Dietary Factors
Some of the listed dietary factors (i.e., L-carnitine, coenzyme Q10, and lipoic acid) can be synthesized by the body.
L-Carnitine
Contents
Summary
- Our body produces L-carnitine from the essential amino acid lysine via a specific biosynthetic pathway. Healthy individuals, including strict vegetarians, generally synthesize enough L-carnitine to prevent deficiency. However, certain conditions like pregnancy may result in increased excretion of L-carnitine, potentially increasing the risk for deficiency. (More information)
- Because of its role in the transport of long-chain fatty acids from the cytosol to the mitochondrial matrix, L-carnitine is critical for mitochondrial fatty acid β-oxidation. (More information)
- L-Carnitine supplementation is indicated for the treatment of primary systemic carnitine deficiency, which is caused by mutations in the gene that codes for the carnitine transporter, OCTN2. (More information)
- L-Carnitine is also approved for the treatment of carnitine deficiencies secondary to inherited diseases, such as propionyl-CoA carboxylase deficiency and medium chain acyl-CoA dehydrogenase deficiency, and in patients with end-stage renal disease undergoing hemodialysis. (More information)
- Evidence from randomized controlled trials suggests that L-carnitine or acylcarnitine esters may be useful adjuncts to standard medical treatment in individuals with cardiovascular disease. (More information)
- Routine administration of L-carnitine to people with end-stage renal disease undergoing hemodialysis is not recommended unless it is to treat carnitine deficiency. (More information)
- Acetyl-L-carnitine (ALCAR) may help reduce the severity of chemotherapy-induced peripheral neuropathy. High-quality evidence is needed to evaluate whether ALCAR may benefit the treatment of peripheral neuropathies associated with diabetes or caused by antiretroviral therapy. (More information)
- There is some low-quality evidence to suggest that supplemental L-carnitine or ALCAR may be beneficial as adjuncts to standard medical therapy of depression, Alzheimer's disease, and hepatic encephalopathy. (More information)
- There is little evidence that L-carnitine supplementation improves cancer-related fatigue, low fertility, or overall physical health. (More information)
- If you choose to take L-carnitine supplements, the Linus Pauling Institute recommends acetyl-L-carnitine at a daily dose of 0.5 to 1 g. Note that supplemental L-carnitine (doses, 0.6-7.0 g) is less efficiently absorbed compared to smaller amounts in food. (More information)
Introduction
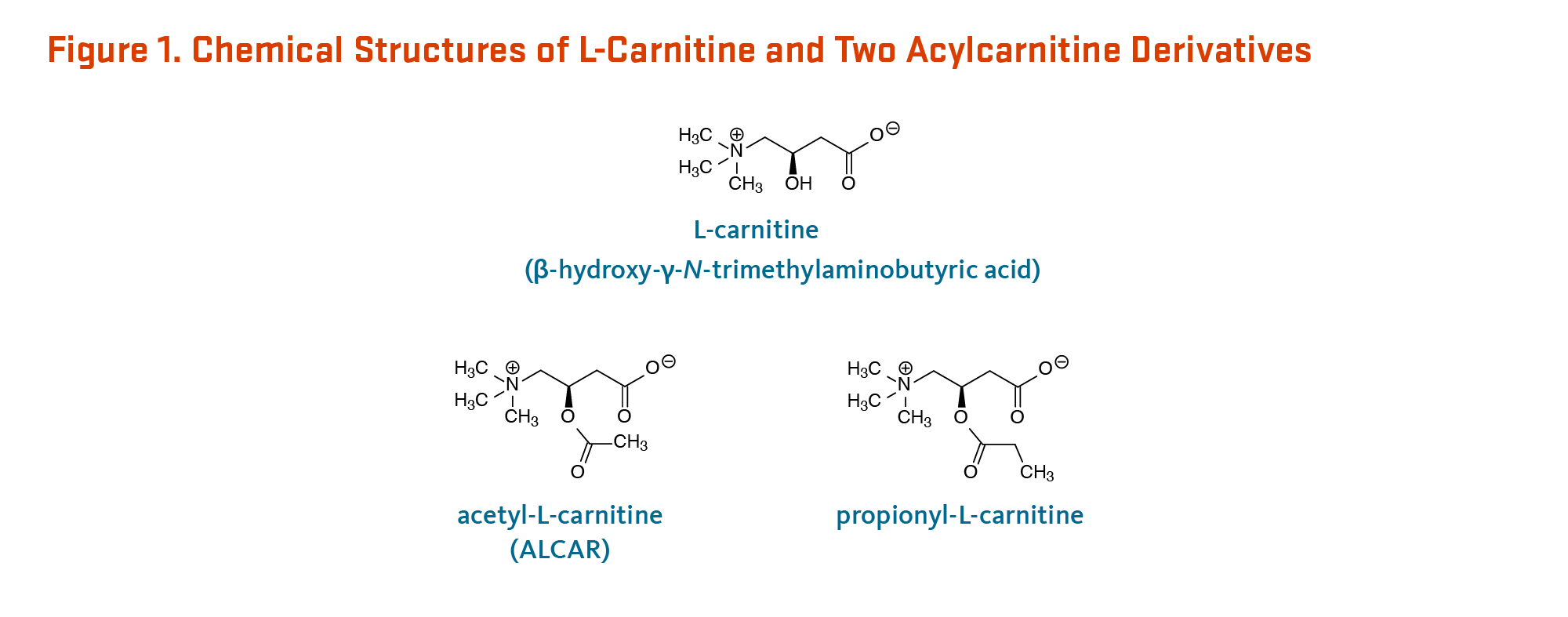
L-Carnitine (β-hydroxy-γ-N-trimethylaminobutyric acid) is a derivative of the amino acid, lysine (Figure 1). It was first isolated from meat (carnus in Latin) in 1905. Only the L-isomer of carnitine is biologically active (1). L-Carnitine appeared to act as a vitamin in the mealworm (Tenebrio molitor) and was therefore termed vitamin BT (2). Vitamin BT, however, is a misnomer because humans and other higher organisms can synthesize L-carnitine (see Metabolism and Bioavailability). Under certain conditions, the demand for L-carnitine may exceed an individual's capacity to synthesize it, making it a conditionally essential nutrient (3, 4).
Metabolism and Bioavailability
In healthy people, carnitine homeostasis is maintained through endogenous biosynthesis of L-carnitine, absorption of carnitine from dietary sources, and reabsorption of carnitine by the kidneys (5).
Endogenous biosynthesis
Humans can synthesize L-carnitine from the amino acids lysine and methionine in a multi-step process occurring across several cell compartments (cytosol, lysosomes, and mitochondria) (reviewed in 6). Across different organs, protein-bound lysine is methylated to form ε-N-trimethyllysine in a reaction catalyzed by specific lysine methyltransferases that use S-adenosyl-methionine (derived from methionine) as a methyl donor. ε-N-Trimethyllysine is released for carnitine synthesis by protein hydrolysis. Four enzymes are involved in endogenous L-carnitine biosynthesis (Figure 2). They are all ubiquitous except γ-butyrobetaine hydroxylase is absent from cardiac and skeletal muscle. This enzyme is, however, highly expressed in human liver, testes, and kidney (7).
L-carnitine is primarily synthesized in the liver and transported via the bloodstream to cardiac and skeletal muscle, which rely on L-carnitine for fatty acid oxidation yet cannot synthesize it (8). The rate of L-carnitine biosynthesis in humans was studied in strict vegetarians (i.e., in people who consume very little dietary carnitine) and estimated to be 1.2 µmol/kg of body weight/day (9). The rate of L-carnitine synthesis depends on the extent to which peptide-linked lysines are methylated and the rate of protein turnover. There is some indirect evidence to suggest that excess lysine in the diet may increase endogenous L-carnitine synthesis; however, changes in dietary carnitine intake level or in renal reabsorption do not appear to affect the rate of endogenous L-carnitine synthesis (6).

Absorption of exogenous L-carnitine
Dietary L-carnitine
The bioavailability of L-carnitine from food can vary depending on dietary composition. For instance, one study reported that bioavailability of L-carnitine in individuals adapted to low-carnitine diets (i.e., vegetarians) was higher (66%-86%) than in those adapted to high-carnitine diets (i.e., regular red meat eaters; 54%-72%) (10). The remainder is degraded by colonic bacteria.
L-carnitine supplements
While bioavailability of L-carnitine from the diet is quite high (see Dietary L-carnitine), absorption from oral L-carnitine supplements is considerably lower. The bioavailability of L-carnitine from oral supplements (doses, 0.6 to 7 g) ranges between 5% and 25% of the total dose (5). Less is known regarding the metabolism of the acetylated form of L-carnitine, acetyl-L-carnitine (ALCAR); however, the bioavailability of ALCAR is thought to be higher than that of L-carnitine. The results of in vitro experiments suggested that ALCAR might be partially hydrolyzed upon intestinal absorption (11). In humans, administration of 2 g/day of ALCAR for 50 days increased plasma ALCAR concentrations by 43%, suggesting that either ALCAR was absorbed without prior hydrolysis or that L-carnitine was re-acetylated in the enterocytes (5).
Elimination and reabsorption
L-Carnitine and short-chain acylcarnitine derivatives (esters of L-carnitine; see Figure 1) are excreted by the kidneys. Renal reabsorption of free L-carnitine is normally very efficient; in fact, an estimated 95% is thought to be reabsorbed by the kidneys (1). Therefore, carnitine excretion by the kidney is usually very low. However, several conditions can decrease the efficiency of carnitine reabsorption and, correspondingly, increase carnitine excretion. Such conditions include high-fat, low-carbohydrate diets; high-protein diets; pregnancy; and certain disease states (see Primary systemic carnitine deficiency) (12). In addition, when circulating L-carnitine concentration increases, as in the case of oral supplementation, renal reabsorption of L-carnitine may become saturated, resulting in increased urinary excretion of L-carnitine (5). Dietary or supplemental L-carnitine that is not absorbed by enterocytes is degraded by colonic bacteria to form two principal products, trimethylamine and γ-butyrobetaine. γ-Butyrobetaine is eliminated in the feces; trimethylamine is efficiently absorbed and metabolized to trimethylamine-N-oxide, which is excreted in the urine (13).
Biological Activities
Mitochondrial oxidation of long-chain fatty acids
L-Carnitine is synthesized primarily in the liver but also in the kidneys and then transported to other tissues. It is most concentrated in tissues that use fatty acids as their primary fuel, such as skeletal and cardiac muscle. In this regard, L-carnitine plays an important role in energy production by conjugating to fatty acids for transport from the cytosol into the mitochondria (6).
L-Carnitine is required for mitochondrial β-oxidation of long-chain fatty acids for energy production (1). Long-chain fatty acids must be esterified to L-carnitine (acylcarnitine) in order to enter the mitochondrial matrix where β-oxidation occurs (Figure 3). On the outer mitochondrial membrane, CPTI (carnitine-palmitoyl transferase I) catalyzes the transfer of medium/long-chain fatty acids esterified to coenzyme A (CoA) to L-carnitine. This reaction is a rate-controlling step for the β-oxidation of fatty acid (13). A transport protein called CACT (carnitine-acylcarnitine translocase) facilitates the transport of acylcarnitine across the inner mitochondrial membrane. On the inner mitochondrial membrane, CPTII (carnitine-palmitoyl transferase II) catalyzes the transfer of fatty acids from L-carnitine to free CoA. Fatty acyl-CoA is then metabolized through β-oxidation in the mitochondrial matrix, ultimately yielding propionyl-CoA and acetyl-CoA (6). Carnitine is eventually recycled back to the cytosol (Figure 3).

Regulation of energy metabolism through modulation of acyl CoA:CoA ratio
Free (nonesterified) CoA is required as a cofactor for numerous cellular reactions. The flux through pathways that require nonesterified CoA, such as the oxidation of glucose, may be reduced if all the CoA available in a given cell compartment is esterified. Carnitine can increase the availability of nonesterified CoA for these other metabolic pathways (6). Within the mitochondrial matrix, CAT (carnitine acetyl transferase) catalyzes the transesterification of short- and medium-chain fatty acids from CoA to carnitine (Figure 3). The resulting acylcarnitine esters (e.g., acetylcarnitine) can remain in the mitochondrial matrix or be exported back into the cytosol via CACT. Free (nonesterified) CoA can then participate in other reactions, such as the generation of acetyl-CoA from pyruvate in a reaction catalyzed by pyruvate dehydrogenase (14). Acetyl-CoA can then be oxidized to produce energy (ATP) in the citric acid cycle.
Other functions in cellular metabolism
In addition to its importance for energy production, L-carnitine was shown to display direct antioxidant properties in vitro (15). Age-related declines in mitochondrial function and increases in mitochondrial oxidant production are thought to be important contributors to the adverse effects of aging. Tissue L-carnitine concentrations have been found to decline with age in humans and animals (16). The expression of most proteins involved in the transport of carnitine (OCTN2) and the acylcarnitine shuttling system across the mitochondrial membrane (CPTIa, CPTII, and CAT; Figure 3) was also found to be much lower in the white blood cells of healthy older adults than of younger adults (17).
Preclinical studies in rodents showed that supplementation with high doses of acetyl-L-carnitine (ALCAR; Figure 1) reversed a number of age-related changes in liver mitochondrial function yet increased liver mitochondrial oxidant production (18). ALCAR supplementation in rats has also been found to improve or reverse age-related mitochondrial declines in skeletal and cardiac muscular function (19, 20). Co-supplementation of aged rats with L-carnitine and α-lipoic acid blunted age-related increases in reactive oxygen species (ROS), lipid peroxidation, protein carbonylation, and DNA strand breaks in a variety of tissues (heart, skeletal muscle, and brain) (21-30). Co-supplementation for three months improved both the number of total and intact mitochondria and mitochondrial ultrastructure of neurons in the hippocampus (30). Although ALCAR exerts antioxidant activities in rodents, it is not known whether taking high doses of ALCAR will have similar effects in humans.
Deficiency
Nutritional carnitine deficiencies have not been identified in healthy people without metabolic disorders, suggesting that most people can synthesize enough L-carnitine (1). Even strict vegetarians (vegans) show no signs of carnitine deficiency, despite the fact that most dietary carnitine is derived from animal sources (9). Infants, particularly premature infants, are born with low stores of L-carnitine, which could put them at risk of deficiency given their rapid rate of growth. One study reported that infants fed carnitine-free, soy-based formulas grew normally and showed no signs of a clinically relevant carnitine deficiency; however, some biochemical measures related to lipid metabolism differed significantly from infants fed the same formula supplemented with L-carnitine (31). Soy-based infant formulas are now fortified with the amount of L-carnitine normally found in human milk (32).
Carnitine status
Renal filtration maintains plasma concentrations of free carnitine around 40 to 50 micromoles (µmol)/L, while plasma concentrations of acetyl-L-carnitine (ALCAR; the most abundant carnitine ester) are around 3 to 6 µmol/L (8). Regardless of the etiology, plasma concentrations of free carnitine ≤20 µmol/L and increased acylcarnitine/free carnitine ratios (≥0.4) are considered abnormal (6). Low carnitine status is generally due to impaired mitochondrial energy metabolism or to carnitine not being efficiently reabsorbed by the kidneys. The rate of carnitine excretion is not a useful indicator of carnitine status because it can vary with dietary carnitine intake and other physiologic parameters. At present, there is no test that assesses functional carnitine deficiency in humans (6).
Primary systemic carnitine deficiency
Primary systemic carnitine deficiency is a rare, autosomal recessive disorder caused by mutations (including deletions) in the SLC22A5 gene coding for carnitine transporter protein OCTN2 (organic cation transporter novel 2) (33). Individuals with defective carnitine transport have poor intestinal absorption of dietary L-carnitine, impaired L-carnitine reabsorption by the kidneys (i.e., increased urinary loss of L-carnitine), and defective L-carnitine uptake by muscles (4). The clinical presentation can vary widely depending on the type of mutation affecting SLC22A5 and the phenotypic manifestation of the mutation, i.e., age of onset, organ involvement, and severity of symptoms at the time of diagnosis (34). The disorder usually presents in early childhood and is characterized by episodes of hypoketotic hypoglycemia (that can cause encephalopathy), hepatomegaly, elevated liver enzymes (transaminases), and hypoammonemia in infants; progressive cardiomyopathy, elevated creatine kinase, and skeletal myopathy in childhood; or fatigability in adulthood (34, 35). The metabolic and myopathic symptoms in infants and children can be fatal such that treatment should start promptly to prevent irreversible organ damage (34). The diagnosis is established by demonstrating abnormally low plasma free carnitine concentrations, reduced carnitine transport of fibroblasts from skin biopsy, and molecular analysis of the gene coding for OCTN2 (33, 34). Treatment consists of pharmacological doses of L-carnitine that are meant to maintain a normal blood carnitine concentration, thereby preventing the risk of hypoglycemia and correcting metabolic and myopathic manifestations (34).
Secondary carnitine deficiency or depletion
Secondary carnitine deficiency or depletion may result from either genetic or acquired conditions.
Hereditary causes include genetic defects in the metabolism of amino acids, cholesterol, and fatty acids, such as propionyl-CoA carboxylase deficiency (aka propionic acidemia) and medium chain acyl-CoA dehydrogenase deficiency (36). Such inherited disorders lead to a buildup of organic acids, which are subsequently removed from the body via urinary excretion of acylcarnitine esters. Increased urinary losses of carnitine can lead to the systemic depletion of carnitine (6).
Systemic carnitine depletion can also occur in disorders of impaired renal reabsorption. For instance, Fanconi's syndrome is a hereditary or acquired condition in which the proximal tubular reabsorption function of the kidneys is impaired (37). This malfunction consequently results in increased urinary losses of carnitine. Patients with renal disease who undergo hemodialysis are at risk for secondary carnitine deficiency because hemodialysis removes carnitine from the blood (see End-stage renal disease) (38).
One example of an exclusively acquired carnitine deficiency involves chronic use of pivalate-conjugated antibiotics. Pivalate is a branched-chain fatty acid anion that is metabolized to form an acyl-CoA ester, which is transesterified to carnitine and subsequently excreted in the urine as pivaloyl carnitine. Urinary losses of carnitine via this route can be 10-fold greater than the sum of daily carnitine intake and biosynthesis and lead to systemic carnitine depletion (see Drug interactions) (39).
Finally, a number of inherited mutations in genes involved in carnitine shuttling and fatty acid oxidation pathways do not systematically result in carnitine depletion (such that carnitine supplementation may not help mitigate the symptoms) but lead to abnormal profiles of acylcarnitine esters in blood (35, 40).
Nutrient interactions
Endogenous biosynthesis of L-carnitine is catalyzed by the concerted action of four different enzymes (see Metabolism and Bioavailability) (Figure 2). This process requires two essential amino acids (lysine and methionine), iron (Fe2+), vitamin B6 in the form of pyridoxal 5’-phosphate, niacin in the form of nicotinamide adenine dinucleotide (NAD), and may also require vitamin C (ascorbate) (4). One of the earliest symptoms of vitamin C deficiency is fatigue, thought to be related to decreased synthesis of L-carnitine (41).
Disease Treatment
In most studies discussed below, it is important to note that treatment with L-carnitine or acyl-L-carnitine esters (i.e., acetyl-L-carnitine [ALCAR], propionyl-L-carnitine; Figure 1) was generally used as an adjunct to standard medical therapy, not in place of it. It is also important to consider the fact that the bioavailability of L-carnitine and acylcarnitine derivatives administered orally is low (~10-20%) (see Metabolism and Bioavailability). Intravenous administration is more likely to increase plasma carnitine concentration, yet homeostatic mechanisms tightly control blood concentration through metabolism and renal excretion: Up to 90% of 2 g of L-carnitine administered intravenously is excreted into the urine within 12 to 24 hours. Only a fraction of the dose is thought to enter the endogenous carnitine pool — largely found in skeletal muscle (reviewed in 8).
Type 2 diabetes mellitus
Several small clinical trials have explored whether supplemental L-carnitine could improve glucose tolerance in people with impaired glucose metabolism. A potential benefit of L-carnitine in these patients is based on the fact that it can (i) increase the oxidation of long-chain fatty acids which accumulation may contribute to insulin resistance in skeletal muscle, and (ii) enhance glucose utilization by reducing acyl-CoA concentration within the mitochondrial matrix (see Biological Activities) (42). A meta-analysis of five trials in participants with either impaired fasting glucose, type 2 diabetes mellitus, or nonalcoholic steatohepatitis found evidence of an improvement in insulin resistance with supplemental L-carnitine compared to placebo (43). Another meta-analysis of four randomized, placebo-controlled trials found evidence of a reduction in fasting plasma glucose concentration and no improvement of glycated hemoglobin concentration in subjects with type 2 diabetes mellitus supplemented with acetyl-L-carnitine (ALCAR) (44). A third meta-analysis of 16 trials suggested that supplementation with (acyl)-L-carnitine may reduce fasting blood glucose and glycated hemoglobin concentrations, but not resistance to insulin (45). In a recent double-blind, randomized, placebo-controlled trial, the effect of ALCAR was examined in 229 participants treated for type 2 diabetes mellitus, hypertension, and dyslipidemia (46). ALCAR supplementation (1 g/day for 6 months) had no effect on systolic or diastolic blood pressure, markers of kidney function (i.e., glomerular filtration rate and albuminuria), markers of glucose homeostasis (i.e., glucose disposal rate, glycated hemoglobin concentration, and a measure of insulin resistance), and blood lipid profile (i.e., concentrations of triglycerides, lipoprotein (a), LDL-cholesterol, HDL-cholesterol, and total cholesterol) (46).
Cardiovascular disease
In the studies discussed below it is important to note that treatment with L-carnitine or propionyl-L-carnitine was used as an adjunct (in addition) to appropriate medical therapy, not in place of it.
Myocardial infarction
Myocardial infarction (MI) occurs when an atherosclerotic plaque in a coronary artery ruptures and obstructs the blood supply to the heart muscle, causing injury or damage to the heart (see the page on Heart Attack). Several clinical trials have explored whether L-carnitine administration immediately after MI diagnosis could reduce injury to heart muscle resulting from ischemia and improve clinical outcomes. An early trial in 160 men and women diagnosed with a recent MI showed that oral L-carnitine (4 g/day) in addition to standard pharmacological treatment for one year significantly reduced mortality and the occurrence of angina attacks compared to the control (47). In another controlled trial in 96 patients, treatment with intravenous L-carnitine (5 g bolus followed by 10 g/day for three days) following a MI resulted in lower concentrations of creatine kinase-MB and troponin-I, two markers of cardiac injury (48). However, not all clinical trials have found L-carnitine supplementation to be beneficial after MI. For example, in a randomized, double-blind, placebo-controlled trial in 60 participants diagnosed with an acute MI, neither mortality nor echocardiographic measures of cardiac function differed between patients treated with intravenous L-carnitine (6 g/day) for seven days followed by oral L-carnitine (3 g/day) for three months and those given a placebo (49). Another randomized placebo-controlled trial in 2,330 patients with acute MI, L-carnitine therapy (9 g/day intravenously for five days, then 4 g/day orally for six months) did not affect the risk of heart failure and death six months after MI (50).
A 2013 meta-analysis of randomized controlled trials found that L-carnitine therapy in patients who experienced an MI reduced the risks of all-cause mortality (-27%; 11 trials; 3,579 participants), ventricular arrhythmias (-65%; five trials; 229 participants), and angina attacks (-40%; 2 trials; 261 participants), but had no effect on the risks of having a subsequent myocardial infarction or developing heart failure (51). Because about 90% of oral L-carnitine supplements is unlikely to be absorbed, one could ask whether the efficacy is equivalent between protocols using both intravenous and oral administration and those using oral administration only (8). This has not been examined in subgroup analyses.
Heart failure
Heart failure is described as the heart's inability to pump enough blood for all of the body's needs (see the page on Heart Failure). In coronary heart disease, accumulation of atherosclerotic plaque in the coronary arteries may prevent heart regions from getting adequate circulation, ultimately resulting in cardiac damage and impaired pumping ability. Myocardial infarction may also damage the heart muscle, which could potentially lead to heart failure. Further, the diminished heart’s capacity to pump blood in cases of dilated cardiomyopathy may lead to heart failure. Because physical exercise increases the demand on the weakened heart, measures of exercise tolerance are frequently used to monitor the severity of heart failure. Echocardiography is also used to determine the left ventricular ejection fraction (LVEF), an objective measure of the heart's pumping ability. An LVEF of less than 40% is indicative of systolic heart failure (52).
An abnormal acylcarnitine profile and a high acylcarnitine to free carnitine ratio in the blood of patients with heart failure have been linked to disease severity and poor prognosis (53-55). Addition of L-carnitine to standard medical therapy for heart failure has been evaluated in several clinical trials. A 2013 meta-analysis of 17 randomized, placebo-controlled studies in a total of 1,625 participants with heart failure found that oral L-carnitine (1.5-6 g/day for seven days to three years) significantly improved several measures of cardiac functional capacity (including exercise tolerance and markers of the left ventricle function), yet had no impact on all-cause mortality (56).
