Minerales
Los minerales son elementos que se originan en la tierra y no pueden ser producidos por los organismos vivos. Las plantas obtienen minerales desde el suelo, y la mayoría de los minerales en nuestra dieta provienen directamente de las plantas o indirectamente de fuentes animales. Los minerales también están presentes en el agua que bebemos, pero varían según la ubicación geográfica. Los minerales provenientes de las plantas también pueden variar dependiendo el lugar ya que el contenido mineral del suelo varia según la ubicación geográfica.
La información sobre vitaminas y minerales del Centro de Información de Micronutrientes del Instituto Linus Pauling se encuentra ahora disponible en un libro titulado Un acercamiento basado en la evidencia a las vitaminas y minerales: Beneficios para la salud y recomendaciones de consumo (An Evidence-based Approach to Vitamins and Minerals: Health Benefits and Intake Recommendations). El libro esta disponible para la venta en Linus Pauling Institute o Thieme Medical Publishers.
Seleccione un mineral de la lista para más información.
Calcio
Contenido
Resumen
- El calcio es un constituyente importante de los huesos y los dientes y también desempeña un papel esencial como segundo mensajero en las vías de señalización celular. Las concentraciones de calcio circulante están estrechamente controladas por la hormona paratiroidea (PTH) y la vitamina D a expensas del esqueleto cuando las ingestas de calcio son inadecuadas. (Más información)
- La ingesta diaria recomendada (IDR; RDA, por sus siglas en inglés) para el calcio es de 1,000 mg/día-1,200 mg/día para adultos. (Más información)
- El esqueleto es una reserva de calcio dispuesta a mantener niveles de calcio normales en el suero en caso de calcio dietario inadecuado. De esta manera, la suficiencia de calcio es requerida para maximizar el logro de la masa ósea máxima durante el crecimiento y para prevenir la desmineralización progresiva de los huesos más tarde en la vida, lo cual lleva a osteoporosis, fragilidad ósea, y un incremento en el riesgo de fracturas. (Más información)
- Las altas concentraciones de calcio y oxalato en la orina son factores de riesgo para la formación de piedras de oxalato de calcio en los riñones. Debido a que la ingesta de calcio dietética ha sido inversamente asociada con la ocurrencia de cálculos, se piensa que un consumo adecuado de calcio puede reducir la absorción de oxalato dietario, reduciendo así el oxalato urinario y la formación de cálculos en el riñón. (Más información)
- Datos provenientes de estudios observacionales y ensayos controlados aleatorios apoyan la suplementación con calcio en la reducción del riesgo de presión arterial alta y preeclampsia en mujeres embarazadas. La Organización Mundial de la Salud aconseja que todas las mujeres embarazadas en zonas con ingestas bajas de calcio (es decir, países de bajos ingresos con ingestas alrededor de 300 a 600 mg/día) se les proporcione calcio suplementario al iniciar la 20a semana del embarazo. (Más información)
- Estudios de cohorte prospectivos han reportado una asociación entre las altas ingestas de calcio y un riesgo menor de desarrollar cáncer colorrectal; sin embargo, ensayos clínicos de mayor tamaño de la suplementación con calcio son necesarios. (Más información)
- Los datos actuales disponibles sugieren que ingestas adecuadas de calcio pueden desempeñar un papel en la regulación del peso corporal y tener beneficios terapéuticos en el manejo de síntomas premenstruales de moderados a severos. (Más información)
- Una ingesta adecuada de calcio es crítica para el mantenimiento de un esqueleto sano. El calcio se encuentra en una variedad de alimentos, incluyendo productos lácteos, frijoles, y vegetales de la familia de la col rizada. A pesar de todo, el contenido y biodisponibilidad varía entre los alimentos, y ciertas drogas son conocidas por afectar adversamente la absorción de calcio. (Más información)
- La hipercalcemia, una condición con concentraciones altas anormales de calcio en la sangre, es usualmente debida a malignidad o a hiperparatiroidismo primario. Sin embargo, el uso de grandes dosis de calcio suplementario, junto con álcali absorbible, incrementa el riesgo de hipercalcemia, especialmente en mujeres postmenopáusicas. Con frecuencia asociada con trastornos gastrointestinales, la hipercalcemia puede ser fatal si no es tratada. (Más información)
- Altas ingestas de calcio — tanto de productos lácteos o suplementos — han sido asociadas con riesgos incrementados de cáncer de próstata y eventos cardiovasculares en algunos, pero no todos, los estudios de intervención y estudios observacionales. Sin embargo, actualmente no existe evidencia de tales efectos negativos cuando las personas consumen un total de 1,000 a 1,200 mg/día de calcio (combinación de dieta y suplementos), como lo recomienda la Junta de Alimentos y Nutrición del Instituto de Medicina. (Más información)
El calcio es el mineral más abundante en el cuerpo humano. Alrededor del 99% del calcio en el cuerpo se encuentra en huesos y dientes, mientras que el otro 1% se encuentra en la sangre y tejidos blandos. Las concentraciones de calcio en la sangre y en el líquido que rodea las células (fluido extracelular) deben mantenerse dentro de un rango de concentración muy estrecho para el funcionamiento fisiológico normal. Las funciones fisiológicas del calcio son tan vitales para la supervivencia que el cuerpo estimularía la reabsorción del hueso (desmineralización) para mantener concentraciones de calcio sanguíneo normales cuando la ingesta de calcio es insuficiente. De esta manera, una ingesta adecuada de calcio es un factor crítico en el mantenimiento de un esqueleto sano (1).
Función
Estructura
El calcio es un elemento estructural importante en huesos y dientes. El componente mineral del hueso consiste principalmente de cristales de hidroxiapatita [Ca10(PO4)6(OH)2], los cuales contienen grandes cantidades de calcio, fósforo, y oxígeno. El hueso es un tejido dinámico que se remodela a lo largo de la vida. Las células óseas llamadas osteoclastos comienzan el proceso de remodelación al disolver o reabsorber hueso. Las células formadoras de hueso denominadas osteoblastos entonces sintetizan hueso nuevo para reemplazar el hueso que fue reabsorbido. Durante el crecimiento normal, la formación de hueso excede la reabsorción ósea. La osteoporosis podría ocurrir cuando la reabsorción ósea excede crónicamente la formación (1).
Homeostasis del calcio
Las concentraciones de calcio en la sangre y el fluido que rodea a las células están estrechamente controladas con el fin de preservar la función fisiológica normal. Un ligero descenso en los niveles de calcio en la sangre (p. ej., en el caso de una ingesta inadecuada de calcio) es detectado por las glándulas paratiroides, resultando en su incremento de la secreción de la hormona paratiroidea (PTH). En los riñones, la PTH estimula la conversión de la vitamina D en su forma activa (1,25-dihidroxivitamina D; calcitriol), que disminuye rápidamente la excreción urinaria de calcio pero aumenta la excreción urinaria de fósforo. Las elevaciones de la PTH también estimulan la reabsorción ósea, lo que resulta en la liberación de mineral óseo (calcio y fosfato) — acciones que también contribuyen a restaurar las concentraciones en suero de calcio. El aumento de la 1,25-dihidroxivitamina D circulante también desencadena la absorción intestinal de calcio y fósforo. Al igual que la PTH, la 1,25-dihidroxivitamina D estimula la liberación de calcio del hueso mediante la activación de los osteoclastos (células que reabsorben los huesos). Cuando el calcio en la sangre aumenta a niveles normales, las glándulas paratiroides dejan de secretar PTH. Un ligero aumento en la concentración de calcio en la sangre estimula la producción y secreción de la hormona peptídica, la calcitonina, por la glándula tiroides. La calcitonina inhibe la secreción de PTH, disminuye tanto la reabsorción ósea como la absorción intestinal de calcio, y aumenta la excreción urinaria de calcio (Figura 1). Finalmente, cambios agudos en las concentraciones de calcio sanguíneo no parecen suscitar la secreción del factor de crecimiento fibroblástico 23 (FGF-23) de la hormona fosfatúrica, el cual es producido por las células formadoras de hueso (osteoblastos/osteocitos) en respuesta a incrementos de la ingesta de fósforo (véase artículo en Fósforo) (2). Mientras que este sistema complejo permite un rápido y estrecho control de los niveles de calcio sanguíneos, lo hace a expensas del esqueleto (1).
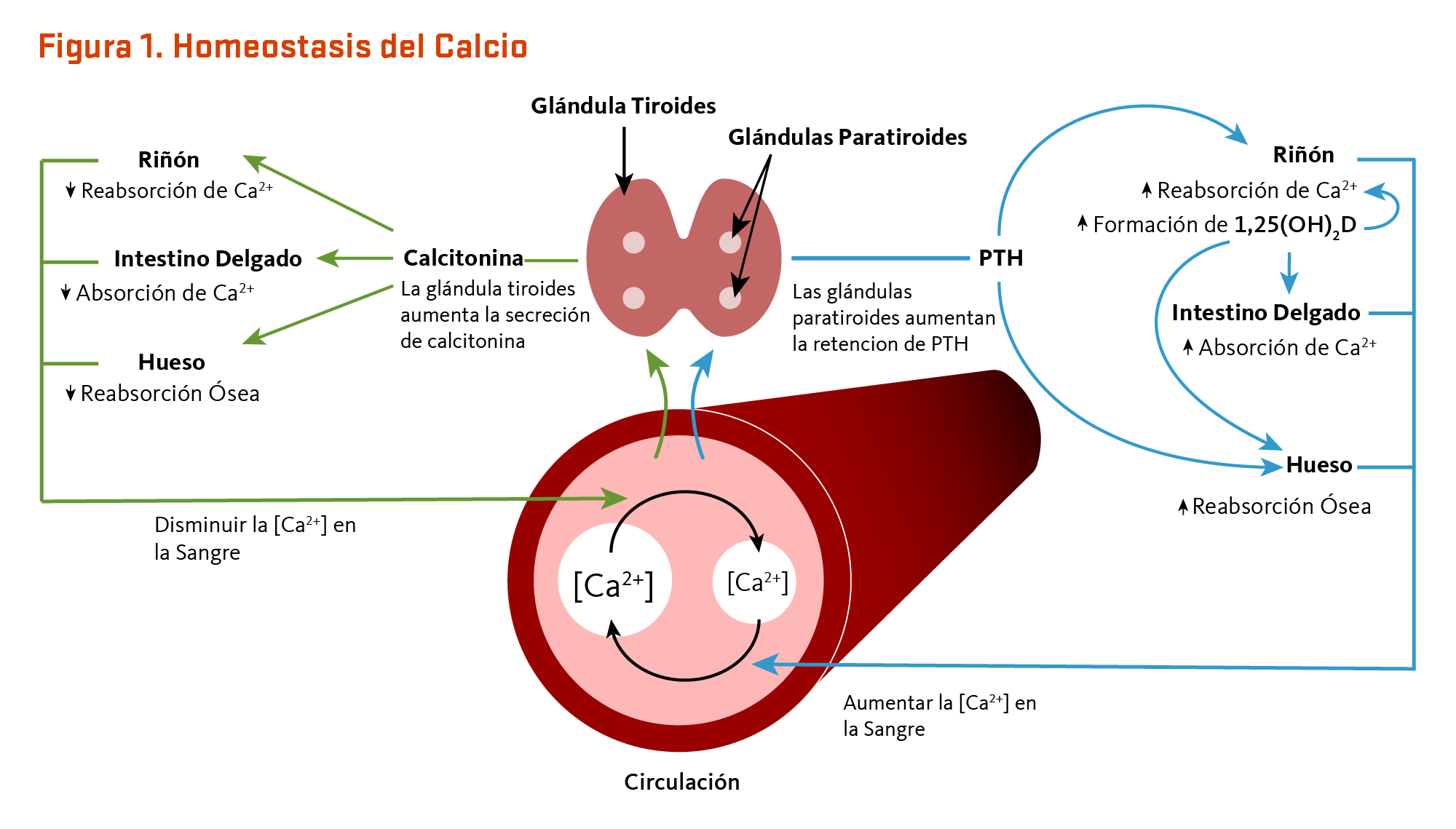
[Figura 1 - Clic para Agrandar]
Señalización celular
El calcio juega un papel en la mediación de la constricción y relajación de los vasos sanguíneos (vasoconstricción y vasodilatación), la transmisión del impulso nervioso, la contracción muscular y la secreción de hormonas como la insulina (1). Las células excitables, tales como las del músculo esquelético y las células nerviosas, contienen canales de calcio voltaje-dependientes de sus membranas celulares que permiten cambios rápidos en las concentraciones de calcio. Por ejemplo, cuando un impulso nervioso estimula una fibra muscular a contraerse, los canales de calcio en la membrana celular se abren para permitir el paso de iones de calcio al interior de la célula muscular. Dentro de la célula estos iones de calcio se unen a proteínas activadoras, las cuales ayudan a liberar un torrente de iones de calcio de las vesículas de almacenaje del retículo endoplásmico (RE) en el interior de la célula. La unión del calcio a la proteína troponina-c inicia una serie de pasos que conducen a la contracción del músculo. La unión del calcio a la proteína calmodulina, activa enzimas que degradan el glicógeno en el músculo para aportar energía a la contracción muscular. Tras completar la acción, el calcio es bombeado fuera de la célula o dentro del RE hasta la siguiente activación (revisado en 3).
Regulación de la función proteínica
El calcio es necesario para estabilizar a un cierto número de proteínas, incluyendo enzimas, optimizando su actividad. La unión de iones de calcio es requerida para la activación de los siete factores "dependientes de vitamina K" en la cascada de la coagulación. El término, "cascada de la coagulación," hace referencia a una serie de eventos, cada uno dependiente del otro que detienen el sangrado a través de la formación de un coágulo (véase el artículo en Vitamina K).
Interacción con nutrientes
Vitamina D
La vitamina D es requerida para la absorción óptima de calcio (véase Función o el artículo sobre Vitamina D). Varios otros nutrientes (y no-nutrientes) influyen en la retención de calcio por el cuerpo y podrían afectar el estado nutricional del calcio.
Sodio
El sodio dietario es un principal determinante de la pérdida urinaria de calcio (1). La ingesta elevada de sodio resulta en una pérdida incrementada de calcio en la orina, posiblemente debido a la competencia entre el sodio y el calcio por la reabsorción en los riñones o por un efecto del sodio sobre la secreción de la hormona paratiroidea (PTH). Se ha encontrado que cada incremento de 1 gramo (g) de sodio (2.5 g de cloruro d sodio; sal NaCl) excretado por el riñón, extrae 26.3 miligramos (mg) de calcio en la orina (1). Un estudio conducido en niñas adolescentes reportó que una dieta alta en sal tuvo un mayor efecto sobre el sodio urinario y la excreción de calcio en niñas de raza blanca en comparación con las de raza negra, sugiriendo diferencias entre grupos étnicos (4). En mujeres adultas, cada gramo extra de sodio consumido por día se proyecta que produce una tasa adicional de pérdida ósea del 1% por año si toda la pérdida de calcio proviniese del esqueleto.
Un cierto número de estudios de intervención y transversales han sugerido que las ingestas altas de sodio son perjudiciales para la salud ósea, especialmente en mujeres mayores (5). Un estudio longitudinal de 2 años en mujeres posmenopáusicas encontró que la excreción urinaria de sodio incrementada (un indicador del incremento de la ingesta de sodio) estaba asociada con una densidad mineral ósea (DMO) disminuida en la cadera (6). Otro estudio en 40 mujeres posmenopáusicas encontró que la adherencia a una dieta baja en sodio (2 g/día) durante seis meses se asoció con reducciones significativas en la excreción de sodio, la excreción de calcio y el propéptido amino-terminal del colágeno tipo I, un biomarcador de la reabsorción ósea. No obstante, estas asociaciones sólo se observaron en mujeres con excreciones urinarias basales elevadas de sodio (7). Finalmente, en un estudio aleatorio, controlado con placebo, en 60 mujeres posmenopáusicas, se ha encontrado que la suplementación con citrato de potasio previene un incremento en la excreción de calcio inducida por el consumo de una dieta alta de sodio (≥5,000 mg/día de sodio elemental) por cuatro semanas (8).
Proteína
El incremento de la ingesta de proteína dietética mejora la absorción intestinal de calcio, como también la excreción urinaria de calcio (9). La Ingesta Diaria Recomendada para la proteína es de 46 gramos (g)/día para mujeres adultas y 56 g/día para hombres adultos; sin embargo, la ingesta promedio de proteínas en los EE.UU. tiende a ser más alta (alrededor de 70 g/día en mujeres adultas y por encima de 100 g por día en hombres adultos) (10). Inicialmente se pensaba que las dietas altas en proteína pueden resultar en un balance negativo del calcio (cuando la suma de la excreción de calcio urinario y fecal se hace mayor que la ingesta de calcio) y así incrementar la pérdida de hueso (11). Sin embargo, la mayoría de los estudios observacionales han reportado tanto ninguna asociación como asociaciones positivas entre la ingesta de proteína y la densidad mineral ósea en niños, adultos, y personas de la tercera edad (revisado en 12). El balance general del calcio parece no cambiar por la alta ingesta dietética de proteína en individuos saludables (13), y evidencia actual sugiere que ingestas de proteína incrementadas en aquellos con suministros adecuados de proteína, calcio, y vitamina D no afectan la DMO o el riesgo de fracturas (14).
Fósforo
El fósforo, el cual es típicamente encontrado en alimentos ricos en proteínas, tiende a incrementar la excreción de calcio en la orina. Se ha encontrado que las dietas con bajas proporciones de calcio a fósforo (Ca:P ≤0.5) incrementan la secreción de la hormona paratiroidea (PTH) y la excreción urinaria de calcio (15, 16). También, la absorción intestinal y la excreción fecal del calcio y fósforo están influenciadas por proporciones de calcio a fósforo de los alimentos ingeridos. De hecho, en el lumen intestinal, las sales de calcio pueden unirse al fósforo para formar complejos que son excretados en las heces. Esto forma las bases para el uso de sales de calcio como quelantes de fósforo para reducir la absorción de fósforo en individuos con insuficiencia renal (17). El incremento de las ingestas de fósforo proveniente de refrescos de cola (altos en ácido fosfórico) y aditivos alimentarios (altos en fosfatos) puede tener efectos adversos en la salud ósea (18). En la actualidad, no hay evidencia convincente de que los niveles de fósforo dietario experimentados en los EE.UU. afectan adversamente la salud ósea. A pesar de todo, la sustitución de la leche u otras fuentes de calcio dietario por grandes cantidades de gaseosas que contienen fósforo puede representar un riesgo serio para la salud ósea en adolescentes y adultos (véase artículo sobre Fósforo).
Cafeína
La exposición a concentraciones de cafeína ≤400 mg/día ha permitido aumentar el contenido de calcio en la orina en dos ensayos controlados aleatorios (19, 20). Sin embargo, la ingesta de cafeína de 400 mg/día no cambió significativamente la excreción de calcio en la orina durante 24 horas en mujeres premenopáusicas en comparación con un placebo (21). Una revisión sistemática de 14 estudios concluyó recientemente que es poco probable que la ingesta diaria de ≤400 mg de cafeína interfieriese con la homeostasis del calcio, impactase negativamente la densidad mineral ósea, o aumente el riesgo de osteoporosis y fractura en individuos con ingestas adecuada de calcio (22).
Deficiencia
Un bajo nivel de calcio sanguíneo (hipocalcemia) usualmente implica una función paratiroidea anormal ya que el esqueleto aporta una gran reserva de calcio para mantener niveles sanguíneos normales, especialmente en el caso de una baja ingesta de calcio. Otras causas de concentraciones bajas de calcio en la sangre incluyen falla renal crónica, deficiencia de vitamina D, y niveles bajos de magnesio sanguíneo frecuentemente observados en casos de alcoholismo severo. La deficiencia de magnesio puede perjudicar la secreción de la hormona paratiroidea (PTH) por las glándulas paratiroideas y disminuir la sensibilidad de los osteoclastos a la PTH. De esta manera, la suplementación con magnesio es requerida para corregir la hipocalcemia en personas con concentraciones bajas de magnesio en el suero (véase artículo sobre Magnesio). Las ingestas de calcio crónicamente bajas en individuos en crecimiento pueden prevenir el logro de una masa ósea máxima óptima. Una vez que se alcanza la masa ósea máxima, la ingesta inadecuada de calcio puede contribuir a la pérdida ósea acelerada y, en última instancia, al desarrollo de la osteoporosis (véase Prevención de Enfermedad) (1).
La Ingesta Diaria Recomendada (IDR)
Recomendaciones actualizadas para la ingesta de calcio basadas en la optimización de la salud ósea fueron dadas a conocer por la Junta de Nutrición y Alimentos (JNA) del Instituto de Medicina en el 2011 (9). La Ingesta Diaria Recomendada (IDR; RDA, por sus siglas en inglés) para el calcio es listada en la Tabla 1 por etapa de vida y género.
| Etapa de la Vida | Edad |
Machos (mg/día) |
Hembras (mg/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 200 (IA) | 200 (IA) |
| Infantes | 6-12 meses | 260 (IA) | 260 (IA) |
|
Niños
|
1-3 años | 700 | 700 |
|
Niños
|
4-8 años | 1,000 | 1,000 |
| Niños | 9-13 años | 1,300 | 1,300 |
| Adolescentes | 14-18 años | 1,300 | 1,300 |
| Adultos | 19-50 años | 1,000 | 1,000 |
| Adultos | 51-70 años | 1,000 | 1,200 |
| Adultos | 71 años y más | 1,200 | 1,200 |
| Embarazo | 14-18 años | - | 1,300 |
| Embarazo | 19-50 años | - | 1,000 |
| Período de lactancia | 14-18 años | - | 1,300 |
| Período de lactancia | 19-50 años | - | 1,000 |
Prevención de Enfermedad
Osteoporosis
La osteoporosis es un trastorno esquelético en el que se compromete la fuerza ósea, resultado en un riesgo de fractura incrementado. Sufrir una fractura de cadera es una de las consecuencias más serias de la osteoporosis. Casi un tercio de aquellos que sufren fracturas osteoporóticas de cadera ingresan a casas de reposo al año siguiente de la fractura, y una de cada cuatro personas muere dentro del año de haber sufrido una fractura osteoporótica de cadera (23). A pesar de ser un diagnóstico común en las mujeres postmenopáusicas, la osteoporosis también afecta de un 4%-6% de los hombres mayores de 50 años (24).
La osteoporosis es un trastorno multifactorial, y la nutrición es solamente uno de los factores contribuyentes a su desarrollo y progresión (25). Otros factores que incrementan el riesgo de desarrollar osteoporosis incluyen, pero sin limitarse, a la edad avanzada, al género femenino, a la deficiencia de estrógeno, al fumar, al alto consumo de alcohol (tres o más bebidas/día), a las enfermedades metabólicas (p. ej., hipertiroidismo), y al uso de ciertos medicamentos (p. ej., corticosteroides y anticonvulsivos) (26). Una predisposición a fracturas osteoporóticas se relaciona con el nivel máximo de masa ósea y con la tasa de pérdida ósea luego de haber alcanzado el nivel máximo de masa ósea. Luego de alcanzada la altura adulta, el esqueleto continúa acumulando hueso hasta la tercera década de la vida. Los factores genéticos ejercen una fuerte influencia sobre el nivel máximo de masa ósea, pero los factores del estilo de vida también pueden jugar un papel significativo. Las estrategias para reducir el riesgo de una fractura osteoporótica incluyen la obtención del nivel máximo de masa ósea y la reducción de la pérdida ósea con la edad. Un cierto número de factores del estilo de vida, incluyendo la dieta (especialmente la ingesta de calcio y proteína) y la actividad física, son susceptibles a intervenciones destinadas a maximizar la masa ósea máxima y limitar el riesgo de fractura osteoporótica (27).
El ejercicio físico es un factor del estilo de vida que ha sido asociado con numerosos beneficios y es probable que contribuya en la prevención de la osteoporosis y fractura osteoporótica. Hay evidencia que sugiere que la actividad física a temprana edad contribuye a la obtención de niveles máximos de masa ósea más altos (27). Por otra parte, la participación continua en actividades físicas en presencia de cantidades adecuadas de calcio y suministros de vitamina D (provenientes de fuentes dietéticas y/o exposición al sol) podrían tener un efecto modesto en la disminución de la tasa de pérdida ósea con la edad (28). Los lineamientos actuales de la Fundación Nacional de Osteoporosis incluyen recomendaciones de estiramientos musculares regulares y ejercicios de resistencia (pesas) para todas las mujeres postmenopáusicas y hombres de 50 años y más (29). Aunque los beneficios en la reducción de la pérdida ósea podrían ser limitados, ejercicios de fortalecimiento muscular, incluyendo levantamiento de pesas y otros ejercicios de resistencia (p. ej., yoga y Pilates) y ejercicios que requieren sostener el propio peso (p. ej., caminar, trotar, y subir escaleras), pueden mejorar la fuerza, postura, balance y coordinación, contribuyendo así a reducir el riesgo de caídas (29). Una compilación de ensayos publicados sobre el calcio, indicó que el efecto beneficioso al esqueleto de la actividad física incrementada se alcanzaba sólo a ingestas de calcio superiores a 1,000 mg/día en mujeres con menopausia tardía (revisado en 28).
La pérdida progresiva de la densidad mineral ósea (DMO) que conduce a la osteopenia (pre-osteoporosis) y la osteoporosis por lo general se evalúa por la absorciometría de rayos X de energía dual (DEXA) en la cadera y espina lumbar (30). Varios ensayos clínicos aleatorios controlados con placebo han evaluado el efecto del calcio suplementario en la preservación de la DMO y la prevención del riesgo de fracturas en hombres y mujeres de 50 años y mayores. Un meta-análisis de 15 ensayos controlados aleatorios, incluyendo a 1,533 hombres y mujeres mayores de 50 años de edad, encontró que el aumento de la ingesta de calcio proveniente de fuentes dietéticas (es decir, leche, leche en polvo, productos lácteos o preparaciones de hidroxiapatita) incrementó la DMO en un 0,6%-1% en la cadera (+ 0,6%) y cuerpo total (+ 1,0%) después de un año y en un 0,7% -1,8% en la columna lumbar (+ 0,7%), cuello femoral (+ 1,8%), cadera total (+ 1,5%), y sitios de cuerpo total (+ 0,9%) después de dos años (31). Un meta-análisis de 51 ensayos controlados aleatorios en 12,257 adultos (>50 años) encontró que la DMO en todos los sitios óseos (columna lumbar, cuello femoral, cadera total, antebrazo) aumentó en un 0,7%-1,4% después de un año y un 0,8%-1,5% después de dos años de calcio suplementario, por sí solo o en combinación con vitamina D (31). Tales aumentos modestos pueden ayudar a limitar la tasa promedio de pérdida de DMO después de la menopausia, pero es poco probable que se traduzcan en reducciones significativas del riesgo de fractura. Un metanálisis de 20 ensayos controlados aleatorios que informaron sobre el riesgo total de fractura encontró una reducción del riesgo del 11% asociada con el suplemento de calcio con o sin vitamina D (32). Sin embargo, no hubo ningún efecto cuando el análisis se limitó a los ensayos más grandes con el menor riesgo de sesgo. Además, no se encontraron reducciones en los riesgos de fracturas de cadera, vertebrales y antebrazos con suplementos de calcio (32). Debido a que la abstinencia de estrógenos perjudica significativamente la absorción intestinal y la reabsorción renal de calcio, el nivel de requerimiento de calcio puede depender de si las mujeres posmenopáusicas reciben terapia de reemplazo hormonal (28).
La Fuerza de Trabajo de Servicios Preventivos de los EE.UU. condujo un meta-análisis de 11 ensayos aleatorios controlados con placebo que incluyó a 52,915 personas mayores (de las cuales el 69% eran mujeres posmenopáusicas) e informó que la suplementación de vitamina D (300-1,000 UI/día) y calcio (500-1,200 mg/día) durante hasta siete años produjo una reducción del 12% en el riesgo de cualquier nueva fractura (33). No hubo un efecto significativo de la vitamina D sin calcio (33). Un meta-análisis recientemente actualizado de ensayos aleatorios controlados con placebo encargado por la Fundación Nacional de Osteoporosis encontró una reducción del 15% en el riesgo de fractura total (8 estudios) y una reducción del 30% en el riesgo de fracturas de cadera (seis estudios) con suplementos de calcio y vitamina D en personas mayores (34). La Fundación Nacional de Osteoporosis recomienda que la ingesta adecuada de calcio (1,000-1,200 mg/día) y vitamina D (800-1,000 UI/día) se incluya en la dieta de todos los hombres y mujeres de mediana edad (35).
El papel y la eficacia de la suplementación con vitamina D en el fortalecimiento óseo y la prevención de fracturas en personas de edad avanzada permanecen siendo temas de controversia. La forma activa de la vitamina D, 1,25-dihidroxivitamina D, estimula la absorción de calcio al promover la síntesis de las proteínas que se unen al calcio en el intestino. Mientras que ninguna cantidad de vitamina D puede compensar la ingesta total inadecuada de calcio, la insuficiencia de vitamina D (definida como concentraciones circulantes de 25-hidroxivitamina D por debajo de los 20 ng/mL [50 nmol/L]) puede llevar a un hiperparatiroidismo secundario y a un riesgo incrementado de osteoporosis (9, 36). Por el contrario, en mujeres postmenopáusicas (edad entre 57-90 años) con ingestas totales adecuadas de calcio (1,400 UI/día), concentraciones en el suero de 25-hidroxivitamina D oscilando de entre 20 ng/mL a 66 ng/mL tuvieron poco efecto en la absorción de calcio (solo un incremento del 6% sobre el intervalo) (37). En un ensayo aleatorio, controlado con placebo, la suplementación con 1,000 UI/día de vitamina D en mujeres postmenopáusicas (edad media, 77.2 años) por un año encontró que esta incrementó significativamente las concentraciones de 25-hidroxivitamina D circulantes en un 34% respecto al valor basal, pero falló en mejorar la absorción de calcio en presencia de altas ingestas totales de calcio (calcio dietario más suplementario correspondiendo a un promedio de 2,100 mg/día) (38). Este estudio también reportó que no hubo diferencias significativas en las medidas de la DMO en la cadera y en el resto del cuerpo entre las mujeres tratadas con placebo y vitamina D. Además, el análisis agrupado de siete ensayos aleatorios controlados, incluyendo 65,517 individuos mayores viviendo en la comunidad o institución, encontró que la vitamina D (400-800 UI/día) puede reducir el riesgo de cualquier fractura únicamente cuando es combinada con calcio (1,000 mg/día) (39). Curiosamente, los resultados de una serie de ensayos incluidos en tres meta-análisis recientes (33, 40, 41) han sugerido que la vitamina D y el calcio suplementarios podrían tener grandes beneficios en la prevención de fracturas en personas de edad avanzada e institucionalizados que también se encuentran en un riesgo incrementado de deficiencia de vitamina C y fracturas en comparación con los que viven en la comunidad (42, 43).
Para más información acerca de la salud ósea y la osteoporosis, visite el sitio web de la Fundación Nacional de la Osteoporosis.
Cálculos renales
Aproximadamente el 6% de las mujeres y el 15% de los hombres en países industrializados tendrán un cálculo renal durante su vida. La mayoría de los cálculos renales están compuestos de oxalato de calcio o fosfato de calcio. Sujetos con un nivel anormal de calcio en la orina (hipercalciuria) están en un riesgo elevado de desarrollar cálculos renales (un proceso llamado nefrolitiasis) (44). Un nivel alto de oxalato urinario es otro factor de riesgo para la formación de cálculos de oxalato de calcio. La mayoría de los sujetos con un historial de cálculos renales y/o hipercalciuria idiopática tienen una absorción intestinal de calcio incrementada (45). Aunque inicialmente se recomendó limitar la ingesta dietética de calcio en estos pacientes, un cierto número de estudios de cohorte prospectivos ha reportado asociaciones entre la baja ingesta total de calcio dietario y un incremento en el riesgo de la incidencia de cálculos renales (46-48). Los análisis prospectivos de tres cohortes de gran tamaño, incluyendo un total de 30,762 hombres y 195,865 mujeres con un seguimiento combinado de 56 años, han indicado que el riesgo de cálculos renales fue significativamente más bajo en individuos en el quintil más alto en comparación con aquellos en el quintil más bajo de la ingesta dietética de calcio proveniente de fuentes lácteas o no lácteas (49). Adicionalmente, un estudio de intervención aleatorio de cinco años que enlistó 120 hombres con hipercalciuria idiopática (edad media, 45 años) reportó que aquellos a los que se les asignó una dieta baja en calcio (aproximadamente 400 mg/día) tuvieron un riesgo de 51% más alto en la recurrencia de cálculos renales en comparación con aquellos con una dieta normal-a-alta de calcio (1,200 mg/día), baja en proteína animal y baja en sal (50).
Los mecanismos por los cuales un incremento del calcio dietario podría reducir el riesgo de la incidencia de cálculos renales no son completamente comprendidos. Una relación inversa se reportó entre la ingesta total de calcio y la absorción intestinal de calcio en el reciente análisis transversal de una cohorte de 5,452 mujeres postmenopáusicas (45). Por otra parte, las mujeres con una ingesta elevada de calcio suplementario y una absorción baja de calcio fueron menos propensas a reportar un historial de cálculos renales (45). La ingesta adecuada de calcio con los alimentos puede reducir la absorción de oxalato en la dieta y disminuir el oxalato urinario a través de la formación de la sal de oxalato de calcio insoluble (51, 52). Un reciente estudio de intervención en 10 adultos jóvenes sin formaciones de cálculos previos observó que la ingesta de grandes cantidades de oxalato no incrementó el riesgo de la ocurrencia de cálculos de oxalato de calcio en presencia de un nivel recomendado de calcio dietario (53).
Sin embargo, un ensayo aleatorio, doble ciego, controlado con placebo en 36,282 mujeres postmenopáusicas reportó que una combinación de calcio suplementario (1,000 mg/día) y vitamina D (400 UI/día) se asoció con un incremento significativo de la incidencia de cálculos renales reportados por los participantes durante un período de tratamiento de siete años. Más ensayos controlados pueden ser necesarios para determinar si el calcio suplementario afecta al riesgo de cálculos renales (54). Sin embargo, una revisión sistemática de estudios observacionales y ensayos controlados aleatorios que reportó principalmente resultados relacionados con los huesos falló en encontrar un efecto de la suplementación con calcio en la incidencia de cálculos (55). Un riesgo potencial de cálculos renales asociado con la suplementación con calcio probablemente dependerá de si el calcio suplementario se ingiere conjuntamente con alimentos que contienen oxalato o si se consume por separado. Se necesita investigación adicional para verificar si los medicamentos para el tratamiento de la osteoporosis (p. ej., los bifosfonatos) en lugar de los suplementos de calcio podrían influir en el riesgo de aparición de cálculos (56).
Datos actuales sugieren que las dietas que proveen de calcio dietario adecuado y bajos niveles de proteína animal, oxalato y sodio podrían beneficiar en la prevención de la recurrencia de cálculos en sujetos con hipercalciuria idiopática (57-59).
Trastornos hipertensivos del embarazo
Los trastornos hipertensivos inducidos por el embarazo, incluyendo hipertensión gestacional, preeclampsia, y eclampsia, complican aproximadamente el 10% de los embarazos y son un mayor riesgo para la salud en mujeres embarazadas y sus descendientes (60). La hipertensión gestacional se define como una presión arterial anormal que usualmente se desarrolla después de la 20a semana del embarazo. La preeclamsia se caracteriza por la escasa perfusión placentaria y una inflamación sistémica que puede involucrar varios sistemas incluyendo el sistema cardiovascular, riñones, hígado y el sistema hematológico (61). Además de la hipertensión gestacional, la preeclampsia está asociada con el desarrollo de hinchazón severa (edema) y la presencia de proteína en la orina (proteinuria). La eclampsia es la ocurrencia de convulsiones en asociación con el síndrome de preeclampsia y es una causa significativa de mortalidad materna y perinatal.
Aunque los casos de preeclampsia están en un alto riesgo de desarrollar eclampsia, un cuarto de las mujeres con eclampsia no exhibe síntomas de preeclampsia al inicio. Factores de riesgo para la preeclampsia incluyen predisposición genética, edad materna avanzada, primeros embarazos, embarazos múltiples (p. ej., gemelos o trillizos), obesidad, diabetes, y algunas enfermedades autoinmunes (61). Mientras que la patogénesis de la preeclamsia no es completamente entendida, parece que la nutrición y especialmente el metabolismo del calcio desempeñan un papel. Datos de estudios epidemiológicos han sugerido una relación inversa entre la ingesta de calcio durante el embarazo y la incidencia de preeclampsia (revisado en 62). El deterioro del metabolismo del calcio cuando la concentración de vitamina D circulante es baja y/o cuando la ingesta de calcio dietario es inadecuada puede contribuir al riesgo de hipertensión durante el embarazo.
El hiperparatiroidismo secundario (un nivel alto de PTH) debido a la deficiencia de vitamina D en mujeres jóvenes embarazadas ha sido asociado con una presión sanguínea materna alta y un riesgo incrementado de preeclampsia (63). Se encontró que el riesgo de una concentración de PTH alta era elevado en mujeres deficientes de vitamina D con bajas ingestas de calcio (<480 mg/día) durante el embarazo cuando se comparó con ingestas adecuadas-a-altas de calcio (≥1,000 mg/día) (64). Además, la deficiencia de vitamina D puede desencadenar la hipertensión a través de la activación inapropiada del sistema renina-angiotensina (véase el artículo sobre Vitamina D).
Efectos potencialmente benéficos en la prevención de la preeclampsia han sido investigados en varios estudios aleatorios controlados con placebo. El meta-análisis más reciente de 13 ensayos en 15,730 mujeres embarazadas encontró que la suplementación con calcio de al menos 1,000 mg/día (mayormente 1,500-2,000 mg/día) de alrededor de 20 semanas de embarazo (34 semanas de embarazo a más tardar) fue asociada con reducciones significantes en el riesgo de presión sanguínea alta, preeclampsia, y nacimiento prematuro (62). Mayores reducciones del riesgo fueron reportadas entre las mujeres embarazadas en alto riesgo de preeclampsia (5 ensayos; 587 mujeres) o con una ingesta dietética baja en calcio (8 ensayos; 10,678 mujeres). Otro meta-análisis de nueve ensayos controlados aleatorios en mujeres de alto riesgo indicó que dosis más bajas de suplementos de calcio (≤800 mg/día), solos o con un tratamiento complementario (es decir, vitamina D, ácido linoleico o antioxidantes), podrían también disminuir el riesgo de preeclampsia en un 62% (65). A pesar de todo, basándose en la revisión sistemática de ensayos aleatorios controlados de alta calidad, los cuales usaron mayormente suplementos de calcio en altas dosis, la Organización Mundial de la Salud (OMS) recientemente recomendó que todas la mujeres embarazadas en áreas con ingestas bajas de calcio (es decir, países de bajos ingresos con ingestas de alrededor de 300-600 mg/día) se les sea dado de entre 1.5 a 2 g (1,500 a 2,000 mg)/día de calcio elemental a partir de la 20a semana de embarazo (66).
Debido a que una suplementación excesiva con calcio puede ser dañina (véase Seguridad), investigación adicional es requerida para verificar si la suplementación con calcio por encima de la recomendación actual del Instituto de Medicina (1,000 mg/día para mujeres embarazadas de entre 19-50 años) proporciona mayores beneficios para las mujeres en alto riesgo de preeclampsia. Por último, la falta de efecto de los suplementos de calcio sobre la proteinuria (reportada en dos ensayos solamente) sugirió que la suplementación con calcio a partir de la mitad del embarazo podría ser demasiado tarde para oponerse a la génesis de la preeclampsia (67, 68). Un estudio aleatorio, doble ciego, controlado con placebo — el Ensayo de la OMS sobre Calcio y Preeclampsia (CAP) — está en curso para evaluar el efecto de la suplementación con calcio de 500 mg/día, iniciando antes del embarazo y hasta la 20a semana de embarazo, en el riesgo de preeclampsia en mujeres de alto riesgo (69, 70).
Cáncer colorrectal
El cáncer colorrectal (CRC) es el cáncer gastrointestinal más común y la segunda causa principal de muertes por cáncer en los EE.UU. (71). El cáncer colorrectal es causado por una combinación de factores genéticos y ambientales, pero el grado en que estos dos tipos de factores influyen en el riesgo de cáncer colorrectal en las personas varía ampliamente. En individuos con poliposis adenomatosa familiar (PAF) o cáncer colorrectal hereditario no asociado a la poliposis, la causa del cáncer es casi completamente genética, mientras que los factores de estilo de vida, incluyendo hábitos dietéticos, el uso de tabaco, y las actividades físicas, influyen en gran manera en el riesgo de cáncer colorrectal esporádico (no hereditario).
Estudios de cohorte prospectivos han reportado consistentemente una asociación inversa entre el consumo de lácteos y el riesgo de cáncer colorrectal. Estudios experimentales en el cultivo celular y modelos en animales han sugerido mecanismos plausibles subyaciendo un papel para el calcio, un nutriente principal en productos lácteos, en la prevención de cáncer colorrectal (72). En el estudio prospectivo multicéntrico de la Investigación Prospectiva Europea sobre el Cáncer y Nutrición (EPIC) de 477,122 individuos, con un seguimiento promedio de 11 años, 4,513 casos de cáncer colorrectal fueron documentados (73). Ingestas de leche, queso, y yogurt, fueron inversamente asociadas con el riesgo de cáncer colorrectal. El quintil más alto de la ingesta total de lácteos frente al más bajo (≥490 g/día vs. <134 g/día) se asoció con un riesgo 23% menor del riesgo de cáncer colorrectal. De igual manera, el riesgo de cáncer colorrectal fue 25% menor en aquellos en la parte superior del quintil en comparación con aquellos en el quintil inferior de la ingesta de calcio de productos lácteos (≥839 mg/día vs. <308 mg/día). El seguimiento de 16 años de 41,403 mujeres (edades de entre 26-46 años al momento de la inclusión) del prospectivo Estudio de Salud de Enfermeras II (NHS II) documentó 2,273 diagnósticos de adenomas colorrectales (pólipos precancerosos). El análisis de la cohorte prospectiva encontró que las mujeres con una ingesta total de calcio de 1,001-1,250 mg/día tuvieron un riesgo 76% menor de desarrollar adenomas avanzados (es decir, adenomas más propensos a convertirse en malignos) en comparación con aquellas con ingestas iguales a o por debajo de los 500 mg/día (74). Además, un análisis dosis-respuesta que usó datos de ocho estudios prospectivos (11,005 casos de cáncer colorrectal) estimó que un incremento de 300 mg/día en la ingesta total de calcio se asoció con un 5% en la reducción del riesgo de cáncer colorrectal (75). La ingesta total diaria de calcio osciló entre 333 a 2,229 mg en los estudios examinados. Además, en el análisis dosis-respuesta de seis estudios prospectivos (8,839 casos de cáncer colorrectal entre 920,837 participantes) se encontró 11% menores probabilidades de adenomas de alto riesgo por cada incremento de 300 mg/día en el calcio total (75).
Sin embargo, el reciente meta-análisis de siete estudios aleatorios, doble ciego, controlados con placebo no encontró evidencia de efecto alguno en la suplementación con calcio (≥500 mg/día) por un periodo medio de 45 meses en el riesgo total de cáncer y en el riesgo de cáncer colorrectal (76). Además, el reciente meta-análisis del ensayo controlado con placebo de la Iniciativa de Salud de las Mujeres falló en demostrar un reducción en el riesgo de cáncer colorrectal en mujeres postmenopáusicas suplementadas con tanto vitamina D (400 UI/día) como calcio (1,000 mg/día) por siete años (77). Finalmente, los resultados del meta-análisis de tres ensayos aleatorios, controlados con placebo han sugerido que la suplementación con calcio (1,200-2,000 mg/día) puede reducir el riesgo de la recurrencia de adenoma en 13% durante tres a cinco años en sujetos con un historial de adenomas (78). Actualmente, no es claro aún si la suplementación con calcio es benéfica en la prevención de cáncer colorrectal. Ensayos de gran tamaño diseñados para evaluar principalmente el efecto de la suplementación a largo plazo en la incidencia de adenomas y/o cáncer colorrectal son necesarios antes de sacar conclusiones.
Toxicidad por plomo
Niños que son expuestos crónicamente al plomo, incluso en pequeñas cantidades, son más propensos a desarrollar problemas de aprendizaje, problemas de conducta, y a tener bajo CI. Déficits en el crecimiento y desarrollo neurológico pueden ocurrir en los infantes de mujeres expuestas al plomo durante el embarazo y lactancia. En los adultos la toxicidad por plomo puede resultar en daño renal y presión sanguínea alta. Aunque en EE.UU. se ha descontinuado el uso de plomo en pinturas, gasolina y latas de alimentos, la toxicidad por plomo continúa siendo un problema de salud significativo, especialmente en niños que viven en áreas urbanas (79).
En el 2012, los Centros para el Control y Prevención de Enfermedades de los Estados Unidos establecieron el valor de referencia para la concentración de plomo en la sangre a 5 microgramos por decilitro (μg/dL) para identificar niños en riesgo (80). A pesar de todo no existe una concentración conocida de plomo en la sangre por debajo de la cual los niños están 100% seguros. Un estudio temprano de más de 300 niños en un vecindario urbano encontró que el 49% de los niños de entre 1 a 8 años tenían niveles de plomo en la sangre por arriba del límite de 10 μg/dL, indicando una exposición excesiva al plomo. En este estudio, solo el 59% de los niños de entre 1 a 3 años y el 41% de los niños de entre 4 a 8 años satisficieron los niveles recomendados para las ingestas de calcio (81).
Una ingesta adecuada de calcio podría tener un efecto protector contra la toxicidad del plomo en al menos dos formas. Se sabe que la ingesta dietética aumentada de calcio disminuye la absorción gastrointestinal del plomo. Una vez que el plomo entra en el cuerpo, el mismo tiende a acumularse en el esqueleto, donde puede permanecer por más de 20 años. La ingesta de calcio adecuada también previene de la exposición al plomo liberado desde el esqueleto durante la desmineralización ósea. Un estudio de las concentraciones de plomo circulante durante el embarazo encontró que las mujeres con una ingesta inadecuada de calcio durante la segunda mitad del embarazo eran más propensas a tener niveles elevados de plomo sanguíneo, probablemente debido al incremento de la desmineralización ósea, llevando a la liberación del plomo acumulado a la sangre (82). El plomo en la sangre de una mujer embarazada es fácilmente transportado a través de la placenta resultando en exposición del feto al plomo en un momento en que el sistema nervioso en desarrollo es altamente vulnerable. En un estudio aleatorio, doble ciego, controlado con placebo en 670 mujeres embarazadas (≤14 semanas de gestación) con ingestas promedio de calcio dietario de 900 mg/día, una suplementación diaria de 1,200 mg de calcio durante el periodo de embarazo resultó en reducciones de 8%-14% en las concentraciones de plomo en la sangre materna (83). Reducciones similares en concentraciones maternas de plomo en la sangre y leche materna de las madres en período de lactancia suplementadas con calcio fueron reportadas en ensayos previos (84, 85). En mujeres posmenopáusicas, los factores que se sabe que disminuyen la desmineralización ósea, incluida la terapia de reemplazo de estrógenos y la actividad física, se han asociado de manera inversa con los niveles de plomo en la sangre (86).
Tratamiento de Enfermedad
Sobrepeso y obesidad
Una ingesta de calcio dietario alta, usualmente asociada con el consumo de productos lácteos, ha sido inversamente relacionada con el peso corporal y la obesidad central en un cierto número de estudios de corte transversal (revisado en 87). Análisis de datos basales transversales de un cierto número de estudios de cohorte prospectivos que no fueron diseñados e impulsados para examinar el efecto de la ingesta de calcio o consumo de lácteos en la obesidad o grasa corporal han proporcionado resultados inconsistentes (87). A pesar de todo, un meta-análisis de 18 estudios de corte transversal y prospectivos pronosticó una reducción en el índice de masa corporal (una medida relativa del peso corporal; IMC) de 1.1 kg/m2 con un incremento en la ingesta de calcio de 400 mg/día a 1,200 mg/día (87). En un estudio de intervención controlado con placebo, 32 sujetos con obesidad fueron asignados aleatoriamente a regímenes de restricción de energía (déficit de 500 kCal/día) por 24 semanas con (1) una dieta estándar que proveía de 400 a 500 mg/día de calcio dietario y un placebo (dieta "baja en calcio"), (2) una dieta estándar y 800 mg/día de calcio suplementario (dieta "alta en calcio"), o (3) una dieta alta en productos lácteos que proveía 1,200 mg/día de calcio dietario y un placebo (88). Las dietas con restricción de energía resultaron en una pérdida significante de peso y grasa corporal en los tres grupos. A pesar de todo, el peso corporal y la pérdida de grasa fueron significativamente más reducidos con la dieta alta en calcio en comparación a la dieta estándar, y reducciones adicionales fueron medidas con la dieta alta en lácteos en comparación a las dietas altas y bajas en calcio. Estos resultados sugieren que mientras la ingesta de calcio puede desempeñar un papel en la regulación del peso corporal, los beneficios adicionales podrían ser atribuidos a otros componentes bioactivos de los productos lácteos, como las proteínas, ácidos grasos, y aminoácidos de cadena ramificada.
A pesar de todo, varios mecanismos han sido propuestos para explicar el potencial impacto del calcio en el peso corporal (revisado en 87). El mecanismo más citado está basado en estudios en el modelo de ratón agutí que muestra que las ingestas bajas de calcio mediante el incremento de la circulación de la hormona paratiroidea (PTH) y vitamina D, pudieron estimular la acumulación de grasa (lipogénesis) en adipocitos (células grasas) (89). Inversamente, altas ingestas de calcio pueden reducir el almacenamiento de grasa, estimular la descomposición de lípidos (lipólisis), e impulsar la oxidación de grasas. Un reciente meta-análisis de ensayos controlados aleatorios estimo que la ingesta alta de calcio (1,300 mg/día) frente a la ingesta baja (488 mg/día) por un mínimo de siete días incrementó la oxidación de grasas en un 11% (90). Sin embargo, un ensayo cruzado, aleatorio, doble ciego, controlado con placebo en 10 individuos con bajo consumo de calcio obesos o con sobrepeso reportó que la suplementación con 800 mg/día de calcio por 5 semanas falló en modificar la expresión de los factores clave que participan en el metabolismo de la grasa (91). Por otra parte, mientras el modelo sugiere un papel de la vitamina D en la lipogénesis (almacenamiento de grasas), estudios en humanos han mostrado que la deficiencia de vitamina D — en lugar de la suficiencia — es frecuentemente asociada con la obesidad, y la vitamina D suplementaria podría ser efectiva en la disminución del peso corporal cuando la restricción calórica es impuesta (92, 93). Otro mecanismo sugiere que las dietas altas en calcio pueden limitar la absorción de grasa dietética en el intestino e incrementar la excreción de grasa fecal. De hecho, en el tracto gastrointestinal, el calcio puede atrapar la grasa dietética en jabones de calcio insolubles de ácidos grasos que son entonces excretados (94). Además, a pesar de la evidencia muy limitada, se ha propuesto que el calcio pudiese estar involucrado en la regulación del apetito y la ingesta de energía (95).
Hasta la fecha, no hay consenso sobre el efecto del calcio en los cambios de peso corporal. Un meta-análisis de 29 ensayos controlados aleatorios en 2,441 participantes (edad media, 41.4 años) encontró que la suplementación con calcio solo se asoció con el peso corporal y la pérdida de grasa en estudios a corto plazo (<1 año) que utilizaron dietas con restricción energética (96). Otro meta-análisis de 41 ensayos controlados aleatorios (4,802 participantes) encontró poco o ningún efecto del aumento de la ingesta de calcio de los suplementos o alimentos lácteos durante >12 semanas en el peso corporal y la composición corporal (97). Finalmente, un meta-análisis de 33 estudios aleatorizados los ensayos controlados (4,733 participantes) no encontró ningún efecto general de la suplementación con calcio (de alimentos o suplementos) durante >12 semanas en los cambios de peso corporal. Sin embargo, otros análisis de subgrupos mostraron reducciones de peso en niños y adolescentes (promedio, -0.26 kg), en adultos (promedio, -0.91 kg) y en aquellos con IMC normal (promedio, -0.53 kg). El calcio suplementario no condujo a la pérdida de peso en mujeres posmenopáusicas o en personas con sobrepeso/obesidad (98). En la actualidad, se justifica la investigación adicional para examinar el efecto de la ingesta de calcio en el metabolismo de la grasa, así como sus beneficios potenciales en el manejo del peso corporal con o sin restricción calórica (99).
Síndrome Premenstrual (SPM)
El SPM hace referencia a un conjunto de síntomas, incluyendo, pero no limitándose a la fatiga, irritabilidad, mal humor/depresión, retención de líquidos, y sensibilidad mamaria, que comienza al poco tiempo luego de la ovulación (mitad de ciclo) y termina con el comienzo de la menstruación (el periodo menstrual) (100). Una forma severa del SPM llamada trastorno disfórico premenstrual (TDPM) ha sido descrito en 3%-8% de las mujeres en edad de procrear. El TDPM interfiere con el funcionamiento normal, afectando las actividades y relaciones diarias (101).
Las ingestas bajas de calcio dietético han sido ligadas al SPM en reportes tempranos, y se ha demostrado que el calcio suplementario disminuye la severidad de los síntomas (102). Un estudio caso-control anidado dentro del Estudio de Salud para Enfermeras II (NHS II) encontró que las mujeres en el quintil más alto de la ingesta dietética de calcio (pero no suplementaria) (mediana de 1,283 mg/día) tenían un riesgo 30% menor de desarrollar SPM en comparación con los del quintil más bajo (mediana de 529 mg/día). De manera similar, las mujeres en el quintil más alto versus el más bajo de la leche descremada o baja en grasa (≥4 porciones/día vs. ≤1 porción/semana) tuvieron un riesgo 46% menor de SPM (103). En un ensayo clínico, aleatorio, doble ciego, controlado con placebo de 466 mujeres con síntomas premenstruales de moderados a graves, el suplemento de calcio (1,200 mg / día) durante tres ciclos menstruales se asoció con una reducción del 48% en las puntuaciones totales de los síntomas, en comparación a una reducción del 30% observada en el grupo placebo (104). Se reportaron efectos positivos similares en los primeros ensayos doble ciego, controlados con placebo, cruzados que administraron 1,000 mg de calcio al día (105, 106). Recientes ensayos controlados aleatorios pequeños también reportaron que el calcio suplementario (400-500 mg/día) durante tres semanas a tres meses redujo la gravedad y/o la frecuencia de los síntomas en mujeres con SPM leve a moderado (107-110). Los datos disponibles actualmente indican que las ingestas diarias de calcio de alimentos y/o suplementos pueden tener beneficios terapéuticos en mujeres diagnosticadas con SPM o TDPM (111, 112).
Hipertensión
La relación entre la ingesta de calcio y la presión arterial se ha investigado ampliamente en las últimas décadas. Un meta-análisis de 23 grandes estudios observacionales realizados en diferentes poblaciones en todo el mundo encontró una reducción en la presión arterial sistólica de 0.34 milímetros de mercurio (mm Hg) por 100 mg de calcio consumido diariamente y una reducción en la presión arterial diastólica de 0.15 mm Hg por 100 mg de calcio (113). En el estudio DASH (Acercamientos Dietarios para Detener la Hipertensión), 549 personas fueron asignadas aleatoriamente a una de tres dietas por ocho semanas: (1) una dieta control baja en frutas, vegetales, y productos lácteos; (2) una dieta rica en frutas (~5 porciones/día) y vegetales (~3 porciones/día); y (3) una dieta combinada rica tanto en frutas y vegetales como en productos lácteos bajos en grasa (~3 porciones/día) (114). La dieta combinada representó un incremento cercano a 800 mg de calcio/día sobre las dietas de control y las dietas ricas en frutas/vegetales por un total cercano a 1,200 mg de calcio/día. En general, la reducción en la presión sanguínea sistólica fue mayor con la dieta combinada que con la dieta de frutas/vegetales o la dieta de control. Entre los participantes diagnosticados con hipertensión, la dieta combinada redujo la presión sanguínea sistólica por 11.4 mm Hg y la presión diastólica por 5.5 mm Hg más que la dieta de control, mientras que la reducción para la dieta de frutas/vegetales fue de 7.2 mm Hg para la presión sanguínea sistólica y 2.8 mm Hg para la presión sanguínea diastólica en comparación a la dieta de control (115). Esta investigación sugiere que la ingesta de calcio al nivel recomendado (1,000-1,200 mg/día) puede ser de ayuda en la prevención y el tratamiento de la hipertensión moderada (116).
A pesar de todo, dos revisiones sistemáticas de gran magnitud y meta-análisis de ensayos controlados aleatorios han examinado el efecto de la suplementación con calcio en la presión sanguínea en comparación con el placebo en tanto individuos normotensos como en hipertensos (117, 118). Ninguno de los análisis reportó algún efecto significativo del calcio suplementario en la presión sanguínea en sujetos normotensos. Se reportó una reducción pequeña pero significativa en la presión arterial sistólica, pero no en la presión arterial diastólica, en participantes con hipertensión. Cabe destacar que la suplementación con calcio en estos ensayos aleatorios controlados osciló entre 400-2,200 mg/día, con 1,000-1,5000 siendo las dosis más comunes. Un meta-análisis más reciente de 13 estudios controlados aleatorios en 485 individuos con presión sanguínea elevada encontró un reducción significativa de 2.5 mm Hg en la presión arterial sistólica pero ningún cambio en la presión arterial diastólica con la suplementación de calcio (119). El efecto modesto del calcio en la presión sanguínea necesita ser confirmado en ensayos de mayor magnitud, alta calidad, y bien controlados antes de que cualquier recomendación sea hecha con respecto al manejo de la hipertensión. Finalmente, una revisión reciente de la literatura en el efecto de la ingesta alta en calcio (dietética o suplementaria) en mujeres postmenopáusicas ha encontrado ya sea ninguna reducción o reducciones leves y transitorias en la presión arterial (120).
Más información acerca de la dieta DASH se encuentra disponible en el Instituto Nacional de Salud (INS).
Fuentes
Fuentes alimenticias
El análisis de los datos de la Encuesta de Evaluación Nacional de Salud y Nutrición (NHANES) 2009-2010 y 2011-2012 encontró ingestas inadecuadas de calcio (definidas como ingestas por debajo del requisito del promedio estimado [REP]) en el 37.7% de los adultos sin suplementos (edades, ≥19 años) y 19.6% de los adultos que toman suplementos multivitamínicos/minerales (121). Los productos lácteos proporcionan el 75% del calcio en la dieta estadounidense. Sin embargo, en general, durante el período más crítico para el desarrollo de la masa ósea máxima, los adolescentes tienden a reemplazar la leche con los refrescos (122). Los productos lácteos representan fuentes ricas y absorbibles de calcio, pero ciertas verduras y granos también proporcionan calcio.
Sin embargo, la biodisponibilidad del calcio debe tomarse en consideración. Mientras que las plantas ricas en calcio en la familia de la col rizada (brócoli, repollo chino, repollo, mostaza, y hojas de nabo) contienen calcio que es tan biodisponible como el de la leche; sin embargo, otros alimentos de origen vegetal contienen componentes que inhiben la absorción de calcio. El ácido oxálico, también conocido como oxalato, es el inhibidor más potente de la absorción del calcio y se encuentra en altas concentraciones en la espinaca y el ruibarbo, y un tanto en camotes y frijoles secos. El ácido fítico (fitato) es un inhibidor menos potente de la absorción del calcio que el oxalato. Las levaduras poseen una enzima (fitasa) que degrada el fitato en los granos durante la fermentación, disminuyendo el contenido de fitato del pan y otros alimentos fermentados. Sólo las fuentes concentradas de fitato, como el salvado de trigo o los frijoles secos, reducen sustancialmente la absorción de calcio (123).
Componentes adicionales de la dieta pueden afectar la absorción de calcio (véase Interacción con nutrientes). La Tabla 2 lista un cierto número de alimentos ricos en calcio, junto con su contenido de calcio. Para más información en el contenido de nutrientes de los alimentos, busque la base de datos de composición de los alimentos de la USDA.
Suplementos
La mayoría de los expertos recomiendan obtener tanto calcio como sea posible de los alimentos debido a que el calcio en los alimentos es acompañado por otros nutrientes importantes que asisten al cuerpo en la utilización del calcio. Sin embargo, los suplementos de calcio pueden ser necesarios para aquellos que tienen dificultad para consumir suficiente calcio proveniente de los alimentos (124). Ninguna tableta multivitamínica/mineral contiene el 100% del valor diario (VD) recomendado para el calcio, ya que es demasiado voluminoso y la píldora resultante sería demasiado grande para ser tragada. El etiquetado de "Información Nutricional," obligatorio en todos los suplementos a la venta en los EE.UU., muestra el contenido de calcio como calcio elemental. Las preparaciones de calcio usadas como suplementos incluyen carbonato de calcio, citrato malato de calcio, lactato de calcio, y gluconato de calcio. Para determinar cuál preparación de calcio es la de su suplemento, tendrá que revisar la lista de ingredientes. Generalmente el carbonato de calcio es el suplemento de calcio más económico. Para maximizar la absorción, no tome más de 500 mg de calcio elemental a la vez. La mayoría de los suplementos de calcio deben tomarse entre comidas, aunque el citrato de calcio y el citrato malato de calcio se pueden tomar a cualquier hora. El citrato de calcio es la formulación de calcio preferida para los individuos que carecen de ácidos estomacales (aclorhidria) o aquellos tratados con drogas que limitan la producción de ácido estomacal (bloqueadores H2 e inhibidores de la bomba de protones) (revisado en 125).
Plomo en los suplementos de calcio
Varias décadas atrás la preocupación aumentó respecto a las concentraciones de plomo en los suplementos de calcio obtenidos de fuentes naturales (concha de ostra, harina de huesos, dolomita) (126). En 1993, investigadores encontraron cantidades medibles de plomo en la mayoría de las 70 preparaciones diferentes que se examinaron (127). Desde entonces, los productores han reducido la cantidad de plomo en los suplementos de calcio a menos de 0.5 microgramos (μg) por cada 1,000 mg de calcio elemental (128). La Administración de Alimentos y Drogas Estadounidense (FDA) ha desarrollado niveles máximos de ingesta tolerable provisionales (PTTI por sus siglas en inglés) del plomo para grupos de edad y género específicos (129). Debido a que el plomo está tan extendido y es de larga duración, nadie puede garantizar suplementos o alimentos enteramente libres de plomo. Un estudio encontró plomo medible en 8 de 21 suplementos, en cantidades que promediaron entre 1 y 2 μg/1,000 mg de calcio elemental, el cual está por debajo del límite tolerable de 7.5 μg/1,000 mg de calcio elemental (130). Una encuesta más reciente de 324 suplementos multivitamínicos/minerales etiquetados para su uso en niños o mujeres encontró que la mayoría de los suplementos resultan en la exposición a plomo oscilando entre 1%-4% de los PTTI (131).
El calcio inhibe la absorción intestinal de plomo, y una ingesta adecuada de calcio es un protector en contra de la toxicidad por plomo, por lo que cantidades trazas de plomo en la suplementación con calcio puede presentar menos riesgo de una exposición excesiva al plomo que un consumo de calcio insuficiente. Mientras que la mayoría de las fuentes de calcio hoy en día son relativamente seguras, busque suplementos aprobados o certificados por pruebas independientes (p. ej., US Pharmacopeia, ConsumerLab.com) (125), siga las instrucciones etiquetadas y evite grandes dosis de calcio suplementario (≥1,500 mg/día).
Seguridad
Toxicidad
La malignidad y el hiperparatiroidismo primario son las causas más comunes de las concentraciones de calcio elevadas en la sangre (hipercalcemia) (132). La hipercalcemia no ha sido asociada con el consumo excesivo de calcio de origen natural proveniente de alimentos. La hipercalcemia ha sido inicialmente reportada con el consumo de grandes cantidades de suplementos de calcio en combinación con antiácidos, particularmente en los días cuando las úlceras pépticas eran tratadas con grandes cantidades de leche, carbonato de calcio (antiácido), y bicarbonato de sodio (un álcali absorbible). Esta condición se denominó síndrome alcalino lácteo (también conocido como síndrome de leche y alcalinos) y se ha reportado con niveles de suplementos de calcio de 1.5 a 16.5 g/día por 2 días hasta 30 años. Dado que el tratamiento para la úlcera péptica ha evolucionado, y debido al uso generalizado de los suplementos de calcio de venta libre, las características demográficas de este síndrome han cambiado y aquellos que están ahora en mayor riesgo son las mujeres posmenopáusicas, mujeres embarazadas, personas receptoras de trasplantes, pacientes con bulimia, y pacientes con diálisis, en lugar de los hombres con úlceras pépticas (revisado en 133). La suplementación con calcio (0.6 g/día-2 g/día por dos a cinco años) se ha asociado con un mayor riesgo de eventos gastrointestinales adversos como estreñimiento, calambres, distensión abdominal, dolor, diarrea (134). La hipercalcemia leve puede presentarse sin síntomas o puede ocasionar pérdida de apetito, náuseas, vómitos, constipación, dolor abdominal, fatiga, micción frecuente (poliuria), e hipertensión (132). Una hipercalcemia más severa puede causar confusión, delirio, coma, y si no es tratada, la muerte (1).
En el 2011, la Junta de Nutrición y Alimentos del Instituto de Medicina actualizó el nivel máximo de ingesta tolerable (NM) para el calcio (9). El (NM) es listado en la Tabla 3 por grupo etario.
Aunque el riesgo de la formación de cálculos renales es elevado en individuos con calcio urinario anormalmente elevado (hipercalciuria), esta condición no se relaciona usualmente con la ingesta de calcio, sino más bien con el incremento en la absorción de calcio en los intestinos o la excreción aumentada de calcio por los riñones (9). En general, la ingesta dietética incrementada de calcio ha sido asociada con la disminución del riesgo de cálculos renales (véase Cálculos renales). Preocupaciones han sido planteadas con respecto a los riesgos de cáncer de próstata y enfermedades vasculares con altas ingestas de calcio.
¿Las ingestas altas de calcio incrementan el riesgo de cáncer de próstata?
El cáncer de próstata es el segundo cáncer más común en hombres en todo el mundo (135). Varios estudios observacionales han expresado la preocupación de que las ingestas altas de lácteos están asociadas con un mayor riesgo de cáncer de próstata (136-138).
El análisis de un estudio de cohorte prospectivo (2,268 hombres seguidos durante casi 25 años) realizado en Islandia, un país con una alta incidencia de cáncer de próstata encontró una asociación positiva entre el consumo de leche (al menos una vez al día) durante la adolescencia y el desarrollo de la próstata cáncer en el futuro (139). Otro gran estudio prospectivo de cohorte en los EE.UU. siguió a 21,660 médicos varones durante 28 años y encontró que los hombres con un consumo diario de leche descremada o baja en grasa de al menos 237 ml (8 oz) tenían un mayor riesgo de desarrollar cáncer de próstata en comparación con consumidores ocasionales (140). El riesgo de cáncer de próstata en etapa temprana de grado bajo se asoció con una mayor ingesta de leche descremada, y el riesgo de desarrollo de cáncer de próstata fatal se relacionó con el consumo regular de leche entera (140). En una cohorte de 3,918 profesionales masculinos de la salud diagnosticados con cáncer de próstata, 229 hombres murieron de cáncer de próstata y 69 desarrollaron cáncer de próstata metastatizado durante una mediana de seguimiento de 7.6 años (141). El riesgo de próstata se encontró que la muerte por cáncer aumentó en los hombres con ingesta alta de leche entera (>4 porciones/semana) en comparación con las bajas (≤3 porciones/mes). Sin embargo, ningún aumento en el riesgo de mortalidad relacionada con el cáncer de próstata se asoció con el consumo de leche descremada y baja en grasa, leche total, productos lácteos bajos en grasa, productos lácteos completos en grasa o productos lácteos totales (141). Un meta-análisis reciente de 32 estudios prospectivos de cohortes encontraron una ingesta alta versus baja de productos lácteos totales (15 estudios), leche total (15 estudios), leche entera (6 estudios), leche baja en grasa (5 estudios), queso (11 estudios) y productos lácteos el calcio (7 estudios) se asoció con aumentos modestos, aunque significativos, en el riesgo de desarrollar cáncer de próstata (142). Sin embargo, no hubo un aumento en el riesgo de cáncer de próstata con el calcio no lácteo (4 estudios) y el calcio de los suplementos (8 estudios). Además, las altas ingestas de productos lácteos no se relacionaron con el cáncer de próstata fatal (142).
Existe cierta evidencia que sugiere que el consumo de leche puede resultar en mayores concentraciones circulantes del factor de crecimiento insulínico tipo I (IGF-I), una proteína conocida por regular la proliferación celular (143). Las concentraciones circulantes de IGF-I se han correlacionado positivamente con el riesgo de desarrollo del cáncer de próstata en un reciente meta-análisis de estudios observacionales (144). El IGF-I transmitido por la leche, así como las proteínas lácteas y el calcio, pueden contribuir a aumentar el IGF-I circulante en los consumidores de leche (143). En el gran estudio EPIC, que examinó el consumo de productos lácteos con relación al cáncer en 142,520 hombres, se encontró que el riesgo de cáncer de próstata es significativamente mayor en aquellos en el quintil superior en comparación con el consumo de proteínas y calcio de los productos lácteos (145). Otro mecanismo subyacente a la posible relación entre la ingesta de calcio y el cáncer de próstata propuso que los niveles altos de calcio en la dieta pueden disminuir las concentraciones circulantes de 1,25-dihidroxivitamina D, la forma activa de la vitamina D, lo que suprime la diferenciación celular mediada por la vitamina D (146). Sin embargo, los estudios hasta la fecha han proporcionado poca evidencia para sugerir que el estatus de la vitamina D puede modificar la asociación entre el calcio lácteo y el riesgo de desarrollo y progresión del cáncer de próstata (147-149).
En un ensayo multicéntrico, doble ciego, controlado con placebo, 672 hombres sanos (edad promedio de 61.8 años) fueron asignados aleatoriamente con una suplementación diaria de calcio (1,200 mg) por cuatro años. Mientras que ningún incremento en el riesgo de cáncer de próstata ha sido reportado durante un período de seguimiento de 10.3 años, la suplementación con calcio resultó en una reducción significativa del riesgo en el período que abarcó desde los dos años después de que el tratamiento fue iniciado hasta los dos años después del que el tratamiento concluyó (150). En una revisión de la literatura publicada en el 2009, la Agencia Estadounidense para la Investigación y Calidad del Cuidado de la Salud indicó que no todos los estudios epidemiológicos encontraron una asociación entre la ingesta de calcio y el cáncer de próstata (151). La revisión reportó que 6 de 11 estudios basados en la observación fallaron en encontrar asociaciones positivas estadísticamente significativas entre el cáncer de próstata y la ingesta de calcio. A pesar de todo, en cinco estudios, se encontró que ingestas diarias de 921 a 2,000 mg de calcio se asociaron con un riesgo incrementado de desarrollar cáncer de próstata cuando se comparó con ingestas que oscilaron entre 455 a 1,000 mg/día (151). Las inconsistencias entre estudios sugieren interacciones complejas entre los factores de riesgo del cáncer de próstata, así como reflejan las dificultades al evaluar el efecto de la ingesta de calcio en individuos con un estilo de vida libre. Por ejemplo, el hecho de que se encontró que individuos con ingestas altas de lácteos y/o calcio son más propensos a formar parte de estilos de vida saludables o más dispuestos a buscar atención médica puede mitigar la importancia estadística de una asociación con el riesgo de cáncer de próstata (152). Hasta que la relación entre el calcio y el cáncer de próstata sea clara, para los hombres es razonable consumir un total de 1,000 a 1,200 mg/día de calcio (combinación de dieta y suplementos), lo cual es recomendado por la Junta de Alimentos y Nutrición del Instituto de Medicina (véase IDR) (9).
¿Incrementan el riesgo de enfermedades cardiovasculares los suplementos de calcio?
Varios estudios observacionales y ensayos controlados aleatorios han planteado preocupaciones con respecto a los potenciales efectos adversos de los suplementos de calcio en el riesgo cardiovascular. El análisis de los datos del estudio prospectivo Factor de Riesgo y Prevención de la Osteoporosis de Kuopio (OSTPRE) encontró que usuarias de suplementos de calcio de entre 10,555 mujeres finlandesas (edades de entre 52-62 años) tuvieron un riesgo 14% mayor de desarrollar una enfermedad coronaria cardíaca en comparación con las no que no eran usuarias de suplementos durante un seguimiento medio de 6.75 años (153). El estudio prospectivo de 23,980 participantes (35-64 años de edad) de la cohorte de Heidelberg de la Investigación Prospectiva Europea del Cáncer y Nutrición de cohortes (EPIC-Heidelberg) observó que la ingesta suplementaria de calcio fue positivamente asociada con el riesgo de infarto al miocardio (ataque al corazón) pero no con el riesgo de accidentes cerebrovasculares o mortalidad relacionada con enfermedades cardiovasculares (ECV) después de seguimiento promedio de 11 años (154). A pesar de todo, el uso de suplementos de calcio (≥400 mg/día vs. 0 mg/día) fue asociado con un riesgo incrementado de la mortalidad relacionada con ECV en 219,059 hombres, pero no en 169,170 mujeres, incluidos en el estudio del Instituto Nacional de Salud (NIH)-AARP Dieta y Salud con un seguimiento promedio de 12 años. Se encontró también que la mortalidad por ECV en hombres era significativamente más alta con ingestas totales (dietéticas más suplementarias) de calcio de 1,500 mg/día y más (155).
Además, los análisis secundarios de dos ensayos controlados aleatorios con placebo diseñados inicialmente para evaluar el efecto del calcio en los resultados de salud ósea también sugirieron un mayor riesgo de ECV en los participantes que recibieron suplementos diarios de 1,000 mg de calcio durante cinco a siete años (156, 157). En el Estudio de Calcio de Auckland de 1,471 mujeres sanas posmenopáusicas (edades ≥55 años), la suplementación con calcio resultó en un mayor riesgo de infarto al miocardio y de un punto final compuesto cardiovascular, que incluye infarto al miocardio, accidente cerebrovascular o muerte súbita (156). El análisis de datos de 36,282 mujeres sanas posmenopáusicas asignadas al azar para recibir una combinación de calcio (1,000 mg/día) y vitamina D (400 UI/día) o un placebo en el estudio de la Iniciativa de Salud de las Mujeres/suplementación con Calcio-Vitamina D (estudio de WHI/CaD) inicialmente no reportó algún efecto adverso en cualquier punto final cardiovascular con calcio (y vitamina D) en comparación con el placebo (158). Se realizó un nuevo análisis con datos de 16,718 mujeres que no tomaron suplementos personales de calcio (fuera del protocolo) durante el estudio de cinco años (157). A pesar de ser criticados por el enfoque adoptado (134, 159), los investigadores estimaron que las mujeres que recibieron suplementos de calcio y vitamina D tenían un riesgo 16% mayor de infarto al miocardio clínico o accidente cerebrovascular y un riesgo 21% mayor de infarto al miocardio en comparación con los que recibieron un placebo (157). Sin embargo, en otro ensayo aleatorizado, doble ciego, controlado con placebo — el estudio de Calcium Intake Fracture Outcome (CAIFOS) — en mujeres de edad avanzada (edad promedio, 75.1 años), no se encontró que la suplementación de 1,200 mg/día de calcio durante cinco años aumentase el riesgo de enfermedad vascular o la mortalidad relacionada (160). El nuevo análisis de los datos de WHI/CaD tampoco mostró un mayor riesgo de mortalidad debido al infarto al miocardio o enfermedad coronaria con terapia con calcio (156). Además, después de un seguimiento adicional de 4.5 años al final del período de tratamiento en el ensayo CAIFOS, los investigadores reportaron menos casos de muertes relacionadas con insuficiencia cardíaca con calcio suplementario en comparación con placebo (160). En otro ensayo aleatorio, controlado con placebo de calcio y/o vitamina D3 (ensayo RECORD), la evaluación del efecto de 1,000 mg/día de calcio (solo o con 800 UI/día de vitamina D) no reportó un incremento significativo en la tasa de mortalidad debido a enfermedad vascular en 5,292 participantes de 70 años y más (161). Un reciente análisis transversal de la Tercera Encuesta de Evaluación Nacional de Salud y Nutrición (NHANES III) evaluaron la asociación entre la ingesta de calcio y la mortalidad cardiovascular en 18,714 adultos sin antecedentes de enfermedad cardíaca. No se observó evidencia de una asociación entre la ingesta de calcio en la dieta, la ingesta de calcio suplementaria o la ingesta de calcio total y la mortalidad cardiovascular en hombres o mujeres (162).
Algunos estudios prospectivos han reportado correlaciones positivas entre las concentraciones altas de calcio en la sangre y el incremento en las tasas de eventos cardiovasculares (163, 164). Debido a que el calcio suplementario puede tener un efecto mayor que el calcio dietario en las concentraciones de calcio circulante (véase Toxicidad), se ha especulado que el uso de suplementos de calcio podría promover la calcificación vascular — un marcador sustituto de la carga de aterosclerosis y un factor de riesgo importante para eventos cardiovasculares — al elevar las concentraciones de calcio del suero. En 1,471 mujeres mayores del Estudio del Calcio de Auckland y 323 hombres mayores saludables de otro ensayo aleatorio controlado con placebo de la suplementación diaria con calcio (600 mg o 1,200 mg) por dos años, se encontró que las concentraciones de calcio en el suero estaban correlacionadas positivamente con la calcificación de la aorta abdominal o calcificación de la arteria coronaria (165). Sin embargo, no hubo efecto alguno de la suplementación con calcio en medidas de puntuaciones de la calcificación vascular en hombres o mujeres (166). No obstante, en el Estudio Multiétnico de Aterosclerosis (MESA), un estudio prospectivo multicéntrico de EE.UU. en 6,814 participantes seguidos durante un promedio de 10 años, el mayor riesgo de desarrollar calcificación de la arteria coronaria se encontró en usuarios de suplementos con la ingesta de calcio total más baja (~306 mg/día de calcio en la dieta y ~91 mg/día de calcio suplementario), en comparación con los usuarios de suplementos con mayor ingesta total de calcio y los no usuarios (167). Finalmente, una evaluación de las lesiones ateroscleróticas en la pared de la arteria carótida de 1,103 participantes en el ensayo de CAIFOS también se realizó después de tres años de suplementación (168). Cuando se comparó con placebo, la suplementación con calcio no mostró ningún efecto sobre el grosor íntima-media carotídeo (GIMC) y la aterosclerosis carotídea. Sin embargo, la aterosclerosis carotídea (pero no la GIMC) se redujo significativamente en mujeres en el tercil más alto en comparación con el tercil más bajo del consumo total de calcio (dieta y suplementos) (≥1,795 mg/día vs. <1,010 mg/día) (168).
El meta-análisis más reciente de 18 ensayos clínicos aleatorios, incluyendo un total de 63,563 mujeres postmenopáusicas, no encontró evidencia alguna de un riesgo incrementado de padecer una enfermedad coronaria cardíaca y mortalidad por cualquier causa con la suplementación de calcio (≥500 mg/día) de por lo menos un año (169). Debido a que estos datos de ensayos clínicos son limitados a análisis de criterios de valoración secundarios, los meta-análisis debiesen ser interpretados con cuidado. Existe una necesidad de estudios diseñados para examinar el efecto de los suplementos de calcio sobre el riesgo de ECV como un resultado primario antes de que conclusiones definitivas puedan ser extraídas. Basados en una revisión actualizada de la literatura que incluyó cuatro ensayos controlados aleatorios, un estudio de caso-control anidado, y 26 estudios de cohorte prospectivos (179), la Fundación Nacional de la Osteoporosis (NOF) y la American Society for Preventive Cardiology (ASPC) concluyeron que el uso de calcio suplementario para individuos generalmente sanos era seguro desde un punto de vista de la salud cardiovascular cuando las ingestas totales de calcio no excedieron el NM (171). La NOF y la ASPC apoyan el uso de suplementos de calcio para corregir las deficiencias en la ingesta de calcio en la dieta y cumplir con las recomendaciones actuales (171).
Interacciones con drogas/fármacos
Tomar suplementos de calcio en combinación con diuréticos tiazídicos (e.g., hidroclorotiazida) incrementa el riesgo de padecer hipercalcemia debido al incremento de la reabsorción de calcio en los riñones. Las dosis altas de calcio suplementario podrían incrementar la probabilidad de ritmos cardíacos anormales en personas que consumen digoxina (Lanoxin) para la falla cardíaca (172). El calcio, cuando se administra intravenosamente, puede disminuir la eficacia de los bloqueadores de canales de calcio (173). No obstante, el calcio dietario y el suplementario no parecen afectar la acción de los bloqueadores de canales de calcio (174). El calcio puede disminuir la absorción de la tetraciclina, de los antibióticos de la clase de las quinilonas, de los bifosfonatos, de sotalol (un β-bloqueador), y de la levotiroxina; por lo tanto, se aconseja separar las dosis de estos medicamentos de alimentos ricos en calcio o suplementos por dos horas antes del calcio o cuatro a seis horas después del calcio (175). El calcio suplementario puede disminuir la concentración de dolutegravir (Tivicay), elvitegravir (Vitekta), y raltegravir (Isentress), tres medicamentos antirretrovirales, en la sangre, por lo que se recomienda a los pacientes que los tomen dos horas antes o después de los suplementos de calcio (175). El calcio intravenoso no debe administrarse dentro de las 48 horas posteriores a la administración intravenosa de ceftriaxona (rocefina), un antibiótico de cefalosporina, ya que puede formarse un precipitado de sal de calcio y ceftriaxona en los pulmones y los riñones y ser una causa de muerte (175). El uso de bloqueadores H2 (p. ej., cimetidina) e inhibidores de bombas de protones (p. ej., omeprazol) puede disminuir la absorción de carbonato de calcio y de fosfato de calcio (revisado en 176, 177), mientras que el litio puede aumentar el riesgo de hipercalcemia en pacientes (175). El uso tópico de calcipotrieno, un análogo de la vitamina D, en el tratamiento de la psoriasis, coloca a los pacientes en riesgo de hipercalcemia si toman suplementos de calcio.
Interacciones calcio-nutrientes
La presencia de calcio disminuye la absorción de hierro proveniente de fuentes no hemo (es decir, la mayoría de los suplementos y fuentes alimenticias además de la carne). Sin embargo, no se ha encontrado que la suplementación con calcio por hasta 12 semanas cambie el estado nutricional del hierro, probablemente debido al incremento compensatorio en la absorción de hierro (1). Los individuos que toman suplementos de hierro deberían tomarlos con dos horas de separación de suplementos o alimentos ricos en calcio para maximizar la absorción de hierro. Aunque no se han asociado las ingestas de calcio elevadas con la reducción de la absorción de zinc o el estado nutricional del zinc, un estudio temprano en 10 hombres y mujeres indicó que 600 mg de calcio consumidos con una comida disminuyó a la mitad la absorción de zinc proveniente de esa comida (véase el artículo sobre Zinc) (178). Se ha encontrado que el calcio suplementario (500 mg de carbonato de calcio) previene la absorción de licopeno (un carotenoide no provitamina A) de la pasta de tomate en 19 adultos sanos al azar en un estudio cruzado (179).
Recomendación del Instituto Linus Pauling
El Instituto Linus Pauling respalda la Ingesta Diaria Recomendada (IDR) establecidos por la Junta de Nutrición y Alimentos del Instituto de Medicina. Seguir estas recomendaciones debería aportar suficiente calcio para promover la salud del esqueleto y también podría disminuir los riesgos de algunas enfermedades crónicas.
Niños y adolescentes (9-18 años)
Para promover la obtención del nivel máximo de masa ósea, los niños y adolescentes deben consumir un total (dieta más suplementos) de 1,300 mg/día de calcio.
Adultos (mujeres entre 19-50 años, hombres entre 19-70 años)
Luego de que se ha alcanzado la altura adulta, el esqueleto continúa acumulando hueso hasta la tercera década de la vida cuando se alcanza el nivel máximo de masa ósea. Para promover la obtención del nivel máximo de masa ósea y minimizar la pérdida ósea con la edad, las mujeres adultas (50 años de edad y menores) y los hombres adultos (70 años de edad y menores) deben consumir un total (dieta más suplementos) de 1,000 mg/día de calcio.
Mujeres de mayor edad (>50 años)
Para minimizar la pérdida ósea, las mujeres posmenopáusicas deben consumir un total (dieta más suplementos) de 1,200 mg/día de calcio. Tomar un suplemento multivitamínico/mineral que contenga al menos 10 μg (400 UI)/día de vitamina D ayudará a asegurar la absorción adecuada del calcio (véase el artículo sobre Vitamina D).
Hombres de mayor edad (>70 años)
Para minimizar la pérdida ósea, los hombres ancianos deberían consumir un total (dieta más suplementos) de 1,200 mg/día de calcio. Tomar un suplemento multivitamínico/mineral que contenga al menos 10 μg (400 UI)/día de vitamina D ayudará a asegurar la absorción adecuada del calcio (véase el artículo sobre Vitamina D).
Mujeres embarazadas y mujeres en período de lactancia
Las adolescentes embarazadas y en amamantamiento (<19 años de edad) deben consumir un total de 1,300 mg/día de calcio, mientras que las mujeres adultas embarazadas y que lactan (≥19 años de edad) deben consumir un total de 1,000 mg/día de calcio.
Autores y Críticos
Escrito originalmente en 2003 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Agosto de 2014 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Mayo 2017 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Septiembre 2017 por:
Connie M. Weaver, Ph.D.
Profesor Distinguido y Director de Alimentos y Nutrición
Universidad de Purdue
Traducido al Español en 2019 por:
Natsumi Then Shimazaki
Instituto Linus Pauling
Universidad Estatal de Oregon
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
La actualización de este artículo en el 2017 fue respaldada por una subvención de Pfizer Inc.
Derechos de autoría 2001-2024 Instituto Linus Pauling
Referencias
1. Weaver CM. Calcium. In: Erdman JJ, Macdonald I, Zeisel S, eds. Present Knowledge in Nutrition. 10th ed: John Wiley & Sons, Inc.; 2012:434-446.
2. Wesseling-Perry K, Wang H, Elashoff R, Gales B, Juppner H, Salusky IB. Lack of FGF23 response to acute changes in serum calcium and PTH in humans. J Clin Endocrinol Metab. 2014;99(10):E1951-E1956. (PubMed)
3. Clapham DE. Calcium signaling. Cell. 2007;131(6):1047-1058. (PubMed)
4. Wigertz K, Palacios C, Jackman LA, et al. Racial differences in calcium retention in response to dietary salt in adolescent girls. Am J Clin Nutr. 2005;81(4):845-850. (PubMed)
5. Frassetto LA, Morris RC, Jr., Sellmeyer DE, Sebastian A. Adverse effects of sodium chloride on bone in the aging human population resulting from habitual consumption of typical American diets. J Nutr. 2008;138(2):419S-422S. (PubMed)
6. Devine A, Criddle RA, Dick IM, Kerr DA, Prince RL. A longitudinal study of the effect of sodium and calcium intakes on regional bone density in postmenopausal women. Am J Clin Nutr. 1995;62(4):740-745. (PubMed)
7. Carbone LD, Barrow KD, Bush AJ, et al. Effects of a low sodium diet on bone metabolism. J Bone Miner Metab. 2005;23(6):506-513. (PubMed)
8. Sellmeyer DE, Schloetter M, Sebastian A. Potassium citrate prevents increased urine calcium excretion and bone resorption induced by a high sodium chloride diet. J Clin Endocrinol Metab. 2002;87(5):2008-2012. (PubMed)
9. Food and Nutrition Board, Institute of Medicine. Dietary Reference Intakes for Calcium and Vitamin D. Washington, D.C.; 2011. (The National Academies Press)
10. Fulgoni VL, 3rd. Current protein intake in America: analysis of the National Health and Nutrition Examination Survey, 2003-2004. Am J Clin Nutr. 2008;87(5):1554S-1557S. (PubMed)
11. Ince BA, Anderson EJ, Neer RM. Lowering dietary protein to U.S. Recommended dietary allowance levels reduces urinary calcium excretion and bone resorption in young women. J Clin Endocrinol Metab. 2004;89(8):3801-3807. (PubMed)
12. Calvez J, Poupin N, Chesneau C, Lassale C, Tome D. Protein intake, calcium balance and health consequences. Eur J Clin Nutr. 2012;66(3):281-295. (PubMed)
13. Kerstetter JE, O'Brien KO, Caseria DM, Wall DE, Insogna KL. The impact of dietary protein on calcium absorption and kinetic measures of bone turnover in women. J Clin Endocrinol Metab. 2005;90(1):26-31. (PubMed)
14. Darling AL, Millward DJ, Torgerson DJ, Hewitt CE, Lanham-New SA. Dietary protein and bone health: a systematic review and meta-analysis. Am J Clin Nutr. 2009;90(6):1674-1692. (PubMed)
15. Grimm M, Muller A, Hein G, Funfstuck R, Jahreis G. High phosphorus intake only slightly affects serum minerals, urinary pyridinium crosslinks and renal function in young women. Eur J Clin Nutr. 2001;55(3):153-161. (PubMed)
16. Kemi VE, Karkkainen MU, Rita HJ, Laaksonen MM, Outila TA, Lamberg-Allardt CJ. Low calcium:phosphorus ratio in habitual diets affects serum parathyroid hormone concentration and calcium metabolism in healthy women with adequate calcium intake. Br J Nutr. 2010;103(4):561-568. (PubMed)
17. Heaney RP. Phosphorus. In: Erdman JJ, Macdonald I, Zeisel S, eds. Present Knowledge in Nutrition. 10th ed. Ames: John Wiley & Sons, Inc.; 2012:447-458.
18. Calvo MS, Moshfegh AJ, Tucker KL. Assessing the health impact of phosphorus in the food supply: issues and considerations. Adv Nutr. 2014;5(1):104-113. (PubMed)
19. Heaney RP, Rafferty K. Carbonated beverages and urinary calcium excretion. Am J Clin Nutr. 2001;74(3):343-347. (PubMed)
20. Ribeiro-Alves MA, Trugo LC, Donangelo CM. Use of oral contraceptives blunts the calciuric effect of caffeine in young adult women. J Nutr. 2003;133(2):393-398. (PubMed)
21. Barger-Lux MJ, Heaney RP, Stegman MR. Effects of moderate caffeine intake on the calcium economy of premenopausal women. Am J Clin Nutr. 1990;52(4):722-725. (PubMed)
22. Wikoff D, Welsh BT, Henderson R, et al. Systematic review of the potential adverse effects of caffeine consumption in healthy adults, pregnant women, adolescents, and children. Food Chem Toxicol. 2017;Apr 21. pii: S0278-6915(17)30170-9. doi: 10.1016/j.fct. 2017.04.002. [Epub ahead of print]. (PubMed)
23. Haleem S, Lutchman L, Mayahi R, Grice JE, Parker MJ. Mortality following hip fracture: trends and geographical variations over the last 40 years. Injury. 2008;39(10):1157-1163. (PubMed)
24. Kaufman JM, Reginster JY, Boonen S, et al. Treatment of osteoporosis in men. Bone. 2013;53(1):134-144. (PubMed)
25. Heaney RP. Calcium, dairy products and osteoporosis. J Am Coll Nutr. 2000;19(2 Suppl):83S-99S. (PubMed)
26. Crandall CJ, Newberry SJ, Diamant A, et al. Treatment to prevent fractures in men and women with low bone density or osteoporosis: update of a 2007 report. Rockville (MD); 2012. (PubMed)
27. Rizzoli R, Bianchi ML, Garabedian M, McKay HA, Moreno LA. Maximizing bone mineral mass gain during growth for the prevention of fractures in the adolescents and the elderly. Bone. 2010;46(2):294-305. (PubMed)
28. Borer KT. Physical activity in the prevention and amelioration of osteoporosis in women : interaction of mechanical, hormonal and dietary factors. Sports Med. 2005;35(9):779-830. (PubMed)
29. National Osteoporosis Foundation. Clinician's Guide to Prevention and Treatment of Osteoporosis. Washington, D.C. 2014.
30. Levis S, Theodore G. Summary of AHRQ's comparative effectiveness review of treatment to prevent fractures in men and women with low bone density or osteoporosis: update of the 2007 report. J Manag Care Pharm. 2012;18(4 Suppl B):S1-15; discussion S13. (PubMed)
31. Tai V, Leung W, Grey A, Reid IR, Bolland MJ. Calcium intake and bone mineral density: systematic review and meta-analysis. BMJ. 2015;351:h4183. (PubMed)
32. Bolland MJ, Leung W, Tai V, et al. Calcium intake and risk of fracture: systematic review. BMJ. 2015;351:h4580. (PubMed)
33. Chung M, Lee J, Terasawa T, Lau J, Trikalinos TA. Vitamin D with or without calcium supplementation for prevention of cancer and fractures: an updated meta-analysis for the U.S. Preventive Services Task Force. Ann Intern Med. 2011;155(12):827-838. (PubMed)
34. Weaver CM, Alexander DD, Boushey CJ, et al. Calcium plus vitamin D supplementation and risk of fractures: an updated meta-analysis from the National Osteoporosis Foundation. Osteoporos Int. 2016;27(1):367-376. (PubMed)
35. Cosman F, de Beur SJ, LeBoff MS, et al. Clinician's Guide to Prevention and Treatment of Osteoporosis. Osteoporos Int. 2014;25(10):2359-2381. (PubMed)
36. Lips P, van Schoor NM. The effect of vitamin D on bone and osteoporosis. Best Pract Res Clin Endocrinol Metab. 2011;25(4):585-591. (PubMed)
37. Gallagher JC, Yalamanchili V, Smith LM. The effect of vitamin D on calcium absorption in older women. J Clin Endocrinol Metab. 2012;97(10):3550-3556. (PubMed)
38. Zhu K, Bruce D, Austin N, Devine A, Ebeling PR, Prince RL. Randomized controlled trial of the effects of calcium with or without vitamin D on bone structure and bone-related chemistry in elderly women with vitamin D insufficiency. J Bone Miner Res. 2008;23(8):1343-1348. (PubMed)
39. Dipart Group. Patient level pooled analysis of 68 500 patients from seven major vitamin D fracture trials in US and Europe. BMJ. 2010;340:b5463. (PubMed)
40. Avenell A, Mak JC, O'Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev. 2014;4:CD000227. (PubMed)
41. Bischoff-Ferrari HA, Willett WC, Orav EJ, et al. A pooled analysis of vitamin D dose requirements for fracture prevention. N Engl J Med. 2012;367(1):40-49. (PubMed)
42. Aspray TJ, Francis RM. Fracture prevention in care home residents: is vitamin D supplementation enough? Age Ageing. 2006;35(5):455-456. (PubMed)
43. Murad MH, Elamin KB, Abu Elnour NO, et al. Clinical review: The effect of vitamin D on falls: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2011;96(10):2997-3006. (PubMed)
44. Lerolle N, Lantz B, Paillard F, et al. Risk factors for nephrolithiasis in patients with familial idiopathic hypercalciuria. Am J Med. 2002;113(2):99-103. (PubMed)
45. Sorensen MD, Eisner BH, Stone KL, et al. Impact of calcium intake and intestinal calcium absorption on kidney stones in older women: the study of osteoporotic fractures. J Urol. 2012;187(4):1287-1292. (PubMed)
46. Curhan GC, Willett WC, Knight EL, Stampfer MJ. Dietary factors and the risk of incident kidney stones in younger women: Nurses' Health Study II. Arch Intern Med. 2004;164(8):885-891. (PubMed)
47. Curhan GC, Willett WC, Rimm EB, Stampfer MJ. A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med. 1993;328(12):833-838. (PubMed)
48. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol. 2004;15(12):3225-3232. (PubMed)
49. Taylor EN, Curhan GC. Dietary calcium from dairy and nondairy sources, and risk of symptomatic kidney stones. J Urol. 2013;190(4):1255-1259. (PubMed)
50. Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med. 2002;346(2):77-84. (PubMed)
51. Hess B, Jost C, Zipperle L, Takkinen R, Jaeger P. High-calcium intake abolishes hyperoxaluria and reduces urinary crystallization during a 20-fold normal oxalate load in humans. Nephrol Dial Transplant. 1998;13(9):2241-2247. (PubMed)
52. Liebman M, Chai W. Effect of dietary calcium on urinary oxalate excretion after oxalate loads. Am J Clin Nutr. 1997;65(5):1453-1459. (PubMed)
53. Lange JN, Wood KD, Mufarrij PW, et al. The impact of dietary calcium and oxalate ratios on stone risk. Urology. 2012;79(6):1226-1229. (PubMed)
54. Jackson RD, LaCroix AZ, Gass M, et al. Calcium plus vitamin D supplementation and the risk of fractures. N Engl J Med. 2006;354(7):669-683. (PubMed)
55. Heaney RP. Calcium supplementation and incident kidney stone risk: a systematic review. J Am Coll Nutr. 2008;27(5):519-527. (PubMed)
56. Candelas G, Martinez-Lopez JA, Rosario MP, Carmona L, Loza E. Calcium supplementation and kidney stone risk in osteoporosis: a systematic literature review. Clin Exp Rheumatol. 2012;30(6):954-961. (PubMed)
57. Escribano J, Balaguer A, Roque i Figuls M, Feliu A, Ferre N. Dietary interventions for preventing complications in idiopathic hypercalciuria. Cochrane Database Syst Rev. 2014;2:CD006022. (PubMed)
58. Heilberg IP, Goldfarb DS. Optimum nutrition for kidney stone disease. Adv Chronic Kidney Dis. 2013;20(2):165-174. (PubMed)
59. Prezioso D, Strazzullo P, Lotti T, et al. Dietary treatment of urinary risk factors for renal stone formation. A review of CLU Working Group. Arch Ital Urol Androl. 2015;87(2):105-120. (PubMed)
60. Duley L. The global impact of pre-eclampsia and eclampsia. Semin Perinatol. 2009;33(3):130-137. (PubMed)
61. Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet. 2010;376(9741):631-644. (PubMed)
62. Hofmeyr GJ, Lawrie TA, Atallah AN, Duley L, Torloni MR. Calcium supplementation during pregnancy for preventing hypertensive disorders and related problems. Cochrane Database Syst Rev. 2014;6:CD001059. (PubMed)
63. Scholl TO, Chen X, Stein TP. Vitamin D, secondary hyperparathyroidism, and preeclampsia. Am J Clin Nutr. 2013;98(3):787-793. (PubMed)
64. Scholl TO, Chen X, Stein TP. Maternal calcium metabolic stress and fetal growth. Am J Clin Nutr. 2014;99(4):918-925. (PubMed)
65. Hofmeyr GJ, Belizan JM, von Dadelszen P, Calcium, Pre-eclampsia Study G. Low-dose calcium supplementation for preventing pre-eclampsia: a systematic review and commentary. BJOG. 2014;121(8):951-957. (PubMed)
66. World Health Organization. Calcium supplementation in pregnant women; 2013.
67. Hofmeyr GJ, Mlokoti Z, Nikodem VC, et al. Calcium supplementation during pregnancy for preventing hypertensive disorders is not associated with changes in platelet count, urate, and urinary protein: a randomized control trial. Hypertens Pregnancy. 2008;27(3):299-304. (PubMed)
68. Villar J, Abdel-Aleem H, Merialdi M, et al. World Health Organization randomized trial of calcium supplementation among low calcium intake pregnant women. Am J Obstet Gynecol. 2006;194(3):639-649. (PubMed)
69. Hofmeyr GJ, Novikova N, Singata M, et al. Protocol 11PRT/4028: Long term calcium supplementation in women at high risk of pre-eclampsia: a randomised, placebo-controlled trial (PACTR201105000267371). The Lancet; 2011.
70. Hofmeyr GJ, Seuc AH, Betran AP, et al. The effect of calcium supplementation on blood pressure in non-pregnant women with previous pre-eclampsia: An exploratory, randomized placebo controlled study. Pregnancy Hypertens. 2015;5(4):273-279. (PubMed)
71. US Centers for Disease Control and Prevention. Colorectal Cancer Statistics. Available at: http://www.cdc.gov/cancer/colorectal/statistics/. Accessed 5/29/17.
72. Whitfield JF. Calcium, calcium-sensing receptor and colon cancer. Cancer Lett. 2009;275(1):9-16. (PubMed)
73. Murphy N, Norat T, Ferrari P, et al. Consumption of dairy products and colorectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC). PLoS One. 2013;8(9):e72715. (PubMed)
74. Massa J, Cho E, Orav EJ, Willett WC, Wu K, Giovannucci EL. Total calcium intake and colorectal adenoma in young women. Cancer Causes Control. 2014;25(4):451-460. (PubMed)
75. Keum N, Lee DH, Greenwood DC, Zhang X, Giovannucci EL. Calcium intake and colorectal adenoma risk: dose-response meta-analysis of prospective observational studies. Int J Cancer. 2015;136(7):1680-1687. (PubMed)
76. Bristow SM, Bolland MJ, MacLennan GS, et al. Calcium supplements and cancer risk: a meta-analysis of randomised controlled trials. Br J Nutr. 2013;110(8):1384-1393. (PubMed)
77. Bolland MJ, Grey A, Gamble GD, Reid IR. Calcium and vitamin D supplements and health outcomes: a reanalysis of the Women's Health Initiative (WHI) limited-access data set. Am J Clin Nutr. 2011;94(4):1144-1149. (PubMed)
78. Bonovas S, Fiorino G, Lytras T, Malesci A, Danese S. Calcium supplementation for the prevention of colorectal adenomas: A systematic review and meta-analysis of randomized controlled trials. World J Gastroenterol. 2016;22(18):4594-4603. (PubMed)
79. Mielke HW, Gonzales C, Powell E, Mielke PW. Evolving from reactive to proactive medicine: community lead (Pb) and clinical disparities in pre- and post-Katrina New Orleans. Int J Environ Res Public Health. 2014;11(7):7482-7491. (PubMed)
80. Centers for Disease Control and Prevention. New blood lead level information. Available at: http://www.cdc.gov/nceh/lead/acclpp/blood_lead_levels.htm, 15 August 2014.
81. Bruening K, Kemp FW, Simone N, Holding Y, Louria DB, Bogden JD. Dietary calcium intakes of urban children at risk of lead poisoning. Environ Health Perspect. 1999;107(6):431-435. (PubMed)
82. Hertz-Picciotto I, Schramm M, Watt-Morse M, Chantala K, Anderson J, Osterloh J. Patterns and determinants of blood lead during pregnancy. Am J Epidemiol. 2000;152(9):829-837. (PubMed)
83. Ettinger AS, Lamadrid-Figueroa H, Tellez-Rojo MM, et al. Effect of calcium supplementation on blood lead levels in pregnancy: a randomized placebo-controlled trial. Environ Health Perspect. 2009;117(1):26-31. (PubMed)
84. Ettinger AS, Tellez-Rojo MM, Amarasiriwardena C, et al. Influence of maternal bone lead burden and calcium intake on levels of lead in breast milk over the course of lactation. Am J Epidemiol. 2006;163(1):48-56. (PubMed)
85. Hernandez-Avila M, Gonzalez-Cossio T, Hernandez-Avila JE, et al. Dietary calcium supplements to lower blood lead levels in lactating women: a randomized placebo-controlled trial. Epidemiology. 2003;14(2):206-212. (PubMed)
86. Muldoon SB, Cauley JA, Kuller LH, Scott J, Rohay J. Lifestyle and sociodemographic factors as determinants of blood lead levels in elderly women. Am J Epidemiol. 1994;139(6):599-608. (PubMed)
87. Dougkas A, Reynolds CK, Givens ID, Elwood PC, Minihane AM. Associations between dairy consumption and body weight: a review of the evidence and underlying mechanisms. Nutr Res Rev. 2011;24(1):72-95. (PubMed)
88. Zemel MB, Thompson W, Milstead A, Morris K, Campbell P. Calcium and dairy acceleration of weight and fat loss during energy restriction in obese adults. Obes Res. 2004;12(4):582-590. (PubMed)
89. Zemel MB, Shi H, Greer B, Dirienzo D, Zemel PC. Regulation of adiposity by dietary calcium. Faseb J. 2000;14(9):1132-1138. (PubMed)
90. Gonzalez JT, Rumbold PL, Stevenson EJ. Effect of calcium intake on fat oxidation in adults: a meta-analysis of randomized, controlled trials. Obes Rev. 2012;13(10):848-857. (PubMed)
91. Bortolotti M, Rudelle S, Schneiter P, et al. Dairy calcium supplementation in overweight or obese persons: its effect on markers of fat metabolism. Am J Clin Nutr. 2008;88(4):877-885. (PubMed)
92. Gallagher JC, Yalamanchili V, Smith LM. The effect of vitamin D supplementation on serum 25(OH)D in thin and obese women. J Steroid Biochem Mol Biol. 2013;136:195-200. (PubMed)
93. Pathak K, Soares MJ, Calton EK, Zhao Y, Hallett J. Vitamin D supplementation and body weight status: a systematic review and meta-analysis of randomized controlled trials. Obes Rev. 2014;15(6):528-537. (PubMed)
94. Christensen R, Lorenzen JK, Svith CR, et al. Effect of calcium from dairy and dietary supplements on faecal fat excretion: a meta-analysis of randomized controlled trials. Obes Rev. 2009;10(4):475-486. (PubMed)
95. Tordoff MG. Calcium: taste, intake, and appetite. Physiol Rev. 2001;81(4):1567-1597. (PubMed)
96. Chen M, Pan A, Malik VS, Hu FB. Effects of dairy intake on body weight and fat: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2012;96(4):735-747. (PubMed)
97. Booth AO, Huggins CE, Wattanapenpaiboon N, Nowson CA. Effect of increasing dietary calcium through supplements and dairy food on body weight and body composition: a meta-analysis of randomised controlled trials. Br J Nutr. 2015;114(7):1013-1025. (PubMed)
98. Li P, Fan C, Lu Y, Qi K. Effects of calcium supplementation on body weight: a meta-analysis. Am J Clin Nutr. 2016;104(5):1263-1273. (PubMed)
99. Soares MJ, Pathak K, Calton EK. Calcium and vitamin D in the regulation of energy balance: where do we stand? Int J Mol Sci. 2014;15(3):4938-4945. (PubMed)
100. Freeman EW. Premenstrual syndrome and premenstrual dysphoric disorder: definitions and diagnosis. Psychoneuroendocrinology. 2003;28 Suppl 3:25-37. (PubMed)
101. Pearlstein T, Steiner M. Premenstrual dysphoric disorder: burden of illness and treatment update. J Psychiatry Neurosci. 2008;33(4):291-301. (PubMed)
102. Bendich A. The potential for dietary supplements to reduce premenstrual syndrome (PMS) symptoms. J Am Coll Nutr. 2000;19(1):3-12. (PubMed)
103. Bertone-Johnson ER, Hankinson SE, Bendich A, Johnson SR, Willett WC, Manson JE. Calcium and vitamin D intake and risk of incident premenstrual syndrome. Arch Intern Med. 2005;165(11):1246-1252. (PubMed)
104. Thys-Jacobs S, Starkey P, Bernstein D, Tian J. Calcium carbonate and the premenstrual syndrome: effects on premenstrual and menstrual symptoms. Premenstrual Syndrome Study Group. Am J Obstet Gynecol. 1998;179(2):444-452. (PubMed)
105. Thys-Jacobs S, Ceccarelli S, Bierman A, Weisman H, Cohen MA, Alvir J. Calcium supplementation in premenstrual syndrome: a randomized crossover trial. J Gen Intern Med. 1989;4(3):183-189. (PubMed)
106. Alvir JM, Thys-Jacobs S. Premenstrual and menstrual symptom clusters and response to calcium treatment. Psychopharmacol Bull. 1991;27(2):145-148. (PubMed)
107. Bharati M. Comparing the Effects of Yoga & Oral Calcium Administration in Alleviating Symptoms of Premenstrual Syndrome in Medical Undergraduates. J Caring Sci. 2016;5(3):179-185. (PubMed)
108. Masoumi SZ, Ataollahi M, Oshvandi K. Effect of combined use of calcium and vitamin B6 on premenstrual syndrome symptoms: a randomized clinical trial. J Caring Sci. 2016;5(1):67-73. (PubMed)
109. Shehata NA. Calcium versus oral contraceptive pills containing drospirenone for the treatment of mild to moderate premenstrual syndrome: a double blind randomized placebo controlled trial. Eur J Obstet Gynecol Reprod Biol. 2016;198:100-104. (PubMed)
110. Shobeiri F, Araste FE, Ebrahimi R, Jenabi E, Nazari M. Effect of calcium on premenstrual syndrome: A double-blind randomized clinical trial. Obstet Gynecol Sci. 2017;60(1):100-105. (PubMed)
111. Nevatte T, O'Brien PM, Backstrom T, et al. ISPMD consensus on the management of premenstrual disorders. Arch Womens Ment Health. 2013;16(4):279-291. (PubMed)
112. Whelan AM, Jurgens TM, Naylor H. Herbs, vitamins and minerals in the treatment of premenstrual syndrome: a systematic review. Can J Clin Pharmacol. 2009;16(3):e407-429. (PubMed)
113. Cappuccio FP, Elliott P, Allender PS, Pryer J, Follman DA, Cutler JA. Epidemiologic association between dietary calcium intake and blood pressure: a meta-analysis of published data. Am J Epidemiol. 1995;142(9):935-945. (PubMed)
114. Appel LJ, Moore TJ, Obarzanek E, et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N Engl J Med. 1997;336(16):1117-1124. (PubMed)
115. Conlin PR, Chow D, Miller ER, 3rd, et al. The effect of dietary patterns on blood pressure control in hypertensive patients: results from the Dietary Approaches to Stop Hypertension (DASH) trial. Am J Hypertens. 2000;13(9):949-955. (PubMed)
116. Miller GD, DiRienzo DD, Reusser ME, McCarron DA. Benefits of dairy product consumption on blood pressure in humans: a summary of the biomedical literature. J Am Coll Nutr. 2000;19(2 Suppl):147S-164S. (PubMed)
117. Allender PS, Cutler JA, Follmann D, Cappuccio FP, Pryer J, Elliott P. Dietary calcium and blood pressure: a meta-analysis of randomized clinical trials. Ann Intern Med. 1996;124(9):825-831. (PubMed)
118. Bucher HC, Cook RJ, Guyatt GH, et al. Effects of dietary calcium supplementation on blood pressure. A meta-analysis of randomized controlled trials. JAMA. 1996;275(13):1016-1022. (PubMed)
119. Dickinson HO, Nicolson DJ, Cook JV, et al. Calcium supplementation for the management of primary hypertension in adults. Cochrane Database Syst Rev. 2006(2):CD004639. (PubMed)
120. Challoumas D, Cobbold C, Dimitrakakis G. Effects of calcium intake on the cardiovascular system in postmenopausal women. Atherosclerosis. 2013;231(1):1-7. (PubMed)
121. Blumberg JB, Frei BB, Fulgoni VL, Weaver CM, Zeisel SH. Impact of frequency of multi-vitamin/multi-mineral supplement intake on nutritional adequacy and nutrient deficiencies in US adults. Nutrients. 2017;9(8). (PubMed)
122. Kit BK, Fakhouri TH, Park S, Nielsen SJ, Ogden CL. Trends in sugar-sweetened beverage consumption among youth and adults in the United States: 1999-2010. Am J Clin Nutr. 2013;98(1):180-188. (PubMed)
123. Zhu K, Prince RL. Calcium and bone. Clin Biochem. 2012;45(12):936-942. (PubMed)
124. Bailey RL, Dodd KW, Goldman JA, et al. Estimation of total usual calcium and vitamin D intakes in the United States. J Nutr. 2010;140(4):817-822. (PubMed)
125. Straub DA. Calcium supplementation in clinical practice: a review of forms, doses, and indications. Nutr Clin Pract. 2007;22(3):286-296. (PubMed)
126. Roberts HJ. Potential toxicity due to dolomite and bonemeal. South Med J. 1983;76(5):556-559. (PubMed)
127. Bourgoin BP, Evans DR, Cornett JR, Lingard SM, Quattrone AJ. Lead content in 70 brands of dietary calcium supplements. Am J Public Health. 1993;83(8):1155-1160. (PubMed)
128. Scelfo GM, Flegal AR. Lead in calcium supplements. Environ Health Perspect. 2000;108(4):309-313. (PubMed)
129. Carrington CD, Bolger PM. An assessment of the hazards of lead in food. Regul Toxicol Pharmacol. 1992;16(3):265-272. (PubMed)
130. Ross EA, Szabo NJ, Tebbett IR. Lead content of calcium supplements. Jama. 2000;284(11):1425-1429. (PubMed)
131. Mindak WR, Cheng J, Canas BJ, Bolger PM. Lead in women's and children's vitamins. J Agric Food Chem. 2008;56(16):6892-6896. (PubMed)
132. Moe SM. Disorders involving calcium, phosphorus, and magnesium. Prim Care. 2008;35(2):215-237, v-vi. (PubMed)
133. Patel AM, Goldfarb S. Got calcium? Welcome to the calcium-alkali syndrome. J Am Soc Nephrol. 2010;21(9):1440-1443. (PubMed)
134. Lewis JR, Zhu K, Prince RL. Adverse events from calcium supplementation: relationship to errors in myocardial infarction self-reporting in randomized controlled trials of calcium supplementation. J Bone Miner Res. 2012;27(3):719-722. (PubMed)
135. World Cancer Research Fund International. Cancer facts and figures - worldwide data. 2012. Available at: http://www.wcrf.org/int/cancer-facts-figures/worldwide-data. Accessed 4/29/17.
136. Gonzalez CA, Riboli E. Diet and cancer prevention: Contributions from the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Eur J Cancer. 2010;46(14):2555-2562. (PubMed)
137. Kurahashi N, Inoue M, Iwasaki M, Sasazuki S, Tsugane AS, Japan Public Health Center-Based Prospective Study G. Dairy product, saturated fatty acid, and calcium intake and prostate cancer in a prospective cohort of Japanese men. Cancer Epidemiol Biomarkers Prev. 2008;17(4):930-937. (PubMed)
138. Raimondi S, Mabrouk JB, Shatenstein B, Maisonneuve P, Ghadirian P. Diet and prostate cancer risk with specific focus on dairy products and dietary calcium: a case-control study. Prostate. 2010;70(10):1054-1065. (PubMed)
139. Torfadottir JE, Steingrimsdottir L, Mucci L, et al. Milk intake in early life and risk of advanced prostate cancer. Am J Epidemiol. 2012;175(2):144-153. (PubMed)
140. Song Y, Chavarro JE, Cao Y, et al. Whole milk intake is associated with prostate cancer-specific mortality among U.S. male physicians. J Nutr. 2013;143(2):189-196. (PubMed)
141. Pettersson A, Kasperzyk JL, Kenfield SA, et al. Milk and dairy consumption among men with prostate cancer and risk of metastases and prostate cancer death. Cancer Epidemiol Biomarkers Prev. 2012;21(3):428-436. (PubMed)
142. Aune D, Navarro Rosenblatt DA, Chan DS, et al. Dairy products, calcium, and prostate cancer risk: a systematic review and meta-analysis of cohort studies. Am J Clin Nutr. 2015;101(1):87-117. (PubMed)
143. Qin LQ, He K, Xu JY. Milk consumption and circulating insulin-like growth factor-I level: a systematic literature review. Int J Food Sci Nutr. 2009;60 Suppl 7:330-340. (PubMed)
144. Rowlands MA, Gunnell D, Harris R, Vatten LJ, Holly JM, Martin RM. Circulating insulin-like growth factor peptides and prostate cancer risk: a systematic review and meta-analysis. Int J Cancer. 2009;124(10):2416-2429. (PubMed)
145. Allen NE, Key TJ, Appleby PN, et al. Animal foods, protein, calcium and prostate cancer risk: the European Prospective Investigation into Cancer and Nutrition. Br J Cancer. 2008;98(9):1574-1581. (PubMed)
146. Moreno J, Krishnan AV, Peehl DM, Feldman D. Mechanisms of vitamin D-mediated growth inhibition in prostate cancer cells: inhibition of the prostaglandin pathway. Anticancer Res. 2006;26(4A):2525-2530. (PubMed)
147. Brandstedt J, Almquist M, Manjer J, Malm J. Vitamin D, PTH, and calcium in relation to survival following prostate cancer. Cancer Causes Control. 2016;27(5):669-677. (PubMed)
148. Brandstedt J, Almquist M, Ulmert D, Manjer J, Malm J. Vitamin D, PTH, and calcium and tumor aggressiveness in prostate cancer: a prospective nested case-control study. Cancer Causes Control. 2016;27(1):69-80. (PubMed)
149. Rowland GW, Schwartz GG, John EM, Ingles SA. Protective effects of low calcium intake and low calcium absorption vitamin D receptor genotype in the California Collaborative Prostate Cancer Study. Cancer Epidemiol Biomarkers Prev. 2013;22(1):16-24. (PubMed)
150. Baron JA, Beach M, Wallace K, et al. Risk of prostate cancer in a randomized clinical trial of calcium supplementation. Cancer Epidemiol Biomarkers Prev. 2005;14(3):586-589. (PubMed)
151. Chung M, Balk EM, Brendel M, et al. Vitamin D and calcium: a systematic review of health outcomes. Evid Rep Technol Assess (Full Rep). 2009(183):1-420. (PubMed)
152. Huncharek M, Muscat J, Kupelnick B. Dairy products, dietary calcium and vitamin D intake as risk factors for prostate cancer: a meta-analysis of 26,769 cases from 45 observational studies. Nutr Cancer. 2008;60(4):421-441. (PubMed)
153. Pentti K, Tuppurainen MT, Honkanen R, et al. Use of calcium supplements and the risk of coronary heart disease in 52-62-year-old women: The Kuopio Osteoporosis Risk Factor and Prevention Study. Maturitas. 2009;63(1):73-78. (PubMed)
154. Li K, Kaaks R, Linseisen J, Rohrmann S. Associations of dietary calcium intake and calcium supplementation with myocardial infarction and stroke risk and overall cardiovascular mortality in the Heidelberg cohort of the European Prospective Investigation into Cancer and Nutrition study (EPIC-Heidelberg). Heart. 2012;98(12):920-925. (PubMed)
155. Xiao Q, Murphy RA, Houston DK, Harris TB, Chow WH, Park Y. Dietary and supplemental calcium intake and cardiovascular disease mortality: the National Institutes of Health-AARP diet and health study. JAMA Intern Med. 2013;173(8):639-646. (PubMed)
156. Bolland MJ, Barber PA, Doughty RN, et al. Vascular events in healthy older women receiving calcium supplementation: randomised controlled trial. BMJ. 2008;336(7638):262-266. (PubMed)
157. Bolland MJ, Grey A, Avenell A, Gamble GD, Reid IR. Calcium supplements with or without vitamin D and risk of cardiovascular events: reanalysis of the Women's Health Initiative limited access dataset and meta-analysis. BMJ. 2011;342:d2040. (PubMed)
158. Hsia J, Heiss G, Ren H, et al. Calcium/vitamin D supplementation and cardiovascular events. Circulation. 2007;115(7):846-854. (PubMed)
159. Abrahamsen B, Sahota O. Do calcium plus vitamin D supplements increase cardiovascular risk? BMJ. 2011;342:d2080. (PubMed)
160. Lewis JR, Calver J, Zhu K, Flicker L, Prince RL. Calcium supplementation and the risks of atherosclerotic vascular disease in older women: results of a 5-year RCT and a 4.5-year follow-up. J Bone Miner Res. 2011;26(1):35-41. (PubMed)
161. Avenell A, MacLennan GS, Jenkinson DJ, et al. Long-term follow-up for mortality and cancer in a randomized placebo-controlled trial of vitamin D(3) and/or calcium (RECORD trial). J Clin Endocrinol Metab. 2012;97(2):614-622. (PubMed)
162. Van Hemelrijck M, Michaelsson K, Linseisen J, Rohrmann S. Calcium intake and serum concentration in relation to risk of cardiovascular death in NHANES III. PLoS One. 2013;8(4):e61037. (PubMed)
163. Foley RN, Collins AJ, Ishani A, Kalra PA. Calcium-phosphate levels and cardiovascular disease in community-dwelling adults: the Atherosclerosis Risk in Communities (ARIC) Study. Am Heart J. 2008;156(3):556-563. (PubMed)
164. Lutsey PL, Alonso A, Michos ED, et al. Serum magnesium, phosphorus, and calcium are associated with risk of incident heart failure: the Atherosclerosis Risk in Communities (ARIC) Study. Am J Clin Nutr. 2014;100(3):756-764. (PubMed)
165. Wang TK, Bolland MJ, van Pelt NC, et al. Relationships between vascular calcification, calcium metabolism, bone density, and fractures. J Bone Miner Res. 2010;25(12):2777-2785. (PubMed)
166. Samelson EJ, Booth SL, Fox CS, et al. Calcium intake is not associated with increased coronary artery calcification: the Framingham Study. Am J Clin Nutr. 2012;96(6):1274-1280. (PubMed)
167. Anderson JJ, Kruszka B, Delaney JA, et al. Calcium intake from diet and supplements and the risk of coronary artery calcification and its progression among older adults: 10-year follow-up of the Multi-Ethnic Study of Atherosclerosis (MESA). J Am Heart Assoc. 2016;5(10). (PubMed)
168. Lewis JR, Zhu K, Thompson PL, Prince RL. The effects of 3 years of calcium supplementation on common carotid artery intimal medial thickness and carotid atherosclerosis in older women: an ancillary study of the CAIFOS randomized controlled trial. J Bone Miner Res. 2014;29(3):534-541. (PubMed)
169. Lewis JR, Radavelli-Bagatini S, Rejnmark L, et al. The effects of calcium supplementation on verified coronary heart disease hospitalization and death in postmenopausal women: a collaborative meta-analysis of randomized controlled trials. J Bone Miner Res. 2015;30(1):165-175. (PubMed)
170. Chung M, Tang AM, Fu Z, Wang DD, Newberry SJ. Calcium intake and cardiovascular disease risk: an updated systematic review and meta-analysis. Ann Intern Med. 2016;165(12):856-866. (PubMed)
171. Kopecky SL, Bauer DC, Gulati M, et al. Lack of evidence linking calcium with or without vtamin D supplementation to cardiovascular disease in generally healthy adults: a clinical guideline from the National Osteoporosis Foundation and the American Society for Preventive Cardiology. Ann Intern Med. 2016;165(12):867-868. (PubMed)
172. Vella A, Gerber TC, Hayes DL, Reeder GS. Digoxin, hypercalcaemia, and cardiac conduction. Postgrad Med J. 1999;75(887):554-556. (PubMed)
173. Moser LR, Smythe MA, Tisdale JE. The use of calcium salts in the prevention and management of verapamil-induced hypotension. Ann Pharmacother. 2000;34(5):622-629. (PubMed)
174. Bania TC, Blaufeux B, Hughes S, Almond GL, Homel P. Calcium and digoxin vs. calcium alone for severe verapamil toxicity. Acad Emerg Med. 2000;7(10):1089-1096. (PubMed)
175. Natural Medicines. Calcium - Professional handout/Drug interactions. Available at: https://naturalmedicines.therapeuticresearch.com/. Accessed 5/31/17.
