Niacin
Contents
Summary
- Dietary precursors of nicotinamide adenine dinucleotide (NAD), including nicotinic acid, nicotinamide, and nicotinamide riboside, are collectively referred to as niacin or vitamin B3. The essential amino acid tryptophan can also be converted into NAD via the kynurenine pathway. (More information)
- NAD can be phosphorylated (NADP) and reduced (NADH and NADPH). NAD functions in oxidation-reduction (redox) reactions and non-redox reactions. (More information)
- Pellagra is the disease of severe niacin deficiency. It is characterized by symptoms affecting the skin, the digestive system, and the nervous system; pellagra can lead to death if left untreated. (More information)
- Causes of niacin deficiency include inadequate oral intake, poor bioavailability from unlimed grains, defective tryptophan absorption, metabolic disorders, and the long-term use of chemotherapeutic treatments. (More information)
- Dietary intake requirements for niacin are based on the urinary excretion of niacin metabolites. (More information)
- NAD is the sole substrate for PARP enzymes and sirtuins involved in DNA repair activities; thus, NAD is critical for genome stability. Several studies, mostly using in vitro and animal models, suggest a possible role for niacin in cancer prevention. In a recent phase III trial, a daily pharmacologic dose of nicotinamide was found to reduce the rate of premalignant skin lesions and nonmelanoma cancers in high-risk subjects. (More information)
- Despite promising initial results, nicotinamide administration has failed to prevent or delay the onset of type 1 diabetes mellitus in high-risk relatives of type 1 diabetic patients. Future research might explore the use of nicotinamide in combined therapy and evaluate activators of NAD-dependent enzymes. (More information)
- At pharmacologic doses, nicotinic acid improved lipid profiles of patients with a history of vascular disease yet failed to reduce recurrent cardiovascular events or mortality. (More information)
- Elevated tryptophan breakdown in the kynurenine pathway and niacin deficiency have been reported in HIV-positive people. However, at present, a better understanding of the role of kynurenine pathway and other NAD biosynthetic pathways during HIV infection is needed to establish whether this population could benefit from niacin supplementation. (More information)
- Most adverse effects of niacin (nicotinic acid and nicotinamide) have been reported with pharmacological doses of nicotinic acid. The tolerable upper intake level (UL) for niacin is based on preventing skin flushing, nicotinic acid’s most prominent side effect. The co-administration of laropiprant — a prostaglandin D2 receptor-1 antagonist — helps reduce nicotinic acid-induced skin flushing. (More information)
Niacin or vitamin B3 is a water-soluble vitamin used by the body to form the nicotinamide coenzyme, NAD+. The term ‘niacin’ is often used to refer to nicotinic acid (pyridine-3-carboxylic acid) only, although other vitamers with a pyridine ring, including nicotinamide (pyridine-3-carboxamide) and nicotinamide riboside, also contribute to NAD+ formation (1). None of the vitamers are related to the nicotine found in tobacco, although their names are similar. Likewise, nicotine — but not nicotinic acid — is an agonist of the nicotinic receptors that respond to the neurotransmitter, acetylcholine.
Metabolism
Essential to all forms of life, the nicotinamide coenzyme NAD+ is synthesized in the body from four precursors that are provided in the diet: nicotinic acid, nicotinamide, nicotinamide riboside, and tryptophan (Figure 1).
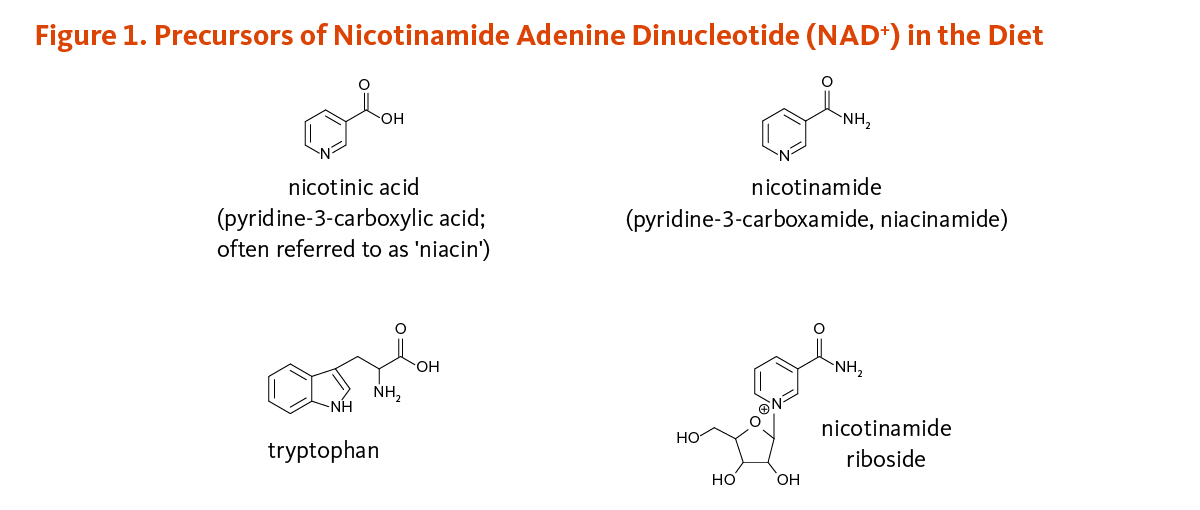
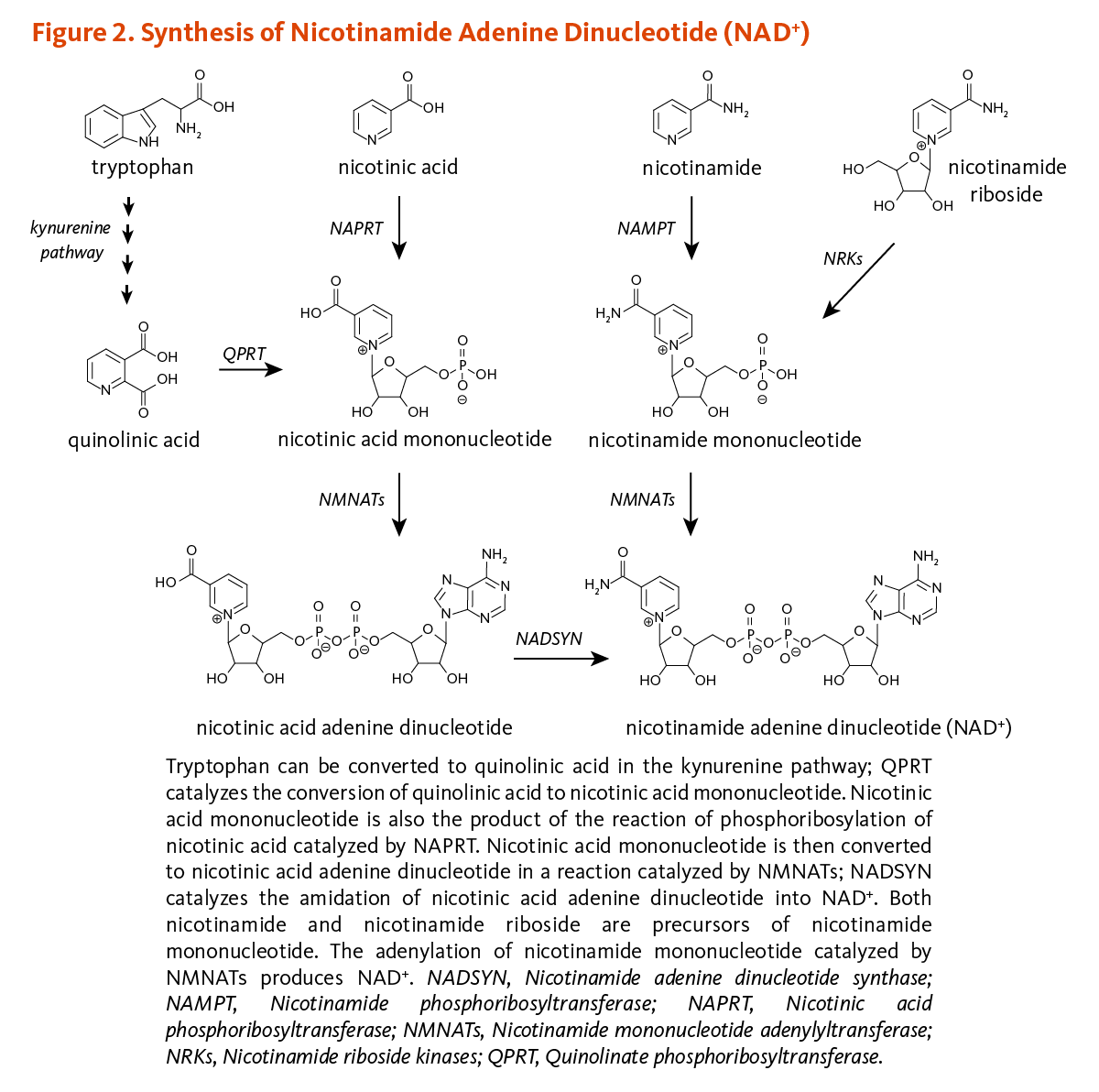
Figure 2 illustrates the separate biosynthetic pathways that lead to NAD+ production from the various dietary precursors. NAD+ is synthesized from nicotinamide and nicotinamide riboside via two enzymatic reactions, while the pathway that yields NAD+ from nicotinic acid - known as the Preiss-Handler pathway — includes three steps. The kynurenine pathway is the longest NAD+ biosynthetic pathway: the catabolism of tryptophan through kynurenine produces quinolinic acid, which is then converted to nicotinic acid mononucleotide, an intermediate in NAD+ metabolism. NAD+ is then synthesized from nicotinic acid mononucleotide in the Preiss-Handler pathway (2).
All pathways generate intermediary mononucleotides — either nicotinic acid mononucleotide or nicotinamide mononucleotide. Specific enzymes, known as phosphoribosyltransferases, catalyze the addition of a phosphoribose moiety onto nicotinic acid or quinolinic acid to produce nicotinic acid mononucleotide or onto nicotinamide to generate nicotinamide mononucleotide. Nicotinamide mononucleotide is also generated by the phosphorylation of nicotinamide riboside, catalyzed by nicotinamide riboside kinases (NRKs). Further, adenylyltransferases catalyze the adenylation of these mononucleotides to form either nicotinic acid adenine dinucleotide or NAD+. Nicotinic acid adenine dinucleotide is then converted to NAD+ by glutamine-dependent NAD+ synthetase (NADSYN), which uses glutamine as an amide group donor (Figure 2) (2). Of note, nicotinic acid adenine dinucleotide has been reported to form following the administration of high-dose nicotinamide riboside, suggesting that a potential deamidation could occur to convert NAD+ to nicotinic acid adenine dinucleotide when the pool of NAD+ is high (1).