Angina pectoris
Angina pectoris is chest pain that occurs when the coronary blood supply is insufficient to meet the metabolic needs of the heart muscle (as with ischemic heart disease; see the page on Angina Pectoris) (57). In a prospective cohort study in over 4,000 participants with suspected angina pectoris, elevated concentrations of certain acylcarnitine intermediates in blood were associated with fatal and non-fatal acute myocardial infarction (58). In early studies, the addition of oral L-carnitine or propionyl-L-carnitine to pharmacologic therapy for chronic stable angina modestly improved exercise tolerance and decreased electrocardiographic signs of ischemia during exercise testing in some angina patients (59-61). Another early study examined hemodynamic and angiographic variables before, during, and after administering intravenous propionyl-L-carnitine (15 mg/kg body weight) in men with myocardial dysfunction and angina pectoris (62). In this study, propionyl-L-carnitine decreased myocardial ischemia, evidenced by significant reductions in ST-segment depression and left ventricular end-diastolic pressure (62). No recent and/or large-scale studies have been conducted to further examine the potential benefit of L-carnitine or propionyl-L-carnitine in the management of angina pectoris.
Intermittent claudication in peripheral arterial disease
In peripheral arterial disease, atherosclerosis of the arteries that supply the lower extremities may diminish blood flow to the point that the metabolic needs of exercising muscles are not sufficiently met, thereby leading to ischemic leg or hip pain known as claudication (see the page on Intermittent Claudication in Peripheral Arterial Disease) (63). Several clinical trials have found that treatment with propionyl-L-carnitine improves exercise tolerance in some patients with intermittent claudication. In a double-blind, placebo-controlled, dose-titration study, 1 to 3 g/day of oral propionyl-L-carnitine for 24 weeks was well tolerated and improved maximal walking distance in patients with intermittent claudication (64). In a randomized, placebo-controlled study of 495 patients with intermittent claudication, 2 g/day of propionyl-L-carnitine for 12 months significantly increased maximal walking distance and the distance walked prior to the onset of claudication in patients whose initial maximal walking distance was less than 250 meters (m), but no effect was seen in patients who had an initial maximal walking distance greater than 250 m (65). More recent trials have associated oral propionyl-L-carnitine supplementation (2 g/day for several months) with improved walking distance and claudication onset time (66, 67), as well as with higher pain-free walking distance and higher ankle-brachial index (a diagnostic measure of peripheral arterial disease) (68). Two 2013 systematic reviews of interventions concluded that the modest benefit of propionyl-L-carnitine on walking performance was equivalent or superior to that obtained with drugs approved for claudication in the US (e.g., pentoxyphylline, cilostazol) yet inferior to improvements seen with supervised exercise interventions (69, 70).
End-stage renal disease
L-Carnitine and short/medium-chain acylcarnitine molecules are removed from the circulation during hemodialysis. Both L-carnitine loss into the dialysate and impaired synthesis by the kidneys contribute to a progressive carnitine deficiency in patients with end-stage renal disease (ESRD) undergoing hemodialysis (71). The low clearance of long-chain acylcarnitine molecules leads to a high acylcarnitine-to-L-carnitine ratio that has been associated with a higher risk of cardiovascular mortality (72). Carnitine depletion in patients undergoing hemodialysis may lead to various conditions, such as muscle weakness and fatigue, plasma lipid abnormalities, and refractory anemia. A 2014 systematic review and meta-analysis of 31 randomized controlled trials in a total of 1,734 patients with ESRD found that L-carnitine treatment — administered either orally or intravenously — resulted in reductions of serum C-reactive protein (a marker of inflammation and predictor of mortality in patients undergoing hemodialysis) and LDL-cholesterol, although the latter was not deemed to be clinically relevant (73). There was no effect of L-carnitine on other serum lipids (i.e., total and HDL-cholesterol, triglycerides) and anemia-related indicators (i.e., hemoglobin concentration, hematocrit, albumin, and required dose of recombinant erythropoietin) (73).
The US National Kidney Foundation did not recommend routine administration of L-carnitine to subjects undergoing dialysis yet encouraged the development of trials in patients with select symptoms that do not respond to standard therapy, i.e., persistent muscle cramps or hypotension during dialysis, severe fatigue, skeletal muscle weakness or myopathy, cardiomyopathy, and anemia requiring large doses of erythropoietin (74, 75).
Finally, the use of L-carnitine (10-20 mg/kg body weight given as a slow bolus injection) is approved by the US FDA to treat L-carnitine deficiency in subjects with ESRD undergoing hemodialysis (76).
Peripheral neuropathy
Antiretroviral-related peripheral neuropathy
The use of certain antiretroviral agents (nucleoside analogs) has been associated with an increased risk of developing peripheral neuropathy in HIV-positive individuals (77, 78). Small, uncontrolled, open-label intervention studies have suggested a beneficial effect of acetyl-L-carnitine (ALCAR) in patients with painful neuropathies. An early uncontrolled study found that 10 out of 16 HIV-positive subjects with painful neuropathy reported improvement after three weeks of intravenous or intramuscular ALCAR treatment (79). Results from another uncontrolled intervention in 21 HIV-positive patients suggested that long-term (two to four years) oral ALCAR supplementation (1.5 g/day) may be a beneficial adjunct to antiretroviral therapy to improve neuropathic symptoms in some HIV-infected individuals (80, 81). Additionally, two small studies in participants presenting with antiretroviral-induced neuropathy found significant reduction in subjects' mean pain intensity with oral ALCAR (1-3 g/day) for 4 to 24 weeks, but no effect on objective neurophysiological parameters was found (82, 83).
A double-blind, placebo-controlled trial in 90 HIV-positive patients with symptomatic distal symmetrical polyneuropathy found no benefit of intramuscular injection with 1 g/day of ALCAR for two weeks in the intention-to-treat analysis, but there was some pain relief in the group of 66 patients who completed the trial (84). Large-scale, controlled studies are needed before any conclusions can be drawn.
Diabetic peripheral neuropathy
Peripheral nerve dysfunction occurs in about 50% of people with diabetes mellitus, and chronic neuropathic pain is present in about one-third of people with diabetic peripheral neuropathy (85). Advanced stages of diabetic peripheral neuropathy can lead to recurrent foot ulcers and infections, and eventually amputations (86). A 2019 systematic review (87) identified three placebo-controlled interventions that examined the effect of oral supplementation with acetyl-L-carnitine (ALCAR; 1.5-3.0 g/day for one year) in individuals with diabetic peripheral neuropathy (3, 88). Low-quality evidence suggested a lower level of pain with ALCAR, as measured with a visual scale analog. Low-quality evidence from another trial that compared the effect of ALCAR (1.5 g/day) with that of methylcobalamin (1.5 mg/day) for 24 weeks suggested no difference between treatments regarding the extent of functional disability (using the Neuropathy Disability Scale) or measures of symptom quality and severity (using the Neuropathy Symptom Scale) (89).
Chemotherapy-induced peripheral neuropathy
A few randomized, double-blind, placebo-controlled trials have examined whether ALCAR might help prevent or treat chemotherapy-induced peripheral neuropathy. A trial in 150 participants with either ovarian cancer or castration-resistant prostate cancer found no evidence that ALCAR (1g every 3 days) given with the anticancer drug sagopilone (for up to six cycles of treatment) reduced the overall risk of peripheral neuropathy compared to a placebo (90). However, ALCAR reduced the risk of high-grade sagopilone-induced neuropathy (90). In contrast, another trial in 409 women with breast cancer found that ALCAR (3 g/day for 24 weeks) increased anticancer drug (taxanes)-induced peripheral neuropathy and decreased measures of functional status compared to placebo (91). A follow-up study reported that the negative impact of 24-week treatment with ALCAR was still observed at week 52; however, no differences between ALCAR and placebo were apparent at week 104 (92). Finally, one trial in 239 participants already suffering from chemotherapy-induced peripheral neuropathy reported a reduction in the severity of neuropathic symptoms with ALCAR (3 g/day for eight weeks) compared to placebo (93). Improvements in electrophysiological parameters were also observed with ALCAR treatment (93).
The results from these trials are conflicting and thus difficult to interpret. The efficacy of ALCAR in the prevention and treatment of chemotherapy-induced peripheral neuropathy remains to be established (94).
Depression
A 2018 meta-analysis identified 12 randomized controlled trials, including 791 participants, that examined the effect of ALCAR on symptoms of depression (95). Evidence from nine trials suggested a reduction in depressive symptoms with ALCAR (3 g/day for a median 8 weeks) compared to a placebo. Three trials that compared ALCAR treatment (1-3 g/day for 7-12 weeks) and antidepressant medications found ALCAR was as effective as antidepressants in treating depressive symptoms (95). Another meta-analysis of trials that compared the safety profile of antidepressants found evidence of fewer adverse effects, and consequently, better adherence to treatment with ALCAR compared to placebo and in contrast to classical pharmaceutical agents (96).
Alzheimer's disease
The metabolomic profiling of acylcarnitine molecules showed variations in serum concentrations of subjects along the continuum from cognitively healthy to affected by Alzheimer's disease (97). Changes in the blood concentrations of specific acylcarnitines in subjects with either subjective memory complaints, mild cognitive impairment, or Alzheimer's disease, compared to cognitively healthy peers may reflect changes in the transport of fatty acids into the mitochondria and/or impairments in energy production. Several clinical trials conducted in the 1990s examined the effect of acetyl-L-carnitine (ALCAR) treatment on the cognitive performance of patients clinically diagnosed with Alzheimer's disease. Early, small trials suggested a beneficial effect of ALCAR with respect to cognitive decline (98-100), whereas later, larger trials found little-to-no effect compared to placebo (101-103). However, a 2003 systematic review highlighted differences in methodologies between early and later studies that make it difficult to compare results (104). Nevertheless, the pooled analysis of 16 trials suggested improvements in the summary measure of patients' global functioning (assessed with the Clinical Global Impressions [CGI-I] scale) after 12 and 24 weeks of (but not after 52 weeks) of ALCAR treatment (1-3 g/day) and in cognitive performance (assessed with the Mini-Mental State Examination [MMSE] scale) after 24 weeks (but not at 12 or 52 weeks) (104). A 2003 meta-analysis of 21 trials found that ALCAR was superior to placebo in several psychometric tests assessing global patient functioning, attention, memory, and some intellectual functions (105).
Hepatic encephalopathy
Hepatic encephalopathy refers to the occurrence of a spectrum of neuropsychiatric signs or symptoms in individuals with acute or chronic liver disease (106). Subclinical hepatic encephalopathy may not feature any symptoms beyond abnormal behavior on psychometric tests or symptoms that are nonspecific in nature. In contract, overt hepatic encephalopathy can present with disorientation, obvious personality change, inappropriate behavior, somnolence, stupor, confusion, and coma (106). Changes in mental status are thought to be caused by the liver failing to detoxify neurotoxic compounds like ammonia. A 2019 systematic review of five placebo-controlled trials conducted by one group of investigators examined the effect of acetyl-L-carnitine (ALCAR) in 398 participants with cirrhosis and portal hypertension (high blood pressure in the portal vein) and presenting with either subclinical or overt hepatic encephalopathy. ALCAR was either administered orally (4 g/day for 90 days) in four trials or intravenously (4 g/day for three days) in one trial. ALCAR was found to significantly reduce blood ammonium concentration compared to placebo. However, none of the trials reported on serious adverse outcomes, including mortality. Additionally, the evidence was too limited to assess the impact on quality of life or mental and physical fatigue.
Cancer-related fatigue
Fatigue is not uncommon in people who have undergone chemotherapy and survived cancer, with fatigue symptoms depending on the specific type of cancer and treatment. Cancer-related fatigue can persist well beyond the end of chemotherapy and be associated with cognitive and functional decline, insomnia, depression, and a reduction in the quality of life (107). A 2017 systematic review and meta-analysis identified 12 intervention studies that assessed the effect of L-carnitine or ALCAR on cancer-related fatigue (reported as a primary or secondary outcome) in cancer survivors (108). Three studies had no control arm, eight studies were open-label, and eight studies included fewer than 100 participants. Overall, only three studies were deemed of good quality. The meta-analysis of these three randomized, double-blind, placebo-controlled trials found no effect of L-carnitine (0.25-4 g/day for 1 week to 3 months) or ALCAR (3 g/day for 6 months) on the level of cancer-related fatigue (108).
Infertility
L-Carnitine is concentrated in the epididymis, where sperm mature and acquire their motility (109). An early cross-sectional study of 101 fertile and infertile men found that L-carnitine concentrations in semen were positively correlated with the number of sperm, the percentage of motile sperm, and the percentage of normal appearing sperm in the sample (110), suggesting that L-carnitine may play an important role in male fertility. One placebo-controlled, double-blind, cross-over trial in 86 subjects with fertility issues found that supplementation with L-carnitine (2 g/day) for two months improved sperm quality, as evidenced by increases in sperm concentration and motility (111). In another placebo-controlled trial conducted by the same group, similar improvements in sperm motility were observed in participants supplemented with 2g/day of L-carnitine and 1 g/day of acetyl-L-carnitine (ALCAR) for six months (112). In both trials, the effect of carnitine was greater in the most severe cases of asthenozoospermia (reduced sperm motility) at baseline (111, 112). Another placebo-controlled, double-blind, randomized study in 44 men with idiopathic asthenozoospermia found an increase in sperm motility in those given ALCAR alone (3 g/day) or ALCAR (1 g/day) plus L-carnitine (2 g/day) compared to those given L-carnitine alone (2 g/day) or a placebo (113). However, a pooled analysis of the two trials that employed ALCAR found no significant effect of ALCAR and L-carnitine on sperm concentration, motility, and morphology (114). Evidence from larger scale clinical trials is still needed to determine whether L-carnitine and ALCAR could play a role in the treatment of male infertility.
Physical health
Frailty
Frailty is a syndrome prevalent among geriatric populations and characterized by a functional decline and a loss of independence to perform the activities of daily living. Frailty in individuals may include at least three of the following symptoms: unintentional weight loss, exhaustion (poor endurance), weakness (low grip strength), slowness, and physical inactivity (115). It is believed that early stages of frailty are amenable to interventions that could avert adverse outcomes, including the increased risk of hospitalization and premature death (116). The suggestion that carnitine deficiency may lead to frailty through mitochondrial dysfunction (117) has been examined in one trial. This randomized, double-blind, placebo-controlled trial in 58 older adults identified as "pre-frail" found an decrease in a Frailty Index score and an improvement in the hand grip test in individuals supplemented with L-carnitine (1.5 g/day) over 10 weeks but not in those given a placebo (118). However, there was no difference in Frailty Index and hand grip test scores between supplemental L-carnitine and placebo groups.
Skeletal muscle wasting
Loss of skeletal muscle mass is associated with a decrease in muscle strength and occurs with aging (119), as well as in several pathological conditions (120-122). Based on preclinical studies, it has been suggested that L-carnitine supplementation could limit the imbalance between protein anabolism (synthesis) and catabolism (degradation) that leads to skeletal muscle wasting (42). A randomized, double-blind, placebo-controlled trial in 28 older women (ages, 65-70 years) found no effect of L-carnitine supplementation (1.5 g/day for 24 weeks) on serum pro-inflammatory cytokine concentrations, body mass and composition (lean [fat-free] mass and skeletal muscle mass), or measures of skeletal muscle strength (123). In contrast, a retrospective cohort study in patients with cirrhosis found a reduced rate of skeletal muscle loss over at least six months in those who were administered L-carnitine (N=35; mean dose, 1.02 g/day) compared to those who were not (124). Of note, supplementation with L-carnitine was given in patients with cirrhosis to control hyperammonemia (N=27), to reduce muscle cramps (N=6), or to prevent carnitine deficiency (N=2). One major limitation of this study beyond its retrospective design is that patients who received L-carnitine had a significantly different clinical presentation; in particular, liver dysfunction was significantly more severe in these patients than in those who were not supplemented (124).
Muscle cramps
Muscle cramps are involuntary and painful contractions of skeletal muscles. Two uncontrolled studies conducted in participants with cirrhosis found that L-carnitine supplementation was safe to use at doses of 0.9 to 1.2 g/day for eight weeks (125) and 1 g/day for 24 weeks (126) and might be considered to control the frequency of cramps. However, whether supplemental L-carnitine can be efficacious to limit the incidence of muscle cramps in patients with cirrhosis remains unknown. An open-label, non-randomized trial in 69 patients with either type 1 or type 2 diabetes mellitus found a reduction in the incidence of muscle cramps and an improvement in the quality of life of those prescribed 0.6 g/day of L-carnitine for four months compared to controls (127). In contrast, there is little evidence to date to suggest that supplemental L-carnitine could reduce muscle cramps in patients undergoing hemodialysis (128). Well-designed trials are necessary to examine whether L-carnitine could be helpful in the management of cramps.
Physical performance
Interest in the potential of L-carnitine supplementation to improve athletic performance is related to its important roles in energy metabolism. A number of small, poorly controlled studies have reported that either acute (dose given one hour before exercise bout) or short-term (two to three weeks) supplementation with L-carnitine (2 to 4 g/day) supported energy production, cardiorespiratory fitness, and endurance capacity during physical exercise (reviewed in 129). However, in a double-blind, placebo-controlled trial in 32 healthy adults, propionyl-L-carnitine (1 g/day or 3 g/day) for eight weeks did not improve aerobic or anaerobic exercise performance (130). An intervention study compared the effect of L-carnitine supplementation (2 g/day for 12 weeks) on plasma and skeletal muscle carnitine concentrations and physical performance between 16 vegetarian and 8 omnivorous male participants (131). At baseline, plasma carnitine concentration was about 10% lower in vegetarian compared to omnivorous participants. However, the content carnitine in skeletal muscle, phosphocreatine, ATP, glycogen, and lactate, as well as measures of physical performance during exercise were equivalent between vegetarians and omnivores. While L-carnitine supplementation normalized plasma carnitine concentration in vegetarians to that observed in omnivores, there was no effect on energy metabolism and physical performance compared to no supplementation and between vegetarians and omnivores (131).
Sources
Biosynthesis
The normal rate of L-carnitine biosynthesis in humans ranges from 0.16 to 0.48 mg/kg of body weight/day (4). Thus, a 70 kg (154 1b) person would synthesize between 11 and 34 mg of carnitine per day. This rate of synthesis, combined with efficient (95%) L-carnitine reabsorption by the kidneys, is sufficient to prevent deficiency in generally healthy people, including strict vegetarians (132).
Food sources
Meat, poultry, fish, and dairy products are the richest sources of L-carnitine, while fruit, vegetables, and grains contain relatively little L-carnitine. Omnivorous diets have been found to provide 23 to 135 mg/day of L-carnitine for an average 70 kg person, while strict vegetarian diets may provide as little as 1 mg/day for a 70 kg person (8). Between 54% and 86% of L-carnitine from food is absorbed, compared to 5%-25% from oral supplements (0.6-7 g/day) (13). Non-milk-based infant formulas (e.g., soy formulas) should be fortified so that they contain 11 mg/L of L-carnitine. Some carnitine-rich foods and their carnitine content in milligrams (mg) are listed in Table 1.
Supplements
Intravenous L-carnitine
Intravenous L-carnitine is available by prescription only for the treatment of primary and secondary L-carnitine deficiencies (76).
Oral L-carnitine
Oral L-carnitine is available by prescription for the treatment of primary and secondary L-carnitine deficiencies (76). It is also available without a prescription as a nutritional supplement; supplemental doses usually range from 0.5 to 2 g/day.
Acetyl-L-carnitine
Acetyl-L-carnitine (ALCAR) is available without a prescription as a nutritional supplement. In addition to providing L-carnitine, it provides acetyl groups that may be used in the formation of the neurotransmitter, acetylcholine. Supplemental doses usually range from 0.5 to 2 g/day (133).
Propionyl-L-carnitine
Propionyl-L-carnitine is not approved by the US FDA for use as a drug to prevent or treat any condition. It is, however, available without prescription as a nutritional supplement.
See Figure 1 for the chemical structures of L-carnitine, acetyl-L-carnitine, and propionyl-L-carnitine.
Safety
Adverse effects
In general, L-carnitine appears to be well tolerated; no toxic effects have been reported in relation to intakes of high doses of L-carnitine. L-Carnitine supplementation may cause mild gastrointestinal symptoms, including nausea, vomiting, abdominal cramps, and diarrhea. Supplements providing more than 3 g/day may cause a "fishy" body odor. Acetyl-L-carnitine (ALCAR) has been reported to increase agitation in some Alzheimer's disease patients (133). Despite claims that L-carnitine or ALCAR might increase seizures in some individuals with seizure disorders (133), these are not supported by any scientific evidence (134). Only the L-isomer of carnitine is biologically active; the D-isomer may actually compete with L-carnitine for absorption and transport, thereby increasing the risk of L-carnitine deficiency (4). Supplements containing a mixture of the D- and L-isomers (D,L-carnitine) have been associated with muscle weakness in patients with kidney disease. Long-term studies examining the safety of ALCAR supplementation in pregnant and breast-feeding women are lacking (133).
Drug interactions
Pivalic acid combines with L-carnitine and is excreted in the urine as pivaloylcarnitine, thereby increasing L-carnitine losses (see also Secondary carnitine deficiency). Consequently, prolonged use of pivalic acid-containing antibiotics, including pivampicillin, pivmecillinam, pivcephalexin, and cefditoren pivoxil (Spectracef), can lead to secondary L-carnitine deficiency (135). The anticonvulsant valproic acid (Depakene) interferes with L-carnitine biosynthesis in the liver and forms with L-carnitine a valproylcarnitine ester that is excreted in the urine. However, L-carnitine supplements are necessary only in a subset of patients taking valproic acid. Risk factors for L-carnitine deficiency with valproic acid include young age (<2 years), severe neurological problems, use of multiple antiepileptic drugs, poor nutrition, and consumption of a ketogenic diet (135). There is insufficient evidence to suggest that nucleoside analogs used in the treatment of HIV infection (i.e., zidovudine [AZT], didanosine [ddI], zalcitabine [ddC], and stavudine [d4T] or certain cancer chemotherapy agents (i.e., ifosfamide, cisplatin) increase the risk of secondary L-carnitine deficiency (135).
Authors and Reviewers
Originally written in 2002 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2012 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in July 2019 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in December 2019 by:
Tory M. Hagen, Ph.D.
Principal Investigator, Linus Pauling Institute
Professor, Dept. of Biochemistry and Biophysics
Helen P. Rumbel Professor for Healthy Aging Research
Oregon State University
Copyright 2002-2024 Linus Pauling Institute
References
1. Rebouche CJ. Carnitine. In: Shils ME, Shike M, Ross AC, Caballero B, Cousins RJ, eds. Modern Nutrition in Health and Disease. 10th ed. Philadelphia: Lippincott, Williams & Wilkins; 2006:537-544.
2. Fraenkel G, Friedman S. Carnitine. Vitam Horm. 1957;15:73-118. (PubMed)
3. De Grandis D, Minardi C. Acetyl-L-carnitine (levacecarnine) in the treatment of diabetic neuropathy. A long-term, randomised, double-blind, placebo-controlled study. Drugs R D. 2002;3(4):223-231. (PubMed)
4. Seim H, Eichler K, Kleber H. L(-)-Carnitine and its precursor, gamma-butyrobetaine. In: Kramer K, Hoppe P, Packer L, eds. Nutraceuticals in Health and Disease Prevention. New York: Marcel Dekker, Inc.; 2001:217-256.
5. Rebouche CJ. Kinetics, pharmacokinetics, and regulation of L-carnitine and acetyl-L-carnitine metabolism. Ann N Y Acad Sci. 2004;1033:30-41. (PubMed)
6. Rebouche CJ. Carnitine. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore; 2014:440-446.
7. Rebouche CJ. Ascorbic acid and carnitine biosynthesis. Am J Clin Nutr. 1991;54(6 Suppl):1147S-1152S. (PubMed)
8. Evans AM, Fornasini G. Pharmacokinetics of L-carnitine. Clin Pharmacokinet. 2003;42(11):941-967. (PubMed)
9. Lombard KA, Olson AL, Nelson SE, Rebouche CJ. Carnitine status of lactoovovegetarians and strict vegetarian adults and children. Am J Clin Nutr. 1989;50(2):301-306. (PubMed)
10. Rebouche CJ, Chenard CA. Metabolic fate of dietary carnitine in human adults: identification and quantification of urinary and fecal metabolites. J Nutr. 1991;121(4):539-546. (PubMed)
11. Gross CJ, Henderson LM, Savaiano DA. Uptake of L-carnitine, D-carnitine and acetyl-L-carnitine by isolated guinea-pig enterocytes. Biochim Biophys Acta. 1986;886(3):425-433. (PubMed)
12. Rebouche CJ, Lombard KA, Chenard CA. Renal adaptation to dietary carnitine in humans. Am J Clin Nutr. 1993;58(5):660-665. (PubMed)
13. Rebouche CJ. L-carnitine. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. ILSI/Wiley-Blackwell; 2012:391-404.
14. McGrane MM. Carbohydrate metabolism--synthesis and oxidation. In: Stipanuk MH, ed. Biochemical and Physiological Aspects of Human Nutrition. Philadelphia: W.B. Saunders Co; 2000:158-210.
