkers Prev. 2013;22(12):2202-2211. (PubMed)
130. Basili S, Tanzilli G, Mangieri E, et al. Intravenous ascorbic acid infusion improves myocardial perfusion grade during elective percutaneous coronary intervention: relationship with oxidative stress markers. JACC Cardiovasc Interv. 2010;3(2):221-229. (PubMed)
131. Wang ZJ, Hu WK, Liu YY, et al. The effect of intravenous vitamin C infusion on periprocedural myocardial injury for patients undergoing elective percutaneous coronary intervention. Can J Cardiol. 2014;30(1):96-101. (PubMed)
132. Rodrigo R, Hasson D, Prieto JC, et al. The effectiveness of antioxidant vitamins C and E in reducing myocardial infarct size in patients subjected to percutaneous coronary angioplasty (PREVEC Trial): study protocol for a pilot randomized double-blind controlled trial. Trials. 2014;15:192. (PubMed)
133. Milei J, Forcada P, Fraga CG, et al. Relationship between oxidative stress, lipid peroxidation, and ultrastructural damage in patients with coronary artery disease undergoing cardioplegic arrest/reperfusion. Cardiovasc Res. 2007;73(4):710-719. (PubMed)
134. Lassnigg A, Punz A, Barker R, et al. Influence of intravenous vitamin E supplementation in cardiac surgery on oxidative stress: a double-blinded, randomized, controlled study. Br J Anaesth. 2003;90(2):148-154. (PubMed)
135. Spoelstra-de Man AME, Elbers PWG, Oudemans-van Straaten HM. Making sense of early high-dose intravenous vitamin C in ischemia/reperfusion injury. Crit Care. 2018;22(1):70. (PubMed)
136. Dingchao H, Zhiduan Q, Liye H, Xiaodong F. The protective effects of high-dose ascorbic acid on myocardium against reperfusion injury during and after cardiopulmonary bypass. Thorac Cardiovasc Surg. 1994;42(5):276-278. (PubMed)
137. Sisto T, Paajanen H, Metsa-Ketela T, Harmoinen A, Nordback I, Tarkka M. Pretreatment with antioxidants and allopurinol diminishes cardiac onset events in coronary artery bypass grafting. Ann Thorac Surg. 1995;59(6):1519-1523. (PubMed)
138. Ramos C, Brito R, Gonzalez-Montero J, et al. Effects of a novel ascorbate-based protocol on infarct size and ventricle function in acute myocardial infarction patients undergoing percutaneous coronary angioplasty. Arch Med Sci. 2017;13(3):558-567. (PubMed)
139. Valls N, Gormaz JG, Aguayo R, et al. Amelioration of persistent left ventricular function impairment through increased plasma ascorbate levels following myocardial infarction. Redox Rep. 2016;21(2):75-83. (PubMed)
140. Hemila H, Suonsyrja T. Vitamin C for preventing atrial fibrillation in high risk patients: a systematic review and meta-analysis. BMC Cardiovasc Disord. 2017;17(1):49. (PubMed)
141. Hu X, Yuan L, Wang H, et al. Efficacy and safety of vitamin C for atrial fibrillation after cardiac surgery: A meta-analysis with trial sequential analysis of randomized controlled trials. Int J Surg. 2017;37:58-64. (PubMed)
142. Polymeropoulos E, Bagos P, Papadimitriou M, Rizos I, Patsouris E, Tauoumpoulis I. Vitamin C for the prevention of postoperative atrial fibrillation after cardiac surgery: a meta-analysis. Adv Pharm Bull. 2016;6(2):243-250. (PubMed)
143. Lagowska-Lenard M, Stelmasiak Z, Bartosik-Psujek H. Influence of vitamin C on markers of oxidative stress in the earliest period of ischemic stroke. Pharmacol Rep. 2010;62(4):751-756. (PubMed)
144. Johansen JS, Harris AK, Rychly DJ, Ergul A. Oxidative stress and the use of antioxidants in diabetes: linking basic science to clinical practice. Cardiovasc Diabetol. 2005;4:5. (PubMed)
145. Balbi ME, Tonin FS, Mendes AM, et al. Antioxidant effects of vitamins in type 2 diabetes: a meta-analysis of randomized controlled trials. Diabetol Metab Syndr. 2018;10:18. (PubMed)
146. Khodaeian M, Tabatabaei-Malazy O, Qorbani M, Farzadfar F, Amini P, Larijani B. Effect of vitamins C and E on insulin resistance in diabetes: a meta-analysis study. Eur J Clin Invest. 2015;45(11):1161-1174. (PubMed)
147. Gillani SW, Sulaiman SAS, Abdul MIM, Baig MR. Combined effect of metformin with ascorbic acid versus acetyl salicylic acid on diabetes-related cardiovascular complication; a 12-month single blind multicenter randomized control trial. Cardiovasc Diabetol. 2017;16(1):103. (PubMed)
148. Levy AP, Friedenberg P, Lotan R, et al. The effect of vitamin therapy on the progression of coronary artery atherosclerosis varies by haptoglobin type in postmenopausal women. Diabetes Care. 2004;27(4):925-930. (PubMed)
149. Asleh R, Levy AP. Divergent effects of alpha-tocopherol and vitamin C on the generation of dysfunctional HDL associated with diabetes and the Hp 2-2 genotype. Antioxid Redox Signal. 2010;12(2):209-217. (PubMed)
150. Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31(4):1250-1256. (PubMed)
151. Mann EA, Baun MM, Meininger JC, Wade CE. Comparison of mortality associated with sepsis in the burn, trauma, and general intensive care unit patient: a systematic review of the literature. Shock. 2012;37(1):4-16. (PubMed)
152. Carr AC, Rosengrave PC, Bayer S, Chambers S, Mehrtens J, Shaw GM. Hypovitaminosis C and vitamin C deficiency in critically ill patients despite recommended enteral and parenteral intakes. Crit Care. 2017;21(1):300. (PubMed)
153. Pravda J. Metabolic theory of septic shock. World J Crit Care Med. 2014;3(2):45-54. (PubMed)
154. Fowler AA, 3rd, Syed AA, Knowlson S, et al. Phase I safety trial of intravenous ascorbic acid in patients with severe sepsis. J Transl Med. 2014;12:32. (PubMed)
155. Zabet MH, Mohammadi M, Ramezani M, Khalili H. Effect of high-dose Ascorbic acid on vasopressor's requirement in septic shock. J Res Pharm Pract. 2016;5(2):94-100. (PubMed)
156. Marik PE, Khangoora V, Rivera R, Hooper MH, Catravas J. Hydrocortisone, vitamin C, and thiamine for the treatment of severe sepsis and septic shock: a retrospective before-after study. Chest. 2017;151(6):1229-1238. (PubMed)
157. Spoelstra-de Man AME, Elbers PWG, Oudemans-Van Straaten HM. Vitamin C: should we supplement? Curr Opin Crit Care. 2018;24(4):248-255. (PubMed)
158. Cameron E, Pauling L. Supplemental ascorbate in the supportive treatment of cancer: Prolongation of survival times in terminal human cancer. Proc Natl Acad Sci U S A. 1976;73(10):3685-3689. (PubMed)
159. Ohno S, Ohno Y, Suzuki N, Soma G, Inoue M. High-dose vitamin C (ascorbic acid) therapy in the treatment of patients with advanced cancer. Anticancer Res. 2009;29(3):809-815. (PubMed)
160. Chen Q, Espey MG, Krishna MC, et al. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci U S A. 2005;102(38):13604-13609. (PubMed)
161. Chen Q, Espey MG, Sun AY, et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc Natl Acad Sci U S A. 2007;104(21):8749-8754. (PubMed)
162. Chen Q, Espey MG, Sun AY, et al. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci U S A. 2008;105(32):11105-11109. (PubMed)
163. Gao P, Zhang H, Dinavahi R, et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12(3):230-238. (PubMed)
164. Kuiper C, Molenaar IG, Dachs GU, Currie MJ, Sykes PH, Vissers MC. Low ascorbate levels are associated with increased hypoxia-inducible factor-1 activity and an aggressive tumor phenotype in endometrial cancer. Cancer Res. 2010;70(14):5749-5758. (PubMed)
165. Vissers MCM, Das AB. Potential mechanisms of action for vitamin C in cancer: reviewing the evidence. Front Physiol. 2018;9:809. (PubMed)
166. Carr AC, Cook J. Intravenous vitamin C for cancer therapy - identifying the current gaps in our knowledge. Front Physiol. 2018;9:1182. (PubMed)
167. Hoffer LJ, Levine M, Assouline S, et al. Phase I clinical trial of i.v. ascorbic acid in advanced malignancy. Ann Oncol. 2008;19(11):1969-1974. (PubMed)
168. Monti DA, Mitchell E, Bazzan AJ, et al. Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PLoS One. 2012;7(1):e29794. (PubMed)
169 Riordan HD, Casciari JJ, Gonzalez MJ, et al. A pilot clinical study of continuous intravenous ascorbate in terminal cancer patients. P R Health Sci J. 2005;24(4):269-276. (PubMed)
170. Stephenson CM, Levin RD, Spector T, Lis CG. Phase I clinical trial to evaluate the safety, tolerability, and pharmacokinetics of high-dose intravenous ascorbic acid in patients with advanced cancer. Cancer Chemother Pharmacol. 2013;72(1):139-146. (PubMed)
171. Carr AC, Vissers MC, Cook JS. The effect of intravenous vitamin C on cancer- and chemotherapy-related fatigue and quality of life. Front Oncol. 2014;4:283. (PubMed)
172. Klingelhoeffer C, Kammerer U, Koospal M, et al. Natural resistance to ascorbic acid induced oxidative stress is mainly mediated by catalase activity in human cancer cells and catalase-silencing sensitizes to oxidative stress. BMC Complement Altern Med. 2012;12:61. (PubMed)
173. Hong SW, Lee SH, Moon JH, et al. SVCT-2 in breast cancer acts as an indicator for L-ascorbate treatment. Oncogene. 2013;32(12):1508-1517. (PubMed)
174. Fritz H, Flower G, Weeks L, et al. Intravenous vitamin C and cancer: a systematic review. Integr Cancer Ther. 2014;13(4):280-300. (PubMed)
175. Jacobs C, Hutton B, Ng T, Shorr R, Clemons M. Is there a role for oral or intravenous ascorbate (vitamin C) in treating patients with cancer? A systematic review. Oncologist. 2015;20(2):210-223. (PubMed)
176. Cabanillas F. Vitamin C and cancer: what can we conclude--1,609 patients and 33 years later? P R Health Sci J. 2010;29(3):215-217. (PubMed)
177. Pauling LC. Vitamin C and the Common Cold. San Francisco: W.H. Freeman; 1970.
178. Hemila H, Chalker E. Vitamin C for preventing and treating the common cold. Cochrane Database Syst Rev. 2013(1):Cd000980. (PubMed)
179. Hemila H. Vitamin C and common cold-induced asthma: a systematic review and statistical analysis. Allergy Asthma Clin Immunol. 2013;9(1):46. (PubMed)
180. Milan SJ, Hart A, Wilkinson M. Vitamin C for asthma and exercise-induced bronchoconstriction. Cochrane Database Syst Rev. 2013(10):Cd010391. (PubMed)
181. Cheng Y, Willett WC, Schwartz J, Sparrow D, Weiss S, Hu H. Relation of nutrition to bone lead and blood lead levels in middle-aged to elderly men. The Normative Aging Study. Am J Epidemiol. 1998;147(12):1162-1174. (PubMed)
182. Simon JA, Hudes ES. Relationship of ascorbic acid to blood lead levels. JAMA. 1999;281(24):2289-2293. (PubMed)
183. Lee DH, Lim JS, Song K, Boo Y, Jacobs DR, Jr. Graded associations of blood lead and urinary cadmium concentrations with oxidative-stress-related markers in the U.S. population: results from the third National Health and Nutrition Examination Survey. Environ Health Perspect. 2006;114(3):350-354. (PubMed)
184. Dawson EB, Evans DR, Harris WA, Teter MC, McGanity WJ. The effect of ascorbic acid supplementation on the blood lead levels of smokers. J Am Coll Nutr. 1999;18(2):166-170. (PubMed)
185. Abam E, Okediran BS, Odukoya OO, Adamson I, Ademuyiwa O. Reversal of ionoregulatory disruptions in occupational lead exposure by vitamin C. Environ Toxicol Pharmacol. 2008;26(3):297-304. (PubMed)
186. Carr AC, Vissers MC. Synthetic or food-derived vitamin C--are they equally bioavailable? Nutrients. 2013;5(11):4284-4304. (PubMed)
187. Johnston CS, Luo B. Comparison of the absorption and excretion of three commercially available sources of vitamin C. J Am Diet Assoc. 1994;94(7):779-781. (PubMed)
188. Austria R, Semenzato A, Bettero A. Stability of vitamin C derivatives in solution and topical formulations. J Pharm Biomed Anal. 1997;15(6):795-801. (PubMed)
189. DeRitter E, Cohen N, Rubin SH. Physiological availability of dehydro-L-ascorbic acid and palmitoyl-L-ascorbic acid. Science. 1951;113:628-631. (PubMed)
190. Davis JL, Paris HL, Beals JW, et al. Liposomal-encapsulated ascorbic acid: influence on vitamin C bioavailability and capacity to protect against ischemia-reperfusion injury. Nutr Metab Insights. 2016;9:25-30. (PubMed)
191. Massey LK, Liebman M, Kynast-Gales SA. Ascorbate increases human oxaluria and kidney stone risk. J Nutr. 2005;135(7):1673-1677. (PubMed)
192. Traxer O, Huet B, Poindexter J, Pak CY, Pearle MS. Effect of ascorbic acid consumption on urinary stone risk factors. J Urol. 2003;170(2 Pt 1):397-401. (PubMed)
193. Auer BL, Auer D, Rodgers AL. The effect of ascorbic acid ingestion on the biochemical and physicochemical risk factors associated with calcium oxalate kidney stone formation. Clin Chem Lab Med. 1998;36(3):143-147. (PubMed)
194. Liebman M, Chai WW, Harvey E, Boenisch L. Effect of supplemental ascorbate and orange juice on urinary oxalate. Nutr Res. 1997;17(3):415-425.
195. Wandzilak TR, D'Andre SD, Davis PA, Williams HE. Effect of high dose vitamin C on urinary oxalate levels. J Urol. 1994;151(4):834-837. (PubMed)
196. Curhan GC, Willett WC, Rimm EB, Stampfer MJ. A prospective study of the intake of vitamins C and B6, and the risk of kidney stones in men. J Urol. 1996;155(6):1847-1851. (PubMed)
197. Curhan GC, Willett WC, Speizer FE, Stampfer MJ. Intake of vitamins B6 and C and the risk of kidney stones in women. J Am Soc Nephrol. 1999;10(4):840-845. (PubMed)
198. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol. 2004;15(12):3225-3232. (PubMed)
199. Thomas LD, Elinder CG, Tiselius HG, Wolk A, Akesson A. Ascorbic acid supplements and kidney stone incidence among men: a prospective study. JAMA Intern Med. 2013;173(5):386-388. (PubMed)
200. Natural Medicines. Vitamin C/Professional handout/Interactions with drugs. Available at: https://naturalmedicines.therapeuticresearch.com. Accessed 7/2/18.
201. Hendler SS, Rorvik DM. PDR for Nutritional Supplements. 2nd ed. Montvale: Thomson Reuters; 2008.
202 Brown BG, Zhao XQ, Chait A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345(22):1583-1592. (PubMed)
203. Collins R, Peto R, Armitage J. The MRC/BHF Heart Protection Study: preliminary results. Int J Clin Pract. 2002;56(1):53-56. (PubMed)
204. Lee SH, Oe T, Blair IA. Vitamin C-induced decomposition of lipid hydroperoxides to endogenous genotoxins. Science. 2001;292(5524):2083-2086. (PubMed)
205. Podmore ID, Griffiths HR, Herbert KE, Mistry N, Mistry P, Lunec J. Vitamin C exhibits pro-oxidant properties. Nature. 1998;392(6676):559. (PubMed)
206. Carr A, Frei B. Does vitamin C act as a pro-oxidant under physiological conditions? Faseb J. 1999;13(9):1007-1024. (PubMed)
207. Brubacher D, Moser U, Jordan P. Vitamin C concentrations in plasma as a function of intake: a meta-analysis. Int J Vitam Nutr Res. 2000;70(5):226-237. (PubMed)
208. Michels AJ, Joisher N, Hagen TM. Age-related decline of sodium-dependent ascorbic acid transport in isolated rat hepatocytes. Arch Biochem Biophys. 2003;410(1):112-120. (PubMed)
Supplemental Forms
The Bioavailability of Different Forms of Vitamin C (Ascorbic Acid)
In the rapidly expanding market of dietary supplements, it is possible to find vitamin C in many different forms with many claims regarding its efficacy or bioavailability. Bioavailability refers to the degree to which a nutrient (or drug) becomes available to the target tissue after it has been administered. We reviewed the literature for the results of scientific research on the bioavailability of different forms of vitamin C.
Natural vs. synthetic ascorbic acid
Natural and synthetic L-ascorbic acid are chemically identical, and there are no known differences in their biological activity. The possibility that the bioavailability of L-ascorbic acid from natural sources might differ from that of synthetic ascorbic acid was investigated in at least two human studies, and no clinically significant differences were observed. A study of 12 males (6 smokers and 6 nonsmokers) found the bioavailability of synthetic ascorbic acid (powder administered in water) to be slightly superior to that of orange juice, based on blood levels of ascorbic acid, and not different based on ascorbic acid in leukocytes (white blood cells) (1). A study in 68 male nonsmokers found that ascorbic acid consumed in cooked broccoli, orange juice, orange slices, and as synthetic ascorbic acid tablets are equally bioavailable, as measured by plasma ascorbic acid levels (2, 3).
Different forms of ascorbic acid
The gastrointestinal absorption of ascorbic acid occurs through an active transport process, as well as through passive diffusion. At low gastrointestinal concentrations of ascorbic acid active transport predominates, while at high gastrointestinal concentrations active transport becomes saturated, leaving only passive diffusion. In theory, slowing down the rate of stomach emptying (e.g., by taking ascorbic acid with food or taking a slow-release form of ascorbic acid) should increase its absorption. While the bioavailability of ascorbic acid appears equivalent whether it is in the form of powder, chewable tablets, or non-chewable tablets, the bioavailability of ascorbic acid from slow-release preparations is less certain.
A study of three men and one woman found 1 gram of ascorbic acid to be equally well absorbed from solution, tablets, and chewable tablets, but the absorption from a timed-release capsule was 50% lower. Absorption was assessed by measuring urinary excretion of ascorbic acid after an intravenous dose of ascorbic acid and then comparing it to urinary excretion after the oral dosage forms (4).
A more recent study examined the plasma levels of ascorbic acid in 59 male smokers supplemented for two months with either 500 mg/day of slow-release ascorbic acid, 500 mg/day of plain ascorbic acid, or a placebo. After two months of supplementation no significant differences in plasma ascorbic acid levels between the slow-release and plain ascorbic acid groups were found (5). A second placebo-controlled trial also evaluated plain ascorbic acid versus slow-release ascorbic acid in 48 male smokers (6). Participants were supplemented with either 250 mg plain ascorbic acid, 250 mg slow-release ascorbic acid, or placebo twice daily for four weeks. No differences were observed in the change in plasma ascorbate concentration or area under the curve following ingestion of either formulation.
Mineral ascorbates
Mineral salts of ascorbic acid (mineral ascorbates) are less acidic, and therefore, considered "buffered." Thus, mineral ascorbates are often recommended to people who experience gastrointestinal problems (upset stomach or diarrhea) with plain ascorbic acid. There appears to be little scientific research to support or refute the claim that mineral ascorbates are less irritating to the gastrointestinal tract. When mineral salts of ascorbic acid are taken, both the ascorbic acid and the mineral appear to be well absorbed, so it is important to consider the dose of the mineral accompanying the ascorbic acid when taking large doses of mineral ascorbates. For the following discussion, it should be noted that 1 gram (g)= 1,000 milligrams (mg) and 1 milligram (mg) = 1,000 micrograms (μg). Mineral ascorbates are available in the following forms:
- Sodium ascorbate: 1,000 mg of sodium ascorbate generally contains 111 mg of sodium. Individuals following low-sodium diets (e.g., for high blood pressure) are generally advised to keep their total dietary sodium intake to less than 2,500 mg/day. Thus, megadoses of vitamin C in the form of sodium ascorbate could significantly increase sodium intake (see Sodium Chloride).
- Calcium ascorbate: Calcium ascorbate generally provides 90-110 mg of calcium (890-910 mg of ascorbic acid) per 1,000 mg of calcium ascorbate. Calcium in this form appears to be reasonably well absorbed. The recommended dietary calcium intake for adults is 1,000 to 1,200 mg/day. Total calcium intake should not exceed the UL, which is 2,500 mg/day for adults aged 19-50 years and 2,000 mg/day for adults older than 50 years (see Calcium).
The following mineral ascorbates are more likely to be found in combination with other mineral ascorbates, as well as other minerals. It's a good idea to check the labels of dietary supplements for the ascorbic acid dose as well as the dose of each mineral. Recommended dietary intakes and maximum upper levels of intake (when available) are listed after the individual mineral ascorbates below:
- Potassium ascorbate: The minimal requirement for potassium is thought to be between 1.6 and 2.0 g/day. Fruit and vegetables are rich sources of potassium, and a diet rich in fruit and vegetables may provide as much as 8 to 11 g/day. Acute and potentially fatal potassium toxicity (hyperkalemia) is thought to occur at a daily intake of about 18 g/day of potassium in adults. Individuals taking potassium-sparing diuretics and those with renal insufficiency (kidney failure) should avoid significant intake of potassium ascorbate. The purest form of commercially available potassium ascorbate contains 0.175 grams (175 mg) of potassium per gram of ascorbate (see Potassium).
- Magnesium ascorbate: The recommended dietary allowance (RDA) for magnesium is 400-420 mg/day for adult men and 310-320 mg/day for adult women. The upper level (UL) of intake for magnesium from supplements should not exceed 350 mg/day (see Magnesium).
- Zinc ascorbate: The RDA for zinc is 11 mg/day for adult men and 8 mg/day for adult women. The upper level (UL) of zinc intake for adults should not exceed 40 mg/day (see Zinc).
- Molybdenum ascorbate: The RDA for molybdenum is 45 micrograms (μg)/day for adult men and women. The upper level (UL) of molybdenum intake for adults should not exceed 2,000 μg (2 mg)/day (see Molybdenum).
- Chromium ascorbate: The recommended dietary intake (AI) for chromium is 30-35 μg/day for adult men and 20-25 μg/day for adult women. A maximum upper level (UL) of intake has not been determined by the US Food and Nutrition Board (see Chromium).
- Manganese ascorbate: The recommended dietary intake (AI) for manganese is 2.3 mg/day for adult men and 1.8 mg/day for adult women. The upper level (UL) of intake for manganese for adults should not exceed 11 mg/day. Manganese ascorbate is found in some preparations of glucosamine and chondroitin sulfate, and following the recommended dose on the label of such supplements could result in a daily intake exceeding the upper level for manganese (see Manganese).
Vitamin C with bioflavonoids
Bioflavonoids or flavonoids are polyphenolic compounds found in plants. Vitamin C-rich fruit and vegetables, especially citrus fruit, are often rich sources of flavonoids as well. The effect of bioflavonoids on the bioavailability of ascorbic acid has been recently reviewed (7).
Results from the 10 clinical studies comparing the absorption of vitamin C alone or vitamin C in flavonoid-containing foods showed no appreciable differences in bioavailability of ascorbic acid. Only one study, which included five men and three women, found that a 500-mg supplement of synthetic ascorbic acid, given in a natural citrus extract containing bioflavonoids, proteins, and carbohydrates, was more slowly absorbed and 35% more bioavailable than synthetic ascorbic acid alone, when based on plasma levels of ascorbic acid (8). The remaining studies showed either no change or slightly lower plasma ascorbate levels in subjects who consumed vitamin C with flavonoids compared to flavonoids alone (7).
Another assessment of vitamin C bioavailability is measuring urinary ascorbate levels to approximate rates of vitamin C excretion. One study in six young Japanese males (22-26 years old) showed a significant reduction in urinary excretion of ascorbic acid in the presence of acerola juice, a natural source of both vitamin C and flavonoids (9). However, three separate studies showed that urinary levels of vitamin C were increased after consumption of kiwifruit (10), blackcurrant juice (11), or orange juice (1). Overall, the impact of flavonoids on the bioavailability of vitamin C seems to be negligible; however, there is a need for carefully controlled studies using specific flavonoid extracts (7).
Ascorbate and vitamin C metabolites (Ester-C®)
Ester-C® contains mainly calcium ascorbate, but also contains small amounts of the vitamin C metabolites, dehydroascorbic acid (oxidized ascorbic acid), calcium threonate, and trace levels of xylonate and lyxonate. In their literature, the manufacturers state that the metabolites, especially threonate, increase the bioavailability of the vitamin C in this product, and they indicate that they have performed a study in humans that demonstrates the increased bioavailability of vitamin C in Ester-C®. This study has not been published in a peer-reviewed journal. A small published study of vitamin C bioavailability in eight women and one man found no difference between Ester-C® and commercially available ascorbic acid tablets with respect to the absorption and urinary excretion of vitamin C (12). Ester-C® should not be confused with ascorbyl palmitate, which is also marketed as "vitamin C ester" (see below).
Ascorbyl palmitate
Ascorbyl palmitate is a fat-soluble antioxidant used to increase the shelf life of vegetable oils and potato chips (13). It is an amphipathic molecule, meaning one end is water-soluble and the other end is fat-soluble. This dual solubility allows it to be incorporated into cell membranes. When incorporated into the cell membranes of human red blood cells, ascorbyl palmitate has been found to protect them from oxidative damage and to protect α-tocopherol (a fat-soluble antioxidant) from oxidation by free radicals (14). However, the protective effects of ascorbyl palmitate on cell membranes have only been demonstrated in the test tube. Taking ascorbyl palmitate orally probably doesn't result in any significant incorporation into cell membranes because most of it appears to be hydrolyzed (broken apart into palmitate and ascorbic acid) in the human digestive tract before it is absorbed. The ascorbic acid released by the hydrolysis of ascorbyl palmitate appears to be as bioavailable as ascorbic acid alone (15). The presence of ascorbyl palmitate in oral supplements contributes to the ascorbic acid content of the supplement and probably helps protect fat-soluble antioxidants in the supplement. The roles of vitamin C in promoting collagen synthesis and as an antioxidant have generated interest in its use on the skin (see the article, Vitamin C and Skin Health). Ascorbyl palmitate is frequently used in topical preparations because it is more stable than some aqueous (water-soluble) forms of vitamin C (16). Ascorbyl palmitate is also marketed as vitamin C ester," which should not be confused with Ester-C® (see above).
D-Isoascorbic acid (Erythorbic acid)
Erythorbic acid is an isomer of ascorbic acid. Isomers are compounds that have the same kinds and numbers of atoms, but different molecular arrangements. The difference in molecular arrangement among isomers may result in different chemical properties. Erythorbic acid is used in the US as an antioxidant food additive and is generally recognized as safe. It has been estimated that more than 200 mg erythorbic acid per capita is introduced daily into the US food system. Unlike ascorbic acid, erythorbic acid does not appear to exert vitamin C activity, for example, it did not prevent scurvy in guinea pigs (one of the few animal species other than humans that does not synthesize ascorbic acid). However, guinea pig studies also indicated that increased erythorbic acid intake reduced the bioavailability of ascorbic acid by up to 50%. In contrast, a series of studies in young women found that up to 1,000 mg/day of erythorbic acid for as long as 40 days was rapidly cleared from the body and had little effect on the bioavailability of ascorbic acid, indicating that erythorbic acid does not diminish the bioavailability of ascorbic acid in humans at nutritionally relevant levels of intake (17).
Other formulations of vitamin C
PureWay-C® is composed of vitamin C and lipid metabolites. Two cell culture studies using PureWay-C® have been published by the same investigators (18, 19), but in vivo data are currently lacking. A small study in healthy adults found that serum levels of vitamin C did not differ when a single oral dose (1 gram) of either PureWay-C® or ascorbic acid was administered (20).
Another formulation of vitamin C, liposomal-encapsulated vitamin C (e.g., Lypo-spheric™ vitamin C) is now commercially available. One report suggested that liposomal-encapsulated vitamin C may be better absorbed than the vitamin in a non-encapsulated form (21).
Large-scale, pharmacokinetic studies are needed to determine how the bioavailability of these vitamin C formulations compares to that of ascorbic acid.
References
1. Pelletier, O. & Keith, M.O. Bioavailability of synthetic and natural ascorbic acid. Journal of the American Dietetic Association. 1974; 64: 271-275
2. Mangels, A.R. et al. The bioavailability to humans of ascorbic acid from oranges, orange juice, and cooked broccoli is similar to that of synthetic ascorbic acid. Journal of Nutrition. 1993; volume 123: pages 1054-1061. (PubMed)
3. Gregory, J.F. Ascorbic acid bioavailability in foods and supplements. Nutrition Reviews. 1993; volume 51: pages 301-309. (PubMed)
4. Yung, S. et al. Ascorbic acid absorption in humans: a comparison among several dosage forms. Journal of Pharmaceutical Sciences. 1982; volume 71: pages 282-285. (PubMed)
5. Nyyssonen, K. et al. Effect of supplementation of smoking men with plain or slow release ascorbic acid on lipoprotein oxidation. European Journal of Clinical Nutrition. 1997; volume 51: pages 154-163. (PubMed)
6. Viscovich M, Lykkesfeldt J, Poulsen HE. Vitamin C pharmacokinetics of plain and slow release formulations in smokers. Clinical nutrition. 2004;23(5):1043-1050. (PubMed)
7. Carr AC, Vissers MC. Synthetic or food-derived vitamin C-are they equally bioavailable? Nutrients. 2013;5(11):4284-4304. (PubMed)
8. Vinson, J.A. & Bose, P. Comparative bioavailability to humans of ascorbic acid alone or in a citrus extract. American Journal of Clinical Nutrition. 1988; volume 48: pages 501-604. (PubMed)
9. Uchida E, Kondo Y, Amano A, et al. Absorption and excretion of ascorbic acid alone and in acerola (Malpighia emarginata) juice: comparison in healthy Japanese subjects. Biol Pharm Bull. 2011;34(11):1744-1747. (PubMed)
10. Carr AC, Bozonet SM, Pullar JM, Simcock JW, Vissers MC. A randomized steady-state bioavailability study of synthetic versus natural (kiwifruit-derived) vitamin C. Nutrients. 2013;5(9):3684-3695. (PubMed)
11. Jones E, Hughes RE. The influence of bioflavonoids on the absorption of vitamin C. IRCS Med Sci. 1984;12:320.
12. Johnston, C.S. & Luo, B. Comparison of the absorption and excretion of three commercially available sources of vitamin C. Journal of the American Dietetic Association. 1994; volume 94: pages 779-781.
13. Cort, W.M. Antioxidant activity of tocopherols, ascorbyl palmitate, and ascorbic acid and their mode of action. Journal of the American Oil Chemists’ Society. 1974; volume 51: pages 321-325.
14. Ross, D. et al. Ascorbate 6-palmitate protects human erythrocytes from oxidative damage. Free Radical Biology and Medicine. 1999; volume 26: pages 81-89. (PubMed)
15. DeRitter, E. et al. Physiologic availability of dehydro-L-ascorbic acid and palmitoyl-L-ascorbic acid. Science. 1951; volume 113: pages 628-631.
16. Austria R. et al. Stability of vitamin C derivatives in solution and in topical formulations. Journal of Pharmacology and Biomedical Analysis. 1997; volume 15: pages 795-801. (PubMed)
17. Sauberlich, H.E. et al. Effects of erythorbic acid on vitamin C metabolism in young women. American Journal of Clinical Nutrition. 1996; volume 64: pages 336-346. (PubMed)
18. Weeks BS, Perez PP. Absorption rates and free radical scavenging values of vitamin C-lipid metabolites in human lymphoblastic cells. Med Sci Monit. 2007;13(10):BR205-210. (PubMed)
19. Weeks BS, Perez PP. A novel vitamin C preparation enhances neurite formation and fibroblast adhesion and reduces xenobiotic-induced T-cell hyperactivation. Med Sci Monit. 2007;13(3):BR51-58. (PubMed)
20. Pancorbo D, Vazquez C, Fletcher MA. Vitamin C-lipid metabolites: uptake and retention and effect on plasma C-reactive protein and oxidized LDL levels in healthy volunteers. Med Sci Monit. 2008;14(11):CR547-551. (PubMed)
21. Davis JL, Paris HL, Beals JW, et al. Liposomal-encapsulated ascorbic scid: influence on vitamin C bioavailability and capacity to protect against ischemia-reperfusion injury. Nutr Metab Insights. 2016;9:25-30. (PubMed)
Copyright 2000-2024 Linus Pauling Institute
Pauling Recommendation
Difference Between Dr. Linus Pauling's Recommendations and the LPI's Recommendation for Vitamin C Intake
Dr. Pauling, for whom the Linus Pauling Institute has great respect, based his own recommendations for vitamin C largely on theoretical arguments. In developing his recommendations, he used cross-species comparisons, evolutionary arguments, the concept of biochemical individuality, and the amount of vitamin C likely consumed in a raw plant food diet. Using this approach, Dr. Pauling suggested in the early 1970s that the optimum daily intake may be about 2,000 milligrams of vitamin C and that everyone should get at least 200 to 250 mg/day. In a 1974 radio interview, he noted that "the first 250 mg is more important than any later 250 mg. The first 250 mg leads you up to the level where the blood is saturated. You can achieve a higher volume [concentration] in the blood by a larger intake, but you get much better improvement for the first 250 mg than for additional grams." Dr. Pauling significantly increased his recommendation in his 1986 book How To Live Longer and Feel Better. At the Linus Pauling Institute, we have based our vitamin C recommendations on the current body of scientific evidence, which is significantly greater than it was at Pauling's time but remains incomplete owing to the many diverse functions of vitamin C in the human body that have yet to be fully understood.
In this context, it is important to note that data from the National Institutes of Health (NIH) have indicated that vitamin C levels in plasma and circulating cells become fully saturated at intakes of about 400 mg/day in young, healthy nonsmokers. These observations are consistent with other data that intakes of about 400 mg/day are associated with reduced risk of heart disease. While these NIH studies are the best we currently have regarding the pharmacokinetics of vitamin C in the human body, they have numerous limitations, including the fact that they are based on a small number of young, healthy men and women. We currently do not know how much vitamin C is required to achieve saturation of cells and tissues in children, older adults, and diseased or stressed individuals. A meta-analysis of 36 studies on the relationship between vitamin C intake and plasma concentrations found that the elderly require a substantially higher daily intake of vitamin C to attain plasma concentrations that younger adults achieve at a lower intake. Additionally, work by Linus Pauling Institute investigators has shown that cellular uptake of vitamin C declines with age, supporting the notion that vitamin C requirements are increased in the elderly.
Therefore, the Linus Pauling Institute's intake recommendation of 400 mg/day of vitamin C for generally healthy adults takes into account the currently available epidemiological, biochemical, and clinical evidence, while acknowledging the extremely low toxicity of vitamin C and the incomplete information regarding optimum intake. It should also be noted that the Linus Pauling Institute's recommendation is strictly directed towards prevention of disease in healthy individuals, not treatment of disease. Thus, individuals suffering from certain diseases may require substantially larger amounts of vitamin C to achieve optimum body levels or derive therapeutic benefits, areas that were of great interest to Linus Pauling and need to be further explored.
Vitamin D
Contents
Summary
- Vitamin D can be synthesized in the skin upon exposure to sunlight and is then metabolized in the liver and kidney to the metabolically active form called 1α,25-dihydroxyvitamin D. Through binding to the vitamin D receptor (VDR), 1α,25-dihydroxyvitamin D can regulate the expression of hundreds of genes involved in skeletal and other biological functions. (More information)
- Vitamin D is essential for maintenance of bone mineralization through the regulation of calcium and phosphorus homeostasis. Vitamin D also exhibits many non-skeletal effects, particularly on the immune, endocrine, and cardiovascular systems. (More information)
- Vitamin D is important for normal bone development and maintenance. Severe vitamin D deficiency causes rickets in children and osteomalacia in adults. (More information)
- Secondary hyperparathyroidism due to vitamin D insufficiency can increase bone breakdown and precipitate osteoporosis. Randomized clinical trials indicate that supplementation with at least 800 IU/day of vitamin D may reduce the risk of falls and fractures in older individuals. (More information)
- Vitamin D can regulate cell differentiation and growth by binding to the vitamin D receptor found in most body cells. Observational studies have reported associations between low sun exposure, poor vitamin D status, and increased risk of developing colorectal and breast cancer. Randomized controlled trials are needed to evaluate whether cancer prevention may benefit from vitamin D supplementation. (More information)
- Various observational studies have reported inverse associations between vitamin D status and the susceptibility or severity of autoimmune diseases, including type 1 diabetes mellitus, multiple sclerosis, rheumatoid arthritis, and systemic lupus erythematosus. (More information)
- Current evidence from observational studies suggests an inverse relationship between circulating vitamin D concentrations and risk of type 2 diabetes mellitus. It is not yet known whether correcting vitamin D deficiency in individuals with glucose intolerance can decrease the risk of progression to type 2 diabetes mellitus. (More information)
- Randomized clinical trials are currently investigating whether vitamin D supplementation can limit cognitive deterioration and disease progression in subjects with neurodegenerative disease. (More information)
- Vitamin D insufficiency in pregnant women may be associated with several adverse effects for the mother and newborn. Safety and benefits of vitamin D supplementation during pregnancy both need to be evaluated in clinical trials. (More information)
- Observational studies have documented an association between vitamin D deficiency and increased incidence and severity of the coronavirus disease, COVID-19. (More information)
- Preliminary studies have shown that vitamin D supplementation may offer promising improvements in the management of atopic dermatitis (eczema) and Crohn’s disease. (More information)
Vitamin D is a fat-soluble vitamin that regulates calcium homeostasis and is vital for bone health (1). While it can also be obtained from dietary sources or supplements, vitamin D3 (cholecalciferol) is synthesized in the human skin from 7-dehydrocholesterol upon exposure to ultraviolet-B (UVB) radiation from sunlight (see the separate article on Vitamin D and Skin Health). Vitamin D2 (ergocalciferol) is a vitamin D analog photosynthesized in plants, mushrooms, and yeasts; vitamin D2 is also sometimes used in vitamin D food fortification (2). When vitamin D3 in skin is inadequate due to insufficient exposure to UVB radiation, oral intake of vitamin D is necessary to meet vitamin D requirements.
Function
Vitamin D metabolism
Cholecalciferol and ergocalciferol are biologically inactive precursors of vitamin D and must be converted to biologically active forms in the liver and kidneys (Figure 1). Indeed, following dietary intake or synthesis in the epidermis of skin after UVB exposure, both forms of vitamin D enter the circulation and are transported to the liver by the vitamin D-binding protein (and to a lesser extent by albumin). In hepatocytes (liver cells), vitamin D is hydroxylated to form 25-hydroxyvitamin D (calcidiol; calcifediol). Exposure to sunlight or dietary intake of vitamin D increases serum concentrations of 25-hydroxyvitamin D. 25-Hydroxyvitamin D constitutes the major circulating form of vitamin D, and the sum of 25-hydroxyvitamin D2 and 25-hydroxyvitamin D3 concentrations in serum is used as an indicator of vitamin D nutritional status (3). The renal 25-hydroxyvitamin D3-1α-hydroxylase enzyme (also known as CYP27B1) eventually catalyzes a second hydroxylation that converts 25-hydroxyvitamin D to 1α,25-dihydroxyvitamin D (calcitriol). The production of 1α,25-dihydroxyvitamin D in the kidneys is regulated by several factors, including serum phosphorus, calcium, parathyroid hormone (PTH), fibroblast growth factor-23 (FGF-23), and 1α,25-dihydroxyvitamin D itself. While the kidney is the main source of 1α-hydroxylase activity, extra-renal production of 1α,25-dihydroxyvitamin D has also been demonstrated in a variety of tissues, including skin, parathyroid gland, breast, colon, prostate, as well as cells of the immune system and bone cells (2). Most of the physiological effects of vitamin D in the body are related to the activity of 1α,25-dihydroxyvitamin D (4). Various forms of vitamin D are listed in Figure 1.
Mechanisms of action
Most, if not all, actions of vitamin D are mediated through a nuclear transcription factor known as the vitamin D receptor (VDR) (Figure 2) (5). Upon entering the nucleus of a cell, 1α,25-dihydroxyvitamin D binds to the VDR and recruits another nuclear receptor known as retinoid X receptor (RXR). In the presence of 1α,25-dihydroxyvitamin D, the VDR/RXR complex binds small sequences of DNA known as vitamin D response elements (VDREs) and initiates a cascade of molecular interactions that modulate the transcription of specific genes. Thousands of VDREs have been identified throughout the genome, and VDR activation by 1α,25-dihydroxyvitamin D is thought to directly and/or indirectly regulate 100 to 1,250 genes (6).
![Figure 2. Conversion to the Active Form of Vitamin D and VDR-mediated Gene Regulation. Mechanism of action. 25-hydroxyvitamin D (25(OH)D) is the main form of vitamin D in the circulation. Most 25(OH)D and 1α,25-dihydroxyvitamin D (1,25(OH)2D) molecules are transported bound to the vitamin D-binding protein and they enter cells via the megalin/tubulin complex. In kidney cells, 1α-hydroxylase (CYP27B1) catalyzes the conversion of 25(OH)D into 1,25(OH)2D. Parathyroid hormone (PTH), estradiol, and low phosphorus concentration ([P]) stimulate this reaction, whereas 1,25(OH)2D, fibroblast growth factor-23 (FGF-23), and high calcium concentration ([Ca]) inhibit it. 1,25(OH)2D enters the circulation and is transported to extra-renal target tissues where it can regulate gene expression. In the nucleus of target cells, 1,25(OH)2D binds to the vitamin D receptor (VDR), which heterodimerizes with the retinoid X receptor (RXR). VDR-RXR binds to vitamin D response elements (VDRE) in the promoter of vitamin D target genes. 25(OH)D, 25-hydroxyvitamin D; 1,25(OH)2D, 1α,25-dihydroxyvitamin D; [Ca], calcium concentration; CYP2R1, 1α-hydroxylase; DBP, vitamin D-binding protein; FGF-23, fibroblast growth factor-23; PTH, parathyroid hormone; [P], phosphorus concentration; RNA Pol II, RNA polymerase II; RXR, retinoid X receptor; VDR, vitamin D nuclear receptor; VDRE, vitamin D response element.](/sites/lpi.oregonstate.edu/files/vitamin-d-figure-2-1200px.png)
Calcium balance
Maintenance of serum calcium concentrations within a narrow range is vital for normal functioning of the nervous system, as well as for bone growth and maintenance of bone density. Vitamin D is essential for the efficient utilization of calcium by the body (1). The parathyroid glands sense serum calcium concentrations and secrete parathyroid hormone (PTH) if calcium concentrations decrease below normal (Figure 3). Elevations in PTH stimulate the activity of the 25-hydroxyvitamin D3-1α-hydroxylase enzyme in the kidney, resulting in the increased production of 1α,25-dihydroxyvitamin D. The active vitamin D form, 1α,25-dihydroxyvitamin D, is released into the circulation and transported to target tissues. Within target cells, 1α,25-dihydroxyvitamin D binds to and induces the activation of VDR, which leads to changes in gene expression that normalize serum calcium by (1) increasing the intestinal absorption of dietary calcium, (2) increasing the reabsorption of calcium filtered by the kidneys, and (3) mobilizing calcium from bone when there is insufficient dietary calcium to maintain normal serum calcium concentrations (7).
![Figure 3. Regulation of Calcium and Phosphorus Homeostasis. Calcium and phosphorus homeostasis. A slight drop in blood calcium concentration ([Ca2+]) results in the secretion of parathyroid hormone (PTH) by the parathyroid glands. PTH simulates the activity of CYP27B1 (25-hydroxyvitamin D-1α-hydroxylase enzyme) that catalyzes the hydroxylation of 25-hydroxyvitamin D into 1α,25-dihydroxyvitamin D. This active form of vitamin D (i) increases the reabsorption of calcium filtered by the kidneys and stimulates phosphorus excretion, (ii) increases the intestinal absorption of both calcium and phosphorus, and (iii) mobilizes calcium (and phosphorus) from bone when dietary calcium intake is insufficient. PTH also stimulates bone resorption that releases calcium and phosphorus. 1α,25-dihydroxyvitamin D inhibits its own production, as well as PTH synthesis, via negative feedback loops. FGF-23 is secreted by bone-forming cells (osteoblasts) in response to an increase in phosphorus intake. FGF-23 inhibits the synthesis of 1α,25-dihydroxyvitamin D and promotes phosphorus excretion in the urine. [Ca2+], calcium concentration; FGF-23, fibroblast growth factor-23; PTH, parathyroid hormone.](/sites/lpi.oregonstate.edu/files/vitamin-d-figure-3-1200px.png)
Phosphorus balance
The regulations of calcium and phosphorus homeostasis are closely related, and the calciotropic hormones, PTH and 1α,25-dihydroxyvitamin D, can also control serum phosphorus. Specifically, 1α,25-dihydroxyvitamin D increases intestinal phosphorus absorption by stimulating the expression of a sodium-phosphate cotransporter in the small intestine. While PTH increases urinary excretion of phosphorus by reducing reabsorption in the kidney, it is not yet clear whether 1α,25-dihydroxyvitamin D can directly regulate renal phosphorus transport. The phosphaturic hormone fibroblast growth factor-23 (FGF-23), secreted by osteoblasts (bone-forming cells), limits the production of 1α,25-dihydroxyvitamin D by inhibiting 25-hydroxyvitamin D-1α-hydroxylase (Figure 3) (reviewed in 8).