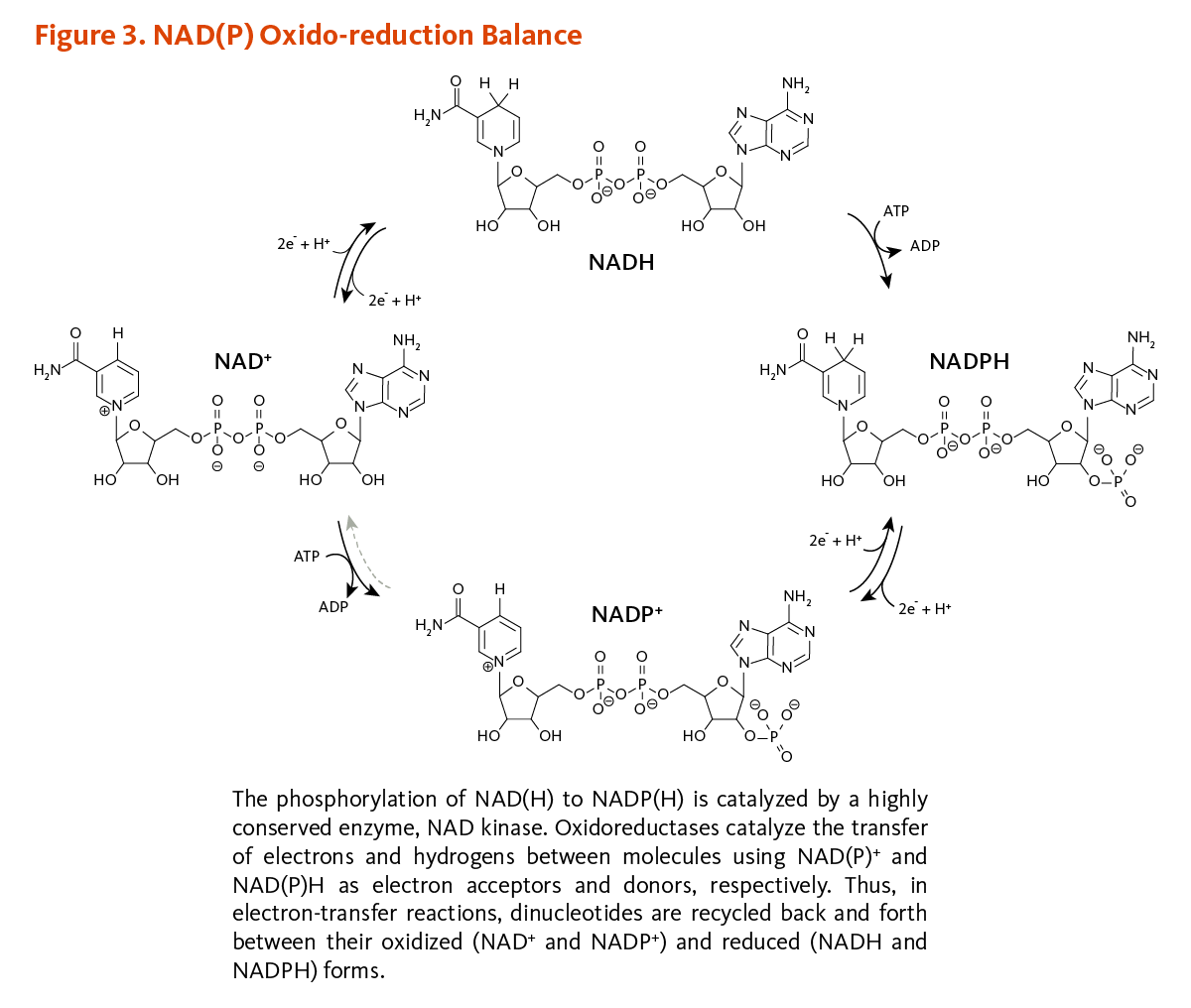
NAD kinase catalyzes the phosphorylation of NAD into NADP using adenosyl triphosphate (ATP) as the phosphoryl donor (3). The oxidation-reduction (redox) properties of the dinucleotide are not affected by the phosphorylation such that the redox pairs NAD+/NADH and NADP+/NADPH show similar redox potentials (4). Oxidation and reduction of the C-4 position of the nicotinamide moiety of NADand its phosphorylated form are essential for electron-transfer reactions supporting vital metabolic and bioenergetic functions in all cells (see Function). Thus, NAD and NADP are recycled back and forth between oxidized (NAD+ and NADP+) and reduced forms (NADH and NADPH), as shown in Figure 3.
Function
NAD as a coenzyme in electron-transfer reactions
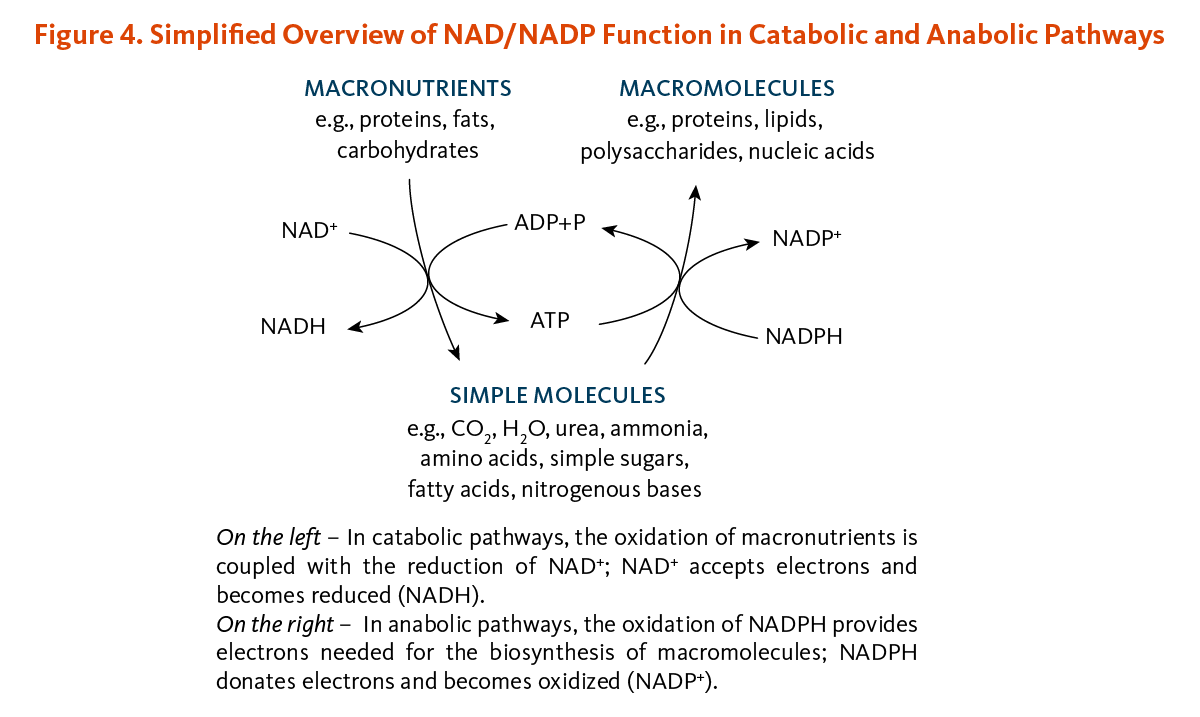
Living organisms derive most of their energy from redox reactions, which are processes involving the transfer of electrons. Over 400 enzymes require the niacin coenzymes, NAD and NADP, mainly to accept or donate electrons for redox reactions (5). NAD and NADP appear to support distinct functions (Figure 4). NAD functions most often in energy-producing reactions involving the degradation (catabolism) of carbohydrates, fats, proteins, and alcohol. NADP generally serves in biosynthetic (anabolic) reactions, such as in the synthesis of fatty acids, steroids (e.g., cholesterol, bile acids, and steroid hormones), and building blocks of other macromolecules (4). NADP is also essential for the regeneration of components of detoxification and antioxidant systems (4). To support these functions, the cell maintains NAD in a largely oxidized state (NAD+) to serve as oxidizing agent for catabolic reactions, while NADP is kept largely in a reduced state (NADPH) to readily donate electrons for reductive cellular processes (4, 6).
NAD as a substrate for NAD-consuming enzymes
The niacin coenzyme, NAD, is the substrate (reactant) for at least four classes of enzymes. Two classes of enzymes with mono adenosine diphosphate (ADP)-ribosyltransferase and/or poly (ADP-ribose) polymerase activities catalyze ADP-ribosyl transfer reactions. Silent information regulator-2 (Sir2)-like proteins (sirtuins) catalyze the removal of acetyl groups from acetylated proteins, utilizing ADP-ribose from NAD as an acceptor for acetyl groups. Finally, ADP-ribosylcyclases are involved in the regulation of intracellular calcium signaling.
ADP-ribosylation
Enzymes with ADP-ribosyltransferase activities were formerly divided between mono ADP-ribosyltransferases (ARTs) and poly (ADP-ribose) polymerases (PARPs). ARTs were first discovered in certain pathogenic bacteria — like those causing cholera or diphtheria — where they mediate the actions of toxins. These enzymes transfer an ADP-ribose residue moiety from NAD to a specific amino acid of a target protein, with the creation of an ADP-ribosylated protein and the release of nicotinamide.
Because most PARPs have been found to exhibit only mono ADP-ribosyltransferase activities, a new nomenclature was proposed for enzymes catalyzing ADP-ribosylation: A family of mono ADP-ribosyltransferases with homology to bacterial diphteria toxins was named ARTD, while enzymes with either mono or poly ADP-ribosyltransferase activities and related to C2 and C3 clostridial toxins were included in the ARTC family (7, 8).
- ARTCs are extracellular enzymes that catalyze the mono ADP-ribosylation of membrane or secreted proteins involved in innate immunity and cell communication (2).
- ARTDs are intracellular enzymes with either mono or poly ADP-ribosyltransferase activities. At least 18 ARTDs have been identified. All ARTDs possess a diphtheria toxin-like catalytic domain that binds NAD+. Only ARTDs 1, 2, 5, and 6 catalyze poly (ADP-ribose) transfers; the others have mono ADP-ribosyltransferase activities. ARTDs were shown to be involved in DNA repair and stress responses, cell signaling, transcription regulation, apoptosis, cell differentiation, maintenance of genomic integrity, and antiviral defense (reviewed in 8).
NAD-dependent deacetylation
Seven sirtuins (SIRT 1-7) have been identified in humans. Sirtuins are a class of NAD-dependent deacetylase enzymes that remove acetyl groups from the acetylated lysine residues of target proteins. During the deacetylation process, the acetyl group is transferred onto the ADP-ribose moiety cleaved off NAD, producing O-acetyl-ADP-ribose. Nicotinamide can exert feedback inhibition to the deacetylation reaction (9). Like ADP-ribosylation, acetylation is a post-translational modification that affects the function of target proteins. The initial interest in sirtuins followed the discovery that their activation could mimic caloric restriction, which has been shown to increase lifespan in lower organisms. Such a role in mammals is controversial, although sirtuins are energy-sensing regulators involved in signaling pathways that could play important roles in delaying the onset of age-related diseases (e.g., cardiovascular disease, cancer, dementia, arthritis). To date, the spectrum of their biological functions includes gene silencing, DNA damage repair, cell cycle regulation, and cell differentiation (10).
Calcium mobilization
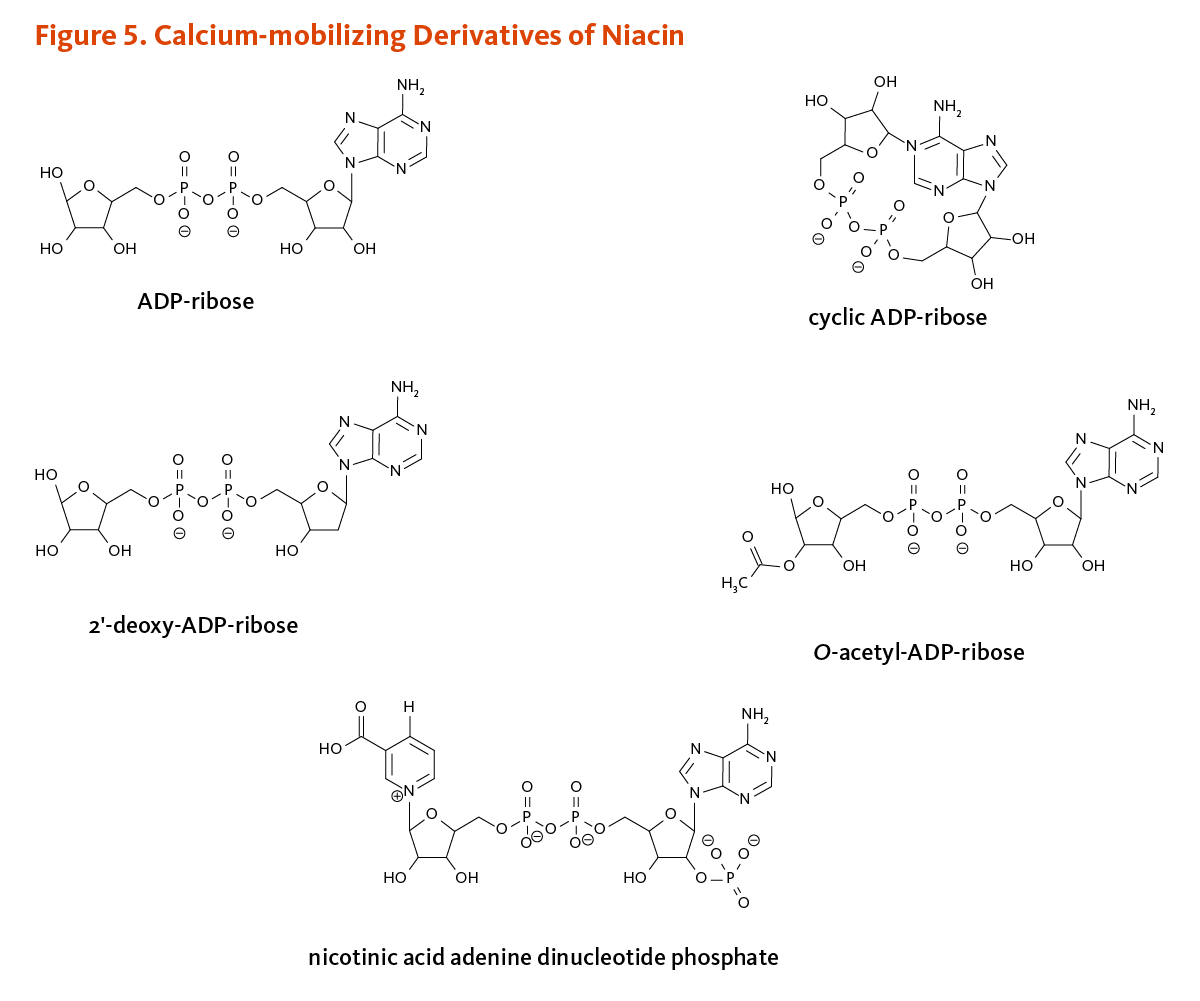
In humans, CD38 and CD157 belong to a family of NAD+ glycohydrolases/ADP-ribosylcyclases. These enzymes catalyze the formation of key regulators of calcium signaling, namely (linear) ADP-ribose, cyclic ADP-ribose, and nicotinic acid adenine dinucleotide phosphate (Figure 5). Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate works within cells to provoke the release of calcium ions from internal storage sites (i.e., endoplasmic reticulum, lysosomes, mitochondria), whereas ADP-ribose stimulates extracellular calcium entry through cell membrane TRPM2 cation channels (2). Another TRPM2 agonist, 2’-deoxy-ADP-ribose, was recently identified in vitro. CD38 was found to catalyze the synthesis of 2’-deoxy-ADP-ribose from nicotinamide mononucleotide and 2’-deoxy-ATP (11). O-acetyl-ADP-ribose generated by the activity of sirtuins also controls calcium entry through TRPM2 channels (6). Intracellular calcium-mediated signal transduction is regulated by transient calcium entry into the cell or release of calcium from intracellular stores. Calcium signaling is critically involved in processes like neurotransmission, insulin release from pancreatic β-cells, muscle cell contraction, and T-lymphocyte activation (6).