15. Solarska K, Lewinska A, Karowicz-Bilinska A, Bartosz G. The antioxidant properties of carnitine in vitro. Cell Mol Biol Lett. 2010;15(1):90-97. (PubMed)
16. Costell M, O'Connor JE, Grisolia S. Age-dependent decrease of carnitine content in muscle of mice and humans. Biochem Biophys Res Commun. 1989;161(3):1135-1143. (PubMed)
17. Karlic H, Lohninger A, Laschan C, et al. Downregulation of carnitine acyltransferases and organic cation transporter OCTN2 in mononuclear cells in healthy elderly and patients with myelodysplastic syndromes. J Mol Med (Berl). 2003;81(7):435-442. (PubMed)
18. Hagen TM, Ingersoll RT, Wehr CM, et al. Acetyl-L-carnitine fed to old rats partially restores mitochondrial function and ambulatory activity. Proc Natl Acad Sci U S A. 1998;95(16):9562-9566. (PubMed)
19. Pesce V, Fracasso F, Cassano P, Lezza AM, Cantatore P, Gadaleta MN. Acetyl-L-carnitine supplementation to old rats partially reverts the age-related mitochondrial decay of soleus muscle by activating peroxisome proliferator-activated receptor gamma coactivator-1alpha-dependent mitochondrial biogenesis. Rejuvenation Res. 2010;13(2-3):148-151. (PubMed)
20. Gomez LA, Heath SH, Hagen TM. Acetyl-l-carnitine supplementation reverses the age-related decline in carnitine palmitoyltransferase 1 (CPT1) activity in interfibrillar mitochondria without changing the l-carnitine content in the rat heart. Mech Ageing Dev. 2012;133(2-3):99-106. (PubMed)
21. Muthuswamy AD, Vedagiri K, Ganesan M, Chinnakannu P. Oxidative stress-mediated macromolecular damage and dwindle in antioxidant status in aged rat brain regions: role of L-carnitine and DL-alpha-lipoic acid. Clin Chim Acta. 2006;368(1-2):84-92. (PubMed)
22. Kumaran S, Panneerselvam KS, Shila S, Sivarajan K, Panneerselvam C. Age-associated deficit of mitochondrial oxidative phosphorylation in skeletal muscle: role of carnitine and lipoic acid. Mol Cell Biochem. 2005;280(1-2):83-89. (PubMed)
23. Kumaran S, Subathra M, Balu M, Panneerselvam C. Supplementation of L-carnitine improves mitochondrial enzymes in heart and skeletal muscle of aged rats. Exp Aging Res. 2005;31(1):55-67. (PubMed)
24. Savitha S, Panneerselvam C. Mitochondrial membrane damage during aging process in rat heart: potential efficacy of L-carnitine and DL alpha lipoic acid. Mech Ageing Dev. 2006;127(4):349-355. (PubMed)
25. Savitha S, Sivarajan K, Haripriya D, Kokilavani V, Panneerselvam C. Efficacy of levo carnitine and alpha lipoic acid in ameliorating the decline in mitochondrial enzymes during aging. Clin Nutr. 2005;24(5):794-800. (PubMed)
26. Sethumadhavan S, Chinnakannu P. Carnitine and lipoic Acid alleviates protein oxidation in heart mitochondria during aging process. Biogerontology. 2006;7(2):101-109. (PubMed)
27. Sundaram K, Panneerselvam KS. Oxidative stress and DNA single strand breaks in skeletal muscle of aged rats: role of carnitine and lipoicacid. Biogerontology. 2006;7(2):111-118. (PubMed)
28. Sethumadhavan S, Chinnakannu P. L-carnitine and alpha-lipoic acid improve age-associated decline in mitochondrial respiratory chain activity of rat heart muscle. J Gerontol A Biol Sci Med Sci. 2006;61(7):650-659. (PubMed)
29. Tamilselvan J, Jayaraman G, Sivarajan K, Panneerselvam C. Age-dependent upregulation of p53 and cytochrome c release and susceptibility to apoptosis in skeletal muscle fiber of aged rats: role of carnitine and lipoic acid. Free Radic Biol Med. 2007;43(12):1656-1669. (PubMed)
30. Aliev G, Liu J, Shenk JC, et al. Neuronal mitochondrial amelioration by feeding acetyl-L-carnitine and lipoic acid to aged rats. J Cell Mol Med. 2009;13(2):320-333. (PubMed)
31. Olson AL, Nelson SE, Rebouche CJ. Low carnitine intake and altered lipid metabolism in infants. Am J Clin Nutr. 1989;49(4):624-628. (PubMed)
32. American Academy of Pediatrics, Committee on Nutrition. Soy protein-based formulas: recommendations for use in infant feeding. Pediatrics. 1998;101(1):148-152.
33. Frigeni M, Balakrishnan B, Yin X, et al. Functional and molecular studies in primary carnitine deficiency. Hum Mutat. 2017;38(12):1684-1699. (PubMed)
34. Magoulas PL, El-Hattab AW. Systemic primary carnitine deficiency: an overview of clinical manifestations, diagnosis, and management. Orphanet J Rare Dis. 2012;7:68. (PubMed)
35. Knottnerus SJG, Bleeker JC, Wust RCI, et al. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev Endocr Metab Disord. 2018;19(1):93-106. (PubMed)
36. Pons R, De Vivo DC. Primary and secondary carnitine deficiency syndromes. J Child Neurol. 1995;10 Suppl 2:S8-24. (PubMed)
37. Gregory MJ, Schwartz GJ. Diagnosis and treatment of renal tubular disorders. Semin Nephrol. 1998;18(3):317-329. (PubMed)
38. Calvani M, Benatti P, Mancinelli A, et al. Carnitine replacement in end-stage renal disease and hemodialysis. Ann N Y Acad Sci. 2004;1033:52-66. (PubMed)
39. Stanley CA. Carnitine deficiency disorders in children. Ann N Y Acad Sci. 2004;1033:42-51. (PubMed)
40. El-Gharbawy A, Vockley J. Inborn errors of metabolism with myopathy: defects of fatty acid oxidation and the carnitine shuttle system. Pediatr Clin North Am. 2018;65(2):317-335. (PubMed)
41. Food and Nutrition Board, Institute of Medicine. Vitamin C. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids. Washington D.C.: National Academy Press; 2000:95-185. (National Academy Press)
42. Ringseis R, Keller J, Eder K. Mechanisms underlying the anti-wasting effect of L-carnitine supplementation under pathologic conditions: evidence from experimental and clinical studies. Eur J Nutr. 2013;52(5):1421-1442. (PubMed)
43. Xu Y, Jiang W, Chen G, et al. L-carnitine treatment of insulin resistance: A systematic review and meta-analysis. Adv Clin Exp Med. 2017;26(2):333-338. (PubMed)
44. Vidal-Casariego A, Burgos-Pelaez R, Martinez-Faedo C, et al. Metabolic effects of L-carnitine on type 2 diabetes mellitus: systematic review and meta-analysis. Exp Clin Endocrinol Diabetes. 2013;121(4):234-238. (PubMed)
45. Asadi M, Rahimlou M, Shishehbor F, Mansoori A. The effect of l-carnitine supplementation on lipid profile and glycaemic control in adults with cardiovascular risk factors: A systematic review and meta-analysis of randomized controlled clinical trials. Clin Nutr. 2019; [Epub ahead of print]. (PubMed)
46. Parvanova A, Trillini M, Podesta MA, et al. Blood pressure and metabolic effects of acetyl-l-carnitine in type 2 diabetes: DIABASI randomized controlled trial. J Endocr Soc. 2018;2(5):420-436. (PubMed)
47. Davini P, Bigalli A, Lamanna F, Boem A. Controlled study on L-carnitine therapeutic efficacy in post-infarction. Drugs Exp Clin Res. 1992;18(8):355-365. (PubMed)
48. Xue YZ, Wang LX, Liu HZ, Qi XW, Wang XH, Ren HZ. L-carnitine as an adjunct therapy to percutaneous coronary intervention for non-ST elevation myocardial infarction. Cardiovasc Drugs Ther. 2007;21(6):445-448. (PubMed)
49. Iyer R, Gupta A, Khan A, Hiremath S, Lokhandwala Y. Does left ventricular function improve with L-carnitine after acute myocardial infarction? J Postgrad Med. 1999;45(2):38-41. (PubMed)
50. Tarantini G, Scrutinio D, Bruzzi P, Boni L, Rizzon P, Iliceto S. Metabolic treatment with L-carnitine in acute anterior ST segment elevation myocardial infarction. A randomized controlled trial. Cardiology. 2006;106(4):215-223. (PubMed)
51. DiNicolantonio JJ, Lavie CJ, Fares H, Menezes AR, O'Keefe JH. L-carnitine in the secondary prevention of cardiovascular disease: systematic review and meta-analysis. Mayo Clin Proc. 2013;88(6):544-551. (PubMed)
52. Trupp RJ, Abraham WT. Congestive heart failure. In: Rakel RE, Bope ET, eds. Conn's Current Therapy. 54th ed. New York: W.B. Sunders Company; 2002:306-313.
53. Ruiz M, Labarthe F, Fortier A, et al. Circulating acylcarnitine profile in human heart failure: a surrogate of fatty acid metabolic dysregulation in mitochondria and beyond. Am J Physiol Heart Circ Physiol. 2017;313(4):H768-h781. (PubMed)
54. Ueland T, Svardal A, Oie E, et al. Disturbed carnitine regulation in chronic heart failure--increased plasma levels of palmitoyl-carnitine are associated with poor prognosis. Int J Cardiol. 2013;167(5):1892-1899. (PubMed)
55. Yoshihisa A, Watanabe S, Yokokawa T, et al. Associations between acylcarnitine to free carnitine ratio and adverse prognosis in heart failure patients with reduced or preserved ejection fraction. ESC Heart Fail. 2017;4(3):360-364. (PubMed)
56. Song X, Qu H, Yang Z, Rong J, Cai W, Zhou H. Efficacy and safety of L-carnitine treatment for chronic heart failure: a meta-analysis of randomized controlled trials. Biomed Res Int. 2017;2017:6274854. (PubMed)
57. US Department of Health & Human Services, National Heart Lung and Blood Institute. Angina. Available at: https://www.nhlbi.nih.gov/health-topics/angina. Accessed 7/1/19.
58. Strand E, Pedersen ER, Svingen GF, et al. Serum acylcarnitines and risk of cardiovascular death and acute myocardial infarction in patients with stable angina pectoris. J Am Heart Assoc. 2017;6(2). (PubMed)
59. Cacciatore L, Cerio R, Ciarimboli M, et al. The therapeutic effect of L-carnitine in patients with exercise-induced stable angina: a controlled study. Drugs Exp Clin Res. 1991;17(4):225-235. (PubMed)
60. Cherchi A, Lai C, Angelino F, et al. Effects of L-carnitine on exercise tolerance in chronic stable angina: a multicenter, double-blind, randomized, placebo controlled crossover study. Int J Clin Pharmacol Ther Toxicol. 1985;23(10):569-572. (PubMed)
61. Iyer RN, Khan AA, Gupta A, Vajifdar BU, Lokhandwala YY. L-carnitine moderately improves the exercise tolerance in chronic stable angina. J Assoc Physicians India. 2000;48(11):1050-1052. (PubMed)
62. Bartels GL, Remme WJ, Pillay M, Schonfeld DH, Kruijssen DA. Effects of L-propionylcarnitine on ischemia-induced myocardial dysfunction in men with angina pectoris. Am J Cardiol. 1994;74(2):125-130. (PubMed)
63. Mills JL. Peripheral arterial disease. In: Rakel RE, Bope ET, eds. Conn's Current Therapy. 54th ed. New York: W.B. Sunders Company; 2002:340-343.
64. Brevetti G, Perna S, Sabba C, Martone VD, Condorelli M. Propionyl-L-carnitine in intermittent claudication: double-blind, placebo-controlled, dose titration, multicenter study. J Am Coll Cardiol. 1995;26(6):1411-1416. (PubMed)
65. Brevetti G, Diehm C, Lambert D. European multicenter study on propionyl-L-carnitine in intermittent claudication. J Am Coll Cardiol. 1999;34(5):1618-1624. (PubMed)
66. Hiatt WR. Carnitine and peripheral arterial disease. Ann N Y Acad Sci. 2004;1033:92-98. (PubMed)
67. Luo T, Li J, Li L, et al. A study on the efficacy and safety assessment of propionyl-L-carnitine tablets in treatment of intermittent claudication. Thromb Res. 2013;132(4):427-432. (PubMed)
68. Santo SS, Sergio N, Luigi DP, et al. Effect of PLC on functional parameters and oxidative profile in type 2 diabetes-associated PAD. Diabetes Res Clin Pract. 2006;72(3):231-237. (PubMed)
69. Brass EP, Koster D, Hiatt WR, Amato A. A systematic review and meta-analysis of propionyl-L-carnitine effects on exercise performance in patients with claudication. Vasc Med. 2013;18(1):3-12. (PubMed)
70. Delaney CL, Spark JI, Thomas J, Wong YT, Chan LT, Miller MD. A systematic review to evaluate the effectiveness of carnitine supplementation in improving walking performance among individuals with intermittent claudication. Atherosclerosis. 2013;229(1):1-9. (PubMed)
71. Hatanaka Y, Higuchi T, Akiya Y, et al. Prevalence of carnitine deficiency and decreased carnitine levels in patients on hemodialysis. Blood Purif. 2019;47 Suppl 2:38-44. (PubMed)
72. Kalim S, Clish CB, Wenger J, et al. A plasma long-chain acylcarnitine predicts cardiovascular mortality in incident dialysis patients. J Am Heart Assoc. 2013;2(6):e000542. (PubMed)
73. Chen Y, Abbate M, Tang L, et al. L-Carnitine supplementation for adults with end-stage kidney disease requiring maintenance hemodialysis: a systematic review and meta-analysis. Am J Clin Nutr. 2014;99(2):408-422. (PubMed)
74. National Kidney Foundation; Kidney Disease Outcomes Quality Initiative. Clinical practice guidelines for nutrition in chronic renal failure. Am J Kidney Dis. 2000;35(6 Suppl 2):S1-140. (PubMed)
75. National Kidney Foundation; Kidney Disease Outcomes Quality Initiative. Clinical practice guidelines and clinical practice recommendations for anemia in chronic kidney disease. Am J Kidney Dis. 2006;47(5 Suppl 3):S11-145. (PubMed)
76. Natural Medicines. Carnitine: professional handout/administration and dosing. Available at: https://naturalmedicines-therapeuticresearch-com. Accessed 7/1/19.
77. Margolis AM, Heverling H, Pham PA, Stolbach A. A review of the toxicity of HIV medications. J Med Toxicol. 2014;10(1):26-39. (PubMed)
78. Moyle GJ, Sadler M. Peripheral neuropathy with nucleoside antiretrovirals: risk factors, incidence and management. Drug Saf. 1998;19(6):481-494. (PubMed)
79. Scarpini E, Sacilotto G, Baron P, Cusini M, Scarlato G. Effect of acetyl-L-carnitine in the treatment of painful peripheral neuropathies in HIV+ patients. J Peripher Nerv Syst. 1997;2(3):250-252. (PubMed)
80. Hart AM, Wilson AD, Montovani C, et al. Acetyl-l-carnitine: a pathogenesis based treatment for HIV-associated antiretroviral toxic neuropathy. Aids. 2004;18(11):1549-1560. (PubMed)
81. Herzmann C, Johnson MA, Youle M. Long-term effect of acetyl-L-carnitine for antiretroviral toxic neuropathy. HIV Clin Trials. 2005;6(6):344-350. (PubMed)
82. Osio M, Muscia F, Zampini L, et al. Acetyl-l-carnitine in the treatment of painful antiretroviral toxic neuropathy in human immunodeficiency virus patients: an open label study. J Peripher Nerv Syst. 2006;11(1):72-76. (PubMed)
83. Valcour V, Yeh TM, Bartt R, et al. Acetyl-l-carnitine and nucleoside reverse transcriptase inhibitor-associated neuropathy in HIV infection. HIV Med. 2009;10(2):103-110. (PubMed)
84. Youle M, Osio M. A double-blind, parallel-group, placebo-controlled, multicentre study of acetyl L-carnitine in the symptomatic treatment of antiretroviral toxic neuropathy in patients with HIV-1 infection. HIV Med. 2007;8(4):241-250. (PubMed)
85. Tesfaye S, Selvarajah D. Advances in the epidemiology, pathogenesis and management of diabetic peripheral neuropathy. Diabetes Metab Res Rev. 2012;28 Suppl 1:8-14. (PubMed)
86. Dy SM, Bennett WL, Sharma R, et al. AHRQ Comparative Effectiveness Reviews. Preventing complications and treating symptoms of diabetic peripheral neuropathy. Rockville (MD): Agency for Healthcare Research and Quality (US); Rockville (MD): Agency for Healthcare Research and Quality (US); Mar 2017. Report No.: 17-EHC005-EF. (PubMed)
87. Rolim LC, da Silva EM, Flumignan RL, Abreu MM, Dib SA. Acetyl-L-carnitine for the treatment of diabetic peripheral neuropathy. Cochrane Database Syst Rev. 2019;6:Cd011265. (PubMed)
88. Sima AA, Calvani M, Mehra M, Amato A. Acetyl-L-carnitine improves pain, nerve regeneration, and vibratory perception in patients with chronic diabetic neuropathy: an analysis of two randomized placebo-controlled trials. Diabetes Care. 2005;28(1):89-94. (PubMed)
89. Li S, Chen X, Li Q, et al. Effects of acetyl-L-carnitine and methylcobalamin for diabetic peripheral neuropathy: A multicenter, randomized, double-blind, controlled trial. J Diabetes Investig. 2016;7(5):777-785. (PubMed)
90. Campone M, Berton-Rigaud D, Joly-Lobbedez F, et al. A double-blind, randomized phase II study to evaluate the safety and efficacy of acetyl-L-carnitine in the prevention of sagopilone-induced peripheral neuropathy. Oncologist. 2013;18(11):1190-1191. (PubMed)
91. Hershman DL, Unger JM, Crew KD, et al. Randomized double-blind placebo-controlled trial of acetyl-L-carnitine for the prevention of taxane-induced neuropathy in women undergoing adjuvant breast cancer therapy. J Clin Oncol. 2013;31(20):2627-2633. (PubMed)
92. Hershman DL, Unger JM, Crew KD, et al. Two-year trends of taxane-induced neuropathy in women enrolled in a randomized trial of acetyl-L-carnitine (SWOG S0715). J Natl Cancer Inst. 2018;110(6):669-676. (PubMed)
93. Sun Y, Shu Y, Liu B, et al. A prospective study to evaluate the efficacy and safety of oral acetyl-L-carnitine for the treatment of chemotherapy-induced peripheral neuropathy. Exp Ther Med. 2016;12(6):4017-4024. (PubMed)
94. van Dam DG, Beijers AJ, Vreugdenhil G. Acetyl-L-carnitine undervalued in the treatment of chemotherapy-induced peripheral neuropathy? Acta Oncol. 2016;55(12):1495-1497. (PubMed)
95. Veronese N, Stubbs B, Solmi M, Ajnakina O, Carvalho AF, Maggi S. Acetyl-L-carnitine supplementation and the treatment of depressive symptoms: a systematic review and meta-analysis. Psychosom Med. 2018;80(2):154-159. (PubMed)
96. Meister R, von Wolff A, Mohr H, et al. Comparative safety of pharmacologic treatments for persistent depressive disorder: a systematic review and network meta-analysis. PLoS One. 2016;11(5):e0153380. (PubMed)
97. Cristofano A, Sapere N, La Marca G, et al. Serum levels of acyl-carnitines along the continuum from normal to Alzheimer's dementia. PLoS One. 2016;11(5):e0155694. (PubMed)
98. Pettegrew JW, Klunk WE, Panchalingam K, Kanfer JN, McClure RJ. Clinical and neurochemical effects of acetyl-L-carnitine in Alzheimer's disease. Neurobiol Aging. 1995;16(1):1-4. (PubMed)
99. Spagnoli A, Lucca U, Menasce G, et al. Long-term acetyl-L-carnitine treatment in Alzheimer's disease. Neurology. 1991;41(11):1726-1732. (PubMed)
100. Sano M, Bell K, Cote L, et al. Double-blind parallel design pilot study of acetyl levocarnitine in patients with Alzheimer's disease. Arch Neurol. 1992;49(11):1137-1141. (PubMed)
101. Thal LJ, Carta A, Clarke WR, et al. A 1-year multicenter placebo-controlled study of acetyl-L-carnitine in patients with Alzheimer's disease. Neurology. 1996;47(3):705-711. (PubMed)
102. Thal LJ, Calvani M, Amato A, Carta A. A 1-year controlled trial of acetyl-l-carnitine in early-onset AD. Neurology. 2000;55(6):805-810. (PubMed)
103. Brooks JO, 3rd, Yesavage JA, Carta A, Bravi D. Acetyl L-carnitine slows decline in younger patients with Alzheimer's disease: a reanalysis of a double-blind, placebo-controlled study using the trilinear approach. Int Psychogeriatr. 1998;10(2):193-203. (PubMed)
104. Hudson S, Tabet N. Acetyl-L-carnitine for dementia. Cochrane Database Syst Rev. 2003(2):Cd003158. (PubMed)
105. Montgomery SA, Thal LJ, Amrein R. Meta-analysis of double blind randomized controlled clinical trials of acetyl-L-carnitine versus placebo in the treatment of mild cognitive impairment and mild Alzheimer's disease. Int Clin Psychopharmacol. 2003;18(2):61-71. (PubMed)
106. Wijdicks EF. Hepatic encephalopathy. N Engl J Med. 2016;375(17):1660-1670. (PubMed)
107. Inglis JE, Lin PJ, Kerns SL, et al. Nutritional interventions for treating cancer-related fatigue: a qualitative review. Nutr Cancer. 2019;71(1):21-40. (PubMed)
108. Marx W, Teleni L, Opie RS, et al. Efficacy and effectiveness of carnitine supplementation for cancer-related fatigue: a systematic literature review and meta-analysis. Nutrients. 2017;9(11). (PubMed)
109. Jeulin C, Lewin LM. Role of free L-carnitine and acetyl-L-carnitine in post-gonadal maturation of mammalian spermatozoa. Hum Reprod Update. 1996;2(2):87-102. (PubMed)
110. Matalliotakis I, Koumantaki Y, Evageliou A, Matalliotakis G, Goumenou A, Koumantakis E. L-carnitine levels in the seminal plasma of fertile and infertile men: correlation with sperm quality. Int J Fertil Womens Med. 2000;45(3):236-240. (PubMed)
111. Lenzi A, Lombardo F, Sgro P, et al. Use of carnitine therapy in selected cases of male factor infertility: a double-blind crossover trial. Fertil Steril. 2003;79(2):292-300. (PubMed)
112. Lenzi A, Sgro P, Salacone P, et al. A placebo-controlled double-blind randomized trial of the use of combined l-carnitine and l-acetyl-carnitine treatment in men with asthenozoospermia. Fertil Steril. 2004;81(6):1578-1584. (PubMed)
113. Balercia G, Regoli F, Armeni T, Koverech A, Mantero F, Boscaro M. Placebo-controlled double-blind randomized trial on the use of L-carnitine, L-acetylcarnitine, or combined L-carnitine and L-acetylcarnitine in men with idiopathic asthenozoospermia. Fertil Steril. 2005;84(3):662-671. (PubMed)
114. Buhling K, Schumacher A, Eulenburg CZ, Laakmann E. Influence of oral vitamin and mineral supplementation on male infertility: a meta-analysis and systematic review. Reprod Biomed Online. 2019;39(2):269-279. (PubMed)
115. Fried LP, Ferrucci L, Darer J, Williamson JD, Anderson G. Untangling the concepts of disability, frailty, and comorbidity: implications for improved targeting and care. J Gerontol A Biol Sci Med Sci. 2004;59(3):255-263. (PubMed)
116. Vermeiren S, Vella-Azzopardi R, Beckwee D, et al. Frailty and the prediction of negative health outcomes: a meta-analysis. J Am Med Dir Assoc. 2016;17(12):1163.e1161-1163.e1117. (PubMed)
117. Crentsil V. Mechanistic contribution of carnitine deficiency to geriatric frailty. Ageing Res Rev. 2010;9(3):265-268. (PubMed)
118. Badrasawi M, Shahar S, Zahara AM, Nor Fadilah R, Singh DK. Efficacy of L-carnitine supplementation on frailty status and its biomarkers, nutritional status, and physical and cognitive function among prefrail older adults: a double-blind, randomized, placebo-controlled clinical trial. Clin Interv Aging. 2016;11:1675-1686. (PubMed)
119. Dhillon RJ, Hasni S. Pathogenesis and management of sarcopenia. Clin Geriatr Med. 2017;33(1):17-26. (PubMed)
120. Ebadi M, Montano-Loza AJ. Clinical relevance of skeletal muscle abnormalities in patients with cirrhosis. Dig Liver Dis. 2019;51(11):1493-1499. (PubMed)
121. Kim SH, Shin MJ, Shin YB, Kim KU. Sarcopenia associated with chronic obstructive pulmonary disease. J Bone Metab. 2019;26(2):65-74. (PubMed)
122. Melenovsky V, Hlavata K, Sedivy P, et al. Skeletal muscle abnormalities and iron deficiency in chronic heart failure: An Exercise (31)P Magnetic Resonance Spectroscopy Study of Calf Muscle. Circ Heart Fail. 2018;11(9):e004800. (PubMed)
123. Sawicka AK, Hartmane D, Lipinska P, Wojtowicz E, Lysiak-Szydlowska W, Olek RA. L-carnitine supplementation in older women. A pilot study on aging skeletal muscle mass and function. Nutrients. 2018;10(2). (PubMed)
124. Ohara M, Ogawa K, Suda G, et al. L-Carnitine suppresses loss of skeletal muscle mass in patients with liver cirrhosis. Hepatol Commun. 2018;2(8):906-918. (PubMed)
125. Nakanishi H, Kurosaki M, Tsuchiya K, et al. L-carnitine reduces muscle cramps in patients with cirrhosis. Clin Gastroenterol Hepatol. 2015;13(8):1540-1543. (PubMed)
126. Hiraoka A, Kiguchi D, Ninomiya T, et al. Can L-carnitine supplementation and exercise improve muscle complications in patients with liver cirrhosis who receive branched-chain amino acid supplementation? Eur J Gastroenterol Hepatol. 2019;31(7):878-884. (PubMed)
127. Imbe A, Tanimoto K, Inaba Y, et al. Effects of L-carnitine supplementation on the quality of life in diabetic patients with muscle cramps. Endocr J. 2018;65(5):521-526. (PubMed)
128. Lynch KE, Feldman HI, Berlin JA, Flory J, Rowan CG, Brunelli SM. Effects of L-carnitine on dialysis-related hypotension and muscle cramps: a meta-analysis. Am J Kidney Dis. 2008;52(5):962-971. (PubMed)
129. Fielding R, Riede L, Lugo JP, Bellamine A. L-carnitine supplementation in recovery after exercise. Nutrients. 2018;10(3). (PubMed)
130. Smith WA, Fry AC, Tschume LC, Bloomer RJ. Effect of glycine propionyl-L-carnitine on aerobic and anaerobic exercise performance. Int J Sport Nutr Exerc Metab. 2008;18(1):19-36. (PubMed)
131. Novakova K, Kummer O, Bouitbir J, et al. Effect of L-carnitine supplementation on the body carnitine pool, skeletal muscle energy metabolism and physical performance in male vegetarians. Eur J Nutr. 2016;55(1):207-217. (PubMed)
132. Rebouche CJ. Carnitine. In: Shils ME, Olson JA, Shike M, Ross AC, eds. Nutrition in Health and Disease. 9th ed. Baltimore: Lippincott Williams & Wilkins; 1999:505-512.