Cell differentiation
Cells that are dividing rapidly are said to be proliferating. Differentiation results in the specialization of cells for specific functions. In general, differentiation of cells leads to a decrease in proliferation. While cellular proliferation is essential for growth and wound healing, uncontrolled proliferation of cells with certain mutations may lead to cancer. The active form of vitamin D, 1α,25-dihydroxyvitamin D, inhibits proliferation and stimulates the differentiation of cells through binding to the VDR (1).
Immunity
Acting through the VDR, 1α,25-dihydroxyvitamin D is a potent immune system modulator. The VDR is expressed by most cells of the immune system, including regulatory T cells and antigen-presenting cells, such as dendritic cells and macrophages (9). Under specific circumstances, monocytes, macrophages, and T cells can express the 25-hydroxyvitamin D3-1α-hydroxylase enzyme and produce 1α,25-dihydroxyvitamin D, which acts locally to regulate the immune response (10, 11). There is considerable scientific evidence that 1α,25-dihydroxyvitamin D has a variety of effects on immune system function, which may enhance innate immunity and inhibit the development of autoimmunity (12). Conversely, vitamin D deficiency may compromise the integrity of the immune system and lead to inappropriate immune responses (see Autoimmune diseases).
Insulin secretion
The VDR is expressed by insulin-secreting cells of the pancreas, and the results of animal studies suggest that 1α,25-dihydroxyvitamin D plays a role in insulin secretion under conditions of increased insulin demand (13, 14). Cross-sectional and prospective studies suggest that insufficient vitamin D status may have an adverse effect on insulin secretion and glucose tolerance in type 2 diabetes mellitus (noninsulin-dependent diabetes mellitus) (reviewed in 15).
Blood pressure regulation
The renin-angiotensin system plays an important role in the regulation of blood pressure (16). Renin is an enzyme that catalyzes the cleavage (splitting) of a small peptide (angiotensin I) from a larger protein (angiotensinogen) produced in the liver. Angiotensin-converting enzyme (ACE) catalyzes the cleavage of angiotensin I to form angiotensin II, a peptide that can increase blood pressure by inducing the constriction of small arteries and by increasing sodium and water retention. The rate of angiotensin II synthesis is dependent on renin (17). Research in mice lacking the gene encoding the VDR indicates that 1α,25-dihydroxyvitamin D decreases the expression of the gene encoding renin through its interaction with the VDR (18). Since inappropriate activation of the renin-angiotensin system can contribute to the development of hypertension, achieving adequate vitamin D status may be important for decreasing the risk of high blood pressure (see Hypertension).
Deficiency
In vitamin D deficiency, calcium absorption cannot be increased enough to satisfy the body’s calcium needs (4). Consequently, PTH production by the parathyroid glands is increased and calcium is mobilized from the skeleton to maintain normal serum calcium concentrations — a condition known as secondary hyperparathyroidism. Although it has long been known that severe vitamin D deficiency has serious consequences for bone health, research suggests that less obvious states of vitamin D deficiency are common and increase the risk of osteoporosis and various other health problems (see Disease Prevention).
Severe vitamin D deficiency
Rickets
In infants and children, severe vitamin D deficiency results in the failure of bone to mineralize. The process of mineralization, which involves the production of crystals of calcium phosphate by bone-forming cells, determines the hardness and strength of bones. Vitamin D deficiency severely affects rapidly growing bones. The growth plates of bones continue to enlarge, but in the absence of adequate mineralization, weight-bearing limbs (arms and legs) become bowed. In infants, rickets may result in delayed closure of the fontanels (soft spots) in the skull, and the rib cage may become deformed due to the pulling action of the diaphragm. In severe cases, low serum calcium concentrations (hypocalcemia) may cause seizures. Although fortification of food has led to complacency regarding vitamin D deficiency, nutritional rickets is still being reported throughout the world (19, 20).
Osteomalacia
Although adult bones are no longer growing, they are in a constant state of turnover, or "remodeling." In adults with severe vitamin D deficiency, the collagenous bone matrix is preserved, but bone mineral is progressively lost, resulting in a softening of bones (osteomalacia), bone pain, and increased risk of osteoporosis (21).
Muscle weakness and pain
Vitamin D deficiency causes muscle weakness and pain in children and adults. Muscle pain and weakness were prominent symptoms of vitamin D deficiency in a study of Arab and Danish Muslim women living in Denmark (22). In a cross-sectional study of 150 consecutive patients referred to a clinic in Minnesota for the evaluation of persistent, nonspecific musculoskeletal pain, 93% had serum 25-hydroxyvitamin D concentrations equal to or below 20 ng/mL, with a mean concentration of 12.1 ng/mL, which is indicative of vitamin D insufficiency (23). Loss of muscle strength greatly contributes to increased risk of falling and bone fractures, especially in older people (24). In addition, long-term vitamin D insufficiency may be a contributing factor to osteoporosis in the elderly (see Osteoporosis).
Risk factors for vitamin D deficiency
Both environmental factors and cultural practices result in variations in vitamin D status:
- Environmental conditions: Geographical locations, including latitude and altitudes, and atmospheric conditions (e.g., air pollution, presence of clouds) greatly influence the intensity of UVB radiation that reaches the ground. Seasonal changes also affect the quality and quantity of UVB rays and thus vitamin D production in skin (25-27).
- Concealed clothing style: In a study of 2,032 Middle Eastern women, who wore a headscarf or covered all skin for religious or cultural reasons, 96% had serum 25-hydroxyvitamin D concentrations less than 20 ng/mL, and 60% had vitamin D concentrations below 12 ng/mL (28). Rickets and osteomalacia are not uncommon in the Middle East and North African regions where children and women cover the majority or all of their skin whenever outside (29).
- Sun safety measures: Sun protection practices, including limiting sun exposure, wearing protective clothing and hats, and applying sunscreens, hinder skin exposure to sunlight and thus result in lower vitamin D3 production and circulating vitamin D metabolites unless there is adequate oral intake. Of note, the application of sunscreen (2 mg/cm2) with a sun protection factor (SPF) of 10 reduces UVB radiation by 90% (30).
- Exclusively breast-fed infants: Infants who are exclusively breast-fed and do not receive vitamin D supplementation are at high risk for vitamin D deficiency, particularly if they have dark skin and/or receive little sun exposure (19). Human milk generally provides 10 to 80 IU of vitamin D per liter (L), which corresponds to 0.2 to 1.5 g/day (8 to 60 IU/day) when using an average daily milk intake of 0.75 L (25 oz) (31). The American Academy of Pediatrics recommends that all breast-fed and partially breast-fed infants be given an oral vitamin D supplement of 400 IU/day (19). Maternal vitamin D supplementation during breast-feeding may contribute to improved vitamin D status of the breast-fed infant, especially in populations with a high prevalence of vitamin D deficiency (32). Older infants and toddlers exclusively fed milk substitutes (e.g., soy-based formulas) and weaning foods that are not vitamin D fortified are at risk for vitamin D deficiency (33).
The efficiency of vitamin D synthesis, absorption, and metabolism also depends on a variety of biological factors:
- Skin pigmentation: People with a dark complexion synthesize less vitamin D on exposure to sunlight than those with light-colored skin (34). A national US survey reported average serum 25-hydroxyvitamin D concentrations of 28.1 ng/mL, 21.6 ng/mL, and 16.9 ng/mL in Caucasian, Mexican American, and African American adults aged ≥20 years old, respectively (25).
- Genetic variations: An international, multicenter, genome-wide association study (GWAS) of 15 cohorts, including ~30,000 participants of European descent — known as the SUNLIGHT [Study of Underlying Genetic Determinants of Vitamin D and Highly Related Traits] consortium — identified common variations (called polymorphisms) in genes involved in cholesterol synthesis, hydroxylation, and vitamin D transport that influence vitamin D status (35). While genetic determinants of low vitamin D status are being identified in populations of European (36, 37) and Asian descent (38, 39), genome-wide association studies are needed in populations of African descent.
- Older age: The elderly have reduced capacity to synthesize vitamin D in skin when exposed to UVB radiation and are more likely to stay indoors or use sunscreen, which prevents vitamin D synthesis. It has been estimated that across Canada, the US, and Europe, the prevalence of vitamin D deficiency ranges between 20%-100% in free-living elderly (40). Moreover, institutionalized adults who are not supplemented with vitamin D are at extremely high risk of vitamin D deficiency (41, 42).
- Chronic kidney disease (CKD): Vitamin D deficiency in patients with impaired renal function is due to a reduced synthesis of 1α,25-dihydroxyvitamin D and an increased loss of 25-hydroxyvitamin D in urine (43).
- Fat malabsorption syndromes: Vitamin D deficiency is common among people with cystic fibrosis and both cholestatic and non-cholestatic liver diseases due to decreased absorption of dietary vitamin D and impaired conversion of vitamin D to 25-hydroxyvitamin D (reviewed in 44).
- Inflammatory bowel disease: People with inflammatory bowel disease like Crohn’s disease appear to be at increased risk of vitamin D deficiency, especially those who have had small bowel resections (45).
- Obesity: Obesity (body mass index ≥30 kg/m2) increases the risk of vitamin D deficiency (46). Once vitamin D is synthesized in the skin or ingested, it can be sequestered in body fat stores, making it less bioavailable to people with higher body fat mass. Moreover, vitamin D supplementation trials have shown that obese people reached much lower serum 25-hydroxyvitamin D concentrations compared to normal weight (BMI <25 kg/m2) participants with equivalent oral dosages (47).
- Magnesium deficiency: Recent findings suggest that high magnesium intakes may reduce the risk of vitamin D insufficiency. Magnesium regulates the activity of critical enzymes in vitamin D metabolism, which would explain how magnesium deficiency negatively affects vitamin D status (48).
Assessing vitamin D nutritional status
Growing awareness that vitamin D insufficiency has serious health consequences beyond rickets and osteomalacia highlights the need for accurate assessment of vitamin D nutritional status. It is currently agreed that the measurement of total serum 25-hydroxyvitamin D concentration (1 ng/mL corresponding to 2.5 nmol/L) is the best indicator to evaluate vitamin D status. However, high-quality evidence is still needed to ensure that the current cutoff values are optimal to define states of insufficiency and deficiency (40). Moreover, only recently, efforts have been made toward the standardization of commercially and laboratory-developed 25-hydroxyvitamin D assays, such that guidelines have been developed using largely unstandardized research data (49). Although laboratory reference values for sufficient vitamin D status were initially based on serum 25-hydroxyvitamin D concentrations from cohorts of healthy individuals, additional studies have suggested that health-based cutoff values aimed at preventing secondary hyperparathyroidism and bone loss should be considerably higher. Indeed, while it is considered that serum 25-hydroxyvitamin D concentrations less than 8 to 10 ng/mL (20 to 25 nmol/L) indicate severe deficiency associated with rickets and osteomalacia, several studies have observed that PTH concentrations (50, 51) and calcium absorption (52) were not optimized with serum 25-hydroxyvitamin D concentrations below 32 ng/mL (80 nmol/L).
Yet, more recent studies have failed to find threshold values of serum 25-hydroxyvitamin D concentrations in relation to PTH suppression and optimal calcium absorption. On the one hand, a cross-sectional analysis of 312,962 clinical samples did not find any evidence of threshold for PTH suppression in the well-fitted curve displaying the inverse association between paired measurements of serum PTH and 25-hydroxyvitamin D, even with 25-hydroxyvitamin D concentrations beyond 70 ng/mL (175 nmol/L) (53). This contradicted an analysis of the US National Health and Nutrition Examination Survey (NHANES 2003-2006) that estimated maximum PTH suppression for 25-hydroxyvitamin D concentrations of 40 ng/mL (100 nmol/L) and above (54). In addition, both studies identified evidence of mild hyperparathyroidism (serum PTH >65 pg/mL) in individuals with serum 25-hydroxyvitamin D concentrations well beyond 20 ng/mL (50 nmol/L), questioning the use of serum PTH as a sensible indicator of vitamin D insufficiency (53, 54). On the other hand, a randomized, placebo-controlled trial in postmenopausal women with vitamin D insufficiency (serum 25-hydroxyvitamin D <20 ng/mL) supplemented with daily vitamin D3 doses from 400 to 4,800 IU found little change (6%) in calcium absorption over a normal 25-hydroxyvitamin D concentration range of 20 to 66 ng/mL (55).
The current cutoffs proposed by the Institute of Medicine (IOM) are as follows: deficiency as serum 25-hydroxyvitamin D values <12 ng/mL (<30 nmol/L), insufficiency as serum 25-hydroxyvitamin D values of 12 to 19 ng/mL (30 to 49 nmol/L), and sufficiency as serum 25-hydroxyvitamin D values of 20 to 50 ng/mL (50 to 125 nmol/L) (56). The dietary reference intakes (EAR, RDA) set by the IOM are based on achieving circulating 25-hydroxyvitamin D concentrations (20 to 50 ng/mL) that are adequate to maintain bone health and optimal calcium absorption (57).
Yet, considering the potential role of circulating concentrations lower than 30 ng/mL in the burden of many chronic diseases (6), the US Endocrine Society has suggested to define cutoff values as follows: deficiency as serum 25-hydroxyvitamin D values ≤20 ng/mL (≤50 nmol/L), insufficiency as serum 25-hydroxyvitamin D values of 21 to 29 ng/mL (51 to 74 nmol/L), and sufficiency as serum 25-hydroxyvitamin D values of 30 to 100 ng/mL (75 to 250 nmol/L) (40). This alternate target range is supported by some observational studies, but it is not based on data from randomized controlled trials (see Disease Prevention) (47). With these latter cutoff values, studies from across the world have estimated that hypovitaminosis D is widespread and that children and adults of all ages are equally at risk of insufficiency and deficiency (58). Data from supplementation studies indicate that vitamin D intakes of at least 800 to 1,000 IU/day are required by adults living in temperate latitudes to achieve serum 25-hydroxyvitamin D concentrations of at least 30 ng/mL (75 nmol/L) (40).
Finally, total serum 25-hydroxyvitamin D concentrations may not always adequately reflect vitamin D bioavailability (59), and additional evidence is needed to improve the determination of vitamin D status in different ethnic populations.
The Recommended Dietary Allowance (RDA)
In 2010, the Food and Nutrition Board (FNB) of the IOM set a Recommended Dietary Allowance (RDA) based on the amount of vitamin D needed for bone health. While the RDA was increased from the adequate intake (AI) set in 1997, the optimal levels of recommended intakes and serum 25-hydroxyvitamin D to minimize hyperthyroidism and maximize bone health in the general population remain controversial (40). The RDA for vitamin D is listed in Table 1 by life stage and gender.
| Life Stage | Age | Males | Females | ||
|---|---|---|---|---|---|
| μg/day | IU/day | μg/day | IU/day | ||
| Infants (AI) | 0-6 months |
10
|
400
|
10
|
400
|
| Infants (AI) | 6-12 months |
10
|
400
|
10
|
400
|
| Children | 1-3 years |
15
|
600
|
15
|
600
|
| Children | 4-8 years |
15
|
600
|
15
|
600
|
| Children | 9-13 years |
15
|
600
|
15
|
600
|
| Adolescents | 14-18 years |
15
|
600
|
15
|
600
|
| Adults | 19-70 years |
15
|
600
|
15
|
600
|
| Adults | 71 years and older |
20
|
800
|
20
|
800
|
| Pregnancy | all ages |
-
|
-
|
15
|
600
|
| Breast-feeding | all ages |
-
|
-
|
15
|
600
|
Disease Prevention
Mortality
In a nine-year follow-up analysis of the Third US National Health and Nutrition Examination Survey (NHANES III) that included 15,099 participants (of which 77% were Caucasians), serum concentrations of 25-hydroxyvitamin D — standardized as per the methodology developed by the Vitamin D Standardization Program [VDSP] — were examined in relation to mortality. The analysis suggested an increase in all-cause mortality with decreasing serum 25-hydroxyvitamin D concentrations <16 ng/mL (60). In contrast, the risk of all-cause mortality varied little for baseline serum 25-hydroxyvitamin D concentrations in the range of 16 to 40 ng/mL (60). Similar results were obtained in a meta-analysis of eight prospective cohort studies that considered the relationship between standardized 25-hydroxyvitamin D concentrations and mortality during a median follow-up period of 10.5 years. The risk of death was found to be 19% higher with 25-hydroxyvitamin D concentrations between 12 and 15.99 ng/mL and 56% higher with concentrations <12 ng/mL compared to the risk associated with concentrations between 30 and 39.99 ng/mL (61). A meta-analysis of 73 prospective cohort studies, including >800,000 participants, found that the lowest versus highest tertile of serum 25-hydroxyvitamin D concentrations was associated with greater risks of all-cause mortality (+35%), mortality due to cardiovascular disease (+35%), and mortality due to cancer (+14%) (62). Yet, a Mendelian randomization analysis — which limits bias due to confounding and reverse causation (63) — of data from three large Danish cohorts of 95,766 adults found a significant association of genetically low plasma 25-hydroxyvitamin D concentrations with all-cause and cancer-related mortality, but not with cardiovascular disease-related mortality (64). Finally, two meta-analyses of randomized controlled trials have suggested a modest reduction in all-cause mortality in older people supplemented with vitamin D and calcium, but not vitamin D alone (62, 65). Additional placebo-controlled trials need to examine further whether supplementation with vitamin D alone or in combination with calcium might help prevent premature death in replete individuals.
Osteoporosis
Vitamin D status, osteoporosis, and risk of fracture
Although the causes of osteoporosis are multifactorial, vitamin D insufficiency can be an important etiological factor in older adults. Osteoporosis affects one-third of women aged 60 to 70 years and two-thirds of women aged 80 years and above (66). A multinational (18 different countries with latitudes ranging from 64 degrees north to 38 degrees south) survey of more than 2,600 postmenopausal women with osteoporosis revealed that 31% of subjects had 25-hydroxyvitamin D concentrations <20 ng/mL (50 nmol/L) (67). In addition, a case-control study that included 111 hip fracture patients and 73 controls (median age, 83 years) found that lower serum concentrations of both 25-hydroxyvitamin D and vitamin K1 in patients compared to controls were associated with an increased risk of hip fracture (68). Without sufficient vitamin D from sun exposure or dietary intake, intestinal calcium absorption can be significantly reduced. This increases PTH secretion by the parathyroid glands; sustained PTH elevation may result in increased bone resorption, which in turn may increase the risk of osteoporotic fracture (69).
Vitamin D supplementation and bone mineral density
The progressive loss of bone mineral density (BMD) leading to osteopenia (pre-osteoporosis) and osteoporosis is commonly observed in older adults, especially the elderly. The results of a meta-analysis of 23 randomized controlled trials with more than 4,000 participants (mean age, 59 years) showed little evidence for an effect of vitamin D supplementation on BMD at any of the five skeletal sites examined, including lumbar spine, femoral neck, trochanter, forearm, and total body. A significant increase in BMD was reported only at the femoral neck (70). It was, however, suggested that individuals in this age group would have adequate calcium intake and thus normal bone metabolism, explaining the lack of an effect of vitamin D in strengthening bone mass (71). Conversely, in older individuals, vitamin D supplementation is essential to correct and maintain adequate concentrations of serum 25-hydroxyvitamin D and to prevent secondary hyperparathyroidism and BMD loss (72).
Vitamin D supplementation and risk of fracture
A prospective cohort study that followed more than 72,000 postmenopausal women in the US for 18 years found that those who consumed at least 600 IU/day of vitamin D from diet and supplements had a 37% lower risk of osteoporotic hip fracture than women who consumed less than 140 IU/day of vitamin D (73). However, daily supplementation with 400 IU of vitamin D3, in combination with 1,000 mg calcium, did not significantly reduce risk of hip fracture compared to a placebo in 36,282 postmenopausal women from the Women's Health Initiative trial (74), suggesting that there might be a threshold of vitamin D intake that is necessary to observe reductions in fracture risk. Results of a genetic analysis of data from this trial also suggested that beneficial effects of vitamin D and calcium supplementation on fracture risk might be limited to women with the lowest genetic risk of low BMD (75). Yet, this trial has been questioned for reasons that include poor adherence and the fact that participants were allowed to take additional vitamin D and calcium supplements that might have confounded the results. In addition, use of hormone replacement therapy was not considered in the study of the effect of vitamin D and calcium on skeletal health in postmenopausal women despite being a major confounding factor in this population (57, 76).
Another trial, the Randomised Evaluation of Calcium Or vitamin D (RECORD) study, reported that oral supplemental vitamin D3 (800 IU/day) alone, or in combination with calcium (1,000 mg/day), did not prevent the occurrence of osteoporotic fractures in elderly adults who had already experienced a low-trauma, osteoporotic fracture (77). In this latter study as well, a number of limitations, including poor adherence and/or the fact that vitamin D supplementation did not raise serum 25-hydroxyvitamin D concentrations to a level that would protect against fractures, might explain the lack of an effect (78). Despite high adherence to treatment, the incidence of non-vertebral fracture was similar in postmenopausal women supplemented with vitamin D3 (initial dose of 200,000 IU followed by 100,000/month) or placebo for over three years in the Vitamin D Assessment (ViDA) trial (79).
Nevertheless, the US Preventive Services Task Force that conducted the meta-analysis of 11 randomized, placebo-controlled trials, including 52,915 older people (of whom 69% were postmenopausal women), found that the supplementation of vitamin D (300-1,000 IU/day) and calcium (500-1,200 mg/day) for up to seven years resulted in a 12% reduction in the risk of any new fracture (80). Another meta-analysis of 11 randomized, double-blind, placebo-controlled trials on the effect of vitamin D supplementation in 31,022 individuals (91% women) 65 years and older indicated that those with the highest vitamin D intake (792-2,000 IU/day) had a 30% lower risk of hip fracture and a 14% lower risk of any non-spine fracture (81). Finally, a third meta-analysis of trials that examined the effect of combined vitamin D and calcium in preventing fractures in older men and postmenopausal women also concluded that the risk of new fractures, including hip fractures, was significantly reduced in those supplemented compared to controls (82). Interestingly, the three meta-analyses have found that the prevention of fractures by supplemental vitamin D and calcium was limited to institutionalized, older people. Indeed, the risk of fracture was not significantly reduced by vitamin D in community-dwelling seniors (80-82).
Vitamin D supplementation and postural balance, muscle strength, and risk of fall
A meta-analysis of seven observational studies in 840 fallers and 1,330 non-fallers found significantly lower serum 25-hydroxyvitamin D concentrations in fallers than in non-fallers (83). Moreover, another meta-analysis of four cohorts from three observational studies reported a modest yet significant inverse association between vitamin D status and the risk of fall (83). Several randomized controlled trials have examined the impact of vitamin D supplementation on muscle strength, postural balance, or risk of fall in older subjects. A meta-analysis of these trials found limited evidence of an effect of vitamin D supplementation on muscle strength and mobility, based on only one type of test for each outcome. Nevertheless, in a recent randomized, double-blind, placebo-controlled study in 160 postmenopausal women (ages, 50-65 years) with suboptimal vitamin D status (mean serum 25-hydroxyvitamin D concentration <20 ng/mL), supplementation with 1,000 IU/day of vitamin D3 significantly improved vitamin D status, as well as upper and lower limb muscle strength and postural balance parameters (84, 85). The risks of fall and recurrent falls were found to be two- to three-fold greater in women in the control group than in those supplemented with vitamin D3 (85). In contrast, another 12-month randomized controlled study in 200 older adults (of which 58% had a baseline serum 25-hydroxyvitamin D concentration <20 ng/mL) showed no benefits regarding lower extremity function or odds of falling in those supplemented with 2,000 IU/day (+/- 10 µg of calcidiol) compared to those who received 800 IU/day (86). The recently published post-hoc analysis of the ViDA trial found no differences in odds of falling and number of falls reported by 5,108 community-dwelling participants (ages, 50-84 years) regardless of whether they were randomized to receive supplemental vitamin D (100,000 IU/month, i.e. ~3,350 IU/day) or a placebo for a mean 3.4 years (79). Most ViDA participants had serum 25-hydroxyvitamin D concentrations ≥20 ng/mL, which might at least partly explain the lack of an effect of vitamin D on falls (87).
Overall, the current evidence suggests that vitamin D3 supplements of 800-1,000 IU/day may be helpful in reducing falls and fracture rates in older adults. In order for vitamin D supplementation to be effective in preserving bone health, adequate dietary calcium (1,000 to 1,200 mg/day) should be consumed (see the article on Calcium) (88).
Cancer
Ecologic studies first suggested an association between Northern latitudes, vitamin D deficiency, and cancer incidence (89). Since the 1980s, several prospective cohort studies have examined the association of vitamin D intake or status and various types of cancer. A 2013 systematic review and meta-analysis of 16 prospective studies, including 137,567 subjects, reported an 11% reduction in total cancer incidence and a 17% reduction in cancer mortality with each 20 ng/mL (50 nmol/L) increase in circulating 25-hydroxyvitamin D concentrations. Yet, a sex-based subgroup analysis of eight studies found an inverse association between circulating vitamin D and cancer mortality in women, but not in men (90). In addition, increasing evidence suggests that a few variations in the gene coding for the vitamin D receptor (VDR) might influence individual vitamin D status and subsequently modify the susceptibility to site-specific cancers (91) and influence cancer survival (92). Finally, many malignant tumors have been found to express the VDR, including breast, lung, skin (melanoma), colon, and bone (93), suggesting that they might be susceptible to the effects of vitamin D. Numerous experimental studies have demonstrated that biologically active forms of vitamin D, such as 1α,25-dihydroxyvitamin D and its analogs, upon binding to the VDR, can control cell fate by inhibiting proliferation and/or inducing cell differentiation or death (apoptosis) of a number of cancerous cell types (94).
Colorectal cancer
The geographic distribution of colon cancer mortality resembles the historical geographic distribution of rickets (95), providing circumstantial evidence that decreased sunlight exposure and diminished vitamin D nutritional status may be related to an increased risk of colon cancer. Evidence from observational studies has largely supported this hypothesis. A recent meta-analysis of four prospective cohort studies, four cross-sectional studies, and seven case-control studies found an inverse relationship between circulating vitamin D and incidence of colorectal adenoma — a benign tumor that may transform to become malignant (96). The analysis identified a 32% risk reduction between top versus bottom quantiles of serum 25-hydroxyvitamin D concentrations (96). Additionally, there is strong evidence from meta-analyses of prospective cohort studies to suggest that higher vitamin D intakes and serum 25-hydroxyvitamin D concentrations are associated with reductions in colorectal cancer risk (97-99). The most recent meta-analysis of four prospective cohort, 17 nested case-control, and three case-control studies found a 38% reduced risk of colorectal cancer with high versus low quantiles of circulating 25-hydroxyvitamin D concentrations (100). A dose-response analysis estimated that serum 25-hydroxyvitamin D concentrations of ~20 to 30 ng/mL (compared to ≤12 ng/mL) were associated with a 17% lower risk of colorectal cancer, and the risk was even lower (-35%) with a serum concentration of 55 ng/mL (100). An earlier dose-response analysis based on five nested case-control studies had estimated that serum 25-hydroxyvitamin D concentrations ≥33 ng/mL (compared to ≤12 ng/mL) were associated with a 50% lower risk of colorectal cancer (101).
However, in a seven-year, randomized, double-blind, placebo-controlled trial in 36,282 postmenopausal women participating in the Women's Health Initiative study, a combination of supplemental vitamin D3 (400 IU/day) and calcium (1,000 mg/day) did not lower incidence of colorectal cancer (102). Another randomized controlled trial of vitamin D3 supplementation (1,000 IU/day), with or without calcium supplementation (1,200 mg/day), found no reduction in the risk of colorectal adenoma recurrence over a three-to-five year period, compared to placebo, after initial adenoma removal in participants (103). Whether these daily vitamin D doses are too low to detect any effect on cancer incidence is not clear (101, 104). Additional randomized clinical trials are needed to assess whether vitamin D supplementation could help prevent colorectal cancer. Moreover, it is uncertain whether genetic variations (polymorphisms) in the sequence of genes involved in vitamin D metabolism and function can influence the relationship between vitamin D status and risk of colorectal adenoma or colorectal cancer (105-107).
Finally, growing evidence suggests that adequate vitamin D status may be linked to better survival of colorectal cancer patients. A meta-analysis of five prospective studies found a 35% reduced risk of colorectal cancer-specific mortality in cancer patients with higher serum 25-hydroxyvitamin D concentrations. A dose-response analysis estimated that every 8 ng/mL increase in 25-hydroxyvitamin D concentration was associated with a 10% decrease in colorectal cancer mortality (108).
Breast cancer
Although ecologic evidence suggests that breast cancer mortality rises with increasing latitudes and decreasing sunlight exposure (109), the most current observational data provide little support for an association between vitamin D nutritional status and risk of breast cancer. An early prospective study of women who participated in the First US National Health and Nutrition Examination Survey (NHANES I) found that Caucasian women with adequate sunlight exposure and dietary vitamin D intake had a significantly reduced risk of breast cancer 20 years later (110). Nonetheless, when this study was included in a meta-analysis with nine more recent prospective studies, there was no significant difference in the risk of developing breast cancer between the highest and lowest levels of vitamin D intakes (111). Moreover, whether an association exists between circulating vitamin D concentrations and risk of breast cancer is unclear. One meta-analysis of 14 observational studies (9,110 cases and 16,244 controls) reported an overall risk reduction of 16% when the highest quantile of serum 25-hydroxyvitamin D concentrations was compared to the lowest. This inverse association was statistically significant in postmenopausal women but not in premenopausal women (112). Yet, another meta-analysis that included a similar set of 14 prospective studies (two studies were different) found no overall association (111). Additionally, a meta-analysis of studies conducted in patients in the early stage of breast cancer identified associations between inadequate vitamin D status and increased risks of recurrence and death (113). Evidence from randomized controlled trials is currently too limited to conclude whether vitamin D supplementation may reduce breast cancer incidence (reviewed in 114).
Nonetheless, three meta-analyses have found an inverse association between circulating vitamin D concentrations and breast cancer-related mortality (111, 115, 116). In one meta-analysis of one retrospective and five prospective cohort studies, the highest versus lowest categories of serum vitamin D concentrations was associated with a 33% reduction in mortality; a dose-response analysis found a 12% reduction per 8 ng/mL increase in serum vitamin D (115).
Finally, current evidence does not suggest that specific genetic variations in the gene coding for the VDR may influence the risk of breast cancer (117, 118).
Other types of cancer
Evidence associating vitamin D status with other types of cancer is currently limited. While incidence of prostate cancer appears to be inversely associated with the availability of sunlight, prospective cohort studies have not generally found significant relationships between serum 25-hydroxyvitamin D concentrations and subsequent risk of developing prostate cancer (119, 120). In fact, some studies have suggested an increased risk of prostate cancer with higher circulating vitamin D concentrations. For example, a nested case-control study of men (622 cases and 1,451 controls) from Scandinavia found a U-shaped relationship between serum 25-hydroxyvitamin D concentrations and prostate cancer risk. In that study, serum 25-hydroxyvitamin D concentrations of 7.6 ng/mL or lower, or 32 ng/mL or higher, were associated with increased prostate cancer risk (121). A meta-analysis of 17 nested case-control studies, three prospective cohort studies, and one retrospective cohort study found a 17% increased risk of prostate cancer in individuals in the highest versus lowest categories of blood 25-hydroxyvitamin D concentrations (122). Potential confounding factors that might explain the detection of a slight increase in prostate cancer cases in men with high circulating vitamin D concentrations have been highlighted in a recent publication (123).
Finally, recent meta-analyses of observational studies found an inverse relationship between vitamin D status and risk of lung cancer (124, 125) and bladder cancer (126, 127). Yet, in the few and often heterogeneous studies published to date, serum 25-hydroxyvitamin D concentrations were not associated with other cancer types, including non-Hodgkin’s lymphoma (128), ovarian cancer (129), gastric cancer (130), or skin cancers (131).
Autoimmune diseases
Insulin-dependent diabetes mellitus (type 1 diabetes mellitus), multiple sclerosis (MS), rheumatoid arthritis (RA), and systemic lupus erythematosus (SLE) are examples of autoimmune diseases. Autoimmune diseases occur when the body mounts an immune response against its own tissue, rather than a foreign pathogen. In type 1 diabetes mellitus, insulin-producing β-cells of the pancreas are the target of an inappropriate immune response. In MS, the targets are the myelin-producing cells of the central nervous system, and in RA, the targets are the collagen-producing cells of the joints (132). SLE is characterized by the presence of a large spectrum of autoantibodies resulting in potential damage to multiple tissues (133). Autoimmune responses are mediated by immune cells called T cells. The biologically active form of vitamin D, 1α,25-dihydroxyvitamin D, has been found to modulate T cell responses, such that the autoimmune responses are diminished. Ecologic studies have found that the prevalence of autoimmune diseases (particularly for MS; 134) increases as latitude increases, suggesting that lower exposure to UVB radiation and associated decreases in skin vitamin D synthesis may play a role in the pathology of these diseases. Results of several prospective cohort studies also suggest that adequate vitamin D status at different ages (including in utero, early childhood, and during adolescence) could possibly decrease the risk of autoimmune diseases.
Type 1 diabetes mellitus
Lower levels of circulating vitamin D have been reported in patients newly diagnosed with type 1 diabetes mellitus compared to age- and sex-matched non-diabetic subjects (135, 136). A greater prevalence of vitamin D insufficiency and deficiency has also been observed in prediabetic children who developed multiple islet autoantibodies (antibodies against insulin-secreting pancreatic cells) compared to autoantibody-negative children. However, a prospective study that followed the cohort of prediabetic children found that their vitamin D status, defined as either insufficient, deficient, or sufficient, was not associated with rate of progression to type 1 diabetes after 5 or 10 years of follow up (137). An earlier prospective cohort study of children born in Finland during the year 1966 and followed for 30 years found that children supplemented with vitamin D during the first year of life had an 88% lower risk of developing type 1 diabetes compared to those receiving no supplementation. Moreover, children suspected of having had rickets (severe vitamin D deficiency) during the first year of life showed a significantly higher risk of developing type 1 diabetes (138). Thus, vitamin D supplementation appears protective against type 1 diabetes onset, and suboptimal vitamin D status in infancy may have long-term effects on immune responses later in life.
There are also limited data suggesting that maternal vitamin D insufficiency during pregnancy may influence the risk of type 1 diabetes in offspring. In a recent case-control study, the risk of childhood onset of type 1 diabetes was more than two-fold greater in children whose mothers had serum 25-hydroxyvitamin D concentrations <21.6 ng/mL (54 nmol/L) during the last trimester of pregnancy compared to children born from women with serum 25-hydroxyvitamin D >35.6 ng/mL (89 nmol/L) (139). Other case-control studies have found that vitamin D supplementation during pregnancy was associated with a lower risk of their children developing diabetes-related autoantibodies (140, 141). However, a larger study conducted in mothers of children at increased genetic risk for diabetes reported no association between the appearance of islet autoantibodies and/or diabetes onset in offspring in the first year of life and maternal vitamin D intake during pregnancy (142). Another case-control study failed to observe a relationship between serum 25-dihydroxyvitamin D during early pregnancy and type 1 diabetes diagnosis in offspring (143). Large prospective studies are needed to establish whether maternal vitamin D status during pregnancy can influence the risk of type 1 diabetes in offspring.
Finally, the relationship of polymorphisms in vitamin D metabolism-related genes and type 1 diabetes is currently under investigation. It has been proposed that specific polymorphisms in genes, such as CYP27B1 (coding for 25-hydroxyvitamin D3-1α-hydroxylase) and VDR, may be functionally relevant to the action of vitamin D and may thus affect disease susceptibility. In a study conducted in 8,517 children and adolescents with type 1 diabetes and 7,320 control subjects, polymorphisms in genes involved in cholesterol synthesis and vitamin D hydroxylation were linked to circulating vitamin D concentrations and diabetic status (26).
Multiple sclerosis
Low levels of sun exposure and vitamin D deficiency appear to be associated with the development of multiple sclerosis (MS). Poor vitamin D status may compromise the function of specific immune cells critical in the regulation of various immune responses and help trigger autoimmunity in MS (144). Genetic determinants of low vitamin D status have been recently linked to an increased susceptibility to adult-onset MS in a Mendelian randomization analysis of data from the Multiple Sclerosis Genetics Consortium (145). This echoed the results of several observational studies that suggested an association between vitamin D sufficiency and decreased MS risk. A retrospective study of levels of ambient UV radiation and cases of MS conducted in Australia revealed that MS incidence in offspring was inversely correlated to maternal exposure to UV during early pregnancy (146). Sun exposure was also used as a surrogate marker for vitamin D exposure in a recent case-control study that included 1,660 MS patients and 3,050 controls. The authors found that infrequent outdoor activities and the use of sunscreen during early childhood and adolescence were associated with an increased risk of developing MS later in life (147). In a cross-sectional study, sun exposure and intake of cod liver oil (rich in vitamin D) during childhood were linked to later symptom onset among veterans with relapsing MS (148). Additionally, a case-control study in US military personnel, including 257 cases of diagnosed MS, found that Caucasian subjects in the highest quintile of serum 25-hydroxyvitamin D (>39.6 ng/mL) had a 62% lower risk of developing MS compared to the lowest quintile (<25.3 ng/mL) (149). Further, in two large cohorts of over 187,000 US women followed for at least 10 years, vitamin D supplement use (≥400 IU/day) was associated with a 41% reduction in the risk of developing MS (150). Another prospective, uncontrolled study monitored incidence of relapse in relation to vitamin D status in 156 patients with relapsing-remitting MS before and after they were given supplemental vitamin D (100,000 IU/month; 6-42 months, median of 31 months), in addition to first-line immunomodulatory therapy (151). Each 4 ng/mL increase in serum 25-hydroxyvitamin D concentration was associated with a 14.9% decrease in incidence of relapse (151). In a multicenter study conducted in patients newly diagnosed with a clinically isolated syndrome (CIS) and treated with interferon (IFN)-β, vitamin D status was predictive of MS disease activity and progression. Higher serum 25-hydroxyvitamin D concentrations (≥20 ng/mL or ≥50 nmol/L) in the first year following CIS diagnosis predicted a longer time to MS diagnosis, lower number of new lesions, and lower changes in lesion and brain volume during the subsequent four years of follow-up (152). However, a retrospective study suggested that vitamin D status in patients with relapsing-remitting MS had no predictive value regarding the time to conversion to secondary progressive MS, which is characterized by a worsening of disability (153).
Clinical trials have failed to demonstrate any benefit of vitamin D supplementation, alone or in combination with IFN-β treatment, with respect to relapse rates and disability-related symptoms in MS patients (154, 155). In other trials, supplemental vitamin D3 also failed to demonstrate immunomodulary activities (156-159). In a recent randomized, placebo-controlled trial in 53 IFN-β-treated patients with relapsing-remitting MS, supplementation with vitamin D3 (7,000 IU/day for four weeks, followed by 14,000 IU/day until week 48) showed little effect on the proportion of some regulatory T and B lymphocytes over the 48-week study period. Vitamin D3 only appeared to help maintain the proportion of anti-inflammatory CD4+ T cells — which decreased in patients given placebo — but failed to enhance their reactivity when stimulated with 1,25-dihydroxyvitamin D in vitro (157). In another trial, supplementation with vitamin D3 (10,400 IU/day for three months) to patients with relapsing-remitting MS was found to reduce the proportion of pro-inflammatory IL-17-producing CD4+ T cells, which are thought to play a central role in MS development (160).
Rheumatoid arthritis
Vitamin D deficiency may also be implicated in the etiology and/or progression of rheumatoid arthritis (RA), although evidence is mainly from animal studies. The absence of vitamin D receptors (VDR) in genetically modified mice has been linked to higher levels of inflammation and increased susceptibility to autoimmunity (161). When transgenic mice that spontaneously develop inflammatory arthritis are also deficient in VDR, they develop a more aggressive form of chronic arthritis (162). Also, specific polymorphisms in the VDR gene have been linked to an increased susceptibility to RA in certain populations, although how these genetic variants influence vitamin D functionality is not fully understood (163-165). The current data, however, point to a role for vitamin D in modulating the inflammatory process that underlies many chronic diseases, including RA. Several cross-sectional studies in individuals with moderate-to-high levels of inflammation have reported either no association or an inverse association between circulating 25-hydroxyvitamin D concentration and markers of inflammation. Nonetheless, there is a lack of intervention trials to show whether vitamin D supplementation could limit inflammation and reduce disease risk (including RA) in subjects with high inflammation levels (166).
At this time, it remains unclear whether the prevalence of vitamin D deficiency is linked to RA incidence. In a large cohort study of nearly 30,000 postmenopausal US women, subjects with the highest total vitamin D intakes (≥467.7 IU/day) had a 33% lower risk of developing RA after 11 years of follow-up than those with the lowest intakes (<221.4 IU/day) (167). Yet, more recent analyses of two large cohorts of nearly 200,000 US women followed for several decades found no association between reported vitamin D dietary intakes (using food frequency questionnaires) during adolescence or adulthood and incidence of RA later in life (168, 169). Moreover, several studies that explored the relationship between circulating vitamin D and disease activity in RA patients have reported mixed results (reviewed in 170). Yet, two recent meta-analyses of observational studies found an inverse relationship between vitamin D status and disease activity in RA patients, assessed using the Disease Activity Score 28 (DAS28) (171, 172). Finally, there is a dearth of studies exploring the effect of vitamin D supplementation on disease activity in arthritis subjects. A small randomized, double-blind, placebo-controlled study in 22 RA patients failed to demonstrate improvements in disease activity and inflammation level in subjects supplemented with calcium (1,500 mg/day) and high doses of vitamin D2 (ergocalciferol; averaging over 4,500 IU/day) for a year compared to placebo (173). Another three-month, randomized controlled trial in 41 women with early RA found no additional benefits of supplemental vitamin D3 (one bolus dose of 300,000 IU) to standard care (methotrexate and glucocorticoids) regarding T-helper lymphocyte enumeration, cytokine production, or clinical parameters including disease activity (174). Supplemental vitamin D also failed to reduce disease recurrence rate in RA patients enrolled in two small randomized controlled trials (175, 176). Since these studies have several limitations, including small sample size, additional research is warranted.
Systemic lupus erythematosus
More prevalent and severe in non-Caucasian populations (Hispanics, African descendants, and Asians) (177), systemic lupus erythematosus (SLE) is an autoimmune disease with heterogeneous clinical manifestations. The disease can potentially affect most tissues and organs, including skin (skin rash and photosensitivity), kidneys (nephritis), and joints (arthritis). There is evidence of a role for vitamin D in the prevention of SLE in animal models (178). Interestingly, a recent meta-analysis of 11 case-control studies found that specific VDR polymorphisms were linked to SLE in Asians particularly (179). However, the functional relevance of such genetic variants is not known (180). Analyses of two large prospective cohort studies of nearly 200,000 US women failed to show an association between dietary vitamin D intake (measured by food frequency questionnaire) during adolescence or adulthood and incidence of SLE later in life (168, 169).
Yet, a suboptimal vitamin D status is commonly observed in subjects with SLE, and this is partly explained by the lack of sunlight exposure, which tends to aggravate disease symptoms (181, 182). Serum concentrations of 25-hydroxyvitamin D were inversely correlated with measures of disease activity in a cohort of 378 patients with SLE (183). The correction of vitamin D insufficiency with high levels of vitamin D3 (100,000 IU/week for one month followed by 100,000 IU/month for six months) in 20 subjects with SLE was linked to a reduction in signs of immune imbalance and in levels of autoantibodies typically detected in SLE, suggesting a therapeutic value for vitamin D in disease treatment (184). Another prospective study conducted in 52 vitamin D-deficient patients with cutaneous lupus erythematosus (a type of lupus with skin disorders only) reported a reduction in disease severity in the group supplemented with vitamin D3 (1,400 IU/day initially, followed by 800 IU/day) and calcium for one year compared to untreated patients (185). Supplementation with vitamin D3 (200 IU/day for one year) was also able to reduce the level of inflammatory cytokines in a randomized, placebo-controlled study conducted in 267 patients with SLE (186). In another randomized, placebo-controlled trial, supplementation with vitamin D3 (50,000 IU/week for six months) improved SLE disease activity index (SLEDAI) and European Consensus Lupus Activity Measurement (ECLAM) scores, as well as some measures of fatigue in young adults with juvenile-onset SLE (187). However, in two other recent studies, supplementation with vitamin D3 (weekly/monthly bolus doses equivalent to ~800 to 7,000 IU/day for 6 to 24 months) improved vitamin D status in SLE patients but failed to show any benefit regarding disease activity (188, 189). While oral vitamin D administration to SLE patients is well tolerated, its efficacy remains questionable and deserves further investigation in clinical trials.
Summary
Thus, evidence from human epidemiological studies suggests that while it cannot yet be concluded that vitamin D supplementation is beneficial in prevention or treatment of autoimmune disease, it is reasonable to assume that correcting vitamin D insufficiency and maintaining sufficient levels could possibly help decrease disease risk (190).
Cardiovascular disease
Hypertension (high blood pressure)
Hypertension is a well-known risk factor for cardiovascular disease (CVD) (191). The results of observational and clinical studies suggest a role for vitamin D in lowering blood pressure, which may be partly explained by the fact that 1α,25-dihydroxyvitamin D inhibits renin synthesis (see Function). Thus, vitamin D deficiency and subsequent upregulation of the renin-angiotensin system may contribute to high blood pressure and CVD risk. It has also been suggested that elevated PTH concentrations may increase the risk of hypertension and CVD (6). Yet, in a recent prospective cohort study of 3,002 individuals (mean age, 59 years at baseline), the incidence of hypertension, which affected 41% of participants during the nine year follow-up period, was not higher in those with serum 25-hydroxyvitamin D concentrations lower than 20 ng/mL and was only marginally associated with elevated PTH concentrations (192). Nevertheless, a meta-analysis of seven prospective studies, including a total of 48,633 participants with nearly 5,000 incident hypertension cases, found a 30% lower risk of hypertension in those in the top versus bottom tertiles of serum 25-hydroxyvitamin D concentrations. The dose-response analysis estimated that every 10 ng/mL increase in serum 25-hydroxyvitamin D concentration was associated with a 12% lower risk of hypertension (193). Another meta-analysis of four prospective and 14 cross-sectional studies also reported an inverse relationship between circulating 25-hydroxyvitamin D and hypertension (194).