176. Ito T, Jensen RT. Association of long-term proton pump inhibitor therapy with bone fractures and effects on absorption of calcium, vitamin B12, iron, and magnesium. Curr Gastroenterol Rep. 2010;12(6):448-457. (PubMed)
177. Wright MJ, Proctor DD, Insogna KL, Kerstetter JE. Proton pump-inhibiting drugs, calcium homeostasis, and bone health. Nutr Rev. 2008;66(2):103-108. (PubMed)
178. Wood RJ, Zheng JJ. High dietary calcium intakes reduce zinc absorption and balance in humans. Am J Clin Nutr. 1997;65(6):1803-1809. (PubMed)
179. Borel P, Desmarchelier C, Dumont U, et al. Dietary calcium impairs tomato lycopene bioavailability in healthy humans. Br J Nutr. 2016;116(12):2091-2096. (PubMed)
Cobre
Contenido
Resumen
- El cobre es un cofactor esencial para las reacciones de oxidación y reducción que implican oxidasas que contienen cobre. Las enzimas de cobre regulan varias vías fisiológicas, como la producción de energía, el metabolismo del hierro, maduración del tejido conjuntivo, y la neurotransmisión. (Más información)
- La deficiencia de cobre puede resultar de la malnutrición, malabsorción, o una ingesta excesiva de zinc y puede ser adquirida o heredada. Síntomas incluyen deficiencias en las células sanguíneas, anormalidades en el tejido óseo y conjuntivo, y desórdenes neurológicos. (Más información)
- Un desequilibrio del cobre marginal ha sido ligado a una alteración de la función inmune, desmineralización de los huesos, y un riesgo incrementado de enfermedades cardiovasculares y neurodegenerativas. Sin embargo el uso de indicadores más precisos del estado nutricional del cobre necesitan ser considerados para investigaciones futuras. (Más información)
- Viseras, mariscos, frutos secos, semillas, cereales de salvado de trigo, y productos de grano entero son buenas fuentes de cobre. (Más información)
- La toxicidad del cobre es rara y la mayoría de las veces es asociada con defectos genéticos de metabolismo del cobre. (Más información)
El cobre (Cu) es un elemento traza esencial para los seres humanos y animales. En el cuerpo, el cobre se desplaza entre sus formas cuproso (Cu1+) y cúprico (Cu2+), aunque la mayoría del cobre en el organismo se encuentra en forma (Cu2+). La habilidad del cobre para aceptar y donar electrones fácilmente explica la importancia de este mismo en las reacciones de oxidación y reducción (redox) y la actividad de barrido de radicales libres (1). Aunque se dice que Hipócrates prescribía compuestos de cobre para tratar enfermedades desde principios del 400 A.C. (2), científicos están aun descubriendo nueva información acerca de las funciones del cobre en el cuerpo humano (3).
Función
El cobre es un componente crítico funcional de varias enzimas esenciales conocidas como cuproenzimas (4). Algunas de las funciones fisiológicas conocidas por ser cobre-dependientes son explicadas a continuación.
Producción de energía
La enzima cobre-dependiente, llamada citocromo C oxidasa, juega un papel crítico en la producción de energía celular. Al catalizar la reducción del oxigeno molecular (O2) a agua (H2O), la citocromo c oxidasa genera un gradiente eléctrico usado por la mitocondria para crear la molécula de almacenaje energético vital, el ATP (5).
Formación del tejido conjuntivo
Otra cuproenzima, la lisil oxidasa, es necesaria para el entrecruzamiento del colágeno y la elastina, los cuales son esenciales para la formación de un tejido conjuntivo fuerte y flexible. La acción de la lisil oxidasa ayuda a mantener la integridad del tejido conjuntivo en el corazón y los vasos sanguíneos, y también participa en la formación ósea (2).
Metabolismo del hierro
Cuatro enzimas que contienen cobre, conocidas como oxidasas multicobre o ferroxidasas tienen la capacidad de oxidar el hierro ferroso (Fe2+) en hierro férrico (Fe3+), la forma del hierro que puede ser cargada en la proteína transferrina para su transporte al sitio de formación de glóbulos rojos. La familia de oxidasas multicobre comprende la ceruloplasmina circulante (la cual representa ~90% del cobre en el plasma), la ceruloplasmina unida a la membrana (llamada ceruloplasmina GPI) y dos proteínas llamadas Hefaestina y Zyklopen encontradas en los intestinos y la placenta respectivamente (6). Interesantemente, ratones que no expresan ceruloplasmina (Cp-/-) tienen un metabolismo normal del cobre pero una acumulación anormal de hierro en el hígado (7, 8). Similarmente, individuos que necesitan ceruloplasmina muestran una sobrecarga de hierro en ciertos tejidos, incluyendo el hígado, cerebro, y la retina (9). Esto apoya la idea de que la actividad ferroxidasa de la ceruloplasmina es esencial para el flujo de hierro en el cuerpo. Por otra parte, el hecho de que la movilización del hierro de los lugares de almacenamiento se altera en la deficiencia de cobre apoya el papel de las oxidasas multicobre en el metabolismo del hierro (10).
Sistema nervioso central
Una serie de reacciones esenciales para el funcionamiento normal del cerebro y el sistema nervioso son catalizadas por cuproenzimas.
Síntesis de neurotransmisores
La dopamina β hidroxilasa cataliza la conversión de la dopamina para el neurotransmisor norepinefrina (11).
Formación y mantenimiento de la mielina
La envoltura de la mielina está hecha de fosfolípidos cuyas síntesis dependen de la actividad de la citocromo c oxidasa (2).
Formación de la melanina
La cuproenzima, tirosinasa, es requerida para la formación del pigmento melanina. La melanina se forma en células llamadas melanocitos y juega un papel en la pigmentación del cabello, la piel y los ojos (2).
Funciones de antioxidantes
Superóxido dismutasa
La enzima superóxido dismutasa (SOD) funciona como un antioxidante al catalizar la conversión de radicales superóxido (radicales libres) a peróxido de hidrógeno, el cual subsecuentemente puede ser reducido a agua por otras enzimas antioxidantes (12). Dos formas de la SOD contienen cobre: (1) la SOD con cobre/zinc se encuentra dentro de la mayoría de las células del cuerpo, incluyendo eritrocitos; y (2) la SOD extracelular que es una enzima contenedora de cobre encontrada en altos niveles en los pulmones y en bajos niveles de plasma sanguíneo (2).
Ceruloplasmina
La ceruloplasmina puede funcionar como un antioxidante de dos diferentes maneras. Iones libres de cobre y hierro son poderosos catalizadores del daño por radicales libres. Al unir el cobre, la ceruloplasmina previene que los iones libres de cobre catalicen el daño oxidativo. La actividad ferroxidasa de la ceruloplasmina (oxidación del hierro ferroso) facilita la carga del hierro en su proteína transportadora, la transferrina, y puede prevenir que los iones libres de hierro (Fe2+) participen en las reacciones dañinas que producen radicales libres (12).
Regulación de la expresión de genes
Los niveles celulares del cobre pueden afectar la síntesis de las proteínas al potenciar o inhibir la transcripción de genes específicos. El cobre puede regular la expresión de los genes incrementando el nivel de estrés oxidativo intracelular. Un cierto número de vías de transducción de señales son activadas en respuesta al estrés oxidativo y pueden conducir al incremento en la expresión de genes envueltos en la desintoxicación de especies reactivas de oxigeno (13).
Interacción con nutrientes
Hierro
Un adecuado estatus nutricional del cobre es necesario para un metabolismo normal del hierro y para la formación de glóbulos rojos. La anemia es una señal clínica de la deficiencia del cobre, y el hierro ha sido encontrado acumulado en los hígados de animales con una deficiencia de cobre, indicando que el cobre (a través de la ceruloplasmina) es requerido para el transporte del hierro a la medula ósea para la formación de glóbulos rojos (ver Metabolismo del hierro) (2). La conexión entre la disponibilidad del cobre y el metabolismo del hierro ha sido también establecida en humanos; una deficiencia de cobre puede llevar a una deficiencia secundaria de ceruloplasmina y una sobrecarga de hierro hepático y/o cirrosis (10). La suplementación oral de cobre restauro los niveles de normales de ceruloplasmina y la actividad plasmática ferroxidasa y corrigió el desorden del metabolismo del hierro en un sujeto con deficiencia de cobre (14). Por otra parte, infantes alimentados con fórmulas altas en hierro absorbieron menos cobre que los infantes alimentados con fórmulas con bajos niveles de hierro, sugiriendo que altas ingestas de hierro en infantes pueden interferir con la absorción de cobre (15).
Zinc
Altas ingestas suplementarias de zinc de 50 mg/día o más por periodos extensos de tiempo puede resultar en una deficiencia de cobre. Una alta ingesta dietaría de zinc incrementa la síntesis de una proteína celular llamada metalotioneína, la cual se une a ciertos metales y previene su absorción atrapándolos en las células intestinales. La metalotioneína tiene una afinidad superior al cobre que al zinc, es por ello que altos niveles of metalotioneína inducidos por un exceso de zinc causan un descenso en la absorción del cobre. En contraste, altas ingestas de cobre no se ha encontrado que afecten el estatus nutricional del zinc (2, 15). La suplementación con zinc (10 mg/día por 8 semanas) fue capaz de restaurar las proporciones normales de cobre/zinc del plasma en 65 sujetos con hemodiálisis a largo plazo que inicialmente exhibieron bajos niveles de zinc y elevados niveles de cobre. Si una mejora del estatus del zinc y el cobre en los pacientes de hemodiálisis puede impactar sus resultados clínicos aún necesita ser evaluada (16).
Fructosa
Dietas altas en fructosa han exacerbado la deficiencia de cobre en ratas pero no en cerdos los cuales poseen un sistema gastrointestinal más parecido al de los seres humanos. Muy altos niveles de fructuosa dietaría (20% del total de las calorías) no resultaron en una reducción de cobre en humanos, sugiriendo que la ingesta de fructosa no resulta en una reducción del cobre en niveles relevantes de dietas normales (2, 15).
Vitamina C
Aunque los suplementos de vitamina C han producido deficiencia de cobre en conejillos de indias (17), el efecto de los suplementos de vitamina C en el estado nutricional del cobre en seres humanos es menos claro. Dos estudios menores en hombres adultos jóvenes sanos indican que la actividad oxidasa de la ceruloplasmina puede verse deteriorada por dosis relativamente altas de vitamina C suplementaria. En un estudio, la suplementación con vitamina C de 1,500 mg/día por dos meses resultó en una disminución significativa de la actividad oxidasa de la ceruloplasmina (18). En el otro estudio, los suplementos de 605 mg de vitamina C/día por tres semanas provocaron una disminución en la actividad oxidasa de la ceruloplasmina, aunque la absorción de cobre no disminuyo (19). Ninguno de estos estudios encontró que la suplementación de vitamina C afectara adversamente el estatus nutricional del cobre.
Deficiencia
La deficiencia franca del cobre dietario o clínicamente evidente es relativamente poco común. Los niveles del cobre en el suero y la ceruloplasmina pueden caer hasta un 30% de lo normal en casos de severa deficiencia del cobre. La hipocupremia (bajo contenido de cobre en la sangre) es también observada en desordenes genéticos debido al metabolismo del cobre como la aceruloplasminemia y paradójicamente en la enfermedad de Wilson, los cuales no están ligados a una deficiencia dietaría del cobre. Uno de los signos clínicos más comunes de la deficiencia del cobre es una anemia que no responde a la terapia con hierro pero es corregida con una suplementación de cobre. Aunque se piensa que la anemia es el resultado defectuoso de la movilización del hierro debido a la disminución de la actividad de la ceruloplasmina, la ausencia de ceruloplasmina en individuos con aceruloplasminemia hereditaria no está siempre asociada con la anemia manifiesta (20). La deficiencia del cobre puede conducir también a anormales bajos números de glóbulos blancos conocidos con neutrófilos (neutropenia), una condición que puede ser acompañada por un incremento en la susceptibilidad a infecciones. Estudios de repleción de cobre han sugerido que la reducción en la disponibilidad del cobre puede afectar el linaje de las células eritroides y mieloides, apoyando el papel del cobre en la regulación de la renovación de células sanguíneas (21, 22). Más investigación es claramente necesaria para descubrir los mecanismos subyacentes de la deficiencia inducida del cobre en la anemia y neutropenia (4, 23). La osteoporosis y otras anormalidades del desarrollo óseo relacionados con la deficiencia del cobre son los más comunes en lactantes de bajo peso al nacer con deficiencia de cobre y niños pequeños. Características menos comunes de la deficiencia de cobre puede incluir perdida de la pigmentación, síntomas neurológicos, y problemas de crecimiento (2, 5).
Individuos en riesgo de deficiencia
La leche de vaca es relativamente baja en cobre, y se han reportado casos de deficiencia de cobre de alto riesgo en infantes y niños alimentados sólo con fórmula de leche de vaca (24). Individuos de alto riesgo incluyen infantes prematuros (especialmente aquellos con bajo peso al nacer), infantes con diarrea prolongada, infantes y niños en recuperación de malnutrición, e individuos con síndromes de malabsorción, incluyendo enfermedad celiaca, esprúe, y síndrome del intestino corto debido a una remoción quirúrgica de una gran porción del intestino. Los individuos que reciben nutrición parenteral total intravenosa carente de cobre u otras dietas restringidas, pueden también necesitar de la suplementación con cobre y otros elementos trazas (2, 5). La deficiencia de cobre en infantes con colestasis (reducida excreción biliar del cobre) ha sido ligada a la nutrición parental a largo plazo carente de cobre (25). Reportes de casos indican que los pacientes con fibrosis quística pueden también estar en mayor riesgo de una deficiencia de cobre (26). Finalmente, una ingesta excesiva de zinc ha llevado a una deficiencia de cobre secundaria en individuos al usar suplementos de zinc o cremas dentales enriquecidas con zinc (27, 28).
Deficiencia de cobre adquirida
Un síndrome neurológico ha sido descrito en adultos con una deficiencia de cobre adquirida (29). Los síntomas incluyen desmielinización, mielopatía, y polineuropatia del sistema nervioso central y una inflamación del nervio óptico. La etiología es desconocida en ausencia de factores de riesgo prominentes (vea factores de riesgo en la siguiente sección: Individuos en riesgo de deficiencia); reportes de casos describen un incremento del contenido de cobre en los intestinos sugiriendo un síndrome de mal absorción como la enfermedad de Menkes, pero las mutaciones en el gen ATP7 no estuvieron ligadas a la condición (vea Deficiencia de cobre hereditaria en la parte de abajo) (30). El remplazo oral de cobre (2 mg/día de cobre elemental) normaliza las concentraciones de cobre y ceruloplasmina en el suero, estabiliza la condición y mejora significativamente la calidad de vida de los sujetos afectados. Sin embargo, la duración de la suplementación con cobre no ha sido establecida aun, y la incrementación de la dosis puede ser requerida en caso de una recaída (29).
Deficiencia de cobre hereditaria
El trafico del cobre dentro de la mayoría de la células excepto por los hepatocitos (células del hígado) esta facilitado por el transporte de Cu1+- y ATPasa llamada ATP7A. Mutaciones en el gen ATP7A perjudican el transporte de cobre intracelular, el cual se acumula en el citosol de los enterocitos y las células endoteliales vasculares (31). Esto resulta en una deficiencia sistémica de cobre y disminuye la actividad de las cuproenzimas. El transporte del cobre en el cerebro es también afectado, llevando a una acumulación de cobre en la barrera hematoencefálica del cerebro y a una disminución de la actividad de las cuproenzimas en las neuronas. Los individuos afectados son diagnosticados con la enfermedad de Menkes o con una forma más leve de la enfermedad llamada síndrome del cuerno occipital (OHS por sus siglas en Inglés). Las características clínicas de la enfermedad de Menkes (MD) incluyen convulsiones intratables, desordenes del tejido conjuntivo, hemorragia subdural, y anormalidades del cabello ("cabello ensortijado"). Los pacientes con OHS exhiben hipotonía muscular y anormalidades del tejido conjuntivo, incluyendo exostosis en los huesos occipitales. Inyecciones subcutáneas de cobre-histidina son usadas para evitar la absorción intestinal defectuosa y mejorar las funciones metabólicas del cobre en los pacientes. Sin embargo, la entrada de cobre en el cerebro sigue siendo limitada (revisado en 32).
La Ingesta Diaria Recomendada (IDR)
Una variedad de indicadores fue usada para establecer la IDR para el cobre, incluyendo la concentración de cobre en el plasma, la actividad de ceruloplasmina plasmática, la actividad de la superóxido dismutasa en los eritrocitos, y las concentraciones de cobre plaquetario (15). La IDR de cobre refleja los resultados de estudios de eliminación y reposición, y está basada en la prevención de una deficiencia.
| Etapa de la Vida | Edad | Machos (μg/día) | Hembras (μg/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 200 (IA) | 200 (IA) |
| Infantes | 7-12 meses | 220 (IA) | 220 (IA) |
| Niños | 1-3 años | 340 | 340 |
| Niños | 4-8 años | 440 | 440 |
| Niños | 9-13 años | 700 | 700 |
| Adolescentes | 14-18 años | 890 | 890 |
| Adultos | 19 años y más | 900 | 900 |
| Embarazo | Todas las edades | - | 1,000 |
| Período de lactancia | Todas las edades | - | 1,300 |
Prevención de Enfermedades
Enfermedad cardiovascular
Mientras está claro que la severa deficiencia de cobre resulta en anormalidades y daño cardíaco (cardiomiopatía) en algunas especies de animales, la patología difiere de la enfermedad cardiovascular aterosclerótica que prevalece en seres humanos (15). Estudios en humanos han producido resultados inconsistentes, y su interpretación se dificulta por la falta de un marcador confiable del estado nutricional del cobre. Fuera del cuerpo, el cobre que se encuentra libre es conocido por ser un pro-oxidante y es frecuentemente usado para producir la oxidación de lipoproteínas de baja densidad (LDL) en tubos de ensayos. Se ha encontrado que la proteína contenedora de cobre, la ceruloplasmina estimula la oxidación de proteínas de baja densidad en tubos de ensayo (33), llevando a algunos científicos a proponer que el incremento en los niveles de cobre podría incrementar el riesgo de aterosclerosis al promover la oxidación de LDL. Sin embargo, hay poca evidencia de que el cobre o la ceruloplasmina promuevan la oxidación de LDL en el cuerpo humano. Además, las cuproenzimas superóxido dismutasa y ceruloplasmina, son conocidas por tener propiedades antioxidantes, llevando a que algunos expertos propongan que más bien es la deficiencia de cobre en vez del exceso la que incrementa el riesgo de enfermedades cardiovasculares (34).
Estudios epidemiológicos
Varios estudios epidemiológicos han encontrado que el incremento en los niveles de cobre en el plasma se asocia con un incremento del riesgo de padecer de enfermedades cardiovasculares. Un estudio de cohorte prospectivo en los EE. UU. examinó los niveles de cobre en el plasma en más de 4,500 hombres y mujeres de entre 30 o más años de edad (35). Durante 16 años de seguimiento, 151 participantes murieron de enfermedad coronaria. Después del ajuste por otros factores de riesgo de enfermedades cardíacas, aquellos con niveles de cobre plasmático en los dos cuartiles más altos tuvieron un riesgo significativamente mayor de morir de una enfermedad coronaria. Otros tres estudios de caso y control llevados a cabo en Europa tuvieron hallazgos similares. Un estudio menor en 60 pacientes con falla cardíaca crónica o enfermedad cardíaca isquémica reportó que el cobre del suero era un predictor de resultados a corto plazo (36). Otro estudio de cohorte prospectivo en 4,035 hombres de mediana edad reportó que altos niveles de cobre en el suero estaban significativamente relacionados con un incremento del 50% en la mortalidad general; sin embargo, el cobre plasmático no estuvo asociado significativamente con la mortalidad cardiovascular en este estudio (37). Adicionalmente, se ha encontrado que el cobre en el suero es elevado en pacientes con cardiopatía reumática (38).
Es importante notar que el cobre en el suero refleja en gran medida la ceruloplasmina y no es un indicador sensible del estado nutricional del cobre (39). Los niveles de ceruloplasmina plasmática son conocidos por incrementar en un 50% o más, bajo ciertas condiciones de estrés físico, como traumas, inflamación o enfermedades. Debido a que más del 90% del cobre plasmático es llevado en la ceruloplasmina, la cual se incrementa en condiciones inflamatorias, el cobre plasmático elevado puede ser simplemente un marcador de la inflamación que acompaña la aterosclerosis. En contraste a los hallazgos epidemiológicos que vinculan el incremento de los niveles de cobre en el suero con la enfermedad cardíaca, dos estudios de autopsias encontraron que los niveles de cobre en el músculo cardíaco eran en realidad más bajos en pacientes que murieron de enfermedad coronaria que en aquellos que murieron por otras causas (40). Además, el contenido de cobre en los glóbulos blancos ha sido positivamente correlacionado con el grado de permeabilidad de las arterias coronarias en pacientes con enfermedad coronaria (41, 42). Aún más, los pacientes con historia de infarto al miocardio (IM) tuvieron concentraciones de superóxido dismutasa extracelular más bajas que aquellos sin un historial de IM (43). Así, debido a la carencia de un biomarcador confiable del estado nutricional del cobre, no es claro si el cobre está o no relacionado con las enfermedades cardiovasculares.
Estudios de intervención/experimentales
Mientras que algunos estudios en un número muy reducido de adultos, alimentados con dietas experimentales bajas en cobre, han demostrado cambios adversos en los niveles de colesterol sanguíneo, incluyendo un incremento total en los niveles de colesterol LDL, y una disminución en los niveles de colesterol HDL (44), otros estudios no han confirmado estos resultados (45). La suplementación de cobre de 2-3 mg/día por cuatro u ocho semanas no resulto en cambios clínicos significantes en los niveles de colesterol (34, 46, 47). Además, la investigación ha fallado en encontrar evidencia que el incremento en la ingesta de cobre incremente el estrés oxidativo. En una prueba multicéntrica controlada con placebo, la suplementación con cobre de 3 y 6 mg/día por seis semanas no resultó en un incremento de la susceptibilidad a la oxidación de LDL, inducida fuera del cuerpo (ex vivo) por cobre o peroxinitrito (una especie reactiva del nitrógeno) (48). Más aún, la suplementación con 3 y 6 mg/día de cobre disminuyó la oxidabilidad in vitro de glóbulos rojos (49), indicando que las ingestas de cobre relativamente altas no incrementan la susceptibilidad a de LDL o de glóbulos rojos a la oxidación.
Resumen
Aunque el cobre libre y la ceruloplasmina pueden promover la oxidación de LDL en tubos de ensayo, hay poca evidencia de que un incremento de cobre dietario aumente el estrés oxidativo en el cuerpo humano. El incremento en los niveles de cobre en el suero han sido asociados con un incremento del riesgo de enfermedades cardiovasculares, pero la importancia de estos hallazgos no está clara debido a la asociación entre los niveles de ceruloplasmina en la sangre y las condiciones inflamatorias. La aclaración de las relaciones entre el estado nutricional del cobre, los niveles de ceruloplasmina y el riesgo de enfermedades cardiovasculares, requiere de más investigación.
Notablemente, se sugirió que las concentraciones elevadas de cobre en el plasma pudieran estar ligadas a niveles altos de homocisteína en individuos con enfermedades vasculares (50, 51). Los niveles incrementados de homocisteína están asociados con lesiones de la pared arterial y un incremento en el riesgo de padecer enfermedades cardiovasculares (52). La interacción entre la homocisteína y el cobre fue ligada a una alteración en la función endotelial vascular en modelos animales (53, 54). Sin embargo, aunque la restricción de cobre en animales ha mostrados algunos efectos benéficos en los niveles de homocisteína y lesiones aterogénicas (55, 56), aun no se conoce si el desequilibrio del cobre contribuye al efecto aterogénico de la homocisteína en seres humanos.
Función del sistema inmune
Se sabe que el cobre juega un papel importante en el desarrollo y mantenimiento de la función del sistema inmune, pero el mecanismo exacto de su acción aún se desconoce. La neutropenia (cantidad anormalmente baja de los glóbulos blancos conocidos como neutrófilos) es un signo clínico de la deficiencia de cobre en seres humanos. Los efectos adversos por insuficiencia de cobre en el sistema inmune parecen ser más pronunciados en infantes. Infantes con síndrome de Menkes, un desorden genético que resulta en una deficiencia de cobre severa, sufren de frecuentes infecciones severas (57, 58). En un estudio de 11 niños malnutridos con evidente deficiencia de cobre, la habilidad de ciertos glóbulos blancos de fagocitar patógenos incrementó significativamente después de un mes de suplementación con cobre (59). Por otra parte, 11 hombres con una dieta baja en cobre (0.66 mg de cobre/día por 24 días y 0.38 mg de cobre/día por otros 40 días) mostraron una disminución de la respuesta proliferaría cuando glóbulos blancos denominados monocitos fueron aislados de su sangre, y presentados con un reto inmunológico en cultivo celular (60). Recientes estudios mecanicistas apoyan el papel del cobre en la innata respuesta inmune contra infecciones bacterianas (examinado en 61). Mientras que una severa deficiencia de cobre tiene efectos adversos en la función inmune, los efectos de la insuficiencia de cobre marginal en seres humanos no están todavía claros.
Osteoporosis
La pérdida progresiva de la densidad mineral ósea (DMO) llevando a una osteopenia (pre-osteoporosis) y osteoporosis es comúnmente observada en la población de adultos mayores. Las mujeres son más afectadas por la osteoporosis que los hombres (ej. una razón de 5:1 en blancos no hispánicos) (62), principalmente debido a la reducción de la producción de estrógeno en la postmenopausia que es esencial para el mantener la fuerza en músculos, huesos, y tejido conectivo (63). La osteoporosis está asociada con un incremento del riesgo de caídas, fracturas de huesos, y mortalidad en individuos mayores de 65 años de edad (64). La osteoporosis también ha sido reportada en infantes con una severa deficiencia de cobre (65, 66) pero no está completamente claro si la deficiencia de cobre marginal durante la adultez contribuye a desarrollar osteoporosis. Mientras que un incremento en la resorción ósea fue observada en 11 adultos masculinos sanos con una ingesta de cobre marginal de 0.7 mg/día por 6 semanas (67), la suplementación de 3 a 6 mg/día de cobre por seis semanas no tuvo efecto en los marcadores bioquímicos de la resorción ósea o formación ósea en dos estudios en hombres y mujeres adultos sanos (68, 69). Sin embargo, es posible que una reducción de la ingesta de cobre y absorción en adultos mayores reduzca la actividad de la enzima cobre-dependiente lisil oxidasa, la cual es requerida en la maduración del colágeno — un elemento clave en la matriz orgánica del hueso.
En general, la investigación con respecto al papel del estado nutricional del cobre en la osteoporosis senil es limitada. Un estudio temprano encontró que los niveles de cobre en el suero en 46 pacientes mayores con fracturas en la cadera fueron significativamente más bajos que aquellos de control emparejado (70). Sin embargo, otro estudio no encontró diferencias en los niveles de cobre en el suero entre mujeres postmenopáusicas con una DMO normal (N=40), osteopenia (N=40) u osteoporosis (N=40) (71). Un pequeño estudio en mujeres perimenopáusicas, que consumieron diariamente un promedio de 1 mg de cobre dietario, reportó una disminución en la pérdida de densidad mineral ósea (DMO) de la columna lumbar luego de la suplementación con cobre de 3 mg/día por dos años (72). Adicionalmente, un ensayo doble ciego controlado con placebo de dos años, en 59 mujeres postmenopáusicas, encontró que la combinación de calcio suplementario y minerales trazas, incluyendo 2.5 mg diarios de cobre, resultó en la mantención de la densidad ósea de la columna dorsal, mientras que el calcio suplementario o los minerales trazas por sí solos, no fueron efectivos en la prevención de la pérdida de la densidad ósea (73). Sin embargo, un estudio aleatorio, doble ciego controlado con placebo más reciente el cual inicialmente se llevó a cabo en 224 mujeres postmenopáusicas sanas de entre 51 a 80 años de edad encontró que la suplementación diaria de 600 mg de calcio, 2 mg de cobre, y 12 mg de zinc por 2 años disminuyo la densidad mineral ósea (DMO) de todo el cuerpo comparada con la suplementación de calcio solo. Más aun, aunque la DMO fue claramente reducida en sujetos con ingestas dietarías de cobre debajo de la IDR (0.9 mg/día), el cobre suplementario no previno la pérdida progresiva de la DMO así como el régimen de calcio solo (74). Finalmente varios estudios han sugerido que la perdida de dientes puede estar relacionada a un DMO sistémico pobre (75, 76). Cuando se comparó con 20 controles sanos emparejados, 50 pacientes (con un promedio de 47.5 años) con una DMO baja en la espina dorsal y un desgate dental avanzado fueron encontrados con un significativo contenido bajo de cobre en el esmalte de los dientes. Sin embargo, a pesar de que evidencia sugiere una desmineralización ósea, los niveles de cobre en el suero en esta población fueron similares a aquellos del grupo sano (77). Más investigación es requerida para llegar a una conclusión con respecto a los efectos de la deficiencia de cobre marginal, y la suplementación de cobre en el metabolismo óseo y la osteoporosis senil.
Enfermedades neurodegenerativas
Enfermedad de Alzheimer
El deterioro cognitivo en individuos con la enfermedad de Alzheimer está ligado a la presencia de placas de β-amiloide y agregados anormales de proteínas formadoras de Tau. La posibilidad de que el desequilibrio del cobre está envuelto en el comienzo del Alzheimer está actualmente siendo investigado. En primer lugar parece que la fracción de cobre libre (no unido a la ceruloplasmina) esta aumentada en trastornos de la homeostasis de cobre, como también en los individuos con Alzheimer (78, 79). Más aun, pacientes con Alzheimer parecen tener altos niveles de cobre plasmático comparados con los que tiene un control sano (80). Entre las muchas hipótesis que apoyan el papel del cobre en el comienzo o progresión del Alzheimer, se sugirió que el cobre podía estar involucrado en la formación de placas seniles a través de la hipermetilación de los péptidos β-amiloides posiblemente llevando a una depleción del zinc, un mayor estrés oxidativo e incluso daño cerebral (81, 82). Investigaciones recientes han identificado también variaciones genéticas (polimorfismo) en el gen ATP7B que puede modificar el riesgo de desarrollar Alzheimer (83). La proteína, ATP7B, es responsable por la excreción de cobre hepático en el tracto biliar, y su deterioro en la enfermedad de Wilson resulta en un incremento en el nivel de cobre “libre” en la sangre y la acumulación de cobre en el cerebro y el hígado.
Más investigaciones son necesarias para indagar si las variantes genéticas pudieran influenciar la susceptibilidad de una exposición ambiental a altos niveles de cobre. La adición de cobre al agua potable ha sido asociada con características patológicas mejoradas en modelos animales con Alzheimer (84, 85). Un estudio en un conejo reporto que combinando una alta dieta de alto colesterol y cobre (0.12 mg/L en agua bebible) pudo poner en peligro la cognición (84). Un estudio de cohorte prospectivo en 3,718 participantes de edad avanzada del Chicago Health and Aging Project, con un seguimiento de 5.5 años, evaluó el impacto de las ingestas de ácidos grasos y cobre usando cuestionarios de frecuencia alimentaria y varias evaluaciones cognitivas. Para los individuos un consumo alto de ácidos grasos saturados y ácidos grasos trans el deterioro cognitivo fue mayor para aquellos en el quintil más elevado del total del consumo de cobre comparado con aquellos en el quintil más bajo (ingesta promedio de 2.75 vs 0.88 mg/día) (86).
Aunque un metabolismo de cobre disfuncional es sugerido como un factor de riesgo para el Alzheimer, podría ser también un síntoma de la enfermedad. Por otra parte, no es claro aun si la suplementación o restricción del cobre podría retrasar la progresión del Alzheimer. Una pequeña prueba contralada doble ciega con placebo en 68 individuos con Alzheimer en un estado leve encontró que la suplementación de 8 mg/día de cobre por un año retrasó la disminución del péptido β-amiloide Aβ42 en el fluido cerebroespinal; una disminución en Aβ42 ha sido ligado a un deterioro cognitivo (87). Sin embargo, este retraso no estuvo asociado con una mejora en el rendimiento cognitivo (88). Basado en el utilización de agentes quelantes de cobre en la enfermedad de Wilson, el reciente uso de acetato de zinc de liberación lenta (150 mg/día por 6 meses) en un estudio aleatorio controlado con placebo en 60 pacientes con leve y Alzheimer moderado resulto en una disminución de cobre libre en el suero y la estabilización de los déficits de la cognición (81). Estudios adicionales en humanos son necesarios para clarificar el papel del cobre en el comienzo y progresión del Alzheimer, y evaluar si el cobre dietario puede ayudar a prevenir Alzheimer en individuos con alto riesgo o controlar la enfermedad en pacientes con Alzheimer.
La enfermedad de Parkinson
Tanto la aparición neurológica de la enfermedad de Wilson y la aceruloplasminemia hereditaria ambas están caracterizadas por la acumulación de cobre en el cerebro, resultando en síntomas neurológicos (distonía y deterioro cognitivo) que se parecen a la enfermedad de Parkinson (89). El nivel de cobre es disminuido en regiones cerebrales de perdida neuronal en pacientes con Parkinson (90). Sim embargo, el reciente metaánalisis de estudios que midieron los niveles de cobre en el suero, plasma y liquido cefalorraquídeo no encontraron ninguna diferencia entre pacientes con Parkinson y sujetos de edad avanzada sanos (91).
Fuentes
Fuentes alimenticias
El cobre se encuentra en una amplia variedad de alimentos y es más abundante en vísceras, mariscos, frutos secos, y semillas. Los cereales de salvado de trigo y los productos de grano entero son también buenas fuentes de cobre. De acuerdo a encuestas nacionales, la ingesta dietética de cobre promedio en los EE.UU. es aproximadamente de 1.0 a 1.1 mg (1,000 a 1,100 μg)/día para mujeres adultas y de 1.2 a 1.6 mg (1,200 a 1,600 μg)/día para hombres adultos (15). El contenido de cobre de algunos alimentos que son relativamente ricos en cobre se muestra en microgramos (μg) en la tabla a continuación. Para mayor información sobre el contenido de nutrientes de los alimentos, revise base de datos de composición de los alimentos de la USDA.
| Alimento | Porción | Cobre (μg) |
|---|---|---|
| Hígado (res), cocido, frito | 1 onza | 4,128 |
| Moluscos, ostras, cocidas | 6 ostras medianas | 2,397 |
| Carne de cangrejo, cocida | 3 onzas | 1,005 |
| Carne de cangrejo, azul, cocida, asada | 3 onzas | 692 |
| Moluscos, almejas, especies mixtas, cocidas, asadas | 3 onzas | 585 |
| Castaña de cajú | 1 onza | 622 |
| Semillas de girasol, secas, tostadas | 1 onza | 519 |
| Avellanas, secas tostadas | 1 onza | 496 |
| Almendras | 1 onza | 292 |
| Mantequilla de maní, sin sal | 2 cucharadas | 185 |
| Lentejas, semillas maduras, cocidas, hervidas, sin sal | 1 taza | 497 |
| Champiñones, blancos, crudos | 1 taza (rebanados) | 223 |
| Cereal de trigo molido | 2 galletas | 167 |
| Chocolate (semidulce) | 1 onza | 198 |
Suplementos
Suplementos de cobre están disponibles como oxido cúprico, gluconato de cobre, sulfato de cobre, quelatos aminoácidos de cobre (92).
Seguridad
Toxicidad
La toxicidad del cobre es poco común en la población general. El envenenamiento agudo con cobre ha ocurrido a través de la contaminación de bebidas que son almacenadas en contenedores con contenido de cobre, así como de suministros de agua contaminada (93). En los EE.UU. la pauta sanitaria para la máxima concentración de cobre en el agua potable es impuesta por la Agencia de Protección Medioambiental Estadounidense (1.3 mg/litro) y por la Organización Mundial de la Salud (OMS) (2 mg/litro) (94). Los síntomas de toxicidad aguda por cobre incluyen dolor abdominal, nauseas, vómitos y diarrea, los cuales ayudan a prevenir una ingesta y absorción adicional de cobre. Signos más graves de toxicidad aguda por cobre incluyen daño hepático severo, falla renal, coma, y muerte. Más preocupante desde un punto de vista nutricional, es la posibilidad de un daño hepático como resultado de una exposición a largo plazo a dosis de cobre más bajas. En individuos generalmente sanos, las dosis de hasta 10,000 μg (10 mg) diarios no han resultado en un daño hepático. Por esta razón, la Junta de Nutrición y Alimentos de los EE.UU. estableció el nivel máximo de ingesta tolerable (NM) de cobre de alimentos y suplementos en 10 mg/día (15). Se debe destacar que los individuos con desórdenes genéticos que afectan al metabolismo del cobre (ej., enfermedad de Wilson, cirrosis infantil de la India, y toxicosis por cobre idiopática) pueden estar en riesgo de efectos adversos de la toxicidad crónica por cobre a niveles de ingesta significativamente más bajos. Existe un poco de preocupación de que la NM de 10 mg/día pueda ser muy alta. En particular, en un estudio hombres consumieron 7.8 mg/día de cobre por 147 días. Ellos acumularon cobre durante ese lapso de tiempo, y algunos índices de la función inmune y antioxidante sugieren que esas funciones fueron adversamente afectadas por las altas ingestas de cobre (95, 96). Sin embargo, otro estudio no reporto algún efecto adverso en individuos suplementados con 8 mg/día de cobre por 6 meses (88).
| Grupo Etario | NM (μg/día) |
|---|---|
| Infantes 0-12 meses | Imposible de establecer* |
| Niños 1-3 años | 1,000 |
| Niños 4-8 años | 3,000 |
| Niños 9-13 años | 5,000 |
| Adolescentes 14-18 años | 8,000 |
| Adultos 19 años y más | 10,000 |
| *La fuente de la ingesta debiera ser sólo de alimentos y fórmula. | |
Interacción con drogas
Relativamente poco es lo que se conoce acerca de la interacción del cobre con drogas. La penicilamina se utiliza para unir cobre y aumentar su eliminación en la enfermedad de Wilson, un desorden genético que provoca una sobrecarga de cobre hepático. Debido a que la penicilamina incrementa dramáticamente la excreción urinaria de cobre, los individuos que toman este medicamento por razones distintas a la sobrecarga de cobre, pueden tener un incremento del requerimiento de cobre. Adicionalmente, los antiácidos pueden intervenir con la absorción del cobre cuando se utilizan en muy altas cantidades (2).
Recomendación del Instituto Linus Pauling
La IDR del cobre (900 μg/día en adultos) es suficiente para prevenir una deficiencia, pero la falta de indicadores claros del estado nutricional del cobre en seres humanos, hace difícil determinar el nivel de ingesta de cobre más adecuado para promover una salud óptima o prevenir enfermedades crónicas. Una dieta variada debería aportar suficiente cobre a la mayoría de las personas. Para aquellos preocupados de que su dieta pueda no aportar el cobre suficiente, un suplemento multivitamínico/mineral generalmente aportará, al menos, la IDR de cobre.
Adultos mayores (>50 años)
Debido a que el envejecimiento no se ha asociado con cambios significativos en los requerimientos de cobre, nuestra recomendación para los adultos mayores es la misma que para los adultos de 50 años o menos (97).
Autores y Críticos
Escrito en Abril de 2003 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Julio de 2007 by:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Diciembre de 2013 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Enero de 2014 por:
Joseph R. Prohaska, Ph.D.
Profesor de Bioquímica, Emérito
Universidad de Minnesota Escuela de Medicina Duluth
Traducido al Español en 2015 por:
Silvia Vazquez Lima
Instituto Linus Pauling
Universidad Estatal de Oregon
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Derechos de autoría 2001-2024 Instituto Linus Pauling
Referencias
1. Linder MC, Hazegh-Azam M. Copper biochemistry and molecular biology. Am J Clin Nutr. 1996;63(5):797S-811S. (PubMed)
2. Turnlund JR. Copper. In: Shils ME, Shike M, Ross AC, Caballero B, Cousins RJ, eds. Modern Nutrition in Health and Disease. 10th ed. Philadelphia: Lippincott Williams & Wilkins; 2006:286-299.
3. Prohaska JR. Copper. In: Erdman JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Ames: Wiley-Blackwell; 2012:540-553.
4. Prohaska JR. Impact of copper limitation on expression and function of multicopper oxidases (ferroxidases). Adv Nutr. 2011;2(2):89-95. (PubMed)
5. Uauy R, Olivares M, Gonzalez M. Essentiality of copper in humans. Am J Clin Nutr. 1998;67(5 Suppl):952S-959S. (PubMed)
6. Vashchenko G, MacGillivray RT. Multi-copper oxidases and human iron metabolism. Nutrients. 2013;5(7):2289-2313. (PubMed)
7. Meyer LA, Durley AP, Prohaska JR, Harris ZL. Copper transport and metabolism are normal in aceruloplasminemic mice. J Biol Chem. 2001;276(39):36857-36861. (PubMed)
8. Harris ZL, Durley AP, Man TK, Gitlin JD. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc Natl Acad Sci U S A. 1999;96(19):10812-10817. (PubMed)
9. Kono S. Aceruloplasminemia. Curr Drug Targets. 2012;13(9):1190-1199. (PubMed)
10. Thackeray EW, Sanderson SO, Fox JC, Kumar N. Hepatic iron overload or cirrhosis may occur in acquired copper deficiency and is likely mediated by hypoceruloplasminemia. J Clin Gastroenterol. 2011;45(2):153-158. (PubMed)
11. Harris ED. Copper. In: O'Dell BL, Sunde RA, eds. Handbook of nutritionally essential minerals. New York: Marcel Dekker, Inc; 1997:231-273.
12. Johnson MA, Fischer JG, Kays SE. Is copper an antioxidant nutrient? Crit Rev Food Sci Nutr. 1992;32(1):1-31.
13. Mattie MD, McElwee MK, Freedman JH. Mechanism of copper-activated transcription: activation of AP-1, and the JNK/SAPK and p38 signal transduction pathways. J Mol Biol. 2008;383(5):1008-1018. (PubMed)
14. Videt-Gibou D, Belliard S, Bardou-Jacquet E, et al. Iron excess treatable by copper supplementation in acquired aceruloplasminemia: a new form of secondary human iron overload? Blood. 2009;114(11):2360-2361. (PubMed)
15. Food and Nutrition Board, Institute of Medicine. Copper. Dietary referencia intakes for vitamin A, vitamin K, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. Washington, D.C.: National Academy Press; 2001:224-257. (National Academy Press)
16. Guo CH, Wang CL. Effects of zinc supplementation on plasma copper/zinc ratios, oxidative stress, and immunological status in hemodialysis patients. Int J Med Sci. 2013;10(1):79-89. (PubMed)
17. Milne DB, Omaye ST. Effect of vitamin C on copper and iron metabolism in the guinea pig. Int J Vitam Nutr Res. 1980;50(3):301-308. (PubMed)
18. Finley EB, Cerklewski FL. Influence of ascorbic acid supplementation on copper status in young adult men. Am J Clin Nutr. 1983;37(4):553-556. (PubMed)
19. Jacob RA, Skala JH, Omaye ST, Turnlund JR. Effect of varying ascorbic acid intakes on copper absorption and ceruloplasmin levels of young men. J Nutr. 1987;117(12):2109-2115. (PubMed)
20. Harris ZL, Klomp LW, Gitlin JD. Aceruloplasminemia: an inherited neurodegenerative disease with impairment of iron homeostasis. Am J Clin Nutr. 1998;67(5 Suppl):972S-977S. (PubMed)
21. Bustos RI, Jensen EL, Ruiz LM, et al. Copper deficiency alters cell bioenergetics and induces mitochondrial fusion through up-regulation of MFN2 and OPA1 in erythropoietic cells. Biochem Biophys Res Commun. 2013;437(3):426-432. (PubMed)
22. Peled T, Landau E, Prus E, Treves AJ, Nagler A, Fibach E. Cellular copper content modulates differentiation and self-renewal in cultures of cord blood-derived CD34+ cells. Br J Haematol. 2002;116(3):655-661. (PubMed)
23. Lazarchick J. Update on anemia and neutropenia in copper deficiency. Curr Opin Hematol. 2012;19(1):58-60. (PubMed)
24. Shaw JC. Copper deficiency and non-accidental injury. Arch Dis Child. 1988;63(4):448-455. (PubMed)
25. Blackmer AB, Bailey E. Management of copper deficiency in cholestatic infants: review of the literature and a case series. Nutr Clin Pract. 2013;28(1):75-86. (PubMed)
26. Best K, McCoy K, Gemma S, Disilvestro RA. Copper enzyme activities in cystic fibrosis before and after copper supplementation plus or minus zinc. Metabolism. 2004;53(1):37-41. (PubMed)
27. Rowin J, Lewis SL. Copper deficiency myeloneuropathy and pancytopenia secondary to overuse of zinc supplementation. J Neurol Neurosurg Psychiatry. 2005;76(5):750-751. (PubMed)
28. Nations SP, Boyer PJ, Love LA, et al. Denture cream: an unusual source of excess zinc, leading to hypocupremia and neurologic disease. Neurology. 2008;71(9):639-643. (PubMed)
29. Prodan CI, Bottomley SS, Holland NR, Lind SE. Relapsing hypocupraemic myelopathy requiring high-dose oral copper replacement. J Neurol Neurosurg Psychiatry. 2006;77(9):1092-1093. (PubMed)
30. Kumar N, Gross JB, Jr. Mutation in the ATP7A gene may not be responsible for hypocupraemia in copper deficiency myelopathy. Postgrad Med J. 2006;82(968):416. (PubMed)
31. Tumer Z. An overview and update of ATP7A mutations leading to Menkes disease and occipital horn syndrome. Hum Mutat. 2013;34(3):417-429. (PubMed)
32. Kodama H, Fujisawa C, Bhadhprasit W. Inherited copper transport disorders: biochemical mechanisms, diagnosis, and treatment. Curr Drug Metab. 2012;13(3):237-250. (PubMed)
33. Fox PL, Mazumder B, Ehrenwald E, Mukhopadhyay CK. Ceruloplasmin and cardiovascular disease. Free Radic Biol Med. 2000;28(12):1735-1744. (PubMed)
34. Jones AA, DiSilvestro RA, Coleman M, Wagner TL. Copper supplementation of adult men: effects on blood copper enzyme activities and indicators of cardiovascular disease risk. Metabolism. 1997;46(12):1380-1383. (PubMed)
35. Ford ES. Serum copper concentration and coronary heart disease among US adults. Am J Epidemiol. 2000;151(12):1182-1188. (PubMed)
36. Malek F, Jiresova E, Dohnalova A, Koprivova H, Spacek R. Serum copper as a marker of inflammation in prediction of short term outcome in high risk patients with chronic heart failure. Int J Cardiol. 2006;113(2):e51-53. (PubMed)
37. Leone N, Courbon D, Ducimetiere P, Zureik M. Zinc, copper, and magnesium and risks for all-cause, cancer, and cardiovascular mortality. Epidemiology. 2006;17(3):308-314. (PubMed)
38. Kosar F, Sahin I, Acikgoz N, Aksoy Y, Kucukbay Z, Cehreli S. Significance of serum trace element status in patients with rheumatic heart disease: a prospective study. Biol Trace Elem Res. 2005;107(1):1-10. (PubMed)
39. Bertinato J, Zouzoulas A. Considerations in the development of biomarkers of copper status. J AOAC Int. 2009;92(5):1541-1550. (PubMed)
40. Klevay LM. Cardiovascular disease from copper deficiency--a history. J Nutr. 2000;130(2S Suppl):489S-492S. (PubMed)
41. Mielcarz G, Howard AN, Mielcarz B, et al. Leucocyte copper, a marker of copper body status is low in coronary artery disease. J Trace Elem Med Biol. 2001;15(1):31-35. (PubMed)
42. Kinsman GD, Howard AN, Stone DL, Mullins PA. Studies in copper status and atherosclerosis. Biochem Soc Trans. 1990;18(6):1186-1188. (PubMed)
43. Wang XL, Adachi T, Sim AS, Wilcken DE. Plasma extracellular superoxide dismutase levels in an Australian population with coronary artery disease. Arterioscler Thromb Vasc Biol. 1998;18(12):1915-1921. (PubMed)
44. Klevay LM. Lack of a recommended dietary allowance for copper may be hazardous to your health. J Am Coll Nutr. 1998;17(4):322-326. (PubMed)
45. Milne DB, Nielsen FH. Effects of a diet low in copper on copper-status indicators in postmenopausal women. Am J Clin Nutr. 1996;63(3):358-364. (PubMed)
46. Medeiros DM, Milton A, Brunett E, Stacy L. Copper supplementation effects on indicators of copper status and serum cholesterol in adult males. Biol Trace Elem Res. 1991;30(1):19-35. (PubMed)
47. DiSilvestro RA, Joseph EL, Zhang W, Raimo AE, Kim YM. A randomized trial of copper supplementation effects on blood copper enzyme activities and parameters related to cardiovascular health. Metabolism. 2012;61(9):1242-1246. (PubMed)
48. Turley E, McKeown A, Bonham MP, et al. Copper supplementation in humans does not affect the susceptibility of low density lipoprotein to in vitro induced oxidation (FOODCUE project). Free Radic Biol Med. 2000;29(11):1129-1134. (PubMed)
49. Rock E, Mazur A, O'Connor J M, Bonham MP, Rayssiguier Y, Strain JJ. The effect of copper supplementation on red blood cell oxidizability and plasma antioxidants in middle-aged healthy volunteers. Free Radic Biol Med. 2000;28(3):324-329. (PubMed)
50. Mansoor MA, Bergmark C, Haswell SJ, et al. Correlation between plasma total homocysteine and copper in patients with peripheral vascular disease. Clin Chem. 2000;46(3):385-391. (PubMed)
51. Celik C, Bastu E, Abali R, et al. The relationship between copper, homocysteine and early vascular disease in lean women with polycystic ovary syndrome. Gynecol Endocrinol. 2013;29(5):488-491. (PubMed)
52. Gerhard GT, Duell PB. Homocysteine and atherosclerosis. Curr Opin Lipidol. 1999;10(5):417-428. (PubMed)
53. Emsley AM, Jeremy JY, Gomes GN, Angelini GD, Plane F. Investigation of the inhibitory effects of homocysteine and copper on nitric oxide-mediated relaxation of rat isolated aorta. Br J Pharmacol. 1999;126(4):1034-1040. (PubMed)
54. Shukla N, Angelini GD, Jeremy JY. Interactive effects of homocysteine and copper on angiogenesis in porcine isolated saphenous vein. Ann Thorac Surg. 2007;84(1):43-49. (PubMed)
55. Uthus EO, Reeves PG, Saari JT. Copper deficiency decreases plasma homocysteine in rats. J Nutr. 2007;137(6):1370-1374. (PubMed)
56. Wei H, Zhang WJ, McMillen TS, Leboeuf RC, Frei B. Copper chelation by tetrathiomolybdate inhibits vascular inflammation and atherosclerotic lesion development in apolipoprotein E-deficient mice. Atherosclerosis. 2012;223(2):306-313. (PubMed)
57. Failla ML, Hopkins RG. Is low copper status immunosuppressive? Nutr Rev. 1998;56(1 Pt 2):S59-64.
58. Percival SS. Copper and immunity. Am J Clin Nutr. 1998;67(5 Suppl):1064S-1068S. (PubMed)
59. Heresi G, Castillo-Duran C, Munoz C, Arevalo M, Schlesinger L. Phagocytosis and immunoglobulin levels in hypocupremic children. Nutr Res. 1985;5:1327-1334.
60. Kelley DS, Daudu PA, Taylor PC, Mackey BE, Turnlund JR. Effects of low-copper diets on human immune response. Am J Clin Nutr. 1995;62(2):412-416. (PubMed)
61. Hodgkinson V, Petris MJ. Copper homeostasis at the host-pathogen interface. J Biol Chem. 2012;287(17):13549-13555. (PubMed)
62. Looker AC, Melton LJ, 3rd, Harris TB, Borrud LG, Shepherd JA. Prevalence and trends in low femur bone density among older US adults: NHANES 2005-2006 compared with NHANES III. J Bone Miner Res. 2010;25(1):64-71. (PubMed)
63. Tiidus PM, Lowe DA, Brown M. Estrogen replacement and skeletal muscle: mechanisms and population health. J Appl Physiol. 2013;115(5):569-578. (PubMed)
64. Cauley JA. Public Health Impact of Osteoporosis. J Gerontol A Biol Sci Med Sci. 2013;68(10):1243-1251. (PubMed)
65. Kanumakala S, Boneh A, Zacharin M. Pamidronate treatment improves bone mineral density in children with Menkes disease. J Inherit Metab Dis. 2002;25(5):391-398. (PubMed)
66. Marquardt ML, Done SL, Sandrock M, Berdon WE, Feldman KW. Copper deficiency presenting as metabolic bone disease in extremely low birth weight, short-gut infants. Pediatrics. 2012;130(3):e695-698. (PubMed)
67. Baker A, Harvey L, Majask-Newman G, Fairweather-Tait S, Flynn A, Cashman K. Effect of dietary copper intakes on biochemical markers of bone metabolism in healthy adult males. Eur J Clin Nutr. 1999;53(5):408-412. (PubMed)
68. Baker A, Turley E, Bonham MP, et al. No effect of copper supplementation on biochemical markers of bone metabolism in healthy adults. Br J Nutr. 1999;82(4):283-290. (PubMed)
69. Cashman KD, Baker A, Ginty F, et al. No effect of copper supplementation on biochemical markers of bone metabolism in healthy young adult females despite apparently improved copper status. Eur J Clin Nutr. 2001;55(7):525-531. (PubMed)
70. Conlan D, Korula R, Tallentire D. Serum copper levels in elderly patients with femoral-neck fractures. Age Ageing. 1990;19(3):212-214. (PubMed)
71. Mutlu M, Argun M, Kilic E, Saraymen R, Yazar S. Magnesium, zinc and copper status in osteoporotic, osteopenic and normal post-menopausal women. J Int Med Res. 2007;35(5):692-695. (PubMed)
72. Eaton-Evans J, Mellwrath EM, Jackson WE, McCartney H, Strain JJ. Copper supplementation and the maintenance of bone mineral density in middle-aged women. J Trace Elem Exp Med. 1996;9:87-94.
73. Strause L, Saltman P, Smith KT, Bracker M, Andon MB. Spinal bone loss in postmenopausal women supplemented with calcium and trace minerals. J Nutr. 1994;124(7):1060-1064. (PubMed)
74. Nielsen FH, Lukaski HC, Johnson LK, Roughead ZK. Reported zinc, but not copper, intakes influence whole-body bone density, mineral content and T score responses to zinc and copper supplementation in healthy postmenopausal women. Br J Nutr. 2011;106(12):1872-1879. (PubMed)
75. Sidiropoulou-Chatzigiannis S, Kourtidou M, Tsalikis L. The effect of osteoporosis on periodontal status, alveolar bone and orthodontic tooth movement. A literature review. J Int Acad Periodontol. 2007;9(3):77-84. (PubMed)
76. Darcey J, Horner K, Walsh T, Southern H, Marjanovic EJ, Devlin H. Tooth loss and osteoporosis: to assess the association between osteoporosis status and tooth number. Br Dent J. 2013;214(4):E10. (PubMed)
77. Sierpinska T, Konstantynowicz J, Orywal K, Golebiewska M, Szmitkowski M. Copper deficit as a potential pathogenic factor of reduced bone mineral density and severe tooth wear. Osteoporos Int. 2013 [Epub ahead of print]. (PubMed)
78. Squitti R, Barbati G, Rossi L, et al. Excess of nonceruloplasmin serum copper in AD correlates with MMSE, CSF [β]-amyloid, and h-τ. Neurology. 2006;67(1):76-82. (PubMed)
79. Arnal N, Cristalli DO, de Alaniz MJ, Marra CA. Clinical utility of copper, ceruloplasmin, and metallothionein plasma determinations in human neurodegenerative patients and their first-degree relatives. Brain Res. 2010;1319:118-130. (PubMed)
80. Ventriglia M, Bucossi S, Panetta V, Squitti R. Copper in Alzheimer's disease: a meta-analysis of serum, plasma, and cerebrospinal fluid studies. J Alzheimers Dis. 2012;30(4):981-984. (PubMed)
81. Brewer GJ. Copper excess, zinc deficiency, and cognition loss in Alzheimer's disease. Biofactors. 2012;38(2):107-113. (PubMed)
82. Squitti R, Polimanti R. Copper phenotype in Alzheimer's disease: dissecting the pathway. Am J Neurodegener Dis. 2013;2(2):46-56. (PubMed)
83. Squitti R, Polimanti R. Copper hypothesis in the missing hereditability of sporadic Alzheimer's disease: ATP7B gene as potential harbor of rare variants. J Alzheimers Dis. 2012;29(3):493-501. (PubMed)
84. Sparks DL, Schreurs BG. Trace amounts of copper in water induce β-amyloid plaques and learning deficits in a rabbit model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2003;100(19):11065-11069. (PubMed)
85. Kitazawa M, Cheng D, Laferla FM. Chronic copper exposure exacerbates both amyloid and tau pathology and selectively dysregulates cdk5 in a mouse model of AD. J Neurochem. 2009;108(6):1550-1560. (PubMed)
86. Morris MC, Evans DA, Tangney CC, et al. Dietary copper and high saturated and trans fat intakes associated with cognitive decline. Arch Neurol. 2006;63(8):1085-1088. (PubMed)
87. Kessler H, Pajonk FG, Bach D, et al. Effect of copper intake on CSF parameters in patients with mild Alzheimer's disease: a pilot phase 2 clinical trial. J Neural Transm. 2008;115(12):1651-1659. (PubMed)
88. Kessler H, Bayer TA, Bach D, et al. Intake of copper has no effect on cognition in patients with mild Alzheimer's disease: a pilot phase 2 clinical trial. J Neural Transm. 2008;115(8):1181-1187. (PubMed)
89. Skjorringe T, Moller LB, Moos T. Impairment of interrelated iron- and copper homeostatic mechanisms in brain contributes to the pathogenesis of neurodegenerative disorders. Front Pharmacol. 2012;3:169. (PubMed)
90. Akatsu H, Hori A, Yamamoto T, et al. Transition metal abnormalities in progressive dementias. Biometals. 2012;25(2):337-350. (PubMed)
91. Mariani S, Ventriglia M, Simonelli I, et al. Fe and Cu do not differ in Parkinson's disease: a replication study plus meta-analysis. Neurobiol Aging. 2013;34(2):632-633. (PubMed)
92. Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. Montvale: Medical Economics Company, Inc; 2001.
93. Bremner I. Manifestations of copper excess. Am J Clin Nutr. 1998;67(5 Suppl):1069S-1073S. (PubMed)
94. Fitzgerald DJ. Safety guidelines for copper in water. Am J Clin Nutr. 1998;67(5 Suppl):1098S-1102S. (PubMed)
95. Turnlund JR, Jacob RA, Keen CL, et al. Long-term high copper intake: effects on indexes of copper status, antioxidant status, and immune function in young men. Am J Clin Nutr. 2004;79(6):1037-1044. (PubMed)
96. Turnlund JR, Keyes WR, Kim SK, Domek JM. Long-term high copper intake: effects on copper absorption, retention, and homeostasis in men. Am J Clin Nutr. 2005;81(4):822-828. (PubMed)
97. Wood RJ, Suter PM, Russell RM. Mineral requirements of elderly people. Am J Clin Nutr. 1995;62(3):493-505. (PubMed)
Cromo
Contenido
Resumen
- El cromo (Cr0) es un elemento traza ubicuo. La forma predomínante del cromo en el cuerpo es el cromo trivalente (Cr3+) el cual pudiese jugar un papel en la función normal de la insulina. (Más información)
- El cromo trivalente se ha propuesto para ser el cofactor de un oligopéptido llamado cromodulina. La cromodulina puede ser capaz de potenciar la acción de la insulina, y por lo tanto mejorar la sensibilidad del tejido a la insulina y facilitar el transporte de la glucosa a las células. (Más información)
- Casos de una potencial deficiencia de cromo han sido asociados con síntomas parecidos a los de la diabetes mellitus: Intolerancia a la glucosa y un incremento en los requerimientos de insulina. (Más información)
- La falta de una medida precisa del estado nutricional del cromo previene la identificación de individuos que pudiesen ser susceptibles a una deficiencia de cromo. En el 2001, el Instituto de Medicina Estadounidense estableció la ingesta adecuada (IA) del cromo en 20-35 μg/día para adultos. (Más información)
- Ensayos controlados aleatorios han fallado en proveer alguna evidencia de los beneficios de la suplementación con cromo en la prevención del tratamiento de la intolerancia a la glucosa y la diabetes mellitus tipo 2. (Más información)
- Una dieta bien balanceada que incluya frutas, verduras, carne, pescado, y granos debería fácilmente cubrir las necesidades dietarías de cromo. (Más información)
- Pocos efectos adversos de la suplementación con cromo han sido reportados. (Más información)
El cromo fue descubierto por primera vez en 1797. El estado de oxidación del cromo más estable en sistemas biológicos es el cromo trivalente (Cr3+), el cual forma complejos relativamente inertes con las proteínas y los ácidos nucleicos (1). La esencialidad del cromo trivalente es cuestionada, y su presunta función en el cuerpo permanece siendo pobremente entendida. Otra forma común y estable del cromo en el medio ambiente es el cromo hexavalente (Cr6+). El cromo hexavalente es derivado del cromo trivalente al calentarse a un pH alcalino y es usado como una fuente de cromo para propósitos industriales. El cromo hexavalente es altamente toxico y está clasificado como un carcinógeno humano al ser inhalado (2). En el ambiente acido del estómago, el cromo hexavalente puede ser fácilmente reducido a cromo trivalente por substancias reductoras presentes en los alimentos, los cuales limitan la ingestión del cromo hexavalente (3-5).
Función
El cromo trivalente ha sido propuesto como el cofactor de una molécula bilógicamente activa que podría aumentar los efectos de la insulina en tejidos diana. La insulina es secretada por células especializadas en el páncreas en respuesta a niveles incrementados de glucosa en la sangre, tal como después de una comida. La insulina se une a los receptores de insulina en la superficie de la célula, activando los receptores y estimulando la asimilación de glucosa por las células. A través de su interacción con los receptores de insulina, la insulina provee a las células con glucosa para generar energía y a mantener a la glucosa dentro de un estrecho rango de concentraciones. Además de sus efectos sobre el metabolismo de los carbohidratos (glucosa), la insulina también tiene efectos en el metabolismo de las grasas y las proteínas (6). Juntas, una disminución de la respuesta a la insulina o una disminución a la sensibilidad a la insulina en tejidos periféricos (tejido adiposo, músculos, y el hígado) y un defecto progresivo en la secreción de la insulina puede resultar en una intolerancia a la glucosa, la cual frecuentemente conduce a que una diabetes mellitus tipo 2 se manifieste. El cuerpo inicialmente incrementa la secreción de insulina por células pancreáticas especializadas para superar la disminución de la sensibilidad a la insulina. Sin embargo, el páncreas eventualmente falla al producir suficiente insulina para mantener niveles normales de concentraciones de glucosa en la sangre. Los individuos con diabetes tipo 2 están en un riesgo incrementado de padecer enfermedades cardiovasculares (7).
Posibles mecanismos de acción
La estructura precisa de la forma biológicamente activa del cromo no es aun conocida. El modelo actual postula que el cromo trivalente podría ser el cofactor de una sustancia de bajo peso molecular ligada al cromo conocida como LMWCr o cromodulina (8). Se piensa que la cromodulina mejora la cascada de eventos de señalización inducida por la unión de la insulina a la subunidad alfa extracelular del receptor de insulina (RI). Tras la unión de la insulina, el dominio de la tirosina quinasa en la subunidad beta del RI se activa y causa la fosforilación de los residuos de la tirosina en la subunidad beta misma. Subsecuentemente, la activación del receptor de insulina desencadena una serie de reacciones de fosforilación rápidas que activan varios efectores, eventualmente resultando en un incremento en la asimilación y almacenamiento de la glucosa (8).
En cuanto al efecto del cromo trivalente en la señalización de la insulina, un modelo temprano sugirió que la unión de la insulina al IR podría estimular el movimiento del cromo dentro de las células y resultar en la unión del cromo a la apocromodulina una forma de la cromodulina que carece de cromo (9, 10). La cromodulina pudiera entonces unirse al RI y regular las moléculas de señalización de la insulina, por ultimo incrementar la translocación de los transportadores de glucosa (GLUT-4) de vesículas citosólicas a la membrana celular (11-13). Algunos aunque no todos los estudios conducidos en modelos basados en células y modelos animales de resistencia a la insulina y diabetes han encontrado que el cromo inhibe la actividad de la proteína tirosina fosfatasa 1B (PTP-1B) y otros reguladores negativos del señalamiento de la insulina, sugiriendo que el cromo podría mejorar la sensibilidad de la insulina bajo condiciones resistentes a la insulina (revisado en 8). Un reciente estudio en ratones diabéticos también sugirió que el cromo puede reducir el aclaramiento de la insulina y mejorar la señalización de la insulina al inhibir la proteólisis (degradación) de la insulina y algunos efectores (14). Adicionalmente mecanismos que pueden subyacer el efecto del cromo en la sensibilidad a la insulina, como la reducción de marcadores de éstres oxidativo e inflamación conocidos por contribuir a la resistencia de la insulina, están bajo investigación (revisado en 8, 10).
Interacciones con nutrientes
Hierro
El cromo compite por uno de los sitios de unión de la proteína transportadora de hierro, la transferrina. Sin embargo, la suplementación de 925 μg/día de cromo en hombres mayores por 12 semanas no afecto significativamente las medidas del estado nutricional del hierro (15). Un estudio en hombres más jóvenes encontró una disminución significativa en la saturación de la transferrina con el hierro después de una suplementación de 200 μg/día de cromo por ocho semanas, pero estudios a largo plazo no han abordado este asunto (16). En un ensayo controlado aleatorio de 12 semanas de duración, la suplementación con picolinato de cromo (200 μg/día) no afecto el estado nutricional del hierro en mujeres premenopáusicas cuando se compara con el ácido picolínico o placebo (17). La sobrecarga de hierro en la hemocromatosis hereditaria puede interferir con el transporte del cromo al competir con la unión con la transferrina. Se ha plantado la hipótesis de que la disminución en el transporte del cromo podría contribuir a la patogénesis de la diabetes mellitus en pacientes con hemocromatosis hereditaria (3).
Vitamina C
La asimilación de cromo en animales es aumentada cuando se administra al mismo tiempo que la vitamina C (5). En un estudio en tres mujeres, la administración de 100 μg de vitamina C junto a 1 mg de cromo dio como resultado niveles plasmáticos de cromo más altos que la de 1 mg de cromo sin vitamina C (3).
Carbohidratos
En comparación a dietas ricas en carbohidratos complejos (ej. granos enteros), las dietas altas en azucares simples (ej. sacarosa) resultan en un incremento en la excreción urinaria de cromo en adultos. Este efecto puede estar relacionado a la secreción de insulina incrementada en respuesta al consumo de azúcares simples en comparación con los carbohidratos complejos (3).
Deficiencia
Potenciales casos de deficiencia de cromo han sido descritos en unos pocos pacientes con alimentación intravenosa (nutrición parenteral) a largo plazo que no recibieron cromo suplementario en sus soluciones intravenosas. Los sujetos desarrollaron una utilización anormal de la glucosa e incrementaron los requerimientos de insulina que respondieron a la suplementación con cromo (18). Sin embargo, debido a que las soluciones intravenosas proporcionan cromo en dosis por arriba de los niveles dietarios, se ha sugerido que el cromo pudiese producir efectos biológicos solo a dosis farmacológicas (10). Ya que el cromo parece mejorar la acción de la insulina y la deficiencia de cromo se ha propuesto que resulta en una intolerancia alterada a la glucosa, se ha creado la hipótesis de que la deficiencia de cromo contribuye como un factor al desarrollo de diabetes tipo 2 (3, 19). Sin embargo, la evidencia para esto permanece ambigua.
La pérdida urinaria de cromo se reporta es incrementada por el ejercicio de resistencia en corredores masculinos, sugiriendo que las necesidades de cromo pueden ser mayores en individuos que se ejercitan regularmente (20). En un estudio, el levantamiento de pesas (ejercicio de resistencia) se encontró que incrementa la excreción urinaria de cromo en hombres mayores. Sin embargo, la absorción de cromo también aumento, dando lugar a poca o ninguna pérdida neta de cromo como resultado de ejercicios de resistencia (21).
En la actualidad, investigaciones de los efectos de la ingesta potencialmente inadecuada de cromo y los factores de riesgo para la insuficiencia de cromo es limitada debido a la falta de herramientas analíticas para determinar el estatus nutricional del cromo (3, 5). Más aun, la ausencia de modelos animales para la deficiencia de cromo dificulta estudiar las posibles anormalidades bioquímicas, fisiológicas, y funcionales asociadas con una ingesta inadecuada de cromo (22).
La Ingesta Adecuada (IA)
Debido a que no hay suficiente información para establecer un requerimiento estimado promedio (REP), la junta de Alimentos y Nutrición (FNB) del Instituto de Medicina estadounidense (IOM) estableció una ingesta adecuada (IA) basada en el cromo contenido en dietas sanas (3).
| Etapa de la Vida | Edad | Machos (μg/día) | Hembras (μg/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 0.2 | 0.2 |
| Infantes | 7-12 meses | 5.5 | 5.5 |
| Niños | 1-3 años | 11 | 11 |
| Niños | 4-8 años | 15 | 15 |
| Niños | 9-13 años | 25 | 21 |
| Adolescentes | 14-18 años | 35 | 24 |
| Adultos | 19-50 años | 35 | 25 |
| Adultos | 51 años y más | 30 | 20 |
| Embarazo | 18 años o menos | - | 29 |
| Embarazo | 19 años y más | - | 30 |
| Período de lactancia | 18 años o menos | - | 44 |
| Período de lactancia | 19 años y más | - | 45 |
El caso de la esencialidad del cromo trivalente ha sido cuestionada tanto en animales como en humanos en las últimas décadas, y la Autoridad de Alimentos y Seguridad Europea – la cual provee guías alimentarias para la comunidad estadounidense – recientemente concluyo que los requerimientos para el cromo no pudieron ser establecidos (22). La IOM actualmente no está considerando la IA para el cromo.
Prevención de Enfermedades
Intolerancia a la glucosa y diabetes mellitus tipo 2
Recientes estudios controlados en pacientes con intolerancia a la glucosa han reportado que la suplementación con cromo mejoro en cierta medida la utilización de la glucosa o tuvo efectos beneficiales en los perfiles de lípidos sanguíneos (23). La intolerancia a la glucosa se refiere al estado pre-diabético y es actualmente definido por la presencia de glucosa en ayunas alterada (concentraciones de glucosa en ayunas en el plasma 110-125 mg/dL) y el estado de intolerancia a la glucosa (concentraciones de glucosa plasmática de 140-199 mg/dL durante una prueba de tolerancia a la glucosa de 2 horas con un carga oral de glucosa de 75-g) (24). La intolerancia a la glucosa está asociada con incrementos modestos en el riesgo de enfermedades cardiovasculares, como también a otras complicaciones microvasculares tradicionales de la diabetes (25). Las estimaciones actuales sugieren que más del 70% de individuos con intolerancia a la glucosa pueden eventualmente desarrollar diabetes tipo 2 (26).
En un reciente estudio aleatorio, doble ciego, controlado con placebo en 56 sujetos con riesgo de padecer diabetes tipo 2, seis meses de suplementación diaria con picolinato de cromo (500 μg o 1,000 μg) no tuvo efecto en las concentraciones de glucosa e insulina, en la sensibilidad a la insulina y en los perfiles de lípidos sanguíneos (27). Otro ensayo aleatorio, controlado con placebo en 31 individuos no diabéticos reporto una gran variabilidad en las concentraciones de cromo en el suero y orina en respuesta a una diaria suplementación con 1,000 μg de picolinato de cromo por 16 semanas. También el grupo suplementado con cromo, los participantes con mayores niveles vs. menores concentraciones de cromo en el suero (>3.1 μg/L vs. ≤3.1 μg/L) exhibieron un decline en la sensibilidad a la insulina que no pudo ser explicado por los cambios en la expresión de los genes implicados en la señalización de la insulina (28). Adicionalmente un meta-análisis de nueve ensayos clínicos aleatorios publicados entre 1992 y 2010 reportaron que el cromo en dosis de 200-1,000 μg/día por 8-16 semanas no tuvieron efecto en las concentraciones de glucosa en ayunas en 309 individuos no diabéticos (29). Aunque más evidencia es necesaria para apoyar la suplementación con cromo, existe la necesidad de una medida precisa del estado nutricional del cromo para identificar a los individuos que podrían padecer una carencia y así poder ser más beneficiados de la suplementación con cromo (29).
Enfermedad cardiovascular
La intolerancia a la glucosa y la diabetes tipo 2 están asociadas con cambios adversos en los perfiles lipídicos y con un riesgo incrementado de enfermedades cardiovasculares. Los estudios que examinaron los efectos de la suplementación con cromo en los perfiles lipídicos han proporcionado resultados inconsistentes. Mientras que algunos estudios han observado reducciones en los niveles plasmáticos de colesterol total, colesterol LDL y triglicéridos o incrementos en los niveles de colesterol HDL, otros estudios no observaron ningún efecto. Tales respuestas inconsistentes de los niveles de lípidos y lipoproteínas en la suplementación con cromo pueden reflejar las diferencias en el estado nutricional del cromo. Es posible que solo aquellos individuos con una ingesta dietaría insuficiente de cromo experimenten los efectos benéficos sobre los perfiles lipídicos después de la suplementación con cromo (4, 5, 30).
Declaraciones de propiedades saludables
Incrementa la masa muscular
Declaraciones de que la suplementación con cromo incrementa la masa corporal magra y disminuye la grasa corporal se basan en la relación entre el cromo y la acción de la insulina (vea Función). Además de regular el metabolismo de la glucosa, se sabe que la insulina afecta el metabolismo de la grasa y de las proteínas (6). Al menos 12 estudios controlados con placebo han comparado el efecto de la suplementación con cromo (172-1,000 μg/día de picolinato de cromo) con o sin un programa de ejercicio sobre la masa corporal magra y las medidas de grasa corporal (revisado en 31). En general, aquellos estudios que utilizaron los métodos de medición de grasa corporal y masa magra más sensibles y precisos (absorciometría de rayos X de energía dual o DEXA e hidrodensiometría o pesaje bajo el agua) no encontraron algún efecto beneficioso de la suplementación con cromo en la composición corporal (4, 30).
Promueve la pérdida de peso
Estudios controlados de la suplementación con cromo han demostrado poco o ningún efecto beneficioso en la pérdida de peso o grasa, y las declaraciones sobre la pérdida de peso en humanos parecen ser exageradas. En 1996, la Comisión Federal de Comercio (CFC) de los EE.UU. estableció que no existían bases científicas para declarar que el picolinato de cromo podría promover la pérdida de peso y la pérdida de grasa en seres humanos (32). Recientemente, un meta-análisis de 11 ensayos aleatorizados, doble ciego controlados con placebo en 866 sujetos con sobrepeso u obesos encontró una significante reducción 0.59 kilogramos (1.10 libras) con cromo suplementario (mas exclusivamente en la forma de picolinato de cromo) en dosis de entre 137 μg/día y 1,000 μg/día por 8-24 semanas (33). Sin embargo, tal pequeño cambio no alcanzo una pérdida de peso clínicamente significante de ≥5% del peso corporal inicial (34). Reportes recientes han sugerido que el cromo suplementario pudiese reducir el antojo e ingesta de comida, en mujeres con sobrepeso y obesidad (35, 36). A pesar de todo, datos actuales disponibles permanecen siendo insuficientes para apoyar el uso de suplementos de cromo como estrategias de pérdida de peso (37).
Tratamiento de Enfermedades
Diabetes mellitus tipo 2
La diabetes mellitus tipo 2 está caracterizada por una hiperglicemia crónica (una concentración elevada de glucosa en la sangre) y una resistencia a la insulina. Debido a que la resistencia a la insulina esta usualmente asociada con un aumento compensatorio en la secreción de insulina, las concentraciones de insulina circulante en los sujetos con diabetes tipo 2 puede ser más elevada que en individuos sanos. A pesar de todo, la resistencia de los tejidos periféricos (especialmente el hígado y musculo esquelético) a la insulina también implica que los efectos fisiológicos de la insulina se ven reducidos.
Desde que el cultivo de células y modelos de roedores para la diabetes han implicado al cromo en la regulación de la sensibilidad a la insulina y los niveles de glucosa sanguínea, la relación entre el estatus nutricional del cromo y la diabetes mellitus tipo 2 ha generado un considerable interés científico. Reportes tempranos observaron que individuos con diabetes tipo 2 evidente por más de 2 años, mostraron tasas más altas de pérdida de cromo urinario que en individuos sanos (38). Estudios menores y bien diseñados sobre la suplementación con cromo en individuos con diabetes tipo 2 no mostraron algún avance en el control de la glucosa sanguínea, aunque estos proporcionaron evidencia en la reducción de las concentraciones de insulina y mejoramiento de los perfiles lípidos sanguíneos (39). En 1997, los resultados de un ensayo controlado con placebo conducido en China indico que la suplementación con cromo pudiese ser beneficial en el tratamiento de la diabetes tipo 2 (40). Ciento ochenta participantes fueron asignados al azar para recibir tanto placebo o suplementos de cromo en la forma de picolinato de cromo a 200 μg/día o 1,000 μg/día. Después de cuatro meses de tratamiento, se encontró que las concentraciones de glucosa sanguínea en ayunas fueron de entre 15% a 19% más bajas en aquellos que tomaron 1,000 μg/día de cromo en comparación con aquellos que tomaron el placebo. A pesar de todo, los niveles de glucosa sanguínea en aquellos que tomaron 200 μg/día no difirieron significativamente de aquellos que tomaron el placebo. El picolato de cromo en dosis de 200 μg/día o 1,000 μg/día fue también asociado con concentraciones de insulina reducidas en comparación al placebo. El nivel de hemoglobina glucosilada Alc (HbA1c), una medida del control glucémico, fue también significativamente reducido en ambos grupos suplementados con cromo. Sin embargo cierto número de limitaciones hicieron difícil extrapolar los resultados a la población de los EE.UU. (41). Además, el estudio fue excluido de recientes meta-análisis de ensayo controlados aleatorios debido a la insuficiencia en la calidad de los datos (29, 42).
Un meta-análisis reciente de siete estudios controlados aleatorios indico que la ingesta de cromo de por lo menos 250 μg/día por no menos de tres meses redujo significativamente las concentraciones de glucosa en ayunas en sujetos diabéticos pero no tuvo efecto en los niveles de HbAIc (42). Otro meta-análisis de nueve ensayos controlados aleatorios, que incluyeron un total de 440 participantes con diabetes, fallo en descubrir una reducción significante en las concentraciones de glucosa en ayunas con una monoterapia con cromo (200-1,000 μg/día) (29). Sin embargo, ensayos controlados aleatorios a grande escala de la suplementación con cromo son necesarios para demostrar si el cromo es efectivo en el tratamiento de la diabetes mellitus tipo 2. Adicionalmente, sugerencias recientes han sido propuestas acerca de que dosis más elevadas de cromo podrían ser requeridas para observar efectos benéficos de la suplementación con cromo (10).
Diabetes gestacional
Pocos estudios han examinado los efectos de la suplementación con cromo sobre la diabetes gestacional, una condición que se estima afecta de entre el 4.6% y 9.2% de las mujeres embarazadas en los Estados Unidos (43). La ocurrencia de la diabetes gestacional durante el embarazo está asociada con una secreción insuficiente de insulina y una intolerancia a la glucosa de gravedad variable (44). La resistencia periferia a la insulina usualmente se incrementa en el segundo y tercer trimestre de embarazo. Debido a que elevadas concentraciones de glucosa materna sanguínea pueden conllevar a efectos adversos en el desarrollo del feto, las mujeres con diabetes gestacional están en un riesgo elevado de experimentar complicaciones en el embarazo (45). Luego del parto, la intolerancia a la glucosa generalmente se ve invertida y la tolerancia a la glucosa vuelve a ser normal. Sin embargo, una de cada tres mujeres que han padecido de diabetes gestacional desarrollan una intolerancia a la glucosa después del parto (prediabetes o diabetes tipo 2) (46, 47).
Un estudio basado en la observacional en mujeres embarazadas no encontró niveles de cromo en el suero asociados con medidas de la tolerancia a la glucosa o resistencia a la insulina al final del embarazo (48). Sin embargo, se desconoce si las medidas de los niveles del cromo en el suero verdaderamente reflejan los niveles de cromo en los tejidos y el estatus del cromo durante el embarazo. Un estudio prospectivo más reciente que dio seguimiento a 425 mujeres embarazadas también fallo en encontrar una correlación entre las concentraciones de cromo en el suero y la incidencia de diabetes gestacional (49). Un estudio de corte transversal de 90 mujeres embarazadas en el sur de la India encontró que aquellas con diabetes gestacional tuvieron concentraciones de cromo de cromo significativamente más bajas en comparación con las mujeres que no padecían de diabetes gestacional. En vista de los resultados mixtos anteriores se debe notar que diferentes métodos fueron usados para medir las concentraciones circulantes de cromo en cada estudio. También, es posible que el estatus nutricional del cromo pueda variar entre poblaciones étnicamente distintas por lo que los estudios que incluyeron mujeres embarazadas con diferentes orígenes étnicos y/o áreas geográficas darían resultados diferentes.
Además, la evidencia existente es insuficiente para poder evaluar el efecto del cromo suplementario en la diabetes gestacional. Las mujeres con diabetes gestacional cuyas dietas fueron suplementadas con 4 μg de cromo por kilogramo de peso corporal diariamente en la forma de picolinato de cromo por ocho semanadas tuvieron una disminución en las concentraciones de glucosa sanguínea en ayunas e insulina en comparación con aquellas que tomaron un placebo. Sin embargo, la terapia de insulina en lugar del picolinato de cromo fue requerida para normalizar los niveles gravemente elevados de glucosa en la sangre (4, 50).
Fuentes
Fuentes alimenticias
La cantidad de cromo en los alimentos es variable y se ha medido de manera precisa en relativamente pocos alimentos. Actualmente, no hay una gran base de datos para el contenido de cromo de los alimentos. Las carnes procesadas, los productos de grano entero, los cereales ricos en salvado, los ejotes, el brócoli, las nueces, y la yema de huevo son ricas fuentes de cromo. Los alimentos con un alto contenido de azúcares simples, como sacarosa y fructosa, son usualmente bajos en cromo y podrían promover la excreción de cromo (4). Las ingestas de cromo promedio en los EE.UU. varían entre los 23 y 29 μg/día en mujeres adultas, y entre 39 y 54 μg/día en hombres adultos (3). El contenido de cromo de algunos alimentos se muestra a continuación expresado en microgramos (μg) (51). Debido a que el contenido de cromo varía significativamente en diferentes lotes de un mismo alimento, la información en la tabla a continuación debiese servir solo como una guía del contenido de cromo en los alimentos.
| Alimento | Porción | Cromo (μg) |
|---|---|---|
| Brócoli | ½ taza | 11.0 |
| Ejotes | ½ taza | 1.1 |
| Papas | 1 taza, molida | 2.7 |
| Jugo de uva | 8 onzas fluidas | 7.5 |
| Jugo de naranja | 8 onzas fluidas | 2.2 |
| Carne de vacuno | 3 onzas | 2.0 |
| Pechuga de pavo | 3 onzas | 1.7 |
| Jamón de pavo (procesado) | 3 onzas | 10.4 |
| Waffle | 1 (~2.5 onzas) | 6.7 |
| Bagel | 1 | 2.5 |
| Muffin Inglés | 1 | 3.6 |
| Manzana con cáscara | 1 mediana | 1.4 |
| Plátano | 1 mediana | 1.0 |
Suplementos
Un reciente estudio de corte transversal índico que el 19% de la población de los EE. UU. usa suplementos dietarios que contienen cromo (es decir, suplementos de un solo nutriente y suplementos con múltiples ingredientes), con la proporción más alta de usuarios (29%) encontrada en adultos mayores de 50 años (52). El cromo trivalente está disponible como suplemento en varias formas, incluyendo cloruro de cromo, nicotinato de cromo, picolinato de cromo, y levadura rica en cromo. Éstas se encuentran disponibles como suplementos independientes o en productos combinados. Las dosis comúnmente varían de 50 a 200 μg de cromo elemental (53). En mucha de la investigación sobre la intolerancia a la glucosa y la diabetes tipo 2, el picolinato de cromo fue la fuente de cromo, aunque recientes investigaciones sugieren que su biodisponibilidad podría no ser más mayor que la de el cromo dietario (54). Algunas preocupaciones han sido planteadas sobre la seguridad a largo plazo de la suplementación con picolinato de cromo (ver Seguridad).
Seguridad
Toxicidad
El cromo hexavalente (cromo VI; Cr6+) es un reconocido carcinógeno. La exposición al cromo hexavalente en polvo ha sido asociada con una incidencia incrementada de cáncer de pulmón y se sabe que provoca inflamación de la piel (dermatitis).
En contraste, existe poca evidencia de que el cromo trivalente (cromo III; Cr3+) es toxico para los seres humanos. La toxicidad de ingestas orales es considerada baja porque el cromo ingerido es pobremente absorbido, y la mayoría del cromo absorbido es rápidamente excretado en la orina (55). Debido a que no se han asociado convincentemente efectos adversos con la ingesta en exceso de cromo III de alimentos o suplementos, la Junta de Nutrición y Alimentos (JNA) del Instituto de Medicina no estableció un nivel máximo de ingesta tolerable (NM) para el cromo. Dado que la información es escasa, la JNA reconoce la posibilidad de efectos negativos de una ingesta elevada de cromo trivalente suplementario a la salud y aconseja precaución (3).
Picolinato de cromo
La mayoría de las preocupaciones en relación a la seguridad a largo plazo de la suplementación con cromo trivalente provienen de varios estudios en cultivos celulares, que sugieren que el cromo III, especialmente en la forma de picolinato de cromo, pudiese incrementar el daño al ADN (56-58). Actualmente, no hay evidencia de que el cromo trivalente incremente el daño al ADN en organismos vivos (3), y un estudio en 10 mujeres que tomaron 400 μg/día de cromo como picolinato de cromo, no encontró evidencia de un incremento del daño oxidativo al ADN, medido a través de anticuerpos contra bases de ADN oxidado (59).
Varios estudios han demostrado la seguridad de dosis diarias de hasta 1,000 μg de cromo por varios meses (40, 60). Sin embargo, ha habido algunos reportes aislados de reacciones adversas serias al picolinato de cromo. Se reportó una falla renal cinco meses después de un ciclo de seis semanas de 600 μg de cromo/día en la forma de picolinato de cromo (61), mientras una falla renal y deterioro de la función hepática fueron reportados luego del uso de 1,200-2,400 μg/día de cromo en la forma de picolinato de cromo por un periodo de cuatro a cinco meses (62). Adicionalmente, un hombre sano de 24 años de edad según se informa, desarrolló una falla renal aguda reversible luego de consumir suplementos que contenían picolinato de cromo por dos semanas (63). Los individuos con enfermedades renales o hepáticas preexistentes pueden estar en riesgo incrementado de efectos adversos y debiesen limitar la ingesta de cromo suplementario (3).
Interacción con drogas
Poco se sabe acerca de las interacciones del cromo con drogas en los seres humanos. Las grandes dosis de antiácidos que contienen carbonato de calcio o hidróxido de magnesio, disminuyeron la absorción de cromo en ratas. En contraste, drogas anti-inflamatorias no esteroideas, como lo es la aspirina y la indometacina pueden incrementar la absorción de cromo en ratas (5).
Recomendación del Instituto Linus Pauling
La falta de indicadores sensibles del estado nutricional del cromo en humanos hace difícil el determinar el nivel de ingesta de cromo más apropiado para promover una salud óptima. Siguiendo la recomendación del Instituto Linus Pauling de tomar un suplemento multivitamínico/mineral que contenga el 100% de los valores diarios (VD) de la mayoría de los nutrientes, generalmente aportará de entre 60 a 120 μg/día de cromo, muy por encima del nivel de ingesta adecuada de 20 a 25 μg/día para mujeres adultas y 30 a 35 μg para hombres adultos.
Adultos mayores (>50 años)
Aunque se desconoce si los requerimientos de cromo son más altos para adultos mayores, un estudio encontró que las concentraciones de cromo en el cabello, sudor, y orina disminuían con la edad (64). Siguiendo la recomendación del Instituto Linus Pauling de tomar un suplemento multivitamínico/mineral que contenga el 100% de los valores diarios (VD) de la mayoría de los nutrientes debiese aportar cromo suficiente a la mayoría de los adultos mayores.
Debido a que la intolerancia a la glucosa y la diabetes tipo 2 están asociadas con problemas de salud serios, los individuos que consideren la suplementación con cromo en altas dosis para tratar cualquiera de las dos condiciones, debieran hacerlo en colaboración con un profesional de la salud calificado.
Autores y Críticos
Escrito en Abril de 2003 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Septiembre de 2007 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Septiembre de 2007 por:
Richard A. Anderson, Ph.D.
Plomo Científico
Centro de Investigación de Nutrición Humana de Beltsville
Beltsville, Maryland
Actualizado en Octubre de 2014 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Octubre de 2014 por:
John B. Vincent, Ph.D.
Profesor del Departamento de Química
La Universidad de Alabama
Tuscaloosa, Alabama
Traducido al Español en 2014 por:
Silvia Vazquez Lima
Instituto Linus Pauling
Universidad Estatal de Oregon
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Derechos de autoría 2001-2023 Instituto Linus Pauling
Referencias
1. Vaidyanathan VG, Asthana Y, Nair BU. Importance of ligand structure in DNA/protein binding, mutagenicity, excision repair and nutritional aspects of chromium(III) complexes. Dalton Trans. 2013;42(7):2337-2346. (PubMed)
2. US Environmental Protection Agency. Chromium compounds; 2000. Available at: http://www.epa.gov/ttn/atw/hlthef/chromium.html. Accessed 9/18/14.
3. Food and Nutrition Board, Institute of Medicine. Chromium. Dietary reference intakes for vitamin A, vitamin K, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. Washington, D.C.: National Academy Press; 2001:197-223. (National Academy Press)
4. Lukaski HC. Chromium as a supplement. Annu Rev Nutr. 1999;19:279-302. (PubMed)
5. Stoecker B. Chromium. In: Shils M, Shike M, Ross A, Caballero B, Cousins R, eds. Modern Nutrition in Health and Disease. Philadelphia: Lippincott, Williams & Wilkins; 2006:332-337.
6. Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799-806. (PubMed)
7. Martin-Timon I, Sevillano-Collantes C, Segura-Galindo A, Del Canizo-Gomez FJ. Type 2 diabetes and cardiovascular disease: Have all risk factors the same strength? World J Diabetes. 2014;5(4):444-470. (PubMed)
8. Hua Y, Clark S, Ren J, Sreejayan N. Molecular mechanisms of chromium in alleviating insulin resistance. J Nutr Biochem. 2012;23(4):313-319. (PubMed)
9. Vincent JB. Elucidating a biological role for chromium at a molecular level. Acc Chem Res. 2000;33(7):503-510. (PubMed)
10. Vincent JB. Chromium: Is It essential, pharmacologically relevant, or toxic? In: Sigel A, Sigel H, Sigel R, eds. Interrelations between Essential Metal Ions and Human Diseases: Springer Science+Business Media Dordrecht; 2013:171-198.
11. Chen G, Liu P, Pattar GR, et al. Chromium activates glucose transporter 4 trafficking and enhances insulin-stimulated glucose transport in 3T3-L1 adipocytes via a cholesterol-dependent mechanism. Mol Endocrinol. 2006;20(4):857-870. (PubMed)
12. Pattar GR, Tackett L, Liu P, Elmendorf JS. Chromium picolinate positively influences the glucose transporter system via affecting cholesterol homeostasis in adipocytes cultured under hyperglycemic diabetic conditions. Mutat Res. 2006;610(1-2):93-100. (PubMed)
13. Wang H, Kruszewski A, Brautigan DL. Cellular chromium enhances activation of insulin receptor kinase. Biochemistry. 2005;44(22):8167-8175. (PubMed)
14. Wang ZQ, Yu Y, Zhang XH, Komorowski J. Chromium-Insulin Reduces Insulin Clearance and Enhances Insulin Signaling by Suppressing Hepatic Insulin-Degrading Enzyme and Proteasome Protein Expression in KKAy Mice. Front Endocrinol (Lausanne). 2014;5:99. (PubMed)
15. Campbell WW, Beard JL, Joseph LJ, Davey SL, Evans WJ. Chromium picolinate supplementation and resistive training by older men: effects on iron-status and hematologic indexes. Am J Clin Nutr. 1997;66(4):944-949. (PubMed)
16. Lukaski HC, Bolonchuk WW, Siders WA, Milne DB. Chromium supplementation and resistance training: effects on body composition, strength, and trace element status of men. Am J Clin Nutr. 1996;63(6):954-965. (PubMed)
17. Lukaski HC, Siders WA, Penland JG. Chromium picolinate supplementation in women: effects on body weight, composition, and iron status. Nutrition. 2007;23(3):187-195. (PubMed)
18. Rech M, To L, Tovbin A, Smoot T, Mlynarek M. Heavy metal in the intensive care unit: a review of current literature on trace element supplementation in critically ill patients. Nutr Clin Pract. 2014;29(1):78-89. (PubMed)
19. Jeejeebhoy KN. The role of chromium in nutrition and therapeutics and as a potential toxin. Nutr Rev. 1999;57(11):329-335. (PubMed)
20. Lukaski HC. Magnesium, zinc, and chromium nutriture and physical activity. Am J Clin Nutr. 2000;72(2 Suppl):585S-593S. (PubMed)
21. Rubin MA, Miller JP, Ryan AS, et al. Acute and chronic resistive exercise increase urinary chromium excretion in men as measured with an enriched chromium stable isotope. J Nutr. 1998;128(1):73-78. (PubMed)
22. European Food Safety Authority (EFSA) Panel on Dietetic Products NaA. Scientific Opinion on Dietary Reference Values for chromium. EFSA Journal. 2014;12(10). Available at: http://www.efsa.europa.eu/en/efsajournal/pub/3845.htm. Accessed 10/17/14.
23. Mertz W. Chromium in human nutrition: a review. J Nutr. 1993;123(4):626-633. (PubMed)
24. World Health Organization Diabetes Programme. Definition and diagnosis of diabetes mellitus and intermediate hyperglycemia: Report of a World Health Organization/International Diabetes Foundation Consultation. Geneva, Switzerland 2006. Available at http://www.who.int/diabetes/publications/diagnosis_diabetes2006/en/. Accessed 10/17/14.
25. Singleton JR, Smith AG, Russell JW, Feldman EL. Microvascular complications of impaired glucose tolerance. Diabetes. 2003;52(12):2867-2873. (PubMed)
26. Nathan DM, Davidson MB, DeFronzo RA, et al. Impaired fasting glucose and impaired glucose tolerance: implications for care. Diabetes Care. 2007;30(3):753-759. (PubMed)
27. Ali A, Ma Y, Reynolds J, Wise JP, Sr., Inzucchi SE, Katz DL. Chromium effects on glucose tolerance and insulin sensitivity in persons at risk for diabetes mellitus. Endocr Pract. 2011;17(1):16-25. (PubMed)
28. Masharani U, Gjerde C, McCoy S, et al. Chromium supplementation in non-obese non-diabetic subjects is associated with a decline in insulin sensitivity. BMC Endocr Disord. 2012;12:31. (PubMed)
29. Bailey CH. Improved meta-analytic methods show no effect of chromium supplements on fasting glucose. Biol Trace Elem Res. 2014;157(1):1-8. (PubMed)
30. Kobla HV, Volpe SL. Chromium, exercise, and body composition. Crit Rev Food Sci Nutr. 2000;40(4):291-308. (PubMed)
31. Vincent JB. The potential value and toxicity of chromium picolinate as a nutritional supplement, weight loss agent and muscle development agent. Sports Med. 2003;33(3):213-230. (PubMed)
32. Federal Trade Commission. Companies Advertising Popular Dietary Supplement "Chromium picolinate" Can't Substantiate Weight Loss and Health Benefit Claims, Says FTC; 1996. Available at https://www.ftc.gov/news-events/press-releases/1996/11/companies-advertising-popular-dietary-supplement-chromium. Accessed 10/17/14.
33. Onakpoya I, Posadzki P, Ernst E. Chromium supplementation in overweight and obesity: a systematic review and meta-analysis of randomized clinical trials. Obes Rev. 2013;14(6):496-507. (PubMed)
34. Wing RR, Pinto AM, Crane MM, Kumar R, Weinberg BM, Gorin AA. A statewide intervention reduces BMI in adults: Shape Up Rhode Island results. Obesity (Silver Spring). 2009;17(5):991-995. (PubMed)
35. Anton SD, Morrison CD, Cefalu WT, et al. Effects of chromium picolinate on food intake and satiety. Diabetes Technol Ther. 2008;10(5):405-412. (PubMed)
36. Brownley KA, Von Holle A, Hamer RM, La Via M, Bulik CM. A double-blind, randomized pilot trial of chromium picolinate for binge eating disorder: results of the Binge Eating and Chromium (BEACh) study. J Psychosom Res. 2013;75(1):36-42. (PubMed)
37. Tian H, Guo X, Wang X, et al. Chromium picolinate supplementation for overweight or obese adults. Cochrane Database Syst Rev. 2013;11:CD010063. (PubMed)
38. Morris BW, MacNeil S, Hardisty CA, Heller S, Burgin C, Gray TA. Chromium homeostasis in patients with type II (NIDDM) diabetes. J Trace Elem Med Biol. 1999;13(1-2):57-61. (PubMed)
39. Hellerstein MK. Is chromium supplementation effective in managing type II diabetes? Nutr Rev. 1998;56(10):302-306. (PubMed)
40. Anderson RA, Cheng N, Bryden NA, et al. Elevated intakes of supplemental chromium improve glucose and insulin variables in individuals with type 2 diabetes. Diabetes. 1997;46(11):1786-1791. (PubMed)
41. Althuis MD, Jordan NE, Ludington EA, Wittes JT. Glucose and insulin responses to dietary chromium supplements: a meta-analysis. Am J Clin Nutr. 2002;76(1):148-155. (PubMed)
42. Abdollahi M, Farshchi A, Nikfar S, Seyedifar M. Effect of chromium on glucose and lipid profiles in patients with type 2 diabetes; a meta-analysis review of randomized trials. J Pharm Pharm Sci. 2013;16(1):99-114. (PubMed)
43. DeSisto CL, Kim SY, Sharma AJ. Prevalence estimates of gestational diabetes mellitus in the United States, Pregnancy Risk Assessment Monitoring System (PRAMS), 2007-2010. Prev Chronic Dis. 2014;11:E104. (PubMed)
44. American Diabetes Association. Gestational diabetes mellitus. Diabetes Care. 2004;27 Suppl 1:S88-90. (PubMed)
45. Mitanchez D. Foetal and neonatal complications in gestational diabetes: perinatal mortality, congenital malformations, macrosomia, shoulder dystocia, birth injuries, neonatal complications. Diabetes Metab. 2010;36(6 Pt 2):617-627. (PubMed)
46. Bellamy L, Casas JP, Hingorani AD, Williams D. Type 2 diabetes mellitus after gestational diabetes: a systematic review and meta-analysis. Lancet. 2009;373(9677):1773-1779. (PubMed)
47. Retnakaran R, Qi Y, Sermer M, Connelly PW, Hanley AJ, Zinman B. Glucose intolerance in pregnancy and future risk of pre-diabetes or diabetes. Diabetes Care. 2008;31(10):2026-2031. (PubMed)
48. Gunton JE, Hams G, Hitchman R, McElduff A. Serum chromium does not predict glucose tolerance in late pregnancy. Am J Clin Nutr. 2001;73(1):99-104. (PubMed)
49. Woods SE, Ghodsi V, Engel A, Miller J, James S. Serum chromium and gestational diabetes. J Am Board Fam Med. 2008;21(2):153-157. (PubMed)
50. Jovanovic-Peterson L, Peterson CM. Vitamin and mineral deficiencies which may predispose to glucose intolerance of pregnancy. J Am Coll Nutr. 1996;15(1):14-20. (PubMed)
51. Anderson RA, Bryden NA, Polansky MM. Dietary chromium intake. Freely chosen diets, institutional diet, and individual foods. Biol Trace Elem Res. 1992;32:117-121. (PubMed)
52. Bailey RL, Gahche JJ, Lentino CV, et al. Dietary supplement use in the United States, 2003-2006. J Nutr. 2011;141(2):261-266. (PubMed)
53. Hendler S, Rorvik D, eds. PDR (Physicians' Desk Reference) for Nutritional Supplements. 2nd edition ed. Montvale: Thomson Reuters; 2008.
54. Laschinsky N, Kottwitz K, Freund B, Dresow B, Fischer R, Nielsen P. Bioavailability of chromium(III)-supplements in rats and humans. Biometals. 2012;25(5):1051-1060. (PubMed)
55. Nielsen FH. Manganese, Molybdenum, Boron, Chromium, and other trace elements. In: Erdman JJ, Macdonald I, Zelssel S, eds. Present Knowledge of Nutrition: John Wiley & Sons, Inc.; 2012.
56. Blasiak J, Kowalik J. A comparison of the in vitro genotoxicity of tri- and hexavalent chromium. Mutat Res. 2000;469(1):135-145. (PubMed)
57. Speetjens JK, Collins RA, Vincent JB, Woski SA. The nutritional supplement chromium(III) tris(picolinate) cleaves DNA. Chem Res Toxicol. 1999;12(6):483-487. (PubMed)
58. Stearns DM, Wise JP, Sr., Patierno SR, Wetterhahn KE. Chromium(III) picolinate produces chromosome damage in Chinese hamster ovary cells. FASEB J. 1995;9(15):1643-1648. (PubMed)
59. Kato I, Vogelman JH, Dilman V, et al. Effect of supplementation with chromium picolinate on antibody titers to 5-hydroxymethyl uracil. Eur J Epidemiol. 1998;14(6):621-626. (PubMed)
60. Hathcock JN. Vitamins and minerals: efficacy and safety. Am J Clin Nutr. 1997;66(2):427-437. (PubMed)
61. Wasser WG, Feldman NS, D'Agati VD. Chronic renal failure after ingestion of over-the-counter chromium picolinate. Ann Intern Med. 1997;126(5):41. (PubMed)
62. Cerulli J, Grabe DW, Gauthier I, Malone M, μgoldrick MD. Chromium picolinate toxicity. Ann Pharmacother. 1998;32(4):428-431. (PubMed)
63. Wani S, Weskamp C, Marple J, Spry L. Acute tubular necrosis associated with chromium picolinate-containing dietary supplement. Ann Pharmacother. 2006;40(3):563-566. (PubMed)
64. Davies S, McLaren Howard J, Hunnisett A, Howard M. Age-related decreases in chromium levels in 51,665 hair, sweat, and serum samples from 40,872 patients--implications for the prevention of cardiovascular disease and type II diabetes mellitus. Metabolism. 1997;46(5):469-473. (PubMed)
Fluoruro
Contenido
Resumen
- El fluoruro es la forma iónica del elemento de origen natural conocido como flúor. El anión incrementa la estabilidad estructural de los dientes y huesos a través de interacciones con fosfatos de calcio. (Más información)
- Las recomendaciones de ingesta diaria para el fluoruro están basadas en las ingestas más seguras y efectivas para prevenir la caries dental. (Más información)
- El uso de productos dentales fluorurados e ingestas adecuadas de fluoruro reducen la ocurrencia de caries durante toda la vida al promover la mineralización dental y re-mineralización. Estudios exhaustivos aleatorios controlados con placebo son requeridos para evaluar si la aplicación tópica de agentes fluorados pudiesen también prevenir la erosión dental. (Más información)
- Evidencia clínica y epidemiológica es actualmente limitada para apoyar el papel de la fluoración del agua en la prevención de la osteoporosis y fracturas óseas. (Más información)
- Ensayos terapéuticos han encontrado un efecto dependiente de la dosis de fluoruro en el riesgo de fracturas en pacientes osteoporóticos. Sin embargo, la ocurrencia de numerosos efectos secundarios justifica estudios adicionales para garantizar que dosis seguras y efectivas puedan ser usadas solas o en combinación con terapias actuales. (Más información)
- Las fuentes principales de fluoruro sistémico y tópico son el agua potable, alimentos y bebidas hechas con agua fluorada, fórmulas para infantes, y productos de cuidado oral que contienen fluoruro. La sal y leche fluoradas están actualmente disponibles fuera de los Estados Unidos en Europa, Latinoamérica, y el Sureste de Asia. (Más información)
- Aunque el aumento de la exposición al fluoruro ha llevado a una disminución en la caries dental, la prevalencia de motas blancas o manchas permanentes en los dientes conocidos como fluorosis, ha incrementado. La homeostasis del tejido óseo puede también ser afectada por la excesiva ingesta de fluoruro. (Más información)
El fluoruro se origina naturalmente como el ion cargado negativamente, fluoruro (F-). El fluoruro es considerado un elemento traza porque solo pequeñas cantidades están presentes en el cuerpo (alrededor de 2.6 gramos en adultos), y porque el requerimiento diario para el mantenimiento de la salud dental es solo unos pocos miligramos al día. Alrededor del 95% del fluoruro total del cuerpo es encontrado en los huesos y dientes (1). Aunque su papel en la prevención de la caries dental, está bien establecido, el fluoruro no es generalmente considerado con un elemento mineral esencial porque los humanos no lo requieren para el crecimiento o para sustentar la vida (2). Sin embargo, si uno considera la prevención de enfermedades crónicas (caries dental) como un criterio importante en la determinación de la esencialidad, entonces el fluoruro podría considerarse como un elemento traza esencial (3).
Función
El fluoruro es absorbido en el estómago y en el intestino delgado. Una vez en el torrente sanguíneo penetra rápidamente en los tejidos mineralizados (huesos y dientes en desarrollo). A niveles de ingesta usuales, el fluoruro no se acumula en los tejidos blandos. Los elementos minerales predominantes en el hueso son cristales de calcio y fosfato, conocidos como cristales de hidroxiapatita. La alta reactividad química y el pequeño radio del fluoruro le permiten desplazar al ion hidroxilo (-OH) más grande, en el cristal de hidroxiapatita formando fluorapatita, o incrementar la densidad cristalina al entrar en los espacios dentro del cristal de hidroxiapatita. La fluorapatita endurece el esmalte dental y estabiliza el mineral óseo (4).
Interacción con nutrientes
Tanto el calcio como el magnesio forman complejos insolubles con el fluoruro y son capaces de disminuir significativamente la absorción de fluoruro cuando están presentes en la misma comida. Sin embargo, la absorción de fluoruro en la forma de monofluorofosfato (a diferencia del fluoruro de sodio) no es alterada por el calcio. También se ha encontrado que una dieta baja en cloruro (sal) incrementa la retención de fluoruro al reducir la excreción urinaria de fluoruro (1).
Deficiencia
En los seres humanos, el único efecto claro de una ingesta inadecuada de fluoruro es el riesgo incrementado de caries dental (caries) en individuos de todas las edades. Investigaciones epidemiologias de los patrones de consumo de agua y la prevalencia de caries dental a través de varias regiones de los EE.UU. con diferentes concentraciones de fluoruro, condujeron al desarrollo de un rango óptimo recomendado de la concentración de fluoruro de 0.7-1.2 mg/L o partes por millón (ppm); la concentración más baja fue recomendada para climas más cálidos donde el consumo de agua es más alto, y la concentración más alta fue recomendada para climas más fríos. Recientemente, el Departamento de Salud y Servicios Humanos de los EE.UU. recomendó que todos los sistemas de agua de uso comunitario se ajustara a la concentración de fluoruro de 0.7 mg/L, agregando "datos recientes no muestran una relación convincente entre la ingesta del líquido y la temperatura ambiente del aire" (5). Esta recomendación fue hecha en un esfuerzo por reducir el riesgo de fluorosis dental y en vista de la amplia disponibilidad de flúor a través de otras fuentes, incluyendo productos del cuidado oral fluorados (6). Un numero de estudios conducidos antes de la introducción de las pastas dentales con fluoruro demostraron que la prevalencia de caries dentales fue de un 40% a 60% más bajo en comunidades con concentraciones optimas de fluoruro en el agua que en comunidades con bajas concentraciones de fluoruro en el agua (7).
La Ingesta Adecuada (IA)
La Junta de Nutrición y Alimentos (JNA) del Instituto de Medicina estadounidense actualizó sus recomendaciones para la ingesta de fluoruro en 1997. Debido a que los datos fueron insuficientes para establecer una Ingesta Diaria Recomendada ( IDR); en cambio, los niveles de Ingesta Adecuada (IA) fueron establecidos basados en las ingestas estimadas (0.05 mg/kg de peso) que demostraron reducir de forma más efectiva la ocurrencia de caries dental sin causar el indeseable efecto secundario de manchas en el esmalte, conocido como fluorosis dental (7). Vea la siguiente sección sobre Seguridad para un análisis de la fluorosis dental.
| Etapa de la Vida | Edad | Machos (mg/día) | Hembras (mg/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 0.01 | 0.01 |
| Infantes | 7-12 meses | 0.5 | 0.5 |
| Niños | 1-3 años | 0.7 | 0.7 |
| Niños | 4-8 años | 1.0 | 1.0 |
| Niños | 9-13 años | 2.0 | 2.0 |
| Adolescentes | 14-18 años | 3.0 | 3.0 |
| Adultos | 19 años y más | 4.0 | 3.0 |
| Embarazo | Todas las edades | - | 3.0 |
| Período de lactancia | Todas las edades | - | 3.0 |
Prevención de Enfermedades
Caries dental (cavidades y dientes picados)
Bacterias cariogénicas (causantes de las cavidades) especificas (principalmente Streptococcus mutans y Streptococcus sobrinus) encontradas en la placa dental son capaces de metabolizar carbohidratos fermentables (azúcares) y convertirlos en ácidos orgánicos que pueden disolver el esmalte dental susceptible. Si no se controla, las bacterias pueden penetrar capas más profundas del diente y progresar hasta el tejido pulpar blando en el centro. La caries sin tratamiento puede llevar a dolor severo, infecciones locales, pérdida o extracción de dientes, problemas nutricionales, e infecciones sistémicas graves en individuos susceptibles (8). Estudios recientes han sugerido una relación entre la inflamación sistémica en individuos con infección periodontal (encías) y la resistencia a la insulina (9), diabetes tipo 2 (10) y la hipertensión (11). Más aun, una salud oral pobre puede constituir un factor de riesgo para enfermedades cardiovasculares (12, 13).
Efectos sistémicos de fluoruro en los dientes
Una exposición incrementada al fluoruro, más comúnmente a través de la fluoración del agua, se ha encontrado que disminuye la incidencia de caries dental en niños y adultos (14). Entre 1976 y 1987 estudios clínicos en varios países diferentes demostraron que la adición de fluoruro a los suministros de agua comunitarios (0.7-1.2 ppm) redujo la caries en un 30%-60% en dientes primarios (de leche) y un 15%-35% en dientes permanentes (15). El fluoruro consumido en el agua parece tener un efecto sistémico en niños antes de la erupción de los dientes típicamente hasta los 12 años de edad. El fluoruro es incorporado en el esmalte en desarrollo de los dientes e incrementa la resistencia a las caries. Dado que el efecto preventivo de las caries del fluoruro es también tópico (superficie) en los niños después de la erupción de los dientes y en adultos, la protección optima alcanzada por el agua fluorada es más probable a ocurrir a través de tanto la exposición sistémica antes y después de la erupción del diente y la exposición tópica después de la erupción.
Efectos tópicos del fluoruro en los dientes
Investigaciones han indicado que la acción primaria del fluoruro ocurre tópicamente después de la erupción de los dientes en el interior de la boca. El fluoruro ingerido es secretado en la saliva y contribuye a la protección tópica. Cuando el esmalte es parcialmente desmineralizado por ácidos orgánicos, el fluoruro en la saliva puede mejorar la remineralización del esmalte a través de sus interacciones con el calcio y el fosfato. El fluoruro que contiene esmalte remineralizado es más resistente al ataque de ácidos y a la desmineralización. En concentraciones salivales asociadas con una ingesta optima de fluoruro, se ha encontrado que el fluoruro inhibe las enzimas bacterianas, resultando una reducción de la producción de ácido por parte de las bacterias cariogénicas (8, 14). Por otra parte, el uso de productos fluorados aplicados tópicamente incluyendo la pasta de dientes, gel, esmalte, y enjuague bucal se piensa que han contribuido a un decline substancial en la prevalencia de caries en las últimas décadas (16). Un reciente meta-análisis de intervenciones de fluoruro en niños y adolescentes (hasta 16 años de edad) encontró que la aplicación de esmalte de fluoruro durante al menos un año estuvo asociada con un reducción del 37% en caries, perdida, llenado de superficies dentales en superficies dentales cariadas de los dientes de leche; el efecto anti-caries en los dientes permanentes corresponde a una disminución del 43% en comparación con ningún tratamiento o placebo (17). Otro meta-análisis de 67 ensayos controlados con placebo llevadas a cabo en niños y adolescentes demostraron un reducción del 23% en caries, perdida y llenado de las superficies dentales en dentición mixta y permanente con pastas dentales que contenían por lo menos 1,000ppm de fluoruro. La disminución alcanzo un 36% con pastas dentales fluoradas con concentraciones de 2,400-2,800 ppm, mientras que no hubo diferencia entre los niveles de fluoruro por debajo de las 600 ppm y el placebo (18).
Erosión dental (desgaste de los dientes)
El ataque del tejido duro dental por los ácidos distintos de los producidos por la placa bacteriana puede conducir a la pérdida del esmalte dental, también conocida como erosión dental. Los factores involucrados en la erosión dental incluyen alimentos y bebidas acidas (ej. bebidas carbonatadas) y reflujo acido (19). El efecto protector de los agentes fluorurados contra la erosión dental ha sido principalmente observado en estudios in vitro (revisado en 19). Sin embargo, un reciente meta-análisis de cuatro ensayos aleatorios que examinaron el efecto del fluoruro en la pasta dental, esmalte, y saliva en la erosión dental no encontró ningún beneficio en general en comparación con el placebo (20). Estudios clínicos más exhaustivos son necesarios para evaluar si las aplicaciones tópicas de fluoruro pueden prevenir la erosión dental y/o reducir la progresión de las lesiones erosivas existentes.
Osteoporosis
Aunque el fluoruro en dosis farmacológicas ha mostrado ser un agente terapéutico potente para la masa ósea de la columna (vea Tratamiento de Enfermedades) existe poca evidencia de que la fluoruración del agua a niveles óptimos para la prevención de la caries dental sea de ayuda en la prevención de la osteoporosis. La mayoría de los estudios realizados hasta la fecha han fallado en encontrar diferencias clínicamente significativas en la densidad mineral ósea (DMO) o en la incidencia de fracturas cuando se comparó a los residentes de áreas con depósitos de agua fluorurada, con los residentes de áreas sin depósitos de agua fluorurada (21). Sin embargo, dos estudios encontraron que la fluoruración del agua potable estaba asociada con una incidencia disminuida de fractura de cadera en adultos mayores. Además, un estudio en Italia encontró un riesgo significativamente mayor de fracturas femorales (cadera) en hombres y mujeres habitantes de un área con baja fluoruración del agua (0.05 ppm), en comparación con el riesgo de una población similar cuyos suministros de agua eran fluorurados naturalmente (1.45 ppm) a niveles más altos que los óptimos para la prevención de la caries dental (22). Otro estudio en Alemania no encontró diferencias significativas en la densidad mineral ósea entre los residentes de una comunidad cuyos suministros de agua habían sido fluorurados de manera óptima por 30 años (1 ppm) comparados con aquellos que residían en una comunidad sin agua fluorurada. No obstante, este estudio reportó que la incidencia de fractura de cadera en hombres y mujeres de 85 años y más, era significativamente más bajo en la comunidad con agua fluorurada en comparación con la comunidad con agua no fluorurada, a pesar de los niveles más altos de calcio en los suministros de agua no fluorurada (23). Otro estudio basado en comunidad en 1,300 mujeres encontró que las elevadas concentraciones de fluoruro en el suero no se relacionaban con la densidad mineral ósea o la incidencia de fractura osteoporótica (24). Finalmente, un estudio de cohorte nacional en Suecia no encontró alguna asociación entre la exposición crónica a el agua fluorurada y la incidencia de fractura de cadera (25).
Tratamiento de Enfermedades
Osteoporosis
La osteoporosis se caracteriza por una disminución en la densidad mineral ósea (DMO) y un incremento en la fragilidad ósea y la susceptibilidad a fracturarse. En general, la DMO disminuida se asocia con un riesgo de fractura incrementado. Sin embargo, la relación habitual entre la DMO y el riesgo de fractura no siempre se mantiene precisa cuando se utilizan dosis de fluoruro elevadas (farmacológicas) para tratar la osteoporosis. La mayoría de las terapias disponibles para la osteoporosis (ej. estrógeno, calcitonina y bifosfonatos) disminuyen la pérdida de hueso (reabsorción), resultando en pequeños incrementos en la DMO. Las dosis farmacológicas de fluoruro son capaces de producir grandes incrementos en la DMO de la columna lumbar. En general, las pruebas terapéuticas del fluoruro en pacientes con osteoporosis no han logrado demostrar consistentemente disminuciones significativas en la ocurrencia de fracturas vertebrales a pesar de los dramáticos incrementos en la DMO de la columna lumbar (26). Un meta-análisis de 11 estudios controlados, incluyendo 1,429 pacientes encontró que el tratamiento con fluoruro causó un aumento de la DMO de la columna lumbar, pero que no fue asociado con un menor riesgo de fracturas vertebrales (27). Este meta-análisis también encontró que las concentraciones más altas de fluoruro se asociaban con un riesgo incrementado de fracturas no vertebrales después de cuatro años de tratamiento. Los primeros estudios que utilizaron dosis elevadas de fluoruro (>20 mg/día) pueden haber inducido la rápida mineralización ósea en ausencia de una ingesta suficiente de calcio y vitamina D, dando como resultado huesos más densos que no eran mecánicamente más fuertes (28, 29). El análisis de la arquitectura ósea también ha ayudado a esclarecer los efectos inconsistentes de la terapia con fluoruro en la reducción de fracturas vertebrales. La investigación ha indicado que la osteoporosis puede estar asociada con un cambio irreversible en la arquitectura del hueso, conocido como conectividad trabecular disminuida. El hueso normal consiste de unas series de placas interconectadas por barras gruesas. Los huesos severamente osteoporóticos tienen pocas placas y los bastones pueden estar fracturados o desconectados (conectividad trabecular disminuida) (30). A pesar de que la terapia con fluoruro incremente la densidad ósea, ésta probablemente no pueda restaurar la conectividad en los pacientes con una pérdida de hueso severa. De esta manera, la terapia con fluoruro puede ser menos efectiva en los individuos con osteoporosis que ya han tenido una pérdida sustancial de la conectividad trabecular (26, 31).