133. Hendler SS, Rorvik DR. Acetyl-L-carnitine. PDR for Nutritional Supplements. 2nd ed. Montvale: Thomson Reuters; 2008:13-16.
134. Zeiler FA, Sader N, Gillman LM, West M. Levocarnitine induced seizures in patients on valproic acid: A negative systematic review. Seizure. 2016;36:36-39. (PubMed)
135. Natural Medicines. Carnitine: professional handout/drug interactions. Available at: https://naturalmedicines-therapeuticresearch-com. Accessed 7/1/19.
Coenzyme Q10
Contents
Summary
- Coenzyme Q10 is a fat-soluble compound that is synthesized by the body and can be obtained from the diet. (More information)
- Coenzyme Q10 plays a central role in mitochondrial oxidative phosphorylation and the production of adenosine triphosphate (ATP). It also functions as an antioxidant in cell membranes and lipoproteins. (More information)
- Endogenous synthesis and dietary intake provide sufficient coenzyme Q10 to prevent deficiency in healthy people, although coenzyme Q10 concentrations in tissues decline with age. (More information)
- Oral supplementation of coenzyme Q10 increases coenzyme Q10 concentrations in plasma and lipoproteins, but it is unclear whether concentrations in peripheral tissues are increased, especially in healthy individuals. (More information)
- Oral high-dose coenzyme Q10 is usually effective to treat mitochondrial disorders that are caused by mutations in coenzyme Q10 biosynthetic genes. (More information)
- There is some evidence to suggest that coenzyme Q10 supplementation may be a useful adjunct to conventional medical therapy for congestive heart failure and in patients undergoing coronary artery bypass graft surgery. (More information)
- There are currently no proven therapeutic benefits of coenzyme Q10 supplementation in diabetes mellitus, neurodegenerative diseases, inherited ataxias, or breast cancer. (More information)
- Coenzyme Q10 supplementation does not appear to improve athletic performance. (More information)
- Although coenzyme Q10 supplements are relatively safe, they may decrease the anticoagulant efficacy of warfarin. (More information)
- The use of cholesterol-lowering medications called statins can decrease circulating coenzyme Q10 concentrations. However, there is no evidence that this causes any adverse side effects in statin-treated patients. (More information)
Introduction
Coenzyme Q10 is a member of the ubiquinone family of compounds. All animals, including humans, can synthesize ubiquinones, hence, coenzyme Q10 is not considered a vitamin (1). The name ubiquinone refers to the ubiquitous presence of these compounds in living organisms and their chemical structure, which contains a functional group known as a benzoquinone. Ubiquinones are fat-soluble molecules with anywhere from 1 to 12 isoprene (5-carbon) units. The ubiquinone found in humans, ubidecaquinone or coenzyme Q10, has a "tail" of 10 isoprene units (a total of 50 carbon atoms) attached to its benzoquinone "head" (Figure 1) (1).
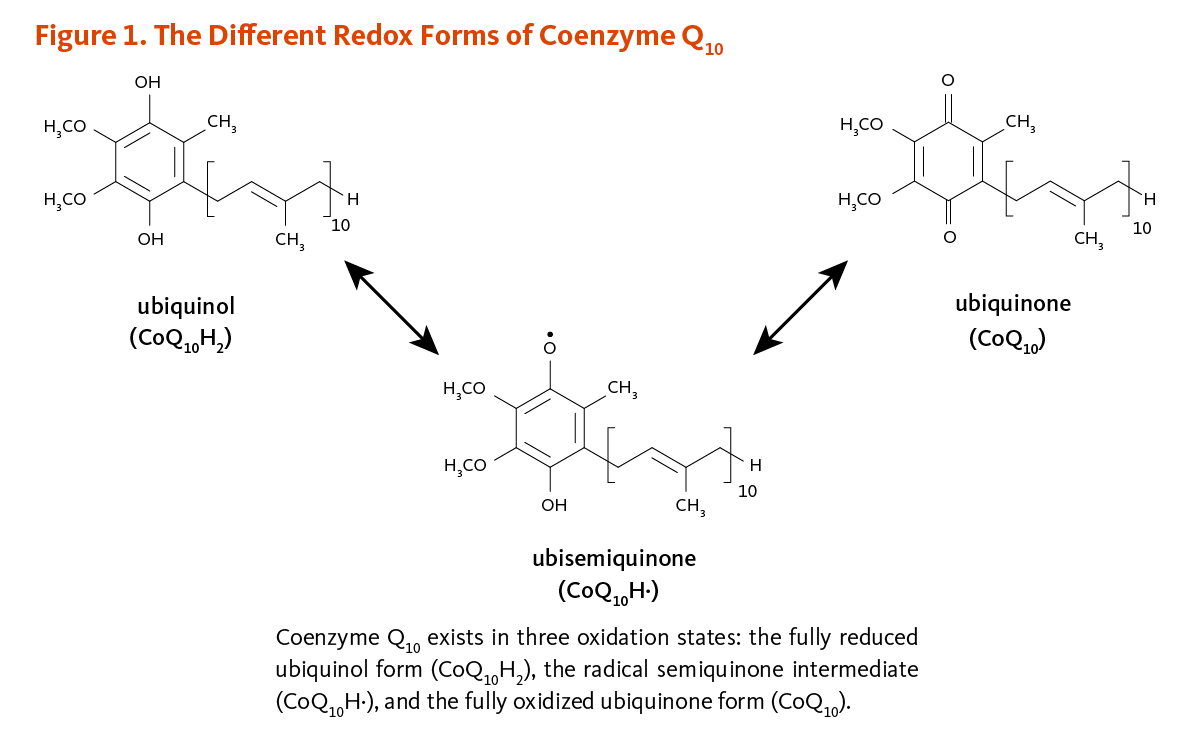
Biological Activities
Coenzyme Q10 is soluble in lipids (fats) and is found in virtually all cell membranes, including mitochondrial membranes. The ability of the benzoquinone head group of coenzyme Q10 to accept and donate electrons is a critical feature to its function. Coenzyme Q10 can exist in three oxidation states (Figure 1): (i) the fully reduced ubiquinol form, CoQ10H2; (ii) the radical semiquinone intermediate, CoQ10H·; and (iii) the fully oxidized ubiquinone form, CoQ10.
Mitochondrial ATP synthesis
The conversion of energy from carbohydrates and fats to ATP, the form of energy used by cells, requires the presence of coenzyme Q10 in the inner mitochondrial membrane. As part of the mitochondrial electron transport chain, coenzyme Q10 accepts electrons from reducing equivalents generated during fatty acid and glucose metabolism and then transfers them to electron acceptors. At the same time, coenzyme Q10 contributes to transfer protons (H+) from the mitochondrial matrix to the intermembrane space, creating a proton gradient across the inner mitochondrial membrane. The energy released when the protons flow back into the mitochondrial interior is used to form ATP (Figure 2) (1). In addition to its role in ATP synthesis, mitochondrial coenzyme Q10 mediates the oxidation of dihydroorotate to orotate in the de novo pyrimidine synthesis.
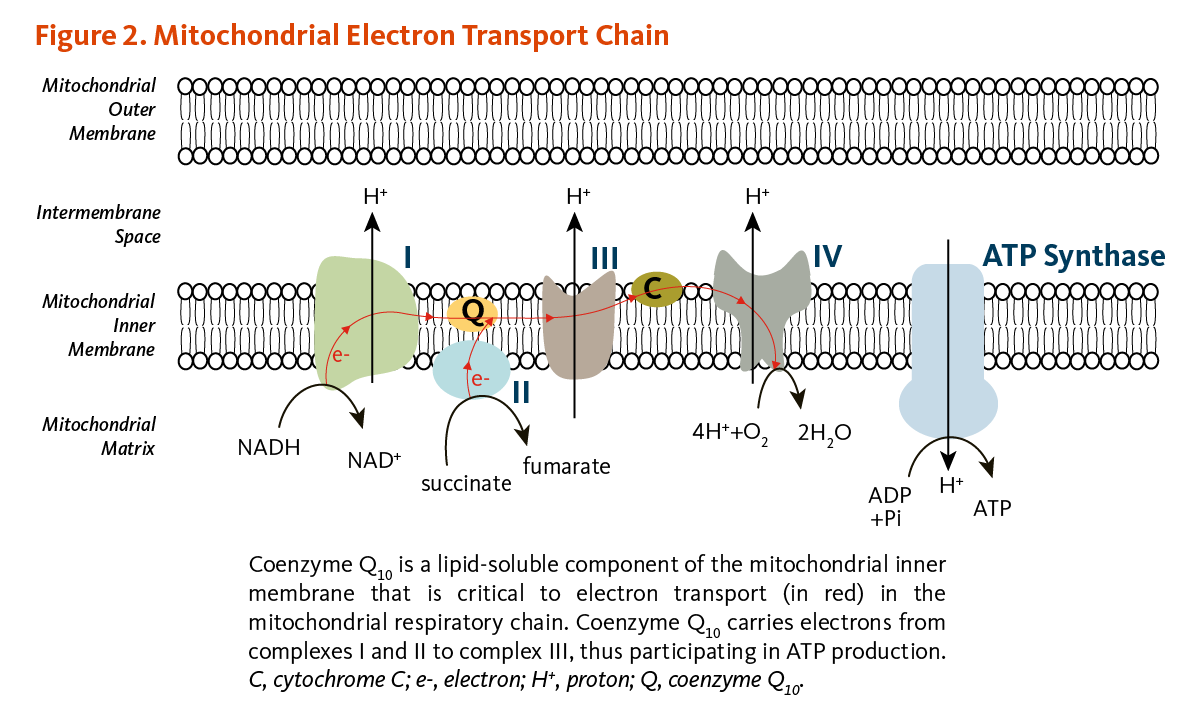
Lysosomal function
Lysosomes are organelles within cells that are specialized for the digestion of cellular debris. The digestive enzymes within lysosomes function optimally at an acidic pH, meaning they require a permanent supply of protons. The lysosomal membranes that separate those digestive enzymes from the rest of the cell contain relatively high concentrations of coenzyme Q10. Research suggests that coenzyme Q10 plays an important role in the transport of protons across lysosomal membranes to maintain the optimal pH (2, 3).
Antioxidant functions
In its reduced form (CoQ10H2), coenzyme Q10 is an effective fat-soluble antioxidant that protects cell membranes and lipoproteins from oxidation. The presence of a significant amount of CoQ10H2 in cell membranes, along with enzymes capable of reducing oxidized CoQ10 back to CoQ10H2 (i.e., NAD(P)H oxidoreductases), supports the idea that CoQ10H2 is an important cellular antioxidant (4). CoQ10H2 has been found to inhibit lipid peroxidation when cell membranes and low-density lipoproteins (LDL) are exposed to oxidizing conditions. When LDL is oxidized, CoQ10H2 is the first antioxidant consumed. In isolated mitochondria, coenzyme Q10 can protect membrane proteins and mitochondrial DNA from the oxidative damage that accompanies lipid peroxidation (5). Moreover, when present, CoQ10H2 was found to limit the formation of oxidized lipids and the consumption of α-tocopherol (a form of vitamin E with antioxidant properties) (6). Indeed, in addition to neutralizing free radicals directly, CoQ10H2 is capable of regenerating antioxidants like α-tocopherol and ascorbate (vitamin C) (4). Finally, the role of coenzyme Q10 as an antioxidant is also exemplified by recent evidence showing that mitochondrial coenzyme Q10 deficiency causes an increased production of mitochondrial superoxide radical anion (O2•–) which might be driving insulin resistance in adipose and muscle tissues (7).
Nutrient interactions
Vitamin E
α-Tocopherol (vitamin E) and coenzyme Q10 are the principal fat-soluble antioxidants in membranes and lipoproteins. When α-tocopherol (α-TOH) neutralizes a free radical, such as a lipid peroxyl radical (LOO·), it becomes oxidized itself, forming α-TO·, which can in turn promote the oxidation of lipoproteins under certain conditions in the test tube, thus propagating a chain reaction. However, when the reduced form of coenzyme Q10 (CoQ10H2) reacts with α-TO·, α-TOH is regenerated and the semiquinone radical (CoQ10H·) is formed. It is possible for CoQ10H· to react with oxygen (O2) to produce superoxide anion radical (O2·-), which is a less reactive pro-oxidant than LOO·. However, CoQ10H· can also reduce α-TO· back to α-TOH, resulting in the formation of fully oxidized coenzyme Q10 (CoQ10), which does not react with O2 to form O2·- (Figure 3) (6, 8).
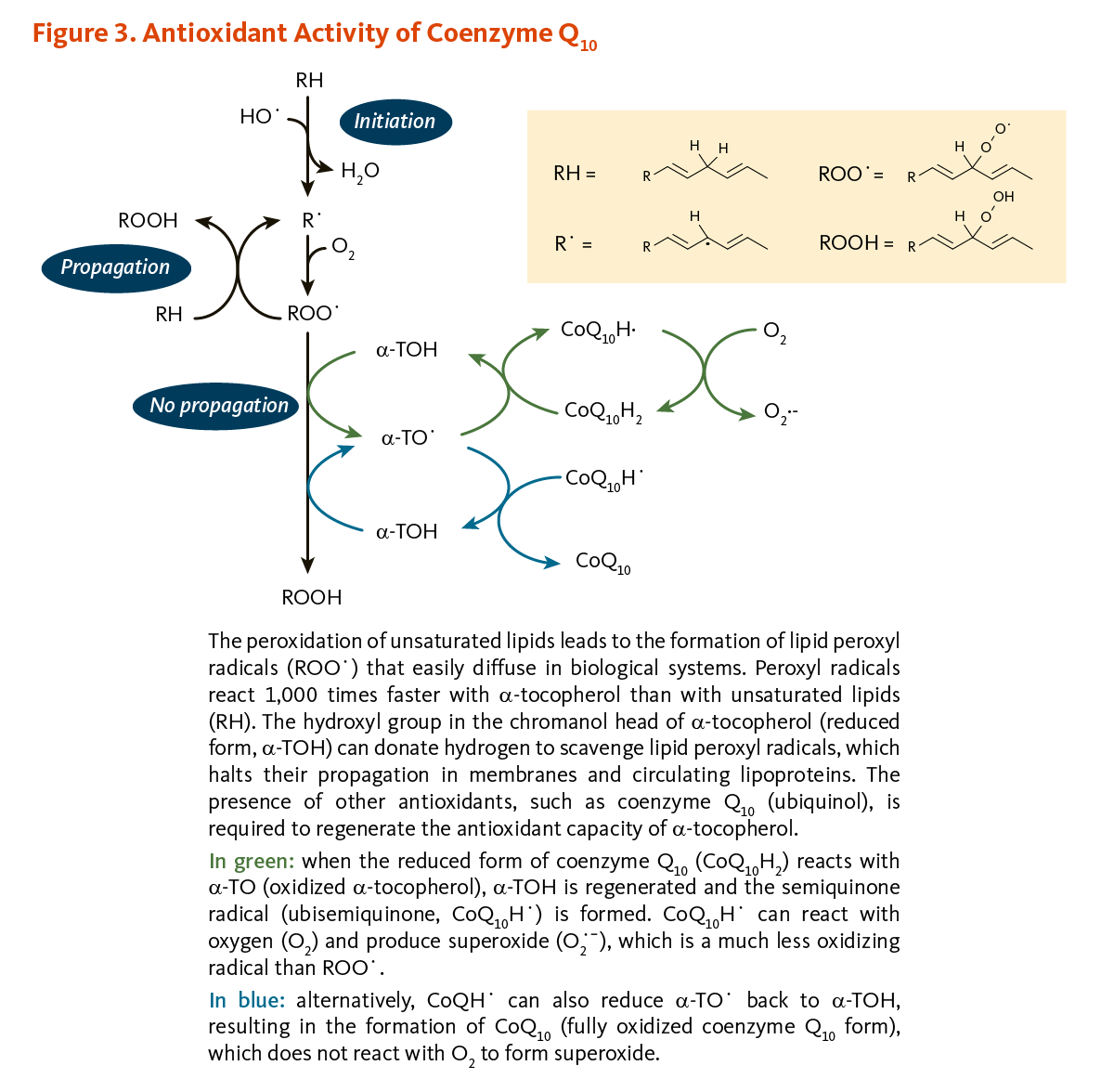
Deficiency
Coenzyme Q10 deficiency has not been described in the general population, so it is generally assumed that normal biosynthesis, with or without a varied diet, provides sufficient coenzyme Q10 to sustain energy production in healthy individuals (9).
Primary coenzyme Q10 deficiency is a rare genetic disorder caused by mutations in genes involved in coenzyme Q10 biosynthetic pathway. To date, mutations in at least nine of these genes have been identified (1). As a result, primary coenzyme Q10 deficiency is a clinically heterogeneous disorder that includes five major phenotypes: (i) severe infantile multi-systemic disease, (ii) encephalomyopathy, (iii) cerebellar ataxia, (iv) isolated myopathy, and (v) nephrotic syndrome. Whereas most mitochondrial respiratory chain disorders are hardly amenable to treatments, oral coenzyme Q10 supplementation has been shown to improve muscular symptoms in some (yet not all) patients with primary coenzyme Q10 deficiency (10). Neurological symptoms in patients with cerebellar ataxia are only partially relieved by coenzyme Q10 (CoQ10H2) supplementation (10).
Secondary coenzyme Q10 deficiency results from mutations or deletions in genes that are not directly related to coenzyme Q10 biosynthetic pathway. Evidence of secondary coenzyme Q10 deficiency has been reported in several mitochondrial disorders, such as mitochondrial DNA depletion syndrome, Kearns-Sayre syndrome, or multiple acyl-CoA dehydrogenase deficiency (MADD) (10). Secondary coenzyme Q10 deficiency has also been identified in non-mitochondrial disorders, such as cardiofaciocutaneous syndrome and Niemann-Pick-type C disease (11). Because the therapeutic potential of supplemental coenzyme Q10 is limited to its capacity to restore electron transfer in a defective mitochondrial respiratory chain and/or to increase antioxidant defense, patients with secondary coenzyme Q10 deficiency may fail to respond to supplementation (see Disease Treatment).
Coenzyme Q10 concentrations have been found to decline gradually with age in a number of different tissues (5, 12), but it is unclear whether this age-associated decline constitutes a deficiency (see Disease Prevention) (13). Decreased plasma concentrations of coenzyme Q10 have been observed in individuals with diabetes mellitus, cancer, and congestive heart failure (see Disease Treatment). Lipid-lowering medications that inhibit the activity of 3-hydroxy-3-methylglutaryl (HMG)-coenzyme A (CoA) reductase (statins), a critical enzyme in both cholesterol and coenzyme Q10 biosynthesis, decrease plasma coenzyme Q10 concentrations (see HMG-CoA reductase inhibitors [statins]), although it remains unproven that this has any clinical implications.
Disease Prevention
Aging
According to the free radical and mitochondrial theories of aging, oxidative damage of cell structures by reactive oxygen species (ROS) plays an important role in the functional declines that accompany aging (14). ROS are generated by mitochondria as a byproduct of ATP production. If not neutralized by antioxidants, ROS may damage mitochondria over time, causing them to function less efficiently and to generate more damaging ROS in a self-perpetuating cycle. Coenzyme Q10 plays an important role in mitochondrial ATP synthesis and functions as an antioxidant in mitochondrial membranes (see Biological Activities). One of the hallmarks of aging is a decline in energy metabolism in many tissues, especially liver, heart, and skeletal muscle. Tissue concentrations of coenzyme Q10 have been found to decline with age, thereby accompanying age-related declines in energy metabolism (12). Early animal studies have not been able to demonstrate an effect of lifelong dietary supplementation with coenzyme Q10 on the lifespan of rats or mice (15-17). Nonetheless, more recent studies have suggested that supplemental coenzyme Q10 could promote mitochondrial biogenesis and respiration (18, 19) and delay senescence in transgenic mice (19). Presently, there is limited scientific evidence to suggest that coenzyme Q10 supplementation prolongs life or prevents age-related functional declines in humans. In a small randomized controlled trial, elderly individuals (>70 years) who received a combination of selenium (100 µg/day) and coenzyme Q10 (200 mg/day) for four years reported an improvement in vitality, physical performance, and quality of life (20). Further, a 12-year follow-up of these participants showed a reduction in cardiovascular mortality with supplemental selenium and coenzyme Q10 compared to placebo (21).
Atherosclerosis
Oxidative modification of low-density lipoproteins (LDL) in arterial walls is thought to represent an early event leading to the development of atherosclerosis. Reduced coenzyme Q10 (CoQ10H2) inhibits the oxidation of LDL in the test tube (in vitro) and works together with α-tocopherol (α-TOH) to inhibit LDL oxidation by regenerating α-TO· back to α-TOH. In the absence of a co-antioxidant, such as CoQ10H2 or vitamin C, α-TO· can, under certain conditions, promote the oxidation of LDL in vitro (6). Supplementation with coenzyme Q10 increases the concentration of CoQ10H2 in human LDL (22). Studies in apolipoprotein E-deficient mice, an animal model of atherosclerosis, found that coenzyme Q10 supplementation with supra-pharmacological amounts of coenzyme Q10 inhibited lipoprotein oxidation in the blood vessel wall and the formation of atherosclerotic lesions (23). Interestingly, co-supplementation of these mice with α-TOH and coenzyme Q10 was more effective in inhibiting atherosclerosis than supplementation with either α-TOH or coenzyme Q10 alone (24).
Another important step in the development of atherosclerosis is the recruitment of immune cells known as monocytes into the blood vessel walls. This recruitment is dependent in part on monocyte expression of cell adhesion molecules (integrins). Supplementation of 10 healthy men and women with 200 mg/day of coenzyme Q10 for 10 weeks resulted in significant decreases in monocyte expression of integrins, suggesting another potential mechanism for the inhibition of atherosclerosis by coenzyme Q10 (25). Although coenzyme Q10 supplementation shows promise as an inhibitor of LDL oxidation and atherosclerosis, more research is needed to determine whether coenzyme Q10 supplementation can inhibit the development or progression of atherosclerosis in humans.
Disease Treatment
Primary and secondary coenzyme Q10 deficiencies
Inherited coenzyme Q10 deficiencies are rare diseases that are clinically and genetically heterogeneous (see Deficiency). In individuals with primary coenzyme Q10 deficiency, early treatment with high-dose coenzyme Q10 supplementation (10–30 mg/kg/day in children and 1.2–3.0 g/day in adults) may improve the pathological phenotype, yet the effectiveness depends on the type of mutations affecting the coenzyme Q10 biosynthetic pathway (1, 26). Early treatment with pharmacological doses of coenzyme Q10 is essential to limit irreversible organ damage in coenzyme Q10-responsive deficiencies (1).