Endothelial dysfunction
Vascular endothelium dysfunction, which contributes to an increased risk of cardiovascular disease (CVD), is common in patients with chronic kidney disease (CKD) (195). In CKD patients, abnormal endothelial function is associated with low values of flow-mediated dilation (FMD) of the brachial artery, a surrogate marker of vascular health. In a recent study conducted in subjects with mild-to-moderate CKD, serum 25-hydroxyvitamin D concentrations were positively associated with FMD values, suggesting a link between suboptimal vitamin D status and endothelial dysfunction (196). In a preliminary intervention study, 26 patients with moderate CKD and vitamin D insufficiency (mean value, 17.2 ng/mL) were supplemented twice with 300,000 IU of vitamin D3 (at weeks 1 and 8) and followed for a total of 16 weeks. Vitamin D supplementation nearly doubled serum 25-hydroxyvitamin D concentrations and decreased PTH concentrations by 68.5%; improved vitamin D status was accompanied by increased FMD values and reduced levels of endothelial dysfunction markers (197). A recent meta-analysis of 12 small randomized controlled trials in participants at high risk for CVD found a significant increase in FMD with vitamin D supplementation (daily doses, 2,500-5,000 IU; weekly dose, 50,000 IU; monthly dose, 60,000 IU; single-bolus doses, 100,000-200,000 IU) for eight weeks to six months (198).
Cardiovascular events in observational studies and clinical trials
To date, the many epidemiological studies investigating the relationship between vitamin D and outcomes of CVD have provided mixed results (reviewed in 199). Recent Mendelian randomization studies found no association between genetically low serum 25-hydroxyvitamin D concentrations and risks of coronary heart disease, ischemic heart disease, or myocardial infarction (200, 201), suggesting that associations reported in observational studies may be due to confounding or reverse causation. In the RECORD trial in 5,292 older people (see Osteoporosis), supplementation with 800 IU/day of vitamin D3 (± calcium) reduced the risk of first cardiac failure but had no effect on the risk of myocardial infarction and stroke compared to supplementation with calcium alone or placebo (202). Data on the effect of vitamin D supplementation on cardiovascular events were collected from 21 randomized controlled studies (including the RECORD trial) in 13,033 participants (≥60 years old) and combined in a meta-analysis (202). No effect of vitamin D (including vitamin D analogs) was found for major cardiovascular events, including heart failure, myocardial infarction, and stroke over follow-up periods of 1 to 6.2 years (202). However, caution is advised when interpreting these results since the trials were initially designed to evaluate the effect of vitamin D on bone health, and cardiovascular outcomes were not primary endpoints. Several randomized controlled trials exploring the effect of vitamin D supplementation on CVD risk are currently underway (203), including two large trials, the Vitamin D and Omega-3 Trial (VITAL) in the US (204) and the D-Health trial in Australia (205). The results of one randomized controlled trial, the Vitamin D Assessment (ViDA) trial in New Zealand, were recently published. The total number of CVD events and time to first CVD event during follow-up did not differ between those supplemented with vitamin D3 (initial dose of 200,000 IU for the first month followed by monthly doses of 100,000 IU) and those given a placebo for a median 3.3 years (206).
Type 2 diabetes mellitus
People with metabolic syndrome are at increased risk for type 2 diabetes mellitus (noninsulin-dependent diabetes mellitus) and cardiovascular disease (CVD). Metabolic syndrome refers to several metabolic disorders, including dyslipidemia, hypertension, insulin resistance, and obesity. A recent study found that the prevalence of type 2 diabetes was associated with low levels of serum 25-hydroxyvitamin D (<30 ng/mL) in 1,801 patients with metabolic syndrome. During an eight-year follow-up period, lower risks of all-cause mortality (72% lower risk) and CVD-specific mortality (64% lower risk) were reported in individuals with serum 25-hydroxyvitamin D concentrations over 30 ng/mL (75 nmol/L) when compared to those with concentrations below 10 ng/mL (25 nmol/L) (207).
In healthy people, vitamin D sufficiency is positively correlated with insulin sensitivity and adequate pancreatic β-cell function. Conversely, vitamin D deficiency might affect glucose homeostasis and cause impaired glucose tolerance and insulin resistance (208). In a cross-sectional study conducted in 12,719 adults, of whom 4,057 had prediabetes (i.e., an increased risk of developing type 2 diabetes), the prevalence of prediabetes was associated with lower concentrations of serum 25-hydroxyvitamin D (≤32.4 ng/mL). Subjects with the lowest concentrations of serum 25-hydroxyvitamin D (≤17.7 ng/mL) were more likely to be current smokers, obese, and have hypertension (209). Vitamin D insufficiency in high-risk individuals may accelerate the progression to overt diabetes. In a prospective study of 2,378 middle-aged men and women followed for 8 to 10 years, the risk for progression to type 2 diabetes from prediabetes was 62% lower in women and 60% lower in men in the highest compared to the lowest quartile of circulating vitamin D (>28.4 ng/mL vs. <18.5 ng/mL). A dose-response analysis measured an average 23% reduction in the risk of progression to type 2 diabetes for every 4 ng/mL (10 nmol/L) increment in serum 25-hydroxyvitamin D concentration (210). A recent review and meta-analysis of 18 prospective cohort studies, including over 210,000 participants followed for a median period of 10 years, found that individuals in the top third of vitamin D levels (reported as either circulating vitamin D or dietary intakes) had lower risks of developing type 2 diabetes (19% lower risk) and metabolic syndrome (14% lower risk) compared to those in the bottom third (211). In another meta-analysis of nine prospective studies, including 28,258 older people (mean age, 67.7 years), lower versus higher circulating vitamin D concentrations at baseline were found to be associated with a 17% higher risk of developing type 2 diabetes over a median follow-up period of 7.3 years (212). Currently, limited evidence suggests that vitamin D supplementation may improve insulin sensitivity in individuals with glucose intolerance or manifest type 2 diabetes (213-216). There is a need for well-designed clinical trials to examine whether maintaining adequate vitamin D levels can prevent adverse metabolic outcomes in healthy and at-risk individuals.
Neurodegenerative diseases
Cognitive impairment and Alzheimer's disease
Alzheimer’s disease (AD) is the most common form of dementia, characterized by the presence of extra-neuronal β-amyloid plaques and intra-neuronal Tau protein aggregates (known as neurofibrillary tangles) in the brain. Mechanistic models currently investigated in animal research suggest that vitamin D deficiency or disorders of vitamin D metabolism and/or the disruption of the vitamin D-VDR pathway in the cerebral regions of the cortex and hippocampus may be involved in the degeneration of neurons and loss of cognitive functions (217). Experimental evidence supporting a role for vitamin D in calcium channel regulation, neuroprotection, and immunomodulation in the central nervous system also implies that low vitamin D status may precede or contribute to cognitive dysfunction with age (218).
A number of observational studies have examined cognitive decline and degenerative brain disease in the elderly in relation to dietary intake of vitamin D and serum 25-hydroxyvitamin D concentration. In a large French cohort study on osteoporosis and hip fractures in postmenopausal women, impairments in global cognitive performance, assessed with the Pfeiffer Short Portable Mental State Questionnaire (SPMSQ), were associated with lower dietary intakes of vitamin D (<1,400 IU/week vs. ≥1,400 IU/week) in 5,596 elderly women (mean age, 80.5 years) (219). A seven-year follow-up study of a subgroup of 498 women indicated that the risk of Alzheimer’s disease (but not other types of dementia) was 77% lower in those in the highest versus the lowest quintiles of vitamin D dietary intakes at baseline (220). Some, but not all, observational studies have found an association between low serum 25-hydroxyvitamin D concentrations and mild cognitive impairment in older adults (219, 221, 222). The cross-sectional and longitudinal analysis of two prospective studies, which included 1,604 men (223) and 6,257 women (224) aged 65 and over, reported a 60% greater odds of cognitive impairment at baseline and a 58% increased risk of cognitive decline during a four-year follow-up period in women, but not in men, with vitamin D deficiency (circulating 25-hydroxyvitamin D <10 ng/mL vs. ≥30 ng/mL). In the nested case-control, multiethnic, Singapore Kidney Eye Study, which included 2,273 individuals (mean age, 70.4 years), serum 25-hydroxyvitamin D concentrations were inversely correlated with cognitive deficits affecting retrograde episodic memory, semantic memory, and orientation in time, as assessed by the Abbreviated Mental Test (AMT) (225).
Yet, systematic reviews and meta-analyses of observational studies have given mixed results regarding the association of vitamin D status with cognitive performance and AD (226-230). Moreover, the recent analysis of data from 1,182 men followed for 18 years in the Uppsala Longitudinal Study of Adult Men (ULSAM) failed to find associations of genetic determinants of vitamin D synthesis, vitamin D intakes, and plasma 25-hydroxyvitamin D concentrations with risks of cognitive impairments, AD, vascular dementia, or all-cause dementia (231). In contrast, another Mendelian randomization study associated genetic determinants of low vitamin D status to higher risk of AD in the International Genomics of Alzheimer’s Project dataset (17,008 AD cases and 37,154 healthy cases) (232).
Nevertheless, the prevalence of vitamin D insufficiency/deficiency ranges between 70% and 90% in older adults, and correcting low concentrations of serum 25-hydroxyvitamin D may help improve cognitive processes, in particular executive functions (233). In a small, non-randomized controlled study in an outpatient clinic, global cognitive function was assessed at baseline and after 16 months in 20 patients supplemented with 800 IU/day (or 100,000 IU/month) of vitamin D and in 24 control subjects. The supplementation of outpatients with vitamin D resulted in the correction of low vitamin D status (average serum 25-hydroxyvitamin D concentration was 16.8 ng/mL at baseline and 30 ng/mL at 16 months) and was associated with a significantly improved scoring in cognition tests compared to the non-supplemented group (234). In a small randomized, placebo-controlled clinical trial in 32 mild-to-moderate AD patients receiving nasal insulin, high doses of vitamin D2 supplementation for eight weeks (up to 36,000 IU/day) did not significantly improve cognitive performance compared to low doses (1,000 IU/day) (235). More research is needed to investigate a causal relationship between vitamin D repletion and potential long-term cognitive benefits in older adults. Further, it is of great importance to evaluate whether correcting vitamin D deficiency in cognitively impaired subjects can improve the impact of anti-dementia therapy (236).
Parkinson's disease
Parkinson’s disease (PD) has been associated with a high prevalence of vitamin D insufficiency among patients, especially those with greater mobility problems (237). A case-control study of 296 outpatients with a mean age of 65 years indicated that 23% of PD subjects had serum 25-hydroxyvitamin D concentrations lower than 20 ng/mL compared to 16% and 10% of AD and healthy individuals, respectively (238). In a prospective cohort study conducted among 3,173 men and women aged 50-79 years and free of PD at baseline, individuals in the highest quartile of serum 25-hydroxyvitamin D (≥20 ng/mL for women and ≥22.8 ng/mL for men) had a 67% lower risk of PD compared to those in the lowest quartile (≤10 ng/mL for women and ≤11.2 ng/mL for men) (239). Meta-analyses pooling data from observational studies all showed that vitamin D inadequacy was more likely reported in subjects with PD than in healthy controls (240-242).
In a randomized, double-blind, placebo-controlled study, 112 PD patients (mean age, 72 years) on standard PD treatment were supplemented with 1,200 IU/day of vitamin D or a placebo for 12 months. Vitamin D supplementation nearly doubled serum 25-hydroxyvitamin D concentration (from mean of 22.5 ng/mL to 41.7 ng/mL) in supplemented subjects and limited the progression of PD, as indicated by a greater proportion of patients who showed no worsening (as assessed by the Hoehn and Yahr stage and the United Parkinson Disease Rating Scale part II) in the supplemented group compared to the placebo group (243). It is not known whether vitamin D insufficiency has a role in the pathogenesis of the disease, but the repletion of vitamin D may provide health benefits that go beyond the prevention and/or the treatment of PD. For example, vitamin D deficiency may contribute to the increased risk of osteoporosis and bone fracture in individuals with neurologic disorders, including PD and multiple sclerosis (244-246). Interestingly, sunlight exposure was found to be associated with improved vitamin D status, higher bone mineral density of the second metacarpal bone, and lower incidence of hip fracture in a prospective study conducted in 324 elderly people with PD (247).
Adverse pregnancy outcomes
A systematic review and meta-analysis of 31 observational studies on maternal vitamin D status and pregnancy outcomes indicated that vitamin D insufficiency may be associated with gestational diabetes mellitus, preeclampsia, and bacterial vaginosis in pregnant women. Low maternal serum vitamin D during pregnancy was also linked to an increased risk for small-for-gestational age infants and low-birth-weight infants, but not for Cesarean section (248). However, the number of intervention trials is currently too limited to draw conclusions as to whether vitamin D supplementation during pregnancy might reduce the incidence of the above-mentioned adverse outcomes (249).
Gestational diabetes mellitus
Abnormal hyperglycemia due to pancreatic β-cell dysfunction characterizes the onset of gestational diabetes mellitus (GDM) in pregnant women without known type 2 diabetes mellitus. This condition is associated with serious adverse maternal outcomes, including preeclampsia, high risk of Cesarean delivery, and life-long increased risk of developing metabolic syndrome and type 2 diabetes mellitus. GDM may also contribute to increased risks of fetal macrosomia (excessive birth weight), neonatal hypoglycemia, infant respiratory distress, and increased life-long risk for obesity, glucose intolerance, type 2 diabetes mellitus, and cardiovascular disease in the offspring (reviewed in 250).
A recent prospective study conducted in 655 pregnant women found that the mean serum 25-hydroxyvitamin D concentration during the first trimester of pregnancy was significantly lower in 54 women who developed incident GDM compared to the rest of the cohort (23 ng/mL vs. 25.4 ng/mL). After multiple adjustments for confounding factors of vitamin D status and GDM risk (including overweight/obesity and prior history of type 2 diabetes and GDM), the study found each 7.5 ng/mL decrease in serum 25-hydroxyvitamin D concentration during early pregnancy was associated with a 48% higher risk of developing GDM (251). Low serum 25-hydroxyvitamin D concentrations (<29.4 ng/mL) during the second trimester of pregnancy were also associated with GDM incidence in a nested case-control study of 118 women with GDM and 219 matched control subjects (252). Five meta-analyses (248, 253-256), including observational studies of moderate-to-high quality, also reported that maternal serum vitamin D concentrations during pregnancy were inversely related to the risk of developing GDM despite evidence of bias amongst studies, such as the use of different methods for serum 25-hydroxyvitamin D measurement, measures done in different trimesters, and different criteria to assess GDM (reviewed in 257).
Further, evidence for the role of vitamin D in glucose regulation during pregnancy was reported in a small randomized, double-blind, placebo-controlled trial in 54 pregnant women diagnosed with GDM. The supplementation with 50,000 IU of vitamin D3 twice during a six-week period (at day 1 and day 21) resulted in significantly lower fasting plasma glucose and serum insulin concentration, reduced insulin resistance, and improved insulin sensitivity compared to placebo (258). This suggests that vitamin D deficiency may adversely affect glucose tolerance during pregnancy and contribute to the onset of GDM. Yet, the potential benefits of vitamin D supplementation in the prevention of glucose intolerance and GDM during pregnancy have not been assessed. A multicentered, randomized controlled trial (DALI) is ongoing in Europe to evaluate the effects of vitamin D and lifestyle interventions (healthy eating and physical activity) on the metabolic status of pregnant women at risk of GDM (inclusion criteria: pre-pregnancy BMI ≥29 kg/m2) (259). Preliminary findings suggest that healthy eating and physical activity can help lower gestational weight gain, when compared to standard-of-care; however, these lifestyle changes are unlikely to reduce the risk of GDM among obese pregnant women (260). The results regarding the effect of vitamin D supplementation in the DALI study are yet to be published.
Health outcomes in offspring
During pregnancy, increased intestinal calcium absorption and mobilization of calcium from the skeleton allows accretion of calcium within the fetal skeleton. Yet, observational studies that examined the relationship between maternal vitamin D status and measures of fetal bone growth have not provided consistent results (261, 262). In addition, recent data from the Maternal Vitamin D Osteoporosis Study (MAVIDOS) suggested no difference in whole-body bone mineral content (BMC) of newborns from mothers randomized to daily supplementation with either vitamin D3 (1,000 IU) or placebo from <17 weeks’ gestation until delivery (263). Further, the risk of fracture in Danish children ages 10 to 18 years was similar regardless of whether their mothers were exposed to extra vitamin D from fortification during pregnancy (264). While maternal vitamin D supplementation during pregnancy effectively prevents the neonate’s risk of vitamin D deficiency at birth (265), there is little evidence that neonatal vitamin D status influences the risk of fracture later during childhood (266).
A few observational studies have given rather weak evidence in support of a relationship between maternal vitamin D sufficiency during pregnancy and incidence of respiratory conditions and allergies in children (267). A randomized controlled trial found that the supplementation of 108 pregnant women in the third trimester (at week 27 of gestation until delivery) with either 800 IU/day or a bolus dose of 200,000 IU of vitamin D3 did not decrease the risk of wheezing, allergic rhinitis, food allergy diagnosis, lower respiratory tract infections, or eczema in offspring at three years of age compared to placebo (N=50) (268). A more recent double-blind, randomized controlled trial found that vitamin D3 supplementation of 295 Danish pregnant women, from weeks 22 to 26 of gestation until delivery, with 2,800 IU/day (70 μg/day) — compared to 400 IU/day (10 μg/day) of vitamin D3 (i.e., the current recommendation in Denmark) (N=286) — reduced the risk of troublesome episodes of lung symptoms by 17% in offspring during the first three years of life (269). However, no differences were reported regarding the risk of persistent wheeze, asthma, allergic sensitization, respiratory tract infections, or eczema between treatment and control groups (269). In a similar randomized, double-blind, controlled trial — the Vitamin D Antenatal Asthma Reduction Trial — conducted in 777 US pregnant women with a history of asthma, allergic rhinitis, or eczema (or whose partner had such an history), supplementation with 2,400 IU/day (60 μg/day) or 400 IU/day (10 μg/day) did not result in differences in the risk of developing asthma or recurrent wheezing in their children at age three years (270). Despite the lack of significance reported in individual studies, the pooled analysis of the three trials found a 19% reduction in the risk of recurrent wheeze in children whose mothers received high-dose versus low-dose vitamin D supplementation during pregnancy (271). In a cohort of 378 mother-child pairs, high serum 25-hydroxyvitamin D concentrations measured in the 34th week of pregnancy were associated with an increase in food allergy of the child during the first two years of life, warranting careful evaluation of the safety of vitamin D supplementation during pregnancy (272).
Since vitamin D insufficiency has been linked to autoimmunity (see Autoimmune diseases), it has also been proposed that poor maternal vitamin D status during pregnancy may contribute to an increased risk of autoimmune diabetes (insulin-dependent type 1 diabetes mellitus) in the offspring. Yet, the results of a study of 3,723 children at high genetic risk for type 1 diabetes and followed for a mean 4.3 year-period found that maternal intake of vitamin D (from food and/or supplements) during the third trimester of pregnancy (assessed through food frequency questionnaires) was not associated with advanced β-cell autoimmunity or clinical diabetes (142). In a nested case-control study, there was no difference in mean serum concentration of 25-hydroxyvitamin D during the first trimester of pregnancy between 343 mothers of children with type 1 diabetes and 343 control mothers (143). A follow-up study suggested that specific maternal VDR polymorphisms, rather than vitamin D status, may be linked to an increased susceptibility to developing type 1 diabetes in children (273). Another nested case-control study (119 mothers of children with type 1 diabetes and 129 control mothers) found an inverse association between maternal vitamin D-binding protein — but not 25-hydroxyvitamin D — concentration during the third trimester of pregnancy and the risk of type 1 diabetes in children (274). At present, there is no established causality between maternal vitamin D status during pregnancy and risk of autoimmune disease in offspring.
Acute respiratory infections
More than 200 viruses are responsible for causing familiar infections of the upper respiratory tract (URT), known as the common cold, resulting in symptoms of nasal congestion and discharge, cough, sore throat, and sneezing (275). The analysis of cross-sectional data from 18,883 participants (ages 12 years and older) of the Third US National Health and Nutrition Examination Survey (NHANES III) reported an inverse relationship between serum 25-hydroxyvitamin D concentrations and recent (self-reported) URT infection (URTI). Compared to levels of circulating vitamin D of 30 ng/mL or above, the risk of URTI was 24% higher in individuals with concentrations between 10 and 29 ng/mL and 36% higher in those with levels below 10 ng/mL (276). A subgroup analysis indicated that low concentrations of serum 25-hydroxyvitamin D in subjects with asthma and chronic obstructive pulmonary disease (COPD) were linked to a greater susceptibility to URTI when compared to people without pulmonary disease.
In a randomized, double-blind, placebo-controlled trial conducted in 322 healthy adults (ages ≥18 years), supplementation with monthly doses of vitamin D3 (200,000 IU for the first two months and 100,000 IU for the following 16 months) significantly raised the mean serum 25-hydroxyvitamin D concentration (from 29 ng/mL to 48 ng/mL) in the intervention group but did not decrease the occurrence of URTI compared to placebo (277). Moreover, in a larger, multicenter, four-arm clinical trial in 2,259 subjects (aged 45-75 years) with a history of colorectal adenoma, daily vitamin D3 supplementation of 1,000 IU did not reduce the number or the duration of URTI episodes during winter or the rest of the year, even among participants with the lowest serum 25-hydroxyvitamin D concentrations at baseline (278). In addition, the post-hoc analysis of data from a randomized, placebo-controlled trial in 644 individuals (ages 60-84 years) found that monthly supplementation with 30,000 IU or 60,000 IU of vitamin D3 for a maximum period of one year did not significantly decrease the rate of antibiotic prescriptions for bacterial airway infections. The stratified analysis, however, found that doses of 60,000 IU/month reduced the risk of using antibiotics in participants ≥70 years old by 47% (279). In addition, compared to placebo, the supplementation of pregnant women with vitamin D3 (with 2,000 IU/day) for three months until birth followed by the supplementation of their infants (800 IU/day) from birth to six months of age significantly reduced the number of acute respiratory infections after the intervention period in children 6 to 18 months old (280). Interestingly, despite significant heterogeneity among trials, the pooled analysis of these data with that of 21 additional trials suggested an overall 12% reduction in incidence of URTI with vitamin D3 given as bolus doses (every week, month, or every three months), daily doses, or a combination of bolus and daily doses (281). Subgroup analyses revealed a 42% reduction in URTI risk with vitamin D3 supplementation in subjects with baseline serum 25-hydroxyvitamin D concentrations <10 ng/mL, while there was no protective effect of vitamin D3 in those with concentrations ≥10 ng/mL. Moreover, large bolus doses (≥30,000 IU) were found to be ineffective compared to daily or weekly doses such that, in subgroup analyses that excluded bolus doses, daily and weekly vitamin D3 regimens appeared to be protective against URTI regardless of baseline vitamin D status. Finally, the effect of vitamin D3 supplementation on URTI did not appear to vary with age, BMI, the presence of asthma or COPD, and flu vaccination status (281).
The findings of a large clinical trial in New Zealand, the Vitamin D Assessment (ViDA) Study, confirmed the ineffectiveness of large bolus doses of vitamin D in reducing the risk of acute respiratory infections (282). In this double-blind, placebo-controlled trial in 5,110 older adults (ages 50-84 at baseline), supplementation with an initial dose of 200,000 IU of vitamin D3 followed by 100,000 IU/month of vitamin D3 for a median of 3.3 years had no effect on the incidence of (self-reported) acute respiratory infections (HR, 1.01; 95% CI, 0.94-1.07). Upon data stratification by vitamin D status, a similar null result (HR, 1.08; 95% CI, 0.95-1.23) was seen in study participants with blood vitamin D concentrations ≤20 ng/mL (≤50 nmol/L; 282).
The recently completed VITAL (Vitamin D and Omega-3 Trial) may provide additional evidence of an effect of daily dosing (2,000 IU/day of vitamin D) on risk of airway infections in older adults, although the prevention of infectious disease is a secondary outcome in the trial (204, 283, 284).
In a trial in preschool-age children, vitamin D supplementation with 2,000 IU/day for at least four months did not decrease the incidence (285) or severity (286) of URTI in winter compared to supplementation with 400 IU/day for at least four months. Supplementation with 2,000 IU/day led to significantly higher mean serum 25-hydroxyvitamin D concentrations compared to the lower dose (48.7 ng/mL vs. 36.8 ng/mL), but the 400 IU dose may have been sufficient to prevent URTI in these young children (285). In a placebo-controlled trial in 1,300 healthy children and adolescents in rural Vietnam (baseline 25-hydroxyvitamin D concentrations of 26 ng/mL for both the control and intervention group), supplementation with 14,000 IU/week of vitamin D for eight months significantly reduced the incidence of non-influenza viral respiratory infections by 24%, but not the incidence of influenza A or B infection (287).
Coronavirus disease, COVID-19
The coronavirus disease, COVID-19, is caused by infection with the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). This virus originated in Wuhan, China in late 2019 and rapidly spread throughout the world causing a global pandemic. Flu-like symptoms, including cough, fever, fatigue, and difficulty breathing, as well as several other diverse symptoms characterize COVID-19 (see the websites of the CDC and the WHO). The effects of the disease vary widely, with the most severe resulting in pneumonia, respiratory distress, and death. Some individuals infected with SARS-CoV-2, however, are asymptomatic but can transmit virus to others (288). Moreover, SARS-CoV-2 infected individuals can spread disease before they experience symptoms (i.e., presymptomatic transmission) (289), placing importance on public health measures like hand washing, face mask wearing, social distancing, and testing and contact tracing to curtail the pandemic.
A number of observational studies have examined the correlation of vitamin D status and COVID-19 incidence, with most — but not all (290-292) — finding vitamin D deficiency or insufficiency associated with an increased risk of SARS-CoV-2 infection. In a retrospective cohort study of more than 191,000 US residents administered a SARS-CoV-2 test during a three-month period in spring 2020, SARS-CoV-2 positivity was strongly associated with a lower serum 25-hydroxyvitamin D concentration, measured at a single point in the 12 months prior to the viral test (293). In this study, each 1 ng/mL increment of serum 25-hydroxyvitamin D was linked to a 1.6% lower risk of SARS-CoV-2 positivity, with the risk being the lowest at serum concentrations of 55 ng/mL and above (293). In a retrospective, population-based study among 7,807 people in Israel, including 782 SARS-CoV-2 positive cases, plasma 25-hydroxyvitamin D concentrations below 30 ng/mL were associated with a 50% higher risk of SARS-CoV-2 infection compared to higher vitamin D concentrations (294). The vitamin D status assessment in this study was done prior to the viral testing, but details on the time frame are lacking. The temporal relationship of the vitamin D status measure and COVID diagnostic test is also a concern in an analysis of a large dataset from the UK Biobank, which found vitamin D status was not associated with SARS-CoV-2 infection: the study utilized vitamin D status assessments done 10 to 14 years prior to the COVID-19 diagnostic test (291). Consistent with findings of most available studies, a small retrospective cohort study (295) and five case-control studies (296-300) reported an inverse association of vitamin D status and SARS-CoV-2 infection. A 2021 meta-analysis of 10 observational studies, including 361,934 participants, reported an association between vitamin D insufficiency or deficiency and increased risk of COVID-19 (OR, 1.43; 95% CI: 1.00-2.05), although heterogeneity across studies was high (301). Further, in an analysis of data from the UK Biobank, regular use of vitamin D supplements was found to be associated with a 34% lower risk of SARS-CoV-2 infection compared nonusers of vitamin D supplements (292).
Additionally, most of the available observational research suggests an association between low vitamin D status and COVID-19 severity, with studies finding vitamin D deficiency linked to an increased risk of disease severity, as measured by need for hospitalization, intensive care unit admission (302), stage of pneumonia (in men but not in women; 303), need for non-invasive ventilation (304), need for invasive mechanical ventilation (305), or a combination of these and other indicators (297). Another study found that insufficient vitamin D status, defined as plasma 25-hydroxyvitamin D concentrations below 30 ng/mL, was associated with an increased likelihood of COVID-related hospitalization, but the association failed to reach statistical significance (p=0.061) except in a subanalysis of individuals over 50 years of age (OR, 2.71; 95% CI: 1.55-4.78; p<0.001) (294). In contrast to most of the evidence to date, one case-control study found vitamin D deficiency was not linked to COVID-19 severity (298). Further, a few studies have looked at the relationship of vitamin D status and COVID-19 mortality, with three finding vitamin D deficiency to be associated with an increased risk of death (303, 305, 306) and two finding no association (304, 307), although one of the studies finding no association used vitamin D status measurements more than 10 years prior to the COVID-19 diagnostic testing (307).
Since there are several known risk factors for severe illness from COVID-19, including advanced age, obesity, and preexisting type 2 diabetes or cardiovascular disease (308), it is important for observational studies to control for potential confounders. Two quasi-experimental studies reported that bolus supplementation with vitamin D3 (either 50,000 IU per month or 80,000-100,000 IU every two to three months) prior to or during SARS-CoV-2 infection was associated with a less severe illness and improved survival in frail elderly COVID-19 patients (309, 310). A number of randomized controlled trials of vitamin D supplementation in COVID-19 prevention and treatment are currently underway; the results of these trials will inform on the causality of the association. Nevertheless, the currently available data indicate that improving vitamin D status through supplementation represents a modifiable risk factor for COVID-19.
Disease Treatment
Atopic dermatitis
Atopic dermatitis or eczema is particularly prevalent in industrialized countries, affecting 10%-20% of children and 1%-3% of adults. Atopic dermatitis is a chronic inflammatory skin disorder characterized by dry and pruritus (itchy) areas of the skin in affected subjects. Local skin inflammation and immune dysfunction can damage the epidermal barrier and increase the susceptibility to skin infections and atopic reactions in affected individuals. The disease is often associated with other atopic diseases, including food allergies, asthma, and allergic rhinitis (311).
While the etiology of the disease is not fully elucidated, it has been suggested that vitamin D deficiency might contribute to the onset and/or the severity of the disease (312). Recently, using large-scale datasets from Caucasian people of European descent, including the UK Biobank resource (313) and the SUNLIGHT (35), the GABRIEL asthma (314), and EAGLE eczema (315) consortia, a Mendelian randomization study found no association between genetically low serum 25-hydroxyvitamin D concentrations and risks of atopic dermatitis, asthma, or high serum immunoglobulin (Ig)-E concentrations (316).
A number of randomized controlled studies have examined whether vitamin D might be an effective adjunct tool in disease management, possibly through regulating local inflammatory reactions and stimulating antimicrobial activities in the skin. Moreover, the beneficial effect of phototherapy observed in specific cases of atopic dermatitis may be partly mediated by the action of vitamin D (311). In a small randomized, double-blind, placebo-controlled study in 45 patients with atopic dermatitis and low vitamin D status (70% of subjects had serum 25-hydroxyvitamin D concentrations <20 ng/mL), daily administration of 1,600 IU of oral vitamin D3, alone or together with 600 IU/day of vitamin E, for a period of 60 days significantly reduced the extent and intensity of eczema, as assessed by the SCORAD (SCORing Atopic Dermatitis) score (317). Vitamin D3 (1,600 IU/day for 60 days) also improved vitamin D status and reduced disease severity in 53 patients with atopic dermatitis in another small randomized trial (318). More recently, vitamin D3 (1,000 IU/day for one month) improved the severity of winter-related atopic dermatitis in Mongolian children, as shown by changes in Eczema Area and Severity Index (EASI) and Investigator’s Global Assessment (IGA) scores (319). A meta-analysis of four small trials (including those cited above) confirmed that supplemental vitamin D can lead to measurable clinical improvements in affected individuals (320). Larger trials are needed to strengthen these preliminary findings and determine the most appropriate and effective supplementation regimen. Of note, topical treatment of psoriasis with vitamin D analogs has been approved by the US Food and Drug Administration (FDA) and may be effective in the management of other skin disorders (321).
Inflammatory bowel diseases
Several ill-defined environmental and genetic factors are thought to contribute to the development of the inappropriate immune response to the intestinal microbiota that causes ulcerative colitis (UC) and Crohn’s disease (CD). While specific VDR polymorphisms may be linked to an increased susceptibility to developing UC and CD (322), higher vitamin D intakes and predicted circulating levels were found to be associated with a reduced incidence of UC and CD in a large cohort of 72,719 women (323). A meta-analysis of six observational studies found an inverse association between vitamin D status and severity of CD (324). Three studies have investigated whether vitamin D3 could benefit patients with CD, possibly through reducing intestinal inflammation. In one multicenter, double-blind, placebo-controlled study, the relapse rate in CD patients in remission after one year of treatment was significantly lower in those supplemented daily with 1,200 IU of vitamin D3 and 1,200 mg of calcium compared to those who received calcium alone (13% vs. 29%) (325). In a second pilot study, incremental daily doses of vitamin D3, from 1,000 IU up to 5,000 IU, were administrated over a 24-week period to 18 CD patients in order to achieve circulating 25-hydroxyvitamin D concentrations >40 ng/mL. Although half of the patients failed to achieve 40 ng/mL, the mean 25-hydroxyvitamin D concentration was raised to 45 ng/mL (from a baseline mean of 16 ng/mL), and the overall improvement in vitamin D status was associated with a significant decrease in disease severity as assessed by Crohn’s Disease Activity (CDAI) scores (326). In a three-month, randomized, double-blind, placebo-controlled trial in 27 CD patients in remission, daily supplementation with vitamin D3 (2,000 IU) improved vitamin D status but had no significant effect on intestinal permeability ('leaky gut') or measures of inflammation and disease activity (327). Yet, the study suggested that achieving serum 25-hydroxyvitamin D concentrations ≥30 ng/mL might help reduce intestinal inflammation and improve patients’ quality of life. Additional studies are needed to confirm the therapeutic efficacy of vitamin D in inflammatory bowel diseases.
Cardiovascular disease
The prospective analysis of 41,504 electronic medical records in the Intermountain Heart Collaborative study found that only one-third of patients had adequate serum 25-hydroxyvitamin D concentrations (>30 ng/mL); vitamin D insufficiency (serum 25-hydroxyvitamin D concentrations ≤30 ng/mL) was associated with increased prevalence and incidence of many cardiovascular conditions, including hypertension, coronary artery disease, heart failure, and stroke (328). Suboptimal vitamin D status has also been linked to arterial stiffness and vascular endothelial dysfunction — strong determinants of incident hypertension and adverse cardiovascular outcomes (329).
Hypertension
Several intervention studies have evaluated the effect of vitamin D supplementation on blood pressure. An early controlled clinical trial in 18 men and women with untreated mild hypertension living in the Netherlands found that exposure to UVB radiation three times weekly for six weeks during the winter increased serum 25-hydroxyvitamin D concentrations by 162%, lowered PTH concentrations by 15%, and decreased 24-hour ambulatory systolic and diastolic blood pressure measurements by an average of 6 mm Hg (330). A recent meta-analysis of 16 randomized controlled trials involving 1,879 participants, either healthy or with pre-existing cardiometabolic conditions (including hypertension), found no significant reduction in systolic and diastolic blood pressures with vitamin D supplementation (800-8,571 IU/day for five weeks to one year). However, a subgroup analysis of six trials found a significant reduction of 1.31 mm Hg in diastolic blood pressure in individuals with preexisting conditions. While improvements in blood pressure may be expected in cases of vitamin D insufficiency/deficiency, the authors noted that suboptimal vitamin D status in participants was not exclusively observed in those with cardiometabolic conditions (331). In contrast, in two recent intervention studies — the Styrian vitamin D hypertension trial (332) and the Vitamin D therapy in individuals at high risk of hypertension trial [DAYLIGHT] (333) — subjects with (pre)hypertension supplemented with vitamin D3 (400 to 4,000 IU/day for two to six months) showed no evidence of blood pressure lowering, regardless of their baseline vitamin D status (insufficient or adequate, according to the IOM’s current cutoffs).
Conditions that decrease vitamin D synthesis in the skin, such as having dark-colored skin, living in temperate latitudes, and aging, are associated with increased prevalence of hypertension (334), suggesting that vitamin D may reduce blood pressure levels in selected groups of individuals. In the above-mentioned meta-analysis, one four-arm, double-blind, placebo-controlled clinical trial was conducted in 283 African Americans randomized to receive daily vitamin D3 supplements of 1,000 IU, 2,000 IU, or 4,000 IU for a period of three months. Systolic blood pressure was decreased by 0.66 mm Hg with 1,000 IU/day, 3.4 mm Hg with 2,000 IU/day, and by 4 mm Hg with 4,000 IU/day while it increased by 1.7 mm Hg in the placebo group when compared to baseline. A significant reduction of 0.2 mm Hg in systolic blood pressure was detected per 1 ng/mL incremental increase in 25-hydroxyvitamin D concentration. However, there was no statistical difference on three-month change in blood pressure between vitamin D3 and placebo (335). Another randomized, placebo-controlled study in 150 elderly participants (mean age, 77 years) showed that supplementation with 100,000 IU of vitamin D3 every three months for one year did not significantly lower blood pressure compared to placebo (336).
Further research is needed to determine whether vitamin D supplementation is helpful in the prevention or management of hypertension.
Congestive heart failure
Congestive heart failure (also called cardiac insufficiency) is characterized by increased heart rate and subsequent hypertrophy of the left heart ventricle. Cardiac insufficiency is associated with a reduced left ventricular ejection fraction (LVEF), assessed by echocardiography. Inhibitors of angiotensin-converting enzyme (ACE) (see Blood pressure regulation) are currently used as first-line therapy for patients with heart failure. In a cross-sectional study in healthy patients who underwent coronary angiography, serum 25-hydroxyvitamin D concentrations <30 ng/mL were associated with poorer coronary flow rates (337). Suboptimal vitamin D status has also been linked to poorer prognosis in patients with heart failure (338). Over the past several years, a number of intervention studies have examined the effect of vitamin D supplementation in those with cardiac insufficiency. In a 12-week randomized, double-blind, placebo-controlled study, daily supplementation with 1,200 IU of vitamin D3 in children with chronic congestive heart failure led to a significant increase in vitamin D status accompanied by an improved heart muscle performance (increased LVEF), as well as by lower levels of PTH and pro-inflammatory cytokines (339). In another randomized, double-blind, placebo-controlled trial in 64 elderly patients with heart failure, participants receiving 800 mg/day of calcium and 50,000 IU/week of vitamin D3 did not perform significantly better at physical performance tasks (used as proxy to assess aerobic capacity and skeletal muscle strength) compared to those supplemented with calcium only (340). A recent meta-analysis of seven small randomized placebo-controlled trials in 573 subjects with heart failure showed that vitamin D supplementation (from 1,000 IU/day to 50,000 IU/week) for six weeks to nine months could reduce serum concentrations of PTH, tumor necrosis factor-α (TNF-α), and C-reactive protein (CRP). Yet, there were no significant differences in LVEF, circulating interleukin-10 (IL-10) concentration, and renin concentration between patients treated with vitamin D and those given a placebo (341). Finally, in the EVITA (Effect of vitamin D on all-cause mortality) trial in patients with end-stage heart failure and inadequate vitamin D status (baseline serum 25-hydroxyvitamin D values, 8.6-19.7 ng/mL), supplementation with 4,000 IU/day for three years did not reduce the risk of mortality compared to placebo (342).
Sources
Sunlight
Solar ultraviolet-B radiation (UVB; wavelengths of 290 to 315 nanometers) stimulates the production of vitamin D3 in the epidermis of the skin (343). Sunlight exposure can provide most people with their entire vitamin D requirement. Children and young adults who spend a short time outside two or three times a week will generally synthesize all the vitamin D they need to prevent deficiency. One study reported that serum vitamin D concentrations following exposure to one minimal erythemal dose of simulated sunlight (the amount required to cause a slight pinkness of the skin) to the whole body was equivalent to ingesting approximately 10,000 to 25,000 IU of vitamin D (344). People with dark-colored skin synthesize markedly less vitamin D on exposure to sunlight than those with lighter complexion (34). Additionally, older adults have diminished capacity to synthesize vitamin D from sunlight exposure and frequently use sunscreen or protective clothing in order to prevent skin cancer and sun damage. The application of sunscreen with an SPF factor of 10 reduces production of vitamin D by 90% (30). In latitudes around 40 degrees north or 40 degrees south (Boston is 42 degrees north), there is insufficient UVB radiation available for vitamin D synthesis from November to early March. Ten degrees farther north or south (Edmonton, Canada), the "vitamin D winter" extends from mid-October to mid-March. It has been estimated that up to 15 minutes of daily sun exposure on the hands, arms, and face around 12 pm throughout the year at 25 degrees latitude (Miami, FL) and during the spring, summer, and fall at 42 degrees (Boston, MA) latitude may provide a light-skinned individual with 1,000 IU of vitamin D (345).
Food sources
Vitamin D is found naturally in only a few foods, such as some fatty fish (mackerel, salmon, sardines), fish liver oils, eggs from hens that have been fed vitamin D, and mushrooms exposed to sunlight or UV light. In the US, milk and infant formula are fortified with vitamin D so that they contain 400 IU (10 μg) per quart. However, other dairy products, such as cheese and yogurt, are not always fortified with vitamin D. Some cereal, bread, and fruit juices may also be fortified with vitamin D. Accurate estimates of average dietary intakes of vitamin D are difficult because of the high variability of the vitamin D content of fortified foods (346). The vitamin D content (sum of vitamin D2 and vitamin D3) of some vitamin D-rich foods is listed in Table 2 in both international units (IU) and micrograms (μg). For more information on the nutrient content of specific foods, search USDA's FoodData Central. The 25-hydroxyvitamin D metabolite is also present at low levels is certain foods, including meats, dairy products, and eggs (347, 348).
Supplements
Most vitamin D supplements available without a prescription contain cholecalciferol (vitamin D3). Multivitamin supplements generally provide 400 IU-1,000 IU (10 μg-25 μg) of vitamin D2 or vitamin D3. Single-ingredient vitamin D supplements may provide 400 to 50,000 IU of vitamin D3, but 400 IU is the most commonly available dose (66). A number of calcium supplements may also provide vitamin D. A meta-analysis of randomized controlled trials suggested that bolus doses of vitamin D2 (ergocalciferol) may not always be as effective as vitamin D3 in raising serum 25-hydroxyvitamin D concentrations, yet no difference in efficacy was found with daily supplementation with vitamin D2 or vitamin D3 (349). Nonetheless, a 25-week, randomized, double-blind, placebo-controlled trial found daily supplementation with 1,000 IU of vitamin D3 initiated at the end of summer to be more efficacious than vitamin D2 in maintaining summertime concentrations of 25-hydroxyvitamin D during fall and winter months (350).
There is growing interest in using the hydroxylated form of cholecalciferol, 25-hydroxyvitamin D3 (calcidiol; calcifediol), as a supplement. This vitamin D metabolite is synthesized by the liver of humans and animals from cholecalciferol produced in skin or consumed in the diet, and is present in some foods (e.g., meats, milk, eggs) at low levels (347, 348). In general, clinical studies have found calcifediol is two to five times more potent at increasing blood concentrations of 25-hydroxyvitamin D compared to supplementation with equivalent doses of cholecalciferol (351-359). Supplementation with calcifediol may thus represent a means to improve vitamin D status rapidly and consistently with lower doses than cholecalciferol and may benefit those individuals with conditions that decrease intestinal absorption of cholecalciferol (359). However, the 25-hydroxyvitamin D3 form is presently not available as an over-the-counter supplement in the United States.
Safety
Toxicity
Vitamin D toxicity (hypervitaminosis D) has not been observed to result from sun exposure. The reason is that excessive sunlight exposure generates a number of biologically inert photoproducts from 7-dehydrocholesterol and cholecalciferol (3). Vitamin D toxicity induces abnormally high serum calcium concentration (hypercalcemia), which could result in bone loss, kidney stones, and calcification of organs like the heart and kidneys if untreated over a long period of time. Hypercalcemia has been observed following daily doses of greater than 50,000 IU of vitamin D (360). Overall, research suggests that vitamin D toxicity is very unlikely in healthy people at intake levels lower than 10,000 IU/day (361-363). However, the Food and Nutrition Board of the IOM conservatively set the tolerable upper intake level (UL) at 4,000 IU/day (100 μg/day) for all adults (Table 3). Certain medical conditions can increase the risk of hypercalcemia in response to vitamin D, including primary hyperparathyroidism, sarcoidosis, tuberculosis, and lymphoma (361). People with these conditions may develop hypercalcemia in response to any increase in vitamin D nutrition and should consult a qualified health care provider regarding any increase in vitamin D intake.
Drug interactions
The following medications should not be taken at the same time as vitamin D because they can decrease the intestinal absorption of vitamin D: cholestyramine (Questran), colestipol (Colestid), orlistat (Xenical), and mineral oil (364, 365). The following medications increase the metabolism of vitamin D and may decrease serum 25-hydroxyvitamin D concentrations: phenytoin (Dilantin), fosphenytoin (Cerebyx), phenobarbital (Luminal), carbamazepine (Tegretol), and rifampin (Rimactane) (6). Cimetidine, a H2 blocker that suppresses stomach acid secretion, inhibits the hydroxylation of vitamin D in the liver (366). Treating acid reflux, gastroesophageal reflux disease (GERD), or ulcers with proton-pump inhibitors (omeprazole, lansoprazole) might interfere with calcium absorption and increase the risk of fracture such that patients are advised to take calcium and vitamin D supplements (367). The oral antifungal medication, ketoconazole, inhibits the 25-hydroxyvitamin D3-1α-hydroxylase enzyme and has been found to reduce serum 1α,25-dihydroxyvitamin D concentrations in healthy men (368). The Endocrine Society also recommends monitoring vitamin D status of patients on glucocorticoids and HIV treatment drugs because these medications increase the catabolism of 25-hydroxyvitamin D (40). The use of some cytostatic agents (cell growth inhibitors) may also increase the degradation of 25-hydroxyvitamin D and 1α,25-dihydroxyvitamin D in cancer patients under chemotherapy (6). The induction of hypercalcemia by toxic levels of vitamin D may precipitate cardiac arrhythmia in patients on digoxin (Lanoxin) (366). Hypercalcemia may also reduce the effectiveness of verapamil (Calan) and diltiazem (Cardizem) in atrial fibrillation (366).