Por otra parte ensayos aleatorios controlados que usaron dosis de fluoruro más bajas (≤20 md/día) con horarios intermitentes de las dosis, o formulación de liberación lenta (fluoruro de sodio con recubrimiento entérico) han demostrado una reducción en incidentes de fracturas vertebrales y no vertebrales junto con un incremento en la densidad ósea de la espina lumbar (32). Mas sin embargo biopsias óseas de mujeres posmenopáusicas osteoporóticas tratadas con 20 mg/día de fluoruro mostraron evidencia de una mineralización ósea anormal a pesar de suplementación de calcio y vitamina D (33). Adicionalmente, un reciente estudio aleatorio, doble ciego controlado con placebo no encontró ningún incremento en la DMO de la espina lumbar en 180 mujeres posmenopáusicas con osteopenia (osteoporosis temprana) a las cuales se les suplemento diariamente con hasta 10 mg/día de fluoruro por un año (34). Estudios adicionales son requeridos para evaluar si una dosis segura de fluoruro puede ser encontrada para maximizar la formación ósea y al mismo tiempo prevenir los defectos de la mineralización.
Seguridad de la terapia con fluoruro para la osteoporosis
Efectos secundarios serios han sido asociados con altas dosis de fluoruro usado para tratar la osteoporosis (32). Estos incluyen irritación gastrointestinal, dolor articular en extremidades inferiores, y el desarrollo de deficiencia de calcio y fracturas por estrés. Las razones para la ocurrencia de dolor en las articulaciones de las extremidades inferiores y para las fracturas por estrés en pacientes que toman fluoruro para la osteoporosis aún no están claros, pero pueden estar relacionados a incrementos rápidos en la formación ósea sin el calcio suficiente para soportar tal aumento (26). Actualmente, el fluoruro de sodio con recubrimiento entérico o las preparaciones de monofluorofosfato ofrecen un menor perfil de efectos secundarios que las dosis elevadas de fluoruro de sodio utilizadas en los previos estudios. Adicionalmente, suficiente calcio y vitamina D deberían ser provistos para sostener la formación ósea inducida por el fluoruro. Aunque la terapia con fluoruro puede ser beneficiosa para el tratamiento de la osteoporosis en individuos elegidos apropiadamente y monitoreados estrechamente, la incertidumbre respecto a su seguridad y beneficios en la reducción de fracturas ha evitado que la Administración de Drogas y Alimentos (FDA) apruebe la terapia con fluoruro para la osteoporosis (35). Las combinaciones de dosis más bajas de fluoruro con agentes anti-reabsorción ósea como estrógenos o bifosfonatos, pueden mejorar los resultados terapéuticos mientras que minimizan los efectos secundarios (36, 37). Sin embargo, recientes estudios aleatorios han mostrado que el riesgo de fracturas permanece sin ser alterado si los tratamientos incluyen fluoruro, agentes anti-reabsorción ósea o ambos (32, 33). Estudios adicionales son requeridos para determinar si cualquier combinación de tratamientos podría proveer beneficios terapéuticos substanciales sobre la monoterapia.
Fuentes
Fluoración del agua
La mayor fuente de fluoruro dietético en la dieta de los EE.UU. es el agua potable. Una adición controlada de fluoruro para el agua es usada por comunidades como una medida de salud pública para ajustar la concentración de fluoruro en el agua potable a un nivel óptimo de 0.7 a 1.2 miligramos (mg) por litro, que corresponde a 0.7-1.2 ppm. Se ha encontrado que este rango de concentración disminuye la incidencia de caries dental mientras que minimiza el riesgo de fluorosis dental y otros efectos adversos. El Departamento de Salud y Servicios Humanos de los EE. UU. ha recomendado recientemente que la concentración optima en el agua potable se fija en 0.7 ppm (vea Seguridad) (6). Aproximadamente el 74% de la población en los EE.UU. recibe agua con el fluoruro suficiente para la prevención de la caries dental (38). La ingesta promedio de fluoruro para los adultos que viven en comunidades fluoruradas varía de 1.4 a 3.4 mg/día en comparación a 0.3 a 1 mg/día en áreas no fluoruradas (7). Debido a que el agua de pozo puede variar ampliamente en su contenido de fluoruro, las personas que consumen agua de pozo debieran hacer medir el contenido de fluoruro de su agua por el distrito local de aguas o por el departamento de salud. La medición de fluoruro en el agua también se justifica en los hogares que utilizan grandes sistemas de tratamientos de agua. Mientras que los ablandadores de agua no están diseñados para cambiar los niveles de fluoruro en el agua, se ha encontrado que los sistemas de osmosis inversa, las unidades de destilación y algunos filtros de agua remueven cantidades significativas de fluoruro del agua. Sin embargo, los filtros tipo Brita no remueven el fluoruro (7, 35).
Las ventas de agua embotellada han aumentado exponencialmente en los EE.UU. en los últimos años y estudios han encontrado que la mayoría de las aguas embotelladas contienen niveles sub-óptimos de fluoruro, aunque existe una variación considerable (39). Por ejemplo, un estudio de 105 diferentes productos de agua embotellada en el área metropolitana de Greater Huston encontró que un poco más del 80% tenían concentraciones de fluoruro de menos de 0.4 ppm; solo un 5% de los productos probados tuvieron concentraciones dentro del rango recomendado (40). Varios otros estudios han reportado hallazgos similares, con la mayoría de aguas embotelladas relativamente bajas en fluoruro, pero muy pocas en el rango optimo o más alto (41-43). La cita aprobada por la FDA "beber agua fluorurada puede reducir el riesgo de caries dental" es solo usada por embotelladores cuando el agua contiene entre 0.6 ppm y 1.0 ppm de fluoruro. Sin embargo, a los embotelladores no se les requiere proveer la concentración de fluoruro en el agua embotellada a menos que el fluoruro fuese agregado (44).
Fórmulas para infantes
Mientras que el consumo de fluoruro del agua presenta muy pocos riesgos de efectos adversos en adultos, excepto en circunstancias extremas (véase Seguridad), el consumo de cantidades relativamente grandes de agua mezclada con fórmulas concentradas, parece incrementar el riesgo del desarrollo de fluorosis dental en infantes (45, 47). Un estudio encontró que, en promedio, al menos la mitad de todo el fluoruro ingerido por infantes de 6 meses y más jóvenes, provenía del agua mezclada con concentrados de formula (48). El estudio de 49 fórmulas para infantes disponibles en la región de Chicago mostro que las formulas listas para tomar, liquidas concentradas, y formulas en polvo (reconstituidas con agua desionizada) tenían significantes concentraciones de fluoruro de 0.15 ppm, 0.27 ppm, y 0.12 ppm, respectivamente (49). El contenido de fluoruro fue significantemente más alto en formulas liquidas concentradas a base de soya que las a base de leche (0.50 ppm vs 0.27 ppm). Usando pesos corporales promedio y el total de la ingesta de fórmulas durante el primer año de vida, los autores estimaron que el riesgo de exceder el nivel de ingesta máxima tolerable para la ingesta de fluoruro era mínimo cuando las formulas liquidas concentradas y en polvo eran reconstituidas con agua con un contenido de menos de 0.5 ppm de fluoruro, pero el riesgo era máximo con 1.0 ppm de agua fluorurada. Agua libre de fluoración o baja en fluoruro denominada como "desionizada," "purificada," "desmineralizada," o "producida a través de osmosis inversa" pueden ser utilizas con el fin de minimizar el riesgo de una fluorosis leve (44). Sin embargo infantes de entre 6 y 12 años de edad pueden no alcanzar la adecuada ingesta de fluoruro si son alimentados con fórmulas listas para tomar o formulas reconstituidas con agua que contenga menos de 0.4 ppm (49).
Fuentes alimenticias y bebidas
El contenido de fluoruro de la mayoría de los alimentos es bajo (menos de 0.05 mg/100 gramos o 0.5 ppm). Las fuentes altas en fluoruro incluyen el té, el cual concentra fluoruro en sus hojas, y los peces marinos que se consumen con huesos (ej. sardinas). Alimentos hechos con pollo mecánicamente separado (deshuesado), como carnes enlatadas, salchichas, y comida para bebes, también aportan fluoruro a la dieta (50). Además, ciertos jugos de frutas, particularmente de uva, tienen a menudo altas concentraciones de fluoruro (51). Por lo general, los alimentos sólo aportan de 0.3-0.6 mg de la ingesta diaria de fluoruro. Un hombre adulto residente en una comunidad con agua fluorurada tiene una ingesta que varía de 1 a 3 mg/día. La ingesta en áreas no fluoruradas es menor a 1 mg/día (2). La tabla entrega un rango del contenido de fluoruro de algunos alimentos ricos en fluoruro. Para más información en el contenido de fluoruro de alimentos y bebidas, busque en la base de datos nacional del fluoruro USDA.
| Alimentos | Porción | Fluoruro (mg) | Fluoruro (ppm)* |
|---|---|---|---|
| Jugo de frutas | 100 mL (3.5 onzas de fluido) | 0.02-0.21 | 0.2-2.1 |
| Cangrejo (enlatado) | 100 g (3.5 onzas) | 0.21 | 2.1 |
| Arroz (cocido) | 100 g (3.5 onzas) | 0.04 | 0.4 |
| Pescado (cocido) | 100 g (3.5 onzas) | 0.02 | 0.2 |
| Pollo | 100 g (3.5 onzas) | 0.015 | 0.15 |
| *1.0 partes por millón (ppm) = 1 miligramo/litro (mg/L) | |||
Suplementos de fluoruro
Los suplementos de fluoruro — disponibles sólo por prescripción en los EE.UU — pretenden ser para infantes de 6 meses en adelante y niños de hasta 16 años de edad que viven en áreas con menores concentraciones de fluoruro en el agua, con el propósito de llevar su ingesta hasta 1 mg/día aproximadamente (7). La Asociación Dental Americana en Asuntos Científicos recomienda la prescripción de suplementos de fluoruro solo para aquellos niños que se encuentran en un alto riesgo de desarrollar caries dental (52). El esquema de la dosis suplementaria de fluoruro en la tabla a continuación fue recomendado por la Asociación Dental Americana, la Academia Americana de Odontología Pediátrica y la Academia Americana de Pediatras (52, 53). Es necesario el conocimiento de la concentración de fluoruro del agua potable local, así como el de otras posibles fuentes de ingesta de fluoruro. Para información más detallada en relación al fluoruro y la prevención de la caries dental, visite el sitio web de la Asociación Dental Americana.
|
Edad |
Nivel de Ion Fluoruro en Agua Potable (ppm)* |
||
|---|---|---|---|
|
<0.3 ppm |
0.3-0.6 ppm |
>0.6 ppm |
|
| Nacimiento - 6 meses | Ninguno | Ninguno | Ninguno |
| 6 meses - 3 años | 0.25 mg/día** | Ninguno | Ninguno |
|
3 años - 6 años |
0.50 mg/día |
0.25 mg/día |
Ninguno |
|
6 años -16 años |
1.0 mg/día |
0.50 mg/día |
Ninguno |
|
*1.0 partes por millón (ppm) = 1 miligramo/litro (mg/L) **2.2 mg de fluoruro de sodio contiene 1 mg de ion fluoruro. |
|||
Pasta dental
Las pastas de dientes fluoruradas son bastante efectivas en la prevención de caries detales pero también aportan considerablemente a la ingesta de fluoruro de los niños, especialmente en niños pequeños, los que son más propensos a tragarse la pasta dental. Los investigadores estiman que los niños por debajo de los 6 años de edad ingieren un promedio de 0.3 mg de fluoruro de la pasta dental con cada cepillado. Los niños menores de 6 años de edad que ingieren más de dos a tres veces la ingesta recomendada de fluoruro, se encuentran en un riesgo incrementado de tener dientes permanentes moteados o con manchas blancas, conocido como fluorosis dental. La principal fuente de ingesta de fluoruro en exceso en este grupo etario proviene de tragar pasta dental con fluoruro. Para prevenir la fluorosis dental a la vez que se provee una protección óptima contra la caries dental, se recomienda que los padres supervisen a los niños menores de 6 años de edad cuando estos se cepillan los dientes con pastas fluoruradas. Además, para desalentar la deglución de la pasta dental, los niños deben ser supervisados durante el cepillado y niños jóvenes deben ser estimulados a usar cantidades muy pequeñas de pasta dental-(una capa delgada de pasta dental que cubra menos de la mitad de la superficie cerdada de un cepillo dental para niños) para niños menores de 3 años, y para niños de entre 3 y 6 años de edad la cantidad de pasta debe ser del tamaño de un guisante (54, 55). Curiosamente, se ha sugerido que el manejo del riesgo de fluorosis dental en infantes que ingieren pasta dental fluorurada podría incluir el uso de la formulación de pasta dental que reduce la absorción gastrointestinal y biodisponibilidad del fluoruro (56).
Fluoruración de la sal
La fluoruración de la sal ha sido implementada en varios países alrededor del mundo como una alternativa a la fluoruración del agua para promover el consumo de fluoruro y mejorar el cuidado oral. Debido a que la fluoruración del agua es extensivamente llevada a cabo en los EE. UU., el fluoruro no es agregado a la sal. Estudios epidemiológicos han mostrado que la incidencia de dientes con caries disminuyo dramáticamente en las regiones donde programas de fluoruración de la sal fueron desarrollados. Mientras que las preocupaciones alrededor de la hipertensión y el control de la ingesta de la población deberían ser abordadas, no se han reportado efectos adversos relacionados a la fluoruración de la sal (revisado en 57). De acuerdo la Organización Mundial de la Salud (OMS), la fluoruración de la sal, en menor medida, la fluoruración de la leche son alternativas accesibles para mejorar la higiene oral en áreas donde el acceso a servicios de la salud oral es limitado y la fluoruración del agua pública no es factible (58).
Seguridad
Efectos adversos
La fluoruración del agua potable pública en los EE.UU. se inició hace más de 70 años. Desde entonces, se le han atribuido una serie de efectos adversos a la fluoruración del agua. Sin embargo, investigaciones científicas exhaustivas no han descubierto evidencia de riesgos incrementados de cáncer, enfermedad del corazón, enfermedad renal, enfermedad hepática, Alzheimer, defectos de nacimiento, o síndrome de Down (6, 59, 60). Un numero de estudios epidemiológicos, mayormente publicados en diarios Chinos, han investigado la asociación entre el contenido del fluoruro en el agua potable y el desarrollo neurológico de los niños. Un meta-análisis de 27 estudios, mayormente llevados a cabo en China, encontró coeficientes intelectuales (IQ) más bajos en niños expuestos a concentraciones de fluoruro que van de 1.8 mg/L a 11 mg/L del agua potable (61). Limitaciones serias, incluyendo heterogeneidad significativa entre los estudios y la co-ocurrencia de otros agentes neurotóxicos en el agua potable, obstaculiza la fuerza del hallazgo y su aplicación a la configuración de los EE.UU. La Academia de Nutrición y Dietética ha estimado recientemente que solo evidencia limitada apoya una asociación entre el contenido de fluoruro en el agua y el IQ de los niños (44). Finalmente, un reciente estudio prospectivo en Nueva Zelanda basado en una cohorte poblacional con un seguimiento de aproximadamente cuatro décadas no encontró asociación alguna entre la exposición al fluoruro en el contexto de los programas de fluoruración del agua comunitaria y los coeficientes intelectuales medidos durante la niñez y a la edad de 38 años (62).
Toxicidad aguda
El fluoruro es tóxico cuando es consumido en cantidades excesivas, por lo que los productos de fluoruro concentrado debiesen utilizarse y almacenarse con precaución para prevenir la posibilidad de un envenenamiento agudo por fluoruro, especialmente en niños y otros individuos vulnerables. Se considera que la dosis más baja que puede ocasionar síntomas adversos es de 5 mg/kg de peso corporal, y considerando la dosis letal más baja en 15 mg/kg de peso corporal. Náuseas, dolor abdominal y vómito casi siempre acompañan a la toxicidad aguda por fluoruro. Otros síntomas como la diarrea, la salivación y lagrimeo excesivo, sudoración, y la debilidad generalizada también pueden aparecer (60). A fin de prevenir el envenenamiento agudo por fluoruro, la Asociación Dental Americana ha recomendado que no se dispensen más de 120 mg de fluoruro (224 mg de fluoruro de sodio) en cada ocasión (35). El uso de altas dosis de fluoruro para tratar la osteoporosis ha sido asociado con algunos efectos adversos, los cuales son discutidos en la sección Tratamiento de Enfermedad.
Fluorosis dental
La forma más leve de fluorosis dental sólo la detecta un observador entrenado y se caracteriza por pequeñas manchas o puntos blancos sobre el esmalte de los dientes. La fluorosis dental moderada está caracterizada por manchas y tinciones leves en los dientes y la fluorosis dental severa deriva en tinciones marcadas y picaduras en los dientes. En sus formas moderada y severa, la fluorosis dental se vuelve una preocupación cosmética cuando afecta a los incisivos y caninos (dientes delanteros). La fluorosis dental es el resultado de un exceso de ingesta de fluoruro previa la erupción de los primeros dientes permanentes (generalmente antes de los 8 años). También es una condición que depende de la dosis, donde las ingestas de fluoruro más altas se asocian con efectos más pronunciados sobre los dientes. La incidencia de fluorosis dental leve o moderada ha incrementado significativamente en los últimos años, mayormente debido a una ingesta incrementada de fluoruro de fórmulas reconstituidas para infantes y pastas dentales, aunque el uso inapropiado de suplementos de fluoruro pueden también contribuir (47). De acuerdo con un sondeo nacional de los EE. UU., la Encuesta Nacional de Salud y Nutrición, 1999-2004, un 23% de las personas de entre 6 a 49 años de edad tuvieron algún grado de fluorosis dental (63). En 1997, la Junta de Alimentos y Nutrición Estadounidense (FNB) del Instituto de Medicina estableció el nivel máximo de ingesta tolerable (NM) para el fluoruro basado en la prevención de la fluorosis dental moderada (7).
| Grupo Etario | NM (mg/día) |
|---|---|
| Infantes 0-6 meses | 0.7 |
| Infantes 7-12 meses | 0.9 |
| Niños 1-3 años | 1.3 |
| Niños 4-8 años | 2.2 |
| Niños 9-13 años | 10.0 |
| Adolescentes 14-18 años | 10.0 |
| Adultos 19 años o más | 10.0 |
Siguiendo las recomendaciones del Consejo Nacional de Investigación Científica, el EPA estadounidense está actualmente reevaluando el nivel máximo tolerable del fluoruro en agua potable (establecido en 4 mg/L) para asegurarse que protege a niños de desarrollar una fluorosis dental severa (44, 59). La EPA también ha establecido una norma estándar no exigible del nivel de fluoruro de 2 mg/L para prevenir la fluorosis dental moderada (64).
Fluorosis esquelética
Una ingesta de fluoruro a niveles excesivos por prolongados periodos de tiempo pueden llevar a cambios en la estructura ósea conocidos como fluorosis esquelética. Las etapas tempranas de la fluorosis esquelética están caracterizadas por un incremento en la masa ósea, detectable por rayos-X. Si muy altas ingestas de fluoruro persisten por muchos años, dolor en las articulaciones y rigidez pueden resultar de los cambios esqueléticos. La forma más severa de fluorosis esquelética es conocida como fluorosis esquelética paralizante, la cual puede resultar en la calcificación de ligamentos, inmovilidad, desgastamiento muscular, y problemas neurológicos relacionados a la compresión de la medula espinal. Mientras que la fluorosis esquelética es endémica en muchas regiones del mundo con concentraciones altamente naturales de fluoruro en el agua potable, la fluorosis esquelética paralizante puede ocurrir solo cuando la ingesta de fluoruro exceda 10 mg/día por lo menos durante un periodo de 10 años (7, 65). Raros casos de fluorosis esquelética en los EE. UU. han sido observados en consumidores de grandes cantidades de té (66-69). Debido al riesgo potencial de fluorosis esquelética, la EPA, la cual regula la fluoruración del agua bajo el Acta de Agua Potable Segura (Safe Drinking Water Act), está actualmente revisando el máximo nivel de fluoruro permitido en el agua potable- un nivel actualmente establecido a 4 mg/L (44, 59).
Interacciones con drogas/fármacos
Los suplementos de calcio, como también antiácidos que contienen calcio y aluminio, pueden disminuir la absorción de fluoruro. Lo más recomendable es tomar estos productos 2 horas antes o después de tomar suplementos de fluoruro (70).
Recomendación del Instituto Linus Pauling
La seguridad y los beneficios a la salud públicos de agua óptimamente fluorurados para la prevención de la caries dental en las personas de todas las edades ha sido bien establecido. El Instituto Linus Pauling apoya las recomendaciones de la Asociación Dental Americana y los Centros para el Control y Prevención de Enfermedades, que incluyen agua óptimamente fluorurada y el uso de pasta dental fluorurada, enjuague bucal fluorurado, esmalte fluorurado, y cuando es necesario, suplementación con fluoruro. Debido al riesgo de fluorosis, cualquier suplementación con fluoruro debiese ser prescrita y estrechamente monitoreada por un dentista o doctor.
Autores y Críticos
Escrito en Febrero de 2001 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Septiembre de 2007 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Noviembre de 2013 por
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Enero de 2014 por:
John J. Warren, D.D.S., M.S.
Profesor
Preventiva y Comunidad de Odontología
Colegio de Odontología
La Universidad de Iowa
Traducido al Español en 2015 por:
Silvia Vazquez Lima
Instituto Linus Pauling
Universidad Estatal de Oregon
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Última actualización 4/29/15 Derechos de autoría 2001-2024 Instituto Linus Pauling
Referencias
1. Cerklewski FL. Fluoride bioavailability--nutritional and clinical aspects. Nutr Res. 1997;17:907-929.
2. Nielsen FH. Ultratrace minerals. In: Shils M, Olson JA, Shike M, Ross AC, eds. Modern Nutrition in Health and Disease. 9th ed. Baltimore: Williams & Wilkins; 1999:283-303.
3. Cerklewski FL. Fluoride--essential or just beneficial. Nutrition. 1998;14(5):475-476.
4. Cerklewski FL. Fluorine. In: O'Dell BL, Sunde RA, eds. Handbook of nutritionally essential minerals. New York: Marcel Dekker, Inc; 1997:583-602.
5. Press release. 2011. EPA and HHS Announce New Scientific Assessments and Actions on Fluoride / Agencies working together to maintain benefits of preventing tooth decay while preventing excessive exposure.
6. US Department of Health and Human Services Federal Panel on Community Water Fluoridation. US Public health service recommendation for fluoride concentration in drinking water for the prevention of dental caries. Public Health Reports. Vol 130, 2015. Available at: http://www.publichealthreports.org/documents/PHS_2015_Fluoride_Guidelines.pdf. Accessed 4/29/15.
7. Food and Nutrition Board, Institute of Medicine. Fluoride. Dietary Reference Intakes: Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington D.C.: National Academy Press; 1997:288-313. (National Academy Press)
8. Centers for Disease Control. Achievements in public health, 1900-1999: fluoridation of drinking water to prevent dental caries. MMWR. 1999;48:933-940.
9. Demmer RT, Squillaro A, Papapanou PN, et al. Periodontal infection, systemic inflammation, and insulin resistance: results from the continuous National Health and Nutrition Examination Survey (NHANES) 1999-2004. Diabetes Care. 2012;35(11):2235-2242. (PubMed)
10. Demmer RT, Jacobs DR, Jr., Desvarieux M. Periodontal disease and incident type 2 diabetes: results from the First National Health and Nutrition Examination Survey and its epidemiologic follow-up study. Diabetes Care. 2008;31(7):1373-1379. (PubMed)
11. Desvarieux M, Demmer RT, Jacobs DR, Jr., et al. Periodontal bacteria and hypertension: the oral infections and vascular disease epidemiology study (INVEST). J Hypertens. 2010;28(7):1413-1421. (PubMed)
12. Demmer RT, Desvarieux M. Periodontal infections and cardiovascular disease: the heart of the matter. J Am Dent Assoc. 2006;137 Suppl:14S-20S; quiz 38S. (PubMed)
13. Zoellner H. Dental infection and vascular disease. Semin Thromb Hemost. 2011;37(3):181-192. (PubMed)
14. DePaola DP. Nutrition in relation to dental medicine. In: Shils M, Olson JA, Shike M, Ross AC, eds. Modern Nutrition in Health and Disease. 9th ed. Baltimore: Williams & Wilkins; 1999:1099-1124.
15. Newbrun E. Effectiveness of water fluoridation. J Public Health Dent. 1989;49(5 Spec No):279-289. (PubMed)
16. Dye BA, Tan S, Smith V, Lewis BG, Barker LK, Thornton-Evans G. Trends in oral health status: United States, 1988-1994 and 1999-2004. National Center for Health Statistics. Vital Health Stat 11(248), 2007. Available at: http://www.cdc.gov/nchs/data/series/sr_11/sr11_248.pdf. Accessed 1/15/14.
17. Marinho VC, Worthington HV, Walsh T, Clarkson JE. Fluoride varnishes for preventing dental caries in children and adolescents. Cochrane Database Syst Rev. 2013;7:CD002279. (PubMed)
18. Walsh T, Worthington HV, Glenny AM, Appelbe P, Marinho VC, Shi X. Fluoride toothpastes of different concentrations for preventing dental caries in children and adolescents. Cochrane Database Syst Rev. 2010(1):CD007868. (PubMed)
19. Magalhaes AC, Wiegand A, Rios D, Honorio HM, Buzalaf MA. Insights into preventive measures for dental erosion. J Appl Oral Sci. 2009;17(2):75-86. (PubMed)
20. Zini A, Krivoroutski Y, Vered Y. Primary prevention of dental erosion by calcium and fluoride: a systematic review. Int J Dent Hyg. 2013; doi: 10.1111/idh.12049. [Epub ahead of print] (PubMed)
21. Krall EA, Dawson-Hughes B. Osteoporosis. In: Shils M, Olson JA, Shike M, Ross AC, eds. Modern Nutrition in Health and Disease. 9th ed. Baltimore: Williams & Wilkins; 1999:1353-1364.
22. Fabiani L, Leoni V, Vitali M. Bone-fracture incidence rate in two Italian regions with different fluoride concentration levels in drinking water. J Trace Elem Med Biol. 1999;13(4):232-237. (PubMed)
23. Lehmann R, Wapniarz M, Hofmann B, Pieper B, Haubitz I, Allolio B. Drinking water fluoridation: bone mineral density and hip fracture incidence. Bone. 1998;22(3):273-278. (PubMed)
24. Sowers M, Whitford GM, Clark MK, Jannausch ML. Elevated serum fluoride concentrations in women are not related to fractures and bone mineral density. J Nutr. 2005;135(9):2247-2252. (PubMed)
25. Nasman P, Ekstrand J, Granath F, Ekbom A, Fored CM. Estimated drinking water fluoride exposure and risk of hip fracture: a cohort study. J Dent Res. 2013;92(11):1029-1034. (PubMed)
26. Cesar Libanati K-H. Fluoride therapy for osteoporosis. In: Marcus R, ed. Osteoporosis. San Diego: Academic Press; 1996:1259-1277.
27. Haguenauer D, Welch V, Shea B, Tugwell P, Adachi JD, Wells G. Fluoride for the treatment of postmenopausal osteoporotic fractures: a meta-analysis. Osteoporos Int. 2000;11(9):727-738. (PubMed)
28. Riggs BL, Hodgson SF, O'Fallon WM, et al. Effect of fluoride treatment on the fracture rate in postmenopausal women with osteoporosis. N Engl J Med. 1990;322(12):802-809. (PubMed)
29. Lundy MW, Stauffer M, Wergedal JE, et al. Histomorphometric analysis of iliac crest bone biopsies in placebo-treated versus fluoride-treated subjects. Osteoporos Int. 1995;5(2):115-129. (PubMed)
30. Fields AJ, Keaveny TM. Trabecular architecture and vertebral fragility in osteoporosis. Curr Osteoporos Rep. 2012;10(2):132-140. (PubMed)
31. Balena R, Kleerekoper M, Foldes JA, et al. Effects of different regimens of sodium fluoride treatment for osteoporosis on the structure, remodeling and mineralization of bone. Osteoporos Int. 1998;8(5):428-435. (PubMed)
32. Vestergaard P, Jorgensen NR, Schwarz P, Mosekilde L. Effects of treatment with fluoride on bone mineral density and fracture risk--a meta-analysis. Osteoporos Int. 2008;19(3):257-268. (PubMed)
33. Reid IR, Cundy T, Grey AB, et al. Addition of monofluorophosphate to estrogen therapy in postmenopausal osteoporosis: a randomized controlled trial. J Clin Endocrinol Metab. 2007;92(7):2446-2452. (PubMed)
34. Grey A, Garg S, Dray M, et al. Low-dose fluoride in postmenopausal women: a randomized controlled trial. J Clin Endocrinol Metab. 2013;98(6):2301-2307. (PubMed)
35. American Dietetic Association. Position of the American Dietetic Association: the impact of fluoride on health. J Am Diet Assoc. 2001;101(1):126-132. (PubMed)
36. Murray TM, Ste-Marie LG. Prevention and management of osteoporosis: consensus statements from the Scientific Advisory Board of the Osteoporosis Society of Canada. 7. Fluoride therapy for osteoporosis. CMAJ.1996;155(7):949-954. (PubMed)
37. Alexandersen P, Riis BJ, Christiansen C. Monofluorophosphate combined with hormone replacement therapy induces a synergistic effect on bone mass by dissociating bone formation and resorption in postmenopausal women: a randomized study. J Clin Endocrinol Metab. 1999;84(9):3013-3020. (PubMed)
38. National Center for Chronic Disease Prevention and Health Promotion, Division of Oral Health. Community water fluoridation: 2010 water fluoridation statistics. Available at: http://www.cdc.gov/fluoridation/statistics/2010stats.htm. Accessed 1/15/14.
39. Cutrufelli R, Pehrsson P, Haytowitz D, Patterson K, Holden J. USDA National Fluoride Database of Selected Beverages and Foods, Release 2. Nutrient Data Laboratory, Beltsville Human Nutrition Research Center, Agricultural Research Service, US Department of Agriculture; 2005. Available at: http://www.ars.usda.gov/SP2UserFiles/Place/12354500/Data/Fluoride/F02.pdf. Accessed 1/15/14.
40. Quock RL, Chan JT. Fluoride content of bottled water and its implications for the general dentist. Gen Dent. 2009;57(1):29-33. (PubMed)
41. Van Winkle S, Levy SM, Kiritsy MC, Heilman JR, Wefel JS, Marshall T. Water and formula fluoride concentrations: significance for infants fed formula. Pediatr Dent. 1995;17(4):305-310. (PubMed)
42. Tate WH, Chan JT. Fluoride concentrations in bottled and filtered waters. Gen Dent. 1994;42(4):362-366.
43. McGuire S. Fluoride content of bottled water. N Engl J Med. 1989;321(12):836-837.
44. Palmer CA, Gilbert JA, Academy of N, Dietetics. Position of the Academy of Nutrition and Dietetics: the impact of fluoride on health. J Acad Nutr Diet. 2012;112(9):1443-1453. (PubMed)
45. Marshall TA, Levy SM, Warren JJ, Broffitt B, Eichenberger-Gilmore JM, Stumbo PJ. Associations between Intakes of fluoride from beverages during infancy and dental fluorosis of primary teeth. J Am Coll Nutr. 2004;23(2):108-116. (PubMed)
46. Pendrys DG. Risk of enamel fluorosis in nonfluoridated and optimally fluoridated populations: considerations for the dental professional. J Am Dent Assoc. 2000;131(6):746-755. (PubMed)
47. Levy SM, Broffitt B, Marshall TA, Eichenberger-Gilmore JM, Warren JJ. Associations between fluorosis of permanent incisors and fluoride intake from infant formula, other dietary sources and dentifrice during early childhood. J Am Dent Assoc. 2010;141(10):1190-1201. (PubMed)
48. Levy SM, Kohout FJ, Guha-Chowdhury N, Kiritsy MC, Heilman JR, Wefel JS. Infants' fluoride intake from drinking water alone, and from water added to formula, beverages, and food. J Dent Res. 1995;74(7):1399-1407. (PubMed)
49. Siew C, Strock S, Ristic H, et al. Assessing a potential risk factor for enamel fluorosis: a preliminary evaluation of fluoride content in infant formulas. J Am Dent Assoc. 2009;140(10):1228-1236. (PubMed)
50. Fein NJ, Cerklewski FL. Fluoride content of foods made with mechanically separated chicken. J Agric Food Chem. 2001;49(9):4284-4286. (PubMed)
51. Kiritsy MC, Levy SM, Warren JJ, Guha-Chowdhury N, Heilman JR, Marshall T. Assessing fluoride concentrations of juices and juice-flavored drinks. J Am Dent Assoc. 1996;127(7):895-902. (PubMed)
52. Rozier RG, Adair S, Graham F, et al. Evidence-based clinical recommendations on the prescription of dietary fluoride supplements for caries prevention: a report of the American Dental Association Council on Scientific Affairs. J Am Dent Assoc. 2010;141(12):1480-1489. (PubMed)
53. Centers for Disease Control and Prevention. Recommendations for using fluoride to prevent and control dental caries in the United States. MMWR Recomm Rep. 2001;50(RR-14):1-42. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5014a1.htm
54. Section on Pediatric Dentistry and Oral Health. Preventive oral health intervention for pediatricians. Pediatrics. 2008;122(6):1387-1394. (PubMed)
55. American Dental Association Council on Scientific Affairs. Fluoride toothpaste use for young children. J Am Dent Assoc. 2014;145(2):190-191. (PubMed)
56. Falcao A, Tenuta LM, Cury JA. Fluoride gastrointestinal absorption from Na2FPO3/CaCO3- and NaF/SiO2-based toothpastes. Caries Res. 2013;47(3):226-233. (PubMed)
57. Pollick HF. Salt fluoridation: a review. J Calif Dent Assoc. 2013;41(6):395-397, 400-394. (PubMed)
58. Marthaler TM, Petersen PE. Salt fluoridation--an alternative in automatic prevention of dental caries. Int Dent J. 2005;55(6):351-358. (PubMed)
59. Committee on Fluoride in Drinking Water NRC. 2006. Fluoride in drinking water: a scientific review of EPA's Standards. Washington D.C.: National Academies Press.
60. Whitford GM. Acute toxicity of ingested fluoride. Monogr Oral Sci. 2011;22:66-80. (PubMed)
61. Choi AL, Sun G, Zhang Y, Grandjean P. Developmental fluoride neurotoxicity: a systematic review and meta-analysis. Environ Health Perspect. 2012;120(10):1362-1368. (PubMed)
62. Broadbent JM, Thomson WM, Ramrakha S, et al. Community Water Fluoridation and Intelligence: Prospective Study in New Zealand. Am J Public Health. 2015;105(1):72-76. (PubMed)
63. Beltrán-Aguilar ED, Barker L, Dye BA. Prevalence and severity of dental fluorosis in the United States, 1999-2004. NCHS data brief, no 53. Hyattsville, MD: National Center for Health Statistics; 2010. Available at: http://www.cdc.gov/nchs/data/databriefs/db53.pdf
64. Agency. UEP. New fluoride risk assessment and relative source contribution documents. 2011. Available at: http://water.epa.gov/action/advisories/drinking/upload/fluoridefactsheet.pdf
65. Whitford GM. The metabolism and toxicity of fluoride. Vol 13. Basel: S. Karger AG; 1996.
66. Hallanger Johnson JE, Kearns AE, Doran PM, Khoo TK, Wermers RA. Fluoride-related bone disease associated with habitual tea consumption. Mayo Clin Proc. 2007;82(6):719-724. (PubMed)
67. Whyte MP, Totty WG, Lim VT, Whitford GM. Skeletal fluorosis from instant tea. J Bone Miner Res. 2008;23(5):759-769. (PubMed)
68. Izuora K, Twombly JG, Whitford GM, Demertzis J, Pacifici R, Whyte MP. Skeletal fluorosis from brewed tea. J Clin Endocrinol Metab. 2011;96(8):2318-2324. (PubMed)
69. Kakumanu N, Rao SD. Images in clinical medicine. Skeletal fluorosis due to excessive tea drinking. N Engl J Med. 2013;368(12):1140. (PubMed)
70. Minerals. Drug Facts and Comparisons. St. Louis: Facts and Comparisons; 2000:27-51.
Fósforo
Contenido
Resumen
- El fósforo es un componente estructural esencial de las membranas celulares y ácidos nucleicos pero también está relacionado en varios procesos biológicos, incluyendo la mineralización ósea, la producción de energía, la señalización celular a través de las reacciones de fosforilación, y la regulación de la homeostasis acido-básica. (Más información)
- La deficiencia de fósforo dietario es poco común y a menudo solo es observada en casos de inanición casi total o en trastornos hereditarios raros que implican la pérdida de fósforo renal. Los síntomas incluyen pérdida del apetito, debilidad muscular, fragilidad ósea, entumecimiento en las extremidades, y raquitismo en niños. (Más información)
- La ingesta diaria recomendada (IDR), de 700 mg/día de fósforo en adultos saludables, es destinada a mantener las concentraciones de fósforo en el suero dentro del rango fisiológico de 2.5 a 4.5 mg/dL. (Más información)
- El fósforo se encuentra en la mayoría de las fuentes alimenticias y es un componente comúnmente usado en los aditivos alimentarios. La biodisponibilidad del fósforo proveniente de los alimentos es usualmente bastante alta con la excepción del fitato de fósforo en fuentes vegetales, como granos, legumbres, y semillas, las cuales son pobremente digeridas. (Más información)
- Estimados de las ingestas de fósforo dietario en los EE.UU. es probable que sean inexactos debido a que las cantidades de aditivos alimentarios basados en fósforo usados en alimentos procesados no son siempre incluidas en la base de datos del contenido nutricional usada para calcular las ingestas de nutrientes. (Más información)
- Las concentraciones altas de fósforo en el suero han sido asociadas con un incremento en las tasas de enfermedades cardiovasculares y mortalidad en sujetos con o sin enfermedad renal. Una deposición anormal de fosfato de calcio en los tejidos blandos puede predisponer a personas a una disfunción vascular y enfermedad cardiovascular. (Más información)
- La hiperfosfatemia, la cual es común en individuos con insuficiencia renal, se caracteriza por ser una condición en la cual existe una acumulación anormalmente alta de fósforo en la sangre, porque los riñones no son capaces de excretarlo efectivamente. (Más información)
- El nivel máximo de ingesta tolerable (NM) para el fósforo es de 4,000 mg/día para adultos generalmente saludables. A pesar de que las ingestas de fósforo que exceden la IDR han sido ligadas a un riesgo incrementado de mortalidad por todas las causas en individuos saludables. (Más información)
- Estudios basados en la observación sugieren que una ingesta con una proporción baja de calcio a fósforo puede ser perjudicial para la salud ósea, especialmente en mujeres con un riesgo incrementado de osteoporosis. (Más información)
El fósforo es un mineral esencial requerido por cada célula del cuerpo para una función normal (1). Ligado al oxígeno en todos los sistemas biológicos, el fósforo se encuentra como fosfato (PO43-) en el cuerpo. Aproximadamente el 85% del fósforo corporal se encuentra en los huesos y dientes (2).
Función
El fósforo es uno de los principales componentes estructurales del hueso en la forma de una sal de fosfato de calcio denominada hidroxiapatita. Los fosfolípidos (p.ej., fosfatidilcolina) son importantes componentes estructurales de las membranas celulares. Toda la producción y almacenaje de energía depende de compuestos fosforilados, como el adenosin trifosfato (ATP) y la creatina fosfato. Los ácidos nucléicos (ADN y ARN), los cuales son responsables del almacenaje y transmisión de la información genética, son largas cadenas de moléculas fosforadas. Un cierto número de enzimas, hormonas, y moléculas de señalización celular, dependen de la fosforilación para su activación. El fósforo también ayuda a mantener el balance ácido-base (pH) normal al actuar como uno de los buffers más importantes del organismo. Además, el 2,3-difosfoglicerato (2,3-DPG), una molécula fosforada, se une a la hemoglobina en los glóbulos rojos y regula la distribución de oxígeno a los tejidos del cuerpo (1).
Regulación
Eje hormona paratiroidea-vitamina D y FGF-23-endocrino
El fósforo dietario es fácilmente absorbido en el intestino delgado, y en individuos saludables, el exceso de fósforo es excretado por los riñones bajo la acción regulatoria de las hormonas endocrinas: hormona paratiroidea (HPT), vitamina D, y el factor de crecimiento de fibroblastos 23 (FGF-23). La aguda regulación de las concentraciones de calcio y fósforo en la sangre es controlada a través de las acciones de la HPT y la forma activa de la vitamina D. Un ligero descenso en los niveles de calcio en la sangre (p. ej., en el caso de una ingesta inadecuada de calcio) es detectado por las glándulas paratiroideas, resultando en la secreción incrementada de la HPT, la cual rápidamente disminuye la excreción urinaria de calcio pero incrementa la excreción urinaria de fósforo y estimula la reabsorción ósea. Esto da como resultado la liberación de mineral óseo (calcio y fosfato) — acciones que restauran las concentraciones de calcio en el suero. Aunque la acción no es inmediata, la HPT también estimula la conversión de vitamina D a su forma activa (1,25-dihidroxivitamina D; calcitriol) en los riñones. El incremento de la 1,25-dihidroxivitamina D circulante a su vez estimula la absorción intestinal incrementada de tanto calcio como fósforo. Una tercera hormona, el FGF-23, desempeña una papel central en la homeostasis del fósforo. El FGF-23 es secretado por las células formados de hueso (osteoblastos/osteocitos) en respuesta a incrementos en la ingesta de fósforo. En un bucle de retroalimentación negativa, el FGF-23 inhibe la producción y estimula la degradación de 1,25-dihidroxivitamina D, como también promueve un incremento en la excreción urinaria de fósforo independientemente de la HPT y 1,25-dihidroxivitamina D (3).
Deficiencia
Una ingesta inadecuada de fósforo resulta en niveles anormalmente bajos de fósforo en el suero (hipofosfatemia) porque la reabsorción renal del fósforo se incrementa para compensar por la disminución de la ingesta. Los efectos de la hipofosfatemia de moderada a severa pueden incluir pérdida del apetito, anemia, debilidad muscular, dolor de huesos, raquitismo (en niños), osteomalasia (en adultos), mayor susceptibilidad a infecciones, entumecimiento y hormigueo de las extremidades, dificultad para caminar e insuficiencia respiratoria. La hipofosfatemia severa puede causar la muerte. Ya que el fósforo se encuentra ampliamente distribuido en los alimentos, la deficiencia de fósforo dietario usualmente sólo se ve en casos cercanos a la inanición total. Entre otros individuos en riesgo de hipofosfatemia se incluye a los alcohólicos, diabéticos en recuperación de un episodio de cetoacidosis diabética, pacientes con alcalosis respiratoria, y pacientes famélicos o anoréxicos en regímenes de realimentación con alto contenido calórico pero muy bajo en fósforo (revisado en 4). La hipofosfatemia causada por trastornos hereditarios de la homeostasis del fósforo (trastornos de la perdida de fósforo) ha sido ligada a la excreción urinaria elevada o la reabsorción renal deteriorada del fósforo en sujetos afectados (revisado en 5).
La Ingesta Diaria Recomendada (IDR)
La ingesta diaria recomendada (IDR) para el fósforo está basada en el mantenimiento de niveles normales de fósforo en el suero en adultos (2.5-4.5 miligramos/decilitro [mg/dL]) y se cree que representa las ingestas adecuadas de fósforo para satisfacer las necesidades celulares y de la formación ósea (6; Tabla 1). La IDR, la cual es la ingesta diaria promedio que satisface los requerimientos del 97.5% de los individuos saludables en una específica etapa de vida y género, está basada en el requerimiento estimado promedio (REP; 580 mg/día de fósforo para adultos) — la ingesta de nutrientes que satisface el requerimiento del 50% de individuos saludables en una particular etapa de vida y género.
| Etapa de la Vida | Edad |
Machos (mg/día) |
Hembras (mg/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 100 (IA) | 100 (IA) |
| Infantes | 7-12 meses | 275 (IA) | 275 (IA) |
| Niños | 1-3 años | 460 | 460 |
| Niños | 4-8 años | 500 | 500 |
| Niños | 9-13 años | 1,250 | 1,250 |
| Adolescentes | 14-18 años | 1,250 | 1,250 |
| Adultos | 19 años y más | 700 | 700 |
| Embarazo | 18 años o menos | - | 1,250 |
| Embarazo | 19 años y más | - | 700 |
| Período de lactancia | 18 años o menos | - | 1,250 |
| Período de lactancia | 19 años y más | - | 700 |
Fuentes
Fuentes alimenticias
El fósforo se encuentra en la mayoría de los alimentos porque es un componente fundamental de todos los organismos vivos. Los productos lácteos, productos de cereales, la carne y el pescado son fuentes particularmente ricas en fósforo (7). El fósforo también es un componente de muchos aditivos alimenticios que son usados en el procesamiento de alimentos y se encuentra en la mayoría de las bebidas gaseosas como ácido fosfórico (8). En la encuesta representativa a nivel nacional NHANES, las ingestas de fósforo estuvieron muy por encima de la REP e IDR con ingestas diarias promedio de 1,602 mg en hombres y 1,128 mg en mujeres (8). El fósforo dietario derivado de los aditivos alimentarios no está siempre incluido en la bases de datos de composición de nutrientes de los alimentos, de esta manera la cantidad total de fósforo consumido por una persona promedio en los EE.UU. puede ser subestimada por más de un 20% (9). Algunos piensan que segmentos de la población de los EE.UU. que consumen más alimentos altamente procesados y cuyas ingestas de fósforo se acercan al nivel máximo de ingesta tolerable de 4,000 mg/día se encuentran en un alto riesgo de desarrollar resultados adversos a la salud (véase Seguridad) (7, 9).
Biodisponibilidad
El fósforo en las semillas vegetales (frijoles, guisantes, cereales, y nueces) se encuentra presente en una forma de almacenaje de fosfato denominada ácido fítico o fitato. Sólo alrededor del 50% del fósforo en los fitatos se encuentra disponible para los seres humanos debido a que carecemos de las enzimas (fitasas) que liberan al fósforo del fitato (10). Las levaduras poseen fitasas, por lo que los granos enteros incorporados al pan con levadura tienen mayor biodisponibilidad del fósforo que los granos enteros incorporados en los cereales del desayuno o en los panes sin levadura (6). Debido a que la reducción de la absorción dietaría de fósforo puede beneficiar individuos con insuficiencia renal que están en riesgo de hiperfosfatemia (fósforo en el suero en o por encima del rango normal-alto), las fuentes de proteína del fósforo en dietas vegetarianas a base de granos pueden ser preferidos sobre las dietas a base de carne (11). La Tabla 2 lista un cierto número de alimentos ricos en fósforo, junto con su contenido en miligramos (mg). Para más información en el contenido nutricional de los alimentos, busque en la base de datos de composición de los alimentos de la USDA.
| Alimento | Porción | Fósforo (mg) |
|---|---|---|
| Salmón (chinook, cocido) | 3 onzas* | 315 |
| Yogurt (natural, sin grasa) | 8 onzas | 306 |
| Leche (descremada) | 8 onzas | 247 |
| Hipogloso (Atlántico o Pacifico, cocido) | 3 onzas | 244 |
| Pavo (carne blanca, cocido) | 3 onzas | 217 |
| Pollo (carne blanca, cocido) | 3 onzas | 135-196 |
| Res (bistec, cocido) | 3 onzas | 179 |
| Lentejas# (cocidas) | ½ taza | 178 |
| Almendras# | 1 onza (23 piezas) | 136 |
| Queso, mozzarella (parcialmente descremado) | 1 onza | 131 |
| Cacahuates# | 1 onza | 108 |
| Huevos (hervidos) | 1 grande | 86 |
| Pan, trigo entero | 1 rebanda | 68 |
| Bebida de cola carbonatada | 12 onzas | 41 |
| Pan, blanco enriquecido | 1 rebanda | 25 |
|
*Una porción de 3 onzas es del tamaño de una baraja de cartas. #El fósforo de los frutos secos, de las semillas y los granos es un 50% menos biodisponible que el fósforo de otras fuentes (12). |
||
Suplementos
El contenido de fósforo de suplementos multivitamínicos/minerales (MVM) varía; una encuesta nacional estadounidense encontró que los suplementos MVM contribuyen en promedio 108 mg de la ingesta diaria de fósforo (10). Las sales de fosfato de sodio y fosfato de potasio son usadas para el tratamiento de la hipofosfatemia que ocurre en los trastornos hereditarios de la pérdida de fosfato, y su uso requiere de supervisión médica. Las sales de calcio de fosfato son algunas veces usadas como suplementos de calcio (13). Drogas de venta libre y con prescripción comúnmente usadas también contribuyen a ingestas de fósforo a niveles aún por definirse (10).
Seguridad
Toxicidad
Varios trastornos caracterizados por niveles de fósforo en el suero por encima de lo normal (hiperfosfatemia) han sido descritos, incluyendo aquellos que resultaron de una absorción incrementada de sales de fosfato tomadas oralmente o por absorción colónica de las sales de fosfato en enemas (1). A pesar de todo, la disrupción de la homeostasis del fósforo esta frecuentemente más asociada con una insuficiencia de la excreción en pacientes con enfermedad crónica renal (ERC) o enfermedad renal en etapa terminal (ERC avanzada). Cuando la función renal esta solo al 20% de lo normal, incluso las ingestas de fósforo dentro del rango recomendado pueden conducir a una hiperfosfatemia. La hiperfosfatemia puede también afectar individuos con niveles inapropiadamente bajos de la hormona paratiroidea (HPT) (hipoparatiroidismo) ya que carecen de estimulación de la HPT de la excreción renal del fosfato y fallan en estimular la síntesis de 1,25-dehidroxivitamina D (la forma activa de la vitamina D). Estos individuos no pueden excretar el exceso de fósforo en la ausencia de ambas hormonas (14). Las concentraciones elevadas de fósforo séricas han sido asociadas con una progresión acelerada de la enfermedad en individuos con insuficiencia renal y han sido ligadas al incremento del riesgo de resultados adversos para la salud en la población general (9, 15).
Altas concentraciones de fósforo en el suero en la población general
El fósforo alto en el suero dentro del rango normal (2.5-4.5 mg/dL) recientemente ha sido asociado con un incremento en la incidencia de enfermedades cardiovasculares (ECV) en individuos con una función renal normal. Dos estudios conducidos en la población general y en individuos con ECV previas han ligado las concentraciones altas-normales de fósforo sérico (≥3.5 mg/dL) a un riesgo cardiovascular mayor (16, 17). Estudios basados en la observación adicionales encontraron que las concentraciones de fósforo en el suero que igualan o están por encima de los 4 mg/dL se asociaron con una duplicación del riesgo de desarrollar ERC incidental y enfermedad renal en etapa terminal en individuos libres de enfermedad renal al inicio del estudio (18). En un estudio de cohorte prospectivo, el cual dio seguimiento a 4,005 adultos jóvenes saludables por más de 15 años, el fósforo más alto en el suero dentro del rango normal fue también asociado con hipertrofia ventricular izquierda, una condición frecuentemente ligada a resultados adversos cardiovasculares (19). En otro estudio de 3,088 participantes saludables de mediana edad con un seguimiento de 17 años, las concentraciones de fósforo en el suero en el cuartil superior del rango normal fueron asociadas con un riesgo dos veces mayor de insuficiencia cardiaca en comparación con el cuartil más bajo (≥3.5 mg/dL vs. <2.9 mg/dL) (16). Se piensa que la calcificación vascular, la cual explica la relación entre el fósforo alto y el riesgo de enfermedades cardiovasculares en pacientes con ERC (véase Hiperfosfatemia en sujetos con enfermedad renal), contribuye a su asociación en individuos con una función renal normal, incluso cuando su fósforo sérico está dentro del rango normal y sus ingestas están por debajo del nivel máximo de ingesta tolerable (NM) (20, 21).
La homeostasis del fósforo está estrechamente regulada por el eje HPT/vitamina D/FGF-23 en los individuos con una función renal normal (véase Regulación). El incremento en la secreción de la HPT y el FGF-23 ayuda a mantener las concentraciones de fósforo en el suero en el rango normal (2.5-4.5 mg/dL) incluso en el contexto de una ingesta alta de fósforo (9). Esto contribuye a que el fósforo sérico sea solo correlacionado débilmente al consumo de fósforo (22). Es de destacar que los incrementos sostenidos en el FGF-23 y la HPT son comúnmente observados durante la ERC a fin de mantener concentraciones normales de fósforo sérico a pesar de una reducción en la excreción de fósforo urinario (23). Un FGF-23 elevado, en lugar del fósforo sérico, parece ser un indicador temprano del trastorno de la homeostasis del fósforo y un vaticinador de resultados adversos a la salud en pacientes en etapa temprana de ECR (23, 24). Por lo tanto, es razonable asumir que la medición del fósforo sérico en personas con una función renal normal no puede adecuadamente reflejar perturbaciones tempranas en el metabolismo del fósforo debido al alto consumo de fósforo.
Hiperfosfatemia en sujetos con enfermedad renal
Estudios basados en la observación han reportado altas tasas de mortalidad y eventos cardiovasculares en asociación con altos niveles de fósforo en la sangre en sujetos con ERC. Un meta-análisis de 13 estudios de cohorte perspectivos, conducidos en más de 90,000 pacientes con ERC, encontró un incremento del 18% en la mortalidad por todas las causas por cada incremento de un 1 mg/dL de la concentración de fósforo en el suero por arriba de los 3.5 mg/dL. Un incremento del 10% del riesgo de muerte relacionada con enfermedades cardiovasculares (ECV) también fue calculado por cada 1 mg/dL mayor de concentración en el meta-análisis de tres estudios (25). Aunque no se ha establecido la causalidad entre la deficiencia de vitamina D y el riesgo de ECV, se ha sugerido que el fracaso al producir 1,25-dihidroxivitamina D en individuos hiperfosfatemicos puede modificar el riesgo de desarrollar enfermedad cardiovascular y renal, como también empeorar la insuficiencia renal en pacientes con ERC (26). Otro mecanismo plausible para la disfunción cardiovascular inducida por la hiperfosfatemia es la deposición de fosfato de calcio en tejidos no esqueléticos, especialmente el sistema vascular (27). Ciertamente, las concentraciones altas de fósforo pueden estimular la expresión de marcadores óseos específicos en las células formadoras de vasos sanguíneos, resultando en el cambio en sus funciones; este proceso, llamado diferenciación osteocondrogénica, transforma células musculares lisas vasculares (CVLM) en células óseas. Se encontró que el cultivo de CVLM de la aorta humana en condiciones hiperfosfatémicas resulto en la mineralización del medio extracelular, imitando la calcificación vascular in vivo (28). La calcificación vascular ha sido asociada con al menos un incremento triple del riesgo de eventos cardiovasculares y mortalidad; el riesgo de eventos cardiovasculares es el doble de alto (es decir, un riesgo incrementado seis veces) en individuos con insuficiencia renal (29).
En pacientes con ECR, trastornos en la remodelación ósea puede resultar en la liberación en exceso de fósforo y calcio a la sangre, la cual exacerba la hiperfosfatemia y la calcificación vascular y acelera el declive de la función renal. Actualmente, la restricción de fósforo dietario es recomendada para normalizar las concentraciones de suero en pacientes con ECR, aunque el impacto en los riesgos de ECV y mortalidad es desconocido.
El Nivel Máximo de Ingesta Tolerable (NM)
Para evitar los efectos adversos de la hiperfosfatemia, la Junta de Alimentos y Nutrición de los EE.UU. estableció un nivel máximo de ingesta tolerable (NM) para el fósforo oral en individuos generalmente sanos (6; Tabla 3). El NM menor para individuos de más de 70 años de edad, en comparación con grupos de edad más jóvenes, refleja el aumento de la probabilidad de insuficiencia renal en personas de edad avanzada. El NM no se aplica a individuos con una significante insuficiencia renal u otras condiciones de salud que sean conocidas por incrementar el riesgo de hiperfosfatemia.
Resultados adversos a la salud han sido asociados con concentraciones normales de fósforo en el suero, sugiriendo que en individuos con una función renal adecuada, la medición de niveles de fósforo en el suero estrechamente controlados puede tergiversar el efecto perjudicial de la alta ingesta de fósforo en la dieta (véase Concentraciones altas de fósforo en el suero en la población general). Mientras que las ingestas de fósforo por debajo del NM de 4,000 mg/día no deberían resultar en hiperfosfatemia o en un riesgo cardiovascular en adultos sanos de entre 19-70 años, un estudio reciente encontró que las ingestas diarias de fósforo de más del doble de la IDR (es decir, >1,400 mg/día) fueron significantemente asociadas con un riesgo incrementado de mortalidad por todas las causas (30).
¿Es la ingesta alta de fósforo perjudicial para la salud ósea?
Algunos investigadores están preocupados acerca del incremento de las cantidades de fósforo en la dieta, las cuales se atribuyen en gran medida al ácido fosfórico en algunas bebidas gaseosas y el uso creciente de aditivos de fosfato en alimentos procesados (31, 32). El fósforo alto en el suero se ha mostrado que perjudica la síntesis de la forma activa de la vitamina D (1,25-dihidroxivitamina D) en los riñones, reduce el calcio en la sangre, y conduce a un incremento de la liberación de la HPT por las glándulas paratiroideas (8). La estimulación de la HPT entonces resulta en la disminución de la excreción de calcio urinario y en el incremento de la reabsorción ósea; ambos contribuyen a que las concentraciones de calcio en el suero regresen a lo normal (8). Si se mantienen, los niveles elevados de HPT podrían tener un efecto adverso en el contenido mineral óseo, pero este efecto parece ser observado con dietas que son altas en fósforo y bajas es calcio, lo que subraya la importancia de una balanceada proporción dietaría de calcio a fósforo. En un estudio de corte transversal de menor tamaño, en el cual participaron 147 mujeres premenopáusicas con ingestas de calcio adecuadas, las participantes con ingestas menores de calcio a fósforo (Ca:P) (proporciones ≤0.5) tuvieron niveles significantemente más altos de HPT en el suero y una excreción urinaria de calcio que aquellas con proporciones de Ca:P más elevadas (proporción >0.5) (33). Un ensayo controlado en 10 mujeres jóvenes no encontró efectos adversos de una dieta rica en fósforo (3,000 mg/día) en hormonas relacionadas con los huesos y marcadores bioquímicos de la reabsorción ósea cuando las ingestas de calcio dietario se mantuvieron a casi 2,000 mg/día (Ca:P =0.66), demostrando de nuevo la importancia del balance entre el calcio y fósforo dietario (34).
Un estudio de corte transversal conducido en 2,344 hombres y mujeres brasileños (edad media, 58 años) no mostro asociación alguna entre las ingestas altas de fósforo y el riesgo incrementado de fractura. Sin embargo, las ingestas de otros minerales y vitaminas relevantes a la salud ósea, como el calcio, magnesio, y la vitamina D, estuvieron por debajo de la IDR en esta población, mientras que las ingestas de fósforo estuvieron cerca de la IDR (35). Mientras que pareciere que los trastornos hormonales y del calcio podrían ser prevenidos por una adecuada ingesta de la proporción de calcio a fósforo, no existe evidencia convincente de que los niveles de fósforo dietario experimentados en los EE.UU. afectan adversamente la densidad mineral ósea. No obstante, la sustitución de bebidas gaseosas que contiene fosfato y bocadillos por la leche y otros alimentos ricos en calcio puede representar un grave riesgo a la salud ósea (vea el artículo en Calcio) (36).
Interacción con drogas/fármacos
Los antiácidos con aluminio reducen la absorción de fósforo dietario al formar fosfato de aluminio, el cual no se absorbe. Cuando se consume en altas dosis, los antiácidos con aluminio pueden producir niveles de fosfato sanguíneo anormalmente bajos (hipofosfatemia) como también agravar la deficiencia de fósforo debida a otras causas (37). La reducción de la acidez estomacal por los inhibidores de la bomba de protones puede también limitar la eficacia de la terapia de quelantes de fosfato en pacientes con insuficiencia renal (38). Las dosis excesivamente altas de 1,25-dihidroxivitamina D, la forma activa de la vitamina D o sus análogos, pueden resultar en hiperfosfatemia (6).
Los suplementos de potasio o los diuréticos ahorradores de potasio tomados junto con suplementos de fósforo pueden provocar niveles sanguíneos elevados de potasio (hiperkalemia). La hiperkalemia puede ser un problema grave resultando en anomalías del ritmo cardíaco (arritmias) potencialmente mortales. Las personas que consumen tal combinación deben informar a su médico tratante y medir sus niveles de potasio en el suero regularmente (37).
Adicionalmente, la prevención de la desmineralización ósea por la terapia de reemplazamiento de hormonas en mujeres postmenopáusicas está asociada con la alta excreción urinaria de fósforo y niveles bajos de fósforo en el suero en mujeres tratadas a comparación de las no tratadas (39, 40).
Recomendación del Instituto Linus Pauling
El Instituto Linus Pauling respalda la IDR para el fósforo (700 mg/día para adultos). Aunque algunos suplementos multivitamínicos/minerales contienen más del 15% de la actual IDR para el fósforo, una dieta variada debería aportar fácilmente el fósforo suficiente a la mayoría de las personas.
Adultos mayores (>50 años)
En la actualidad, no existe evidencia de que los requerimientos de fósforo en adultos mayores difieran de los de un adulto joven, y una dieta variada debiera aportar fácilmente la actual IDR (700 mg/día) del fósforo para aquellos mayores de 50 años de edad.
Autores y Críticos
Escrito originalmente en 2001 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Abril de 2003 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Agosto de 2007 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Junio de 2014 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Junio de 2014 por:
Mona S. Calvo, Ph.D.
Profesor Clínico, Emérito
Oficina de Investigación Aplicada y Evaluación de Seguridad
Centro para la Seguridad Alimentaria y Nutrición Aplicada
Administración de Alimentos y Drogas de los EE.UU.
Traducido al Español en 2016 por:
Silvia Vazquez Lima
Instituto Linus Pauling
Universidad Estatal de Oregon
Los hallazgos y conclusiones de este crítico no necesariamente representan las opiniones y puntos de vista de la Administración de Alimentos y Drogas de los EE.UU.
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Derechos de autoría 2001-2024 Instituto Linus Pauling
Referencias
1. Knochel JP. Phosphorus. In: Shils ME, Shike M, Ross AC, Caballero B, Cousins RJ, eds. Modern Nutrition in Health and Disease. 10th ed. Baltimore: Lippincott Williams & Wilkins; 2006:211-222.
2. Heaney RP. Phosphorus. In: Erdman Jr. JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Ames: Wiley-Blackwell; 2012; 447-458.
3. Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev. 2012;92(1):131-155. (PubMed)
4. Amanzadeh J, Reilly RF, Jr. Hypophosphatemia: an evidence-based approach to its clinical consequences and management. Nat Clin Pract Nephrol. 2006;2(3):136-148. (PubMed)
5. Alizadeh Naderi AS, Reilly RF. Hereditary disorders of renal phosphate wasting. Nat Rev Nephrol. 2010;6(11):657-665. (PubMed)
6. Food and Nutrition Board, Institute of Medicine. Phosphorus. Dietary Reference Intakes: Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington D.C.: National Academy Press; 1997:146-189. (National Academy Press)
7. Takeda E, Yamamoto H, Yamanaka-Okumura H, Taketani Y. Dietary phosphorus in bone health and quality of life. Nutr Rev. 2012;70(6):311-321. (PubMed)
8. Calvo MS, Moshfegh AJ, Tucker KL. Assessing the health impact of phosphorus in the food supply: issues and considerations. Adv Nutr. 2014;5(1):104-113. (PubMed)
9. Calvo MS, Uribarri J. Public health impact of dietary phosphorus excess on bone and cardiovascular health in the general population. Am J Clin Nutr. 2013;98(1):6-15. (PubMed)
10. Calvo MS, Uribarri J. Contributions to total phosphorus intake: all sources considered. Semin Dial. 2013;26(1):54-61. (PubMed)
11. Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol. 2011;6(2):257-264. (PubMed)
12. National Research Council, Food and Nutrition Board. Recommended Dietary Allowances. 10th ed. Washington, D.C.: National Academy Press; 1989:184-187.
13. Phosphorus. In: Hendler SS, Rorvik DM, eds., eds. PDR for Nutritional Supplements. 2nd ed. Montvale: Physicians' Desk Reference; 2008:494-497.
14. Al-Azem H, Khan AA. Hypoparathyroidism. Best Pract Res Clin Endocrinol Metab. 2012;26(4):517-522. (PubMed)
15. Menon MC, Ix JH. Dietary phosphorus, serum phosphorus, and cardiovascular disease. Ann N Y Acad Sci. 2013; 1301:21-26. (PubMed)
16. Dhingra R, Sullivan LM, Fox CS, et al. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med. 2007;167(9):879-885. (PubMed)
17. Tonelli M, Sacks F, Pfeffer M, et al. Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation. 2005;112(17):2627-2633. (PubMed)
18. O'Seaghdha CM, Hwang SJ, Muntner P, Melamed ML, Fox CS. Serum phosphorus predicts incident chronic kidney disease and end-stage renal disease. Nephrol Dial Transplant. 2011;26(9):2885-2890. (PubMed)
19. Foley RN, Collins AJ, Herzog CA, Ishani A, Kalra PA. Serum phosphate and left ventricular hypertrophy in young adults: the coronary artery risk development in young adults study. Kidney Blood Press Res. 2009;32(1):37-44. (PubMed)
20. Shuto E, Taketani Y, Tanaka R, et al. Dietary phosphorus acutely impairs endothelial function. J Am Soc Nephrol. 2009;20(7):1504-1512. (PubMed)
21. Tuttle KR, Short RA. Longitudinal relationships among coronary artery calcification, serum phosphorus, and kidney function. Clin J Am Soc Nephrol. 2009;4(12):1968-1973. (PubMed)
22. de Boer IH, Rue TC, Kestenbaum B. Serum phosphorus concentrations in the third National Health and Nutrition Examination Survey (NHANES III). Am J Kidney Dis. 2009;53(3):399-407. (PubMed)
23. Isakova T, Wahl P, Vargas GS, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79(12):1370-1378. (PubMed)
24. Isakova T, Xie H, Yang W, et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA. 2011;305(23):2432-2439. (PubMed)
25. Palmer SC, Hayen A, Macaskill P, et al. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: a systematic review and meta-analysis. JAMA. 2011;305(11):1119-1127. (PubMed)
26. Li YC. Vitamin D: roles in renal and cardiovascular protection. Curr Opin Nephrol Hypertens. 2012;21(1):72-79. (PubMed)
27. Hruska KA, Mathew S, Lund R, Qiu P, Pratt R. Hyperphosphatemia of chronic kidney disease. Kidney Int. 2008;74(2):148-157. (PubMed)
28. Jono S, McKee MD, Murry CE, et al. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000;87(7):E10-17. (PubMed)
29. Rennenberg RJ, Kessels AG, Schurgers LJ, van Engelshoven JM, de Leeuw PW, Kroon AA. Vascular calcifications as a marker of increased cardiovascular risk: a meta-analysis. Vasc Health Risk Manag. 2009;5(1):185-197. (PubMed)
30. Chang AR, Lazo M, Appel LJ, Gutierrez OM, Grams ME. High dietary phosphorus intake is associated with all-cause mortality: results from NHANES III. Am J Clin Nutr. 2014;99(2):320-327. (PubMed)
31. Calvo MS, Park YK. Changing phosphorus content of the US diet: potential for adverse effects on bone. J Nutr. 1996;126(4 Suppl):1168S-1180S. (PubMed)
32. Calvo MS. Dietary considerations to prevent loss of bone and renal function. Nutrition. 2000;16(7-8):564-566. (PubMed)
33. Kemi VE, Karkkainen MU, Rita HJ, Laaksonen MM, Outila TA, Lamberg-Allardt CJ. Low calcium:phosphorus ratio in habitual diets affects serum parathyroid hormone concentration and calcium metabolism in healthy women with adequate calcium intake. Br J Nutr. 2010;103(4):561-568. (PubMed)
34. Grimm M, Muller A, Hein G, Funfstuck R, Jahreis G. High phosphorus intake only slightly affects serum minerals, urinary pyridinium crosslinks and renal function in young women. Eur J Clin Nutr. 2001;55(3):153-161. (PubMed)
35. Pinheiro MM, Schuch NJ, Genaro PS, Ciconelli RM, Ferraz MB, Martini LA. Nutrient intakes related to osteoporotic fractures in men and women--the Brazilian Osteoporosis Study (BRAZOS). Nutr J. 2009;8:6. (PubMed)
36. Calvo MS, Tucker KL. Is phosphorus intake that exceeds dietary requirements a risk factor in bone health? Ann N Y Acad Sci. 2013; 1301:29-35. (PubMed)
37. Minerals. Drug Facts and Comparisons. St. Louis: Facts and Comparisons; 2000:27-51.
38. Cervelli MJ, Shaman A, Meade A, Carroll R, McDonald SP. Effect of gastric acid suppression with pantoprazole on the efficacy of calcium carbonate as a phosphate binder in haemodialysis patients. Nephrology (Carlton). 2012;17(5):458-465. (PubMed)
39. Zhang D, Maalouf NM, Adams-Huet B, Moe OW, Sakhaee K. Effects of sex and postmenopausal estrogen use on serum phosphorus levels: a cross-sectional study of the National Health and Nutrition Examination Survey (NHANES) 2003-2006. Am J Kidney Dis. 2014;63(2):198-205. (PubMed)
40. Bansal N, Katz R, de Boer IH, et al. Influence of estrogen therapy on calcium, phosphorus, and other regulatory hormones in postmenopausal women: the MESA study. J Clin Endocrinol Metab. 2013;98(12):4890-4898. (PubMed)
Hierro
Contenido
Resumen
- El hierro es un componente esencial de cientos de proteínas y enzimas que soportan funciones biológicas esenciales, como el transporte de oxígeno, la producción de energía, y la síntesis de ADN. La hemoglobina, mioglobina, los citocromos y las peroxidasas requieren un grupo hemo que contenga hierro como grupo prostético para sus actividades biológicas. (Más información)
- Debido a que el cuerpo excreta muy poco hierro, el metabolismo del hierro está estrechamente regulado. En particular, la hormona regulatoria del hierro, hepcidina, bloquea la absorción de hierro dietario, promueve la captación celular de hierro, y reduce la biodisponibilidad del hierro cuando las reservas de hierro del cuerpo son suficientes para cumplir con los requisitos. (Más información)
- El estatus del hierro puede ser evaluado en hombres sanos, y mujeres no embarazadas usando exámenes de laboratorio que miden la ferritina del suero (proteína que almacena hierro), el hierro del suero, la capacidad total de fijación del hierro, la saturación de transferrina (el transportador de hierro principal sanguíneo), y del receptor de transferrina soluble. (Más información)
- La deficiencia de hierro resulta debido a un suministro inadecuado de hierro a las células después del agotamiento de las reservas del cuerpo. La anemia microcítica ocurre cuando las reservas de hierro del cuerpo son tan bajas que la síntesis de la hemoglobina y la formación de glóbulos rojos están severamente deterioradas. (Más información)
- La deficiencia de hierro es la deficiencia nutricional más común mundialmente, afectando principalmente niños, mujeres en edad de procrear, mujeres embarazadas, donantes de sangre frecuentes, e individuos con ciertas condiciones médicas. (Más información)
- Mucho de nuestro requerimiento de hierro es satisfecho a través del reciclaje de glóbulos rojos senescentes. La ingesta diaria recomendada (IDR) del hierro es de 8 mg/día para hombres y mujeres postmenopáusicas, 18 mg/día para mujeres premenopáusicas, y 27 mg/día para mujeres embarazadas. (Más información)
- La deficiencia de hierro con o sin anemia en niños ha sido asociada con un desarrollo cognitivo pobre, un rendimiento escolar pobre, y patrones de comportamiento anormales. Evidencia limitada sugiere que la suplementación con hierro no tiene efecto en el desarrollo psicomotor y la función cognitiva de infantes anémicos deficientes de hierro menores de tres años, pero puede mejorar atención y concentración en niños mayores, adolescentes, y mujeres con anemia y/o deficiencia de hierro. (Más información)
- El hierro hemo proviene de la hemoglobina y mioglobina en fuentes alimenticias de origen animal y representa el 10%-15% de la ingesta dietaría total del hierro en consumidores de carne. Sin embargo, debido a que es mejor absorbido que el hierro no hemo encontrado en ambas fuentes de origen animal y vegetal, el hierro hemo contribuye hasta un 40% del hierro total absorbido. (Más información)
- La deposición tóxica de hierro en órganos vitales en pacientes afectados por hemocromatosis hereditaria ha sido asociada con numerosas condiciones crónicas, incluyendo cáncer de hígado y diabetes mellitus tipo 2. La ingesta incrementada de hierro hemo y/o la perdida de la homeostasis del hierro podrían también incrementar el riesgo de enfermedades crónicas en individuos sin desordenes genéticos. (Más información)
- La suplementación con hierro puede causar irritación gastrointestinal, nausea, vomito, diarrea, o constipación, e interfiere con la absorción y eficacia de ciertos medicamentos, incluyendo antibióticos y fármacos usados para tratar la osteoporosis, hipotiroidismo, o los síntomas de la enfermedad de Parkinson. (Más información)
El hierro es el cuarto elemento más abundante en la corteza terrestre y uno de los micronutrientes mejor estudiados en la ciencia de la nutrición (1, 2). Es un elemento clave en el metabolismo de todos los organismos vivos. El hierro existe en dos estados de oxidación biológicamente relevantes: la forma ferrosa (Fe2+) y la forma férrica (Fe3+). El hierro es un componente esencial de cientos de proteínas y enzimas que soportan funciones biológicas esenciales, como el transporte de oxígeno, la producción de energía, la síntesis de ADN, y la replicación y crecimiento celular.
Función
El grupo hemo es un compuesto que contiene hierro encontrado en un cierto número de moléculas biológicamente importantes (Figura 1). Algunas, pero no todas las proteínas dependientes de hierro son proteínas que contienen un grupo hemo (también llamadas hemoproteínas). Las proteínas dependientes de hierro que llevan a cabo un amplio rango de actividades biológicas pueden ser clasificadas de la siguiente manera (1, 3):
- Globina-hemo: proteínas no enzimáticas involucradas en el transporte y almacenamiento de oxigeno (p.ej., hemoglobina, mioglobina, neuroglobina)
- Enzimas hemo involucradas en el transporte de electrones (p.ej., citocromos a, b, f; citocromo c oxidasa) y/o con actividad oxidasa (p.ej., sulfito oxidasa, citocromo P450 oxidasas, mieloperoxidasa, peroxidasas, catalasa, óxido nítrico sintasa endotelial, ciclooxigenasa)
- Proteínas hierro-azufre (Fe-S) agrupadas con actividades oxidorreductasas involucradas en la producción de energía (p.ej., succinato deshidrogenasa, isocitrato deshidrogenasa, NADH deshidrogenasa, aconitasa, xantina oxidasa, ferredoxina-1) o involucradas en la replicación y reparación del ADN (ADN polimerasas, ADN helicasas)
- Enzimas no hemo que requieren hierro como un cofactor para sus actividades catalíticas (p.ej., fenilalanina, tirosina, triptófano, y lisina hidroxilasas; factor inducible por hipoxia (HIF) prolil y las asparaginil hidroxilasas; ribonucleótido reductasa)
- Las proteínas no hemo responsables del transporte y almacenamiento de hierro (p.ej., ferritina, transferrina, haptoglobina, hemopexina, lactoferrina).
Las proteínas que contienen hierro soportan un cierto número de funciones, algunas de las cuales son listadas abajo.
[Figura 1 - Clic para Agrandar]
Transporte y almacenamiento de oxígeno
Las proteínas globina-hemo son proteínas que contienen el grupo hemo involucradas en el transporte y almacenamiento de oxígeno y, en menor medida, pueden actuar como captadores de radicales libres (1). La hemoglobina es la proteína principal encontrada en los glóbulos rojos y representa alrededor de dos-tercios del hierro del cuerpo (3). El papel vital de la hemoglobina en el transporte de oxigeno desde los pulmones al resto del cuerpo proviene de su habilidad única de captar rápidamente al oxígeno durante el corto periodo de tiempo que permanece en contacto con los pulmones, y de liberar este oxígeno según sea necesario durante su circulación a través de los tejidos. La mioglobina actúa en el transporte y almacenaje de oxígeno a corto plazo en las células musculares, ayudando a igualar el suministro y la demanda de oxígeno de los músculos en funcionamiento (1). Una tercera globina llamada neuroglobina es preferencialmente expresada en el sistema nervioso central, pero su función no está bien entendida (4).
Transporte de electrones y metabolismo energético
Los citocromos son enzimas que contienen el grupo hemo y que desempeñan papeles importantes en el transporte de electrones mitocondrial requerido para la producción de energía celular y así la vida. Especialmente, los citocromos sirven como transportadores de electrones durante la síntesis de ATP, el principal compuesto de almacenaje de energía en las células. El citocromo P450 (CYP) es una familia de enzimas involucradas en el metabolismo de una serie de moléculas biológicas importantes (incluyendo ácidos orgánicos: ácidos grasos; prostaglandinas; esteroides; esteroles; y vitaminas A, D, y K), así como en la detoxificación y metabolismo de drogas y contaminantes. Enzimas no hemo que contienen hierro en el ciclo del ácido cítrico, como la NADH deshidrogenasa y la succinato deshidrogenasa, también son fundamentales para el metabolismo energético (1).
Funciones antioxidantes y pro-oxidantes beneficiales
Catalasa y algunas peroxidasas son enzimas que contienen el grupo hemo y que protegen a las células contra la acumulación de peróxido de hidrogeno, una especia reactiva de oxigeno (ERO) potencialmente dañina al catalizar una reacción que convierte al peróxido de hidrógeno en agua y oxígeno. Como parte de la respuesta inmune, algunos glóbulos blancos engullen bacterias y las exponen a las ERO con el objetivo de eliminarlas. La síntesis de una de estas ERO, el ácido hipocloroso, por porte de los neutrófilos, es catalizada por la enzima que contiene un grupo hemo mieloperoxidasa (1).
Además, en la glándula tiroides, la peroxidasa tiroidea que contiene un grupo hemo cataliza la yodación de la tiroglobulina para la producción de hormonas tiroideas tal que el metabolismo tiroideo puede verse afectado en la deficiencia de hierro y en la anemia por deficiencia de hierro (véase Interacción con Nutrientes).
Detección de oxígeno
La insuficiencia de oxígeno (hipoxia), como la que experimentan los que viven en altitudes elevadas o los que sufren de enfermedades pulmonares crónicas, induce respuestas fisiológicas compensatorias, incluyendo un incremento en la formación de glóbulos rojos (eritropoyesis), un incremento del crecimiento de vasos sanguíneos (angiogénesis), y un incremento en la producción de enzimas utilizadas en el metabolismo anaeróbico. La hipoxia es también observada en condiciones patológicas como isquemia/accidente cerebrovascular y trastornos inflamatorios. Bajo condiciones hipóxicas, los factores de transcripción conocidos como factores inducibles por hipoxia (HIF) se unen a elementos de respuesta en los genes que codifican para varias proteínas involucradas en las respuestas compensatorias a la hipoxia e incrementan su síntesis. Enzimas dependientes de hierro de la familia dioxigenasa, HIF prolil hidroxilasas y asparaginil hidroxilasa (factor inhibidor de HIF-1 [FIH-1]), han sido implicadas en la regulación de los HIF. Cuando la tensión de oxígeno celular es adecuada, subunidades HIF-α recién sintetizadas (HIF-1α, HIF-2α, HIF-3α) son modificadas por la enzima prolil hidroxilasa en un proceso dependiente de hierro/2-oxoglutarato que tiene por objetivo la rápida degradación del HIF-α. La hidroxilación de asparaginil inducida por el FIH-1 del HIF-α perjudica el reclutamiento de co-activadores para el complejo transcripcional HIF-α y por lo tanto previene la actividad transcripcional de HIF-α. Cuando la tensión de oxígeno celular cae por debajo del umbral crítico, la prolil hidroxilasa ya no puede marcar a HIF-α para su degradación, permitiendo a la HIF-α unirse a la HIF-1β y formar un factor de transcripción capaz de entrar en el núcleo y unirse a elementos de respuesta hipóxica (ERH) específicos en los genes diana como el gen de la eritropoyetina (EPO) (5).
Replicación y reparación del ADN
Las ribonucleótido reductasas (RNR) son enzimas dependientes de hierro que catalizan la síntesis de desoxirribonucleótidos necesaria para la replicación del ADN. Las RNR también facilitan la reparación del ADN en respuesta al daño del ADN. Otras enzimas esenciales para la síntesis y reparación del ADN, como las ADN polimerasas y ADN helicasas, son proteínas Fe-S agrupadas. Aunque los mecanismos subyacentes aún no están claros, se encontró que el agotamiento de hierro intracelular inhibe la progresión del ciclo, crecimiento y división celular. La inhibición de la síntesis hemo también indujo la detención del ciclo celular en células de cáncer de mama (6).
El hierro es requerido para un cierto número de funciones vitales adicionales incluyendo, el crecimiento, reproducción, curación y función inmune.
Regulación
Regulación sistémica de la homeostasis del hierro
Mientras que el hierro es un mineral esencial, es potencialmente toxico debido a que el hierro libre dentro de la célula puede llevar a la generación de radicales libres causando estrés oxidativo y daño celular. Por lo tanto, es importante para el cuerpo el regular sistemáticamente la homeostasis del hierro. El cuerpo regula estrechamente el transporte de hierro a lo largo de varios compartimientos corporales, tales como los glóbulos rojos en desarrollo (eritroblastos), macrófagos circulantes, células hepáticas (hepatocitos) que almacenan hierro, y otros tejidos (7). Las concentraciones de hierro intracelulares son reguladas de acuerdo con las necesidades de hierro del cuerpo (véase abajo), pero las señales extracelulares también regulan la homeostasis del hierro en el cuerpo a través de la acción de la hepcidina.
La hepcidina, una hormona peptídica principalmente sintetizada por las células del hígado es el regulador clave de la homeostasis sistémica del hierro. La hepcidina puede inducir la internalización y degradación de la proteína de eflujo de hierro, ferroportina-1; la ferroportina-1 regula la liberación de hierro proveniente de ciertas células, como los enterocitos, hepatocitos, y macrófagos recicladores de hierro, al plasma (8). Cuando la concentración de hierro es baja y en situaciones de anemia por deficiencia de hierro, la expresión de hepcidina es mínima, permitiendo la absorción de hierro de la dieta y la movilización de hierro de reservas corporales. En contraste, cuando existen reservas de hierro suficientes o en el caso de una sobrecarga de hierro, la hepcidina inhibe la absorción dietaría de hierro, promueve el secuestro de hierro celular, y reduce la biodisponibilidad del hierro. La expresión de hepcidina es positivamente regulada en condiciones de inflamación y estrés en el retículo endoplásmico y es negativamente regulada en la hipoxia (9). En la hemocromatosis tipo 2B, la deficiencia en hepcidina debido a mutaciones en el gen de la hepcidina, HAMP, causa una acumulación de hierro anormal en los tejidos (véase Sobrecarga de Hierro). Debe notarse que también se piensa que la hepcidina tiene un papel antimicrobiano importante en la respuesta inmune innata al limitar la disponibilidad del hierro a los microorganismos invasores (véase Defensa de retención de hierro durante la infección) (10).
Regulación de hierro intracelular
Los elementos de respuesta al hierro (IRE) son secuencias cortas de nucleótidos encontradas en los ARN mensajeros (ARNm) que codifican para las proteínas clave en la regulación del almacenamiento, transporte y utilización del hierro. Las proteínas reguladoras del hierro (IRP: IRP-1, IRP-2) pueden unirse a los IRE y controlar la estabilidad y traducción del ARNm, de este modo regulando la síntesis de proteínas específicas, como la ferritina (proteína de almacenamiento del hierro) y el receptor-1 de la transferrina (TfR; controla la absorción de hierro celular) (1, 2).
Cuando el suministro de hierro es bajo, el hierro no se hace disponible para su almacenamiento o liberación al plasma. Menos hierro se une a las IRP, permitiendo la unión de IRP a los IRE. La unión de IRP a los IRE localizada en el extremo 5’ de los ARNm que codifican para la ferritina y ferroportina-1 (proteína de eflujo de hierro) inhibe la traducción y la síntesis proteica del ARNm. La traducción del ARNm que codifica para la enzima reguladora clave y la síntesis del grupo hemo en glóbulos rojos inmaduros es también reducida para conservar hierro. En contraste, la unión de IRP a IRE en el extremo 3’ de los ARNm que codifican para el TfR y el transportador de metal divalente-1 (DMT1) estimula la síntesis de transportadores de hierro, incrementando por lo tanto la absorción de hierro en las células (1, 2).
Cuando el suministro de hierro es alto, más hierro se une a las IRP, previniendo por lo tanto la unión de IRP a los IRE en los ARNm. Esto permite una síntesis incrementada de proteínas involucradas en el almacenamiento (ferritina) y eflujo (ferroportina-1) de hierro y una síntesis disminuida de transportadores de hierro (TfR y DMT1) tal que la absorción de hierro es limitada (2). En el cerebro, se evita también que las IRP se unan al extremo 5’ del ARNm de la proteína precursora amiloidea (APP), permitiendo la expresión de APP. La APP estimula el eflujo de hierro de las neuronas mediante la estabilización de ferroportina-1. En la enfermedad de Parkinson (EP), la expresión de APP es suprimida inapropiadamente, resultando en la acumulación de hierro en las neuronas dopaminérgicas (11, 12).
Defensa de retención de hierro durante la infección
El hierro es requerido por los agentes más infecciosos para crecer y esparcirse, como también por el huésped infectado a fin de montar una respuesta inmune efectiva. Hierro suficiente es crítico para la diferenciación y proliferación de linfocitos T y para la generación de especies reactivas de oxigeno (ERO) requerida para matar los patógenos (13). Durante la infección e inflamación, la síntesis de hepcidina es regulada positivamente, las concentraciones de hierro del suero disminuyen, y las concentraciones de ferritina (la proteína que almacena el hierro) incrementan, apoyando la idea de que el secuestro de hierro a partir de los patógenos es un mecanismo de defensa importante del huésped (2).
Reciclaje de hierro
El contenido total de hierro del cuerpo en adultos se estima es de 2.3 g en mujeres y 3.8 g en hombres (2). El cuerpo excreta muy poco hierro, las perdidas basales, la pérdida de sangre menstrual, y la necesidad de hierro para la síntesis de nuevos tejidos son compensados por la absorción diaria de una pequeña proporción de hierro dietario (1 a 2 mg/día). El hierro del cuerpo es principalmente encontrado en los glóbulos rojos, los cuales contienen 3.5 mg de hierro por g de hemoglobina. Los glóbulos rojos senescentes son engullidos por los macrófagos en el bazo, y alrededor de 20 mg de hierro pueden ser recuperados diariamente del reciclado del grupo hemo. El hierro liberado es depositado en la ferritina de los macrófagos del bazo o bien exportado por la ferroportina-1 (proteína de eflujo del hierro) hacia la transferrina (el transportador de hierro sanguíneo principal) que suministra hierro a otros tejidos. El reciclaje de hierro es muy eficiente, con alrededor de 35 mg reciclados diariamente (1).
Evaluación del estatus del hierro
Medidas de los suministros de hierro, hierro circulante, y parámetros hematológicos pueden ser usados para evaluar el estatus del hierro de personas saludables en la ausencia de trastornos inflamatorios, infección parasitaria, y obesidad. Biomarcadores del hierro comúnmente usados incluyen la ferritina del suero (proteína almacenadora de hierro), el hierro del suero, la capacidad total de fijación del hierro (TIBC), y la saturación de transferrina (el principal transportador de hierro sanguíneo; TSAT). El receptor soluble de transferrina (sTfR) es también un indicador del estatus del hierro cuando los suministros de hierro son agotados. En la deficiencia de hierro y en la anemia por deficiencia de hierro, la abundancia de receptores de transferrina en la superficie de células que unen la transferrina diférrica es incrementada para maximizar la absorción de hierro disponible. Por lo tanto, la concentración de sTfR generada por la escisión de los receptores de transferrina unidos a las células es incrementada en la deficiencia de hierro. Los marcadores hematológicos, incluyendo la concentración de hemoglobina, la concentración de hemoglobina corpuscular media, el volumen de glóbulos rojos promedio, y el contenido de hemoglobina reticulocitaria pueden ayudar a detectar anomalías si la anemia está presente (9, 14).
Debe notarse, que la ferritina del suero es una proteína reactiva de fase aguda que es positivamente regulada por la inflamación. Curiosamente, la concentración de hepcidina del suero es también incrementada por la inflamación para limitar la disponibilidad del hierro a los patógenos. Por lo tanto, es importante incluir marcadores de la inflamación (p.ej., proteína-C reactiva, fibrinógeno) cuando se evalúe el estatus del hierro para descartar inflamación (14).
Interacción con Nutrientes
Vitamina A
La deficiencia de vitamina A frecuentemente coexiste con la deficiencia de hierro y puede exacerbar la anemia por deficiencia de hierro al alterar el metabolismo del hierro (15). Se ha mostrado que la suplementación con vitamina A tiene efectos beneficiales en la anemia por deficiencia de hierro y mejora el estatus nutricional del hierro entre niños y mujeres embarazadas (15, 16). La combinación de vitamina A y hierro parece reducir la anemia más efectivamente que el hierro o la vitamina A suplementarios por separado (17). La vitamina A puede facilitar la movilización del hierro de sitios de almacenaje hacia los glóbulos rojos en desarrollo para su incorporación en la hemoglobina (15, 16). Además, estudios en ratas han mostrado que la deficiencia de hierro altera los niveles de vitamina A del plasma e hígado (18, 19).
Cobre
Un estatus del cobre nutricional adecuado es necesario para el metabolismo normal del hierro y la formación de glóbulos rojos. La anemia es un signo clínico de la deficiencia de cobre, y se ha encontrado que el hierro se acumula en los hígados de animales deficientes de cobre, indicando que el cobre (vía ceruloplasmina que contiene cobre) es requerido para el transporte de hierro a la médula ósea para la formación de glóbulos rojos (20). La conexión entre la disponibilidad del cobre y el metabolismo del hierro ha sido también establecida en los humanos; la deficiencia de cobre puede llevar a la deficiencia secundaria de ceruloplasmina y a la sobrecarga de hierro hepático y/o cirrosis (21). La suplementación oral de cobre restaura los niveles normales de ceruloplasmina y la actividad de la ferroxidasa del plasma y corrige el trastorno del metabolismo del hierro en sujetos deficientes de cobre (22). Además, los infantes alimentados con formula alta en hierro absorben menos cobre que los infantes alimentados con formula baja en hierro, sugiriendo que las ingestas de hierro altas pueden interferir con la absorción de cobre en infantes (23).
Zinc
El zinc es esencial para mantener una eritropoyesis adecuada. Cuando la deficiencia de zinc coexiste con la deficiencia de hierro, esta puede exacerbar la anemia por deficiencia de hierro (24). Por otra parte, altas dosis de suplementos de hierro, tomados junto con suplementos de zinc en ayunas, pueden inhibir la absorción de zinc. Cuando son tomados con alimentos, el hierro suplementario parece no inhibir la absorción del zinc. Se ha encontrado que los alimentos fortificados con hierro perjudican la absorción del zinc (25, 26).
Calcio
La presencia de calcio disminuye la absorción de hierro de tanto fuentes no-hemo (es decir, la mayoría de los suplementos y fuentes alimenticias aparte de carnes, aves de corral, y mariscos) como de fuentes hemo (27). Sin embargo, la suplementación con calcio hasta por 12 semanas no se ha encontrado que cambie el estatus nutricional del hierro, probablemente debido a un incremento compensatorio en la absorción del hierro (28). Individuos que toman suplementos de hierro deben tomarlos con dos horas de diferencia de alimentos o suplementos ricos en calcio para maximizar la absorción del hierro.
Yodo
La anemia por deficiencia de hierro severa puede perjudicar el metabolismo tiroideo de las siguientes maneras: (1) al alterar la respuesta de la hormona estimulante de la tiroides de la glándula pituitaria; (2) al reducir la actividad de la peroxidasa tiroidea que cataliza la yodación de la tiroglobulina para la producción de las hormonas tiroideas; y (3) en el hígado al limitar la conversión de T4 a T3 incrementado la rotación de T3 y disminuyendo la unión de T3 a los receptores nucleares (29). Se estima que el bocio y la anemia por deficiencia de hierro coexiste en hasta un 25% de los niños en edad escolar en el oeste y norte de África (30). Un estudio controlado aleatorio en niños deficientes de hierro con bocio mostró una reducción mayor en el tamaño de la tiroides tras el consumo de sal yodada junto con 60 mg/día de hierro cuatro veces por semana en comparación al placebo (31). Intervenciones adicionales han confirmado que corrigiendo la anemia por deficiencia de hierro mejoró la eficacia de la suplementación con yodo para mitigar los desórdenes tiroideos (revisado en 29, 30).
Deficiencia
Niveles de deficiencia de hierro
La deficiencia de hierro es la más común de las deficiencias nutricionales en los EE.UU. y el mundo. Los niveles de deficiencia de hierro están listados a continuación de menos a más severa.
Agotamiento del suministro de hierro
Las reservas de hierro son agotadas, pero el suministro funcional del hierro no es limitado.
Deficiencia de hierro funcional temprana
Antes del desarrollo de la anemia aparente, el suministro funcional de hierro a los tejidos incluyendo la médula ósea, es inadecuado tanto como para deteriorar la eritropoyesis.
Anemia por deficiencia de hierro
Por definición, la anemia está presente cuando las concentraciones de hemoglobina individual caen por debajo de dos desviaciones estándar de distribución del promedio para la hemoglobina en una población sana del mismo sexo y edad y que viven a la misma altitud (32). En el 2013, la anemia por deficiencia de hierro fue la causa principal de años vividos con discapacidades en niños y adolescentes en los 50 países más poblados. Los países con la prevalencia más alta de anemia por deficiencia de hierro en individuos menores de 19 años fueron Afganistán (41%) y Yemen (39.8%); India contribuyó con el mayor número de casos de anemia (147.9 millones). Se estimó la prevalencia en los EE.UU. en un 19.3% con cerca de 16 millones de casos de anemia por deficiencia de hierro en niños y adolescentes (33).
La anemia por deficiencia de hierro ocurre cuando existe hierro inadecuado para soportar la formación de glóbulos rojos normal. La anemia de la deficiencia de hierro es caracterizada usualmente como microcítica e hipocrómica, es decir que los glóbulos rojos son considerablemente más pequeños que los normales y su contenido de hemoglobina es disminuido tal que son más pálidos de lo normal. En esta etapa de la deficiencia de hierro, los síntomas pueden ser el resultado de un insuficiente aporte de oxígeno a causa de la anemia y/o a una función subóptima de las enzimas dependientes de hierro. Cambios en los parámetros hematológicos son usados en el diagnóstico clínico de la anemia por deficiencia de hierro (véase Evaluación del estatus del hierro). Es importante recordar que la deficiencia de hierro no es la única causa de anemia y que el diagnóstico y tratamiento de la deficiencia de hierro únicamente sobre las bases de la anemia puede conducir a un mal diagnóstico o a un tratamiento inapropiado de la causa subyacente (34). Véase también los artículos en Folato y Vitamina B12 para información sobre otras causas nutricionales de la anemia.
Síntomas de la deficiencia de hierro
La mayoría de los síntomas de la deficiencia de hierro son el resultado de la anemia asociada y puede incluir fatiga, ritmo cardíaco acelerado, palpitaciones y respiración acelerada al realizar esfuerzo. La deficiencia de hierro perjudica el desempeño atlético y la capacidad de realizar trabajo físico en varias formas. En la anemia por deficiencia de hierro, el contenido de hemoglobina reducido de los glóbulos rojos resulta en un menor suministro de oxígeno a los tejidos activos. Los niveles disminuidos de mioglobina en las células musculares limitan la cantidad de oxígeno que puede ser entregado a las mitocondrias para el metabolismo oxidativo. El agotamiento de hierro puede también disminuir la capacidad oxidativa del musculo al disminuir el contenido mitocondrial de citocromos y otras enzimas dependientes de hierro requeridas para el transporte de electrones y la síntesis de ATP (véase Función) (35).
Una función tiroidea pobre y una síntesis de la hormona tiroidea deteriorada son propensas a interrumpir la habilidad para mantener una temperatura corporal normal al exponerse al frío en individuos con deficiencia de hierro (véase Función). La deficiencia de hierro puede también alterar a la fagocitosis de neutrófilos y la actividad microbiana y las respuestas proliferativas de linfocitos-T a la infección (1). La anemia por deficiencia de hierro severa puede resultar en uñas quebradizas con forma de cuchara, llagas en las comisuras de la boca, atrofia de las papilas gustativas y una lengua adolorida. En raros casos, la anemia por deficiencia de hierro avanzada puede causar dificultad al tragar debido a la formación de membranas de tejido en la garganta y esófago debido a la degradación de los músculos faríngeos (36). El desarrollo de membranas esofágicas, también conocido como síndrome Plummer-Vinson, puede requerir de una predisposición genética además de la deficiencia de hierro. Se ha mostrado que la deficiencia de hierro y la anemia por deficiencia de hierro en la niñez temprana perjudican el desarrollo psicomotor e inducen alteraciones cognitivas y de comportamiento a largo y corto plazo (revisado en 37). Además, la pica, un trastorno del comportamiento caracterizado por el consumo de objetos no alimenticios, puede ser un síntoma y una causa de la deficiencia de hierro (38).
Individuos en un riesgo incrementado de deficiencia de hierro
Grupos de etapas de la vida con mayores requisitos
Neonatos e infantes de hasta seis meses de edad: Suministros corporales maternos de hierro inadecuados y la anemia durante el embarazo pueden reducir la duración del periodo de gestación y el peso al nacer; recién nacidos prematuros y/o con bajo peso al nacer están en un riesgo mayor de padecer anemia por deficiencia de hierro (14). Las complicaciones del embarazo, incluyendo la preeclampsia y la diabetes mellitus gestacional, pueden llevar también a suministros de hierro bajos en infantes prematuros y a término (14).
La mayoría de los 150 a 250 mg de hierro presentes en un recién nacido de termino completo saludable son acumulados durante el tercer trimestre del embarazo y son suficientes para los primeros cuatro a seis meses de vida (34). Los suministros de hierro son esenciales para los infantes de menos de seis meses de edad porque la leche materna es relativamente pobre en hierro (0.2 mg/L-04 mg/L), y la absorción intestinal del hierro permanece bajo hasta los seis meses de edad. Los requerimientos altos de hierro durante este periodo de índices de crecimiento rápido y sostenido pueden empeorar el déficit de hierro del cuerpo en infantes prematuros (14). Además, una revisión de ensayos controlados aleatorios sugirió que los infantes con una sujeción temprana del cordón umbilical (≤1 min después del nacimiento) son por lo menos dos veces más propensos a ser deficientes de hierro a los tres o seis meses en comparación con aquellos con una sujeción del cordón tardía (39). Sin embargo, recién nacidos saludables de termino completo tienen menos necesidad de fuentes externas de hierro antes de los seis meses de edad (1).
Infantes y niños con edades de entre 6 meses y 3 años: Los suministros de hierro de un infante de termino completo son usualmente suficientes para durar por los primeros meses de vida, pero existe un riesgo incrementado de deficiencia de hierro para los infantes mayores de seis meses (1). Dada la necesidad sostenida de hierro para aumentar la masa de tejido, el volumen de sangre y reponer las reservas de hierro, la ingesta diaria recomendada (IDR) para el hierro es de 11 mg/día para infantes de entre siete a 12 meses de edad, establecida por el Instituto de Medicina de los EE.UU. (véase Tabla 1).
La IDR del hierro es de 7 mg/día para niños de entre 1 y 3 años de edad. Basada en datos de la Encuesta de Examinación Nacional de Salud y Nutrición de EE.UU. (NHANES) de 1999-2002, la prevalencia de una deficiencia de hierro en niños de 12 a 15 meses de edad varia de un 6.6%-15.2%, y la prevalencia de la anemia por deficiencia de hierro es de 0.9%-4.4%, dependiendo de la etnicidad y estatus económico (14).
Debe notarse que la Organización Mundial de la Salud (OMS) y la Academia Americana de Pediatras recomiendan una detección universal para la anemia a la edad de un año. Sin embargo, un reporte reciente por la Fuerza de Trabajo de Servicios Preventivos de los EE. UU. (USPSTF por sus siglas en inglés (US Preventive Services Task Force)) declaró que existe insuficiente evidencia para evaluar los beneficios contra los perjuicios de la detección (34, 40).
Adolescentes: El inicio de la adolescencia es un periodo de crecimiento rápido. La pérdida de sangre que acompaña a la menstruación en mujeres adolescentes se suma al requerimiento incrementado de hierro de la adolescencia (1). La IDR del hierro es de 11 mg/día y 15 mg/día para adolescentes varones y mujeres respectivamente (véase Tabla 1).
Mujeres no embarazadas en edad de procrear: Basado en datos del NHANES del 2003-2006, el porcentaje de mujeres estadounidenses con dos o tres marcadores del estatus de hierro (es decir, hemoglobina, ferritina, y % de saturación de transferrina) por debajo de los valores de corte para la deficiencia fue 9.8% en mujeres no embarazadas (41).
El uso de anticonceptivos orales disminuye la pérdida de sangre menstrual y está por lo tanto asociado con el mejoramiento del estatus del hierro en comparación a los dispositivos intrauterinos (bobina de cobre) (1).
El amamantamiento está asociado con necesidades dietarías menores de hierro, permitiendo la repleción de las reservas de hierro agotadas durante el embarazo y parto. Sin embargo, la repleción de hierro puede ser incompleto en mujeres con partos frecuentes las cuales están por lo tanto en un riesgo incrementado de deficiencia de hierro (41).
Mujeres embarazadas: El requerimiento de hierro es significantemente incrementado durante el embarazo debido a un incremento en la utilización de hierro por el feto y placenta en desarrollo, como también por la expansión del volumen sanguíneo (42). Un análisis de datos del NHANES 2005-2006 encontró que el 18.1% de las mujeres embarazadas (edad promedio, 27.5 años) estaban deficientes de hierro, evaluado por la relación logarítmica del receptor de transferrina soluble a la ferritina del suero (43). La prevalencia de la deficiencia de hierro fue mayor durante el segundo (20.7%) y tercer (29.7%) trimestre en comparación con el primer trimestre (4.5%) de gestación. Además, se encontró que la deficiencia de hierro en el embarazo era más prevalente en mexicanas (23.6%) y afroamericanas (29.6%) que en americanas caucásicas no hispanas (13.9%) (43).
Individuos con pérdida crónica de sangre
El sangramiento crónico o la pérdida aguda de sangre puede resultar en deficiencia de hierro. Un milímetro (ml) de sangre con una concentración de hemoglobina de 150 g/L contiene 0.5 mg de hierro. Es así como la pérdida crónica de pequeñas cantidades de sangre puede derivar en una deficiencia de hierro.
Infestación parasítica: Una causa común de la pérdida crónica de sangre y de la deficiencia de hierro en países en vías de desarrollo es la infección con parásitos intestinales (44).
Donación frecuente de sangre: Las personas que donan sangre con frecuencia, especialmente las mujeres en menstruación pudiesen necesitar incrementar su ingesta de hierro para prevenir su deficiencia ya que cada 500 ml de sangre donada contienen entre 200 y 250 mg de hierro (45, 46).
Ejercicio regular intenso: Se ha encontrado que las pérdidas diarias de hierro son mayores en atletas involucrados en entrenamientos de resistencia intensos. Esto puede deberse a la expansión de la masa de células sanguíneas y masa muscular, al incremento en el sangrado microscópico del tracto gastrointestinal (con el uso regular de fármacos antiinflamatorios) o al aumento en la fragilidad y hemólisis de glóbulos rojos (47). La Junta de Nutrición y Alimentos estima que el requerimiento promedio de hierro puede ser un 30% más alto para aquellos que realizan ejercicio intenso con regularidad (25).
Individuos con absorción incrementada de hierro
Enfermedad celiaca: La enfermedad celiaca (celiaquía) es un trastorno autoinmune el cual se estima ocurre en 1% de la población. Cuando las personas con enfermedad celiaca consumen alimentos o productos que contienen gluten, la respuesta del sistema inmune daña la mucosa intestinal, lo que puede resultar en malabsorción de nutrientes y anemia por deficiencia de hierro (48).
Gastritis atrófica: Esta condición esta usualmente asociada con la presencia de anticuerpos dirigidos hacia las células estomacales y ha sido implicada en la anemia perniciosa (véase el artículo en Vitamina B12). La gastritis atrófica simultáneamente perjudica la absorción de vitamina B12 y hierro; sin embargo, en mujeres que menstrúan, la deficiencia de hierro puede ocurrir años antes del agotamiento de las reservas corporales de vitamina B12 (47).
Infección con Helicobacter pylori: La infección con H. pylori está asociada con la anemia por deficiencia de hierro, especialmente en niños, incluso en la ausencia de sangrado gastrointestinal. Datos del NHANES 2000-2001 en individuos mayores de tres años mostro que la presencia de deficiencia de hierro (basada en las concentraciones de ferritina del suero) era 40% más prevalente en aquellos infectados con H. pylori que en los individuos sin H. pylori (49). El sangrado intestinal oculto y la competición por hierro dietario de parte de las bacterias puede explicar la deficiencia de hierro en individuos infectados. Además, la infección con Helicobacter pylori puede también jugar un papel en la patogénesis de la gastritis atrófica (47).
Enfermedades intestinales inflamatorias (EII): la anemia por deficiencia de hierro es comúnmente reportada entre pacientes con EII (p.ej., la colitis ulcerosa, enfermedad de Crohn), probablemente debido a tanto la absorción intestinal alterada del hierro como a la pérdida de sangre de la mucosa ulcerada (50).
Cirugía de derivación gástrica: algunos tipos de cirugía de derivación (bariátrica) gástrica incrementan el riesgo de la deficiencia de hierro al causar malabsorción de hierro, entre otros nutrientes (51).
Obesidad: Una asociación inversa entre el peso corporal y el estatus del hierro ha sido reportada en varios estudios basados en la observación en niños y adultos (52, 53). Una mayor expresión de la hepcidina en personas obesas puede perjudicar la absorción de hierro a pesar de una ingesta dietaría adecuada de hierro. La pérdida de peso podría disminuir la concentración de hepcidina del suero y mejorar el estatus del hierro en individuos obesos (9).
Anemia por enfermedad crónica: la inflamación crónica y aguda puede conducir a concentraciones circulantes de hierro anormalmente bajas y al desarrollo de la anemia. Este tipo de anemia por inflamación, también conocida como anemia por enfermedad crónica (AEC), es comúnmente observada en desordenes inflamatorios, cáncer, enfermedad crítica, trauma, infección crónica, e infestación parasítica. Se piensa que la anemia se desarrolla porque la absorción de hierro dietario y la movilización del hierro de las reservas del cuerpo son inhibidas por la regulación positiva de la hepcidina inducida por la inflamación (véase también Regulación sistémica de la homeostasis del hierro) (9).
Otras causas de la deficiencia de hierro
Dieta vegetariana con fuentes inadecuadas de hierro: Debido a que el hierro de origen vegetal (hierro no hemo) es menos eficientemente absorbido que aquel de origen animal (véase Fuentes), la Junta de Nutrición y Alimentos (JNA) del Instituto de Medicina (IOM) de los EE.UU. estimó que la biodisponibilidad del hierro proveniente de dietas vegetarianas fue de solo 10% en comparación al 18% proveniente de una dieta occidental mixta. Por lo tanto, la ingesta diaria recomendada (IDR) del hierro para los individuos que consumen una dieta vegetariana completamente puede ser 1.8 veces mayor que la IDR para los no vegetarianos (25). Sin embargo, una dieta vegetariana no parece estar asociada con un riesgo incrementado de deficiencia de hierro cuando esta incluye granos enteros, legumbres, nueces, semillas, frutos secos, cereal fortificado con hierro, y vegetales de hoja verde (véase Fuentes) (54).
Enfermedad renal crónica (ERC): Las pérdidas de hierro en pacientes con ERC son debido a la significante pérdida de sangre gastrointestinal (1.2 L de sangre perdida/año correspondiente a ~400 mg de hierro/año) en comparación con individuos con una función renal normal (0.83 mL de sangre perdida/año correspondiente a ~100 mg hierro/año). Las pérdidas de sangre estimadas son incluso mayores en pacientes en hemodiálisis, y las pérdidas de hierro pueden ser de entre 1,000 a 2,000 mg/año o más. La inflamación persistente en pacientes con ERC puede también contribuir al suministro inadecuado de hierro para la formación de glóbulos rojos a pesar de existir reservas de hierro adecuadas (55).
La Ingesta Diaria Recomendada (IDR)
La IDR fue revisada en el 2001 y se basa en la prevención de una deficiencia de hierro y en la mantención de depósitos de hierro adecuados, en individuos que consumen una dieta mixta (Tabla 1; 25).
| Etapa de la Vida | Edad | Machos (mg/día) | Hembras (mg/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 0.27 (IA) | 0.27 (IA) |
| Infantes | 7-12 meses | 11 | 11 |
| Niños | 1-3 años | 7 | 7 |
| Niños | 4-8 años | 10 | 10 |
| Niños | 9-13 años | 8 | 8 |
| Adolescentes | 14-18 años | 11 | 15 |
| Adultos | 19-50 años | 8 | 18 |
| Adultos | 51 años y más | 8 | 8 |
| Embarazo | Todas las edades | - | 27 |
| Período de lactancia | 18 años y menores | - | 10 |
| Período de lactancia | 19 años y más | - | 9 |
Prevención de Enfermedades
La prevención o alivio de la deficiencia de hierro o de la anemia por deficiencia de hierro puede limitar el impacto de la insuficiencia de hierro y la eritropoyesis defectuosa en las condiciones de salud y enfermedades siguientes.
Deterioro del desarrollo psicomotor, cognitivo, e intelectual en los niños
El hierro es crítico para el desarrollo del sistema nervioso central, y se piensa que la deficiencia de hierro es especialmente perjudicial durante los períodos prenatal y postnatal temprano. Las enzimas dependientes de hierro son requeridas para la mielinización nerviosa, la síntesis de neurotransmisores, y el metabolismo energético neuronal normal (56). La mayoría de los estudios basados en la observación han encontrado relaciones entre la deficiencia de hierro — con o sin anemia — en niños y el desarrollo cognitivo pobre, el rendimiento escolar escaso y patrones de comportamiento anormales (revisado en 37). Si los déficits psicomotores y mentales pueden ser atribuidos a la falta de hierro, solo, o al efecto combinado de la deficiencia de hierro y bajas concentraciones de hemoglobina — como en la anemia por deficiencia de hierro y la anemia por inflamación — en la niñez temprana permanece sin estar claro (14).
Una revisión sistemática reciente de seis ensayos controlados con placebo de menor magnitud (publicados entre 1978 y 1989) en niños con anemia por deficiencia de hierro menores de 27 meses no encontró evidencia convincente de que la terapia con hierro (por menos de 11 días) tuviera algún efecto consistente sobre las mediciones del desarrollo psicomotor y mental en los 30 días posteriores al inicio del tratamiento (57). Solo un ensayo aleatorio, doble ciego, en infantes anémicos deficientes de hierro examinó el impacto de la terapia con hierro por cuatro meses y encontró un beneficio significante en los índices de desarrollo cognitivo que necesitan ser confirmado (58). Una revisión de cinco ensayos controlados aleatorios en infantes deficientes de hierro no anémicos (0-9 meses de edad) sugirió un mejoramiento en el desarrollo psicomotor (pero no mental) durante los primeros 18 meses de vida (59). La suplementación en la infancia temprana (4 a 6 meses) también falló en demostrar algún efecto a largo plazo en el rendimiento cognitivo y en el rendimiento escolar a la edad de 9 años en comparación al placebo (60). En la actualidad, evidencia que apoye algún beneficio de la terapia con hierro sobre los resultados del neurodesarrollo en infantes con deficiencia de hierro, con o sin anemia, permanece limitada.
La terapia con hierro podría ser más efectiva en mejorar los resultados cognitivos en niños mayores con anemia y/o deficiencia de hierro. Una revisión sistemática de 17 ensayos controlados aleatorios encontró que la suplementación con hierro no tuvo efecto en el desarrollo mental de niños menores de 27 meses, pero modestamente mejoró las puntuaciones del desarrollo mental en niños mayores de siete años de edad (61). Un meta-análisis más reciente de ensayos controlados aleatorios en niños mayores de seis años, adolescentes, y mujeres con deficiencia de hierro, anemia, y anemia por deficiencia de hierro sugirió que el hierro suplementario pudo mejorar la atención y concentración independientemente del estatus de hierro de los participantes (62). Una mejora potencial en las puntuaciones del coeficiente intelectual con la terapia de hierro fue también reportada en participantes anémicos independientemente de sus estatus de hierro. No se observaron beneficios adicionales con respecto a las medidas del rendimiento de la memoria, la función psicomotora, y logros escolares.
Alteraciones en las funciones cerebrales debido a la deficiencia de hierro son más propensas a ser resistentes a la terapia con hierro cuando estas ocurren en la niñez temprana. Consecuencias a largo plazo de la deficiencia de hierro en la vida temprana pueden incluir logros socioeconómicos pobres y un riesgo incrementado de ciertas psicopatologías, incluyendo ansiedad, depresión, y esquizofrenia (56).
Resultados adversos del embarazo
Estudios epidemiológicos aportan fuerte evidencia de una asociación entre la anemia severa en mujeres embarazadas y los resultados adversos del embarazo, como el bajo peso al nacer, nacimiento prematuro y la mortalidad materna y neonatal (63). Aunque la deficiencia de hierro puede ser un factor contribuyente principal de la anemia severa, evidencia de que la anemia por deficiencia de hierro cause resultados pobres en el embarazo es aún escaza. Además, se demostró que la suplementación con hierro durante el embarazo mejoró el estatus del hierro y los parámetros hematológicos en mujeres, pero falló en reducir significantemente los resultados adversos del embarazo, incluyendo peso bajo al nacer y/o nacimiento prematuro, muerte neonatal, y anomalías congénitas (64). Además, la suplementación rutinaria durante el embarazo no tuvo efecto en la duración de la gestación o en los puntajes Apgar (40). Sin embargo, la mayoría de los expertos consideran al control de la anemia materna como una parte importante del cuidado de salud neonatal, y el Instituto de Medicina recomienda someterse a la detección de anemia en cada trimestre del embarazo (65).
El requerimiento de hierro se incrementa mayormente en el segundo y tercer trimestre, y la IDR para mujeres embarazadas es de 27 mg/día de hierro (véase La Ingesta Diaria Recomendada) (25). El Colegio Americano de Obstetricias y Ginecólogos recomienda la detección de anemia en todas las mujeres embarazadas y recomienda la suplementación de hierro cuando es necesaria (66). Sin embargo, la Fuerza de Trabajo de Servicios Preventivos de los EE. UU. (40) y la Academia Americana de Médicos de Familia (67) consideran que la evidencia para evaluar los perjuicios y beneficios de la detección de anemia por deficiencia de hierro y de la suplementación con hierro durante el embarazo es escaza.
En regiones donde la malaria es endémica, sin embargo, la suplementación con hierro puede mejorar los resultados del embarazo cuando es proporcionada en conjunto con medidas de prevención y manejo de la malaria. Dos ensayos aleatorios controlados con placebo recientes fallaron en encontrar un incremento del riesgo de infección con malaria en tanto mujeres embarazadas deficientes de hierro como aquellas repletas de hierro suplementadas con hierro, apoyando el uso de la suplementación universal con hierro en países donde la malaria es endémica los cuales adoptan tratamientos intermitentes preventivos para la malaria (68, 69).
Toxicidad por plomo
Los niños crónicamente expuestos al plomo, incluso en pequeñas cantidades, son más propensos a desarrollar dificultades de aprendizaje, problemas de comportamiento, y tienen bajos cocientes intelectuales. Déficits en el crecimiento y desarrollo neurológico pueden ocurrir en los infantes de mujeres expuestas al plomo durante el embarazo y periodo de lactancia. En los adultos, la toxicidad por plomo puede resultar en daño renal y presión sanguínea alta. Aunque el uso de plomo en pinturas, gasolina, y latas para alimentos ha sido descontinuado en los EE.UU., la toxicidad por plomo continúa siendo un problema de salud importante, especialmente en niños que viven en el centro de las ciudades (70). En el 2012, el Centro para el Control y Prevención de Enfermedades Estadounidense estableció el valor de referencia para la concentración de plomo en la sangre en 5 microgramos por decilitro (μg/dL) para identificar niños en riesgo. Sin embargo, no existe una concentración de plomo en la sangre conocida por debajo de la cual niños pueden estar 100% seguros (71).
La deficiencia de hierro y el envenenamiento por plomo comparten un cierto número de los mismos factores de riesgo, incluyendo un estatus socioeconómico bajo, grupos étnicos minoritarios y la residencia en áreas urbanas. La deficiencia de hierro puede incrementar el riesgo de envenenamiento por plomo en niños, especialmente al incrementar la absorción intestinal de plomo a través del transportador intestinal DMT1 (72). Sin embargo, el uso de la suplementación con hierro en el envenenamiento por plomo podría ser reservado para niños verdaderamente deficientes de hierro o para niños repletos de hierro con exposición crónica al plomo (p.ej., aquellos que viven en una vivienda expuesta al plomo) (72).
Tratamiento de Enfermedades
Síndrome de piernas inquietas
El síndrome de piernas inquietas (SPI; también llamado la enfermedad de Willis-Ekbom) es un trastorno de movimiento neurológico de etiología desconocida. Las personas con SPI experimentan sensaciones desagradables las que resultan en una irresistible urgencia por mover sus piernas y un alivio transitorio con el movimiento. Estas sensaciones son más comunes al descasar y con frecuencia interfieren con el sueño (73). La prevalencia del SPI es más alta en mujeres que en hombres e incrementa con la edad (74). Este síndrome parece ser heredado en aproximadamente el 50% de los pacientes, pero también ha sido relacionado con la insuficiencia renal crónica (73). La deficiencia de hierro puede estar involucrada en el desarrollo de SPI, posiblemente al afectar la actividad de la tirosina hidroxilasa, una enzima dependiente de hierro que limita la velocidad en la síntesis del neurotransmisor, dopamina (74). El manejo del SPI incluye la terapia con hierro y el uso de fármacos como los agonistas dopaminérgicos (73). Evidencia clínica reciente es insuficiente para evaluar si la terapia con hierro puede ayudar a aliviar alguno de los síntomas del SPI (74). Sin embargo, la Junta Asesora Médica de la Fundación del Síndrome de la Enfermedad de Willis-Ekbom sugiere que el estatus del hierro debe ser evaluado en todos los pacientes con SPI, y que la terapia son hierro debe intentarse caso por caso en aquellos que podrían beneficiarse de ella (73).
Fuentes
Fuentes alimenticias
La cantidad de hierro de los alimentos o suplementos que es absorbida y usada por el cuerpo es influenciada por el estatus nutricional del hierro del individuo y si el hierro está o no en la forma hemo. Debido a que es absorbido por un mecanismo diferente al del hierro no hemo, el hierro hemo es absorbido más rápido y su absorción es menos afectada por otros factores dietarios (2). En un intento por mejorar el estatus de hierro del cuerpo, la absorción del hierro es mayor en individuos que están anémicos o deficientes de hierro en comparación con individuos repletos de hierro.