NAD as a ligand
NAD+ has been identified as an endogenous agonist of purinergic membrane receptors of the P2Y subclass. In particular, NAD was found to bind to P2Y1 receptor and act as an inhibitory neurotransmitter at neuromuscular junctions in visceral smooth muscles (12). Extracellular NAD+ was also found to behave like a proinflammatory cytokine, triggering the activation of isolated granulocytes. NAD+ binding to the P2Y11 receptor at the granulocyte surface activated a signaling cascade involving cyclic ADP-ribose and the rise of intracellular calcium, eventually stimulating superoxide generation and chemotaxis (13). Similar observations were made with lipopolysaccharide-activated monocytes (14). Extracellular NAADP+ and ADP-ribose might also bind to P2Y receptors and trigger intracellular NAADP+- and ADP-ribose-dependent calcium mobilization (see Calcium mobilization) (15, 16).
Lipid-lowering effects with pharmacologic doses of nicotinic acid
For over half a century, pharmacologic doses of nicotinic acid, but not nicotinamide, have been known to reduce serum cholesterol (see Disease Treatment) (17). However, the exact mechanisms underlying the lipid-lowering effect of nicotinic acid remain speculative. Two G-protein-coupled membrane receptors, GPR109A and GPR109B, bind nicotinic acid with high and low affinity, respectively. These nicotinic acid receptors are primarily expressed in adipose tissue and immune cells (but not lymphocytes). They are also found in retinal pigmented and colonic epithelial cells, keratinocytes, breast cells, microglia, and possibly at low levels in the liver (18). Thus, lipid-modifying effects of nicotinic acid are likely to be mediated by receptor-independent mechanisms in major tissues of lipid metabolism like liver and skeletal muscle. Early in vitro data suggested that nicotinic acid could impair very-low-density lipoprotein (VLDL) secretion by inhibiting triglyceride synthesis and triggering ApoB lipoprotein degradation in hepatocytes (19). In another study, nicotinic acid affected the hepatic uptake of ApoAI lipoprotein, thereby reducing high-density lipoprotein (HDL) removal from the circulation (reviewed in 20). In adipocytes, the binding of nicotinic acid to GPR109A was found to initiate a signal transduction cascade resulting in reductions in free fatty acid production via the inhibition of hormone-sensitive lipase involved in triglyceride lipolysis (21). Nonetheless, recent observations have suggested that the lipid-lowering effect of nicotinic acid was not due to its anti-lipolytic activity (22). Trials showed that synthetic agonists of GPR109A acutely lowered free fatty acids yet failed to affect serum lipids (22). Aside from its impact on HDL and other plasma lipids, nicotinic acid has exhibited anti-atherosclerotic activities in cultured monocytes, macrophages, or vascular endothelial cells, by modulating inflammation and oxidative stress and regulating cell adhesion, migration, and differentiation (reviewed in 18).
Deficiency
Pellagra
Causes
The late stage of severe niacin deficiency is known as pellagra. Early records of pellagra followed the widespread cultivation of corn in Europe in the 1700s (23). The disease is generally associated with poorer social classes whose chief dietary staple consisted of cereal like corn or sorghum. Pellagra was also common in the southern United States during the early 1900s where income was low and corn products were a major dietary staple (24). Interestingly, pellagra was not known in Mexico, where corn was also an important dietary staple and much of the population was also poor. In fact, if corn contains appreciable amounts of niacin, it is present in a bound form that is not nutritionally available to humans. The traditional preparation of corn tortillas in Mexico involves soaking the corn in a lime (calcium oxide) solution, prior to cooking. Heating the corn in an alkaline solution results in the release of bound niacin, increasing its bioavailability (25). Pellagra epidemics were also unknown to Native Americans who consumed immature corn that contains predominantly unbound (bioavailable) niacin (24).
Niacin deficiency or pellagra may result from inadequate dietary intake of NAD precursors, including tryptophan. Niacin deficiency — often associated with malnutrition — is observed in the homeless population, in individuals suffering from anorexia nervosa or obesity, and in consumers of diets high in maize and poor in animal protein (26-29). Deficiencies of other B vitamins and some trace minerals may aggravate niacin deficiency (30, 31). Malabsorptive disorders that can lead to pellagra include Crohn’s disease and megaduodenum (32, 33). Patients with Hartnup’s disease, a hereditary disorder resulting in defective tryptophan absorption, have developed pellagra (see Niacin-responsive genetic disorders). Carcinoid syndrome, a condition of increased secretion of serotonin and other catecholamines by carcinoid tumors, may also result in pellagra due to increased utilization of dietary tryptophan for serotonin rather than niacin synthesis. Further, prolonged treatment with the anti-tuberculosis drug isoniazid has resulted in niacin deficiency (34). Other pharmaceutical agents, including the immunosuppressive drugs azathioprine (Imuran) and 6-mercaptopurine, the anti-cancer drug 5-fluorouracil (5-FU, Adrucil), and levodopa/carbidopa (Sinemet; two drugs given to people with Parkinson’s disease), are known to increase the reliance on dietary niacin by interfering with the tryptophan-kynurenine-niacin pathway (35). Finally, other populations at risk for niacin deficiency include dialysis patients, cancer patients (36, 37), individuals suffering from chronic alcoholism (38), and people with HIV (see HIV/AIDS below). Further, chronic alcohol intake can lead to severe niacin deficiency through reducing dietary niacin intake and interfering with the tryptophan-to-NAD conversion (30).
Symptoms of deficiency
The most common symptoms of niacin deficiency involve the skin, the digestive system, and the nervous system. The symptoms of pellagra are commonly referred to as the three "Ds": sun-sensitive dermatitis, diarrhea, and dementia. A fourth "D," death, occurs if pellagra is left untreated (5). In the skin, a thick, scaly, darkly pigmented rash develops symmetrically in areas exposed to sunlight. In fact, the word "pellagra" comes from "pelle agra," the Italian phrase for rough skin. Symptoms related to the digestive system include inflammation of the mouth and tongue ("bright red tongue"), vomiting, constipation, abdominal pain, and ultimately, diarrhea. Gastrointestinal disorders and diarrhea contribute to the ongoing malnourishment of the patients. Neurologic symptoms include headache, apathy, fatigue, depression, disorientation, and memory loss and are more consistent with delirium than with the historically described dementia (38). Disease presentations vary in appearance since the classic triad rarely presents in its entirety. The absence of dermatitis, for example, is known as pellagra sine pellagra.
Treatment
To treat pellagra, the World Health Organization (WHO) recommends administering nicotinamide to avoid the flushing commonly caused by nicotinic acid (see Safety). Treatment guidelines suggest using 300 mg/day of oral nicotinamide in divided doses, or 100 mg/day administered parenterally in divided doses, for three to four weeks (37, 39). Because patients with pellagra often display additional vitamin deficiencies, administration of a vitamin B-complex preparation is advised (39).
The Recommended Dietary Allowance (RDA)
Niacin equivalent (NE)
The term "niacin equivalent" (NE) is used to describe the contribution to dietary intake of all the forms of niacin that are available to the body. In healthy individuals, less than 2% of dietary tryptophan is converted to NAD in the kynurenine pathway (40). The synthesis of NAD from tryptophan is fairly inefficient and depends on enzymes requiring vitamin B6 and riboflavin, as well as a heme (iron)-containing enzyme. Nonetheless, tryptophan is essential as a precursor for NAD+. Inherited defects in tryptophan transport and metabolism result in severe clinical disorders attributed to NAD+ depletion (see Niacin-responsive genetic disorders). On average, 60 milligrams (mg) of tryptophan are considered to correspond to 1 mg of niacin or 1 mg of NE.
Daily recommended intakes
The recommended dietary allowance (RDA) for niacin is based on the prevention of deficiency. Pellagra can be prevented by about 11 mg NE/day, but 12 mg to 16 mg NE/day has been found to normalize the urinary excretion of niacin metabolites (breakdown products) in healthy young adults. Because pellagra represents severe deficiency, the Food and Nutrition Board (FNB) of the US Institute of Medicine chose to use the excretion of niacin metabolites as an indicator of niacin nutritional status rather than symptoms of pellagra (41). However, it has been argued that cellular NAD and NADP content may be more relevant indicators of niacin nutritional status (24).
| Life Stage | Age | Males (mg NE*/day) | Females (mg NE/day) |
|---|---|---|---|
| Infants | 0-6 months | 2 (AI) | 2 (AI) |
| Infants | 7-12 months | 4 (AI) | 4 (AI) |
| Children | 1-3 years | 6 | 6 |
| Children | 4-8 years | 8 | 8 |
| Children | 9-13 years | 12 | 12 |
| Adolescents | 14-18 years | 16 | 14 |
| Adults | 19 years and older | 16 | 14 |
| Pregnancy | all ages | - | 18 |
| Breast-feeding | all ages | - | 17 |
| *NE, niacin equivalent: 1 mg NE = 60 mg of tryptophan = 1 mg niacin | |||
Disease Prevention
Cancer
Studies of cultured cells (in vitro) provide evidence that NAD content influences mechanisms that maintain genomic stability. Loss of genomic stability, characterized by a high rate of damage to DNA and chromosomes, is a hallmark of cancer (42). The current understanding is that the pool of NAD is decreased during niacin deficiency and that it affects the activity of NAD-consuming enzymes rather than redox and metabolic functions (43). Among NAD-dependent reactions, poly ADP-ribosylations catalyzed by PARP enzymes (ARTDs) are critical for the cellular response to DNA injury. After DNA damage, PARPs are activated; the subsequent poly ADP-ribosylations of a number of signaling and structural molecules by PARPs were shown to facilitate DNA repair at DNA strand breaks (44). Cellular depletion of NAD has been found to decrease levels of the tumor suppressor protein p53, a target for poly ADP-ribosylation, in human breast, skin, and lung cells (45). The expression of p53 was also altered by niacin deficiency in rat bone marrow cells (46). Impairment of DNA repair caused by niacin deficiency could lead to genomic instability and drive tumor development in rat models (47, 48). Both PARPs and sirtuins have been recently involved in the maintenance of heterochromatin, a chromosomal domain associated with genome stability, as well as in transcriptional gene silencing, telomere integrity, and chromosome segregation during cell division (49, 50). Neither the cellular NAD content nor the dietary intake of NAD precursors necessary for optimizing protective responses following DNA damage has been determined, but both are likely to be higher than that required for the prevention of pellagra.