Linus Pauling Institute Recommendation
The Linus Pauling Institute recommends that generally healthy adults take 2,000 IU (50 μg) of supplemental vitamin D daily. Most multivitamins contain 400 IU (10 μg) of vitamin D, and single-ingredient vitamin D supplements are available for additional supplementation. Sun exposure, diet, skin color, and body mass index (BMI) have variable, substantial impact on body vitamin D levels. To adjust for individual differences and ensure adequate body vitamin D status, the Linus Pauling Institute recommends aiming for a serum 25-hydroxyvitamin D concentration of at least 30 ng/mL (75 nmol/L). Observational studies suggest that serum 25-hydroxyvitamin D concentrations between 30 ng/mL and 60 ng/mL are associated with lower risks of adverse health outcomes, including cancers and autoimmune diseases.
The American Academy of Pediatrics currently suggests that all infants, children, and adolescents receive 400 IU of supplemental vitamin D daily (19). Consistent with the recommendations of the Endocrine Society (40), the Linus Pauling Institute recommends daily intakes of 400 to 1,000 IU (10 to 25 μg) of vitamin D in infants and 600 to 1,000 IU (15 to 25 μg) of vitamin D in children and adolescents. Given the average vitamin D content of breast milk, infant formula, and the diets of children and adolescents, supplementation may be necessary to meet these recommendations.
Older adults (>50 years)
Daily supplementation with 2,000 IU (50 μg) of vitamin D is especially important for older adults because aging is associated with a reduced capacity to synthesize vitamin D in the skin upon sun exposure.
Authors and Reviewers
Originally written in 2000 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in March 2003 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in March 2004 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in January 2008 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in July 2014 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
The 2014 update of this article was supported by a grant from Bayer Consumer Care AG, Basel, Switzerland.
Updated in July 2017 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in October 2017 by:
Adrian F. Gombart, Ph.D.
Principal Investigator, Linus Pauling Institute
Associate Professor, Department of Biochemistry and Biophysics
Oregon State University
The 2017 update of this article was supported by a grant from Pfizer Inc.
Last updated 2/11/21 Copyright 2000-2024 Linus Pauling Institute
References
1. Holick MF. Vitamin D: importance in the prevention of cancers, type 1 diabetes, heart disease, and osteoporosis. Am J Clin Nutr. 2004;79(3):362-371. (PubMed)
2. Bikle DD. Vitamin D metabolism, mechanism of action, and clinical applications. Chem Biol. 2014;21(3):319-329. (PubMed)
3. Volmer DA, Mendes LR, Stokes CS. Analysis of vitamin D metabolic markers by mass spectrometry: Current techniques, limitations of the "gold standard" method, and anticipated future directions. Mass Spectrom Rev. 2015;34(1):2-23. (PubMed)
4. Holick MF. Vitamin D: A millenium perspective. J Cell Biochem. 2003;88(2):296-307. (PubMed)
5. Sutton AL, MacDonald PN. Vitamin D: more than a "bone-a-fide" hormone. Mol Endocrinol. 2003;17(5):777-791. (PubMed)
6. Grober U, Spitz J, Reichrath J, Kisters K, Holick MF. Vitamin D: Update 2013: From rickets prophylaxis to general preventive healthcare. Dermatoendocrinol. 2013;5(3):331-347. (PubMed)
7. Lieben L, Carmeliet G. The delicate balance between vitamin D, calcium and bone homeostasis: lessons learned from intestinal- and osteocyte-specific VDR null mice. J Steroid Biochem Mol Biol. 2013;136:102-106. (PubMed)
8. Fukumoto S. Phosphate metabolism and vitamin D. Bonekey Rep. 2014;3:497. (PubMed)
9. Lin R, White JH. The pleiotropic actions of vitamin D. Bioessays. 2004;26(1):21-28. (PubMed)
10. Edfeldt K, Liu PT, Chun R, et al. T-cell cytokines differentially control human monocyte antimicrobial responses by regulating vitamin D metabolism. Proc Natl Acad Sci U S A. 2010;107(52):22593-22598. (PubMed)
11. Smolders J, Thewissen M, Damoiseaux J. Control of T cell activation by vitamin D. Nat Immunol. 2011;12(1):3; author reply 3-4. (PubMed)
12. Aranow C. Vitamin D and the immune system. J Investig Med. 2011;59(6):881-886. (PubMed)
13. Zeitz U, Weber K, Soegiarto DW, Wolf E, Balling R, Erben RG. Impaired insulin secretory capacity in mice lacking a functional vitamin D receptor. Faseb J. 2003;17(3):509-511. (PubMed)
14. Bourlon PM, Billaudel B, Faure-Dussert A. Influence of vitamin D3 deficiency and 1,25 dihydroxyvitamin D3 on de novo insulin biosynthesis in the islets of the rat endocrine pancreas. J Endocrinol. 1999;160(1):87-95. (PubMed)
15. Heer M, Egert S. Nutrients other than carbohydrates: their effects on glucose homeostasis in humans. Diabetes Metab Res Rev. 2015;31(1):14-35. (PubMed)
16. Sheng H-W. Sodium, chloride, and potassium. In: Stipanuk M, ed. Biochemical and Physiological Aspects of Human Nutrition. Philadelphia: W.B. Saunders Company; 2000:686-710.
17. Sigmund CD. Regulation of renin expression and blood pressure by vitamin D(3). J Clin Invest. 2002;110(2):155-156. (PubMed)
18. Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110(2):229-238. (PubMed)
19. Wagner CL, Greer FR, American Academy of Pediatrics Section on B, American Academy of Pediatrics Committee on N. Prevention of rickets and vitamin D deficiency in infants, children, and adolescents. Pediatrics. 2008;122(5):1142-1152. (PubMed)
20. Goldacre M, Hall N, Yeates DG. Hospitalisation for children with rickets in England: a historical perspective. Lancet. 2014;383(9917):597-598. (PubMed)
21. Jones AN, Hansen KE. Recognizing the musculoskeletal manifestations of vitamin D deficiency. J Musculoskelet Med. 2009;26(10):389-396. (PubMed)
22. Bringhurst FR, Demay MB, Kronenberg HM. Mineral Metabolism. In: Larson PR, Kronenberg HM, Melmed S, Polonsky KS, eds. Williams Textbook of Endocrinology. 10th ed. Philadelphia: Saunders Book Company; 2003:1317-1320.
23. Plotnikoff GA, Quigley JM. Prevalence of severe hypovitaminosis D in patients with persistent, nonspecific musculoskeletal pain. Mayo Clin Proc. 2003;78(12):1463-1470. (PubMed)
24. Deandrea S, Lucenteforte E, Bravi F, Foschi R, La Vecchia C, Negri E. Risk factors for falls in community-dwelling older people: a systematic review and meta-analysis. Epidemiology. 2010;21(5):658-668. (PubMed)
25. Al-Khalidi B, Kimball SM, Rotondi MA, Ardern CI. Standardized serum 25-hydroxyvitamin D concentrations are inversely associated with cardiometabolic disease in US adults: a cross-sectional analysis of NHANES, 2001-2010. Nutr J. 2017;16(1):16. (PubMed)
26. Cooper JD, Smyth DJ, Walker NM, et al. Inherited variation in vitamin D genes is associated with predisposition to autoimmune disease type 1 diabetes. Diabetes. 2011;60(5):1624-1631. (PubMed)
27. Webb AR, Kline L, Holick MF. Influence of season and latitude on the cutaneous synthesis of vitamin D3: exposure to winter sunlight in Boston and Edmonton will not promote vitamin D3 synthesis in human skin. J Clin Endocrinol Metab. 1988;67(2):373-378. (PubMed)
28. Nichols EK, Khatib IM, Aburto NJ, et al. Vitamin D status and determinants of deficiency among non-pregnant Jordanian women of reproductive age. Eur J Clin Nutr. 2012;66(6):751-756. (PubMed)
29. Bassil D, Rahme M, Hoteit M, Fuleihan Gel H. Hypovitaminosis D in the Middle East and North Africa: prevalence, risk factors and impact on outcomes. Dermatoendocrinol. 2013;5(2):274-298. (PubMed)
30. Balk SJ, Council on Environmental H, Section on D. Ultraviolet radiation: a hazard to children and adolescents. Pediatrics. 2011;127(3):e791-817. (PubMed)
31. Dawodu A, Tsang RC. Maternal vitamin D status: effect on milk vitamin D content and vitamin D status of breastfeeding infants. Adv Nutr. 2012;3(3):353-361. (PubMed)
32. Thiele DK, Senti JL, Anderson CM. Maternal vitamin D supplementation to meet the needs of the breastfed infant: a systematic review. J Hum Lact. 2013;29(2):163-170. (PubMed)
33. Wharton B, Bishop N. Rickets. Lancet. 2003;362(9393):1389-1400. (PubMed)
34. Chen TC, Chimeh F, Lu Z, et al. Factors that influence the cutaneous synthesis and dietary sources of vitamin D. Arch Biochem Biophys. 2007;460(2):213-217. (PubMed)
35. Wang TJ, Zhang F, Richards JB, et al. Common genetic determinants of vitamin D insufficiency: a genome-wide association study. Lancet. 2010;376(9736):180-188. (PubMed)
36. Ahn J, Yu K, Stolzenberg-Solomon R, et al. Genome-wide association study of circulating vitamin D levels. Hum Mol Genet. 2010;19(13):2739-2745. (PubMed)
37. Wang W, Ingles SA, Torres-Mejia G, et al. Genetic variants and non-genetic factors predict circulating vitamin D levels in Hispanic and non-Hispanic White women: the Breast Cancer Health Disparities Study. Int J Mol Epidemiol Genet. 2014;5(1):31-46. (PubMed)
38. Elkum N, Alkayal F, Noronha F, et al. Vitamin D insufficiency in Arabs and South Asians positively associates with polymorphisms in GC and CYP2R1 genes. PLoS One. 2014;9(11):e113102. (PubMed)
39. Zhang Y, Yang S, Liu Y, Ren L. Relationship between polymorphisms in vitamin D metabolism-related genes and the risk of rickets in Han Chinese children. BMC Med Genet. 2013;14:101. (PubMed)
40. Holick MF, Binkley NC, Bischoff-Ferrari HA, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96(7):1911-1930. (PubMed)
41. Harris SS, Soteriades E, Coolidge JA, Mudgal S, Dawson-Hughes B. Vitamin D insufficiency and hyperparathyroidism in a low income, multiracial, elderly population. J Clin Endocrinol Metab. 2000;85(11):4125-4130. (PubMed)
42. Allain TJ, Dhesi J. Hypovitaminosis D in older adults. Gerontology. 2003;49(5):273-278. (PubMed)
43. Doorenbos CR, van den Born J, Navis G, de Borst MH. Possible renoprotection by vitamin D in chronic renal disease: beyond mineral metabolism. Nat Rev Nephrol. 2009;5(12):691-700. (PubMed)
44. Pappa HM, Bern E, Kamin D, Grand RJ. Vitamin D status in gastrointestinal and liver disease. Curr Opin Gastroenterol. 2008;24(2):176-183. (PubMed)
45. Jahnsen J, Falch JA, Mowinckel P, Aadland E. Vitamin D status, parathyroid hormone and bone mineral density in patients with inflammatory bowel disease. Scand J Gastroenterol. 2002;37(2):192-199. (PubMed)
46. Arunabh S, Pollack S, Yeh J, Aloia JF. Body fat content and 25-hydroxyvitamin D levels in healthy women. J Clin Endocrinol Metab. 2003;88(1):157-161. (PubMed)
47. Gallagher JC, Yalamanchili V, Smith LM. The effect of vitamin D supplementation on serum 25(OH)D in thin and obese women. J Steroid Biochem Mol Biol. 2013;136:195-200. (PubMed)
48. Deng X, Song Y, Manson JE, et al. Magnesium, vitamin D status and mortality: results from US National Health and Nutrition Examination Survey (NHANES) 2001 to 2006 and NHANES III. BMC Med. 2013;11:187. (PubMed)
49. Sempos CT, Durazo-Arvizu RA, Binkley N, Jones J, Merkel JM, Carter GD. Developing vitamin D dietary guidelines and the lack of 25-hydroxyvitamin D assay standardization: The ever-present past. J Steroid Biochem Mol Biol. 2016;164:115-119. (PubMed)
50. Chapuy MC, Preziosi P, Maamer M, et al. Prevalence of vitamin D insufficiency in an adult normal population. Osteoporos Int. 1997;7(5):439-443. (PubMed)
51. Thomas MK, Lloyd-Jones DM, Thadhani RI, et al. Hypovitaminosis D in medical inpatients. N Engl J Med. 1998;338(12):777-783. (PubMed)
52. Heaney RP, Dowell MS, Hale CA, Bendich A. Calcium absorption varies within the reference range for serum 25-hydroxyvitamin D. J Am Coll Nutr. 2003;22(2):142-146. (PubMed)
53. Valcour A, Blocki F, Hawkins DM, Rao SD. Effects of age and serum 25-OH-vitamin D on serum parathyroid hormone levels. J Clin Endocrinol Metab. 2012;97(11):3989-3995. (PubMed)
54. Ginde AA, Wolfe P, Camargo CA, Jr., Schwartz RS. Defining vitamin D status by secondary hyperparathyroidism in the US population. J Endocrinol Invest. 2012;35(1):42-48. (PubMed)
55. Gallagher JC, Yalamanchili V, Smith LM. The effect of vitamin D on calcium absorption in older women. J Clin Endocrinol Metab. 2012;97(10):3550-3556. (PubMed)
56. Looker AC, Johnson CL, Lacher DA, Pfeiffer CM, Schleicher RL, Sempos CT. Vitamin D status: United States, 2001-2006. NCHS Data Brief. 2011(59):1-8. (PubMed)
57. Food and Nutrition Board, Institute of Medicine. Dietary Reference Intakes for Calcium and Vitamin D. Washington, D.C.: The National Academies Press; 2011. (The National Academies Press)
58. Mithal A, Wahl DA, Bonjour JP, et al. Global vitamin D status and determinants of hypovitaminosis D. Osteoporos Int. 2009;20(11):1807-1820. (PubMed)
59. Powe CE, Evans MK, Wenger J, et al. Vitamin D-binding protein and vitamin D status of black Americans and white Americans. N Engl J Med. 2013;369(21):1991-2000. (PubMed)
60. Durazo-Arvizu RA, Dawson-Hughes B, Kramer H, et al. The reverse J-shaped association between serum total 25-hydroxyvitamin D concentration and all-cause mortality: the impact of assay standardization. Am J Epidemiol. 2017;185(8):720-726. (PubMed)
61. Gaksch M, Jorde R, Grimnes G, et al. Vitamin D and mortality: Individual participant data meta-analysis of standardized 25-hydroxyvitamin D in 26916 individuals from a European consortium. PLoS One. 2017;12(2):e0170791. (PubMed)
62. Chowdhury R, Kunutsor S, Vitezova A, et al. Vitamin D and risk of cause specific death: systematic review and meta-analysis of observational cohort and randomised intervention studies. BMJ. 2014;348:g1903. (PubMed)
63. Gupta V, Walia GK, Sachdeva MP. 'Mendelian randomization': an approach for exploring causal relations in epidemiology. Public Health. 2017;145:113-119. (PubMed)
64. Afzal S, Brondum-Jacobsen P, Bojesen SE, Nordestgaard BG. Genetically low vitamin D concentrations and increased mortality: Mendelian randomisation analysis in three large cohorts. BMJ. 2014;349:g6330. (PubMed)
65. Bjelakovic G, Gluud LL, Nikolova D, et al. Vitamin D supplementation for prevention of mortality in adults. Cochrane Database Syst Rev. 2014(1):Cd007470. (PubMed)
66. Wacker M, Holick MF. Vitamin D - effects on skeletal and extraskeletal health and the need for supplementation. Nutrients. 2013;5(1):111-148. (PubMed)
67. Lips P, Hosking D, Lippuner K, et al. The prevalence of vitamin D inadequacy amongst women with osteoporosis: an international epidemiological investigation. J Intern Med. 2006;260(3):245-254. (PubMed)
68. Torbergsen AC, Watne LO, Wyller TB, et al. Vitamin K1 and 25(OH)D are independently and synergistically associated with a risk for hip fracture in an elderly population: A case control study. Clin Nutr. 2015;34(1):101-106. (PubMed)
69. Lips P, van Schoor NM. The effect of vitamin D on bone and osteoporosis. Best Pract Res Clin Endocrinol Metab. 2011;25(4):585-591. (PubMed)
70. Reid IR, Bolland MJ, Grey A. Effects of vitamin D supplements on bone mineral density: a systematic review and meta-analysis. Lancet. 2014;383(9912):146-155. (PubMed)
71. Rosen CJ. Vitamin D supplementation: bones of contention. Lancet. 2014;383(9912):108-110. (PubMed)
72. Mocanu V, Vieth R. Three-year follow-up of serum 25-hydroxyvitamin D, parathyroid hormone, and bone mineral density in nursing home residents who had received 12 months of daily bread fortification with 125 mug of vitamin D3. Nutr J. 2013;12:137. (PubMed)
73. Feskanich D, Willett WC, Colditz GA. Calcium, vitamin D, milk consumption, and hip fractures: a prospective study among postmenopausal women. Am J Clin Nutr. 2003;77(2):504-511. (PubMed)
74. Jackson RD, LaCroix AZ, Gass M, et al. Calcium plus vitamin D supplementation and the risk of fractures. N Engl J Med. 2006;354(7):669-683. (PubMed)
75. Wang Y, Wactawski-Wende J, Sucheston-Campbell LE, et al. The influence of genetic susceptibility and calcium plus vitamin D supplementation on fracture risk. Am J Clin Nutr. 2017;105(4):970-979. (PubMed)
76. Gurney EP, Nachtigall MJ, Nachtigall LE, Naftolin F. The Women's Health Initiative trial and related studies: 10 years later: a clinician's view. J Steroid Biochem Mol Biol. 2014;142:4-11. (PubMed)
77. Grant AM, Avenell A, Campbell MK, et al. Oral vitamin D3 and calcium for secondary prevention of low-trauma fractures in elderly people (Randomised Evaluation of Calcium Or vitamin D, RECORD): a randomised placebo-controlled trial. Lancet. 2005;365(9471):1621-1628. (PubMed)
78. Bischoff-Ferrari HA, Giovannucci E, Willett WC, Dietrich T, Dawson-Hughes B. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84(1):18-28. (PubMed)
79. Khaw KT, Stewart AW, Waayer D, et al. Effect of monthly high-dose vitamin D supplementation on falls and non-vertebral fractures: secondary and post-hoc outcomes from the randomised, double-blind, placebo-controlled ViDA trial. Lancet Diabetes Endocrinol. 2017;5(6):438-447. (PubMed)
80. Chung M, Lee J, Terasawa T, Lau J, Trikalinos TA. Vitamin D with or without calcium supplementation for prevention of cancer and fractures: an updated meta-analysis for the US Preventive Services Task Force. Ann Intern Med. 2011;155(12):827-838. (PubMed)
81. Bischoff-Ferrari HA, Willett WC, Orav EJ, et al. A pooled analysis of vitamin D dose requirements for fracture prevention. N Engl J Med. 2012;367(1):40-49. (PubMed)
82. Avenell A, Mak JC, O'Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev. 2014;4:CD000227. (PubMed)
83. Annweiler C, Beauchet O. Questioning vitamin D status of elderly fallers and nonfallers: a meta-analysis to address a 'forgotten step'. J Intern Med. 2015;277(1):16-44. (PubMed)
84. Cangussu LM, Nahas-Neto J, Orsatti CL, Bueloni-Dias FN, Nahas EA. Effect of vitamin D supplementation alone on muscle function in postmenopausal women: a randomized, double-blind, placebo-controlled clinical trial. Osteoporos Int. 2015;26(10):2413-2421. (PubMed)
85. Cangussu LM, Nahas-Neto J, Orsatti CL, et al. Effect of isolated vitamin D supplementation on the rate of falls and postural balance in postmenopausal women fallers: a randomized, double-blind, placebo-controlled trial. Menopause. 2016;23(3):267-274. (PubMed)
86. Bischoff-Ferrari HA, Dawson-Hughes B, Orav EJ, et al. Monthly high-dose vitamin D treatment for the prevention of functional decline: a randomized clinical trial. JAMA Intern Med. 2016;176(2):175-183. (PubMed)
87. Murad MH, Elamin KB, Abu Elnour NO, et al. Clinical review: The effect of vitamin D on falls: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2011;96(10):2997-3006. (PubMed)
88. Boonen S, Lips P, Bouillon R, Bischoff-Ferrari HA, Vanderschueren D, Haentjens P. Need for additional calcium to reduce the risk of hip fracture with vitamin d supplementation: evidence from a comparative metaanalysis of randomized controlled trials. J Clin Endocrinol Metab. 2007;92(4):1415-1423. (PubMed)
89. Grant WB. Update on evidence that support a role of solar ultraviolet-B irradiance in reducing cancer risk. Anticancer Agents Med Chem. 2013;13(1):140-146. (PubMed)
90. Yin L, Ordonez-Mena JM, Chen T, Schottker B, Arndt V, Brenner H. Circulating 25-hydroxyvitamin D serum concentration and total cancer incidence and mortality: a systematic review and meta-analysis. Prev Med. 2013;57(6):753-764. (PubMed)
91. Gandini S, Gnagnarella P, Serrano D, Pasquali E, Raimondi S. Vitamin D receptor polymorphisms and cancer. Adv Exp Med Biol. 2014;810:69-105. (PubMed)
92. Vaughan-Shaw PG, O'Sullivan F, Farrington SM, et al. The impact of vitamin D pathway genetic variation and circulating 25-hydroxyvitamin D on cancer outcome: systematic review and meta-analysis. Br J Cancer. 2017;116(8):1092-1110. (PubMed)
93. Gombart AF, Luong QT, Koeffler HP. Vitamin D compounds: activity against microbes and cancer. Anticancer Res. 2006;26(4A):2531-2542. (PubMed)
94. Thorne J, Campbell MJ. The vitamin D receptor in cancer. Proc Nutr Soc. 2008;67(2):115-127. (PubMed)
95. Garland CF, Garland FC, Gorham ED. Calcium and vitamin D. Their potential roles in colon and breast cancer prevention. Ann N Y Acad Sci. 1999;889:107-119. (PubMed)
96. Choi YJ, Kim YH, Cho CH, Kim SH, Lee JE. Circulating levels of vitamin D and colorectal adenoma: A case-control study and a meta-analysis. World J Gastroenterol. 2015;21(29):8868-8877. (PubMed)
97. Gandini S, Boniol M, Haukka J, et al. Meta-analysis of observational studies of serum 25-hydroxyvitamin D levels and colorectal, breast and prostate cancer and colorectal adenoma. Int J Cancer. 2011;128(6):1414-1424. (PubMed)
98. Ma Y, Zhang P, Wang F, Yang J, Liu Z, Qin H. Association between vitamin D and risk of colorectal cancer: a systematic review of prospective studies. J Clin Oncol. 2011;29(28):3775-3782. (PubMed)
99. Touvier M, Chan DS, Lau R, et al. Meta-analyses of vitamin D intake, 25-hydroxyvitamin D status, vitamin D receptor polymorphisms, and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev. 2011;20(5):1003-1016. (PubMed)
100. Ekmekcioglu C, Haluza D, Kundi M. 25-Hydroxyvitamin D status and risk for colorectal cancer and type 2 diabetes mellitus: a systematic review and meta-analysis of epidemiological studies. Int J Environ Res Public Health. 2017;14(2). (PubMed)
101. Gorham ED, Garland CF, Garland FC, et al. Optimal vitamin D status for colorectal cancer prevention: a quantitative meta analysis. Am J Prev Med. 2007;32(3):210-216. (PubMed)
102. Cauley JA, Chlebowski RT, Wactawski-Wende J, et al. Calcium plus vitamin D supplementation and health outcomes five years after active intervention ended: the Women's Health Initiative. J Womens Health (Larchmt). 2013;22(11):915-929. (PubMed)
103. Baron JA, Barry EL, Mott LA, et al. A trial of calcium and vitamin D for the prevention of colorectal adenomas. N Engl J Med. 2015;373(16):1519-1530. (PubMed)
104. Holick MF. Calcium plus vitamin D and the risk of colorectal cancer. N Engl J Med. 2006;354(21):2287-2288; author reply 2287-2288. (PubMed)
105. Barry EL, Peacock JL, Rees JR, et al. Vitamin D receptor genotype, vitamin D3 supplementation, and risk of colorectal adenomas: a randomized clinical trial. JAMA Oncol. 2017;3(5):628-635. (PubMed)
106. Hiraki LT, Joshi AD, Ng K, et al. Joint effects of colorectal cancer susceptibility loci, circulating 25-hydroxyvitamin D and risk of colorectal cancer. PLoS One. 2014;9(3):e92212. (PubMed)
107. Vidigal VM, Silva TD, de Oliveira J, Pimenta CAM, Felipe AV, Forones NM. Genetic polymorphisms of vitamin D receptor (VDR), CYP27B1 and CYP24A1 genes and the risk of colorectal cancer. Int J Biol Markers. 2017;32(2):e224-e230. (PubMed)
108. Maalmi H, Ordonez-Mena JM, Schottker B, Brenner H. Serum 25-hydroxyvitamin D levels and survival in colorectal and breast cancer patients: Systematic review and meta-analysis of prospective cohort studies. 2014;50(8):1510-1521. (PubMed)
109. Mohr SB, Garland CF, Gorham ED, Grant WB, Garland FC. Relationship between low ultraviolet B irradiance and higher breast cancer risk in 107 countries. Breast J. 2008;14(3):255-260. (PubMed)
110. John EM, Schwartz GG, Dreon DM, Koo J. Vitamin D and breast cancer risk: the NHANES I Epidemiologic follow-up study, 1971-1975 to 1992. National Health and Nutrition Examination Survey. Cancer Epidemiol Biomarkers Prev. 1999;8(5):399-406. (PubMed)
111. Kim Y, Je Y. Vitamin D intake, blood 25(OH)D levels, and breast cancer risk or mortality: a meta-analysis. Br J Cancer. 2014;110(11):2772-2784. (PubMed)
112. Wang D, Velez de-la-Paz OI, Zhai JX, Liu DW. Serum 25-hydroxyvitamin D and breast cancer risk: a meta-analysis of prospective studies. Tumour Biol. 2013;34(6):3509-3517. (PubMed)
113. Rose AA, Elser C, Ennis M, Goodwin PJ. Blood levels of vitamin D and early stage breast cancer prognosis: a systematic review and meta-analysis. Breast Cancer Res Treat. 2013;141(3):331-339. (PubMed)
114. Sperati F, Vici P, Maugeri-Sacca M, et al. Vitamin D supplementation and breast cancer prevention: a systematic review and meta-analysis of randomized clinical trials. PLoS One. 2013;8(7):e69269. (PubMed)
115. Hu K, Callen DF, Li J, Zheng H. Circulating vitamin D and overall survival in breast cancer patients: a dose-response meta-analysis of cohort studies. Integr Cancer Ther. 2017:1534735417712007. (PubMed)
116. Mohr SB, Gorham ED, Kim J, Hofflich H, Garland CF. Meta-analysis of vitamin D sufficiency for improving survival of patients with breast cancer. Anticancer Res. 2014;34(3):1163-1166. (PubMed)
117. Lu D, Jing L, Zhang S. Vitamin D receptor polymorphism and breast cancer risk: a meta-analysis. Medicine (Baltimore). 2016;95(18):e3535. (PubMed)
118. Mun MJ, Kim TH, Hwang JY, Jang WC. Vitamin D receptor gene polymorphisms and the risk for female reproductive cancers: A meta-analysis. Maturitas. 2015;81(2):256-265. (PubMed)
119. Gilbert R, Martin RM, Beynon R, et al. Associations of circulating and dietary vitamin D with prostate cancer risk: a systematic review and dose-response meta-analysis. Cancer Causes Control. 2011;22(3):319-340. (PubMed)
120. van der Rhee H, Coebergh JW, de Vries E. Is prevention of cancer by sun exposure more than just the effect of vitamin D? A systematic review of epidemiological studies. Eur J Cancer. 2013;49(6):1422-1436. (PubMed)
121. Tuohimaa P, Tenkanen L, Ahonen M, et al. Both high and low levels of blood vitamin D are associated with a higher prostate cancer risk: a longitudinal, nested case-control study in the Nordic countries. Int J Cancer. 2004;108(1):104-108. (PubMed)
122. Xu Y, Shao X, Yao Y, et al. Positive association between circulating 25-hydroxyvitamin D levels and prostate cancer risk: new findings from an updated meta-analysis. J Cancer Res Clin Oncol. 2014;140(9):1465-1477. (PubMed)
123. Grant WB, Karras SN, Bischoff-Ferrari HA, et al. Do studies reporting 'U'-shaped serum 25-hydroxyvitamin D-health outcome relationships reflect adverse effects? Dermatoendocrinol. 2016;8(1):e1187349. (PubMed)
124. Wu X, Cheng J, Yang K. Vitamin D-related gene polymorphisms, plasma 25-hydroxy-vitamin D, cigarette smoke and non-small cell lung cancer (NSCLC) Risk. Int J Mol Sci. 2016;17(10). (PubMed)
125. Zhang L, Wang S, Che X, Li X. Vitamin D and lung cancer risk: a comprehensive review and meta-analysis. Cell Physiol Biochem. 2015;36(1):299-305. (PubMed)
126. Liao Y, Huang JL, Qiu MX, Ma ZW. Impact of serum vitamin D level on risk of bladder cancer: a systemic review and meta-analysis. Tumour Biol. 2015;36(3):1567-1572. (PubMed)
127. Zhang H, Zhang H, Wen X, Zhang Y, Wei X, Liu T. Vitamin D deficiency and increased risk of bladder carcinoma: a meta-analysis. Cell Physiol Biochem. 2015;37(5):1686-1692. (PubMed)
128. Lu D, Chen J, Jin J. Vitamin D status and risk of non-Hodgkin lymphoma: a meta-analysis. Cancer Causes Control. 2014;25(11):1553-1563. (PubMed)
129. Prescott J, Bertrand KA, Poole EM, Rosner BA, Tworoger SS. Surrogates of long-term vitamin d exposure and ovarian cancer risk in two prospective cohort studies. Cancers (Basel). 2013;5(4):1577-1600. (PubMed)
130. Khayatzadeh S, Feizi A, Saneei P, Esmaillzadeh A. Vitamin D intake, serum Vitamin D levels, and risk of gastric cancer: A systematic review and meta-analysis. J Res Med Sci. 2015;20(8):790-796. (PubMed)
131. Gandini S, Raimondi S, Gnagnarella P, Dore JF, Maisonneuve P, Testori A. Vitamin D and skin cancer: a meta-analysis. Eur J Cancer. 2009;45(4):634-641. (PubMed)
132. Deluca HF, Cantorna MT. Vitamin D: its role and uses in immunology. Faseb J. 2001;15(14):2579-2585. (PubMed)
133. Agmon-Levin N, Mosca M, Petri M, Shoenfeld Y. Systemic lupus erythematosus one disease or many? Autoimmun Rev. 2012;11(8):593-595. (PubMed)
134. Goodin DS. The epidemiology of multiple sclerosis: insights to disease pathogenesis. Handb Clin Neurol. 2014;122:231-266. (PubMed)
135. Littorin B, Blom P, Scholin A, et al. Lower levels of plasma 25-hydroxyvitamin D among young adults at diagnosis of autoimmune type 1 diabetes compared with control subjects: results from the nationwide Diabetes Incidence Study in Sweden (DISS). Diabetologia. 2006;49(12):2847-2852. (PubMed)
136. Pozzilli P, Manfrini S, Crino A, et al. Low levels of 25-hydroxyvitamin D3 and 1,25-dihydroxyvitamin D3 in patients with newly diagnosed type 1 diabetes. Horm Metab Res. 2005;37(11):680-683. (PubMed)
137. Raab J, Giannopoulou EZ, Schneider S, et al. Prevalence of vitamin D deficiency in pre-type 1 diabetes and its association with disease progression. Diabetologia. 2014;57(5):902-908. (PubMed)
138. Hypponen E, Laara E, Reunanen A, Jarvelin MR, Virtanen SM. Intake of vitamin D and risk of type 1 diabetes: a birth-cohort study. Lancet. 2001;358(9292):1500-1503. (PubMed)
139. Sorensen IM, Joner G, Jenum PA, Eskild A, Torjesen PA, Stene LC. Maternal serum levels of 25-hydroxy-vitamin D during pregnancy and risk of type 1 diabetes in the offspring. Diabetes. 2012;61(1):175-178. (PubMed)
140. Brekke HK, Ludvigsson J. Vitamin D supplementation and diabetes-related autoimmunity in the ABIS study. Pediatr Diabetes. 2007;8(1):11-14. (PubMed)
141. Fronczak CM, Baron AE, Chase HP, et al. In utero dietary exposures and risk of islet autoimmunity in children. Diabetes Care. 2003;26(12):3237-3242. (PubMed)
142. Marjamaki L, Niinisto S, Kenward MG, et al. Maternal intake of vitamin D during pregnancy and risk of advanced beta cell autoimmunity and type 1 diabetes in offspring. Diabetologia. 2010;53(8):1599-1607. (PubMed)
143. Miettinen ME, Reinert L, Kinnunen L, et al. Serum 25-hydroxyvitamin D level during early pregnancy and type 1 diabetes risk in the offspring. Diabetologia. 2012;55(5):1291-1294. (PubMed)
144. Smolders J, Thewissen M, Peelen E, et al. Vitamin D status is positively correlated with regulatory T cell function in patients with multiple sclerosis. PLoS One. 2009;4(8):e6635. (PubMed)
145. Mokry LE, Ross S, Ahmad OS, et al. Vitamin D and risk of multiple sclerosis: a Mendelian randomization study. PLoS Med. 2015;12(8):e1001866. (PubMed)
146. Staples J, Ponsonby AL, Lim L. Low maternal exposure to ultraviolet radiation in pregnancy, month of birth, and risk of multiple sclerosis in offspring: longitudinal analysis. BMJ. 2010;340:c1640. (PubMed)
147. Bjornevik K, Riise T, Casetta I, et al. Sun exposure and multiple sclerosis risk in Norway and Italy: The EnvIMS study. Mult Scler. 2014; 20(8):1042-1049. (PubMed)
148. McDowell TY, Amr S, Culpepper WJ, et al. Sun exposure, vitamin D and age at disease onset in relapsing multiple sclerosis. Neuroepidemiology. 2011;36(1):39-45. (PubMed)
149. Munger KL, Levin LI, Hollis BW, Howard NS, Ascherio A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA. 2006;296(23):2832-2838. (PubMed)
150. Munger KL, Zhang SM, O'Reilly E, et al. Vitamin D intake and incidence of multiple sclerosis. Neurology. 2004;62(1):60-65. (PubMed)
151. Pierrot-Deseilligny C, Rivaud-Pechoux S, Clerson P, de Paz R, Souberbielle JC. Relationship between 25-OH-D serum level and relapse rate in multiple sclerosis patients before and after vitamin D supplementation. Ther Adv Neurol Disord. 2012;5(4):187-198. (PubMed)
152. Ascherio A, Munger KL, White R, et al. Vitamin d as an early predictor of multiple sclerosis activity and progression. JAMA Neurol. 2014;71(3):306-314. (PubMed)
153. Muris AH, Smolders J, Rolf L, et al. Vitamin D status does not affect disability progression of patients with multiple sclerosis over three year follow-up. PLoS One. 2016;11(6):e0156122. (PubMed)
154. Kampman MT, Steffensen LH, Mellgren SI, Jorgensen L. Effect of vitamin D3 supplementation on relapses, disease progression, and measures of function in persons with multiple sclerosis: exploratory outcomes from a double-blind randomised controlled trial. Mult Scler. 2012;18(8):1144-1151. (PubMed)
155. Soilu-Hanninen M, Aivo J, Lindstrom BM, et al. A randomised, double blind, placebo controlled trial with vitamin D3 as an add on treatment to interferon beta-1b in patients with multiple sclerosis. J Neurol Neurosurg Psychiatry. 2012;83(5):565-571. (PubMed)
156. Mrad MF, El Ayoubi NK, Esmerian MO, Kazan JM, Khoury SJ. Effect of vitamin D replacement on immunological biomarkers in patients with multiple sclerosis. Clin Immunol. 2017;181:9-15. (PubMed)
157. Muris AH, Smolders J, Rolf L, Thewissen M, Hupperts R, Damoiseaux J. Immune regulatory effects of high dose vitamin D3 supplementation in a randomized controlled trial in relapsing remitting multiple sclerosis patients receiving IFNbeta; the SOLARIUM study. J Neuroimmunol. 2016;300:47-56. (PubMed)
158. O'Connell K, Sulaimani J, Basdeo SA, et al. Effects of vitamin D3 in clinically isolated syndrome and healthy control participants: A double-blind randomised controlled trial. Mult Scler J Exp Transl Clin. 2017;3(3):2055217317727296. (PubMed)
159. Rosjo E, Steffensen LH, Jorgensen L, et al. Vitamin D supplementation and systemic inflammation in relapsing-remitting multiple sclerosis. J Neurol. 2015;262(12):2713-2721. (PubMed)
160. Sotirchos ES, Bhargava P, Eckstein C, et al. Safety and immunologic effects of high- vs low-dose cholecalciferol in multiple sclerosis. Neurology. 2016;86(4):382-390. (PubMed)
161. Bruce D, Whitcomb JP, August A, McDowell MA, Cantorna MT. Elevated non-specific immunity and normal Listeria clearance in young and old vitamin D receptor knockout mice. Int Immunol. 2009;21(2):113-122. (PubMed)
162. Zwerina K, Baum W, Axmann R, et al. Vitamin D receptor regulates TNF-mediated arthritis. Ann Rheum Dis. 2011;70(6):1122-1129. (PubMed)
163. Hitchon CA, Sun Y, Robinson DB, et al. Vitamin D receptor polymorphism rs2228570 (Fok1) is associated with rheumatoid arthritis in North American natives. J Rheumatol. 2012;39(9):1792-1797. (PubMed)
164. Lee YH, Bae SC, Choi SJ, Ji JD, Song GG. Associations between vitamin D receptor polymorphisms and susceptibility to rheumatoid arthritis and systemic lupus erythematosus: a meta-analysis. Mol Biol Rep. 2011;38(6):3643-3651. (PubMed)
165. Mosaad YM, Hammad EM, Fawzy Z, et al. Vitamin D receptor gene polymorphism as possible risk factor in rheumatoid arthritis and rheumatoid related osteoporosis. Hum Immunol. 2014;75(5):452-461. (PubMed)
166. Zanetti M, Harris SS, Dawson-Hughes B. Ability of vitamin D to reduce inflammation in adults without acute illness. Nutr Rev. 2014;72(2):95-98. (PubMed)
167. Merlino LA, Curtis J, Mikuls TR, Cerhan JR, Criswell LA, Saag KG. Vitamin D intake is inversely associated with rheumatoid arthritis: results from the Iowa Women's Health Study. Arthritis Rheum. 2004;50(1):72-77. (PubMed)
168. Costenbader KH, Feskanich D, Holmes M, Karlson EW, Benito-Garcia E. Vitamin D intake and risks of systemic lupus erythematosus and rheumatoid arthritis in women. Ann Rheum Dis. 2008;67(4):530-535. (PubMed)
169. Hiraki LT, Munger KL, Costenbader KH, Karlson EW. Dietary intake of vitamin D during adolescence and risk of adult-onset systemic lupus erythematosus and rheumatoid arthritis. Arthritis Care Res (Hoboken). 2012;64(12):1829-1836. (PubMed)
170. Sen D, Ranganathan P. Vitamin D in rheumatoid arthritis: panacea or placebo? Discov Med. 2012;14(78):311-319. (PubMed)
171. Lee YH, Bae SC. Vitamin D level in rheumatoid arthritis and its correlation with the disease activity: a meta-analysis. Clin Exp Rheumatol. 2016;34(5):827-833. (PubMed)
172. Lin J, Liu J, Davies ML, Chen W. Serum vitamin D level and rheumatoid arthritis disease activity: review and meta-analysis. PLoS One. 2016;11(1):e0146351. (PubMed)
173. Hansen KE, Bartels CM, Gangnon RE, Jones AN, Gogineni J. An evaluation of high-dose vitamin D for rheumatoid arthritis. J Clin Rheumatol. 2014;20(2):112-114. (PubMed)
174. Buondonno I, Rovera G, Sassi F, et al. Vitamin D and immunomodulation in early rheumatoid arthritis: A randomized double-blind placebo-controlled study. PLoS One. 2017;12(6):e0178463. (PubMed)
175. Dehghan A, Rahimpour S, Soleymani-Salehabadi H, Owlia MB. Role of vitamin D in flare ups of rheumatoid arthritis. Z Rheumatol. 2014;73(5):461-464. (PubMed)
176. Yang J, Liu L, Zhang Q, Li M, Wang J. Effect of vitamin D on the recurrence rate of rheumatoid arthritis. Exp Ther Med. 2015;10(5):1812-1816. (PubMed)
177. Gonzalez LA, Toloza SM, McGwin G, Jr., Alarcon GS. Ethnicity in systemic lupus erythematosus (SLE): its influence on susceptibility and outcomes. Lupus. 2013;22(12):1214-1224. (PubMed)
178. Hsieh CC, Lin BF. Dietary factors regulate cytokines in murine models of systemic lupus erythematosus. Autoimmun Rev. 2011;11(1):22-27. (PubMed)
179. Mao S, Huang S. Association between vitamin D receptor gene BsmI, FokI, ApaI and TaqI polymorphisms and the risk of systemic lupus erythematosus: a meta-analysis. Rheumatol Int. 2014;34(3):381-388. (PubMed)
180. Monticielo OA, Teixeira Tde M, Chies JA, Brenol JC, Xavier RM. Vitamin D and polymorphisms of VDR gene in patients with systemic lupus erythematosus. Clin Rheumatol. 2012;31(10):1411-1421. (PubMed)
181. Ruiz-Irastorza G, Egurbide MV, Olivares N, Martinez-Berriotxoa A, Aguirre C. Vitamin D deficiency in systemic lupus erythematosus: prevalence, predictors and clinical consequences. Rheumatology (Oxford). 2008;47(6):920-923. (PubMed)
182. Toloza SM, Cole DE, Gladman DD, Ibanez D, Urowitz MB. Vitamin D insufficiency in a large female SLE cohort. Lupus. 2010;19(1):13-19. (PubMed)
183. Amital H, Szekanecz Z, Szucs G, et al. Serum concentrations of 25-OH vitamin D in patients with systemic lupus erythematosus (SLE) are inversely related to disease activity: is it time to routinely supplement patients with SLE with vitamin D? Ann Rheum Dis. 2010;69(6):1155-1157. (PubMed)
184. Terrier B, Derian N, Schoindre Y, et al. Restoration of regulatory and effector T cell balance and B cell homeostasis in systemic lupus erythematosus patients through vitamin D supplementation. Arthritis Res Ther. 2012;14(5):R221. (PubMed)
185. Cutillas-Marco E, Marquina-Vila A, Grant W, Vilata-Corell J, Morales-Suarez-Varela M. Vitamin D and cutaneous lupus erythematosus: effect of vitamin D replacement on disease severity. Lupus. 2014;23(7):615-623. (PubMed)
186. Abou-Raya A, Abou-Raya S, Helmii M. The effect of vitamin D supplementation on inflammatory and hemostatic markers and disease activity in patients with systemic lupus erythematosus: a randomized placebo-controlled trial. J Rheumatol. 2013;40(3):265-272. (PubMed)
187. Lima GL, Paupitz J, Aikawa NE, Takayama L, Bonfa E, Pereira RM. Vitamin D supplementation in adolescents and young adults With juvenile systemic lupus erythematosus for improvement in disease activity and fatigue scores: a randomized, double-blind, placebo-controlled trial. Arthritis Care Res (Hoboken). 2016;68(1):91-98. (PubMed)
188. Andreoli L, Dall'Ara F, Piantoni S, et al. A 24-month prospective study on the efficacy and safety of two different monthly regimens of vitamin D supplementation in pre-menopausal women with systemic lupus erythematosus. Lupus. 2015;24(4-5):499-506. (PubMed)
189. Karimzadeh H, Shirzadi M, Karimifar M. The effect of Vitamin D supplementation in disease activity of systemic lupus erythematosus patients with Vitamin D deficiency: A randomized clinical trial. J Res Med Sci. 2017;22:4. (PubMed)
190. Antico A, Tampoia M, Tozzoli R, Bizzaro N. Can supplementation with vitamin D reduce the risk or modify the course of autoimmune diseases? A systematic review of the literature. Autoimmun Rev. 2012;12(2):127-136. (PubMed)
191. Wang TJ, Pencina MJ, Booth SL, et al. Vitamin D deficiency and risk of cardiovascular disease. Circulation. 2008;117(4):503-511. (PubMed)
192. van Ballegooijen AJ, Kestenbaum B, Sachs MC, et al. Association of 25-Hydroxyvitamin D and parathyroid hormone with incident hypertension: MESA (Multi-Ethnic Study of Atherosclerosis). J Am Coll Cardiol. 2014;63(12):1214-1222. (PubMed)
193. Kunutsor SK, Apekey TA, Steur M. Vitamin D and risk of future hypertension: meta-analysis of 283,537 participants. Eur J Epidemiol. 2013;28(3):205-221. (PubMed)
194. Burgaz A, Orsini N, Larsson SC, Wolk A. Blood 25-hydroxyvitamin D concentration and hypertension: a meta-analysis. J Hypertens. 2011;29(4):636-645. (PubMed)
195. Moody WE, Edwards NC, Madhani M, et al. Endothelial dysfunction and cardiovascular disease in early-stage chronic kidney disease: cause or association? Atherosclerosis. 2012;223(1):86-94. (PubMed)
196. Chitalia N, Recio-Mayoral A, Kaski JC, Banerjee D. Vitamin D deficiency and endothelial dysfunction in non-dialysis chronic kidney disease patients. Atherosclerosis. 2012;220(1):265-268. (PubMed)
197. Chitalia N, Ismail T, Tooth L, et al. Impact of vitamin d supplementation on arterial vasomotion, stiffness and endothelial biomarkers in chronic kidney disease patients. PLoS One. 2014;9(3):e91363. (PubMed)