Hierro hemo
El hierro hemo proviene principalmente de la hemoglobina y mioglobina en la carne, aves de corral y pescado. Aunque el hierro hemo representa sólo entre el 10% y 15% del hierro encontrado en la dieta, puede aportar hasta un tercio del hierro dietario total absorbido (54). La absorción del hierro hemo esta menos influenciada por otros factores dietarios que el hierro no hemo (27).
Hierro no hemo
Las plantas, productos lácteos, la carne y las sales de hierro agregadas a los alimentos y suplementos son todas fuentes de hierro no hemo. La absorción del hierro no hemo está fuertemente influenciada por potenciadores e inhibidores presentes en una misma comida (27).
Potenciadores de la absorción de hierro no hemo
- Vitamina C (ácido ascórbico): La vitamina C potencia fuertemente la absorción del hierro no hemo al reducir el hierro férrico dietario (Fe3+) a hierro ferroso (Fe2+), y al formar un complejo absorbible de hierro-ácido ascórbico (75).
- Otros ácidos orgánicos: Los ácidos cítrico, málico, tartárico y láctico tienen algunos efectos estimulantes sobre la absorción de hierro no hemo (1).
- Carne, aves de corral, y pescado: Además de aportar hierro hemo altamente absorbible, la carne, el pescado y las aves de corral también estimulan la absorción de hierro no hemo. El mecanismo para esta estimulación de la absorción del hierro no hemo aún no está claro (1, 25).
Inhibidores de la absorción de hierro no hemo
- Acido fítico (fitato): El ácido fítico, presente en legumbres, granos enteros, nueces y semillas, inhibe la absorción de hierro no hemo, probablemente al unirse a él. Pequeñas cantidades de ácido fítico (5 a 10 mg) pueden reducir la absorción de hierro no hemo en un 50%. Se ha demostrado que la absorción de hierro de legumbres, como frijoles de soya, frijoles negros, lentejas, frijoles mung, y guisantes, es tan baja como 2% (25). La preparación de alimentos, incluyendo el remojo, germinación, fermentación y cocción, puede ayudar a eliminar o degradar el ácido fítico (27).
- Compuestos polifenólicos:Los compuestos polifenólicos en el café, té negro, y te de hiervas pueden inhibir marcadamente la absorción del hierro no hemo (76). Este efecto puede ser reducido por la presencia de vitamina C (27, 77).
- Proteína de soya: La proteína de soya, como la encontrada en el tofu, tiene un efecto inhibidor sobre la absorción de hierro que es solo parcialmente relacionada con su contenido de ácido fítico (27, 77).
- Calcio: El calcio parece afectar la absorción de hierro de tanto fuentes hemo como no hemo. Sin embargo, su efecto parece ser limitado cuando uno consume una amplia variedad de alimentos con niveles variados de promotores e inhibidores de la absorción de hierro (27).
Encuestas nacionales en los EE.UU. han indicado que la ingesta dietaría promedio de hierro es de 16 a 18 mg/día en hombres, 12 mg/día en mujeres pre- y postmenopáusicas, y alrededor de 15 mg/día en mujeres embarazadas (25). De esta manera, la mayoría de las mujeres premenopáusicas y embarazadas en los EE.UU. consumen menos que la IDR del hierro, y muchos hombres consumen más que la IDR (véase La Ingesta Diaria Recomendada). En los EE.UU., la mayoría de los productos con granos se fortifican con hierro no hemo. El contenido de hierro de algunos alimentos relativamente ricos en hierro se muestra en miligramos (mg) en la Tabla 2. Para más información sobre el contenido de nutrientes de alimentos específicos, revise la base de datos de composición de los alimentos de la USDA.
Suplementos
Los suplementos de hierro están indicados para la prevención y tratamiento de la deficiencia de hierro y anemia por deficiencia de hierro. Los individuos que no están en riesgo de una deficiencia de hierro (p.ej., hombres adultos y mujeres postmenopáusicas) no deben consumir suplementos de hierro sin la evaluación médica apropiada. Una serie de suplementos de hierro se encuentran disponibles, y diferentes formas aportan distintas proporciones de hierro elemental. El sulfato ferroso heptahidrato es 20% hierro elemental; el sulfato ferroso monohidrato es un 33% hierro elemental; el gluconato ferroso es un 12% hierro elemental; y el fumarato ferroso es un 33% hierro elemental. Salvo que se indique lo contrario, el hierro discutido en este artículo es hierro elemental.
Sobrecarga de Hierro
La desregulación de la absorción intestinal de hierro resulta en una sobrecarga de hierro debido a que el cuerpo no puede excretar el exceso de hierro (2). Sin embargo, la sobrecarga de hierro debido a la suplementación de hierro es muy rara en individuos sanos sin una predisposición genética. Varios trastornos genéticos pueden llevar a la acumulación patológica del hierro en el cuerpo a pesar de la ingesta normal de hierro. La suplementación en individuos que no son deficientes de hierro debe ser evitada debido a la frecuencia de enfermedades hereditarias no detectadas y a preocupaciones recientes con respecto a los efectos más sutiles de la ingesta crónica de hierro en exceso (véase Enfermedades asociadas con la sobrecarga de hierro).
Enfermedades hereditarias de la sobrecarga de hierro
Hemocromatosis hereditaria
La hemocromatosis hereditaria (HH) hace referencia a trastornos autosómicos recesivos de aparición tardía del metabolismo del hierro que resultan en una acumulación de hierro en el hígado, corazón, otros tejidos. Estos trastornos pueden llevar a cirrosis hepática, diabetes mellitus, cardiomiopatía (daño del músculo cardíaco), hipogonadismo, artropatía (problemas en las articulaciones), y un incremento en la pigmentación de la piel (revisado en 78). Existen cuatro tipos principales de HH, los que se clasifican de acuerdo al gen específico que se encuentra mutado. El tipo más común de HH, denominado Tipo 1 o HH ligada a HFE, es el resultado de mutaciones en el gen HFE (79, 80). La mayoría de los casos de HH Tipo 1 son homocigotos para la mutación C282Y G>A (rs1800560) en el gen HFE. Otra mutación encontrada en el 4% de los pacientes con HH Tipo 1 es H63D C>G (rs1799945) en el gen HFE. Se piensa que la proteína codificada por el gen HFE juega un papel en la regulación de la absorción intestinal de hierro dietario y en la detección de las reservas de hierro del cuerpo (81). Las mutaciones del gen HFE están asociadas con una absorción celular incrementada de hierro. Con un inicio típico de la enfermedad antes de los 30 años, la hemocromatosis juvenil (HH Tipo 2) es mas mucho más rara que la HH Tipo 1 y resulta de mutaciones genéticas que afectan la función de la hemojuvelina (Tipo 2A) o de la hepcidina (Tipo 2B) (82). La HH Tipo 3 resulta de mutaciones en el gen del receptor 2 de la transferrina (TFR2), y la HH Tipo 4 (también llamada enfermedad de la ferroportina) resulta de mutaciones en el gen que codifica para la ferroportina-1 (SLC40A1), una proteína importante en la exportación de hierro de las células (véase Regulación). La HH Tipo 4 es el segundo trastorno de sobrecarga de hierro más común después de la HH Tipo 1 (78).
La sobrecarga de hierro en la HH es tratada por flebotomía, con la remoción de 500 ml de sangre cada vez, en intervalos determinados según la severidad de la sobrecarga de hierro. La terapia de quelación es una opción alterna para agotar el hierro en pacientes con HH que no pueden someterse a un tratamiento de flebotomía. A los individuos con HH se les aconseja que eviten el hierro suplementario, aunque generalmente no se les recomienda evitar alimentos ricos en hierro. Regímenes de altas dosis de vitamina C pueden empeorar la sobrecarga de hierro en pacientes con HH (75). El consumo de alcohol se desaconseja fuertemente debido al incremento en el riesgo de cirrosis hepática (83). Pruebas genéticas, las cuales requieren de una muestra de sangre, están disponibles para quienes puedan estar en riesgo de HH, como, por ejemplo, los individuos con historial familiar de hemocromatosis.
Otras condiciones hereditarias
Otros trastornos genéticos que conducen a la sobrecarga de hierro incluyen la aceruloplasminemia, hipotransferrinemia, la ataxia de Friedreich y la porfiria cutánea tardía (2).
Enfermedades de sobrecarga de hierro adquirida
La sobrecarga de hierro puede desarrollarse en individuos con varias anemias hereditarias que no son causadas por la deficiencia de hierro. La absorción dietaría excesiva de hierro puede ocurrir en respuesta a los continuos esfuerzos del cuerpo para formar glóbulos rojos. La beta-talasemia está caracterizada por la síntesis defectuosa de hemoglobina A debido a mutaciones en el gen de la β-globina. Pacientes afectados por la talasemia intermedia no requieren transfusión de sangre como lo hacen aquellos afectados por la forma más severa de la enfermedad (llamada talasemia mayor), sin embargo, desarrollan una sobrecarga de hierro debido a la absorción intestinal incrementada de hierro (84). Otros pacientes anémicos en riesgo de una sobrecarga de hierro incluyen aquellos con anemia sideroblástica, anemia hemolítica, deficiencia de piruvato quinasa y talasemia mayor, especialmente porque son tratados con numerosas transfusiones. Pacientes con esferocitosis hereditaria y talasemia menor usualmente no desarrollan sobrecargas de hierro a menos que sean mal diagnosticadas como si tuvieran una deficiencia de hierro y sean tratados con altas dosis de hierro por muchos años. La sobrecarga de hierro ha sido también asociada con la hemodiálisis y las enfermedades hepáticas crónicas (metabólicas, virales, y alcohólicas) (2).
Enfermedades asociadas con la sobrecarga de hierro
La deposición toxica de hierro en órganos vitales en la hemocromatosis hereditaria (HH) ha sido asociada con una incidencia incrementada de cáncer de hígado, diabetes mellitus tipo 2, y enfermedades neurodegenerativas. La sobrecarga de hierro podría también incrementar el riesgo de enfermedades crónicas en individuos sin HH. A pesar de todo, aún no se conoce completamente si la acumulación de tejido de hierro en aquellos que no se ven afectados por trastornos genéticos se debe a la ingesta alta de hierro en la dieta. (1).
Cáncer
La hemocromatosis hereditaria que está caracterizada por la acumulación hepática anormal de hierro es un factor de riesgo para el cáncer de hígado (carcinoma hepatocelular; CHC). Se piensa que la acumulación de hierro funciona como un carcinógeno al incrementar el estrés oxidativo que causa daño a los lípidos, proteínas, y al ADN. Un meta-análisis de nueve estudios basados en la observación encontró un riesgo de CHC incrementado con la mutación C282Y en el gen HFE de los participantes saludables y pacientes con enfermedad hepática crónica (véase Sobrecarga de Hierro) (85). Otros meta-análisis han reportado asociaciones entre las mutaciones C282Y y H63D del gen HFE y riesgos incrementados de cáncer en general (86, 87). Sin embargo, estudios que reporten sobre las mutaciones del gen HFE y el riesgo de cáncer en sitios extrahepáticos son escasos y/o inconsistentes. Algunos, pero no todos, los estudios basados en la observación encontraron asociaciones significantes entre la mutación C282Y y el riesgo de cáncer colorrectal (88), de mama (88, 89), y el cáncer epitelial de ovario (90). La presencia de la mutación H63D en el gen HFE fue ligada a un riesgo incrementado de leucemia (91, 92) y cáncer gástrico (93).
También se ha investigado si el hierro dietario alto podría incrementar el riesgo de cáncer en individuos sin hemocromatosis. El consumo de carnes rojas o procesadas (pero no carne blanca), ricas en hierro hemo, ha sido ligado a un riesgo incrementado de cáncer colorrectal (CCR) (94). La exposición a compuestos cancerígenos (llamados aminas heterocíclicas) generados cuando la carne es cocida a altas temperaturas y a los compuestos cancerígenos N-nitroso formados en el tracto gastrointestinal después del consumo de carne roja y procesada puede explicar tal asociación (95). Varios meta-análisis de estudios basados en la observación han también sugerido una asociación potencial del hierro hemo en la carne roja y el CCR (96-98). Esto ha sido explicado por una exposición incrementada de las células del colon a los compuestos N-nitroso potencialmente dañinos y a los productos finales de la peroxidación lipídica derivados de las reacciones hemo catalizadas por el hierro (99). Además, resultados recientes del estudio de Investigación Prospectiva Europea sobre el Cáncer y Nutrición (EPIC) de gran magnitud sugirieron un riesgo mayor de adenocarcinoma esofágico con altas ingestas de carnes rojas/procesadas y hierro hemo (100).
Enfermedades cardiovasculares
Estudios experimentales han sugerido un papel del estrés oxidativo inducido por el hierro en el daño a la pared de los vasos sanguíneos y el desarrollo de la aterosclerosis, que subyacen a la mayoría de las formas de enfermedades cardiovasculares (101). Sin embargo, estudios epidemiológicos del estatus de hierro nutricional y las enfermedades cardiovasculares en humanos han proporcionado resultados conflictivos. Una revisión sistemática reciente y meta-análisis de 17 estudios de cohorte prospectivos en 156,427 participantes (9,236 casos de enfermedad coronaria cardíaca [ECC] o infarto al miocardio [IM]) no encontraron evidencia para apoyar la existencia de asociaciones fuertes entre un cierto número de mediciones diferentes del estatus del hierro y ECC/MI (102). Solo los individuos en el tercil más alto de la saturación de transferrina del suero versus aquellos en el más bajo exhibieron una incidencia 18% menor de ECC/MI (102). Otro meta-análisis de 21 estudios prospectivos encontró que la saturación de transferrina del suero y el hierro del suero estaban inversamente asociados con el riesgo de ECC. Sin embargo, los autores notaron que la mayoría de los estudios no se ajustaron a los efectos de confusión de la inflamación (103). La revisión también reportó una asociación inversa entre la incidencia de ECC y la ingesta dietaría de hierro total, pero el hierro hemo dietario fue positivamente asociado con la incidencia de ECC (103). Aunque la relación entre los suministros de hierro y ECC/MI requiere de mayor clarificación, sería prudente para aquellos que no están en riesgo de deficiencia de hierro (p.ej., hombres adultos y mujeres postmenopáusicas) evitar la ingesta de hierro en exceso.
Diabetes mellitus tipo 2 y síndrome metabólico
Los individuos con hemocromatosis hereditaria (HH) son conocidos por estar en un riesgo mayor de desarrollar diabetes mellitus tipo 2 (104). Evidencia creciente también sugiere un papel del hierro en exceso en la patogénesis de la diabetes tipo 2 independiente de la hemocromatosis. Estudios de cohorte prospectivos, de corte transversal, y de caso-control han reportado un riesgo incrementado de diabetes tipo 2 (105) y de síndrome metabólico (106) con concentraciones altas de ferritina frente a las concentraciones bajas (que reflejan los suministros de hierro del cuerpo) después de ajustarse por la inflamación. Actualmente no está claro como otros índices del estatus del hierro se relacionan con el riesgo de diabetes tipo 2 (107-110). Se piensa que el estrés oxidativo inducido por la sobrecarga de hierro en pacientes con HH daña las células β pancreáticas y altera la secreción de insulina. En los sujetos sin HH, el exceso de hierro podría dañar el hígado, interfiriendo con el metabolismo de la glucosa y desencadenando resistencia a la insulina, en lugar de perjudicar la función de las células β (111, 112). Se ha mostrado que la eliminación de hierro por flebotomía mejora los parámetros metabólicos en sujetos con diabetes tipo 2 (113) y con síndrome metabólico (114). Ensayos controlados aleatorios adicionales son necesarios para determinar si la disminución de las reservas corporales de hierro ayudara en la prevención de diabetes tipo 2 y del síndrome metabólico.
Enfermedades neurodegenerativas
El hierro es requerido para la función cerebral y nerviosa normal a través de su participación en el metabolismo celular, como también en la síntesis de neurotransmisores y de mielina. La desregulación de la homeostasis del hierro ha sido observada en un cierto número de enfermedades neurodegenerativas, incluyendo la enfermedad de Alzheimer, la enfermedad de Parkinson, y la esclerosis lateral amiotrófica (ELA; la enfermedad de Lou Gehrig) (115-117). La acumulación anormal de hierro en el cerebro no parece ser una consecuencia del incremento de hierro dietario, sino más bien una interrupción en el complejo proceso de regulación celular del hierro (117). La acumulación cerebral de hierro puede resultar en un incremento en el estrés oxidativo, y el cerebro es particularmente susceptible al daño oxidativo. Los mecanismos detrás de la disrupción de la homeostasis del hierro en el cerebro de pacientes afectados por enfermedades neurodegenerativas son activamente investigados. Por ejemplo, estudios que usan modelos de ratones genéticamente modificados indicaron que la supresión de la expresión de la proteína precursora amiloide (APP) por la elevación en dirección 5’ del óxido nítrico (NO) (11) o por la perdida de la proteína tau (12) pudo perjudicar la exportación neuronal de hierro y resultar en la acumulación de hierro en regiones específicas del cerebro afectadas por la enfermedad de Parkinson. Un ensayo piloto, doble ciego, controlado con placebo en pacientes con la enfermedad de Parkinson en etapa temprana demostró que la administración oral del quelante de hierro, deferiprona, por 12 meses redujo la deposición de hierro en la parte del cerebro llamada sustancia negra y mejoró el desempeño motor sin comprometer la homeostasis sistémica del hierro (118, 119).
Seguridad
Toxicidad
Sobredosis
La sobredosis accidental de productos que contienen hierro es la mayor causa individual de fatalidades por envenenamiento en niños menores de seis años de edad. Aunque la dosis oral letal de hierro elemental es aproximadamente de 180 a 250 mg/kg de peso corporal, considerablemente menos ha sido fatal. Los síntomas de toxicidad aguda pueden ocurrir con dosis de hierro de 20 a 60 mg/kg de peso corporal. La sobredosis de hierro es una situación de emergencia debido a que la severidad de la toxicidad del hierro se relaciona con la cantidad de hierro elemental absorbido. El envenenamiento agudo por hierro produce síntomas en cuatro etapas: (1) Entre una a seis horas de la ingesta, los síntomas pueden incluir náuseas, vómitos, dolor abdominal, heces de color negro, letargo, pulso acelerado y débil, baja presión sanguínea, fiebre, dificultad para respirar, y coma; (2) Si los síntomas no resultan inmediatamente fatales, los síntomas pueden desaparecer durante aproximadamente 24 horas; (3) Los síntomas pueden regresar de 12 a 48 horas después de la ingesta de hierro y pueden incluir signos severos de una falla en los siguientes sistemas de órganos: cardiovascular, renal, hepático, hematológico (sangre), y el sistema nervioso central; y (4) El daño a largo plazo al sistema nervioso central, al hígado (cirrosis) y estómago puede aparecer de dos a seis semanas después de la ingesta (25, 120).
Efectos adversos
En niveles terapéuticos usados para tratar la deficiencia de hierro, los suplementos de hierro pueden causar irritación gastrointestinal, náuseas, vómitos, diarrea, o constipación. Las heces con frecuencia aparecerán de color más oscuro. Los líquidos con hierro pueden teñir los dientes de manera temporal, pero diluyendo los líquidos ayuda a prevenir este efecto (120). Consumir suplementos de hierro con alimentos en vez de con el estómago vacío puede aliviar los efectos gastrointestinales (37). La Junta de Nutrición y Alimentos (JNA) del Instituto de Medicina basó el nivel máximo de ingesta tolerable (NM) para el hierro en la prevención de las molestias gastrointestinales (Tabla 3). El NM para adolescentes (14-18 años de edad) y adultos, incluyendo mujeres embarazadas y en amamantamiento, es de 45 mg/día. Se debe destacar que el NM no está pensado para ser aplicado a individuos en tratamiento con hierro bajo supervisión médica estricta. Los individuos con hemocromatosis hereditaria u otras condiciones de sobrecarga de hierro, así como los individuos con cirrosis alcohólica y otras enfermedades hepáticas, pueden experimentar efectos adversos con niveles de ingesta de hierro por debajo del NM (25).
| Grupo Etario | NM (mg/día) |
|---|---|
| Infantes 0-12 meses | 40 |
| Niños 1-13 años | 40 |
| Adolescentes 14-18 años | 45 |
| Adultos 19 años y más | 45 |
Interacción con drogas/fármacos
Los medicamentos que disminuyen la acidez del estómago, como los antiácidos, los antagonistas del receptor de histamina (H2) (p.ej., cimetidina, ranitidina), y los inhibidores de la bomba de protones (p.ej., omeprazole, lanzoprazole), pueden deteriorar la absorción de hierro. Tomar suplementos de hierro al mismo tiempo que los siguientes medicamentos puede resultar en una disminución en la absorción y la eficacia del medicamento: carbidopa y levodopa (Sinemet), levotiroxina (Synthroid, Levoxyl), metildopa (Aldomet), penicilamina (Cuprimine, Depen), quinilonas, tetraciclinas, y bifosfonatos (120). Por lo tanto, es mejor tomar estos medicamentos con dos horas de diferencia de los suplementos de hierro. La colestiramina (Questran) y el colestipol (Colestid), utilizados para disminuir los niveles de colesterol sanguíneo, también deben tomarse con al menos cuatro horas de diferencia de los suplementos de hierro ya que pueden interferir con la absorción de hierro (121). El alopurinol (Zyloprim), un medicamento utilizado para tratar la gota puede incrementar el almacenaje de hierro en el hígado y no debe utilizarse en combinación con suplementos de hierro.
¿Incrementa la suplementación con hierro el riesgo de malaria en regiones donde la malaria es endémica?
A pesar de las funciones criticas del hierro en el sistema inmune, la naturaleza de la relacione entre el estatus del hierro y la susceptibilidad a la infección, especialmente con respecto a la malaria, ha sido controversial. Debido a que la retención de hierro es un mecanismo de defensa reconocido contra los patógenos (véase Defensa de retención de hierro durante la infección) preocupaciones han sido planteadas con respecto a la seguridad de la suplementación con hierro, especialmente en niños repletos de hierro que viven en regiones donde la malaria es endémica (122).
La suplementación con hierro de niños que residen en los trópicos ha sido asociada con un riesgo incrementado de malaria clínica y otras infecciones como la neumonía (123, 124). Un ensayo controlado aleatorio en 24,076 niños (edades, 1-35 meses) viviendo en una región de malaria endémica del este de África (Tanzania) investigó los efectos del hierro y ácido fólico suplementarios, con o sin zinc, en comparación a los efectos del zinc solo o un placebo, en la mortalidad por todas las causas y las admisiones al hospital (125). Se encontró que la administración de hierro, ácido fólico, y/o zinc incrementaba el riesgo de efectos adversos serios, admisión al hospital, y muerte, y por lo tanto fue detenido prematuramente. Análisis subsecuentes del ensayo revelaron que los niños repletos de hierro fueron más propensos que los niños deficientes de hierro (con o sin anemia) a estar en riesgo de padecer efectos adversos después de la suplementación con hierro (125). Tal riesgo potencial de efectos adversos con una suplementación rutinaria de hierro no fue observado en niños de preescolar en lugares sin malaria (al sur de Nepal) (126).
Una revisión reciente de 35 ensayos indicó que la suplementación con hierro no incremento el riesgo de malaria clínica o de otras enfermedades parasitarias, infecciones, y mortalidad por todas las causas en niños viviendo en regiones de malaria endémica en las cuales la prevención y manejo de la malaria están disponibles (127). Además, un análisis agrupado de tres ensayos de alta cualidad demostró que el hierro suplementario en combinación con el tratamiento anti-malaria protegió a los niños contra la malaria clínica y mejoró los parámetros hematológicos (127). La Organización Mundial de la Salud (OMS) actualmente recomienda la provisión de la suplementación con hierro en infantes y niños, junto con las medidas de prevención de la malaria, diagnosis, y tratamiento en áreas de malaria endémica (128).
Recomendación del Instituto Linus Pauling
El seguimiento de la IDR de hierro debiera aportar suficiente hierro para prevenir una deficiencia sin causar efectos adversos en la mayoría de los individuos. Aunque se puede obtener hierro suficiente a través de una dieta variada, una cantidad considerable de personas no consume hierro adecuado para prevenir una deficiencia. Un suplemento multivitamínico/mineral que contenga el 100% del valor diario (VD) de hierro aporta 18 mg de hierro elemental. Mientras que esta cantidad de hierro puede ser beneficiosa para mujeres premenopáusicas, está muy por encima de la IDR para hombres y mujeres postmenopáusicas.
Hombres adultos y mujeres postmenopáusicas
Ya que la hemocromatosis hereditaria es relativamente común y los efectos a largo plazo del hierro dietético en exceso sobre el riesgo de enfermedades crónicas aún no está claro, los hombres y mujeres postmenopáusicas que no se encuentran en riesgo de una deficiencia de hierro debieran tomar un suplemento multivitamínico/mineral sin hierro. Una serie de multivitamínicos formulados específicamente para hombres o personas por encima de los 50 años de edad, no contienen hierro.
Adultos mayores (>50 años)
Los depósitos de hierro moderadamente elevados podrían ser mucho más comunes que la deficiencia de hierro en individuos de mediana edad y mayores (129). Por lo tanto, los adultos mayores no debieran por lo general tomar suplementos nutricionales que contengan hierro a menos que hayan sido diagnosticados con deficiencia de hierro. Más aún, es extremadamente importante que se determine la causa subyacente de la deficiencia de hierro, más que simplemente tratarla con suplementos de hierro (véase La Ingesta Diaria Recomendada).
Autores y Críticos
Originalmente escrito en 2001 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Marzo de 2003 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Enero de 2006 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Agosto de 2009 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Agosto de 2016 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Mayo de 2016 por:
Marianne Wessling-Resnick, Ph.D.
Profesor de Bioquímica Nutricional
Departamento de Genética y Enfermedades Complejas
Harvard T.H. Chan Escuela de Salud Publica
Traducido al Español en 2018 por:
Silvia Vazquez Lima
Instituto Linus Pauling
Universidad Estatal de Oregon
La actualización del articulo en el 2016 fue suscrita, en parte, por una subvención de Bayer Consumer Care AG, Basilea, Suiza.
Derechos de autoría 2001-2024 Instituto Linus Pauling
Referencias
1. Aggett PJ. Iron. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Ames: Wiley-Blackwell; 2012:506-520.
2. Wessling-Resnick M. Iron. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed: Lippincott Williams & Wilkins; 2014:176-188.
3. Winter WE, Bazydlo LA, Harris NS. The molecular biology of human iron metabolism. Lab Med. 2014;45(2):92-102. (PubMed)
4. Burmester T, Hankeln T. What is the function of neuroglobin? J Exp Biol. 2009;212(Pt 10):1423-1428. (PubMed)
5. Salminen A, Kauppinen A, Kaarniranta K. 2-Oxoglutarate-dependent dioxygenases are sensors of energy metabolism, oxygen availability, and iron homeostasis: potential role in the regulation of aging process. Cell Mol Life Sci. 2015;72(20):3897-3914. (PubMed)
6. Zhang C. Essential functions of iron-requiring proteins in DNA replication, repair and cell cycle control. Protein Cell. 2014;5(10):750-760. (PubMed)
7. Anderson GJ, Darshan D, Wilkins SJ, Frazer DM. Regulation of systemic iron homeostasis: how the body responds to changes in iron demand. Biometals. 2007;20(3-4):665-674. (PubMed)
8. Fleming MD. The regulation of hepcidin and its effects on systemic and cellular iron metabolism. Hematology Am Soc Hematol Educ Program. 2008:151-158. (PubMed)
9. Tussing-Humphreys L, Pusatcioglu C, Nemeth E, Braunschweig C. Rethinking iron regulation and assessment in iron deficiency, anemia of chronic disease, and obesity: introducing hepcidin. J Acad Nutr Diet. 2012;112(3):391-400. (PubMed)
10. Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090-2093. (PubMed)
11. Ayton S, Lei P, Hare DJ, et al. Parkinson's disease iron deposition caused by nitric oxide-induced loss of beta-amyloid precursor protein. J Neurosci. 2015;35(8):3591-3597. (PubMed)
12. Lei P, Ayton S, Finkelstein DI, et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med. 2012;18(2):291-295. (PubMed)
13. Bhaskaram P. Immunobiology of mild micronutrient deficiencies. Br J Nutr. 2001;85 Suppl 2:S75-80. (PubMed)
14. Baker RD, Greer FR, Committee on Nutrition American Academy of Pediatrics. Diagnosis and prevention of iron deficiency and iron-deficiency anemia in infants and young children (0-3 years of age). Pediatrics. 2010;126(5):1040-1050. (PubMed)
15. Semba RD, Bloem MW. The anemia of vitamin A deficiency: epidemiology and pathogenesis. Eur J Clin Nutr. 2002;56(4):271-281. (PubMed)
16. Allen LH. Iron supplements: scientific issues concerning efficacy and implications for research and programs. J Nutr. 2002;132(4 Suppl):813S-819S. (PubMed)
17. Suharno D, West CE, Muhilal, Karyadi D, Hautvast JG. Supplementation with vitamin A and iron for nutritional anaemia in pregnant women in West Java, Indonesia. Lancet. 1993;342(8883):1325-1328. (PubMed)
18. Jang JT, Green JB, Beard JL, Green MH. Kinetic analysis shows that iron deficiency decreases liver vitamin A mobilization in rats. J Nutr. 2000;130(5):1291-1296. (PubMed)
19. Rosales FJ, Jang JT, Pinero DJ, Erikson KM, Beard JL, Ross AC. Iron deficiency in young rats alters the distribution of vitamin A between plasma and liver and between hepatic retinol and retinyl esters. J Nutr. 1999;129(6):1223-1228. (PubMed)
20. Turnlund JR. Copper. In: Shils ME, Shike M, Ross AC, Caballero B, Cousins RJ, eds. Modern Nutrition in Health and Disease. 10th ed. Philadelphia: Lippincott Williams & Wilkins; 2006:286-299.
21. Thackeray EW, Sanderson SO, Fox JC, Kumar N. Hepatic iron overload or cirrhosis may occur in acquired copper deficiency and is likely mediated by hypoceruloplasminemia. J Clin Gastroenterol. 2011;45(2):153-158. (PubMed)
22. Videt-Gibou D, Belliard S, Bardou-Jacquet E, et al. Iron excess treatable by copper supplementation in acquired aceruloplasminemia: a new form of secondary human iron overload? Blood. 2009;114(11):2360-2361. (PubMed)
23. Food and Nutrition Board, Institute of Medicine. Copper. Dietary Reference Intakes for Vitamin A, Vitamin K, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Washington, D.C.: National Academy Press; 2001:224-257. (National Academy Press)
24. Kelkitli E, Ozturk N, Aslan NA, et al. Serum zinc levels in patients with iron deficiency anemia and its association with symptoms of iron deficiency anemia. Ann Hematol. 2016;95(5):751-756. (PubMed)
25. Food and Nutrition Board, Institute of Medicine. Iron. Dietary Reference Intakes for Vitamin A, Vitamin K, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Washington, D.C.: National Academy Press; 2001:290-393. (National Academy Press)
26. Lynch SR. Interaction of iron with other nutrients. Nutr Rev. 1997;55(4):102-110. (PubMed)
27. Hurrell R, Egli I. Iron bioavailability and dietary reference values. Am J Clin Nutr. 2010;91(5):1461S-1467S. (PubMed)
28. Weaver CM. Calcium. In: Erdman JJ, Macdonald I, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed: John Wiley & Sons, Inc.; 2012:434-446.
29. Zimmermann MB. The influence of iron status on iodine utilization and thyroid function. Annu Rev Nutr. 2006;26:367-389. (PubMed)
30. Hess SY. The impact of common micronutrient deficiencies on iodine and thyroid metabolism: the evidence from human studies. Best Pract Res Clin Endocrinol Metab. 2010;24(1):117-132. (PubMed)
31. Hess SY, Zimmermann MB, Adou P, Torresani T, Hurrell RF. Treatment of iron deficiency in goitrous children improves the efficacy of iodized salt in Cote d'Ivoire. Am J Clin Nutr. 2002;75(4):743-748. (PubMed)
32. World Health Organization, United Nations Children's Fund, United Nations University. Iron deficiency anaemia: assessment, prevention and control - A guide for programme managers 2001.
33. Global Burden of Disease Pediatrics C, Kyu HH, Pinho C, et al. Global and National Burden of Diseases and Injuries Among Children and Adolescents Between 1990 and 2013: Findings From the Global Burden of Disease 2013 Study. JAMA Pediatr. 2016;170(3):267-287. (PubMed)
34. Wang M. Iron deficiency and other types of anemia in infants and children. Am Fam Physician. 2016;93(4):270-278. (PubMed)
35. Beard JL. Iron biology in immune function, muscle metabolism and neuronal functioning. J Nutr. 2001;131(2S-2):568S-579S; discussion 580S. (PubMed)
36. Changela K, Haeri NS, Krishnaiah M, Reddy M. Plummer-Vinson syndrome with proximal esophageal web. J Gastrointest Surg. 2015;20(5):1074-1075. (PubMed)
37. Jauregui-Lobera I. Iron deficiency and cognitive functions. Neuropsychiatr Dis Treat. 2014;10:2087-2095. (PubMed)
38. Lee GR. Disorders of iron metabolism and heme synthesis. In: Lee GR, Foerster J, Paraskevas F, Greer JP, Rogers GM, eds. Wintrobe's Clinical Hematology. Baltimore: Williams and Wilkins; 1999:979-1070.
39. McDonald SJ, Middleton P, Dowswell T, Morris PS. Effect of timing of umbilical cord clamping of term infants on maternal and neonatal outcomes. Evid Based Child Health. 2014;9(2):303-397. (PubMed)
40. Siu AL, Force USPST. Screening for iron deficiency anemia in young children: USPSTF recommendation statement. Pediatrics. 2015;136(4):746-752. (PubMed)
41. Miller EM. Iron status and reproduction in US women: National Health and Nutrition Examination Survey, 1999-2006. PLoS One. 2014;9(11):e112216. (PubMed)
42. Brody T. Nutritional Biochemistry. 2nd ed. San Diego: Academic Press; 1999.
43. Mei Z, Cogswell ME, Looker AC, et al. Assessment of iron status in US pregnant women from the National Health and Nutrition Examination Survey (NHANES), 1999-2006. Am J Clin Nutr. 2011;93(6):1312-1320. (PubMed)
44. Khuroo MS, Khuroo MS, Khuroo NS. Trichuris dysentery syndrome: a common cause of chronic iron deficiency anemia in adults in an endemic area (with videos). Gastrointest Endosc. 2010;71(1):200-204. (PubMed)
45. Brittenham GM. Iron deficiency in whole blood donors. Transfusion. 2011;51(3):458-461. (PubMed)
46. Li H, Condon F, Kessler D, et al. Evidence of relative iron deficiency in platelet- and plasma-pheresis donors correlates with donation frequency. J Clin Apher. 2016; doi: 10.1002/jca.21448. [Epub ahead of print]. (PubMed)
47. Hershko C, Skikne B. Pathogenesis and management of iron deficiency anemia: emerging role of celiac disease, helicobacter pylori, and autoimmune gastritis. Semin Hematol. 2009;46(4):339-350. (PubMed)
48. Mahadov S, Green PH. Celiac disease: a challenge for all physicians. Gastroenterol Hepatol (N Y). 2011;7(8):554-556. (PubMed)
49. Cardenas VM, Mulla ZD, Ortiz M, Graham DY. Iron deficiency and Helicobacter pylori infection in the United States. Am J Epidemiol. 2006;163(2):127-134. (PubMed)
50. Dignass AU, Gasche C, Bettenworth D, et al. European consensus on the diagnosis and management of iron deficiency and anaemia in inflammatory bowel diseases. J Crohns Colitis. 2015;9(3):211-222. (PubMed)
51. Aron-Wisnewsky J, Verger EO, Bounaix C, et al. Nutritional and Protein Deficiencies in the Short Term following Both Gastric Bypass and Gastric Banding. PLoS One. 2016;11(2):e0149588. (PubMed)
52. Lecube A, Carrera A, Losada E, Hernandez C, Simo R, Mesa J. Iron deficiency in obese postmenopausal women. Obesity (Silver Spring). 2006;14(10):1724-1730. (PubMed)
53. Nead KG, Halterman JS, Kaczorowski JM, Auinger P, Weitzman M. Overweight children and adolescents: a risk group for iron deficiency. Pediatrics. 2004;114(1):104-108. (PubMed)
54. Saunders AV, Craig WJ, Baines SK, Posen JS. Iron and vegetarian diets. Med J Aust. 2013;199(4 Suppl):S11-16. (PubMed)
55. Macdougall IC, Bircher AJ, Eckardt KU, et al. Iron management in chronic kidney disease: conclusions from a "Kidney Disease: Improving Global Outcomes" (KDIGO) Controversies Conference. Kidney Int. 2016;89(1):28-39. (PubMed)
56. Doom JR, Georgieff MK. Striking while the iron is hot: Understanding the biological and neurodevelopmental effects of iron deficiency to optimize intervention in early childhood. Curr Pediatr Rep. 2014;2(4):291-298. (PubMed)
57. Wang B, Zhan S, Gong T, Lee L. Iron therapy for improving psychomotor development and cognitive function in children under the age of three with iron deficiency anaemia. Cochrane Database Syst Rev. 2013;6:CD001444. (PubMed)
58. Idjradinata P, Pollitt E. Reversal of developmental delays in iron-deficient anaemic infants treated with iron. Lancet. 1993;341(8836):1-4. (PubMed)
59. Szajewska H, Ruszczynski M, Chmielewska A. Effects of iron supplementation in nonanemic pregnant women, infants, and young children on the mental performance and psychomotor development of children: a systematic review of randomized controlled trials. Am J Clin Nutr. 2010;91(6):1684-1690. (PubMed)
60. Pongcharoen T, DiGirolamo AM, Ramakrishnan U, Winichagoon P, Flores R, Martorell R. Long-term effects of iron and zinc supplementation during infancy on cognitive function at 9 y of age in northeast Thai children: a follow-up study. Am J Clin Nutr. 2011;93(3):636-643. (PubMed)
61. Sachdev H, Gera T, Nestel P. Effect of iron supplementation on mental and motor development in children: systematic review of randomised controlled trials. Public Health Nutr. 2005;8(2):117-132. (PubMed)
62. Falkingham M, Abdelhamid A, Curtis P, Fairweather-Tait S, Dye L, Hooper L. The effects of oral iron supplementation on cognition in older children and adults: a systematic review and meta-analysis. Nutr J. 2010;9:4. (PubMed)
63. Burke RM, Leon JS, Suchdev PS. Identification, prevention and treatment of iron deficiency during the first 1000 days. Nutrients. 2014;6(10):4093-4114. (PubMed)
64. Pena-Rosas JP, De-Regil LM, Garcia-Casal MN, Dowswell T. Daily oral iron supplementation during pregnancy. Cochrane Database Syst Rev. 2015;7:CD004736. (PubMed)
65. Institute of Medicine Committee on Preventive Services for Women; Board on Population Health and Public Health Practice. Clinical prevention services for women - closing the gaps: The National Academies Press; 2011. (The National Academies Press)
66. American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 95: anemia in pregnancy. Obstet Gynecol. 2008;112(1):201-207. (PubMed)
67. American Academy of Family Physicians. Clinical preventive service recommendation: iron deficiency anemia. http://www.aafp.org/patient-care/clinical-recommendations/all/iron-deficiency-anemia.html. Accessed 4/17/16.
68. Etheredge AJ, Premji Z, Gunaratna NS, et al. Iron supplementation in iron-replete and nonanemic pregnant women in Tanzania: a randomized clinical trial. JAMA Pediatr. 2015;169(10):947-955. (PubMed)
69. Mwangi MN, Roth JM, Smit MR, et al. Effect of daily antenatal iron supplementation on plasmodium infection in Kenyan women: a randomized clinical trial. JAMA. 2015;314(10):1009-1020. (PubMed)
70. Mielke HW, Gonzales C, Powell E, Mielke PW. Evolving from reactive to proactive medicine: community lead (Pb) and clinical disparities in pre- and post-Katrina New Orleans. Int J Environ Res Public Health. 2014;11(7):7482-7491. (PubMed)
71. Centers for Disease Control and Prevention. New blood lead level information. [Web page]. Available at: http://www.cdc.gov/nceh/lead/acclpp/blood_lead_levels.htm. Accessed 6/1/16.
72. Kwong WT, Friello P, Semba RD. Interactions between iron deficiency and lead poisoning: epidemiology and pathogenesis. Sci Total Environ. 2004;330(1-3):21-37. (PubMed)
73. Silber MH, Becker PM, Earley C, Garcia-Borreguero D, Ondo WG, Medical Advisory Board of the Willis-Ekbom Disease F. Willis-Ekbom Disease Foundation revised consensus statement on the management of restless legs syndrome. Mayo Clin Proc. 2013;88(9):977-986. (PubMed)
74. Trotti LM, Bhadriraju S, Becker LA. Iron for restless legs syndrome. Cochrane Database Syst Rev. 2012;5:CD007834. (PubMed)
75. Johnston CS. Vitamin C. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Ames: Wiley-Blackwell; 2012:248-260.
76. Morck TA, Lynch SR, Cook JD. Inhibition of food iron absorption by coffee. Am J Clin Nutr. 1983;37(3):416-420. (PubMed)
77. Natural Medicines. Iron: Interactions with Herbs & Supplements [professional monograph]; 2016. Available at: https://naturalmedicines.therapeuticresearch.com/. Accessed 6/1/16.
78. Liu J, Pu C, Lang L, Qiao L, Abdullahi MA, Jiang C. Molecular pathogenesis of hereditary hemochromatosis. Histol Histopathol. 2016:11762. [Epub ahead of print]. (PubMed)
79. Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13(4):399-408. (PubMed)
80. Franchini M, Veneri D. Recent advances in hereditary hemochromatosis. Ann Hematol. 2005;84(6):347-352. (PubMed)
81. Ayonrinde OT, Milward EA, Chua AC, Trinder D, Olynyk JK. Clinical perspectives on hereditary hemochromatosis. Crit Rev Clin Lab Sci. 2008;45(5):451-484. (PubMed)
82. Wallace DF, Subramaniam VN. Non-HFE haemochromatosis. World J Gastroenterol. 2007;13(35):4690-4698. (PubMed)
83. Powell LW, Seckington RC, Deugnier Y. Haemochromatosis. Lancet. 2016; pii: S0140-6736(15)01315-X. doi: 10.1016/S0140-6736(15)01315-X. [Epub ahead of print]. (PubMed)
84. Oikonomidou PR, Casu C, Rivella S. New strategies to target iron metabolism for the treatment of beta thalassemia. Ann N Y Acad Sci. 2016; 1368(1):162-168. (PubMed)
85. Jin F, Qu LS, Shen XZ. Association between C282Y and H63D mutations of the HFE gene with hepatocellular carcinoma in European populations: a meta-analysis. J Exp Clin Cancer Res. 2010;29:18. (PubMed)
86. Shen LL, Gu DY, Zhao TT, Tang CJ, Xu Y, Chen JF. Implicating the H63D polymorphism in the HFE gene in increased incidence of solid cancers: a meta-analysis. Genet Mol Res. 2015;14(4):13735-13745. (PubMed)
87. Zhang M, Xiong H, Fang L, et al. Meta-Analysis of the Association between H63D and C282Y Polymorphisms in HFE and Cancer Risk. Asian Pac J Cancer Prev. 2015;16(11):4633-4639. (PubMed)
88. Osborne NJ, Gurrin LC, Allen KJ, et al. HFE C282Y homozygotes are at increased risk of breast and colorectal cancer. Hepatology. 2010;51(4):1311-1318. (PubMed)
89. Liu X, Lv C, Luan X, Lv M. C282Y polymorphism in the HFE gene is associated with risk of breast cancer. Tumour Biol. 2013;34(5):2759-2764. (PubMed)
90. Gannon PO, Medelci S, Le Page C, et al. Impact of hemochromatosis gene (HFE) mutations on epithelial ovarian cancer risk and prognosis. Int J Cancer. 2011;128(10):2326-2334. (PubMed)
91. Kennedy AE, Kamdar KY, Lupo PJ, et al. Examination of HFE associations with childhood leukemia risk and extension to other iron regulatory genes. Leuk Res. 2014;38(9):1055-1060. (PubMed)
92. Viola A, Pagano L, Laudati D, et al. HFE gene mutations in patients with acute leukemia. Leuk Lymphoma. 2006;47(11):2331-2334. (PubMed)
93. Agudo A, Bonet C, Sala N, et al. Hemochromatosis (HFE) gene mutations and risk of gastric cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Carcinogenesis. 2013;34(6):1244-1250. (PubMed)
94. Larsson SC, Wolk A. Meat consumption and risk of colorectal cancer: a meta-analysis of prospective studies. Int J Cancer. 2006;119(11):2657-2664. (PubMed)
95. Cross AJ, Ferrucci LM, Risch A, et al. A large prospective study of meat consumption and colorectal cancer risk: an investigation of potential mechanisms underlying this association. Cancer Res. 2010;70(6):2406-2414. (PubMed)
96. Bastide NM, Pierre FH, Corpet DE. Heme iron from meat and risk of colorectal cancer: a meta-analysis and a review of the mechanisms involved. Cancer Prev Res (Phila). 2011;4(2):177-184. (PubMed)
97. Fonseca-Nunes A, Jakszyn P, Agudo A. Iron and cancer risk--a systematic review and meta-analysis of the epidemiological evidence. Cancer Epidemiol Biomarkers Prev. 2014;23(1):12-31. (PubMed)
98. Qiao L, Feng Y. Intakes of heme iron and zinc and colorectal cancer incidence: a meta-analysis of prospective studies. Cancer Causes Control. 2013;24(6):1175-1183. (PubMed)
99. Bastide NM, Chenni F, Audebert M, et al. A central role for heme iron in colon carcinogenesis associated with red meat intake. Cancer Res. 2015;75(5):870-879. (PubMed)
100. Jakszyn P, Lujan-Barroso L, Agudo A, et al. Meat and heme iron intake and esophageal adenocarcinoma in the European Prospective Investigation into Cancer and Nutrition study. Int J Cancer. 2013;133(11):2744-2750. (PubMed)
101. de Valk B, Marx JJ. Iron, atherosclerosis, and ischemic heart disease. Arch Intern Med. 1999;159(14):1542-1548. (PubMed)
102. Das De S, Krishna S, Jethwa A. Iron status and its association with coronary heart disease: systematic review and meta-analysis of prospective studies. Atherosclerosis. 2015;238(2):296-303. (PubMed)
103. Hunnicutt J, He K, Xun P. Dietary iron intake and body iron stores are associated with risk of coronary heart disease in a meta-analysis of prospective cohort studies. J Nutr. 2014;144(3):359-366. (PubMed)
104. Swaminathan S, Fonseca VA, Alam MG, Shah SV. The role of iron in diabetes and its complications. Diabetes Care. 2007;30(7):1926-1933. (PubMed)
105. Orban E, Schwab S, Thorand B, Huth C. Association of iron indices and type 2 diabetes: a meta-analysis of observational studies. Diabetes Metab Res Rev. 2014;30(5):372-394. (PubMed)
106. Abril-Ulloa V, Flores-Mateo G, Sola-Alberich R, Manuel-y-Keenoy B, Arija V. Ferritin levels and risk of metabolic syndrome: meta-analysis of observational studies. BMC Public Health. 2014;14:483. (PubMed)
107. Huth C, Beuerle S, Zierer A, et al. Biomarkers of iron metabolism are independently associated with impaired glucose metabolism and type 2 diabetes: the KORA F4 study. Eur J Endocrinol. 2015;173(5):643-653. (PubMed)
108. Montonen J, Boeing H, Steffen A, et al. Body iron stores and risk of type 2 diabetes: results from the European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam study. Diabetologia. 2012;55(10):2613-2621. (PubMed)
109. Podmore C, Meidtner K, Schulze MB, et al. The association of multiple biomarkers of iron metabolism and type 2 diabetes: the EPIC-InterAct study. Diabetes Care. 2016; 39(4):572-581. (PubMed)
110. Yeap BB, Divitini ML, Gunton JE, et al. Higher ferritin levels, but not serum iron or transferrin saturation, are associated with Type 2 diabetes mellitus in adult men and women free of genetic haemochromatosis. Clin Endocrinol (Oxf). 2015;82(4):525-532. (PubMed)
111. Fernandez-Real JM, McClain D, Manco M. Mechanisms linking glucose homeostasis and iron metabolism toward the onset and progression of type 2 diabetes. Diabetes Care. 2015;38(11):2169-2176. (PubMed)
112. Huang J, Karnchanasorn R, Ou HY, et al. Association of insulin resistance with serum ferritin and aminotransferases-iron hypothesis. World J Exp Med. 2015;5(4):232-243. (PubMed)
113. Fernandez-Real JM, Penarroja G, Castro A, Garcia-Bragado F, Hernandez-Aguado I, Ricart W. Blood letting in high-ferritin type 2 diabetes: effects on insulin sensitivity and β-cell function. Diabetes. 2002;51(4):1000-1004. (PubMed)
114. Houschyar KS, Ludtke R, Dobos GJ, et al. Effects of phlebotomy-induced reduction of body iron stores on metabolic syndrome: results from a randomized clinical trial. BMC Med. 2012;10:54. (PubMed)
115. Belaidi AA, Bush AI. Iron neurochemistry in Alzheimer's disease and Parkinson's disease: targets for therapeutics. J Neurochem. 2015; doi: 10.1111/jnc.13425. [Epub ahead of print]. (PubMed)
116. Kwan JY, Jeong SY, Van Gelderen P, et al. Iron accumulation in deep cortical layers accounts for MRI signal abnormalities in ALS: correlating 7 tesla MRI and pathology. PLoS One. 2012;7(4):e35241. (PubMed)
117. Wong BX, Duce JA. The iron regulatory capability of the major protein participants in prevalent neurodegenerative disorders. Front Pharmacol. 2014;5:81. (PubMed)
118. Devos D, Moreau C, Devedjian JC, et al. Targeting chelatable iron as a therapeutic modality in Parkinson's disease. Antioxid Redox Signal. 2014;21(2):195-210. (PubMed)
119. Grolez G, Moreau C, Sablonniere B, et al. Ceruloplasmin activity and iron chelation treatment of patients with Parkinson's disease. BMC Neurol. 2015;15:74. (PubMed)
120. Hendler SS, Rorvik DM. PDR for Nutritional Supplements. 2nd ed. Montvale: Thomson Reuters; 2008.
121. Natural Medicines. Iron: Interactions with Drugs [professional monograph]; 2016. Available at: https://naturalmedicines.therapeuticresearch.com/. Accessed 6/1/16.
122. Wander K, Shell-Duncan B, McDade TW. Evaluation of iron deficiency as a nutritional adaptation to infectious disease: an evolutionary medicine perspective. Am J Hum Biol. 2009;21(2):172-179. (PubMed)
123. Oppenheimer SJ. Iron and its relation to immunity and infectious disease. J Nutr. 2001;131(2S-2):616S-633S; discussion 633S-635S. (PubMed)
124. van den Hombergh J, Dalderop E, Smit Y. Does iron therapy benefit children with severe malaria-associated anaemia? A clinical trial with 12 weeks supplementation of oral iron in young children from the Turiani Division, Tanzania. J Trop Pediatr. 1996;42(4):220-227. (PubMed)
125. Sazawal S, Black RE, Ramsan M, et al. Effects of routine prophylactic supplementation with iron and folic acid on admission to hospital and mortality in preschool children in a high malaria transmission setting: community-based, randomised, placebo-controlled trial. Lancet. 2006;367(9505):133-143. (PubMed)
126. Tielsch JM, Khatry SK, Stoltzfus RJ, et al. Effect of routine prophylactic supplementation with iron and folic acid on preschool child mortality in southern Nepal: community-based, cluster-randomised, placebo-controlled trial. Lancet. 2006;367(9505):144-152. (PubMed)
127. Neuberger A, Okebe J, Yahav D, Paul M. Oral iron supplements for children in malaria-endemic areas. Cochrane Database Syst Rev. 2016;2:CD006589. (PubMed)
128. World Health Organization. Guideline: daily iron supplementation in infants and children. Geneva: World Health Organization; 2016.
129. Fairweather-Tait SJ, Wawer AA, Gillings R, Jennings A, Myint PK. Iron status in the elderly. Mech Ageing Dev. 2014;136-137:22-28. (PubMed)
Magnesio
Contenido
Resumen
- El magnesio es un mineral esencial y un cofactor para cientos de enzimas. El magnesio está involucrado en muchas vías fisiológicas, incluida la producción de energía, la síntesis de ácidos nucleicos y proteínas, el transporte de iones, la señalización celular y también tiene funciones estructurales. (Más información)
- La deficiencia severa de magnesio puede impedir la homeostasis de la vitamina D y el calcio. Ciertos individuos son más susceptibles a la deficiencia de magnesio, especialmente aquellos con trastornos gastrointestinales o renales, aquellos que sufren de alcoholismo crónico y personas mayores. (Más información)
- La ingesta dietética inadecuada y/o las bajas concentraciones séricas de magnesio se han asociado con un mayor riesgo de enfermedad cardiovascular, osteoporosis y trastornos metabólicos, incluidos el síndrome metabólico, la hipertensión y la diabetes mellitus tipo 2. Los estudios preliminares han demostrado que el magnesio mejoró la sensibilidad a la insulina en individuos con riesgo de diabetes mellitus tipo 2. Los ensayos aleatorios controlados también han investigado el papel de los suplementos de magnesio en la prevención de complicaciones después de un accidente cerebrovascular o cirugía cardíaca. (Más información)
- El sulfato de magnesio se usa en la atención obstétrica para la prevención de convulsiones en mujeres embarazadas con preeclampsia o eclampsia. Los estudios de observación y los ensayos aleatorios controlados también respaldan el papel del magnesio en la prevención del daño cerebral en los bebés prematuros. (Más información)
- El uso de suplementos de magnesio se está explorando actualmente en el tratamiento de diversas afecciones, como hipertensión, enfermedad cardiovascular, diabetes mellitus tipo 2, asma, y dolor. (Más información)
- Aproximadamente la mitad de la población adulta de EE.UU. puede tener una ingesta insuficiente de magnesio para respaldar la adecuación nutricional. Las fuentes dietéticas ricas en magnesio incluyen verduras de hoja verde, granos sin refinar, legumbres, frijoles, y nueces. (Más información)
-
El nivel máximo de ingesta tolerable (NM) para el magnesio suplementario es de 350 mg/día. Una ingesta excesiva de magnesio suplementario puede resultar en efectos adversos, especialmente en individuos con insuficiencia renal. (Más información)
El magnesio desempeña papeles importantes en la estructura y la función del cuerpo humano. El cuerpo humano adulto contiene aproximadamente 25 gramos (g) de magnesio. Alrededor del 50 al 60% de todo el magnesio en el cuerpo se encuentra en el esqueleto y el resto se encuentra en los tejidos blandos, principalmente en el músculo. El magnesio es el segundo catión intracelular más abundante después del potasio. La sangre contiene menos del 1% del magnesio corporal total. Sólo la forma libre e ionizada de magnesio (Mg2+) es fisiológicamente activa. El magnesio quelado y unido a proteínas sirve para tamponar el conjunto de magnesio ionizado libre (1).
Función
El magnesio participa en más de 300 reacciones metabólicas esenciales, algunas de las cuales se analizan a continuación (2).
Producción de energía
El metabolismo de carbohidratos y grasas para producir energía necesita de numerosas reacciones químicas dependientes de magnesio. El magnesio es requerido por la proteína sintetizadora de adenosina trifosfato (ATP) en la mitocondria. El ATP, es la molécula que aporta energía a casi todos los procesos metabólicos, y que encontramos principalmente en un complejo con magnesio (MgATP) (3).
Síntesis de moléculas esenciales
El metabolismo de carbohidratos y grasas para producir energía necesita de numerosas reacciones químicas dependientes de magnesio. El magnesio es requerido por la proteína sintetizadora de adenosina trifosfato (ATP) en la mitocondria. El ATP, es la molécula que aporta energía a casi todos los procesos metabólicos, y que encontramos principalmente en un complejo con magnesio (MgATP) (3).
Papeles estructurales
El magnesio desempeña un papel estructural en el hueso, en las membranas celulares, y en los cromosomas (3).
Transporte de iones a través de las membranas celulares
El magnesio es necesario para el transporte activo de iones como el potasio y el calcio a través de las membranas celulares. A través de su papel en los sistemas de transporte de iones, el magnesio altera la conducción de los impulsos nerviosos, la contracción muscular, y el ritmo cardíaco normal (3).
Señalización celular
La señalización celular requiere MgATP para la fosforilación de proteínas y la formación de la molécula de señalización celular, monofosfato de adenosina cíclico (cAMP). El cAMP está involucrado en muchos procesos, incluida la secreción de hormona paratiroidea (PTH) de las glándulas paratiroides (consulte los artículos sobre Vitamina D y Calcio para obtener información sobre el papel de la PTH en el cuerpo) (3).
Migración celular
Los niveles de calcio y magnesio en el fluido que rodea a las células afecta la migración de una serie de distintos tipos celulares. Tales efectos en la migración celular pueden ser importantes en la sanación de heridas (3).
Interacción con nutrientes
Zinc
Altas dosis de zinc en forma suplementaria aparentemente interfieren con la absorción de magnesio. Un estudio informó que los suplementos de zinc de 142 mg/día (muy por encima del nivel máximo de consumo tolerable (NM) de 40 mg/día para zinc) en hombres adultos sanos disminuyeron significativamente la absorción de magnesio y alteraron el equilibrio de magnesio (la diferencia entre la ingesta de magnesio y el magnesio pérdida) (4).
Fibra
En estudios experimentales se encontró que grandes incrementos en la ingesta de fibra dietética disminuyen la utilización del magnesio. Sin embargo, la medida en la que la fibra dietética afecta el estatus nutricional del magnesio en individuos con una dieta variada fuera del laboratorio aún no está clara (2, 3).
Proteína
La ingesta de proteínas en la dieta puede afectar la absorción de magnesio. Un estudio en niños adolescentes encontró que la absorción de magnesio estaba directamente relacionada con la ingesta de proteínas, siendo la absorción de magnesio la más baja cuando la ingesta de proteínas era inferior a 30 g/día (5).
Vitamina D y calcio
La forma activa de la vitamina D (calcitriol) puede aumentar ligeramente la absorción intestinal de magnesio (6). Sin embargo, no está claro si la absorción de magnesio depende del calcitriol como lo es la absorción de calcio y fosfato. En la mayoría de los estudios no se ha encontrado que la alta ingesta de calcio afecte el balance del magnesio. Se sabe que los niveles insuficientes de magnesio en la sangre resultan en bajos niveles de calcio en la sangre, resistencia a la acción de la hormona paratiroidea (PTH), y en resistencia a algunos de los efectos de la vitamina D (2, 3).
Deficiencia
Factores de riesgo
La deficiencia de magnesio en individuos sanos que consumen una dieta balanceada es bastante rara, ya que el magnesio es abundante tanto en alimentos animales como vegetales y debido a que los riñones son capaces de limitar la excreción urinaria de magnesio cuando la ingesta es baja. Las siguientes condiciones aumentan el riesgo de una deficiencia de magnesio (7):
- Desorden gastrointestinal: La diarrea prolongada, la enfermedad de Crohn, los síndromes de malabsorción, la enfermedad celíaca, la extirpación quirúrgica de una porción del intestino y la inflamación intestinal causada por radiación pueden conducir a una disminución del magnesio.
- Trastornos renales (pérdida de magnesio): La diabetes mellitus y el uso a largo plazo de ciertos diuréticos (véase Interacción con drogas) pueden resultar en un incremento en la pérdida urinaria de magnesio. Muchos otros medicamentos pueden provocar también una pérdida renal de magnesio (3).
- Trastornos endocrinos y metabólicos: Varias afecciones, como la diabetes mellitus, los trastornos de las glándulas paratiroides, el agotamiento de fosfato, el aldosteronismo primario, e incluso la lactancia excesiva, pueden conducir al agotamiento de magnesio.
La mala ingesta dietética, los problemas gastrointestinales, y el aumento de la pérdida urinaria de magnesio pueden contribuir al agotamiento del magnesio en personas que sufren de alcoholismo. Los adultos mayores tienen ingestas dietéticas de magnesio relativamente bajas (8, 9). La absorción intestinal de magnesio tiende a disminuir con la edad, y la excreción urinaria de magnesio tiende a aumentar con la edad; por lo tanto, la ingesta subóptima de magnesio en la dieta puede aumentar el riesgo de agotamiento de magnesio en los ancianos (2).
Signos y síntomas
Aunque la deficiencia severa de magnesio es poco común, se ha inducido experimentalmente. Cuando se indujo la deficiencia de magnesio en humanos, el primer signo fue una disminución en la concentración sérica de magnesio. La hipomagnesemia generalmente describe concentraciones séricas de magnesio inferiores a 0.74 milimoles/litro (mmol/L) o 1.40 miliequivalentes/litro (mEq/L) o 1.70 miligramos/decilitro (mg/dL). Con el tiempo, la concentración sérica de calcio también comenzó a disminuir (hipocalcemia) a pesar del calcio en la dieta adecuado. La hipocalcemia persistió a pesar del aumento de la secreción de la hormona paratiroidea (PTH), que regula la homeostasis del calcio. Por lo general, el aumento de la secreción de PTH produce rápidamente la movilización de calcio del hueso y la normalización de la concentración de calcio en la sangre. A medida que avanzaba el agotamiento de magnesio, la secreción de PTH disminuía a bajas concentraciones. Además de la hipomagnesemia, los signos de deficiencia severa de magnesio incluyeron hipocalcemia, bajas concentraciones séricas de potasio (hipocalemia), retención de sodio, bajas concentraciones de PTH circulante, síntomas neurológicos y musculares (temblor, espasmos musculares, tetania), pérdida de apetito, náuseas, vómitos, y cambios de personalidad (3).
Si bien la deficiencia leve de magnesio puede no provocar síntomas clínicos, puede estar asociada con un mayor riesgo de desarrollar enfermedades crónicas (ver Prevención de Enfermedad) (1).
Evaluar el estatus de magnesio
Actualmente no hay un indicador confiable del estatus del magnesio. La prueba de tolerancia al magnesio, que básicamente determina la retención de magnesio (mediante la recolección de orina de 24-h) después de la administración intravenosa de magnesio, se considera el estándar de oro (1). Si este método es un buen indicador de hipomagnesemia en adultos, parece ser poco sensible a los cambios en el estatus del magnesio en personas sanas. Además, el método es invasivo e inconveniente, y por lo tanto difícil de usar de forma rutinaria (10). Otro método para evaluar el estatus del magnesio es a través de mediciones de magnesio ionizado en plasma, que representa la forma fisiológicamente activa de magnesio. Sin embargo, se desconoce si el magnesio ionizado por plasma refleja las reservas corporales (10).
En la práctica, el estatus del magnesio generalmente se determina mediante evaluaciones de la ingesta dietética de magnesio, la concentración sérica de magnesio y/o la concentración de magnesio en la orina (10). Sin embargo, cada uno de estos indicadores tiene limitaciones. Aunque se usa predominantemente en estudios epidemiológicos y es el único indicador disponible para los clínicos, se ha encontrado que la concentración séricas de magnesio responde pobremente a la suplementación con magnesio. Con respecto a las ingestas dietéticas de magnesio, se absorbe aproximadamente del 30 al 40% del magnesio ingerido, pero la absorción varía con la cantidad de magnesio ingerida y con la composición de la matriz alimentaria. Finalmente, un estatus de deficiencia de magnesio no se ha asociado a una clara concentración límite de magnesio en la orina. La concentración de magnesio en la orina fluctúa rápidamente con la ingesta dietética, pero las mediciones de magnesio en la orina de 24-horas se pueden usar además de otros indicadores para evaluar el estatus de la población. Actualmente, se puede utilizar una combinación de los tres marcadores — magnesio dietético, suero y orina — para obtener una evaluación válida del estatus del magnesio (revisado en 10).
La Ingesta Diaria Recomendada (IDR)
En 1997, la Junta de Alimentos y Nutrición del Instituto de Medicina incrementó la ingesta diaria recomendada (IDR) de magnesio, en base a los resultados de estudios de balance estrictamente controlados que utilizaron métodos más precisos para medir el magnesio (Tabla 1) (2). Los estudios de equilibrio son útiles para determinar la cantidad de un nutriente que evitará la deficiencia; sin embargo, tales estudios proporcionan poca información sobre la cantidad de un nutriente requerido para la prevención de enfermedades crónicas o una salud óptima.
| Grupo Etario | Edad | Machos (mg/día) | Hembras (mg/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 30 (IA*) | 30 (IA) |
| Infantes | 7-12 meses | 75 (IA) | 75 (IA) |
| Niños | 1-3 años | 80 | 80 |
| Niños | 4-8 años | 130 | 130 |
| Niños | 9-13 años | 240 | 240 |
| Adolescentes | 14-18 años | 410 | 360 |
| Adultos | 19-30 años | 400 | 310 |
| Adultos | 31 años y más | 420 | 320 |
| Embarazo | 18 años y menos | - | 400 |
| Embarazo | 19-30 años | - | 350 |
| Embarazo | 31 años y más | - | 360 |
| Período de lactancia | 18 años y menos | - | 360 |
| Período de lactancia | 19-30 años | - | 310 |
| Período de lactancia | 31 años y más | - | 320 |
| *Ingesta Adecuada |
Prevención de Enfermedad
Síndrome metabólico
El síndrome metabólico se refiere a la presentación concomitante de varios trastornos metabólicos en un individuo, que incluyen dislipidemia, hipertensión, resistencia a la insulina, y obesidad (11). Las personas con síndrome metabólico tienen un mayor riesgo de desarrollar diabetes mellitus tipo 2, enfermedad cardiovascular, y algunos tipos de cáncer (12-14). Un análisis de datos de 2015 de la Encuesta Nacional de Salud y Examen Nutricional de los EE. UU (NHANES 2001-2010) en 9,148 adultos (edad media, 50 años) encontró un riesgo 32% menor de síndrome metabólico en aquellos en el cuantil más alto versus más bajo de ingesta de magnesio (≥355 mg/día versus <197 mg/día) (15). Varios meta-análisis de principalmente estudios transversales también han informado una asociación inversa entre la ingesta dietética de magnesio y el riesgo de síndrome metabólico (16-18). Además, se han informado concentraciones séricas más bajas de magnesio en individuos con síndrome metabólico en comparación con los controles (18, 19). Sin embargo, el magnesio circulante representa solo el 1% de las reservas corporales totales y está estrictamente regulado; por lo tanto, las concentraciones séricas de magnesio no reflejan mejor el estatus de magnesio (1). En la actualidad, se necesita evidencia adicional de estudios diseñados prospectivamente para informar la posible relación entre el magnesio dietético y circulante y el riesgo de síndrome metabólico.
La inflamación sistémica, que contribuye al desarrollo de trastornos metabólicos, se ha correlacionado inversamente con la ingesta de magnesio en un estudio transversal de 11,686 mujeres (≥45 años). En este estudio, la prevalencia más baja de síndrome metabólico se encontró en el grupo de mujeres en el quintil más alto de ingestas de magnesio (ingesta mediana, 422 mg/día) (20). Varios ensayos aleatorios controlados también informaron una reducción en la proteína C reactiva circulante (PCR) — un marcador de inflamación — después de la administración de suplementos de magnesio por vía oral (21). Esto podría constituir un mecanismo potencial a través del cual el magnesio podría desempeñar un papel en la prevención de trastornos metabólicos.
Enfermedad cardiovascular
Hipertensión (presión sanguínea alta)
Grandes estudios de cohorte prospectivos han examinado la relación entre el magnesio y la presión arterial. Sin embargo, el hecho de que los alimentos con alto contenido de magnesio (frutas, verduras, granos integrales) con frecuencia tengan un alto contenido de potasio y fibra dietética ha dificultado la evaluación del efecto independiente del magnesio sobre la presión arterial. Hallazgos de grandes cohortes, incluido Estudio de Seguimiento de Profesionales de la Salud (HPFS, por sus siglas en inglés) (22), el Estudio de Salud de Enfermeras (NHS) (23), el estudio del Riesgo de Aterosclerosis en Comunidades (RAEC) (24), y el estudio de Desarrollo de Arterias Coronarias en Adulto Jóvenes (CARDIA, por sus siglas en inglés) (25), se han resumido en un meta-análisis reciente (26). El análisis agrupado de siete estudios prospectivos mostró un riesgo 8% menor de hipertensión con ingestas dietéticas de magnesio más altas o más bajas (26). En uno de estos estudios, los datos de 5,511 hombres y mujeres seguidos durante un período medio de 7.6 años encontraron que las concentraciones más altas de magnesio en la orina correspondían a una reducción del 25% en el riesgo de hipertensión, mientras que no hubo asociación entre las concentraciones en plasma de magnesio y el riesgo de hipertensión (27). Tampoco hubo evidencia de una asociación entre las concentraciones circulantes de magnesio y el riesgo de hipertensión en un meta-análisis de tres estudios de cohorte prospectivos (26).
La relación entre la ingesta de magnesio y el riesgo de hipertensión sugiere que mejorar la calidad de la dieta o usar suplementos de magnesio podría desempeñar un papel en la prevención de la hipertensión en aquellos con ingestas dietéticas inadecuadas.
Calcificación vascular
La acumulación de placa dentro de las paredes arteriales — un proceso llamado aterosclerosis — es un evento temprano en el desarrollo de enfermedades cardiovasculares. La calcificación de las placas ateroscleróticas que ocurre con la progresión de la aterosclerosis se ha asociado con un aumento de tres a cuatro veces en el riesgo de eventos cardiovasculares y mortalidad (28).
Individuos con enfermedad renal crónica (ERC): Las anomalías en el metabolismo mineral y óseo no son infrecuentes en personas con insuficiencia renal y se han asociado con un mayor riesgo de enfermedad cardiovascular y mortalidad (29, 30). En particular, se cree que la concentración elevada de fósforo en la sangre y la mayor deposición de fosfato de calcio dentro de la vasculatura promueven la calcificación vascular. Dado que el magnesio puede funcionar como un antagonista del calcio, se ha sugerido que podría utilizarse para ralentizar o revertir la calcificación de los vasos observados en pacientes con ERC. En un estudio transversal en pacientes con ERC que no requieren diálisis, las concentraciones séricas más altas de magnesio se asociaron con puntuaciones más bajas de densidad de calcificación de la arteria coronaria en aquellos en el extremo más alto de las concentraciones séricas normales de fósforo (es decir, ≥3.4 mg/dL) (31). Un pequeño ensayo aleatorizado, controlado con placebo en participantes con ERC que no requieren diálisis examinó el efecto del hidróxido de magnesio oral de liberación lenta sobre la propensión a la calcificación del suero midiendo el tiempo necesario para que las partículas primarias de calciproteína (que contienen fosfato de calcio amorfo) se transformen en partículas secundarias de calciproteína (que contienen hidroxiapatita cristalina) (32). El aumento de la propensión a la calcificación del suero se ha asociado con un mayor riesgo de mortalidad en pacientes con funciones renales deterioradas (33, 34). El ensayo encontró un aumento en la concentración sérica de magnesio y una reducción en la propensión a la calcificación del suero — es decir, un aumento en el tiempo necesario para que ocurra el 50% de la transformación (T50) — con 720 mg/día de magnesio suplementario durante ocho semanas en comparación con placebo (32). La propensión a la calcificación del suero también se redujo cuando se aumentó la concentración de magnesio (de 1 a 2 mEq/L durante 28 días) en el dialisato de pacientes con insuficiencia renal establecida (35). Se está realizando un ensayo aleatorio controlado más grande en pacientes con ERC que no requieren diálisis, para examinar más a fondo el efecto del magnesio oral en los marcadores de calcificación vascular, marcadores del metabolismo mineral y óseo, incidencia de eventos cardiovasculares, y deterioro de la función renal (36).
Individuos con función renal normal: El análisis transversal de los datos de 2,695 participantes de mediana edad en el Framingham Heart Study mostró que las probabilidades de tener calcificación de la arteria coronaria fueron 58% más bajas en aquellos en el cuartil más alto versus el más bajo de ingestas totales de magnesio (valores medios, 427 mg/día versus 259 mg/día) (37). La concentración sérica de magnesio también se encontró inversamente asociada con la calcificación vascular en estudios transversales recientes basados en la población (38-40). Ninguna investigación ha examinado aún si mejorar el estatus de magnesio de personas generalmente sanas podría desempeñar un papel en la prevención de la aterosclerosis.
Riesgo de enfermedad cardiovascular
Ingestas dietéticas de magnesio: Varios grandes estudios de cohorte prospectivos, incluido el Estudio de Seguimiento de Profesionales de la Salud (HPFS, por sus siglas en inglés) y el Estudio de Salud de Enfermeras (NHS), han examinado la ingesta de magnesio con relación a los resultados cardiovasculares. En el análisis más reciente del NHS, que siguió a cerca de 90,000 enfermeras durante 28 años, las que se encontraban en el quintil más alto de ingesta de magnesio tenían un riesgo 39% menor de infarto al miocardio mortal (pero no enfermedad coronaria no mortal [EAC]) en comparación con aquellos en el quintil más bajo (>342 mg/día versus <246 mg/día) (41). Un meta-análisis de nueve estudios de cohorte prospectivos, realizado principalmente en participantes sin enfermedad cardiovascular en la base, reportó un riesgo 22% menor de EAC por ingesta dietética incremental de 200 mg/día en magnesio (42). Un meta-análisis más reciente de Fang et al. (43) incluyó seis estudios e informó un riesgo 10% menor de EAC con ingestas dietéticas de magnesio más altas versus más bajas.
Las ingestas más altas de magnesio se asociaron con una reducción del 8 al 11% en el riesgo de accidente cerebrovascular en dos meta-análisis de estudios prospectivos, cada uno con más de 240,000 participantes (44, 45). El análisis agrupado más reciente de 14 estudios encontró un riesgo 12% menor de accidente cerebrovascular con ingestas de magnesio más altas versus más bajas y estimó una reducción del riesgo en 7% de accidente cerebrovascular asociado con cada incremento de 100-mg en la ingesta diaria de magnesio (43).
Sólo dos estudios prospectivos han examinado el riesgo de insuficiencia cardíaca con relación a la ingesta de magnesio. El análisis agrupado sugirió un riesgo 31% menor de insuficiencia cardíaca con ingestas dietéticas de magnesio más altas (43).
Finalmente, un meta-análisis de 13 estudios prospectivos en más de 475,000 participantes reportó que el riesgo de eventos cardiovasculares totales, incluyendo accidente cerebrovascular, infarto al miocardio no mortal y EAC, fue 15% menor en individuos con ingestas más altas de magnesio (46). Sin embargo, en el meta-análisis reciente de ocho estudios de Fang et al. (43), no hubo asociación entre la ingesta dietética de magnesio y el riesgo de enfermedad cardiovascular total.
Es importante tener en cuenta que si bien estos estudios de cohorte prospectivos evaluaron la asociación entre el magnesio dietético y la enfermedad cardiovascular; los estudios no tuvieron en cuenta el uso de magnesio suplementario por una fracción significativa de los participantes.
Concentraciones séricas de magnesio: Un gran estudio prospectivo (casi 14,000 hombres y mujeres) asoció mayores concentraciones séricas de magnesio con un menor riesgo de ECC en mujeres pero no en hombres (47). Este estudio se incluyó en un meta-análisis de cuatro estudios que no mostraron evidencia de algún riesgo disminuido de EAC con el aumento de las concentraciones séricas de magnesio (42). En contraste, un incremento de 0.2 mmol/L en la concentración sérica de magnesio se asoció con un riesgo 30% menor de enfermedad cardiovascular total en un análisis agrupado de ocho estudios de cohorte prospectivos (42). En el recientemente publicado British Regional Heart Study que siguió a 3.523 hombres durante una media de 15 años, no hubo asociación entre la concentración sérica de magnesio y los eventos incidentales de EAC, sin embargo, la concentración sérica de magnesio se asoció inversamente con el riesgo de insuficiencia cardíaca (48).
Mortalidad cardiovascular
Varios estudios iniciales encontraron una menor mortalidad relacionada con enfermedades cardiovasculares en poblaciones que consumen agua "dura" habitualmente. El agua dura (alcalina) generalmente es alta en magnesio, pero también puede contener más calcio y fluoruro que el agua "blanda," lo que hace que los efectos cardioprotectores del agua dura sean difíciles de atribuir al magnesio solamente (49). Además, los meta-análisis de estudios prospectivos no han encontrado asociaciones entre la ingesta de magnesio y las enfermedades cardiovasculares (50) o mortalidad por todas las causas (43). En un análisis prospectivo de los datos del NHANES de 14,353 participantes, seguido por un período medio de 28.6 años, el riesgo de mortalidad por todas las causas y accidente cerebrovascular aumentó significativamente en aquellos con concentraciones séricas de magnesio bajas en lugar de normales (<0.7 mmol/L versus 0.8-0.89 mmol/L) (51). Por el contrario, la hipermagnesemia (concentración sérica de magnesio >0,89 mmol/L) — en lugar de hipomagnesemia — en personas con insuficiencia cardíaca se asoció con un mayor riesgo de mortalidad cardiovascular y por todas las causas (52).
Hemorragia subaracnoidea aneurismática
Se ha reportado la aparición de hipomagnesemia en pacientes que sufrieron una hemorragia subaracnoidea (un tipo de accidente cerebrovascular) causada por la ruptura de un aneurisma cerebral (53). Los malos resultados neurológicos después de una hemorragia subaracnoidea aneurismática (aSAH, en inglés) se han relacionado con una contracción dependiente de calcio inapropiada de las arterias (conocida como vasoespasmo arterial cerebral), lo que lleva a una isquemia cerebral tardía (54). Debido a que el magnesio es un antagonista del calcio y un vasodilatador potente, varios ensayos aleatorios controlados han examinado si las infusiones intravenosas de sulfato de magnesio podrían reducir la incidencia de vasoespasmo después de una aSAH. Un meta-análisis de nueve ensayos aleatorios controlados encontró que la terapia con magnesio después de una aSAH disminuyó significativamente el vasoespasmo, pero no pudo prevenir el deterioro neurológico ni disminuir el riesgo de muerte (55). Otro meta-análisis de 13 ensayos en 2,413 pacientes con aSAH concluyó que la infusión de sulfato de magnesio no tuvo ningún beneficio con respecto al resultado neurológico y la mortalidad, a pesar de una reducción en la incidencia de isquemia cerebral tardía (56). El análisis post-hoc de un pequeño ensayo aleatorio controlado sugirió que mantener la infusión de sulfato de magnesio durante 10 días después de la aSAH o hasta que desaparezcan los signos de vasoespasmo podría proteger contra el infarto cerebral secundario cuando están presentes marcadores de vasoconstricción y reducción de la perfusión cerebral (57, 58). La evidencia actual no respalda el uso de suplementos de magnesio en la práctica clínica para pacientes con aSAH más allá de la normalización del estatus de magnesio.
Complicaciones de la cirugía cardíaca
La arritmia auricular (también llamada fibrilación auricular) es una afección definida como la aparición de anomalías persistentes en la frecuencia cardíaca; tales arritmias a menudo complican la recuperación de los pacientes después de la cirugía cardíaca. El uso de magnesio en la profilaxis de la arritmia auricular postoperatoria después de la cirugía de revascularización coronaria ha sido evaluado como un agente único o adjunto a las moléculas antiarrítmicas clásicas (conocidas como β-bloqueadores y amiodarona) en varios ensayos aleatorios controlados prospectivos. Un meta-análisis de 21 estudios de intervención mostró que las infusiones intravenosas de magnesio pudieron significativamente reducir la arritmia auricular postoperatoria en pacientes tratados en comparación a los no tratados (59). Los resultados de un meta-análisis más reciente de 22 ensayos controlados con placebo sugirieron que el magnesio puede reducir efectivamente la arritmia auricular cuando se administra después de la operación, como un bolo, y durante más de 24 horas (60). Sin embargo, otro meta-análisis de cuatro ensayos encontró que el magnesio no era más efectivo que otros agentes antiarrítmicos (60). Además, el meta-análisis de cinco ensayos aleatorios controlados también sugirió que el magnesio intravenoso agregado al tratamiento con β-bloqueadores no disminuyó el riesgo de arritmia auricular en comparación con el β-bloqueador solo y se asoció con más efectos adversos (bradicardia e hipotensión) (61). Actualmente, todavía faltan pruebas de alta calidad para respaldar el uso de magnesio en la profilaxis de la fibrilación auricular postoperatoria y otras arritmias en pacientes con contraindicaciones a los agentes antiarrítmicos de primera línea (60).
Diabetes mellitus
Las preocupaciones de salud pública con respecto a las epidemias de obesidad y diabetes mellitus tipo 2 y el papel prominente del magnesio en el metabolismo de la glucosa han llevado a los científicos a investigar la relación entre el consumo de magnesio y la diabetes mellitus tipo 2. Un estudio de cohorte prospectivo que siguió a más de 25,000 individuos, de 35 a 65 años de edad, durante siete años no encontró diferencias en la incidencia de diabetes mellitus tipo 2 al comparar el quintil más alto (377 mg/día) de ingesta de magnesio con el quintil más bajo (268 mg/día) (62). Sin embargo, la inclusión de este estudio en un meta-análisis de ocho estudios de cohorte mostró que el riesgo de diabetes tipo 2 estaba inversamente correlacionado con la ingesta de magnesio (62). El meta-análisis más reciente de 25 estudios prospectivos de cohortes, incluidos 637,922 individuos y 26,828 casos nuevos de diabetes mellitus tipo 2, encontró que una mayor ingesta de magnesio se asoció con un riesgo 17% menor de diabetes mellitus tipo 2 (63). Varios meta-análisis realizados hasta la fecha reportaron una disminución del 8 al 15% en el riesgo de desarrollar diabetes mellitus tipo 2 con cada incremento de 100 mg en la ingesta dietética de magnesio (63-66).
La resistencia a la insulina, caracterizada por alteraciones tanto en la secreción de insulina por el páncreas como por la acción de la insulina en los tejidos diana, se ha relacionado con un estatus inadecuado de magnesio. Un análisis transversal del Cohortes para la Investigación del Corazón y el Envejecimiento en el Consorcio de Epidemiología Genómica (CHARGE, en inglés) que incluyó 15 cohortes con un total de 52,684 participantes sin diabetes, mostró que las ingestas de magnesio estaban inversamente asociadas con las concentraciones de insulina en ayunas después de múltiples ajustes, incluyendo varios factores de estilo de vida, índice de masa corporal (IMC), consumo de cafeína y consumo de fibra (65). Se piensa que las células pancreáticas β, que secretan insulina, podrían ser menos responsivo a los cambios en la sensibilidad a la insulina en individuos con deficiencia de magnesio (67). Un ensayo aleatorizado, doble ciego, controlado con placebo que inscribió a 97 adultos sanos con hipomagnesemia significativa (concentración sérica de magnesio ≤0.70 mmol/L) mostró que el consumo diario de 638 mg de magnesio (de una solución de cloruro de magnesio) durante tres meses mejoró la función de las células pancreáticas β, lo que resulta en concentraciones más bajas de glucosa e insulina en ayunas (68). En un ensayo aleatorio controlado de seguimiento, la administración de 382 mg/día de magnesio durante cuatro meses a los participantes (edad media, 42 años) con hipomagnesemia (concentración sérica de magnesio <0,74 mmoles/L) y glucosa en ayunas deteriorada mejoró las concentraciones séricas de magnesio, así como las concentraciones de glucosa en ayunas y poscarga (69). Otros marcadores metabólicos, incluidos los triglicéridos en suero, el colesterol HDL, y una medida de resistencia a la insulina, también mejoraron en individuos tratados con magnesio versus placebo (69). Además, se han reportado mejoras metabólicas similares después de la suplementación de magnesio (382 mg/día durante cuatro meses) a los participantes que eran tanto hipomagnesémicos como delgados pero metabólicamente obesos (es decir, con trastornos metabólicos generalmente asociados con la obesidad) (70). En otro estudio, la suplementación con 365 mg/día de magnesio (del hidrocloruro de aspartato de magnesio) durante seis meses disminuyó la resistencia a la insulina en 27 personas con sobrepeso con valores normales de suero y magnesio intracelular (71). Este último estudio sugiere que el magnesio podría tener efectos adicionales sobre la tolerancia a la glucosa y la sensibilidad a la insulina que van más allá de la normalización de las concentraciones séricas de magnesio en individuos con hipomagnesemia.
Osteoporosis
Aunque la disminución de la densidad mineral ósea (DMO) es la característica principal de la osteoporosis, otros cambios osteoporóticos en la matriz de colágeno y la composición mineral ósea pueden dar como resultado huesos que son frágiles y más susceptibles a fracturas (72). Alrededor del 60% del magnesio corporal total se almacena en el esqueleto y se sabe que influye tanto en la matriz ósea como en el metabolismo mineral óseo. El magnesio en la superficie de los huesos también está disponible para el intercambio dinámico con la sangre (73). A medida que disminuye el contenido de magnesio en el mineral óseo, los cristales de hidroxiapatita del hueso pueden hacerse más grandes y más frágiles. Algunos estudios han encontrado un menor contenido de magnesio y cristales de hidroxiapatita más grandes en los huesos de las mujeres con osteoporosis en comparación con las mujeres sin la enfermedad (74). Es sabido que concentraciones séricas inadecuadas de magnesio resultan en bajas concentraciones séricas de calcio, resistencia a la acción de la hormona paratiroidea (PTH, en inglés), y en resistencia a algunos de los efectos de la vitamina D (calcitriol), cada uno de los cuales puede conducir a un incremento en la pérdida ósea (véase los artículos sobre Vitamina D y Calcio). Las concentraciones séricas de magnesio más bajas pueden no ser inusuales en mujeres posmenopáusicas con osteoporosis (75), y la hipomagnesemia es un efecto adverso del uso del medicamento recetado teriparatida (Forsteo) en el tratamiento de la osteoporosis (76).
Las ingestas dietéticas de magnesio más altas se han asociado con un aumento de la DMO (77) específica del sitio y del cuerpo en estudios basados en la observación, incluyendo estudios de adultos mayores. Más recientemente, un gran estudio de cohorte realizado en casi dos tercios de la población noruega encontró que el nivel de magnesio en el agua potable está inversamente asociado con el riesgo de fractura de cadera (79). En el estudio de la Iniciativa de Salud de las Mujeres, el análisis de datos de 4,778 participantes (edad media, 63 años) seguido durante aproximadamente siete años mostró que las ingestas de magnesio más altas se asociaron con una DMO más alta de la cadera y de todo el cuerpo, pero no con una reducción de la cadera o fracturas totales (80). Además, el quintil más alto versus el más bajo de la ingesta total de magnesio se asoció con un riesgo 23% mayor de fracturas en la parte inferior del brazo y la muñeca (80). En un estudio de cohorte de casos anidado dentro de la Investigación Prospectiva Europea sobre el Cáncer y Nutrición (EPIC)-Norfolk, que incluyó a 5.319 individuos, se descubrió que las ingestas totales de magnesio y potasio estaban inversamente asociadas con la medición de la atenuación de ultrasonido de banda ancha (BUA, en inglés) del hueso del talón (calcáneo) — que predicen el riesgo de fractura incidental — y con el riesgo de fracturas de cadera (81).
Pocos estudios han abordado el efecto de la suplementación con magnesio sobre la DMO o la osteoporosis en humanos. En un pequeño grupo de mujeres posmenopáusicas con osteoporosis, la suplementación con magnesio de 750 mg/día durante seis meses, seguida de 250 mg/día durante 18 meses más, produjo un aumento de la DMO en la muñeca después de un año, sin un aumento adicional después de dos años de suplementación (82). Un estudio en mujeres posmenopáusicas que estaban tomando terapia de reemplazo de estrógenos y un suplemento multivitamínico encontró que la suplementación con 400 mg/día adicionales de magnesio y 600 mg/día de calcio resultó en un aumento de la DMO en el talón en comparación con las mujeres posmenopáusicas que recibieron solamente terapia de reemplazo de estrógenos (83). Un estudio aleatorio controlado más reciente realizado en 20 mujeres posmenopáusicas con osteoporosis sugirió que la suplementación con dosis altas de citrato de magnesio (1,830 mg/día) durante un mes podría reducir la rápida tasa de pérdida ósea que caracteriza la osteoporosis (84). La evidencia aún no es suficiente para sugerir que el magnesio suplementario en exceso de la IDR podría ser eficaz en la prevención de la osteoporosis a menos que se requiera la normalización de la concentración sérica de magnesio (85).
Sarcopenia
La sarcopenia es una condición caracterizada por una pérdida de masa del músculo esquelético que aumenta la fragilidad y el riesgo de caídas en adultos mayores (86). Varios estudios transversales han reportado una asociación positiva entre la ingesta dietética de magnesio y las medidas indirectas de la masa del músculo esquelético en adultos de mediana edad y adultos mayores (87-90). Un estudio aleatorio controlado de 2014 en 139 mujeres mayores sanas y físicamente activas (edad media, 71.5 años) encontró un impacto escaso o nulo de la suplementación con magnesio (900 mg/día de óxido de magnesio) durante 12 semanas en la composición corporal y la fuerza muscular, aún así, se mejoró el puntaje de la prueba de Short Physical Performance Battery [SPPB] — un indicador compuesto de la función física (91). Se necesita más investigación para examinar más a fondo el efecto de los suplementos de magnesio en la composición corporal, la fuerza muscular, y el rendimiento físico en adultos mayores, ya sean físicamente activos o sedentarios, y con un estatus de magnesio normal o inadecuado.
Tratamiento de Enfermedad
El uso de dosis farmacológicas de magnesio para tratar enfermedades específicas se discute a continuación. Aunque muchos de los estudios citados usaron magnesio suplementario en dosis considerablemente más altas que el nivel máximo de ingesta tolerable (NM), el cual es de 350 mg/día establecido por la Junta de Nutrición y Alimentos (véase Seguridad), es importante destacar que estos estudios se realizaron bajo supervisión médica. Debido a los potenciales riesgos de las dosis elevadas de magnesio suplementario, especialmente en presencia de una función renal deteriorada, cualquier ensayo de tratamiento de enfermedades que utilice dosis de magnesio más altas que el NM debe realizarse bajo supervisión médica. Además, el magnesio intravenoso se ha utilizado en el tratamiento de varias afecciones.
Complicaciones en el embarazo
Preeclampsia y eclampsia
La preeclampsia y eclampsia del embarazo son condiciones específicas del embarazo que pueden ocurrir en cualquier momento, entre las 20 semanas de gestación y las seis semanas posteriores al nacimiento. La preeclampsia (a veces llamada toxemia del embarazo) afecta aproximadamente al 4% de las mujeres embarazadas en los EE.UU. (92). La preeclampsia se define como la presencia de presión arterial elevada (hipertensión), proteínas en la orina e hinchazón severa (edema) durante el embarazo (93). La eclampsia ocurre con la adición de convulsiones a la tríada de síntomas preeclámpticos y es una causa importante de mortalidad perinatal y materna (93, 94). Aunque los casos de preeclampsia tienen un alto riesgo de desarrollar eclampsia, una cuarta parte de las mujeres eclámpticas inicialmente no presentan síntomas preeclámpticos (95).
Aunque se han reportado concentraciones más bajas de magnesio en la sangre y el cerebro de mujeres con preeclampsia que en mujeres embarazadas sanas, no hay evidencia de que el desequilibrio de magnesio pueda causar eventos adversos en el embarazo. Un meta-análisis de 2014 de 10 ensayos aleatorios controlados no encontró ningún efecto de la administración oral de sal de magnesio durante embarazos normales y en riesgo sobre el riesgo de preeclampsia, mortalidad perinatal, y lactantes pequeños para la edad de gestación (96).
Durante muchos años, el sulfato de magnesio intravenoso en dosis altas ha sido el tratamiento de elección para prevenir las convulsiones eclámpticas que pueden ocurrir en asociación con preeclampsia grave en el embarazo o durante el parto (97, 98). Una revisión sistemática de siete ensayos aleatorios en 1,396 mujeres con eclampsia comparó el efecto de la administración de sulfato de magnesio con el tratamiento con diazepam (un anticonvulsivo conocido) sobre los resultados perinatales. El régimen de magnesio disminuyó significativamente los riesgos de convulsiones recurrentes y muerte materna en comparación con el diazepam. Además, el uso de magnesio para el cuidado de mujeres eclámpticas resultó en recién nacidos con puntajes de Apgar más altos; no hubo diferencias significativas en el riesgo de parto prematuro y mortalidad perinatal (95). Investigaciones adicionales han confirmado que la infusión de sulfato de magnesio siempre debe considerarse en el tratamiento de la preeclampsia y la eclampsia severas para prevenir las convulsiones iniciales y recurrentes (99). Además, la Organización Mundial de la Salud (OMS) recomienda el uso de sulfato de magnesio — administrado por vía intramuscular o intravenosa — como tratamiento de primera línea para la prevención de la eclampsia en mujeres con preeclampsia grave, en lugar de otros anticonvulsivos (100). Se necesita más investigación para evaluar la eficacia de la infusión de sal de magnesio en la profilaxis de la eclampsia en mujeres con preeclampsia leve (101). Además, no está claro si es necesario prolongar el uso de magnesio después del parto en mujeres que presentaron preeclampsia grave durante el embarazo para reducir el riesgo de eclampsia después del parto (102).
Neuroprotección perinatal
Mientras que el sulfato de magnesio intravenoso está incluido en la atención médica de la preeclampsia y eclampsia, el Colegio Americano de Obstetras y Ginecólogos y la Sociedad para la Medicina Materno-Fetal apoyan su uso en dos situaciones adicionales: condiciones específicas de la prolongación a corto plazo del embarazo y la neuroprotección del feto en el parto prematuro (103).
El parto pretérmino, que se define por el parto prematuro de un bebé entre las 20 y 37 semanas de gestación estimada, se asocia con un mayor riesgo de mortalidad perinatal y morbilidad a corto y largo plazo. El Colegio Americano de Obstetras y Ginecólogos y la Sociedad para la Medicina Materno-Fetal aprueba el uso de diferentes clases de medicamentos — conocidos como tocolíticos — que están destinados a retrasar el parto durante el tiempo suficiente para que los corticoides prenatales puedan usarse para acelerar la maduración pulmonar en el feto de mujeres con riesgo inminente de parto pretérmino (104). Un meta-análisis del 2014 de 37 ensayos encontró que la infusión intravenosa de sulfato de magnesio no era más eficaz que los tocolíticos comúnmente utilizados (p. ej., agonistas de los receptores β-adrenérgicos, bloqueadores de los canales de calcio, inhibidores de prostaglandinas) para retrasar el parto o prevenir resultados infantiles graves (105). La evidencia muy limitada también sugirió que la infusión de magnesio en dosis altas versus bajas puede reducir la duración de las hospitalizaciones en recién nacidos ingresados en unidades de cuidados intensivos (106).
La relación entre el sulfato de magnesio y el riesgo de daño cerebral en bebés prematuros se ha evaluado en estudios basados en la observación. Un meta-análisis de seis estudios de caso y control y cinco estudios de cohorte prospectivos mostró que el uso de magnesio disminuyó significativamente el riesgo de parálisis cerebral, así como la mortalidad (107). Sin embargo, el alto grado de heterogeneidad entre los estudios de cohortes y el hecho de que la exposición a corticosteroides (que se sabe que disminuye la mortalidad prenatal) fue mayor en los casos de niños expuestos al magnesio en comparación con los controles implica una interpretación cautelosa de los resultados. Sin embargo, un meta-análisis de cinco ensayos aleatorios controlados, que incluyó a 5,493 mujeres en riesgo de parto prematuro y 6,135 bebés, encontró que la terapia con magnesio administrada a madres que dan a luz antes del término disminuyó el riesgo de parálisis cerebral en un 32% sin causar eventos maternos adversos severos, pero este tratamiento no disminuyó el riesgo de otros trastornos neurológicos o mortalidad en la primera infancia (108). Otro meta-análisis realizado en cinco ensayos aleatorios controlados encontró que la administración intravenosa de magnesio a los recién nacidos que sufrían de asfixia perinatal podría ser beneficiosa en términos de resultados neurológicos a corto plazo, aunque no hubo ningún efecto sobre la mortalidad (109). Se necesitan ensayos adicionales para evaluar los beneficios a largo plazo del magnesio en la atención pediátrica.
Enfermedad cardiovascular
Hipertensión
Si bien los resultados de los estudios de intervención no han sido completamente consistentes (2), la última revisión de los datos destacó un beneficio terapéutico de los suplementos de magnesio en el tratamiento de la hipertensión. Un meta-análisis de 2012 examinó 22 ensayos aleatorios, controlados con placebo de suplementación con magnesio realizados en 1,173 individuos con presión sanguínea normal (normotensiva) o hipertensión (tratados con medicamentos o sin tratamiento). La suplementación oral con magnesio (dosis media de 410 mg/día; rango de 120 a 973 mg/día) durante un período medio de 11.3 meses redujo significativamente la presión sanguínea sistólica en 2 a 3 mm Hg y la presión sanguínea diastólica en 3 a 4 mm Hg (110); se observó un mayor efecto a dosis más altas (≥370 mg/día). Los resultados de 19 de los 22 ensayos incluidos en el meta-análisis se revisaron previamente junto con otros 25 estudios de intervención (111). El examen sistemático de estos 44 ensayos sugirió un efecto reductor de la presión sanguínea asociado con el magnesio suplementario en individuos hipertensos pero no normotensos.
Las dosis de magnesio requeridas para lograr una disminución en la presión sanguínea parecían depender de si los participantes con presión sanguínea alta fueron tratados con medicamentos antihipertensivos, incluyendo los diuréticos. Los ensayos de intervención en participantes tratados mostraron una disminución en la hipertensión con dosis de magnesio de 243 mg/día a 486 mg/día, mientras que los pacientes no tratados requirieron dosis superiores a 486 mg/día para lograr una disminución significativa de la presión sanguínea. Un meta-análisis más reciente de estudios aleatorios controlados con 2,028 participantes encontró que el magnesio suplementario a una dosis media de 368 mg/día (rango: 238-960 mg/día) durante una mediana de tres meses (rango: 3 semanas-6 meses) aumentó la concentración sérica de magnesio en 0.05 mmol/L (27 ensayos) y redujo la presión sanguínea sistólica en 2 mm Hg y la presión sanguínea diastólica en 1.78 mm Hg (37 ensayos) (112). Un meta-análisis de 2017 restringido a ensayos en participantes con condiciones pre-clínicas subyacentes (resistencia a la insulina o prediabetes) o clínicas (diabetes mellitus tipo 2 o enfermedad coronaria) encontró una disminución de 4.18 mm Hg en la presión sanguínea sistólica y una disminución de 2.27 mm Hg en la presión sanguínea diastólica con dosis suplementarias de magnesio que oscilan entre 365 mg/día y 450 mg/día durante uno a seis meses (113).
Mientras que la administración suplementaria de magnesio por vía oral puede ser útil en individuos hipertensos que están agotados de magnesio debido al uso crónico de diuréticos y/o una ingesta dietética inadecuada (7), varios factores dietéticos juegan un papel en la hipertensión. Por ejemplo, la adherencia a la dieta DASH — una dieta rica en frutas, verduras, y productos lácteos bajos en grasa y baja en grasas saturadas y totales — se ha relacionado con reducciones significativas en la presión sanguínea sistólica y diastólica (114).
Aterosclerosis
Las células endoteliales vasculares recubren las paredes arteriales donde están en contacto con la sangre que fluye a través del sistema circulatorio. El endotelio vascular que funciona normalmente promueve la vasodilatación cuando es necesario, por ejemplo, durante el ejercicio, e inhibe la formación de coágulos sanguíneos. Por el contrario, la disfunción endotelial produce vasoconstricción generalizada y anomalías en la coagulación. En la enfermedad cardiovascular, la inflamación crónica se asocia con la formación de placas ateroscleróticas en las arterias. La aterosclerosis deteriora la función endotelial normal, aumentando el riesgo de vasoconstricción y formación de coágulos, lo que puede conducir a un ataque al corazón o accidente cerebrovascular (revisado en 115). Una revisión sistemática reciente identificó seis ensayos aleatorios controlados que examinaron el efecto de las dosis farmacológicas de magnesio oral en la función endotelial vascular (116). Tres de seis ensayos, que incluyeron individuos con enfermedad arterial coronaria (117), diabetes mellitus (118), o hipertensión (119), reportaron una mejora en la dilatación mediada por flujo (DMF) con magnesio suplementario en comparación con el control. En otras palabras, mejoró la respuesta de dilatación normal de la arteria braquial (del brazo) para incrementar el flujo de sangre. Por el contrario, no hubo evidencia de algún efecto de los suplementos de magnesio en la DMF en tres ensayos realizados en pacientes de hemodiálisis (120) o participantes sanos con normal (121) o alto índice de masa corporal (IMC) (122). Un análisis agrupado de los seis ensayos en 262 participantes encontró que la suplementación con 107 a 730 mg/día de magnesio durante uno a seis meses resultó en una mejora general de la DMF, independientemente del estado de salud o las concentraciones basales de magnesio de los participantes (116).
La medición del grosor de las capas internas de las arterias carótidas a veces se usa como un marcador sustituto de la aterosclerosis (123). Mayores concentraciones séricas de magnesio se han asociado con una reducción del grosor íntima-media carotídeo (CIMT, en inglés) en todas las mujeres y en los hombres caucásicos que participan en el estudio de Riesgo de Aterosclerosis en Comunidades (RAEC) (124). Un meta-análisis de cuatro estudios de intervención pequeños (145 participantes en total) y heterogéneos no encontró ningún efecto de la suplementación con magnesio (98.6 a 368 mg/día durante 2 a 6 meses) sobre CIMT (116).
Supervivencia después de infarto al miocardio
Los resultados de varios ensayos aleatorizados, controlados con placebo han sugerido que un magnesio intravenoso administrado temprano después de un presunto infarto al miocardio podría disminuir el riesgo de muerte. El estudio más influyente fue un ensayo aleatorizado, controlado con placebo en 2,316 pacientes que encontró una disminución significativa en la mortalidad en el grupo de pacientes que recibieron sulfato de magnesio intravenoso dentro de las 24 horas de sospecha de infarto de miocardio (7.8% de mortalidad por todas las causas en el grupo experimental vs. 10.3% de mortalidad por todas las causas en el grupo placebo) (125). El seguimiento de uno a cinco años después del tratamiento reveló que la mortalidad por enfermedad cardiovascular fue 21% menor en el grupo tratado con magnesio (126). Sin embargo, un ensayo controlado con placebo más grande en más de 58,000 pacientes no encontró una disminución significativa en la mortalidad de cinco semanas en pacientes tratados con sulfato de magnesio intravenoso dentro de las 24 horas de sospecha de infarto al miocardio, lo que generó controversia con respecto a la eficacia de este tratamiento (127). Una encuesta de EE.UU. sobre el tratamiento de más de 173,000 individuos con infarto al miocardio agudo encontró que sólo el 5% recibió magnesio intravenoso en las primeras 24 horas después del infarto y que la mortalidad fue mayor en este grupo en comparación con el grupo de pacientes no tratados con magnesio (128). Una revisión sistemática de 2007 de 26 ensayos clínicos, incluyendo 73,363 participantes, concluyó que la administración intravenosa de magnesio no parece disminuir la mortalidad por infarto al miocardio y, por lo tanto, no debe utilizarse como tratamiento (129). Por lo tanto, el uso de sulfato de magnesio intravenoso en la terapia del infarto al miocardio agudo sigue siendo controvertido.
Diabetes mellitus
El agotamiento de magnesio se ha asociado con diabetes mellitus tipo 1 (dependiente de insulina) y tipo 2, así como con diabetes gestacional. Se han reportado bajas concentraciones séricas de magnesio (hipomagnesemia) en 13.5 a 47.7% de las personas con diabetes mellitus tipo 2 (130). Una de las causas del agotamiento puede ser una mayor pérdida urinaria de magnesio causada por una mayor excreción urinaria de glucosa que acompaña a la diabetes mal controlada. Se ha demostrado que el agotamiento del magnesio aumenta la resistencia a la insulina en algunos estudios y puede afectar negativamente el control de la glucosa en sangre en la diabetes mellitus (véase también Diabetes mellitus) (131). Un pequeño estudio en nueve personas con diabetes mellitus tipo 2 informó que el magnesio suplementario (300 mg/día durante 30 días), en forma de una solución de sal líquida que contiene magnesio, mejoró la insulina en ayunas pero no las concentraciones de glucosa en ayunas (132). Un estudio aleatorizado, doble ciego, controlado con placebo en 63 individuos con diabetes mellitus tipo 2 e hipomagnesemia encontró que aquellos que tomaban una solución oral de cloruro de magnesio (638 mg/día de magnesio elemental) durante 16 semanas habían mejorado las medidas de sensibilidad a la insulina y control glucémico en comparación con los que tomaban un placebo (133). El meta-análisis más reciente de nueve ensayos aleatorizados, doble ciego, y controlados, concluyó que el magnesio suplementario oral redujo las concentraciones plasmáticas de glucosa en ayunas en individuos con diabetes (134). Sin embargo, la suplementación con magnesio no mejoró otros marcadores de homeostasis de la glucosa, como la concentración de hemoglobina glicosilada (HbA1c), las concentraciones de insulina en ayunas y después de la carga de glucosa, y las medidas de resistencia a la insulina (134). Otro meta-análisis de ensayos que incluyó participantes con riesgo de diabetes mellitus o con diabetes mellitus sugirió que la evidencia para respaldar un beneficio de la suplementación con magnesio en las medidas de resistencia a la insulina fue más fuerte en sujetos con deficiencia de magnesio que en aquellos con concentraciones séricas normales de magnesio (135). La corrección de las deficiencias de magnesio existentes puede mejorar el metabolismo de la glucosa y la sensibilidad a la insulina en sujetos con diabetes, pero sigue siendo incierto si la suplementación con magnesio podría tener algún beneficio terapéutico en pacientes con un estatus adecuado de magnesio.
Asma
La aparición de hipomagnesemia puede ser mayor en pacientes con asma que en personas sin asma (136). Varios ensayos clínicos han examinado el efecto de las infusiones intravenosas de magnesio en los ataques agudo de asma en niños o adultos que no respondieron al tratamiento inicial en la sala de emergencias. De hecho, el magnesio puede promover la broncodilatación en sujetos con asma al interferir con mecanismos como la activación de los receptores de N-metil D-aspartato (NMDA) que desencadenan la broncoconstricción al facilitar la entrada de calcio en las células del músculo liso de las vías respiratorias (137). En un meta-análisis de seis ensayos aleatorios controlados (cuasi) en 325 niños con asma aguda tratados con un agonista del receptor β2-adrenérgico de acción corta (p. ej., salbutamol) y esteroides sistémicos, el tratamiento con sulfato de magnesio intravenoso mejoró las mediciones de la función respiratoria y redujo el riesgo de ingreso hospitalario en un 30% en comparación con el control (138). Otro meta-análisis de ensayos aleatorios controlados realizado principalmente en adultos con exacerbaciones del asma indicó que las infusiones únicas de 1.2 a 2 g de sulfato de magnesio durante 15 a 30 minutos podrían reducir el riesgo de ingreso hospitalario y mejorar la función pulmonar después de que los tratamientos iniciales fallaran (es decir, oxígeno, agonista β2 de acción corta, y esteroides) (139).
También se ha investigado el uso de magnesio inhalado nebulizado para tratar el asma. Una revisión sistemática reciente de 25 ensayos aleatorios controlados, incluidos adultos, niños, o ambos, encontró poca evidencia de que el sulfato de magnesio inhalado solo o junto con un agonista del receptor adrenérgico β2 y/o un anticolinérgico muscarínico (p. ej., ipratropio) podría mejorar la función pulmonar. en pacientes con asma aguda (140). Además, la suplementación oral con magnesio no tiene ningún valor conocido en el tratamiento del asma crónica (141-143).
Manejo del dolor
El potencial efecto analgésico del magnesio se atribuye en particular a su capacidad para bloquear los receptores NMDA, que se encuentran en el cerebro y la médula espinal y están involucrados en la transducción del dolor (144).
Dolor postoperatorio
Varios estudios de intervención han examinado el papel del magnesio en el control del dolor y el requerimiento analgésico en pacientes durante el período inmediato posterior a la cirugía.
Después de una cesárea: Las estrategias de manejo del dolor después de una cesárea generalmente implican la inyección de un analgésico en el espacio epidural (para la analgesia epidural) o en el espacio subaracnoideo (para la analgesia espinal [intratecal]). Un meta-análisis reciente de nueve ensayos aleatorios controlados resumió la evidencia sobre el uso potencial de sulfato de magnesio para controlar o aliviar el dolor postoperatorio en 827 mujeres que se sometieron a cesárea (145). Todos los ensayos evaluaron el efecto de un régimen analgésico de primera línea (es decir, bupivacaína o lidocaína, con o sin opioides) con y sin la adición de sulfato de magnesio. Los resultados sugirieron que la anestesia (8 estudios) y el bloqueo sensorial (6 estudios) duraron más en las mujeres que recibieron el sulfato de magnesio adicional. El uso de sulfato de magnesio también resultó en una puntuación de dolor más baja (3 estudios) y en un menor consumo postoperatorio de analgésicos (4 estudios). Además, no hubo diferencias en la aparición de efectos secundarios entre los regímenes (145). Un reciente ensayo aleatorio controlado en 60 mujeres sanas sometidas a cesárea electiva confirmó que la adición de sulfato de magnesio a un régimen de bupivacaína/opioide aumentó la duración de la anestesia espinal y redujo el nivel de dolor, pero no mejoró la potencia de la bupivacaína (146). En otro estudio en mujeres con preeclampsia leve que recibieron una inyección epidural de ropivacaína después de una cesárea, la infusión espinal de sulfato de magnesio aumentó la duración del bloqueo sensorial y motor, así como el tiempo antes de que los pacientes solicitaran un analgésico, en comparación con el midazolam (147).
Después de una variedad de otras cirugías: La eficacia del magnesio intravenoso también se ha examinado para el control del dolor local, regional, o sistémico, después de una variedad de cirugías diferentes. Una revisión de cuatro pequeños estudios aleatorios controlados sugirió que, cuando se agrega a los analgésicos locales, la infusión de magnesio a pacientes sometidos a tonsilectomía podría disminuir el dolor y la incidencia de laringoespasmo, extender el tiempo hasta el primer requerimiento analgésico postoperatorio, y disminuir el número de solicitudes de analgésicos postoperatorio (148). Se reportaron observaciones similares en dos meta-análisis adicionales, pero hubo discrepancias con respecto a la capacidad del magnesio para aliviar el dolor (149, 150). De hecho, la revisión de ocho ensayos por Xie et al. (150), de los cuales solo dos usaron la misma escala para puntuar el dolor, no mostraron alguna disminución en el dolor con el magnesio en comparación con el control. Finalmente, ambos meta-análisis no reportaron ninguna reducción en el riesgo de náuseas y vómitos postoperatorios con la administración intravenosa de magnesio (148, 150). Un meta-análisis de 2018 de cuatro ensayos aleatorios controlados en 263 pacientes también sugirió que la infusión de sulfato de magnesio puede ayudar a reducir las puntuaciones de dolor a las 2 y 8 horas (pero no 24 horas) después de la colecistectomía laparoscópica (151). Estudios recientes han examinado el uso de sulfato de magnesio para el control del dolor después de otras cirugías, incluidas la histerectomía (152, 153), la cirugía de columna (154, 155), o durante la endoscopia nasal (156) o la cirugía de implantación coclear (157). A pesar de los resultados contradictorios o los informes de beneficios limitados del magnesio, se necesitan más investigaciones antes de poder llegar a conclusiones.
Dolor neuropático
El efecto del magnesio sobre el dolor neuropático ha sido examinado en algunos estudios clínicos. Se encontró que la administración intravenosa de sulfato de magnesio alivia parcial o completamente el dolor en pacientes con neuralgia posherpética, un tipo de dolor neuropático causado por la infección por herpes zoster (culebrilla) (158, 159). En un ensayo aleatorio controlado más reciente en 45 pacientes con neuralgia posherpética o dolor neuropático de origen traumático o quirúrgico, la suplementación oral de magnesio no logró mejorar las medidas de dolor y la calidad de vida en comparación con un placebo (160). Se está realizando otro ensayo para examinar el impacto del magnesio intravenoso con ketamina en el dolor neuropático (161).
Dolores de cabeza por migrañas
Se han reportado concentraciones más bajas de magnesio intracelular (tanto en glóbulos rojos como en glóbulos blancos) en individuos que sufren de dolores de cabeza por migraña recurrentes en comparación con individuos sin migraña (162). Además, la incidencia de hipomagnesemia también parece ser mayor en las mujeres que experimentan migrañas con la menstruación en comparación con las mujeres sin migrañas menstruales (163).
Algunos estudios de intervención han examinado si un aumento en la concentración intracelular de magnesio con magnesio suplementario (oral) podría ayudar a disminuir la frecuencia y la gravedad de las migrañas en las personas afectadas. Dos ensayos tempranos controlados con placebo demostraron disminuciones modestas en la frecuencia de dolores de cabeza por migraña después de la suplementación con 600 mg/día de magnesio (162, 164). Otro ensayo controlado con placebo en 86 niños con frecuentes migrañas encontró que el óxido de magnesio oral (9 mg/kg de peso corporal/día) disminuyó la frecuencia de dolor de cabeza durante la intervención de 16 semanas (165). Sin embargo, no hubo reducción en la frecuencia de dolores de cabeza por migraña con 485 mg/día de magnesio en otro estudio controlado con placebo realizado en 69 adultos que sufrían ataques de migraña (166). La eficiencia de la absorción de magnesio varía con el tipo de complejo de magnesio oral, y esto podría explicar los resultados contradictorios. Aunque no se observaron efectos adversos graves durante estos ensayos de dolor de cabeza por migraña, del 19 al 40% de las personas que tomaron los suplementos de magnesio reportaron diarrea e irritación gástrica.
La eficacia de las infusiones de magnesio también se investigó en un ensayo cruzado aleatorio, simple ciego, controlado con placebo, de 30 pacientes con migraña (167). La administración de 1 gramo de sulfato de magnesio intravenoso puso fin a los ataques, abolió los síntomas asociados, y evitó la recurrencia en 24 horas en casi el 90% de los sujetos. Mientras que este resultado prometedor se confirmó en otro ensayo (168), dos estudios aleatorizados adicionales, controlados con placebo, encontraron que el sulfato de magnesio era menos efectivo que otras moléculas (p. ej., metoclopramida) en el tratamiento de las migrañas (169, 170). El meta-análisis más reciente de cinco ensayos aleatorizados, doble ciego, y controlados, no reportó ningún efecto beneficioso de la infusión de magnesio para la migraña en adultos (171). Otros dos estudios de intervención más recientes sugirieron que la infusión de sulfato de magnesio podría ser más efectiva y más rápida que la dexametasona/metoclopramida (172) o el citrato de cafeína (173) para aliviar el dolor en pacientes con migraña aguda.
La eficacia del magnesio debe examinarse en estudios más amplios que consideren el estatus del magnesio en quienes padecen migraña (174).
Lesión o enfermedad crítica
La hipomagnesemia no es infrecuente en pacientes ingresados en unidades de cuidados intensivos (UCI). Dos meta-análisis recientes de estudios de cohorte prospectivos y retrospectivos reportaron concentraciones séricas de magnesio ≤0.75 mmol/L en pacientes de la UCI al ingreso o dentro de las 24 horas posteriores al ingreso, lo que se asocia con una mayor necesidad de ventilación mecánica, mayor estadía en la UCI, y mayor riesgo de mortalidad hospitalaria (175, 176). Un análisis agrupado de tres estudios también sugirió un mayor riesgo de sepsis en pacientes con hipomagnesemia en la UCI (175). Un estudio prospectivo reciente realizado en pacientes ingresados con lesión grave en la cabeza encontró mejores resultados neurológicos después de seis meses en aquellos que presentaron concentraciones séricas normales de magnesio al ingreso en comparación con aquellos con hipomagnesemia (concentraciones séricas de magnesio <0.65 mmol/L) (177). Sin embargo, actualmente no hay evidencia disponible que sugiera que la administración de magnesio podría mejorar los resultados en pacientes críticos o con lesiones graves (178).
Fuentes
Fuentes alimenticias
El análisis de los datos de la encuesta nacional de nutrición de EE.UU. (NHANES 2003-2006) mostró una ingesta promedio de magnesio en adultos (edades ≥19 años) de 278 mg/día cuando sólo se consideraron fuentes de alimentos no fortificados (179). Considerando todas las fuentes de ingestas de magnesio (es decir, alimentos y suplementos no fortificados y fortificados), la ingesta promedio en adultos se estimó en alrededor de 330 mg/día — un valor cercano a los requerimientos estimados promedio (REP) de magnesio — lo que sugiere que aproximadamente la mitad de la población adulta puede estar en riesgo de insuficiencia de magnesio (179). Sin embargo, las consecuencias a largo plazo de la ingesta alimentaria inadecuada siguen sin estar claras (1).
Ya que el magnesio es parte de la clorofila, el pigmento verde en las plantas, los vegetales de hojas verdes son ricos en magnesio. Los granos sin refinar (granos enteros) y las nueces también tienen un alto contenido de magnesio. Las carnes y la leche tienen un contenido de magnesio intermedio, mientras que los alimentos refinados por lo general presentan el contenido de magnesio más bajo. El agua es una fuente de ingesta variable; el agua más dura usualmente tiene concentraciones más altas de sales de magnesio (2). Algunos alimentos que son relativamente ricos en magnesio están listados en la Tabla 2, junto a su contenido de magnesio en miligramos (mg). Para mayor información sobre el contenido de nutrientes de los alimentos, revise la base de datos de composición de los alimentos de la USDA.
Suplementos
Los suplementos de magnesio están disponibles como óxido de magnesio, hidróxido de magnesio, gluconato de magnesio, cloruro de magnesio, y sales de citrato de magnesio, así como una serie de quelatos de aminoácidos como el aspartato de magnesio. Las sales de hidróxido de magnesio, óxido, o trisilicato, se usan como antiácidos para mitigar la hiperacidez gástrica y los síntomas de la enfermedad de reflujo gastroesofágico (180).
Seguridad
Toxicidad
No se han identificado efectos adversos del magnesio que ocurre naturalmente en los alimentos. Sin embargo, se han observado efectos adversos del exceso de magnesio con la ingesta de varias sales de magnesio (es decir, magnesio suplementario) (6). El síntoma inicial del exceso de suplementos de magnesio es la diarrea — un efecto secundario bien conocido del magnesio que se usa terapéuticamente como laxante. Las personas con una función renal deteriorada tienen un mayor riesgo de efectos adversos de los suplementos de magnesio, y se han presentado síntomas de toxicidad por magnesio en personas con una función renal deteriorada que toman dosis moderadas de laxantes o antiácidos que contienen magnesio. Las concentraciones séricas elevadas de magnesio (hipermagnesemia) pueden provocar una caída de la presión sanguínea (hipotensión). Algunos de los efectos posteriores de la toxicidad del magnesio, como el letargo, la confusión, las alteraciones del ritmo cardíaco normal, y el deterioro de la función renal, están relacionados con la hipotensión grave. A medida que progresa la hipermagnesemia, puede ocurrir debilidad muscular y dificultad para respirar. La hipermagnesemia severa puede provocar paro cardíaco (2, 3). La Junta de Nutrición y Alimentos del Instituto de Medicina (JNA) estableció el nivel máximo de ingesta tolerable (NM) para el magnesio en 350 mg/día; este NM representa el nivel más alto de ingesta de magnesio suplementario diario que probablemente no representa un riesgo de diarrea o trastornos gastrointestinales en casi todos los individuos. La JNA advierte que los individuos con deterioro renal se encuentran en un riesgo más alto de efectos adversos a causa de la ingesta de magnesio suplementario en exceso. Sin embargo, la JNA también destaca que hay algunas condiciones que pueden justificar dosis más altas de magnesio bajo supervisión médica (2).