Bone marrow
Cancer patients often suffer from bone marrow suppression following chemotherapy, given that bone marrow is one of the most proliferative tissues in the body and thus a primary target for chemotherapeutic agents. Niacin deficiency was found to decrease bone marrow NAD and poly-ADP-ribose levels and increase the risk of chemically induced leukemia in rats (51). Conversely, a pharmacologic dose of either nicotinic acid or nicotinamide was able to increase NAD and poly ADP-ribose in bone marrow and decrease the development of leukemia in rats (52). It has been suggested that niacin deficiency often observed in cancer patients could sensitize bone marrow tissue to the suppressive effect of chemotherapy. However, little is known regarding cellular NAD levels and the prevention of DNA damage or cancer in humans. One study in two healthy individuals involved elevating NAD levels in blood lymphocytes by supplementation with 100 mg/day of nicotinic acid for eight weeks. Compared to non-supplemented individuals, the supplemented individuals had reduced DNA strand breaks in lymphocytes exposed to free radicals in a test tube assay (53). However, nicotinic acid supplementation of up to 100 mg/day for 14 weeks in 21 healthy smokers failed to provide any evidence of a decrease in cigarette smoke-induced genetic damage in blood lymphocytes compared to placebo (54). More recently, the frequency of chromosome translocation was used to evaluate DNA damage in peripheral blood lymphocytes of 82 pilots chronically exposed to ionizing radiation, a known human carcinogen. In this observational study, the rate of chromosome aberrations was significantly lower in subjects with higher (28.4 mg/day) compared to lower (20.5 mg/day) dietary niacin intake (55). Higher availability of NAD+ in X-irradiated peripheral blood lymphocytes was found to favor DNA repair by enhancing survival, particularly through SIRT-mediated p53 deacetylation (56).
Upper digestive tract
Generally, relationships between dietary factors and cancer are established first in epidemiological studies and followed up by basic cancer research at the cellular level. In the case of niacin, research on biochemical and cellular aspects of DNA repair has stimulated an interest in the relationship between niacin intake and cancer risk in human populations (57). A large case-control study found increased consumption of niacin, along with antioxidant nutrients, to be associated with decreased incidence of oral (mouth), pharyngeal (throat), and esophageal cancers in northern Italy and Switzerland. An increase in daily niacin intake of 6.2 mg was associated with about a 40% decrease in cases of cancers of the mouth and throat, while a 5.2 mg increase in daily niacin intake was associated with a similar decrease in cases of esophageal cancer (58, 59).
Skin
Niacin deficiency can lead to severe sunlight sensitivity in exposed skin. Given the implication of NAD-dependent enzymes in DNA repair, there has been some interest in the effect of niacin on skin health. In vitro and animal experiments have helped gather information, but human data on niacin/NAD status and skin cancer are very limited. One study reported that niacin supplementation decreased the risk of ultraviolet light (UV)-induced skin cancers in mice, despite the fact that mice convert tryptophan to NAD more efficiently than rats and humans and thus do not get severely deficient (60). Hyper-proliferation and impaired differentiation of skin cells can alter the integrity of the skin barrier and increase the occurrence of pre-malignant and malignant skin conditions. A protective effect of niacin was suggested by topical application of myristyl nicotinate, a niacin derivative, which successfully increased the expression of epidermal differentiation markers in subjects with photodamaged skin (61). The activation of the nicotinic acid receptors, GPR109A and GPR109B, by pharmacologic doses of niacin could be involved in improving skin barrier function. Conversely, differentiation defects in skin cancer cells were linked to the abnormal cellular localization of defective nicotinic acid receptors (62). Nicotinamide restriction with subsequent depletion of cellular NAD was shown to increase oxidative stress-induced DNA damage in a precancerous skin cell model, implying a protective role of NAD-dependent pathways in cancer (63). Altered NAD availability also affects sirtuin expression and activity in UV-exposed human skin cells. Along with PARPs, NAD-consuming sirtuins could play an important role in the cellular response to photodamage and skin homeostasis (64).
A pooled analysis of two large US prospective cohort studies that followed 41,808 men and 72,308 women for up to 26 years suggested that higher versus lower intake of niacin (from diet and supplements) might be protective against squamous-cell carcinoma but not against basal-cell carcinoma and melanoma (65). A phase III, randomized, double-blind, placebo-controlled trial in 386 subjects with a history of nonmelanoma skin cancer recently examined the effect of daily nicotinamide supplementation (1 g) for 12 months on skin cancer recurrence at three-month intervals over an 18-month period (66). Nicotinamide effectively reduced the rate of premalignant actinic keratose (-11%), squamous-cell carcinoma (-30%), and basal-cell carcinoma (-20%) compared to placebo after 12 months, yet this protection was not sustained during the six-month post-supplementation period (66). Larger trials are needed to assess whether nicotinamide could reduce the risk of melanomas, which are not as common as other skin cancer but are more deadly (67).
Type 1 diabetes mellitus
Type 1 diabetes mellitus in children is caused by the autoimmune destruction of insulin-secreting β-cells in the pancreas. Prior to the onset of symptomatic diabetes, specific antibodies, including islet cell autoantibodies (ICA), can be detected in the blood of high-risk individuals (68). In an experimental animal model of diabetes, high levels of nicotinamide are administered to protect β-cells from damage caused by streptozotocin (69).
Yet, pharmacologic doses of nicotinamide (up to 3 g/day) have not been found to be effective in delaying or preventing the onset of type 1 diabetes in at-risk subjects. An analysis of 10 trials, of which five were placebo-controlled, found evidence of improved β-cell function after one year of treatment with nicotinamide, but the analysis failed to find any clinical evidence of improved glycemic control (70). A large, multicenter randomized controlled trial of nicotinamide in ICA-positive siblings (ages, 3-12 years) of type 1 diabetic patients also failed to find a difference in the incidence of type 1 diabetes after three years (70). A randomized, double-blind, placebo-controlled multicenter trial of nicotinamide (maximum of 3 g/day) was conducted in 552 ICA-positive relatives of patients with type 1 diabetes. The proportion of relatives who developed type 1 diabetes within five years was comparable whether they were treated with nicotinamide or placebo (71). Nicotinamide could reduce inflammation-related parameters in these high-risk subjects yet was ineffective to prevent disease onset (72). More recently, case reports of the combined use of nicotinamide (25 mg/kg/day) and acetyl-L-carnitine (50 mg/kg/day) in children at risk for type 1 diabetes showed promising results, warranting further investigation (73).
Disease Treatment
Niacin supplements at pharmacologic doses (i.e., doses much larger than those needed to prevent deficiency) have been used in an attempt to treat a range of conditions, some of which are discussed below.
Niacin-responsive genetic disorders
Congenital NAD deficiency-related disorders can result from mutations in genes involved in the uptake and transport of the various dietary NAD+ precursors or in the distinct metabolic pathways leading to NAD+ production (see Metabolism). Some of these disorders might respond to niacin supplementation. For example, defective transport of tryptophan into cells results in Hartnup disease, which features signs of severe niacin deficiency (74). Hartnup disease is due to mutations in the SLCA19 gene, which codes for a sodium-dependent neutral amino acid transporter expressed primarily in the kidneys and intestine. Disease management involves supplementation with nicotinic acid or nicotinamide (75). Recessive mutations in genes coding for enzymes of the kynurenine pathway — namely kynureninase and 3-hydroxyanthranilic-acid 3,4-dioxygenase — lead to combined vertebral, anal, cardiac, tracheo-esophageal, renal, and limb (VACTERL) congenital malformations (76). Depletion of NAD+, rather than accumulation of intermediate metabolites in the kynurenine pathway, was found to be responsible for these malformations. Niacin supplementation throughout pregnancy ensured adequate levels of NAD+ and prevented congenital anomalies in mice with kynurenine pathway mutations (76). In humans, the dose of NAD+ precursors necessary to avert NAD deficiency-induced congenital VACTERL malformations has yet to be defined (77).
Nicotinamide may also rescue NAD+ depletion secondary to an ultra-rare inborn error of glutamine metabolism (78). Glutamine is required for the conversion of nicotinic acid adenine dinucleotide to NAD+ catalyzed by NAD+ synthetase (Figure 2). Thus, inherited glutamine synthetase deficiency specifically affects the synthesis of NAD+ from the NAD+ precursors, tryptophan and nicotinic acid. If the combined deficiencies of glutamine and NAD+ are responsible for the severe clinical phenotype of subjects with inherited glutamine synthetase deficiency, it is likely that supplementation with both glutamine and nicotinamide would provide some relief (78).
Finally, many inborn errors of metabolism result from genetic mutations decreasing cofactor binding affinity and, subsequently, enzyme efficiency (79). In many cases, the administration of high doses of the vitamins serving as precursors of cofactors can restore enzymatic activity — at least partially — and lessen signs of the genetic diseases (79). Given the large number of enzymes requiring NAD, it is speculated that many of the conditions due to defective enzymes might be rescued by niacin supplementation (5).
Cardiovascular disease
Cardiovascular risk factors
Nicotinic acid is a well-known lipid-lowering agent: Nicotinic acid therapy markedly increases high-density lipoprotein (HDL) cholesterol concentrations, decreases serum lipoprotein(a) concentrations, and shifts small, dense low-density lipoprotein (LDL) particles to large, buoyant LDL particles. All of these changes in the blood lipid profile are considered cardioprotective. Low concentrations of HDL-cholesterol are one major risk factor for coronary heart disease (CHD), and an increase in HDL concentrations is associated with a reduction of that risk (80). Because of the adverse side effects associated with high doses of nicotinic acid (see Safety), nicotinic acid has most often been used in combination with other lipid-lowering medications at slightly lower doses (17). In particular, low-dose nicotinic acid is often co-administered with 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors (statins), the cornerstone of treatment of hyperlipidemia, a major risk factor for CHD. A placebo-controlled study in 39 patients taking statins (cerivastatin, atorvastatin, or simvastatin) found that a very low dose of nicotinic acid (100 mg/day) increased HDL-cholesterol by only 2.1 mg/dL, and the combination had no effect on LDL-cholesterol, total cholesterol, or triglyceride concentrations (81).
The Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2 study — a double-blind, placebo-controlled trial — investigated the incremental effect of adding nicotinic acid (1 g/day) to statin therapy in 167 patients with known CHD and low HDL concentrations on carotid intima-media thickness (CIMT) (82), a surrogate endpoint for the development of atherosclerosis. The addition of extended-release nicotinic acid to simvastatin prevented the increase in CIMT compared to simvastatin monotherapy. A post-hoc analysis of data from ARBITER2 showed that the blockade of atherosclerotic progression was related to the increase in HDL concentrations in patients with normal glycemic status. However, in the presence of additional risk factors, such as impaired fasting glucose or diabetes mellitus, the increase in HDL concentrations was not predictive of CIMT reduction and atherosclerotic retardation (83). A comparative efficacy trial (ARBITER6) also showed a significant reduction of baseline CIMT with extended-release nicotinic acid (2 g/day for 14 months), as opposed to ezetimibe (a cholesterol-lowering drug), in patients taking statins (84).
Additional studies have examined the impact of nicotinic acid on endothelium-dependent brachial flow-mediated dilation (FMD) in patients at risk of CHD or with established CHD. The measurement of FMD is often used as a surrogate marker of endothelial function; FMD values are inversely correlated with the risk of future cardiovascular events (85). A meta-analysis of seven randomized controlled trials, including 441 participants, showed a significant, 2% increase with nicotinic acid (1-2 g/day) administered for 12 weeks to one year (86).
Cardiovascular events
Several randomized, placebo-controlled, multicenter trials have investigated the efficacy and safety of nicotinic acid therapy, alone or in combination with other lipid-lowering agents, on outcomes of cardiovascular disease (CVD). Specifically, the Coronary Drug Project (CDP) followed over 8,000 men with a previous myocardial infarction for six years (87). Compared to the placebo group, patients who took 3 g of immediate-release nicotinic acid daily experienced an average 10% reduction in total blood cholesterol, a 26% decrease in triglycerides, a 27% reduction in recurrent nonfatal myocardial infarction, and a 26% reduction in cerebrovascular events (stroke and transient ischemic attacks). In addition, nine-year follow-up post-trial revealed a 10% reduction in total deaths with nicotinic acid treatment.