198. Mazidi M, Karimi E, Rezaie P, Vatanparast H. The impact of vitamin D supplement intake on vascular endothelial function; a systematic review and meta-analysis of randomized controlled trials. Food Nutr Res. 2017;61(1):1273574. (PubMed)
199. Messa P, Curreri M, Regalia A, Alfieri CM. Vitamin D and the cardiovascular system: an overview of the recent literature. Am J Cardiovasc Drugs. 2014;14(1):1-14. (PubMed)
200. Brondum-Jacobsen P, Benn M, Afzal S, Nordestgaard BG. No evidence that genetically reduced 25-hydroxyvitamin D is associated with increased risk of ischaemic heart disease or myocardial infarction: a Mendelian randomization study. Int J Epidemiol. 2015;44(2):651-661. (PubMed)
201. Manousaki D, Mokry LE, Ross S, Goltzman D, Richards JB. Mendelian randomization studies do not support a role for vitamin D in coronary artery disease. Circ Cardiovasc Genet. 2016;9(4):349-356. (PubMed)
202. Ford JA, MacLennan GS, Avenell A, Bolland M, Grey A, Witham M. Cardiovascular disease and vitamin D supplementation: trial analysis, systematic review, and meta-analysis. Am J Clin Nutr. 2014;100(3):746-755. (PubMed)
203. Chin K, Appel LJ, Michos ED. Vitamin D, calcium, and cardiovascular disease: a"D"vantageous or "D"etrimental? An era of uncertainty. Curr Atheroscler Rep. 2017;19(1):5. (PubMed)
204. Pradhan AD, Manson JE. Update on the Vitamin D and OmegA-3 trial (VITAL). J Steroid Biochem Mol Biol. 2016;155(Pt B):252-256. (PubMed)
205. Neale RE, Armstrong BK, Baxter C, et al. The D-Health Trial: A randomized trial of vitamin D for prevention of mortality and cancer. Contemp Clin Trials. 2016;48:83-90. (PubMed)
206. Scragg R, Stewart AW, Waayer D, et al. Effect of monthly high-dose vitamin D supplementation on cardiovascular disease in the vitamin D assessment study: a randomized clinical trial. JAMA Cardiol. 2017;2(6):608-616. (PubMed)
207. Thomas GN, o Hartaigh B, Bosch JA, et al. Vitamin D levels predict all-cause and cardiovascular disease mortality in subjects with the metabolic syndrome: the Ludwigshafen Risk and Cardiovascular Health (LURIC) Study. Diabetes Care. 2012;35(5):1158-1164. (PubMed)
208. Chiu KC, Chu A, Go VL, Saad MF. Hypovitaminosis D is associated with insulin resistance and beta cell dysfunction. Am J Clin Nutr. 2004;79(5):820-825. (PubMed)
209. Shankar A, Sabanayagam C, Kalidindi S. Serum 25-hydroxyvitamin D levels and prediabetes among subjects free of diabetes. Diabetes Care. 2011;34(5):1114-1119. (PubMed)
210. Deleskog A, Hilding A, Brismar K, Hamsten A, Efendic S, Ostenson CG. Low serum 25-hydroxyvitamin D level predicts progression to type 2 diabetes in individuals with prediabetes but not with normal glucose tolerance. Diabetologia. 2012;55(6):1668-1678. (PubMed)
211. Khan H, Kunutsor S, Franco OH, Chowdhury R. Vitamin D, type 2 diabetes and other metabolic outcomes: a systematic review and meta-analysis of prospective studies. Proc Nutr Soc. 2013;72(1):89-97. (PubMed)
212. Lucato P, Solmi M, Maggi S, et al. Low vitamin D levels increase the risk of type 2 diabetes in older adults: A systematic review and meta-analysis. Maturitas. 2017;100:8-15. (PubMed)
213. George PS, Pearson ER, Witham MD. Effect of vitamin D supplementation on glycaemic control and insulin resistance: a systematic review and meta-analysis. Diabet Med. 2012;29(8):e142-150. (PubMed)
214. Gulseth HL, Wium C, Angel K, Eriksen EF, Birkeland KI. Effects of vitamin D supplementation on insulin sensitivity and insulin secretion in subjects with type 2 diabetes and vitamin D deficiency: a randomized controlled trial. Diabetes Care. 2017;40(7):872-878. (PubMed)
215. Lee CJ, Iyer G, Liu Y, et al. The effect of vitamin D supplementation on glucose metabolism in type 2 diabetes mellitus: A systematic review and meta-analysis of intervention studies. J Diabetes Complications. 2017;31(7):1115-1126. (PubMed)
216. Talaei A, Mohamadi M, Adgi Z. The effect of vitamin D on insulin resistance in patients with type 2 diabetes. Diabetol Metab Syndr. 2013;5(1):8. (PubMed)
217. Gezen-Ak D, Yilmazer S, Dursun E. Why vitamin d in Alzheimer's disease? The hypothesis. J Alzheimers Dis. 2014;40(2):257-269. (PubMed)
218. Landel V, Annweiler C, Millet P, Morello M, Feron F. Vitamin D, cognition and Alzheimer's disease: the therapeutic benefit is in the D-tails. J Alzheimers Dis. 2016;53(2):419-444. (PubMed)
219. Annweiler C, Schott AM, Rolland Y, Blain H, Herrmann FR, Beauchet O. Dietary intake of vitamin D and cognition in older women: a large population-based study. Neurology. 2010;75(20):1810-1816. (PubMed)
220. Annweiler C, Rolland Y, Schott AM, et al. Higher vitamin D dietary intake is associated with lower risk of Alzheimer's disease: a 7-year follow-up. J Gerontol A Biol Sci Med Sci. 2012;67(11):1205-1211. (PubMed)
221. Annweiler C, Fantino B, Schott AM, Krolak-Salmon P, Allali G, Beauchet O. Vitamin D insufficiency and mild cognitive impairment: cross-sectional association. Eur J Neurol. 2012;19(7):1023-1029. (PubMed)
222. Hooshmand B, Lokk J, Solomon A, et al. Vitamin D in relation to cognitive impairment, cerebrospinal fluid biomarkers, and brain volumes. J Gerontol A Biol Sci Med Sci. 2014;69(9):1132-1138. (PubMed)
223. Slinin Y, Paudel ML, Taylor BC, et al. 25-Hydroxyvitamin D levels and cognitive performance and decline in elderly men. Neurology. 2010;74(1):33-41. (PubMed)
224. Slinin Y, Paudel M, Taylor BC, et al. Association between serum 25(OH) vitamin D and the risk of cognitive decline in older women. J Gerontol A Biol Sci Med Sci. 2012;67(10):1092-1098. (PubMed)
225. Annweiler C, Milea D, Whitson HE, et al. Vitamin D insufficiency and cognitive impairment in Asians: a multi-ethnic population-based study and meta-analysis. J Intern Med. 2016;280(3):300-311. (PubMed)
226. Annweiler C, Llewellyn DJ, Beauchet O. Low serum vitamin D concentrations in Alzheimer's disease: a systematic review and meta-analysis. J Alzheimers Dis. 2013;33(3):659-674. (PubMed)
227. Balion C, Griffith LE, Strifler L, et al. Vitamin D, cognition, and dementia: a systematic review and meta-analysis. Neurology. 2012;79(13):1397-1405. (PubMed)
228. Lopes da Silva S, Vellas B, Elemans S, et al. Plasma nutrient status of patients with Alzheimer's disease: Systematic review and meta-analysis. Alzheimers Dement. 2014;10(4):485-502. (PubMed)
229. Shen L, Ji HF. Vitamin D deficiency is associated with increased risk of Alzheimer's disease and dementia: evidence from meta-analysis. Nutr J. 2015;14:76. (PubMed)
230. Sommer I, Griebler U, Kien C, et al. Vitamin D deficiency as a risk factor for dementia: a systematic review and meta-analysis. BMC Geriatr. 2017;17(1):16. (PubMed)
231. Olsson E, Byberg L, Karlstrom B, et al. Vitamin D is not associated with incident dementia or cognitive impairment: an 18-y follow-up study in community-living old men. Am J Clin Nutr. 2017;105(4):936-943. (PubMed)
232. Mokry LE, Ross S, Morris JA, Manousaki D, Forgetta V, Richards JB. Genetically decreased vitamin D and risk of Alzheimer disease. Neurology. 2016;87(24):2567-2574. (PubMed)
233. Annweiler C, Montero-Odasso M, Llewellyn DJ, Richard-Devantoy S, Duque G, Beauchet O. Meta-analysis of memory and executive dysfunctions in relation to vitamin D. J Alzheimers Dis. 2013;37(1):147-171. (PubMed)
234. Annweiler C, Fantino B, Gautier J, Beaudenon M, Thiery S, Beauchet O. Cognitive effects of vitamin D supplementation in older outpatients visiting a memory clinic: a pre-post study. J Am Geriatr Soc. 2012;60(4):793-795. (PubMed)
235. Stein MS, Scherer SC, Ladd KS, Harrison LC. A randomized controlled trial of high-dose vitamin D2 followed by intranasal insulin in Alzheimer's disease. J Alzheimers Dis. 2011;26(3):477-484. (PubMed)
236. Annweiler C, Karras SN, Anagnostis P, Beauchet O. Vitamin D supplements: a novel therapeutic approach for Alzheimer patients. Front Pharmacol. 2014;5:6. (PubMed)
237. Sato Y, Kikuyama M, Oizumi K. High prevalence of vitamin D deficiency and reduced bone mass in Parkinson's disease. Neurology. 1997;49(5):1273-1278. (PubMed)
238. Evatt ML, Delong MR, Khazai N, Rosen A, Triche S, Tangpricha V. Prevalence of vitamin D insufficiency in patients with Parkinson disease and Alzheimer disease. Arch Neurol. 2008;65(10):1348-1352. (PubMed)
239. Knekt P, Kilkkinen A, Rissanen H, Marniemi J, Saaksjarvi K, Heliovaara M. Serum vitamin D and the risk of Parkinson disease. Arch Neurol. 2010;67(7):808-811. (PubMed)
240. Lv Z, Qi H, Wang L, et al. Vitamin D status and Parkinson's disease: a systematic review and meta-analysis. Neurol Sci. 2014;35(11):1723-1730. (PubMed)
241. Shen L, Ji HF. Associations between vitamin D status, supplementation, outdoor work and risk of Parkinson's disease: a meta-analysis assessment. Nutrients. 2015;7(6):4817-4827. (PubMed)
242. Zhao Y, Sun Y, Ji HF, Shen L. Vitamin D levels in Alzheimer's and Parkinson's diseases: a meta-analysis. Nutrition. 2013;29(6):828-832. (PubMed)
243. Suzuki M, Yoshioka M, Hashimoto M, et al. Randomized, double-blind, placebo-controlled trial of vitamin D supplementation in Parkinson disease. Am J Clin Nutr. 2013;97(5):1004-1013. (PubMed)
244. Dobson R, Yarnall A, Noyce AJ, Giovannoni G. Bone health in chronic neurological diseases: a focus on multiple sclerosis and parkinsonian syndromes. Pract Neurol. 2013;13(2):70-79. (PubMed)
245. Torsney KM, Noyce AJ, Doherty KM, Bestwick JP, Dobson R, Lees AJ. Bone health in Parkinson's disease: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2014;85(10):1159-66. (PubMed)
246. van den Bos F, Speelman AD, Samson M, Munneke M, Bloem BR, Verhaar HJ. Parkinson's disease and osteoporosis. Age Ageing. 2013;42(2):156-162. (PubMed)
247. Sato Y, Iwamoto J, Honda Y. Amelioration of osteoporosis and hypovitaminosis D by sunlight exposure in Parkinson's disease. Parkinsonism Relat Disord. 2011;17(1):22-26. (PubMed)
248. Aghajafari F, Nagulesapillai T, Ronksley PE, Tough SC, O'Beirne M, Rabi DM. Association between maternal serum 25-hydroxyvitamin D level and pregnancy and neonatal outcomes: systematic review and meta-analysis of observational studies. BMJ. 2013;346:f1169. (PubMed)
249. Perez-Lopez FR, Pasupuleti V, Mezones-Holguin E, et al. Effect of vitamin D supplementation during pregnancy on maternal and neonatal outcomes: a systematic review and meta-analysis of randomized controlled trials. Fertil Steril. 2015;103(5):1278-1288.e1274. (PubMed)
250. Alzaim M, Wood RJ. Vitamin D and gestational diabetes mellitus. Nutr Rev. 2013;71(3):158-167. (PubMed)
251. Lacroix M, Battista MC, Doyon M, et al. Lower vitamin D levels at first trimester are associated with higher risk of developing gestational diabetes mellitus. Acta Diabetol. 2014;51(4):609-616. (PubMed)
252. Parlea L, Bromberg IL, Feig DS, Vieth R, Merman E, Lipscombe LL. Association between serum 25-hydroxyvitamin D in early pregnancy and risk of gestational diabetes mellitus. Diabet Med. 2012;29(7):e25-32. (PubMed)
253. Lu M, Xu Y, Lv L, Zhang M. Association between vitamin D status and the risk of gestational diabetes mellitus: a meta-analysis. Arch Gynecol Obstet. 2016;293(5):959-966. (PubMed)
254. Poel YH, Hummel P, Lips P, Stam F, van der Ploeg T, Simsek S. Vitamin D and gestational diabetes: a systematic review and meta-analysis. Eur J Intern Med. 2012;23(5):465-469. (PubMed)
255. Wei SQ, Qi HP, Luo ZC, Fraser WD. Maternal vitamin D status and adverse pregnancy outcomes: a systematic review and meta-analysis. J Matern Fetal Neonatal Med. 2013;26(9):889-899. (PubMed)
256. Zhang MX, Pan GT, Guo JF, Li BY, Qin LQ, Zhang ZL. Vitamin D deficiency increases the risk of gestational diabetes mellitus: a meta-analysis of observational studies. Nutrients. 2015;7(10):8366-8375. (PubMed)
257. Triunfo S, Lanzone A, Lindqvist PG. Low maternal circulating levels of vitamin D as potential determinant in the development of gestational diabetes mellitus. J Endocrinol Invest. 2017;40(10):1049-1059. (PubMed)
258. Asemi Z, Hashemi T, Karamali M, Samimi M, Esmaillzadeh A. Effects of vitamin D supplementation on glucose metabolism, lipid concentrations, inflammation, and oxidative stress in gestational diabetes: a double-blind randomized controlled clinical trial. Am J Clin Nutr. 2013;98(6):1425-1432. (PubMed)
259. Jelsma JG, van Poppel MN, Galjaard S, et al. DALI: Vitamin D and lifestyle intervention for gestational diabetes mellitus (GDM) prevention: an European multicentre, randomised trial - study protocol. BMC Pregnancy Childbirth. 2013;13:142. (PubMed)
260. Simmons D, Devlieger R, van Assche A, et al. Effect of Physical Activity and/or Healthy Eating on GDM Risk: The DALI Lifestyle Study. J Clin Endocrinol Metab. 2017;102(3):903-913. (PubMed)
261. Galthen-Sorensen M, Andersen LB, Sperling L, Christesen HT. Maternal 25-hydroxyvitamin D level and fetal bone growth assessed by ultrasound: a systematic review. Ultrasound Obstet Gynecol. 2014;44(6):633-640. (PubMed)
262. Shor DB, Barzel J, Tauber E, Amital H. The effects of maternal vitamin D on neonatal growth parameters. Eur J Pediatr. 2015;174(9):1169-1174. (PubMed)
263. Cooper C, Harvey NC, Bishop NJ, et al. Maternal gestational vitamin D supplementation and offspring bone health (MAVIDOS): a multicentre, double-blind, randomised placebo-controlled trial. Lancet Diabetes Endocrinol. 2016;4(5):393-402. (PubMed)
264. Handel MN, Frederiksen P, Osmond C, Cooper C, Abrahamsen B, Heitmann BL. Prenatal exposure to vitamin D from fortified margarine and risk of fractures in late childhood: period and cohort results from 222 000 subjects in the D-tect observational study. Br J Nutr. 2017;117(6):872-881. (PubMed)
265. Rodda CP, Benson JE, Vincent AJ, Whitehead CL, Polykov A, Vollenhoven B. Maternal vitamin D supplementation during pregnancy prevents vitamin D deficiency in the newborn: an open-label randomized controlled trial. Clin Endocrinol (Oxf). 2015;83(3):363-368. (PubMed)
266. Handel MN, Frederiksen P, Cohen A, Cooper C, Heitmann BL, Abrahamsen B. Neonatal vitamin D status from archived dried blood spots and future risk of fractures in childhood: results from the D-tect study, a population-based case-cohort study. Am J Clin Nutr. 2017;106(1):155-161. (PubMed)
267. Bountouvi E, Douros K, Papadopoulou A. Can getting enough vitamin D during pregnancy reduce the risk of getting asthma in childhood? Front Pediatr. 2017;5:87. (PubMed)
268. Goldring ST, Griffiths CJ, Martineau AR, et al. Prenatal vitamin D supplementation and child respiratory health: a randomised controlled trial. PLoS One. 2013;8(6):e66627. (PubMed)
269. Chawes BL, Bonnelykke K, Stokholm J, et al. Effect of vitamin D3 supplementation during pregnancy on rsk of persistent wheeze in the offspring: a randomized clinical trial. JAMA. 2016;315(4):353-361. (PubMed)
270. Litonjua AA, Carey VJ, Laranjo N, et al. Effect of prenatal supplementation with vitamin D on asthma or eecurrent wheezing in offspring by age 3 years: the VDAART randomized clinical trial. JAMA. 2016;315(4):362-370. (PubMed)
271. Vahdaninia M, Mackenzie H, Helps S, Dean T. Prenatal intake of vitamins and allergic outcomes in the offspring: a systematic review and meta-analysis. J Allergy Clin Immunol Pract. 2017;5(3):771-778.e775. (PubMed)
272. Weisse K, Winkler S, Hirche F, et al. Maternal and newborn vitamin D status and its impact on food allergy development in the German LINA cohort study. Allergy. 2013;68(2):220-228. (PubMed)
273. Miettinen ME, Smart MC, Kinnunen L, et al. Maternal VDR variants rather than 25-hydroxyvitamin D concentration during early pregnancy are associated with type 1 diabetes in the offspring. Diabetologia. 2015;58(10):2278-2283. (PubMed)
274. Sorensen IM, Joner G, Jenum PA, et al. Vitamin D-binding protein and 25-hydroxyvitamin D during pregnancy in mothers whose children later developed type 1 diabetes. Diabetes Metab Res Rev. 2016;32(8):883-890. (PubMed)
275. Makela MJ, Puhakka T, Ruuskanen O, et al. Viruses and bacteria in the etiology of the common cold. J Clin Microbiol. 1998;36(2):539-542. (PubMed)
276. Ginde AA, Mansbach JM, Camargo CA, Jr. Association between serum 25-hydroxyvitamin D level and upper respiratory tract infection in the Third National Health and Nutrition Examination Survey. Arch Intern Med. 2009;169(4):384-390. (PubMed)
277. Murdoch DR, Slow S, Chambers ST, et al. Effect of vitamin D3 supplementation on upper respiratory tract infections in healthy adults: the VIDARIS randomized controlled trial. JAMA. 2012;308(13):1333-1339. (PubMed)
278. Rees JR, Hendricks K, Barry EL, et al. Vitamin D3 supplementation and upper respiratory tract infections in a randomized, controlled trial. Clin Infect Dis. 2013;57(10):1384-1392. (PubMed)
279. Tran B, Armstrong BK, Ebeling PR, et al. Effect of vitamin D supplementation on antibiotic use: a randomized controlled trial. Am J Clin Nutr. 2014;99(1):156-161. (PubMed)
280. Grant CC, Kaur S, Waymouth E, et al. Reduced primary care respiratory infection visits following pregnancy and infancy vitamin D supplementation: a randomised controlled trial. Acta Paediatr. 2015;104(4):396-404. (PubMed)
281. Martineau AR, Jolliffe DA, Hooper RL, et al. Vitamin D supplementation to prevent acute respiratory tract infections: systematic review and meta-analysis of individual participant data. BMJ. 2017;356:i6583. (PubMed)
282. Scragg R, Waayer D, Stewart AW, et al. The Vitamin D Assessment (ViDA) study: design of a randomized controlled trial of vitamin D supplementation for the prevention of cardiovascular disease, acute respiratory infection, falls and non-vertebral fractures. J Steroid Biochem Mol Biol. 2016;164:318-325. (PubMed)
283. Manson JE, Bassuk SS, Buring JE, Group VR. Principal results of the VITamin D and OmegA-3 TriaL (VITAL) and updated meta-analyses of relevant vitamin D trials. J Steroid Biochem Mol Biol. 2020;198:105522. (PubMed)
284. Gold DR, Litonjua AA, Carey VJ, et al. Lung VITAL: Rationale, design, and baseline characteristics of an ancillary study evaluating the effects of vitamin D and/or marine omega-3 fatty acid supplements on acute exacerbations of chronic respiratory disease, asthma control, pneumonia and lung function in adults. Contemp Clin Trials. 2016;47:185-195. (PubMed)
285. Aglipay M, Birken CS, Parkin PC, et al. Effect of high-dose vs standard-dose wintertime vitamin D supplementation on viral upper respiratory tract infections in young healthy children. JAMA. 2017;318(3):245-254. (PubMed)
286. Hueniken K, Aglipay M, Birken CS, et al. Effect of high-dose vitamin D supplementation on upper respiratory tract infection symptom severity in healthy children. Pediatr Infect Dis J. 2019;38(6):564-568. (PubMed)
287. Loeb M, Dang AD, Thiem VD, et al. Effect of Vitamin D supplementation to reduce respiratory infections in children and adolescents in Vietnam: A randomized controlled trial. Influenza Other Respir Viruses. 2019;13(2):176-183. (PubMed)
288. Kronbichler A, Kresse D, Yoon S, Lee KH, Effenberger M, Shin JI. Asymptomatic patients as a source of COVID-19 infections: A systematic review and meta-analysis. Int J Infect Dis. 2020;98:180-186. (PubMed)
289. Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA. 2020;324(8):782-793. (PubMed)
290. Ferrari D, Locatelli M. No significant association between vitamin D and COVID-19. A retrospective study from a northern Italian hospital. Int J Vitam Nutr Res. 2020:1-4. (PubMed)
291. Hastie CE, Mackay DF, Ho F, et al. Vitamin D concentrations and COVID-19 infection in UK Biobank. Diabetes Metab Syndr. 2020;14(4):561-565. (PubMed)
292. Ma H, Zhou T, Heianza Y, Qi L. Habitual use of vitamin D supplements and risk of coronavirus disease 2019 (COVID-19) infection: a prospective study in UK Biobank. Am J Clin Nutr. 2021;113(5):1275-1281. (PubMed)
293. Kaufman HW, Niles JK, Kroll MH, Bi C, Holick MF. SARS-CoV-2 positivity rates associated with circulating 25-hydroxyvitamin D levels. PLoS One. 2020;15(9):e0239252. (PubMed)
294. Merzon E, Tworowski D, Gorohovski A, et al. Low plasma 25(OH) vitamin D level is associated with increased risk of COVID-19 infection: an Israeli population-based study. FEBS J. 2020;287(17):3693-3702. (PubMed)
295. Meltzer DO, Best TJ, Zhang H, Vokes T, Arora V, Solway J. Association of vitamin D status and other clinical characteristics with COVID-19 test results. JAMA Netw Open. 2020;3(9):e2019722. (PubMed)
296. D'Avolio A, Avataneo V, Manca A, et al. 25-Hydroxyvitamin D concentrations are lower in patients with positive PCR for SARS-CoV-2. Nutrients. 2020;12(5):1359. (PubMed)
297. Ye K, Tang F, Liao X, et al. Does serum vitamin D level affect COVID-19 infection and its severity?-a case-control study. J Am Coll Nutr. 2020:1-8. (PubMed)
298. Hernandez JL, Nan D, Fernandez-Ayala M, et al. Vitamin D status in hospitalized patients with SARS-CoV-2 infection. J Clin Endocrinol Metab. 2020;106(3):e1343-e1353. (PubMed)
299. Im JH, Je YS, Baek J, Chung MH, Kwon HY, Lee JS. Nutritional status of patients with COVID-19. Int J Infect Dis. 2020;100:390-393. (PubMed)
300. Abdollahi A, Kamali Sarvestani H, Rafat Z, et al. The association between the level of serum 25(OH) vitamin D, obesity, and underlying diseases with the risk of developing COVID-19 infection: A case-control study of hospitalized patients in Tehran, Iran. J Med Virol. 2020;93(4):2359-2364. (PubMed)
301. Liu N, Sun J, Wang X, Zhang T, Zhao M, Li H. Low vitamin D status is associated with coronavirus disease 2019 outcomes: a systematic review and meta-analysis. Int J Infect Dis. 2021;104:58-64. (PubMed)
302. Panagiotou G, Tee SA, Ihsan Y, et al. Low serum 25-hydroxyvitamin D (25[OH]D) levels in patients hospitalized with COVID-19 are associated with greater disease severity. Clin Endocrinol (Oxf). 2020;93(4):508-511. (PubMed)
303. De Smet D, De Smet K, Herroelen P, Gryspeerdt S, Martens GA. Serum 25(OH)D level on hospital admission associated with COVID-19 stage and mortality. Am J Clin Pathol. 2020;155(3):381-388. (PubMed)
304. Baktash V, Hosack T, Patel N, et al. Vitamin D status and outcomes for hospitalised older patients with COVID-19. Postgrad Med J. 2020. [Epub ahead of print] (PubMed)
305. Radujkovic A, Hippchen T, Tiwari-Heckler S, Dreher S, Boxberger M, Merle U. Vitamin D deficiency and outcome of COVID-19 patients. Nutrients. 2020;12(9): 2757. (PubMed)
306. Carpagnano GE, Di Lecce V, Quaranta VN, et al. Vitamin D deficiency as a predictor of poor prognosis in patients with acute respiratory failure due to COVID-19. J Endocrinol Invest. 2020;(4):765-771. (PubMed)
307. Hastie CE, Pell JP, Sattar N. Vitamin D and COVID-19 infection and mortality in UK Biobank. Eur J Nutr. 2020;60(1):545-548. (PubMed)
308. Centers for Disease Control and Prevention. Assessing Risk Factors for Severe COVID-19 Illness. Available at: https://www.cdc.gov/coronavirus/2019-ncov/covid-data/investigations-discovery/assessing-risk-factors.html. Accessed 1/22/21.
309. Annweiler G, Corvaisier M, Gautier J, et al. Vitamin D supplementation associated to better survival in hospitalized frail elderly COVID-19 patients: the GERIA-COVID quasi-experimental study. Nutrients. 2020;12(11):3377. (PubMed)
310. Annweiler C, Hanotte B, Grandin de l'Eprevier C, Sabatier JM, Lafaie L, Celarier T. Vitamin D and survival in COVID-19 patients: A quasi-experimental study. J Steroid Biochem Mol Biol. 2020;204:105771. (PubMed)
311. Mesquita Kde C, Igreja AC, Costa IM. Atopic dermatitis and vitamin D: facts and controversies. An Bras Dermatol. 2013;88(6):945-953. (PubMed)
312. Lee SA, Hong S, Kim HJ, Lee SH, Yum HY. Correlation between serum vitamin D level and the severity of atopic dermatitis associated with food sensitization. Allergy Asthma Immunol Res. 2013;5(4):207-210. (PubMed)
313. Sudlow C, Gallacher J, Allen N, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12(3):e1001779. (PubMed)
314. Moffatt MF, Gut IG, Demenais F, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363(13):1211-1221. (PubMed)
315. Paternoster L, Zhurov AI, Toma AM, et al. Genome-wide association study of three-dimensional facial morphology identifies a variant in PAX3 associated with nasion position. Am J Hum Genet. 2012;90(3):478-485. (PubMed)
316. Manousaki D, Paternoster L, Standl M, et al. Vitamin D levels and susceptibility to asthma, elevated immunoglobulin E levels, and atopic dermatitis: A Mendelian randomization study. PLoS Med. 2017;14(5):e1002294. (PubMed)
317. Javanbakht MH, Keshavarz SA, Djalali M, et al. Randomized controlled trial using vitamins E and D supplementation in atopic dermatitis. J Dermatolog Treat. 2011;22(3):144-150. (PubMed)
318. Amestejani M, Salehi BS, Vasigh M, et al. Vitamin D supplementation in the treatment of atopic dermatitis: a clinical trial study. J Drugs Dermatol. 2012;11(3):327-330. (PubMed)
319. Camargo CA, Jr., Ganmaa D, Sidbury R, Erdenedelger K, Radnaakhand N, Khandsuren B. Randomized trial of vitamin D supplementation for winter-related atopic dermatitis in children. J Allergy Clin Immunol. 2014;134(4):831-835.e831. (PubMed)
320. Kim G, Bae JH. Vitamin D and atopic dermatitis: A systematic review and meta-analysis. Nutrition. 2016;32(9):913-920. (PubMed)
321. Wat H, Dytoc M. Off-label uses of topical vitamin d in dermatology: a systematic review. J Cutan Med Surg. 2014;18(2):91-108. (PubMed)
322. Xue LN, Xu KQ, Zhang W, Wang Q, Wu J, Wang XY. Associations between vitamin D receptor polymorphisms and susceptibility to ulcerative colitis and Crohn's disease: a meta-analysis. Inflamm Bowel Dis. 2013;19(1):54-60. (PubMed)
323. Ananthakrishnan AN, Khalili H, Higuchi LM, et al. Higher predicted vitamin D status is associated with reduced risk of Crohn's disease. Gastroenterology. 2012;142(3):482-489. (PubMed)
324. Sadeghian M, Saneei P, Siassi F, Esmaillzadeh A. Vitamin D status in relation to Crohn's disease: meta-analysis of observational studies. Nutrition. 2016;32(5):505-514. (PubMed)
325. Jorgensen SP, Agnholt J, Glerup H, et al. Clinical trial: vitamin D3 treatment in Crohn's disease - a randomized double-blind placebo-controlled study. Aliment Pharmacol Ther. 2010;32(3):377-383. (PubMed)
326. Yang L, Weaver V, Smith JP, Bingaman S, Hartman TJ, Cantorna MT. Therapeutic effect of vitamin d supplementation in a pilot study of Crohn's patients. Clin Transl Gastroenterol. 2013;4:e33. (PubMed)
327. Raftery T, Martineau AR, Greiller CL, et al. Effects of vitamin D supplementation on intestinal permeability, cathelicidin and disease markers in Crohn's disease: Results from a randomised double-blind placebo-controlled study. United European Gastroenterol J. 2015;3(3):294-302. (PubMed)
328. Anderson JL, May HT, Horne BD, et al. Relation of vitamin D deficiency to cardiovascular risk factors, disease status, and incident events in a general healthcare population. Am J Cardiol. 2010;106(7):963-968. (PubMed)
329. Al Mheid I, Patel R, Murrow J, et al. Vitamin D status is associated with arterial stiffness and vascular dysfunction in healthy humans. J Am Coll Cardiol. 2011;58(2):186-192. (PubMed)
330. Krause R, Buhring M, Hopfenmuller W, Holick MF, Sharma AM. Ultraviolet B and blood pressure. Lancet. 1998;352(9129):709-710. (PubMed)
331. Kunutsor SK, Burgess S, Munroe PB, Khan H. Vitamin D and high blood pressure: causal association or epiphenomenon? Eur J Epidemiol. 2014;29(1):1-14. (PubMed)
332. Pilz S, Gaksch M, Kienreich K, et al. Effects of vitamin D on blood pressure and cardiovascular risk factors: a randomized controlled trial. Hypertension. 2015;65(6):1195-1201. (PubMed)
333. Arora P, Song Y, Dusek J, et al. Vitamin D therapy in individuals with prehypertension or hypertension: the DAYLIGHT trial. Circulation. 2015;131(3):254-262. (PubMed)
334. Rostand SG. Ultraviolet light may contribute to geographic and racial blood pressure differences. Hypertension. 1997;30(2 Pt 1):150-156. (PubMed)
335. Forman JP, Scott JB, Ng K, et al. Effect of vitamin D supplementation on blood pressure in blacks. Hypertension. 2013;61(4):779-785. (PubMed)
336. Witham MD, Price RJ, Struthers AD, et al. Cholecalciferol treatment to reduce blood pressure in older patients with isolated systolic hypertension: the VitDISH randomized controlled trial. JAMA Intern Med. 2013;173(18):1672-1679. (PubMed)
337. Oz F, Cizgici AY, Oflaz H, et al. Impact of vitamin D insufficiency on the epicardial coronary flow velocity and endothelial function. Coron Artery Dis. 2013;24(5):392-397. (PubMed)
338. Liu LC, Voors AA, van Veldhuisen DJ, et al. Vitamin D status and outcomes in heart failure patients. Eur J Heart Fail. 2011;13(6):619-625. (PubMed)
339. Shedeed SA. Vitamin D supplementation in infants with chronic congestive heart failure. Pediatr Cardiol. 2012;33(5):713-719. (PubMed)
340. Boxer RS, Kenny AM, Schmotzer BJ, Vest M, Fiutem JJ, Pina IL. A randomized controlled trial of high dose vitamin D3 in patients with heart failure. JACC Heart Fail. 2013;1(1):84-90. (PubMed)
341. Jiang WL, Gu HB, Zhang YF, Xia QQ, Qi J, Chen JC. Vitamin D supplementation in the treatment of chronic heart failure: a meta-analysis of randomized controlled trials. Clin Cardiol. 2016;39(1):56-61. (PubMed)
342. Zittermann A, Ernst JB, Prokop S, et al. Effect of vitamin D on all-cause mortality in heart failure (EVITA): a 3-year randomized clinical trial with 4000 IU vitamin D daily. Eur Heart J. 2017;38(29):2279-2286. (PubMed)
343. Norman AW, Henry HH. Vitamin D. In: Bowman BA, Russell RM, eds. Present Knowledge in Nutrition. 9th ed. Washington, D.C.: ILSI Press; 2006:198-210.
344. Holick MF. Vitamin D: the underappreciated D-lightful hormone that is important for skeletal and cellular health. Curr Opin Endocrinol Diabetes. 2002;9:87-98.
345. Terushkin V, Bender A, Psaty EL, Engelsen O, Wang SQ, Halpern AC. Estimated equivalency of vitamin D production from natural sun exposure versus oral vitamin D supplementation across seasons at two US latitudes. J Am Acad Dermatol. 2010;62(6):929 e921-929. (PubMed)
346. Food and Nutrition Board, Institute of Medicine. Vitamin D. Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington, D.C.: National Academies Press; 1999:250-287. (National Academy Press)
347. Ovesen L, Brot C, Jakobsen J. Food contents and biological activity of 25-hydroxyvitamin D: a vitamin D metabolite to be reckoned with? Ann Nutr Metab. 2003;47(3-4):107-113. (PubMed)
348. Jakobsen J, Christensen T. Natural vitamin D in food: to what degree does 25-hydroxyvitaminn D contribute to the vitamin D activity in food? JMBR Plus. 2020: doi: 10.1002/jbm1004.10453.
349. Tripkovic L, Lambert H, Hart K, et al. Comparison of vitamin D2 and vitamin D3 supplementation in raising serum 25-hydroxyvitamin D status: a systematic review and meta-analysis. Am J Clin Nutr. 2012;95(6):1357-1364. (PubMed)
350. Logan VF, Gray AR, Peddie MC, Harper MJ, Houghton LA. Long-term vitamin D3 supplementation is more effective than vitamin D2 in maintaining serum 25-hydroxyvitamin D status over the winter months. Br J Nutr. 2013;109(6):1082-1088. (PubMed)
351. Barger-Lux MJ, Heaney RP, Dowell S, Chen TC, Holick MF. Vitamin D and its major metabolites: serum levels after graded oral dosing in healthy men. Osteoporos Int. 1998;8(3):222-230. (PubMed)
352. Cashman KD, Seamans KM, Lucey AJ, et al. Relative effectiveness of oral 25-hydroxyvitamin D3 and vitamin D3 in raising wintertime serum 25-hydroxyvitamin D in older adults. Am J Clin Nutr. 2012;95(6):1350-1356. (PubMed)
353. Bischoff-Ferrari HA, Dawson-Hughes B, Stocklin E, et al. Oral supplementation with 25(OH)D3 versus vitamin D3: effects on 25(OH)D levels, lower extremity function, blood pressure, and markers of innate immunity. J Bone Miner Res. 2012;27(1):160-169. (PubMed)
354. Jetter A, Egli A, Dawson-Hughes B, et al. Pharmacokinetics of oral vitamin D(3) and calcifediol. Bone. 2014;59:14-19. (PubMed)
355. Navarro-Valverde C, Sosa-Henriquez M, Alhambra-Exposito MR, Quesada-Gomez JM. Vitamin D3 and calcidiol are not equipotent. J Steroid Biochem Mol Biol. 2016;164:205-208. (PubMed)
356. Shieh A, Ma C, Chun RF, et al. Effects of cholecalciferol vs calcifediol on total and free 25-hydroxyvitamin D and parathyroid hormone. J Clin Endocrinol Metab. 2017;102(4):1133-1140. (PubMed)
357. Vaes AMM, Tieland M, de Regt MF, Wittwer J, van Loon LJC, de Groot L. Dose-response effects of supplementation with calcifediol on serum 25-hydroxyvitamin D status and its metabolites: A randomized controlled trial in older adults. Clin Nutr. 2018;37(3):808-814. (PubMed)
358. Graeff-Armas LA, Bendik I, Kunz I, Schoop R, Hull S, Beck M. Supplemental 25-hydroxycholecalciferol is more effective than cholecalciferol in raising serum 25-hydroxyvitamin D concentrations in older adults. J Nutr. 2020;150(1):73-81. (PubMed)
359. Quesada-Gomez JM, Bouillon R. Is calcifediol better than cholecalciferol for vitamin D supplementation? Osteoporos Int. 2018;29(8):1697-1711. (PubMed)
360. Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266-281. (PubMed)
361. Vieth R. Vitamin D supplementation, 25-hydroxyvitamin D concentrations, and safety. Am J Clin Nutr. 1999;69(5):842-856. (PubMed)
362. Heaney RP, Davies KM, Chen TC, Holick MF, Barger-Lux MJ. Human serum 25-hydroxycholecalciferol response to extended oral dosing with cholecalciferol. Am J Clin Nutr. 2003;77(1):204-210. (PubMed)
363. Vieth R, Chan PC, MacFarlane GD. Efficacy and safety of vitamin D3 intake exceeding the lowest observed adverse effect level. Am J Clin Nutr. 2001;73(2):288-294. (PubMed)
364. Knodel LC, Talbert RL. Adverse effects of hypolipidaemic drugs. Med Toxicol. 1987;2(1):10-32. (PubMed)
365. McDuffie JR, Calis KA, Booth SL, Uwaifo GI, Yanovski JA. Effects of orlistat on fat-soluble vitamins in obese adolescents. Pharmacotherapy. 2002;22(7):814-822. (PubMed)
366. Natural Medicines. Vitamin D. Professional handout/Drug interactions. Available at: https://naturalmedicines.therapeuticresearch.com. Accessed 6/11/17.
367. Panday K, Gona A, Humphrey MB. Medication-induced osteoporosis: screening and treatment strategies. Ther Adv Musculoskelet Dis. 2014;6(5):185-202. (PubMed)
368. Glass AR, Eil C. Ketoconazole-induced reduction in serum 1,25-dihydroxyvitamin D. J Clin Endocrinol Metab. 1986;63(3):766-769. (PubMed)
Vitamin E
Contents
Read the Nutrition Information Brief on vitamin E by Maret G. Traber, Ph.D.
Summary
- Naturally occurring vitamin E includes eight fat-soluble isoforms: α-, β-, γ-, and δ-tocopherol and α-, β-, γ-, and δ-tocotrienol. Yet, the body preferentially uses α-tocopherol, and only α-tocopherol supplementation can reverse vitamin E deficiency symptoms. (More information)
- α-Tocopherol functions as a chain-breaking antioxidant, preventing the propagation of free radicals in membranes and plasma lipoproteins. α-Tocopherol is also likely involved in strengthening certain aspects of cell-mediated immunity. (More information)
- Vitamin E deficiency can be caused by fat malabsorption disorders or by genetic abnormalities that affect vitamin E transport. Severe deficiency symptoms include vitamin E deficiency-induced ataxia, peripheral neuropathy, muscle weakness, and damage to the retina of the eye. (More information)
- The current recommended dietary allowance (RDA) is 15 mg/day of α-tocopherol. It is estimated that more than 90% of Americans adults do not meet the estimated average requirement (EAR) of 12 mg/day of α-tocopherol. (More information)
- Randomized controlled trials investigating primary and/or secondary prevention of chronic diseases, such as cardiovascular disease, cancer, and cataracts, do not currently support a preventative effect of supplemental α-tocopherol. (More information)
- Limited clinical evidence suggests that vitamin E supplementation may be beneficial for managing age-related macular degeneration and fatty liver diseases secondary to type 2 diabetes mellitus. (More information)
- Supplementation with α-tocopherol was found to slow cognitive decline or loss of functional abilities in cognitively impaired subjects in some, but not all, clinical studies. (More information)
- Plant seeds, especially sunflower seeds, almonds, and hazelnuts, are rich sources of α-tocopherol such that many vegetable oils (e.g., olive oil and canola oil) also contain α-tocopherol. Other sources include tomato, avocado, spinach, asparagus, Swiss chard, and broccoli. (More information)
- High doses of supplemental α-tocopherol may interfere with the vitamin K-dependent blood clotting cascade and increase the risk of bleeding in individuals taking anticoagulant drugs. A tolerable upper intake level (UL) for α-tocopherol in adults is set at 1,000 mg/day and applies to all possible stereoisomers of α-tocopherol. (More information)
The term vitamin E describes a family of eight fat-soluble molecules with antioxidant activities: four tocopherol isoforms (α-, β-, γ-, and δ-tocopherol) and four tocotrienol isoforms (α-, β-, γ-, and δ-tocotrienol) (Figure 1). Only one form, α-tocopherol, meets human vitamin E requirements (see The RDA). In the human liver, α-tocopherol is the form of vitamin E that is preferentially bound to α-tocopherol transfer protein (α-TTP) and incorporated into lipoproteins that transport α-tocopherol in the blood for delivery to extrahepatic tissues. Therefore, it is the predominant form of vitamin E found in the blood and tissues (1). In addition, α-tocopherol appears to be the form of vitamin E with the greatest nutritional significance, such that it will be the primary topic of the following discussion.
Function
α-Tocopherol
Natural versus synthetic α-tocopherol
Natural α-tocopherol made by plants found in food has an RRR-configuration at the 2, 4’, and 8’-position of the α-tocopherol molecule (wrongly referred to as d-α-tocopherol) (see Figure 1). Chemically synthesized all-rac-α-tocopherol (all-racemic-α-tocopherol; incorrectly labeled dl-α-tocopherol) is a mixture of eight stereoisomers of α-tocopherol, which arose from the three chiral carbons at the 2, 4’, and 8’-positions: RRR-, RSR-, RRS-, RSS-, SRR-, SSR-, SRS-, and SSS-α-tocopherol (see Figure 1). While all stereoisomers have equal in vitro antioxidant activity, only the forms in the R-conformation at position 2 (noted 2R) meet the vitamin E requirements in humans (2).
Antioxidant activity
The main function of α-tocopherol in humans is that of a fat-soluble antioxidant. Fats, which are an integral part of all cell membranes, are vulnerable to damage through lipid peroxidation by free radicals. α-Tocopherol is uniquely suited to intercept peroxyl radicals and thus prevent a chain reaction of lipid oxidation (Figure 2). When a molecule of α-tocopherol neutralizes a free radical, it is oxidized and its antioxidant capacity is lost. Other antioxidants, such as vitamin C, are capable of regenerating the antioxidant capacity of α-tocopherol (Figure 2) (reviewed in 1).
Aside from maintaining the integrity of cell membranes throughout the body, α-tocopherol protects the fats in low-density lipoproteins (LDLs) from oxidation. Lipoproteins are particles composed of lipids and proteins that transport fats through the bloodstream. LDLs specifically transport cholesterol from the liver to the tissues of the body. Oxidized LDLs have been implicated in the development of cardiovascular disease (3).

Effects on cell-mediated immunity
Other functions of α-tocopherol are likely to be related to its antioxidant capacity (1). For instance, α-tocopherol can protect the physiological properties of lipid bilayer membranes and may influence the activity of membrane proteins and enzymes (4). In cell culture studies, α-tocopherol was found to improve the formation of an adhesive junction (known as immune synapse) between naïve T lymphocytes and antigen-presenting cells (APC), which eventually prompted T cell activation and proliferation (see Disease Prevention) (5, 6).
γ-Tocopherol and tocotrienols
Vitamin E forms other than α-tocopherol are also known to be potent antioxidants. Tocotrienols and γ-tocopherol are thought to be better scavengers of peroxyl radicals and reactive nitrogen species, respectively, than α-tocopherol (7). Yet, in the body, (1) α-tocopherol is preferentially retained in the liver by the binding to α-tocopherol transfer protein (α-TTP), which incorporates α-tocopherol into lipoproteins for delivery to extrahepatic tissues; and (2) forms of vitamin E other than α-tocopherol are actively metabolized and excreted. Hence, while γ-tocopherol is the most common form of vitamin E in the American diet (8), its plasma and tissue concentrations are generally significantly lower than those of α-tocopherol, and more γ-tocopherol is excreted in urine than α-tocopherol, suggesting less γ-tocopherol is needed for use by the body (1).