It is not clear to what extent coenzyme Q10 supplementation might have therapeutic benefit in patients with inherited secondary Q10 deficiencies. For example, multiple acyl-CoA dehydrogenase deficiency (MADD), caused by mutations in genes that impair the activity of enzymes involved in the transfer of electrons from acyl-CoA to coenzyme Q10, is usually responsive to riboflavin monotherapy yet patients with low coenzyme Q10 concentrations might also benefit from co-supplementation with coenzyme Q10 and riboflavin (27). Another study suggested clinical improvements in secondary coenzyme Q10 deficiency with supplemental coenzyme Q10 in patients presenting with ataxia (28). Because the cause of secondary coenzyme Q10 in inherited conditions is generally unknown, it is difficult to predict whether improving coenzyme Q10 status with supplemental coenzyme Q10 would lead to clinical benefits for the patients.
Finally, coenzyme Q10 deficiency can be secondary to the inhibition of HMG-CoA reductase by statin drugs (see Deficiency). A 2015 meta-analysis of six small, randomized controlled trials found no reduction in statin-induced muscle pain with 100 to 400 mg/day of supplemental coenzyme Q10 for one to three months (29). The trials failed to establish a diagnosis of relative coenzyme Q10 deficiency before the intervention started, hence limiting the conclusion of the meta-analysis. While statin therapy may not necessary lead to a reduction in circulating coenzyme Q10 concentrations, further research needs to examine whether secondary coenzyme Q10 deficiency might be predisposing patients to statin-induced myalgia (30).
Cardiovascular disease
Congestive heart failure
Impairment of the heart's ability to pump enough blood for all of the body's needs is known as congestive heart failure. In coronary heart disease (CHD), accumulation of atherosclerotic plaque in the coronary arteries may prevent parts of the cardiac muscle from getting adequate blood supply, ultimately resulting in heart damage and impaired pumping ability. Heart failure can also be caused by myocardial infarction, hypertension, diseases of the heart valves, cardiomyopathy, and congenital heart diseases. Because physical exercise increases the demand on the weakened heart, measures of exercise tolerance are frequently used to monitor the severity of heart failure. Echocardiography is also used to determine the left ventricular ejection fraction, an objective measure of the heart's pumping ability (31).
A study of 1,191 heart failure patients found that low plasma coenzyme Q10 concentration was a good biomarker of advanced heart disease (32). A number of small intervention trials that administered supplemental coenzyme Q10 to congestive heart failure patients have been conducted. A 2014 literature review identified seven small randomized controlled trials examining the effect of coenzyme Q10 supplementation (60-200 mg/day for ≤3 months in most trials) in heart failure patients (33). Pooling data from some of the trials showed an increase in serum coenzyme Q10 concentrations (three studies) but no effect on left ventricular ejection fraction (two studies) or exercise capacity (two studies) (33). However, a recent meta-analysis of 14 randomized, placebo-controlled trials in 2,149 participants with heart failure found that supplemental coenzyme Q10 (30-300 mg/day) resulted in a 39% reduction in mortality (seven studies), improved exercise capacity (four studies), but had no effect on left ventricular ejection fraction (nine studies) compared to placebo (34).
A trial is presently being conducted to assess the value of supplemental coenzyme Q10 and/or D-ribose in the treatment of congestive heart failure in patients with normal left ventricular ejection fraction (35).
Ischemia-reperfusion injury
The heart muscle may become oxygen-deprived (ischemic) as the result of myocardial infarction or during cardiac surgery. Increased generation of reactive oxygen species (ROS) when the heart muscle's oxygen supply is restored (reperfusion) might be an important contributor to myocardial damage occurring during ischemia-reperfusion (36). Pretreatment of animals with coenzyme Q10 has been found to preserve myocardial function following ischemia-reperfusion injury by increasing ATP concentration, enhancing antioxidant capacity and limiting oxidative damage, regulating autophagy, and reducing cardiomyocyte apoptosis (37). Another potential source of ischemia-reperfusion injury is aortic clamping during some types of cardiac surgery, such as coronary artery bypass graft (CABG) surgery. Early placebo-controlled trials found that coenzyme Q10 pretreatment (60-300 mg/day for 7-14 days prior to surgery) provided some benefit in short-term outcome measures after CABG surgery (38, 39). In a small randomized controlled trial in 30 patients, oral administration of coenzyme Q10 for 7 to 10 days before CABG surgery reduced the need for mediastinal drainage, platelet transfusion, and positive inotropic drugs (e.g. dopamine) and the risk of arrhythmia within 24 hours post-surgery (40). In one trial that did not find preoperative coenzyme Q10 supplementation to be of benefit, patients were treated with 600 mg of coenzyme Q10 12 hours prior to surgery (41), suggesting that preoperative coenzyme Q10 treatment may need to commence at least one week prior to CABG surgery to improve surgical outcomes. The combined administration of coenzyme Q10, lipoic acid, omega-3 fatty acids, magnesium orotate, and selenium at least two weeks before CABG surgery and four weeks after was examined in a randomized, placebo-controlled trial in 117 patients with heart failure (42). The treatment resulted in lower concentration of troponin-I (a marker of cardiac injury), shorter length of hospital stay, and reduced risk of postoperative transient cardiac dysfunction compared to placebo (42).
Although trials have included relatively few people and examined mostly short-term, post-surgical outcomes, the results are promising (43).
Periprocedural myocardial injury
Coronary angioplasty (also called percutaneous coronary intervention) is a nonsurgical procedure for treating obstructive coronary heart disease, including unstable angina pectoris, acute myocardial infarction, and multivessel coronary heart disease. Angioplasty involves temporarily inserting and inflating a tiny balloon into the clogged artery to help restore the blood flow to the heart. Periprocedural myocardial injury that occurs in up to one-third of patients undergoing otherwise uncomplicated angioplasty increases the risk of morbidity and mortality at follow-up.
A prospective cohort study followed 55 patients with acute ST segment elevation myocardial infarction (a type of heart attack characterized by the death of some myocardial tissue) who underwent angioplasty (44). Plasma coenzyme Q10 concentration one month after angioplasty was positively correlated with less inflammation and oxidative stress and predicted favorable left ventricular end-systolic volume remodeling at six months (44). One randomized controlled trial has examined the effect of coenzyme Q10 supplementation on periprocedural myocardial injury in patients undergoing coronary angioplasty (45). The administration of 300 mg of coenzyme Q10 12 hours before the angioplasty to 50 patients reduced the concentration of C-reactive protein ([CRP]; a marker of inflammation) within 24 hours following the procedure compared to placebo. However, there was no difference in concentrations of two markers of myocardial injury (creatine kinase and troponin-I) or in the incidence of major adverse cardiac events one month after angioplasty between active treatment and placebo (45). Additional trials are needed to examine whether coenzyme Q10 therapy can improve clinical outcomes in patients undergoing coronary angioplasty.
Angina pectoris
Myocardial ischemia may also lead to chest pain known as angina pectoris. People with angina pectoris often experience symptoms when the demand for oxygen exceeds the capacity of the coronary circulation to deliver it to the heart muscle, e.g., during exercise. Five small placebo-controlled studies have examined the effects of oral coenzyme Q10 supplementation (60-600 mg/day) in addition to conventional medical therapy in patients with chronic stable angina (46). In most of the studies, coenzyme Q10 supplementation improved exercise tolerance and reduced or delayed electrocardiographic changes associated with myocardial ischemia compared to placebo. However, only two of the studies found significant decreases in symptom frequency and use of nitroglycerin with coenzyme Q10 supplementation. Presently, there is only limited evidence suggesting that coenzyme Q10 supplementation would be a useful adjunct to conventional angina therapy.
Hypertension
Very few high-quality trials have examined the potential therapeutic benefit of coenzyme Q10 supplementation in the treatment of primary hypertension (47). A systematic review identified two small randomized, double-blind, placebo-controlled trials that found little evidence of a reduction in systolic or diastolic blood pressure following the administration of coenzyme Q10 (100-200 mg/day) for three months (47). In contrast, a meta-analysis that used less stringent selection criteria included 17 small trials and found evidence of a blood pressure-lowering effect of coenzyme Q10 in patients with cardiovascular disease or metabolic disorders (48). The effect of coenzyme Q10 on blood pressure needs to be examined in large, well-designed clinical trials.
Cardiovascular risk factors
Endothelial dysfunction: Normally functioning vascular endothelium promotes blood vessel relaxation (vasodilation) when needed (for example, during exercise) and inhibits the formation of blood clots. Atherosclerosis is associated with impairment of vascular endothelial function, thereby compromising vasodilation and normal blood flow. Endothelium-dependent vasodilation is impaired in individuals with elevated serum cholesterol concentrations, as well as in patients with coronary heart disease or diabetes mellitus. A 2012 meta-analysis examining the results of five small randomized controlled trials in 194 subjects in total found that supplemental coenzyme Q10 (150-300 mg/day for 4 to 12 weeks) resulted in a clinically significant, 1.7% increase in flow-dependent endothelial-mediated dilation (49). Evidence from larger studies is needed to further establish the effect of coenzyme Q10 on endothelium-dependent vasodilation.
Inflammation: Several small randomized controlled trials in patients at increased cardiovascular disease risk or with established cardiovascular disease have examined the effect of supplemental coenzyme Q10 for ≤3 months on circulating inflammation markers i.e., CRP, interleukin-6, and/or tumor necrosis factor-α. Recently published pooled analyses of these trials have given mixed results (50-52). Larger studies are needed to examine the effect of coenzyme Q10 supplementation on low-grade inflammation.
Blood lipids: Elevated plasma lipoprotein(a) concentration is an independent risk factor for cardiovascular disease. A meta-analysis of six controlled trials (of which five were randomized) in 409 participants found a reduction in plasma lipoprotein(a) concentration with coenzyme Q10 supplementation (100-300 mg/day for 4-12 weeks) (53). Other effects of coenzyme Q10 on blood lipids have not been reported (51, 53, 54).
A therapeutic approach combining coenzyme Q10 with other antioxidants might prove to be more effective to target co-existing metabolic disorders in individuals at risk for cardiovascular disease (55).
Diabetes mellitus
Diabetes mellitus is a condition of increased oxidative stress and impaired energy metabolism. Plasma concentrations of reduced coenzyme Q10 (CoQ10H2) have been found to be lower in diabetic patients than healthy controls after normalization to plasma cholesterol concentrations (56, 57). Randomized controlled trials that examined the effect of coenzyme Q10 supplementation found little evidence of benefits on glycemic control in patients with diabetes mellitus. A meta-analysis of six trials in participants with type 2 diabetes and one trial in participants with type 1 diabetes found that 100 to 200 mg/day of coenzyme Q10 for three to six months lowered neither fasting plasma glucose nor levels of glycated hemoglobin ([HbA1c]; a measure of glycemic control). Maternally inherited diabetes mellitus-deafness syndrome (MIDD) is caused by a mutation in mitochondrial DNA, which is inherited exclusively from one's mother. MIDD accounts for less than 1% of all cases of diabetes. Some early evidence suggested that long-term coenzyme Q10 supplementation (150 mg/day) may improve insulin secretion and prevent progressive hearing loss in these patients (58, 59).
Of note, the pathogenesis of type 2 diabetes mellitus involves the early onset of glucose intolerance and hyperinsulinemia associated with the progressive loss of tissue responsiveness to insulin. Recent experimental studies tied insulin resistance to a decrease in coenzyme Q10 expression and showed that supplementation with coenzyme Q10 could restore insulin sensitivity (7). Coenzyme Q10 supplementation might thus be a more useful tool for the primary prevention of type 2 diabetes rather than for its management.
Neurodegenerative diseases
Parkinson's disease
Parkinson's disease is a degenerative neurological disorder characterized by tremors, muscular rigidity, and slow movements. It is estimated to affect approximately 1% of Americans over the age of 65. Mitochondrial dysfunction and oxidative damage in a part of the brain called the substantia nigra may play a role in the development of the disease (60). Decreased ratios of reduced-to-oxidized coenzyme Q10 have been found in platelets of individuals with Parkinson's disease (61, 62). One study also found higher concentrations of oxidized coenzyme Q10 in the cerebrospinal fluid of patients with untreated Parkinson’s disease compared to healthy controls (63). Additionally, a study in postmortem Parkinson’s disease patients found lower concentrations of total coenzyme Q10 in the cortex region of the brain compared to age-matched controls, but no differences were seen in other brain areas, including the striatum, substantia nigra, and cerebellum (64).
A 16-month randomized, placebo-controlled phase II clinical trial evaluated the safety and efficacy of 300, 600, or 1,200 mg/day of coenzyme Q10 in 80 people with early Parkinson's disease (65). Coenzyme Q10 supplementation was well tolerated at all doses and resulted in a slower deterioration of function in Parkinson's disease patients in the group taking 1,200 mg/day. A phase III clinical trial was then designed to further examine the effect of high-dose coenzyme Q10 (1,200-2,400 mg/day) and vitamin E (1,200 IU/day) supplementation on both motor and non-motor symptoms associated with Parkinson’s disease. This trial was prematurely terminated because it was unlikely that such a treatment was effective in treating Parkinson’s disease (66). A smaller placebo-controlled trial showed that oral administration of 300 mg/day of coenzyme Q10 for 48 to 96 months moderately improved motor symptoms in treated patients (with Levodopa) with re-emerging symptoms but not in patients at an early stage of the disease (67). Two recent meta-analyses of randomized, placebo-controlled trials found no evidence that coenzyme Q10 improved motor-related symptoms or delayed the progression of the disease when compared to placebo (68, 69).
Huntington's disease
Huntington's disease is an inherited neurodegenerative disorder characterized by selective degeneration of nerve cells known as striatal spiny neurons. Symptoms, such as movement disorders and impaired cognitive function, typically develop in the fourth decade of life and progressively deteriorate over time. Animal models indicate that impaired mitochondrial function and glutamate-mediated neurotoxicity may be involved in the pathology of Huntington's disease. Some, but not all, studies in mouse models of Huntington’s disease have suggested that coenzyme Q10 supplementation could improve motor performance, overall survival, and various hallmarks of Huntington's disease, i.e., brain atrophy, ventricular enlargement, and striatal neuronal atrophy (70, 71). Interestingly, co-administration of coenzyme Q10 with remacemide (an NMDA receptor antagonist), the antibiotic minocycline, or creatine led to greater improvements in most biochemical and behavioral parameters (70-72).
To date, only two clinical trials have examined whether coenzyme Q10 might be efficacious in human patients with Huntington's disease. A 30-month, randomized, placebo-controlled trial of coenzyme Q10 (600 mg/day), remacemide, or both in 347 patients with early Huntington's disease found that neither coenzyme Q10 nor remacemide significantly altered the decline in total functional capacity, although coenzyme Q10 supplementation (with or without remacemide) resulted in a nonsignificant trend toward a slower decline (73). A 20-week pilot trial examined the safety and tolerability of increasing dosages of coenzyme Q10 (1,200 mg/day, 2,400 mg/day, and 3,600 mg/day) in eight healthy subjects and in 20 patients with Huntington’s disease; 22 of the subjects completed the study (74). All dosages were generally well tolerated, with gastrointestinal symptoms being the most frequently reported adverse effect. Blood concentrations of coenzyme Q10 at the end of the study were maximized with the daily dose of 2,400 mg (74). This dose was tested in a multicenter phase III clinical trial in 609 participants with early-stage Huntington’s disease. Participants were randomized to receive either 2,400 mg/day of coenzyme Q10 or placebo for five years (75). The trial was prematurely halted because it appeared unlikely to demonstrate any health benefit in supplemented patients — about one-third of participants completed the trial at the time of study termination (75). Although coenzyme Q10 is generally well tolerated, there is no evidence that supplementation can improve functional and cognitive symptoms in Huntington's disease patients.
Inherited ataxias
Friedreich's ataxia (FRDA): FRDA is an autosomal recessive neurodegenerative disease caused by mutations in the gene FXN that encodes for the mitochondrial protein, frataxin. Frataxin is needed for the making of iron-sulfur clusters (ISC). ISC-containing subunits are especially important for the mitochondrial respiratory chain and for the synthesis of heme-containing proteins (76). Frataxin deficiency is associated with imbalances in iron-sulfur containing proteins, mitochondrial respiratory chain dysfunction and lower ATP production, and accumulation of iron in the mitochondria, which increases oxidative stress and oxidative damage to macromolecules of the respiratory chain (77). Clinically, FRDA is a progressive disease characterized by ataxia, areflexia, speech disturbance (dysarthria), sensory loss, motor dysfunction, cardiomyopathy, diabetes, and scoliosis (77). A pilot study administering coenzyme Q10 (200 mg/day) and vitamin E (2,100 IU/day) to 10 FDRA patients found that energy metabolism of cardiac and skeletal muscle was improved after only three months of therapy (78). Follow-up assessments at 47 months indicated that cardiac and skeletal muscle improvements were maintained and that FRDA patients showed significant increases in fractional shortening, a measure of cardiac function. Moreover, the therapy was effective at preventing the progressive decline of neurological function (79). Another study reported both coenzyme Q10 and vitamin E deficiencies among FRDA patients and suggested that co-supplementation with both compounds, at doses as low as 30 mg/day of coenzyme Q10 and 4 IU/day of vitamin E, might improve disease symptoms (80). Large-scale, randomized controlled trials are necessary to determine whether coenzyme Q10, in conjunction with vitamin E, has therapeutic benefit in FRDA. At present, about one-half of patients use coenzyme Q10 and vitamin E supplements despite the lack of proven therapeutic benefit (77).
Spinocerebellar ataxias (SCAs): SCAs are a group of rare autosomal dominant neurodegenerative diseases characterized by gait difficulty, loss of hand dexterity, dysarthria, and cognitive decline. SCA1, 2, 3, and 6 are the most common SCAs (81). In vitro coenzyme Q10 treatment of forearm skin fibroblasts isolated from patients with SCA2 was found to reduce oxidative stress and normalize complex I and II-III activity of the mitochondrial respiratory chain (82). A multicenter prospective cohort study that followed 319 patients with SCAs (≥15 years) found no difference in the rate of disease progression over two years between those taking supplemental coenzyme Q10 (median dose, 600 mg/day) and nonusers (81).
Cancer
Early interest in coenzyme Q10 as a potential therapeutic agent in cancer was stimulated by an observational study that found that individuals with lung, pancreas, and especially breast cancer were more likely to have low plasma coenzyme Q10 concentrations than healthy controls (83). Two randomized controlled trials have explored the effect of coenzyme Q10 as an adjunct to conventional therapy for breast cancer. Supplementation with coenzyme Q10 failed to improve measures of fatigue and quality of life in patients newly diagnosed with breast cancer (84) and in patients receiving chemotherapy (85).
Performance
Athletic performance
There is little evidence that supplementation with coenzyme Q10 improves athletic performance in healthy individuals. A few placebo-controlled trials have examined the effects of 100 to 150 mg/day of supplemental coenzyme Q10 for three to eight weeks on physical performance in trained and untrained men. Most did not find significant differences between the group taking coenzyme Q10 and the group taking placebo with respect to measures of aerobic exercise performance, such as maximal oxygen consumption (VO2 max) and exercise time to exhaustion (86-90). One study found the maximal cycling workload to be slightly (4%) increased after eight weeks of coenzyme Q10 supplementation compared to placebo, although measures of aerobic power were not increased (91). Two studies actually found significantly greater improvement in measures of anaerobic (87) and aerobic (86) exercise performance with a placebo than with supplemental coenzyme Q10. More recent studies have suggested that coenzyme Q10 could help reduce both muscle damage-associated oxidative stress and low-grade inflammation induced by strenuous exercise (92-95). Studies on the effect of supplementation on physical performance in women are lacking, but there is little reason to suspect a gender difference in the response to coenzyme Q10 supplementation.
Sources
Biosynthesis
Coenzyme Q10 is synthesized in most human tissues. The biosynthesis of coenzyme Q10 involves three major steps: (1) synthesis of the benzoquinone structure from 4-hydroxybenzoate derived from either tyrosine or phenylalanine, two amino acids; (2) synthesis of the polyisoprenoid side chain from acetyl-coenzyme A (CoA) via the mevalonate pathway; and (3) the joining (condensation) of these two structures to form coenzyme Q10. In the mevalonate pathway, the enzyme 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase, which converts HMG-CoA into mevalonate, is common to the biosynthetic pathways of both coenzyme Q10 and cholesterol and is inhibited by statins (cholesterol-lowering drugs; see Drug interactions) (1).
Of note, pantothenic acid (formerly vitamin B5) is the precursor of coenzyme A, and pyridoxine (vitamin B6), in the form of pyridoxal-5'-phosphate, is required for the conversion of tyrosine to 4-hydroxyphenylpyruvic acid that constitutes the first step in the biosynthesis of the benzoquinone structure of coenzyme Q10. It is not known to what extent the coenzyme Q10 biosynthetic pathway may be affected by inadequate pantothenic acid and/or vitamin B6 nutritional status.
Food sources
It has been estimated that dietary consumption contributes to about 25% of plasma coenzyme Q10, but there are currently no specific dietary intake recommendations for coenzyme Q10 from the US National Academy of Medicine (formerly the Institute of Medicine) or other agencies (96). The extent to which dietary consumption contributes to tissue coenzyme Q10 concentrations is not clear.
Based on studies employing food frequency questionnaires, the average dietary intake of coenzyme Q10 is about 3 to 6 mg/day (97). Rich sources of dietary coenzyme Q10 include mainly meat, poultry, and fish. Other good sources include soybean, corn, olive, and canola oils; nuts; and seeds. Fruit, vegetables, eggs, and dairy products are moderate sources of coenzyme Q10 (97). Some dietary sources are listed in Table 1.
| Food | Serving | Coenzyme Q10 (mg) |
|---|---|---|
| Beef, fried | 3 ounces* | 2.6 |
| Herring, marinated | 3 ounces | 2.3 |
| Chicken, fried | 3 ounces | 1.4 |
| Soybean oil | 1 tablespoon | 1.3 |
| Canola oil | 1 tablespoon | 1.0 |
| Rainbow trout, steamed | 3 ounces | 0.9 |
| Peanuts, roasted | 1 ounce | 0.8 |
| Sesame seeds, roasted | 1 ounce | 0.7 |
| Pistachio nuts, roasted | 1 ounce | 0.6 |
| Broccoli, boiled | ½ cup, chopped | 0.5 |
| Cauliflower, boiled | ½ cup, chopped | 0.4 |
| Orange | 1 medium | 0.3 |
| Strawberries | ½ cup | 0.1 |
| Egg, boiled | 1 medium | 0.1 |
| *A three-ounce serving of meat or fish is about the size of a deck of cards. | ||
Supplements
Coenzyme Q10 is available without a prescription as a dietary supplement in the US. Doses in supplements for adults range from 30 to 100 mg/day, which are considerably higher than typically estimated dietary coenzyme Q10 intakes. Coenzyme Q10 is fat-soluble and is best absorbed with fat in a meal. Doses higher than 100 mg/day are generally divided into two or three doses throughout the day (101). Less than 5% of orally administered coenzyme Q10 is thought to reach the circulation (102). Therefore, pharmacological doses of coenzyme Q10 as high as 1,200 to 3,000 mg/day for adults and 30 mg/kg/day for children are usually needed to relieve symptoms in patients with coenzyme Q10 deficiency (26).
Does oral coenzyme Q10 supplementation increase tissue concentrations?
Oral supplementation with coenzyme Q10 is known to increase blood and lipoprotein concentrations of coenzyme Q10 in humans (2, 15, 22). Plasma coenzyme Q10 appears to reach a plateau following supplementation with a dose of 2,400 mg/day (103, 104). Yet, under normal circumstances, uptake of supplemental coenzyme Q10 from peripheral tissues/organs is likely limited because coenzyme Q10 is ubiquitously synthesized (105). Nonetheless, under certain physiological circumstances (e.g., aging) or in pathologies, coenzyme Q10 status might be compromised and it is then presumed that supplementation might increase coenzyme Q10 concentrations in tissues that are deficient (106). For example, a study in 24 older adults supplemented with 300 mg/day of coenzyme Q10 or placebo for at least seven days prior to cardiac surgery found that the coenzyme Q10 content of atrial tissue was significantly increased in those taking coenzyme Q10, especially in patients greater than 70 years of age (38). In another study of patients with left ventricular dysfunction, supplementation with 150 mg/day of coenzyme Q10 for four weeks before cardiac surgery increased coenzyme Q10 concentrations in the heart but not in skeletal muscle (107). Finally, a 2007 review of the literature highlighted that plasma coenzyme Q10 concentrations higher than ‘normal’ were likely needed to promote coenzyme Q10 uptake by peripheral tissues and different tissues may indeed require different plasma thresholds for the uptake of coenzyme Q10 (102).
Safety
Toxicity
There have been no reports of significant adverse side effects of oral coenzyme Q10 supplementation at doses as high as 3,000 mg/day for up to eight months (103), 1,200 mg/day for up to 16 months (65), and 600 mg/day for up to 30 months (73). According to the observed safe level (OSL) risk assessment method, evidence of safety is strong with doses up to 1,200 mg/day of coenzyme Q10 (108). Some people have experienced gastrointestinal symptoms, such as nausea, diarrhea, appetite suppression, heartburn, and abdominal discomfort, especially with daily doses ≥200 mg (109). These adverse effects may be minimized if daily doses >100 mg are divided into two or three daily doses (101). During pregnancy, the use of coenzyme Q10 supplements (100 mg twice daily) from 20 weeks' gestation was found to be safe (110). Because reliable data in lactating women are not available, supplementation should be avoided during breast-feeding (110).