Interacción con drogas/fármacos
El magnesio interfiere con la absorción de la digoxina (un medicamento para el corazón), la nitrofurantoína (un antibiótico), y con ciertas drogas contra la malaria, lo que podría potencialmente disminuir la eficacia del medicamento. Los bifosfonatos (p. ej., alendronato y etidronato), los cuales son drogas utilizadas para tratar la osteoporosis, y el magnesio deben ser tomados con dos horas de separación para que la absorción de los bifosfonatos no se inhiba (181, 182). También se ha encontrado que el magnesio reduce la eficacia de la clorpromazina (un tranquilizante), la penicilamina, los anticoagulantes orales, y las clases de antibióticos de quinolona y tetraciclina (181, 182). El magnesio intravenoso podría inhibir la entrada de calcio en las células musculares lisas y provocar hipotensión y debilidad muscular si se administra con bloqueadores de los canales de calcio (p. ej., nifedipina, nicardipina) (182). Debido a que el magnesio intravenoso ha incrementado los efectos de ciertos medicamentos relajantes musculares usados durante la anestesia, se le aconseja informar al personal médico si usted se encuentra tomando suplementos de magnesio, laxantes, o antiácidos, previo a procedimientos quirúrgicos. Además, el uso a largo plazo (tres meses o más) de inhibidores de la bomba de protones (drogas/fármacos utilizados para reducir la cantidad de ácido estomacal) puede aumentar el riesgo de hipomagnesemia (183, 184). Si se toman dosis elevadas de furosemida (Lasix) y de algunos diuréticos tiazídicos (p. ej., hidroclorotiazida) por períodos de tiempo extendidos, esta puede interferir con la reabsorción de magnesio en los riñones y provocar el agotamiento de magnesio (182). Muchos otros medicamentos pueden también resultar en una pérdida renal de magnesio (3).
Recomendación del Instituto Linus Pauling
El Instituto Linus Pauling respalda la IDR más reciente para la ingesta de magnesio (400-420 mg/día para hombres y 310-320 mg/día para mujeres). A pesar de que el magnesio es abundante en los alimentos, se considera un nutriente deficiente. Al seguir la recomendación del Instituto Linus Pauling de consumir un suplemento multivitamínico/mineral a diario, se puede asegurar una ingesta de al menos 100 mg/día de magnesio. Pocos suplementos multivitamínicos/minerales contienen más de 100 mg de magnesio debido a su masa. Ya que el magnesio es abundante en los alimentos, consumir una dieta variada que aporte verduras verdes, granos enteros, y nueces, a diario, ayudará a satisfacer el resto del requerimiento de magnesio de un individuo.
Adultos mayores (>50 años)
Los adultos mayores son menos propensos que los adultos más jóvenes a consumir suficiente magnesio para satisfacer sus necesidades y, por lo tanto, deben tener cuidado de comer alimentos ricos en magnesio además de tomar un suplemento multivitamínico/mineral a diario. Dado que los adultos mayores también tienen más probabilidades de tener una función renal deteriorada, deben evitar tomar más de 350 mg/día de magnesio suplementario sin consulta médica (véase Seguridad).
Autores y Críticos
Originally written in 2001 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2003 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in August 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in October 2013 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in November 2018 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in February 2019 by:
Stella L. Volpe, Ph.D., RDN, ACSM-CEP, FACSM
Professor and Chair
Department of Nutrition Sciences
Drexel University
Traducido al Español en 2019 por:
Natsumi Then Shimazaki
Instituto Linus Pauling
Universidad Estatal de Oregon
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Copyright 2001-2024 Linus Pauling Institute
Referencias
1. Volpe SL. Magnesium. In: Erdman Jr. JW, Macdonald IA, Ziegler EE, eds. Present Knowledge in Nutrition. 10th ed: ILSI Press; 2012:459-474.
2. Food and Nutrition Board, Institute of Medicine. Magnesium. Dietary Reference Intakes: Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington, D.C.: National Academy Press; 1997:190-249. (National Academy Press)
3. Rude RK, Shils ME. Magnesium. In: Shils ME, Shike M, Ross AC, Caballero B, Cousins RJ, eds. Modern Nutrition in Health and Disease. 10th ed. Baltimore: Lippincott Williams & Wilkins; 2006:223-247.
4. Spencer H, Norris C, Williams D. Inhibitory effects of zinc on magnesium balance and magnesium absorption in man. J Am Coll Nutr. 1994;13(5):479-484. (PubMed)
5. Schwartz R, Walker G, Linz MD, MacKellar I. Metabolic responses of adolescent boys to two levels of dietary magnesium and protein. I. Magnesium and nitrogen retention. Am J Clin Nutr. 1973;26(5):510-518. (PubMed)
6. Navarro-Gonzalez JF, Mora-Fernandez C, Garcia-Perez J. Clinical implications of disordered magnesium homeostasis in chronic renal failure and dialysis. Semin Dial. 2009;22(1):37-44. (PubMed)
7. Rude RK. Magnesium. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. China: Williams & Wilkins; 2014:159-175.
8. Moshfegh A, Goldman J, Ahuja J, Rhodes D, LaComb R. What We Eat in America, NHANES 2005-2006: Usual Nutrient Intakes from Food and Water Compared to 1997 Dietary Reference Intakes for Vitamin D, Calcium, Phosphorus, and Magnesium. 2009.
9. Sebastian RS, Cleveland LE, Goldman JD, Moshfegh AJ. Older adults who use vitamin/mineral supplements differ from nonusers in nutrient intake adequacy and dietary attitudes. J Am Diet Assoc. 2007;107(8):1322-1332. (PubMed)
10. Costello RB, Nielsen F. Interpreting magnesium status to enhance clinical care: key indicators. Curr Opin Clin Nutr Metab Care. 2017;20(6):504-511. (PubMed)
11. Grundy SM, Cleeman JI, Daniels SR, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112(17):2735-2752. (PubMed)
12. Esposito K, Chiodini P, Colao A, Lenzi A, Giugliano D. Metabolic syndrome and risk of cancer: a systematic review and meta-analysis. Diabetes Care. 2012;35(11):2402-2411. (PubMed)
13. Ninomiya JK, L'Italien G, Criqui MH, Whyte JL, Gamst A, Chen RS. Association of the metabolic syndrome with history of myocardial infarction and stroke in the Third National Health and Nutrition Examination Survey. Circulation. 2004;109(1):42-46. (PubMed)
14. Sung KC, Lee MY, Kim YH, et al. Obesity and incidence of diabetes: Effect of absence of metabolic syndrome, insulin resistance, inflammation and fatty liver. Atherosclerosis. 2018;275:50-57. (PubMed)
15. Moore-Schiltz L, Albert JM, Singer ME, Swain J, Nock NL. Dietary intake of calcium and magnesium and the metabolic syndrome in the National Health and Nutrition Examination (NHANES) 2001-2010 data. Br J Nutr. 2015;114(6):924-935. (PubMed)
16. Dibaba DT, Xun P, Fly AD, Yokota K, He K. Dietary magnesium intake and risk of metabolic syndrome: a meta-analysis. Diabet Med. 2014;31(11):1301-1309. (PubMed)
17. Ju SY, Choi WS, Ock SM, Kim CM, Kim DH. Dietary magnesium intake and metabolic syndrome in the adult population: dose-response meta-analysis and meta-regression. Nutrients. 2014;6(12):6005-6019. (PubMed)
18. Sarrafzadegan N, Khosravi-Boroujeni H, Lotfizadeh M, Pourmogaddas A, Salehi-Abargouei A. Magnesium status and the metabolic syndrome: A systematic review and meta-analysis. Nutrition. 2016;32(4):409-417. (PubMed)
19. La SA, Lee JY, Kim DH, Song EL, Park JH, Ju SY. Low magnesium levels in adults with metabolic syndrome: a meta-analysis. Biol Trace Elem Res. 2016;170(1):33-42. (PubMed)
20. Song Y, Ridker PM, Manson JE, Cook NR, Buring JE, Liu S. Magnesium intake, C-reactive protein, and the prevalence of metabolic syndrome in middle-aged and older U.S. women. Diabetes Care. 2005;28(6):1438-1444. (PubMed)
21. Dibaba DT, Xun P, He K. Dietary magnesium intake is inversely associated with serum C-reactive protein levels: meta-analysis and systematic review. Eur J Clin Nutr. 2014;68(4):510-516. (PubMed)
22. Ascherio A, Rimm EB, Giovannucci EL, et al. A prospective study of nutritional factors and hypertension among US men. Circulation. 1992;86(5):1475-1484. (PubMed)
23. Ascherio A, Hennekens C, Willett WC, et al. Prospective study of nutritional factors, blood pressure, and hypertension among US women. Hypertension. 1996;27(5):1065-1072. (PubMed)
24. Peacock JM, Folsom AR, Arnett DK, Eckfeldt JH, Szklo M. Relationship of serum and dietary magnesium to incident hypertension: the Atherosclerosis Risk in Communities (ARIC) Study. Ann Epidemiol. 1999;9(3):159-165. (PubMed)
25. He K, Liu K, Daviglus ML, et al. Magnesium intake and incidence of metabolic syndrome among young adults. Circulation. 2006;113(13):1675-1682. (PubMed)
26. Han H, Fang X, Wei X, et al. Dose-response relationship between dietary magnesium intake, serum magnesium concentration and risk of hypertension: a systematic review and meta-analysis of prospective cohort studies. Nutr J. 2017;16(1):26. (PubMed)
27. Joosten MM, Gansevoort RT, Mukamal KJ, et al. Urinary magnesium excretion and risk of hypertension: the prevention of renal and vascular end-stage disease study. Hypertension. 2013;61(6):1161-1167. (PubMed)
28. Rennenberg RJ, Kessels AG, Schurgers LJ, van Engelshoven JM, de Leeuw PW, Kroon AA. Vascular calcifications as a marker of increased cardiovascular risk: a meta-analysis. Vasc Health Risk Manag. 2009;5(1):185-197. (PubMed)
29. Major RW, Cheng MRI, Grant RA, et al. Cardiovascular disease risk factors in chronic kidney disease: A systematic review and meta-analysis. PLoS One. 2018;13(3):e0192895. (PubMed)
30. Palmer SC, Hayen A, Macaskill P, et al. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: a systematic review and meta-analysis. JAMA. 2011;305(11):1119-1127. (PubMed)
31. Sakaguchi Y, Hamano T, Nakano C, et al. Association between density of coronary artery calcification and serum magnesium levels among patients with chronic kidney disease. PLoS One. 2016;11(9):e0163673. (PubMed)
32. Bressendorff I, Hansen D, Schou M, et al. Oral magnesium supplementation in chronic kidney disease stages 3 and 4: efficacy, safety, and effect on serum calcification propensity-a prospective randomized double-blinded placebo-controlled clinical trial. Kidney Int Rep. 2017;2(3):380-389. (PubMed)
33. Pasch A, Block GA, Bachtler M, et al. Blood calcification propensity, cardiovascular events, and survival in patients receiving hemodialysis in the EVOLVE trial. Clin J Am Soc Nephrol. 2017;12(2):315-322. (PubMed)
34. Smith ER, Ford ML, Tomlinson LA, et al. Serum calcification propensity predicts all-cause mortality in predialysis CKD. J Am Soc Nephrol. 2014;25(2):339-348. (PubMed)
35. Bressendorff I, Hansen D, Schou M, Pasch A, Brandi L. The effect of increasing dialysate magnesium on serum calcification propensity in subjects with end stage kidney disease: a randomized, controlled clinical trial. Clin J Am Soc Nephrol. 2018;13(9):1373-1380. (PubMed)
36. Bressendorff I, Hansen D, Schou M, Kragelund C, Brandi L. The effect of magnesium supplementation on vascular calcification in chronic kidney disease-a randomised clinical trial (MAGiCAL-CKD): essential study design and rationale. BMJ Open. 2017;7(6):e016795. (PubMed)
37. Hruby A, O'Donnell CJ, Jacques PF, Meigs JB, Hoffmann U, McKeown NM. Magnesium intake is inversely associated with coronary artery calcification: the Framingham Heart Study. JACC Cardiovasc Imaging. 2014;7(1):59-69. (PubMed)
38. Hisamatsu T, Miura K, Fujiyoshi A, et al. Serum magnesium, phosphorus, and calcium levels and subclinical calcific aortic valve disease: A population-based study. Atherosclerosis. 2018;273:145-152. (PubMed)
39. Lee SY, Hyun YY, Lee KB, Kim H. Low serum magnesium is associated with coronary artery calcification in a Korean population at low risk for cardiovascular disease. Nutr Metab Cardiovasc Dis. 2015;25(11):1056-1061. (PubMed)
40. Posadas-Sanchez R, Posadas-Romero C, Cardoso-Saldana G, et al. Serum magnesium is inversely associated with coronary artery calcification in the Genetics of Atherosclerotic Disease (GEA) study. Nutr J. 2016;15:22. (PubMed)
41. Chiuve SE, Sun Q, Curhan GC, et al. Dietary and plasma magnesium and risk of coronary heart disease among women. J Am Heart Assoc. 2013;2(2):e000114. (PubMed)
42. Del Gobbo LC, Imamura F, Wu JH, de Oliveira Otto MC, Chiuve SE, Mozaffarian D. Circulating and dietary magnesium and risk of cardiovascular disease: a systematic review and meta-analysis of prospective studies. Am J Clin Nutr. 2013;98(1):160-173. (PubMed)
43. Fang X, Wang K, Han D, et al. Dietary magnesium intake and the risk of cardiovascular disease, type 2 diabetes, and all-cause mortality: a dose-response meta-analysis of prospective cohort studies. BMC Med. 2016;14(1):210. (PubMed)
44. Larsson SC, Orsini N, Wolk A. Dietary magnesium intake and risk of stroke: a meta-analysis of prospective studies. Am J Clin Nutr. 2012;95(2):362-366. (PubMed)
45. Nie ZL, Wang ZM, Zhou B, Tang ZP, Wang SK. Magnesium intake and incidence of stroke: meta-analysis of cohort studies. Nutr Metab Cardiovasc Dis. 2013;23(3):169-176. (PubMed)
46. Qu X, Jin F, Hao Y, et al. Magnesium and the risk of cardiovascular events: a meta-analysis of prospective cohort studies. PLoS One. 2013;8(3):e57720. (PubMed)
47. Liao F, Folsom AR, Brancati FL. Is low magnesium concentration a risk factor for coronary heart disease? The Atherosclerosis Risk in Communities (ARIC) Study. Am Heart J. 1998;136(3):480-490. (PubMed)
48. Wannamethee SG, Papacosta O, Lennon L, Whincup PH. Serum magnesium and risk of incident heart failure in older men: The British Regional Heart Study. Eur J Epidemiol. 2018;33(9):873-882. (PubMed)
49. Catling LA, Abubakar I, Lake IR, Swift L, Hunter PR. A systematic review of analytical observational studies investigating the association between cardiovascular disease and drinking water hardness. J Water Health. 2008;6(4):433-442. (PubMed)
50. Xu T, Sun Y, Xu T, Zhang Y. Magnesium intake and cardiovascular disease mortality: A meta-analysis of prospective cohort studies. Int J Cardiol. 2013;167(6):3044-3047. (PubMed)
51. Zhang X, Xia J, Del Gobbo LC, Hruby A, Dai Q, Song Y. Serum magnesium concentrations and all-cause, cardiovascular, and cancer mortality among U.S. adults: Results from the NHANES I Epidemiologic Follow-up Study. Clin Nutr. 2018;37(5):1541-1549. (PubMed)
52. Angkananard T, Anothaisintawee T, Eursiriwan S, et al. The association of serum magnesium and mortality outcomes in heart failure patients: A systematic review and meta-analysis. Medicine (Baltimore). 2016;95(50):e5406. (PubMed)
53. van den Bergh WM, Algra A, van der Sprenkel JW, Tulleken CA, Rinkel GJ. Hypomagnesemia after aneurysmal subarachnoid hemorrhage. Neurosurgery. 2003;52(2):276-281; discussion 281-272. (PubMed)
54. Chen T, Carter BS. Role of magnesium sulfate in aneurysmal subarachnoid hemorrhage management: A meta-analysis of controlled clinical trials. Asian J Neurosurg. 2011;6(1):26-31. (PubMed)
55. Yarad EA, Hammond NE, Winner ABNRPsbE. Intravenous magnesium therapy in adult patients with an aneurysmal subarachnoid haemorrhage: A systematic review and meta-analysis. Aust Crit Care. 2013;26(3):105-117. (PubMed)
56. Golan E, Vasquez DN, Ferguson ND, Adhikari NK, Scales DC. Prophylactic magnesium for improving neurologic outcome after aneurysmal subarachnoid hemorrhage: systematic review and meta-analysis. J Crit Care. 2013;28(2):173-181. (PubMed)
57. Kunze E, Lilla N, Stetter C, Ernestus RI, Westermaier T. Magnesium protects in episodes of critical perfusion after aneurysmal SAH. Transl Neurosci. 2018;9:99-105. (PubMed)
58. Westermaier T, Stetter C, Vince GH, et al. Prophylactic intravenous magnesium sulfate for treatment of aneurysmal subarachnoid hemorrhage: a randomized, placebo-controlled, clinical study. Crit Care Med. 2010;38(5):1284-1290. (PubMed)
59. Arsenault KA, Yusuf AM, Crystal E, et al. Interventions for preventing post-operative atrial fibrillation in patients undergoing heart surgery. Cochrane Database Syst Rev. 2013;1:CD003611. (PubMed)
60. Fairley JL, Zhang L, Glassford NJ, Bellomo R. Magnesium status and magnesium therapy in cardiac surgery: A systematic review and meta-analysis focusing on arrhythmia prevention. J Crit Care. 2017;42:69-77. (PubMed)
61. Wu X, Wang C, Zhu J, Zhang C, Zhang Y, Gao Y. Meta-analysis of randomized controlled trials on magnesium in addition to beta-blocker for prevention of postoperative atrial arrhythmias after coronary artery bypass grafting. BMC Cardiovasc Disord. 2013;13:5. (PubMed)
62. Schulze MB, Schulz M, Heidemann C, Schienkiewitz A, Hoffmann K, Boeing H. Fiber and magnesium intake and incidence of type 2 diabetes: a prospective study and meta-analysis. Arch Intern Med. 2007;167(9):956-965. (PubMed)
63. Fang X, Han H, Li M, et al. Dose-response relationship between dietary magnesium intake and risk of type 2 diabetes mellitus: a systematic review and meta-regression analysis of prospective cohort studies. Nutrients. 2016;8(11). (PubMed)
64. Dong JY, Xun P, He K, Qin LQ. Magnesium intake and risk of type 2 diabetes: meta-analysis of prospective cohort studies. Diabetes Care. 2011;34(9):2116-2122. (PubMed)
65. Hruby A, Ngwa JS, Renstrom F, et al. Higher magnesium intake is associated with lower fasting glucose and insulin, with no evidence of interaction with select genetic loci, in a meta-analysis of 15 CHARGE Consortium Studies. J Nutr. 2013;143(3):345-353. (PubMed)
66. Larsson SC, Wolk A. Magnesium intake and risk of type 2 diabetes: a meta-analysis. J Intern Med. 2007;262(2):208-214. (PubMed)
67. Simental-Mendia LE, Rodriguez-Moran M, Guerrero-Romero F. Failure of beta-cell function for compensate variation in insulin sensitivity in hypomagnesemic subjects. Magnes Res. 2009;22(3):151-156. (PubMed)
68. Guerrero-Romero F, Rodriguez-Moran M. Magnesium improves the beta-cell function to compensate variation of insulin sensitivity: double-blind, randomized clinical trial. Eur J Clin Invest. 2011;41(4):405-410. (PubMed)
69. Guerrero-Romero F, Simental-Mendia LE, Hernandez-Ronquillo G, Rodriguez-Moran M. Oral magnesium supplementation improves glycaemic status in subjects with prediabetes and hypomagnesaemia: A double-blind placebo-controlled randomized trial. Diabetes Metab. 2015;41(3):202-207. (PubMed)
70. Rodriguez-Moran M, Guerrero-Romero F. Oral magnesium supplementation improves the metabolic profile of metabolically obese, normal-weight individuals: a randomized double-blind placebo-controlled trial. Arch Med Res. 2014;45(5):388-393. (PubMed)
71. Mooren FC, Kruger K, Volker K, Golf SW, Wadepuhl M, Kraus A. Oral magnesium supplementation reduces insulin resistance in non-diabetic subjects - a double-blind, placebo-controlled, randomized trial. Diabetes Obes Metab. 2011;13(3):281-284. (PubMed)
72. Castiglioni S, Cazzaniga A, Albisetti W, Maier JA. Magnesium and osteoporosis: current state of knowledge and future research directions. Nutrients. 2013;5(8):3022-3033. (PubMed)
73. Vormann J. Magnesium: Nutrition and Homeostasis. AIMS Public Health. 2016;3(2):329-340. (PubMed)
74. Sojka JE, Weaver CM. Magnesium supplementation and osteoporosis. Nutr Rev. 1995;53(3):71-74. (PubMed)
75. Zheng J, Mao X, Ling J, He Q, Quan J, Jiang H. Association between serum level of magnesium and postmenopausal osteoporosis: a meta-analysis. Biol Trace Elem Res. 2014;159(1-3):8-14. (PubMed)
76. Begin MJ, Ste-Marie LG, Coupal L, Ethier J, Rakel A. Hypomagnesemia during teriparatide treatment in osteoporosis: incidence and determinants. J Bone Miner Res. 2018;33(8):1444-1449. (PubMed)
77. Tucker KL, Hannan MT, Chen H, Cupples LA, Wilson PW, Kiel DP. Potassium, magnesium, and fruit and vegetable intakes are associated with greater bone mineral density in elderly men and women. Am J Clin Nutr. 1999;69(4):727-736. (PubMed)
78. Ryder KM, Shorr RI, Bush AJ, et al. Magnesium intake from food and supplements is associated with bone mineral density in healthy older white subjects. J Am Geriatr Soc. 2005;53(11):1875-1880. (PubMed)
79. Dahl C, Sogaard AJ, Tell GS, et al. Nationwide data on municipal drinking water and hip fracture: Could calcium and magnesium be protective? A NOREPOS study. Bone. 2013;57(1):84-91. (PubMed)
80. Orchard TS, Larson JC, Alghothani N, et al. Magnesium intake, bone mineral density, and fractures: results from the Women's Health Initiative observational study. Am J Clin Nutr. 2014;99(4):926-933. (PubMed)
81. Hayhoe RP, Lentjes MA, Luben RN, Khaw KT, Welch AA. Dietary magnesium and potassium intakes and circulating magnesium are associated with heel bone ultrasound attenuation and osteoporotic fracture risk in the EPIC-Norfolk cohort study. Am J Clin Nutr. 2015;102(2):376-384. (PubMed)
82. Stendig-Lindberg G, Tepper R, Leichter I. Trabecular bone density in a two year controlled trial of peroral magnesium in osteoporosis. Magnes Res. 1993;6(2):155-163. (PubMed)
83. Abraham GE, Grewal H. A total dietary program emphasizing magnesium instead of calcium. Effect on the mineral density of calcaneous bone in postmenopausal women on hormonal therapy. J Reprod Med. 1990;35(5):503-507. (PubMed)
84. Aydin H, Deyneli O, Yavuz D, et al. Short-term oral magnesium supplementation suppresses bone turnover in postmenopausal osteoporotic women. Biol Trace Elem Res. 2010;133(2):136-143. (PubMed)
85. Nieves JW. Bone. Maximizing bone health--magnesium, BMD and fractures. Nat Rev Endocrinol. 2014;10(5):255-256. (PubMed)
86. Landi F, Liperoti R, Russo A, et al. Sarcopenia as a risk factor for falls in elderly individuals: results from the ilSIRENTE study. Clin Nutr. 2012;31(5):652-658. (PubMed)
87. Hayhoe RPG, Lentjes MAH, Mulligan AA, Luben RN, Khaw KT, Welch AA. Cross-sectional associations of dietary and circulating magnesium with skeletal muscle mass in the EPIC-Norfolk cohort. Clin Nutr. 2019;38(1):317-323. (PubMed)
88. Scott D, Blizzard L, Fell J, Giles G, Jones G. Associations between dietary nutrient intake and muscle mass and strength in community-dwelling older adults: the Tasmanian Older Adult Cohort Study. J Am Geriatr Soc. 2010;58(11):2129-2134. (PubMed)
89. Welch AA, Kelaiditi E, Jennings A, Steves CJ, Spector TD, MacGregor A. Dietary magnesium is positively associated with skeletal muscle power and indices of muscle mass and may attenuate the association between circulating C-reactive protein and muscle mass in women. J Bone Miner Res. 2016;31(2):317-325. (PubMed)
90. Welch AA, Skinner J, Hickson M. Dietary magnesium may be protective for aging of bone and skeletal muscle in middle and younger older age men and women: cross-sectional findings from the UK Biobank cohort. Nutrients. 2017;9(11). (PubMed)
91. Veronese N, Berton L, Carraro S, et al. Effect of oral magnesium supplementation on physical performance in healthy elderly women involved in a weekly exercise program: a randomized controlled trial. Am J Clin Nutr. 2014;100(3):974-981. (PubMed)
92. Bibbins-Domingo K, Grossman DC, Curry SJ, et al. Screening for preeclampsia: US Preventive Services Task Force recommendation statement. JAMA. 2017;317(16):1661-1667. (PubMed)
93. Jeyabalan A. Epidemiology of preeclampsia: impact of obesity. Nutr Rev. 2013;71 Suppl 1:S18-25. (PubMed)
94. Duley L. The global impact of pre-eclampsia and eclampsia. Semin Perinatol. 2009;33(3):130-137. (PubMed)
95. Duley L, Henderson-Smart DJ, Chou D. Magnesium sulphate versus phenytoin for eclampsia. Cochrane Database Syst Rev. 2010(10):CD000128. (PubMed)
96. Makrides M, Crosby DD, Bain E, Crowther CA. Magnesium supplementation in pregnancy. Cochrane Database Syst Rev. 2014(4):Cd000937. (PubMed)
97. Sibai BM. Diagnosis, prevention, and management of eclampsia. Obstet Gynecol. 2005;105(2):402-410. (PubMed)
98. Altman D, Carroli G, Duley L, et al. Do women with pre-eclampsia, and their babies, benefit from magnesium sulphate? The Magpie Trial: a randomised placebo-controlled trial. Lancet. 2002;359(9321):1877-1890. (PubMed)
99. McDonald SD, Lutsiv O, Dzaja N, Duley L. A systematic review of maternal and infant outcomes following magnesium sulfate for pre-eclampsia/eclampsia in real-world use. Int J Gynaecol Obstet. 2012;118(2):90-96. (PubMed)
100. World Health Organization. WHO recommendations for prevention and treatment of pre-eclampsia and eclampsia. Implications and actions. Available at: http://www.who.int/reproductivehealth/publications/maternal_perinatal_health/program-action-eclampsia/en/. Accessed 10/9/18.
101. Berhan Y, Berhan A. Should magnesium sulfate be administered to women with mild pre-eclampsia? A systematic review of published reports on eclampsia. J Obstet Gynaecol Res. 2015;41(6):831-842. (PubMed)
102. Vigil-DeGracia P, Ludmir J, Ng J, et al. Is there benefit to continue magnesium sulphate postpartum in women receiving magnesium sulphate before delivery? A randomised controlled study. Bjog. 2018;125(10):1304-1311. (PubMed)
103. American College of Obstetricians and Gynecologists Committee. Committee opinion no. 573: magnesium sulfate use in obstetrics. Obstet Gynecol. 2013;122(3):727-728. (PubMed)
104. Roberts D, Brown J, Medley N, Dalziel SR. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst Rev. 2017;3:Cd004454. (PubMed)
105. Crowther CA, Brown J, McKinlay CJ, Middleton P. Magnesium sulphate for preventing preterm birth in threatened preterm labour. Cochrane Database Syst Rev. 2014(8):Cd001060. (PubMed)
106. McNamara HC, Crowther CA, Brown J. Different treatment regimens of magnesium sulphate for tocolysis in women in preterm labour. Cochrane Database Syst Rev. 2015(12):Cd011200. (PubMed)
107. Wolf HT, Hegaard HK, Greisen G, Huusom L, Hedegaard M. Treatment with magnesium sulphate in pre-term birth: a systematic review and meta-analysis of observational studies. J Obstet Gynaecol. 2012;32(2):135-140. (PubMed)
108. Crowther CA, Middleton PF, Voysey M, et al. Assessing the neuroprotective benefits for babies of antenatal magnesium sulphate: An individual participant data meta-analysis. PLoS Med. 2017;14(10):e1002398. (PubMed)
109. Tagin M, Shah PS, Lee KS. Magnesium for newborns with hypoxic-ischemic encephalopathy: a systematic review and meta-analysis. J Perinatol. 2013;33(9):663-669. (PubMed)
110. Kass L, Weekes J, Carpenter L. Effect of magnesium supplementation on blood pressure: a meta-analysis. Eur J Clin Nutr. 2012;66(4):411-418. (PubMed)
111. Rosanoff A. Magnesium supplements may enhance the effect of antihypertensive medications in stage 1 hypertensive subjects. Magnes Res. 2010;23(1):27-40. (PubMed)
112. Zhang X, Li Y, Del Gobbo LC, et al. Effects of magnesium supplementation on blood pressure: a meta-analysis of randomized double-blind placebo-controlled trials. Hypertension. 2016;68(2):324-333. (PubMed)
113. Dibaba DT, Xun P, Song Y, Rosanoff A, Shechter M, He K. The effect of magnesium supplementation on blood pressure in individuals with insulin resistance, prediabetes, or noncommunicable chronic diseases: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2017;106(3):921-929. (PubMed)
114. Appel LJ, Moore TJ, Obarzanek E, et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N Engl J Med. 1997;336(16):1117-1124. (PubMed)
115. Maier JA. Endothelial cells and magnesium: implications in atherosclerosis. Clin Sci (Lond). 2012;122(9):397-407. (PubMed)
116. Darooghegi Mofrad M, Djafarian K, Mozaffari H, Shab-Bidar S. Effect of magnesium supplementation on endothelial function: A systematic review and meta-analysis of randomized controlled trials. Atherosclerosis. 2018;273:98-105. (PubMed)
117. Shechter M, Sharir M, Labrador MJ, Forrester J, Silver B, Bairey Merz CN. Oral magnesium therapy improves endothelial function in patients with coronary artery disease. Circulation. 2000;102(19):2353-2358. (PubMed)
118. Barbagallo M, Dominguez LJ, Galioto A, Pineo A, Belvedere M. Oral magnesium supplementation improves vascular function in elderly diabetic patients. Magnes Res. 2010;23(3):131-137. (PubMed)
119. Cunha AR, D'El-Rei J, Medeiros F, et al. Oral magnesium supplementation improves endothelial function and attenuates subclinical atherosclerosis in thiazide-treated hypertensive women. J Hypertens. 2017;35(1):89-97. (PubMed)
120. Mortazavi M, Moeinzadeh F, Saadatnia M, Shahidi S, McGee JC, Minagar A. Effect of magnesium supplementation on carotid intima-media thickness and flow-mediated dilatation among hemodialysis patients: a double-blind, randomized, placebo-controlled trial. Eur Neurol. 2013;69(5):309-316. (PubMed)
121. Cosaro E, Bonafini S, Montagnana M, et al. Effects of magnesium supplements on blood pressure, endothelial function and metabolic parameters in healthy young men with a family history of metabolic syndrome. Nutr Metab Cardiovasc Dis. 2014;24(11):1213-1220. (PubMed)
122. Joris PJ, Plat J, Bakker SJ, Mensink RP. Effects of long-term magnesium supplementation on endothelial function and cardiometabolic risk markers: A randomized controlled trial in overweight/obese adults. Sci Rep. 2017;7(1):106. (PubMed)
123. Mookadam F, Moustafa SE, Lester SJ, Warsame T. Subclinical atherosclerosis: evolving role of carotid intima-media thickness. Prev Cardiol. 2010;13(4):186-197. (PubMed)
124. Ma J, Folsom AR, Melnick SL, et al. Associations of serum and dietary magnesium with cardiovascular disease, hypertension, diabetes, insulin, and carotid arterial wall thickness: the ARIC study. Atherosclerosis Risk in Communities Study. J Clin Epidemiol. 1995;48(7):927-940. (PubMed)
125. Woods KL, Fletcher S, Roffe C, Haider Y. Intravenous magnesium sulphate in suspected acute myocardial infarction: results of the second Leicester Intravenous Magnesium Intervention Trial (LIMIT-2). Lancet. 1992;339(8809):1553-1558. (PubMed)
126. Woods KL, Fletcher S. Long-term outcome after intravenous magnesium sulphate in suspected acute myocardial infarction: the second Leicester Intravenous Magnesium Intervention Trial (LIMIT-2). Lancet. 1994;343(8901):816-819. (PubMed)
127. Fourth International Study of Infarct Survival (ISIS-4) Collaborative Group. A randomised factorial trial assessing early oral captopril, oral mononitrate, and intravenous magnesium sulphate in 58,050 patients with suspected acute myocardial infarction. Lancet. 1995;345(8951):669-685. (PubMed)
128. Ziegelstein RC, Hilbe JM, French WJ, Antman EM, Chandra-Strobos N. Magnesium use in the treatment of acute myocardial infarction in the United States (observations from the Second National Registry of Myocardial Infarction). Am J Cardiol. 2001;87(1):7-10. (PubMed)
129. Li J, Zhang Q, Zhang M, Egger M. Intravenous magnesium for acute myocardial infarction. Cochrane Database Syst Rev. 2007(2):CD002755. (PubMed)
130. Pham PC, Pham PM, Pham SV, Miller JM, Pham PT. Hypomagnesemia in patients with type 2 diabetes. Clin J Am Soc Nephrol. 2007;2(2):366-373. (PubMed)
131. Takaya J, Higashino H, Kobayashi Y. Intracellular magnesium and insulin resistance. Magnes Res. 2004;17(2):126-136. (PubMed)
132. Yokota K, Kato M, Lister F, et al. Clinical efficacy of magnesium supplementation in patients with type 2 diabetes. J Am Coll Nutr. 2004;23(5):506S-509S. (PubMed)
133. Rodriguez-Moran M, Guerrero-Romero F. Oral magnesium supplementation improves insulin sensitivity and metabolic control in type 2 diabetic subjects: a randomized double-blind controlled trial. Diabetes Care. 2003;26(4):1147-1152. (PubMed)
134. Veronese N, Watutantrige-Fernando S, Luchini C, et al. Effect of magnesium supplementation on glucose metabolism in people with or at risk of diabetes: a systematic review and meta-analysis of double-blind randomized controlled trials. Eur J Clin Nutr. 2016;70(12):1354-1359. (PubMed)
135. Simental-Mendia LE, Sahebkar A, Rodriguez-Moran M, Guerrero-Romero F. A systematic review and meta-analysis of randomized controlled trials on the effects of magnesium supplementation on insulin sensitivity and glucose control. Pharmacol Res. 2016;111:272-282. (PubMed)
136. Hashimoto Y, Nishimura Y, Maeda H, Yokoyama M. Assessment of magnesium status in patients with bronchial asthma. J Asthma. 2000;37(6):489-496. (PubMed)
137. Irazuzta JE, Chiriboga N. Magnesium sulfate infusion for acute asthma in the emergency department. J Pediatr (Rio J). 2017;93 Suppl 1:19-25. (PubMed)
138. Su Z, Li R, Gai Z. Intravenous and nebulized magnesium sulfate for treating acute asthma in children: a systematic review and meta-analysis. Pediatr Emerg Care. 2018;34(6):390-395. (PubMed)
139. Kew KM, Kirtchuk L, Michell CI. Intravenous magnesium sulfate for treating adults with acute asthma in the emergency department. Cochrane Database Syst Rev. 2014(5):Cd010909. (PubMed)
140. Knightly R, Milan SJ, Hughes R, et al. Inhaled magnesium sulfate in the treatment of acute asthma. Cochrane Database Syst Rev. 2017;11:Cd003898. (PubMed)
141. Monteleone CA, Sherman AR. Nutrition and asthma. Arch Intern Med. 1997;157(1):23-34. (PubMed)
142. Beasley R, Aldington S. Magnesium in the treatment of asthma. Curr Opin Allergy Clin Immunol. 2007;7(1):107-110. (PubMed)
143. Fogarty A, Lewis SA, Scrivener SL, et al. Oral magnesium and vitamin C supplements in asthma: a parallel group randomized placebo-controlled trial. Clin Exp Allergy. 2003;33(10):1355-1359. (PubMed)
144. Na HS, Ryu JH, Do SH. The role of magnesium in pain. In: Vink R, Nechifor M, eds. Magnesium in the Central Nervous System. Adelaide (AU): University of Adelaide Press; 2011. (PubMed)
145. Wang SC, Pan PT, Chiu HY, Huang CJ. Neuraxial magnesium sulfate improves postoperative analgesia in Cesarean section delivery women: A meta-analysis of randomized controlled trials. Asian J Anesthesiol. 2017;55(3):56-67. (PubMed)
146. Xiao F, Xu W, Feng Y, et al. Intrathecal magnesium sulfate does not reduce the ED50 of intrathecal hyperbaric bupivacaine for cesarean delivery in healthy parturients: a prospective, double blinded, randomized dose-response trial using the sequential allocation method. BMC Anesthesiol. 2017;17(1):8. (PubMed)
147. Paleti S, Prasad PK, Lakshmi BS. A randomized clinical trial of intrathecal magnesium sulfate versus midazolam with epidural administration of 0.75% ropivacaine for patients with preeclampsia scheduled for elective cesarean section. J Anaesthesiol Clin Pharmacol. 2018;34(1):23-28. (PubMed)
148. Vlok R, Melhuish TM, Chong C, Ryan T, White LD. Adjuncts to local anaesthetics in tonsillectomy: a systematic review and meta-analysis. J Anesth. 2017;31(4):608-616. (PubMed)
149. Cho HK, Park IJ, Yoon HY, Hwang SH. Efficacy of adjuvant magnesium for posttonsillectomy morbidity in children: a meta-analysis. Otolaryngol Head Neck Surg. 2018;158(1):27-35. (PubMed)
150. Xie M, Li XK, Peng Y. Magnesium sulfate for postoperative complications in children undergoing tonsillectomies: a systematic review and meta-analysis. J Evid Based Med. 2017;10(1):16-25. (PubMed)
151. Chen C, Tao R. The impact of magnesium sulfate on pain control after laparoscopic cholecystectomy: a meta-analysis of randomized controlled studies. Surg Laparosc Endosc Percutan Tech. 2018;28(6):349-353. (PubMed)
152. Abd-Elsalam KA, Fares KM, Mohamed MA, Mohamed MF, El-Rahman AMA, Tohamy MM. Efficacy of magnesium sulfate added to local anesthetic in a transversus abdominis plane block for analgesia following total abdominal hysterectomy: a randomized trial. Pain Physician. 2017;20(7):641-647. (PubMed)
153. Imani F, Rahimzadeh P, Faiz HR, Abdullahzadeh-Baghaei A. An evaluation of the adding magnesium sulfate to ropivacaine on ultrasound-guided transverse abdominis plane block after abdominal hysterectomy. Anesth Pain Med. 2018;8(4):e74124. (PubMed)
154. Martin DP, Samora WP, 3rd, Beebe AC, et al. Analgesic effects of methadone and magnesium following posterior spinal fusion for idiopathic scoliosis in adolescents: a randomized controlled trial. J Anesth. 2018;32(5):702-708. (PubMed)
155. Srivastava VK, Mishra A, Agrawal S, Kumar S, Sharma S, Kumar R. Comparative evaluation of dexmedetomidine and magnesium sulphate on propofol consumption, haemodynamics and postoperative recovery in spine surgery: a prospective, randomized, placebo controlled, double-blind study. Adv Pharm Bull. 2016;6(1):75-81. (PubMed)
156. Hamed MA. Comparative study between magnesium sulfate and lidocaine for controlled hypotension during functional endoscopic sinus surgery: a randomized controlled study. Anesth Essays Res. 2018;12(3):715-718. (PubMed)
157. Hassan PF, Saleh AH. Dexmedetomidine versus magnesium sulfate in anesthesia for cochlear implantation surgery in pediatric patients. Anesth Essays Res. 2017;11(4):1064-1069. (PubMed)
158. Brill S, Sedgwick PM, Hamann W, Di Vadi PP. Efficacy of intravenous magnesium in neuropathic pain. Br J Anaesth. 2002;89(5):711-714. (PubMed)
159. Tanaka M, Shimizu S, Nishimura W, et al. Relief of neuropathic pain with intravenous magnesium [Article in Japanese]. Masui. 1998;47(9):1109-1113. (PubMed)
160. Pickering G, Morel V, Simen E, et al. Oral magnesium treatment in patients with neuropathic pain: a randomized clinical trial. Magnes Res. 2011;24(2):28-35. (PubMed)
161. Delage N, Morel V, Picard P, Marcaillou F, Pereira B, Pickering G. Effect of ketamine combined with magnesium sulfate in neuropathic pain patients (KETAPAIN): study protocol for a randomized controlled trial. Trials. 2017;18(1):517. (PubMed)
162. Mauskop A, Altura BM. Role of magnesium in the pathogenesis and treatment of migraines. Clin Neurosci. 1998;5(1):24-27. (PubMed)
163. Mauskop A, Altura BT, Altura BM. Serum ionized magnesium levels and serum ionized calcium/ionized magnesium ratios in women with menstrual migraine. Headache. 2002;42(4):242-248. (PubMed)
164. Peikert A, Wilimzig C, Kohne-Volland R. Prophylaxis of migraine with oral magnesium: results from a prospective, multi-center, placebo-controlled and double-blind randomized study. Cephalalgia. 1996;16(4):257-263. (PubMed)
165. Wang F, Van Den Eeden SK, Ackerson LM, Salk SE, Reince RH, Elin RJ. Oral magnesium oxide prophylaxis of frequent migrainous headache in children: a randomized, double-blind, placebo-controlled trial. Headache. 2003;43(6):601-610. (PubMed)
166. Pfaffenrath V, Wessely P, Meyer C, et al. Magnesium in the prophylaxis of migraine--a double-blind placebo-controlled study. Cephalalgia. 1996;16(6):436-440. (PubMed)
167. Demirkaya S, Vural O, Dora B, Topcuoglu MA. Efficacy of intravenous magnesium sulfate in the treatment of acute migraine attacks. Headache. 2001;41(2):171-177. (PubMed)
168. Bigal ME, Bordini CA, Tepper SJ, Speciali JG. Intravenous magnesium sulphate in the acute treatment of migraine without aura and migraine with aura. A randomized, double-blind, placebo-controlled study. Cephalalgia. 2002;22(5):345-353. (PubMed)
169. Corbo J, Esses D, Bijur PE, Iannaccone R, Gallagher EJ. Randomized clinical trial of intravenous magnesium sulfate as an adjunctive medication for emergency department treatment of migraine headache. Ann Emerg Med. 2001;38(6):621-627. (PubMed)
170. Cete Y, Dora B, Ertan C, Ozdemir C, Oktay C. A randomized prospective placebo-controlled study of intravenous magnesium sulphate vs. metoclopramide in the management of acute migraine attacks in the Emergency Department. Cephalalgia. 2005;25(3):199-204. (PubMed)
171. Choi H, Parmar N. The use of intravenous magnesium sulphate for acute migraine: meta-analysis of randomized controlled trials. Eur J Emerg Med. 2014; 21(1):2-9. (PubMed)
172. Shahrami A, Assarzadegan F, Hatamabadi HR, Asgarzadeh M, Sarehbandi B, Asgarzadeh S. Comparison of therapeutic effects of magnesium sulfate vs. dexamethasone/metoclopramide on alleviating acute migraine headache. J Emerg Med. 2015;48(1):69-76. (PubMed)
173. Baratloo A, Mirbaha S, Delavar Kasmaei H, Payandemehr P, Elmaraezy A, Negida A. Intravenous caffeine citrate vs. magnesium sulfate for reducing pain in patients with acute migraine headache; a prospective quasi-experimental study. Korean J Pain. 2017;30(3):176-182. (PubMed)
174. Mauskop A, Varughese J. Why all migraine patients should be treated with magnesium. J Neural Transm. 2012;119(5):575-579. (PubMed)
175. Jiang P, Lv Q, Lai T, Xu F. Does hypomagnesemia impact on the outcome of patients admitted to the intensive care unit? A systematic review and meta-analysis. Shock. 2017;47(3):288-295. (PubMed)
176. Upala S, Jaruvongvanich V, Wijarnpreecha K, Sanguankeo A. Hypomagnesemia and mortality in patients admitted to intensive care unit: a systematic review and meta-analysis. Qjm. 2016;109(7):453-459. (PubMed)
177. Nayak R, Attry S, Ghosh SN. Serum magnesium as a marker of neurological outcome in severe traumatic brain injury patients. Asian J Neurosurg. 2018;13(3):685-688. (PubMed)
178. Ardehali SH, Dehghan S, Baghestani AR, Velayati A, Vahdat Shariatpanahi Z. Association of admission serum levels of vitamin D, calcium, Phosphate, magnesium and parathormone with clinical outcomes in neurosurgical ICU patients. Sci Rep. 2018;8(1):2965. (PubMed)
179. Fulgoni VL, 3rd, Keast DR, Bailey RL, Dwyer J. Foods, fortificants, and supplements: Where do Americans get their nutrients? J Nutr. 2011;141(10):1847-1854. (PubMed)
180. Natural Medicines. Magnesium. Professional handout/Effectiveness. Available at: https://naturalmedicines.therapeuticresearch.com/. Accessed 10/29/18.
181. Hendler SS, Rorvik DM. PDR for Nutritional Supplements. Montvale: Thomson Reuters; 2008.
182. Natural Medicines. Magnesium. Professional handout/Drug Interactions. Available at: https://naturalmedicines.therapeuticresearch.com/. Accessed 10/19/18.
183. Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6(4):443-451. (PubMed)
184. US Food and Drug Administration. Proton pump inhibitor drugs (PPIs): drug safety communication - low magnesium levels can be associated with long-term use. 08/04/2017 Available at: https://www.fda.gov/Drugs/DrugSafety/ucm245011.htm. Accessed 10/22/18.
Manganeso
Contenido
El manganeso es un elemento mineral que es nutricionalmente esencial y a la vez potencialmente tóxico. La derivación de su nombre de la palabra griega para magia sigue siendo apropiada, ya que los científicos aún siguen trabajando para comprender los diversos efectos de la deficiencia y de la toxicidad del manganeso en los seres vivos (1).
Función
El manganeso juega un papel importante en una serie de procesos fisiológicos, como constituyente de múltiples enzimas y como activador de otras (2).
Función antioxidante
La superóxido dismutasa de manganeso (MnSOD) es la principal enzima antioxidante en la mitocondria. Debido a que la mitocondria consume cerca del 90% del oxígeno usado en las células, son especialmente vulnerables al estrés oxidativo. El radical superóxido es una de las especies reactivas del oxígeno producidas en la mitocondria durante la síntesis de ATP. La MnSOD cataliza la conversión de los radicales superóxido en peróxido de hidrógeno, el que puede ser reducido en agua por otras enzimas antioxidantes (3).
Metabolismo
Una serie de enzimas activadas por manganeso desempeñan labores importantes en el metabolismo de los carbohidratos, aminoácidos, y colesterol (4). La piruvato carboxilasa, una enzima con manganeso, y la fosfoenolpiruvato carbokinasa (FEPCK), una enzima activada por manganeso, son fundamentales en la gluconeogénesis — la producción de glucosa a partir de precursores que no son carbohidratos. La arginasa, otra enzima con manganeso, es necesaria para el ciclo de la úrea en el hígado, un proceso que detoxifica el amoniaco generado durante el metabolismo de los aminoácidos (3). En el cerebro, la enzima activada por manganeso, glutamina sintetasa, convierte al aminoácido glutamato en glutamina. El glutamato es un transmisor excitotóxico y un precursor del ácido γ-aminobutírico (GABA), un neurotransmisor inhibitorio (5,6).
Desarrollo óseo
La deficiencia de manganeso provoca un desarrollo anormal del esqueleto en una serie de especies animales. El manganeso es el cofactor preferido de las enzimas denominadas glicosiltransferasas; estas enzimas son necesarias para la síntesis de los proteoglicanos necesarios para la formación de cartílago y huesos sanos (7).
Curación de heridas
La curación de heridas es un proceso complejo que requiere de un incremento en la producción de colágeno. El manganeso es necesario para la activación de la prolidasa, una enzima cuya función es la de aportar el aminoácido prolina para la formación de colágeno en las células epiteliales humanas (8). Un trastorno genético conocido como deficiencia de prolidasa resulta entre otros problemas, en la curación anormal de las heridas, y se caracteriza por un metabolismo anormal del manganeso (7). La síntesis de glicosaminoglicanos, la que necesita de glicosiltransferasas activadas por manganeso, puede también jugar un papel importante en la curación de heridas (9).
Interacción con nutrientes
Hierro
Aunque los mecanismos específicos para la absorción y el transporte del manganeso no han sido determinados, cierta evidencia sugiere que el hierro y el manganeso pueden compartir vías de absorción y transporte comunes (10). La absorción de manganeso desde una comida disminuye si el contenido de hierro de la comida aumenta (7). La suplementación con hierro (60 mg/día por cuatro meses) se asoció con niveles disminuidos de manganeso en la sangre y con una menor actividad de la MnSOD en glóbulos blancos, indicando una reducción en el estado nutricional del manganeso (11). Además, el estado nutricional del hierro en los individuos puede alterar la biodisponibilidad del manganeso. La absorción intestinal de manganeso se incrementa durante la deficiencia de hierro, y el incremento en los depósitos de hierro (niveles de ferritina) se asocia con una absorción disminuida del manganeso (12). Los hombres por lo general absorben menos manganeso que las mujeres; esto puede estar relacionado al hecho de que generalmente los hombres poseen depósitos de hierro más altos (13). Más aún, se ha demostrado que la deficiencia de hierro incrementa el riesgo de acumulación de manganeso en el cerebro (14).
Magnesio
El magnesio suplementario (200 mg/día) ha demostrado disminuir levemente la biodisponibilidad del manganeso en adultos sanos, ya sea disminuyendo su absorción o incrementando su excreción (15).
Calcio
En un grupo de estudios, el calcio suplementario (500 mg/día) disminuyó levemente la biodisponibilidad del manganeso en adultos sanos. La leche como fuente de calcio tiene un efecto menor, mientras que el carbonato de calcio y el fosfato de calcio tienen un mayor efecto (15). Varios otros estudios han encontrado efectos mínimos de la suplementación con calcio sobre el metabolismo del manganeso (16).
Deficiencia
La deficiencia de manganeso ha sido observada en una serie de especies animales. Los signos de la deficiencia de manganeso incluyen retraso del crecimiento, deterioro de la función reproductiva, trastornos esqueletales, tolerancia a la glucosa deteriorada, y alteración del metabolismo de lípidos y carbohidratos. En seres humanos, la demostración del síndrome de deficiencia de manganeso es menos evidente (2, 7). Un niño bajo nutrición parenteral total (NPT) a largo plazo sin manganeso sufrió de desmineralización ósea y retraso del crecimiento, los que fueron corregidos por la suplementación con manganeso (17). Los hombres jóvenes que fueron alimentados con una dieta baja en manganeso presentaron niveles disminuidos de colesterol plasmático y una erupción cutánea transitoria (18). Los niveles sanguíneos de calcio, fósforo, y fosfatasa alcalina también se elevaron, lo que podría indicar un incremento de la remodelación ósea como una consecuencia del insuficiente manganeso en la dieta. Las mujeres jóvenes alimentados con una dieta pobre en manganeso presentaron una tolerancia anormal moderada a la glucosa en respuesta a la infusión intravenosa (IV) de glucosa (16). Por lo general, la deficiencia de manganeso no es común, y existe más preocupación por la toxicidad relacionada a la sobreexposición a manganeso (véase Seguridad).
La Ingesta Adecuada (IA)
Ya que no hay suficiente información acerca los requerimientos de manganeso como para establecer una Ingesta Diaria Recomendada (IDR), la Junta de Nutrición y Alimentos (JNA) del Instituto de Medicina estableció un nivel de ingesta adecuada (IA). Como no se ha documentado una deficiencia de manganeso evidente con el consumo de dietas naturales, la JNA basó la IA en las ingestas dietéticas de manganeso promedio, determinadas por el Estudio de Dieta Total — una encuesta anual sobre el contenido mineral de las dietas Americanas representativas (4). Los valores IA para el manganeso se muestran en miligramos (mg)/día en la tabla a continuación según edad y género. Los requerimientos de manganeso aumentan en el embarazo y la lactancia (4).
| Etapa de la Vida | Edad |
Machos (mg/día) |
Hembras (mg/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 0.003 | 0.003 |
| Infantes | 7-12 meses | 0.6 | 0.6 |
| Niños | 1-3 años | 1.2 | 1.2 |
| Niños | 4-8 años | 1.5 | 1.5 |
| Niños | 9-13 años | 1.9 | 1.6 |
| Adolescentes | 14-18 años | 2.2 | 1.6 |
| Adultos | 19 años y más | 2.3 | 1.8 |
| Embarazo | Todas las edades | - | 2.0 |
| Período de lactancia | Todas las edades | - | 2.6 |
Prevención de Enfermedades
Los niveles bajos de manganeso dietético o los niveles bajos de manganeso en la sangre o los tejidos se han asociado con varias enfermedades crónicas. Aunque actualmente se piensa que la insuficiencia de manganeso no es la causa de las enfermedades presentadas a continuación, puede ser necesaria más investigación para determinar si un estado nutricional de manganeso bajo el óptimo contribuye a los procesos de ciertas enfermedades.
Osteoporosis
Se ha encontrado que las mujeres con osteoporosis presentan niveles disminuidos de manganeso en el suero o en el plasma, y también un aumento de la respuesta plasmática frente a una dosis oral de manganeso (19, 20), sugiriendo que podrían tener un estado de manganeso más bajo que las mujeres sin osteoporosis. Aún así, un estudio más reciente en mujeres postmenopáusicas con y sin osteoporosis no encontró ninguna diferencia en los niveles plasmáticos de manganeso (21). Un estudio en mujeres postmenopáusicas sanas encontró que un suplemento con manganeso (5 mg/día), cobre (2.5 mg/día), y zinc (15 mg/día) en combinación con un suplemento de calcio (1,000 mg/día), era más efectivo que el suplemento de calcio por si solo en la prevención de la pérdida de hueso espinal, durante un periodo de dos años (22). Sin embargo, la presencia de otros elementos traza en los suplementos hace imposible el determinar si la suplementación con manganeso era o no un agente beneficioso en la mantención de la densidad mineral ósea.
Diabetes mellitus
La deficiencia de manganeso provoca una intolerancia a la glucosa similar a la de la diabetes mellitus en algunas especies animales, pero los estudios que examinaron el estado del manganeso en seres humanos diabéticos han generado resultados contradictorios. En un estudio, los niveles de manganeso en el total de la sangre, no difirieron significativamente entre los 57 diabéticos y los 28 controles no diabéticos (23). No obstante, la excreción de manganeso urinario tendió a ser levemente más alta en 185 diabéticos comparados con 185 controles no diabéticos (24). Un estudio de caso y control de 250 individuos diabéticos y no diabéticos, encontró que los individuos diabéticos tipo 2 tenían niveles de manganeso plasmático más altos que los no diabéticos (25). Sin embargo, un estudio más reciente en 257 diabéticos tipo 2 y 166 controles no diabéticos, encontró niveles sanguíneos más bajos de manganeso en los pacientes diabéticos (26). Adicionalmente, un estudio del estado de manganeso funcional encontró que la actividad de la MnSOD, una enzima antioxidante, era más baja en los glóbulos blancos de los diabéticos que en los no diabéticos (27). En diabéticos y controles no diabéticos, ni 15 mg ni 30 mg de manganeso oral mejoraron la tolerancia a la glucosa, cuando se administraron al mismo tiempo que un examen de glucosa oral (28). Aunque el manganeso parece jugar un papel en el metabolismo de la glucosa, hay poca evidencia de que la suplementación con manganeso mejore la tolerancia a la glucosa en los individuos diabéticos y no diabéticos.
Epilepsia (trastornos convulsivos)
Las ratas deficientes de manganeso son más susceptibles a las convulsiones que las ratas con manganeso suficiente, y las ratas que son genéticamente propensas a la epilepsia tienen niveles más bajos de manganeso en la sangre y en el cerebro. Se dice que ciertos subgrupos de humanos con epilepsia tienen niveles más bajos de manganeso en la sangre total que los controles no epilépticos. Un estudio encontró que los niveles de manganeso sanguíneo de los individuos con epilepsia de origen desconocido eran más bajos que en los individuos con epilepsia inducida por trauma (e.g., herida en la cabeza) o por enfermedad, sugiriendo una posible relación genética entre la epilepsia y el metabolismo anormal del manganeso. Mientras que la deficiencia de manganeso no parece ser una causa de epilepsia en seres humanos, la relación entre el metabolismo del manganeso y la epilepsia amerita mayor investigación (7, 29).
Fuentes
Fuentes alimenticias
En los EE.UU., se estima que las ingestas de magnesio dietético promedio oscilan entre 2.1-2.3 mg/día para hombres y entre 1.6-1.8 mg/día para mujeres. Las personas que consumen dietas vegetarianas y dietas de tipo occidental tienen ingestas de manganeso de hasta 10.9 mg/día (4). Las fuentes ricas en manganeso incluyen los granos enteros, las nueces, los vegetales con hojas y los tés. Los alimentos ricos en ácido fítico, como los frijoles, semillas, nueces, granos enteros, y los productos de soya, o los alimentos altos en ácido oxálico como el repollo, la espinaca, y los camotes, pueden inhibir levemente la absorción de manganeso. Aunque los tés son ricas fuentes de manganeso, los taninos presentes en el té pueden reducir moderadamente la absorción de manganeso (15). Se ha encontrado que la ingesta de otros minerales que incluyen al hierro, al calcio y al fósforo, limita la retención de manganeso (4). El contenido de manganeso de algunos alimentos ricos en manganeso se muestra en miligramos en la tabla siguiente. Para más información sobre el contenido de nutrientes de los alimentos, revise la base de datos de composición de los alimentos de la USDA (30).
| Alimento | Porción | Manganeso (mg) |
|---|---|---|
| Piña, cruda | ½ taza, en trozos | 0.77 |
| Jugo de piña | ½ taza (4 fl. oz.) | 0.63 |
| Pacanas | 1 onza (19 mitades) | 1.28 |
| Almendras | 1 onza (23 almendras enteras) | 0.65 |
| Maní | 1 onza | 0.55 |
| Avena instantánea (preparada con agua) | 1 paquete | 0.99 |
| Cereal de salvado con pasas | 1 taza | 0.78-3.02 |
| Arroz integral, cocido | ½ taza | 1.07 |
| Pan de trigo entero | 1 rebanada | 0.60 |
| Frijoles pinto, cocidos | ½ taza | 0.39 |
| Habas, cocidas | ½ taza | 0.49 |
| Frijoles blancos, cocidos | ½ taza | 0.48 |
| Espinaca, cocida | ½ taza | 0.84 |
| Camote, cocido | ½ taza, molido | 0.44 |
| Té (verde) | 1 taza (8 onzas) | 0.41-1.58 |
| Té (negro) | 1 taza (8 onzas) | 0.18-0.77 |
Leche materna y fórmulas infantiles
Los infantes se encuentran expuestos a cantidades variables de manganeso dependiendo de su fuente de nutrición. Las concentraciones de manganeso en la leche materna, en la fórmula en base a leche de vaca, y en la fórmula en base a leche de soya varían entre 3 y 10 μg/litro, 30-50 μg/litro, y 200-300 μg/litro, respectivamente. Sin embargo, la biodisponibilidad del manganeso desde la leche materna es más alto que desde las fórmulas infantiles, y no se han reportado deficiencias de manganeso en los infantes amamantados ni toxicidad en los infantes alimentados con fórmula (31).
Agua
Las concentraciones de manganeso en el agua potable van desde 1 a 100 microgramos (μg)/litro, pero la mayoría de las fuentes contienen menos de 10 μg/litro (32). La Agencia de Protección Ambiental (APA) de los EE.UU. recomienda 0.05 mg (50 μg)/litro como la máxima concentración de manganeso permitida en el agua potable (33).
Suplementos
El manganeso lo encontramos en suplementos en varias formas diferentes, incluyendo gluconato de manganeso, sulfato de manganeso, ascorbato de manganeso, y quelatos aminoacídicos de manganeso. El manganeso se encuentra disponible como suplemento independiente o en productos combinados (34). Se pueden encontrar niveles relativamente altos de ascorbato de manganeso en los productos de salud de los huesos y articulaciones que contienen sulfato de condroitina y clorhidrato de glucosamina (véase Seguridad).
Seguridad
Toxicidad
Manganeso inhalado
La toxicidad por manganeso puede causar múltiples problemas neurológicos y es un reconocido peligro sanitario para las personas que inhalan polvo de manganeso, como soldadores y fundidores (1, 4). A diferencia del manganeso ingerido, el manganeso inhalado es transportado directamente al cerebro antes de que pueda ser metabolizado en el hígado (35). Los síntomas de la toxicidad por manganeso por lo general aparecen lentamente en un periodo de meses y hasta años. En su peor forma, la toxicidad por manganeso puede derivar en un trastorno neurológico permanente con síntomas similares a los de la enfermedad de Parkinson, incluyendo temblores, dificultad para caminar, y espasmos de los músculos faciales. Este síndrome, con frecuencia denominado manganismo, algunas veces se ve precedido por síntomas psiquiátricos como irritabilidad, agresividad, e incluso alucinaciones (36, 37). Además, la inhalación ambiental o laboral de manganeso puede causar una respuesta inflamatoria en los pulmones (38). Los síntomas clínicos de los efectos en el pulmón incluyen tos, bronquitis aguda, y disminución de la función pulmonar (39).
Tricarbonil Metilciclopentadienil Manganeso (MMT)
El MMT es un compuesto con manganeso utilizado en la gasolina como un aditivo antidetonante. Aunque en Canadá se ha utilizado con este propósito por más de 20 años, la incertidumbre respecto a los efectos adversos en la salud por la inhalación de los gases emitidos, impidió a los EE.UU. aprobar su uso en la gasolina sin plomo. En 1995, un fallo de la corte de los EE.UU. permitió que el MMT estuviera disponible para uso generalizado en la gasolina sin plomo (35). Un estudio en Montreal, donde el MMT había sido usado por más de 10 años, encontró que los niveles de manganeso en el aire eran similares a aquellos en las áreas donde no utilizaba MMT (40). Un estudio Canadiense más reciente encontró concentraciones más altas de manganeso respirable en un área urbana versus una rural, pero las concentraciones promedio en ambas áreas estaban por debajo del nivel seguro establecido por la APA de los EE.UU. (41). Sin embargo, el impacto de la exposición a largo plazo a bajos niveles de productos de combustión de MMT aún no ha sido evaluado exhaustivamente y requerirá de estudios adicionales (42).
Manganeso ingerido
Evidencia limitada sugiere que las ingesta elevadas manganeso desde el agua potable puede estar asociada con síntomas neurológicos similares a los de la enfermedad de Parkinson. Varios síntomas neurológicos fueron reportados en 25 personas que bebieron por un periodo de dos a tres meses, el agua contaminada con manganeso, y probablemente, con otros de los contaminantes provenientes de pilas secas (43). Se encontró que los niveles de manganeso en el agua eran de 14 mg/litro después de dos meses del inicio de los síntomas, niveles que ya pudieron haber estado disminuyendo (1). Un estudio de adultos mayores en Grecia encontró una alta prevalencia de síntomas neurológicos en los expuestos a niveles de manganeso en el agua de 1.8-2.3 mg/litro (44), mientras que un estudio en Alemania no encontró evidencia de un incremento en los síntomas neurológicos en las personas que bebieron agua con niveles de manganeso entre 0.3-2.2 mg/litro, en comparación con las que bebieron agua que contenía menos de 0.05 mg/litro (45). El manganeso en el agua potable puede ser más biodisponible que el manganeso en los alimentos. No obstante, ninguno de los estudios midió el manganeso dietético, por lo que la ingesta total de manganeso en estos casos es desconocida. En los EE.UU., la APA recomienda 0.05 mg/litro como la concentración máxima de manganeso disponible en el agua potable (33).
Además, estudios más recientes han demostrado que los niños expuestos a niveles elevados de manganeso a través del agua potable experimentaron déficit cognitivos y conductuales (46). Por ejemplo, un estudio de corte transversal en 142 niños de 10 años que fueron expuestos a concentraciones promedio de manganeso en el agua de 0.8 mg/litro, encontró que los niños expuestos a niveles más altos de manganeso tenían puntajes significativamente más bajos en tres pruebas de capacidad intelectual (47). Otro estudio asoció los niveles elevados de manganeso en el agua corriente con los trastornos de conducta hiperactiva en niños (48). Estos y otros reportes recientes han aumentado la preocupación sobre los efectos neuroconductuales de la exposición a manganeso en niños (46).
Un solo caso de toxicidad por manganeso fue reportado en una persona que tomó grandes cantidades de suplementos minerales durante años (49), mientras que se reportó otro caso como resultado de una persona que consumía un suplemento herbal Chino (36). La toxicidad por manganeso provocada sólo por alimentos no ha sido reportada en seres humanos, aunque ciertas dietas vegetarianas podrían aportar hasta 20 mg/día de manganeso (4, 32).
Manganeso intravenoso
Se ha observado neurotoxicidad por manganeso en individuos que recibían nutrición parenteral total, tanto como resultado de un exceso de manganeso en la solución o como un contaminante incidental (50). Los neonatos son especialmente vulnerables a la neurotoxicidad asociada al manganeso (51). Los infantes que reciben NPT con manganeso pueden estar expuestos a concentraciones de manganeso 100 veces más altas que los infantes amamantados (31). Debido a su potencial toxicidad, algunos argumentan en contra de incluir al manganeso en la nutrición parenteral (52).
Individuos con mayor susceptibilidad a la toxicidad por manganeso
- Enfermedad hepática crónica: El manganeso se elimina del cuerpo principalmente en la bilis. Así, un deterioro de la función hepática puede llevar a una disminución de la excreción de manganeso. La acumulación de manganeso en individuos con cirrosis o falla hepática puede contribuir a problemas neurológicos y a síntomas parecidos a los de la enfermedad de Parkinson (1, 34).
- Recién nacidos: Los recién nacidos pueden ser más susceptibles a la toxicidad por manganeso debido a una mayor expresión de receptores para la proteína transportadora de manganeso (transferrina) en las células nerviosas en desarrollo y a la inmadurez del sistema de eliminación de bilis del hígado (4).
- Niños: En comparación a los adultos, los infantes y niños tienen una absorción intestinal de manganeso más alta, así como una excreción biliar de manganeso más baja (46). Por lo tanto, los niños son especialmente susceptibles a cualquier efecto neurotóxico negativo del manganeso. De hecho, varios estudios recientes en niños en edad escolar reportaron efectos deletéreos cognitivos y conductuales posterior a la exposición excesiva a manganeso (47, 48, 53).
- Poblaciones deficientes de hierro: Se ha demostrado que la deficiencia de hierro incrementa el riesgo de acumulación de manganeso en el cerebro (14).
Debido a las graves consecuencias de la neurotoxicidad por manganeso, la Junta de Nutrición y Alimentos del Instituto de Medicina estableció un nivel máximo de ingesta tolerable (NM) muy conservador para el manganeso; los NMs se muestran en la tabla a continuación según edad (4).
| Grupo Etario | NM (mg/día) |
|---|---|
| Infantes 0-12 meses | Imposible de determinar* |
| Niños 1-3 años | 2 |
| Niños 4-8 años | 3 |
| Niños 9-13 años | 6 |
| Adolescentes 14-18 años | 9 |
| Adultos 19 años y más | 11 |
| *La fuente de la ingesta debiera provenir sólo de alimentos y fórmula. | |
Interacción con drogas
Los antiácidos y los laxantes con magnesio, y el medicamento antibiótico tetraciclina, pueden disminuir la absorción de manganeso si se consumen junto con alimentos o suplementos que contengan manganeso (34).
Niveles elevados de manganeso en suplementos comerciales para la salud ósea/articular
Dos estudios han encontrado que los suplementos que contienen una combinación de clorhidrato de glucosamina, sulfato de condroitina, y ascorbato de manganeso, son beneficiosos en el alivio del dolor provocado por la osteoartritis leve o moderada de rodilla, cuando se comparó con un placebo (54, 55). En un estudio, la dosis de manganeso elemental aportado por los suplementos fue de 30 mg/día por ocho semanas (55) y de 40 mg/día por seis meses en el otro (54). Durante ninguno de los estudios se reportaron efectos adversos, y los niveles de manganeso en la sangre no fueron medidos. Ninguno de los estudios comparó el tratamiento con ascorbato de manganeso con un tratamiento que contuviera clorhidrato de glucosamina y sulfato de condroitina sin ascorbato de manganeso, por lo que es imposible determinar si el suplemento hubiera provocado el mismo beneficio sin las dosis elevadas de manganeso.
Recomendación del Instituto Linus Pauling
La ingesta adecuada (IA) para el manganeso (2.3 mg/día para hombres adultos y 1.8 mg/día para mujeres adultas) parece ser suficiente como para prevenir una deficiencia en la mayoría de los individuos. La ingesta diaria de manganeso con la mayor probabilidad de promover una salud óptima es desconocida. Siguiendo la recomendación del Instituto Linus Pauling de consumir un suplemento multivitamínico/mineral que contenga el 100% de los valores diarios (VD) de la mayoría de los nutrientes, generalmente aportará 2 mg/día de manganeso en adición al de los alimentos. Debido a la potencial toxicidad y a la falta de información respecto a sus beneficios, no se recomienda la suplementación con manganeso más allá del 100% del VD (2 mg/día). Actualmente no hay evidencia de que una dieta en base a vegetales ricos en manganeso provoque toxicidad por manganeso.
Adultos mayores (>50 años)
Se desconoce si el requerimiento de manganeso es más alto para adultos mayores. Sin embargo, las enfermedades hepáticas son más comunes en adultos mayores y pueden incrementar el riesgo de toxicidad por manganeso al disminuir la eliminación del manganeso desde el cuerpo (véase Toxicidad). La suplementación con manganeso más allá del 100% del VD (2 mg/día) no se recomienda.
Autores y Críticos
Escrito en 2001 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Junio de 2007 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Marzo de 2010 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Marzo de 2010 por:
Michael Aschner, Ph.D.
Jefe del Departamento de Nutrición
Gray E.B. Stahlman Profesor de Neurociencia
Profesor de Pediatría
Profesor de Farmacología
Vanderbilt University Medical Center
Traducido al Español en 2012 por:
Guillermo Sandoval, Facultad de Odontologia, Universidad de Chile;
Revisado y editado en Diciembre 2012 por:
Andrew F.G. Quest, Ph.D. y Lisette Leyton, Ph.D.,
Profesores Titulares del Instituto de Ciencias Biomédicas,
Facultad de Medicina, Universidad de Chile,
en el marco del proyecto Anillo #ACT1111, grupo NEMESIS.
La traducción de el MIC del Inglés al Español fue asegurado, en parte, por una subvención de Bayer Consumer Care AG, Basel, Switzerland.
Derechos de autoría 2001-2024 Instituto Linus Pauling
Referencias
1. Keen CL, Ensunsa JL, Watson MH, et al. Nutritional aspects of manganese from experimental studies. Neurotoxicology. 1999;20(2-3):213-223. (PubMed)
2. Nielsen FH. Ultratrace minerals. In: Shils M, Olson JA, Shike M, Ross AC, eds. Modern Nutrition in Health and Disease. 9th ed. Baltimore: Williams & Wilkins; 1999:283-303.
3. Leach RM, Harris ED. Manganese. In: O'Dell BL, Sunde RA, eds. Handbook of nutritionally essential minerals. New York: Marcel Dekker, Inc; 1997:335-355.
4. Food and Nutrition Board, Institute of Medicine. Manganese. Dietary reference intakes for vitamin A, vitamin K, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. Washington, D.C.: National Academy Press; 2001:394-419. (National Academy Press)
5. Wedler FC. Biochemical and nutritional role of manganese: an overview. In: Klimis-Tavantzis DJ (ed). Manganese in health and disease. Boca Raton: CRC Press, Inc.; 1994:1-37.
6. Albrecht J, Sonnewald U, Waagepetersen HS, Schousboe A. Glutamine in the central nervous system: function and dysfunction. Front Biosci. 2007;12:332-343. (PubMed)
7. Keen CL, Zidenberg-Cherr S. Manganese. In: Ziegler EE, Filer LJ, eds. Present Knowledge in Nutrition. 7th ed. Washington D.C.: ILSI Press; 1996:334-343.
8. Muszynska A, Palka J, Gorodkiewicz E. The mechanism of daunorubicin-induced inhibition of prolidase activity in human skin fibroblasts and its implication to impaired collagen biosynthesis. Exp Toxicol Pathol. 2000;52(2):149-155. (PubMed)
9. Shetlar MR, Shetlar CL. The role of manganese in wound healing. In: Klimis-Tavantzis DL, ed. Manganese in health and disease. Boca Raton: CRC Press, Inc.; 1994:145-157.
10. Fitsanakis VA, Zhang N, Garcia S, Aschner M. Manganese (Mn) and Iron (Fe): Interdependency of Transport and Regulation. Neurotox Res. 2009. (PubMed)
11. Davis CD, Greger JL. Longitudinal changes of manganese-dependent superoxide dismutase and other indexes of manganese and iron status in women. Am J Clin Nutr. 1992;55(3):747-752. (PubMed)
12. Finley JW. Manganese absorption and retention by young women is associated with serum ferritin concentration. Am J Clin Nutr. 1999;70(1):37-43. (PubMed)
13. Finley JW, Johnson PE, Johnson LK. Sex affects manganese absorption and retention by humans from a diet adequate in manganese. Am J Clin Nutr. 1994;60(6):949-955. (PubMed)
14. Aschner M, Dorman DC. Manganese: pharmacokinetics and molecular mechanisms of brain uptake. Toxicol Rev. 2006;25(3):147-154. (PubMed)
15. Kies C. Bioavailability of manganese. In: Klimis-Tavantzis DL, ed. Manganese in health and disease. Boca Raton: CRC Press, Inc; 1994:39-58.
16. Johnson PE, Lykken GI. Manganese and calcium absorption and balance in young women fed diets with varying amounts of manganese and calcium. J Trace Elem Exp Med. 1991;4:19-35.
17. Norose N, Terai M, Norose K. Manganese deficiency in a child with very short bowel syndrome receiving long-term parenteral nutrition. J Trace Elem Exp Med. 1992;5:100-101 (abstract).
18. Friedman BJ, Freeland-Graves JH, Bales CW, et al. Manganese balance and clinical observations in young men fed a manganese-deficient diet. J Nutr. 1987;117(1):133-143. (PubMed)
19. Freeland-Graves J, Llanes C. Models to study manganese deficiency. In: Klimis-Tavantzis DL, ed. Manganese in health and disease. Boca Raton: CRC Press, Inc; 1994;59-86.
20. Reginster JY, Strause LG, Saltman P, Franchimont P. Trace elements and postmenopausal osteoporosis: a preliminary study of decreased serum manganese. Med Sci Res. 1988;16:337-338.
21. Odabasi E, Turan M, Aydin A, Akay C, Kutlu M. Magnesium, zinc, copper, manganese, and selenium levels in postmenopausal women with osteoporosis. Can magnesium play a key role in osteoporosis? Ann Acad Med Singapore. 2008;37(7):564-567. (PubMed)
22. Strause L, Saltman P, Smith KT, Bracker M, Andon MB. Spinal bone loss in postmenopausal women supplemented with calcium and trace minerals. J Nutr. 1994;124(7):1060-1064. (PubMed)
23. Walter RM, Jr., Uriu-Hare JY, Olin KL, et al. Copper, zinc, manganese, and magnesium status and complications of diabetes mellitus. Diabetes Care. 1991;14(11):1050-1056. (PubMed)
24. el-Yazigi A, Hannan N, Raines DA. Urinary excretion of chromium, copper, and manganese in diabetes mellitus and associated disorders. Diabetes Res. 1991;18(3):129-134. (PubMed)
25. Ekin S, Mert N, Gunduz H, Meral I. Serum sialic acid levels and selected mineral status in patients with type 2 diabetes mellitus. Biol Trac Elem Res. 2003;94:193-201. (PubMed)
26. Kazi TG, Afridi HI, Kazi N, et al. Copper, chromium, manganese, iron, nickel, and zinc levels in biological samples of diabetes mellitus patients. Biol Trace Elem Res. 2008;122(1):1-18. (PubMed)
27. Nath N, Chari SN, Rathi AB. Superoxide dismutase in diabetic polymorphonuclear leukocytes. Diabetes. 1984;33(6):586-589. (PubMed)
28. Walter RM, Aoki TT, Keen CL. Acute oral manganese does not consistently affect glucose tolerance in non diabetic and type II diabetic humans. J Trace Elem Exp Med. 1991;4:73-79.
29. Carl GF, Gallagher BB. Manganese and epilepsy. In: Klimis-Tavantzis DL, ed. Manganese in health and disease. Boca Raton: CRC Press, Inc; 1994:133-157.
30. U.S. Department of Agriculture, Agricultural Research Service. USDA National Nutrient Database for Standard Reference, Release 22. 2009. Available at: http://ndb.nal.usda.gov//. Accessed 3/3/10.
31. Aschner JL, Aschner M. Nutritional aspects of manganese homeostasis. Mol Aspects Med. 2005;26(4-5):353-362. (PubMed)
32. Keen CL, Zidenberg-Cherr S. Manganese toxicity in humans and experimental animals. In: Klimis-Tavantzis DL, ed. Manganese in health and disease. Boca Raton: CRC Press, Inc; 1994:193-205.
33. EPA Office of Water. Current Drinking Water Standards. Environmental Protection Agency, [Web page]. http://www.epa.gov/safewater/mcl.html. Accessed 9/14/06.
34. Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. Montvale: Medical Economics Company, Inc; 2001.
35. Davis JM. Methylcyclopentadienyl manganese tricarbonyl: health risk uncertainties and research directions. Environ Health Perspect. 1998;106 Suppl 1:191-201. (PubMed)
36. Pal PK, Samii A, Calne DB. Manganese neurotoxicity: a review of clinical features, imaging and pathology. Neurotoxicology. 1999;20(2-3):227-238. (PubMed)
37. Aschner M, Aschner JL. Manganese neurotoxicity: cellular effects and blood-brain barrier transport. Neurosci Biobehav Rev. 1991;15(3):333-340. (PubMed)
38. Han J, Lee JS, Choi D, et al. Manganese (II) induces chemical hypoxia by inhibiting HIF-prolyl hydroxylase: implication in manganese-induced pulmonary inflammation. Toxicol Appl Pharmacol. 2009;235(3):261-267. (PubMed)
39. Roels H, Lauwerys R, Buchet JP, et al. Epidemiological survey among workers exposed to manganese: effects on lung, central nervous system, and some biological indices. Am J Ind Med. 1987;11(3):307-327. (PubMed)
40. Zayed J, Thibault C, Gareau L, Kennedy G. Airborne manganese particulates and methylcyclopentadienyl manganese tricarbonyl (MMT) at selected outdoor sites in Montreal. Neurotoxicology. 1999;20(2-3):151-157. (PubMed)
41. Bolte S, Normandin L, Kennedy G, Zayed J. Human exposure to respirable manganese in outdoor and indoor air in urban and rural areas. J Toxicol Environ Health A. 2004;67(6):459-467. (PubMed)
42. Aschner M. Manganese: brain transport and emerging research needs. Environ Health Perspect. 2000;108 Suppl 3:429-432. (PubMed)
43. Kawamura R. Intoxication by manganese in well water. Kisasato Archives of Experimental Medicine. 1941;18:145-169.
44. Kondakis XG, Makris N, Leotsinidis M, Prinou M, Papapetropoulos T. Possible health effects of high manganese concentration in drinking water. Arch Environ Health. 1989;44(3):175-178. (PubMed)
45. Vieregge P, Heinzow B, Korf G, Teichert HM, Schleifenbaum P, Mosinger HU. Long term exposure to manganese in rural well water has no neurological effects. Can J Neurol Sci. 1995;22(4):286-289. (PubMed)
46. Ljung K, Vahter M. Time to re-evaluate the guideline value for manganese in drinking water? Environ Health Perspect. 2007;115(11):1533-1538. (PubMed)
47. Wasserman GA, Liu X, Parvez F, et al. Water manganese exposure and children's intellectual function in Araihazar, Bangladesh. Environ Health Perspect. 2006;114(1):124-129. (PubMed)
48. Bouchard M, Laforest F, Vandelac L, Bellinger D, Mergler D. Hair manganese and hyperactive behaviors: pilot study of school-age children exposed through tap water. Environ Health Perspect. 2007;115(1):122-127. (PubMed)
49. Keen C, Zidenberg-Cherr S. Manganese toxicity in humans and experimental animals. In: Klimis-Tavantzis D (ed). Manganese in health and disease. Boca Raton: CRC Press, Inc.; 1994.
50. Dobson AW, Erikson KM, Aschner M. Manganese neurotoxicity. Ann NY Acad Sci. 2004;1012:115-128. (PubMed)
51. Erikson KM, Thompson K, Aschner J, Aschner M. Manganese neurotoxicity: a focus on the neonate. Pharmacol Ther. 2007;113(2):369-377. (PubMed)
52. Hardy IJ, Gillanders L, Hardy G. Is manganese an essential supplement for parenteral nutrition? Curr Opin Clin Nutr Metab Care. 2008;11(3):289-296. (PubMed)
53. Wright RO, Amarasiriwardena C, Woolf AD, Jim R, Bellinger DC. Neuropsychological correlates of hair arsenic, manganese, and cadmium levels in school-age children residing near a hazardous waste site. Neurotoxicology. 2006;27(2):210-216. (PubMed)
54. Das A, Jr., Hammad TA. Efficacy of a combination of FCHG49 glucosamine hydrochloride, TRH122 low molecular weight sodium chondroitin sulfate and manganese ascorbate in the management of knee osteoarthritis. Osteoarthritis Cartilage. 2000;8(5):343-350. (PubMed)
55. Leffler CT, Philippi AF, Leffler SG, Mosure JC, Kim PD. Glucosamine, chondroitin, and manganese ascorbate for degenerative joint disease of the knee or low back: a randomized, double-blind, placebo-controlled pilot study. Mil Med. 1999;164(2):85-91. (PubMed)
Molibdeno
Contenido
Resumen
- El átomo de molibdeno es parte del cofactor de molibdeno en el sitio activo de cuatro enzimas en los seres humanos: sulfito oxidasa, xantina oxidasa, aldehído oxidasa y la amidoxima mitocondrial componente reductor. (Más información)
- El exceso de consumo de molibdeno causa enfermedades fatales por deficiencia de cobre en animales de pastoreo. Su rumen es el sitio de la generación de sulfuro alto, y la interacción de molibdeno con resultados de sulfuro en la formación de tiomolibdatos. Tetratiomolibdato, un tiomolibdato con cuatro átomos, puede formar complejos con el cobre previniendo su absorción y bloqueando la actividad de enzimas dependiente de cobre. (Más información)
- En los seres humanos, la terapia tetratiomolibdato ha sido desarrollada para la enfermedad de Wilson, una enfermedad genética en la cual la acumulación de cobre en los tejidos conduce al daño de hígado y cerebro. Recientemente, el uso de tetratiomolibdato ha sido explorado para el tratamiento de cáncer y enfermedades inflamatorias. (Más información)
- Las mutaciones en el cofactor de molibdeno ruta biocinética conduce a la deficiencia de todas las enzimas dependientes de molibdeno. La deficiencia del cofactor molibdeno Tipo A se debe a las mutaciones en el gen MOCS1, mientras que deficiencia Tipo 2 es causado por la mutaciones en MOCS2. Ambos Tipo A y Tipo B resultan de la deficiencia en la perdida de la actividad de sulfito oxidasa, también observado en la deficiencia de sulfito oxidasa aislada y caracterizada por anormalidades neurológicas graves en los pacientes afectados. (Más información)
- Se está considerando nuevas opciones de tratamiento para la deficiencia de cofactor de molibdeno. Suplementación de monofosfato pyranopterin cíclico a los pacientes con deficiencia Tipo A puede corregir el desorden metabólico y prevenir el deterioro neurológico. Pacientes con deficiencia Tipo B no carecen de esta molécula y por lo tanto, no pueden beneficiarse de este tratamiento. Sin embargo un estudio recientes mostro que la suplementación con piridoxina es estos pacientes podría aliviar el sufrimiento mediante la supresión de convulsiones. (Más información)
- El contenido de molibdeno de los alimentos depende en el contenido de molibdeno de los suelos, lo cual puede variar considerablemente. Variación en la incidencia mundial de cáncer de esófago se ha relacionado al contenido de molibdeno en suelos y alimentos. Observaciones similares se han realizado con el fin de identificar los factores asociados con la duración de la esperanza de vida de una población. (Más información)
Molibdeno es un oligoelemento esencial para casi todas las formas de vida. Funciona como un cofactor para una seria de enzimas que catalizan importantes transformaciones químicas en el carbono global, nitrógeno, azufre y los ciclos de azufre (1). Por lo tanto, enzimas molibdeno dependiente no son necesarias para la salud humana, sino para la salud de nuestro ecosistema.
Función
La forma biológica del átomo de molibdeno es una molécula orgánica conocida como el cofactor molibdeno (Moco) presente en el sitio activo de las enzimas que contienen Moco (enzima molibdeno) (2). En seres humanos, molibdeno es conocido por funcionar como un factor de cuatro enzimas:
- Sulfito oxidasa cataliza la transformación de sulfito a sulfato, una reacción que es necesaria para el metabolismo de los aminoácidos que contiene azufre (metionina y cisteína).
- Xantina oxidasa cataliza la descomposición de los nucleótidos (precursores de ADN y ARN) para formar ácido úrico, lo que contribuye a la capacidad antioxidante de la plasma de la sangre.
- Aldehido oxidasa y la xantina oxidase catalizan reacciones de hidroxilacion que implican un numero de moléculas diferentes con estructuras químicas similares. Xantina oxidase y aldehído oxidase también juegan un rol en el metabolismo de drogas y toxinas (3).
- Amidoxina mitocondrial componente reductor (mARC) fue descrito solo recientemente (4), y su función precisa está bajo investigación. Los estudios iniciales mostraron que mARC forma un sistema enzimáticos de tres componentes con citocromo b5 y NADH citocromo b5 redactase que cataliza la desintoxicación de bases muta génicos N-hidoxilados (5).
De estas enzimas, sulfito oxidasa se conoce ser crucial para la salud de los seres humanos. Xantinuria hereditario, caracterizado por la deficiencia en xantina oxidase (Tipo 1) o por la deficiencia tanto en la xantina oxidasa y aldehído oxidasa (Tipo 2), puede se asintomática (6). Sin embargo, en menos de la mitad de los casos, los individuos afectados presentan una serie de problemas de salud de severa variable (7, 8).
Interacción con nutrientes
Cobre
Un estudio anterior reporto que la ingesta de molibdeno de 500 μg/día y 1,500 μg/día de sorgo aumenta la excreción urinaria de cobre (2). Sin embargo, los resultados más recientes, de un estudio mejor controlado indicaron que una ingesta dietética alta de molibdeno (hasta 1,500 μg/día) no afectó adversamente el estado nutricional en ocho hombres jóvenes sanos (9).
Tetratiomolibdato
El exceso de molibdeno en la dieta se ha encontrado ser el resultado en la deficiencia de cobre en los animales de pastoreo (rumiantes). En el tracto digestivo de los rumiantes, la formación de compuestos que contienen azufre y molibdeno, conocido como timolibdatos, previene la absorción de cobre y puede causar desordenes fatales de dependencia de cobre (10). Tetratiomolibdato (TM) es una molécula que puede formar complejos de alta afinidad con cobre, controlando cobre libre (cobre que no está unido a la ceruloplasmina) e inhibición de chaperones de cobre y enzimas que contienen cobre (11, 12). La habilidad de TM para reducir los niveles de cobre libre es explotara en el tratamiento de la enfermedad de Wilson, un desorden genético caracterizado por acumulación de cobre en los tejidos responsables por la hepatitis y desórdenes neurológicos. Empeoramiento neurológico se ha relacionado a niveles tóxicos de cobre libre en el suero de pacientes neurológicamente presentes. La terapia TM parecer ser capaz de estabilizar el estado neurológico y prevenir el deterioro neurológico en estos pacientes, en comparación a con la opción de tratamiento inicial estándar (13).
Cobre también es requerido como cofactor de enzimas envueltas en la inflamación y angiogénesis, conocidos para acelerar la progresión y metástasis de cáncer. Estudios de depleción de cobre que emplean TM se han iniciado en pacientes con neoplasias avanzadas con el fin de prevenir la progresión de la enfermedad o recaída. Estos ensayos pilotos mostraron un resultado prometedor en personas con cáncer de riñón metastásico (14), cancer colorrectal metastásico (15), y cáncer de mama con alto riesgo de recaída (16). TM fue relativamente bien tolerado y estabilizo la enfermedad o previno la recaída en correlación con el agotamiento de cobre. La eficaz de TM es también investigado en modelos de animales con enfermedades inflamatorias y relacionado a lo inmune (17, 18), y, en este momento, se necesitan estudios clínicos para evaluar si la depleción de cobre puede estabilizar la enfermedad y mejorar la supervivencia en los seres humanos, tal como lo sugiera un estudio de terapia TM con los pacientes con cirrosis biliar (19).
Deficiencia
La deficiencia dietética de molibdeno nunca se ha observada en personas saludables (2).
Deficiencia de molibdeno adquirida
El único casa documentado de deficiencia de molibdeno adquirida ocurrió en pacientes con la enfermedad de Crohn en la nutrición parenteral total a largo plazo (NPT) sin molibdeno añadido a la NPT solución (20). El paciente desarrollo un ritmo cardiaco y respiratorio rápido, dolores de cabeza, y ceguera nocturna, y en la última instancia se convirtió en estado de coma. El paciente fue diagnosticado con defectos en la producción de ácido úrico y metabolismo de aminoácidos de azufre. La condición clínica de pacientes mejoro y la intolerancia de aminoácidos desapareció cuando se suspendió la solución de NPT fue descontinuada y en su lugar fue suplementada con molibdeno en la forma de molibdato de amonio (160 μg/día) (20).
Deficiencia de cofactor de molibdeno heredada
Dado a las funciones de molibdeno solo en la forma de Moco en los seres humanos, cualquier perturbación del metabolismo de Moco puede perturbar la función de todas las enzimas molibdeno. La comprensión actual de la esencialidad de molibdeno en humanos se basa principalmente en los estudios de individuos con trastornos metabólicos congénitos muy raros causados por la deficiencia en Moco. Moco se sintetiza de novo por una vía metabólica de etapas múltiples que implican cuatro genes: MOCS1, MOCS2, MOCS3, and GPHN (Figura 1). Hasta la fecha, más de 60 mutaciones afectan la mayoría de MOCS1 y MOCS2 se han identificado (21).

La ausencia de un Moco funcional tiene un impacto directo sobre la actividad de las enzimas de molibdeno. Desordenes metabólicos especialmente asociados con la deficiencia de actividad de sulfito oxidasa incluye una acumulación de sulfito, taurina, S-sulfocisteina y tiosulfato (Figura 2). Este perfil metabólico es idéntico a la observada en la deficiencia aislada de sulfito oxidase (ISOD) una condición hereditaria causada por mutaciones en el gene SUOX que codifica para sulfito oxidasa (22). En comparación con ISOD, la deficiencia de moco (MocoD) también afecta la vía de xantina y conduce a la acumulación de hipoxantina y xantina, y baja el ácido úrico en la sangre a niveles indetectables (Figura 3). MocoD y ISOD se han diagnosticado en más de 100 individuos en todo el mundo. Sin embargo la incidencia global de MocoD es probable que sea subestimada como resultado de una falta de diagnósticos o reportes (21, 23, 24). Ambos desordenes derivan de rasgos recesivos, lo que significa que solo las personas que heredan dos copias de genes anormales (uno de cada padre) desarrollan la enfermedad. Los individuos que heredan solo una copia del gen anormal son conocidos como portadores de los rasgos pero no presentan ningún síntoma. ISOD y MocoD pueden diagnosticarse relativamente temprano en el embarazo (10-14 semanas de gestación) mediante ensayos de actividad enzimática utilizando células amnióticas y muestras de vellosidades coriónicas y mediante pruebas genéticas (23, 25).
MocoD e ISOD normalmente ocurren en los primeros días de vida, aunque algunos casos de MocoD con presentaciones tardías se han descrito (26-28). La pérdida de actividad de sulfito oxidasa en ISOD y MocoD conduce a severa disfunción neurológica caracterizada por el atrofio cerebral, retraso mental, convulsiones intratables, y la dislocación de los lentes oculares. En la actualidad, no esta claro si los efectos neurológicos son el resultado de la acumulación de metabolito toxico, como el sulfito, o inadecuada producción de sulfate. Pacientes con ISOD y MocoD también se encontraron con elevada excreción de semialdehido adipico a-amino (a-AASA) (29). Acumulación de a-AASA es la firma metabólica de la deficiencia en a-AASA deshidrogenada observada en pacientes con epilepsia dependiente piridoxida. La deficiencia enzimática en estos individuos provoca aumento en a-AASA y en su forma cíclica piperideina 6 carboxilato (P6C). P6C puede atrapar piridoxal 5 fosfatos (PLP), la forma activa de vitamina B6 (piridoxina), lo que lleva a una deficiencia en PLP, lo cual se corregí con suplemento de piridoxina. Una disminución en PLP también se ha observado en el líquido cefalorraquideo de pacientes ISOD y MocoD (30). No es claro si sulfito es responsable por la acumulación de a-AASA y la deficiencia en PLP en pacientes ISOD y MocoD. Sin embargo la suplementación de piridoxina y ácido fólico en pacientes con MocoD normalizaron con éxito el nivel PLP y abolieron las convulsiones en dos pacientes con mutaciones en MOCS2 (MocoD Tipo B) (31). Aunque los medicamentos anticonvulsivo y las restricciones dietéticas de aminoácidos que contienes sulfuro pueden ser beneficiosos en algunos casos (32), no hay tratamientos opciones para pacientes con mutaciones en MOCS2, GPHN (MocoD Tipo C), o genes SUOX. Suplementos de piridoxina es una nueva opción de ser considerado para aliviar características clínicas especificas en pacientes.
Un tratamiento exitoso usa monofosfato ciclico pyranopterin (CPMP) ha sido descrito para los pacientes con mutaciones en los genes MOCS1, y un ensayo clínico con enfoque retrospectivo esta en marcha para avaluar su seguridad. El gene MOCS1 controla el paso inicial en la ruta biocinética de Moco, catalizando la conversión de trifosfato de guanosina en cPMP. Por lo tanto, los pacientes con mutaciones en los genes MOCS1 carecen cPMP. La administración diaria de cPMP a pacientes resuelve todas las anormalidades metabólicas asociadas con la defectuosidad de sulfito oxidasa y vías xantinas y previene nuevos signos de deterioro neurológico (33, 34). Un diagnostico temprano y la iniciación de tratamiento son esenciales para asegurar el éxito (34). Dado que la terapia de remplazo cPMP solo puede beneficiar MocoD Tipo A, se requiere métodos de tratamientos adicionales.
La Ingesta Diaria Recomendada (IDR)
La IDR para molibdeno fue revisado recientemente en enero, 2001 (2). Se basa en los resultados de los estudios de balance nutricional en ocho hombres saludables bajo condiciones de laboratorio controlado (35, 36). Los valores de IDR para molibdeno están enlistados en la tabla siguiente en microgramos (μg)/día por año y genero. Se establecieron los niveles de ingesta adecuada (IA) para los infantes basado en el promedio de ingesta molibdeno de la leche humana, exclusiva.
| Etapa de la Vida | Edad | Machos (μg/día) | Hembras (μg/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 2 (IA) | 2 (IA) |
| Infantes | 7-12 meses | 3 (IA) | 3 (IA) |
| Niños | 1-3 años | 17 | 17 |
| Niños | 4-8 años | 22 | 22 |
| Niños | 9-13 años | 34 | 34 |
| Adolescentes | 14-18 años | 43 | 43 |
| Adultos | 19 años o más | 45 | 45 |
| Embarazo | Todas las edades | - | 50 |
| Período de lactancia | Todas las edades | - | 50 |
Prevención de Enfermedades
Cáncer de esófago
Linxian es una pequeña región en el norte de China donde el incidente de cáncer del esófago y estomago es muy alta (10 veces mas que el promedio en China y 100 veces mas que el promedio en los U.S.). La tierra en esta región es baja en molibdeno y otros elementos minerales: por lo tanto, ingesta dietética de molibdeno es también bajo. Estudios conducidos en otras áreas de baja y alta incidencia de cáncer de esófago mostraron que el contenido de molibdeno y zinc en el cabello y uñas es significativamente menor en habitantes de las regiones de alto riesgo en comparación con los puntos fríos. Por otra parte, los pacientes con cáncer de esófago muestra un contenido reducido de los elementos traza en comparación con los parientes sanos (37, 38).
El aumento de nitrosaminas, los cuales son conocidos como carcinógenos, puede ser uno de una serie de factores dietéticos y ambientales que contribuye al desarrollo de cáncer de esófago en residentes de las regiones con alto riesgo. Adición de molibdato de amonio al suelo puede disminuir el riesgo de cáncer de esófago, limitando la exposición de nitrosamina. No esta claro si la suplementación dietética de molibdeno suplementario es beneficial en la disminución del riesgo de cáncer de esófago. Los ensayos de intervención conducidos en áreas de Linxian con suplementación de minerales y vitaminas, incluyendo molibdeno (30 μg/día), no lograron disminuir las tasas de mortalidad de cáncer de esófago u otros tipos de cáncer y la incidencia en un periodo de cinco años (revisado en 39).
Longevidad
Rugao es un condado en la providencia de Jiangsu (China), famoso por la longevidad de sus habitantes. La longevidad extendida difícilmente puede atribuirse a diferencia significativas en las tradiciones, estilos de vida, o hábitos de alimentación entre los residentes, y las personas mas longevas no están relacionados entre sin, limitando la posibilidad de influenza genética. Sin embargo, el condado tiene un gran numero de suelos cuyas composiciones podrían afectar la traza de distribución de oligoelementos en el agua y cultivos y podrían ser vinculado con la salud humana y longevidad.
Se encontraron correlaciones significativamente entre la proporción de personas mayores de 90 años de edad por cada 100,000 habitantes y oligoelementos, incluyendo molibdeno, en suelos, agua potable, y arroz, lo cual constituye elementos fundamentales de su medio natural (40). El porcentaje de personas de larga vida (>80 años) en Zhongxiang (provincia de Hubei) también positivamente vinculado con el contenido de molibdeno en sus alimentos básicos, arroz (41). En estas regiones, es probable que las combinaciones de oligoelementos contribuyan a la salud optima y longevidad en comparación con el único efecto de molibdeno.
Fuentes
Fuentes alimenticias
El Estudio de la Dieta Total, una encuesta anual de contenido de minerales en una dieta típica estadounidense, indica que la ingesta dietética de los promedios de molibdeno es 76 μg/día para mujeres y 109 μg/día para hombres. Por lo tanto, la ingestas usual de molibdeno son superiores a las dosis diarias recomendadas por la IDR para el molibdeno. Las legumbres, tales como los frijoles, lentejas, y chicharos, son las fuentes mas ricas en molibdeno. Los productos de granos y nueces están considerados como buenas fuentes, mientras que los productos de animales, frutas y muchas verduras son generalmente bajos en molibdeno (2). Debido al contenido de molibdeno de plantas depende del contenido de molibdeno de los suelos y otras condiciones ambientales, el contenido de molibdeno de los alimentos puede variar considerablemente (38, 42).
Suplementos
Molibdeno en suplementos nutricionales es generalmente en la forma de molibdato de sodio o molibdato de amonio (43).
Seguridad
Toxicidad
La toxicidad de compuestos de molibdeno parecer ser relativamente baja en humanos. El aumento de los niveles séricos de ácido úricos y ceruloplasmina (una enzima de oxidante de hierro) se ha reportado en trabajadores expuestos en una planta de tostación de molibdeno (44). Los síntomas de seudogotosa se ha reportado en la población Armenia que consume 10 a 15 miligramos (mg) de molibdeno diaria (45). En otros estudios, la sangre y los niveles de acido úrico no se elevaron por la ingesta de molibdeno hasta 1.5 mg/día (2). Solo ha reportado un caso de toxicidad aguda relacionada a molibdeno de un suplemento dietético: un varón adulto reporto consumir un total de 13.5 mg de molibdeno durante un periodo de 18 días (300-800 μg/día) y desarrollo psicosis aguda con alucinaciones, convulsiones, y otros síntomas neurológicos (46). Sin embargo, un estudio controlado en cuatro, hombres jóvenes saludables encontraron que la ingesta de molibdeno, que va dese 22 μg/día a 1,490 μg/día (casi 1.5mg/día) no provoco efectos adversos graves cuando molibdeno fue dada durante 24 días (35).
La Junta de Alimentos y Nutricionales (JAN/FNB) del Instituto de Medicina encontró poca evidencia que el exceso de molibdeno se asocia con resultados adversos para la salud de las personas generalmente sanas. Para determinar la tolerancia del nivel máximo de ingesta tolerable (NM), la JAN/FNB selecciono efectos adversos sobre la reproducción en ratas como el índice más sensible de toxicidad y aplico un factor de incertidumbre grande por que los datos de animales fueron usados (2). La NM de molibdeno esta enlistada por edad y en la siguiente tabla.
| Grupo Etario | NM (μg/día) |
|---|---|
| Infantes 0-12 meses | No es posible establizar* |
| Niños 1-3 años | 300 |
| Niños 4-8 años | 600 |
| Niños 9-13 años | 1,100 (1.1 mg/día) |
| Adolescentes 14-18 años | 1,700 (1.7 mg/día) |
| Adultos 19 años y más | 2,000 (2.0 mg/día) |
| *La fuente de ingesta debería ser solo de alimentos y formula. | |
Interacción con drogas
Se ha encontrado que dosis altas de molibdeno inhiben el metabolismo de paracetamol en ratas (47); sin embargo, no fue encontrado si esto ocurre a dosis clínicamente relevantes en humanos.
Recomendación del Instituto Linus Pauling
La IDR para molibdeno (45 μg/día para adultos) es suficiente para prevenir la deficiencia. Aunque la ingesta de molibdeno para la salud optima se desconoce, actualmente no hay evidencia de que una ingesta mayor que la IDR sea beneficioso. La mayoría de las personas en los EE.UU. consumen mas molibdeno de los suficiente en sus dietas, lo que hace la suplementación innecesaria. Siguiendo las recomendaciones generales del Instituto de Linus Pauling de tomar un suplemento multivitamínico/mineral que contenga 100% de los valores diarios (VD) para la mayoría de los nutrientes es probable que proporcione 75 μg/día de molibdeno porque la VD para molibdeno no ha sido revisada para reflejar la IDR reciente. Aunque la cantidad de molibdeno encontrada reciente en la mayoría de los suplementos multivitamínicos/minerales es superior a la dosis de IDR, es muy por debajo de las niveles máximos de ingesta tolerable (NM) de 2,000 μg/día y debería ser seguro para los adultos.
Adultos mayores (>50 años)
Debido a que el envejecimiento no se ha asociado con cambios significativos en el requisito de molibdeno (2), nuestras recomendaciones para adultos mayores es la misma que para los adultos de 50 años o menores.
Autores y Críticos
Escrito en 2001 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Abril de 2007 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Junio de 2013 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Julio de 2013 por:
Ralf R. Mendel, Ph.D.
Profesor de Biologia Vegetal y Director
Departamento Biologia Vegetal
Universidad de Braunschweig de Tecnologia
Braunschweig, Alemania
Traducido al Español en 2012 por:
Guillermo Sandoval, Facultad de Odontologia, Universidad de Chile;
Revisado y editado en Diciembre 2012 por:
Andrew F.G. Quest, Ph.D. y Lisette Leyton, Ph.D.,
Profesores Titulares del Instituto de Ciencias Biomédicas,
Facultad de Medicina, Universidad de Chile,
en el marco del proyecto Anillo #ACT1111, grupo NEMESIS.
La traducción de el MIC del Inglés al Español fue asegurado, en parte, por una subvención de Bayer Consumer Care AG, Basel, Switzerland.
Derechos de autoría 2001-2024 Instituto Linus Pauling
Referencias
1. Wuebbens MM, Liu MT, Rajagopalan K, Schindelin H. Insights into molybdenum cofactor deficiency provided by the crystal structure of the molybdenum cofactor biosynthesis protein MoaC. Structure Fold Des. 2000;8(7):709-718. (PubMed)
2. Food and Nutrition Board, Institute of Medicine. Molybdenum. In: Dietary reference intakes for vitamin A, vitamin K, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. Washington, D.C.: National Academy Press; 2001:420-441. (National Academy Press)
3. Eckhert C. Other trace elements In: Shils ME, Shike M, Ross AC, Caballero B, Cousins RJ, eds. Modern Nutrition in Health and Disease. 10th ed. Philadelphia: Lippincott, Williams & Wilkins; 2006:338-350.
4. Wahl B, Reichmann D, Niks D, et al. Biochemical and spectroscopic characterization of the human mitochondrial amidoxime reducing components hmARC-1 and hmARC-2 suggests the existence of a new molybdenum enzyme family in eukaryotes. J Biol Chem. 2010;285(48):37847-37859. (PubMed)
5. Plitzko B, Ott G, Reichmann D, et al. The Involvement of Mitochondrial Amidoxime Reducing Components 1 and 2 and Mitochondrial Cytochrome b5 in N-reductive Metabolism in Human Cells. J Biol Chem. 2013;288(28):20228-20237. (PubMed)
6. Beedham C. Molybdenum hydroxylases as drug-metabolizing enzymes. Drug Metab Rev. 1985;16(1-2):119-156. (PubMed)
7. Zannolli R, Micheli V, Mazzei MA, et al. Hereditary xanthinuria type II associated with mental delay, autism, cortical renal cysts, nephrocalcinosis, osteopenia, and hair and teeth defects. J Med Genet. 2003;40(11):e121. (PubMed)
8. Fujiwara Y, Kawakami Y, Shinohara Y, Ichida K. A case of hereditary xanthinuria type 1 accompanied by bilateral renal calculi. Intern Med. 2012;51(14):1879-1884. (PubMed)
9. Turnlund JR, Keyes WR. Dietary molybdenum: Effect on copper absorption, excretion, and status in young men. In: Roussel AM, ed. Trace Elements in Man and Animals. Vol 10. New York: Kluwer Academic Press.; 2000:951-953.
10. Suttle NF. Copper imbalances in ruminants and humans: unexpected common ground. Adv Nutr. 2012;3(5):666-674. (PubMed)
11. Helz GR, Erickson BE. Extraordinary stability of copper(I)-tetrathiomolybdate complexes: possible implications for aquatic ecosystems. Environ Toxicol Chem. 2011;30(1):97-102. (PubMed)
12. Alvarez HM, Xue Y, Robinson CD, et al. Tetrathiomolybdate inhibits copper trafficking proteins through metal cluster formation. Science. 2010;327(5963):331-334. (PubMed)
13. Brewer GJ, Askari F, Dick RB, et al. Treatment of Wilson's disease with tetrathiomolybdate: V. Control of free copper by tetrathiomolybdate and a comparison with trientine. Transl Res. 2009;154(2):70-77. (PubMed)
14. Redman BG, Esper P, Pan Q, et al. Phase II trial of tetrathiomolybdate in patients with advanced kidney cancer. Clin Cancer Res. 2003;9(5):1666-1672. (PubMed)
15. Gartner EM, Griffith KA, Pan Q, et al. A pilot trial of the anti-angiogenic copper lowering agent tetrathiomolybdate in combination with irinotecan, 5-flurouracil, and leucovorin for metastatic colorectal cancer. Invest New Drugs. 2009;27(2):159-165. (PubMed)
16. Jain S, Cohen J, Ward MM, et al. Tetrathiomolybdate-associated copper depletion decreases circulating endothelial progenitor cells in women with breast cancer at high risk of relapse. Ann Oncol. 2013;24(6):1491-1498. (PubMed)
17. Hou G, Abrams GD, Dick R, Brewer GJ. Efficacy of tetrathiomolybdate in a mouse model of multiple sclerosis. Transl Res. 2008;152(5):239-244. (PubMed)
18. Wei H, Zhang WJ, McMillen TS, Leboeuf RC, Frei B. Copper chelation by tetrathiomolybdate inhibits vascular inflammation and atherosclerotic lesion development in apolipoprotein E-deficient mice. Atherosclerosis. 2012;223(2):306-313. (PubMed)
19. Askari F, Innis D, Dick RB, et al. Treatment of primary biliary cirrhosis with tetrathiomolybdate: results of a double-blind trial. Transl Res. 2010;155(3):123-130. (PubMed)
20. Abumrad NN, Schneider AJ, Steel D, Rogers LS. Amino acid intolerance during prolonged total parenteral nutrition reversed by molybdate therapy. Am J Clin Nutr. 1981;34(11):2551-2559. (PubMed)
21. Reiss J, Hahnewald R. Molybdenum cofactor deficiency: Mutations in GPHN, MOCS1, and MOCS2. Hum Mutat. 2011;32(1):10-18. (PubMed)
22. Tan WH, Eichler FS, Hoda S, et al. Isolated sulfite oxidase deficiency: a case report with a novel mutation and review of the literature. Pediatrics. 2005;116(3):757-766. (PubMed)
23. Shalata A, Mandel H, Dorche C, et al. Prenatal diagnosis and carrier detection for molybdenum cofactor deficiency type A in northern Israel using polymorphic DNA markers. Prenat Diagn. 2000;20(1):7-11. (PubMed)
24. Kikuchi K, Hamano S, Mochizuki H, Ichida K, Ida H. Molybdenum cofactor deficiency mimics cerebral palsy: differentiating factors for diagnosis. Pediatr Neurol. 2012;47(2):147-149. (PubMed)
25. Johnson JL. Prenatal diagnosis of molybdenum cofactor deficiency and isolated sulfite oxidase deficiency. Prenat Diagn. 2003;23(1):6-8. (PubMed)
26. Hughes EF, Fairbanks L, Simmonds HA, Robinson RO. Molybdenum cofactor deficiency-phenotypic variability in a family with a late-onset variant. Dev Med Child Neurol. 1998;40(1):57-61. (PubMed)
27. Vijayakumar K, Gunny R, Grunewald S, et al. Clinical neuroimaging features and outcome in molybdenum cofactor deficiency. Pediatr Neurol. 2011;45(4):246-252. (PubMed)
28. Alkufri F, Harrower T, Rahman Y, et al. Molybdenum cofactor deficiency presenting with a parkinsonism-dystonia syndrome. Mov Disord. 2013;28(3):399-401. (PubMed)
29. Mills PB, Footitt EJ, Ceyhan S, et al. Urinary AASA excretion is elevated in patients with molybdenum cofactor deficiency and isolated sulphite oxidase deficiency. J Inherit Metab Dis. 2012;35(6):1031-1036. (PubMed)
30. Footitt EJ, Heales SJ, Mills PB, Allen GF, Oppenheim M, Clayton PT. Pyridoxal 5'-phosphate in cerebrospinal fluid; factors affecting concentration. J Inherit Metab Dis. 2011;34(2):529-538. (PubMed)
31. Struys EA, Nota B, Bakkali A, Al Shahwan S, Salomons GS, Tabarki B. Pyridoxine-dependent epilepsy with elevated urinary alpha-amino adipic semialdehyde in molybdenum cofactor deficiency. Pediatrics. 2012;130(6):e1716-1719. (PubMed)
32. Johnson JL, Duran M. Molybdenum cofactor deficiency and isolated sulfite deficiency. In: Scriver RC, ed. Metabolic and molecular bases of inherited disease. New York: μgraw-Hill; 2001:3163-3177.
33. Veldman A, Santamaria-Araujo JA, Sollazzo S, et al. Successful treatment of molybdenum cofactor deficiency type A with cPMP. Pediatrics. 2010;125(5):e1249-1254. (PubMed)
34. Hitzert MM, Bos AF, Bergman KA, et al. Favorable outcome in a newborn with molybdenum cofactor type A deficiency treated with cPMP. Pediatrics. 2012;130(4):e1005-1010. (PubMed)
35. Turnlund JR, Keyes WR, Peiffer GL. Molybdenum absorption, excretion, and retention studied with stable isotopes in young men at five intakes of dietary molybdenum. Am J Clin Nutr. 1995;62(4):790-796. (PubMed)
36. Turnlund JR, Keyes WR, Peiffer GL, Chiang G. Molybdenum absorption, excretion, and retention studied with stable isotopes in young men during depletion and repletion. Am J Clin Nutr. 1995;61(5):1102-1109. (PubMed)
37. Nouri M, Chalian H, Bahman A, et al. Nail molybdenum and zinc contents in populations with low and moderate incidence of esophageal cancer. Arch Iran Med. 2008;11(4):392-396. (PubMed)
38. Ray SS, Das D, Ghosh T, Ghosh AK. The levels of zinc and molybdenum in hair and food grain in areas of high and low incidence of esophageal cancer: a comparative study. Glob J Health Sci. 2012;4(4):168-175. (PubMed)
39. Goodman M, Bostick RM, Kucuk O, Jones DP. Clinical trials of antioxidants as cancer prevention agents: past, present, and future. Free Radic Biol Med. 2011;51(5):1068-1084. (PubMed)
40. Huang B, Zhao Y, Sun W, et al. Relationships between distributions of longevous population and trace elements in the agricultural ecosystem of Rugao County, Jiangsu, China. Environ Geochem Health. 2009;31(3):379-390. (PubMed)
41. Lv J, Wang W, Krafft T, Li Y, Zhang F, Yuan F. Effects of several environmental factors on longevity and health of the human population of Zhongxiang, Hubei, China. Biol Trace Elem Res. 2011;143(2):702-716. (PubMed)
42. Mills CF, Davis GK. Molybdenum. In: Mertz W, ed. Trace elements in human and animal nutrition. 5th ed. San Diego: Academic Press; 1987:429-463.
43. Molybdenum. In: Hendler S, Rorvik D, eds. PDR for Nutritional Supplements. 2nd ed. Montvale: Physicians' Desk Reference Inc.; 2008:425-429.
44. Walravens PA, Moure-Eraso R, Solomons, CC, Chapell, R, Bentley G. Biochemical abnormalities in workers exposed to molybdenum dust. Arch Environ Health. 1979;34(5):302-308. (PubMed)
45. Vyskocil A, Viau C. Assessment of molybdenum toxicity in humans. J Appl Toxicol. 1999;19(3):185-192. (PubMed)
46. Momcilovic B. A case report of acute human molybdenum toxicity from a dietary molybdenum supplement--a new member of the "Lucor metallicum" family. Arh Hig Rada Toksikol. 1999;50(3):289-297. (PubMed)
47. Boles JW, Klaassen CD. Effects of molybdate and pentachlorophenol on the sulfation of acetaminophen. Toxicology. 2000;146(1):23-35. (PubMed)
Potasio
Contenido
Resumen
- El potasio es considerado un "nutriente que preocupa a la salud pública" de acuerdo con las Pautas Dietéticas 2015-2020 para los estadounidenses ya que su subconsumo en la población de los EE. UU. se asocia con efectos adversos para la salud (hipertensión y enfermedad cardiovascular). (Más información)
- La función normal del cuerpo depende de la regulación estricta de las concentraciones de potasio tanto dentro como fuera de las células. (Más información)
- La baja concentración de potasio en la sangre (hipokalemia) puede causar parálisis muscular o ritmos cardíacos anormales y puede ser fatal. La hipokalemia se debe generalmente a la pérdida excesiva de potasio, así como es con vómitos o diarrea prolongados, el uso diuréticos o con enfermedad renal. (Más información)
- La hipertensión crónica daña el corazón, los vasos sanguíneos y los riñones, lo que aumenta el riesgo de enfermedad cardiovascular. El aumento de la ingesta de potasio dietario puede ayudar a disminuir la presión arterial en individuos normotensos e hipertensos. (Más información)
- Los resultados de estudios observacionales reportaron que un mayor consumo de potasio en la dieta se asoció con menores riesgos de accidente cerebrovascular y formación de cálculos renales. La evidencia del papel de las ingestas de potasio en la promoción de la salud ósea sigue siendo débil. (Más información)
- La ingesta adecuada (IA) de potasio es de 2,600 mg/día para las mujeres y 3,400 mg/día para los hombres. La IA para cada grupo de edad/etapa de la vida se estableció según el nivel de ingesta reportado en poblaciones aparentemente sanas. (Más información)
- Buenas fuentes dietéticas de potasio incluyen frutas y verduras, algunas nueces y semillas, y productos lácteos. (Más información)
- Las preocupaciones de seguridad con el consumo de potasio son limitadas en personas sanas porque los riñones ajustan la excreción urinaria de potasio a la ingesta de potasio. El uso concomitante de suplementos de potasio con ciertas drogas/fármacos puede aumentar el riesgo de toxicidad por potasio. (Más información)
El potasio es un mineral dietético esencial y electrolito. El término electrolito se refiere a una sustancia que se disocia en iones (partículas cargadas) en solución, lo que la hace capaz de conducir electricidad. La función normal del cuerpo depende de la regulación estricta de las concentraciones de potasio tanto dentro como fuera de las células (1).
Función
Mantenimiento del potencial de membrana
El potasio (K+) es el principal ion cargado positivamente (catión) en el líquido dentro de las células, mientras que el sodio (Na+) es el catión principal en el líquido extracelular. Las concentraciones de potasio son aproximadamente 30 veces más altas dentro que fuera de las células, mientras que las concentraciones de sodio son más de 10 veces más bajas dentro que fuera de las células. Las diferencias de concentración entre el potasio y el sodio a través de las membranas celulares crean un gradiente electroquímico conocido como potencial de membrana. El potencial de membrana de una célula se mantiene mediante bombas de iones en la membrana celular, especialmente las bombas de Na+/K+-ATPasa. Estas bombas utilizan ATP (energía) para bombear el sodio fuera de la célula a cambio de potasio (Figura 1). Su actividad se ha estimado representa del 20%-40% del gasto de energía en reposo en un adulto típico. La mayor proporción de energía dedicada al mantenimiento de los gradientes de concentración de sodio/potasio enfatiza la importancia de esta función en el sustento de la vida. El estrecho control del potencial de membrana de la célula es crítico para la transmisión del impulso nervioso, la contracción muscular, y la función cardíaca (2-4).
[Figura 1 - Clic para Agrandar]
Cofactor para enzimas
Un número limitado de enzimas requiere la presencia de potasio para su actividad. La activación de Na+/K+-ATPasa requiere la presencia de sodio y potasio. La presencia de potasio también es necesaria para la actividad del piruvato quinasa, una enzima importante en el metabolismo de los carbohidratos (5).
Deficiencia
Una concentración anormalmente baja de potasio plasmático se conoce como hipokalemia. La hipokalemia es más comúnmente un resultado de la pérdida excesiva de potasio, por ejemplo, por vómitos o diarrea prolongados, el uso de algunos diuréticos y otros medicamentos (véase Interacciones con drogas/fármacos), algunas formas de enfermedad renal o trastornos metabólicos. Los síntomas de la hipokalemia están relacionados con alteraciones en el potencial de membrana y el metabolismo celular (1). Incluyen fatiga, debilidad muscular y calambres, y parálisis intestinal, que puede provocar hinchazón, estreñimiento y dolor abdominal. La hipokalemia crónica está asociada a la hipertensión y a la formación de cálculos renales (véase Prevención de Enfermedades y Tratamiento de Enfermedades). La hypokalemia severa puede resultar en parálisis muscular o ritmos cardíacos anormales (arritmias cardíacas) que pueden ser fatales (1, 6).
Condiciones que incrementan el riesgo de hipokalemia (véase también Interacciones con drogas/fármacos; 1):
- El uso de diuréticos que desperdician potasio (p. ej., diuréticos tiazídicos o furosemida)
- Vómito prolongado o diarrea
- Uso excesivo o abuso de laxantes
- Anorexia nervosa o bulimia
- Sudoración excesiva
- Nefropatías
- Poliuria
- Producción anormalmente alta de aldosterona (hiperaldosteronismo)
- Agotamiento de magnesio
- Recuperación de la desnutrición prolongada
La ingesta baja de potasio sola por lo general no produce hipokalemia. Sin embargo, una cantidad insuficiente de potasio dietario en pacientes con riesgo de hipokalemia puede precipitar la hipokalemia (1).
Raramente el consumo habitual de grandes cantidades de regaliz negro ha provocado hipokalemia (7, 8). El regaliz contiene un compuesto (es decir, ácido glicirrícico) con efectos fisiológicos similares a los de la aldosterona, una hormona que aumenta la excreción urinaria de potasio.