Studies conducted in vitro and in animals have indicated that γ-tocopherol and its major metabolite, γ-carboxyethyl hydroxychroman (γ-CEHC), may play a role in protecting the body from free radical-induced damage in various conditions of oxidative stress and inflammation (reviewed in 7). Limited intervention studies (highlighted in 7) have not convincingly demonstrated a potential anti-inflammatory effect of γ-tocopherol in humans. Yet, in two recent randomized, placebo-controlled studies, the supplementation of smokers with γ-tocopherol potentiated short-term benefits of smoking cessation (with or without nicotine replacement therapy) on vascular endothelial function (9, 10).
Numerous preclinical studies have also suggested that tocotrienols might be beneficial in the prevention of chronic diseases (11). For instance, tocotrienols (especially δ-tocotrienol) have shown greater anti-proliferative and pro-apoptotic effects than tocopherols in malignant cell lines (12). However, a number of factors, including dose, formulation, and type of study population, affect the bioavailability of tocotrienols and may undermine their putative efficacy in humans (13). There are currently no data available on the effectiveness of supplemental tocotrienols in humans (11).
Nutrient interactions
Dietary and circulating fatty acids
The mechanism of vitamin E digestion and uptake into intestinal cells (enterocytes) is unclear but requires bile acids and pancreatic enzymes, and the packaging along with dietary fat into chylomicrons. The efficiency of vitamin E absorption increases with the amount of fat in ingested food, such that vitamin E absorption from supplements is likely to be minimal with low-fat meals (14, 15).
In the circulation, all lipoproteins (i.e., VLDLs, LDLs, and HDLs) are involved in the transport and tissue distribution of α-tocopherol (1). Increased concentrations of lipids (cholesterol and triglycerides) in the blood have been correlated to higher serum α-tocopherol concentrations. However, if a high blood concentration of lipids is associated with a slower turnover of lipoproteins, then the distribution of α-tocopherol to tissues may be substantially altered (16).
Vitamin C
A few human studies using conditions of oxidative stress have demonstrated the importance of vitamin C (ascorbic acid) in the recycling of oxidized α-tocopherol back to its reduced state (see Figure 2). Oxidative stress caused by cigarette smoking accelerates the depletion of plasma α-tocopherol in smokers compared to nonsmokers (17). In a double-blind, placebo-controlled trial in 11 smokers and 13 nonsmokers given α-tocopherol and γ-tocopherol that was labeled with deuterium (hence traceable), supplementation with vitamin C reduced the rate of vitamin E loss in plasma, most probably by regenerating tocopheryl radicals back to nonoxidized forms (18).
Vitamin K
One study in adults with normal coagulation (clotting) status found that daily supplementation with 1,000 IU (670 mg) of RRR-α-tocopherol for 12 weeks decreased γ-carboxylation of prothrombin, a vitamin K-dependent factor in the coagulation cascade (19). Individuals taking anticoagulant drugs like warfarin and those who are vitamin K deficient should not take vitamin E supplements without medical supervision because of the increased risk of bleeding (see Safety) (20).
Deficiency
Causes
Severe vitamin E deficiency rarely occurs in humans but has been observed as a result of malnutrition (21). Severe vitamin E deficiency has been associated with specific genetic defects affecting the transport of α-tocopherol by α-tocopherol transfer protein (α-TTP) and lipoproteins. Vitamin E deficiency has also been observed in individuals with fat malabsorption syndromes, which impair the absorption of dietary fats and therefore fat-soluble vitamins like vitamin E (see Nutrient interactions) (21).
Symptoms
Severe vitamin E deficiency results mainly in neurologic symptoms, including impaired balance and coordination (spinocerebellar ataxia), injury to the sensory nerves (peripheral neuropathy), muscle weakness (myopathy), and damage to the retina of the eye (retinopathy). For this reason, people who develop peripheral neuropathy, ataxia, or retinitis pigmentosa (RP) of unknown causes should be screened for vitamin E deficiency (21). The results of one randomized controlled trial in 601 patients with common forms of RP indicated that daily supplementation with 400 IU of all-rac-α-tocopherol (180 mg of RRR-α-tocopherol) modestly but significantly increased the loss of retinal function (22). In contrast, daily supplementation with 15,000 IU of vitamin A (4,500 μg RAE) significantly slowed the loss of retinal function over a period of four to six years, suggesting that patients with common forms of RP may benefit from long-term vitamin A supplementation but should avoid high-dose supplemental vitamin E.
Inherited defects in α-TTP are associated with a characteristic syndrome called AVED (Ataxia with Vitamin E Deficiency). A recent case study reported that visual impairment in a middle-age patient with AVED was caused by both RP and early-onset macular degeneration (23). Supplementation with high-dose vitamin E (800-1,200 mg/day) is used to prevent neurologic deterioration in AVED subjects (21).
Moreover, the developing nervous system appears to be especially vulnerable to vitamin E deficiency. For instance, children with severe vitamin E deficiency at birth rapidly experience irreversible neurologic symptoms if not treated with vitamin E. In contrast, individuals who develop gastrointestinal disorders affecting vitamin E absorption in adulthood may not develop neurologic symptoms for 10-20 years (21). It should also be noted that neurologic symptoms caused by vitamin E deficiency have not been reported in healthy individuals who consume diets low in vitamin E.
Marginal deficiency
Although frank vitamin E deficiency is rare, marginal intake of vitamin E is relatively common. Between 1988 and 1994, the US National Health and Nutrition Examination Survey III (NHANES III) examined the dietary intake and blood concentrations of α-tocopherol in 16,295 adults. The study reported that about one-third of all participants had blood concentrations of α-tocopherol below 20 micromoles/liter (μmol/L) — a cutoff value chosen because of its initial association with an increased risk for cardiovascular disease (24). More recent data from 18,063 participants in NHANES 2003-2006 indicated an average dietary intake of α-tocopherol from food (including enriched and fortified sources) among Americans adults of 7.2 mg/day (25). This intake is well below the current recommended dietary allowance of 15 mg/day (see the RDA). At this level of dietary intake, more than 93% of American adults do not meet the estimated average requirement (EAR) of 12 mg/day for vitamin E (25). In addition, a recent nested case-control study in Bangladeshi women suggested that inadequate vitamin E status during early pregnancy may be associated with an increased risk of miscarriage (26).
Cigarette smoking is thought to increase the utilization of α-tocopherol such that smokers might be at increased risk of deficiency compared with nonsmokers (17). Also, the 19-year follow-up analysis of the Alpha-Tocopherol, Beta-Carotene cancer (ATBC) trial in older, male smokers indicated that participants in the highest versus lowest quintile of serum α-tocopherol concentrations (>31 μmol/L vs. <23 μmol/L) at baseline had reduced risks of total and cause-specific mortality (27).
It is not known whether marginal vitamin E deficiency increases the risk of chronic disease (1).
The Recommended Dietary Allowance (RDA)
The RDA for vitamin E was last revised by the Food and Nutrition Board of the US Institute of Medicine in 2000 (Table 1) (2). The RDA is based largely on the results of studies done in the 1950s in men fed vitamin E-deficient diets. In a test-tube analysis, vitamin E suppresses the breakdown of red blood cells (known as hemolysis) induced by hydrogen peroxide. Because hemolysis has also been reported in children with severe vitamin E deficiency, the preventive effect of vitamin E against oxidative damage-induced hemolysis was considered to be a clinically relevant in vitro analysis to assess vitamin E status. Importantly, this means that the latest RDA for vitamin E continues to be based on the prevention of deficiency symptoms rather than on health promotion and prevention of chronic disease.
The forms of α-tocopherol that meet the recommended intakes are RRR-α-tocopherol — the only naturally occurring form of vitamin E — and the three synthetic isomers, RRS-, RSR-, and RSS-α-tocopherol, which are found in nutritional supplements and fortified food.
Table 1 lists the RDA for α-tocopherol expressed in both milligrams (mg) and international units (IU).
| Life Stage | Age | Males | Females | ||
|---|---|---|---|---|---|
| mg/day | IU/day | mg/day | IU/day | ||
| Infants (AI) | 0-6 months |
4
|
6
|
4
|
6
|
| Infants (AI) | 7-12 months |
5
|
7.5
|
5
|
7.5
|
| Children | 1-3 years |
6
|
9
|
6
|
9
|
| Children | 4-8 years |
7
|
10.5
|
7.5
|
10.5
|
| Children | 9-13 years |
11
|
16.5
|
11
|
16.5
|
| Adolescents | 14-18 years |
15
|
22.5
|
15
|
22.5
|
| Adults | 19 years and older |
15
|
22.5
|
15
|
22.5
|
| Pregnancy | all ages |
-
|
-
|
15
|
22.5
|
| Breast-feeding | all ages |
-
|
-
|
19
|
28.5
|
|
*These recommended intakes are limited to 2R-stereoisomeric forms of α-tocopherol. |
|||||
Disease Prevention
Age-related deterioration of immune function
The natural age-related decline of the immune function is accompanied by an increased susceptibility to infections, a poorer response to immunization, and higher risks of developing cancers and autoimmune diseases. α-Tocopherol has been shown to enhance specifically the T cell-mediated immune response that declines with advancing age (reviewed in 28). T cell impaired response has been partly associated with a reduced capacity of naïve T cells to be activated during antigen presentation, and to produce interleukin-2 (IL-2) and proliferate as a result (6). However, very few studies have addressed the potential association between α-tocopherol and immune function in humans (28). In a small intervention study in older adults (mean age, 70 years), supplementation with 200 mg/day of all-rac-α-tocopherol (equivalent to 100 mg of RRR-α-tocopherol) for three months significantly improved natural killer (NK) cytotoxic activity, neutrophil chemotaxis, phagocytic response, and enhanced mitogen-induced lymphocyte proliferation and interleukin-2 (IL-2) production compared to baseline (29). In an earlier trial, daily supplementation of healthy older adults (≥65 years of age) with 200 mg of all-rac-α-tocopherol for 235 days also improved T lymphocyte-mediated immunity — as measured with the delayed-type hypersensitivity (DTH) skin test — and increased the production of antibodies in response to hepatitis B and tetanus vaccines (30).
Lower α-tocopherol doses failed to improve the DTH response compared to a placebo in another study in healthy participants (ages, 65-80 years) (31). A randomized, placebo-controlled trial in 617 nursing home residents (≥65 years of age) reported that daily supplementation with 200 IU of synthetic α-tocopherol (90 mg of RRR-α-tocopherol) for one year significantly lowered the risk of contracting upper respiratory tract infections, especially the common cold, but had no effect on lower respiratory tract (lung) infections (32). More research is needed to examine whether supplemental vitamin E might enhance immune function and reduce risk of infection in older adults.
Cardiovascular disease
Primary prevention: in healthy adults
Observational studies: Results of several large observational studies in both men and women have suggested an inverse relationship between vitamin E consumption and risk of myocardial infarction or death from heart disease. Each study had a prospective design that measured vitamin E intake in generally healthy people who were followed over a period of time to determine the onset of cardiovascular events and analyze the association between the exposure and the outcome(s). In two of the studies, individuals who consumed more than 7 mg/day of dietary α-tocopherol were 35% less likely to die from heart disease than those who consumed less than 3-5 mg/day of α-tocopherol (33, 34). Two other large studies observed a significantly reduced risk of heart disease only in women and men who consumed at least 100 IU (67 mg)/day of supplemental RRR-α-tocopherol (35, 36).
Intervention studies: A randomized, placebo-controlled, intervention trial in 39,876 women (aged ≥45 years) participating in the Women's Health Study (WHS) found that supplementation with 600 IU (400 mg) of RRR-α-tocopherol every other day for 10 years resulted in a 34% reduction in nonfatal myocardial infarction and a 49% reduction in cardiovascular-related deaths but only in women aged at least 65 years at baseline (representing 10% of study participants) (37). Further analyses of WHS data showed that women in the vitamin E arm of the study experienced a 21% reduction in risk of venous thromboembolism (VTE) compared to placebo: the reduction was of 12% in women younger than 55 years old, 26% in women aged 65 years and older, and 44% in women with a history of VTE (38, 39). Another large randomized controlled trial — the Physicians’ Health Study II (PHSII) — conducted in healthy middle-aged men found no significant effect of 400 IU of synthetic α-tocopherol (180 mg of RRR-α-tocopherol), given every other day for eight years, on the risk of major cardiovascular events in the entire cohort and in subgroup analyses (40). Besides, concerns were raised regarding a possible harmful effect of high-dose vitamin E supplementation on the risk of hemorrhagic stroke in this cohort (40).
Secondary prevention: in individuals with or at risk of cardiovascular disease
Conventional risk factors for cardiovascular disease (CVD) include cigarette smoking, physical inactivity, hypertension, dyslipidemia, and being overweight or obese. Other factors such as oxidative stress and inflammation are also thought to contribute to increasing CVD risk, especially in patients with chronic conditions like type 2 diabetes mellitus and chronic kidney disease. Although trials do not appear to support any cardiovascular benefit in healthy middle-aged and older subjects, vitamin E supplementation might help improve cardiovascular health and/or lower the risk of CVD in specific, higher risk subjects.
Observational studies: The presence of atherosclerotic plaques in arterial walls is one of the hallmarks of cardiovascular disease. Plaque rupture that causes blood clot formation is the usual cause of myocardial and cerebrovascular infarctions. The cross-sectional Asymptomatic Carotid Atherosclerosis Disease In Manfredonia (ACADIM) study in 640 at-risk individuals reported an inverse association between carotid intima-media thickness (CIMT) — a marker of atherosclerosis — and circulating concentrations of antioxidants, including vitamin E (41). However, other observational studies found no association between plasma vitamin E concentrations and CIMT (reviewed in 42).
Intervention studies: A small randomized controlled study assessing the effect of lipid-lowering drugs in men who had previously undergone a coronary artery bypass surgery found that the use of at least 100 IU/day (compared to less than 100 IU/day) of supplemental α-tocopherol (45 mg of RRR-α-tocopherol) was associated with reduced CIMT progression over a two-year period but only among participants in the placebo arm of the study (i.e., those who did not receive lipid-lowering drugs) (43). However, a recent meta-analysis of seven small placebo-controlled trials found little evidence that vitamin E supplementation may improve flow-mediated vascular dilation (FMD) of the brachial artery, a marker of vascular endothelial health that is adversely affected by CVD risk factors (44). In the Cambridge Heart AntiOxidant Study (CHAOS), a randomized, placebo-controlled intervention trial in 2,002 patients with coronary heart disease, daily supplementation with either 400 IU or 800 IU of synthetic α-tocopherol (180 mg or 360 mg of RRR-α-tocopherol) for a median 18 months dramatically reduced the occurrence of nonfatal myocardial infarctions by 77%. However, vitamin E supplementation did not significantly reduce total or cardiovascular-related deaths (45).
Another small trial in patients with renal failure — the Secondary Prevention with Antioxidants of cardiovascular disease in End-stage renal disease (SPACE) — found that supplementation with 800 IU (536 mg)/day of RRR-α-tocopherol for an average of 1.4 years significantly reduced the risk of myocardial infarction compared to placebo (46). A more recent randomized controlled study suggested that vitamin E supplementation may benefit a subgroup of patients with type 2 diabetes mellitus. The multicenter study by Milman et al. (47) was conducted in 1,434 type 2 diabetics (≥55 years old) carrying a specific variant of the haptoglobin protein (Hp), Hp2-2, which has a lower efficacy to bind and remove pro-oxidant, free hemoglobin from plasma, compared to Hp1-1 and Hp1-2 variants. The daily supplementation with 400 IU (268 mg) of RRR-α-tocopherol for 18 months resulted in a lower risk of myocardial infarction compared to placebo (47).
Other larger intervention trials conducted in cigarette smokers (the Alpha-Tocopherol, Beta-Carotene cancer prevention [ATBC] study (48)), in individuals at-risk of CVD (the Heart Outcomes Prevention Evaluation [HOPE]-The Ongoing Outcomes [HOPE-TOO study] (49)), or in patients who have suffered a myocardial infarction (Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico-GISSI-prevenzione trial (50)) failed to find significant CVD risk reductions with α-tocopherol supplementation. Besides, potentially harmful effects of supplemental vitamin E were reported on the risk of hemorrhagic stroke in the ATBC trial and on the risk of heart failure in the HOPE and GISSI trials (see Safety) (48-50).
Cancer
Oxidative damage to DNA by free radicals can lead to mutations that may contribute to causing cancer (51). Because of its ability to neutralize free radicals, vitamin E has been suggested to possess anticancer activity by protecting cells against oxidative damage. Yet, several large prospective cohort studies have failed to find significant associations between vitamin E intake and the incidence of lung or breast cancer (2). More recently, the VITamins And Lifestyle (VITAL) study prospectively assessed the association between long-term use of supplemental vitamins (10-year intake) and the risk of lung cancer in a cohort of 77,126 men and women (52). No relationships were reported between intake of multivitamins, vitamin C, vitamin E, or folate and the risk of lung cancer. However, the use of supplemental vitamin E in current but not in former smokers was associated with an 11% increased risk of lung cancer for every 100 mg/day increase, and intakes greater than 215 mg/day were specifically linked to a 29% increase in risk for non-small cell lung cancer (52).
To date, most clinical trials have failed to find any beneficial effects of vitamin E supplementation on the risk of various cancers. A randomized, placebo-controlled trial (RCT) in 39,876 women participating in the Women's Health Study found that supplementation with 600 IU (400 mg) of RRR-α-tocopherol every other day for 10 years had no effect on overall cancer incidence, tissue-specific cancer incidence, or cancer-related deaths (37). Yet, the results of a few large randomized controlled trials have suggested that vitamin E supplementation might affect the risk of prostate cancer. The Alpha-Tocopherol, Beta-Carotene cancer (ATBC) prevention study was a four-arm, randomized, double-blind, placebo-controlled trial designed to investigate the effect of α-tocopherol supplementation on lung cancer development in 29,133 male smokers. The study found a 32% reduction in the incidence of prostate cancer in participants given daily supplements of 50 mg of synthetic α-tocopherol (equivalent to 25 mg of RRR-α-tocopherol) alone or in combination with β-carotene compared to those given β-carotene alone or a placebo (53). However, no differences in the incidence of prostate cancer were found between α-tocopherol recipients and nonrecipients during the 18-year post-intervention period (54). In the Physicians’ Health Study II (PHS II), which followed 14,641 healthy men aged 50 years and older, supplementation with 400 IU of synthetic vitamin E (equivalent to 180 mg of RRR-α-tocopherol) every other day for eight years had no effect on the risk of prostate cancer, other site-specific cancers, or total cancer (55). The supplementation of vitamin E (equivalent to 180 mg/day of RRR-α-tocopherol), alone or in combination with selenium, in the multicenter, randomized, placebo-controlled SELECT trial (SELenium and vitamin E Cancer prevention Trial) was halted because there was no evidence of benefit in preventing prostate cancer in 35,533 healthy men aged 50 years and older (56). After a median of seven years’ follow-up, the risk of prostate cancer was found to be significantly increased by 17% in participants supplemented with vitamin E alone during the trial period — but not when vitamin E was combined with selenium — compared to placebo (57). A study of cases versus subcohort individuals drawn from the SELECT study assessed the effect of vitamin E and/or selenium supplementation on prostate cancer risk in relation to the selenium status of participants at baseline (58). Supplemental selenium with or without vitamin E was associated with a significant increase in the risk of advanced prostate cancer in individuals with higher versus lower selenium status. In addition, the risks of total and advanced prostate cancer were significantly elevated with vitamin E supplementation in subjects with low versus high selenium status (58). Recent investigations have suggested that sequence variations (polymorphisms) in vitamin E-related genes and genes coding for antioxidant enzymes, including selenoproteins, might modify the impact of high-dose vitamin E and selenium on the risk of prostate cancer (59-61).
Cataracts
Age-related cataracts appear to be the result of protein oxidation in the lens of the eye; antioxidants like α-tocopherol may protect the lens against oxidative damage from reactive oxygen species. In a recent cross-sectional study, vitamin E concentrations were found to be significantly lower in the lens and blood of subjects with age-related nuclear, but not cortical, cataracts when compared with an age-matched control group (62). However, earlier studies reported higher vitamin E concentrations in the lenses and blood of patients with cataracts (63, 64).
The results of several observational studies that examined the association between vitamin E consumption and the incidence or severity of cataracts are also mixed. Some reported that increased vitamin E intake protected against cataract development, while others found no association (65). Yet, a meta-analysis of eight studies, including 15,021 participants, found a 17% reduction in the risk of age-related cataract in subjects in the highest versus lowest quantile of dietary vitamin E intake (66). A recent prospective cohort study of 31,120 Swedish men followed for a mean of 8.4 years observed a greater risk of developing cataract in occasional and regular users of high-dose (about 100 mg/day) vitamin E supplements only, when compared with non-supplement users (67). However, the use of supplemental high-dose vitamin E with additional supplements or the use of low-dose vitamin E-containing multivitamin supplements was not found to be associated with an elevated cataract risk. A meta-analysis based on data from over 350,000 participants in 10 studies — including the above-cited study by Zheng Selin et al. (67) — found no association between supplemental vitamin E and risk of cataract (66).
In clinical settings, the supplementation of high-dose vitamin E — alone or in addition to other supplements — was found to be safe, yet the benefits regarding cataract risk or progression were limited. An early intervention trial found that a daily supplement of 50 mg of synthetic α-tocopherol (equivalent to 25 mg of RRR-α-tocopherol) did not alter the incidence of cataract surgery in male smokers (68). A randomized, placebo-controlled intervention trial in 4,629 men and women found that a daily antioxidant supplement containing 500 mg of vitamin C, 400 IU of all-rac-α-tocopheryl acetate (equivalent to 180 mg of RRR-α-tocopherol), and 15 mg of β-carotene did not affect development and progression of age-related cataracts over a seven-year period (69). Similarly, a four-armed, randomized, placebo-controlled study of 11,267 men from the Selenium and Vitamin E Cancer prevention trial (SELECT) failed to observe a reduction in cataract incidence with 400 IU/day of supplemental all-rac-α-tocopheryl acetate (180 mg/day of RRR-α-tocopherol), alone or in combination with selenium (200 μg/day), during a mean 5.6 years of follow-up (70). Daily antioxidant supplementation with 500 mg of vitamin C, 400 IU (268 mg) of RRR-α-tocopherol, and 15 mg of β-carotene did not limit the progression of cataract in a five-year intervention trial (71). Another four-year randomized, placebo-controlled trial reported that supplements containing 500 IU/day (335 mg/day) of RRR-α-tocopherol did not reduce the incidence or progression of cataract in older adults (72). Current available data from clinical trials do not support a preventative effect of vitamin E on cataracts.
Disease Treatment
Age-related macular degeneration
A recent pooled analysis of four randomized controlled trials in 62,520 subjects found that supplemental vitamin E or β-carotene did not reduce the risk of developing age-related macular degeneration (AMD), a multifactorial disease affecting the central area of the retina (73). However, a review of currently available data suggested that supplements of antioxidants plus zinc may reduce the progression of AMD and vision loss in affected individuals (74). The main evidence came from the Age-Related Eye Disease Study (AREDS). In this clinical trial, participants with borderline to advanced age-related macular degeneration (AMD) were randomized to receive (1) placebo; (2) antioxidant vitamins (15 mg/day of β-carotene, 500 mg/day of ascorbic acid, and 400 IU/day of all-rac-α-tocopheryl acetate); (3) zinc (80 mg/day) and copper (2 mg/day); or (4) both antioxidant vitamins and zinc and copper (75). The five-year results indicated that the risk of developing advanced AMD was significantly reduced in those taking zinc with or without antioxidant vitamins. Antioxidant vitamins alone failed to prevent the progression to advanced AMD, even in individuals at higher risk. It was concluded from this study that a combination of antioxidant vitamins and minerals may benefit people with intermediate AMD or advanced AMD in one eye (74, 76).
Type 2 diabetes mellitus
Oxidative stress contributes to the progression of type 2 diabetes mellitus and causes damage to many organs and tissues, including the pancreas, brain, eyes, peripheral nerves, and kidneys. Evidence from animal studies suggests that vitamin E supplementation could mitigate the role of oxidative damage in the occurrence of diabetes complications (reviewed in 77). In the Alpha-Tocopherol Beta-Carotene cancer prevention (ATBC) trial in male smokers, supplementation with 50 mg/day of synthetic α-tocopherol (25 mg/day of RRR-α-tocopherol) had no effect on the risk of incident type 2 diabetes mellitus during the 19-year post-intervention follow-up. Likewise, supplemental vitamin E intake during the trial made no difference on the incidence of macrovascular complications or mortality in participants with established type 2 diabetes (78). In addition, a meta-analysis of 14 heterogeneous randomized controlled trials, including 714 type 2 diabetic individuals, found that supplementation with vitamin E (200-1,800 IU/day for 6-27 weeks) had no effect on markers of glycemic control, including glycated hemoglobin A1c (HbA1c) level and measures of fasting glucose and fasting insulin concentrations (79). Further subgroup analyses indicated that higher doses of vitamin E (>400 IU/day) supplemented for longer periods (>12 weeks) significantly reduced HbA1c level and fasting insulin concentration, suggesting that vitamin E could possibly enhance insulin action and glucose disposal in type 2 diabetic individuals (79). Another recent meta-analysis of randomized controlled trials found that endothelial function in normal-weight and overweight — but not obese — patients with type 2 diabetes was significantly improved by supplementation with vitamin E and/or vitamin C (80). Although there is reason to suspect that vitamin E supplementation may have utility in the management of type 2 diabetes, evidence for benefit from large, well-controlled clinical trials is still lacking.
Fatty liver diseases
The increasing incidence of nonalcoholic fatty liver disease (NAFLD) in children and adults in industrialized countries is mainly attributed to the ongoing epidemic of obesity and type 2 diabetes mellitus. NAFLD results from the abnormal accumulation of fat (steatosis) in the liver in the absence of heavy alcohol consumption. Although the condition is considered to be largely benign, NAFLD can progress to a more severe disease called nonalcoholic steatohepatitis (NASH) with increased risks of cirrhosis, hepatocellular carcinoma (liver cancer), and cardiovascular disease (81). Oxidative stress is thought to be one of the possible mechanisms responsible for prompting inflammatory processes that can lead to the progression of NAFLD to NASH.
There is currently no established treatment for NAFLD and NASH other than interventions that encourage lifestyle changes and the use of medicines to control or treat metabolic disorders (82). In the multicenter PIVENS (PIoglitazone versus Vitamin E versus placebo for the treatment of Nonalcoholic Steatohepatitis) trial, 247 nondiabetic subjects with NASH were randomized to receive 30 mg/day of pioglitazone (an insulin-sensitizing drug), 800 IU/day (536 mg/day) of RRR-α-tocopherol, or a placebo for 96 weeks (83). Only vitamin E supplementation significantly increased the overall rate of improvement in histological abnormalities that characterize NASH on liver biopsies (i.e., hepatocellular ballooning, steatosis, and lobular inflammation) (84). Both active treatments improved some markers of liver function (i.e., alanine aminotransferase and aspartate aminotransferase) (84). Yet, results from another two-year, randomized controlled trial — called TONIC for Treatment Of Nonalcoholic fatty liver disease In Children — in 173 children (ages, 8-17 years) with NAFLD failed to observe any significant reduction in blood concentrations of alanine and aspartate aminotransferases either with supplemental vitamin E (536 mg/day of RRR-α-tocopherol) or with metformin (an anti-diabetic drug; 1,000 mg/day) compared to placebo (85). However, vitamin E supplementation significantly improved the overall disease activity score — used to quantify the severity of the disease. In addition, a recent meta-analysis of another six trials found that vitamin E significantly lowered circulating aminotransferase concentrations in NAFLD and NASH patients, suggesting liver function improvements (86). Finally, in a small nonrandomized, unblinded, controlled study in 42 obese children (mean age, 8 years) with NAFLD, lifestyle recommendations combined with 600 mg/day of supplemental RRR-α-tocopheryl acetate for six months reduced markers of oxidative stress and liver dysfunction and improved insulin sensitivity and the profile of lipid in the blood, when compared to baseline. No such changes in markers of oxidative stress, liver function, and glucose utilization were reported in the lifestyle intervention only group (87). Further randomized and well-controlled studies are needed to confirm these preliminary findings.
Cognitive deterioration and Alzheimer's disease
Mitochondrial dysfunction and oxidative stress are thought to contribute to the onset and/or progression of several neurodegenerative diseases, especially Alzheimer's disease (AD) (88). The progressive degeneration of neuronal cells that accompanies the decline of memory and other cognitive functions in subjects with Alzheimer’s disease is associated with an intracellular aggregation of Tau fibrils, an extraneuronal accumulation of β-amyloid peptides into senile plaques, and an oxidation-reduction (redox) imbalance of complex etiology. In the brain of patients with mild cognitive impairment (MCI) and those with AD, the level of markers of oxidative damage to DNA, proteins, and lipids is increased, while the expression and activities of glutathione and antioxidant enzymes are reduced (reviewed in 88). In addition, a recent meta-analysis reported that circulating concentrations of vitamins, including vitamin A, vitamin C, and vitamin E, were significantly lower in AD patients than in cognitively healthy individuals (89). Other studies have documented low concentrations of vitamin E in cerebrospinal fluid of cognitively impaired patients (reviewed in 90).
Because a reduction in oxidative stress may help maintain cognitive status and/or prevent deterioration, the effects of vitamin E supplementation have been assessed in a few intervention studies. An early multicenter, randomized, placebo-controlled study in individuals with AD of moderate severity found that supplementation with 2,000 IU/day of all-rac-α-tocopherol (equivalent to 900 mg/day of RRR-α-tocopherol) for two years significantly delayed cognitive decline, slowed disease progression, and increased median survival (91). However, a placebo-controlled trial in 769 patients with MCI found that the same dosage of vitamin E did not affect the probability of progression from MCI to AD over a three-year period (92). In another double-blind, placebo-controlled trial, an improvement in cognitive performance — measured by the Mini Mental State Examination (MMSE) scoring system — was reported in AD patients randomized to receive 800 IU/day of all-rac-α-tocopherol (360 mg/day of RRR-α-tocopherol) for six months only when the treatment effectively reduced oxidative stress (as assessed by the measure of total glutathione and markers of lipid peroxidation in the blood) (93). Conversely, a failure to reduce oxidative stress resulted in supplemental vitamin E being more detrimental to the cognitive function of AD patients than placebo. In the most recent multicenter, randomized, double-blind, placebo-controlled study, supplemental vitamin E (2,000 IU/day; form of vitamin E not mentioned in the publication) for over two years significantly delayed functional decline — determined by the (in)ability to perform basic activities of daily living — and reduced the annual mortality rate in mild and moderate AD patients (94). Yet, vitamin E failed to affect cognitive performance measured with MMSE scores and other cognitive ability tests.
While there is currently little evidence to suggest that long-term supplementation of vitamin E provides any cognitive benefits in healthy older adults (95), additional research needs to confirm whether vitamin E supplementation could benefit the management of patients with mild-to-moderate cognitive impairments.
Sources
Food sources
Major sources of α-tocopherol in the American diet include vegetable oils (olive, sunflower, and safflower oils), nuts, whole grains, and green leafy vegetables. All eight forms of vitamin E (α-, β-, γ-, and δ-tocopherols and α-, β-, γ-, and δ-tocotrienols) occur naturally in mostly plant-based foods but in varying amounts. Table 2 lists the content of α-tocopherol and γ-tocopherol (in milligrams) in some rich sources of vitamin E. For more information on the vitamin E content of foods, search USDA's FoodData Central.
In the US, the average intake of α-tocopherol from food (including enriched and fortified sources) for adults (≥19 years of age) is 7.2 mg/day (25); this level is well below the RDA of 15 mg/day of α-tocopherol (see Table 1). While it appears feasible for individuals to meet the RDA from food only, Americans would have to depart from their current dietary practices and include greater intakes of nuts, seeds, fruit, and vegetables without increasing fat intake above recommended levels (96).
Supplements
RRR-α-tocopherol is the only stereoisomeric form of α-tocopherol found in unfortified foods. The same is not always true for nutritional supplements. Vitamin E supplements generally contain 100 IU to 1,000 IU of α-tocopherol. Supplements made from entirely natural sources contain only RRR-α-tocopherol (wrongly labeled d-α-tocopherol). RRR-α-tocopherol is the most bioavailable form of α-tocopherol in the body. Synthetic α-tocopherol, which is often found in fortified food and nutritional supplements and usually labeled all-rac-α-tocopherol or dl-α-tocopherol, include all eight possible stereoisomers of α-tocopherol (see Function). Because half of the isomers present as a mixture in synthetic α-tocopherol are not usable by the body, synthetic α-tocopherol is less bioavailable than natural α-tocopherol (see Figure 1). To calculate the amount (in milligrams) of α-tocopherol bioavailable in a supplement, the conversion factors are as follows:
-
Natural vitamin E (RRR-α-tocopherol) containing supplements:
IU of RRR-α-tocopherol x 0.67 = mg of RRR-α-tocopherol
Example: 100 IU of natural vitamin E provides 67 mg of RRR-α-tocopherol
-
Synthetic vitamin E (all-rac-α-tocopherol) containing supplements:
IU of all-rac-α-tocopherol x 0.45 = mg of RRR-α-tocopherol
Example: 100 IU of synthetic vitamin E provides 45 mg of 2R-α-tocopherol
In addition, vitamin E-fortified foods often contain synthetic α-tocopherol, and amounts of vitamin E are provided as a percentage of the daily value (DV) of 30 IU (approximately 20 mg of RRR-α-tocopherol).
α-Tocopheryl esters
α-Tocopheryl succinate and α-tocopheryl acetate are the main esterified forms of vitamin E in nutritional supplements. Tocopherol esters are more resistant to oxidation during storage than unesterified tocopherols (1). When taken orally, the succinate or acetate moieties are removed from α-tocopherol in the intestine. The bioavailability of α-tocopherol from α-tocopheryl succinate and α-tocopheryl acetate is equivalent to that of free α-tocopherol (97). Hence, the conversion factors used to determine the amount of bioavailable α-tocopherol provided by α-tocopheryl succinate and α-tocopheryl acetate are the same as those used for α-tocopherol (see above) (2). Cell culture studies indicated that the vitamin E ester, α-tocopheryl succinate, could inhibit proliferation and induce apoptosis in a number of cancer cell lines (12). Limited data from animal models of cancer found that α-tocopheryl succinate administered by injection inhibited tumor growth (98). There is currently no evidence in humans that taking oral α-tocopheryl succinate supplements delivers α-tocopheryl succinate to tissues. Of note, current research investigates nanomedicines to increase α-tocopheryl succinate bioavailability before exploring putative benefits in clinical settings (98).
α-Tocopheryl nicotinate is another α-tocopherol ester formed from synthetic α-tocopherol and nicotinic acid (niacin). While α-tocopheryl nicotinate can be prescribed as a lipid-lowering agent in Europe and Japan, it is marketed as a supplement only in the US (99).
α-Tocopheryl phosphates (Ester-E®)
There is currently no published evidence that supplements containing α-tocopheryl phosphates are more efficiently absorbed or have greater bioavailability in humans than those containing α-tocopherol (99).
Other supplemental forms
Supplements containing γ-tocopherol, mixed tocopherols, or tocotrienols are also commercially available (99). The amounts of α- and γ-tocopherol in mixed tocopherol supplements vary, so it is important to read the label to determine the amount of each tocopherol form present in a capsule.
Safety
Toxicity
Few side effects have been noted in adults taking supplements of less than 2,000 mg of α-tocopherol daily (either natural or synthetic vitamin E). However, most studies assessing safety issues or toxicity of α-tocopherol supplementation have lasted only a few weeks to a few months, and side effects associated with long-term α-tocopherol supplementation have not been adequately studied. The most worrisome possibility is that of impaired blood clotting, which increases the likelihood of hemorrhage in some individuals. A meta-analysis of randomized controlled trials found that daily vitamin E supplementation — equivalent to 25 to 536 mg/day of RRR-α-tocopherol — for several years resulted in a significant, 10% reduction in the risk of ischemic stroke (five trials, 91,393 participants) and a nonsignificant trend towards an increased risk of hemorrhagic stroke (five trials, 100,748 participants) (100).
A tolerable upper intake level (UL) for any form of supplemental α-tocopherol (all possible stereoisomers) has been established by the Food and Nutrition Board of the Institute of Medicine to avoid the potential risk of bleeding (Table 3). Specifically, the UL of 1,000 mg/day of α-tocopherol in any supplemental form (equivalent to 1,500 IU/day of RRR-α-tocopherol or 1,100 IU/day of all-rac-α-tocopherol) corresponds to the highest dose unlikely to result in hemorrhage in almost all adults (2). Although only certain isomers of α-tocopherol are retained in the circulation, all forms are absorbed and metabolized by the liver. Hence, the rationale for a UL that refers to all stereoisomers of α-tocopherol is based on the fact that any form of α-tocopherol (natural or synthetic) can be absorbed and thus be potentially harmful.
| Age Group | mg/day# |
|---|---|
| Infants 0-12 months | Not possible to establish## |
| Children 1-3 years | 200 |
| Children 4-8 years | 300 |
| Children 9-13 years | 600 |
| Adolescents 14-18 years | 800 |
| Adults 19 years and older | 1,000 |
|
*The UL for α-tocopherol applies to all stereoisomers of α-tocopherol (natural and synthetic) found in supplements and fortified food. |
|
Some physicians recommend discontinuing high-dose vitamin E supplementation two to four weeks before elective surgery — including dental procedures — to decrease the risk of hemorrhage (99).
Because dietary vitamin E is essential to prevent vitamin E deficiency in the newborn, vitamin E must be supplied in parenteral nutrition solutions in infants who cannot be given enteral feeding, such as prematurely born infants. Yet, preterm infants appear to be especially vulnerable to adverse effects of α-tocopherol supplementation, and supplemental vitamin E should be administered only under controlled supervision by a pediatrician (101).
Finally, the results of only one randomized controlled trial in 601 patients with common forms of retinitis pigmentosa (RP) indicated that supplementation with 400 IU/day of synthetic vitamin E (equivalent to 180 mg/day of RRR-α-tocopherol) modestly but significantly accelerated the loss of retinal function compared to placebo (22). Patients with common forms of RP should therefore avoid taking high-dose vitamin E supplements if they are not deficient in vitamin E (see Deficiency).
Does vitamin E supplementation increase the risk of all-cause mortality?
A prospective observational study in over 4,000 participants of the Framingham Heart Study and the Framingham Offspring Study — with or without preexisting cardiovascular disease — found no statistically significant association between vitamin E supplement intake and cardiovascular or all-cause mortality after a 10-year follow-up period (102). However, in addition to reports of increased risk of hemorrhage and heart failure with supplemental vitamin E in several randomized controlled studies (see Cardiovascular disease), a meta-analysis by Miller et al. (103) suggested an increased risk of death with the use of large doses of vitamin E, yet lower than the UL. Specifically, this meta-analysis combined the results of 19 clinical trials of vitamin E supplementation that mostly focused on secondary prevention and, as such, included subjects with pre-existing conditions including heart disease, end-stage renal failure, and Alzheimer's disease. The study found that daily supplementation with at least 400 IU of synthetic vitamin E (equivalent to 180 mg of RRR-α-tocopherol) resulted in a 4% increase in risk of death from any cause compared to placebo (103). However, further dose-response analysis and adjustment for intake of other vitamin and mineral supplements indicated that all-cause mortality risk was significantly increased by 7% only at a dose of 2,000 IU/day, which is notably higher than the UL for adults (1,100 IU/day of synthetic tocopherol or 1,500 IU/day of natural tocopherol; see Table 3). Additionally, a more recent meta-analysis of 46 randomized trials, including 171,244 participants, found that supplemental vitamin E, singly or in combination with other antioxidants, did not significantly alter the risk of all-cause mortality (104). At present, there is no convincing evidence that vitamin E supplementation below the UL increases the risk of death from cardiovascular disease or other causes, especially in generally healthy subjects. Yet, individuals with pre-existing conditions may be at increased risk of serious adverse effects (including death) if one considers the possibility that large doses of supplemental vitamin E might interfere with medications, and possibly lower their efficacy or increase their toxicity (1).
Nutrient interactions
Large doses of vitamin E may inhibit vitamin K-dependent carboxylase activity and interfere with the coagulation cascade (see the article on Vitamin K) (19). Hence, the use of vitamin E supplements may increase the risk of bleeding in individuals taking anticoagulant drugs (blood thinners), such as heparin and the vitamin K antagonist, warfarin (Coumadin); antiplatelet drugs, such as clopidogrel (Plavix), ticlopidine (Ticlid), tirofiban (Aggrastat), and dipyridamole (Aggrenox); and non-steroidal anti-inflammatory drugs (NSAIDs), including aspirin, ibuprofen, and others. In addition, individuals who may be vitamin K deficient due to liver failure, those with a propensity to bleed (e.g., bleeding peptic ulcers), and those with inherited bleeding disorders (e.g., hemophilia) or a history of hemorrhagic stroke, should not take α-tocopherol supplements without close medical supervision because of the increased risk of hemorrhage (20, 99). Finally, it cannot be excluded that vitamin E would potentiate the antithrombotic activity of supplemental fish oils and herbal products, such as garlic, curcumin, or Ginkgo biloba (99).
Drug interactions
A number of cholesterol-lowering medications (like cholestyramine and colestipol), as well as orlistat, sucralfate, mineral oil, and the fat substitute, olestra, which interfere with fat absorption, may theoretically decrease the absorption of fat-soluble vitamins, including vitamin E. The anticonvulsant drugs phenobarbital, phenytoin (Dilantin), and carbamazepine (Tegretol), may also lower plasma vitamin E concentrations in individuals with epilepsy (105).
Antioxidants and statins (3-hydroxy-3-methylglutary-coenzyme A reductase inhibitors)
A three-year randomized controlled trial in 160 patients with coronary heart disease (CHD) and low high-density-lipoprotein (HDL) levels found that a combination of simvastatin (Zocor) and niacin increased the HDL2 subfraction level (considered the most cardioprotective), inhibited the progression of coronary artery stenosis (narrowing), and decreased the frequency of cardiovascular events, such as myocardial infarction and stroke (106). Surprisingly, when an antioxidant combination of 1,000 mg of vitamin C, 800 IU (536 mg) of RRR-α-tocopherol, 100 μg of selenium, and 25 mg of β-carotene daily, was taken with the simvastatin-niacin combination, the protective effects were diminished. However, in a much larger randomized controlled trial of simvastatin and an antioxidant combination of 600 mg of all-rac-α-tocopherol (297 mg of RRR-α-tocopherol), 250 mg of vitamin C, and 20 mg of β-carotene daily in more than 20,000 men and women with CHD or diabetes, the antioxidant combination did not adversely affect the cardioprotective effects of simvastatin therapy over a five-year period (107). These contradictory findings indicate that further research is needed on potential interactions between antioxidant supplementation and cholesterol-lowering agents like statins.
Linus Pauling Institute Recommendation
The Recommended Dietary Allowance (RDA) for vitamin E for adult men and women is 15 mg (22.5 IU) per day. Notably, more than 90% of individuals two years of age and older in the US do not meet the daily requirement for vitamin E from food sources alone. Therefore, LPI recommends that generally healthy adults (aged 19 years and older) take a daily multivitamin/mineral (MVM) supplement, which usually contains 30 IU of synthetic vitamin E — equivalent to 13.5 mg of RRR-α-tocopherol and 90% of the RDA.
Older adults (>50 years)
The Linus Pauling Institute’s recommendation to take a daily multivitamin/mineral (MVM) supplement containing vitamin E is also appropriate for generally healthy older adults. MVMs typically contain 30 IU of synthetic vitamin E, covering 90% of the RDA.
Authors and Reviewers
Originally written in 2000 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in November 2004 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in June 2008 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in May 2015 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in October 2015 by:
Maret G. Traber, Ph.D.
Helen P. Rumbel Professor for Micronutrient Research, Linus Pauling Institute
Professor, College of Public Health and Human Sciences
Oregon State University
Copyright 2000-2024 Linus Pauling Institute
References
1. Traber MG. Vitamin E. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Washington, D.C.: Wiley-Blackwell; 2012:214-229.