Drug interactions
Warfarin
Concomitant use of warfarin (Coumadin) and coenzyme Q10 supplements has been reported to decrease the anticoagulant effect of warfarin in a few cases (111). An individual on warfarin should not begin taking coenzyme Q10 supplements without consulting the health care provider who is managing his or her anticoagulant therapy. If warfarin and coenzyme Q10 are to be used concomitantly, blood tests to assess clotting time (prothrombin time; PT/INR) should be monitored frequently, especially in the first two weeks.
HMG-CoA reductase inhibitors (statins)
HMG-CoA reductase is an enzyme that catalyzes a biochemical reaction that is common to both cholesterol and coenzyme Q10 biosynthetic pathways (see Biosynthesis). Statins are HMG-CoA reductase inhibitors that are widely used as cholesterol-lowering medications. Statins can thus also reduce the endogenous synthesis of coenzyme Q10. Therapeutic use of statins, including simvastatin (Zocor), pravastatin (Pravachol), lovastatin (Mevacor, Altocor, Altoprev), rosuvastatin (Crestor), and atorvastatin (Lipitor), has been shown to decrease circulating coenzyme Q10 concentrations (112-121). However, because coenzyme Q10 circulates with lipoproteins, plasma coenzyme Q10 concentration is influenced by the concentration of circulating lipids (122, 123). It is likely that circulating coenzyme Q10 concentrations are decreased because statins reduce circulating lipids rather than because they inhibit coenzyme Q10 synthesis (124). In addition, very few studies have examined coenzyme Q10 concentrations in tissues other than blood such that the extent to which statin therapy affects coenzyme Q10 concentrations in the body's tissues is unknown (118, 120, 125). Finally, there is currently little evidence to suggest that secondary coenzyme Q10 deficiency is responsible for statin-associated muscle symptoms in treated patients. In addition, supplementation with coenzyme Q10 failed to relieve myalgia in statin-treated patients (see Disease Treatment) (126, 127).
Authors and Reviewers
Originally written in 2003 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in February 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in March 2012 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2018 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in May 2018 by:
Roland Stocker, Ph.D.
Centre for Vascular Research
School of Medical Sciences (Pathology) and
Bosch Institute
Sydney Medical School
The University of Sydney
Sydney, New South Wales, Australia
Copyright 2003-2024 Linus Pauling Institute
References
1. Acosta MJ, Vazquez Fonseca L, Desbats MA, et al. Coenzyme Q biosynthesis in health and disease. Biochim Biophys Acta. 2016;1857(8):1079-1085. (PubMed)
2. Crane FL. Biochemical functions of coenzyme Q10. J Am Coll Nutr. 2001;20(6):591-598. (PubMed)
3. Nohl H, Gille L. The role of coenzyme Q in lysosomes. In: Kagan VEQ, P. J., ed. Coenzyme Q: Molecular Mechanisms in Health and Disease. Boca Raton: CRC Press; 2001:99-106.
4. Navas P, Villalba JM, de Cabo R. The importance of plasma membrane coenzyme Q in aging and stress responses. Mitochondrion. 2007;7 Suppl:S34-40. (PubMed)
5. Ernster L, Dallner G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim Biophys Acta. 1995;1271(1):195-204. (PubMed)
6. Thomas SR, Stocker R. Mechanisms of antioxidant action of ubiquinol-10 for low-density lipoprotein. In: Kagan VE, Quinn PJ, eds. Coenzyme Q: Molecular Mechanisms in Health and Disease. Boca Raton: CRC Press; 2001:131-150.
7. Fazakerley DJ, Chaudhuri R, Yang P, et al. Mitochondrial CoQ deficiency is a common driver of mitochondrial oxidants and insulin resistance. Elife. 2018;7. (PubMed)
8. Kagan VE, Fabisak JP, Tyurina YY. Independent and concerted antioxidant functions of coenzyme Q. In: Kagan VE, Quinn PJ, eds. Coenzyme Q: Molecular Mechanisms in Health and Disease. Boca Raton: CRC Press; 2001:119-130.
9. Overvad K, Diamant B, Holm L, Holmer G, Mortensen SA, Stender S. Coenzyme Q10 in health and disease. Eur J Clin Nutr. 1999;53(10):764-770. (PubMed)
10. Hargreaves IP. Coenzyme Q10 as a therapy for mitochondrial disease. Int J Biochem Cell Biol. 2014;49:105-111. (PubMed)
11. Fragaki K, Chaussenot A, Benoist JF, et al. Coenzyme Q10 defects may be associated with a deficiency of Q10-independent mitochondrial respiratory chain complexes. Biol Res. 2016;49:4. (PubMed)
12. Kalén A, Appelkvist EL, Dallner G. Age-related changes in the lipid compositions of rat and human tissues. Lipids. 1989;24(7):579-584. (PubMed)
13. Hernandez-Camacho JD, Bernier M, Lopez-Lluch G, Navas P. Coenzyme Q10 Supplementation in Aging and Disease. Front Physiol. 2018;9:44. (PubMed)
14. Beckman KB, Ames BN. Mitochondrial aging: open questions. Ann N Y Acad Sci. 1998;854:118-127. (PubMed)
15. Singh RB, Niaz MA, Kumar A, Sindberg CD, Moesgaard S, Littarru GP. Effect on absorption and oxidative stress of different oral Coenzyme Q10 dosages and intake strategy in healthy men. Biofactors. 2005;25(1-4):219-224. (PubMed)
16. Sohal RS, Kamzalov S, Sumien N, et al. Effect of coenzyme Q10 intake on endogenous coenzyme Q content, mitochondrial electron transport chain, antioxidative defenses, and life span of mice. Free Radic Biol Med. 2006;40(3):480-487. (PubMed)
17. Lapointe J, Hekimi S. Early mitochondrial dysfunction in long-lived Mclk1+/- mice. J Biol Chem. 2008;283(38):26217-26227. (PubMed)
18. Schmelzer C, Kubo H, Mori M, et al. Supplementation with the reduced form of coenzyme Q10 decelerates phenotypic characteristics of senescence and induces a peroxisome proliferator-activated receptor-alpha gene expression signature in SAMP1 mice. Mol Nutr Food Res. 2010;54(6):805-815. (PubMed)
19. Tian G, Sawashita J, Kubo H, et al. Ubiquinol-10 supplementation activates mitochondria functions to decelerate senescence in senescence-accelerated mice. Antioxid Redox Signal. 2014;20(16):2606-2620. (PubMed)
20. Johansson P, Dahlstrom O, Dahlstrom U, Alehagen U. Improved health-related quality of life, and more days out of hospital with supplementation with selenium and coenzyme Q10 combined. Results from a double-blind, placebo-controlled prospective study. J Nutr Health Aging. 2015;19(9):870-877. (PubMed)
21. Alehagen U, Aaseth J, Alexander J, Johansson P. Still reduced cardiovascular mortality 12 years after supplementation with selenium and coenzyme Q10 for four years: A validation of previous 10-year follow-up results of a prospective randomized double-blind placebo-controlled trial in elderly. PLoS One. 2018;13(4):e0193120. (PubMed)
22. Mohr D, Bowry VW, Stocker R. Dietary supplementation with coenzyme Q10 results in increased levels of ubiquinol-10 within circulating lipoproteins and increased resistance of human low-density lipoprotein to the initiation of lipid peroxidation. Biochim Biophys Acta. 1992;1126(3):247-254. (PubMed)
23. Witting PK, Pettersson K, Letters J, Stocker R. Anti-atherogenic effect of coenzyme Q10 in apolipoprotein E gene knockout mice. Free Radic Biol Med. 2000;29(3-4):295-305. (PubMed)
24. Thomas SR, Leichtweis SB, Pettersson K, et al. Dietary cosupplementation with vitamin E and coenzyme Q(10) inhibits atherosclerosis in apolipoprotein E gene knockout mice. Arterioscler Thromb Vasc Biol. 2001;21(4):585-593. (PubMed)
25. Turunen M, Wehlin L, Sjoberg M, et al. beta2-Integrin and lipid modifications indicate a non-antioxidant mechanism for the anti-atherogenic effect of dietary coenzyme Q10. Biochem Biophys Res Commun. 2002;296(2):255-260. (PubMed)
26. Rahman S, Clarke CF, Hirano M. 176th ENMC International Workshop: diagnosis and treatment of coenzyme Q(1)(0) deficiency. Neuromuscul Disord. 2012;22(1):76-86. (PubMed)
27. Gempel K, Topaloglu H, Talim B, et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain. 2007;130(Pt 8):2037-2044. (PubMed)
28. Pineda M, Montero R, Aracil A, et al. Coenzyme Q(10)-responsive ataxia: 2-year-treatment follow-up. Mov Disord. 2010;25(9):1262-1268. (PubMed)
29. Banach M, Serban C, Sahebkar A, et al. Effects of coenzyme Q10 on statin-induced myopathy: a meta-analysis of randomized controlled trials. Mayo Clin Proc. 2015;90(1):24-34. (PubMed)
30. Potgieter M, Pretorius E, Pepper MS. Primary and secondary coenzyme Q10 deficiency: the role of therapeutic supplementation. Nutr Rev. 2013;71(3):180-188. (PubMed)
31. Trupp RJ, Abraham WT. Congestive heart failure. In: Rakel RE, Bope ET, eds. Rakel: Conn's Current Therapy 2002. 54th ed. New York: W. B. Saunders Company; 2002:306-313.
32. McMurray JJ, Dunselman P, Wedel H, et al. Coenzyme Q10, rosuvastatin, and clinical outcomes in heart failure: a pre-specified substudy of CORONA (controlled rosuvastatin multinational study in heart failure). J Am Coll Cardiol. 2010;56(15):1196-1204. (PubMed)
33. Madmani ME, Yusuf Solaiman A, Tamr Agha K, et al. Coenzyme Q10 for heart failure. Cochrane Database Syst Rev. 2014(6):Cd008684. (PubMed)
34. Lei L, Liu Y. Efficacy of coenzyme Q10 in patients with cardiac failure: a meta-analysis of clinical trials. BMC Cardiovasc Disord. 2017;17(1):196. (PubMed)
35. Pierce JD, Mahoney DE, Hiebert JB, et al. Study protocol, randomized controlled trial: reducing symptom burden in patients with heart failure with preserved ejection fraction using ubiquinol and/or D-ribose. BMC Cardiovasc Disord. 2018;18(1):57. (PubMed)
36. Milei J, Forcada P, Fraga CG, et al. Relationship between oxidative stress, lipid peroxidation, and ultrastructural damage in patients with coronary artery disease undergoing cardioplegic arrest/reperfusion. Cardiovasc Res. 2007;73(4):710-719. (PubMed)
37. Liang S, Ping Z, Ge J. Coenzyme Q10 regulates antioxidative stress and autophagy in acute myocardial ischemia-reperfusion injury. Oxid Med Cell Longev. 2017;2017:9863181. (PubMed)
38. Rosenfeldt FL, Pepe S, Linnane A, et al. The effects of ageing on the response to cardiac surgery: protective strategies for the ageing myocardium. Biogerontology. 2002;3(1-2):37-40. (PubMed)
39. Langsjoen PH, Langsjoen AM. Overview of the use of CoQ10 in cardiovascular disease. Biofactors. 1999;9(2-4):273-284. (PubMed)
40. Makhija N, Sendasgupta C, Kiran U, et al. The role of oral coenzyme Q10 in patients undergoing coronary artery bypass graft surgery. J Cardiothorac Vasc Anesth. 2008;22(6):832-839. (PubMed)
41. Taggart DP, Jenkins M, Hooper J, et al. Effects of short-term supplementation with coenzyme Q10 on myocardial protection during cardiac operations. Ann Thorac Surg. 1996;61(3):829-833. (PubMed)
42. Leong JY, van der Merwe J, Pepe S, et al. Perioperative metabolic therapy improves redox status and outcomes in cardiac surgery patients: a randomised trial. Heart Lung Circ. 2010;19(10):584-591. (PubMed)
43. Celik T, Iyisoy A. Coenzyme Q10 and coronary artery bypass surgery: what we have learned from clinical trials. J Cardiothorac Vasc Anesth. 2009;23(6):935-936. (PubMed)
44. Huang CH, Kuo CL, Huang CS, et al. High plasma coenzyme Q10 concentration is correlated with good left ventricular performance after primary angioplasty in patients with acute myocardial infarction. Medicine (Baltimore). 2016;95(31):e4501. (PubMed)
45. Aslanabadi N, Safaie N, Asgharzadeh Y, et al. The randomized clinical trial of coenzyme Q10 for the prevention of periprocedural myocardial injury following elective percutaneous coronary intervention. Cardiovasc Ther. 2016;34(4):254-260. (PubMed)
46. Tran MT, Mitchell TM, Kennedy DT, Giles JT. Role of coenzyme Q10 in chronic heart failure, angina, and hypertension. Pharmacotherapy. 2001;21(7):797-806. (PubMed)
47. Ho MJ, Li EC, Wright JM. Blood pressure lowering efficacy of coenzyme Q10 for primary hypertension. Cochrane Database Syst Rev. 2016;3:Cd007435. (PubMed)
48. Tabrizi R, Akbari M, Sharifi N, et al. The effects of coenzyme Q10 supplementation on blood pressures among patients with metabolic diseases: a systematic review and meta-analysis of randomized controlled trials. High Blood Press Cardiovasc Prev. 2018;25(1):41-50. (PubMed)
49. Gao L, Mao Q, Cao J, Wang Y, Zhou X, Fan L. Effects of coenzyme Q10 on vascular endothelial function in humans: a meta-analysis of randomized controlled trials. Atherosclerosis. 2012;221(2):311-316. (PubMed)
50. Fan L, Feng Y, Chen GC, Qin LQ, Fu CL, Chen LH. Effects of coenzyme Q10 supplementation on inflammatory markers: A systematic review and meta-analysis of randomized controlled trials. Pharmacol Res. 2017;119:128-136. (PubMed)
51. Mazidi M, Kengne AP, Banach M. Effects of coenzyme Q10 supplementation on plasma C-reactive protein concentrations: A systematic review and meta-analysis of randomized controlled trials. Pharmacol Res. 2018;128:130-136. (PubMed)
52. Zhai J, Bo Y, Lu Y, Liu C, Zhang L. Effects of coenzyme Q10 on markers of inflammation: a systematic review and meta-analysis. PLoS One. 2017;12(1):e0170172. (PubMed)
53. Sahebkar A, Simental-Mendia LE, Stefanutti C, Pirro M. Supplementation with coenzyme Q10 reduces plasma lipoprotein(a) concentrations but not other lipid indices: A systematic review and meta-analysis. Pharmacol Res. 2016;105:198-209. (PubMed)
54. Suksomboon N, Poolsup N, Juanak N. Effects of coenzyme Q10 supplementation on metabolic profile in diabetes: a systematic review and meta-analysis. J Clin Pharm Ther. 2015;40(4):413-418. (PubMed)
55. Shargorodsky M, Debby O, Matas Z, Zimlichman R. Effect of long-term treatment with antioxidants (vitamin C, vitamin E, coenzyme Q10 and selenium) on arterial compliance, humoral factors and inflammatory markers in patients with multiple cardiovascular risk factors. Nutr Metab (Lond). 2010;7:55. (PubMed)
56. McDonnell MG, Archbold GP. Plasma ubiquinol/cholesterol ratios in patients with hyperlipidaemia, those with diabetes mellitus and in patients requiring dialysis. Clin Chim Acta. 1996;253(1-2):117-126. (PubMed)
57. Lim SC, Tan HH, Goh SK, et al. Oxidative burden in prediabetic and diabetic individuals: evidence from plasma coenzyme Q(10). Diabet Med. 2006;23(12):1344-1349. (PubMed)
58. Alcolado JC, Laji K, Gill-Randall R. Maternal transmission of diabetes. Diabet Med. 2002;19(2):89-98. (PubMed)
59. Suzuki S, Hinokio Y, Ohtomo M, et al. The effects of coenzyme Q10 treatment on maternally inherited diabetes mellitus and deafness, and mitochondrial DNA 3243 (A to G) mutation. Diabetologia. 1998;41(5):584-588. (PubMed)
60. Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol. 2008;4(11):600-609. (PubMed)
61. Gotz ME, Gerstner A, Harth R, et al. Altered redox state of platelet coenzyme Q10 in Parkinson's disease. J Neural Transm. 2000;107(1):41-48. (PubMed)
62. Shults CW, Haas RH, Passov D, Beal MF. Coenzyme Q10 levels correlate with the activities of complexes I and II/III in mitochondria from parkinsonian and nonparkinsonian subjects. Ann Neurol. 1997;42(2):261-264. (PubMed)
63. Isobe C, Abe T, Terayama Y. Levels of reduced and oxidized coenzyme Q-10 and 8-hydroxy-2'-deoxyguanosine in the cerebrospinal fluid of patients with living Parkinson's disease demonstrate that mitochondrial oxidative damage and/or oxidative DNA damage contributes to the neurodegenerative process. Neurosci Lett. 2010;469(1):159-163. (PubMed)
64. Hargreaves IP, Lane A, Sleiman PM. The coenzyme Q10 status of the brain regions of Parkinson's disease patients. Neurosci Lett. 2008;447(1):17-19. (PubMed)
65. Shults CW, Oakes D, Kieburtz K, et al. Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol. 2002;59(10):1541-1550. (PubMed)
66. Beal MF, Oakes D, Shoulson I, et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: no evidence of benefit. JAMA Neurol. 2014;71(5):543-552. (PubMed)
67. Yoritaka A, Kawajiri S, Yamamoto Y, et al. Randomized, double-blind, placebo-controlled pilot trial of reduced coenzyme Q10 for Parkinson's disease. Parkinsonism Relat Disord. 2015;21(8):911-916. (PubMed)
68. Negida A, Menshawy A, El Ashal G, et al. Coenzyme Q10 for patients with Parkinson's disease: a systematic review and meta-analysis. CNS Neurol Disord Drug Targets. 2016;15(1):45-53. (PubMed)
69. Zhu ZG, Sun MX, Zhang WL, Wang WW, Jin YM, Xie CL. The efficacy and safety of coenzyme Q10 in Parkinson's disease: a meta-analysis of randomized controlled trials. Neurol Sci. 2017;38(2):215-224. (PubMed)
70. Ferrante RJ, Andreassen OA, Dedeoglu A, et al. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington's disease. J Neurosci. 2002;22(5):1592-1599. (PubMed)
71. Stack EC, Smith KM, Ryu H, et al. Combination therapy using minocycline and coenzyme Q10 in R6/2 transgenic Huntington's disease mice. Biochim Biophys Acta. 2006;1762(3):373-380. (PubMed)
72. Yang L, Calingasan NY, Wille EJ, et al. Combination therapy with coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson's and Huntington's diseases. J Neurochem. 2009;109(5):1427-1439. (PubMed)
73. The Huntington Study Group. A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington's disease. Neurology. 2001;57(3):397-404. (PubMed)
74. Hyson HC, Kieburtz K, Shoulson I, et al. Safety and tolerability of high-dosage coenzyme Q10 in Huntington's disease and healthy subjects. Mov Disord. 2010;25(12):1924-1928. (PubMed)
75. McGarry A, McDermott M, Kieburtz K, et al. A randomized, double-blind, placebo-controlled trial of coenzyme Q10 in Huntington disease. Neurology. 2017;88(2):152-159. (PubMed)
76. Burk K. Friedreich Ataxia: current status and future prospects. Cerebellum Ataxias. 2017;4:4. (PubMed)
77. Strawser C, Schadt K, Hauser L, et al. Pharmacological therapeutics in Friedreich ataxia: the present state. Expert Rev Neurother. 2017;17(9):895-907. (PubMed)
78. Lodi R, Hart PE, Rajagopalan B, et al. Antioxidant treatment improves in vivo cardiac and skeletal muscle bioenergetics in patients with Friedreich's ataxia. Ann Neurol. 2001;49(5):590-596. (PubMed)
79. Hart PE, Lodi R, Rajagopalan B, et al. Antioxidant treatment of patients with Friedreich ataxia: four-year follow-up. Arch Neurol. 2005;62(4):621-626. (PubMed)
80. Cooper JM, Korlipara LV, Hart PE, Bradley JL, Schapira AH. Coenzyme Q10 and vitamin E deficiency in Friedreich's ataxia: predictor of efficacy of vitamin E and coenzyme Q10 therapy. Eur J Neurol. 2008;15(12):1371-1379. (PubMed)
81. Lo RY, Figueroa KP, Pulst SM, et al. Coenzyme Q10 and spinocerebellar ataxias. Mov Disord. 2015;30(2):214-220. (PubMed)
82. Cornelius N, Wardman JH, Hargreaves IP, et al. Evidence of oxidative stress and mitochondrial dysfunction in spinocerebellar ataxia type 2 (SCA2) patient fibroblasts: Effect of coenzyme Q10 supplementation on these parameters. Mitochondrion. 2017;34:103-114. (PubMed)
83. Folkers K, Osterborg A, Nylander M, Morita M, Mellstedt H. Activities of vitamin Q10 in animal models and a serious deficiency in patients with cancer. Biochem Biophys Res Commun. 1997;234(2):296-299. (PubMed)
84. Lesser GJ, Case D, Stark N, et al. A randomized, double-blind, placebo-controlled study of oral coenzyme Q10 to relieve self-reported treatment-related fatigue in newly diagnosed patients with breast cancer. J Support Oncol. 2013;11(1):31-42. (PubMed)
85. Iwase S, Kawaguchi T, Yotsumoto D, et al. Efficacy and safety of an amino acid jelly containing coenzyme Q10 and L-carnitine in controlling fatigue in breast cancer patients receiving chemotherapy: a multi-institutional, randomized, exploratory trial (JORTC-CAM01). Support Care Cancer. 2016;24(2):637-646. (PubMed)
86. Laaksonen R, Fogelholm M, Himberg JJ, Laakso J, Salorinne Y. Ubiquinone supplementation and exercise capacity in trained young and older men. Eur J Appl Physiol Occup Physiol. 1995;72(1-2):95-100. (PubMed)
87. Malm C, Svensson M, Ekblom B, Sjodin B. Effects of ubiquinone-10 supplementation and high intensity training on physical performance in humans. Acta Physiol Scand. 1997;161(3):379-384. (PubMed)
88. Weston SB, Zhou S, Weatherby RP, Robson SJ. Does exogenous coenzyme Q10 affect aerobic capacity in endurance athletes? Int J Sport Nutr. 1997;7(3):197-206. (PubMed)
89. Porter DA, Costill DL, Zachwieja JJ, et al. The effect of oral coenzyme Q10 on the exercise tolerance of middle-aged, untrained men. Int J Sports Med. 1995;16(7):421-427. (PubMed)
90. Braun B, Clarkson PM, Freedson PS, Kohl RL. Effects of coenzyme Q10 supplementation on exercise performance, VO2max, and lipid peroxidation in trained cyclists. Int J Sport Nutr. 1991;1(4):353-365. (PubMed)
91. Bonetti A, Solito F, Carmosino G, Bargossi AM, Fiorella PL. Effect of ubidecarenone oral treatment on aerobic power in middle-aged trained subjects. J Sports Med Phys Fitness. 2000;40(1):51-57. (PubMed)
92. Abdizadeh L, Jafari A, Armanfar M. Effects of short-term coenzyme Q10 supplementation on markers of oxidative stress and inflammation after downhill running in male mountaineers. Science & Sports. 2015;30(6):328-334.
93. Díaz-Castro J, Guisado R, Kajarabille N, et al. Coenzyme Q(10) supplementation ameliorates inflammatory signaling and oxidative stress associated with strenuous exercise. Eur J Nutr. 2012;51(7):791-799. (PubMed)
94. Leelarungrayub D, Rawattikanon A, Klaphajone J, Pothong-sunan P, Bloomer RJ. Coenzyme Q10 supplementation decreases oxidative stress and improves physical performance in young swimmers Open Sports Med J 2010;4(1):1-8.
95. Ostman B, Sjodin A, Michaelsson K, Byberg L. Coenzyme Q10 supplementation and exercise-induced oxidative stress in humans. Nutrition. 2012;28(4):403-417. (PubMed)
96. Weber C. Dietary intake and absorption of coenzyme Q. In: Kagan VE, Quinn PJ, eds. Coenzyme Q: Molecular Mechanisms in Health and Disease. Boca Raton: CRC Press; 2001:209-215.
97. Pravst I, Zmitek K, Zmitek J. Coenzyme Q10 contents in foods and fortification strategies. Crit Rev Food Sci Nutr. 2010;50(4):269-280. (PubMed)
98. Mattila P, Kumpulainen J. Coenzymes Q9 and Q10: Contents in foods and dietary intake. J Food Comp Anal. 2001;14(4):409-417.
99. Kamei M, Fujita T, Kanbe T, et al. The distribution and content of ubiquinone in foods. Int J Vitam Nutr Res. 1986;56(1):57-63. (PubMed)
100. Weber C, Bysted A, Holmer G. Coenzyme Q10 in the diet--daily intake and relative bioavailability. Mol Aspects Med. 1997;18 Suppl:S251-254. (PubMed)
101. Natural Medicines. Coenzyme Q10. Professional handout/Adverse effects. Available at: https://naturalmedicines.therapeuticresearch.com. Accessed 4/23/18.