La Ingesta Adecuada (IA)
Las Ingestas Dietéticas Recomendadas para el potasio han sido revisadas recientemente por la Junta de Nutrición y Alimentos (JNA) de la Academia Nacional de Medicina. La JNA no encontró suficiente evidencia para determinar un Requerimiento Estimado Promedio (REP) y derivar una Ingesta Diaria Recomendada (RDA, por sus siglas en inglés); en cambio, establecieron una ingesta adecuada (IA) basada en ingestas medias en personas generalmente sanas (Tabla 1; 9). La JNA no encontró evidencia suficiente de estudios en humanos los cuales examinaron las ingestas de potasio en relación con la enfermedad crónica y la mortalidad (revisado recientemente por la Agencia para la Investigación y la Calidad del Cuidado de la Salud; 10) para informar a las Ingestas Dietéticas Recomendadas para el potasio (11).
Prevención de Enfermedades
Las dietas de las personas que residen en los países industrializados occidentales son muy diferentes de las que se consumían antes de la revolución agrícola y el cambio hacia el consumo de alimentos procesados altamente refinados (12). Entre otras diferencias, la ingesta diaria de cloruro de sodio (sal) en las dietas modernas es aproximadamente tres veces más alta que la ingesta diaria de potasio en una base molar, mientras que la ingesta de sal en cultivos primitivos es aproximadamente siete veces más baja que la ingesta de potasio (13). La deficiencia relativa de potasio dietario en la dieta moderna y una mayor proporción de sodio a potasio pueden contribuir al desarrollo de algunas enfermedades crónicas.
Accidente cerebrovascular
Estudios observacionales han reportado consistentemente un mayor riesgo de enfermedad cardiovascular con ingestas elevadas de sodio dietario (14, 15). Varios estudios de cohorte prospectivo también han hallado una asociación inversa entre la ingesta de potasio y el riesgo de accidente cerebrovascular. Un meta-análisis de nueve estudios de cohorte prospectivo mostró que las ingestas diarias de potasio que oscilaban entre 3,510 mg y 4,680 mg se asociaron con un riesgo reducido en 30% de accidente vascular (16). No se encontraron asociaciones con enfermedad coronaria cardíaca o enfermedad total cardiovascular. En un meta-análisis más reciente de 16 estudios, se encontró que la ingesta de potasio más alta versus la más baja se asoció con un riesgo 13% menor de accidente cerebrovascular después de múltiples ajustes (incluso para la presión arterial) (17). El riesgo más bajo de accidente cerebrovascular correspondió a ingestas diarias de potasio alrededor de 3,500 mg. Los análisis de subgrupos mostraron un riesgo reducido de accidente cerebrovascular isquémico, pero no un accidente cerebrovascular hemorrágico. Finalmente, en un reciente meta-análisis de 16 estudios observacionales, cada aumento de 1 unidad en la proporción de sodio a potasio dietario se encontró estaba asociado con un riesgo 22% mayor de accidente cardiovascular (12).
Cálculos renales
El calcio urinario anormalmente alto (hipercalciuria) aumenta el riesgo de desarrollar cálculos renales. En individuos con antecedentes de desarrollar cálculos renales que contienen calcio, el aumento de la carga de ácido dietario ha sido significativamente asociado con el incremento en la excreción del calcio urinario (18). Aumentando la ingesta de potasio dietario (y álcali) al aumentar la ingesta de frutas y verduras o al tomar suplementos de bicarbonato de sodio (KHCO3) se encontró que disminuyen la excreción de calcio urinario. Por otro lado, se encontró que la privación de potasio aumenta la excreción de calcio urinario (19, 20).
Tres grandes estudios de cohorte prospectivo de EE. UU. — el Estudio de Seguimiento de Profesionales de la Salud y los Estudios de Salud de Enfermeras I y II — que incluyeron 193,676 participantes, examinaron la ingesta de potasio dietario y la proporción de proteína a potasio (un marcador de la carga de ácido en la dieta) en la dieta en relación al riesgo de desarrollar cálculos renales (21). En las tres cohortes, la ingesta de potasio dietario se derivó casi en su totalidad de alimentos ricos en potasio, como las frutas y verduras. En las tres cohortes, se encontró que individuos en el quintil más alto de la ingesta de potasio tienen un 33%-56% menos probabilidad de desarrollar cálculos renales sintomáticos que aquellos en el quintil más bajo de la ingesta. Adicionalmente, un análisis conjunto de los datos de las tres cohortes mostró que aquellos con la proporción de proteína animal a potasio más alta versus la más baja tenían un 41% más probabilidades de desarrollar cálculos renales (21).
La alcalinización urinaria con citrato de potasio suplementario se usa en los formadores de piedra para reducir el riesgo de formación recurrente de piedras (revisado en 22). Sin embargo, la terapia con citrato de potasio sólo debe iniciarse bajo la supervisión de un proveedor médico.
Osteoporosis
En un estudio de cohorte de casos de 2015 anidado en el Estudio Prospectivo Europeo sobre Dieta y Cáncer (EPIC), un estudio de Norfolk, que incluyó a 5,319 individuos, se encontró que la ingesta dietética de potasio (sola o combinada con ingestas de magnesio) se encontró está inversamente asociada con las mediciones de la atenuación por ultrasonido de la banda ancha (BUA, por sus siglas en inglés) del hueso del talón (calcáneo) (un factor predictivo del riesgo de fractura incidental) y riesgo de fractura de cadera en mujeres pero no en hombres (23). Más recientemente, un estudio transversal en adultos mayores coreanos reportó una densidad mineral ósea (DMO) más alta de la cadera y del cuello del fémur en las personas que se encuentran en el tercil más alto versus más bajo de las ingestas de potasio (24). Aunque estos estudios observacionales sugieren una relación entre la ingesta de potasio y la salud ósea, no se puede establecer si existe una relación causa y efecto.
Los mecanismos por los cuales el potasio podría influir en la salud ósea son pocos conocidos. Las dietas modernas (occidentales) tienden a ser relativamente bajas en fuentes de álcali (frutas y verduras) y altas en fuentes de ácido (pescado, carne y queso) (25). Cuando la cantidad de iones de bicarbonato son insuficientes para mantener el pH normal, el cuerpo es capaz de movilizar sales de calcio alcalinas desde los huesos para neutralizar los ácidos consumidos en la dieta o generados por el metabolismo (26). Debido a que las frutas y verduras son ricos en potasio y en precursores de iones de bicarbonato, aumentando su consumo puede ayudar a reducir el contenido neto de ácido de la dieta y preservar el calcio en los huesos, que de otro modo podrían ser movilizados para mantener un pH normal.
Alternativamente, la suplementación de bicarbonato de potasio podría disminuir la excreción de ácido urinario y calcio e influir en el recambio óseo — un ensayo pequeño en mujeres posmenopáusicas halló que la suplementación con bicarbonato de potasio aumenta los biomarcadores de la formación ósea y una disminución de los biomarcadores de la reabsorción ósea (27). Un ensayo controlado de dos años, aleatorio, doble-ciego, en 201 adultos mayores sin osteoporosis (edad media, 69 años) encontró evidencia de aumento de la columna lumbar, cadera, cuello femoral, y DMO total del cuerpo, así como DMO trabecular del radio y tibia, con citrato de potasio suplementario (2,340 mg/day) en comparación con el placebo (28). También se encontró que el citrato de potasio aumenta la concentración sérica del propéptido N-terminal del procolágeno tipo I (PINP) — un marcador de la formación ósea — y reduce la concentración urinaria del telopéptido N del colágeno tipo I (NTX) — un marcador de reabsorción ósea (28). Otro ensayo de tres meses, aleatorio, controlado con placebo en 244 adultos (≥50 años) examinó el efecto del bicarbonato de potasio oral, ya sea a 39 mg/kg/día o 58.5 mg/kg/día, en marcadores de recambio óseo (29). Ambos regímenes de dosificación llevaron a reducciones de la concentración de PINP en suero y en la concentración de NTX en la orina, pero la evidencia de algún efecto fue más fuerte con la dosis más baja (dosis media administrada, 3,160 mg/día) en lugar de con la dosis más alta (dosis media administrada, 4,760 mg/día). En contraste, un ensayo controlado aleatorio de dos años encontró que ni la suplementación con citrato de potasio (721 mg/día or 2,165 mg/día) ni un aumento en el consumo de frutas y verduras (300 mg/día) tuvo un impacto en los marcadores de recambio óseo o aumento de la DMO en mujeres posmenopáusicas (30). Un meta-análisis de estudios de intervención encontró que el citrato o el bicarbonato de potasio podrían reducir la excreción de ácido neto y calcio urinario, pero la evidencia de apoyar algún efecto en marcadores de recambio óseo y densidad ósea fue débil (31). El ensayo controlado, aleatorio, doble ciego, más reciente en 40 mujeres posmenopáusicas con osteopenia no encontró diferencias en los marcadores de recambio óseo durante un período de seis meses entre los que recibieron suplementos con citrato de potasio y los que tomaron un placebo (32). Los autores destacaron la posibilidad de que un efecto del suplemento de potasio podría tener un impacto en la salud ósea en un subconjunto de sujetos con ingesta baja de potasio y/o signos de acidosis de bajo grado. En este estudio, todos los participantes recibieron suplementos diarios de carbonato de calcio (500 mg/día) y vitamina D (10 µg/día).
En general, el hecho de que consumir frutas y verduras ricas en potasio pueda influir en la salud ósea y ayudar a disminuir el riesgo de osteoporosis sigue siendo incierto.
Tratamiento de Enfermedades
Hipertensión
Cuarenta y cinco por ciento de los adultos estadounidenses tienen hipertensión (niveles de presión sanguíneas ≥130/80 mm Hg) (33). La hipertensión crónica daña el corazón, los vasos sanguíneos y los riñones, lo que aumenta el riesgo de enfermedad cardíaca e infarto cerebrovascular, así como la enfermedad renal hipertensiva (34, 35). Se reconoce que las dietas modernas, que son altas en sodio y bajas en potasio, se reconocen contribuyen en gran medida a la alta prevalencia de hipertensión (véase el artículo sobre Sodio). A diferencia de los recordatorios dietéticos de 24 horas, las recolecciones de orina de 24 horas proporcionan estimaciones exactas de ingestas dietéticas de sodio y potasio (36). Un análisis de la Encuesta de Evaluación Nacional de Salud y Nutrición (NHANES) estadounidense de 2014 mostró un aumento en la presión arterial sistólica al aumentar la excreción de sodio y el aumentar la proporción de sodio a potasio en la orina (37). En este estudio, el cuartil más alto versus el más bajo de la excreción urinaria de potasio (valores medios, 3,043 mg/día versus 1,484 mg/día) se asoció con un riesgo 62% menor de hipertensión (37).
El ensayo de los Enfoques Dietéticos para Detener la Hipertensión (DASH) proporcionó evidencia del efecto reductor de la presión arterial de una dieta rica en potasio y calcio, modestamente alta en proteína, y baja en grasas totales, grasas saturadas, colesterol, carnes rojas, dulces, y bebidas con azúcar comparada con la dieta típica estadounidense (38). De hecho, en comparación con la dieta de control que proporcionó sólo 3.5 porciones/día de frutas y verduras y 1,700 mg/día de potasio, la adherencia a la dieta DASH que incluyó 8.5 porciones/día de frutas y vegetales y 4,100 mg/día de potasio redujo la presión arterial sistólica/diastólica por un promedio de 11.4/5.5 mm Hg en individuos con hipertensión y de 3.5/2.1 mm Hg en aquellos sin hipertensión (38). Un meta-análisis del 2014 de 17 ensayos controlados aleatorios que examinó el efecto de la dieta DASH en comparación con una dieta de control en un total de 2,561 adultos encontró reducciones generales en la presión arterial sistólica y diastólica en 6.7 mm Hg y 3.5 mm Hg, respectivamente (39). Por muy efectiva que sea la dieta DASH, los efectos de disminución de la presión arterial difícilmente pueden atribuirse solamente a las ingestas de potasio (40).
Un meta-análisis del 2015 de 15 ensayos controlados aleatorios, que incluyó 917 individuos, evaluó los efectos del aumento de la ingesta de potasio, principalmente en forma de suplementos de cloruro de potasio (KCl) en la presión arterial (41). Trece estudios incluyeron participantes hipertensos que no estuvieran tomando medicamentos antihipertensiva y dos estudios incluyeron sujetos normotensos o en riesgo. La mayoría de los estudios utilizaron dosis suplementarias de potasio entre 2,340 y 2,535 mg/día (60-65 mmol/L). El aumento de la ingesta de potasio dio lugar a reducciones generales de la presión arterial sistólica en 4.7 mm Hg y la presión arterial sistólica en 3.5 mm Hg. El efecto reductor de la presión arterial del potasio suplementario fue más pronunciado cuando el análisis estaba limitado a individuos con hipertensión: la presión sistólica y diastólica se redujeron en 6.8 mm Hg y 4.6 mm Hg, respectivamente (41). Dos meta-análisis adicionales publicados en el 2017 también confirmaron un efecto reductor de la presión arterial del potasio suplementario. Los hallazgos sugirieron cierta evidencia de un mayor efecto cuando la ingesta basal de potasio era inferior a 3,510 mg/día (vs. ≥3,510 mg/day [90 mmol]) (42). Los meta-análisis también informaron una relación dosis-respuesta entre la ingesta de potasio y la disminución de la presión arterial (42, 43).
El potasio suplementario puede ayudar a disminuir la presión arterial, pero los suplementos de potasio sólo deben usarse en consulta con un proveedor médico (véase Suplementos). Incrementando la ingesta de potasio a los niveles recomendados (véase La Ingesta Adecuada) al consumir una dieta rica en frutas y verduras puede ayudar a disminuir la presión arterial y puede tener beneficios adicionales para la salud. La presión arterial es un marcador de riesgo cardiovascular confiable (44). Sin embargo, aunque el reducir el consumo de sodio al mismo tiempo que el incrementar la ingesta de potasio ayuda a disminuir la presión arterial (45), la evidencia actual sugiere que el asesoramiento dietético y las intervenciones de apoyo pueden no ser suficientes para brindar beneficios cardiovasculares en individuos con hipertensión (46).
Fuentes
Fuentes alimenticias
Las fuentes más ricas de potasio son las frutas y verduras. Las nueces, semillas, y productos lácteos también son buenas fuentes de potasio. Una encuesta dietética en los EE.UU. indicó que la ingesta promedio de potasio en la dieta fue de 2,408 mg/día para mujeres adultas y de 3,172 mg/día para hombres adultos (47). Debido a que muchos individuos en la población consumen potasio en cantidades muy por debajo de la IA y debido a que el subconsumo de potasio está vinculado a efectos adversos para la salud, el potasio ha sido reconocido como un "nutriente de preocupación de la salud pública" en las Directrices Dietarías para los Americanos del 2015-2020. En el 2016, la Administración de Drogas y Alimentos (FDA) requirió que los fabricantes mostraran el contenido de potasio de los alimentos en la etiqueta de Información Nutricional de los alimentos (48).
Algunas fuentes dietéticas de potasio relativamente buenas se enumeran en la Tabla 2, junto con su contenido de potasio en miligramos (mg). Para más información sobre el contenido nutricional de los alimentos, busque en la base de datos de composición de alimentos del USDA (49).
Suplementos
Los suplementos multivitamínicos y minerales en los EE.UU. no contienen más de 99 mg de potasio por porción (50). Un miliequivalente (mEq) o un milimol (mmol) corresponde a aproximadamente 39 mg de potasio. Las dosis más altas de potasio suplementario generalmente se prescriben para prevenir y tratar el agotamiento del potasio y la hipokalemia. El uso de suplementos de potasio más potentes en la deficiencia de potasio requiere una estrecha supervisión de las concentraciones séricas de potasio. El potasio está disponible en diferentes formas suplementarias, incluyendo el cloruro de potasio, citrato de potasio, gluconato de potasio, bicarbonato de potasio, aspartato de potasio y orotato de potasio (50). Debido a la posibilidad de graves efectos secundarios, uno debe buscar consulta médica antes de decidir usar un suplemento de potasio (véase Seguridad). La mejor manera de aumentar la ingesta de potasio de uno es aumentando el consumo de alimentos y bebidas ricos en potasio (50).
Finalmente, muchos sustitutos de sal contienen cloruro de potasio, y el acesulfamo de potasio (Ace-K) es un endulcorante de uso general aprobado por la FDA.
Seguridad
Toxicidad
Las concentraciones séricas de potasio anormalmente elevadas se conocen como hiperkalemia. La hiperkalemia ocurre cuando la ingesta de potasio excede la capacidad de los riñones para eliminarlo. La insuficiencia renal aguda o crónica, el uso de diuréticos ahorradores de potasio y la insuficiente secreción de aldosterona (hipoaldosteronismo) puede resultar en la acumulación de potasio debido a una disminución de la excreción urinaria de potasio. Las dosis orales de potasio >18 g tomadas al mismo tiempo en individuos que no están acostumbrados a ingestas elevadas pueden provocar hiperkalemia severa, incluso en aquellos con función renal normal (6, 50). La hiperkalemia también puede deberse a un cambio de potasio intracelular en la circulación, que puede ocurrir con la ruptura de glóbulos rojos (hemólisis) o daño tisular (p. ej., trauma o quemaduras graves). Los síntomas de hiperkalemia pueden incluir hormigueo en las manos y los pies, debilidad muscular y parálisis temporal. La complicación más seria de hiperkalemia es el desarrollo de un ritmo anormal cardíaco (arritmia cardíaca), que puede provocar un paro cardíaco (51). Un meta-análisis de estudios controlados aleatorios mostró que la frecuencia cardíaca en adultos sanos era poco probable que se viera afectada por el uso crónico de potasio suplementario con una dosis de 2 a 3 g/día (52).
Véase Interacciones con drogas/fármacos para una discusión de los medicamentos que aumentan el riesgo de hiperkalemia.
Reacciones adversas a los suplementos de potasio
Los síntomas gastrointestinales son los efectos secundarios más comunes de los suplementos de potasio, como náusea, vómito, molestias abdominales y diarrea. La ulceración intestinal ha sido reportada después del uso de tabletas de cloruro de potasio con recubrimiento entérico. Tomar potasio con las comidas o tomar una forma microencapsulada de potasio puede reducir los efectos secundarios gastrointestinales (50). Ocasionalmente pueden aparecer erupciones. La reacción adversa más severa a la suplementación con potasio es la hiperkalemia, pero es poco frecuente en sujetos con función renal normal (véase Toxicidad). Los individuos con función renal anormal y aquellos que toman medicamentos ahorradores de potasio (véase Interacciones con drogas/fármacos) deben ser monitoreados de cerca para prevenir la hiperkalemia (50, 53).
Interacciones con drogas/fármacos
La Tabla 3 enumera las clases de medicamentos que se sabe que aumentan el riesgo de hiperkalemia (potasio sérico elevado) en pacientes que también usan suplementos de potasio (50, 51, 54).
Se sabe que varias clases de medicamentos inducen hipokalemia (potasio sérico bajo; Tabla 4; 55). En la ausencia de tratamiento, la hipokalemia puede tener complicaciones graves e incluso ser mortal (véase Deficiencia). Varios mecanismos explican cómo ciertos medicamentos pueden llevar al agotamiento de potasio. Por ejemplo, los diuréticos tanto de asa como de tiazida aumentan la excreción urinaria de potasio. Los corticoides causan retención de sodio que conduce a un aumento compensatorio en la excreción urinaria de potasio. Las penicilinas formuladas como sales de sodio también estimulan la excreción de potasio. Varios medicamentos, incluidos los aminoglucósidos, los agentes antifúngicos (anfotericina B, fluconazol), y el cisplatino, pueden dañar el epitelio tubular renal y provocar una pérdida severa de potasio. Antibióticos de tetraclicina obsoletos se han relacionado con trastornos electrolitos.
Recomendación del Instituto Linus Pauling
Existe evidencia sustancial que sugiere que una dieta rica en alimentos y bebidas ricas en potasio pueden estar asociadas con menores riesgos de accidente cerebrovascular, hipertensión, cálculos renales y posiblemente osteoporosis. Sin embargo, actualmente no hay pruebas suficientes para establecer una relación causal entre las ingestas de potasio y el riesgo de estas condiciones crónicas (10). Como consecuencia, las ingestas medias de potasio observadas en personas aparentemente sanas se utilizaron para establecer ingestas adecuadas (IA) por edad/etapa de la vida en la reciente revisión de las Ingestas Dietéticas de Referencia para el potasio. Los valores de la IA son 2.6 g/día para las mujeres y 3.4 g/día para los hombres (véase La Ingesta Adecuada).
Las frutas y verduras se encuentran entre las fuentes más ricas de potasio dietario, y una gran cantidad de evidencia respalda la asociación de un mayor consumo de frutas y verduras con un riesgo reducido de enfermedad cardiovascular. El Instituto Linus Pauling recomienda el consumo de una dieta rica en alimentos ricos en potasio (véase Fuentes), especialmente frutas, verduras, nueces y productos lácteos para asegurar una ingesta adecuada de potasio.
Adultos mayores (>50 años)
Una dieta rica en frutas y verduras que ofrezca 2.6-3.4 g/día de potasio (véase La Ingesta Adecuada) debe contribuir a mantener un bajo riesgo de enfermedad crónica en adultos mayores generalmente saludables. Esta recomendación no se aplica a las personas a las que se les ha recomendado que limiten el consumo de potasio por parte de un profesional de la salud (véase Seguridad).
Autores y Críticos
Originalmente escrito en 2001 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Febrero de 2004 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Diciembre de 2010 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Abril de 2019 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Abril de 2019 por:
Connie M. Weaver, Ph.D.
Profesor Distinguido y Director de Alimentos y Nutrición
Universidad de Purdue
Traducido al Español en 2019 por:
Natsumi Then Simazaki
Instituto Linus Pauling
Universidad Estatal de Oregon
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Derechos de autoría 2001-2024 Instituto Linus Pauling
Referencias
1. Bailey JL, Sands JM, Franch HA. Water, electrolytes, and acid — Base Metabolism In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease: Lippincott Williams & Wilkins; 2014:102-132.
2. Clausen T. Quantification of Na+,K+ pumps and their transport rate in skeletal muscle: functional significance. J Gen Physiol. 2013;142(4):327-345. (PubMed)
3. Larsen BR, Stoica A, MacAulay N. Managing brain extracellular K(+) during neuronal activity: the physiological role of the Na(+)/K(+)-ATPase subunit isoforms. Front Physiol. 2016;7:141. (PubMed)
4. Shattock MJ, Ottolia M, Bers DM, et al. Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J Physiol. 2015;593(6):1361-1382. (PubMed)
5. Sheng H-W. Sodium, chloride and potassium. In: Stipanuk M, ed. Biochemical and Physiological Aspects of Human Nutrition. Philadelphia: W.B. Saunders Company; 2000:686-710.
6. Food and Nutrition Board, Institute of Medicine. Potassium. Dietary Reference Intakes for Water, Potassium, Sodium, Chloride, and Sulfate. Washington, D.C.: National Academies Press; 2005:186-268. (The National Academies Press)
7. Mumoli N, Cei M. Licorice-induced hypokalemia. Int J Cardiol. 2008;124(3):e42-44. (PubMed)
8. Walker BR, Edwards CR. Licorice-induced hypertension and syndromes of apparent mineralocorticoid excess. Endocrinol Metab Clin North Am. 1994;23(2):359-377. (PubMed)
9. Food and Nutrition Board, National Academy of Medicine. Dietary Reference Intakes for Sodium and Potassium - uncorrected proofs. The National Academies of Sciences, Engineering, and Medicine. Washington, D.C.: The National Academies Press; 2019. (The National Academies Press)
10. Newberry SJ, Chung M, Anderson CAM, et al. AHRQ Comparative Effectiveness Reviews. Sodium and potassium intake: effects on chronic disease outcomes and risks. Rockville (MD): Agency for Healthcare Research and Quality (US); 2018. (PubMed)
11. Food and Nutrition Board, National Academy of Medicine. Potassium: Dietary Reference Intakes based on chronic disease. Dietary Reference Intakes for Sodium and Potassium - uncorrected proofs. The National Academies of Sciences, Engineering, and Medicine. Washington, D.C.: National Academy Press; 2019:121-154. (The National Academies Press)
12. Weaver CM. Potassium and health. Adv Nutr. 2013;4(3):368s-377s. (PubMed)
13. Young DB, Lin H, McCabe RD. Potassium's cardiovascular protective mechanisms. Am J Physiol. 1995;268(4 Pt 2):R825-837. (PubMed)
14. Aburto NJ, Ziolkovska A, Hooper L, Elliott P, Cappuccio FP, Meerpohl JJ. Effect of lower sodium intake on health: systematic review and meta-analyses. BMJ. 2013;346:f1326. (PubMed)
15. Jayedi A, Ghomashi F, Zargar MS, Shab-Bidar S. Dietary sodium, sodium-to-potassium ratio, and risk of stroke: A systematic review and nonlinear dose-response meta-analysis. Clin Nutr. 2018; doi: 10.1016/j.clnu.2018.05.017. [Epub ahead of print]. (PubMed)
16. Aburto NJ, Hanson S, Gutierrez H, Hooper L, Elliott P, Cappuccio FP. Effect of increased potassium intake on cardiovascular risk factors and disease: systematic review and meta-analyses. BMJ. 2013;346:f1378. (PubMed)
17. Vinceti M, Filippini T, Crippa A, de Sesmaisons A, Wise LA, Orsini N. Meta-analysis of potassium intake and the risk of stroke. J Am Heart Assoc. 2016;5(10). (PubMed)
18. Trinchieri A, Zanetti G, Curro A, Lizzano R. Effect of potential renal acid load of foods on calcium metabolism of renal calcium stone formers. Eur Urol. 2001;39 Suppl 2:33-36; discussion 36-37. (PubMed)
19. Lemann J, Jr., Pleuss JA, Gray RW. Potassium causes calcium retention in healthy adults. J Nutr. 1993;123(9):1623-1626. (PubMed)
20. Morris RC, Jr., Schmidlin O, Tanaka M, Forman A, Frassetto L, Sebastian A. Differing effects of supplemental KCl and KHCO3: pathophysiological and clinical implications. Semin Nephrol. 1999;19(5):487-493. (PubMed)
21. Ferraro PM, Mandel EI, Curhan GC, Gambaro G, Taylor EN. Dietary protein and potassium, diet-dependent net acid load, and risk of incident kidney stones. Clin J Am Soc Nephrol. 2016;11(10):1834-1844. (PubMed)
22. Suarez M, Youssef RF. Potassium citrate: treatment and prevention of recurrent calcium nephrolithiasis. J Clin Nephrol Res. 2015;2(1).
23. Hayhoe RP, Lentjes MA, Luben RN, Khaw KT, Welch AA. Dietary magnesium and potassium intakes and circulating magnesium are associated with heel bone ultrasound attenuation and osteoporotic fracture risk in the EPIC-Norfolk cohort study. Am J Clin Nutr. 2015;102(2):376-384. (PubMed)
24. Kong SH, Kim JH, Hong AR, Lee JH, Kim SW, Shin CS. Dietary potassium intake is beneficial to bone health in a low calcium intake population: the Korean National Health and Nutrition Examination Survey (KNHANES) (2008-2011). Osteoporos Int. 2017;28(5):1577-1585. (PubMed)
25. Fenton TR, Eliasziw M, Lyon AW, Tough SC, Hanley DA. Meta-analysis of the quantity of calcium excretion associated with the net acid excretion of the modern diet under the acid-ash diet hypothesis. Am J Clin Nutr. 2008;88(4):1159-1166. (PubMed)
26. Morris RC, Jr., Frassetto LA, Schmidlin O, Forman A, Sabastian A. Expression of osteoporosis as determined by diet-disordered electrolyte and acid-base metabolism. In: Burckhardt P, Dawson-Hughes B, Heaney R, eds. Nutritional Aspects of Osteoporosis. San Diego: Academic Press; 2001:357-378.
27. Sebastian A, Harris ST, Ottaway JH, Todd KM, Morris RC, Jr. Improved mineral balance and skeletal metabolism in postmenopausal women treated with potassium bicarbonate. N Engl J Med. 1994;330(25):1776-1781. (PubMed)
28. Jehle S, Hulter HN, Krapf R. Effect of potassium citrate on bone density, microarchitecture, and fracture risk in healthy older adults without osteoporosis: a randomized controlled trial. J Clin Endocrinol Metab. 2013;98(1):207-217. (PubMed)
29. Dawson-Hughes B, Harris SS, Palermo NJ, et al. Potassium bicarbonate supplementation lowers bone turnover and calcium excretion in older men and women: a randomized dose-finding trial. J Bone Miner Res. 2015;30(11):2103-2111. (PubMed)
30. Macdonald HM, Black AJ, Aucott L, et al. Effect of potassium citrate supplementation or increased fruit and vegetable intake on bone metabolism in healthy postmenopausal women: a randomized controlled trial. Am J Clin Nutr. 2008;88(2):465-474. (PubMed)
31. Lambert H, Frassetto L, Moore JB, et al. The effect of supplementation with alkaline potassium salts on bone metabolism: a meta-analysis. Osteoporos Int. 2015;26(4):1311-1318. (PubMed)
32. Granchi D, Caudarella R, Ripamonti C, et al. Potassium citrate supplementation decreases the biochemical markers of bone loss in a group of osteopenic women: the results of a randomized, double-blind, placebo-controlled pilot study. Nutrients. 2018;10(9). (PubMed)
33. Centers for Disease Control and Prevention. High Blood Pressure Facts. November 2016. Available at: https://www.cdc.gov/bloodpressure/facts.htm. Accessed 11/30/18.
34. Mente A, O'Donnell M, Rangarajan S, et al. Associations of urinary sodium excretion with cardiovascular events in individuals with and without hypertension: a pooled analysis of data from four studies. Lancet. 2016;388(10043):465-475. (PubMed)
35. Sanghavi S, Vassalotti JA. Dietary sodium: a therapeutic target in the treatment of hypertension and CKD. J Ren Nutr. 2013;23(3):223-227. (PubMed)
36. Cogswell ME, Loria CM, Terry AL, et al. Estimated 24-hour urinary sodium and potassium excretion in US adults. JAMA. 2018;319(12):1209-1220. (PubMed)
37. Jackson SL, Cogswell ME, Zhao L, et al. Association between urinary sodium and potassium excretion and blood pressure among adults in the United States: National Health and Nutrition Examination Survey, 2014. Circulation. 2018;137(3):237-246. (PubMed)
38. Appel LJ, Moore TJ, Obarzanek E, et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N Engl J Med. 1997;336(16):1117-1124. (PubMed)
39. Saneei P, Salehi-Abargouei A, Esmaillzadeh A, Azadbakht L. Influence of Dietary Approaches to Stop Hypertension (DASH) diet on blood pressure: a systematic review and meta-analysis on randomized controlled trials. Nutr Metab Cardiovasc Dis. 2014;24(12):1253-1261. (PubMed)
40. Weaver CM, Stone MS, Lobene AJ, Cladis DP, Hodges JK. What is the evidence base for a potassium requirement? Nutr Today. 2018;53(5):184-195. (PubMed)
41. Binia A, Jaeger J, Hu Y, Singh A, Zimmermann D. Daily potassium intake and sodium-to-potassium ratio in the reduction of blood pressure: a meta-analysis of randomized controlled trials. J Hypertens. 2015;33(8):1509-1520. (PubMed)
42. Filippini T, Violi F, D'Amico R, Vinceti M. The effect of potassium supplementation on blood pressure in hypertensive subjects: A systematic review and meta-analysis. Int J Cardiol. 2017;230:127-135. (PubMed)
43. Poorolajal J, Zeraati F, Soltanian AR, Sheikh V, Hooshmand E, Maleki A. Oral potassium supplementation for management of essential hypertension: A meta-analysis of randomized controlled trials. PLoS One. 2017;12(4):e0174967. (PubMed)
44. Viera AJ. Screening for hypertension and lowering blood pressure for prevention of cardiovascular disease events. Med Clin North Am. 2017;101(4):701-712. (PubMed)
45. Appel LJ, Giles TD, Black HR, et al. ASH position paper: dietary approaches to lower blood pressure. J Am Soc Hypertens. 2010;4(2):79-89. (PubMed)
46. Adler AJ, Taylor F, Martin N, Gottlieb S, Taylor RS, Ebrahim S. Reduced dietary salt for the prevention of cardiovascular disease. Cochrane Database Syst Rev. 2014(12):Cd009217. (PubMed)
47. Hoy MK, Goldman JD. Potassium Intake of the US Population: What We Eat In America, NHANES 2009-2010. 2012.
48. Food and Drug Administration. Food Labeling: Revision of the Nutrition and Supplement Facts Labels. Available at: https://www.regulations.gov/document?D=FDA-2012-N-1210-0875. Accessed 12/12/18.
49. US Department of Agriculture ARS. USDA National Nutrient Database for Standard Reference, Release 22. 2009. Available at: http://www.nal.usda.gov/fnic/foodcomp/search/. Accessed 3/15/10.
50. Hendler SS, Rorvik DR. PDR for Nutrititional Supplements. Montvale: Thomson Reuters; 2008.
51. Mandal AK. Hypokalemia and hyperkalemia. Med Clin North Am. 1997;81(3):611-639. (PubMed)
52. Gijsbers L, Molenberg FJ, Bakker SJ, Geleijnse JM. Potassium supplementation and heart rate: A meta-analysis of randomized controlled trials. Nutr Metab Cardiovasc Dis. 2016;26(8):674-682. (PubMed)
53. Gennari FJ. Hypokalemia. N Engl J Med. 1998;339(7):451-458. (PubMed)
54. Natural Medicines. Potassium/Professional Monograph/Interactions with Drugs. June 26, 2018. Available at: https://naturalmedicines.therapeuticresearch.com/. Accessed 11/19/18.
55. Natural Medicines. Potassium/Professional Monograph/Nutrient Depletion. June 26, 2018. Available at: https://naturalmedicines.therapeuticresearch.com/. Accessed 11/19/18.
Selenio
Contenido
Resumen
- El selenio ejerce varias funciones biológicas principalmente como parte del aminoácido, selenocisteína, el cual se encuentra en por lo menos 25 proteínas que contienen selenocisteína (selenoproteínas). (Más información)
- Cinco glutatión peroxidasas, tres tiorredoxina reductasas, tres iodotironina deiodinasas y una metionina sulfóxido reductasa B1 están entre las selenoproteínas mejor caracterizadas con funciones conocidas. (Más información)
- La protección antioxidante deteriorada en los individuos deficientes de selenio puede afectar las respuestas fisiológicas al estrés. La cardiomiopatía de Keshan y la osteoartropatía de Kashin-Beck son enfermedades que ocurren específicamente en áreas con deficiencia de selenio en Asia. (Más información)
- La ingesta diaria recomendada actual (IDR) establecida por el Instituto de Medicina de los EE.UU. es de 55 μg/día para adolescentes y adultos de todas las edades. (Más información)
- Estudios basados en la observación tempranos han encontrado tanto asociaciones nulas o inversas entre la exposición al selenio y el riesgo de canceres de sitio especifico. Sin embargo, evidencia actual proveniente de ensayos de intervención en participantes repletos de selenio no apoya un efecto protector de la suplementación con selenio contra el cáncer. (Más información)
- Evidencia preliminar de ensayos controlados aleatorios sugiere que la suplementación con selenio podría prevenir la progresión de la carga viral e incrementar el conteo de células inmunes en pacientes VIH-positivos. (Más información)
- Los niveles y formas químicas del selenio en alimentos de origen vegetal varían de acuerdo a la composición y contenido de selenio del suelo en el cual las plantas son cultivadas. Fuentes alimenticias ricas en selenio incluyen nuez de Brasil, granos, mariscos, vísceras, aves de corral, y productos lácteos. (Más información)
- El nivel máximo de ingesta tolerable (NM) para el selenio es de 400 μg/día para adolescentes y adultos e incluye ambos selenio de alimentos, el cual promedia alrededor de 100 μg/día para los adultos en los EE.UU., y selenio proveniente de suplementos. (Más información)
- Debido a que cierta evidencia sugiere que las concentraciones altas de selenio en el suero pueden tener efectos adversos en el control glicémico, individuos con un estatus de selenio alto y/o aquellos en riesgo de diabetes mellitus tipo 2 deberían evitar tomar suplementos de selenio. (Más información)
El selenio es un elemento traza que es esencial en pequeñas cantidades, pero como todos los elementos esenciales, el selenio puede ser tóxico en niveles elevados. A diferencia de las plantas, la mayoría de los animales — incluyendo a los seres humanos — requieren selenio para el funcionamiento apropiado de un cierto número de enzimas dependientes de selenio conocidas como selenoproteínas. Durante la síntesis de proteínas (traducción) el aminoácido selenocisteína es incorporado en proteínas elongadas en ubicaciones muy específicas en la secuencia de aminoácidos con el objetivo de formar selenoproteínas funcionales. Aunque plantas superiores no parecen necesitar selenio para su sobrevivencia, estas pueden incorporar lo de manera no específica en moléculas que contienen sulfuro cuando el mineral está presente en el suelo (1). Debe notarse que, en los animales, el aminoácido selenometionina puede ser incorporado de manera no especifica en las proteínas en lugar de metionina (2). Sin embargo, solo las proteínas que contienen selenocisteína son consideradas como selenoproteínas (Figura 1).

[Figura 1 - Clic para Agrandar]
Función
Selenoproteínas
Veinticinco genes que codifican para las selenoproteínas han sido identificados en los humanos (3). La inserción de selenocisteína en las selenoproteínas durante la traducción es dirigida por la presencia de una secuencia de inserción de selenocisteína (SECIS) dentro de los ARNm de la selenoproteína. Brevemente, el reconocimiento de SECIS por la maquinaria de traducción resulta en el reclutamiento de factores de traducción específicos que decodifican codones UGA en marco al insertar selenocisteína dentro de selenoproteínas elongadas (4).
La investigación esta gradualmente descubriendo las funciones metabólicas de todas las selenoproteínas humanas, incluyendo variantes de empalme (3). Algunas de las selenoproteínas con una función identificada incluyen:
Glutatión peroxidasas
Se han identificado cinco enzimas glutatión peroxidasas (GPx1-4 y GPx6) que contienen selenio: la GPx1 (GPx citosólica), la GPx2 (GPx especifica de las células epiteliales expresada en el recubrimiento intestinal y pulmones) la GPx3 (altamente expresada en las glándulas tiroideas y rinones), la GPx4 (GPx fosfolípido-hidroperóxido; PHGPx) y la Gpx6 (expresada en el epitelio olfativo) (4). Las isoenzimas GPx son todas enzimas antioxidantes que reducen a las especies reactivas de oxígeno (ERO) potencialmente dañinas, como el peróxido de hidrógeno y los hidroperóxidos lipídicos, en productos inocuos como el agua y los alcoholes al acoplar su reducción con la oxidación de glutatión (Figura 2). La espermatogénesis y la fertilidad masculina son altamente dependientes de GPx4 y de la selenoproteína P (SEPP1; véase abajo). En los testículos, la GPx4 reduce los hidroperóxidos fosfolípidos, por consiguiente, protegiendo espermatozoides inmaduros contra el estrés oxidativo. La GPx4 es también una proteína estructural principal de la capsula que incorpora la espiral mitocondrial del espermatozoide maduro involucrada en la motilidad del esperma. La SEPP1es esencial para el suministro de selenio a los testículos, y modelos animales carentes del gen SEPP1 son infértiles debido a la biodisponibilidad pobre de selenio en los tejidos, a la síntesis defectuosa de GPx4, y a la maduración deteriorada del esperma (5).

[Figura 2 - Clic para Agrandar]
Tiorredoxina reductasa
En los mamíferos, tres isoenzimas tiorredoxina reductasas que contienen selenocisteína (TrxR) han sido identificadas en el sistema tiorredoxina: TrxR1 citosólica, TrxR3 mitocondrial, y tiorredoxina glutatión reductasa testículo-especifica TGR. Las TrxRs son enzimas homodiméricas, y cada monómero contiene dominios de unión FAD- y NADPH- y un sitio catalítico que contiene selenocisteína. Las TrxRs catalizan la reducción de un amplio rango de sustratos, incluyendo la tiorredoxina y la proteína disulfuro isomerasa (PDI) (véase Figura 2 arriba). TrxRs también sirven como donadores de electrones para la regeneración de antioxidantes pequeños, reciclando posiblemente el ácido ascórbico (vitamina C), ácido α-lipoico, α-tocoferol (vitamina E) y la coenzima Q10 de sus formas oxidadas (6). El mantenimiento de la tiorredoxina en una forma reducida por las TrxRs es importante para la regulación del crecimiento celular y la supervivencia. La proteína tiorredoxina, junto con TrxR1 (o TrxR3), NADPH, y FAD, constituyen el sistema antioxidante tiorredoxina involucrado en la reducción de enzimas antioxidantes (p. ej., peroxirredoxinas, metionina sulfóxido reductasas y ribonucleótido reductasa) y de muchas proteínas de señalización sensibles a la oxidación/reducción (redox) (7). La TrxR1 es una de las selenoproteínas más investigadas y es considerada una de las principales enzimas antioxidantes y reguladoras del proceso redox en células mamíferas.
Iodotironina deiodinasas (deiodinasas de la hormona tiroidea)
La glándula tiroides libera cantidades muy pequeñas de la forma biológicamente activa de la hormona tiroidea (triyodotironina o T3) y grandes cantidades de la hormona tiroidea en su forma inactiva (precursor de la T3: tiroxina o T4) en la circulación. La mayoría de la T3 biológicamente activa en la circulación y dentro de las células es generada a través de la remoción de un átomo de yodo de la T4 en una reacción catalizada por enzimas iodotironina deiodinasas dependientes de selenio. Dos diferentes iodotironina deiodinasas dependientes de selenio (DIO tipo 1 y 2) pueden desiodar la T4, incrementando así la T3 circulante, mientras que una tercera iodotironina deiodinasa (DIO tipo 3) puede convertir ambas T3 y T4 a metabolitos inactivos (Figura 3) (8). Debe notarse que, la inactivación de genes que codifican para las DIO en modelos de roedores ha revelado un papel para la DIO tipo 1 en la homeostasis del yodo y la importancia de las DIO tipo 2 y 3 en la maduración de los sistemas auditorio y visual durante el desarrollo fetal (8). Así, la importancia del selenio en el desarrollo, crecimiento y metabolismo normal no está limitado a su papel en la regulación de la función de la glándula tiroides.

[Figura 3 - Clic para Agrandar]
Selenoproteína P
La selenoproteína P (SEPP1) es predominantemente producida por el hígado, un sitio principal de almacenamiento para el selenio, y secretado en el plasma. La glicoproteína en toda su longitud contiene un dominio rico en selenio con nueve residuos de selenocisteína, como también un sitio catalítico similar a la tiorredoxina con un residuo de selenocisteína. La SEPP1 constituye la principal forma de transporte de selenio a los tejidos periféricos (9). SEPP1 también funciona como un antioxidante que protege a las células del daño oxidativo al permitir la completa actividad de las tiorredoxina reductasas y glutatión peroxidasas a través de un suministro adecuado de selenio a los tejidos extrahepáticos (véase Glutatión peroxidasas). SEPP1 parece ser especialmente crítica para la homeostasis del selenio en el cerebro y los testículos donde el receptor 2 de apolipoproteína E (apoER2) facilita la absorción de SEPP1. La megalina es otro receptor lipoproteico SEPP1-especifico que ayuda a limitar la perdida urinaria de selenio a través de la reabsorción de SEPP1 por los riñones (10). Además, la SEPP1 ha sido recientemente implicada en la regulación del metabolismo de la glucosa y la sensibilidad a la insulina (11).
Selenoproteína W
La selenoproteína W (SEPW o SelW) existe en diferentes isoformas (homólogos) y es altamente conservada entre especies. En los humanos, la SEPW es expresada en numerosos tejidos, encontrando los niveles más altos en el musculo esquelético y corazón (12). SEPW contiene un residuo de selenocisteína y un residuo de cisteína que se une a una molécula de glutatión, sugiriendo un papel en la regulación del proceso redox (13). La expresión de SEPW esta correlacionada con el estatus del selenio y parece ser sensible a bajos suministros de selenio (14, 15). Se ha encontrado que la expresión de SEPW en el cerebro confiere protección contra la muerte de células neuronales inducida por el estrés oxidativo (16). La SEPW parece también ser un regulador negativo para las proteínas 14-3-3. Ciertamente, se encontró que la inhibición de 14-3-3 por la SEPW en las células del cáncer de seno incremento la proliferación celular y la supervivencia celular a través del incremento en la resistencia al estrés genotóxico (17). En las células del músculo esquelético, se demostró que SEPW redujo la unión de 14-3-3 a TAZ, permitiendo la translocación de TAZ al núcleo y la subsecuente activación de los genes de diferenciación celular del musculo (18). Finalmente, se encontró que la SEPW previene la degradación del receptor del factor de crecimiento epidérmico (EGFR) en células epiteliales del seno y próstata en cultivo. El EGFR es constitutivamente activado en muchos tumores, y evidencia de un papel para la SEPW en la activación y señalización del EGFR podría ayudar a arrojar luz sobre la relación entre el estatus del selenio y el riesgo de cáncer (19).
Selenofosfato sintetasa 2
No existe un suministro libre del aminoácido selenocisteína en las células tal que la síntesis de selenocisteína tome lugar en un ARNt especializado durante la traducción del ARNm de la selenoproteína. La reacción es catalizada por la L-seril-ARNtSec selenio transferasa dependiente de piridoxal-5'-fosfato (PLP) y usa selenofosfato (monoselenio fosfato) como donante de selenio (Figura 4) (20). La selenofosfato sintetasa 2 es una selenoenzima que cataliza la síntesis dependiente de ATP del selenofosfato a partir del seleniuro de hidrógeno (Figura 4) (3).
![Figura 4. Síntesis de la Selenocisteína. La síntesis de selenocisteína (Sec) toma lugar en un ARNt especial inicialmente aminoacetilado (cargado) con el aminoácido serina (Ser). Ser-ARNt[Ser]Sec es entonces fosforilado para producir pSer-ARNt[Ser]Sec. La L-seril-ARNtSec selenio transferasa dependiente de piridoxal-5'-fosfato (PLP) cataliza la conversión de Ser-ARNt[Ser]Sec a Sec-ARNt[Ser]Sec usando selenofosfato como donante de selenio. El selenofosfato es sintetizado a partir del seleniuro de hidrógeno en una reacción catalizada por la selenoenzima selenofosfato sintetasa 2. La selenocisteína es incorporada en las proteinas elongadas durante la traducción. Se, selenio; Ser-ARNt, L-seril-ARNt; Sec-ARNt, L-selenocisteinilo-ARNt.](/sites/lpi.oregonstate.edu/files/selenio-figure-4-1200px.png)
[Figura 4 - Clic para Agrandar]
Metionina-R-sulfóxido reductasa B1 (previamente llamada selenoproteína R)
El sistema de reducción de la metionina sulfóxido está involucrado en la protección contra el estrés oxidativo y es especialmente crítico para la regeneración de proteínas dañadas por las especies reactivas de oxigeno (ERO). Ciertamente, las ERO pueden oxidar los residuos de metionina (sulfóxidos de metionina) dentro de las proteínas y potencialmente deteriorar sus actividades. En los humanos, dos familias estereospecíficas de las metionina sulfóxido reductasas (MsrA y MsrB) son codificadas por un único gen MSRA y tres genes MSRB (MSRB1-3). La MsrA cataliza la reducción de la forma-S de la metionina sulfóxido; la forma-R de la metionina sulfóxido es reducida por la MsrB, 2 y 3. Solo la MsrB1 ha sido caracterizada como una selenoproteína con un residuo de selenocisteína en su sitio catalítico. La MsrB1 parece estar involucrada en la regulación redox de ciertas proteínas. En los macrófagos, la reorganización de la actina del citoesqueleto necesaria para la quimiotaxis y fagocitosis requiere la reducción dependiente de MsrB1 de los residuos de metionina-R-sulfóxido dentro de la actina (21). Estudios que usaron la inactivación del gen MSR en ratones han mostrado que la reducción del sulfóxido de metionina está implicada en la regulación del ciclo de la metionina (revisado en 22). Se ha encontrado que tanto el sistema antioxidante de la tiorredoxina (Trx) como el de la glutarredoxina depediente de GSH (Grx) reducen las metionina sulfóxido reductasas in vitro y/o in vivo (véase Figura 2 arriba) (22).
Selenoproteína de 15 kDa
La selenoproteína de 15 kDa (selenoproteína 15; SEP15) es altamente expresada en varios tejidos, incluyendo la próstata, riñones, hígado, y cerebro (23). Aunque su función no es aun conocida, se encontró que la SEP15 interactúa con la UDP-glucosa:glicoproteína glucosiltransferasa (UGGT) del retículo endoplásmico, una enzima involucrada en el control de calidad del plegamiento de las glicoproteínas (24, 25). Debido a que la SEP15 tiene un sitio catalítico como la tiorredoxina, se piensa que la SEP15 regula ya sea la actividad de la UGGT o el estado redox de los sustratos de UGGT (26). Se encontró que los ratones carentes de una SEP15 funcional desarrollaron cataratas nucleares (opacificación del cristalino) a una edad temprana sugiriendo que la SEP15 puede ser critica para el sistema de control de calidad del plegamiento de proteínas en el cristalino (27). La SEP15 puede también estar implicada en los mecanismos anticancerígenos (revisado en 28).
Selenoproteína S
La selenoproteína mamífera (conocida como SEPS1, SelS, o selenoproteína de membrana que interactúa con VCP [VIMP]) es una proteína de membrana del retículo endoplasmático (RE). La SEPS1 está involucrada en la respuesta celular al estrés del RE (degradación asociada al RE; DARE) activada por la detección de proteínas mal plegadas. La SEP1 contribuye a la remoción y transferencia (retrotranslocalización) de las proteínas mal plegadas provenientes del lumen del RE al citosol donde las proteínas son marcadas con ubiquitina antes de ser degradadas. Se encontró que un polimorfismo o variación en la secuencia dentro de un elemento de respuesta al RE localizado en el promotor del SEPS1 resulta en la actividad disminuida del promotor del SEPS1 y de la expresión de genes (29). El polimorfismo correspondiente a la sustitución de una guanina (G) por una adenina (A) en el nucleótido -105 (-105G>A) ha sido asociado con niveles incrementados de citoquinas proinflamatorias en el plasma. Además, un estudio de caso y control recientemente reportó que el alelo A no era más prevalente en individuos afectados por la tiroiditis de Hashimoto (TH) — una enfermedad autoinmune mediada por células T resultando en la destrucción de las células tiroideas — que en los controles saludables (30). Otras asociaciones entre el polimorfismo del SEPS1 (incluyendo -105G>A) y la susceptibilidad a varias condiciones, como la preeclampsia, la enfermedad coronaria cardíaca, o los canceres gastrointestinales, sugieren fuertemente un papel para esta selenoproteína en la regulación de las respuestas inflamatorias e inmunes (31-34).
Otras selenoproteínas menos bien caracterizadas, que también son localizadas en el lumen del RE y/o la membrana, incluyen las selenoproteínas K, M, N y T (35).
Interacciones con nutrientes
Nutrientes antioxidantes
La importancia del selenio para los sistemas biológicos, y específicamente para el balance redox celular (pro-oxidante/antioxidante), se deriva de su presencia como selenocisteína en el sitio catalítico de las selenoproteínas (véase Función). Otros minerales que son componentes críticos de las enzimas antioxidantes incluyen el cobre (como superóxido dismutasa), zinc (como superóxido dismutasa), y hierro (como catalasa). El selenio actúa en sinergia con las vitaminas antioxidantes, la vitamina C (ácido ascórbico) y la vitamina E (α-tocoferol), al regenerarlos a partir de sus formas oxidadas y promoviendo su protección antioxidante máxima (36-38).
Yodo
Mientras que el yodo es un componente esencial de las hormonas tiroideas, las iodotironina deiodinasas que contienen selenio (DIO) son enzimas requeridas para la conversión de tiroxina (T4) a la hormona tiroidea biológicamente activa triyodotironina (T3) (véase Función). La actividad de la DIO1 puede también estar involucrada en la regulación de la homeostasis del yodo (39). Las selenoenzimas, glutatión peroxidasas, también desempeñan un papel crítico en la función tiroidea porque catalizan la degradación de peróxidos generados durante la síntesis de la hormona tiroidea (8). La epidemiologia de las deficiencias coexistentes de yodo y selenio en África central, pero no en China, ha sido ligada a la prevalencia de cretinismo mixedematoso, una forma severa de hipotiroidismo congénito acompañado por retraso mental y físico. La deficiencia de selenio puede ser solo uno de varios factores indeterminados que podrían exacerbar los efectos perjudiciales de la deficiencia de yodo (40). Curiosamente, se encontró que la deficiencia de selenio en roedores tiene poco impacto en las actividades de DIO ya que parece que el selenio se suministra en prioridad para la adecuada síntesis de las DIO a expensas de otras selenoenzimas (8).
Deficiencia
La ingesta insuficiente de selenio puede afectar negativamente la actividad de varias enzimas sensibles al selenio, incluyendo las glutatión peroxidasas (GPx1 y GPx3), iodotironina deiodinasas, selenoproteínas W, y metionina-R-sulfóxido reductasa B1 (MsrB1). Incluso cuando es severa, la deficiencia aislada de selenio usualmente no deriva en una enfermedad clínica evidente. Sin embargo, a comparación de los sujetos con un estatus adecuado de selenio, los individuos con deficiencia de selenio podrían ser más susceptibles a estrés fisiológico adicional (41). La deficiencia prolongada de selenio puede contribuir a las enfermedades de Keshan y Kashin-Beck (véase abajo).
Individuos en riesgo incrementado de deficiencia de selenio
Se ha reportado deficiencia de selenio en pacientes crónicamente enfermos que recibieron nutrición parenteral total (NPT) sin selenio añadido por periodos de tiempo prolongados. Debilidad muscular, pérdida muscular, y cardiomiopatía (inflamación y daño al músculo cardíaco) han sido observados en estos pacientes. Hoy en día, las soluciones de NPT son rutinariamente suplementadas con selenio. El riesgo de una deficiencia de selenio puede incrementarse después de una cirugía bariátrica o en condiciones gastrointestinales severas, como la enfermedad de Crohn. Algunas dietas medicas especializadas como aquellas usadas para tratar ciertos desordenes metabólicos, incluyendo la fenilcetonuria, homocistinuria, y la enfermedad de la orina con olor a jarabe de arce, necesitan ser suplementadas con selenio para asegurar un estatus óptimo de selenio en los pacientes (42).
Enfermedad de Keshan
La enfermedad de Keshan es una forma fatal de cardiomiopatía dilatada que fue descrita primero en niños y mujeres jóvenes de una región en China con deficiencia de selenio. La forma aguda de esta enfermedad está caracterizada por la aparición repentina de insuficiencia cardíaca, mientras que la forma crónica resulta en un agrandamiento cardíaco de moderado a severo con grados variables de deficiencia cardíaca (43). La incidencia de la enfermedad de Keshan se asocia estrechamente con ingestas dietarías muy bajas de selenio y con un pobre estado nutricional del selenio. Se encontró que la suplementación con selenio (en la forma de selenito de sodio; Na2SeO3) protegió a las personas de desarrollar la enfermedad de Keshan pero no pudo revertir el daño al musculo cardiaco una vez que había ocurrido (43). Un reciente estudio de caso y control reporto que la actividad de la glutatión peroxidasa 1 sensible al selenio (GPx1) era significativamente menor en pacientes con Keshan en comparación a individuos saludables. Curiosamente, un polimorfismo de GPX1 específico que resulta en una transición de prolina a leucina en la posición 198 (Pro198Leu) está asociado con una reducción en la actividad de la GPx1 y fue encontrado más prevalente en los pacientes con Keshan. Este polimorfismo del GPX1 podría conferir una susceptibilidad mayor a la enfermedad de Keshan en portadores con un estatus nutricional de selenio bajo (44).
Mientras que la deficiencia de selenio es un factor etiológico fundamental de la enfermedad de Keshan, las variaciones estacionales y anuales en su ocurrencia sugieren que hay otros factores, especialmente un agente infeccioso podría estar involucrado además de la deficiencia de selenio (45). El Coxsackievirus B3 es un tipo de virus que ha sido aislado de pacientes con Keshan, y los estudios en animales han mostrado que este virus es capaz de causar una inflamación del corazón (miocarditis) en ratones deficientes de selenio. Los estudios en ratones también indicaron que el estrés oxidativo inducido por la deficiencia de selenio pudo resultar en cambios en el genoma viral; estos siendo capaces de convertir a una cepa relativamente inocua de coxsackievirus B3 en una cepa causante de miocarditis (43). Aunque aún sin ser probado en la enfermedad de Keshan, es posible que la deficiencia de selenio pueda incrementar la virulencia de los virus con el potencial de invadir y dañar el musculo cardíaco (46).
Enfermedad de Kashin-Beck
La enfermedad de Kashin-Beck (KBD) es otra condición endémica que afecta un estimado de 2.5 millones de personas en el Tíbet, norte y centro de China, Corea del Norte, y Sureste de Siberia (47). La KBD se caracteriza por una degeneración del cartílago articular entre las articulaciones (osteoartritis) que puede resultar en deformidades de las articulaciones y enanismo en las formas más severas de la enfermedad. La enfermedad afecta niños tan jóvenes como de dos años de edad. Al igual que con la enfermedad de Keshan, la KBD es prevalente en provincias deficientes de selenio y así generalmente afecta personas con ingestas muy bajas de selenio (47). Estudios recientes han sugerido que la susceptibilidad incrementada de KBD es las poblaciones deficientes de selenio podría resultar de una protección antioxidante reducida asociada con polimorfismos en los genes GPX (48, 49). A pesar de todo, la etiología parece ser multifactorial, ya que un cierto número de otros factores causantes han sido sugeridos para la KBD, incluyendo toxinas fúngicas en granos, deficiencia de yodo, y agua potable contaminada (43).
Meta-análisis recientes de unos pocos ensayos de menor magnitud y de estudios de cohorte prospectivos han indicado que el mejoramiento del estatus nutricional del selenio en niños que viven en áreas endémicas puede ayudar a reducir la incidencia de KBD (50). También, existe evidencia limitada que sugiere que la suplementación con selenio podría ser útil en el tratamiento de pacientes con KBD. Un meta-análisis de 10 ensayos controlados aleatorios reportó un incremento significante en la tasa de reparación de lesiones oseas en niños con KBD suplementados con selenito de sodio por al menos un año (51). Ensayos más amplios de mejor calidad son necesarios para evaluar si la suplementación con selenio podría resultar en remisión de la enfermedad.
La Ingesta Diaria Recomendada (IDR)
La ingesta diaria recomendada (IDR) para el selenio fueron revisadas por última vez en el 2000 por la Junta de Nutrición y Alimentos (JNA) del Instituto de Medicina de los EE.UU. La IDR más reciente está basada en el requerimiento estimado promedio (REP) necesario para maximizar la actividad de la enzima antioxidante glutatión peroxidasa (GPx) en el plasma (52).
| Etapa de la Vida | Edad | Machos (μg/día) | Hembras (μg/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 15 (IA) | 15 (IA) |
| Infantes | 7-12 meses | 20 (IA) | 20 (IA) |
| Niños | 1-3 años | 20 | 20 |
| Niños | 4-8 años | 30 | 30 |
| Niños | 9-13 años | 40 | 40 |
| Adolescentes | 14-18 años | 55 | 55 |
| Adultos | 19 años y más | 55 | 55 |
| Embarazo | Todas las edades | - | 60 |
| Período de lactancia | Todas las edades | - | 70 |
Debe notarse que la tercera Encuesta Nacional de Salud y Examinación de Nutrición (NHANES III) reportó que más del 99% de los participantes estadounidenses tuvieron concentraciones de selenio en el suero consistentes con los requerimientos de selenio requeridos (52), sugiriendo que la suplementación con selenio no es necesaria para los americanos.
Prevención de Enfermedades
Cáncer
Estudios en animales
Ha habido investigación considerable sobre el efecto de la suplementación con selenio en la incidencia de cáncer en animales. Mas de dos tercios de los más de 100 estudios publicados en 20 modelos diferentes de animales de canceres espontáneos, virales e inducidos químicamente encontró que la suplementación con selenio (a por lo menos niveles de ingesta adecuados) reduce significativamente la incidencia de tumores, especialmente en comparación a dietas deficientes de selenio (53). Evidencia de los efectos inhibidores de cáncer del selenio ha provisto de una sólida justificación para la investigación de asociaciones potenciales entre la ingesta de selenio y el riesgo de cáncer en humanos.
Estudios basados en la observación
La mayoría de la evidencia epidemiológica temprana proveniente de estudios caso-control anidados y de caso y control sugirieron tanto asociaciones nulas como inversas entre la exposición al selenio y el riesgo de canceres de sitio especifico (54). Marcadores de la exposición al selenio incluyen el contenido de selenio sanguíneo y de las uñas del pie, como también la actividad de glutatión peroxidasa (GPx) en el plasma. Sin embargo, no está claro si reflejan adecuadamente la exposición al selenio proveniente de fuentes dietarías y suplementarias (véase Fuentes) o la distribución de selenio en los tejidos y órganos que podrían ser afectados por el cáncer. En el estudio prospectivo Danish Prospective Diet, Cancer, and Health, que dio seguimiento a más de 57,000 hombres y mujeres por 14 años, se encontró que el riesgo de cáncer rectal era 42% mayor en fumadores actuales en comparación a los no fumadores. Ninguna diferencia entre fumadores y no fumadores con respecto a las ingestas suplementarias y dietarías de micronutrientes antioxidantes, incluyendo selenio, que contribuyera a la asociación entre el fumar y el cáncer rectal fue encontrada (55). Sin embargo, debido a que estudios consistentemente han reportado bajas concentraciones de selenio en la sangre y bajas actividades de la GPx en los fumadores en comparación a los no fumadores (revisado en 56), el estimado de las ingestas de selenio podría no ser un marcador fidedigno de la exposición al selenio en esta población. También, las formas químicas del selenio encontradas en los alimentos son variadas (véase Fuentes) y pueden tener efectos biológicos y toxicológicos muy diferentes (57, 58).
Una revisión Cochrane reciente incluyó 55 estudios basados en la observación completados — mayormente con un diseño de caso-control anidado — publicados durante tres décadas (54). Un meta-análisis de 16 de estos estudios basados en la observación, incluyendo más de 144,000 individuos, reportó que el estatus de selenio más alto frente al más bajo estaba asociado con un riesgo 31% más bajo de cáncer en cualquier sitio y un riesgo 40% más bajo de mortalidad relacionada al cáncer. Un riesgo significantemente menor fue reportado para el cáncer de vejiga (5 estudios) y el cáncer de próstata (17 estudios); sin embargo, el estatus alto de selenio no estuvo inversamente relacionado con los riesgos de cáncer de seno (8 estudios), cáncer pulmonar (12 estudios), cáncer colorrectal (5 estudios), y cáncer gástrico (5 estudios) (54). Otro meta-análisis de 16 estudios basados en la observación reportó una relación inversa entre el cáncer de seno y las concentraciones de selenio en el suero (59). Las diferencias en género en la susceptibilidad al cáncer han sido reportadas en algunos estudios, aunque evidencia consistente de efectos diferentes en hombres y mujeres parece ser carente.
Variaciones de un solo nucleótido (polimorfismos) en la secuencia de los genes pueden modificar el nivel de expresión de genes y la estabilidad y actividad de las proteínas sintetizadas. Por ejemplo, una transición de prolina a leucina causada por un polimorfismo especifico en el gen GPX1 (rs1050450 C>T) está asociada con la actividad enzimática reducida de la GPx1. La descripción de varios polimorfismos en los genes que codifican para las selenoproteínas ha llevado a la evolución de posibles asociaciones con el estatus de selenio y la incidencia de cáncer. Notablemente, ciertos polimorfismos en los genes que codifican para las selenoproteínas han sido asociados con riesgos incrementados de canceres colorrectales y gástricos (revisado en 60). Adicionalmente, un cierto número de estudios han investigado el efecto de los polimorfismos de la selenoproteína en la relación entre el estatus del selenio y el riesgo de cáncer de próstata. Un estudio caso-control anidado dentro de la cohorte EPIC-Heidelberg ha combinado la genotipificación para varias variantes de la selenoproteína con marcadores del estatus del selenio (61). Brevemente, el estudio mostró que un polimorfismo del gen GPX1 (rs1050450 C>T) afectó la asociación entre las concentraciones de selenio y el riesgo de cáncer de próstata. Específicamente, se encontró que las concentraciones de selenio estaban inversamente asociadas con el riesgo de cáncer de próstata solo entre portadores del alelo GPX1. Variantes adicionales en los genes de la selenoproteína podrían mitigar el efecto del estatus del selenio en el riesgo de cáncer de próstata (62, 63). En otro estudio caso-control anidado dentro del Physicians’ Health Study (PHS), se encontró que los individuos en el cuartil más alto de las concentraciones de selenio frente aquellos en el cuartil más bajo tenían un riesgo reducido de mortalidad relacionada al cáncer de próstata a menos que portaran una variante especifica en el gen de la selenoproteína de 15kDa (SEP15 rs561104 G>A) (64). Mas investigación es necesaria para desentrañar aún más los mecanismos que subyacen la influencia de las interacciones genes-dieta en el riesgo de desarrollar cáncer.
Ensayos de intervención
Estudios basados en la comunidad: Un ensayo de intervención muy temprano de la suplementación con selenio se llevó a cabo en China entre una población general de 130,471 individuos viviendo en una zona con alto riesgo de infección por hepatitis B viral y cáncer de hígado. El ensayo proveyó sal de mesa enriquecida con selenito de sodio a la población de uno de los municipios (20,847 personas) usando a otros cuatro municipios como controles. Durante un periodo de seguimiento de ocho años, la incidencia promedio de cáncer de hígado se redujo en un 35% en la población enriquecida con selenio, mientras que no se encontró ninguna reducción en las poblaciones de control. En un ensayo clínico en la misma región, 226 individuos con evidencia de infección por hepatitis B crónica fueron suplementados diariamente con 200 μg de selenio en la forma de tabletas de levadura enriquecidas con selenio o con un placebo de tabletas de levadura. Durante el periodo de seguimiento de 4 años, 7 de 113 de los individuos que tomaban el placebo desarrollaron cáncer hepático primario, mientras que ninguno de los 113 sujetos suplementados con selenio desarrolló cáncer de hígado (65).
Ensayos controlados aleatorios: El estudio doble ciego, controlado con placebo para la Prevención Nutricional del Cáncer (NPC) en 1,312 adultos mayores con historial de cáncer de piel del tipo no melanoma, encontró que la suplementación con 200 μg/día de levadura enriquecida con selenio (levadura selenizada) por un promedio de 7.4 años resulto en una disminución del 52% en la incidencia de cáncer de próstata en hombres (revisado en 66). El efecto protector de la suplementación con selenio fue mayor en los hombres con niveles basales del plasma más bajos de selenio y del antígeno prostático específico (APE). Una incidencia reducida de cáncer total, pulmonar, y colorrectal fue asociada con la suplementación de 200 μg/día (67) pero no con 400 μg/día de levadura enriquecida con selenio (68). Además, la suplementación con selenio incremento el riesgo de un tipo de cáncer de piel (carcinoma celular escamoso) en un 25%. Un ensayo aleatorio de intervención, controlado con placebo de mayor escala (estudio SELECT) en más de 35,000 hombres de mediana edad y repletos de selenio, a los cuales se les asignó aleatoriamente una suplementación con selenio (en la forma de selenometionina, 200 μg/día) y/o vitamina E, fue detenido debido a preocupaciones con respecto a un riesgo incrementado de diabetes tipo 2 con el selenio y un riesgo incrementado de cáncer de próstata con la vitamina E (69, 70). Además, la suplementación con selenio, únicamente o junto con la vitamina E, no mostro ningún beneficio con respecto al riesgo de canceres de próstata, pulmón, o colorrectal después de un seguimiento de 5.5 años (71, 72). La carencia de un efecto beneficial de la suplementación con selenio fue apoyada en un reciente meta-análisis de ensayos controlados aleatorios (54).
Enfermedades cardiovasculares
Los niveles de actividad bajos de las selenoenzimas, glutatión peroxidasas (GPx), han sido reportados en enfermedades relacionadas al estrés oxidativo, incluyendo las enfermedades cardiovasculares (ECV) (73). En teoría, el mantenimiento de un estatus óptimo de selenio tiene el potencial de proteger contra el estrés oxidativo (incluyendo la peroxidación lipídica) y podría eventualmente prevenir la inflamación crónica y los trastornos cardiovasculares. Sin embargo, análisis de datos de corte transversal provenientes de 13,887 adultos estadounidenses incluidos en la Tercera Encuesta Nacional de Salud y Examinación de Nutrición (NHANES III, 1988-1994) fallaron en mostrar alguna asociación significante entre las concentraciones de selenio y la mortalidad por ECV, enfermedad coronaria cardiaca (ECC), o por accidente cerebrovascular (74). Además, mientras que individuos con insuficiencia renal están en un riesgo más alto de desarrollar ECC en comparación con aquellos con una función renal normal, tal riesgo no se encontró que fuese mayor con bajas en lugar de concentraciones normales de selenio en el suero (≤98 ng/mL vs. >98 ng/mL) (75).
Un estudio de corte transversal basado en los datos de NHANES 2003-2004 proveniente de 2,638 participantes de 40 años y más encontró que el riesgo de presión sanguínea alta (hipertensión), un factor contribuyente principal para las ECV era 73% más alto en individuos en el quintil más alto versus aquellos en el quintil más bajo de las concentraciones de selenio del suero (≥150 ng/mL vs. <122 ng/mL) (76). Sin embargo, una reciente revisión sistemática de la literatura falló en encontrar evidencia suficiente para apoyar alguna relación entre las concentraciones de selenio en el suero y la hipertensión (77). Además, unos pocos estudios basados en la observación han también reportado asociaciones entre el estatus normal a alto del selenio y los niveles de lípidos elevados en el suero en las poblaciones repletas de selenio, especulando que el selenio podría interferir con el metabolismo de las grasas y adversamente afectar la salud cardiovascular (78, 79). Hoy en día, ensayos controlados aleatorios no han provisto de resultados consistentes con respecto al efecto de la suplementación con selenio en los niveles lipídicos ni tampoco han demostrado algún beneficio cardiovascular del selenio en los individuos con ingestas óptimas o subóptimas de selenio (80).
Tratamiento de Enfermedades
Disfunción inmune
La deficiencia de selenio ha sido asociada con una inmunidad deteriorada e inflamación crónica (81). Una cantidad considerable de investigación conducida en cultivo de células y modelos animales indica que el selenio juega papeles esenciales en la regulación de la migración, proliferación, diferenciación, activación y función optima de las células inmunes, influenciando así la inmunidad innata, la producción de anticuerpos dependientes de células B y la inmunidad mediada por células T (revisado en 82). Evidencia reciente en el papel del selenio y selenoproteínas en la producción de mediadores lipídicos (llamados eicosanoides) involucrados en las respuestas inflamatorias sugiere que la suplementación con selenio podría mitigar las respuestas inflamatorias disfuncionales que contribuyen a la patogénesis de muchas condiciones de salud crónicas (83). Hoy en día, ensayos controlados aleatorios son necesarios para evaluar los beneficios potenciales de la suplementación con selenio en los desórdenes inflamatorios, tales como el asma (84) y la enfermedad intestinal inflamatoria (85).
Enfermedades infecciosas
VIH/SIDA
En áreas de malnutrición generalizada, las deficiencias de micronutrientes (incluyendo el selenio) son comunes en los individuos infectados con el virus de inmunodeficiencia humana (VIH) que causa el síndrome de inmunodeficiencia adquirida (SIDA). Antes de que la terapia antirretroviral (TAR) fuese estándar para el tratamiento del VIH, estudios basados en la observación habían consistentemente reportado asociaciones entre las concentraciones bajas de selenio en el suero y la infección de VIH en sujetos bien nutridos (86). El estatus pobre de selenio ha sido ligado a riesgos incrementados de cardiomiopatía dilatada y mortalidad en niños y adultos infectados con VIH, como también transmisión de VIH de madre a hijo y mortalidad perinatal (revisado en 87). Estudios de laboratorio tempranos han sugerido que el VIH podría interrumpir las defensas antioxidantes normales en las células T infectadas al reducir los niveles de selenoproteínas, es decir, tiorredoxina reductasas y glutatión peroxidasas (87). Curiosamente, un estudio de corte transversal encontró que los individuos VIH-seropositivos que recibían TAR por más de dos años tenían cargas virales indetectables en el plasma, conteos de linfocitos T CD4 más altos, y concentraciones de selenio adecuadas en el suero en comparación a sujetos no tratados previamente con TAR (88). Debido a que la actividad antioxidante de las selenoproteínas puede interferir con la replicación viral de las células inmunes infectadas con VIH (89, 90), se ha sugerido que la suplementación con selenio podría servir como un potencial adyuvante de la TAR para los pacientes con VIH.
Unos pocos ensayos de la suplementación con selenio en individuos infectados con VIH han sido conducidos. Un ensayo aleatorio, doble ciego, controlado con placebo en 186 adultos VIH positivos inicialmente encontró que la suplementación con 200 μg/día de selenio por dos años significativamente disminuyo la tasa de admisiones al hospital (91). Otro ensayo aleatorio, doble ciego, controlado con placebo en 174 individuos VIH positivos reportó que la suplementación con 200 μg/día de selenio (en la forma de levadura enriquecida con selenio) por nueve meses incremento las concentraciones de selenio del suero, mejoró el conteo de linfocitos T CD4, y previno cualquier progresión de la carga viral del VIH (92). En un tercer ensayo doble ciego en Tanzania, 913 mujeres embarazadas de entre 12 y 27 semanas de gestación fueron aleatoriamente asignadas a recibir 200 μg/día de selenio (como selenometionina) o un placebo hasta los seis meses después del nacimiento. La suplementación con selenio no tuvo efecto en el conteo de células T CD4, CD8, y CD3 y en la carga viral del VIH, pero significantemente disminuyó el riesgo de diarrea aguda o persistente (93, 94). Además, el riesgo de muerte entre las seis semanas y seis meses después del parto fue significantemente reducido en infantes y madres suplementados con selenio en comparación al placebo (94).
Un reciente ensayo de cuatro brazos en Botswana aleatoriamente asigno 878 adultos VIH positivos en etapa temprana de la infección a recibir tanto un tratamiento con placebo, multivitaminas (vitamina B, C, y E), 200 μg/día de selenio, o ambos multivitaminas y selenio por 24 meses (95). A diferencia del selenio solo, la suplementación con multivitaminas (con o sin selenio) disminuyó el riesgo de declive inmunológico al significantemente incrementar el tiempo antes de que la iniciación de TAR fuese necesaria (es decir, cuando el conteo de células T CD4 cayera por debajo de las 251 células/mcL) en comparación al placebo. En el estudio, un resultado combinado de (1) conteo de células T CD4 cayendo por debajo de las 251 células/mcL; (2) la ocurrencia de condiciones que definen el SIDA; y (3) muerte relacionada al SIDA — cualquiera que ocurriese primero — fue usado para evaluar la progresión de la enfermedad en los diferentes brazos del tratamiento. En comparación al placebo, hubo un periodo de tiempo más largo a partir de la aleatorización hasta la fecha del resultado compuesto en individuos suplementados con multivitaminas más selenio, pero no en aquellos que recibieron multivitaminas o selenio únicamente (95). Más investigación es necesaria para replicar estos resultados preliminares, especialmente en condiciones y comunidades donde la malnutrición acelera la progresión de la infección del VIH y el acceso a la terapia retroviral pueda ser limitada.
Sepsis
El síndrome de respuesta inflamatoria sistémica (SIRS) resulta de una respuesta inflamatoria sistémica que puede ocurrir debido a una infección (sepsis) (96). La sepsis severa y el choque séptico — definido como una baja presión sanguínea inducida por la sepsis persistente — están asociados con tasas elevadas de mortalidad en pacientes en estado crítico (96, 97). Debido a que las respuestas inflamatorias sistémicas involucran estrés oxidativo excesivo, se ha sugerido que el proporcionar nutrientes antioxidantes como el selenio puede mejorar el resultado de pacientes en estado crítico en unidades de cuidado intensivo. Dos meta-análisis recientes de ensayos controlados aleatorios encontró que la suplementación intravenosa con selenio (como selenito de sodio) en pacientes en estado crítico padeciendo de SIRS, sepsis, o choque séptico resulto en un riesgo significantemente reducido de mortalidad en un 17% a 27% (98, 99). Más ensayos son necesarios para identificar el programa apropiado de la administración de selenio (en términos de dosis, ruta, y duración del tratamiento) y para evaluar los resultados adicionales (p. ej., complicaciones infecciosas y duración de la estadía en el hospital).
Enfermedades autoinmunes de la tiroides
La tiroiditis de Hashimoto (TH; tiroiditis autoinmune crónica) es una enfermedad autoinmune caracterizada por la infiltración de células T en la glándula tiroides y autoanticuerpos circulantes (predominantemente contra la peroxidasa tiroidea), causando inflamación prolongada, daño a los tejidos, e hipotiroidismo (8). Mientras que la función de la glándula tiroides de los individuos saludables esta usualmente protegida de variaciones en el suministro del selenio, la deficiencia de selenio y los polimorfismos genéticos que afectan la actividad de las selenoproteínas podrían ser factores contribuyentes potenciales para las enfermedades autoinmunes de la tiroides. Una reciente revisión sistemática (100) identificó cuatro ensayos controlados aleatorios que evaluaron el efecto de la suplementación con selenio como un tratamiento adyuvante para la terapia de remplazo de T4 (levotiroxina) en pacientes con TH (101-104). Mientras que tres de cuatro estudios sugirieron una reducción en los niveles de autoanticuerpos circulantes, ninguno de ellos proporciono información sobre si el selenio puede mejorar los síntomas relacionados a la salud y el estado de ánimo para permitir una dosis disminuida de levotiroxina. Otro ensayo controlado aleatorio encontró que la suplementación con selenio mejoró el bienestar de los pacientes afectados por otra enfermedad autoinmune de la tiroides que conduce al hipertiroidismo (enfermedad de Graves) (105). Los resultados de dos ensayos aleatorios, controlados con placebo en proceso — el ensayo CATALYST en pacientes con TH y el ensayo GRASS en pacientes con enfermedad de Graves — podrían proporcionar una visión en un efecto del selenio en el criterio de la calidad de vida específica a la tiroides e informar los criterios de toma de decisiones clínicas (106, 107).
Fuentes
Fuentes alimenticias
Las fuentes alimenticias más ricas de selenio son las vísceras y los mariscos, seguidos de las carnes con músculo. El agua potable no se considera como una fuente significante de selenio en Norteamérica. Sin embargo, en áreas, donde los niveles altos de selenio en el suelo contribuyen al contenido de selenio del agua, niveles más altos de selenio pueden ser encontrados en pozos usados para el agua potable (108). En general, existe una amplia variación en el contenido de selenio de plantas y granos, especialmente debido a que algunas plantas incluyendo el ajo, las nueces de Brasil, y múltiples Brassica especies, tienden a acumular selenio ("acumuladores de selenio"), mientras otras asimilan selenio en menor grado ("no-acumuladores"). La asimilación de selenio por las plantas también depende del contenido de selenio del suelo. Las nueces de Brasil que crecen en áreas de Brasil con suelos ricos en selenio pueden proporcionar más de 100 μg de selenio en una nuez, mientras que aquellas que crecen en suelos carentes de selenio proporcionan 10 veces menos (109). En los EE.UU., los granos son una buena fuente de selenio, pero las frutas y verduras tienden a ser fuentes de selenio relativamente pobres.
Varias formas químicas (especies) de selenio se encuentran en acumuladores de selenio, incluyendo el selenato (selenio inorgánico), la selenometionina, la selenocisteína, la selenio-metil-selenocisteína, y la γ-glutamil-selenio-metil-selenocisteína. Aunque los últimos dos compuestos son predominantes en plantas de las familias Allium y Brassicaceae (las cuales incluyen a los ajos, cebollas, y brócoli), el trigo, otros granos (incluyendo las nueces de Brasil), y la soya son ricos en selenometionina y contienen pequeñas cantidades de selenocisteína y selenato. Se conoce menos acerca de las especies de selenio y la distribución en fuentes dietarías de origen animal. La nutrición animal y las condiciones de crecimiento ciertamente contribuyen a las especies de selenio formadas y sus cantidades relativas, y se asume que la ruta metabólica del selenio dietario en animales es similar a la de los humanos. La selenocisteína es predominantemente formada en animales que se alimentan de selenio inorgánico, mientras que la selenometionina se deriva de las fuentes dietarías de origen vegetal (revisado en 110).
En los EE.UU., el cuestionario nacional NHANES III reportó ingestas dietarías promedio oscilando entre los 100.5 μg/día y 158.5 μg/día en adultos de entre 19-50 años de edad (52). La Tabla 2 lista algunas fuentes alimenticias ricas en selenio y su contenido de selenio en microgramos (μg). Para más información en el contenido de selenio de alimentos específicos, revise la base de datos de composición de los alimentos de la USDA.
| Alimento | Porción | Selenio (μg) |
|---|---|---|
| Nueces de Brasil (de suelo rico en selenio) | 1 onza (6 unidades) | 543.5* |
| Atún (aleta amarilla, cocido, seco al calor) | 3 onzas | 92.0 |
| Ostras (Pacifico, crudas) | 3 onzas | 65.4 |
| Almejas (mixtas, cocidas, al vapor) | 3 onzas | 54.4 |
| Hipogloso (Atlántico y Pacífico, cocido, seco al calor) | 3 onzas | 47.1 |
| Camarón (cocido, al vapor) | 3 onzas | 42.1 |
| Salmón (Chinook, cocido, seco al calor) | 3 onzas | 39.8 |
| Fideos (de huevo, cocido, enriquecidos) | 1 taza | 38.2 |
| Cangrejo (reina, cocido, al vapor) | 3 onzas | 37.7 |
| Puerco (carne magra, lomo, cocido, rostizado) | 3 onzas | 32.5 |
| Carne de res (magra, bistec, cocida, a la parrilla) | 3 onzas | 30.6 |
| Pollo (carne blanca, cocida, rostizada) | 3 onzas | 25.8 |
| Arroz (integral, grano grande, cocido) | 1 taza | 19.1 |
| Semillas de girasol (secas) | ¼ taza | 18.6 |
| Pan de trigo entero | 2 rebandas | 16.4 |
| Leche (sin grasa o descremada) | 8 onzas fluidas (1 taza) | 7.6 |
| *Por encima del nivel máximo de ingesta tolerable (NM) de 400 μg/día. | ||
Suplementos
Los suplementos de selenio están disponibles en varias formas. El selenito y el selenato de sodio son formas inorgánicas del selenio. El selenato de sodio es absorbido casi completamente, pero se excreta una cantidad significativa en la orina antes de que pueda ser incorporado a las proteínas. El selenito de sodio sólo se absorbe en un 50% pero es retenido de mejor manera que el selenato una vez que es absorbido. La selenometionina es absorbida en aproximadamente un 90% (52); sin embargo, solo alrededor de un 34% puede ser actualmente convertida en selenometionina libre (111). La selenometionina y la levadura enriquecida con selenio, los cuales aportan principalmente selenometionina, también se encuentran disponibles como suplementos. El consumidor debería estar al tanto de que algunas formas de levaduras enriquecidas con selenio disponibles contienen levadura más formas de selenio principalmente inorgánicas.
Aunque tanto las formas orgánicas como las inorgánicas del selenio pueden ser metabolizadas a selenocisteína por el cuerpo y ser incorporadas en las selenoenzimas, podrían no contribuir igualmente al mantenimiento de un estatus de selenio adecuado. En ensayos de intervención, la suplementación con una forma orgánica de selenio (selenometionina) incremento más efectivamente las concentraciones de selenio en la sangre en comparación a la suplementación con formas inorgánicas (es decir, el selenito de sodio y selenato de sodio) (110). Sin embargo, las formas inorgánicas pueden incrementar la actividad de la glutatión peroxidasa (GPx) en el plasma más efectivamente que las formas orgánicas (revisado en 112). También se ha sugerido que la incorporación de selenometionina en lugar de metionina en las proteínas de los tejidos puede asegurar que el selenio esté disponible tras el recambio proteico (110).
Alimentos enriquecidos con selenio
Los alimentos enriquecidos con selenio han sido de interés para los científicos, especialmente debido a la insinuación de que algunas de las formas químicas del selenio producidas por las plantas podrían ser modificadores del riesgo de cáncer más potentes que aquellas disponibles actualmente en suplementos. Aunque no existe evidencia actual de los beneficios a largo plazo de la salud asociados con el consumo de alimentos enriquecidos con selenio, resultados de estudios en animales y ensayos de intervención sugieren que las fuentes basadas en proteína de selenio son más efectivas en incrementar la actividad de GPx que la levadura enriquecida con selenio y selenometionina (112). La fortificación de alimentos puede también representar una estrategia rentable para mejorar el estatus nutricional de selenio en poblaciones en riesgo de insuficiencia (113).
Seguridad
Toxicidad
Aunque el selenio es requerido para la salud, altas dosis de selenio pueden ser toxicas. La toxicidad aguda y fatal ha ocurrido por la ingesta accidental o suicida de cantidades en gramos de selenio. Se reportó toxicidad por selenio clínicamente significativa en 13 individuos luego de que tomaran suplementos que contenían 27.3 mg (27,300 μg) por tableta debido a una falla de fabricación. La toxicidad crónica por selenio (selenosis) puede ocurrir con dosis de selenio más pequeñas por largos periodos de tiempo. Los síntomas de selenosis reportados con mayor frecuencia son la pérdida y fragilidad de uñas y cabello. Otros síntomas podrían incluir molestias gastrointestinales, erupciones cutáneas, aliento con olor a ajo, fatiga, irritabilidad, y trastornos neurológicos. En un área de China con una alta prevalencia de selenosis, los efectos tóxicos ocurrieron con una mayor frecuencia cuando las concentraciones de selenio sanguíneo alcanzaban un nivel correspondiente a una ingesta de 850 μg/día.
La Junta de Nutrición y Alimentos (JNA) del Instituto de Medicina de los EE.UU. estableció el nivel máximo de ingesta tolerable (NM) para el selenio en 400 μg/día para adultos basado en la prevención de la pérdida y fragilidad de uñas y cabello y de signos iniciales de la toxicidad crónica por selenio (52). El NM de 400 μg/día para los adultos incluyen tanto el selenio obtenido de alimentos como el selenio de suplementos (Tabla 3).
¿Incrementan el riesgo de diabetes mellitus tipo 2 los suplementos de selenio?
Unos pocos estudios han examinado la relación entre el estatus de selenio y la diabetes mellitus tipo 2. En el análisis de corte transversal de datos del NHANES III (1988-1994) provenientes de 8,876 participantes adultos, el quintil más alto frente al más bajo de las concentraciones de selenio en el suero (≥137 ng/mL vs. <111 ng/mL) se asoció con un riesgo incrementado de diabetes tipo 2 (114). Análisis de datos de 917 participantes (≥40 años de edad) del NHANES 2003-2004 también indicaron un incremento en la prevalencia de diabetes tipo 2 en el cuartil más alto de las concentraciones de selenio en el suero frente al cuartil más bajo (≥147 ngm/L vs. <124 ng/mL). Los individuos en el cuartil más alto frente aquellos en el más bajo de las concentraciones de selenio del suero tuvieron también niveles más altos de glucosa en el plasma y hemoglobina glicosilada, sugiriendo un control glicémico pobre (115). El estudio aleatorio, doble ciego, controlado con placebo en 1,312 participantes en el ensayo de Prevención Nutricional del Cáncer (NPC) encontró que la suplementación con selenio (200 μg/día; seguimiento promedio de 7.7 años) significantemente incremento el riesgo de diabetes tipo 2 en participantes en el tercil más alto de las concentraciones de selenio basales del plasma (116). Además, en el ensayo SELECT (Selenium and Vitamin E Cancer Prevention Trial), más casos de diabetes tipo 2 fueron encontrados en el grupo de selenio (200 μg/día; seguimiento medio de 5.5 años) en comparación al grupo de placebo, pero esto solo fue una tendencia y no fue estadísticamente significante (72).
En la actualidad, los mecanismos detrás de estas observaciones no son bien entendidos. Un incremento en la sensibilidad a la insulina ha sido reportado en individuos con deficiencia congénita (de nacimiento) de la mayoría de selenoproteínas (117). Los resultados de varios estudios en animales también indicaron que la suplementación con selenio y las selenoproteínas pueden interferir con la acción de la insulina y con la homeostasis de la glucosa (revisado en 118). Por otra parte, estudios recientes han encontrado que el metabolismo deteriorado de la glucosa en pacientes con diabetes tipo 2 puede afectar la expresión de SEPP1 y la homeostasis del selenio (11, 119, 120). Mientras que más investigación es necesaria para completamente entender la interacción entre el metabolismo de los carbohidratos y la homeostasis del selenio, el uso de suplementos de selenio es considerado menos propenso a incrementar el riesgo de diabetes tipo 2 en individuos saludables, pero debe ser desalentado en aquellos con un estatus alto de selenio y/o en un riesgo incrementado de desarrollar diabetes tipo 2 (118).
Interacción con drogas/fármacos
En la actualidad, pocas interacciones entre el selenio y medicamentos han sido reportadas (121). Por ejemplo, el medicamento anticonvulsivo ácido valpróico y el agente quimioterapéutico cisplatino pueden disminuir las concentraciones de selenio circulante en sujetos tratados (122, 123). También se ha encontrado en estudios en animales que el selenito de sodio reduce la toxicidad del antibiótico nitrofuratoina y del herbicida paraquat (124).
Suplementos antioxidantes y estatinas
Un ensayo aleatorizado controlado de tres años en 160 pacientes con enfermedad coronaria cardíaca (ECC) documentada y bajos niveles de lipoproteínas de alta densidad (HDL) encontró que una combinación de simvastatina (Zocor) y niacina incrementó los niveles subfracción de HDL2, inhibió la progresión de estenosis (estrechamiento) arterial coronaria, y disminuyó la frecuencia de eventos cardiovasculares (125). Sorprendentemente, cuando se tomó una combinación antioxidante (1,000 mg de vitamina C, 800 UI de vitamina E (d-α-tocoferol), 100 μg de selenio, y 25 mg de β-caroteno diariamente) junto a la combinación de simvastatina-niacina, los efectos protectores fueron disminuidos. Sin embargo, la contribución individual del selenio no se puede determinar, y otros estudios han reportado que las vitaminas antioxidantes solas pudieron interferir con la acción de los medicamentos que elevan los HDL incluyendo las estatinas (126).
Recomendación del Instituto Linus Pauling
Se estima que la dieta Americana promedio aporta cerca de 100 μg/día de selenio, una cantidad bastante superior a la actual IDR (55 μg/día) y que parece ser suficiente para optimizar la actividad de la glutatión peroxidasa (GPx) celular y del plasma. Un reciente estudio controlado aleatorio de 10 semanas en adultos británicos saludables (edades, 50-64 años) estimó que una ingesta total de selenio de alrededor de 105 μg/día era requerida para maximizar las concentraciones de selenoproteínas P (SEPP1), otro biomarcador útil del estatus del selenio (127). Sin embargo, un ensayo similar en una cohorte americana con altas concentraciones basales de selenio en el plasma no encontró efecto alguno de la suplementación con selenio en las concentraciones de SEPP1 (128). Mientras que la cantidad de selenio varía considerablemente en los suplementos multivitamínicos/minerales (MVM), estos suplementos raramente aportan más que el Valor Diario (VD) de 70 μg. Consumir una dieta variada y tomar un suplemento MVM diariamente debiera aportar selenio suficiente a la mayoría de las personas en los EE.UU. y ayudaría a mejorar el estatus de selenio en las poblaciones con ingestas menores de selenio fuera de los EE.UU.
Hombres
En la actualidad, el efecto de la suplementación con selenio en el riesgo de cáncer no es lo suficiente clara para apoyar una recomendación general para un suplemento extra de selenio, especialmente en hombres con concentraciones de suero consistentes con ingestas adecuadas de selenio. El ensayo SELECT encontró que 200 μg/día de selenio suplementario no disminuyó el riesgo de cáncer de próstata en hombres (72), refutando los resultados del ensayo NPC (véase Cáncer). Otro estudio multicéntrico, aleatorio, doble ciego, controlado con placebo (the Negative Biopsy Trial) en 699 hombres en alto riesgo de cáncer de próstata no encontró efecto alguno de 200 μg/día o 400 μg/día de selenio en el riesgo de cáncer de próstata durante un seguimiento promedio de 36 meses (129). Además, debido a que evidencia actual sugiere una relación en forma de U entre el estatus de selenio y el riesgo de cáncer de próstata (130), los hombres debiesen evitar tomar selenio suplementario que sobrepase 200 μg/día.
Mujeres
En la actualidad, no existe evidencia clínica que demuestre que la suplementación con selenio por arriba de los niveles recomendados disminuya el riesgo de cáncer de seno aunque algunos, pero no todos, estudios basados en la observación han encontrado una relación inversa entre el estatus del selenio y el cáncer de seno en mujeres (59).
Adultos mayores (>50 años)
El envejecimiento no se ha asociado con cambios significativos en el requerimiento de selenio. Un ensayo aleatorio, doble ciego, controlado con placebo de cinco años en personas danesas mayores saludables (edades de inclusión, 60-74 años) encontró que la suplementación con selenio (100-300 μg/día) tuvo poco a no impacto en los niveles de enzimas antioxidantes, incluyendo GPx (131).
Autores y Críticos
Originalmente escrito en 2001 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Octubre de 2003 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Noviembre de 2007 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Noviembre de 2014 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Junio de 2015 por:
Petra A. Tsuji, Ph.D., M.P.H.
Profesor Asistente
Departamento de Ciencias Biológicas
Universidad de Towson
Traducido al Español en 2017 por:
Silvia Vazquez Lima
Instituto Linus Pauling
Universidad Estatal de Oregon
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Derechos de autoría 2001-2024 Instituto Linus Pauling
Referencias
1. Rayman MP. The importance of selenium to human health. Lancet. 2000;356(9225):233-241. (PubMed)
2. Mangiapane E, Pessione A, Pessione E. Selenium and selenoproteins: an overview on different biological systems. Curr Protein Pept Sci. 2014;15(6):598-607. (PubMed)
3. Mariotti M, Ridge PG, Zhang Y, et al. Composition and evolution of the vertebrate and mammalian selenoproteomes. PLoS One. 2012;7(3):e33066. (PubMed)
4. Terry EN, Diamond JA. Selenium. In: Erdman Jr J, Macdonald J, Zeisel S, eds. Present Knowledge in Nutrition. 10th ed. Ames: Wiley-Blackwell; 2012:568-585.
5. Boitani C, Puglisi R. Selenium, a key element in spermatogenesis and male fertility. Adv Exp Med Biol. 2008;636:65-73. (PubMed)
6. Arner ES. Focus on mammalian thioredoxin reductases--important selenoproteins with versatile functions. Biochim Biophys Acta. 2009;1790(6):495-526. (PubMed)
7. Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med. 2014;66:75-87. (PubMed)
8. Schomburg L. Selenium, selenoproteins and the thyroid gland: interactions in health and disease. Nat Rev Endocrinol. 2012;8(3):160-171. (PubMed)
9. Hill KE, Wu S, Motley AK, et al. Production of selenoprotein P (Sepp1) by hepatocytes is central to selenium homeostasis. J Biol Chem. 2012;287(48):40414-40424. (PubMed)
10. Olson GE, Winfrey VP, Hill KE, Burk RF. Megalin mediates selenoprotein P uptake by kidney proximal tubule epithelial cells. J Biol Chem. 2008;283(11):6854-6860. (PubMed)
11. Misu H, Takamura T, Takayama H, et al. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab. 2010;12(5):483-495. (PubMed)
12. Whanger PD. Selenoprotein expression and function-selenoprotein W. Biochim Biophys Acta. 2009;1790(11):1448-1452. (PubMed)
13. Jeong D, Kim TS, Chung YW, Lee BJ, Kim IY. Selenoprotein W is a glutathione-dependent antioxidant in vivo. FEBS Lett. 2002;517(1-3):225-228. (PubMed)
14. Reeves MA, Hoffmann PR. The human selenoproteome: recent insights into functions and regulation. Cell Mol Life Sci. 2009;66(15):2457-2478. (PubMed)
15. Reszka E, Jablonska E, Gromadzinska J, Wasowicz W. Relevance of selenoprotein transcripts for selenium status in humans. Genes Nutr. 2012;7(2):127-137. (PubMed)
16. Chung YW, Jeong D, Noh OJ, et al. Antioxidative role of selenoprotein W in oxidant-induced mouse embryonic neuronal cell death. Mol Cells. 2009;27(5):609-613. (PubMed)
17. Jeon YH, Park YH, Kwon JH, Lee JH, Kim IY. Inhibition of 14-3-3 binding to Rictor of mTORC2 for Akt phosphorylation at Ser473 is regulated by selenoprotein W. Biochim Biophys Acta. 2013;1833(10):2135-2142. (PubMed)
18. Jeon YH, Park YH, Lee JH, Hong JH, Kim IY. Selenoprotein W enhances skeletal muscle differentiation by inhibiting TAZ binding to 14-3-3 protein. Biochim Biophys Acta. 2014;1843(7):1356-1364. (PubMed)
19. Alkan Z, Duong FL, Hawkes WC. Selenoprotein W controls epidermal growth factor receptor surface expression, activation and degradation via receptor ubiquitination. Biochim Biophys Acta. 2015;1853(5):1087-1095. (PubMed)
20. Ganichkin OM, Xu XM, Carlson BA, et al. Structure and catalytic mechanism of eukaryotic selenocysteine synthase. J Biol Chem. 2008;283(9):5849-5865. (PubMed)
21. Lee BC, Peterfi Z, Hoffmann FW, et al. MsrB1 and MICALs regulate actin assembly and macrophage function via reversible stereoselective methionine oxidation. Mol Cell. 2013;51(3):397-404. (PubMed)
22. Kim HY. The methionine sulfoxide reduction system: selenium utilization and methionine sulfoxide reductase enzymes and their functions. Antioxid Redox Signal. 2013;19(9):958-969. (PubMed)
23. Kumaraswamy E, Malykh A, Korotkov KV, et al. Structure-expression relationships of the 15-kDa selenoprotein gene. Possible role of the protein in cancer etiology. J Biol Chem. 2000;275(45):35540-35547. (PubMed)
24. Korotkov KV, Kumaraswamy E, Zhou Y, Hatfield DL, Gladyshev VN. Association between the 15-kDa selenoprotein and UDP-glucose:glycoprotein glucosyltransferase in the endoplasmic reticulum of mammalian cells. J Biol Chem. 2001;276(18):15330-15336. (PubMed)
25. Labunskyy VM, Ferguson AD, Fomenko DE, Chelliah Y, Hatfield DL, Gladyshev VN. A novel cysteine-rich domain of Sep15 mediates the interaction with UDP-glucose:glycoprotein glucosyltransferase. J Biol Chem. 2005;280(45):37839-37845. (PubMed)
26. Labunskyy VM, Hatfield DL, Gladyshev VN. The Sep15 protein family: roles in disulfide bond formation and quality control in the endoplasmic reticulum. IUBMB Life. 2007;59(1):1-5. (PubMed)
27. Kasaikina MV, Fomenko DE, Labunskyy VM, et al. Roles of the 15-kDa selenoprotein (Sep15) in redox homeostasis and cataract development revealed by the analysis of Sep 15 knockout mice. J Biol Chem. 2011;286(38):33203-33212. (PubMed)
28. Hatfield DL, Tsuji PA, Carlson BA, Gladyshev VN. Selenium and selenocysteine: roles in cancer, health, and development. Trends Biochem Sci. 2014;39(3):112-120. (PubMed)
29. Curran JE, Jowett JB, Elliott KS, et al. Genetic variation in selenoprotein S influences inflammatory response. Nat Genet. 2005;37(11):1234-1241. (PubMed)
30. Santos LR, Duraes C, Mendes A, et al. A polymorphism in the promoter region of the selenoprotein S gene (SEPS1) contributes to Hashimoto's thyroiditis susceptibility. J Clin Endocrinol Metab. 2014;99(4):E719-723. (PubMed)
31. Alanne M, Kristiansson K, Auro K, et al. Variation in the selenoprotein S gene locus is associated with coronary heart disease and ischemic stroke in two independent Finnish cohorts. Hum Genet. 2007;122(3-4):355-365. (PubMed)
32. Cox AJ, Lehtinen AB, Xu J, et al. Polymorphisms in the Selenoprotein S gene and subclinical cardiovascular disease in the Diabetes Heart Study. Acta Diabetol. 2013;50(3):391-399. (PubMed)
33. Moses EK, Johnson MP, Tommerdal L, et al. Genetic association of preeclampsia to the inflammatory response gene SEPS1. Am J Obstet Gynecol. 2008;198(3):336 e331-335. (PubMed)
34. Shibata T, Arisawa T, Tahara T, et al. Selenoprotein S (SEPS1) gene -105G>A promoter polymorphism influences the susceptibility to gastric cancer in the Japanese population. BMC Gastroenterol. 2009;9:2. (PubMed)
35. Shchedrina VA, Zhang Y, Labunskyy VM, Hatfield DL, Gladyshev VN. Structure-function relations, physiological roles, and evolution of mammalian ER-resident selenoproteins. Antioxid Redox Signal. 2010;12(7):839-849. (PubMed)
36. Li X, Hill KE, Burk RF, May JM. Selenium spares ascorbate and alpha-tocopherol in cultured liver cell lines under oxidant stress. FEBS Lett. 2001;508(3):489-492. (PubMed)
37. May JM, Mendiratta S, Hill KE, Burk RF. Reduction of dehydroascorbate to ascorbate by the selenoenzyme thioredoxin reductase. J Biol Chem. 1997;272(36):22607-22610. (PubMed)
38. Murer SB, Aeberli I, Braegger CP, et al. Antioxidant supplements reduced oxidative stress and stabilized liver function tests but did not reduce inflammation in a randomized controlled trial in obese children and adolescents. J Nutr. 2014;144(2):193-201. (PubMed)
39. Schneider MJ, Fiering SN, Thai B, et al. Targeted disruption of the type 1 selenodeiodinase gene (Dio1) results in marked changes in thyroid hormone economy in mice. Endocrinology. 2006;147(1):580-589. (PubMed)
40. Hess SY. The impact of common micronutrient deficiencies on iodine and thyroid metabolism: the evidence from human studies. Best Pract Res Clin Endocrinol Metab. 2010;24(1):117-132. (PubMed)
41. Thomson CD. Assessment of requirements for selenium and adequacy of selenium status: a review. Eur J Clin Nutr. 2004;58(3):391-402. (PubMed)
42. Cooper A, Mones RL, Heird WC. Nutritional management of infants and children with specific diseases and other conditions. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore: Lippincott Williams & Wilkins; 2012:988-1005.
43. Sunde RA. Selenium. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore: Lippincott Williams & Wilkins; 2014:265-276.
44. Lei C, Niu X, Wei J, Zhu J, Zhu Y. Interaction of glutathione peroxidase-1 and selenium in endemic dilated cardiomyopathy. Clin Chim Acta. 2009;399(1-2):102-108. (PubMed)
45. Chen J. An original discovery: selenium deficiency and Keshan disease (an endemic heart disease). Asia Pac J Clin Nutr. 2012;21(3):320-326. (PubMed)
46. Harthill M. Review: micronutrient selenium deficiency influences evolution of some viral infectious diseases. Biol Trace Elem Res. 2011;143(3):1325-1336. (PubMed)
47. Stone R. Diseases. A medical mystery in middle China. Science. 2009;324(5933):1378-1381. (PubMed)
48. Du XH, Dai XX, Xia Song R, et al. SNP and mRNA expression for glutathione peroxidase 4 in Kashin-Beck disease. Br J Nutr. 2012;107(2):164-169. (PubMed)
49. Xiong YM, Mo XY, Zou XZ, et al. Association study between polymorphisms in selenoprotein genes and susceptibility to Kashin-Beck disease. Osteoarthritis Cartilage. 2010;18(6):817-824. (PubMed)
50. Zou K, Liu G, Wu T, Du L. Selenium for preventing Kashin-Beck osteoarthropathy in children: a meta-analysis. Osteoarthritis Cartilage. 2009;17(2):144-151. (PubMed)
51. Jirong Y, Huiyun P, Zhongzhe Y, et al. Sodium selenite for treatment of Kashin-Beck disease in children: a systematic review of randomised controlled trials. Osteoarthritis Cartilage. 2012;20(7):605-613. (PubMed)
52. Food and Nutrition Board, Institute of Medicine. Selenium. Dietary reference intakes for vitamin C, vitamin E, selenium, and carotenoids. Washington, D.C.: National Academy Press; 2000:284-324. (National Academy Press)
53. Combs GF, Jr., Gray WP. Chemopreventive agents: selenium. Pharmacol Ther. 1998;79(3):179-192. (PubMed)
54. Vinceti M, Dennert G, Crespi CM, et al. Selenium for preventing cancer. Cochrane Database Syst Rev. 2014;3:CD005195. (PubMed)
55. Hansen RD, Albieri V, Tjonneland A, Overvad K, Andersen KK, Raaschou-Nielsen O. Effects of smoking and antioxidant micronutrients on risk of colorectal cancer. Clin Gastroenterol Hepatol. 2013;11(4):406-415 e403. (PubMed)
56. Northrop-Clewes CA, Thurnham DI. Monitoring micronutrients in cigarette smokers. Clin Chim Acta. 2007;377(1-2):14-38. (PubMed)
57. Hazane-Puch F, Champelovier P, Arnaud J, et al. Long-term selenium supplementation in HaCaT cells: importance of chemical form for antagonist (protective versus toxic) activities. Biol Trace Elem Res. 2013;154(2):288-298. (PubMed)
58. Weekley CM, Harris HH. Which form is that? The importance of selenium speciation and metabolism in the prevention and treatment of disease. Chem Soc Rev. 2013;42(23):8870-8894. (PubMed)
59. Babaknejad N, Sayehmiri F, Sayehmiri K, et al. The relationship between selenium levels and breast cancer: a systematic review and meta-analysis. Biol Trace Elem Res. 2014;159(1-3):1-7. (PubMed)
60. Meplan C, Hesketh J. The influence of selenium and selenoprotein gene variants on colorectal cancer risk. Mutagenesis. 2012;27(2):177-186. (PubMed)
61. Steinbrecher A, Meplan C, Hesketh J, et al. Effects of selenium status and polymorphisms in selenoprotein genes on prostate cancer risk in a prospective study of European men. Cancer Epidemiol Biomarkers Prev. 2010;19(11):2958-2968. (PubMed)
62. Gerstenberger JP, Bauer SR, Van Blarigan EL, et al. Selenoprotein and antioxidant genes and the risk of high-grade prostate cancer and prostate cancer recurrence. Prostate. 2015;75(1):60-69. (PubMed)
63. Meplan C, Rohrmann S, Steinbrecher A, et al. Polymorphisms in thioredoxin reductase and selenoprotein K genes and selenium status modulate risk of prostate cancer. PLoS One. 2012;7(11):e48709. (PubMed)
64. Penney KL, Schumacher FR, Li H, et al. A large prospective study of SEP15 genetic variation, interaction with plasma selenium levels, and prostate cancer risk and survival. Cancer Prev Res (Phila). 2010;3(5):604-610. (PubMed)
65. Yu SY, Zhu YJ, Li WG. Protective role of selenium against hepatitis B virus and primary liver cancer in Qidong. Biol Trace Elem Res. 1997;56(1):117-124. (PubMed)
66. Vinceti M, Crespi CM, Malagoli C, Del Giovane C, Krogh V. Friend or foe? The current epidemiologic evidence on selenium and human cancer risk. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2013;31(4):305-341. (PubMed)
67. Clark LC, Combs GF, Jr., Turnbull BW, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA. 1996;276(24):1957-1963. (PubMed)
68. Reid ME, Duffield-Lillico AJ, Slate E, et al. The nutritional prevention of cancer: 400 μg per day selenium treatment. Nutr Cancer. 2008;60(2):155-163. (PubMed)
69. National Cancer Institute. Review of Prostate Cancer Prevention Study Shows No Benefit for Use of Selenium and Vitamin E Supplements. [Web page]. Available at: http://www.cancer.gov/newscenter/pressreleases/SELECTresults2008. Accessed 10/28/08.
70. Hatfield DL, Gladyshev VN. The Outcome of Selenium and Vitamin E Cancer Prevention Trial (SELECT) reveals the need for better understanding of selenium biology. Mol Interv. 2009;9(1):18-21. (PubMed)
71. Klein EA, Thompson IM, Jr., Tangen CM, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2011;306(14):1549-1556. (PubMed)
72. Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2009;301(1):39-51. (PubMed)
73. Flores-Mateo G, Carrillo-Santisteve P, Elosua R, et al. Antioxidant enzyme activity and coronary heart disease: meta-analyses of observational studies. Am J Epidemiol. 2009;170(2):135-147. (PubMed)
74. Bleys J, Navas-Acien A, Guallar E. Serum selenium levels and all-cause, cancer, and cardiovascular mortality among US adults. Arch Intern Med. 2008;168(4):404-410. (PubMed)
75. Eaton CB, Abdul Baki AR, Waring ME, Roberts MB, Lu B. The association of low selenium and renal insufficiency with coronary heart disease and all-cause mortality: NHANES III follow-up study. Atherosclerosis. 2010;212(2):689-694. (PubMed)
76. Laclaustra M, Navas-Acien A, Stranges S, Ordovas JM, Guallar E. Serum selenium concentrations and hypertension in the US Population. Circ Cardiovasc Qual Outcomes. 2009;2(4):369-376. (PubMed)
77. Kuruppu D, Hendrie HC, Yang L, Gao S. Selenium levels and hypertension: a systematic review of the literature. Public Health Nutr. 2014;17(6):1342-1352. (PubMed)
78. Laclaustra M, Stranges S, Navas-Acien A, Ordovas JM, Guallar E. Serum selenium and serum lipids in US adults: National Health and Nutrition Examination Survey (NHANES) 2003-2004. Atherosclerosis. 2010;210(2):643-648. (PubMed)
79. Stranges S, Laclaustra M, Ji C, et al. Higher selenium status is associated with adverse blood lipid profile in British adults. J Nutr. 2010;140(1):81-87. (PubMed)
80. Rees K, Hartley L, Day C, Flowers N, Clarke A, Stranges S. Selenium supplementation for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2013;1:CD009671. (PubMed)
81. McKenzie RC, Beckett GJ, Arthur JR. Effects of selenium on immunity and aging. In: Hatfield DL, Berry MJ, Gladyshev VN, eds. Selenium: Its Molecular Biology and Role in Human Health. 2nd ed. New York: Springer; 2006:311-323.
82. Huang Z, Rose AH, Hoffmann PR. The role of selenium in inflammation and immunity: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2012;16(7):705-743. (PubMed)
83. Mattmiller SA, Carlson BA, Sordillo LM. Regulation of inflammation by selenium and selenoproteins: impact on eicosanoid biosynthesis. J Nutr Sci. 2013;2:e28. (PubMed)
84. Norton RL, Hoffmann PR. Selenium and asthma. Mol Aspects Med. 2012;33(1):98-106. (PubMed)
85. Speckmann B, Steinbrenner H. Selenium and selenoproteins in inflammatory bowel diseases and experimental colitis. Inflamm Bowel Dis. 2014;20(6):1110-1119. (PubMed)
86. Drain PK, Kupka R, Mugusi F, Fawzi WW. Micronutrients in HIV-positive persons receiving highly active antiretroviral therapy. Am J Clin Nutr. 2007;85(2):333-345. (PubMed)
87. Stone CA, Kawai K, Kupka R, Fawzi WW. Role of selenium in HIV infection. Nutr Rev. 2010;68(11):671-681. (PubMed)
88. de Menezes Barbosa EG, Junior FB, Machado AA, Navarro AM. A longer time of exposure to antiretroviral therapy improves selenium levels. Clin Nutr. 2015;34(2):248-251. (PubMed)
89. Baum MK, Miguez-Burbano MJ, Campa A, Shor-Posner G. Selenium and interleukins in persons infected with human immunodeficiency virus type 1. J Infect Dis. 2000;182 Suppl 1:S69-73. (PubMed)
90. Kalantari P, Narayan V, Natarajan SK, et al. Thioredoxin reductase-1 negatively regulates HIV-1 transactivating protein Tat-dependent transcription in human macrophages. J Biol Chem. 2008;283(48):33183-33190. (PubMed)
91. Burbano X, Miguez-Burbano MJ, McCollister K, et al. Impact of a selenium chemoprevention clinical trial on hospital admissions of HIV-infected participants. HIV Clin Trials. 2002;3(6):483-491. (PubMed)
92. Hurwitz BE, Klaus JR, Llabre MM, et al. Suppression of human immunodeficiency virus type 1 viral load with selenium supplementation: a randomized controlled trial. Arch Intern Med. 2007;167(2):148-154. (PubMed)
93. Kupka R, Mugusi F, Aboud S, Hertzmark E, Spiegelman D, Fawzi WW. Effect of selenium supplements on hemoglobin concentration and morbidity among HIV-1-infected Tanzanian women. Clin Infect Dis. 2009;48(10):1475-1478. (PubMed)
94. Kupka R, Mugusi F, Aboud S, et al. Randomized, double-blind, placebo-controlled trial of selenium supplements among HIV-infected pregnant women in Tanzania: effects on maternal and child outcomes. Am J Clin Nutr. 2008;87(6):1802-1808. (PubMed)
95. Baum MK, Campa A, Lai S, et al. Effect of micronutrient supplementation on disease progression in asymptomatic, antiretroviral-naive, HIV-infected adults in Botswana: a randomized clinical trial. JAMA. 2013;310(20):2154-2163. (PubMed)
96. Bone RC, Balk RA, Cerra FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101(6):1644-1655. (PubMed)
97. Mann EA, Baun MM, Meininger JC, Wade CE. Comparison of mortality associated with sepsis in the burn, trauma, and general intensive care unit patient: a systematic review of the literature. Shock. 2012;37(1):4-16. (PubMed)
98. Alhazzani W, Jacobi J, Sindi A, et al. The effect of selenium therapy on mortality in patients with sepsis syndrome: a systematic review and meta-analysis of randomized controlled trials. Crit Care Med. 2013;41(6):1555-1564. (PubMed)
99. Huang TS, Shyu YC, Chen HY, et al. Effect of parenteral selenium supplementation in critically ill patients: a systematic review and meta-analysis. PLoS One. 2013;8(1):e54431. (PubMed)
100. van Zuuren EJ, Albusta AY, Fedorowicz Z, Carter B, Pijl H. Selenium Supplementation for Hashimoto's Thyroiditis: Summary of a Cochrane Systematic Review. Eur Thyroid J. 2014;3(1):25-31. (PubMed)
101. Karanikas G, Schuetz M, Kontur S, et al. No immunological benefit of selenium in consecutive patients with autoimmune thyroiditis. Thyroid. 2008;18(1):7-12. (PubMed)
102. Krysiak R, Okopien B. The effect of levothyroxine and selenomethionine on lymphocyte and monocyte cytokine release in women with Hashimoto's thyroiditis. J Clin Endocrinol Metab. 2011;96(7):2206-2215. (PubMed)
103. Negro R, Greco G, Mangieri T, Pezzarossa A, Dazzi D, Hassan H. The influence of selenium supplementation on postpartum thyroid status in pregnant women with thyroid peroxidase autoantibodies. J Clin Endocrinol Metab. 2007;92(4):1263-1268. (PubMed)
104. Turker O, Kumanlioglu K, Karapolat I, Dogan I. Selenium treatment in autoimmune thyroiditis: 9-month follow-up with variable doses. J Endocrinol. 2006;190(1):151-156. (PubMed)
105. Marcocci C, Kahaly GJ, Krassas GE, et al. Selenium and the course of mild Graves' orbitopathy. N Engl J Med. 2011;364(20):1920-1931. (PubMed)
106. Watt T, Cramon P, Bjorner JB, et al. Selenium supplementation for patients with Graves' hyperthyroidism (the GRASS trial): study protocol for a randomized controlled trial. Trials. 2013;14:119. (PubMed)
107. Winther KH, Watt T, Bjorner JB, et al. The chronic autoimmune thyroiditis quality of life selenium trial (CATALYST): study protocol for a randomized controlled trial. Trials. 2014;15:115. (PubMed)
108. Peplow D, Edmonds R. Health risks associated with contamination of groundwater by abandoned mines near Twisp in Okanogan County, Washington, USA. Environ Geochem Health. 2004;26(1):69-79. (PubMed)
109. Chang JC, Gutenmann WH, Reid CM, Lisk DJ. Selenium content of Brazil nuts from two geographic locations in Brazil. Chemosphere. 1995;30(4):801-802. (PubMed)
110. Rayman MP, Infante HG, Sargent M. Food-chain selenium and human health: spotlight on speciation. Br J Nutr. 2008;100(2):238-253. (PubMed)
111. Reyes LH, Encinar JR, Marchante-Gayon JM, Alonso JI, Sanz-Medel A. Selenium bioaccessibility assessment in selenized yeast after "in vitro" gastrointestinal digestion using two-dimensional chromatography and mass spectrometry. J Chromatogr A. 2006;1110(1-2):108-116. (PubMed)
112. Bermingham EN, Hesketh JE, Sinclair BR, Koolaard JP, Roy NC. Selenium-Enriched Foods Are More Effective at Increasing Glutathione Peroxidase (GPx) Activity Compared with Selenomethionine: A Meta-Analysis. Nutrients. 2014;6(10):4002-4031. (PubMed)
113. Giacosa A, Faliva MA, Perna S, Minoia C, Ronchi A, Rondanelli M. Selenium fortification of an Italian rice cultivar via foliar fertilization with sodium selenate and its effects on human serum selenium levels and on erythrocyte glutathione peroxidase activity. Nutrients. 2014;6(3):1251-1261. (PubMed)
114. Bleys J, Navas-Acien A, Guallar E. Serum selenium and diabetes in U.S. adults. Diabetes Care. 2007;30(4):829-834. (PubMed)
115. Laclaustra M, Navas-Acien A, Stranges S, Ordovas JM, Guallar E. Serum selenium concentrations and diabetes in U.S. adults: National Health and Nutrition Examination Survey (NHANES) 2003-2004. Environ Health Perspect. 2009;117(9):1409-1413. (PubMed)
116. Stranges S, Marshall JR, Natarajan R, et al. Effects of Long-Term Selenium Supplementation on the Incidence of Type 2 Diabetes: A Randomized Trial. Ann Intern Med. 2007;147(4):217-223. (PubMed)
117. Schoenmakers E, Agostini M, Mitchell C, et al. Mutations in the selenocysteine insertion sequence-binding protein 2 gene lead to a multisystem selenoprotein deficiency disorder in humans. J Clin Invest. 2010;120(12):4220-4235. (PubMed)
118. Steinbrenner H. Interference of selenium and selenoproteins with the insulin-regulated carbohydrate and lipid metabolism. Free Radic Biol Med. 2013;65:1538-1547. (PubMed)
119. Kaur P, Rizk NM, Ibrahim S, et al. iTRAQ-based quantitative protein expression profiling and MRM verification of markers in type 2 diabetes. J Proteome Res. 2012;11(11):5527-5539. (PubMed)
120. Yang SJ, Hwang SY, Choi HY, et al. Serum selenoprotein P levels in patients with type 2 diabetes and prediabetes: implications for insulin resistance, inflammation, and atherosclerosis. J Clin Endocrinol Metab. 2011;96(8):E1325-1329. (PubMed)
121. Azrak RG, Cao S, Pendyala L, et al. Efficacy of increasing the therapeutic index of irinotecan, plasma and tissue selenium concentrations is methylselenocysteine dose dependent. Biochem Pharmacol. 2007;73(9):1280-1287. (PubMed)
122. Graf WD, Oleinik OE, Glauser TA, Maertens P, Eder DN, Pippenger CE. Altered antioxidant enzyme activities in children with a serious adverse experience related to valproic acid therapy. Neuropediatrics. 1998;29(4):195-201. (PubMed)
123. Vernie LN, de Goeij JJ, Zegers C, de Vries M, Baldew GS, McVie JG. Cisplatin-induced changes of selenium levels and glutathione peroxidase activities in blood of testis tumor patients. Cancer Lett. 1988;40(1):83-91. (PubMed)
124. Flodin NW. Micronutrient supplements: toxicity and drug interactions. Prog Food Nutr Sci. 1990;14(4):277-331. (PubMed)
125. Brown BG, Zhao XQ, Chait A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345(22):1583-1592. (PubMed)
126. Tousoulis D, Antoniades C, Stefanadis C. Statins and antioxidant vitamins: should co-administration be avoided? J Am Coll Cardiol. 2006;47(6):1237; author reply 1237-1238. (PubMed)
127. Hurst R, Armah CN, Dainty JR, et al. Establishing optimal selenium status: results of a randomized, double-blind, placebo-controlled trial. Am J Clin Nutr. 2010;91(4):923-931. (PubMed)
128. Burk RF, Norsworthy BK, Hill KE, Motley AK, Byrne DW. Effects of chemical form of selenium on plasma biomarkers in a high-dose human supplementation trial. Cancer Epidemiol Biomarkers Prev. 2006;15(4):804-810. (PubMed)
129. Algotar AM, Stratton MS, Ahmann FR, et al. Phase 3 clinical trial investigating the effect of selenium supplementation in men at high-risk for prostate cancer. Prostate. 2013;73(3):328-335. (PubMed)
130. Chiang EC, Shen S, Kengeri SS, et al. Defining the Optimal Selenium Dose for Prostate Cancer Risk Reduction: Insights from the U-Shaped Relationship between Selenium Status, DNA Damage, and Apoptosis. Dose Response. 2009;8(3):285-300. (PubMed)
131. Ravn-Haren G, Krath BN, Overvad K, et al. Effect of long-term selenium yeast intervention on activity and gene expression of antioxidant and xenobiotic metabolising enzymes in healthy elderly volunteers from the Danish Prevention of Cancer by Intervention by Selenium (PRECISE) pilot study. Br J Nutr. 2008;99(6):1190-1198. (PubMed)
Sodio
Contenido
Resumen
- El sodio y el cloro — electrolitos principales del compartimiento fluido fuera de las células (es decir, extracelular) — trabajan juntos para controlar el volumen extracelular y la presión sanguínea. Perturbaciones en las concentraciones de sodio en el fluido extracelular están asociadas con trastornos del balance de agua. (Más información)
- Varios mecanismos actúan en el riñón para asegurar que la cantidad de sodio que se pierde a través de la excreción renal sea adecuadamente compensada por la cantidad de sodio consumida manteniendo de este modo la homeostasis del sodio. (Más información)
- La hiponatremia (concentraciones anormalmente bajas de sodio en la sangre) es común entre los adultos mayores y en los individuos con hipertensión, enfermedad renal, y enfermedad del corazón. La hiponatremia también ocurre en hasta un 30% de los pacientes hospitalizados. (Más información)
- La hiponatremia aguda severa puede conducir a un edema cerebral con consecuencias neurológicas y ser letal si no es prontamente diagnosticado y tratado. La hiponatremia crónica leve con efectos a la salud adversos a largo plazo, tales como déficits de atención, inestabilidad al andar, caídas, y pérdida ósea y fracturas, ha sido asociada con la morbilidad y mortalidad cardiovascular. (Más información)
- En el 2019, La Academia Nacional de Medicina de los Estados Unidos estableció una ingesta adecuada (IA) para el sodio de 1.5 gramos (g)/día en los adultos, equivalente a 3.8 g/día de cloruro de sodio (sal). (Más información)
- La Academia Nacional de Medicina estableció una Ingesta para la Disminución del Riesgo de Enfermedad Crónica (CDRR, por sus siglas en inglés) de sodio de 2.3 g/día (5.8 g/día de sal) para los adultos basado en evidencia de posibles beneficios en la salud a largo plazo en la presión sanguínea y riesgo de hipertensión y enfermedades cardiovasculares asociadas con la reducción de ingestas de sodio por debajo de este nivel. (Más información)
- Las ingestas de sodio actuales de la población adulta estadounidense superan por mucho la CDRR. El sodio ha sido identificado como un nutriente de preocupación de salud pública por su consumo excesivo. (Más información)
- El sodio dietario en exceso es un contribuidor principal para la hipertensión, el cual es un factor de riesgo prevenible de las enfermedades cardiovasculares. Estudios controlados aleatorios demostraron que la reducción de sodio dietario (de 1.8 a 3.2 g/día) puede reducir la presión sanguínea en sujetos con presión sanguínea elevada. Sin embargo, evidencia actual falla en apoyar una disminución de la morbilidad y mortalidad cardiovascular con una restricción moderada de sodio en pacientes con hipertensión. (Más información)
-
Resultados adversos de salud adicionales, incluyendo el cáncer gástrico, osteoporosis, y cálculos renales, también han sido ligados al consumo en exceso de sodio. (Más información)
La sal (cloruro de sodio) es esencial para la vida. El sodio total del cuerpo en una persona promedio de 70 kg es alrededor de 4,200 mmol (~100g), de los cuales 40% se encuentra en el hueso y 60% en el fluido dentro y fuera de las células (1). El cloro total del cuerpo promedio es de 2,310 mmol (~82 g), del cual 70% es distribuido en el fluido extracelular y el restante se encuentra en el colágeno del tejido conectivo (1). Múltiples mecanismos trabajan en conjunto para regular estrechamente las concentraciones de sodio y cloruro del cuerpo. Aunque esta revisión enfatiza la función y los requerimientos del sodio, los iones de sodio y cloro trabajan juntos para controlar el volumen extracelular y la presión sanguínea (1).