2. Food and Nutrition Board, Institute of Medicine. Vitamin E. Dietary reference intakes for vitamin C, vitamin E, selenium, and carotenoids. Washington, D.C.: National Academy Press; 2000:186-283. (National Academy Press)
3. Trpkovic A, Resanovic I, Stanimirovic J, et al. Oxidized low-density lipoprotein as a biomarker of cardiovascular diseases. Crit Rev Clin Lab Sci. 2014:1-16. (PubMed)
4. Davis S, Davis BM, Richens JL, et al. α-Tocopherols modify the membrane dipole potential leading to modulation of ligand binding by P-glycoprotein. J Lipid Res. 2015;56(8):1543-1550. (PubMed)
5. Marko MG, Ahmed T, Bunnell SC, et al. Age-associated decline in effective immune synapse formation of CD4(+) T cells is reversed by vitamin E supplementation. J Immunol. 2007;178(3):1443-1449. (PubMed)
6. Molano A, Meydani SN. Vitamin E, signalosomes and gene expression in T cells. Mol Aspects Med. 2012;33(1):55-62. (PubMed)
7. Jiang Q. Natural forms of vitamin E: metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic Biol Med. 2014;72:76-90. (PubMed)
8. Jiang Q, Christen S, Shigenaga MK, Ames BN. γ-Tocopherol, the major form of vitamin E in the US diet, deserves more attention. Am J Clin Nutr. 2001;74(6):714-722. (PubMed)
9. Mah E, Pei R, Guo Y, et al. γ-Tocopherol-rich supplementation additively improves vascular endothelial function during smoking cessation. Free Radic Biol Med. 2013;65:1291-1299. (PubMed)
10. Mah E, Pei R, Guo Y, et al. Greater γ-tocopherol status during acute smoking abstinence with nicotine replacement therapy improved vascular endothelial function by decreasing 8-iso-15(S)-prostaglandin F2α. Exp Biol Med (Maywood). 2015;240(4):527-533. (PubMed)
11. Ahsan H, Ahad A, Iqbal J, Siddiqui WA. Pharmacological potential of tocotrienols: a review. Nutr Metab (Lond). 2014;11(1):52. (PubMed)
12. Constantinou C, Papas A, Constantinou AI. Vitamin E and cancer: An insight into the anticancer activities of vitamin E isomers and analogs. Int J Cancer. 2008;123(4):739-752. (PubMed)
13. Fu JY, Che HL, Tan DM, Teng KT. Bioavailability of tocotrienols: evidence in human studies. Nutr Metab (Lond). 2014;11(1):5. (PubMed)
14. Bruno RS, Leonard SW, Park SI, Zhao Y, Traber MG. Human vitamin E requirements assessed with the use of apples fortified with deuterium-labeled α-tocopheryl acetate. Am J Clin Nutr. 2006;83(2):299-304. (PubMed)
15. Leonard SW, Good CK, Gugger ET, Traber MG. Vitamin E bioavailability from fortified breakfast cereal is greater than that from encapsulated supplements. Am J Clin Nutr. 2004;79(1):86-92. (PubMed)
16. Traber MG, Leonard SW, Bobe G, et al. α-Tocopherol disappearance rates from plasma depend on lipid concentrations: studies using deuterium-labeled collard greens in younger and older adults. Am J Clin Nutr. 2015;101(4):752-759. (PubMed)
17. Leonard SW, Bruno RS, Ramakrishnan R, Bray T, Traber MG. Cigarette smoking increases human vitamin E requirements as estimated by plasma deuterium-labeled CEHC. Ann N Y Acad Sci. 2004;1031:357-360. (PubMed)
18. Bruno RS, Leonard SW, Atkinson J, et al. Faster plasma vitamin E disappearance in smokers is normalized by vitamin C supplementation. Free Radic Biol Med. 2006;40(4):689-697. (PubMed)
19. Booth SL, Golly I, Sacheck JM, et al. Effect of vitamin E supplementation on vitamin K status in adults with normal coagulation status. Am J Clin Nutr. 2004;80(1):143-148. (PubMed)
20. Pastori D, Carnevale R, Cangemi R, et al. Vitamin E serum levels and bleeding risk in patients receiving oral anticoagulant therapy: a retrospective cohort study. J Am Heart Assoc. 2013;2(6):e000364. (PubMed)
21. Traber MG. Vitamin E. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Philadelphia: Lippincott Williams & Wilkins; 2014:293-304.
22. Berson EL, Rosner B, Sandberg MA, et al. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(6):761-772. (PubMed)
23. Iwasa K, Shima K, Komai K, Nishida Y, Yokota T, Yamada M. Retinitis pigmentosa and macular degeneration in a patient with ataxia with isolated vitamin E deficiency with a novel c.717 del C mutation in the TTPA gene. J Neurol Sci. 2014;345(1-2):228-230. (PubMed)
24. Ford ES, Sowell A. Serum α-tocopherol status in the United States population: findings from the Third National Health and Nutrition Examination Survey. Am J Epidemiol. 1999;150(3):290-300. (PubMed)
25. Fulgoni VL, 3rd, Keast DR, Bailey RL, Dwyer J. Foods, fortificants, and supplements: Where do Americans get their nutrients? J Nutr. 2011;141(10):1847-1854. (PubMed)
26. Shamim AA, Schulze K, Merrill RD, et al. First-trimester plasma tocopherols are associated with risk of miscarriage in rural Bangladesh. Am J Clin Nutr. 2015;101(2):294-301. (PubMed)
27. Wright ME, Lawson KA, Weinstein SJ, et al. Higher baseline serum concentrations of vitamin E are associated with lower total and cause-specific mortality in the Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study. Am J Clin Nutr. 2006;84(5):1200-1207. (PubMed)
28. Wu D, Meydani SN. Age-associated changes in immune function: impact of vitamin E intervention and the underlying mechanisms. Endocr Metab Immune Disord Drug Targets. 2014;14(4):283-289. (PubMed)
29. De la Fuente M, Hernanz A, Guayerbas N, Victor VM, Arnalich F. Vitamin E ingestion improves several immune functions in elderly men and women. Free Radic Res. 2008;42(3):272-280. (PubMed)
30. Meydani SN, Meydani M, Blumberg JB, et al. Vitamin E supplementation and in vivo immune response in healthy elderly subjects. A randomized controlled trial. JAMA. 1997;277(17):1380-1386. (PubMed)
31. Pallast EG, Schouten EG, de Waart FG, et al. Effect of 50- and 100-mg vitamin E supplements on cellular immune function in noninstitutionalized elderly persons. Am J Clin Nutr. 1999;69(6):1273-1281. (PubMed)
32. Meydani SN, Leka LS, Fine BC, et al. Vitamin E and respiratory tract infections in elderly nursing home residents: a randomized controlled trial. JAMA. 2004;292(7):828-836. (PubMed)
33. Knekt P, Reunanen A, Jarvinen R, Seppanen R, Heliovaara M, Aromaa A. Antioxidant vitamin intake and coronary mortality in a longitudinal population study. Am J Epidemiol. 1994;139(12):1180-1189. (PubMed)
34. Kushi LH, Folsom AR, Prineas RJ, Mink PJ, Wu Y, Bostick RM. Dietary antioxidant vitamins and death from coronary heart disease in postmenopausal women. N Engl J Med. 1996;334(18):1156-1162. (PubMed)
35. Rimm EB, Stampfer MJ, Ascherio A, Giovannucci E, Colditz GA, Willett WC. Vitamin E consumption and the risk of coronary heart disease in men. N Engl J Med. 1993;328(20):1450-1456. (PubMed)
36. Stampfer MJ, Hennekens CH, Manson JE, Colditz GA, Rosner B, Willett WC. Vitamin E consumption and the risk of coronary disease in women. N Engl J Med. 1993;328(20):1444-1449. (PubMed)
37. Lee IM, Cook NR, Gaziano JM, et al. Vitamin E in the primary prevention of cardiovascular disease and cancer: the Women's Health Study: a randomized controlled trial. JAMA. 2005;294(1):56-65. (PubMed)
38. Glynn RJ, Ridker PM, Goldhaber SZ, Zee RY, Buring JE. Effects of random allocation to vitamin E supplementation on the occurrence of venous thromboembolism: report from the Women's Health Study. Circulation. 2007;116(13):1497-1503. (PubMed)
39. Violi F, Pignatelli P. Letter by Violi and Pignatelli regarding article, "Effects of random allocation to vitamin E supplementation on the occurrence of venous thromboembolism: report from the Women's Health Study". Circulation. 2008;117(15):e312; author reply e313. (PubMed)
40. Sesso HD, Buring JE, Christen WG, et al. Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians' Health Study II randomized controlled trial. JAMA. 2008;300(18):2123-2133. (PubMed)
41. Riccioni G, D'Orazio N, Palumbo N, et al. Relationship between plasma antioxidant concentrations and carotid intima-media thickness: the Asymptomatic Carotid Atherosclerotic Disease In Manfredonia Study. Eur J Cardiovasc Prev Rehabil. 2009;16(3):351-357. (PubMed)
42. Riccioni G, Bazzano LA. Antioxidant plasma concentration and supplementation in carotid intima media thickness. Expert Rev Cardiovasc Ther. 2008;6(5):723-729. (PubMed)
43. Azen SP, Qian D, Mack WJ, et al. Effect of supplementary antioxidant vitamin intake on carotid arterial wall intima-media thickness in a controlled clinical trial of cholesterol lowering. Circulation. 1996;94(10):2369-2372. (PubMed)
44. Joris PJ, Mensink RP. Effects of supplementation with the fat-soluble vitamins E and D on fasting flow-mediated vasodilation in adults: a meta-analysis of randomized controlled trials. Nutrients. 2015;7(3):1728-1743. (PubMed)
45. Stephens NG, Parsons A, Schofield PM, Kelly F, Cheeseman K, Mitchinson MJ. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Lancet. 1996;347(9004):781-786. (PubMed)
46. Boaz M, Smetana S, Weinstein T, et al. Secondary prevention with antioxidants of cardiovascular disease in endstage renal disease (SPACE): randomised placebo-controlled trial. Lancet. 2000;356(9237):1213-1218. (PubMed)
47. Milman U, Blum S, Shapira C, et al. Vitamin E supplementation reduces cardiovascular events in a subgroup of middle-aged individuals with both type 2 diabetes mellitus and the haptoglobin 2-2 genotype: a prospective double-blinded clinical trial. Arterioscler Thromb Vasc Biol. 2008;28(2):341-347. (PubMed)
48. Leppala JM, Virtamo J, Fogelholm R, et al. Controlled trial of α-tocopherol and β-carotene supplements on stroke incidence and mortality in male smokers. Arterioscler Thromb Vasc Biol. 2000;20(1):230-235. (PubMed)
49. Lonn E, Bosch J, Yusuf S, et al. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA. 2005;293(11):1338-1347. (PubMed)
50. Marchioli R, Levantesi G, Macchia A, et al. Vitamin E increases the risk of developing heart failure after myocardial infarction: Results from the GISSI-Prevenzione trial. J Cardiovasc Med (Hagerstown). 2006;7(5):347-350. (PubMed)
51. Dizdaroglu M. Oxidatively induced DNA damage: mechanisms, repair and disease. Cancer Lett. 2012;327(1-2):26-47. (PubMed)
52. Slatore CG, Littman AJ, Au DH, Satia JA, White E. Long-term use of supplemental multivitamins, vitamin C, vitamin E, and folate does not reduce the risk of lung cancer. Am J Respir Crit Care Med. 2008;177(5):524-530. (PubMed)
53. Heinonen OP, Albanes D, Virtamo J, et al. Prostate cancer and supplementation with α-tocopherol and β-carotene: incidence and mortality in a controlled trial. J Natl Cancer Inst. 1998;90(6):440-446. (PubMed)
54. Virtamo J, Taylor PR, Kontto J, et al. Effects of α-tocopherol and β-carotene supplementation on cancer incidence and mortality: 18-year postintervention follow-up of the Alpha-tocopherol, Beta-carotene Cancer Prevention Study. Int J Cancer. 2014;135(1):178-185. (PubMed)
55. Wang L, Sesso HD, Glynn RJ, et al. Vitamin E and C supplementation and risk of cancer in men: posttrial follow-up in the Physicians' Health Study II randomized trial. Am J Clin Nutr. 2014;100(3):915-923. (PubMed)
56. Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2009;301(1):39-51. (PubMed)
57. Klein EA, Thompson IM, Jr., Tangen CM, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2011;306(14):1549-1556. (PubMed)
58. Kristal AR, Darke AK, Morris JS, et al. Baseline selenium status and effects of selenium and vitamin e supplementation on prostate cancer risk. J Natl Cancer Inst. 2014;106(3):djt456. (PubMed)
59. Cheng TY, Barnett MJ, Kristal AR, et al. Genetic variation in myeloperoxidase modifies the association of serum α-tocopherol with aggressive prostate cancer among current smokers. J Nutr. 2011;141(9):1731-1737. (PubMed)
60. Gerstenberger JP, Bauer SR, Van Blarigan EL, et al. Selenoprotein and antioxidant genes and the risk of high-grade prostate cancer and prostate cancer recurrence. Prostate. 2015;75(1):60-69. (PubMed)
61. Major JM, Yu K, Weinstein SJ, et al. Genetic variants reflecting higher vitamin e status in men are associated with reduced risk of prostate cancer. J Nutr. 2014;144(5):729-733. (PubMed)
62. Katta AV, Katkam RV, Geetha H. Lipid peroxidation and the total antioxidant status in the pathogenesis of age related and diabetic cataracts: a study on the lens and blood. J Clin Diagn Res. 2013;7(6):978-981. (PubMed)
63. Ferrigno L, Aldigeri R, Rosmini F, Sperduto RD, Maraini G, Italian-American Cataract Study G. Associations between plasma levels of vitamins and cataract in the Italian-American Clinical Trial of Nutritional Supplements and Age-Related Cataract (CTNS): CTNS Report #2. Ophthalmic Epidemiol. 2005;12(2):71-80. (PubMed)
64. Krepler K, Schmid R. α-Tocopherol in plasma, red blood cells and lenses with and without cataract. Am J Ophthalmol. 2005;139(2):266-270. (PubMed)
65. West AL, Oren GA, Moroi SE. Evidence for the use of nutritional supplements and herbal medicines in common eye diseases. Am J Ophthalmol. 2006;141(1):157-166. (PubMed)
66. Zhang Y, Jiang W, Xie Z, Wu W, Zhang D. Vitamin E and risk of age-related cataract: a meta-analysis. Public Health Nutr. 2015:1-11. (PubMed)
67. Zheng Selin J, Rautiainen S, Lindblad BE, Morgenstern R, Wolk A. High-dose supplements of vitamins C and E, low-dose multivitamins, and the risk of age-related cataract: a population-based prospective cohort study of men. Am J Epidemiol. 2013;177(6):548-555. (PubMed)
68. Teikari JM, Rautalahti M, Haukka J, et al. Incidence of cataract operations in Finnish male smokers unaffected by α-tocopherol or β-carotene supplements. J Epidemiol Community Health. 1998;52(7):468-472. (PubMed)
69. Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E and β-carotene for age-related cataract and vision loss: AREDS report no. 9. Arch Ophthalmol. 2001;119(10):1439-1452. (PubMed)
70. Christen WG, Glynn RJ, Gaziano JM, et al. Age-related cataract in men in the selenium and vitamin E cancer prevention trial eye endpoints study: a randomized clinical trial. JAMA Ophthalmol. 2015;133(1):17-24. (PubMed)
71. Gritz DC, Srinivasan M, Smith SD, et al. The Antioxidants in Prevention of Cataracts Study: effects of antioxidant supplements on cataract progression in South India. Br J Ophthalmol. 2006;90(7):847-851. (PubMed)
72. McNeil JJ, Robman L, Tikellis G, Sinclair MI, McCarty CA, Taylor HR. Vitamin E supplementation and cataract: randomized controlled trial. Ophthalmology. 2004;111(1):75-84. (PubMed)
73. Evans JR, Lawrenson JG. Antioxidant vitamin and mineral supplements for preventing age-related macular degeneration. Cochrane Database Syst Rev. 2012;6:CD000253. (PubMed)
74. Evans JR, Lawrenson JG. Antioxidant vitamin and mineral supplements for slowing the progression of age-related macular degeneration. Cochrane Database Syst Rev. 2012;11:CD000254. (PubMed)
75. Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, β-carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119(10):1417-1436. (PubMed)
76. Hobbs RP, Bernstein PS. Nutrient Supplementation for Age-related Macular Degeneration, Cataract, and Dry Eye. J Ophthalmic Vis Res. 2014;9(4):487-493. (PubMed)
77. Pazdro R, Burgess JR. The role of vitamin E and oxidative stress in diabetes complications. Mech Ageing Dev. 2010;131(4):276-286. (PubMed)
78. Kataja-Tuomola MK, Kontto JP, Mannisto S, Albanes D, Virtamo JR. Effect of α-tocopherol and β-carotene supplementation on macrovascular complications and total mortality from diabetes: results of the ATBC Study. Ann Med. 2010;42(3):178-186. (PubMed)
79. Xu R, Zhang S, Tao A, Chen G, Zhang M. Influence of vitamin E supplementation on glycaemic control: a meta-analysis of randomised controlled trials. PLoS One. 2014;9(4):e95008. (PubMed)
80. Montero D, Walther G, Stehouwer CD, Houben AJ, Beckman JA, Vinet A. Effect of antioxidant vitamin supplementation on endothelial function in type 2 diabetes mellitus: a systematic review and meta-analysis of randomized controlled trials. Obes Rev. 2014;15(2):107-116. (PubMed)
81. Dyson JK, Anstee QM, McPherson S. Non-alcoholic fatty liver disease: a practical approach to diagnosis and staging. Frontline Gastroenterol. 2014;5(3):211-218. (PubMed)
82. Musso G, Cassader M, Rosina F, Gambino R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): a systematic review and meta-analysis of randomised trials. Diabetologia. 2012;55(4):885-904. (PubMed)
83. Chalasani NP, Sanyal AJ, Kowdley KV, et al. Pioglitazone versus vitamin E versus placebo for the treatment of non-diabetic patients with non-alcoholic steatohepatitis: PIVENS trial design. Contemp Clin Trials. 2009;30(1):88-96. (PubMed)
84. Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362(18):1675-1685. (PubMed)
85. Lavine JE, Schwimmer JB, Van Natta ML, et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA. 2011;305(16):1659-1668. (PubMed)
86. Ji HF. Vitamin E therapy on aminotransferase levels in NAFLD/NASH patients. Nutrition. 2015;31(6):899. (PubMed)
87. D'Adamo E, Marcovecchio ML, Giannini C, et al. Improved oxidative stress and cardio-metabolic status in obese prepubertal children with liver steatosis treated with lifestyle combined with Vitamin E. Free Radic Res. 2013;47(3):146-153. (PubMed)
88. Zhao Y, Zhao B. Oxidative stress and the pathogenesis of Alzheimer's disease. Oxid Med Cell Longev. 2013;2013:316523. (PubMed)
89. Lopes da Silva S, Vellas B, Elemans S, et al. Plasma nutrient status of patients with Alzheimer's disease: Systematic review and meta-analysis. Alzheimers Dement. 2014;10(4):485-502. (PubMed)
90. Kontush K, Schekatolina S. Vitamin E in neurodegenerative disorders: Alzheimer's disease. Ann N Y Acad Sci. 2004;1031:249-262. (PubMed)
91. Sano M, Ernesto C, Thomas RG, et al. A controlled trial of selegiline, α-tocopherol, or both as treatment for Alzheimer's disease. The Alzheimer's Disease Cooperative Study. N Engl J Med. 1997;336(17):1216-1222. (PubMed)
92. Petersen RC, Thomas RG, Grundman M, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005;352(23):2379-2388. (PubMed)
93. Lloret A, Badia MC, Mora NJ, Pallardo FV, Alonso MD, Vina J. Vitamin E paradox in Alzheimer's disease: it does not prevent loss of cognition and may even be detrimental. J Alzheimers Dis. 2009;17(1):143-149. (PubMed)
94. Dysken MW, Sano M, Asthana S, et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA. 2014;311(1):33-44. (PubMed)
95. Kang JH, Cook N, Manson J, Buring JE, Grodstein F. A randomized trial of vitamin E supplementation and cognitive function in women. Arch Intern Med. 2006;166(22):2462-2468. (PubMed)
96. Gao X, Wilde PE, Lichtenstein AH, Bermudez OI, Tucker KL. The maximal amount of dietary α-tocopherol intake in U.S. adults (NHANES 2001-2002). J Nutr. 2006;136(4):1021-1026. (PubMed)
97. Cheeseman KH, Holley AE, Kelly FJ, Wasil M, Hughes L, Burton G. Biokinetics in humans of RRR-α-tocopherol: the free phenol, acetate ester, and succinate ester forms of vitamin E. Free Radic Biol Med. 1995;19(5):591-598. (PubMed)
98. Duhem N, Danhier F, Preat V. Vitamin E-based nanomedicines for anti-cancer drug delivery. J Control Release. 2014;182:33-44. (PubMed)
99. Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. 2nd edition ed: Thomson Reuters; 2008.
100. Schurks M, Glynn RJ, Rist PM, Tzourio C, Kurth T. Effects of vitamin E on stroke subtypes: meta-analysis of randomised controlled trials. BMJ. 2010;341:c5702. (PubMed)
101. Brion LP, Bell EF, Raghuveer TS, Soghier L. What is the appropriate intravenous dose of vitamin E for very-low-birth-weight infants? J Perinatol. 2004;24(4):205-207. (PubMed)
102. Dietrich M, Jacques PF, Pencina MJ, et al. Vitamin E supplement use and the incidence of cardiovascular disease and all-cause mortality in the Framingham Heart Study: Does the underlying health status play a role? Atherosclerosis. 2009;205(2):549-553. (PubMed)
103. Miller ER, 3rd, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med. 2005;142(1):37-46. (PubMed)
104. Bjelakovic G, Nikolova D, Gluud C. Meta-regression analyses, meta-analyses, and trial sequential analyses of the effects of supplementation with β-carotene, vitamin A, and vitamin E singly or in different combinations on all-cause mortality: do we have evidence for lack of harm? PLoS One. 2013;8(9):e74558. (PubMed)
105. https://naturalmedicines.therapeuticresearch.com/. Vitamin E professional monograph. Natural Medicines; 2014 Copyright.
106. Brown BG, Zhao XQ, Chait A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345(22):1583-1592. (PubMed)
107. Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360(9326):23-33. (PubMed)
Vitamin K
Contents
Summary
- Naturally occurring forms of vitamin K include phylloquinone (vitamin K1) and a family of molecules called menaquinones (MKs or vitamin K2). (More information)
- With limited vitamin K storage capacity, the body recycles vitamin K in the vitamin K oxidation-reduction cycle in order to reuse it multiple times. (More information)
- Vitamin K is the essential cofactor for the carboxylation of glutamic acid residues in many vitamin K-dependent proteins (VKDPs) that are involved in blood coagulation, bone metabolism, prevention of vessel mineralization, and regulation of various cellular functions. (More information)
- Vitamin K deficiency increases the risk of excessive bleeding (hemorrhage). An injection of vitamin K is recommended to protect all newborns from life-threatening bleeding within the skull. (More information)
- The adequate intake (AI) level for vitamin K is set at 90 μg/day for women and 120 μg/day for men. (More information)
- Vitamin K deficiency may impair the activity of VKDPs and increase the risk of osteoporosis and fractures. Some, but not all, observational studies have found lower vitamin K status to be associated with lower bone mineral density and higher fracture incidence. Overall, intervention trials have been inconclusive regarding the role of supplemental vitamin K (vitamin K1 or vitamin K2) in further reducing bone loss in otherwise calcium- and vitamin D-replete adults. (More information)
- Abnormal mineralization of blood vessels increases with age and is a major risk factor for cardiovascular disease. Vitamin K inadequacy may inactivate several VKDPs that inhibit the formation of calcium precipitates in vessels. There is only limited evidence of a benefit of supplemental vitamin K in the prevention of vessel calcification and cardiovascular events. (More information)
- While the results of a few observational studies suggest that low vitamin K status might have a role in development of osteoarthritis, randomized controlled trials are needed. (More information)
- Phylloquinone is found at high concentrations in green leafy vegetables and certain plant oils, while most menaquinones are usually found in cheese, animal products, and certain fermented foods. (More information)
-
Several drugs, including vitamin K antagonists (e.g., warfarin), are known to interfere with vitamin K absorption and metabolism. (More information)

Function
Vitamin K functions as a cofactor for the enzyme, γ-glutamylcarboxylase, which catalyzes the carboxylation of the amino acid glutamic acid (Glu) to γ-carboxyglutamic acid (Gla). Vitamin K-dependent γ-carboxylation that occurs only on specific glutamic acid residues in identified vitamin K-dependent proteins (VKDP) is critical for their ability to bind calcium (5).
Vitamin K oxidation-reduction cycle
Although vitamin K is a fat-soluble vitamin, the body stores very small amounts that are rapidly depleted without regular dietary intake. Perhaps because of its limited ability to store vitamin K, the body recycles it through a process called the vitamin K-epoxide cycle (Figure 2). The vitamin K cycle allows a small amount of vitamin K to be reused many times for protein carboxylation, thus decreasing the dietary requirement. Briefly, vitamin K hydroquinone (reduced form) is oxidized to vitamin K epoxide (oxidized form). The reaction enables γ-glutamylcarboxylase to carboxylate selective glutamic acid residues on vitamin K-dependent proteins. The recycling of vitamin K epoxide (oxidized form) to hydroquinone (reduced form) is carried out by two reactions that reduce vitamin K epoxide (KO) to vitamin K quinone and then to vitamin K hydroquinone (KH2; Figure 2). Additionally, the enzyme vitamin K-epoxide reductase (VKOR) catalyzes the reduction of KO to vitamin K quinone and may be involved — as well as another yet-to-defined reductase — in the production of KH2 from vitamin K quinone (6, 7). The anticoagulant drug warfarin acts as a vitamin K antagonist by inhibiting VKOR activity, hence preventing vitamin K recycling (see Coagulation).

Coagulation
The ability to bind calcium ions (Ca2+) is required for the activation of the several vitamin K-dependent clotting factors, or proteins, in the coagulation (clotting) cascade. The term ‘coagulation cascade’ refers to a series of events, each dependent on the other, that stops bleeding through clot formation. Vitamin K-dependent γ-carboxylation of specific glutamic acid residues in those proteins makes it possible for them to bind calcium. Factors II (prothrombin), VII, IX, and X make up the core of the coagulation cascade. Protein Z appears to enhance the action of thrombin (the activated form of prothrombin) by promoting its association with phospholipids in cell membranes. Protein C and protein S are anticoagulant proteins that provide control and balance in the coagulation cascade; protein Z also has an anticoagulatory function. Control mechanisms for the coagulation cascade exist since uncontrolled clotting may be as life threatening as uncontrolled bleeding. Vitamin K-dependent coagulation factors are synthesized in the liver. Consequently, severe liver disease results in lower blood levels of vitamin K-dependent clotting factors and an increased risk for uncontrolled bleeding (hemorrhage) (8).
Oral anticoagulant therapy with vitamin K antagonists
Some people are at increased risk of forming clots, which could block the flow of blood in arteries of the heart, brain, or lungs, resulting in myocardial infarction (heart attack), stroke, or pulmonary embolism, respectively. Abnormal clotting is not related to excessive vitamin K intake, and there is no known toxicity associated with vitamin K1 or vitamin K2 (see Toxicity). Some oral anticoagulants, such as warfarin (Jantoven, formerly known as Coumadin), inhibit coagulation by antagonizing the action of vitamin K. Warfarin prevents the recycling of vitamin K by blocking VKOR activity, thus creating a functional vitamin K deficiency (Figure 2). Inadequate γ-carboxylation of vitamin K-dependent coagulation proteins interferes with the coagulation cascade, which inhibits blood clot formation. Large quantities of dietary or supplemental vitamin K can overcome the anticoagulant effect of vitamin K antagonists; thus, patients taking these drugs are cautioned against consuming very large or highly variable quantities of vitamin K (see Drug interactions). Experts now advise a reasonably constant dietary intake of vitamin K that meets current dietary recommendations (90-120 μg/day) for patients taking vitamin K antagonists like warfarin (9, 10).
Skeletal formation and prevention of soft tissue calcification
Vitamin K-dependent γ-carboxylation is essential to several bone-related proteins, including osteocalcin, anticoagulation factor protein S, matrix γ-carboxylated glutamate (Gla) protein (MGP), Gla-rich protein (GRP), and periostin (originally called osteoblast-specific factor-2). Osteocalcin (also known as bone Gla protein) is synthesized by osteoblasts (bone-forming cells); the synthesis of osteocalcin is regulated by the active form of vitamin D, 1,25-dihydroxyvitamin D (calcitriol). The calcium-binding capacity of osteocalcin requires vitamin K-dependent γ-carboxylation of three glutamic acid residues. Although its function in bone mineralization is not fully understood, osteocalcin is required for the growth and maturation of calcium hydroxyapatite crystals (see Osteoporosis) (11).
Protein S appears to play a role in the breakdown of bone mediated by osteoclasts. Individuals with inherited protein S deficiency suffer complications related to increased blood clotting, as well as osteonecrosis (12, 13). Protein S can bind and activate receptors of the TAM family that are involved in phagocytosis. Mutations in TAM receptors can result in visual impairment, defective spermatogenesis, autoimmune disorders, and platelet disorders (14).
MGP has been found in cartilage, bone, and soft tissue, including blood vessel walls, where it is synthesized and secreted by vascular smooth muscle cells. MGP is involved in the inhibition of calcification at various sites, including cartilage, vessel wall, skin elastic fibers, and the trabecular meshwork in the eye (see Vascular calcification) (15, 16). Moreover, several VKDPs, including MGP, have been associated with calcification sites in arteries, skin, kidneys, and eyes in certain inherited conditions, such as pseudoxanthoma elasticum and beta-thalassemia (17, 18).
The vitamin K-dependent proteins, GRP and periostin, are also synthesized in bone tissue, but their roles in bone metabolism are still unclear (19, 20). Expressed in normal human skin and vascular tissues, GRP has been colocalized with abnormal mineral deposits in the extracellular matrix in calcified arteries and calcified skin lesions (21). Expressed in most connective tissues, including skin and bone, periostin was initially associated with cell adhesion and migration. This VKDP also appears to promote angiogenesis (formation of new blood vessels) during cardiac valve degeneration and tumor growth (22, 23).
Current research suggests that reduced γ-glutamylcarboxylase activity and/or lower vitamin K bioavailability may impair the activity of VKDPs and contribute to bone mineralization defects and abnormal soft tissue calcification (see Disease Prevention) (24).
Regulation of cellular functions
Growth arrest-specific gene 6 protein (Gas6) is a vitamin K-dependent protein that was identified in 1993. It has been found throughout the nervous system, as well in the heart, lungs, stomach, kidneys, and cartilage. Identified as a ligand of the TAM family of transmembrane tyrosine kinase receptors, Gas6 appears to be a cellular growth regulation factor with cell-signaling activities. Gas6 has been involved in diverse cellular functions, including phagocytosis, cell adhesion, cell proliferation, and protection against apoptosis (5). It may also play important roles in the developing and aging nervous system (reviewed in 25). Further, Gas6 appears to regulate platelet signaling and vascular hemostasis (26). Expressed in most tissues and involved in many cellular functions, Gas6 has also been linked to several pathological conditions, including clot formation (thrombogenesis), atherosclerosis, chronic inflammation, and cancer growth (27-29).
Deficiency
Overt vitamin K deficiency results in impaired blood clotting, usually demonstrated by laboratory tests that measure clotting time. Symptoms include easy bruising and bleeding that may be manifested as nosebleeds, bleeding gums, blood in the urine, blood in the stool, tarry black stools, or extremely heavy menstrual bleeding. In infants, vitamin K deficiency may result in life-threatening bleeding within the skull (intracranial hemorrhage) (8).
Multiple biomarkers of vitamin K status exist (reviewed in 30), but only impaired blood coagulation is used as a measure of clinical vitamin K deficiency.
Adults
Vitamin K deficiency is uncommon in healthy adults for a number of reasons: (1) vitamin K is widespread in foods (see Food sources); (2) the vitamin K cycle conserves vitamin K (see Vitamin K oxidation-reduction cycle); and (3) bacteria that normally inhabit the large intestine synthesize menaquinones (vitamin K2), although it is unclear whether significant amounts are absorbed and utilized (see Food sources). Adults at risk for vitamin K deficiency include those taking vitamin K antagonists and individuals with significant liver damage or disease (8). Additionally, individuals with fat malabsorption disorders, including inflammatory bowel disease and cystic fibrosis, may be at increased risk of vitamin K deficiency (31-34).
Infants
Newborn babies who are exclusively breast-fed are at increased risk for vitamin K deficiency because human milk is relatively low in vitamin K compared to formula. Newborn infants, in general, have low vitamin K status for the following reasons: (1) vitamin K transport across the placental barrier is limited; (2) liver storage of vitamin K is very low; (3) the vitamin K cycle may not be fully functional in newborns, especially premature infants; (4) the vitamin K content of breast milk is low, and immature gut flora (5, 35). Infants whose mothers are on anticonvulsant medication to prevent seizures are also at risk for vitamin K deficiency. Vitamin K deficiency in newborns may result in a bleeding disorder called vitamin K deficiency bleeding (VKDB) of the newborn (reviewed in 36). Because VKDB is life threatening and easily prevented, the American Academy of Pediatrics and a number of similar international organizations recommend that an intramuscular dose of phylloquinone (vitamin K1) be administered to all newborns shortly after birth (reviewed in 37).
Controversies around vitamin K administration to newborns
Vitamin K and childhood leukemia: In the early 1990s, two retrospective studies were published suggesting a possible association between phylloquinone injections in newborns and the development of childhood leukemia and other forms of childhood cancer. However, two large retrospective studies in the US and Sweden, which reviewed the medical records of 54,000 and 1.3 million children, respectively, found no evidence of a relationship between childhood cancers and phylloquinone injections at birth (38, 39). Moreover, a pooled analysis of six case-control studies, including 2,431 children diagnosed with childhood cancer and 6,338 cancer-free children, found no evidence that phylloquinone injections for newborns increased the risk of childhood leukemia (40). In a policy statement, the American Academy of Pediatrics recommended that routine vitamin K prophylaxis for newborns be continued because VKDB is life threatening and the risks of cancer are unproven and unlikely (41). In the last decade, physicians have reported a rise in late-onset cases of VKDB due to an increasing trend of parental omission or refusal of newborn vitamin K prophylaxis (42, 43).
Lower doses of vitamin K1 for premature infants: The results of two studies of vitamin K levels in premature infants suggest that the standard initial dose of phylloquinone (vitamin K1) for full-term infants (1.0 mg) may be too high for premature infants (44, 45). These findings have led some experts to suggest the use of an initial phylloquinone dose of 0.3 mg/kg for infants with birth weights of less than 1,000 g (2 lbs, 3 oz), and an initial phylloquinone dose of 0.5 mg would probably prevent hemorrhagic disease in newborns (44). Yet, additional studies are needed to determine the best vitamin K prophylaxis in premature infants (46).
The Adequate Intake (AI)
In 2001, the US Food and Nutrition Board (FNB) of the Institute of Medicine (now the National Academy of Medicine) established the adequate intake (AI) level for vitamin K based on phylloquinone intake levels in healthy individuals (Table 1). The AI for infants was based on estimated intake of vitamin K from breast milk (47). The FNB did not consider menaquinone intakes; data on menaquinone consumption are limited (48).
Disease Prevention
Osteoporosis
The discovery of vitamin K-dependent proteins in bone has led to research on the role of vitamin K in maintaining bone health.
Vitamin K and bone health: observational studies
Vitamin K1: Observational studies have found a relationship between phylloquinone (vitamin K1) and age-related bone loss (osteoporosis). The Nurses’ Health Study followed more than 72,000 women for 10 years. In an analysis of this cohort, women whose phylloquinone intakes were lower than 109 micrograms/day (μg/day) had a 30% higher risk for hip fracture compared to women with intakes equal to or above 109 μg/day (49). Another prospective study in over 800 elderly men and women, followed in the Framingham Heart Study for seven years, found that participants with dietary vitamin K intakes in the highest quartile (median, 254 μg/day) had a 65% lower risk of hip fracture than those with intakes in the lowest quartile (median, 56 μg/day) (50). Osteoporotic fractures are often linked to a reduction in bone mineralization. Yet, the investigators found no association between dietary phylloquinone intake and bone mineral density (BMD) in the Framingham subjects (50). While other studies failed to observe associations between dietary phylloquinone intake and measures of bone strength, BMD, or fracture incidence (51, 52), the cross-sectional study of a cohort of 3,199 middle-aged women found that subjects in the highest quartile of dietary phylloquinone intake had significantly greater hip and lumbar spine BMD than those in the lowest quartile (162 μg/day vs. 59 μg/day) (53). Moreover, cross-sectional and case-control studies have reported associations between higher phylloquinone intakes and lower incidence of hip fracture (54, 55).
However, because green leafy vegetables are the primary dietary source of phylloquinone and because they are usually part of a balanced diet, high phylloquinone consumption may simply be an indicator of healthy eating habits, which may — rather than phylloquinone itself — account for all or part of the association reported in observational studies (56).
The few studies that measured plasma phylloquinone generally found that higher circulating levels were associated with lower fracture risk (15, 57, 58). For example, the incidence of vertebral fractures was inversely correlated with lumbar BMD and plasma phylloquinone in a four-year prospective study that included 379 Japanese women ages 30-88 years (57). Yet, observational studies are not designed to making causal inferences, and only randomized controlled trials can evaluate whether phylloquinone may have beneficial effects on bone health (see Vitamin K supplementation studies and osteoporosis).
Vitamin K2: There are few studies on associations between menaquinones (vitamin K2) and bone health, perhaps because of the limited number of dietary sources of menaquinone-4 (MK-4), the main form of vitamin K2 present in Western diets. The Japanese food natto, made of cooked soybeans fermented by Bacillus subtilis natto, is rich in MK-7. In a prospective cohort study that followed 944 Japanese women (ages 20-79 years), total hip BMD at baseline was positively associated with natto intake in postmenopausal women (59). During the three-year follow-up period, the rate of BMD loss at the femoral neck was significantly lower in women consuming natto (>200 μg/day of MK-7) compared to non-consumers. No association was found between natto intake and BMD in premenopausal women (59).
Total hip and femoral neck BMD was also reportedly higher in nearly 2,000 Japanese men aged 65 years and older who regularly consumed at least of one pack per day of natto (≥350 μg/day of MK-7) compared to those consuming less than one pack per week (<50 μg/day of MK-7) (60). Yet, increasing natto consumption also maximizes the intake of other dietary compounds (e.g., soy isoflavones) that have potential benefits for skeletal health; thus, there is need to find reliable measures of vitamin K status. To date, observational studies have failed to unequivocally support an association between circulating menaquinone (MK-7 and MK-4) levels and fracture risk (15, 61).
A meta-analysis that pooled the results of four prospective cohort studies and one nested case-control study found higher total dietary vitamin K intakes to be associated with a lower risk of total fracture (RR, 0.78; 95% CI: 0.56, 0.99) (62). In a recent Japanese cohort study not included in this meta-analysis (i.e., the Murakami Cohort Study), a protective association of dietary vitamin K intake was seen for vertebral fractures (p=0.005); vitamin K intake was not associated with total fracture in women or with either vertebral or total fracture in men (63).
Biomarker of vitamin K status and bone health
Total circulating levels of the bone protein, osteocalcin (OC), have been shown to be sensitive markers of bone formation. Several hormones and growth factors, including vitamin D but not vitamin K, regulate osteocalcin synthesis by osteoblasts. However, vitamin K is an essential cofactor for the γ-carboxylation of three glutamic acid residues in osteocalcin. Undercarboxylation of osteocalcin in human bone and serum has been linked to poor vitamin K status. The degree of osteocalcin γ-carboxylation is responsive to vitamin K nutritional interventions, and thus is used as a relative indicator of vitamin K status (11).
Circulating levels of uncarboxylated osteocalcin (ucOC) were found to be higher in postmenopausal women than premenopausal women and markedly higher in women over the age of 70. Also, high ratios of ucOC to total OC (ucOC/OC) appear to be predictive of hip fracture risk in elderly women (64, 65). Although vitamin K deficiency would seem the most likely cause of elevated blood ucOC/OC ratio, some investigators have documented an inverse relationship between biochemical measures of vitamin D nutritional status and ucOC levels, as well as a significant lowering of ucOC/OC ratio by vitamin D supplementation (66). It has been suggested that increased circulating ucOC/OC ratio could reflect a poor overall nutritional status that would include vitamin D inadequacy, which would explain the above-mentioned observations (67). However, in several randomized, placebo-controlled intervention studies conducted in young girls (67, 68) and postmenopausal women (69), vitamin D supplementation failed to decrease ucOC/OC ratios or show any additive effect on ucOC/OC lowering by supplemental vitamin K.
Vitamin K supplementation studies and osteoporosis
Vitamin K1 supplementation: The review of five randomized clinical trials that assessed the effect of phylloquinone (vitamin K1) supplementation on hip BMD using doses ranging from 200 μg/day to 5,000 μg/day for durations of 12 to 36 months found little promising benefit for bone health (15). Although supplementation with phylloquinone decreased ucOC levels in all five studies, only one study reported an effect of supplemental phylloquinone on BMD (70). In this study, 150 postmenopausal women were randomized to receive a placebo, minerals (500 mg/day of calcium, 150 mg/day of magnesium, and 10 mg/day of zinc) plus vitamin D (320 IU/day), or minerals, vitamin D, and phylloquinone (1,000 μg/day). The rate of BMD loss at the femoral neck, but not at the lumbar spine, was significantly lower in subjects with supplemental phylloquinone compared to the other two groups. Thus, evidence of a putative benefit of phylloquinone on bone health in older adults is considered weak. None of the studies were designed to assess the effect of phylloquinone on osteoporotic-related fractures. Further investigation may seek to evaluate whether phylloquinone supplementation could improve skeletal health in subjects at high-risk for vitamin K inadequacy (e.g., individuals with malabsorption syndromes or cystic fibrosis).
Vitamin K2 supplementation: Pharmacological doses of menaquinone-4 (MK-4; brand name, menatetrenone) are currently used in Japan in the treatment of osteoporosis (71). Accordingly, most intervention trials investigating the effect of high-dose MK-4 on bone loss have been conducted in Japanese postmenopausal women. In a three-year placebo-controlled trial among postmenopausal women with osteopenia, adding a MK-7 supplement (375 μg/day) to combined calcium-vitamin D supplementation did not affect BMD or other bone health parameters despite reductions in serum ucOC (72). At present, the potential role for supplemental menaquinones on bone health still needs to be established in large, randomized, and well-controlled trials.
A 2019 systematic review and meta-analysis of clinical trials in postmenopausal women or women with osteoporosis found vitamin K supplementation — of any form — lowered risk of clinical fracture compared to controls (OR, 0.72; 95% CI, 0.55-0.95; 9 trials varying from 6 months to 2 years: 3 using phylloquinone, 5 using MK-4, and 1 using MK-7) (73). However, no benefit of vitamin K supplementation was seen for vertebral fractures (7 trials) or BMD (22 trials). The authors of this meta-analysis noted that the high heterogeneity of the included trials (i.e., form and dose of vitamin K, use of other supplements and drugs by participants, and treatment length) makes it difficult to inform clinical recommendations (73).
Vitamin K antagonists and bone health
Certain oral anticoagulants, such as warfarin, are known to be antagonists of vitamin K (see Coagulation). Few studies have examined chronic use of warfarin and risk of fracture in older women. One study reported no association between long-term warfarin treatment and fracture risk (74), while another one found a significantly higher risk of rib and vertebral fractures in warfarin users compared to nonusers (75). Additionally, a study in elderly patients with atrial fibrillation reported that long-term warfarin treatment was associated with a significantly higher risk of osteoporotic fracture in men but not in women (76). A meta-analysis of the results of 11 published studies found that oral anticoagulation therapy was associated with a very modest reduction in BMD at the wrist and no change in BMD at the hip or spine (77). The development of new anticoagulants that do not block vitamin K recycling may offer a safer alternative to the use of vitamin K antagonists (78).
Cardiovascular disease
Observational studies examining vitamin K intake in relation to cardiovascular-related mortality have found conflicting results (48). An inverse relationship between vitamin K intake and mortality was reported in a US national survey (NHANES III) of 3,401 participants (79). Adequate vs. inadequate vitamin K intakes (based on sex-specific AI: 90 μg/day for women and 120 μg/day for men) were associated with a 22% lower risk of cardiovascular disease (CVD)-related mortality and a 15% lower risk of all-cause mortality. The report also indicated that, while over two-thirds of individuals with chronic kidney disease had vitamin K intakes below the AI, the risk of CVD mortality was 41% lower in those with adequate compared to suboptimal intakes (79). However, higher vitamin K intakes were not associated with lower CVD mortality in a prospective study that followed 7,216 older adults at risk for developing CVD (80). This study associated higher intakes of phylloquinone, but not of menaquinones, with lower risk of all-cause mortality. More recently, in a prospective study that followed a cohort of 33,289 Dutch men and women for an average of 16.8 years (the EPIC-Netherlands), neither phylloquinone intake nor menaquinone intake at baseline was associated with mortality from cardiovascular disease, coronary heart disease, stroke, or all causes (81). Yet, a prospective study of 56,048 men and women participating in the Danish Diet, Cancer, and Health cohort reported higher phylloquinone intakes to be associated with lower risks of both cardiovascular disease-related mortality and all-cause mortality (82). Moreover, a few studies have examined the association of fasting circulating phylloquinone with cardiovascular-related or all-cause mortality. In a recent meta-analysis of individual participant data from three prospective cohort studies (the Multi-Ethnic Study of Atherosclerosis; the Health, Aging, and Body Composition Study, and the Framingham Offspring Study), a blood phylloquinone concentration less than or equal to 0.5 nmol/L (n=1,081) was associated with a 19% higher risk of all-cause mortality compared to a blood concentration >1.0 nmol/L (n=1,698; 83). This analysis did not find circulating phylloquinone to be linked with incident cardiovascular disease (83).
Observational studies offer limited support of an inverse relationship between phylloquinone intake and risk of incident cardiovascular disease, despite high intakes being sometimes regarded as a marker of healthy dietary habits associated with low cardiovascular risk (reviewed in 84). A prospective cohort study of 16,057 Dutch women (ages 49-70 years), followed for a mean period of 8.1 years, found a 9% reduction in risk for coronary heart disease (CHD) per each incremental 10 μg/day increase in menaquinone intake (85). In another earlier Dutch study that examined 4,807 healthy men and women 55 years and older, participants in the highest tertile of menaquinone intake (>32.7 μg/day) had a 41% lower risk of incident CHD and a 26% lower risk of all-cause mortality than those in the lowest tertile (<21.6 μg/day) (86). In addition, menaquinone intake was found to be inversely associated with aortic calcification, a major risk factor for CVD (86). A smaller prospective study among 2,987 Norwegian adults, followed for an average of 10.8 years, reported higher dietary intakes of vitamin K2, but not of vitamin K1, were associated with a lower risk of CHD (87). While these data are interesting, it is important to note that vitamin K2 may solely be a marker of a heart-healthy diet and might not itself be cardioprotective. Most recently, the prospective Danish Diet, Cancer, and Health Study followed 53,372 older adults for an average of 21 years, finding both phylloquinone and menaquinone intake at baseline to be inversely associated with hospitalizations due to atherosclerotic cardiovascular events, including ischemic heart disease, ischemic stroke, and peripheral arterial disease (88).
Large-scale supplementation trials are needed to determine whether vitamin K1 or vitamin K2 reduces the risk of CHD or cardiovascular-related events like myocardial infarction or stroke.