The HDL-Atherosclerosis Treatment Study (HATS), a three-year randomized controlled trial in 160 patients with documented CHD and low HDL concentrations, found that a combination of simvastatin and nicotinic acid (2-3 g/day) increased HDL concentrations, inhibited the progression of coronary artery stenosis (narrowing), and decreased the frequency of cardiovascular events, including myocardial infarction and stroke, compared to placebo (88). A subgroup analysis of the HATS patients with metabolic syndrome showed a reduction in rate of primary clinical events even though glucose and insulin metabolism were moderately impaired by nicotinic acid (89). Moreover, a review of nicotinic acid safety and tolerability among the HATS subjects showed glycemic control in diabetic patients returned to pretreatment values following eight months of disease management with medication and diet (90). Similarly, the cardiovascular benefit of long-term nicotinic acid therapy outweighed the modest increase in risk of newly onset type 2 diabetes in patients from the CDP study (91).
In contrast, the AIM-HIGH (Atherothrombosis intervention in metabolic syndrome with low HDL/high triglycerides: impact on global health outcomes) trial, which examined the incremental effect of extended-release nicotinic acid (1.5-2 g/day) on 3,414 patients who had established CVD and atherogenic dyslipidemia and were treated with simvastatin (+/- ezetimibe), provided disappointing results. Indeed, in these patients who had achieved target concentrations of LDL-cholesterol (<70 mg/dL) before randomization, the HDL-raising effect of nicotinic acid treatment failed to reduce the number of cardiovascular events after a mean follow-up of three years (92, 93). While some limitations, like a greater use of simvastatin and ezetimibe in the control group, may have confounded the results, it was also suggested that low HDL-cholesterol might be a marker of risk rather than a causal risk factor for predicting CVD (93). In addition, a post-hoc analysis of 505 participants with stage 3 chronic kidney disease found an increase in all-cause mortality in those randomized to nicotinic acid compared to those in the placebo group (94).
Although nicotinic acid failed to reduce the number of cardiovascular events in simvastatin-treated patients with low LDL-cholesterol, these results cannot be extrapolated to patients with higher LDL-cholesterol at baseline. A much larger multicenter, randomized, double-blind, placebo-controlled trial — the HPS2-THRIVE (Heart protection study 2: treatment of HDL to reduce the incidence of vascular events) trial — in 25,673 participants with vascular disease examined the incremental effect of extended-release nicotinic acid (2 g/day) and laropiprant (a prostaglandin D2 receptor-1 antagonist; 40 mg/day) on the incidence of vascular events. Compared to placebo, nicotinic acid/laropiprant reduced LDL-cholesterol by an average of 10 mg/dL, decreased triglycerides by 33 mg/dL, and increased HDL-cholesterol by 6 mg/dL after a median 3.9-year follow-up period. Nonetheless, nicotinic acid/laropiprant showed no effect on the incidence of major vascular events and death of any cause (95).
A recent meta-analysis of 23 randomized controlled trials — including the CDP, AIM-HIGH, and HPS2-THRIVE trials — in 39,195 subjects with a history of vascular disease compared the effect of nicotinic acid alone or as an add-on to other lipid-lowering agents. No cardiovascular benefits were associated with nicotinic acid therapy: the number of fatal and non-fatal myocardial infarctions and strokes was not decreased with nicotinic acid supplementation (median dose of 2 g/day for a median period of 11.5 months) (96).
Despite the lack of evidence for a role of nicotinic acid in CVD prevention (96, 97), the use of nicotinic acid therapy has rapidly increased over the years in the US (98).
Friedreich’s ataxia
Friedreich’s ataxia, a common form of inherited ataxia, is an early onset recessive disorder with clinical features that includes progressive ataxia, scoliosis, dysarthria, cardiomyopathy, and diabetes mellitus (99). Most affected subjects carry homozygous guanine-adenine-adenine (GAA) repeat expansions in the first intron of the gene FXN coding for the protein frataxin. These abnormal and unstable GAA repeats trigger gene silencing through heterochromatin formation, leading to significantly reduced frataxin expression (100). Frataxin is a mitochondrial protein needed for the making of iron-sulfur clusters (ISC). ISC-containing subunits are especially important for the mitochondrial respiratory chain and for the synthesis of heme-containing proteins (99).
Predominantly localized in the nucleus, SIRT1 is a NAD+-dependent deacetylase that promotes gene silencing through heterochromatin formation. Nicotinamide has been shown to antagonize heterochromatization of the FXN locus and upregulate frataxin expression in lymphoblastoid cells derived from Friedreich’s ataxia-affected patients, possibly through inhibiting SIRT1 activity (100). In an open-label, dose escalating pilot trial in 10 adult patients with Friedreich’s ataxia, single and repeated doses of nicotinamide (2-8 g) for up to eight weeks were found to be well tolerated (101). Repeated daily doses of 3.5 to 6 g of nicotinamide led to significant increases in frataxin concentration in peripheral white blood cells (101). Yet, no neurological improvements were reported, suggesting that the duration of the treatment was too short and/or the nervous system of the participants was unresponsive to increases in frataxin (102). To our knowledge, there is currently no ongoing trial designed to further investigate the effect of nicotinamide in Friedreich’s ataxia-affected patients.
HIV/AIDS
The first step in the kynurenine pathway is catalyzed by the extrahepatic enzyme, indoleamine 2,3-dioxygenase (IDO), which is responsible for the oxidative cleavage of tryptophan. The chronic stimulation of tryptophan oxidation, mediated by an increased activity of IDO and/or inadequate dietary niacin intakes, is observed with the infection of human immunodeficiency virus (HIV), the virus that causes acquired immunodeficiency syndrome (AIDS). Interferon-gamma (IFN-γ) is a cytokine produced by cells of the immune system in response to infection. Through stimulating the enzyme IDO, IFN-γ increases the breakdown of tryptophan, thus supporting the finding that the average tryptophan concentration in blood is significantly lower in HIV patients compared to uninfected subjects (40). An increased degradation of tryptophan via the kynurenine pathway appears to coexist with intracellular niacin/NAD deficiency in HIV infection (103). An explanatory model for these paradoxical observations incriminates the oxidative stress induced by multiple nutrient deficiencies in HIV patients (103). In particular, the activation of PARP enzymes (ARTDs) by oxidative damage to DNA could be responsible for inducing niacin/NAD depletion (see Function). The breakdown of tryptophan would then be a compensatory response to inadequate niacin/NAD levels.
However, metabolites derived from the oxidation of tryptophan in the kynurenine pathway regulate specific T-lymphocyte subgroups. As mentioned above, circulating IFN-γ, but also viral and bacterial products, can activate IDO during HIV infection. The overstimulation of the tryptophan pathway has been involved in the loss of normal T-lymphocyte function, which characterizes HIV infection (104, 105). The increased IDO activity has been linked to the altered immune response that contributes to the persistence of HIV (104). Antiretroviral therapy (ART) only partially restores normal IDO activity, without normalizing it, yet induces viral suppression and CD4 T-cell recovery (106). In a monkey model for HIV infection, a partial and transient blockade of IDO with IDO inhibitor 1-methyl tryptophan proved ineffective to reduce the viral load in plasma and intestinal tissues beyond the level achieved by ART (107). At present, a better understanding of the role of kynurenine pathway and other NAD biosynthetic pathways during HIV infection is needed before the relevance and clinical implications of niacin supplementation in HIV treatment could be considered.
Nonetheless, pharmacologic doses of nicotinic acid have been shown to be well tolerated in HIV patients with hyperlipidemia (108). Abnormal lipid profiles observed in patients have been attributed to the HIV infection and to the highly active antiretroviral treatment (HAART) (109). Moreover, insulin resistance has been detected together with dyslipidemia in ART-treated patients (110). Cardiovascular disease (CVD) is the second most frequent cause of deaths in the HIV population, and the rate of CVD is predicted to increase further as patients are living longer due to successful antiretroviral therapies. As for the general population, statin-based therapy appears to benefit HIV patients in terms of atherogenic protection and CVD risk reduction, although contraindications exist due to drug interactions with ART. Other first-line treatments include lipid-lowering fibrates, which are preferred to nicotinic acid due to the increased risk of glucose intolerance and insulin resistance (111). Nevertheless, an unblinded, controlled pilot study showed that extended-release nicotinic acid (0.5-1.5 g/day for 12 weeks) could effectively improve endothelial function of the brachial artery in ART-treated HIV subjects with low HDL-cholesterol and no history of CVD (112). In addition, a combined treatment of fibrates, extended-release nicotinic acid (0.5-2 g/day), and lifestyle changes (low-fat diet and exercise) for 24 weeks was effective in normalizing lipid parameters in a cohort of 191 ART-treated patients. Increased risk of liver dysfunction was detected in subjects receiving both fibrates and niacin, but insulin sensitivity was not affected by nicotinic acid treatment given alone or when combined with fibrates (113). Another 24-week, open-label, uncontrolled trial in 99 ART-treated patients found that randomization to extended-release nicotinic acid (0.5-2 g/day) or fenofibrates increased blood HDL-cholesterol but did not reduce inflammatory markers or improve endothelial function when compared to baseline (114).
Schizophrenia
Schizophrenia is a neurologic disorder with unclear etiology that is diagnosed purely from its clinical presentation. Because neurologic disorders associated with pellagra resemble acute schizophrenia, niacin-based therapy for the condition was investigated during the 1950s-70s (reviewed in 115). The adjunctive use of nutrients like niacin to correct deficiencies associated with neurologic symptoms is called orthomolecular psychiatry (116). Such an approach has not been included in psychiatric practice; practitioners have instead relied solely on antipsychotic drugs to eliminate the clinical symptoms of schizophrenia. Nevertheless, recent scientific advances and new hypotheses on the benefit of nutrient supplementation in the treatment of psychiatric disorders have suggested the re-assessment of orthomolecular medicine by the medical community (117, 118).
Skin flushing is one major side effect of the therapeutic use of nicotinic acid and the primary reason for non-adherence to treatment (see Toxicity). Flushing is caused by the activation of phospholipase A2, an enzyme that stimulates the production of a specific lipid from the prostanoid family called prostaglandin D2. Prostaglandin D2, synthesized by antigen-presenting cells of the skin and mucosa (i.e., the Langerhans cells), can induce the dilation of blood vessels and trigger a flushing response. Interestingly, patients with schizophrenia tend not to flush following treatment with nicotinic acid. This blunted skin flushing response suggests abnormal prostanoid signaling in schizophrenic patients (119, 120). An association has been found between the altered niacin sensitivity and greater functional impairment in schizophrenic subjects (121), which supports other findings suggesting that altered lipid metabolism could critically impair brain development and contribute to the disease (122). Interestingly, blunted skin flushing responses are more prevalent in first-degree relatives of people with schizophrenia than in the general population, suggesting that reduced niacin sensitivity is a heritable trait within affected families (123).
Sources
Food sources
Good sources of niacin include yeast, meat, poultry, red fish (e.g., tuna, salmon), cereal (especially fortified cereal), legumes, and seeds. Milk, green leafy vegetables, coffee, and tea also provide some niacin (124). In plants, especially mature cereal grains like corn and wheat, niacin may be bound to sugar molecules in the form of glycosides, which significantly decrease its bioavailability (25).
In the United States, the average dietary intake of niacin is about 30 mg/day for young adult men and 20 mg/day for young adult women. In a sample of adults over the age of 60, men and women were found to have an average dietary intake of 21 mg/day and 17 mg/day, respectively (41). Some foods with substantial amounts of niacin are listed in Table 2, along with their niacin content in milligrams (mg). Food composition tables generally list niacin content without including niacin equivalents (NE) from tryptophan or any adjustment for niacin bioavailability. For more information on the nutrient content of specific foods, search USDA's FoodData Central database; data included in Table 2 are from this database (125).