Función
El sodio (Na+) y cloruro (Cl-) son los principales iones en el compartimiento extracelular, lo cual incluye el plasma sanguíneo, fluido intersticial (fluido entre las células), y el fluido transcelular (p. ej., el fluido cerebroespinal, líquido sinovial). Como tales, desempeñan papeles críticos en una serie de procesos que sustentan la vida.
Mantenimiento del potencial de membrana
El sodio y el cloruro son electrolitos que contribuyen a mantener las diferencias de carga y concentración a través de las membranas celulares. El potasio (K+) es el principal ion (catión) con carga positiva dentro de las células, mientras que el sodio es el principal catión en el fluido extracelular. Las concentraciones de potasio son alrededor de 30 veces más altas dentro que fuera de las células, mientras que las concentraciones de sodio son más de 10 veces inferior dentro que fuera de las células. Las diferencias de concentración entre el sodio y el potasio a través de las membranas celulares crean un gradiente electroquímico conocido como potencial de membrana. El potencial de membrana de una célula es mantenido por bombas de iones en la membrana celular, especialmente las bombas de Na+/K+ ATPasa. Estas bombas utilizan ATP (energía) para bombear el sodio fuera de la célula a cambio de potasio (Figura 1). Su actividad se ha estimado representa del 20% -40% del gasto de energía en reposo en un adulto típico. La mayor proporción de energía dedicada a la mantención de los gradientes de concentración de sodio/potasio enfatizan la importancia de esta función en el sustento de la vida. El estrecho control del potencial de membrana de la célula es crítico para la transmisión del impulso nervioso, la contracción muscular y la función cardíaca (2-4).
[Figura 1 - Clic para Agrandar]
Absorción y transporte de nutrientes
La absorción de sodio en el intestino delgado juega un importante papel en la absorción de cloro, aminoácidos, glucosa y agua. Mecanismos similares se encuentran involucrados en la reabsorción de estos nutrientes luego de que han sido filtrados de la sangre por los riñones. El cloruro, en la forma de ácido clorhídrico (HCl) es también un componente importante del jugo gástrico, el cual ayuda en la digestión y la absorción de muchos nutrientes (5).
Mantenimiento del volumen sanguíneo y presión sanguínea
Debido a que el sodio es el principal determinante del volumen de fluido extracelular, incluyendo al volumen sanguíneo, una serie de mecanismos fisiológicos que regulan el volumen sanguíneo y la presión sanguínea trabajan al ajustar el contenido de sodio del cuerpo. En el sistema circulatorio, los receptores de presión (baroreceptores) perciben cambios en la presión sanguínea y envían señales excitatorias o inhibitorias al sistema nervioso y/o a las glándulas endocrinas para alterar la regulación de sodio por los riñones. En general, la retención de sodio resulta en la retención de agua, y la pérdida de sodio resulta en pérdida de agua (6). A continuación, se describen tres mecanismos que contribuyen a sistema de control homeostático multifactorial más grande que gobierna el volumen sanguíneo y la presión sanguínea a través de la regulación del balance de sodio. Estos mecanismos regulatorios son especialmente importantes para el control del transporte de sodio en varios segmentos de la nefrona (unidad básica estructural del riñón), incluyendo los túbulos contorneados proximales y distales, la porción gruesa ascendente del asa de Henle y el túbulo colector.
Sistema renina-angiotensina-aldosterona
En respuesta a una disminución significativa en el volumen o presión sanguínea (p.ej., pérdida de sangre o deshidratación serias), los riñones liberan renina en la circulación. La renina es una enzima que separa a un péptido pequeño (angiotensina I) de una proteína más grande (angiotensinógeno) producida por el hígado. La angiotensina I es dividida en un péptido más pequeño (angiotensina II) por la enzima convertidora de angiotensina (ECA), una enzima presente en la superficie interna de los vasos sanguíneos y en los pulmones, hígado y riñones. La angiotensina II estimula la contracción de pequeñas arterias, resultando en un incremento en la presión sanguínea. La angiotensina II también es un potente estimulador de la síntesis de aldosterona por las glándulas adrenales. La aldosterona es una hormona esteroide que actúa en los riñones para incrementar la reabsorción de sodio y la excreción de potasio. La retención de sodio por los riñones incrementa la retención de agua, resultando en un incremento en el volumen y presión sanguínea (7).
Hormona antidiurética
La secreción de la hormona antidiurética (ADH; también conocida como arginina vasopresina [AVP]) por la glándula pituitaria posterior es estimulada por un descenso significativo en el volumen o la presión sanguínea. Junto con el sistema renina-angiotensina-aldosterona, la ADH estimula los canales de sodio epiteliales (ENaC) en las membranas celulares apicales a lo largo de los túbulos distales de la nefrona del riñón para incrementar la reabsorción de sodio y agua (8).
Sistema dopaminérgico
La dopamina es producida a partir de la L-DOPA en los túbulos proximales del riñón y actúa en los receptores de dopamina distribuidos a lo largo de los túbulos proximales y la porción gruesa ascendiente del asa de Henle para regular el transporte de sodio. La dopamina promueve la excreción de sodio (natriuresis) al inhibir el intercambiador Na+/H+ y el cotransportador Na+/fosfato (Pi) en las membranas celulares apicales y el cotransportador Na+/bicarbonato (HCO3-) y la ATPasa Na+/K+ en las membranas celulares basolaterales. El efecto inhibidor de la dopamina en la ATPasa Na+/K+ es potenciada por la hormona natriurética, el péptido natriurético atrial (ANP), que es secretado por las células del músculo cardíaco en la circulación (9).
Deficiencia
La hiponatremia, definida como una concentración de sodio en el suero ([Na+]) <136 mmol/litro (mM), puede resultar de un incremento en la retención de fluidos (hiponatremia dilucional) o de una pérdida de sodio incrementada. Las ingestas inadecuadas de sodio raramente resultan en hiponatremia, incluso en aquellos con dietas muy bajas en sal, debido a que los riñones incrementan la excreción de agua a fin de mantener la osmolalidad sérica (es decir, el balance de electrolito-agua). Los resultados de la Encuesta de Evaluación Nacional de Salud y Nutrición (NHANES) estadounidense de 1999-2004 indicaron una prevalencia en general de hiponatremia del 1.9% en una muestra representativa de la población estadounidense de 14,697 participantes de 18 años de edad y mayores (10). Se encontró que la hiponatremia era más prevalente entre individuos de mayor edad (3.1% en aquellos con edades de entre 65 a 84 años) y en aquellos que sufrían de hipertensión (2.9%), diabetes mellitus (3.3%), enfermedad coronaria cardíaca (ECC; 2.6%), accidente cerebrovascular (3.6%), enfermedad pulmonar obstructiva crónica (EPOC; 3.9%), cáncer (3.4%), y trastornos psiquiátricos (2.9%) (10). La hiponatremia es también común en pacientes hospitalizados, con un estimado del 15%-30% con hiponatremia leve, (suero [Na+]: 130-135 mM) y hasta un 7% con hiponatremia moderada a severa (suero [Na+]: <130 mM) (11).
Causas de la hiponatremia
La hiponatremia dilucional puede deberse a una secreción inapropiada de la hormona antidiurética (ADH), lo cual se asocia con trastornos que afectan el sistema nervioso central, y con el uso de ciertas drogas (véase Interacción con drogas/fármacos). En algunos casos, la ingesta excesiva de agua puede conducir también a la hiponatremia dilucional (véase también Hiponatremia asociada al ejercicio). Las condiciones que incrementan la pérdida de sodio y de cloro incluyen diarrea o vómitos severos o prolongados, sudoración excesiva y persistente, el uso de algunos diuréticos, y algunas formas de enfermedad renal. La restricción muy severa de la ingesta de sodio dietario en pacientes renales con hipertensión e insuficiencia cardíaca congestiva podría también resultar en un agotamiento de sodio dañino para el cuerpo (1).
Hiponatremia asociada al ejercicio
La hiponatremia asociada al ejercicio (EAH) es una hiponatremia dilucional que ocurre en individuos que compiten en eventos de ejercicios de resistencia (hasta 6 horas de duración) y ultra resistencia (>6 horas de duración), como maratones, triatlones Ironman, carreras de bicicleta de montaña, caminatas de excursionista, y eventos de natación a distancia en aguas abiertas. Debe notarse que la EAH sintomática ha sido reportada cada vez más en eventos más cortos, tales como medios maratones y triatlones de velocidad. El desarrollo de la hiponatremia durante o hasta 24 horas después de la actividad física intensa y/o sostenida ha sido ligada a la sobrecarga de líquidos debido a las ingestas excesivas de agua, la excreción urinaria de agua alterada debido a la persistente secreción de ADH, y a la temperatura ambiente muy baja o muy alta (12). Los factores de riesgo incluyen hiperhidratación previa al ejercicio, el uso de fármacos antiinflamatorios no esteroideos (AINE), y el ejercicio prolongado (>4 h) (revisado en 12).
Signos y síntomas de la hiponatremia
Los síntomas de hiponatremia incluyen dolor de cabeza, náuseas, vómitos, calambres musculares, fatiga, desorientación, y desmayos. Complicaciones de la hiponatremia severa y en rápido desarrollo pueden incluir edema cerebral (hinchazón del cerebro), convulsiones, coma y daño cerebral. La hiponatremia aguda o severa puede ser fatal sin el tratamiento médico oportuno y apropiado (13).
La hiponatremia crónica leve ha sido asociada con deficiencias en el modo de andar y la atención, caídas, y pérdida ósea y fracturas, especialmente en mujeres y personas de la tercera edad (14, 15). Un reciente estudio de cohorte prospectivo de 11 años de 3,000 hombres libres de enfermedades cardiovasculares (ECV) también reportó riesgos significantemente más altos de accidentes cerebrovasculares, enfermedad coronaria cardíaca, eventos de ECV totales, mortalidad relacionada a ECV, y mortalidad por todas las causas en participantes con hiponatremia leve a severa ([Na+] en el suero <139 mM) en comparación con aquellos con [Na+] en el suero de entre 139 mM y 144 mM (16). Además, en un meta-análisis de 81 estudios basados en la observación en pacientes con condiciones médicas diversas (incluyendo enfermedades cardiovasculares, infecciones pulmonares, y cirrosis), se encontró que el riesgo de mortalidad era casi tres veces mayor en sujetos hiponatrémicos en comparación a sujetos normonatrémicos (17). El mejoramiento o normalización de [Na+] en el suero en sujetos hiponatrémicos se asoció con una disminución en la tasa de mortalidad en pacientes con condiciones clínicas diversas (18).
Interacción con drogas/fármacos
Tabla 1 lista algunos medicamentos que pueden incrementar el riesgo de hiponatremia (10, 19).
La Ingesta Adecuada (IA) por Sodio
En el 2019, la Junta de Nutrición y Alimentos (JNA) de la Academia Nacional de Medicina estadounidense revisó las Ingestas Dietéticas de Referencia para el sodio (20). La JNA no encontró suficiente evidencia para- determinar un Requerimiento Estimado Promedio (REP) y obtener una Ingesta Diaria Recomendada (IDR; RDA, por sus siglas en inglés). En cambio, establecieron una ingesta adecuada (IA) de sodio (Tabla 2; 20). Las consideraciones explicadas por la JNA para establecer una IA para el sodio incluyeron que la evidencia disponible fue insuficiente para identificar los efectos adversos para la salud asociados con un bajo consumo de sodio y que había pruebas sustanciales que sugerían posibles beneficios para la salud a largo plazo asociados con la reducción del consumo habitual de sodio por debajo de 2,300 mg/día (véase Ingesta para la Disminución del Riesgo de Enfermedad Crónica de sodio). La IA no se pudo derivar de las ingestas dietéticas promedio de personas aparentemente sanas en los Estados Unidos ya que los niveles de ingesta promedio están muy por encima de los 2,300 mg/día (véase Fuentes).
Fuentes
La mayoría del sodio y cloro en la dieta proviene de la sal (21). Muy poco sodio se produce naturalmente en los alimentos. En lugar de esto, el sodio es agregado para hacer ciertos alimentos no perecederos, y es utilizado de manera ubicua en el suministro de alimentos de los Estados Unidos de manera que todos los grupos de alimentos contribuyan a los niveles de consumo de sodio (21). Se ha estimado que el 75% de la ingesta de sal en EE.UU. se deriva de la sal añadida durante el procesamiento o la elaboración de los alimentos, más que de la sal añadida en la mesa o durante su preparación (22). Las ingestas más bajas de sal se asociaron con dietas que enfatizan alimentos sin procesar, especialmente frutas, vegetales, y legumbres. Datos combinados de las Encuestas de Evaluación Nacional de Salud y Nutrición (NHANES) estadounidense de 2007-2008 y 2009-2010 indicaron ingestas dietéticas de sodio promedio de 3,100 mg/día en niños (edades, 3-18 años), 3,800 mg/día en adultos (edades, 19-50 años), y 3,300 mg/día en adultos mayores (>50 años) (23). Las ingestas habituales se estimaron en 4,400 g/día y 3,100 g/día en hombres y mujeres adultos (edades, 19-50 años), respectivamente. En general, se observó que las ingestas de sodio entre los hombres de todos los grupos de edades eran de 20%-45% más altas que entre las mujeres (23). Estas ingestas están muy por encima de la Ingesta para la Disminución del Riesgo de Enfermedad Crónica de sodio de 2,300 mg/día (véase Ingesta para la Disminución del Riesgo de Enfermedad Crónica de sodio).
La Tabla 3 lista el contenido de sodio (en miligramos [mg]) de algunos alimentos que son altos en sal, y la Tabla 4 lista algunos alimentos que son relativamente bajos en sal. La mayoría del sodio es consumido en la forma de cloruro de sodio (sal). El contenido de sal de los alimentos puede ser calculado multiplicando el contenido de sodio por 2.5.
Ejemplo: 2,000 mg (2 g) de sodio x 2.5 = 5,000 mg (5 g) de sal.
Para más información acerca del contenido de sodio de los alimentos, busque en la base de datos de composición de alimentos de la USDA.
El valor diario (VD) para el sodio es de menos de 2,400 mg. El % del VD incluido en la etiqueta de Información Nutricional de alimentos y bebidas empaquetadas está destinado a ayudar a los consumidores a hacer elecciones informadas y considerar los alimentos con bajo (≤5% del VD por porción) en lugar de alto (≥20% del VD por porción) contenido de sodio (24).
Seguridad
Toxicidad
Las ingestas excesivas de cloruro de sodio conducen a un incremento del volumen de fluido extracelular a medida que el agua es extraída de las células para mantener las concentraciones normales de sodio fuera de las células. Sin embargo, mientras se puedan satisfacer las necesidades de agua, los riñones funcionando normalmente pueden excretar el exceso de sodio y restaurar la normalidad del sistema (1). La ingesta de grandes cantidades de sal puede resultar en náusea, vómito, diarrea, y cólicos abdominales (25). La hipernatremia, definida como concentraciones de sodio en el suero ([Na+]) >145 mM, es mucho menos común que la hiponatremia y es raramente causada por la ingesta excesiva de sodio (p. ej., por la ingesta de grandes cantidades de agua de mar o infusiones intravenosas de solución salina concentrada) (26). La hipernatremia generalmente se desarrolla a partir de la pérdida de agua en exceso (p. ej., quemaduras, infecciones respiratorias, pérdida renal, diarrea osmótica, trastornos hipotalámicos) o de la ingesta reducida de agua, frecuentemente acompañada por un deterioro en el mecanismo de la sed (6). Los síntomas de hipernatremia con evidencia de deshidratación proveniente de la pérdida de agua en exceso pueden incluir mareos o desmayos, baja presión sanguínea, y una producción de orina disminuida. La hipernatremia severa ([Na+] >158 mM) puede resultar en un estatus mental alterado, letargo, irritabilidad, estupor, convulsiones, y coma. La contracción cerebral aguda puede provocar hemorragia intracraneal y subaracnoidea (26).
Las Ingestas Dietéticas de Referencia para el sodio fueron revisadas recientemente por la Junta de Nutrición y Alimentos de la Academia Nacional de Medicina (20). Usando un nuevo modelo ampliado de Ingestas Dietética de Referencia, el Instituto de Medicina no encontró evidencia suficiente de efectos adversos toxicológicos del exceso de consumo de sodio; por lo tanto, no establecieron un nivel máximo de ingesta tolerable (NM) para el sodio (20).
Riesgos a la salud del exceso de sodio dietario
Hipertensión
La presión sanguínea normal es definida por una presión sanguínea sistólica por debajo de los 120 mm Hg y una presión sanguínea diastólica por debajo de los 80 mm Hg, frecuentemente denotada como <120/80 mm Hg (27). En la actualidad, alrededor de un tercio de los adultos estadounidenses padece de hipertensión (niveles de presión sanguínea ≥140/90 mm Hg), y otro tercio tiene presión sanguínea elevada (prehipertensión, correspondiendo a niveles de ≥120/80 mm Hg y <140/90 mm Hg) que los pone en riesgo de hipertensión (28). La hipertensión crónica daña el corazón, vasos sanguíneos, y riñones, incrementando por lo tanto el riesgo de enfermedades cardiacas y accidentes cerebrovasculares, como también enfermedades renales hipertensivas. En un cierto número de estudios clínicos, la ingesta de sal ha sido significantemente correlacionada con la hipertrofia ventricular izquierda, un engrosamiento anormal del músculo cardíaco, lo cual se asocia con un incremento en la mortalidad por enfermedades cardíacas (29). Varias líneas de investigación, conducidas en las ultimas décadas, han proporcionado evidencia de una relación entre el consumo de sodio y resultados a la salud. Por ejemplo, estudios de cohorte basados en la observación, como el buen diseñado Estudio Internacional de la Sal y Presión Sanguínea (INTERSALT; por sus siglas en inglés), ha asociado la ingesta de sodio en exceso con un incremento progresivo de la presión sanguínea con la edad (30, 31). Además, un cierto número de estudios de intervención, incluyendo el Ensayo de Intervenciones No-farmacológicas en los Ancianos (TONE), los Ensayos de Prevención de la Hipertensión (TOHP), y los Enfoques Alimenticios para Detener la Hipertensión (DASH)-sodio, han demostrado que la reducción de sodio dietario puede efectivamente prevenir o mejorar la hipertensión entre subgrupos de poblaciones en riesgo elevado (véase Ensayos clínicos de la presión sanguínea).
Sensibilidad a la sal: Las respuestas de la presión sanguínea a los cambios a corto plazo en la ingesta de sodio son heterogéneas. En efecto, algunos individuos tienen poco a ningún cambio en la presión sanguínea en respuesta a la manipulación del sodio y son identificados como "sal-resistentes." A diferencia, los individuos que experimentan un mayor cambio en la presión sanguínea luego de la manipulación de sodio dietario son llamados "sal-sensibles" (32, 33). La mayoría de los protocolos utilizados en los estudios de sensibilidad a la sal implican manipulaciones extremas de la ingesta de sodio (carga de sodio y agotamiento de sodio) durante un corto período de tiempo de unos pocos días o de hasta una semana. Una típica intervención isocalórica controlada puede incluir una manipulación de dos fases, aleatoria, cruzada de siete días de sodio dietario con una dieta alta en sodio (6.9 a 8.1 g/día) y una dieta muy baja en sodio (0.5 g/día). La resistencia a la sal es frecuentemente definida como un cambio de ≤5 mm Hg en 24 h en la presión arterial media (PAM) entre una dieta alta en sodio y una dieta baja en sodio. Inversamente, la sensibilidad a la sal es definida por cambios de >5 mm Hg en la PAM entre las dietas altas y bajas en sodio (34).
Se estima que alrededor del 26% de los individuos normotensos y 51% de los hipertensos son sal-sensitivos (35). Se ha encontrado que la sensibilidad a la sal entre sujetos normotensos predice la hipertensión futura (36, 37). Estudios de cohorte prospectivos a largo plazo también proporcionaron evidencia sustancial que sugiere que la sensibilidad a la sal puede ser un factor de riesgo independiente para las enfermedades cardiovasculares (revisado en 38). La sensibilidad a la sal involucra una mayor reabsorción del sodio en los túbulos proximales del riñón y una mayor tasa de filtración glomerular (TFG) en una dieta alta en sodio que en la resistencia a la sal. Se piensa que el aumento en la presión sanguínea compensa la retención alta de sodio y fluido al desencadenar una mayor excreción renal. Inversamente, la resistencia a la sal esta asociada con una excreción adecuada del exceso de sodio tal que el consumo de grandes cantidades de sodio no incrementa marcadamente la presión sanguínea (1).
Observaciones tempranas sugirieron que ciertos subgrupos de la población, incluyendo afroamericanos, individuos mayores (>45 años), y pacientes hipertensos, tendían a tener respuestas mayores en la presión sanguínea promedio a los cambios en la ingesta de sodio (38). No obstante, un meta-análisis reciente de ensayos controlados aleatorios encontró diferencias étnicas marginales en la respuesta de la presión arterial a la reducción de sodio (por al menos una semana) en comparación a datos previamente reportados (39). La investigación que examina las bases genéticas de la sensibilidad a la sal podría eventualmente conducir a una mejor y más confiable clasificación de los individuos según su sensibilidad a la sal. Sin embargo, en la actualidad, los análisis de variaciones comunes (conocidos como polimorfismos) en la secuencia de genes específicos involucrados en la retención del sodio por el riñón fallaron en mostrar resultados consistentes (revisado en 40). Un reciente meta-análisis de nueve estudios basados en la observación falló en mostrar asociaciones significantes entre polimorfismos de genes específicos en el sistema renina-angiotensina-aldosterona y la respuesta de la presión sanguínea a la sal (41). Otros polimorfismos en genes como aquellos que codifican para la quinasa de receptor acoplado a proteínas G tipo 4 (GRK4), los canales de sodio epiteliales (ENaC) y reguladores, o α-aducina pueden favorecer la retención del sodio por el riñón, predisponiendo por lo tanto a la sensibilidad a la presión sanguínea a la sal (revisado en 40). Finalmente, además de las predisposiciones genéticas, factores como la calidad de la dieta (p. ej., la dieta DASH) y el peso corporal probablemente influencian la sensibilidad de la presión sanguínea a la sal (38).
Ensayos clínicos de la presión sanguínea: De particular importancia son los resultados de ensayos multicéntricos a largo plazo que son los más relevantes para la práctica clínica y de salud pública, es decir, TONE (42), TOHP (43), y DASH (Enfoques Alimenticios para Detener la Hipertensión)-sodio. TONE demostró que la modesta reducción en la ingesta de sodio de alrededor de 1.0 g/día (~2.5 g/día de sal) resultó en un mejor control de la hipertensión en adultos mayores que inicialmente usaban medicamentos para la presión sanguínea (42). TOHP-Fase II (el segundo de dos ensayos de prevención de la hipertensión) demostró que un nivel similar de reducción del sodio significantemente redujo la presión sanguínea sistólica (pero no la diastólica) en 1.2 mm Hg en participantes con sobrepeso con hipertensión después de tres años y limitó la aparición de la hipertensión en un 18% después de cuatro años en comparación con el cuidado usual (sin intervención dietaría) (43, 44). Se encontró que la adherencia a la dieta DASH, la cual enfatiza frutas, vegetales, granos enteros, aves de corral, pescado, nueces, y productos lácteos bajos en grasa, sustancialmente disminuye la presión sanguínea sistólica/diastólica en 11.4/5.5 mm Hg en personas hipertensas y 3.5/2.1 mm Hg en personas normotensas en comparación a una dieta estadounidense típica (45). La dieta DASH también es marcadamente más alta en potasio y calcio, modestamente más alta en proteína, y más baja en grasas totales, grasas saturadas, colesterol, carnes rojas, azúcares, y bebidas con azúcar que la dieta estadounidense habitual. El ensayo DASH-sodio comparó la dieta DASH con una dieta típica estadounidense (control) en tres niveles de ingesta de sodio: niveles bajo (1.5 g/día, IA actual), intermedio (2.3 g/día; recomendado como el límite superior según las pautas dietéticas de los EE.UU.), y alto (3.2 g/día; la ingesta estadounidense habitual) (46). En cada nivel de ingesta de sodio, las presiones sistólicas y diastólicas fueron sistemáticamente menores en personas (pre)hipertensas (presión sanguínea >120/80 mm Hg) que consumían la dieta DASH en comparación a la dieta de control. La reducción de la ingesta de sodio también significantemente disminuyó la presión sanguínea en consumidores hipertensos de tanto la dieta DASH como de la dieta estadounidense habitual. Sin embargo, en los participantes prehipertensos, el efecto reductor de la presión sanguínea de la disminución de sodio fue solamente significativo en aquellos que consumían la dieta estadounidense habitual; la reducción de sodio falló en reducir la presión sanguínea en todos los participantes prehipertensos asignados a la dieta DASH, con la excepción de los afroamericanos. En comparación a la dieta de control alta en sal, la presión sanguínea promedio en participantes (pre)hipertensos en la dieta DASH baja en sodio disminuyó en 8.9/4.5 mm Hg. Los resultados de los ensayos DASH respaldan la idea de que la reducción de sodio en el contexto de un patrón dietario saludable ofrece un enfoque efectivo para la prevención y el tratamiento de la hipertensión (47).
Una mayoría de ensayos clínicos aleatorios han examinado el efecto de la reducción de sodio dietario en la presión sanguínea en las personas (pre)hipertensas en lugar de en las personas normotensas (presión sanguínea normal). Un reciente meta-análisis evaluó los resultados de la reducción modesta de sodio de 22 ensayos en 990 participantes con hipertensión (presión sanguínea ≥140/90 mm Hg) y 12 ensayos en 2,240 participantes sin hipertensión (presión sanguínea <140/90 mm Hg). Una reducción modesta de sodio de 1.8 g/día (equivalente a 4.4 g/día de sal; basada en excreciones urinarias de sodio de 24-horas) de al menos cuatro semanas disminuyó la presión sanguínea sistólica y diastólica en un promedio de 5.4/2.8 mm Hg en sujetos con hipertensión y 2.4/1.0 mm Hg en aquellos sin hipertensión (48). Otro análisis combinado de ocho estudios controlados aleatorios demostró una relación dosis-respuesta entre la reducción de sodio (de 1.8 a 3.2 g/día) y la presión sanguínea en sujetos con presión sanguínea por encima de 130/80, pero no hubo tal relación en los sujetos cuya presión sanguínea estaba por debajo de 130/80 (49).
Las Directrices Dietarías para los Americanos de 2015-2020 concurrieron con la Directriz de la Asociación Americana del Corazón (AHA)/Colegio Americano de Cardiología (ACC) del 2013 para recomendar que los individuos que se beneficiarían de la reducción de la presión sanguínea, particularmente adultos prehipertensos e hipertensos, deben seguir el patrón de alimentación DASH para disminuir las ingestas de sodio (50). Las recomendaciones incluyen ingestas de sodio de no más de 2.4 g/día, una reducción adicional de la ingesta de sodio de 1.5 g/día para un efecto reductor mayor de la presión sanguínea, o una reducción de la ingesta de sodio de por lo menos 1 g/día si las ingestas de sodio de 1.5 g/día o 2.4 g/día no pueden ser alcanzadas (50, 51).
Más información respecto a la dieta DASH se encuentra disponible en el Instituto Nacional del Corazón, Pulmón, y Sangre (NHLBI) del Instituto Nacional de la Salud (NIH).
Disfunción endotelial
Estudios tempranos en animales y humanos reportaron que la ingesta alta en sal estaba asociada con alteraciones patológicas en la estructura y función de grandes arterias elásticas, independientes de los cambios en la presión sanguínea (revisado en 52). La disfunción endotelial se considera un paso temprano en el desarrollo de la aterosclerosis. Las alteraciones en la estructura y función del endotelio vascular que recubre la superficie interna de todos los vasos sanguíneos están asociadas con la pérdida de la vasodilatación dependiente del endotelio mediada por el óxido nítrico (NO) normal. La disfunción endotelial resulta en la vasoconstricción generalizada y anormalidades de la coagulación. La medición de la dilatación mediada por flujo (DMF) de la arteria braquial es frecuentemente usada como un marcador funcional de la función endotelial; los valores de la DMF están inversamente correlacionados con el riesgo de eventos cardiovasculares futuros (53).
El vínculo entre la ingesta de sodio y las enfermedades cardiovasculares ha tradicionalmente involucrado a la hipertensión. Sin embargo, investigaciones recientes en individuos normotensos sal-resistentes han reportado que la carga de sodio dietario puede alterar la función endotelial independientemente de los cambios en la presión sanguínea (54, 55). Un estudio usando condiciones controladas de sodio dietario también demostró que una dieta alta en sodio (6.9 g/día por una semana) disminuyó la DMF de la arteria braquial en la misma medida en los participantes normotensos sal-sensitivos y sal-resistentes (56). Por otra parte, una ingesta dietaría de sodio de tres semanas de 3 g luego de una ingesta basal de 2.4 g/día de sodio no disminuyó la DMF en 36 adultos pre(hipertensos) sin tratar con valores de DMF basales significativamente menores que aquellos usualmente observados en sujetos normotensos saludables (57). Los marcadores circulantes de la función endotelial y la inflamación de bajo grado se mantuvieron sin cambios; sólo la excreción de sodio y la presión sanguínea sistólica incrementaron (57).
En los adultos normotensos saludables en los cuales la DMF disminuyó significativamente luego de una comida que contenía 1.5 g de sodio, la suplementación con potasio (1.5 g) puede limitar la reducción de la DMF inducida por el sodio alto en el estado postprandial (58, 59). En un estudio cruzado de 12 semanas en 25 sujetos con sobrepeso/obesos y normotensos, una dieta que contenía 2.3 g/día disminuyó la DMF después de dos días y por seis semanas, en comparación con una dieta que contenía 3.5 g/día (60). En otro ensayo aleatorio, cruzado, controlado con placebo, la restricción de sodio dietario (1.2 g/día vs 3.5 g/día) por cinco semanas significativamente disminuyó la presión sanguínea sistólica (pero no la diastólica) en 12 mm Hg e incrementó la DMF en un 68% en 17 sujetos (pre)hipertensos (edad promedio, 62 años) (61). Los mejoramientos en tanto la presión sanguínea como en la función endotelial sugieren que la restricción de sodio tiene un potencial fuerte para reducir el riesgo de ECV al preservar la vasculatura.
Morbilidad y mortalidad cardiovascular
Una alta ingesta dietaría de sodio es un factor de riesgo para enfermedades cardiovasculares. Un análisis combinado de 13 estudios de cohorte prospectivos en 177,025 participantes en seguimiento por 3 a 17 años encontró mayores riesgos de enfermedades cardiovasculares (+17%) y accidentes cerebrovasculares (+23%) con una diferencia promedio de 2 g de sodio (5 g de sal) entre el consumo diario más alto y el más bajo entre los estudios (62). Se encontró que las ingestas más altas de sodio estaban asociadas con un riesgo incrementado de un accidente cerebrovascular (+24%) — pero no con el riesgo de enfermedades cardiovasculares o enfermedad coronaria cardíaca — en un meta-análisis más reciente de 10 estudios de cohorte prospectivos y ensayos controlados aleatorios (63).
Un meta-análisis de estudios controlados aleatorios examinó el efecto de la reducción de sodio dietario en los eventos cardiovasculares y la mortalidad (64). Las intervenciones dietarías destinadas a reducir la ingesta de sodio fallaron en demostrar un efecto en la mortalidad por todas las causas en individuos hipertensos y no-hipertensos. Evidencia actual también falla en apoyar una disminución en la morbilidad y mortalidad cardiovascular en pacientes con hipertensión (64). De la misma manera, el reporte patrocinado por los Centros para el Control y la Prevención de Enfermedades (CDC) sobre Ingesta de Sodio en Poblaciones por el Instituto de Medicina de los Estados Unidos (IOM: rebautizado como Academia Nacional de Medicina) indicó que la evidencia era escasa y de baja calidad para evaluar si las ingestas de sodio por debajo de 2.3 g/día podrían disminuir el riesgo de enfermedad cardíaca, accidente cardiovascular, y mortalidad por todas las causas en la población general estadounidense (65). Además, el comité del IOM no encontró evidencia de beneficios pero algo de evidencia del daño potencial en la reducción de las ingestas de sodio a 1.5 g/día-2.3 g/día en individuos con diabetes mellitus, enfermedad renal, o enfermedad cardiovascular (65). Sin embargo, en una reciente revisión de la literatura, la Agencia para la Investigación y la Calidad del Cuidado de la Salud (AHRQ) identificó evidencia de poca fuerza para sugerir que la disminución de sodio podría reducir el riesgo de una medida compuesta de resultados cardiovasculares (siete ensayos) y el riesgo de morbilidad y mortalidad cardiovascular combinadas (ocho ensayos) (66).
Cáncer gástrico
En el reporte basado en evidencia, Alimentos, Nutrición, Actividad Física, y la Prevención del Cáncer (2007), el Fondo Mundial para la Investigación del Cáncer/Instituto Americano para la Investigación del Cáncer concluyó que la sal era una causa probable del cáncer de estómago (67). Un reciente meta-análisis de siete estudios de cohorte prospectivos en casi 270,000 participantes demostró un riesgo 68% mayor de cáncer gástrico con el nivel de ingesta de sal más alto versus el más bajo (68). Hallazgos de estudios basados en la observación, conducidos principalmente en países asiáticos, también sugirieron un riesgo incrementado del riesgo de cáncer gástrico con ingestas altas de alimentos salados, encurtidos, y productos de carne procesados (68-70). Las ingestas bajas de frutas y vegetales, los cuales protegen contra el cáncer gástrico, en poblaciones con ingestas altas de alimentos salados podrían también contribuir a incrementar el riesgo de cáncer gástrico (69, 71).
Estudios en animales sugieren que altas concentraciones de sal pueden dañar las células que recubren el estómago, potencialmente incrementando el riesgo de infección bacteriana por Helicobacter pylori (72, 73) y el daño genético que promueve el cáncer (74). La colonización por H. pylori es un factor de riesgo reconocido para el cáncer gástrico. Estudios de caso y control que examinaron la interacción potencial entre la ingesta de sal y la infección por H. pylori en el riesgo de cáncer gástrico han proporcionado resultados mixtos (75-78). Aunque existe poca evidencia de que la sal en si sea un carcinógeno, ingestas altas de alimentos salados pueden incrementar el riesgo de cáncer gástrico en individuos infectados con H. pylori o expuestos a carcinógenos gástricos (67). Alimentos salados, como la carne procesada, la carne curada, y el pescado salado, contienen altos niveles de compuestos nitrosados que pueden contribuir al incremento del riesgo de cáncer gástrico (67).
Osteoporosis
La osteoporosis es un trastorno esquelético multifactorial en el que se compromete la resistencia del hueso, resultando en un riesgo incrementado de fractura. La nutrición es uno de los muchos factores que contribuyen al desarrollo y progresión de la osteoporosis. El sodio dietario es un determinante principal de la perdida urinaria de calcio (79). La ingesta alta en sodio resulta en una pérdida incrementada de calcio en la orina, posiblemente debido a la competencia entre el sodio y el calcio para la reabsorción en los riñones o al efecto del sodio en la secreción de la hormona paratiroidea (PTH) (véase el artículo sobre Calcio). Se ha encontrado que por cada incremento de 1-g en sodio (2.5 g de sal) excretado por los riñones, este arrastra cerca de 26.3 mg de calcio a la orina (79). Un estudio conducido en mujeres adolescentes reportó que una dieta alta en sal tuvo un efector mayor en la excreción urinaria de sodio y calcio en mujeres blancas que en mujeres de raza negra, sugiriendo diferencias entre grupos étnicos (80). En mujeres adultas, cada gramo adicional de sodio consumido por día se proyecta produce una tasa adicional de pérdida ósea del 1% por año si toda la pérdida de calcio proviene del esqueleto.
Un cierto número de estudios de intervención y transversales han sugerido que las ingestas altas de sodio son perjudiciales para la salud ósea, especialmente en mujeres mayores (81). En particular, la ingesta alta de sodio en conjunto con la ingesta baja en calcio puede especialmente ser perjudicial para la salud ósea (82-84). Un estudio longitudinal de dos años en mujeres postmenopáusicas encontró que la excreción urinaria incrementada de sodio (un indicador del incremento en la ingesta de sodio) está asociada con una disminución de la densidad mineral ósea (DMO) en la cadera (85). El análisis de regresión lineal estimó que la DMO podría ser mantenida reduciendo la ingesta de sodio a un nivel de 2.3 g/día e incrementando la ingesta de calcio a 1.2 g/día. Un segundo estudio longitudinal en mujeres postmenopáusicas encontró que la ingesta de sodio habitual de aproximadamente 3 g/día no era perjudicial para la DMO sobre tres años de seguimiento (86). Notablemente, la ingesta de calcio promedio en esta población del estudio fue de 1.3 a 1.5 g/día — ligeramente por encima de la IDR para el calcio en mujeres mayores a 50 años. Otro estudio en 40 mujeres postmenopáusicas encontró que la adherencia a una dieta baja en sodio (2 g/día) por seis meses estaba asociada con reducciones significantes en la excreción de sodio, excreción de calcio, y en tripéptido aminoterminal del colágeno tipo I (un biomarcador de la reabsorción ósea). Sin embargo, estas asociaciones fueron solamente observadas en mujeres con excreciones urinarias elevadas de sodio basales (87). Finalmente, en un estudio aleatorio, controlado con placebo en 60 mujeres postmenopáusicas, la suplementación con citrato de potasio previno un aumento en la excreción de calcio inducida por el consumo de una dieta alta en sodio (≥5 g/día de sodio) por cuatro semanas (88).
Cálculos renales
La mayoría de los cálculos renales están compuestos de oxalato de calcio o fosfato de calcio. Los sujetos con un nivel de calcio en la orina anormalmente alto (hipercalciuria) están en un riesgo mayor de desarrollar cálculos renales (un proceso llamado nefrolitiasis) (89). Un estudio de cohorte prospectivo de gran tamaño que siguió a más de 90,000 mujeres por un periodo de 12 años encontró que las mujeres con una ingesta de sodio que promediaba 4.9 g/día (12.6 g/día de sal) tenían un riesgo un 30% más alto de desarrollar cálculos renales sintomáticos que las mujeres que promediaron una ingesta de sodio de 1.5 g/día (3.8 g/día de sal) (90). Debido a que la excreción urinaria de calcio es incrementada por las altas ingestas de sodio (79), la restricción de sodio dietario podría reducir el riesgo de la formación de cálculos, especialmente en pacientes con un historial de cálculos renales, al limitar la excreción de calcio (91). Un estudio de intervención aleatorio de cinco años que inscribió a 120 hombres con hipercalciuria idiopática (edad promedio, 45 años) reportó que aquellos asignados a una dieta normal a alta en calcio (1.2 g/día) y baja en sodio (1.2 g/día) tuvieron una reducción del 49% del riesgo de la recurrencia de cálculos renales en comparación a aquellos con una dieta baja en calcio (~0.4 g/día) (92).
Ingesta para la Disminución del Riesgo de Enfermedad Crónica
En el 2004, la Junta de Nutrición y Alimentos (JNA) de la Academia Nacional de Medicina de los Estados Unidos (anteriormente, el Instituto de Medicina [IOM]) estableció un nivel máximo de ingesta tolerable (NM) de 2.3 g/día de sodio (5.8 g/día de sal) para los adultos, basado en los efectos adversos de las ingestas elevadas de sodio en la presión sanguínea, un factor de riesgo importante para las enfermedades cardiovasculares y renales (93). El reporte del 2015-2020 de las Pautas Dietarías para los estadounidenses también reconoce que el consumo en exceso de sodio posee riesgos potenciales a la salud y enfatiza la reducción de la ingesta de sodio en el contexto de un patrón dietario saludable, siguiendo el NM establecido por el panel del IOM (50).
En una actualización en el 2019 de las Ingestas Dietéticas de Referencia para el sodio, la Academia Nacional de Medicina hizo uso de un nuevo modelo expandido de Ingestas Dietéticas de Referencia (20). Consecuentemente, sólo evidencia de un riesgo toxicológico asociado con el consumo excesivo de sodio fue considerada para establecer un NM para el sodio (20), mientras que anteriormente el NM para el sodio se basaba en la evidencia de cualquier tipo de efectos adversos (93). Además, la evidencia de la relación entre los niveles de ingesta de sodio, presión sanguínea, y enfermedad cardiovascular — considerada hacia el establecimiento de un NM en el reporte anterior — ahora ha sido revisada para establecer una nueva categoría de Ingestas Dietéticas de Referencia, a saber, la Ingesta para la Disminución de Riesgo de Enfermedad Crónica (CDRR, por sus siglas en inglés) de sodio (20).
Utilizando el modelo expandido de Ingestas Dietéticas de Referencia, la Academia Nacional de Medicina no encontró evidencia suficiente de efectos toxicológicos adversos para establecer un NM para el sodio (20).
En contraste, evidencia sustancial de ensayos mostrando una disminución en los riesgos de hipertensión y enfermedad cardiovascular con la disminución de las ingestas de sodio fue usada para establecer una CDRR para el sodio en adultos aparentemente sanos de 2,300 mg/día (20). La CDRR para adultos sanos significa que se espera que la reducción de las ingestas de sodio habituales a por lo menos 2,300 mg/día desde niveles más altos reduzca el riesgo de enfermedades crónicas.
Los valores de la CDRR para otras edades/etapas de la vida se han extrapolado del valor de la CDRR establecida para adultos usando los requerimientos de energía estimados; estas recomendaciones basadas en la CDRR para cada edad/etapa de la vida se muestran en la Tabla 5.
Es de notar que, en su reporte del 2013 sobre la Ingesta de Sodio en las Poblaciones, el comité de la JNA consideró que las recomendaciones para subgrupos de población, incluyendo aquellos que son más sensitivos a los efectos del sodio en la presión sanguínea, como personas mayores (≥51 años), afroamericanos, e individuos con hipertensión, diabetes mellitus, o enfermedad renal crónica, deben ser similares a aquellas para la población estadounidense en general (65). El comité del IOM no encontró evidencia de apoyo para recomendar que estos subgrupos reduzcan su ingesta de sodio a 1.5 g/día o menos (65).
Por el contrario, la Guía sobre el Manejo del Estilo de Vida para Reducir el Riesgo Cardiovascular de la Asociación Americana del Corazón (AHA)/Colegio Americano de Cardiología (ACC) del 2013 recomienda que los adultos con (pre)hipertensión no consuman más de 2.4 g/día de sodio, reduzcan adicionalmente la ingesta de sodio a 1.5 g/día para un mayor efecto reductor de la presión sanguínea, o reduzcan su ingesta diaria en al menos 1 g si las ingestas de sodio de 1.5 g/día o 2.4 g/día no pueden ser alcanzadas (51).
Finalmente, el reporte del 2010 del IOM sobre las Estrategias para Reducir la Ingesta de Sodio en los Estados Unidos sugirió que la Administración de Alimentos y Drogas del los EE.UU. (FDA) revisitara el estatus Generalmente Reconocido Como Seguro (GRAS) de la sal agregada a los alimentos procesados, comida de restaurante, y aditivos alimentarios, a fin de reducir el contenido de sal en el suministro de alimentos y asistir en alcanzar la ingestas de sodio consistentes con las Pautas Dietarías de los Estados Unidos y las recomendaciones del IOM (21). Estas recomendaciones están actualmente siendo revisadas por la FDA (22).
Interacción con drogas/fármacos
La administración de bicarbonato de sodio de manera oral puede reducir la eficacia del antibiótico cefpodoxima y el fármaco antidiabético clorpropamida al limitar la absorción del fármaco o aumentar la excreción urinaria del fármaco. La administración intravenosa de bicarbonato de sodio puede también reducir los efectos de la aspirina y el descongestionante nasal pseudoefedrina. La ingesta excesiva de bicarbonato de sodio puede incrementar el riesgo de hipopotasemia (concentración anormalmente baja de potasio en la sangre) en pacientes que toman fármacos que disminuyen el potasio como diuréticos (p. ej., hidroclorotiazida, furosemida o bumetanida), el agente anti-gota colchicina, antiácidos que contienen calcio o magnesio (94).
Recomendación del Instituto Linus Pauling
Existe evidencia fuerte y consistente de que las dietas relativamente bajas en sodio (2.3 g/día o menos) y altas en potasio se asocian con un riesgo disminuido de presión sanguínea alta y los riesgos asociados con enfermedades cardiovasculares y renales. Además, el ensayo DASH demostró que una dieta que enfatiza frutas, verduras, granos enteros, nueces y productos lácteos bajos en grasa disminuyó sustancialmente la presión sanguínea, un efecto que aumentó al reducir la ingesta de sal a 2.3 g/día de sodio (5.8 g/día de sal). El Instituto Linus Pauling recomienda una dieta rica en frutas y verduras (al menos 9 porciones/día) y que limite los alimentos procesados altos en sal.
Adultos mayores (>50 años)
Debido a que los efectos de la sal sobre la presión sanguínea aumentan con la edad, la reducción de sodio en el contexto de un patrón dietario saludable puede beneficiar especialmente a los adultos mayores, los cuales están en un riesgo incrementado de presión sanguínea alta, enfermedades cardiovasculares, y enfermedad renal.
Autores y Críticos
Originalmente escrito en 2001 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Febrero de 2004 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Noviembre de 2008 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Mayo de 2016 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Diciembre de 2016 por:
Harry G. Preuss, M.D., M.A.C.N., C.N.S.
Profesor de Bioquímica, Fisiología, Medicina, y Patología
Centro Médico de la Universidad de Georgetown
Traducido al Español en 2018 por:
Silvia Vazquez Lima
Instituto Linus Pauling
Universidad Estatal de Oregon
Última actualización en 4/22/19 Derechos de autoría 2001-2024 Instituto Linus Pauling
References
1. Preuss HG, Clouatre DL. Sodium, chloride, and potassium. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. John Wiley & Sons; 2012:475-492.
2. Clausen T. Quantification of Na+,K+ pumps and their transport rate in skeletal muscle: functional significance. J Gen Physiol. 2013;142(4):327-345. (PubMed)
3. Larsen BR, Stoica A, MacAulay N. Managing brain extracellular K(+) during neuronal activity: the physiological role of the Na(+)/K(+)-ATPase subunit isoforms. Front Physiol. 2016;7:141. (PubMed)
4. Shattock MJ, Ottolia M, Bers DM, et al. Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J Physiol. 2015;593(6):1361-1382. (PubMed)
5. Sullivan S, Alpers D, Klein S. Nutritional physiology of the alimentary tract. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Lippincott Williams & Wilkins; 2014:540-573.
6. Bailey JL, Sands JM, Franch HA. Water, electrolytes, and acid-base metabolism. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Lippincott Williams & Wilkins; 2014:102-132.
7. Kopple JD. Nutrition, diet, and the kidney. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Lippincott Williams & Wilkins; 2014:1330-1371.
8. Kortenoeven ML, Pedersen NB, Rosenbaek LL, Fenton RA. Vasopressin regulation of sodium transport in the distal nephron and collecting duct. Am J Physiol Renal Physiol. 2015;309(4):F280-299. (PubMed)
9. Rayner B, Ramesar R. The importance of G protein-coupled receptor kinase 4 (GRK4) in pathogenesis of salt sensitivity, salt sensitive hypertension and response to antihypertensive treatment. Int J Mol Sci. 2015;16(3):5741-5749. (PubMed)
10. Mohan S, Gu S, Parikh A, Radhakrishnan J. Prevalence of hyponatremia and association with mortality: results from NHANES. Am J Med. 2013;126(12):1127-1137. (PubMed)
11. Hoorn EJ, Lindemans J, Zietse R. Development of severe hyponatraemia in hospitalized patients: treatment-related risk factors and inadequate management. Nephrol Dial Transplant. 2006;21(1):70-76. (PubMed)
12. Urso C, Brucculeri S, Caimi G. Physiopathological, epidemiological, clinical and therapeutic aspects of exercise-associated hyponatremia. J Clin Med. 2014;3(4):1258-1275. (PubMed)
13. Giuliani C, Peri A. Effects of hyponatremia on the brain. J Clin Med. 2014;3(4):1163-1177. (PubMed)
14. Holm JP, Amar AO, Hyldstrup L, Jensen JE. Hyponatremia, a risk factor for osteoporosis and fractures in women. Osteoporos Int. 2016;27(3):989-1001. (PubMed)
15. Zaino CJ, Maheshwari AV, Goldfarb DS. Impact of mild chronic hyponatremia on falls, fractures, osteoporosis, and death. Am J Orthop (Belle Mead NJ). 2013;42(11):522-527. (PubMed)
16. Wannamethee SG, Shaper AG, Lennon L, Papacosta O, Whincup P. Mild hyponatremia, hypernatremia and incident cardiovascular disease and mortality in older men: A population-based cohort study. Nutr Metab Cardiovasc Dis. 2016;26(1):12-19. (PubMed)
17. Corona G, Giuliani C, Parenti G, et al. Moderate hyponatremia is associated with increased risk of mortality: evidence from a meta-analysis. PLoS One. 2013;8(12):e80451. (PubMed)
18. Corona G, Giuliani C, Verbalis JG, Forti G, Maggi M, Peri A. Correction: hyponatremia improvement is associated with a reduced risk of mortality: evidence from a meta-analysis. PLoS One. 2016;11(3):e0152846. (PubMed)
19. Adrogue HJ, Madias NE. Hyponatremia. N Engl J Med. 2000;342(21):1581-1589. (PubMed)
20. Food and Nutrition Board, National Academy of Medicine. Dietary Reference Intakes for Sodium and Potassium -- uncorrected proofs. Washington, D.C.: The National Academies Press; 2019. (The National Academies Press)
21. Institute of Medicine Committee on Strategies to Reduce Sodium Intake. Strategies to reduce sodium intake in the United States. Washington, D.C. 2010.
22. US Food and Drug Administration. Lowering salt in your diet. March 29, 2016. http://www.fda.gov/ForConsumers/ConsumerUpdates/ucm181577.htm. Accessed 5/6/16.
23. Agarwal S, Fulgoni VL, 3rd, Spence L, Samuel P. Sodium intake status in United States and potential reduction modeling: an NHANES 2007-2010 analysis. Food Sci Nutr. 2015;3(6):577-585. (PubMed)
24. US Food and Drug Administration. Sodium in your diet: use the Nutrition Facts label and reduce your intake. May 22, 2016. Available at: http://www.fda.gov/Food/ResourcesForYou/Consumers/ucm315393.htm. Accessed 5/23/16.
25. Minerals. Drug Facts and Comparisons. St. Louis: Facts and Comparisons; 2000:27-51.
26. Reynolds RM, Padfield PL, Seckl JR. Disorders of sodium balance. BMJ. 2006;332(7543):702-705. (PubMed)
27. American Heart Association. Understanding blood pressure readings. March 23, 2016. Available at: http://www.heart.org/HEARTORG/Conditions/HighBloodPressure/AboutHighBloodPressure/Understanding-Blood-Pressure-Readings_UCM_301764_Article.jsp#.VyzuIxJJmM8. Accessed 5/6/16.
28. Centers for Disease Control and Prevention. High blood pressure facts. February 19, 2015. Available at: http://www.cdc.gov/bloodpressure/facts.htm. Accessed 5/6/16.
29. Chrysant GS. High salt intake and cardiovascular disease: is there a connection? Nutrition. 2000;16(7-8):662-664. (PubMed)
30. Intersalt Cooperative Research Group. Intersalt: an international study of electrolyte excretion and blood pressure. Results for 24 hour urinary sodium and potassium excretion. BMJ. 1988;297(6644):319-328. (PubMed)
31. Elliott P, Stamler J, Nichols R, et al. Intersalt revisited: further analyses of 24 hour sodium excretion and blood pressure within and across populations. Intersalt Cooperative Research Group. BMJ. 1996;312(7041):1249-1253. (PubMed)
32. Luft FC, Weinberger MH. Heterogeneous responses to changes in dietary salt intake: the salt-sensitivity paradigm. Am J Clin Nutr. 1997;65(2 Suppl):612S-617S. (PubMed)
33. Weinberger MH. Salt sensitivity of blood pressure in humans. Hypertension. 1996;27(3 Pt 2):481-490. (PubMed)
34. Schmidlin O, Sebastian AF, Morris RC, Jr. What initiates the pressor effect of salt in salt-sensitive humans? Observations in normotensive blacks. Hypertension. 2007;49(5):1032-1039. (PubMed)
35. He FJ, MacGregor GA. Reducing population salt intake worldwide: from evidence to implementation. Prog Cardiovasc Dis. 2010;52(5):363-382. (PubMed)
36. Barba G, Galletti F, Cappuccio FP, et al. Incidence of hypertension in individuals with different blood pressure salt-sensitivity: results of a 15-year follow-up study. J Hypertens. 2007;25(7):1465-1471. (PubMed)
37. Mu J, Zheng S, Lian Q, Liu F, Liu Z. Evolution of blood pressure from adolescents to youth in salt sensitivies: a 18-year follow-up study in Hanzhong children cohort. Nutr J. 2012;11:70. (PubMed)
38. Franco V, Oparil S. Salt sensitivity, a determinant of blood pressure, cardiovascular disease and survival. J Am Coll Nutr. 2006;25(3 Suppl):247S-255S. (PubMed)
39. Graudal N, Jurgens G. The blood pressure sensitivity to changes in sodium intake is similar in Asians, Blacks, and Whites. An analysis of 92 randomized controlled trials. Front Physiol. 2015;6:157. (PubMed)
40. Armando I, Villar VA, Jose PA. Genomics and pharmacogenomics of salt-sensitive hypertension. Curr Hypertens Rev. 2015;11(1):49-56. (PubMed)
41. Sun J, Zhao M, Miao S, Xi B. Polymorphisms of three genes (ACE, AGT and CYP11B2) in the renin-angiotensin-aldosterone system are not associated with blood pressure salt sensitivity: A systematic meta-analysis. Blood Press. 2016;25(2):117-122. (PubMed)
42. Whelton PK, Appel LJ, Espeland MA, et al. Sodium reduction and weight loss in the treatment of hypertension in older persons: a randomized controlled trial of nonpharmacologic interventions in the elderly (TONE). TONE Collaborative Research Group. JAMA. 1998;279(11):839-846. (PubMed)
43. The Trials of Hypertension Prevention Collaborative Research Group. Effects of weight loss and sodium reduction intervention on blood pressure and hypertension incidence in overweight people with high-normal blood pressure. The Trials of Hypertension Prevention, phase II. Arch Intern Med. 1997;157(6):657-667. (PubMed)
44. Kumanyika SK, Cook NR, Cutler JA, et al. Sodium reduction for hypertension prevention in overweight adults: further results from the Trials of Hypertension Prevention Phase II. J Hum Hypertens. 2005;19(1):33-45. (PubMed)
45. Appel LJ, Moore TJ, Obarzanek E, et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N Engl J Med. 1997;336(16):1117-1124. (PubMed)
46. Sacks FM, Svetkey LP, Vollmer WM, et al. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N Engl J Med. 2001;344(1):3-10. (PubMed)
47. Greenland P. Beating high blood pressure with low-sodium DASH. N Engl J Med. 2001;344(1):53-55. (PubMed)
48. He FJ, Li J, Macgregor GA. Effect of longer term modest salt reduction on blood pressure: Cochrane systematic review and meta-analysis of randomised trials. BMJ. 2013;346:f1325. (PubMed)
49. Graudal N, Hubeck-Graudal T, Jurgens G, McCarron DA. The significance of duration and amount of sodium reduction intervention in normotensive and hypertensive individuals: a meta-analysis. Adv Nutr. 2015;6(2):169-177. (PubMed)
50. US Department of Health and Human Services and US Department of Agriculture. 2015-2020 Dietary Guidelines for Americans. Available at: http://health.gov/dietaryguidelines/2015/guidelines/.
51. Eckel RH, Jakicic JM, Ard JD, et al. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 Suppl 2):S76-99. (PubMed)
52. Safar ME, Thuilliez C, Richard V, Benetos A. Pressure-independent contribution of sodium to large artery structure and function in hypertension. Cardiovasc Res. 2000;46(2):269-276. (PubMed)
53. Ras RT, Streppel MT, Draijer R, Zock PL. Flow-mediated dilation and cardiovascular risk prediction: a systematic review with meta-analysis. Int J Cardiol. 2013;168(1):344-351. (PubMed)
54. DuPont JJ, Greaney JL, Wenner MM, et al. High dietary sodium intake impairs endothelium-dependent dilation in healthy salt-resistant humans. J Hypertens. 2013;31(3):530-536. (PubMed)
55. Greaney JL, DuPont JJ, Lennon-Edwards SL, Sanders PW, Edwards DG, Farquhar WB. Dietary sodium loading impairs microvascular function independent of blood pressure in humans: role of oxidative stress. J Physiol. 2012;590(21):5519-5528. (PubMed)
56. Matthews EL, Brian MS, Ramick MG, Lennon-Edwards S, Edwards DG, Farquhar WB. High dietary sodium reduces brachial artery flow-mediated dilation in humans with salt-sensitive and salt-resistant blood pressure. J Appl Physiol (1985). 2015;118(12):1510-1515. (PubMed)
57. Gijsbers L, Dower JI, Schalkwijk CG, et al. Effects of sodium and potassium supplementation on endothelial function: a fully controlled dietary intervention study. Br J Nutr. 2015;114(9):1419-1426. (PubMed)
58. Blanch N, Clifton PM, Petersen KS, Keogh JB. Effect of sodium and potassium supplementation on vascular and endothelial function: a randomized controlled trial. Am J Clin Nutr. 2015;101(5):939-946. (PubMed)
59. Dickinson KM, Clifton PM, Keogh JB. Endothelial function is impaired after a high-salt meal in healthy subjects. Am J Clin Nutr. 2011;93(3):500-505. (PubMed)
60. Dickinson KM, Clifton PM, Keogh JB. A reduction of 3 g/day from a usual 9 g/day salt diet improves endothelial function and decreases endothelin-1 in a randomised cross_over study in normotensive overweight and obese subjects. Atherosclerosis. 2014;233(1):32-38. (PubMed)
61. Jablonski KL, Racine ML, Geolfos CJ, et al. Dietary sodium restriction reverses vascular endothelial dysfunction in middle-aged/older adults with moderately elevated systolic blood pressure. J Am Coll Cardiol. 2013;61(3):335-343. (PubMed)
62. Strazzullo P, D'Elia L, Kandala NB, Cappuccio FP. Salt intake, stroke, and cardiovascular disease: meta-analysis of prospective studies. BMJ. 2009;339:b4567. (PubMed)
63. Aburto NJ, Ziolkovska A, Hooper L, Elliott P, Cappuccio FP, Meerpohl JJ. Effect of lower sodium intake on health: systematic review and meta-analyses. BMJ. 2013;346:f1326. (PubMed)
64. Adler AJ, Taylor F, Martin N, Gottlieb S, Taylor RS, Ebrahim S. Reduced dietary salt for the prevention of cardiovascular disease. Cochrane Database Syst Rev. 2014;12:CD009217. (PubMed)
65. US Institute of Medicine. Sodium intake in populations: Assessment of evidence. Washington, D.C.; 2013. (The National Academies Press)
66. Newberry SJ, Chung M, Anderson CAM, et al. AHRQ Comparative Effectiveness Reviews. Sodium and potassium intake: effects on chronic disease outcomes and risks. Rockville (MD): Agency for Healthcare Research and Quality (US); 2018. (PubMed)
67. World Cancer Research Fund/American Institute for Cancer Research. Food, Nutrition, Physical Activity, and the Prevention of Cancer: a Global Perspective. Washington, D.C. 2007.
68. D'Elia L, Rossi G, Ippolito R, Cappuccio FP, Strazzullo P. Habitual salt intake and risk of gastric cancer: a meta-analysis of prospective studies. Clin Nutr. 2012;31(4):489-498. (PubMed)
69. Fang X, Wei J, He X, et al. Landscape of dietary factors associated with risk of gastric cancer: A systematic review and dose-response meta-analysis of prospective cohort studies. Eur J Cancer. 2015;51(18):2820-2832. (PubMed)
70. Tsugane S. Salt, salted food intake, and risk of gastric cancer: epidemiologic evidence. Cancer Sci. 2005;96(1):1-6. (PubMed)
71. Liu C, Russell RM. Nutrition and gastric cancer risk: an update. Nutr Rev. 2008;66(5):237-249. (PubMed)
72. Bergin IL, Sheppard BJ, Fox JG. Helicobacter pylori infection and high dietary salt independently induce atrophic gastritis and intestinal metaplasia in commercially available outbred Mongolian gerbils. Dig Dis Sci. 2003;48(3):475-485. (PubMed)
73. Gaddy JA, Radin JN, Loh JT, et al. High dietary salt intake exacerbates Helicobacter pylori-induced gastric carcinogenesis. Infect Immun. 2013;81(6):2258-2267. (PubMed)
74. Kato S, Tsukamoto T, Mizoshita T, et al. High salt diets dose-dependently promote gastric chemical carcinogenesis in Helicobacter pylori-infected Mongolian gerbils associated with a shift in mucin production from glandular to surface mucous cells. Int J Cancer. 2006;119(7):1558-1566. (PubMed)
75. Fontham ET, Ruiz B, Perez A, Hunter F, Correa P. Determinants of Helicobacter pylori infection and chronic gastritis. Am J Gastroenterol. 1995;90(7):1094-1101. (PubMed)
76. Machida-Montani A, Sasazuki S, Inoue M, et al. Association of Helicobacter pylori infection and environmental factors in non-cardia gastric cancer in Japan. Gastric Cancer. 2004;7(1):46-53. (PubMed)
77. Peleteiro B, Lopes C, Figueiredo C, Lunet N. Salt intake and gastric cancer risk according to Helicobacter pylori infection, smoking, tumour site and histological type. Br J Cancer. 2011;104(1):198-207. (PubMed)
78. Zhong C, Li KN, Bi JW, Wang BC. Sodium intake, salt taste and gastric cancer risk according to Helicobacter pylori infection, smoking, histological type and tumor site in China. Asian Pac J Cancer Prev. 2012;13(6):2481-2484. (PubMed)
79. Weaver CM. Calcium. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. John Wiley & Sons; 2012:434-446.
80. Wigertz K, Palacios C, Jackman LA, et al. Racial differences in calcium retention in response to dietary salt in adolescent girls. Am J Clin Nutr. 2005;81(4):845-850. (PubMed)
81. Frassetto LA, Morris RC, Jr., Sellmeyer DE, Sebastian A. Adverse effects of sodium chloride on bone in the aging human population resulting from habitual consumption of typical American diets. J Nutr. 2008;138(2):419S-422S. (PubMed)
82. Bedford JL, Barr SI. Higher urinary sodium, a proxy for intake, is associated with increased calcium excretion and lower hip bone density in healthy young women with lower calcium intakes. Nutrients. 2011;3(11):951-961. (PubMed)
83. Heaney RP. Role of dietary sodium in osteoporosis. J Am Coll Nutr. 2006;25(3 Suppl):271S-276S. (PubMed)
84. Park SM, Jee J, Joung JY, et al. High dietary sodium intake assessed by 24-hour urine specimen increase urinary calcium excretion and bone resorption marker. J Bone Metab. 2014;21(3):189-194. (PubMed)
85. Devine A, Criddle RA, Dick IM, Kerr DA, Prince RL. A longitudinal study of the effect of sodium and calcium intakes on regional bone density in postmenopausal women. Am J Clin Nutr. 1995;62(4):740-745. (PubMed)
86. Ilich JZ, Brownbill RA, Coster DC. Higher habitual sodium intake is not detrimental for bones in older women with adequate calcium intake. Eur J Appl Physiol. 2010;109(4):745-755. (PubMed)
87. Carbone LD, Barrow KD, Bush AJ, et al. Effects of a low sodium diet on bone metabolism. J Bone Miner Metab. 2005;23(6):506-513. (PubMed)
88. Sellmeyer DE, Schloetter M, Sebastian A. Potassium citrate prevents increased urine calcium excretion and bone resorption induced by a high sodium chloride diet. J Clin Endocrinol Metab. 2002;87(5):2008-2012. (PubMed)
89. Lerolle N, Lantz B, Paillard F, et al. Risk factors for nephrolithiasis in patients with familial idiopathic hypercalciuria. Am J Med. 2002;113(2):99-103. (PubMed)
90. Curhan GC, Willett WC, Speizer FE, Spiegelman D, Stampfer MJ. Comparison of dietary calcium with supplemental calcium and other nutrients as factors affecting the risk for kidney stones in women. Ann Intern Med. 1997;126(7):497-504. (PubMed)
91. Nouvenne A, Meschi T, Prati B, et al. Effects of a low-salt diet on idiopathic hypercalciuria in calcium-oxalate stone formers: a 3-mo randomized controlled trial. Am J Clin Nutr. 2010;91(3):565-570. (PubMed)
92. Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med. 2002;346(2):77-84. (PubMed)
93. Food and Nutrition Board, Institute of Medicine. Sodium and Chloride. Dietary Reference Intakes for Water, Potassium, Sodium, Chloride, and Sulfate. Washington, D.C.: National Academy Press; 2004:247-392. (The National Academies Press)
94. Natural Medicines. Sodium bicarbonate: interactions with drugs; 2016. Available at: https://naturalmedicines.therapeuticresearch.com/.
Yodo
Contenido
Resumen
- El yodo es un componente clave de las hormonas tiroideas, las cuales son requeridas a lo largo de la vida para el crecimiento, desarrollo neurológico, y el metabolismo normal. (Más información)
- La ingesta insuficiente de yodo perjudica la producción de las hormonas tiroideas, dando como resultado una condición llamada hipotiroidismo. La deficiencia de yodo resulta en un rango de desórdenes adversos a la salud con grados variables de severidad, desde un agrandamiento de la glándula tiroides (bocio) a un retraso físico y mental severo conocido como cretinismo. (Más información)
- El hipotiroidismo inducido por la deficiencia de yodo tiene efectos adversos en todas las etapas del desarrollo pero es más perjudicial para el cerebro en desarrollo. La deficiencia de yodo materna durante el embarazo puede resultar en hipotiroidismo fetal y materno, como también en aborto, nacimiento prematuro, y alteraciones neurológicas en el recién nacido. (Más información)
- Incluso en áreas con programas de yodación voluntarios/mandatorios y en países repletos de yodo, las mujeres embarazadas, madres lactando, e infantes jóvenes están entre los más vulnerables a la deficiencia de yodo debido a sus requerimientos especiales durante estas etapas de la vida. (Mas información)
- La ingesta diaria recomendada (IDR) para el yodo es de 150 microgramos (μg)/día en adultos, 220 (μg)/día en mujeres embarazadas, y 290 (μg)/día en mujeres en período de lactancia. Durante el embarazo y lactancia, el feto e infante dependen totalmente de la ingesta de yodo materna para la síntesis de la hormona tiroidea. (Más información)
- La acumulación en la tiroides de yodo radioactivo (131I) incrementa el riesgo de desarrollar cáncer de tiroides, especialmente en niños. En caso de emergencias por radiación, medidas preventivas actuales incluyen la distribución de dosis farmacológicas de yoduro de potasio que reducirían el riesgo de la absorción de 131I por la glándula tiroides. (Más información)
- Los mariscos son una fuente excelente de yodo dietario. Los productos lácteos, granos, huevos, y aves de corral contribuyen sustancialmente a las ingestas de yodo dietario en los EE.UU. (Más información)
- Más de 120 países alrededor del mundo han introducido programas de fortificación de la sal con yodo a fin de corregir la deficiencia de yodo en poblaciones. (Más información)
- En poblaciones deficientes de yodo, un incremento rápido de la ingesta de yodo puede prevenir hipertiroidismo inducido por el yodo. El riesgo de hipertiroidismo inducido por el yodo es especialmente alto en personas mayores con bocio multinodular. (Más información)
- En adultos suficientes de yodo, la ingesta a largo plazo por arriba del nivel máximo de ingesta tolerable (NM) de 1,100 μg/día podría incrementar el riesgo de trastornos de la tiroides, incluyendo bocio inducido por el yodo e hipotiroidismo. (Más información)
El yodo (I), es un elemento traza no metálico requerido por los humanos para la síntesis de hormonas tiroideas. La deficiencia de yodo es un problema de salud importante en gran parte del mundo. La mayoría del yodo del planeta tierra se encuentra en los océanos, y el contenido de yodo en el suelo varía según la región. Mientras más antigua y más expuesta sea la superficie del suelo, más aumenta la probabilidad de que el yodo haya sido lixiviado por la erosión. Las regiones montañosas como los Himalayas, los Andes, los Alpes y los valles fluviales inundados como la llanura del Rio Ganges en India; y muchas regiones interiores, como África y Asia central, Europa central y oriental, y la región del medio oeste de Norte América se encuentran entre las áreas más severamente deficientes de yodo en el mundo (1).
Función
El yodo es un componente esencial de las hormonas tiroideas, triyodotironina (T3) y tiroxina (T4), y por lo tanto, es esencial para la función normal de la tiroides. Para cumplir con la demanda corporal de hormonas tiroideas, la glándula tiroides captura el yodo proveniente de la sangre y lo incorpora en la glicoproteína extensa (660 kDa) tiroglobulina. La hidrolisis de la tiroglobulina por las enzimas lisosomales da lugar a hormonas tiroideas que son almacenadas y liberadas en la circulación cuando es necesario. En los tejidos diana, tales como el hígado y el cerebro, la T4 (la hormona tiroidea circulante mas abundante) puede ser convertida a T3 por enzimas que contienen selenio conocidas como yodotironina deiodinasas (DIO) (Figura 1; véase también Interacciones con nutrientes). La T3 es la hormona tiroidea fisiológicamente activa que puede unirse a los receptores tiroideos en el núcleo de las células y regular la expresión de genes. De esta manera, las hormonas tiroideas regulan un cierto numero de procesos fisiológicos, incluyendo el crecimiento, desarrollo, metabolismo, y función reproductiva (2).
La regulación de la función tiroidea es un proceso complejo que involucra el hipotálamo y la glándula pituitaria. En respuesta a la secreción de la hormona liberadora de tirotropina (TRH) por parte del hipotálamo, la glándula pituitaria secreta la hormona estimulante de la tiroides (TSH), la cual estimula la captura de yodo, la síntesis y liberación de T3 y T4 por la glándula tiroides. La presencia de T4 y T3 circulantes adecuadas retroalimentan tanto al nivel del hipotálamo como de la glándula pituitaria, disminuyendo la producción de TRH y TSH (Figura 2). Cuando los niveles de T4 circulantes disminuyen, la glándula pituitaria incrementa su secreción de TSH, resultando en un incremento de la captura de yodo, como también en una producción y liberación incrementada de ambas T3 y T4. La deficiencia de yodo da como resultado una producción inadecuada de T4. En respuesta a los niveles disminuidos de T4 sanguíneos, la glándula pituitaria incrementa su volumen de producción de TSH. La persistencia de niveles elevados de TSH pueden conducir a la hipertrofia (agrandamiento) de la glándula tiroides, también conocida como bocio (véase Deficiencia) (3).

Deficiencia
La glándula tiroides de un adulto saludable concentra el 70-80% de un contenido total de yodo en el cuerpo de 15-20 mg y utiliza alrededor de 80 μg de yodo diariamente para sintetizar las hormonas tiroideas. En contraste, la deficiencia de yodo crónica puede resultar en una reducción dramática del contenido de yodo en la tiroides bien por debajo de 1 mg (1). La deficiencia de yodo es reconocida como la causa más común de daño cerebral prevenible en el mundo. El espectro de los trastornos por deficiencia de yodo (TDY) incluye retardo mental, hipotiroidismo, bocio, y grados variables de otras anormalidades del crecimiento y del desarrollo (4). La Organización Mundial de la Salud (OMS) estimó que más del 30% de la población mundial (2 billones de personas) tiene una ingesta de yodo insuficiente, al medirse las concentraciones urinarias medianas de yodo por debajo de los 100 microgramos μg/litro (5). Además, cerca de un tercio de los niños en edad escolar (6-12 años) alrededor del mundo (241 millones de niños en el 2011) tienen una ingesta insuficiente de yodo (6, 7). Esfuerzos internacionales mayores han producido mejoramientos dramáticos en la corrección de la deficiencia de yodo en la década de los 90, principalmente a través del uso de sal yodada en los países deficientes de yodo (4). Aunque alrededor del 70% de los hogares en el mundo ahora tienen acceso a la sal yodada (8), la deficiencia leve a moderada de yodo permanece siendo una preocupación de salud publica en al menos 30 países; no existen datos disponibles de excreción de yodo en otros 42 países, incluyendo Israel, Siria, y Sierra Leona (7). Para más información acerca del esfuerzo para erradicar la deficiencia de yodo, visite la página web de la Red Global de Yodo (previamente conocido como International Council for the Control of Iodine Deficiency Disorders [Consejo Internacional para el Control de Trastornos por Deficiencia de Yodo]) y la OMS.
Biomarcadores del estatus del yodo
Mas del 90% del yodo ingerido es excretado en la orina dentro de un periodo de 24-48 horas tal que las ingestas diarias de yodo en una población pueden ser extrapoladas a partir de medidas del punto mediano de las concentraciones de yodo urinarias (9, 10). De acuerdo con el criterio de la OMS, la deficiencia de yodo en la población es definida por las concentraciones urinarias medianas de yodo menores de 150 microgramos (μg)/L para las mujeres embarazadas y 100 μg/L para todos los otros grupos (Tabla 1). Las ingestas adecuadas corresponden a las concentraciones urinarias medianas de yodo de 100-199 μg/L en niños de edad escolar y 150-249 μg/L en mujeres embarazadas (Tabla 1). Mientras que la concentración urinaria mediana de yodo es un indicador poblacional de la ingesta dietaría reciente de yodo, múltiples colecciones de yodo urinario de 24 horas son preferibles para estimar la ingesta en individuos (9-11).
| Grupo de Población | Mediana/Rango de Concentraciones Urinarias de Yodo (μg/L) | Ingesta de Yodo |
|---|---|---|
| Niños (<2 años) | <100 | Insuficiente |
| ≥100 | Adecuado | |
| Niños (≥6 años), adolescentes, y adultos* | <100 | Insuficiente |
| 100-199 | Adecuado | |
| 200-299 | Más que adecuado | |
| >300 | Excesivo | |
| Mujeres embarazadas | <150 | Insufficient |
| 150-249 | Adecuado | |
| 250-499 | Más que adecuado | |
| ≥500 | Excesivo | |
| Mujeres en período de lactancia# | <100 | Insuficiente |
| ≥100 | Adecuado | |
|
*Excluye mujeres embarazadas o en período de lactancia. |
||
En muchos países, las concentraciones de TSH en el suero son usados en la evaluación para el hipotiroidismo congénito en recién nacidos. La TSH de recién nacidos puede ser usada como un indicador del estatus poblacional del yodo. Sin embargo, en niños mayores y adultos, la TSH del suero no es un indicador sensitivo del estatus de yodo ya que las concentraciones son usualmente mantenidas dentro del rango normal a pesar de la deficiencia franca de yodo (12). La concentración de tiroglobulina del suero en niños de edad escolar es un marcador sensible del estatus del yodo en las poblaciones (13). En áreas de bocio endémico, cambios en el tamaño de la tiroides reflejan la nutrición de yodo a largo plazo (de meses a años). La evaluación de la tasa de bocio en una población es usada para definir la severidad de la deficiencia de yodo, como también monitorear el impacto a largo plazo de programas de yodación de la sal sostenidos (4, 10). Finalmente, las concentraciones de la hormona tiroidea del suero no reflejan adecuadamente la nutrición del yodo en las poblaciones (1).
Trastornos por deficiencia de yodo
Todos los efectos adversos de la deficiencia de yodo en animales y humanos son colectivamente denominados trastornos por deficiencia de yodo (revisado en 1). El agrandamiento de la tiroides, o bocio, es uno de los signos tempranos y mas visibles de la deficiencia de yodo. Es una adaptación fisiológica de la glándula tiroides en respuesta a la estimulación persistente llevada a cabo por la TSH (véase Función). En una deficiencia leve de yodo, el agrandamiento de la tiroides puede ser suficiente para maximizar la absorción de yodo disponible y proveer al cuerpo con suficientes hormonas tiroideas. Sin embargo, bocios grandes pueden obstruir la tráquea y el esófago y dañar los nervios laríngeos recurrentes.
Casos mas severos de deficiencia de yodo resultan en la síntesis alterada de la hormona tiroidea conocido como hipotiroidismo. La ingesta adecuada de yodo generalmente reduce el tamaño de los bocios, pero la reversibilidad de los efectos del hipotiroidismo depende en la etapa de vida del individuo. La deficiencia de yodo que induce el hipotiroidismo tiene efectos adversos en todas las etapas del desarrollo, pero es más dañina al cerebro en desarrollo. Además de regular muchos aspectos del crecimiento y desarrollo, las hormonas tiroideas son importantes para la migración, proliferación, y diferenciación de poblaciones neuronales específicas, la arquitectura en general de la corteza cerebral, la formación de conexiones axonales, y la mielinización del sistema nervioso central, lo cual ocurre antes y poco después del nacimiento (revisado en 14).
Los efectos de la deficiencia de yodo en las diferentes etapas de la vida son discutidos a continuación.
Embarazo y lactancia
Los requerimientos diarios de yodo se incrementan significativamente en mujeres embarazadas y en período de lactancia debido a (1) la producción incrementada de la hormona tiroidea y su transferencia al feto en el embarazo temprano y antes de que la glándula tiroides fetal sea funcional, (2) a la transferencia de yodo hacia el feto durante la gestación tardía, (3) a la excreción de yodo urinario incrementada, y (4) a la trasferencia de yodo al infante a través de la leche materna (véase también La IDR) (12, 15).
Durante el embarazo, el tamaño de la glándula tiroides es incrementado en un 10% en mujeres que residen en regiones insuficientes de yodo e incrementado en un 20%-40% en aquellas viviendo en regiones deficientes de yodo (16). La deficiencia de yodo durante el embarazo puede resultar en hipotiroidismo en mujeres. El hipotiroidismo materno ha sido asociado con un riesgo incrementado de preeclampsia, aborto espontáneo, muerte fetal, nacimiento prematuro, e infantes con un bajo peso al nacer (revisado en 16). Además, la deficiencia severa de yodo durante el embarazo puede resultar en hipotiroidismo congénito y deficiencias cognitivas en la descendencia (véase Desarrollo prenatal) (12).
Las mujeres deficientes de yodo que están en período de lactancia pueden no ser capaces de proveer suficiente yodo a sus infantes los cuales son particularmente vulnerable a los efectos de la deficiencia de yodo (véase Recién nacidos e infantes) (17). Un suplemento prenatal diario de 150 μg de yodo, como lo recomienda la Asociación para la Tiroides Americana (ATA) (16), ayuda a asegurar que las mujeres estadounidenses embarazadas y en período de lactancia consuman suficiente yodo durante estos periodos críticos. En áreas deficientes de yodo donde la sal yodada no está disponible, la Red Global de Yodo (IGN; previamente conocida como Consejo Internacional para el Control de Trastornos por Deficiencia de Yodo), la Organización Mundial de la Salud (OMS), y la UNICEF recomiendan que mujeres lactando reciban una dosis anual de 400 mg de yodo (o 250 μg/día) y que amamanten exclusivamente por al menos seis meses. Cuando el amamantamiento no sea posible, la suplementación directa al infante (<2 años de edad) con una dosis anual de 200 mg de yodo (o 90 μg/día) es recomendado (4). Un ensayo aleatorio y controlado con placebo recientemente demostró que la suplementación materna (con una sola dosis de 400 mg de yodo) mejoró el estatus del yodo en infantes amamantados mas eficientemente que la suplementación directa a los infantes (con una sola dosis de 100 mg de yodo) por un periodo de al menos seis meses (18). Sin embargo, la suplementación de mujeres en período de lactancia fallo en incrementar las concentraciones urinarias de yodo maternas por arriba de los 100 μg/L, sugiriendo que las madres suplementadas permanecieron deficientes de yodo (18).
Desarrollo prenatal
La deficiencia de yodo fetal es causada por la deficiencia de yodo en la madre (véase Embarazo y lactancia). Durante el embarazo, antes de que la glándula tiroides del feto sea funcional a las 16-20 semanas de gestación, la tiroxina (T4) materna atraviesa la placenta para promover el desarrollo fetal y embriónico normal. Por ello, la deficiencia materna de yodo y el hipotiroidismo pueden resultar en complicaciones del embarazo adversas, incluyendo perdida del feto, desprendimiento placentario, preeclampsia, parto prematuro, e hipotiroidismo congénito en la descendencia (16). Los efectos del hipotiroidismo materno en la descendencia dependen en el tiempo y severidad de la deficiencia de yodo in utero. Una forma severa de hipotiroidismo congénito puede llevar al cretinismo, una condición asociada con el retraso mental irreversible. El cuadro clínico del cretinismo neurológico en la descendencia incluye retraso mental y físico severo, sordera, mudez, y espasticidad motora.
Una forma mixedematosa del cretinismo ha sido asociada con la deficiencia coexistente de yodo y selenio en África central (véase Interacción con nutrientes) y se caracteriza por un grado menos severo de retraso mental que en el cretinismo neurológico. Sin embargo, los individuos afectados exhiben todas las características del hipotiroidismo severo, incluyendo retraso severo en el crecimiento, y retraso en la maduración sexual (12). Dos estudios de cohorte longitudinales (uno en el Reino Unido y otro en Australia) recientemente observaron que incluso la deficiencia de yodo de leve a severa durante el embarazo estaba asociada con puntuaciones de IQ y varias mediciones de rendimiento de alfabetización reducidas en niños de 8 a 9 años de edad (19, 20).
Recién nacidos e infantes (hasta un año de edad)
La mortalidad infantil es más alta en áreas de deficiencia severa de yodo que en regiones repletas de yodo, y varios estudios han demostrado un incremento en la supervivencia infantil tras la corrección de la deficiencia de yodo (8, 21, 22). La infancia es un periodo de rápido crecimiento y desarrollo cerebral. Suficiente hormona tiroidea, la cual depende en la ingesta adecuada de yodo, es esencial para el desarrollo normal del cerebro. Incluso en la ausencia de hipotiroidismo congénito, la deficiencia de yodo durante la infancia puede resultar en el desarrollo anormal del cerebro, y, consecuentemente, en un desarrollo intelectual deteriorado (23, 24).
Niños y adolescentes
La deficiencia de yodo en niños y adolescentes es frecuentemente asociada con el bocio. La incidencia del bocio se eleva en la adolescencia y es más común en niñas que en niños. Los niños en edad escolar en áreas deficientes de yodo muestran rendimientos escolares mas pobres, IQ mas bajos, y una incidencia más alta de dificultades del aprendizaje que los grupos coincidentes de áreas con suficiente yodo. Tres meta-análisis de estudios de corte transversal primeramente concluyeron que la deficiencia crónica de yodo estaba asociada con puntuaciones IQ promedio reducidas por 7-13.5 puntos en los participantes (principalmente niños) (25-27). Sin embargo, estos estudios basados en la observación no distinguieron entre la deficiencia de yodo durante el embarazo y durante la niñez, y tales estudios basados en la observación podrían ser confundidos por factores económicos, sociales o educacionales que influencian el desarrollo del niño.
Adultos
La ingesta inadecuada de yodo puede resultar en bocio e hipotiroidismo en adultos. Aunque los efectos del hipotiroidismo son mas sutiles en los cerebros adultos que en los de los niños, investigación sugiere que el hipotiroidismo resulta en logros sociales y económicos pobres debido a baja educabilidad, apatía y productividad laboral reducida (28). Otros síntomas del hipotiroidismo en adultos incluyen fatiga, aumento de peso, intolerancia al frio, y constipación.
Finalmente, debido a que la deficiencia de yodo induce un incremento en la capacidad de captura del yodo de la tiroides, los individuos deficientes de yodo de todas las edades son más susceptibles al cáncer de tiroides inducido por la radiación (véase Prevención de Enfermedades), como también el hipertiroidismo inducido por yodo después de un incremento en las ingestas de yodo (véase Seguridad) (2).
Individuos y poblaciones en riesgo de deficiencia de yodo
Mientras que el riesgo de deficiencia de yodo en las poblaciones que viven en áreas deficientes de yodo sin programas de fortificación de yodo adecuados es bien reconocido, se han planteado preocupaciones de que ciertas subpoblaciones en países considerados suficientes de yodo no consuman suficiente yodo (7, 29). El mayor uso de métodos para evaluar el estatus de yodo (véase Biomarcadores del estatus del yodo) ha mostrado que la deficiencia de yodo también ocurre en áreas donde la prevalencia de bocio es baja, en áreas costeras, en países altamente desarrollados, y en regiones donde la deficiencia de yodo fue previamente eliminada (4).
Los Estados Unidos está considerando como suficiente de yodo. Sin embargo, en años reciente, ingestas dietarías de yodo en la población estadounidense han disminuido. Datos provenientes de la última Encuesta de Examinación Nacional de Salud y Nutrición de EE.UU. (NHANES 2009-2010) indicaron que la concentración urinaria de yodo mediana para la población general era de 144 μg/L en comparación a 164 μg/L reportados en las evaluaciones previas (NHANES 2005-2006 y 2007-2008) (30, 31). Además de las diferencias regionales a través de los EE.UU. se encontraron variaciones étnicas. En todos los grupos de edad, las concentraciones urinarias de yodo medianas mostraron ser más bajas en afroamericanos que en hispanos y caucásicos.
Además, las concentraciones urinarias de yodo medianas en mujeres no embarazadas en edad de procrear y en mujeres embarazadas indicaron que la deficiencia leve de yodo ha reemergido en los Estados Unidos en años recientes (31).
Mujeres no embarazadas
Datos provenientes del NHANES 2007-2010 estadounidense indicaron que el 37.3% de las mujeres no embarazadas (edades de 15-44 años) tuvieron concentraciones urinarias de yodo menores de 100 μg/L, reflejando ingestas de yodo potencialmente insuficientes (véase Biomarcadores del estatus del yodo) (31). Solo un quinto de las mujeres no embarazadas reportó que usaban suplementos que contenían yodo en una NHANES (2001-2006) mas temprana (32). Sin embargo, las ingestas adecuadas de yodo en mujeres en edad de procrear (150 μg/día; véase La IDR) son esenciales para reservas óptimas de yodo, especialmente si están considerando embarazarse. Algunos expertos sugieren un consumo diario de 250 μg/día de yodo antes de la concepción para asegurar una producción de la hormona tiroidea y un suministro de yodo al embrión y feto adecuados durante el embarazo (véase Embarazo y lactancia) (12).
Mujeres embarazadas
No existen estadísticas acerca del peso mundial de la deficiencia de yodo en mujeres embarazadas, pero datos nacionales y regionales sugieren que este grupo es especialmente vulnerable. Dado que los requerimientos de yodo son incrementados durante el embarazo, la concentración urinaria de yodo mediana debe ser de al menos 150 μg/L (Biomarcadores del estatus del yodo). Datos agrupados de NHANES 2005-2010 reportaron que las mujeres estadounidenses embarazadas tuvieron una concentración urinaria de yodo mediana de 129 μg/L, y la concentración mediana mas baja (109 μg/L) fue observada durante el primer trimestre de gestación, cuando el embrión/feto se basa exclusivamente en las hormonas tiroideas maternas (31).
Mujeres que amamantan
Mientras que datos con respecto al estatus de yodo de mujeres que amamantan en los EE.UU. son limitados, las ingestas dietarías que fueron inadecuadas durante el embarazo son más propensas a ser insuficientes en una fracción significativa de las mujeres que amamantan (33, 34). Una revisión sistemática de la literatura recientemente reportó ingestas dietarías de yodo subóptimas en mujeres que amamantan en algunos países con programas de fortificación obligatoria, incluyendo Dinamarca, Australia, e India (35). La Asociación para la Tiroides American (ATA) recomienda que todas las mujeres norteamericanas que están embarazadas o que amamantan suplementen su ingesta dietaría de yodo con 150 μg/día de yodo (36).
Infantes amamantados y en destete
El cuerpo de un recién nacido saludable contiene solo alrededor de 300 μg de yodo, lo que hace a los recién nacidos extremadamente vulnerables a la deficiencia de yodo (28), e infantes que son amamantados son completamente dependientes de las ingestas maternas de yodo de la síntesis de la hormona tiroidea. Incluso en áreas cubiertas por un programa de yodación de sal, infantes en destete están en alto riesgo de deficiencia de yodo, especialmente si estos no están recibiendo formula infantil que contenga yodo (17).
Individuos que consumen dietas especiales
Se ha encontrado que las dietas que excluyen la sal yodada, el pescado, y las algas marinas contienen muy poco yodo (9). Los individuos que consumen alimentos etiquetados para bajar de peso pueden también estar en riesgo de ingestas inadecuadas de yodo (37). Un estudio de corte transversal estadounidense en 78 vegetarianos y 63 veganos reportó concentraciones urinarias de yodo medianas de 147 μg/L y 78.5 μg/L, respectivamente, sugiriendo ingestas de yodo inadecuadas entre veganos (38). Dos casos de bocio y/o hipotiroidismo también han sido recientemente reportados en niños que siguen dietas restrictivas para controlar la inflamación esofágica (esofagitis eosinofílica) (39) o alergias (40).
Pacientes que requieren nutrición parenteral
Aunque el yodo no es usualmente agregado a las soluciones de nutrición parenteral (NP), los desinfectantes tópicos que contienen yodo y otras fuentes adventicias proveen cantidades substanciales de yodo para algunos pacientes con NP tal que la ocurrencia de deficiencia de yodo es poco probable. Sin embargo, la deficiencia podría ocurrir, especialmente en infantes prematuros con reservas corporales limitadas, si los antisépticos a base de clorhexidina remplazan los antisépticos yodados (28, 41).
Interacciones con nutrientes
Deficiencias en selenio, hierro, o vitamina A concurrentes pueden exacerbar los efectos de la deficiencia de yodo (revisado en 42).
Selenio
Mientras que el yodo es un componente esencial de las hormonas tiroideas, las yodotironina deiodinasas que contienen selenio (DIO) son enzimas (o selenoenzimas) requeridas para la conversión de T4 en la hormona tiroidea biológicamente activa, T3 (véase el articulo sobre Selenio). La actividad de la DIO1 puede estar también involucrada en la regulación de la homeostasis del yodo (43). Además, las glutatión peroxidasas son selenoenzimas que protegen la glándula tiroides del daño inducido por el peróxido de hidrogeno durante la síntesis de la hormona tiroidea (44). Un estudio aleatorio controlado con placebo en 151 mujeres embarazadas en riesgo de desarrollar una enfermedad tiroidea autoinmune encontró que la suplementación con selenio (200 μg/día en la forma de selenometionina) a las 12 semanas de gestación hasta 12 semanas después del parto redujo el riesgo de disfunción tiroidea e hipotiroidismo permanente (45). Sin embargo, otro ensayo (el Ensayo de Intervención del Selenio en el Embarazo) no encontró beneficio alguno de la suplementación con selenio (60 μg/día a partir de 12-14 semanas de gestación hasta el parto) sobre el placebo en las concentraciones de autoanticuerpos circulantes en mujeres embarazadas levemente deficientes de yodo (46).
La epidemiologia de las deficiencias de yodo y selenio coexistentes en África central han sido ligadas a la prevalencia de cretinismo mixedematoso, una forma severa de hipotiroidismo congénito acompañada por retraso mental y físico. La deficiencia de selenio puede ser solamente uno de los varios factores indeterminados que podrían exacerbar los efectos perjudiciales de la deficiencia de yodo (42). Por otra parte, los resultados de ensayos controlados aleatorios de intervención han mostrado que la corrección de solo la deficiencia de selenio puede tener un efecto nocivo sobre el metabolismo de la hormona tiroidea en niños en edad escolar con deficiencia de yodo y selenio coexistente (47, 48). Finalmente, se encontró que la deficiencia de selenio en roedores tuvo poco impacto en las actividades de las DIO ya que parece que el selenio es suministrado como prioridad para la síntesis adecuada de las DIO a expensas de otras selenoenzimas (44).
Hierro
La anemia por deficiencia severa de hierro puede deteriorar el metabolismo de la tiroides de las siguientes maneras: (1) al alterar la respuesta de la TSH de la glándula pituitaria (2) al reducir la actividad de la peroxidasa tiroidea que cataliza la yodación de la tiroglobulina para la producción de las hormonas tiroideas; y (3) en el hígado limitando la conversión de la T4 a T3, incrementando la rotación de T3, y disminuyendo la unión de T3 a los receptores nucleares (49). Se estima que el bocio y la anemia por deficiencia de hierro coexisten en hasta un 25% de los niños en edad escolar en África occidental y del norte (42). Un estudio controlado aleatorio en niños deficientes de hierro con bocio mostró una reducción mayor en el tamaño de la tiroides tras el consumo de sal yodada junto con 60 mg/día de hierro cuatro veces por semana en comparación al placebo (50). Intervenciones adicionales han confirmado que la corrección de la anemia por deficiencia de hierro mejoró la eficacia de la suplementación de yodo para mitigar los trastornos de la tiroides (revisado en 42, 49).
Vitamina A
En el norte y oeste de África, la deficiencia de vitamina A y el bocio inducido por la deficiencia de yodo pueden coexistir en hasta un 50% de los niños. El estatus de la vitamina A, como otros factores nutricionales, parece influenciar en la respuesta a la profilaxis con yodo en poblaciones con deficiencia de yodo (51). Se encontró que la deficiencia de vitamina A en modelos animales interfiere con el eje pituitario-tiroideo al (1) incrementar la síntesis y secreción de la hormona estimulante de la tiroides (TSH) por la glándula pituitaria, (2) al incrementar el tamaño de la glándula tiroidea, (3) al reducir la absorción de yodo por la glándula tiroidea y perjudicando la síntesis y yodación de la tiroglobulina, y (4) al incrementar las concentraciones circulantes de las hormonas tiroideas (revisado en 52). Un estudio de corte transversal de 138 niños con deficiencias concurrentes de vitamina A y yodo encontró que la severidad de la deficiencia de vitamina A estaba asociada con un riesgo mas alto de bocio y concentraciones mas altas de TSH circulantes y de hormonas tiroideas (51). Estos niños recibieron sal enriquecida con yodo junto con vitamina A (200,000 UI al inicio del estudio y a los 5 meses) o un placebo en un ensayo aleatorio, doble ciego de 10 meses. La suplementación con vitamina A significantemente disminuyo la concentración de la TSH y el volumen de la tiroides en comparación al placebo (51). En otro ensayo, la suplementación con vitamina A sola, (sin yodo) a niños deficientes de yodo redujo el volumen de la glándula tiroides, como también las concentraciones de TSH y tiroglobulina (53). Sin embargo, la vitamina A suplementaria no tuvo efecto adicional en la función de la tiroides/metabolismo hormonal cuando a los niños también se les administro aceite yodado
Goitrógenos
Algunos alimentos contienen sustancias que intervienen con la utilización del yodo o con la producción de hormonas tiroideas; estas sustancias son llamadas goitrógenos. La ocurrencia del bocio en la República Democrática del Congo ha sido relacionada con el consumo de casava, la cual contiene linamarina, un compuesto que es metabolizado en tiocianato y que bloquea la captación tiroidea de yodo (1). En poblaciones deficientes de yodo, se ha asociado el fumar tabaco con un riesgo incrementado de padecer bocio (54, 55). El cianuro en el humo del tabaco es convertido a tiocianato en el hígado, colocando a los fumadores con baja ingesta de yodo en riesgo de desarrollar bocio. Además, el tiocianato afecta el transporte de yodo hacia la glándula mamaria lactante, llevando a concentraciones bajas de yodo en la leche materna y al deterioro del suministro de yodo a los neonatos/infantes de mujeres que fuman (2). Algunas especies de mijo, camotes, frijoles, y vegetales crucíferos (por ejemplo, repollo, brócoli, coliflor, coles de Bruselas) también contienen goitrógenos (1). Además, se ha encontrado que las isoflavonas de la soya, genisteína y daidzeína, inhiben la síntesis de la hormona tiroidea (56). La mayoría de estos goitrógenos carecen de importancia clínica a menos que se consuman en grandes cantidades o que haya coexistencia de deficiencia de yodo. Contaminantes industriales, como el perclorato (véase Seguridad), resorcinol y ácido ftálico podrían ser también goitrógenicos (1, 57).
La Ingesta Diaria Recomendada (IDR)
La IDR para el yodo fue reevaluada por la Junta de Nutrición y Alimentos (JNA) del Instituto de Medicina (IOM) en el 2001 (Tabla 2). Las cantidades recomendadas fueron calculadas usando varios métodos, incluyendo la medida de acumulación de yodo en las glándulas tiroides de individuos con función tiroidea normal (9). Recomendaciones similares han sido hechas por varias organizaciones, incluyendo la Asociación para la Tiroides Americana (ATA) (16, 58), la Organización Mundial de la Salud (OMS), la Red Global de Yodo (IGN; previamente conocida como Consejo Internacional para el Control de Trastornos por Deficiencia de Yodo), y el Fondo de las Naciones Unidas para la Infancia (UNICEF) (4). Debe notarse que, la OMS, IGN, y la UNICEF recomiendan ingestas diarias de 250 μg de yodo para tanto mujeres embarazadas como en período de lactancia (4).
Prevención de Enfermedades
Cáncer de tiroides inducido por radiación
El yodo radioactivo, especialmente el yodo 131 (131I), puede ser liberado al medio ambiente como resultado de accidentes en reactores nucleares, como el accidente nuclear de Chernóbil en 1986 en Ucrania y el accidente nuclear en 2011 de Fukushima Daiichi en Japón. La acumulación tiroidea de yodo radioactivo incrementa el riesgo de desarrollar cáncer de tiroides, especialmente en niños (59). El incremento en la actividad de captura de yodo de la glándula tiroides en la deficiencia de yodo resulta en un incremento en la acumulación tiroidea de yodo radioactivo (131I). Así, los individuos deficientes de yodo están en un riesgo incrementado de desarrollar cáncer de tiroides inducido por radiación debido a que acumularán mayores cantidades de yodo radioactivo. El yoduro de potasio administrado en dosis farmacológicas (hasta 130 mg para adultos) dentro de las 48 horas previas u ocho horas posteriores a la exposición a la radiación de un accidente en un reactor nuclear, puede reducir significativamente la captación tiroidea de 131I y disminuir el riesgo de cáncer de tiroides inducido por radiación (60). El uso oportuno y generalizado de la profilaxis con yoduro de potasio en Polonia, luego del accidente en el reactor nuclear de Chernóbil en 1986, podría explicar la falta de un incremento significativo en el cáncer de tiroides infantil en comparación con las áreas de lluvia radioactiva en donde la profilaxis con yoduro de potasio no fue utilizada extensamente (61). En los Estados Unidos, la Comisión Reguladora Nuclear (CRN) requiere que se considere al yoduro de potasio como una medida de protección al público general en caso de una liberación importante de radioactividad proveniente de una planta de energía nuclear (62). Véase también la Información del Yoduro de Potasio de la FDA de los EE.UU.
Tratamiento de Enfermedades
Condición mamaria fibroquística
La condición mamaria fibroquística constituye una condición benigna (no-cancerosa) de las mamas, caracterizada por abultamientos e incomodidad en una o ambas mamas. La formación de quistes y cambios fibrosos en la apariencia del tejido mamario ocurre en al menos 50% de las mujeres premenopáusicas y no son usualmente asociados con un riesgo incrementado de cáncer de seno (63). La causa de cambios fibroquísticos es desconocida, pero variaciones en la estimulación hormonal durante los ciclos menstruales pueden desencadenar cambios en el tejido mamario (63).
Unos pocos estudios basados en la observación también sugieren una asociación entre las enfermedades mamarias benignas (incluyendo, pero no limitándose a los cambios fibroquísticos) y los trastornos de la tiroides. Recientemente, un estudio de caso y control pequeño (166 casos vs. 72 controles) mostró que la frecuencia de enfermedades mamarias benignas fue mayor en mujeres con bocio nodular (54.9%) o con tiroiditis de Hashimoto (47.4%) que en controles eutiroideos (29.2%) (64). Inversamente, se encontró que la prevalencia de autoinmunidad antitiroidea e hipotiroidismo era significantemente mas alta en mujeres con enfermedades mamarias benignas en comparación a los controles (65, 66). Curiosamente, se encontró que la corrección del hipotiroidismo con T4 suplementaria mejoró algunos de los síntomas de enfermedades mamarias benignas, incluyendo dolor mamario (mastalgia) y secreción del pezón (65).
En ratones tratados con estrógeno, la deficiencia de yodo conduce a cambios similares a aquellos vistos en mamas fibroquísticas, mientras que la repleción con yodo revierte dichos cambios (67). Un estudio no controlado en 233 mujeres con condición mamaria fibroquística encontró que el tratamiento con yodo molecular acuoso (I2) en una dosis de 0.08 mg de I2/kg de peso corporal a diario por 6 a 18 meses, se asociaba con una mejora en el dolor y otros síntomas en cerca del 70% de los participantes (68). Alrededor del 10% de los participantes del estudio reportaron efectos secundarios que fueron descritos como menores por parte de los investigadores. Un ensayo de doble ciego controlado con placebo de yodo molecular acuoso (0.07-0.09 mg de I2/kg de peso corporal a diario por seis meses) en 56 mujeres con condición mamaria fibroquística, encontró que el 65% de las mujeres que consumieron yodo molecular reportaron una mejora comparado con el 33% de aquellas que tomaron el placebo (68). Un ensayo doble ciego controlado con placebo en 87 mujeres con dolor mamario documentado reporto que el yodo molecular (1.5, 3, o 6 mg/día) por seis meses mejoro el dolor en general (69). En este estudio, 38.5% de las mujeres que recibieron 1.5 mg/día, 37.9% de las que recibieron 3 mg/día, y 51.7% de las que recibieron 6 mg/día reportaron por lo menos una reducción del 50% en el dolor mamario autoevaluado en comparación al 8.3% en el grupo de placebo.
Ensayos clínicos controlados a gran escala son necesarios para determinar el valor terapéutico del yodo molecular en mamas fibroquísticas. Además, las dosis de yodo usadas en estos estudios (1.5 a 6 mg/día para una persona de 60 kg) son más altas que el nivel máximo de ingesta tolerable (NM) recomendado por la Junta de Nutrición y Alimentos (JNA) del Instituto de Medicina y deben ser solamente usadas bajo supervisión médica (véase Seguridad).
Fuentes
Fuentes alimenticias
Datos del Estudio de Dieta Total de los EE.UU. en proceso, el cual monitorea los niveles de algunos contaminantes y nutrientes en productos alimenticios, indica que las ingesta de yodo dietario en adultos oscila entre 138 y 268 microgramos (μg)/día. Ingestas promedio considerablemente más altas (304-353 μg/día de yodo) fueron reportadas en muchachos de 14 a 16 años de edad (70).
Los mariscos son ricos en yodo debido a que los animales marinos pueden concentrar el yodo del agua marina. Ciertos tipos de algas comestibles (p.ej., wakame) son también muy ricos en yodo (71). El contenido de yodo de alimentos que son cultivados y crecidos en un suelo particular depende del contenido de yodo de este suelo. En los Estados Unidos, los productos lácteos contribuyen hasta un 90% de las ingestas de yodo estimadas totales en infantes, por lo menos un 70% en niños (edades 2-10 años), 53%-63% en adolescentes (edades 14-16 años), y cerca del 50% en adultos (70). En el Reino Unido y el norte de Europa, los niveles de yodo en productos lácteos tienden a ser menores en el verano cuando al ganado se le permite pastar en pastizales con bajo contenido de yodo en el suelo (9).
Otras buenas fuentes de yodo dietario incluyen huevos, frutos, productos de granos, y aves de corral (70). Alimentos procesados pueden contribuir a la ingesta de yodo si la sal yodada o aditivos alimentarios, como el yodato de calcio y el yodato de potasio, son agregados durante la producción. Sin embargo, en los EE.UU., virtualmente ninguna sal yodada es utilizada en la fabricación de alimentos procesados y productos de comida rápida, y a la industria alimenticia no se le requiere listar el contenido de yodo en el envasado de alimentos (72). La Tabla 3 lista el contenido de yodo de algunos alimentos ricos en yodo en microgramos (μg). Debido a que el contenido de yodo de los alimentos puede variar considerablemente, estos valores deben ser considerados como aproximados (73).
Suplementos
Suplementos de yodo de venta libre
El yoduro de potasio esta disponible como un suplemento nutricional, típicamente en productos combinados, como en suplementos multivitamínicos/minerales. El yodo compone aproximadamente el 77% del peso total del yoduro de potasio (56). Un suplemento multivitamínico/mineral que contenga el 100% del valor diario (VD) del yodo aporta 150 μg de yodo. Aunque la mayoría de las personas en los EE.UU. consumen suficiente yodo en sus dietas (véase Fuentes) es poco probable que un aporte adicional de 150 μg/día resulte en una ingesta excesiva de yodo. La Asociación para la Tiroides Americana (ATA) recomienda la suplementación prenatal con 150 μg/día de yodo y aconseja contra la ingestión de ≥500 μg/día de yodo proveniente de yodo, yoduro de potasio, y suplementos de algas marinas para los niños y adultos, y durante el embarazo y período de lactancia (véase también Seguridad) (36, 74).
Programas de fortificación de yodo
La fortificación de la sal con yodo es un método factible y de bajo costo para eliminar la deficiencia de yodo, y los programas de yodación de la sal han sido implementados en casi todos los países. En Norteamérica, la fortificación de la sal con yodo es obligatoria en Canadá y algunas partes de México, pero solo es opcional en los Estados Unidos tal que solo un 52% de la sal de mesa en los Estados Unidos es yodada y solo un quinto del total de sal consumida en los Estados Unidos esta yodada (72, 75). El yoduro de potasio (KI), el yoduro cuproso (CuI), y el yodato de potasio (KIO3) son usados para yodar la sal. La Administración de Alimentos y Fármacos de los EE.UU. (FDA) recomienda entre 46 y 76 μg de yodo por gramo de sal en la sal yodada. Sin embargo, el reciente análisis de 88 muestras de sal yodada de calidad alimentaria de los EE.UU. revelo que el contenido de yodo estaba por debajo del rango recomendado en un 52% de las muestras y por arriba del rango en 7% de las muestras (76).
En otros países, la sal comúnmente contiene de 20-50 μg de yodo por gramo de sal, dependiendo de las regulaciones locales (76). En países como Dinamarca (77), Australia (78, 79), y Nueva Zelanda (80), el uso de sal yodada en el proceso de la elaboración del pan es obligatoria. Enfoques adicionales han sido explorados, incluyendo la fortificación del azúcar (81), la fortificación de huevos (82), el uso de sal yodada en la preparación de pescado fermentado y salsa de pescado (83), y el uso de fertilizantes ricos en yodo (84). Además, la fortificación de alimentos para el ganado con yodo y el uso de yodóforos para el saneamiento durante el ordeño contribuyen al incremento del contenido de yodo en los productos lácteos (85). Finalmente, dosis anuales de aceite vegetal yodada son administradas oralmente o intramuscularmente a individuos en poblaciones deficientes de yodo que no tienen acceso a la sal yodada (4, 56).
Seguridad
Toxicidad agud
El envenenamiento agudo con yodo es poco común y usualmente ocurre sólo con dosis de muchos gramos. Los síntomas de envenenamiento agudo con yodo incluyen ardor en la boca, garganta y estómago, fiebre, náuseas, vómitos, diarrea, pulso débil, cianosis y coma (1).
Ingestas de yodo excesivas
Riego de hipertiroidismo inducido por yodo en individuos deficientes de yodo
Los programas de suplementación de yodo en las poblaciones deficientes de yodo han sido asociados con un aumento en la incidencia de hipertiroidismo inducido por yodo (IIH), principalmente en adultos mayores y en aquellos con bocio multinodular (86). Se ha encontrado que las ingestas de yodo de 150-200 μg/día incrementan la incidencia de IIH en poblaciones deficientes de yodo. La deficiencia de yodo incrementa el riesgo de desarrollar nódulos tiroideos autónomos, los que no responden al control de la TSH (véase Función). Estos nódulos autónomos pueden entonces producir en demasía las hormonas tiroideas en respuesta al suministro repentino de yodo. Los síntomas del IIH incluyen pérdida de peso, taquicardia (alta frecuencia de pulso), debilidad muscular, y calor en la piel. El IIH puede ser peligroso en individuos con una enfermedad cardíaca subyacente. Sin embargo, debido a que la principal causa de bocio nodular y IIH es la deficiencia crónica de yodo, el beneficio de los programas de yodación supera en gran medida el riesgo de IIH en poblaciones deficientes de yodo (1).
Riesgo de hipotiroidismo en individuos suficientes de yodo
En individuos suficientes de yodo, el exceso en la ingesta de yodo es más comúnmente asociado con concentraciones sanguíneas elevadas de la hormona estimulante de la tiroides (TSH) que inhibe la producción de la hormona tiroidea, conduciendo al hipotiroidismo y bocio. Una concentración de TSH en el suero ligeramente elevada sin una disminución en el suero de T4 o T3 es el signo mas temprano de una función tiroidea anormal cuando la ingesta de yodo es excesiva. En los adultos yodo-suficientes, se han encontrado niveles elevados de TSH en el suero con ingestas de yodo crónicas de ≥750 μg/día en niños y ≥1,700 μg/día en adultos. Debido a que varias especies comestibles de algas marinas contribuyen sustancialmente a las comidas asiáticas tradicionales, las ingestas dietarías promedio japonesas se estiman oscilan entre 1,000 a 3,000 μg de yodo/día (71). El bocio inducido por el yodo y el hipotiroidismo no son comunes en Japón y pueden revertirse restringiendo la ingesta de algas marinas (71). Ingestas prolongadas de más de 18,000 μg/día (18 mg/día) incrementan la incidencia de bocio en adultos. En recién nacidos, el bocio inducido por el yodo e hipotiroidismo pueden ocurrir debido a tanto ingestas maternas altas como a la exposición alta de antisépticos yodados (87). A fin de minimizar el riesgo de efectos adversos a la salud, la Junta de Nutrición y Alimentos (JNA) del Instituto de Medicina estadounidense estableció un nivel máximo de ingesta tolerable (NM) para el yodo que es probable que sea seguro en casi todos los individuos. Los valores del NM para el yodo están listados en la Tabla 4 por grupo etario; el NM no se aplica en individuos que están siendo tratados con yodo bajo supervisión medica (74).
Individuos con sensibilidad aumentada a la ingesta de yodo en exceso
Los individuos con deficiencia de yodo y aquellos con enfermedades tiroideas preexistentes, incluyendo el bocio nodular, la tiroiditis autoinmune de Hashimoto, la enfermedad de Graves, y un historial de tiroidectomía parcial, pueden ser sensibles a los niveles de ingesta de yodo considerados como seguros para la población general y podrían no estar protegidos por el NM para el yodo (9). Los infantes, ancianos, y mujeres embarazadas y en periodo de lactación pueden también ser más susceptibles al yodo en exceso (véase Suplementos) (74).
¿Ingestas elevadas y/o insuficientes de yodo incrementan el riesgo de cáncer de tiroides?
En las ultimas décadas, la incidencia de cáncer de tiroides ha incrementado mundialmente. En los Estados Unidos, la incidencia de cáncer de tiroides — representando el 4% de todos los cánceres recientemente diagnosticados — ha incrementado de 4.9 casos por persona en 1983 a 14.7 casos por 100,000 personas en 2011, pero la tasa de mortalidad del cáncer de tiroides ha permanecido baja (cerca de 0.5 por 100,000 personas) (88). Representando más del 80% de todos los cánceres de tiroides, el cáncer tiroideo papilar es menos agresivo y tiene un mejor pronóstico que el cáncer tiroideo folicular o cáncer tiroideo anaplásico. La incidencia incrementada de cáncer de tiroides alrededor del mundo es probablemente debido a al menos en parte al mejoramiento de las actividades de detección y diagnóstico. Sin embargo, porque ha coincidido con la introducción de los programas de fortificación con yodo, una posible contribución de las ingestas de yodo incrementadas ha sido hipotetizada. Sin embargo, en los Estados Unidos, la incidencia incrementada de canceres tiroideos (primariamente cáncer papilar) en las últimas décadas fue paralela con una reducción en la ingesta promedio de yodo (89).
Estudios ecológicos también sugirieron que la profilaxis con yodo en poblaciones que fueron previamente deficientes de yodo estaba asociada con una incidencia incrementada de cáncer papilar en lugar del subtipo de cáncer folicular, y con una incidencia reducida del cáncer tiroideo anaplásico mas agresivo (89). Mientras que los cambios en las ingestas de yodo parecen afectar el cáncer tiroideo de tipo histológico, no es aun claro si la deficiencia de yodo y/o el yodo en exceso incrementan el riesgo de cáncer de tiroides (88).
Interacción con drogas/fármacos
La amiodarona, un medicamento utilizado para prevenir la arritmia cardíaca, contiene niveles elevados de yodo y puede afectar la función tiroidea (90). Los medicamentos antitiroideos para tratar el hipertiroidismo, como el propiltiouracilo (PTU), metimazole y carbimazol, pueden incrementar el riesgo de hipotiroidismo. Adicionalmente, el uso a largo plazo de litio para tratar trastornos del estado del ánimo puede incrementar el riesgo de hipotiroidismo (91). Además, el uso de dosis farmacológicas de yoduro de potasio puede disminuir el efecto anticoagulante de la warfarina (cumarina) (92).
Contaminantes
El perclorato es un agente oxidante encontrado en propulsores de cohete, bolsas de aire, fuegos artificiales, herbicidas, y fertilizantes. Principalmente como un resultado de la actividad humana, se ha encontrado que el perclorato contamina el agua potable y muchos alimentos (57). La exposición crónica a concentraciones de perclorato a niveles mayores de 20 μg por kg de peso corporal (pc) por día interfiere con la absorción de yodo por la glándula tiroides y puede conducir al hipotiroidismo (93). La Agencia de Protección Ambiental (EPA) estadounidense recomienda que la exposición oral diaria de perclorato no debe exceder 0.7 μg/kg pc para proteger la población mas sensible, es decir, los fetos de mujeres embarazadas que podrían ser deficientes de yodo y/o hipotiroideos (94). Entre todos los grupos de edad, los niños de dos años de edad tienen las ingestas de perclorato estimadas más altas por día con 0.35-0.39 μg/kg pc/día. Las ingestas promedio estimadas de perclorato en los Estados Unidos oscilan entre 0.08 y 0.11 μg/kg pc/día (70).
Recomendación del Instituto Linus Pauling
La IDR para el yodo es suficiente para asegurar la función tiroidea normal. En la actualidad no existe evidencia de que las ingestas de yodo mas altas que la IDR sean beneficiales. La mayoría de las personas en los EE.UU. consume yodo suficiente en sus dietas, haciendo la suplementación innecesaria.
Mujeres embarazadas y en período de lactancia
Dada la importancia de la suficiencia de yodo durante el desarrollo prenatal e infantil, las mujeres embarazadas y que lactan deben tomar un suplemento que provee 150 μg de yodo por día (véase Deficiencia).
Adultos mayores (>50 años)
Debido a que el envejecimiento no ha sido asociado con cambios significantes en el requerimiento de yodo, la recomendación del ILP para la ingesta de yodo no es diferente para los adultos mayores.
Autores y Críticos
Originalmente escrito en 2001 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Abril de 2003 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Julio de 2007 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Marzo de 2010 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Agosto de 2015 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Agosto de 2015 por:
Elizabeth N. Pearce, M.D., M.Sc.
Profesor Asociado de Medicina
Escuela de Medicina de la Universidad de Boston
Traducido al Español en 2017 por:
Silvia Vazquez Lima
Instituto Linus Pauling
Universidad Estatal de Oregon
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Derechos de autoría 2001-2024 Instituto Linus Pauling
Referencias
2. Laurberg P. Iodine. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed: Lippincott Williams & Wilkins; 2014:217-224.