Vascular calcification
One of the hallmarks of cardiovascular disease is the presence of atherosclerotic plaques in arterial walls. Plaque rupture that causes blood clot formation (thrombogenesis) is the usual cause of a myocardial infarction (heart attack) or stroke. While calcification of the plaques occurs as the atherosclerosis progresses, it is unclear whether calcification increases plaque instability and could predict risk of rupture and thrombogenesis (89). However, calcification may be predictive of future cardiovascular events, especially in those with chronic kidney disease (90). A meta-analysis of 30 prospective cohort studies, including a total of 218,080 participants, found that the presence of vascular calcification was associated with an overall three- to four-fold increased risk of cardiovascular events and mortality (91). An early population-based study of postmenopausal women (ages, 60-79 years) observed that the younger women (60-69 years) with aortic calcifications had lower vitamin K intakes than those without aortic calcifications, but this was not true for older women (70-79 years) (92). A prospective cohort study in 807 men and women, 39-45 years of age, did not find any correlation between dietary phylloquinone intake and coronary artery calcification, as measured non-invasively by computed tomography (93). Additionally, neither phylloquinone nor menaquinone intake was associated with calcification of breast arteries in a cross-sectional study of 1,689 women ages 49-70 years (94). However, in another cross-sectional study, the upper vs. lowest quartile of menaquinone (MK-4 to MK-10) intake (median intakes, 48.5 μg/day vs. 18 μg/day) was found to be associated with a 20% reduced prevalence of coronary artery calcification in 564 postmenopausal women (95).
Research has uncovered possible mechanisms by which vitamin K may inhibit mineralization (calcification) of vessels while promoting bone mineralization. The potential mechanisms, although not yet fully understood, implicate vitamin K-dependent proteins, including matrix Gla protein (MGP), Gla-rich protein (GRP), and Gas6 (96-98). Secreted by various cell types, such as vascular smooth muscle cells (VSMCs) in arterial vessel walls, MGP appears to be important for the prevention of calcification of soft tissues, including cartilage, vasculature, skin, and trabecular meshwork cells in the eye (15, 99). In MGP knockout mice, conversion of VSMCs into bone-like cells and extensive vessel calcification results in large vessel rupture and premature death. In humans, defective MGP gene has been linked to Keutel syndrome, a rare inherited condition characterized in particular by abnormal cartilage calcification and pulmonary artery stenosis (narrowing). Calcification prevention by MGP involved several mechanisms, including the binding to calcium crystals and the inhibition of proteins (bone morphogenic proteins; BMPs) known to promote ectopic bone formation (reviewed in 100).
Calcium-binding activity of MGP is regulated by two types of modifications (known as post-translational modifications since they take place after protein synthesis): the vitamin K-dependent carboxylation of up to five Glu residues and the phosphorylation of serine residues. A variation in the sequence (polymorphism) of the gene for MGP leading to a threonine-to-alanine transition in one of the five Gla domains of the protein may possibly prevent carboxylation and elicit a change in MGP ability to bind calcium. This polymorphism, known as MGPThr83Ala, has been associated with the progression of coronary artery calcification over a mean period of 10.6 years in a community-based prospective study that followed 605 middle-aged men and women (101). This association was only observed among participants without detectable calcification at baseline and not in those who had baseline calcification (101). Interestingly, MGPThr83Ala was also associated with higher risk of myocardial infarction and femoral artery calcification in carriers of the genotype (102).
Additionally, a small study initially found that, while undercarboxylated MGP (ucMGP) was absent from the innermost lining of the carotid arteries in healthy subjects, the majority of MGP in the carotid arterial lining of patients with atherosclerosis was undercarboxylated (103). In another study that examined the association between circulating MGP and incident cardiovascular events in 577 older men and women followed for a mean period of 5.6 years, the risk of cardiovascular disease (i.e., coronary artery disease, peripheral arterial disease, and cerebrovascular disease) was two- to three-fold greater in subjects in the highest vs. lowest tertile of plasma dephosphorylated and undercarboxylated MGP (dp-ucMGP) (104). The results of another prospective study suggested that circulating dp-ucMGP may be predictive of mortality risk in subjects with overt vascular disease (105). Indeed, the risk of cardiovascular-related and all-cause mortality was found to be nearly doubled in subjects with coronary artery disease or stroke in the highest vs. lowest quartile of dp-ucMGP concentrations (105).
Because suboptimal vitamin K nutritional status may limit carboxylation and result in biologically inactive ucMGP, it has been speculated that vitamin K supplementation may protect against vascular calcification. A three-year, double-blind, controlled trial investigated the potential effect of vitamin K on the progression of coronary calcification in 401 older, community-dwelling adults (ages, 60-80 years) free of cardiovascular disease at baseline (106). The participants were randomized to receive a daily multivitamin plus calcium and vitamin D with or without 500 mg of phylloquinone. Using measurements of coronary artery calcification at baseline and follow up, it was found that phylloquinone supplementation was able to limit the progression of vascular calcification and reduce plasma dp-ucMGP compared to control (106, 107). Although circulating dp-ucMGP was correlated to various markers of vitamin K status, no association with measures of coronary artery calcification were found (107). Smaller trials in those at high risk for coronary artery disease (108) or with existing aortic valve calcification (109), type 2 diabetes mellitus (110), or kidney disease (111, 112) have reported no benefit of vitamin K2 supplementation on progression of vascular calcification. A meta-analysis of controlled clinical trials found that vitamin K supplementation reduced dp-ucMGP (7 trials), reduced ucOC (4 trials), and decreased progression of vascular calcification (3 trials) (113). Yet, the authors emphasize that any conclusions from their pooled analysis are limited by the high heterogeneity of the trials with respect to the form and dose of vitamin K utilized, as well as the assays that assessed vascular calcification (113).
Thus, further investigations are necessary to examine the role of other vitamin K-dependent proteins (e.g., GRP, periostin, Gas6) in human atherosclerotic plaque calcification and to evaluate the effect of supplemental vitamin K on the progression of vascular calcification and risk of cardiovascular disease.
Vitamin K antagonists and vascular calcification
Several cross-sectional studies have reported increased vascular calcium scores (a means to quantify vascular calcification) in chronic users of vitamin K antagonists compared to nonusers (reviewed in 114). Warfarin therapy has also been associated with higher circulating concentrations of dp-ucMGP in a prospective study that examined vascular calcification in subjects with cardiovascular disease (105). Newly developed direct inhibitors of coagulation factors that do not interfere with VKDP activity may be more suitable than vitamin K antagonists, especially with regards to vascular calcification (114).
Osteoarthritis
Osteoarthritis, a degenerative joint condition that affects more than 32 million US adults (115), is characterized by the breakdown of articular cartilage (i.e., cartilage within the joint). Because several vitamin K-dependent proteins are present in cartilage and in bone (116), vitamin K inadequacy may have a role in the development of osteoarthritis. A few observational studies have investigated a possible link between vitamin K intake or status and osteoarthritis. A cross-sectional study among 719 Japanese older adults found dietary intake of vitamin K to be inversely associated with knee osteoarthritis (117). In the Framingham Offspring Study (n=672; mean age, 66 years), higher plasma concentrations of phylloquinone were associated with a lower risk of hand, but not knee, osteoarthritis (118). A longitudinal study of 1,180 US adults (mean age, 62 years) found that low plasma concentrations of phylloquinone (≤0.5 nM) at baseline — indicative of a subclinical vitamin K deficiency — were associated with a 56% increase in risk of knee osteoarthritis after 30 months compared to those with higher plasma concentrations (119). In a more recent longitudinal study among 523 older US adults participating in the Health, Aging, and Body Composition Study, those with extremely low plasma concentrations of phylloquinone (<0.2 nM) at baseline had increased progression of knee osteoarthritis over three years, assessed by MRI of articular cartilage and the meniscus; those with higher plasma concentrations experienced no significant progression of knee osteoarthritis (120). Moreover, recent studies have associated use of the vitamin K antagonist drugs with higher risks of osteoarthritis (121) and joint replacement (121) of the knee and hip compared to nonusers.
While these observational data are interesting, randomized controlled trials are needed to determine whether vitamin K supplementation in those with low vitamin K status might help prevent or treat osteoarthritis. In an ancillary study of a double-blind, controlled trial examining the effects of vitamin K supplementation on bone loss and vascular calcification in older adults, no effects of phylloquinone supplementation (500 μg/day) were found on incidence of hand osteoarthritis after three years (122). Study participants were not screened for vitamin K status, and in a subgroup analysis, those with serum phylloquinone ≤1 nM at baseline that reached >1 nM at year 3 had less joint deterioration. These data infer that only those individuals with low vitamin K status benefit from vitamin K supplementation. Unfortunately, no measures were available for knee osteoarthritis, and it is well established that hand and knee osteoarthritis represent different phenotypes. Additional clinical trials that specifically examine the effect of vitamin K supplementation on osteoarthritis development are needed, especially in those with inadequate vitamin K status.
Sources
A US national survey, NHANES 2011-2012, found that average dietary intakes of vitamin K (all forms) vary greatly among individuals, with values ranging from 80 to 195 μg/day of phylloquinone for men and 78 to 223 μg/day of phylloquinone for women (123). Mean intakes for both men and women were 117 μg/day of phylloquinone; however, 57% of men and 37% of women did not meet the Adequate Intake (AI) level (123).
Food sources
Vitamin K1
Phylloquinone (vitamin K1) is the major dietary form of vitamin K in most diets. Green leafy vegetables and some plant oils (soybean, canola, olive, and cottonseed) are major contributors of dietary vitamin K. Mixed dishes have also been found to significantly contribute to vitamin K intake in the US (123). However, phylloquinone bioavailability from green vegetables is lower than from oil or supplements. Also, the phylloquinone content of green vegetables depends on their content in chlorophyll (green pigment), so that outer leaves have more phylloquinone than inner leaves. The efficiency of phylloquinone intestinal absorption varies among plant sources and is increased with the addition of a fat source to a meal. Finally, the hydrogenation of vegetable oils may decrease the absorption and biological effect of dietary phylloquinone (reviewed in 2, 9). If you wish to check foods for their nutrient content, including phylloquinone, search USDA’s FoodData Central. A number of phylloquinone-rich foods are listed in Table 2, with their content in phylloquinone expressed in micrograms (μg).
Vitamin K2
Menaquinones (vitamin K2) are primarily of microbial origins and thus commonly found in fermented foods, such as cheese, curds, and natto (fermented soybeans) (124, 125). MK-4 is the only menaquinone that is not produced by bacteria. MK-4 is formed from menadione (a synthetic form of vitamin K) found in animal feeds or is converted in a tissue-specific way from multiple dietary forms of vitamin K, including phylloquinone and various menaquinones (4, 126). Menaquinone-4 is found in dairy products, including milk, and in some meats (125). Longer chain menaquinones are found in limited fermented food products. The Japanese fermented soybean-based, natto, is rich in MK-7 (998 μg/100 g) and also contains MK-8 (84 μg/100 g). Some cheeses contain MK-8 and MK-9 (2, 125). Additionally, animal livers are a source of long-chain menaquinones (MK-7 to MK-13) (9).
Food composition databases, including USDA’s FoodData Central, have limited data on menaquinone content in foods. Thus, menaquinone contribution to total vitamin K intakes is difficult to estimate and likely to vary between populations with different food consumption practices (2, 125). Bacteria that normally colonize the large intestine (colon) can synthesize menaquinones. It was initially thought that up to 50% of the human vitamin K requirement might be met by bacterial synthesis. However, all forms of vitamin K are absorbed in the small intestine via a mechanism requiring bile salts, while most of the menaquinone production takes place in the colon where bile salts are lacking. Current research suggests that the contribution of bacterial synthesis to vitamin K status is much less than previously thought, although the exact contribution remains unclear (16, 127).
Supplements
In the US, both phylloquinone and menaquinones are available without a prescription in multivitamin, single-nutrient, or multiple-nutrient supplements in varying doses; vitamin K content of multivitamins typically range from 20 to 120 μg per tablet (128). The US Food and Drug Administration (FDA) has not authorized any health claims for any forms of vitamin K.
Safety
Toxicity
Although allergic reactions are possible, there is no known toxicity associated with high doses (dietary or supplemental) of the phylloquinone (vitamin K1) or menaquinone (vitamin K2) forms of vitamin K (47). The same is not true for synthetic menadione (vitamin K3) and its derivatives. Menadione can interfere with the function of glutathione, one of the body's natural antioxidants, resulting in oxidative damage to cell membranes. Menadione given by injection has induced liver toxicity, jaundice, and hemolytic anemia (due to the rupture of red blood cells) in infants; therefore, menadione is no longer used for treatment of vitamin K deficiency (5). No tolerable upper intake level (UL) has been established for vitamin K (47).
Nutrient interactions
Large doses of vitamin A and vitamin E have been found to antagonize vitamin K (8). Excess vitamin A appears to interfere with vitamin K absorption, whereas vitamin E may inhibit vitamin K-dependent carboxylase activity and interfere with the coagulation cascade (129). One study in adults with normal coagulation status found that supplementation with 1,000 IU/day of vitamin E for 12 weeks decreased γ-carboxylation of prothrombin, a vitamin K-dependent protein (130). Individuals taking anticoagulatory drugs like warfarin and those who are vitamin K deficient should not take vitamin E supplements without close medical supervision because of the increased risk of hemorrhage (excessive bleeding) (131).
Drug interactions
The anticoagulant effect of vitamin K antagonists (e.g., warfarin) may be compromised by very high dietary or supplemental vitamin K intake. Moreover, daily phylloquinone supplements of up to 100 μg are considered safe for patients taking warfarin, but therapeutic anticoagulant stability may be undermined by daily doses of MK-7 as low as 10 to 20 μg (132). It is generally recommended that individuals using warfarin try to consume the AI for vitamin K (90-120 μg/day) and avoid large fluctuations in vitamin K intake that might interfere with the adjustment of their anticoagulant dose (9, 10, 133). The prescription of anti-vitamin K anticoagulants, anticonvulsants (e.g., phenytoin), and anti-tuberculosis drugs (e.g., rifampin and isoniazid) to pregnant or breast-feeding women may place the newborn at increased risk of vitamin K deficiency (134).
The drug amiodarone, used in the management of certain cardiac arrhythmias (irregular heartbeat), including atrial fibrillation, can enhance the anticoagulant effect of warfarin and thus increase the risk of hemorrhage (135, 136). Further, the use of cholesterol-lowering medications (like cholestyramine and colestipol), as well as orlistat, mineral oil, and the fat substitute, olestra, may interfere with fat absorption and affect the absorption of fat-soluble vitamins, including vitamin K (137).
Linus Pauling Institute Recommendation
It is not clear whether the AI for vitamin K is enough to optimize the γ-carboxylation of vitamin K-dependent proteins in bone (see Osteoporosis). To consume the amount of vitamin K associated with a decreased risk of hip fracture in the Framingham Heart Study (about 250 μg/day) (50), an individual would need to eat a little more than ½ cup of chopped broccoli or a large salad of mixed greens every day. Though the dietary intake of vitamin K required for optimal function of all vitamin K-dependent proteins is not yet known, the Linus Pauling Institute recommends taking a multivitamin/mineral supplement and eating at least one cup of dark-green leafy vegetables daily. Replacing dietary saturated fats like butter and cheese with monounsaturated fats found in olive oil and canola oil will increase dietary vitamin K intake and may decrease the risk of cardiovascular disease.
Older adults (>50 years)
Because older adults are at increased risk of osteoporosis and hip fracture, the above recommendation for a multivitamin/mineral supplement and at least one cup of dark green leafy vegetables daily is especially relevant.
Authors and Reviewers
Originally written in 2000 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in May 2004 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in May 2008 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in July 2014 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in May 2022 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in July 2022 by:
Sarah L. Booth, Ph.D.
Director, Vitamin K Research Program
Jean Mayer USDA Human Nutrition Research Center on Aging
Tufts University
Copyright 2000-2024 Linus Pauling Institute
References
1. Brody T. Nutritional Biochemistry. 2nd ed. San Diego: Academic Press; 1999.
2. Booth SL. Vitamin K: food composition and dietary intakes. Food Nutr Res. 2012;56. (PubMed)
3. Kidd PM. Vitamins D and K as pleiotropic nutrients: clinical importance to the skeletal and cardiovascular systems and preliminary evidence for synergy. Altern Med Rev. 2010;15(3):199-222. (PubMed)
4. Nakagawa K, Hirota Y, Sawada N, et al. Identification of UBIAD1 as a novel human menaquinone-4 biosynthetic enzyme. Nature. 2010;468(7320):117-121. (PubMed)
5. Ferland G. Vitamin K. In: ISLI, ed. Present Knowledge in Nutrition. 10th ed: John Wiley & Sons; 2012:230-247.
6. Rishavy MA, Hallgren KW, Wilson LA, Usubalieva A, Runge KW, Berkner KL. The vitamin K oxidoreductase is a multimer that efficiently reduces vitamin K epoxide to hydroquinone to allow vitamin K-dependent protein carboxylation. J Biol Chem. 2013;288(44):31556-31566. (PubMed)
7. Tie JK, Jin DY, Straight DL, Stafford DW. Functional study of the vitamin K cycle in mammalian cells. Blood. 2011;117(10):2967-2974. (PubMed)
8. Olson RE. Vitamin K. In: Shils ME, Olson JA, Shike M, Ross AC, eds. Modern Nutrition in Health and Disease. 9th ed. Baltimore: Williams & Wilkins; 1999:363-380.
9. Holmes MV, Hunt BJ, Shearer MJ. The role of dietary vitamin K in the management of oral vitamin K antagonists. Blood Rev. 2012;26(1):1-14. (PubMed)
10. Violi F, GY L, P P, D P. Interaction between dietary vitamin K intake and anticoagulation by vitamin K antagonists: is it really true?: a systematic review. Medicine (Baltimore). 2016;95(10):e2895. (PubMed)
11. Gundberg CM, Lian JB, Booth SL. Vitamin K-dependent carboxylation of osteocalcin: friend or foe? Adv Nutr. 2012;3(2):149-157. (PubMed)
12. Pierre-Jacques H, Glueck CJ, Mont MA, Hungerford DS. Familial heterozygous protein-S deficiency in a patient who had multifocal osteonecrosis. A case report. J Bone Joint Surg Am. 1997;79(7):1079-1084. (PubMed)
13. Rawat RS, Mehta Y, Arora D, Trehan N. Asymptomatic type B right atrial thrombus in a case with protein S deficiency. Ann Card Anaesth. 2014;17(3):237-239. (PubMed)
14. van der Meer JH, van der Poll T, van 't Veer C. TAM receptors, Gas6, and protein S: roles in inflammation and hemostasis. Blood. 2014;123(16):2460-2469. (PubMed)
15. Booth SL. Roles for vitamin K beyond coagulation. Annu Rev Nutr. 2009;29:89-110. (PubMed)
16. Shea MK, Booth SL. Vitamin K. Adv Nutr. 2022;13(1):350-351. (PubMed)
17. Boraldi F, Annovi G, Guerra D, et al. Fibroblast protein profile analysis highlights the role of oxidative stress and vitamin K recycling in the pathogenesis of pseudoxanthoma elasticum. Proteomics Clin Appl. 2009;3(9):1084-1098. (PubMed)
18. Boraldi F, Garcia-Fernandez M, Paolinelli-Devincenzi C, et al. Ectopic calcification in beta-thalassemia patients is associated with increased oxidative stress and lower MGP carboxylation. Biochim Biophys Acta. 2013;1832(12):2077-2084. (PubMed)
19. Coutu DL, Wu JH, Monette A, Rivard GE, Blostein MD, Galipeau J. Periostin, a member of a novel family of vitamin K-dependent proteins, is expressed by mesenchymal stromal cells. J Biol Chem. 2008;283(26):17991-18001. (PubMed)
20. Viegas CS, Simes DC, Laize V, Williamson MK, Price PA, Cancela ML. Gla-rich protein (GRP), a new vitamin K-dependent protein identified from sturgeon cartilage and highly conserved in vertebrates. J Biol Chem. 2008;283(52):36655-36664. (PubMed)
21. Viegas CS, Cavaco S, Neves PL, et al. Gla-rich protein is a novel vitamin K-dependent protein present in serum that accumulates at sites of pathological calcifications. Am J Pathol. 2009;175(6):2288-2298. (PubMed)
22. Hakuno D, Kimura N, Yoshioka M, et al. Periostin advances atherosclerotic and rheumatic cardiac valve degeneration by inducing angiogenesis and MMP production in humans and rodents. J Clin Invest. 2010;120(7):2292-2306. (PubMed)
23. Kudo Y, Siriwardena BS, Hatano H, Ogawa I, Takata T. Periostin: novel diagnostic and therapeutic target for cancer. Histol Histopathol. 2007;22(10):1167-1174. (PubMed)
24. Vanakker OM, Martin L, Schurgers LJ, et al. Low serum vitamin K in PXE results in defective carboxylation of mineralization inhibitors similar to the GGCX mutations in the PXE-like syndrome. Lab Invest. 2010;90(6):895-905. (PubMed)
25. Ferland G. Vitamin K and the nervous system: an overview of its actions. Adv Nutr. 2012;3(2):204-212. (PubMed)
26. Laurance S, Lemarie CA, Blostein MD. Growth arrest-specific gene 6 (gas6) and vascular hemostasis. Adv Nutr. 2012;3(2):196-203. (PubMed)
27. Robins RS, Lemarie CA, Laurance S, Aghourian MN, Wu J, Blostein MD. Vascular Gas6 contributes to thrombogenesis and promotes tissue factor up-regulation after vessel injury in mice. Blood. 2013;121(4):692-699. (PubMed)
28. Rothlin CV, Leighton JA, Ghosh S. Tyro3, Axl, and Mertk receptor signaling in inflammatory bowel disease and colitis-associated cancer. Inflamm Bowel Dis. 2014;20(8):1472-80. (PubMed)
29. Tjwa M, Moons L, Lutgens E. Pleiotropic role of growth arrest-specific gene 6 in atherosclerosis. Curr Opin Lipidol. 2009;20(5):386-392. (PubMed)
30. Card DJ, Gorska R, Harrington DJ. Laboratory assessment of vitamin K status. J Clin Pathol. 2020;73(2):70-75. (PubMed)
31. Jagannath VA, Fedorowicz Z, Thaker V, Chang AB. Vitamin K supplementation for cystic fibrosis. Cochrane Database Syst Rev. 2013;4:CD008482. (PubMed)
32. Nakajima S, Iijima H, Egawa S, et al. Association of vitamin K deficiency with bone metabolism and clinical disease activity in inflammatory bowel disease. Nutrition. 2011;27(10):1023-1028. (PubMed)
33. Nowak JK, Grzybowska-Chlebowczyk U, Landowski P, et al. Prevalence and correlates of vitamin K deficiency in children with inflammatory bowel disease. Sci Rep. 2014;4:4768. (PubMed)
34. Dong R, Wang N, Yang Y, et al. Review on vitamin K deficiency and its biomarkers: focus on the novel application of PIVKA-II in clinical practice. Clin Lab. 2018;64(4):413-424. (PubMed)
35. Araki S, Shirahata A. Vitamin K deficiency bleeding in infancy. Nutrients. 2020;12(3):780. (PubMed)
36. Shearer MJ. Vitamin K deficiency bleeding (VKDB) in early infancy. Blood Rev. 2009;23(2):49-59. (PubMed)
37. Jullien S. Vitamin K prophylaxis in newborns. BMC Pediatr. 2021;21(Suppl 1):350. (PubMed)
38. Klebanoff MA, Read JS, Mills JL, Shiono PH. The risk of childhood cancer after neonatal exposure to vitamin K. N Engl J Med. 1993;329(13):905-908. (PubMed)
39. Ekelund H, Finnstrom O, Gunnarskog J, Kallen B, Larsson Y. Administration of vitamin K to newborn infants and childhood cancer. BMJ. 1993;307(6896):89-91. (PubMed)
40. Roman E, Fear NT, Ansell P, et al. Vitamin K and childhood cancer: analysis of individual patient data from six case-control studies. Br J Cancer. 2002;86(1):63-69. (PubMed)
41. American Academy of Pediatrics Committee on Fetus and Newborn. Controversies concerning vitamin K and the newborn. Pediatrics. 2003;112(1 Pt 1):191-192. (PubMed)
42. Schulte R, Jordan LC, Morad A, Naftel RP, Wellons JC, 3rd, Sidonio R. Rise in late onset vitamin K deficiency bleeding in young infants because of omission or refusal of prophylaxis at birth. Pediatr Neurol. 2014;50(6):564-568. (PubMed)
43. Majid A, Blackwell M, Broadbent RS, et al. Newborn vitamin K prophylaxis: a historical perspective to understand modern barriers to uptake. Hosp Pediatr. 2019;9(1):55-60. (PubMed)
44. Costakos DT, Greer FR, Love LA, Dahlen LR, Suttie JW. Vitamin K prophylaxis for premature infants: 1 mg versus 0.5 mg. Am J Perinatol. 2003;20(8):485-490. (PubMed)
45. Kumar D, Greer FR, Super DM, Suttie JW, Moore JJ. Vitamin K status of premature infants: implications for current recommendations. Pediatrics. 2001;108(5):1117-1122. (PubMed)
46. Ardell S, Offringa M, Ovelman C, Soll R. Prophylactic vitamin K for the prevention of vitamin K deficiency bleeding in preterm neonates. Cochrane Database Syst Rev. 2018;2:CD008342. (PubMed)
47. Food and Nutrition Board, Institute of Medicine. Vitamin K. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Washington, D.C.: National Academy Press; 2001:162-196. (National Academy Press)
48. Shea MK, Berkner KL, Ferland G, Fu X, Holden RM, Booth SL. Perspective: Evidence before enthusiasm-a critical review of the potential cardiovascular benefits of vitamin K. Adv Nutr. 2021;12(3):632-646. (PubMed)
49. Feskanich D, Weber P, Willett WC, Rockett H, Booth SL, Colditz GA. Vitamin K intake and hip fractures in women: a prospective study. Am J Clin Nutr. 1999;69(1):74-79. (PubMed)
50. Booth SL, Tucker KL, Chen H, et al. Dietary vitamin K intakes are associated with hip fracture but not with bone mineral density in elderly men and women. Am J Clin Nutr. 2000;71(5):1201-1208. (PubMed)
51. Rejnmark L, Vestergaard P, Charles P, et al. No effect of vitamin K1 intake on bone mineral density and fracture risk in perimenopausal women. Osteoporos Int. 2006;17(8):1122-1132. (PubMed)
52. McLean RR, Booth SL, Kiel DP, et al. Association of dietary and biochemical measures of vitamin K with quantitative ultrasound of the heel in men and women. Osteoporos Int. 2006;17(4):600-607. (PubMed)
53. Macdonald HM, McGuigan FE, Lanham-New SA, Fraser WD, Ralston SH, Reid DM. Vitamin K1 intake is associated with higher bone mineral density and reduced bone resorption in early postmenopausal Scottish women: no evidence of gene-nutrient interaction with apolipoprotein E polymorphisms. Am J Clin Nutr. 2008;87(5):1513-1520. (PubMed)
54. Apalset EM, Gjesdal CG, Eide GE, Tell GS. Intake of vitamin K1 and K2 and risk of hip fractures: The Hordaland Health Study. Bone. 2011;49(5):990-995. (PubMed)
55. Torbergsen AC, Watne LO, Wyller TB, et al. Vitamin K1 and 25(OH)D are independently and synergistically associated with a risk for hip fracture in an elderly population: A case control study. Clin Nutr. 2015;34(1):101-106. (PubMed)
56. Booth SL, Mayer J. Warfarin use and fracture risk. Nutr Rev. 2000;58(1):20-22. (PubMed)
57. Tsugawa N, Shiraki M, Suhara Y, et al. Low plasma phylloquinone concentration is associated with high incidence of vertebral fracture in Japanese women. J Bone Miner Metab. 2008;26(1):79-85. (PubMed)
58. Moore AE, Kim E, Dulnoan D, et al. Serum vitamin K1 (phylloquinone) is associated with fracture risk and hip strength in post-menopausal osteoporosis: A cross-sectional study. Bone. 2020;141:115630. (PubMed)
59. Ikeda Y, Iki M, Morita A, et al. Intake of fermented soybeans, natto, is associated with reduced bone loss in postmenopausal women: Japanese Population-Based Osteoporosis (JPOS) Study. J Nutr. 2006;136(5):1323-1328. (PubMed)
60. Fujita Y, Iki M, Tamaki J, et al. Association between vitamin K intake from fermented soybeans, natto, and bone mineral density in elderly Japanese men: the Fujiwara-kyo Osteoporosis Risk in Men (FORMEN) study. Osteoporos Int. 2012;23(2):705-714. (PubMed)
61. Kaneki M, Hodges SJ, Hosoi T, et al. Japanese fermented soybean food as the major determinant of the large geographic difference in circulating levels of vitamin K2: possible implications for hip-fracture risk. Nutrition. 2001;17(4):315-321. (PubMed)
62. Hao G, Zhang B, Gu M, et al. Vitamin K intake and the risk of fractures: A meta-analysis. Medicine (Baltimore). 2017;96(17):e6725. (PubMed)
63. Platonova K, Kitamura K, Watanabe Y, et al. Dietary calcium and vitamin K are associated with osteoporotic fracture risk in middle-aged and elderly Japanese women, but not men: the Murakami Cohort Study. Br J Nutr. 2021;125(3):319-328. (PubMed)
64. Szulc P, Chapuy MC, Meunier PJ, Delmas PD. Serum undercarboxylated osteocalcin is a marker of the risk of hip fracture in elderly women. J Clin Invest. 1993;91(4):1769-1774. (PubMed)
65. Vergnaud P, Garnero P, Meunier PJ, Breart G, Kamihagi K, Delmas PD. Undercarboxylated osteocalcin measured with a specific immunoassay predicts hip fracture in elderly women: the EPIDOS Study. J Clin Endocrinol Metab. 1997;82(3):719-724. (PubMed)
66. Shearer MJ. The roles of vitamins D and K in bone health and osteoporosis prevention. Proc Nutr Soc. 1997;56(3):915-937. (PubMed)
67. O'Connor E, Molgaard C, Michaelsen KF, Jakobsen J, Cashman KD. Vitamin D-vitamin K interaction: effect of vitamin D supplementation on serum percentage undercarboxylated osteocalcin, a sensitive measure of vitamin K status, in Danish girls. Br J Nutr. 2010;104(8):1091-1095. (PubMed)
68. Kanellakis S, Moschonis G, Tenta R, et al. Changes in parameters of bone metabolism in postmenopausal women following a 12-month intervention period using dairy products enriched with calcium, vitamin D, and phylloquinone (vitamin K(1)) or menaquinone-7 (vitamin K (2)): the Postmenopausal Health Study II. Calcif Tissue Int. 2012;90(4):251-262. (PubMed)
69. Bolton-Smith C, McMurdo ME, Paterson CR, et al. Two-year randomized controlled trial of vitamin K1 (phylloquinone) and vitamin D3 plus calcium on the bone health of older women. J Bone Miner Res. 2007;22(4):509-519. (PubMed)
70. Braam LA, Knapen MH, Geusens P, et al. Vitamin K1 supplementation retards bone loss in postmenopausal women between 50 and 60 years of age. Calcif Tissue Int. 2003;73(1):21-26. (PubMed)
71. Orimo H, Nakamura T, Hosoi T, et al. Japanese 2011 guidelines for prevention and treatment of osteoporosis--executive summary. Arch Osteoporos. 2012;7:3-20. (PubMed)
72. Ronn SH, Harslof T, Oei L, Pedersen SB, Langdahl BL. The effect of vitamin MK-7 on bone mineral density and microarchitecture in postmenopausal women with osteopenia, a 3-year randomized, placebo-controlled clinical trial. Osteoporos Int. 2021;32(1):185-191. (PubMed)
73. Mott A, Bradley T, Wright K, et al. Effect of vitamin K on bone mineral density and fractures in adults: an updated systematic review and meta-analysis of randomised controlled trials. Osteoporos Int. 2019;30(8):1543-1559. (PubMed)
74. Jamal SA, Browner WS, Bauer DC, Cummings SR. Warfarin use and risk for osteoporosis in elderly women. Study of Osteoporotic Fractures Research Group. Ann Intern Med. 1998;128(10):829-832. (PubMed)
75. Caraballo PJ, Heit JA, Atkinson EJ, et al. Long-term use of oral anticoagulants and the risk of fracture. Arch Intern Med. 1999;159(15):1750-1756. (PubMed)
76. Gage BF, Birman-Deych E, Radford MJ, Nilasena DS, Binder EF. Risk of osteoporotic fracture in elderly patients taking warfarin: results from the National Registry of Atrial Fibrillation 2. Arch Intern Med. 2006;166(2):241-246. (PubMed)
77. Caraballo PJ, Gabriel SE, Castro MR, Atkinson EJ, Melton LJ, 3rd. Changes in bone density after exposure to oral anticoagulants: a meta-analysis. Osteoporos Int. 1999;9(5):441-448. (PubMed)
78. Fusaro M, Crepaldi G, Maggi S, et al. Bleeding, vertebral fractures and vascular calcifications in patients treated with warfarin: hope for lower risks with alternative therapies. Curr Vasc Pharmacol. 2011;9(6):763-769. (PubMed)
79. Cheung CL, Sahni S, Cheung BM, Sing CW, Wong IC. Vitamin K intake and mortality in people with chronic kidney disease from NHANES III. Clin Nutr. 2015;34(2):235-240. (PubMed)
80. Juanola-Falgarona M, Salas-Salvado J, Martinez-Gonzalez MA, et al. Dietary intake of vitamin K is inversely associated with mortality risk. J Nutr. 2014;144(5):743-750. (PubMed)
81. Zwakenberg SR, den Braver NR, Engelen AIP, et al. Vitamin K intake and all-cause and cause specific mortality. Clin Nutr. 2017;36(5):1294-1300. (PubMed)
82. Palmer CR, Bellinge JW, Dalgaard F, et al. Association between vitamin K1 intake and mortality in the Danish Diet, Cancer, and Health cohort. Eur J Epidemiol. 2021;36(10):1005-1014. (PubMed)
83. Shea MK, Barger K, Booth SL, et al. Vitamin K status, cardiovascular disease, and all-cause mortality: a participant-level meta-analysis of 3 US cohorts. Am J Clin Nutr. 2020;111(6):1170-1177. (PubMed)
84. Rees K, Guraewal S, Wong YL, et al. Is vitamin K consumption associated with cardio-metabolic disorders? A systematic review. Maturitas. 2010;67(2):121-128. (PubMed)
85. Gast GC, de Roos NM, Sluijs I, et al. A high menaquinone intake reduces the incidence of coronary heart disease. Nutr Metab Cardiovasc Dis. 2009;19(7):504-510. (PubMed)
86. Geleijnse JM, Vermeer C, Grobbee DE, et al. Dietary intake of menaquinone is associated with a reduced risk of coronary heart disease: the Rotterdam Study. J Nutr. 2004;134(11):3100-3105. (PubMed)
87. Haugsgjerd TR, Egeland GM, Nygard OK, et al. Association of dietary vitamin K and risk of coronary heart disease in middle-age adults: the Hordaland Health Study Cohort. BMJ Open. 2020;10(5):e035953. (PubMed)
88. Bellinge JW, Dalgaard F, Murray K, et al. Vitamin K intake and atherosclerotic cardiovascular disease in the Danish Diet Cancer and Health Study. J Am Heart Assoc. 2021;10(16):e020551. (PubMed)
89. Otsuka F, Sakakura K, Yahagi K, Joner M, Virmani R. Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler Thromb Vasc Biol. 2014;34(4):724-736. (PubMed)
90. Chen J, Budoff MJ, Reilly MP, et al. Coronary artery calcification and risk of cardiovascular disease and death among patients with chronic kidney disease. JAMA Cardiol. 2017;2(6):635-643. (PubMed)
91. Rennenberg RJ, Kessels AG, Schurgers LJ, van Engelshoven JM, de Leeuw PW, Kroon AA. Vascular calcifications as a marker of increased cardiovascular risk: a meta-analysis. Vasc Health Risk Manag. 2009;5(1):185-197. (PubMed)
92. Jie KS, Bots ML, Vermeer C, Witteman JC, Grobbee DE. Vitamin K intake and osteocalcin levels in women with and without aortic atherosclerosis: a population-based study. Atherosclerosis. 1995;116(1):117-123. (PubMed)
93. Villines TC, Hatzigeorgiou C, Feuerstein IM, O'Malley PG, Taylor AJ. Vitamin K1 intake and coronary calcification. Coron Artery Dis. 2005;16(3):199-203. (PubMed)
94. Maas AH, van der Schouw YT, Beijerinck D, et al. Vitamin K intake and calcifications in breast arteries. Maturitas. 2007;56(3):273-279. (PubMed)
95. Beulens JW, Bots ML, Atsma F, et al. High dietary menaquinone intake is associated with reduced coronary calcification. Atherosclerosis. 2009;203(2):489-493. (PubMed)
96. Qiu C, Zheng H, Tao H, et al. Vitamin K2 inhibits rat vascular smooth muscle cell calcification by restoring the Gas6/Axl/Akt anti-apoptotic pathway. Mol Cell Biochem. 2017;433(1-2):149-159. (PubMed)
97. Holden RM, Hetu MF, Li TY, et al. Circulating Gas6 is associated with reduced human carotid atherosclerotic plaque burden in high risk cardiac patients. Clin Biochem. 2019;64:6-11. (PubMed)
98. Viegas CS, Rafael MS, Enriquez JL, et al. Gla-rich protein acts as a calcification inhibitor in the human cardiovascular system. Arterioscler Thromb Vasc Biol. 2015;35(2):399-408. (PubMed)
99. Borras T, Comes N. Evidence for a calcification process in the trabecular meshwork. Exp Eye Res. 2009;88(4):738-746. (PubMed)
100. Schurgers LJ, Uitto J, Reutelingsperger CP. Vitamin K-dependent carboxylation of matrix Gla-protein: a crucial switch to control ectopic mineralization. Trends Mol Med. 2013;19(4):217-226. (PubMed)
101. Cassidy-Bushrow AE, Bielak LF, Levin AM, et al. Matrix gla protein gene polymorphism is associated with increased coronary artery calcification progression. Arterioscler Thromb Vasc Biol. 2013;33(3):645-651. (PubMed)
102. Herrmann SM, Whatling C, Brand E, et al. Polymorphisms of the human matrix gla protein (MGP) gene, vascular calcification, and myocardial infarction. Arterioscler Thromb Vasc Biol. 2000;20(11):2386-2393. (PubMed)
103. Schurgers LJ, Teunissen KJ, Knapen MH, et al. Novel conformation-specific antibodies against matrix gamma-carboxyglutamic acid (Gla) protein: undercarboxylated matrix Gla protein as marker for vascular calcification. Arterioscler Thromb Vasc Biol. 2005;25(8):1629-1633. (PubMed)
104. van den Heuvel EG, van Schoor NM, Lips P, et al. Circulating uncarboxylated matrix Gla protein, a marker of vitamin K status, as a risk factor of cardiovascular disease. Maturitas. 2014;77(2):137-141. (PubMed)
105. Mayer O, Jr., Seidlerova J, Bruthans J, et al. Desphospho-uncarboxylated matrix Gla-protein is associated with mortality risk in patients with chronic stable vascular disease. Atherosclerosis. 2014;235(1):162-168. (PubMed)
106. Shea MK, O'Donnell CJ, Hoffmann U, et al. Vitamin K supplementation and progression of coronary artery calcium in older men and women. Am J Clin Nutr. 2009;89(6):1799-1807. (PubMed)
107. Shea MK, O'Donnell CJ, Vermeer C, et al. Circulating uncarboxylated matrix gla protein is associated with vitamin K nutritional status, but not coronary artery calcium, in older adults. J Nutr. 2011;141(8):1529-1534. (PubMed)
108. Ikari Y, Torii S, Shioi A, Okano T. Impact of menaquinone-4 supplementation on coronary artery calcification and arterial stiffness: an open label single arm study. Nutr J. 2016;15(1):53. (PubMed)
109. Diederichsen ACP, Lindholt JS, Moller S, et al. Vitamin K2 and D in patients with aortic valve calcification: a randomized double-blinded clinical trial. Circulation. 2022;145(18):1387-1397. (PubMed)
110. Zwakenberg SR, de Jong PA, Bartstra JW, et al. The effect of menaquinone-7 supplementation on vascular calcification in patients with diabetes: a randomized, double-blind, placebo-controlled trial. Am J Clin Nutr. 2019;110(4):883-890. (PubMed)
111. Kurnatowska I, Grzelak P, Masajtis-Zagajewska A, et al. Effect of vitamin K2 on progression of atherosclerosis and vascular calcification in nondialyzed patients with chronic kidney disease stages 3-5. Pol Arch Med Wewn. 2015;125(9):631-640. (PubMed)
112. De Vriese AS, Caluwe R, Pyfferoen L, et al. Multicenter randomized controlled trial of vitamin K antagonist replacement by rivaroxaban with or without vitamin K2 in hemodialysis patients with atrial fibrillation: the Valkyrie Study. J Am Soc Nephrol. 2020;31(1):186-196. (PubMed)
113. Lees JS, Chapman FA, Witham MD, Jardine AG, Mark PB. Vitamin K status, supplementation and vascular disease: a systematic review and meta-analysis. Heart. 2019;105(12):938-945. (PubMed)
114. Chatrou ML, Winckers K, Hackeng TM, Reutelingsperger CP, Schurgers LJ. Vascular calcification: the price to pay for anticoagulation therapy with vitamin K-antagonists. Blood Rev. 2012;26(4):155-166. (PubMed)
115. US Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Division of Population Health,. Osteoarthritis (OA). Available at: https://www.cdc.gov/arthritis/basics/osteoarthritis.htm. Accessed 5/19/22.
116. Harshman SG, Shea MK. The role of vitamin K in chronic aging diseases: inflammation, cardiovascular disease, and osteoarthritis. Curr Nutr Rep. 2016;5(2):90-98. (PubMed)
117. Oka H, Akune T, Muraki S, et al. Association of low dietary vitamin K intake with radiographic knee osteoarthritis in the Japanese elderly population: dietary survey in a population-based cohort of the ROAD study. J Orthop Sci. 2009;14(6):687-692. (PubMed)
118. Neogi T, Booth SL, Zhang YQ, et al. Low vitamin K status is associated with osteoarthritis in the hand and knee. Arthritis Rheum. 2006;54(4):1255-1261. (PubMed)
119. Misra D, Booth SL, Tolstykh I, et al. Vitamin K deficiency is associated with incident knee osteoarthritis. Am J Med. 2013;126(3):243-248. (PubMed)
120. Shea MK, Kritchevsky SB, Hsu FC, et al. The association between vitamin K status and knee osteoarthritis features in older adults: the Health, Aging and Body Composition Study. Osteoarthritis Cartilage. 2015;23(3):370-378. (PubMed)
121. Boer CG, Szilagyi I, Nguyen NL, et al. Vitamin K antagonist anticoagulant usage is associated with increased incidence and progression of osteoarthritis. Ann Rheum Dis. 2021;80(5):598-604. (PubMed)
122. Neogi T, Felson DT, Sarno R, Booth SL. Vitamin K in hand osteoarthritis: results from a randomised clinical trial. Ann Rheum Dis. 2008;67(11):1570-1573. (PubMed)
123. Harshman SG, Finnan EG, Barger KJ, et al. Vegetables and mixed dishes are top contributors to phylloquinone intake in US adults: data from the 2011-2012 NHANES. J Nutr. 2017;147(7):1308-1313. (PubMed)
124. Vermeer C, Raes J, van 't Hoofd C, Knapen MHJ, Xanthoulea S. Menaquinone content of cheese. Nutrients. 2018;10(4). (PubMed)
125. Walther B, Karl JP, Booth SL, Boyaval P. Menaquinones, bacteria, and the food supply: the relevance of dairy and fermented food products to vitamin K requirements. Adv Nutr. 2013;4(4):463-473. (PubMed)
126. Ellis JL, Fu X, Karl JP, et al. Multiple dietary vitamin K forms are converted to tissue menaquinone-4 in mice. J Nutr. 2022;152(4):981-993. (PubMed)
127. Beulens JW, Booth SL, van den Heuvel EG, Stoecklin E, Baka A, Vermeer C. The role of menaquinones (vitamin K(2)) in human health. Br J Nutr. 2013;110(8):1357-1368. (PubMed)
128. US Department of Health and Human Services, National Institutes of Health, Office of Dietary Supplements. Dietary Supplement Label Database (DSLD). [Internet]. Accessed 3/7/22. Available from: https://dsld.od.nih.gov/.
129. Traber MG. Vitamin E and K interactions--a 50-year-old problem. Nutr Rev. 2008;66(11):624-629. (PubMed)
130. Booth SL, Golly I, Sacheck JM, et al. Effect of vitamin E supplementation on vitamin K status in adults with normal coagulation status. Am J Clin Nutr. 2004;80(1):143-148. (PubMed)
131. Pastori D, Carnevale R, Cangemi R, et al. Vitamin E serum levels and bleeding risk in patients receiving oral anticoagulant therapy: a retrospective cohort study. J Am Heart Assoc. 2013;2(6):e000364. (PubMed)
132. Shearer MJ, Newman P. Recent trends in the metabolism and cell biology of vitamin K with special reference to vitamin K cycling and MK-4 biosynthesis. J Lipid Res. 2014;55(3):345-362. (PubMed)
133. Chang CH, Wang YW, Yeh Liu PY, Kao Yang YH. A practical approach to minimize the interaction of dietary vitamin K with warfarin. J Clin Pharm Ther. 2014;39(1):56-60. (PubMed)
134. Thorp JA, Gaston L, Caspers DR, Pal ML. Current concepts and controversies in the use of vitamin K. Drugs. 1995;49(3):376-387. (PubMed)
135. Reiffel JA. An important indirect drug interaction between dronedarone and warfarin that may be extrapolated to other drugs that can alter gastrointestinal function. Am Heart J. 2011;161(2):e5; author reply e7. (PubMed)
136. Shirolkar SC, Fiuzat M, Becker RC. Dronedarone and vitamin K antagonists: a review of drug-drug interactions. Am Heart J. 2010;160(4):577-582. (PubMed)
137. Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. Montvale: Medical Economics Company, Inc.; 2001.