Supplements
Niacin supplements are available as nicotinamide or nicotinic acid. Nicotinamide is the form of niacin typically used in nutritional supplements and in food fortification. Nicotinic acid is available over the counter and with a prescription as a cholesterol-lowering agent (126). Nicotinic acid for anti-hyperlipidemic use is available in three formulations: immediate-release (crystalline) nicotinic acid (absorption time, 1-2 hrs), extended-release nicotinic acid (absorption time, 8-12 hrs), and sustained-release nicotinic acid (absorption time, >12 hrs) (127). At the pharmacologic dose required for cholesterol-lowering effects, the use of nicotinic acid should be approached as if it were a drug (see Safety). Individuals should only undertake cholesterol-lowering therapy with nicotinic acid under the supervision of a qualified health care provider in order to minimize potentially adverse effects and maximize therapeutic benefits.
Safety
Toxicity
Nicotinic acid
Common side effects of nicotinic acid include flushing, pruritus (severe itching of the skin), skin rashes, and gastrointestinal disturbances, such as nausea and vomiting (97). Transient episodes of low blood pressure (hypotension) and headache have also been reported. Hepatotoxicity (liver cell damage), including elevated liver enzymes and jaundice, has been observed at intakes as low as 750 mg/day of nicotinic acid (128). Although hepatitis has been observed with extended-release nicotinic acid at dosages as little as 500 mg/day for two months, almost all reports of severe hepatitis have been associated with doses of 3 to 9 g/day used to treat high cholesterol for months or years (41). It is unclear whether immediate-release (crystalline) nicotinic acid is less toxic to the liver than extended-release forms (41). Yet, immediate-release nicotinic acid is often used at higher doses than extended-release forms, and severe liver toxicity has occurred in individuals who substituted extended-release nicotinic acid for immediate-release nicotinic acid at equivalent doses (126). Large doses of nicotinic acid have been observed to impair glucose tolerance, likely because of a decrease in insulin sensitivity. Impaired glucose tolerance in susceptible (pre-diabetic) individuals could result in elevated blood glucose concentrations and clinical type 2 diabetes mellitus. An analysis of the HPS2-THRIVE trial (see Cardiovascular disease), using data from 17,374 participants without type 2 diabetes at baseline, found a significantly higher proportion of newly diagnosed cases among those randomized to nicotinic acid/laropiprant than to placebo (5.7% versus 4.3%) over a 3.9 year-period (95). Likewise, randomization to nicotinic acid/laropiprant significantly increased the risk of serious disturbances in diabetes control (leading to hospitalization) compared to placebo among 8,299 participants with diabetes at baseline (95). Elevated blood concentrations of uric acid, occasionally resulting in attacks of gout in susceptible individuals, have also been observed with high-dose nicotinic acid therapy (126). Niacin at doses of 1.5 to 5 g/day has resulted in a few case reports of blurred vision and other eye problems, which have generally been reversible upon discontinuation (41). People with abnormal liver function or a history of liver disease, diabetes, active peptic ulcer disease, gout, cardiac arrhythmias, inflammatory bowel disease, migraine headaches, or alcoholism may be more susceptible to the adverse effects of excess niacin intake than the general population (41).
Nicotinamide
Nicotinamide is generally better tolerated than nicotinic acid; it does not generally cause flushing (126). However, nausea, vomiting, and signs of liver toxicity (elevated liver enzymes, jaundice) have been observed at very high doses (≥10 g/day) (126).
Nicotinamide riboside
A study in 12 healthy subjects found that nicotinamide riboside at three single doses (100 mg, 300 mg, and 1,000 mg) safely increased blood NAD+. Two of the participants self-reported skin flushing after taking the 300-mg dose, and two others reported feeling hot following intake of 1,000 mg of nicotinamide riboside (1). In a recent randomized, placebo-controlled trial in 120 healthy adults (ages, 60-80 years), daily supplementation with nicotinamide riboside (250 mg or 500 mg) and pterostilbene (a SIRT activator; 50 mg or 100 mg) for eight weeks showed a favorable side effect profile, with no evidence of higher incidence of adverse effects compared to placebo (129). Most recently, a randomized, placebo-controlled trial in 40 obese men (ages, 40-70 years) found that daily supplementation with nicotinamide riboside (2,000 mg/day divided into two daily dosages) for 12 weeks was associated with reports of only minor side effects, including excessive sweating, pruritus, and mild gastrointestinal symptoms like bloating (139).
The tolerable upper intake level (UL)
Flushing of the skin primarily on the face, arms, and chest is a common side effect of nicotinic acid and may occur initially at doses as low as 30 mg/day. Although flushing from nicotinamide is rare, the Food and Nutrition Board set the tolerable upper intake level (UL) for niacin (nicotinic acid and nicotinamide) at 35 mg/day in adults to avoid the adverse effect of flushing (41). Analysis of data from the US National Health and Nutrition Examination Survey (NHANES) 2003-2006 found that 15.8% of children and adolescents (ages 2-18 years) and 8.5% of adults (≥19 years) had total usual niacin intakes exceeding the UL (130). The UL applies to the general population and is not meant to apply to individuals who are being treated with a nutrient under medical supervision (e.g., high-dose nicotinic acid for elevated blood cholesterol concentrations).
Drug interactions
The occurrence of rhabdomyolysis is increased in patients treated with statins (HMG-CoA reductase inhibitors). Rhabdomyolysis is a relatively uncommon condition in which muscle cells are broken down, releasing enzymes and electrolytes into the blood, and sometimes resulting in kidney failure (131). Co-administration of nicotinic acid with a statin seems to enhance the risk of rhabdomyolosis (132). A new drug, laropiprant, blocks prostanoid receptors and reduces nicotinic acid-induced flushing (133). A randomized, placebo-controlled trial was designed to identify possible adverse effects of the niacin/laropiprant combination in over 25,000 simvastatin-treated subjects (134). When added to the statin therapy, niacin/laropiprant increased the risk of myopathy and rhabdomyolosis, particularly in Asian subjects. It is possible that the niacin/laropiprant combination further reduces the poor tolerability to statin treatment observed in certain populations (135).
In the three-year, randomized controlled HATS study, concurrent therapy with antioxidants (1,000 mg/day of vitamin C, 800 IU/day of RRR-α-tocopherol, 100 µg/day of selenium, and 25 mg/day of β-carotene) diminished the protective effects of the simvastatin-nicotinic acid combination (136). Although the mechanism for these effects is not known, the benefit of concurrent antioxidant therapy in patients on lipid-lowering agents has been questioned (137).
Adverse effects of large doses of nicotinic acid may be exacerbated by the concomitant use of certain medications. The risk of myopathy may be further increased in those taking nicotinic acid and bile acid sequestrants (e.g., cholestyramine, colestipol) or the anti-lipidemic drug, gemfibrozil (Lopid), and the risk of hepatotoxicity observed with nicotinic acid might be enhanced by drugs like paracetamol, amiodarone (Cordarone), or carbamazepine (Tegretol) (35). In addition, large doses of nicotinic acid may reduce uric acid excretion, thereby opposing the action of uricosuric agents like probenecid (Probalan) (35).
Several other medications may interact with niacin therapy or with absorption and metabolism of the vitamin (126). Estrogen and estrogen-containing oral contraceptives increase the efficiency of niacin synthesis from tryptophan, resulting in a decreased dietary requirement for niacin (138). Long-term administration of chemotherapy agents has been reported to cause symptoms of pellagra; therefore, niacin supplementation may be needed (see Pellagra causes).
Linus Pauling Institute Recommendation
The optimum intake of niacin for health promotion and chronic disease prevention is not yet known. The RDA (16 mg NE/day for men and 14 mg NE/day for women) is easily obtainable by consuming a varied diet and should prevent deficiency in most people. Following the Linus Pauling Institute recommendation to take a daily multivitamin/mineral supplement, containing 100% of the Daily Value (DV) for niacin, will provide at least 20 mg of niacin daily.
Authors and Reviewers
Originally written in 2000 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in August 2002 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in June 2007 by:
Victoria J. Drake, Ph.D
Linus Pauling Institute
Oregon State University
Updated in July 2013 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in December 2017 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in March 2018 by:
Mirella Meyer-Ficca, Ph.D.
Research Assistant Professor
Utah State University
The 2017 update of this article was supported by a grant from ChromaDex, Inc.
Last updated 8/10/18 Copyright 2000-2024 Linus Pauling Institute
References
1. Trammell SA, Schmidt MS, Weidemann BJ, et al. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun. 2016;7:12948. (PubMed)
2. Nikiforov A, Kulikova V, Ziegler M. The human NAD metabolome: functions, metabolism and compartmentalization. Crit Rev Biochem Mol Biol. 2015;50(4):284-297. (PubMed)
3. Kawai S, Murata K. Structure and function of NAD kinase and NADP phosphatase: key enzymes that regulate the intracellular balance of NAD(H) and NADP(H). Biosci Biotechnol Biochem. 2008;72(4):919-930. (PubMed)
4. Agledal L, Niere M, Ziegler M. The phosphate makes a difference: cellular functions of NADP. Redox Rep. 2010;15(1):2-10. (PubMed)
5. Penberthy WT, Kirkland JB. Niacin. In: Erdman JW, MacDonald I, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Ames: International Life Sciences Institute; 2012:293-306.
6. Kirkland JB. Niacin. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore: Lippincott Williams & Wilkins; 2014:331-340.
7. Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci. 2010;35(4):208-219. (PubMed)
8. Liu C, Yu X. ADP-ribosyltransferases and poly ADP-ribosylation. Curr Protein Pept Sci. 2015;16(6):491-501. (PubMed)
9. Hwang ES, Song SB. Nicotinamide is an inhibitor of SIRT1 in vitro, but can be a stimulator in cells. Cell Mol Life Sci. 2017;74(18):3347-3362. (PubMed)
10. Morris BJ. Seven sirtuins for seven deadly diseases of aging. Free Radic Biol Med. 2013;56:133-171. (PubMed)
11. Fliegert R, Bauche A, Wolf Perez AM, et al. 2'-Deoxyadenosine 5'-diphosphoribose is an endogenous TRPM2 superagonist. Nat Chem Biol. 2017;13(9):1036-1044. (PubMed)
12. Mutafova-Yambolieva VN, Hwang SJ, Hao X, et al. Beta-nicotinamide adenine dinucleotide is an inhibitory neurotransmitter in visceral smooth muscle. Proc Natl Acad Sci U S A. 2007;104(41):16359-16364. (PubMed)
13. Moreschi I, Bruzzone S, Nicholas RA, et al. Extracellular NAD+ is an agonist of the human P2Y11 purinergic receptor in human granulocytes. J Biol Chem. 2006;281(42):31419-31429. (PubMed)
14. Klein C, Grahnert A, Abdelrahman A, Muller CE, Hauschildt S. Extracellular NAD(+) induces a rise in [Ca(2+)](i) in activated human monocytes via engagement of P2Y(1) and P2Y(11) receptors. Cell Calcium. 2009;46(4):263-272. (PubMed)
15. Moreschi I, Bruzzone S, Bodrato N, et al. NAADP+ is an agonist of the human P2Y11 purinergic receptor. Cell Calcium. 2008;43(4):344-355. (PubMed)
16. Huang C, Hu J, Subedi KP, et al. Extracellular adenosine diphosphate ribose mobilizes intracellular Ca2+ via purinergic-dependent Ca2+ pathways in rat pulmonary artery smooth muscle cells. Cell Physiol Biochem. 2015;37(5):2043-2059. (PubMed)
17. Knopp RH. Drug treatment of lipid disorders. N Engl J Med. 1999;341(7):498-511. (PubMed)
18. Graff EC, Fang H, Wanders D, Judd RL. Anti-inflammatory effects of the hydroxycarboxylic acid receptor 2. Metabolism. 2016;65(2):102-113. (PubMed)
19. Jin FY, Kamanna VS, Kashyap ML. Niacin accelerates intracellular ApoB degradation by inhibiting triacylglycerol synthesis in human hepatoblastoma (HepG2) cells. Arterioscler Thromb Vasc Biol. 1999;19(4):1051-1059. (PubMed)
20. Kamanna VS, Ganji SH, Kashyap ML. Recent advances in niacin and lipid metabolism. Curr Opin Lipidol. 2013;24(3):239-245. (PubMed)
21. Carlson LA. Studies on the effect of nicotinic acid on catecholamine stimulated lipolysis in adipose tissue in vitro. Acta Med Scand. 1963;173:719-722. (PubMed)
22. Lauring B, Taggart AK, Tata JR, et al. Niacin lipid efficacy is independent of both the niacin receptor GPR109A and free fatty acid suppression. Sci Transl Med. 2012;4(148):148ra115. (PubMed)
23. Brody T. Nutritional Biochemistry. 2nd ed. San Diego: Academic Press; 1999.
24. Kirkland JB. Niacin. In: Zempleni J, Suttie JW, Gregory III JF, Stover PJ, eds. Handbook of Vitamins. 5th ed. Boca Raton: CRC Press; 2013:149-190.