3. Larsen PR, Davies TF, Hay ID. The thyroid gland. In: Wilson JD, Foster DW, Kronenberg HM, Larsen PR, eds. Williams Textbook of Endocrinology. 9th ed. Philadelphia: W.B. Saunders Company; 1998:389-515.
4. WHO, UNICEF, ICCIDD. Assessment of iodine deficiency disorders and monitoring of their eliminination: a guide for programme managers, 3rd ed. 2007. http://www.who.int/nutrition/publications/micronutrients/iodine_deficiency/9789241595827/en/. Accessed 8/28/15.
5. de Benoist B, McLean E, Andersson M, Rogers L. Iodine deficiency in 2007: global progress since 2003. Food Nutr Bull. 2008;29(3):195-202. (PubMed)
6. Andersson M, Karumbunathan V, Zimmermann MB. Global iodine status in 2011 and trends over the past decade. J Nutr. 2012;142(4):744-750. (PubMed)
7. Pearce EN, Andersson M, Zimmermann MB. Global iodine nutrition: Where do we stand in 2013? Thyroid. 2013;23(5):523-528. (PubMed)
8. United Nations Children's Fund. The State of the World's Children 2007, UNICEF. New York; 2006.
9. Food and Nutrition Board, Institue of Medicine. Iodine. Dietary reference intakes for vitamin A, vitamin K, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanandium, and zinc. Washington, D.C.: National Academy Press; 2001:258-289. (National Academy Press)
10. Zimmermann MB, Andersson M. Assessment of iodine nutrition in populations: past, present, and future. Nutr Rev. 2012;70(10):553-570. (PubMed)
11. Konig F, Andersson M, Hotz K, Aeberli I, Zimmermann MB. Ten repeat collections for urinary iodine from spot samples or 24-hour samples are needed to reliably estimate individual iodine status in women. J Nutr. 2011;141(11):2049-2054. (PubMed)
12. Zimmermann MB. The effects of iodine deficiency in pregnancy and infancy. Paediatr Perinat Epidemiol. 2012;26 Suppl 1:108-117. (PubMed)
13. Ma ZF, Skeaff SA. Thyroglobulin as a biomarker of iodine deficiency: a review. Thyroid. 2014;24(8):1195-1209. (PubMed)
14. Di Liegro I. Thyroid hormones and the central nervous system of mammals (Review). Mol Med Rep. 2008;1(3):279-295. (PubMed)
15. Zimmermann MB. Are weaning infants at risk of iodine deficiency even in countries with established iodized salt programs? Nestle Nutr Inst Workshop Ser. 2012;70:137-146. (PubMed)
16. Stagnaro-Green A, Abalovich M, Alexander E, et al. Guidelines of the American Thyroid Association for the diagnosis and management of thyroid disease during pregnancy and postpartum. Thyroid. 2011;21(10):1081-1125. (PubMed)
17. Andersson M, Aeberli I, Wust N, et al. The Swiss iodized salt program provides adequate iodine for school children and pregnant women, but weaning infants not receiving iodine-containing complementary foods as well as their mothers are iodine deficient. J Clin Endocrinol Metab. 2010;95(12):5217-5224. (PubMed)
18. Bouhouch RR, Bouhouch S, Cherkaoui M, et al. Direct iodine supplementation of infants versus supplementation of their breastfeeding mothers: a double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2(3):197-209. (PubMed)
19. Bath SC, Steer CD, Golding J, Emmett P, Rayman MP. Effect of inadequate iodine status in UK pregnant women on cognitive outcomes in their children: results from the Avon Longitudinal Study of Parents and Children (ALSPAC). Lancet. 2013;382(9889):331-337. (PubMed)
20. Hynes KL, Otahal P, Hay I, Burgess JR. Mild iodine deficiency during pregnancy is associated with reduced educational outcomes in the offspring: 9-year follow-up of the gestational iodine cohort. J Clin Endocrinol Metab. 2013;98(5):1954-1962. (PubMed)
21. Cobra C, Muhilal, Rusmil K, et al. Infant survival is improved by oral iodine supplementation. J Nutr. 1997;127(4):574-578. (PubMed)
22. DeLong GR, Leslie PW, Wang SH, et al. Effect on infant mortality of iodination of irrigation water in a severely iodine-deficient area of China. Lancet. 1997;350(9080):771-773. (PubMed)
23. Hetzel BS. Iodine and neuropsychological development. J Nutr. 2000;130(2S Suppl):493S-495S. (PubMed)
24. Levander OA, Whanger PD. Deliberations and evaluations of the approaches, endpoints and paradigms for selenium and iodine dietary recommendations. J Nutr. 1996;126(9 Suppl):2427S-2434S. (PubMed)
25. Bleichrodt N, Born, M.P. A meta-analysis of research on iodine and its relationship to cognitive development. In: Stanbury JB, ed. The damaged brain of iodine deficiency: cognitive, behavioral, neuromotor, educative aspects. New York: Cognizant Communication Corporation; 1994:195-200.
26. Qian M, Wang D, Watkins WE, et al. The effects of iodine on intelligence in children: a meta-analysis of studies conducted in China. Asia Pac J Clin Nutr. 2005;14(1):32-42. (PubMed)
27. Bougma K, Aboud FE, Harding KB, Marquis GS. Iodine and mental development of children 5 years old and under: a systematic review and meta-analysis. Nutrients. 2013;5(4):1384-1416. (PubMed)
28. Zimmermann MB. Iodine: it's important in patients that require parenteral nutrition. Gastroenterology. 2009;137(5 Suppl):S36-46. (PubMed)
29. Lazarus JH. Iodine status in europe in 2014. Eur Thyroid J. 2014;3(1):3-6. (PubMed)
30. Caldwell KL, Makhmudov A, Ely E, Jones RL, Wang RY. Iodine status of the U.S. population, National Health and Nutrition Examination Survey, 2005-2006 and 2007-2008. Thyroid. 2011;21(4):419-427. (PubMed)
31. Caldwell KL, Pan Y, Mortensen ME, Makhmudov A, Merrill L, Moye J. Iodine status in pregnant women in the National Children's Study and in U.S. women (15-44 years), National Health and Nutrition Examination Survey 2005-2010. Thyroid. 2013;23(8):927-937. (PubMed)
32. Gregory CO, Serdula MK, Sullivan KM. Use of supplements with and without iodine in women of childbearing age in the United States. Thyroid. 2009;19(9):1019-1020. (PubMed)
33. Kirk AB, Martinelango PK, Tian K, Dutta A, Smith EE, Dasgupta PK. Perchlorate and iodide in dairy and breast milk. Environ Sci Technol. 2005;39(7):2011-2017. (PubMed)
34. Pearce EN, Leung AM, Blount BC, et al. Breast milk iodine and perchlorate concentrations in lactating Boston-area women. J Clin Endocrinol Metab. 2007;92(5):1673-1677. (PubMed)
35. Nazeri P, Mirmiran P, Shiva N, Mehrabi Y, Mojarrad M, Azizi F. Iodine nutrition status in lactating mothers residing in countries with mandatory and voluntary iodine fortification programs: an updated systematic review. Thyroid. 2015;25(6):611-620. (PubMed)
36. Becker DV, Braverman LE, Delange F, et al. Iodine supplementation for pregnancy and lactation-United States and Canada: recommendations of the American Thyroid Association. Thyroid. 2006;16(10):949-951. (PubMed)
37. Kuriti M, Pearce EN, Braverman LE, He X, Leung AM. Iodine content of U.S. weight-loss food. Endocr Pract. 2014;20(3):232-235. (PubMed)
38. Leung AM, Lamar A, He X, Braverman LE, Pearce EN. Iodine status and thyroid function of Boston-area vegetarians and vegans. J Clin Endocrinol Metab. 2011;96(8):E1303-1307. (PubMed)
39. Brooks MJ, Post EM. Acquired hypothyroidism due to iodine deficiency in an American child. J Pediatr Endocrinol Metab. 2014;27(11-12):1233-1235. (PubMed)
40. Cheetham T, Plumb E, Callaghan J, Jackson M, Michaelis L. Dietary restriction causing iodine-deficient goitre. Arch Dis Child. 2015;100(8):784-786. (PubMed)
41. Belfort MB, Pearce EN, Braverman LE, He X, Brown RS. Low iodine content in the diets of hospitalized preterm infants. J Clin Endocrinol Metab. 2012;97(4):E632-636. (PubMed)
42. Hess SY. The impact of common micronutrient deficiencies on iodine and thyroid metabolism: the evidence from human studies. Best Pract Res Clin Endocrinol Metab. 2010;24(1):117-132. (PubMed)
43. Schneider MJ, Fiering SN, Thai B, et al. Targeted disruption of the type 1 selenodeiodinase gene (Dio1) results in marked changes in thyroid hormone economy in mice. Endocrinology. 2006;147(1):580-589. (PubMed)
44. Schomburg L. Selenium, selenoproteins and the thyroid gland: interactions in health and disease. Nat Rev Endocrinol. 2012;8(3):160-171. (PubMed)
45. Negro R, Greco G, Mangieri T, Pezzarossa A, Dazzi D, Hassan H. The influence of selenium supplementation on postpartum thyroid status in pregnant women with thyroid peroxidase autoantibodies. J Clin Endocrinol Metab. 2007;92(4):1263-1268. (PubMed)
46. Mao J, Pop VJ, Bath SC, Vader HL, Redman CW, Rayman MP. Effect of low-dose selenium on thyroid autoimmunity and thyroid function in UK pregnant women with mild-to-moderate iodine deficiency. Eur J Nutr. 2014. [Epub ahead of print] (PubMed)
47. Contempre B, Duale NL, Dumont JE, Ngo B, Diplock AT, Vanderpas J. Effect of selenium supplementation on thyroid hormone metabolism in an iodine and selenium deficient population. Clin Endocrinol (Oxf). 1992;36(6):579-583. (PubMed)
48. Contempre B, Dumont JE, Ngo B, Thilly CH, Diplock AT, Vanderpas J. Effect of selenium supplementation in hypothyroid subjects of an iodine and selenium deficient area: the possible danger of indiscriminate supplementation of iodine-deficient subjects with selenium. J Clin Endocrinol Metab. 1991;73(1):213-215. (PubMed)
49. Zimmermann MB. The influence of iron status on iodine utilization and thyroid function. Annu Rev Nutr. 2006;26:367-389. (PubMed)
50. Hess SY, Zimmermann MB, Adou P, Torresani T, Hurrell RF. Treatment of iron deficiency in goitrous children improves the efficacy of iodized salt in Cote d'Ivoire. Am J Clin Nutr. 2002;75(4):743-748. (PubMed)
51. Zimmermann MB, Wegmuller R, Zeder C, Chaouki N, Torresani T. The effects of vitamin A deficiency and vitamin A supplementation on thyroid function in goitrous children. J Clin Endocrinol Metab. 2004;89(11):5441-5447. (PubMed)
52. Zimmermann MB. Interactions of vitamin A and iodine deficiencies: effects on the pituitary-thyroid axis. Int J Vitam Nutr Res. 2007;77(3):236-240. (PubMed)
53. Zimmermann MB, Jooste PL, Mabapa NS, et al. Vitamin A supplementation in iodine-deficient African children decreases thyrotropin stimulation of the thyroid and reduces the goiter rate. Am J Clin Nutr. 2007;86(4):1040-1044. (PubMed)
54. Knudsen N, Brix TH. Genetic and non-iodine-related factors in the aetiology of nodular goitre. Best Pract Res Clin Endocrinol Metab. 2014;28(4):495-506. (PubMed)
55. Rendina D, De Palma D, De Filippo G, et al. Prevalence of simple nodular goiter and Hashimoto's thyroiditis in current, previous, and never smokers in a geographical area with mild iodine deficiency. Horm Metab Res. 2015;47(3):214-219. (PubMed)
56. Hendler SS, Rorvik DM, eds. PDR for Nutritional Supplements. 2nd ed. Montvale: Thomson Reuters; 2008.
57. Council on Environmental Health, Rogan WJ, Paulson JA, et al. Iodine deficiency, pollutant chemicals, and the thyroid: new information on an old problem. Pediatrics. 2014;133(6):1163-1166. (PubMed)
58. Leung AM, Pearce EN, Braverman LE, Stagnaro-Green A. AAP recommendations on iodine nutrition during pregnancy and lactation. Pediatrics. 2014;134(4):e1282. (PubMed)
59. Cardis E, Howe G, Ron E, et al. Cancer consequences of the Chernobyl accident: 20 years on. J Radiol Prot. 2006;26(2):127-140. (PubMed)
60. Zanzonico PB, Becker DV. Effects of time of administration and dietary iodine levels on potassium iodide (KI) blockade of thyroid irradiation by 131I from radioactive fallout. Health Phys. 2000;78(6):660-667. (PubMed)
61. Nauman J, Wolff J. Iodide prophylaxis in Poland after the Chernobyl reactor accident: benefits and risks. Am J Med. 1993;94(5):524-532. (PubMed)
62. Nuclear Regulatory Commission. Consideration of potassium iodide in emergency plans. Nuclear Regulatory Commission. Final rule. Fed Regist. 2001;66(13):5427-5440. (PubMed)
63. Guray M, Sahin AA. Benign breast diseases: classification, diagnosis, and management. Oncologist. 2006;11(5):435-449. (PubMed)
64. Anil C, Guney T, Gursoy A. The prevalence of benign breast diseases in patients with nodular goiter and Hashimoto's thyroiditis. J Endocrinol Invest. 2015;38(9):971-975. (PubMed)
65. Bhargav PR, Mishra A, Agarwal G, Agarwal A, Verma AK, Mishra SK. Prevalence of hypothyroidism in benign breast disorders and effect of thyroxine replacement on the clinical outcome. World J Surg. 2009;33(10):2087-2093. (PubMed)
66. Giustarini E, Pinchera A, Fierabracci P, et al. Thyroid autoimmunity in patients with malignant and benign breast diseases before surgery. Eur J Endocrinol. 2006;154(5):645-649. (PubMed)
67. Eskin BA, Grotkowski CE, Connolly CP, Ghent WR. Different tissue responses for iodine and iodide in rat thyroid and mammary glands. Biol Trace Elem Res. 1995;49(1):9-19. (PubMed)
68. Ghent WR, Eskin BA, Low DA, Hill LP. Iodine replacement in fibrocystic disease of the breast. Can J Surg. 1993;36(5):453-460. (PubMed)
69. Kessler JH. The effect of supraphysiologic levels of iodine on patients with cyclic mastalgia. Breast J. 2004;10(4):328-336. (PubMed)
70. Murray CW, Egan SK, Kim H, Beru N, Bolger PM. US Food and Drug Administration's Total Diet Study: dietary intake of perchlorate and iodine. J Expo Sci Environ Epidemiol. 2008;18(6):571-580. (PubMed)
71. Zava TT, Zava DT. Assessment of Japanese iodine intake based on seaweed consumption in Japan: A literature-based analysis. Thyroid Res. 2011;4:14. (PubMed)
72. Leung AM, Braverman LE, Pearce EN. History of U.S. iodine fortification and supplementation. Nutrients. 2012;4(11):1740-1746. (PubMed)
73. Pennington JAT, Schoen SA, Salmon GD, Young B, Johnson RD, Marts RW. Composition of core foods of the U.S. food supply, 1982-1991. III. Copper, manganese, selenium, iodine. J Food Comp Anal. 1995;8:171-217.
74. Leung AM, Avram AM, Brenner AV, et al. Potential risks of excess iodine ingestion and exposure: statement by the american thyroid association public health committee. Thyroid. 2015;25(2):145-146. (PubMed)
75. Maalouf J, Barron J, Gunn JP, Yuan K, Perrine CG, Cogswell ME. Iodized salt sales in the United States. Nutrients. 2015;7(3):1691-1695. (PubMed)
76. Dasgupta PK, Liu Y, Dyke JV. Iodine nutrition: iodine content of iodized salt in the United States. Environ Sci Technol. 2008;42(4):1315-1323. (PubMed)
77. Rasmussen LB, Ovesen L, Christensen T, et al. Iodine content in bread and salt in Denmark after iodization and the influence on iodine intake. Int J Food Sci Nutr. 2007;58(3):231-239. (PubMed)
78. Charlton KE, Yeatman H, Brock E, et al. Improvement in iodine status of pregnant Australian women 3 years after introduction of a mandatory iodine fortification programme. Prev Med. 2013;57(1):26-30. (PubMed)
79. Clifton VL, Hodyl NA, Fogarty PA, et al. The impact of iodine supplementation and bread fortification on urinary iodine concentrations in a mildly iodine deficient population of pregnant women in South Australia. Nutr J. 2013;12:32. (PubMed)
80. Skeaff SA, Lonsdale-Cooper E. Mandatory fortification of bread with iodised salt modestly improves iodine status in schoolchildren. Br J Nutr. 2013;109(6):1109-1113. (PubMed)
81. Eltom M, Elnagar B, Sulieman EA, et al. The use of sugar as a vehicle for iodine fortification in endemic iodine deficiency. Int J Food Sci Nutr. 1995;46(3):281-289. (PubMed)
82. Charoensiriwatana W, Srijantr P, Teeyapant P, Wongvilairattana J. Consuming iodine enriched eggs to solve the iodine deficiency endemic for remote areas in Thailand. Nutr J. 2010;9:68. (PubMed)
83. Chanthilath B, Chavasit V, Chareonkiatkul S, Judprasong K. Iodine stability and sensory quality of fermented fish and fish sauce produced with the use of iodated salt. Food Nutr Bull. 2009;30(2):183-188. (PubMed)
84. Weng HX, Liu HP, Li DW, Ye M, Pan L, Xia TH. An innovative approach for iodine supplementation using iodine-rich phytogenic food. Environ Geochem Health. 2014;36(4):815-828. (PubMed)
85. Zimmermann MB. Symposium on 'Geographical and geological influences on nutrition': Iodine deficiency in industrialised countries. Proc Nutr Soc. 2010;69(1):133-143. (PubMed)
86. Laurberg P, Nohr SB, Pedersen KM, et al. Thyroid disorders in mild iodine deficiency. Thyroid. 2000;10(11):951-963. (PubMed)
87. Nishiyama S, Mikeda T, Okada T, Nakamura K, Kotani T, Hishinuma A. Transient hypothyroidism or persistent hyperthyrotropinemia in neonates born to mothers with excessive iodine intake. Thyroid. 2004;14(12):1077-1083. (PubMed)
88. Davies L, Morris LG, Haymart M, et al. American Association of Clinical Endocrinologists and American College of Endocrinology Disease State Clinical Review: The Increasing Incidence of Thyroid Cancer. Endocr Pract. 2015;21(6):686-696. (PubMed)
89. Zimmermann MB, Galetti V. Iodine intake as a risk factor for thyroid cancer: a comprehensive review of animal and human studies. Thyroid Res. 2015;8:8. (PubMed)
90. Ahmed S, Van Gelder IC, Wiesfeld AC, Van Veldhuisen DJ, Links TP. Determinants and outcome of amiodarone-associated thyroid dysfunction. Clin Endocrinol (Oxf). 2011;75(3):388-394. (PubMed)
91. McKnight RF, Adida M, Budge K, Stockton S, Goodwin GM, Geddes JR. Lithium toxicity profile: a systematic review and meta-analysis. Lancet. 2012;379(9817):721-728. (PubMed)
92. Kurnik D, Loebstein R, Farfel Z, Ezra D, Halkin H, Olchovsky D. Complex drug-drug-disease interactions between amiodarone, warfarin, and the thyroid gland. Medicine (Baltimore). 2004;83(2):107-113. (PubMed)
93. US NRC. Health Implications of Perchlorate Ingestion The National Academies Press. Available at: http://www.nap.edu/openbook.php?record_id=11202. Accessed 08/11/2015.
94. US EPA. Perchlorate and Perchlorate Salts. 02/18/2005. http://www.epa.gov/iris/subst/1007.htm. Accessed 08/11/2015.
Zinc
Contenido
Resumen
- El zinc es un mineral nutricional esencial para funciones catalíticas, estructurales, y regulatorias en el cuerpo. (Más información)
- La deficiencia severa de zinc es una enfermedad rara y es causada por una condición hereditaria llamada. La deficiencia de zinc adquirida se debe principalmente a síndromes de malabsorción y alcoholismo crónico. (Más información)
- La deficiencia de zinc dietético es bastante común en el mundo en desarrollo, afectando a un estimado de 2 mil millones de personas. El consumo de dietas altas en fitato y que carecen de alimentos de origen animal provocan una deficiencia de zinc en estas poblaciones. (Más información)
- La ingesta diaria recomendada (IDR) para hombres y mujeres adultos es de 11 mg/día y 8 mg/día de zinc, respectivamente. (Más información)
- El consumo a largo plazo de zinc en exceso del nivel máximo de ingesta tolerable (NM; 40 mg/día para adultos) puede provocar deficiencia de cobre. (Más información)
- La deficiencia de zinc dietético se ha asociado con deterioro de crecimiento y desarrollo en niños, complicaciones del embarazo, y disfunción inmune con una mayor susceptibilidad a infecciones. (Más información)
- La suplementación con dosis de zinc superiores a la NM es efectiva para reducir la duración de los síntomas del resfriado común. El uso de zinc en dosis diarias de 50 a 180 mg durante una o dos semanas no ha tenido efectos secundarios graves. (Más información)
- La evidencia actual sugiere que el zinc suplementario puede ser útil en el tratamiento de condiciones crónicas, como la degeneración macular relacionada con la edad, la diabetes mellitus, la enfermedad de Wilson y el VIH/SIDA. (Más información)
-
La biodisponibilidad de zinc es relativamente alta en carne, huevos, y mariscos; el zinc es menos biodisponible en granos enteros y legumbres debido a su alto contenido en fitato que inhibe la absorción de zinc. (Más información)
El zinc es un elemento traza esencial para todas las formas de vida. La deficiencia clínica de zinc en humanos se describió por primera vez en 1961, cuando el consumo de dietas con baja biodisponibilidad de zinc debido al alto contenido de fitato (véase Fuentes alimenticias) se asoció con "enanismo nutricional adolescente" en el Medio Oriente (1). Desde entonces, varios expertos han reconocido la insuficiencia de zinc como un importante problema de salud pública, especialmente en países de bajos recursos (2, 3).
Función
Numerosos aspectos del metabolismo celular dependen del zinc. El zinc juega un papel importante en el crecimiento y el desarrollo, la función inmune, la neurotransmisión, la visión, la reproducción, y el transporte intestinal de iones (4). Utilizando enfoques de minería de datos, se ha estimado que más de 3,000 proteínas en humanos tienen sitios de unión de zinc funcionales (5). A nivel celular, la función del zinc se puede dividir en tres categorías: (1) catalítica, (2) estructural, y (3) regulatoria (3) (6).
Función catalítica
Más de 50 enzimas diferentes dependen del zinc por su capacidad de catalizar reacciones químicas vitales (7). Las enzimas dependientes de zinc se pueden encontrar en las seis clases de enzimas (8), según lo definido por la Unión Internacional de Bioquímica y Biología Molecular (9). Durante las reacciones enzimáticas, el zinc puede tener una función catalítica directa o un rol estructural (por ej., estabilizar la estructura de las enzimas catalíticas; véase más abajo).
Función estructural
El zinc juega un papel esencial en el plegamiento de algunas proteínas. Una estructura con forma de dedo, conocida como motivo dedo de zinc, estabiliza la estructura de varias proteínas. Los ejemplos de proteínas con dedos de zinc incluyen la superfamilia de receptores nucleares que se unen y responden a los esteroides y otras moléculas, como los estrógenos, las hormonas tiroideas, la vitamina D, y la vitamina A (10). Los motivos de dedos de zinc en la estructura de los receptores nucleares les permiten unirse al ADN y actuar como factores de transcripción para regular la expresión de los genes (ver Función regulatoria). Los motivos de dedos de zinc también están involucrados en las interacciones de proteínas con otras proteínas, ribonucleótidos, y lípidos (6). La eliminación de zinc de las proteínas que contienen zinc da como resultado el plegamiento de proteínas y la pérdida de función.
Las metalotioneínas son ejemplos de proteínas con un motivo de unión a zinc. Las metalotioneínas son pequeñas proteínas ricas en cisteína de unión a metales con una alta afinidad por el zinc. Trabajan en concierto con transportadores de zinc, regulando las concentraciones de zinc libre en el citosol (11). Las metalotioneínas también están involucradas en la regulación de la homeostasis de los iones metálicos, la defensa celular contra el estrés oxidativo, y la desintoxicación de metales pesados (11, 12).
La enzima antioxidante, la cobre-zinc superóxido dismutasa 1 (SOD 1), está hecha de dos dímeros idénticos, cada uno de los cuales incluye un sitio activo con un ion catalítico de cobre y un hierro estructural de zinc. La desmetalación de SOD1 se ha implicado en la formación de agregados amiloides en algunas formas de esclerosis lateral amiotrófica (ELA) hereditaria – una enfermedad de la neurona motora que conduce a atrofia muscular y parálisis (13).
Función regulatoria
Se ha descubierto que las proteínas de dedos de zinc regulan la expresión de genes al actuar como factores de transcripción (véase arriba). El zinc también juega un papel en la señalización celular a través del factor de transcripción 1 (MTF1) que se une al elemento de respuesta al metal (MRE); MTF1 tiene un dominio de dedo de zinc que permite su unión a secuencias MRE en el promotor de genes diana y la posterior expresión de genes sensibles al zinc (6). El zinc también puede tener una función regulatoria directa, modulando la actividad de las enzimas de señalización celular y los factores de transcripción (6). El zinc extracelular también puede estimular un receptor sensor de zinc que desencadena la liberación de calcio intracelular, un segundo mensajero en las vías de señalización (14). Se ha encontrado que el zinc influye en la liberación de hormonas (ver Diabetes mellitus tipo 2) (15) y la transmisión del impulso nervioso (16).
Interacción con nutrientes
Cobre
Consumir grandes cantidades de zinc (50 mg/día o más) por un período de semanas puede interferir en la biodisponibilidad del cobre. Las ingestas elevadas de zinc inducen la síntesis intestinal de una proteína que se une al cobre denominada metalotioneína (véase el artículo sobre Cobre). La metalotioneína atrapa al cobre dentro de las células intestinales y evita su absorción sistémica (véase Enfermedad de Wilson). Las ingestas habituales de zinc no alteran la absorción de cobre, y las ingestas elevadas de cobre no afectan la absorción de zinc (17).
Hierro
El hierro y el zinc compiten por las vías de absorción (18). Los niveles de hierro suplementario (38-65 mg/día de hierro elemental) pero no dietético pueden disminuir la absorción de zinc (18, 19). Esta interacción es preocupante en el manejo de la suplementación con hierro durante el embarazo y la lactancia y ha llevado a algunos expertos a recomendar suplementos de zinc para mujeres embarazadas y lactantes que toman suplementos de hierro (20, 21). No se ha demostrado que la fortificación de alimentos con hierro afecte negativamente la absorción de zinc (22). En un estudio controlado con placebo, la suplementación con zinc (10 mg/día) durante tres meses en niños de ocho a nueve años disminuyeron significativamente las concentraciones séricas de hierro, pero no hasta el punto de causar anemia (23). Estudios aleatorios controlados adicionales informaron un empeoramiento del estatis nutricional del hierro con la administración crónica de suplementos de zinc (24, 25).
Calcio
Los altos niveles de calcio dietético perjudican la absorción de zinc en animales, pero no está claro si esto ocurre en humanos (17). Un estudio mostró que el aumento de la ingesta de calcio de las mujeres posmenopáusicas en 890 mg/día en forma de leche o fosfato de calcio (ingesta total de calcio, 1,360 mg/día) redujeron la absorción de zinc y el equilibrio de zinc en mujeres posmenopáusicas (26). Sin embargo, otro estudio encontró que aumentar la ingesta de calcio de las adolescentes en 1,000 mg/día en forma de malato de citrato de calcio (ingesta total de calcio, 1,667 mg/día) no afectó la absorción o el equilibrio de zinc (27). El calcio en combinación con el fitato podría afectar la absorción de zinc, lo que sería particularmente relevante para las personas que con frecuencia consumen tortillas hechas con cal (es decir, óxido de calcio). Un estudio en 10 mujeres sanas (rango de edad, 21-47 años) encontró que la ingesta alta de calcio dietético (~1,800 mg/día) no perjudicaba la absorción de zinc independientemente del contenido de fitato en la dieta (28). Para obtener más información sobre el fitato, ver Fuentes alimenticias.
Folato
La biodisponibilidad del folato dietético (vitamina B9) aumenta por la acción de una enzima dependiente de zinc. En consecuencia, algunos estudios encontraron que la baja ingesta de zinc disminuyó la absorción de folato. También se sugirió que la suplementación con ácido fólico — la forma sintética de ácido fólico — podría afectar la utilización de zinc en individuos con un estatus marginal de zinc (17, 29). Sin embargo, un estudio informó que la suplementación con una dosis relativamente alta de ácido fólico (800 µg/día) durante 25 días no alteró la absorción o el estatus del zinc en un grupo de estudiantes que fueron alimentados con una dieta baja en zinc (3.5 mg/día) (30).
Vitamina A
El zinc y la vitamina A interactúan de varias maneras. El zinc es un componente de la proteína de unión al retinol, una proteína necesaria para transportar la vitamina A en la sangre. El zinc también se requiere para la enzima que convierte el retinol (vitamina A) en retinal. Esta última forma de vitamina A es necesaria para la síntesis de rodopsina, una proteína en el ojo que absorbe la luz y, por lo tanto, participa en la adaptación a la oscuridad. La deficiencia de zinc se ha asociado con una disminución de la liberación de vitamina A del hígado, lo que puede contribuir a los síntomas de ceguera nocturna que se observan con la deficiencia de zinc (31, 32).
Deficiencia
Deficiencia severa de zinc
Gran parte de lo que se sabe sobre la deficiencia severa de zinc se deriva del estudio de individuos nacidos con acrodermatitis enteropática, un trastorno genético resultante de la absorción y el transporte deteriorados de zinc (33). Los síntomas de la deficiencia severa de zinc incluyen la desaceleración o el cese del crecimiento y el desarrollo, maduración sexual retrasada, erupciones cutáneas características, diarrea crónica y grave, deficiencias del sistema inmunitario, cicatrización de heridas deteriorada, disminución del apetito, sensación de sabor alterada, ceguera nocturna, hinchazón y opacamiento de la córnea, y trastornos del comportamiento. Antes de que se conociera la causa de la acrodermatitis enteropática, los pacientes generalmente morían en la infancia. La terapia oral con zinc produce la remisión completa de los síntomas, aunque debe mantenerse indefinidamente en individuos con el trastorno genético (33, 34).
Deficiencia leve de zinc
Ahora se reconoce que la deficiencia leve de zinc contribuye a una serie de problemas de salud, especialmente comunes en niños que viven en países de bajos recursos. Se estima que 2,000 millones de personas en todo el mundo se ven afectadas por la deficiencia de zinc dietético (3). La falta de un indicador sensible y específico de la deficiencia marginal de zinc dificulta el estudio científico de sus implicaciones para la salud (8). Sin embargo, los ensayos controlados de la suplementación moderada de zinc han demostrado que la deficiencia marginal de zinc contribuye al deterioro del desarrollo físico y neuropsicológico y al aumento de la susceptibilidad a infecciones potencialmente mortales en niños pequeños (34). De hecho, se ha estimado que la deficiencia de zinc causa más de 450,000 muertes al año en niños menores de cinco años, lo que representa el 4.4% de las muertes infantiles mundiales (35). Para una discusión más detallada de la relación de la deficiencia de zinc con los problemas de salud, véase la sección de Prevención de Enfermedades.
En los países industrializados, es poco probable que la deficiencia de zinc dietético cause deficiencia severa de zinc en individuos sin algún trastorno genético, malabsorción de zinc o condiciones de mayor pérdida de zinc, como quemaduras severas o diarrea prolongada. También se ha informado de deficiencia severa de zinc en individuos sometidos a nutrición parenteral total sin zinc, en aquellos que abusan del alcohol, y en aquellos que toman ciertos medicamentos como la penicilamina (ver Interacción con drogas/fármacos) (36).
Individuos en riesgo de deficiencia de zinc (6, 36-38):
- Los bebés prematuros y de bajo peso al nacer
- Los bebés mayores lactantes y niños pequeños con una ingesta inadecuada de alimentos complementarios ricos en zinc
- Niños y adolescentes
- Mujeres embarazadas y en lactancia (amamantando), especialmente adolescentes
- Pacientes que reciben nutrición parenteral total (alimentación intravenosa)
- Individuos con malnutrición, incluyendo a aquellos con malnutrición proteíno-energética y anorexia nerviosa
- Individuos con diarrea persistente o severa
- Individuos con síndromes de malabsorción, incluyendo enfermedad celíaca y síndrome de intestino corto
- Individuos con enfermedad inflamatorias del intestino, incluyendo enfermedad de Crohn y colitis ulcerosa
- Individuos con enfermedad hepática alcohólica con una mayor excreción de zinc urinario y bajos niveles hepáticos de zinc
- Individuos con enfermedad crónica renal
- Individuos con anemia de células falciformes
- Las personas que usan medicamentos que disminuyen la absorción de zinc intestinal, aumentan la excreción de zinc, o menoscaben la utilización de zinc (véase Interacción con drogas/fármacos)
- Adultos mayores (mayores de 65 años)
- Vegetarianos estrictos: La necesidad de zinc dietético podría ser hasta un 50% más alta para los vegetarianos estrictos cuya alimento básico principal son granos y legumbres, debido a que los altos niveles de ácido fítico en estos alimentos disminuyen la absorción de zinc (véase Fuentes alimenticias) (29).
Biomarcadores del estatus de zinc
Actualmente, no hay un marcador biológico sensible y específico para detectar la deficiencia de zinc en los seres humanos. Las concentraciones bajas de zinc en plasma o suero se usan típicamente como indicadores del estatus del zinc en poblaciones y en estudios de intervención, pero tienen una serie de limitaciones, incluida la falta de sensibilidad para detectar deficiencia marginal de zinc, variaciones diurnas, y confusión por inflamación, estrés, y hormonas (38, 39).
La Ingesta Diaria Recomendada (IDR)
La ingesta diaria recomendada (IDR) de zinc se enumera por género y grupo de edad en la Tabla 1. Los bebés, niños, adolescentes, y mujeres embarazadas y lactantes tienen un mayor riesgo de deficiencia de zinc. Dado que no se dispone fácilmente de un indicador sensible del estatus nutricional del zinc, la IDR para el zinc se basa en varios indicadores diferentes del estatus nutricional del zinc y representa la ingesta diaria que probablemente prevenga la deficiencia en casi todos los individuos en un grupo específico de edad y género (29).
| Etapa de la Vida | Edad | Machos (mg/día) | Hembras (mg/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 2 (IA) | 2 (IA) |
| Infantes | 7-12 meses | 3 | 3 |
| Niños | 1-3 años | 3 | 3 |
| Niños | 4-8 años | 5 | 5 |
| Niños | 9-13 años | 8 | 8 |
| Adolescentes | 14-18 años | 11 | 9 |
| Adultos | 19 años y más | 11 | 8 |
| Embarazo | 18 años o menos | - | 12 |
| Embarazo | 19 años y más | - | 11 |
| Período de lactancia | 18 años o menos | - | 13 |
| Período de lactancia | 19 años y más | - | 12 |
Prevención de Enfermedades o Condiciones Relacionadas a la Deficiencia de Zinc
Complicaciones del embarazo y resultados adversos del embarazo
Las estimaciones basadas en el suministro nacional de alimentos indican que la ingesta dietética de zinc es probablemente inadecuada en la mayoría de los países de ingresos bajos y medianos, especialmente en África subsahariana y Asia meridional (40). El estatus inadecuado de zinc durante el embarazo interfiere con el desarrollo fetal, y los recién nacidos prematuros de madres con deficiencia de zinc sufren retraso en el crecimiento y dermatitis y corren el riesgo de infecciones, enterocolitis necrotizante, enfermedad pulmonar crónica, y retinopatía del prematuro (4). La deficiencia materna de zinc también se ha asociado con una serie de complicaciones del embarazo y malos resultados. Un reciente estudio de caso y control realizado en un hospital iraní informó mayores probabilidades de malformaciones congénitas en recién nacidos de madres con bajas concentraciones séricas de zinc durante el último mes de embarazo (41). Una revisión de 2016 de 64 estudios observacionales encontró una relación inversa entre el estatus materno de zinc y la gravedad de la preeclampsia, así como entre la ingesta materna de zinc y el riesgo de recién nacidos con bajo peso al nacer (42). No hubo asociaciones aparentes entre el estatus materno de zinc y el riesgo de diabetes mellitus gestacional y parto prematuro. Sin embargo, las conclusiones de este análisis se vieron limitadas por el hecho de que la mayoría de los estudios observacionales se realizaron en mujeres de poblaciones que no estaban en riesgo de deficiencia de zinc (42).
Hasta la fecha, la evidencia disponible de los ensayos de intervención materna de zinc realizados en todo el mundo no respalda la recomendación de la suplementación de zinc de rutina durante el embarazo. Una revisión sistemática de 2015 y un meta-análisis de 21 ensayos aleatorios controlados en más de 17,000 mujeres y sus bebés encontraron una reducción del 14% en los partos prematuros con suplementos de zinc durante el embarazo, principalmente en mujeres de bajos ingresos (43). Sin embargo, este análisis no encontró suplementos de zinc para beneficiar otros indicadores de la salud materna o infantil, incluidos la muerte fetal o neonatal, el bajo peso al nacer, la edad gestacional pequeña, y la hipertensión inducida por el embarazo. Tampoco hubo efecto del zinc suplementario en la hemorragia posparto, las infecciones maternas, las malformaciones congénitas, y los resultados del desarrollo infantil (43). Una revisión reciente de 17 ensayos (de los cuales 15 se realizaron en países de bajos y medianos ingresos) encontró que la suplementación materna con múltiples micronutrientes (que incluyen, entre otros, zinc, hierro, y ácido fólico) redujo el riesgo de parto de recién nacidos con bajo peso y lactantes pequeños para la edad de gestación en comparación con el hierro suplementario con o sin ácido fólico (44). Si bien la suplementación con micronutrientes múltiples probablemente beneficiaría a las mujeres embarazadas con deficiencias de micronutrientes coexistentes en países de bajos y medianos ingresos, no hay evidencia para recomendar suplementación con zinc aisladamente en mujeres embarazadas de cualquier entorno (43, 45).
Deterioro del crecimiento y desarrollo
Retraso del crecimiento
Los retrasos significativos en el crecimiento lineal y el aumento de peso, conocidos como retraso del crecimiento o falta de crecimiento, son características comunes de la deficiencia leve de zinc en los niños. En las décadas de 1970 y 1980, se llevaron a cabo varios estudios aleatorios, controlados con placebo, en Denver, Colorado de suplementos de zinc en niños pequeños con retrasos significativos en el crecimiento. La suplementación moderada de zinc (5.7 mg/día) dio como resultado un aumento en las tasas de crecimiento en comparación con el placebo (46). Varios meta-análisis de datos de crecimiento de ensayos de intervención de zinc han confirmado la ocurrencia generalizada de deficiencia de zinc limitante del crecimiento en niños pequeños, especialmente en países de bajos y medianos ingresos (47-49). Una revisión sistemática y un meta-análisis de 2018 identificaron 54 ensayos que examinaron el impacto de los suplementos de zinc durante la infancia (en promedio, 7.6 mg/día durante 30.9 semanas) o en la infancia (en promedio, 8.5 mg/día durante 38.9 semanas) en las mediciones antropométricas en niños (50). Hubo evidencia de un efecto positivo del zinc suplementario en la estatura, peso y el puntaje Z peso/edad (WAZ, en inglés) de niños, pero tampoco en el puntaje Z de talla/edad (HAZ, en inglés) o el puntaje Z peso/talla (WHZ, en inglés). Además, la suplementación con zinc no disminuyó los riesgos de bajo peso (WAZ <-2 desviación estándar [SD]), emaciación (WHZ <-2 SD) o retraso en el crecimiento (HAZ <-2 SD) en niños (50). Aunque los mecanismos exactos para el efecto limitante del crecimiento de la deficiencia de zinc no se conoce, la investigación indica que la disponibilidad de zinc afecta a los sistemas de señalización celular que coordinan la respuesta a la hormona reguladora del crecimiento, el factor de crecimiento similar a la insulina-1 (IGF-1) (51).
Desarrollo mental y psicomotor retrasado en niños pequeños
Una nutrición adecuada es esencial para el crecimiento y el desarrollo del cerebro, especialmente durante los primeros 1,000 días de vida — un período crítico de desarrollo para todos los órganos y sistemas, desde la concepción hasta los 24 meses de edad (52). Los estudios en animales han establecido que la deficiencia de zinc en los primeros años de vida interfiere con desarrollo cerebral normal y funciones cognitivas (revisado en 53). Los datos sobre el efecto de la suplementación con zinc durante el embarazo en los resultados neurológicos y psicomotores de los bebés son muy limitados. En un ensayo aleatorizado, controlado con placebo en mujeres afroamericanas, la suplementación materna diaria con 25 mg de zinc de aproximadamente 19 semanas de gestación no tuvo efecto en los puntajes de las pruebas de desarrollo neurológico en sus hijos a los cinco años de edad (54).
Varios estudios han informado sobre el efecto de la suplementación de zinc postnatal en el desarrollo mental y motor. Dos ensayos aleatorios controlados tempranos, uno realizado en India y el otro en Guatemala, sugirieron que la suplementación postnatal con 10 mg/día de zinc resultó en que los niños pequeños fueran más vigorosos (55) y funcionalmente activos (56). En un ensayo realizado en recién nacidos brasileños de bajos ingresos familias y con un peso de entre 1,500 g y 2,499 g al nacer, ni la suplementación con zinc durante ocho semanas con 1 mg/día o 5 mg/día mejoró el desarrollo mental y psicomotor a los 6 o 12 meses de edad en comparación con un placebo y se evaluó utilizando las escalas de Bayley del desarrollo infantil (BSID, en inglés) para el Índice de Desarrollo Mental (IDM) y el Índice de Desarrollo Psicomotor (IDP) (57). Además, un ensayo aleatorizado, controlado con placebo, doble ciego en recién nacidos chilenos (pesos al nacer >2,300 g) de familias de bajos ingresos no reportó ningún efecto de la suplementación con zinc (5 mg/día) en los índices de desarrollo mental y psicomotor a los 6 y 12 meses (58). Otros dos ensayos encontraron que el zinc suplementario no logró mejorar el IDM o IDP a los 12 meses de edad cuando se administró zinc (10 mg/día) a bebés de seis meses durante seis meses (59) o al final de la intervención en niños pequeños de 12-18 meses cuando se administró zinc (30 mg/día) durante cuatro meses (60). Una revisión Cochrane de 2012 de ocho ensayos clínicos no encontró evidencia de que la administración de suplementos de zinc postnatal mejoró el desarrollo mental o motor de bebés y niños de poblaciones con un estatus de zinc presumiblemente inadecuado (61).
Función del sistema inmunitario deteriorada
La ingesta adecuada de zinc es esencial para mantener la integridad del sistema inmunitario (62), específicamente para el desarrollo normal y la función de las células que median las respuestas inmunes innatas (neutrófilos, macrófagos, y células natural killer) y adaptativas (linfocitos-B y linfocitos-T) (63). Debido a que los patógenos también requieren que el zinc prospere e invada, un mecanismo de defensa antimicrobiano bien establecido en el cuerpo secuestra el zinc libre de los microbios (64). Otro mecanismo opuesto consiste en intoxicar los microbios intracelulares dentro de los macrófagos con exceso de zinc (65). A través del debilitamiento innato y respuestas inmunes adaptables, la deficiencia de zinc disminuye la capacidad del cuerpo para combatir los patógenos (63, 64). Como consecuencia, las personas con deficiencia de zinc experimentan una mayor susceptibilidad a una variedad de agentes infecciosos (66).
Mayor susceptibilidad a enfermedades infecciosas en niños
Diarrea: el zinc promueve la resistencia de la mucosa a las infecciones al apoyar la actividad de las células inmunes y la producción de anticuerpos contra los patógenos invasores (63, 64, 67). Por lo tanto, una deficiencia de zinc aumenta la susceptibilidad a las infecciones intestinales y constituye un importante contribuyente a las enfermedades diarreicas en niños (66). A su vez, la diarrea persistente contribuye a la deficiencia de zinc y la desnutrición (66). Investigaciones indican que la deficiencia de zinc también puede potenciar los efectos de las toxinas producidas por bacterias que causan diarrea como E. coli (68). Se estima que las enfermedades diarreicas son responsables de muertes anuales de alrededor de 500,000 niños menores de cinco años en países de ingresos bajos y medianos (69). Se ha demostrado que la administración de suplementos de zinc en combinación con la terapia de rehidratación oral reduce significativamente la duración y la gravedad de la diarrea infantil aguda y persistente y aumenta la supervivencia en una serie de ensayos aleatorios controlados (70). Un meta-análisis de 2016 de ensayos aleatorios controlados encontraron que la suplementación con zinc redujo la duración de la diarrea aguda en un día en niños >6 meses que presentaron signos de desnutrición (5 ensayos; 419 niños) (71). Sin embargo, había poca evidencia que sugiriera que el zinc podría ser tan eficaz para disminuir la duración de los episodios diarreicos agudos en niños <6 meses y en niños bien alimentados >6 meses. La administración de suplementos de zinc también disminuyó la duración de la diarrea persistente en niños en más de medio día (5 ensayos; 529 niños) (71).
La Organización Mundial de la Salud (OMS) y el Fondo de las Naciones Unidas para la Infancia (UNICEF) actualmente recomiendan suplementar a los niños pequeños con 10 a 20 mg/día de zinc como parte del tratamiento para los episodios diarreicos agudos y prevenir episodios adicionales en los dos o tres meses. después de la suplementación con zinc (72).
Neumonía: La neumonía — causada por infecciones virales o bacterianas del tracto respiratorio inferior (LRTI, por sus siglas en inglés) — representa casi 1 millón de muertes anuales en niños, principalmente en países de ingresos bajos y medianos (69). Vacunas contra Haemophilus influenzae tipo B, neumococo, tos ferina, y el sarampión pueden ayudar a prevenir la neumonía (73). Según un informe de la OMS de 2009 sobre los factores de riesgo de enfermedad, la deficiencia de zinc puede ser responsable del 13% de todos los casos de LRTI, principalmente neumonía y gripe, en niños menores de 5 años (74). En consecuencia, un meta-análisis de 2016 de seis ensayos encontró que la suplementación con zinc en niños menores de 5 años disminuyó el riesgo de neumonía en un 13% (75). Sin embargo, no está claro si el zinc suplementario, junto con la terapia con antibióticos, es beneficioso en el tratamiento de la neumonía. Un reciente ensayo aleatorio, controlado con placebo realizado en niños gambianos que no tenían deficiencia de zinc no mostró ningún beneficio de la suplementación con zinc (10 mg/día o 20 mg/día [dependiendo de la edad del niño] durante 7 días) administrado junto con antibióticos en el tratamiento de la neumonía severa (76). Un meta-análisis de 2018 de cinco ensayos (1,822 participantes) no encontró mejora cuando se utilizó zinc como un complemento al tratamiento con antibióticos en niños con neumonía (77). Sin embargo, hubo evidencia de que el zinc suplementario disminuyó el riesgo de mortalidad relacionada con la neumonía (3 ensayos; 1,318 participantes) (77).
Malaria: Los primeros estudios han indicado que la suplementación con zinc puede reducir la incidencia de ataques clínicos de malaria en niños (78). Un ensayo controlado con placebo en niños en edad preescolar en Papúa Nueva Guinea encontró que la suplementación con zinc disminuyó la frecuencia de asistencia al centro de salud debido a la malaria de Plasmodium falciparum en un 38% (79). Además, el número de episodios de malaria acompañados de altas concentraciones de parásitos circulantes se redujo en un 68%, lo que sugiere que la suplementación con zinc puede ser beneficiosa para prevenir episodios más graves de malaria. Sin embargo, un ensayo de seis meses en más de 700 niños de África occidental no encontró ninguna diferencia en la frecuencia o gravedad de los episodios de malaria entre los niños suplementados con zinc y los que recibieron un placebo (80). Otro ensayo aleatorio controlado informó que los suplementos de zinc no beneficiaron niños en edad preescolar con malaria aguda, y no complicada (81). También hay poca evidencia que sugiera que la suplementación con zinc podría reducir el riesgo de mortalidad relacionada con la malaria en niños (82). En la actualidad, no hay evidencia suficiente para sugerir un tratamiento profiláctico y/o terapéutico papel del zinc suplementario en el tratamiento de la malaria infantil (48). Un ensayo aleatorizado reciente controlado con placebo no proporcionó evidencia clara de un efecto protector del zinc (25 mg/día) administrado a mujeres de Tanzania durante su primer trimestre de gestación hasta el parto sobre el riesgo de infección por malaria placentaria (83).
Deterioro relacionado con la edad en la función inmune
El estatus inadecuado de zinc en sujetos de edad avanzada no es infrecuente y se cree que exacerba la disminución de la función inmune relacionada con la edad (84). En un estudio, las bajas concentraciones séricas de zinc en los residentes de hogares de ancianos se asociaron con un mayor riesgo de neumonía y relacionado a neumonía y mortalidad por todas las causas (85). Los ensayos que examinan los efectos de la suplementación con zinc en la función inmune en adultos de mediana edad y ancianos han arrojado resultados mixtos (revisado en 86). Algunos estudios mostraron efectos mixtos o ningún efecto de la suplementación con zinc en los parámetros de la función inmune (87-89). Sin embargo, se descubrió que la administración de suplementos de zinc tiene un impacto positivo en ciertos aspectos de la función inmune que se ven afectados por la deficiencia de zinc, como la disminución de la función de las células T (un tipo de linfocito) (90). Por ejemplo, un estudio aleatorio, controlado con placebo en adultos mayores de 65 años encontró que la suplementación con zinc (25 mg/día) durante tres meses aumentó las concentraciones en la sangre de células T colaboradoras y células T citotóxicas (91). Además, un ensayo aleatorio, doble ciego, controlado con placebo en 101 adultos mayores (de 50 a 70 años) con concentraciones normales de zinc en la sangre mostraron que la administración de suplementos de zinc a 15 mg/día durante seis meses mejoró la proporción de células T colaboradoras/células T citotóxicas, que tiende a disminuir con la edad y es un predictor de supervivencia (92). Sin embargo, el estudio también sugirió que una dosis de 30 mg/día de zinc podría reducir la cantidad de linfocitos-B, que juegan un papel central en la inmunidad humoral. Además, la suplementación con zinc no tuvo efecto sobre varios parámetros inmunes, incluidos los marcadores de inflamación, las medidas de capacidad fagocítica de granulocitos y monocitos, o la producción de citocinas por monocitos activados (92).
Un ensayo más reciente examinó el efecto de la suplementación diaria con múltiples micronutrientes, incluidos 5 mg o 30 mg de zinc durante tres meses, sobre el estado del zinc y los marcadores de la función inmune en participantes ancianos institucionalizados (edad media, >80 años) con bajas concentraciones séricas de zinc (93). El estatus de zinc mejoró con la dosis de 30 mg/día, pero no con 5 mg/día — pero las personas con más deficiencia de zinc no lograron alcanzar las concentraciones séricas normales de zinc dentro del período de intervención. El número de células T circulantes también aumentó significativamente en aquellos que tomaron el suplemento de micronutrientes con la dosis más alta versus baja de zinc (93).
Se requiere más investigación antes de que se pueda recomendar la administración de suplementos de zinc a los adultos mayores, especialmente a aquellos sin síntomas de disminución de inmunidad. No obstante, la alta prevalencia de deficiencia de zinc entre los adultos mayores institucionalizados debe abordarse y probablemente mejoraría el rendimiento de sus sistemas inmunes (86).
Diabetes mellitus tipo 2
Existe una estrecha relación entre el zinc y la acción de la insulina. Específicamente, en las células β pancreáticas, el zinc está involucrado en la síntesis de insulina y el almacenamiento en vesículas secretoras. El zinc se libera con la hormona cuando aumentan las concentraciones de glucosa en la sangre (15). También se entiende que el zinc estimula la absorción y el metabolismo de la glucosa por los tejidos sensibles a la insulina al activar la vía de señalización de insulina intracelular (94). Polimorfismos de un solo nucleótido (SNP) en el gen SLC30A8 (miembro 8 de la familia 30 de portadores de solutos), que codifica un transportador de zinc que se localiza junto con la insulina en las células β, se ha asociado con mayores riesgos de diabetes mellitus tipo 1 y tipo 2 (95), aunque se descubrió que el riesgo de diabetes mellitus tipo 2 se redujo con variantes raras de truncamiento de proteínas del gen (96). El primer estudio de cohorte prospectivo para examinar el riesgo de diabetes tipo 2 en relación con la ingesta de zinc — el Estudio de Salud de Enfermeras (NHS) — siguió a 82,297 enfermeras registradas en EE.UU. durante 24 años. El análisis de datos mostró un riesgo 8% menor de diabetes tipo 2 con la ingesta más alta versus más baja de zinc dietético (valores medios, 11.8 mg/día versus 4.9 mg/día) (97). Este hallazgo fue consistente con el resultado del Estudio Longitudinal Australiano sobre la salud de la mujer (ALSWH) que inscribió a 8,921 mujeres durante seis años y mostró un riesgo 50% menor de diabetes con la ingesta más alta versus más baja de zinc dietético ajustado por energía (98). Tanto los estudios NHS como ALSWH también reportaron un riesgo reducido de diabetes con relaciones mayores versus relaciones más bajas de zinc a hierro hemo dietético (97, 98), aunque la importancia no está clara ya que se sabe que el hierro no hemo, en lugar del hierro hemo, interfiere con la absorción de zinc dietético (véase Interacción con nutrientes). El hierro hemo puede ser un indicador del consumo de carne roja, que se ha asociado positivamente con el riesgo de diabetes tipo 2 (99). Sin embargo, otros dos estudios de cohorte prospectivos: el Estudio Multiétnico de Aterosclerosis (MESA; 4,982 participantes) y el Estudio de Dieta y Salud (232,007 participantes) de los Institutos Nacionales de la Asociación Estadounidense de Jubilados (NIH-AARP) — no pudo encontrar evidencia de una asociación entre la ingesta de zinc y el riesgo de diabetes tipo 2 (100, 101). Otro estudio de cohorte prospectivo reciente, la dieta Malmo y el estudio de Cáncer en 26,132 participantes suecos de mediana edad seguidos durante 19 años, encontró un mayor riesgo de diabetes con una mayor ingesta de zinc dietético y un menor riesgo de diabetes en los usuarios de suplementos de zinc (en comparación con los no usuarios) y en aquellos con una mayor proporción de ingesta de zinc a hierro (102). Los autores reportaron una asociación inversa más fuerte entre la proporción de ingesta de zinc a hierro y el riesgo de diabetes entre los participantes obesos que portaban un genotipo SLC30A8 específico (102).
Los resultados de algunos estudios de intervención a corto plazo sugieren que los suplementos de zinc pueden mejorar el manejo de la glucosa en sujetos con prediabetes. Una revisión sistemática de 2015 identificó tres ensayos cortos (de 4 a 12 semanas) realizados en adultos con prediabetes y encontró poca evidencia de una mejora en la resistencia a la insulina con suplementos de zinc (103). Sin embargo, un ensayo aleatorizado, y controlado con placebo de 2016 en 55 bangladesíes con prediabetes mostró que la suplementación diaria con sulfato de zinc (30 mg/día durante 6 meses) mejoró la glucosa en la sangre en ayunas, así como las medidas de la función de las células β y la sensibilidad a la insulina (104). Observaciones similares se realizaron en otro ensayo reciente en 100 individuos de Sri Lanka para recibir, de manera aleatoria, suplementación con zinc diaria (20 mg de zinc elemental) o un placebo durante un año (105). El zinc suplementario mejoró el estatus del zinc y las medidas de control glucémico (105). Se necesitan estudios a gran escala, y a largo plazo para proporcionar conclusiones definitivas sobre el beneficio potencial de la suplementación con zinc en sujetos con riesgo de diabetes tipo 2.
Tratamiento de Enfermedad
Las dosis de zinc suplementario en muchos de los ensayos clínicos mencionados a continuación excedieron el nivel máximo de ingesta tolerable (NM). Tal alto consumo de zinc suplementario puede conducir a efectos adversos para la salud con el uso prolongado (véase Seguridad).
Enfermedad de Wilson
La proteína, ATP7B, es responsable de la excreción de cobre hepático en el tracto biliar, y su deterioro en la enfermedad de Wilson resulta en una mayor concentración de cobre 'libre' (es decir, no unido a la proteína portadora de cobre, ceruloplasmina) en la sangre, una mayor excreción de cobre en la orina (hipercupriuria), la deposición de cobre en parte de la córnea (formando anillos de Kayser-Fleischer), y la acumulación de cobre en el hígado y el cerebro (106). Esta condición hereditaria es progresiva y mortal sin tratamiento. El estándar de atención para pacientes sintomáticos generalmente incluye una fase inicial (alrededor de 2 a 6 meses) de quelación de cobre con agentes como penicilamina o trientina (trietilentetramina) seguida de una terapia de mantenimiento de por vida con penicilamina y/o trientina y/o sales de zinc (107). Los pacientes que se presentan sin síntomas pueden tratarse con dosis terapéuticas de mantenimiento de un agente quelante o con zinc (108). La metalotioneína inducida por zinc en la mucosa intestinal une el cobre y evita su absorción (véase Interacción con nutrientes). Cada vez hay más pruebas que sugieren que las sales de zinc son una alternativa más segura, más barata, y eficaz a los agentes quelantes de metales — que se han asociado con un empeoramiento de los síntomas durante la fase inicial del tratamiento en algunos pacientes (109). El uso de zinc es recomendado como seguro y eficaz en pacientes pediátricos (110, 111) y adultos (112-114).
Resfriado común
Pastillas de zinc
No existe un tratamiento comprobado para el resfriado común (115). Se ha recomendado el uso de pastillas de zinc dentro de las 24 horas posteriores a la aparición de los síntomas del resfriado, y la ingesta continua cada dos o tres horas mientras se está despierto hasta que los síntomas desaparezcan, para reducir la duración del resfriado común (116). Varios ensayos clínicos que examinan el efecto del zinc se han publicado hasta la fecha. Una revisión sistemática y un meta-análisis de 2012 de 13 ensayos aleatorios controlados reportó que la suplementación con zinc en forma de pastillas o jarabe acortó la duración de los síntomas del resfriado, pero hubo una heterogeneidad significativa (efectos inconsistentes en los estudios incluidos) para los resultados primarios (117). Una revisión Cochrane de 2013 confirmó que el zinc oral administrado dentro de las 24 horas posteriores al inicio de los síntomas podría reducir la duración de los síntomas del resfriado (14 ensayos, 1,656 participantes) (118). Los análisis de subgrupos también sugirieron que el zinc oral fue efectivo independientemente de la edad de los participantes (niños o adultos) y el tipo de formulación de zinc (pastillas de gluconato/acetato o jarabe de sulfato). Además, se observaron efectos beneficiosos sobre la duración del resfriado en los ensayos que proporcionaron más de 75 mg/día de zinc, pero no en los ensayos que utilizaron dosis más bajas. El análisis agrupado de cinco ensayos no encontró evidencia de un efecto del zinc oral sobre la gravedad de los síntomas del resfriado. El análisis de los resultados de los ensayos secundarios sugirió una resolución más rápida de los síntomas específicos del resfriado (tos, congestión nasal, drenaje nasal, dolor de garganta) y una menor proporción de participantes que presentaban síntomas del resfriado después de siete días de tratamiento en participantes suplementados con zinc versus placebo (118).
Los hallazgos inconsistentes entre los ensayos se han atribuido en parte a diferentes cantidades de zinc liberado de varias formas utilizadas en las pastillas (particularmente acetato de zinc y gluconato de zinc) (119, 120). Se ha argumentado que el sabor desagradable de los complejos formadores de gluconato de zinc con carbohidratos condujo a un cumplimiento deficiente, lo que explica los resultados negativos de los ensayos (119, 121). Sin embargo, cuando recientemente se realizó un meta-análisis sobre los resultados de siete ensayos (575 participantes) que emplearon pastillas de zinc a dosis >75 mg/día, no hubo evidencia de una diferencia en la eficacia observada entre los ensayos que utilizaron acetato de zinc (3 ensayos) o gluconato de zinc (4 ensayos) (122).
Con numerosos ensayos y meta-análisis bien controlados, la eficacia de las pastillas o jarabe de zinc en el tratamiento de los síntomas del resfriado común ya no es cuestionable. Un meta-análisis de siete ensayos informó recientemente una reducción del 33% en la duración de los síntomas del resfriado con la ingesta de pastillas de zinc (>75 mg/día de zinc elemental) (122). Sin embargo, se descubrió que muchas formulaciones de zinc suplementarias de venta libre liberan iones de zinc cero (es decir, la forma biológicamente activa de zinc) o que contienen aditivos (p. ej., magnesio, ciertos aminoácidos, ácido cítrico) que cancelan el beneficio del zinc o empeoran los síntomas del resfriado (119).
Finalmente, aunque tomar pastillas de zinc para un resfriado cada dos o tres horas mientras se está despierto resultará en ingestas diarias de zinc muy por encima del nivel máximo de ingesta tolerable (NM) de 40 mg/día para adultos (véase Seguridad), el uso de zinc a diario dosis de 50 a 180 mg durante una o dos semanas no han dado lugar a efectos secundarios graves (117). El mal gusto y las náuseas fueron los efectos adversos más frecuentes informados en los ensayos terapéuticos (117). El uso de pastillas de zinc durante períodos prolongados (p. ej., 6-8 semanas) es probable que resulte en deficiencia de cobre (véase Interacción con nutrientes y Seguridad).
Zinc intranasal (geles nasales de zinc y aerosoles nasales)
Las preparaciones intranasales de zinc, diseñadas para aplicarse directamente al epitelio nasal (células que recubren las fosas nasales), se comercializan como remedios de venta libre para el resfriado. Mientras que dos ensayos controlados con placebo encontraron que el gluconato de zinc intranasal acortó moderadamente la duración de los síntomas del resfriado (123, 124), otro encontró que el zinc intranasal no era beneficioso (125). El análisis agrupado de estos tres ensayos no mostró ningún beneficio general del zinc intranasal en el riesgo de seguir experimentando síntomas de resfriado para el día 3 (126). Se ha propuesto la existencia de un circuito eléctrico biológicamente cerrado (BCEC) boca-nariz para explicar la eficacia del suministro de zinc oral en lugar de intranasal (119). Específicamente, se sugiere que el interior de la nariz repele el zinc iónico (Zn2+) de tal manera que el zinc iónico administrado por pastillas para la garganta y que migran de la boca a la nariz son más efectivos contra la infección por rinovirus que los que se administran directamente en la nariz (119). Son motivo de grave preocupación los reportes de casos de individuos que experimentan pérdida del sentido del olfato (anosmia) después de usar zinc intranasal como remedio para el resfriado (127). Dado que la anosmia asociada con zinc puede ser irreversible, se deben evitar las preparaciones intranasales de zinc.
La degeneración macular relacionada con la edad
La degeneración macular relacionada con la edad (DMAE) es una enfermedad degenerativa de la mácula y una de las principales causas de ceguera en personas mayores de 65 años en los EE.UU. (128). La mácula es la porción de la retina en la parte posterior del ojo involucrada con la visión central. Se hipotetiza que el zinc juega un papel en el desarrollo de la DMAE por varias razones: (1) el zinc se encuentra en altas concentraciones en la parte de la retina afectada por la DMAE, (2) se ha demostrado que el contenido de zinc en la retina disminuye con la edad, y (3) se ha demostrado que las actividades de algunas enzimas retinianas dependientes de zinc disminuyen con la edad. Hasta la fecha, los estudios de cohorte prospectivos han mostrado evidencia limitada que sugiere una asociación entre el consumo de zinc dietético y la incidencia de DMAE (129-131).
Sin embargo, un ensayo aleatorio controlado temprano suscitó interés cuando descubrió que 200 mg/día de sulfato de zinc (81 mg/día de zinc elemental) durante dos años limitaron la pérdida de visión en pacientes con DMAE (132). Sin embargo, un ensayo posterior que utilizó la misma dosis y duración no encontró beneficios para los pacientes con una forma más avanzada de DMAE en un ojo (133). Los ensayos pequeños generalmente no reportaron un efecto protector de la suplementación con vitaminas y minerales en la DMAE (134, 135). Sin embargo, un ensayo aleatorizado, doble ciego, controlado con placebo en 74 pacientes con DMAE informó que la suplementación con 50 mg/día de monocisteína de zinc durante seis meses mejoró las medidas de la función macular, incluida la agudeza visual, la sensibilidad al contraste, y la fotorecuperación (136). Un gran ensayo aleatorizado, controlado con placebo de suplementos diarios con antioxidantes (500 mg de vitamina C, 400 UI de vitamina E y 15 mg de β-caroteno) y altas dosis de zinc (80 mg de zinc como óxido de zinc y 2 mg de cobre como óxido cúprico) — Estudio sobre Enfermedad de los Ojos Relacionada con la Edad (AREDS) — descubrió que la administración de altas dosis de zinc solo o con antioxidantes combinados a individuos con signos de degeneración macular moderada a severa redujo significativamente el riesgo de desarrollar degeneración macular avanzada durante un seguimiento medio de 6.3 años (137). Un análisis de seguimiento realizado cuatro años después de la finalización del ensayo en 2001, que incluyó a casi el 85% de los participantes sobrevivientes, encontró que el beneficio de la formulación de AREDS (antioxidantes combinados más zinc) había persistido (138). De hecho, las probabilidades de desarrollar DMAE tardía, especialmente DMAE neovascular, fue menor tanto en los participantes con bajo riesgo de desarrollar DMAE como en aquellos que estaban en riesgo y recomendaron continuar tomando la formulación de AREDS después de que finalizó el ensayo. Sin embargo, no hubo efecto de la formulación de AREDS sobre el riesgo de desarrollar atrofia geográfica central (138). Otro ensayo, AREDS2, examinó el efecto de una formulación de AREDS sin β-caroteno y/o que contiene 25 mg en lugar de 80 mg de zinc (139). El ensayo no mostró diferencias aparentes en el riesgo de desarrollar DMAE avanzada con el uso de formulaciones AREDS que contienen 25 mg u 80 mg de zinc y/o β-caroteno (140). Un meta-análisis reciente de cinco ensayos (incluido el estudio AREDS original) confirmó el efecto protector del zinc suplementario contra la DMAE neovascular y avanzada (141).
En conclusión, la formulación de AREDS que combina antioxidantes y zinc (25 mg u 80 mg) puede retrasar la progresión de la enfermedad en pacientes con DMAE. Se aconseja a los pacientes, especialmente a los fumadores y aquellos con enfermedad vascular, que discutan con su médico los beneficios frente a los daños potenciales que podrían estar asociados con el uso a largo plazo de altas dosis de vitaminas antioxidantes y carotenoides (141).
Diabetes mellitus
Diabetes mellitus tipo 2
El control glucémico deficiente y la micción frecuente en pacientes con diabetes mellitus pueden estar impulsando la pérdida urinaria de zinc y contribuir a la deficiencia marginal de zinc (142, 143). Sin embargo, debido al papel del zinc en la función de las células β y la acción de la insulina (véase Prevención de Enfermedades), varios ensayos aleatorios controlados han examinado si la suplementación con zinc (solo o con otros minerales y vitaminas) podría desempeñar un papel en el control de la diabetes, especialmente al mejorar el control glucémico en personas con diabetes tipo 2 (15). De los 12 ensayos que midieron los estatus basales de zinc de los participantes al inicio del estudio, la suplementación con zinc (20-240 mg/día) durante 4 a 16 semanas mejoró la glucosa en sangre en ayunas en pacientes que presentaron deficiencia de zinc (6 estudios). El zinc suplementario también redujo la proporción de hemoglobina glucosilada (HbA1c) en dos ensayos realizados en participantes con deficiencia de zinc, pero no en cuatro estudios que incluyeron participantes sin deficiencia de zinc (15). Los pacientes con diabetes tipo 2 deben asegurarse de que su dieta proporcione suficiente zinc para cubrir sus necesidades, especialmente si su glucosa en sangre está mal controlada.
Diabetes mellitus gestacional
La diabetes mellitus gestacional se define como la hiperglucemia que se diagnostica por primera vez durante el embarazo. La condición se asocia con un mayor riesgo de resultados adversos del embarazo (144). Un grupo de investigadores en Irán realizó dos pequeños ensayos aleatorios, controlados con placebo para examinar el efecto de los suplementos de zinc en mujeres embarazadas con diabetes gestacional. El zinc suplementario (30 mg/día) durante seis semanas durante el embarazo mejoró el estatus del zinc, redujo la glucosa en sangre en ayunas, y mejoró la sensibilidad a la insulina en mujeres con diabetes gestacional, pero no tuvo impacto en los resultados del embarazo, incluida la necesidad de cesárea, necesidad de terapia con insulina, tamaño de nacimiento del recién nacido, y puntajes de Apgar, o incidencia de hiperbilirrubinemia (145, 146). Se informaron mejoras similares de los marcadores de control glucémico en otro ensayo controlado con placebo que aleatorizó a mujeres embarazadas con diabetes gestacional para recibir zinc (4 mg) junto con magnesio (100 mg), calcio (400 mg), y vitamina D (200 UI) dos veces al día durante seis semanas (147). También hubo alguna evidencia que sugiere que el zinc suplementario podría ayudar a corregir otros trastornos metabólicos (p. ej., perfil anormal de lípidos en sangre) asociados con la gestación diabetes (147, 148).
VIH/SIDA
El zinc suficiente es esencial para mantener la función del sistema inmune, y las personas infectadas por el VIH son particularmente susceptibles a la deficiencia de zinc. En pacientes infectados por el VIH, las bajas concentraciones séricas de zinc se han asociado con la progresión de la enfermedad y una mayor mortalidad (149, 150). En un estudio realizado en pacientes con SIDA, 45 mg/día de zinc durante un mes dieron como resultado una disminución de la incidencia de infecciones oportunistas en comparación con placebo (151). Un ensayo controlado con placebo en 231 adultos VIH positivos con bajo estatus de zinc encontró que la suplementación con zinc (12 mg/día para mujeres y 15 mg/día para hombres) durante 18 meses redujo la incidencia de falla inmunológica (definida por un recuento de CD4+ <200 células/mm3) en un 76% y la tasa de diarrea en un 60% (152). Una revisión sistemática de 2011 que identificó tres ensayos aleatorios controlados en entornos principalmente pobres en recursos concluyó que la suplementación con zinc era segura y eficaz para reducir las infecciones oportunistas en Adultos VIH positivos (153).
La evidencia de los beneficios de la suplementación con zinc en mujeres embarazadas y niños VIH positivos es muy limitada. En un ensayo doble ciego, aleatorizado, controlado con placebo en Tanzania, la administración de zinc (25 mg/día) a mujeres entre 12 y 27 semanas de gestación hasta seis meses después del parto no logró reducir la carga viral materna ni limitar transmisión infantil del VIH (154). Un ensayo controlado aleatorio con placebo de suplementos de zinc (10 mg/día durante 6 meses) en 96 niños VIH positivos (6 meses a 5 años) en Sudáfrica no mostró ningún efecto sobre el recuento de CD4+ y la carga viral (155). Hubo evidencia que mostró una disminución en la incidencia de diarrea acuosa en niños suplementados con zinc en comparación con aquellos que tomaron un placebo, pero no hubo diferencias en la incidencia de neumonía, infección del oído, o infección del tracto respiratorio superior (155). Otro ensayo en Uganda mostró que el zinc suplementario en niños con neumonía severa disminuyó efectivamente la letalidad independientemente del estatus de VIH de los niños (156). Si bien se recomienda la suplementación con zinc durante el embarazo y la lactancia en poblaciones con probabilidad de tener deficiencia de zinc (43, 71, 75), su uso en el manejo de la infección por VIH requiere más investigación (157).
Enfermedad de Alzheimer
Se ha reportado sobre homeostasis anormal de metales traza, en particular cobre y zinc, en individuos afectados por la enfermedad de Alzheimer — la forma más común de demencia. Específicamente, los resultados de estudios de caso y control han mostrado concentraciones séricas de cobre más altas y concentraciones séricas de zinc más bajas en personas con enfermedad de Alzheimer en comparación con los controles cognitivamente saludables (158-160). Sobre la base de la utilización de sales de zinc en la enfermedad de Wilson, se ha propuesto que la suplementación con zinc podría mejorar el estatus de zinc y cobre y limitar el deterioro cognitivo adicional en individuos con enfermedad de Alzheimer. El uso de acetato de zinc de liberación lenta (150 mg/día durante seis meses) en un estudio aleatorio, controlado con placebo de 60 pacientes con enfermedad de Alzheimer de leve a moderada corrigió el bajo nivel de zinc y disminuyó el cobre 'libre' en suero (es decir, no unido a la ceruloplasmina) (161). Además, cuando un análisis post-hoc se restringió a participantes mayores de 70 años (N=29), se descubrió que la suplementación con zinc previno el deterioro de los puntajes cognitivos durante el período de prueba (161). Evidencia adicional es necesitaba confirmar si la suplementación con zinc podría desempeñar un papel en la estabilización de los déficits cognitivos en adultos mayores con demencia.
Depresión
Un análisis de datos de la encuesta de Boston Community Community Health (BACH), que incluyó a 3,708 participantes (edades, 30-79 años), reportó mayores probabilidades de síntomas de depresión en mujeres (pero no en hombres) en los cuartiles más bajos versus más altos del total (mediana valores, 8.7 mg/día frente a 26.8 mg/día) e ingestas de zinc dietético (valores medios, 7.6 mg/día frente a 13.1 mg/día) (162). La posibilidad de que el zinc pueda desempeñar un papel en la prevención o el alivio de la depresión se ha explorado en dos ensayos realizados por un grupo de investigación. Los datos de estos ensayos se analizaron siguiendo un enfoque por protocolo (es decir, restringido a los participantes que completaron los estudios). Un ensayo preliminar aleatorizado, doble ciego, controlado con placebo en 20 sujetos (edad media, 43 años) tratados por depresión mayor mostró que la suplementación con 25 mg/día de zinc redujo los síntomas de depresión a las 6 y 12 semanas según lo evaluado por Escala de Calificación de Depresión de Hamilton (HDRS, en inglés) y del Inventario de Depresión de Beck (BDI, inglés) (163). Un segundo ensayo controlado con placebo en 60 participantes tratados con el antidepresivo imipramina (Tofranil; 100-200 mg/día) evaluó la respuesta terapéutica al zinc suplementario (25 mg/día) usando HDRS, BDI, escala de Clinical Global Impression (CGI) y puntuaciones de la Escala de Depresión de Montgomery-Åsberg (MADRS, en inglés) (164). La suplementación con zinc mejoró las medidas basadas en la puntuación de la respuesta terapéutica y la remisión después de seis semanas, pero sólo cuando el análisis fue restringido a participantes resistentes a la imipramina. Sin embargo, no hubo evidencia de un efecto del zinc después de 12 semanas (164).
Sepsis neonatal
La sepsis es una condición potencialmente mortal que causa disfunción orgánica como consecuencia de la respuesta desregulada del huésped a la infección (165). La sepsis se acompaña de cambios en la homeostasis del zinc caracterizados en particular por una disminución en la concentración sérica de zinc y un aumento en la concentración de zinc en el hígado (166). Se cree que estos cambios en la distribución de zinc son parte del mecanismo de defensa de un huésped por el cual el huésped puede limitar la disponibilidad de zinc a los patógenos, así como estimular el sistema inmunológico. Dicho mecanismo se ha descrito para otros metales de transición, incluidos el hierro y el manganeso (167). Sin embargo, las concentraciones séricas más bajas de zinc en pacientes críticos con alto riesgo de insuficiencia orgánica se han asociado con episodios recurrentes de sepsis y resultados más pobres (168, 169). Una revisión sistemática de 2018 identificó cuatro ensayos que examinaron el efecto de la administración de suplementos de zinc en recién nacidos con sepsis (166). Se encontró que la administración de suplementos de zinc produce una disminución de la inflamación (170) y un mejor desarrollo neurológico (171, 172). Tres de cada cuatro ensayos que examinaron la tasa de mortalidad no mostraron ningún efecto de suplemento de zinc (170, 172, 173).
Fuentes
Fuentes alimenticias
Los mariscos, la carne de res, y otras carnes rojas son fuentes ricas de zinc; las nueces y las legumbres son fuentes vegetales relativamente buenas de zinc. La biodisponibilidad de zinc (la fracción de zinc retenida y utilizada por el cuerpo) es relativamente alta en carne, huevos, y mariscos debido a la relativa ausencia de compuestos que inhiben la absorción de zinc y la presencia de aminoácidos que contienen azufre (cisteína y metionina) que mejora la absorción de zinc. El zinc en los productos integrales y las proteínas vegetales está menos biodisponible debido a su contenido relativamente alto de fitato, que inhibe la absorción de zinc (174). La acción enzimática de la levadura reduce el nivel de fitato en los alimentos; por lo tanto, los panes integrales con levadura tienen más zinc biodisponible que los panes integrales sin levadura.
Las encuestas dietéticas nacionales en los EE.UU. estiman que la ingesta dietética promedio de zinc de alimentos naturales y fortificados es de aproximadamente 12.3 mg/día en adultos, con aproximadamente el 12% de la población adulta en riesgo de ingesta inadecuada (175). El contenido de zinc de algunos alimentos relativamente rico en zinc se enumera en la Tabla 2 en miligramos (mg). Para obtener más información sobre el contenido de nutrientes de alimentos específicos, busque en la base de datos de composición de alimentos del USDA (FoodData Central) (176).
Suplementos
Una serie de suplementos de zinc están disponibles comercialmente, incluyendo acetato de zinc, gluconato de zinc, picolinato de zinc, y sulfato de zinc. El picolinato de zinc se ha promovido como una forma más absorbible de zinc, pero hay pocos datos que respalden esta idea en humanos. El trabajo limitado en animales sugiere que la mayor absorción intestinal de picolinato de zinc puede compensarse con una mayor eliminación (29).
Seguridad
Toxicidad
Toxicidad aguda
Se han producido brotes aislados de toxicidad aguda por zinc como resultado del consumo de alimentos o bebidas contaminados con zinc liberado de envases galvanizados. Los signos de toxicidad aguda por zinc son dolor abdominal, diarrea, náuseas, y vómitos. Dosis únicas de 225 a 450 mg de zinc generalmente inducen vómitos. Se ha reportado un malestar gastrointestinal más leve a dosis de 50 a 150 mg/día de zinc suplementario. Se ha reportado de fiebre por vapores metálicos después de la inhalación de vapores de óxido de zinc. Específicamente, la sudoración profusa, la debilidad, y la respiración rápida pueden desarrollarse dentro de las ocho horas posteriores a la inhalación de óxido de zinc y persistir durante 12 a 24 horas después de que finaliza la exposición (6, 29).
Efectos adversos
La principal consecuencia del consumo a largo plazo de zinc excesivo es la deficiencia de cobre. Se ha encontrado que la ingesta total de zinc de 60 mg/día (50 mg por suplementos y 10 mg de zinc dietético) por hasta 10 semanas produce signos de deficiencia de cobre (29). También se ha reportado deficiencia de cobre después del uso crónico de cantidades excesivas de zinc que contienen cremas para dentaduras postizas (≥2 tubos por semana que contienen 17-34 mg/g de zinc) (177). Para prevenir la deficiencia de cobre, la Junta de Alimentos y Nutrición de EE.UU. estableció el nivel máximo de ingesta tolerable (NM) para adultos en 40 mg/día, incluyendo zinc dietético y suplementario (Tabla 3) (29).
Zinc intranasal
Se sabe que el zinc intranasal causa una pérdida del sentido del olfato (anosmia) en animales de laboratorio (178), y ha habido varios reportes de casos de individuos que desarrollaron anosmia después de usar gluconato de zinc intranasal (127). Dado que la anosmia asociada al zinc puede ser irreversible, se debe evitar el uso de geles y aerosoles nasales de zinc.
Interacción con drogas/fármacos
El uso de suplementos de zinc disminuye la absorción de ciertos medicamentos, incluidos cefalexina (Keplex) y penicilamina (Cuprimina, Depen), así como los medicamentos antirretrovirales atazanavir (Reyataz) y ritonavir (Norvir) (179). La administración concomitante de suplementos de zinc con ciertos medicamentos como la tetraciclina y los antibióticos de quinolona pueden disminuir la absorción tanto del zinc como de los medicamentos, reduciendo potencialmente la eficacia del medicamento. Tomar suplementos de zinc y estos medicamentos con al menos dos horas de diferencia debería prevenir esta interacción.
El uso terapéutico de agentes quelantes de metales, como la penicilamina (usada para tratar la sobrecarga de cobre en la enfermedad de Wilson) y el pentaacetato de dietilentriamina (DTPA; usado para tratar la sobrecarga de hierro), ha resultado en una deficiencia severa de zinc. Los medicamentos anticonvulsivos, especialmente el valproato de sodio, también pueden precipitar la deficiencia de zinc. El uso prolongado de diuréticos puede aumentar la excreción urinaria de zinc, lo que resulta en una mayor pérdida de zinc. Debido a que el zinc suplementario puede reducir la glucosa en la sangre, se aconseja a quienes toman agentes antidiabéticos que usen suplementos de zinc con precaución.
Recomendación del Instituto Linus Pauling
La Ingesta Diaria Recomendada de zinc (8 mg/día para mujeres adultas y 11 mg/día para hombres adultos) parece suficiente para prevenir la deficiencia en la mayoría de las personas, pero la falta de indicadores sensibles del estado nutricional del zinc en humanos hace que sea difícil determinar el nivel de la ingesta de zinc tiene más probabilidades de promover una salud óptima. Seguir la recomendación del Instituto Linus Pauling de tomar un suplemento multivitamínico/mineral generalmente proporcionará al menos la Ingesta Diaria Recomendada de zinc. Las ingestas diarias totales (suplementarias + dietéticas) de zinc no deben exceder el NM (40 mg/día para adultos) para limitar el riesgo de deficiencia de cobre en particular (véase Seguridad).
Adultos mayores (> 50 años)
Aunque no se sabe que el requerimiento de zinc sea mayor para los adultos mayores, muchos tienen una ingesta inadecuada de zinc dietético (180, 181). Una capacidad reducida para absorber zinc, una mayor probabilidad de estados de enfermedad que alteran la utilización de zinc, y un mayor uso de medicamentos que disminuyen la biodisponibilidad de zinc pueden contribuir a un mayor riesgo de deficiencia leve de zinc en adultos mayores. La ingesta adecuada de zinc dietético es esencial para los adultos mayores porque las consecuencias de la deficiencia leve de zinc, como la función del sistema inmunitario deteriorado, son especialmente relevantes para el mantenimiento de su salud.
Autores y Críticos
Escrito originalmente en 2001 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizad en Diciembre de 2003 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizad en Octubre de 2007 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizad en Junio de 2013 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizad en Febrero de 2019 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Mayo 2019 por:
Emily Ho, Ph.D.
Director Dotado, Moore Family Center de Alimentos de Grano Entero
Nutrición y Salud Preventiva
Profesor de la Escuela de Ciencias Biológicas y Salud de la Población
Investigador Principal, Instituto Linus Pauling
Universidad Estatal de Oregon
Traducido al Español en 2019 por:
Natsumi Then Shimazaki
Instituto Linus Pauling
Universidad Estatal de Oregon
Copyright 2001-2022 Instituto Linus Pauling
Referencias
1. Prasad AS, Halsted JA, Nadimi M. Syndrome of iron deficiency anemia, hepatosplenomegaly, hypogonadism, dwarfism and geophagia. Am J Med. 1961;31:532-546. (PubMed)
2. Penny ME. Zinc supplementation in public health. Ann Nutr Metab. 2013;62 Suppl 1:31-42. (PubMed)
3. Prasad AS. Impact of the discovery of human zinc deficiency on health. J Trace Elem Med Biol. 2014;28(4):357-363. (PubMed)
4. Terrin G, Berni Canani R, Di Chiara M, et al. Zinc in early life: a key element in the fetus and preterm neonate. Nutrients. 2015;7(12):10427-10446. (PubMed)
5. Andreini C, Banci L, Bertini I, Rosato A. Counting the zinc-proteins encoded in the human genome. J Proteome Res. 2006;5(1):196-201. (PubMed)
6. King JC, Cousins RJ. Zinc. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore: Lippincott Williams & Wilkins; 2014:189-205.
7. Vallee BL, Falchuk KH. The biochemical basis of zinc physiology. Physiol Rev. 1993;73(1):79-118. (PubMed)
8. King JC. Zinc: an essential but elusive nutrient. Am J Clin Nutr. 2011;94(2):679S-684S. (PubMed)
9. Cornish-Bowden A. Current IUBMB recommendations on enzyme nomenclature and kinetics. Perspectives in Science. 2014;1(1-6):74-87.
10. Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83(6):835-839. (PubMed)
11. Atrian-Blasco E, Santoro A, Pountney DL, Meloni G, Hureau C, Faller P. Chemistry of mammalian metallothioneins and their interaction with amyloidogenic peptides and proteins. Chem Soc Rev. 2017;46(24):7683-7693. (PubMed)
12. Hijova E. Metallothioneins and zinc: their functions and interactions. Bratisl Lek Listy. 2004;105(5-6):230-234. (PubMed)
13. Sirangelo I, Iannuzzi C. The role of metal binding in the amyotrophic lateral sclerosis-related aggregation of copper-zinc superoxide dismutase. Molecules. 2017;22(9). (PubMed)
14. Hershfinkel M, Moran A, Grossman N, Sekler I. A zinc-sensing receptor triggers the release of intracellular Ca2+ and regulates ion transport. Proc Natl Acad Sci U S A. 2001;98(20):11749-11754. (PubMed)
15. Ruz M, Carrasco F, Rojas P, Basfi-Fer K, Hernandez MC, Perez A. Nutritional effects of zinc on metabolic syndrome and type 2 diabetes: mechanisms and main findings in human studies. Biol Trace Elem Res. 2019; 188(1):177-188. (PubMed)
16. Takeda A, Tamano H. The impact of synaptic Zn(2+) dynamics on cognition and its decline. Int J Mol Sci. 2017;18(11). (PubMed)
17. Holt RR, Uriu-Adams JY, Keen CL. Zinc. In: Erdman Jr JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Washington D.C.: ILSI Press; 2012:521-539.
18. Sandstrom B. Micronutrient interactions: effects on absorption and bioavailability. Br J Nutr. 2001;85 Suppl 2:S181-185. (PubMed)
19. Zaman K, McArthur JO, Abboud MN, et al. Iron supplementation decreases plasma zinc but has no effect on plasma fatty acids in non-anemic women. Nutr Res. 2013;33(4):272-278. (PubMed)
20. O'Brien KO, Zavaleta N, Caulfield LE, Wen J, Abrams SA. Prenatal iron supplements impair zinc absorption in pregnant Peruvian women. J Nutr. 2000;130(9):2251-2255. (PubMed)
21. Fung EB, Ritchie LD, Woodhouse LR, Roehl R, King JC. Zinc absorption in women during pregnancy and lactation: a longitudinal study. Am J Clin Nutr. 1997;66(1):80-88. (PubMed)
22. Davidsson L, Almgren A, Sandstrom B, Hurrell RF. Zinc absorption in adult humans: the effect of iron fortification. Br J Nutr. 1995;74(3):417-425. (PubMed)
23. de Brito NJ, Rocha ED, de Araujo Silva A, et al. Oral zinc supplementation decreases the serum iron concentration in healthy schoolchildren: a pilot study. Nutrients. 2014;6(9):3460-3473. (PubMed)
24. Carter RC, Kupka R, Manji K, et al. Zinc and multivitamin supplementation have contrasting effects on infant iron status: a randomized, double-blind, placebo-controlled clinical trial. Eur J Clin Nutr. 2018;72(1):130-135. (PubMed)
25. de Oliveira Kde J, Donangelo CM, de Oliveira AV, Jr., da Silveira CL, Koury JC. Effect of zinc supplementation on the antioxidant, copper, and iron status of physically active adolescents. Cell Biochem Funct. 2009;27(3):162-166. (PubMed)
26. Wood RJ, Zheng JJ. High dietary calcium intakes reduce zinc absorption and balance in humans. Am J Clin Nutr. 1997;65(6):1803-1809. (PubMed)
27. McKenna AA, Ilich JZ, Andon MB, Wang C, Matkovic V. Zinc balance in adolescent females consuming a low- or high-calcium diet. Am J Clin Nutr. 1997;65(5):1460-1464. (PubMed)
28. Hunt JR, Beiseigel JM. Dietary calcium does not exacerbate phytate inhibition of zinc absorption by women from conventional diets. Am J Clin Nutr. 2009;89(3):839-843. (PubMed)
29. Food and Nutrition Board, Institute of Medicine. Zinc. Dietary reference intakes for vitamin A, vitamin K, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. Washington, D.C.: National Academy Press; 2001:442-501. (National Academy Press)
30. Kauwell GP, Bailey LB, Gregory JF, 3rd, Bowling DW, Cousins RJ. Zinc status is not adversely affected by folic acid supplementation and zinc intake does not impair folate utilization in human subjects. J Nutr. 1995;125(1):66-72. (PubMed)
31. Boron B, Hupert J, Barch DH, et al. Effect of zinc deficiency on hepatic enzymes regulating vitamin A status. J Nutr. 1988;118(8):995-1001. (PubMed)
32. Christian P, West KP, Jr. Interactions between zinc and vitamin A: an update. Am J Clin Nutr. 1998;68(2 Suppl):435S-441S. (PubMed)
33. Ciampo I, Sawamura R, Ciampo LAD, Fernandes MIM. Acrodematitis enteropathica: clinical manifestations and pediatric diagnosis. Rev Paul Pediatr. 2018;36(2):238-241. (PubMed)
34. Hambidge M. Human zinc deficiency. J Nutr. 2000;130(5S Suppl):1344S-1349S. (PubMed)
35. Fischer Walker CL, Ezzati M, Black RE. Global and regional child mortality and burden of disease attributable to zinc deficiency. Eur J Clin Nutr. 2009;63(5):591-597. (PubMed)
36. Prasad AS. Discovery of human zinc deficiency: 50 years later. J Trace Elem Med Biol. 2012;26(2-3):66-69. (PubMed)
37. International Zinc Nutrition Consultative Group, Brown KH, Rivera JA, et al. International Zinc Nutrition Consultative Group (IZiNCG) technical document #1. Assessment of the risk of zinc deficiency in populations and options for its control. Food Nutr Bull. 2004;25(1 Suppl 2):S99-203. (PubMed)
38. Krebs NF. Update on zinc deficiency and excess in clinical pediatric practice. Ann Nutr Metab. 2013;62 Suppl 1:19-29. (PubMed)
39. Gibson RS, Hess SY, Hotz C, Brown KH. Indicators of zinc status at the population level: a review of the evidence. Br J Nutr. 2008;99 Suppl 3:S14-23. (PubMed)
40. Wessells KR, Brown KH. Estimating the global prevalence of zinc deficiency: results based on zinc availability in national food supplies and the prevalence of stunting. PLoS One. 2012;7(11):e50568. (PubMed)
41. Moghimi M, Ashrafzadeh S, Rassi S, Naseh A. Maternal zinc deficiency and congenital anomalies in newborns. Pediatr Int. 2017;59(4):443-446. (PubMed)
42. Wilson RL, Grieger JA, Bianco-Miotto T, Roberts CT. Association between maternal zinc status, dietary zinc intake and pregnancy complications: a systematic review. Nutrients. 2016;8(10). (PubMed)
43. Ota E, Mori R, Middleton P, et al. Zinc supplementation for improving pregnancy and infant outcome. Cochrane Database Syst Rev. 2015(2):Cd000230. (PubMed)
44. Haider BA, Bhutta ZA. Multiple-micronutrient supplementation for women during pregnancy. Cochrane Database Syst Rev. 2017;4:Cd004905. (PubMed)
45. Petry N, Olofin I, Boy E, Donahue Angel M, Rohner F. The effect of low dose iron and zinc intake on child micronutrient status and development during the first 1000 days of life: a systematic review and meta-analysis. Nutrients. 2016;8(12). (PubMed)
46. Walravens PA, Hambidge KM, Koepfer DM. Zinc supplementation in infants with a nutritional pattern of failure to thrive: a double-blind, controlled study. Pediatrics. 1989;83(4):532-538. (PubMed)
47. Hambidge M, Krebs N. Zinc and growth. In: Roussell AM, ed. Trace elements in man and animals 10: Proceedings of the tenth international symposium on trace elements in man and animals. New York: Plenum Press; 2000:977-980.
48. Brown KH, Peerson JM, Baker SK, Hess SY. Preventive zinc supplementation among infants, preschoolers, and older prepubertal children. Food Nutr Bull. 2009;30(1 Suppl):S12-40. (PubMed)
49. Imdad A, Bhutta ZA. Effect of preventive zinc supplementation on linear growth in children under 5 years of age in developing countries: a meta-analysis of studies for input to the lives saved tool. BMC Public Health. 2011;11 Suppl 3:S22. (PubMed)
50. Liu E, Pimpin L, Shulkin M, et al. Effect of zinc supplementation on growth outcomes in children under 5 years of age. Nutrients. 2018;10(3). (PubMed)
51. MacDonald RS. The role of zinc in growth and cell proliferation. J Nutr. 2000;130(5S Suppl):1500S-1508S. (PubMed)
52. Thousand day global initiative. Available at: https://thousanddays.org/. Accessed 2/14/19.
53. Bhatnagar S, Taneja S. Zinc and cognitive development. Br J Nutr. 2001;85 Suppl 2:S139-145. (PubMed)
54. Tamura T, Goldenberg RL, Ramey SL, Nelson KG, Chapman VR. Effect of zinc supplementation of pregnant women on the mental and psychomotor development of their children at 5 y of age. Am J Clin Nutr. 2003;77(6):1512-1516. (PubMed)
55. Sazawal S, Bentley M, Black RE, Dhingra P, George S, Bhan MK. Effect of zinc supplementation on observed activity in low socioeconomic Indian preschool children. Pediatrics. 1996;98(6 Pt 1):1132-1137. (PubMed)
56. Bentley ME, Caulfield LE, Ram M, et al. Zinc supplementation affects the activity patterns of rural Guatemalan infants. J Nutr. 1997;127(7):1333-1338. (PubMed)
57. Ashworth A, Morris SS, Lira PI, Grantham-McGregor SM. Zinc supplementation, mental development and behaviour in low birth weight term infants in northeast Brazil. Eur J Clin Nutr. 1998;52(3):223-227. (PubMed)
58. Castillo-Duran C, Perales CG, Hertrampf ED, Marin VB, Rivera FA, Icaza G. Effect of zinc supplementation on development and growth of Chilean infants. J Pediatr. 2001;138(2):229-235. (PubMed)
59. Lind T, Lonnerdal B, Stenlund H, et al. A community-based randomized controlled trial of iron and zinc supplementation in Indonesian infants: effects on growth and development. Am J Clin Nutr. 2004;80(3):729-736. (PubMed)
60. Taneja S, Bhandari N, Bahl R, Bhan MK. Impact of zinc supplementation on mental and psychomotor scores of children aged 12 to 18 months: a randomized, double-blind trial. J Pediatr. 2005;146(4):506-511. (PubMed)
61. Gogia S, Sachdev HS. Zinc supplementation for mental and motor development in children. Cochrane Database Syst Rev. 2012;12:CD007991. (PubMed)
62. Baum MK, Shor-Posner G, Campa A. Zinc status in human immunodeficiency virus infection. J Nutr. 2000;130(5S Suppl):1421S-1423S. (PubMed)
63. Maares M, Haase H. Zinc and immunity: An essential interrelation. Arch Biochem Biophys. 2016;611:58-65. (PubMed)
64. Subramanian Vignesh K, Deepe GS, Jr. Immunological orchestration of zinc homeostasis: The battle between host mechanisms and pathogen defenses. Arch Biochem Biophys. 2016;611:66-78. (PubMed)
65. Subramanian Vignesh K, Landero Figueroa JA, Porollo A, Caruso JA, Deepe GS, Jr. Granulocyte macrophage-colony stimulating factor induced Zn sequestration enhances macrophage superoxide and limits intracellular pathogen survival. Immunity. 2013;39(4):697-710. (PubMed)
66. Fischer Walker C, Black RE. Zinc and the risk for infectious disease. Annu Rev Nutr. 2004;24:255-275. (PubMed)
67. Shankar AH, Prasad AS. Zinc and immune function: the biological basis of altered resistance to infection. Am J Clin Nutr. 1998;68(2 Suppl):447S-463S. (PubMed)
68. Wapnir RA. Zinc deficiency, malnutrition and the gastrointestinal tract. J Nutr. 2000;130(5S Suppl):1388S-1392S. (PubMed)
69. Liu L, Oza S, Hogan D, et al. Global, regional, and national causes of child mortality in 2000-13, with projections to inform post-2015 priorities: an updated systematic analysis. Lancet. 2015;385(9966):430-440. (PubMed)
70. Black RE. Progress in the use of ORS and zinc for the treatment of childhood diarrhea. J Glob Health. 2019;9(1):010101. (PubMed)
71. Lazzerini M, Wanzira H. Oral zinc for treating diarrhoea in children. Cochrane Database Syst Rev. 2016;12:Cd005436. (PubMed)
72. WHO and UNICEF. Clinical management of acute diarrhoea. Available at: https://www.who.int/publications/i/item/WHO_FCH_CAH_04.7. Accessed 3/18/24.
73. World Health Organization. Fact sheets: pneumonia. November 6, 2016. Available at: https://www.who.int/news-room/fact-sheets/detail/pneumonia. Accessed 2/11/19.
74. World Health Organization. Global health risks: mortality and burden of disease attributable to selected major risks. 2009. Available at: https://www.who.int/publications/i/item/9789241563871. Accessed 3/18/24.
75. Lassi ZS, Moin A, Bhutta ZA. Zinc supplementation for the prevention of pneumonia in children aged 2 months to 59 months. Cochrane Database Syst Rev. 2016;12:Cd005978. (PubMed)
76. Howie S, Bottomley C, Chimah O, et al. Zinc as an adjunct therapy in the management of severe pneumonia among Gambian children: randomized controlled trial. J Glob Health. 2018;8(1):010418. (PubMed)
77. Wang L, Song Y. Efficacy of zinc given as an adjunct to the treatment of severe pneumonia: A meta-analysis of randomized, double-blind and placebo-controlled trials. Clin Respir J. 2018;12(3):857-864. (PubMed)
78. Black MM. Zinc deficiency and child development. Am J Clin Nutr. 1998;68(2 Suppl):464S-469S. (PubMed)
79. Shankar AH. Nutritional modulation of malaria morbidity and mortality. J Infect Dis. 2000;182 Suppl 1:S37-53. (PubMed)
80. Müller O, Becher H, van Zweeden AB, et al. Effect of zinc supplementation on malaria and other causes of morbidity in west African children: randomised double blind placebo controlled trial. BMJ. 2001;322(7302):1567. (PubMed)
81. Zinc Against Plasmodium Study Group. Effect of zinc on the treatment of Plasmodium falciparum malaria in children: a randomized controlled trial. Am J Clin Nutr. 2002;76(4):805-812. (PubMed)
82. Sazawal S, Black RE, Ramsan M, et al. Effect of zinc supplementation on mortality in children aged 1-48 months: a community-based randomised placebo-controlled trial. Lancet. 2007;369(9565):927-934. (PubMed)
83. Darling AM, Mugusi FM, Etheredge AJ, et al. Vitamin A and zinc supplementation among pregnant women to prevent placental malaria: a randomized, double-blind, placebo-controlled trial in Tanzania. Am J Trop Med Hyg. 2017;96(4):826-834. (PubMed)
84. Mocchegiani E, Romeo J, Malavolta M, et al. Zinc: dietary intake and impact of supplementation on immune function in elderly. Age (Dordr). 2013;35(3):839-860. (PubMed)
85. Meydani SN, Barnett JB, Dallal GE, et al. Serum zinc and pneumonia in nursing home elderly. Am J Clin Nutr. 2007;86(4):1167-1173. (PubMed)
86. Haase H, Rink L. The immune system and the impact of zinc during aging. Immun Ageing. 2009;6:9. (PubMed)
87. Bogden JD, Oleske JM, Lavenhar MA, et al. Effects of one year of supplementation with zinc and other micronutrients on cellular immunity in the elderly. J Am Coll Nutr. 1990;9(3):214-225. (PubMed)
88. Bogden JD, Oleske JM, Lavenhar MA, et al. Zinc and immunocompetence in elderly people: effects of zinc supplementation for 3 months. Am J Clin Nutr. 1988;48(3):655-663. (PubMed)
89. Provinciali M, Montenovo A, Di Stefano G, et al. Effect of zinc or zinc plus arginine supplementation on antibody titre and lymphocyte subsets after influenza vaccination in elderly subjects: a randomized controlled trial. Age Ageing. 1998;27(6):715-722. (PubMed)
90. Prasad AS. Clinical, immunological, anti-inflammatory and antioxidant roles of zinc. Exp Gerontol. 2008;43(5):370-377. (PubMed)
91. Fortes C, Forastiere F, Agabiti N, et al. The effect of zinc and vitamin A supplementation on immune response in an older population. J Am Geriatr Soc. 1998;46(1):19-26. (PubMed)
92. Hodkinson CF, Kelly M, Alexander HD, et al. Effect of zinc supplementation on the immune status of healthy older individuals aged 55-70 years: the ZENITH Study. J Gerontol A Biol Sci Med Sci. 2007;62(6):598-608. (PubMed)
93. Barnett JB, Dao MC, Hamer DH, et al. Effect of zinc supplementation on serum zinc concentration and T cell proliferation in nursing home elderly: a randomized, double-blind, placebo-controlled trial. Am J Clin Nutr. 2016;103(3):942-951. (PubMed)
94. Norouzi S, Adulcikas J, Sohal SS, Myers S. Zinc stimulates glucose oxidation and glycemic control by modulating the insulin signaling pathway in human and mouse skeletal muscle cell lines. PLoS One. 2018;13(1):e0191727. (PubMed)
95. Gu HF. Genetic, epigenetic and biological effects of zinc transporter (SLC30A8) in type 1 and type 2 diabetes. Curr Diabetes Rev. 2017;13(2):132-140. (PubMed)
96. Flannick J, Thorleifsson G, Beer NL, et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet. 2014;46(4):357-363. (PubMed)
97. Sun Q, van Dam RM, Willett WC, Hu FB. Prospective study of zinc intake and risk of type 2 diabetes in women. Diabetes Care. 2009;32(4):629-634. (PubMed)
98. Vashum KP, McEvoy M, Shi Z, et al. Is dietary zinc protective for type 2 diabetes? Results from the Australian longitudinal study on women's health. BMC Endocr Disord. 2013;13:40. (PubMed)
99. Schwingshackl L, Hoffmann G, Lampousi AM, et al. Food groups and risk of type 2 diabetes mellitus: a systematic review and meta-analysis of prospective studies. Eur J Epidemiol. 2017;32(5):363-375. (PubMed)
100. de Oliveira Otto MC, Alonso A, Lee DH, et al. Dietary intakes of zinc and heme iron from red meat, but not from other sources, are associated with greater risk of metabolic syndrome and cardiovascular disease. J Nutr. 2012;142(3):526-533. (PubMed)
101. Song Y, Xu Q, Park Y, Hollenbeck A, Schatzkin A, Chen H. Multivitamins, individual vitamin and mineral supplements, and risk of diabetes among older U.S. adults. Diabetes Care. 2011;34(1):108-114. (PubMed)
102. Drake I, Hindy G, Ericson U, Orho-Melander M. A prospective study of dietary and supplemental zinc intake and risk of type 2 diabetes depending on genetic variation in SLC30A8. Genes Nutr. 2017;12:30. (PubMed)
103. El Dib R, Gameiro OL, Ogata MS, et al. Zinc supplementation for the prevention of type 2 diabetes mellitus in adults with insulin resistance. Cochrane Database Syst Rev. 2015(5):Cd005525. (PubMed)
104. Islam MR, Attia J, Ali L, et al. Zinc supplementation for improving glucose handling in pre-diabetes: A double blind randomized placebo controlled pilot study. Diabetes Res Clin Pract. 2016;115:39-46. (PubMed)
105. Ranasinghe P, Wathurapatha WS, Galappatthy P, Katulanda P, Jayawardena R, Constantine GR. Zinc supplementation in prediabetes: A randomized double-blind placebo-controlled clinical trial. J Diabetes. 2018;10(5):386-397. (PubMed)
106. Mak CM, Lam CW. Diagnosis of Wilson's disease: a comprehensive review. Crit Rev Clin Lab Sci. 2008;45(3):263-290. (PubMed)
107. Poujois A, Woimant F. Wilson's disease: A 2017 update. Clin Res Hepatol Gastroenterol. 2018;42(6):512-520. (PubMed)
108. Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47(6):2089-2111. (PubMed)
109. Avan A, de Bie RMA, Hoogenraad TU. Wilson's disease should be treated with zinc rather than trientine or penicillamine. Neuropediatrics. 2017;48(5):394-395. (PubMed)
110. Brewer GJ, Dick RD, Johnson VD, Fink JK, Kluin KJ, Daniels S. Treatment of Wilson's disease with zinc XVI: treatment during the pediatric years. J Lab Clin Med. 2001;137(3):191-198. (PubMed)
111. Eda K, Mizuochi T, Iwama I, et al. Zinc monotherapy for young children with presymptomatic Wilson disease: A multicenter study in Japan. J Gastroenterol Hepatol. 2018;33(1):264-269. (PubMed)
112. Gupta P, Choksi M, Goel A, et al. Maintenance zinc therapy after initial penicillamine chelation to treat symptomatic hepatic Wilson's disease in resource constrained setting. Indian J Gastroenterol. 2018;37(1):31-38. (PubMed)
113. Shimizu N, Fujiwara J, Ohnishi S, et al. Effects of long-term zinc treatment in Japanese patients with Wilson disease: efficacy, stability, and copper metabolism. Transl Res. 2010;156(6):350-357. (PubMed)
114. Sinha S, Taly AB. Withdrawal of penicillamine from zinc sulphate-penicillamine maintenance therapy in Wilson's disease: promising, safe and cheap. J Neurol Sci. 2008;264(1-2):129-132. (PubMed)
115. Centers for Disease Control and Prevention. Common colds: protect yourself and others. February 12, 2018. Available at: https://www.cdc.gov/features/rhinoviruses/. Accessed 2/7/19.
116. Rao G, Rowland K. PURLs: Zinc for the common cold--not if, but when. J Fam Pract. 2011;60(11):669-671. (PubMed)
117. Science M, Johnstone J, Roth DE, Guyatt G, Loeb M. Zinc for the treatment of the common cold: a systematic review and meta-analysis of randomized controlled trials. CMAJ. 2012;184(10):E551-561. (PubMed)
118. Singh M, Das RR. Zinc for the common cold. Cochrane Database Syst Rev. 2013(6):Cd001364. (PubMed)
119. Eby GA, 3rd. Zinc lozenges as cure for the common cold--a review and hypothesis. Med Hypotheses. 2010;74(3):482-492. (PubMed)
120. Hemila H. Zinc lozenges may shorten the duration of colds: a systematic review. Open Respir Med J. 2011;5:51-58. (PubMed)
121. Jackson JL, Lesho E, Peterson C. Zinc and the common cold: a meta-analysis revisited. J Nutr. 2000;130(5S Suppl):1512S-1515S. (PubMed)
122. Hemila H. Zinc lozenges and the common cold: a meta-analysis comparing zinc acetate and zinc gluconate, and the role of zinc dosage. JRSM Open. 2017;8(5):2054270417694291. (PubMed)
123. Mossad SB. Effect of zincum gluconicum nasal gel on the duration and symptom severity of the common cold in otherwise healthy adults. QJM. 2003;96(1):35-43. (PubMed)
124. Hirt M, Nobel S, Barron E. Zinc nasal gel for the treatment of common cold symptoms: a double-blind, placebo-controlled trial. Ear Nose Throat J. 2000;79(10):778-780, 782. (PubMed)
125. Belongia EA, Berg R, Liu K. A randomized trial of zinc nasal spray for the treatment of upper respiratory illness in adults. Am J Med. 2001;111(2):103-108. (PubMed)
126. D'Cruze H, Arroll B, Kenealy T. Is intranasal zinc effective and safe for the common cold? A systematic review and meta-analysis. J Prim Health Care. 2009;1(2):134-139. (PubMed)
127. DeCook CA, Hirsch AR. Anosmia due to inhalational zinc: a case report. Chem Senses. 2000;25(5):659.
128. Centers for Disease Control and Prevention. Learn about age-related macular degeneration. July 18, 2018. Available at: https://www.cdc.gov/features/healthyvisionmonth/index.html. Accessed 2/8/19.
129. Cho E, Stampfer MJ, Seddon JM, et al. Prospective study of zinc intake and the risk of age-related macular degeneration. Ann Epidemiol. 2001;11(5):328-336. (PubMed)
130. van Leeuwen R, Boekhoorn S, Vingerling JR, et al. Dietary intake of antioxidants and risk of age-related macular degeneration. JAMA. 2005;294(24):3101-3107. (PubMed)
131. VandenLangenberg GM, Mares-Perlman JA, Klein R, Klein BE, Brady WE, Palta M. Associations between antioxidant and zinc intake and the 5-year incidence of early age-related maculopathy in the Beaver Dam Eye Study. Am J Epidemiol. 1998;148(2):204-214. (PubMed)
132. Newsome DA, Swartz M, Leone NC, Elston RC, Miller E. Oral zinc in macular degeneration. Arch Ophthalmol. 1988;106(2):192-198. (PubMed)
133. Stur M, Tittl M, Reitner A, Meisinger V. Oral zinc and the second eye in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1996;37(7):1225-1235. (PubMed)
134. Evans JR. Antioxidant vitamin and mineral supplements for slowing the progression of age-related macular degeneration. Cochrane Database Syst Rev. 2006(2):CD000254. (PubMed)
135. Evans J. Antioxidant supplements to prevent or slow down the progression of AMD: a systematic review and meta-analysis. Eye (Lond). 2008;22(6):751-760. (PubMed)
136. Newsome DA. A randomized, prospective, placebo-controlled clinical trial of a novel zinc-monocysteine compound in age-related macular degeneration. Curr Eye Res. 2008;33(7):591-598. (PubMed)
137. Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119(10):1417-1436. (PubMed)
138. Chew EY, Clemons TE, Agron E, et al. Long-term effects of vitamins C and E, beta-carotene, and zinc on age-related macular degeneration: AREDS report no. 35. Ophthalmology. 2013;120(8):1604-1611.e1604. (PubMed)
139. Age-Related Eye Disease Study 2 Research Group. Lutein + zeaxanthin and omega-3 fatty acids for age-related macular degeneration: the Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial. JAMA. 2013;309(19):2005-2015. (PubMed)
140. Aronow ME, Chew EY. Age-related Eye Disease Study 2: perspectives, recommendations, and unanswered questions. Curr Opin Ophthalmol. 2014;25(3):186-190. (PubMed)
141. Evans JR, Lawrenson JG. Antioxidant vitamin and mineral supplements for slowing the progression of age-related macular degeneration. Cochrane Database Syst Rev. 2017;7:Cd000254. (PubMed)
142. Blostein-Fujii A, DiSilvestro RA, Frid D, Katz C, Malarkey W. Short-term zinc supplementation in women with non-insulin-dependent diabetes mellitus: effects on plasma 5'-nucleotidase activities, insulin-like growth factor I concentrations, and lipoprotein oxidation rates in vitro. Am J Clin Nutr. 1997;66(3):639-642. (PubMed)
143. Perez A, Rojas P, Carrasco F, et al. Association between zinc nutritional status and glycemic control in individuals with well-controlled type-2 diabetes. J Trace Elem Med Biol. 2018;50:560-565. (PubMed)
144. Billionnet C, Mitanchez D, Weill A, et al. Gestational diabetes and adverse perinatal outcomes from 716,152 births in France in 2012. Diabetologia. 2017;60(4):636-644. (PubMed)
145. Karamali M, Heidarzadeh Z, Seifati SM, et al. Zinc supplementation and the effects on metabolic status in gestational diabetes: A randomized, double-blind, placebo-controlled trial. J Diabetes Complications. 2015;29(8):1314-1319. (PubMed)
146. Karamali M, Heidarzadeh Z, Seifati SM, et al. Zinc supplementation and the effects on pregnancy outcomes in gestational diabetes: a randomized, double-blind, placebo-controlled trial. Exp Clin Endocrinol Diabetes. 2016;124(1):28-33. (PubMed)
147. Karamali M, Bahramimoghadam S, Sharifzadeh F, Asemi Z. Magnesium-zinc-calcium-vitamin D co-supplementation improves glycemic control and markers of cardiometabolic risk in gestational diabetes: a randomized, double-blind, placebo-controlled trial. Appl Physiol Nutr Metab. 2018;43(6):565-570. (PubMed)
148. Ostadmohammadi V, Samimi M, Mobini M, et al. The effect of zinc and vitamin E cosupplementation on metabolic status and its related gene expression in patients with gestational diabetes. J Matern Fetal Neonatal Med. 2018:1-8. (PubMed)
149. Lai H, Lai S, Shor-Posner G, Ma F, Trapido E, Baum MK. Plasma zinc, copper, copper:zinc ratio, and survival in a cohort of HIV-1-infected homosexual men. J Acquir Immune Defic Syndr. 2001;27(1):56-62. (PubMed)
150. Wellinghausen N, Kern WV, Jochle W, Kern P. Zinc serum level in human immunodeficiency virus-infected patients in relation to immunological status. Biol Trace Elem Res. 2000;73(2):139-149. (PubMed)
151. Mocchegiani E, Muzzioli M. Therapeutic application of zinc in human immunodeficiency virus against opportunistic infections. J Nutr. 2000;130(5S Suppl):1424S-1431S. (PubMed)
152. Baum MK, Lai S, Sales S, Page JB, Campa A. Randomized, controlled clinical trial of zinc supplementation to prevent immunological failure in HIV-infected adults. Clin Infect Dis. 2010;50(12):1653-1660. (PubMed)
153. Zeng L, Zhang L. Efficacy and safety of zinc supplementation for adults, children and pregnant women with HIV infection: systematic review. Trop Med Int Health. 2011;16(12):1474-1482. (PubMed)
154. Villamor E, Aboud S, Koulinska IN, et al. Zinc supplementation to HIV-1-infected pregnant women: effects on maternal anthropometry, viral load, and early mother-to-child transmission. Eur J Clin Nutr. 2006;60(7):862-869. (PubMed)
155. Bobat R, Coovadia H, Stephen C, et al. Safety and efficacy of zinc supplementation for children with HIV-1 infection in South Africa: a randomised double-blind placebo-controlled trial. Lancet. 2005;366(9500):1862-1867. (PubMed)
156. Srinivasan MG, Ndeezi G, Mboijana CK, et al. Zinc adjunct therapy reduces case fatality in severe childhood pneumonia: a randomized double blind placebo-controlled trial. BMC Med. 2012;10:14. (PubMed)
157. McHenry MS, Dixit A, Vreeman RC. A systematic review of nutritional supplementation in HIV-infected children in resource-limited settings. J Int Assoc Provid AIDS Care. 2015;14(4):313-323. (PubMed)
158. Li DD, Zhang W, Wang ZY, Zhao P. Serum copper, zinc, and iron levels in patients with Alzheimer's disease: a meta-analysis of case-control studies. Front Aging Neurosci. 2017;9:300. (PubMed)
159. Ventriglia M, Brewer GJ, Simonelli I, et al. Zinc in Alzheimer's disease: a meta-analysis of serum, plasma, and cerebrospinal fluid studies. J Alzheimers Dis. 2015;46(1):75-87. (PubMed)
160. Ventriglia M, Bucossi S, Panetta V, Squitti R. Copper in Alzheimer's disease: a meta-analysis of serum, plasma, and cerebrospinal fluid studies. J Alzheimers Dis. 2012;30(4):981-984. (PubMed)
161. Brewer GJ. Copper excess, zinc deficiency, and cognition loss in Alzheimer's disease. Biofactors. 2012;38(2):107-113. (PubMed)
162. Maserejian NN, Hall SA, McKinlay JB. Low dietary or supplemental zinc is associated with depression symptoms among women, but not men, in a population-based epidemiological survey. J Affect Disord. 2012;136(3):781-788. (PubMed)
163. Nowak G, Siwek M, Dudek D, Zieba A, Pilc A. Effect of zinc supplementation on antidepressant therapy in unipolar depression: a preliminary placebo-controlled study. Pol J Pharmacol. 2003;55(6):1143-1147. (PubMed)
164. Siwek M, Dudek D, Paul IA, et al. Zinc supplementation augments efficacy of imipramine in treatment resistant patients: a double blind, placebo-controlled study. J Affect Disord. 2009;118(1-3):187-195. (PubMed)
165. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus definitions for sepsis and septic shock (sepsis-3). JAMA. 2016;315(8):801-810. (PubMed)
166. Alker W, Haase H. Zinc and Sepsis. Nutrients. 2018;10(8). (PubMed)
167. Hood MI, Skaar EP. Nutritional immunity: transition metals at the pathogen-host interface. Nat Rev Microbiol. 2012;10(8):525-537. (PubMed)
168. Hoeger J, Simon TP, Beeker T, Marx G, Haase H, Schuerholz T. Persistent low serum zinc is associated with recurrent sepsis in critically ill patients - A pilot study. PLoS One. 2017;12(5):e0176069. (PubMed)
169. Saleh NY, Abo El Fotoh WMM. Low serum zinc level: The relationship with severe pneumonia and survival in critically ill children. Int J Clin Pract. 2018;72(6):e13211. (PubMed)
170. Banupriya N, Vishnu Bhat B, Benet BD, Sridhar MG, Parija SC. Efficacy of zinc supplementation on serum calprotectin, inflammatory cytokines and outcome in neonatal sepsis - a randomized controlled trial. J Matern Fetal Neonatal Med. 2017;30(13):1627-1631. (PubMed)
171. Banupriya N, Bhat BV, Benet BD, Catherine C, Sridhar MG, Parija SC. Short term oral zinc supplementation among babies with neonatal sepsis for reducing mortality and improving outcome - a double-blind randomized controlled trial. Indian J Pediatr. 2018;85(1):5-9. (PubMed)
172. Newton B, Bhat BV, Dhas BB, Mondal N, Gopalakrishna SM. Effect of zinc supplementation on early outcome of neonatal sepsis--a randomized controlled trial. Indian J Pediatr. 2016;83(4):289-293. (PubMed)
173. Mehta K, Bhatta NK, Majhi S, Shrivastava MK, Singh RR. Oral zinc supplementation for reducing mortality in probable neonatal sepsis: a double blind randomized placebo controlled trial. Indian Pediatr. 2013;50(4):390-393. (PubMed)
174. Gupta RK, Gangoliya SS, Singh NK. Reduction of phytic acid and enhancement of bioavailable micronutrients in food grains. J Food Sci Technol. 2015;52(2):676-684. (PubMed)
175. Fulgoni VL, 3rd, Keast DR, Bailey RL, Dwyer J. Foods, fortificants, and supplements: Where do Americans get their nutrients? J Nutr. 2011;141(10):1847-1854. (PubMed)
176. US Department of Agriculture, Agricultural Research Service. FoodData Central, 2019. fdc.nal.usda.gov.
177. Nations SP, Boyer PJ, Love LA, et al. Denture cream: an unusual source of excess zinc, leading to hypocupremia and neurologic disease. Neurology. 2008;71(9):639-643. (PubMed)
178. McBride K, Slotnick B, Margolis FL. Does intranasal application of zinc sulfate produce anosmia in the mouse? An olfactometric and anatomical study. Chem Senses. 2003;28(8):659-670. (PubMed)
179. Natural Medicines. Zinc: professional handout/drug interactions. Available at: https://naturalmedicines.therapeuticresearch.com. Accessed 1/28/19.
180. Ervin RB, Kennedy-Stephenson J. Mineral intakes of elderly adult supplement and non-supplement users in the third national health and nutrition examination survey. J Nutr. 2002;132(11):3422-3427. (PubMed)
181. Kvamme JM, Gronli O, Jacobsen BK, Florholmen J. Risk of malnutrition and zinc deficiency in community-living elderly men and women: the Tromso Study. Public Health Nutr. 2015;18(11):1907-1913. (PubMed)