102. Bhagavan HN, Chopra RK. Plasma coenzyme Q10 response to oral ingestion of coenzyme Q10 formulations. Mitochondrion. 2007;7 Suppl:S78-88. (PubMed)
103. Ferrante KL, Shefner J, Zhang H, et al. Tolerance of high-dose (3,000 mg/day) coenzyme Q10 in ALS. Neurology. 2005;65(11):1834-1836. (PubMed)
104. Shults CW, Flint Beal M, Song D, Fontaine D. Pilot trial of high dosages of coenzyme Q10 in patients with Parkinson's disease. Exp Neurol. 2004;188(2):491-494. (PubMed)
105. Svensson M, Malm C, Tonkonogi M, Ekblom B, Sjodin B, Sahlin K. Effect of Q10 supplementation on tissue Q10 levels and adenine nucleotide catabolism during high-intensity exercise. Int J Sport Nutr. 1999;9(2):166-180. (PubMed)
106. Bhagavan HN, Chopra RK. Coenzyme Q10: absorption, tissue uptake, metabolism and pharmacokinetics. Free Radic Res. 2006;40(5):445-453. (PubMed)
107. Keith M, Mazer CD, Mikhail P, Jeejeebhoy F, Briet F, Errett L. Coenzyme Q10 in patients undergoing CABG: Effect of statins and nutritional supplementation. Nutr Metab Cardiovasc Dis. 2008;18(2):105-111. (PubMed)
108. Hathcock JN, Shao A. Risk assessment for coenzyme Q10 (Ubiquinone). Regul Toxicol Pharmacol. 2006;45(3):282-288. (PubMed)
109. Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. Montvale: Thomson Reuters; 2008.
110. Natural Medicines. Coenzyme Q10. Professional handout/Safety. Available at: https://naturalmedicines.therapeuticresearch.com. Accessed 4/23/18.
111. Natural Medicines. Coenzyme Q10. Professional handout/Drug Interactions. Available at: https://naturalmedicines.therapeuticresearch.com. Accessed 4/23/18.
112. Folkers K, Langsjoen P, Willis R, et al. Lovastatin decreases coenzyme Q levels in humans. Proc Natl Acad Sci U S A. 1990;87(22):8931-8934. (PubMed)
113. Colquhoun DM, Jackson R, Walters M, et al. Effects of simvastatin on blood lipids, vitamin E, coenzyme Q10 levels and left ventricular function in humans. Eur J Clin Invest. 2005;35(4):251-258. (PubMed)
114. Mabuchi H, Higashikata T, Kawashiri M, et al. Reduction of serum ubiquinol-10 and ubiquinone-10 levels by atorvastatin in hypercholesterolemic patients. J Atheroscler Thromb. 2005;12(2):111-119. (PubMed)
115. Bargossi AM, Battino M, Gaddi A, et al. Exogenous CoQ10 preserves plasma ubiquinone levels in patients treated with 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors. Int J Clin Lab Res. 1994;24(3):171-176. (PubMed)
116. Watts GF, Castelluccio C, Rice-Evans C, Taub NA, Baum H, Quinn PJ. Plasma coenzyme Q (ubiquinone) concentrations in patients treated with simvastatin. J Clin Pathol. 1993;46(11):1055-1057. (PubMed)
117. Ghirlanda G, Oradei A, Manto A, et al. Evidence of plasma CoQ10-lowering effect by HMG-CoA reductase inhibitors: a double-blind, placebo-controlled study. J Clin Pharmacol. 1993;33(3):226-229. (PubMed)
118. Laaksonen R, Jokelainen K, Laakso J, et al. The effect of simvastatin treatment on natural antioxidants in low-density lipoproteins and high-energy phosphates and ubiquinone in skeletal muscle. Am J Cardiol. 1996;77(10):851-854. (PubMed)
119. Laaksonen R, Ojala JP, Tikkanen MJ, Himberg JJ. Serum ubiquinone concentrations after short- and long-term treatment with HMG-CoA reductase inhibitors. Eur J Clin Pharmacol. 1994;46(4):313-317. (PubMed)
120. Elmberger PG, Kalen A, Lund E, et al. Effects of pravastatin and cholestyramine on products of the mevalonate pathway in familial hypercholesterolemia. J Lipid Res. 1991;32(6):935-940. (PubMed)
121. Ashton E, Windebank E, Skiba M, et al. Why did high-dose rosuvastatin not improve cardiac remodeling in chronic heart failure? Mechanistic insights from the UNIVERSE study. Int J Cardiol. 2011;146(3):404-407. (PubMed)
122. Hughes K, Lee BL, Feng X, Lee J, Ong CN. Coenzyme Q10 and differences in coronary heart disease risk in Asian Indians and Chinese. Free Radic Biol Med. 2002;32(2):132-138. (PubMed)
123. Hargreaves IP, Duncan AJ, Heales SJ, Land JM. The effect of HMG-CoA reductase inhibitors on coenzyme Q10: possible biochemical/clinical implications. Drug Saf. 2005;28(8):659-676. (PubMed)
124. Stocker R, Pollicino C, Gay CA, et al. Neither plasma coenzyme Q10 concentration, nor its decline during pravastatin therapy, is linked to recurrent cardiovascular disease events: a prospective case-control study from the LIPID study. Atherosclerosis. 2006;187(1):198-204. (PubMed)
125. Laaksonen R, Jokelainen K, Sahi T, Tikkanen MJ, Himberg JJ. Decreases in serum ubiquinone concentrations do not result in reduced levels in muscle tissue during short-term simvastatin treatment in humans. Clin Pharmacol Ther. 1995;57(1):62-66. (PubMed)
126. Tan JT, Barry AR. Coenzyme Q10 supplementation in the management of statin-associated myalgia. Am J Health Syst Pharm. 2017;74(11):786-793. (PubMed)
127. Taylor BA. Does coenzyme Q10 supplementation mitigate statin-associated muscle symptoms? Pharmacological and methodological considerations. Am J Cardiovasc Drugs. 2018;18(2):75-82. (PubMed)
Lipoic Acid
Contents
Summary
- Lipoic acid (often called α-lipoic acid), also known as thioctic acid, is a naturally occurring compound that is also synthesized by humans. (More information)
- Endogenously synthesized lipoic acid is bound to protein and functions as a cofactor for several important mitochondrial multienzyme complexes. (More information)
- Supplementation with lipoic acid transiently increases plasma and cellular concentrations of unbound lipoic acid. (More information)
- Lipoic acid is a direct antioxidant, although any increase in radical scavenging activity in vivo is likely transient. Lipoic acid may also trigger antioxidant defense, enhance cellular glucose uptake, and modulate the activity of various cell-signaling molecules and transcription factors. (More information)
- Some evidence from small randomized controlled studies suggests that high-dose lipoic acid may improve measures of glucose utilization in subjects with metabolic disorders. (More information)
- Available evidence suggests that treatment with intravenous or oral lipoic acid may help reduce symptoms of diabetic peripheral neuropathy. It is important to note that many of the studies examining the efficacy of lipoic acid for the treatment of diabetic neuropathy have been conducted by one German research group and funded by the manufacturer of lipoic acid in Germany. (More information)
- Lipoic acid was found to slow disease progression when administered to a mouse model of multiple sclerosis. A two-year clinical trial designed to assess the effect of lipoic acid on loss of mobility and changes in brain volume in patients with progressive multiple sclerosis is ongoing. (More information)
- Current evidence is too limited to suggest whether lipoic acid supplementation could benefit patients with Alzheimer’s disease. (More information)
- Supplementation with lipoic acid may show some benefits in terms of weight control for individuals with high body mass index. (More information)
- Lipoic acid occurs naturally in food covalently bound to protein, whereas supplements contain unbound (free) lipoic acid. (More information)
Introduction
Lipoic acid (often called α-lipoic acid), also known as thioctic acid, is a naturally occurring organosulfur compound that is synthesized by plants and animals, including humans (1, 2). Lipoic acid is covalently bound to certain proteins, which function as part of essential mitochondrial multienzyme complexes involved in energy and amino acid metabolism (see Biological Activities). In addition to the physiological functions of protein-bound lipoic acid, there is increasing scientific and medical interest in potential therapeutic uses of pharmacological doses of free (unbound) lipoic acid (3).
Lipoic acid contains two thiol (sulfur) groups, which may be oxidized or reduced; dihydrolipoic acid is the reduced form of lipoic acid (Figure 1) (4). Lipoic acid also contains an asymmetric carbon, which means that lipoic acid can exist as one of two possible optical isomers, also called enantiomers. These enantiomers are mirror images of each other: R-lipoic acid and S-lipoic acid (Figure 1). Only the R-enantiomer is endogenously synthesized and covalently bound to protein. R-lipoic acid occurs naturally in food (see Food sources). Free (unbound) lipoic acid supplements may contain either R-lipoic acid or a 50:50 (racemic) mixture of R-lipoic acid and S-lipoic acid (see Supplements).
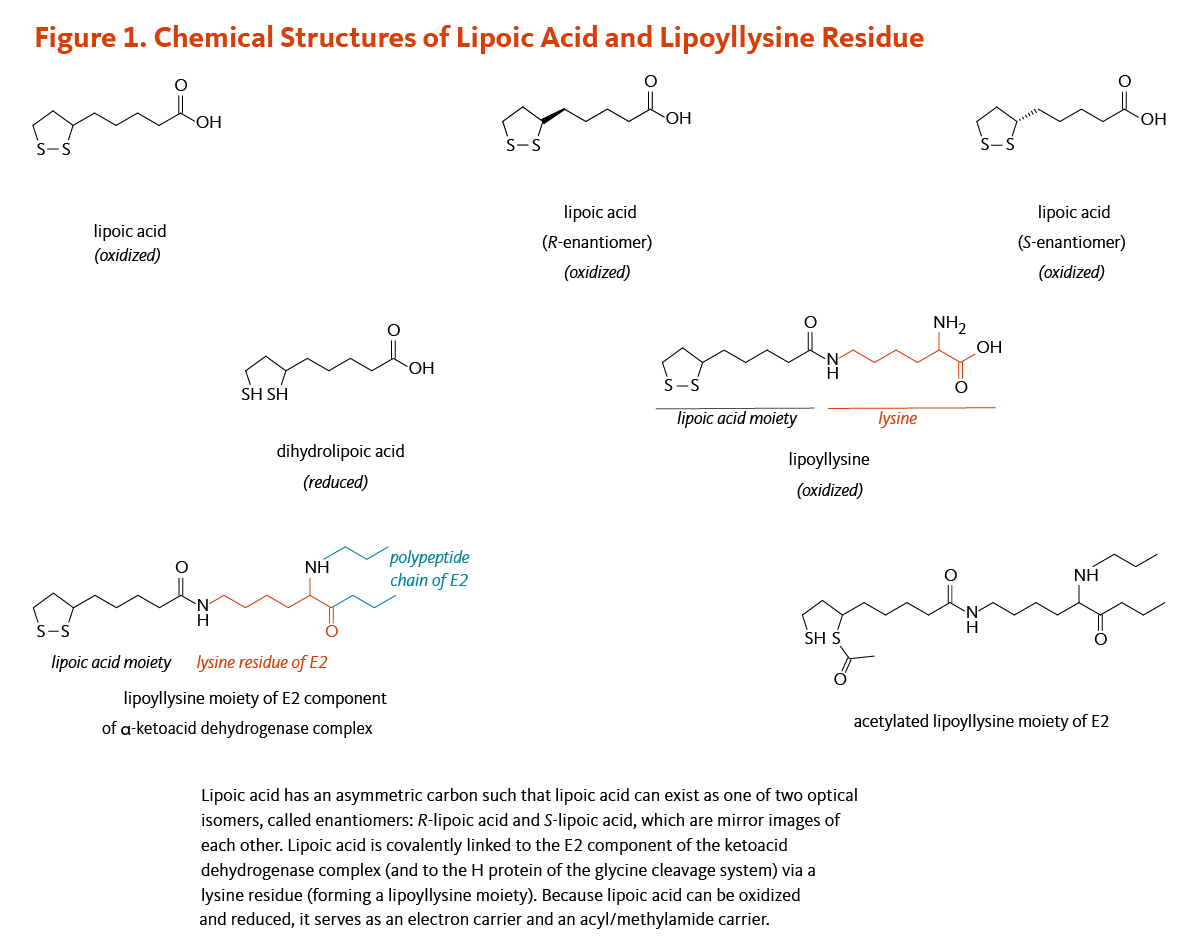
Metabolism and Bioavailability
Endogenous biosynthesis
The synthesis of lipoic acid has been characterized in detail in the yeast Saccharomyces cerevisiae, but not all genes involved in the process have been identified in humans (5). Lipoic acid is synthesized de novo in mitochondria from octanoic acid, an 8-carbon fatty acid (C8:0), bound to the acyl-carrier protein (ACP; see article on Pantothenic Acid) during the process of fatty acid synthesis (Figure 2). An enzyme called lipoyl (octanoyl) transferase 2 catalyzes the transfer of the octanoyl moiety from octanoyl-ACP to a conserved lysine of the H protein of the glycine cleavage system (see also Biological Activities). The next reaction is the insertion of two sulfur atoms at positions 6 and 8 of the protein H-bound octanoyl moiety, thereby producing a dihydrolipoyl moiety. This step is catalyzed by the lipoic acid synthetase (also called lipoyl synthase), an enzyme containing iron-sulfur clusters that act as sulfur donors in the reaction (5). Finally, the enzyme lipoyl transferase 1 catalyzes the transfer of the dihydrolipoyl moiety from the H protein of the glycine cleavage system to conserved lysine residues of the E2 components of the α-ketoacid dehydrogenase multienzyme complexes (5). The oxidation of the dihydrolipoyl moiety is catalyzed by a dihydrolipoamide dehydrogenase (Figure 2).
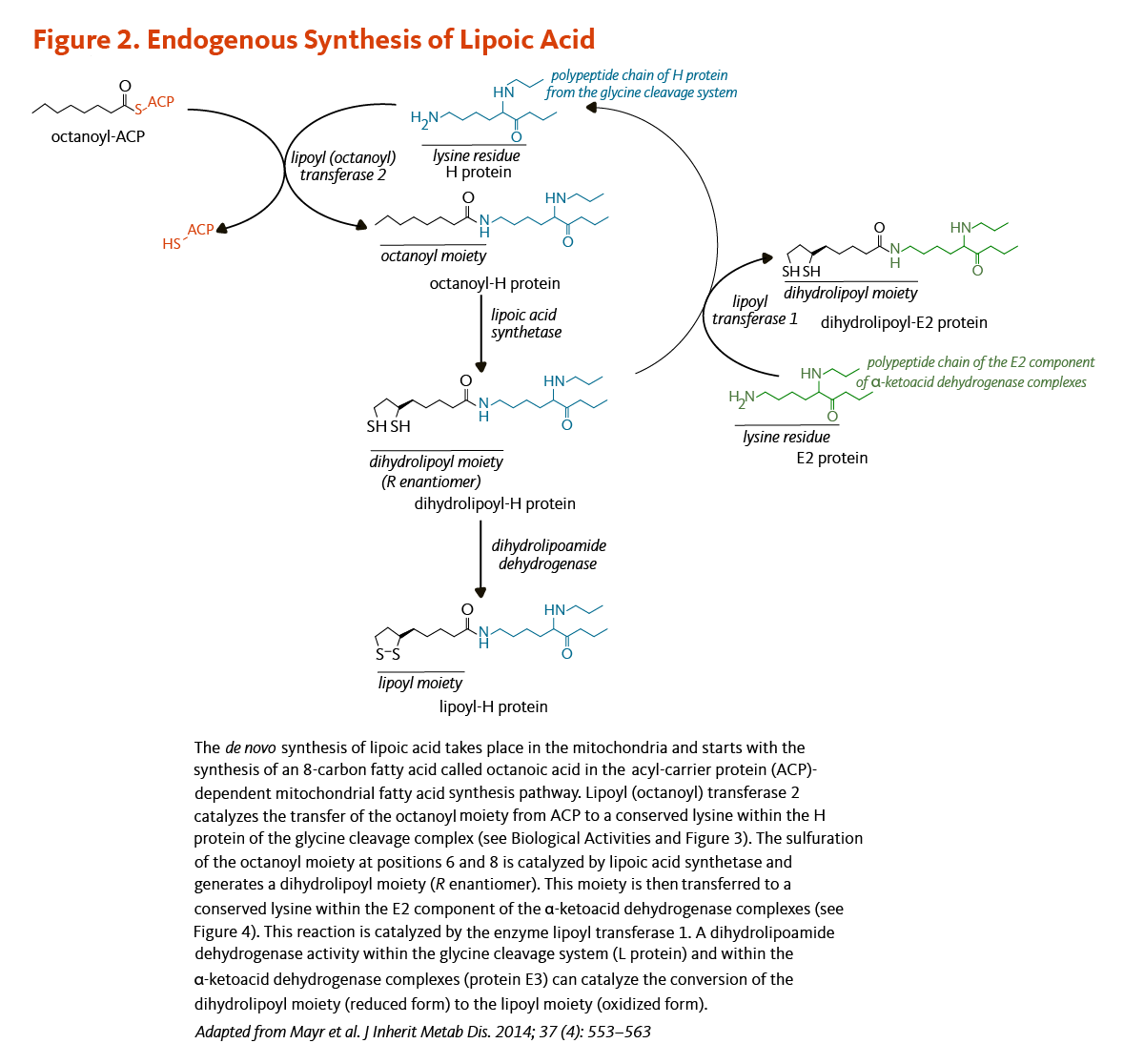
Dietary and supplemental lipoic acid
Consumption of lipoic acid from food has not yet been found to result in detectable increases of free lipoic acid in human plasma or cells (3, 6). In contrast, high oral doses of free lipoic acid (≥50 mg) significantly, yet transiently, increase the concentration of free lipoic acid in plasma and cells. Pharmacokinetic studies in humans have found that about 30%-40% of an oral dose of a racemic mixture of R-lipoic acid and S-lipoic acid is absorbed (6, 7). Oral lipoic acid supplements are better absorbed on an empty stomach than with food: taking lipoic acid with food (versus without food) decreased peak plasma lipoic acid concentrations by about 30% and total plasma lipoic acid concentrations by about 20% (8). A liquid formulation of R-lipoic acid was found to be better absorbed and more stable in the plasma, suggesting that it might be more efficacious than the solid form in the management of a condition like diabetic neuropathy (9, 10).
There may also be differences in bioavailability of the two isomers of lipoic acid. Following single oral doses R,S-lipoic acid (racemic mixture), peak plasma concentrations of R-lipoic acid were found to be 40%-50% higher than S-lipoic acid, suggesting a differential absorption in favor of the R-enantiomer (6, 8, 11). Yet, following oral ingestion, both enantiomers are rapidly metabolized and excreted. Plasma lipoic acid concentrations generally peak within one hour or less and decline rapidly (6, 7, 11, 12). In cells, lipoic acid is swiftly reduced to dihydrolipoic acid, and in vitro studies indicate that dihydrolipoic acid is then rapidly exported from cells (3). Moreover, a pilot study in 19 healthy adults suggested that the bioavailability of R,S-lipoic acid and R-lipoic acid may vary with age and gender (13).
Finally, there is no evidence in humans that exogenous lipoic acid can be 'activated' with ATP or GTP and incorporated into lipoic acid-dependent enzymes by a lipoyl transferase (14). As a consequence, a loss of lipoic acid-dependent enzymatic activity caused by defects in endogenous lipoic acid synthesis (see Deficiency) cannot be rescued by the provision of exogenous lipoic acid (5).
Biological Activities
Protein-bound lipoic acid
Enzyme cofactor
R-lipoic acid is an essential cofactor for several mitochondrial multienzyme complexes that catalyze critical reactions related to the catabolism (breakdown) of amino acids and the production of energy (15). R-lipoic acid is covalently bound to a specific lysine residue in at least one of the proteins in each multienzyme complex. Such a non-protein cofactor is known as a "prosthetic group."
R-lipoic acid functions as a prosthetic group for the biological activity of the following multienzyme complexes:
- The glycine cleavage system that catalyzes the decarboxylation of glycine coupled with the addition of a methylene group (-CH2) to tetrahydrofolate to form 5,10-methylene tetrahydrofolate, an important cofactor in the synthesis of nucleic acids (Figure 3). Within the glycine cleavage system, R-lipoic acid is covalently bound to a conserved lysine of the H protein (Figures 2 and 3).
- Four α-ketoacid dehydrogenase complexes (Figure 4), including:
(i) the pyruvate dehydrogenase complex that catalyzes the conversion of pyruvate to acetyl-coenzyme A (CoA), an important substrate for energy production via the citric acid cycle;
(ii) the α-ketoglutarate dehydrogenase complex that catalyzes the conversion of α-ketoglutarate to succinyl CoA, another important intermediate of the citric acid cycle;
(iii) the branched-chain α-ketoacid dehydrogenase complex that is involved in the decarboxylation of ketoacids in the catabolic pathway of the branched-chain amino acids, namely leucine, isoleucine, and valine;
(iv) the 2-oxoadipate dehydrogenase complex that catalyzes the decarboxylation of 2-oxoadipate to glutaryl-CoA in the catabolic pathway of lysine, hydroxylysine, and tryptophan.
All four α-ketoacid dehydrogenase complexes contain three enzymatic activities, namely E1, E2, and E3. E1 is a thiamin pyrophosphate (TPP)-dependent α-ketoacid dehydrogenase, R-lipoic acid functions as a prosthetic group essential for E2 transacetylase activity, and E3 is a flavin adenine dinucleotide (FAD)-dependent dihydrolipoamide dehydrogenase (Figure 4). R-lipoic acid is also found in the E3-binding protein (protein X component) of the pyruvate dehydrogenase complex (5).

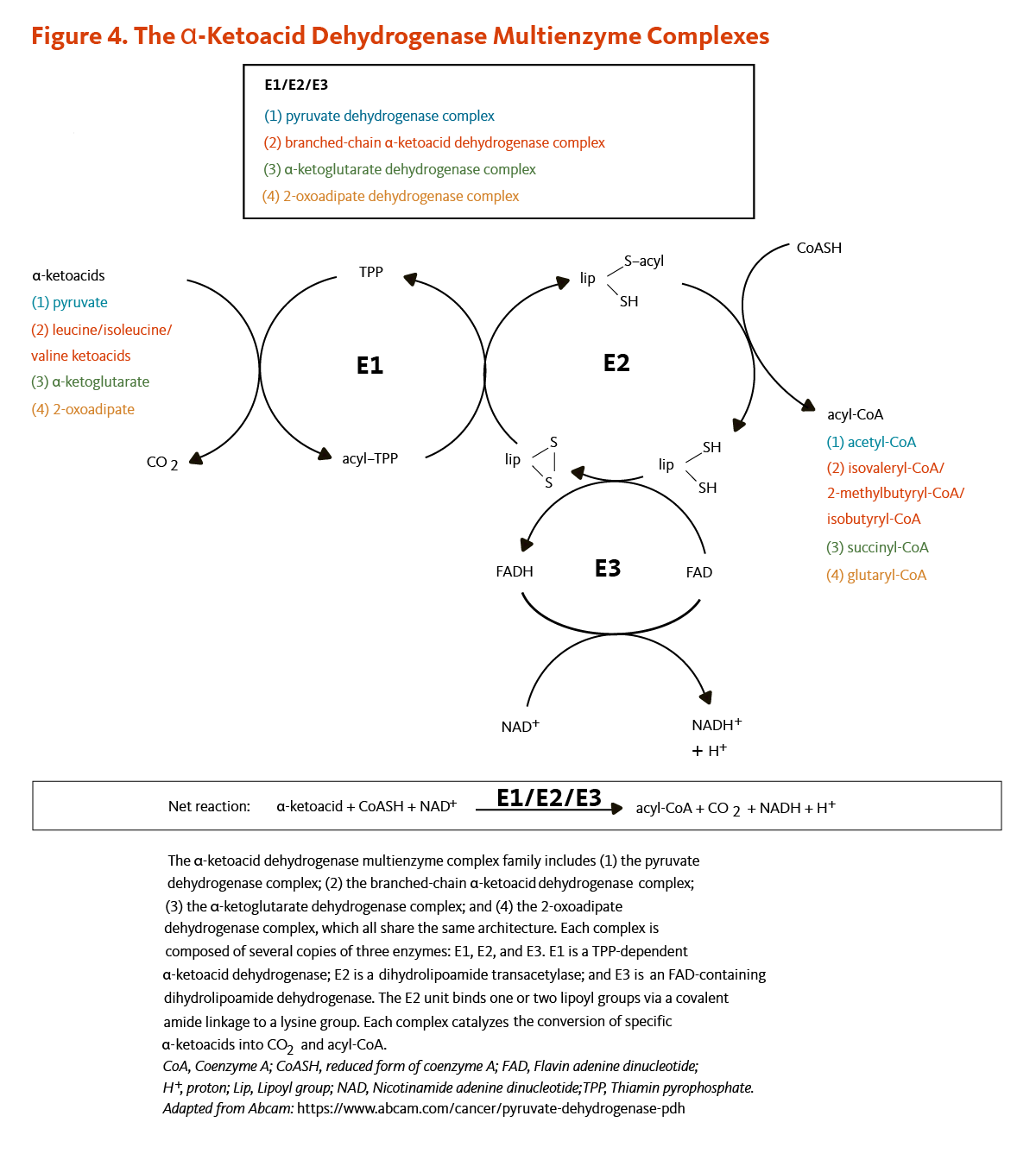
Unbound lipoic acid
When considering the biological activities of supplemental (unbound) lipoic acid, it is important to keep in mind the limited and transient nature of the increases in plasma and tissue lipoic acid (see Metabolism and Bioavailability) (3).