25. Gregory JF, 3rd. Nutritional properties and significance of vitamin glycosides. Annu Rev Nutr. 1998;18:277-296. (PubMed)
26. Dawson B, Favaloro EJ, Taylor J, Aggarwal A. Unrecognized pellagra masquerading as odynophagia. Intern Med J. 2006;36(7):472-474. (PubMed)
27. Jagielska G, Tomaszewicz-Libudzic EC, Brzozowska A. Pellagra: a rare complication of anorexia nervosa. Eur Child Adolesc Psychiatry. 2007;16(7):417-420. (PubMed)
28. Kertesz SG. Pellagra in 2 homeless men. Mayo Clin Proc. 2001;76(3):315-318. (PubMed)
29. Prakash R, Gandotra S, Singh LK, Das B, Lakra A. Rapid resolution of delusional parasitosis in pellagra with niacin augmentation therapy. Gen Hosp Psychiatry. 2008;30(6):581-584. (PubMed)
30. Badawy AA. Pellagra and alcoholism: a biochemical perspective. Alcohol Alcohol. 2014;49(3):238-250. (PubMed)
31. Majewski M, Kozlowska A, Thoene M, Lepiarczyk E, Grzegorzewski WJ. Overview of the role of vitamins and minerals on the kynurenine pathway in health and disease. J Physiol Pharmacol. 2016;67(1):3-19. (PubMed)
32. Rosmaninho A, Sanches M, Fernandes IC, et al. Letter: Pellagra as the initial presentation of Crohn disease. Dermatol Online J. 2012;18(4):12. (PubMed)
33. Zaraa I, Belghith I, El Euch D, et al. A case of pellagra associated with megaduodenum in a young woman. Nutr Clin Pract. 2013;28(2):218-222. (PubMed)
34. Bilgili SG, Karadag AS, Calka O, Altun F. Isoniazid-induced pellagra. Cutan Ocul Toxicol. 2011;30(4):317-319. (PubMed)
35. Natural Medicines. Professional Monograph - Niacin/Interactions with drugs. Available at: https://naturalmedicines.therapeuticresearch.com/. Accessed 8/2/17.
36. Dreizen S, McCredie KB, Keating MJ, Andersson BS. Nutritional deficiencies in patients receiving cancer chemotherapy. Postgrad Med. 1990;87(1):163-167, 170. (PubMed)
37. Nogueira A, Duarte AF, Magina S, Azevedo F. Pellagra associated with esophageal carcinoma and alcoholism. Dermatol Online J. 2009;15(5):8. (PubMed)
38. Oldham MA, Ivkovic A. Pellagrous encephalopathy presenting as alcohol withdrawal delirium: a case series and literature review. Addict Sci Clin Pract. 2012;7(1):12. (PubMed)
39. World Health Organization, United Nations High Commissions for Refugees. Pellagra and its prevention and control in major emergencies. World Health Organization. 2000. Available at: http://www.who.int/nutrition/publications/emergencies/WHO_NHD_00.10/en/. Accessed 6/20/13.
40. Murray MF. Tryptophan depletion and HIV infection: a metabolic link to pathogenesis. Lancet Infect Dis. 2003;3(10):644-652. (PubMed)
41. Food and Nutrition Board, Institute of Medicine. Niacin. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B-6, Vitamin B-12, Pantothenic Acid, Biotin, and Choline. Washington, D.C.: The National Academies Press; 1998:123-149. (The National Academies Press)
42. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11(3):220-228. (PubMed)
43. Kirkland JB. Niacin requirements for genomic stability. Mutat Res. 2012;733(1-2):14-20. (PubMed)
44. Burkle A. Poly(ADP-ribose). The most elaborate metabolite of NAD+. FEBS J. 2005;272(18):4576-4589. (PubMed)
45. Jacobson EL, Shieh WM, Huang AC. Mapping the role of NAD metabolism in prevention and treatment of carcinogenesis. Mol Cell Biochem. 1999;193(1-2):69-74. (PubMed)
46. Spronck JC, Nickerson JL, Kirkland JB. Niacin deficiency alters p53 expression and impairs etoposide-induced cell cycle arrest and apoptosis in rat bone marrow cells. Nutr Cancer. 2007;57(1):88-99. (PubMed)
47. Spronck JC, Kirkland JB. Niacin deficiency increases spontaneous and etoposide-induced chromosomal instability in rat bone marrow cells in vivo. Mutat Res. 2002;508(1-2):83-97. (PubMed)
48. Kostecki LM, Thomas M, Linford G, et al. Niacin deficiency delays DNA excision repair and increases spontaneous and nitrosourea-induced chromosomal instability in rat bone marrow. Mutat Res. 2007;625(1-2):50-61. (PubMed)
49. Dantzer F, Santoro R. The expanding role of PARPs in the establishment and maintenance of heterochromatin. FEBS J. 2013;280(15):3508-3518. (PubMed)
50. El Ramy R, Magroun N, Messadecq N, et al. Functional interplay between Parp-1 and SirT1 in genome integrity and chromatin-based processes. Cell Mol Life Sci. 2009;66(19):3219-3234. (PubMed)
51. Boyonoski AC, Spronck JC, Gallacher LM, et al. Niacin deficiency decreases bone marrow poly(ADP-ribose) and the latency of ethylnitrosourea-induced carcinogenesis in rats. J Nutr. 2002;132(1):108-114. (PubMed)
52. Boyonoski AC, Spronck JC, Jacobs RM, Shah GM, Poirier GG, Kirkland JB. Pharmacological intakes of niacin increase bone marrow poly(ADP-ribose) and the latency of ethylnitrosourea-induced carcinogenesis in rats. J Nutr. 2002;132(1):115-120. (PubMed)
53. Weitberg AB. Effect of nicotinic acid supplementation in vivo on oxygen radical-induced genetic damage in human lymphocytes. Mutat Res. 1989;216(4):197-201. (PubMed)
54. Hageman GJ, Stierum RH, van Herwijnen MH, van der Veer MS, Kleinjans JC. Nicotinic acid supplementation: effects on niacin status, cytogenetic damage, and poly(ADP-ribosylation) in lymphocytes of smokers. Nutr Cancer. 1998;32(2):113-120. (PubMed)
55. Yong LC, Petersen MR. High dietary niacin intake is associated with decreased chromosome translocation frequency in airline pilots. Br J Nutr. 2011;105(4):496-505. (PubMed)
56. Weidele K, Beneke S, Burkle A. The NAD+ precursor nicotinic acid improves genomic integrity in human peripheral blood mononuclear cells after X-irradiation. DNA Repair (Amst). 2017;52:12-23. (PubMed)
57. Jacobson EL. Niacin deficiency and cancer in women. J Am Coll Nutr. 1993;12(4):412-416. (PubMed)
58. Negri E, Franceschi S, Bosetti C, et al. Selected micronutrients and oral and pharyngeal cancer. Int J Cancer. 2000;86(1):122-127. (PubMed)
59. Franceschi S, Bidoli E, Negri E, et al. Role of macronutrients, vitamins and minerals in the aetiology of squamous-cell carcinoma of the oesophagus. Int J Cancer. 2000;86(5):626-631. (PubMed)
60. Gensler HL, Williams T, Huang AC, Jacobson EL. Oral niacin prevents photocarcinogenesis and photoimmunosuppression in mice. Nutr Cancer. 1999;34(1):36-41. (PubMed)
61. Jacobson EL, Kim H, Kim M, et al. A topical lipophilic niacin derivative increases NAD, epidermal differentiation and barrier function in photodamaged skin. Exp Dermatol. 2007;16(6):490-499. (PubMed)
62. Bermudez Y, Benavente CA, Meyer RG, Coyle WR, Jacobson MK, Jacobson EL. Nicotinic acid receptor abnormalities in human skin cancer: implications for a role in epidermal differentiation. PLoS One. 2011;6(5):e20487. (PubMed)
63. Benavente CA, Jacobson EL. Niacin restriction upregulates NADPH oxidase and reactive oxygen species (ROS) in human keratinocytes. Free Radic Biol Med. 2008;44(4):527-537. (PubMed)
64. Benavente CA, Schnell SA, Jacobson EL. Effects of niacin restriction on sirtuin and PARP responses to photodamage in human skin. PLoS One. 2012;7(7):e42276. (PubMed)
65. Park SM, Li T, Wu S, et al. Niacin intake and risk of skin cancer in US women and men. Int J Cancer. 2017;140(9):2023-2031. (PubMed)
66. Chen AC, Martin AJ, Choy B, et al. A phase 3 randomized trial of nicotinamide for skin-cancer chemoprevention. N Engl J Med. 2015;373(17):1618-1626. (PubMed)
67. Minocha R, Damian DL, Halliday GM. Melanoma and nonmelanoma skin cancer chemoprevention: A role for nicotinamide? Photodermatol Photoimmunol Photomed. 2018;34(1):5-12. (PubMed)
68. Orban T, Sosenko JM, Cuthbertson D, et al. Pancreatic islet autoantibodies as predictors of type 1 diabetes in the Diabetes Prevention Trial-Type 1. Diabetes Care. 2009;32(12):2269-2274. (PubMed)
69. Szkudelski T. Streptozotocin-nicotinamide-induced diabetes in the rat. Characteristics of the experimental model. Exp Biol Med (Maywood). 2012;237(5):481-490. (PubMed)
70. Lampeter EF, Klinghammer A, Scherbaum WA, et al. The Deutsche Nicotinamide Intervention Study: an attempt to prevent type 1 diabetes. DENIS Group. Diabetes. 1998;47(6):980-984. (PubMed)
71. Gale EA, Bingley PJ, Emmett CL, Collier T, European Nicotinamide Diabetes Intervention Trial Group. European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet. 2004;363(9413):925-931. (PubMed)
72. Hedman M, Ludvigsson J, Faresjo MK. Nicotinamide reduces high secretion of IFN-gamma in high-risk relatives even though it does not prevent type 1 diabetes. J Interferon Cytokine Res. 2006;26(4):207-213. (PubMed)
73. Fernandez IC, Del Carmen Camberos M, Passicot GA, Martucci LC, Cresto JC. Children at risk of diabetes type 1. Treatment with acetyl-L-carnitine plus nicotinamide - Case reports. J Pediatr Endocrinol Metab. 2013;26(3-4):347-355. (PubMed)
74. Patel AB, Prabhu AS. Hartnup disease. Indian J Dermatol. 2008;53(1):31-32. (PubMed)
75. Oakley A, Wallace J. Hartnup disease presenting in an adult. Clin Exp Dermatol. 1994;19(5):407-408. (PubMed)
76. Shi H, Enriquez A, Rapadas M, et al. NAD deficiency, congenital malformations, and niacin supplementation. N Engl J Med. 2017;377(6):544-552. (PubMed)
77. Vander Heiden MG. Metabolism and congenital malformations - NAD's effects on development. N Engl J Med. 2017;377(6):509-511. (PubMed)
78. Hu L, Ibrahim K, Stucki M, et al. Secondary NAD+ deficiency in the inherited defect of glutamine synthetase. J Inherit Metab Dis. 2015;38(6):1075-1083. (PubMed)
79. Ames BN, Elson-Schwab I, Silver EA. High-dose vitamin therapy stimulates variant enzymes with decreased coenzyme binding affinity (increased K(m)): relevance to genetic disease and polymorphisms. Am J Clin Nutr. 2002;75(4):616-658. (PubMed)
80. Bays HE, Shah A, Lin J, Sisk CM, Dong Q, Maccubbin D. Consistency of extended-release niacin/laropiprant effects on Lp(a), ApoB, non-HDL-C, Apo A1, and ApoB/ApoA1 ratio across patient subgroups. Am J Cardiovasc Drugs. 2012;12(3):197-206. (PubMed)
81. Wink J, Giacoppe G, King J. Effect of very-low-dose niacin on high-density lipoprotein in patients undergoing long-term statin therapy. Am Heart J. 2002;143(3):514-518. (PubMed)
82. Taylor AJ, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation. 2004;110(23):3512-3517. (PubMed)
83. Taylor AJ, Zhu D, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Relationship between glycemic status and progression of carotid intima-media thickness during treatment with combined statin and extended-release niacin in ARBITER 2. Vasc Health Risk Manag. 2007;3(1):159-164. (PubMed)
84. Villines TC, Stanek EJ, Devine PJ, et al. The ARBITER 6-HALTS Trial (Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol 6-HDL and LDL Treatment Strategies in Atherosclerosis): final results and the impact of medication adherence, dose, and treatment duration. J Am Coll Cardiol. 2010;55(24):2721-2726. (PubMed)
85. Ras RT, Streppel MT, Draijer R, Zock PL. Flow-mediated dilation and cardiovascular risk prediction: a systematic review with meta-analysis. Int J Cardiol. 2013;168(1):344-351. (PubMed)
86. Sahebkar A. Effect of niacin on endothelial function: a systematic review and meta-analysis of randomized controlled trials. Vasc Med. 2014;19(1):54-66. (PubMed)
87. Canner PL, Berge KG, Wenger NK, et al. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol. 1986;8(6):1245-1255. (PubMed)
88. Brown BG, Zhao XQ, Chait A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345(22):1583-1592. (PubMed)
89. Vittone F, Chait A, Morse JS, Fish B, Brown BG, Zhao XQ. Niacin plus simvastatin reduces coronary stenosis progression among patients with metabolic syndrome despite a modest increase in insulin resistance: a subgroup analysis of the HDL-Atherosclerosis Treatment Study (HATS). J Clin Lipidol. 2007;1(3):203-210. (PubMed)
90. Zhao XQ, Morse JS, Dowdy AA, et al. Safety and tolerability of simvastatin plus niacin in patients with coronary artery disease and low high-density lipoprotein cholesterol (The HDL Atherosclerosis Treatment Study). Am J Cardiol. 2004;93(3):307-312. (PubMed)
91. Sazonov V, Maccubbin D, Sisk CM, Canner PL. Effects of niacin on the incidence of new onset diabetes and cardiovascular events in patients with normoglycaemia and impaired fasting glucose. Int J Clin Pract. 2013;67(4):297-302. (PubMed)
92. Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255-2267. (PubMed)
93. Michos ED, Sibley CT, Baer JT, Blaha MJ, Blumenthal RS. Niacin and statin combination therapy for atherosclerosis regression and prevention of cardiovascular disease events: reconciling the AIM-HIGH (Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides: Impact on Global Health Outcomes) trial with previous surrogate endpoint trials. J Am Coll Cardiol. 2012;59(23):2058-2064. (PubMed)
94. Kalil RS, Wang JH, de Boer IH, et al. Effect of extended-release niacin on cardiovascular events and kidney function in chronic kidney disease: a post hoc analysis of the AIM-HIGH trial. Kidney Int. 2015;87(6):1250-1257. (PubMed)
95. Landray MJ, Haynes R, Hopewell JC, et al. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371(3):203-212. (PubMed)
96. Schandelmaier S, Briel M, Saccilotto R, et al. Niacin for primary and secondary prevention of cardiovascular events. Cochrane Database Syst Rev. 2017;6:Cd009744. (PubMed)
97. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 Pt B):2889-2934. (PubMed)
98. Jackevicius CA, Tu JV, Ko DT, de Leon N, Krumholz HM. Use of niacin in the United States and Canada. JAMA Intern Med. 2013;173(14):1379-1381. (PubMed)
99. Burk K. Friedreich ataxia: current status and future prospects. Cerebellum Ataxias. 2017;4:4. (PubMed)
100. Chan PK, Torres R, Yandim C, et al. Heterochromatinization induced by GAA-repeat hyperexpansion in Friedreich's ataxia can be reduced upon HDAC inhibition by vitamin B3. Hum Mol Genet. 2013;22(13):2662-2675. (PubMed)
101. Libri V, Yandim C, Athanasopoulos S, et al. Epigenetic and neurological effects and safety of high-dose nicotinamide in patients with Friedreich's ataxia: an exploratory, open-label, dose-escalation study. Lancet. 2014;384(9942):504-513. (PubMed)
102. Lynch DR, Fischbeck KH. Nicotinamide in Friedreich's ataxia: useful or not? Lancet. 2014;384(9942):474-475. (PubMed)
103. Taylor EW. The oxidative stress-induced niacin sink (OSINS) model for HIV pathogenesis. Toxicology. 2010;278(1):124-130. (PubMed)
104. Favre D, Mold J, Hunt PW, et al. Tryptophan catabolism by indoleamine 2,3-dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci Transl Med. 2010;2(32):32ra36. (PubMed)
105. Jenabian MA, Patel M, Kema I, et al. Distinct tryptophan catabolism and Th17/Treg balance in HIV progressors and elite controllers. PLoS One. 2013;8(10):e78146. (PubMed)
106. Chen J, Shao J, Cai R, et al. Anti-retroviral therapy decreases but does not normalize indoleamine 2,3-dioxygenase activity in HIV-infected patients. PLoS One. 2014;9(7):e100446. (PubMed)
107. Dunham RM, Gordon SN, Vaccari M, et al. Preclinical evaluation of HIV eradication strategies in the simian immunodeficiency virus-infected rhesus macaque: a pilot study testing inhibition of indoleamine 2,3-dioxygenase. AIDS Res Hum Retroviruses. 2013;29(2):207-214. (PubMed)
108. Souza SA, Chow DC, Walsh EJ, Ford S, 3rd, Shikuma C. Pilot study on the safety and tolerability of extended release niacin for HIV-infected patients with hypertriglyceridemia. Hawaii Med J. 2010;69(5):122-125. (PubMed)
109. Dube MP, Lipshultz SE, Fichtenbaum CJ, et al. Effects of HIV infection and antiretroviral therapy on the heart and vasculature. Circulation. 2008;118(2):e36-40. (PubMed)
110. Carr A, Samaras K, Burton S, et al. A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS. 1998;12(7):F51-58. (PubMed)
111. Giannarelli C, Klein RS, Badimon JJ. Cardiovascular implications of HIV-induced dyslipidemia. Atherosclerosis. 2011;219(2):384-389. (PubMed)
112. Chow DC, Stein JH, Seto TB, et al. Short-term effects of extended-release niacin on endothelial function in HIV-infected patients on stable antiretroviral therapy. AIDS. 2010;24(7):1019-1023. (PubMed)
113. Balasubramanyam A, Coraza I, Smith EO, et al. Combination of niacin and fenofibrate with lifestyle changes improves dyslipidemia and hypoadiponectinemia in HIV patients on antiretroviral therapy: results of "heart positive," a randomized, controlled trial. J Clin Endocrinol Metab. 2011;96(7):2236-2247. (PubMed)
114. Dube MP, Komarow L, Fichtenbaum CJ, et al. Extended-release niacin versus fenofibrate in HIV-infected participants with low high-density lipoprotein cholesterol: effects on endothelial function, lipoproteins, and inflammation. Clin Infect Dis. 2015;61(5):840-849. (PubMed)
115. Hoffer LJ. Vitamin therapy in schizophrenia. Isr J Psychiatry Relat Sci. 2008;45(1):3-10. (PubMed)
116. Pauling L. Orthomolecular psychiatry. Varying the concentrations of substances normally present in the human body may control mental disease. Science. 1968;160(3825):265-271. (PubMed)
117. Seybolt SE. Is it time to reassess alpha lipoic acid and niacinamide therapy in schizophrenia? Med Hypotheses. 2010;75(6):572-575. (PubMed)
118. Zell M, Grundmann O. An orthomolecular approach to the prevention and treatment of psychiatric disorders. Adv Mind Body Med. 2012;26(2):14-28. (PubMed)
119. Yao JK, Dougherty GG, Jr., Gautier CH, et al. Prevalence and specificity of the abnormal niacin response: a potential endophenotype marker in schizophrenia. Schizophr Bull. 2016;42(2):369-376. (PubMed)
120. Sun L, Yang X, Jiang J, et al. Identification of the niacin-blunted subgroup of schizophrenia patients from mood disorders and healthy individuals in Chinese population. Schizophr Bull. 2017; doi: 10.1093/schbul/sbx150. [Epub ahead of print]. (PubMed)
121. Messamore E. Niacin subsensitivity is associated with functional impairment in schizophrenia. Schizophr Res. 2012;137(1-3):180-184. (PubMed)
122. Horrobin DF. The membrane phospholipid hypothesis as a biochemical basis for the neurodevelopmental concept of schizophrenia. Schizophr Res. 1998;30(3):193-208. (PubMed)
123. Messamore E. The niacin response biomarker as a schizophrenia endophenotype: A status update. Prostaglandins Leukot Essent Fatty Acids. 2017; pii: S0952-3278(16)30249-6. doi: 10.1016/j.plefa.2017.06.014. [Epub ahead of print]. (PubMed)
124. Jacob R, Swenseid M. Niacin. In: Ziegler E, Filer L, eds. Present Knowledge in Nutrition. 7th ed. Washington D.C.: ILSI Press; 1996:185-190.
125. US Department of Agriculture. USDA National Nutrient Database for Standard Reference, Release 25. 2012. https://ndb.nal.usda.gov/ndb/. Accessed 7/30/17.
126. Hendler SS, Rorvik DR. PDR for Nutritional Supplements. 2nd ed. Montvale: Thomson Reuters; 2008.
127. Minto C, Vecchio MG, Lamprecht M, Gregori D. Definition of a tolerable upper intake level of niacin: a systematic review and meta-analysis of the dose-dependent effects of nicotinamide and nicotinic acid supplementation. Nutr Rev. 2017;75(6):471-490. (PubMed)
128. MacKay D, Hathcock J, Guarneri E. Niacin: chemical forms, bioavailability, and health effects. Nutr Rev. 2012;70(6):357-366. (PubMed)
129. Dellinger RW, Santos SR, Morris M, et al. Repeat dose NRPT (nicotinamide riboside and pterostilbene) increases NAD(+) levels in humans safely and sustainably: a randomized, double-blind, placebo-controlled study. NPJ Aging Mech Dis. 2017;3:17. (PubMed)
130. Fulgoni VL, 3rd, Keast DR, Bailey RL, Dwyer J. Foods, fortificants, and supplements: Where do Americans get their nutrients? J Nutr. 2011;141(10):1847-1854. (PubMed)
131. Kar S, Chockalingam A. Statin-associated rhabdomyolysis with acute renal failure complicated by intradialytic NSTEMI: a review of lipid management considerations. Am J Ther. 2013;20(1):57-60. (PubMed)
132. Cziraky MJ, Willey VJ, McKenney JM, et al. Risk of hospitalized rhabdomyolysis associated with lipid-lowering drugs in a real-world clinical setting. J Clin Lipidol. 2013;7(2):102-108. (PubMed)
133. Maccubbin DL, Chen F, Anderson JW, et al. Effectiveness and safety of laropiprant on niacin-induced flushing. Am J Cardiol. 2012;110(6):817-822. (PubMed)
134. Hps Thrive Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J. 2013;34(17):1279-1291. (PubMed)
135. Lewey J, Shrank WH, Bowry AD, Kilabuk E, Brennan TA, Choudhry NK. Gender and racial disparities in adherence to statin therapy: a meta-analysis. Am Heart J. 2013;165(5):665-678, 678 e661. (PubMed)
136. Cheung MC, Zhao XQ, Chait A, Albers JJ, Brown BG. Antioxidant supplements block the response of HDL to simvastatin-niacin therapy in patients with coronary artery disease and low HDL. Arterioscler Thromb Vasc Biol. 2001;21(8):1320-1326. (PubMed)
137. Brown BG, Cheung MC, Lee AC, Zhao XQ, Chait A. Antioxidant vitamins and lipid therapy: end of a long romance? Arterioscler Thromb Vasc Biol. 2002;22(10):1535-1546. (PubMed)
138. Rios-Avila L, Coats B, Chi YY, et al. Metabolite profile analysis reveals association of vitamin B-6 with metabolites related to one-carbon metabolism and tryptophan catabolism but not with biomarkers of inflammation in oral contraceptive users and reveals the effects of oral contraceptives on these processes. J Nutr. 2015;145(1):87-95. (PubMed)
139. Dollerup OL, Christensen B,Svart M, et al. A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: safety, insulin-sensitivity, and lipid-mobilizing effects. Am J Clin Nutr. 2018;108:343-353. (PubMed)