Antioxidant activities
Scavenging reactive oxygen and nitrogen species: Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are highly reactive compounds with the potential to damage DNA, proteins, and lipids in cell membranes. Both lipoic acid and dihydrolipoic acid can directly scavenge (neutralize) physiologically relevant ROS and RNS in the test tube (reviewed in 3). However, whether direct quenching reactions occur in vivo is unknown. The highest tissue concentrations of free lipoic acid likely to be achieved through oral supplementation are at least 10 times lower than those of other intracellular antioxidants, such as vitamin C and glutathione. Moreover, free lipoic acid is rapidly eliminated from cells, so any increases in direct radical scavenging activity are unlikely to be sustained.
Regeneration of other antioxidants: When an antioxidant scavenges a free radical, it becomes oxidized itself and is not able to scavenge additional ROS or RNS until it has been reduced. In the test tube, dihydrolipoic acid is a potent reducing agent with the capacity to reduce the oxidized forms of several important antioxidants, including coenzyme Q10, vitamin C, and glutathione (Figure 5) (16, 17). Dihydrolipoic acid may also reduce the oxidized form of α-tocopherol (vitamin E) directly or indirectly through regenerating oxidized vitamin C (see the article on Vitamin E) (18) or oxidized coenzyme Q10 (see the article on Coenzyme Q10) (19). Whether dihydrolipoic acid effectively regenerates antioxidants under physiological conditions is unclear (3).
Metal chelation: Redox-active metal ions, such as free iron and copper, can induce oxidative damage by catalyzing reactions that generate highly reactive free radicals (20). Compounds that chelate free metal ions in a way that prevents them from generating free radicals offer promise in the treatment of neurodegenerative diseases and other chronic diseases in which metal-induced oxidative damage may play a pathogenic role (21). Both lipoic acid and dihydrolipoic acid have been found to inhibit copper- and iron-mediated oxidative damage in the test tube (22, 23) and to inhibit excess iron and copper accumulation in animal models (24, 25). Lipoic acid may also be helpful as an adjunct treatment against heavy metal toxicity. No clinical trial has examined the use of lipoic acid as a chelating agent in mercury toxicity, yet it has proven to be effective in several mammalian species (26, 27).
Activation of antioxidant signaling pathways: Glutathione is an important intracellular antioxidant that also plays a role in the detoxification and elimination of potential carcinogens and toxins. Reductions in glutathione synthesis and tissue glutathione concentrations in aged animals (compared to younger ones) are suggestive of a potentially lower ability to respond to oxidative stress or toxin exposure (28). Lipoic acid has been found to increase glutathione concentrations in cultured cells and in the tissues of aged animals fed lipoic acid (29, 30). Lipoic acid might be able to increase glutathione synthesis in aged rats by up-regulating the expression of γ-glutamylcysteine ligase (γ-GCL), the rate-limiting enzyme in glutathione synthesis (31), and by increasing cellular uptake of cysteine, an amino acid required for glutathione synthesis (32). Lipoic acid was found to upregulate the expression of γ-GCL and other antioxidant enzymes via the activation of the nuclear factor E2-related factor 2 (Nrf2)-dependent pathway (31, 33).
Briefly, Nrf2 is a transcription factor that is bound to the protein Kelch-like ECH-associated protein 1 (Keap1) in the cytosol. Keap1 responds to oxidative stress signals by freeing Nrf2. Upon release, Nrf2 translocates to the nucleus where it can bind to the antioxidant response element (ARE) located in the promoter region of genes coding for antioxidant enzymes and scavengers. Lipoic acid — but not dihydrolipoic acid — can react with specific sulfhydryl residues of Keap1, causing the release of Nrf2 (34). Nrf2/ARE target genes code for several mediators of the antioxidant response, including γ-GCL, NAD(P)H quinone oxidoreductase 1 (NQO-1), heme oxygenase-1 (HO-1), catalase, and superoxide dismutase (SOD). For example, the upregulation of the Nrf2 pathway by lipoic acid in cultured hepatocytes and in the liver of obese or diabetic rats prevented lipid overload-induced steatosis (35) and cell death (36). Lipoic acid also protected liver from oxidative stress-induced liver injury in methotrexate-treated rats through the activation of Nrf-2 pathway and other anti-inflammatory pathways (37). Pre-treatment and post-treatment with lipoic acid, respectively, prevented and reversed lipopolysaccharide (LPS)-induced lung histopathological alterations in rats through Nrf2-mediated HO-1 upregulation (38).
Inhibition of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX): NOX is a plasma membrane-bound enzymatic complex that catalyzes the production of superoxide from oxygen and NADPH and has been involved in innate immune defense against microbes (39). Lipoic acid prevented NOX-induced superoxide production in a rat model of cerebral ischemia and limited infarct volume and neurological deficiencies through upregulating the insulin-phosphatidylinositide-3 kinase (PI3K)-protein kinase B (PKB/Akt) signaling pathway (40). Treatment of gastric cancer cells with lipoic acid limited NOX-generated ROS production and reduced cancer cell proliferation induced by Helicobacter pylori (H. pylori) infection (41).
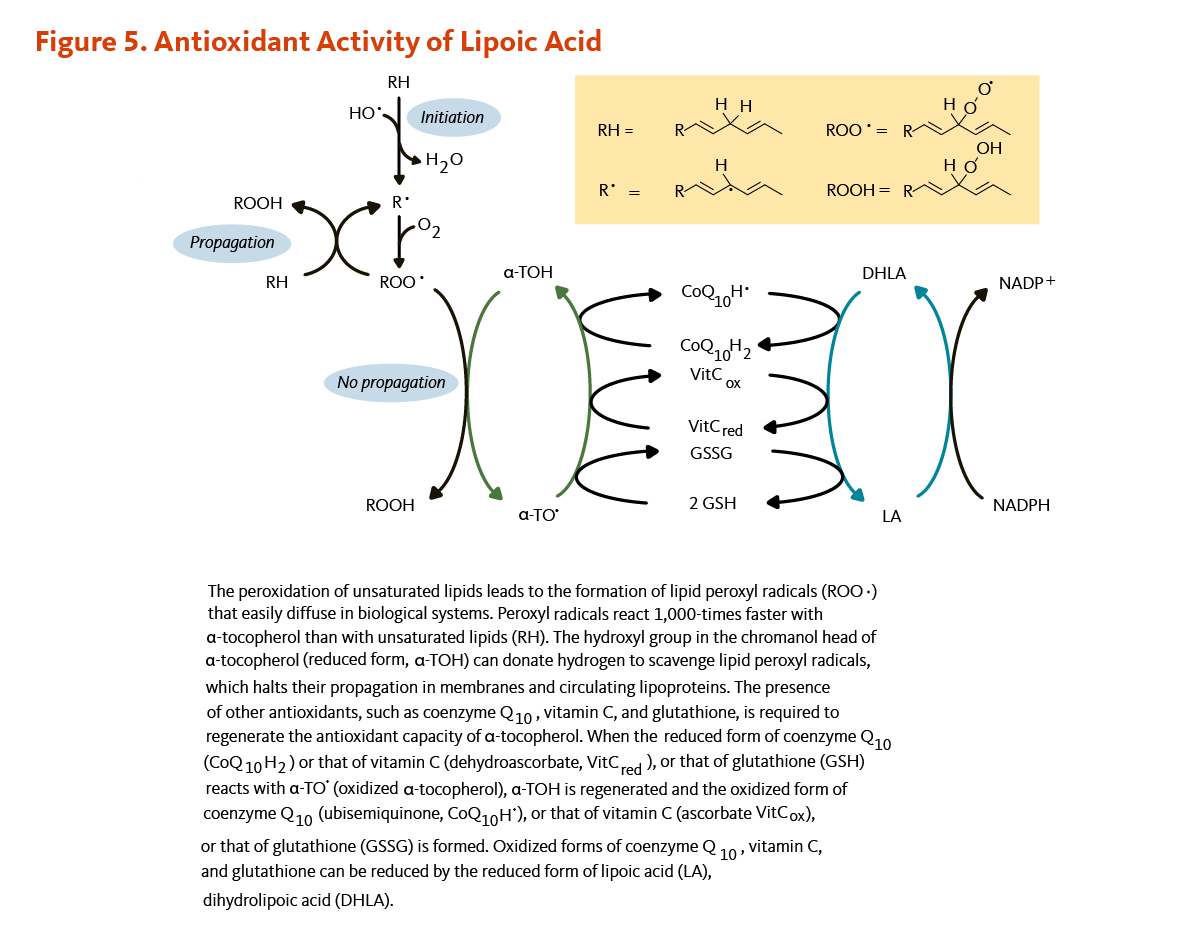
Regulation of cellular glucose uptake
The binding of insulin to the insulin receptor stimulates a cascade of protein phosphorylations leading to the translocation of glucose transporters (GLUT4) to the cell membrane and an increased cellular uptake of glucose (3, 42). Lipoic acid has been found to activate the insulin signaling cascade in cultured cells (3, 42, 43), increase GLUT4 translocation to cell membranes, and increase glucose uptake in cultured adipose and muscle cells (44, 45). A computer modeling study suggested that lipoic acid might bind to the intracellular tyrosine kinase domain of the insulin receptor and stabilize the active form of the enzyme (43).
Regulation of other signaling pathways
In addition to Nrf2 and insulin signaling pathways, lipoic acid was found to target other cell-signaling molecules thereby affecting a variety of cellular processes, including metabolism, stress responses, proliferation, and survival. For example, in cultured endothelial cells, lipoic acid was found to inhibit IKK-β, an enzyme that promotes the translocation of redox-sensitive and pro-inflammatory transcription factor, nuclear factor-kappa B (NFκB) from the cytosol to the nucleus (46). Lipoic acid has also been shown to improve nitric oxide (NO)-dependent vasodilation in aged rats by increasing PKB/Akt-dependent phosphorylation of endothelial NO synthase (eNOS) and eNOS-catalyzed NO production (47). Additionally, lipoic acid increased mitochondrial biogenesis through triggering AMP-activated protein kinase (AMPK)-induced transcription factor PGC-1α activation in skeletal muscle of aged mice (48). Several reviews of the literature have described pathways that are potential targets of lipoic acid in various models and under different experimental conditions (49-52).
Deficiency
Lipoic acid deficiency has been described in rare cases of inherited mutations in the lipoic acid biosynthetic pathway. Mutations identified in patients with defective lipoic acid metabolism affect genes involved in the synthesis of iron-sulfur clusters and genes coding for lipoic acid synthetase (LIAS), lipoyl transferase 1 (LIPT1), and dihydrolipoamide dehydrogenase (E3 component of α-ketoacid dehydrogenase complexes; DLD) (5, 53, 54).
Disease Treatment
Diabetes mellitus
Chronically elevated blood glucose concentration is the hallmark of diabetes mellitus. Type 1 diabetes is caused by the autoimmune destruction of the insulin-producing β-cells of the pancreas, leading to an insufficient production of insulin. Exogenous insulin is required to maintain a normal blood glucose concentration (i.e., fasting blood glucose <100 milligram per deciliter [mg/dL]). In contrast, impaired tissue glucose uptake in response to insulin (a phenomenon called insulin resistance) plays a key role in the development of type 2 diabetes (55). Although patients with type 2 diabetes may eventually require insulin, interventions that enhance insulin sensitivity may be used to maintain normal blood glucose concentrations. The term 'prediabetes' is sometimes used to describe early metabolic abnormalities that place individuals at high risk of developing type 2 diabetes. Of note, these patients are also at high risk for cardiovascular disease. According to the American Diabetes Association, prediabetes can be defined by a condition of impaired fasting glucose, characterized by a fasting blood glucose concentration between 100 mg/dL and 125 mg/dL and/or a condition of impaired glucose tolerance, characterized by a 2-hour blood glucose concentration ≥140 mg/dL following an oral glucose tolerance test (56).
Glucose utilization
The effect of high-dose lipoic acid on glucose utilization has been primarily examined in individuals with type 2 diabetes. An early clinical trial in 13 patients with type 2 diabetes found that a single intravenous infusion of 1,000 mg of lipoic acid improved insulin-stimulated glucose disposal (i.e., insulin sensitivity) by 50% compared to a placebo infusion (57). A placebo-controlled study of 72 patients with type 2 diabetes found that oral administration of lipoic acid at doses of 600 mg/day, 1,200 mg/day or 1,800 mg/day improved insulin sensitivity by 25% after four weeks of treatment (58). There were no significant differences among the three doses of lipoic acid, suggesting that 600 mg/day may be the maximum effective dose (55). However, in a more recent randomized, placebo-controlled study in 102 subjects, daily supplementation with 600 mg of lipoic acid (+/- 800 mg of vitamin E [α-tocopherol]) for 16 weeks had no effect on fasting blood glucose, fasting blood insulin, or a measure of insulin resistance called the homeostatic model assessment of insulin resistance (HOMA-IR) index (59). A 2018 systematic review and meta-analysis identified 20 randomized controlled trials (published between 2007 and 2017) that examined the effect of supplemental lipoic acid on markers of glucose utilization in 1,245 subjects with metabolic disorders (not limited to type 2 diabetes) (60). Administration of lipoic acid (200 to 1,800 mg/day for 2 weeks to 1 year), alone or together with other nutrients, was found to lower fasting blood glucose and insulin concentrations, insulin resistance, and blood HbA1c concentration — a marker of glycemic control over the past few months (60).
Endothelial function
The inner lining of blood vessels, known as the vascular endothelium, plays an important role in the maintenance of cardiovascular health. In particular, nitric oxide (NO) regulates vascular tone and blood flow by promoting the relaxation of all types of blood vessels, including arteries — a phenomenon called vasodilation. Alterations in NO-mediated endothelium-dependent vasodilation results in widespread vasoconstriction and coagulation abnormalities and is considered to be an early step in the development of atherosclerosis. The presence of chronic hyperglycemia, insulin resistance, oxidative stress, and pro-inflammatory mechanisms contribute to endothelial dysfunction in patients with diabetes mellitus (61).
The measurement of brachial flow-mediated dilation (FMD) is often used as a surrogate marker of endothelial function. Two techniques are being used to measure endothelium-dependent vasodilation. One technique measures the forearm blood flow by venous occlusion plethysmography during infusion of acetylcholine. Using this invasive technique, intra-arterial infusion of lipoic acid was found to improve endothelium-dependent vasodilation in 39 subjects with type 2 diabetes but not in 11 healthy controls (62). A more recent randomized, double-blind, placebo-controlled study in 30 patients with type 2 diabetes found that intravenous infusion of 600 mg of lipoic acid improved the response to the endothelium-dependent vasodilator acetylcholine but not to the endothelium-independent vasodilator, glycerol trinitrate (63). Another noninvasive technique using ultrasound to measure flow-mediated vasodilation was used in two additional studies conducted by Xiang et al. (64, 65). The results of these randomized, placebo-controlled studies showed that intravenous lipoic acid could improve endothelial function in patients with impaired fasting glucose (64) or impaired glucose tolerance (65).
One randomized placebo-controlled trial that assessed the effect of oral lipoic acid supplementation in 58 patients diagnosed with metabolic syndrome, a condition characterized by abnormal glucose and lipid metabolism, showed that flow-mediated vasodilation improved by 44% with 300 mg/day of lipoic acid for four weeks (66).
Diabetic neuropathy
Peripheral neuropathy: Up to 50% of diabetic patients develop peripheral neuropathy, a type of nerve damage that may result in pain, loss of sensation, and weakness, particularly in the lower extremities (67). Peripheral neuropathy is also a leading cause of lower limb amputation in diabetic patients (68). Several mechanisms have been proposed to explain chronic hyperglycemia-induced nerve damage, such as intracellular accumulation of sorbitol, glycation reactions, and oxidative and nitrosative stress (reviewed in 69). The results of several large randomized controlled trials indicated that maintaining blood glucose at near normal concentrations was the most important step in limiting the risk of diabetic neuropathy and lower extremity amputation (70-72). However, evidence of the efficacy of enhanced control of glycemia in preventing neuropathy is stronger in patients with type 1 diabetes than in those with type 2 diabetes (73). Moreover, this glucose control intervention increased the risk of hypoglycemic episodes (73).
The efficacy of lipoic acid, administered either intravenously or orally, in the management of neuropathic symptoms has been examined in patients with diabetes. Meta-analyses of randomized controlled trials suggest that infusion of 300 to 600 mg/day of lipoic acid for two to four weeks significantly reduced the symptoms of diabetic neuropathy to a clinically meaningful degree (55, 74). Regarding the efficacy of oral lipoic acid supplementation, an initial short-term study in 24 patients with type 2 diabetes mellitus found that the symptoms of peripheral neuropathy improved in those who took 600 mg of lipoic acid three times a day for three weeks compared to those who took a placebo (75). A larger clinical trial randomly assigned more than 500 patients with type 2 diabetes and symptomatic peripheral neuropathy to one of the following treatments: (i) 600 mg/day of intravenous lipoic acid for three weeks followed by 1,800 mg/day of oral lipoic acid for six months, (ii) 600 mg/day of intravenous lipoic acid for three weeks followed by oral placebo for six months, or (iii) intravenous placebo for three weeks followed by oral placebo for six months (76). Evidence of improvements in sensory and motor deficits — assessed by physicians — could be observed after three weeks of intravenous lipoic acid therapy, yet not at the end of six months of oral lipoic acid therapy. However, another randomized, double-blind, placebo-controlled trial in 181 patients with diabetic neuropathy found that oral supplementation with either 600 mg/day, 1,200 mg/day, or 1,800 mg/day of lipoic acid for five weeks significantly improved neuropathic symptoms (77). In this study, the 600 mg/day dose was as effective as the higher doses. Finally, a four-year, multicenter, clinical trial in 421 diabetic patients with distal symmetric sensorimotor polyneuropathy found no difference between oral administration of 600 mg/day of lipoic and placebo on the primary endpoint, a composite score that assessed neuropathic impairment of the lower limbs and nerve conduction (78). Yet, measures of specific neuropathic impairments (secondary outcomes) improved with lipoic acid supplementation (78). A post-hoc analysis suggested that oral lipoic acid supplementation may reduce neuropathic symptoms particularly in subjects with a high burden of cardiovascular disease, diabetes, and neuropathy yet with normal body mass index (BMI) and blood pressure (79).
Autonomic neuropathy: Another neuropathic complication of diabetes mellitus is cardiac autonomic neuropathy (CAN), which occurs in as many as 25% of diabetic patients (55). CAN is characterized by damage to the nerve fibers that innervate the heart and blood vessels, leading to reduced heart rate variability (variability in the time interval between heartbeats) and increased risk of mortality (80). In a randomized controlled trial of 72 patients with type 2 diabetes and reduced heart rate variability, oral supplementation with 800 mg/day of lipoic acid for four months resulted in significant improvement in two out of four measures of heart rate variability compared to placebo (81).
Summary: Overall, the available research suggests that treatment with intravenous or oral lipoic acid may help reduce symptoms of diabetic peripheral neuropathy. The use of lipoic acid is currently approved for the treatment of diabetic neuropathy in Germany (4). It is important to note that many of the studies that examined the efficacy of lipoic acid in the treatment of diabetic neuropathy have been primarily conducted by one German research group and funded by the manufacturer of lipoic acid in Germany (82).
Diabetic retinopathy
Chronic hyperglycemia can damage blood vessels in the retina and cause a potentially sight-threatening condition called diabetic retinopathy (83). One placebo-controlled study examined the effect of lipoic acid on the visual capability of 80 participants of whom 12 had type 1 diabetes, 48 had type 2 diabetes, and 20 were diabetes-free. The result showed that daily oral administration of 300 mg of lipoic acid for three months prevented the deterioration of contrast sensitivity in patients with diabetes and improved it in healthy patients compared to placebo (84).
Multiple sclerosis
Multiple sclerosis is an autoimmune disease of unknown etiology that is characterized by the progressive destruction of myelin and nerve fibers in the central nervous system, causing neurological symptoms in affected individuals (85). There are four main types of multiple sclerosis defined according to the disease course: (i) clinically isolated syndrome, (ii) relapsing-remitting multiple sclerosis, (iii) secondary progressive multiple sclerosis, and (iv) primary progressive multiple sclerosis (for more information, visit the National Multiple Sclerosis Society website) (86). Lipoic acid was found to effectively slow disease progression when administered either orally (87), intraperitoneally (88), or subcutaneously (89) to mice with experimental autoimmune encephalomyelitis (EAE), a model of multiple sclerosis. In vitro and animal studies have found that lipoic acid exhibits immunomodulatory properties through mechanisms that stimulate the production of cyclic AMP (cAMP) (90, 91) — a central regulator of innate immune functions — and inhibits the migration of immune cells into the brain and spinal cord (92), possibly by decreasing endothelial expression of cell adhesion molecules, inhibiting expression of enzymes like matrix metalloproteinases (MMP), and/or reducing the permeability of the blood-brain barrier (87, 89, 93, 94).
Only a few studies have examined lipoic acid supplementation in humans. A small pilot study designed to evaluate the safety of lipoic acid in 30 people with relapsing or progressive multiple sclerosis found that treatment with 1,200 to 2,400 mg/day of oral lipoic acid for two weeks was generally well tolerated (see Safety) (95). In this study, higher serum concentrations of lipoic acid were associated with the lowest serum concentrations of MMP-9 — a marker of inflammation (95). Another study suggested that an oral dose of 1,200 mg of lipoic acid in subjects with multiple sclerosis could help achieve serum lipoic acid concentrations similar to those found to be therapeutic in mice (96). A randomized, placebo-controlled study in 52 subjects (mean age, 30 years) with relapsing-remitting multiple sclerosis found an increase in total antioxidant capacity in blood with lipoic acid supplementation (1,200 mg/day for 12 weeks) yet not in the activity of specific antioxidant enzymes (superoxide dismutase and glutathione peroxidase) (97). Supplemental lipoic acid also decreased the serum concentrations of some (IFN-γ, ICAM-1, TGF-γ, IL-4), but not all markers (TNF-γ, IL-6, MMP-9), cytokines and other inflammation (98). In addition, lipoic acid supplementation did not reduce the severity of multiple sclerosis symptoms, as assessed by the Expanded Disability Status Scale (EDSS) scoring system (98, 99).
A two-year clinical trial designed to assess the effect of lipoic acid (1,200 mg/day) on loss of mobility and changes in brain volume in patients with progressive multiple sclerosis is ongoing (85).
Cognitive impairments and dementia
Studies in animal models of aging and neurodegenerative disease have indicated that lipoic acid administration might improve measures of spatial memory, learning capacity, and/or motor function (reviewed in 100).
It is not known whether oral lipoic acid supplementation can slow cognitive decline related to aging or pathological conditions in humans. An uncontrolled, open-label trial in nine patients with probable Alzheimer’s disease and related dementias, who were also taking acetylcholinesterase inhibitors, reported that oral supplementation with 600 mg/day of lipoic acid appeared to stabilize cognitive function over a one-year period (101). A subsequent study that followed 43 patients for up to four years found those with mild dementia or moderate-early dementia who took lipoic acid (600 mg/day), in addition to acetylcholinesterase inhibitors, experienced slower cognitive decline compared to the typical cognitive decline of Alzheimer’s patients as reported in the literature (102). However, the significance of these findings is difficult to assess without a control group for comparison. A randomized controlled trial found that oral supplementation with 1,200 mg/day of lipoic acid for 10 weeks was of no benefit in treating HIV-associated cognitive impairment (103). The results of another randomized trial in 39 patients with Alzheimer’s disease suggested that supplementation with fish oil concentrate (high in omega-3 fatty acids) with or without lipoic acid (600 mg/day) for one year could delay the progression in cognitive and functional impairments assessed by the Instrumental Activities of Daily Living (IADL) scoring system compared to placebo (104). Interestingly, patients who took fish oil concentrate together with lipoic acid showed no worsening of global cognitive function (as assessed by the Mini-Mental State Examination [MMSE] score system) over 12 months as opposed to those who took either the fish oil concentrate alone or a placebo (104). Larger trials are needed to confirm these preliminary findings and further evaluate the usefulness of supplemental lipoic acid in the prevention and/or management of neurodegenerative diseases.
Weight management
A 2018 meta-analysis of randomized, placebo-controlled trials found that lipoic acid supplementation in those with high body mass index (BMI) resulted in significant, yet modest, reductions in weight (9 studies) and BMI (11 studies) in the absence of caloric restriction (except in one study) (105). Subgroup analyses revealed that weight loss was greater in overweight versus obese participants, in unhealthy versus healthy participants, with daily doses ≤600 mg, and for intervention period shorter than 10 weeks. There was no reduction in waist circumference with supplemental lipoic acid (5 studies) (105). Substantial weight and BMI reductions with lipoic acid supplementation in overweight or obese subjects were also reported in a prior meta-analysis (106).
Sources
Endogenous biosynthesis
R-lipoic acid is synthesized endogenously by humans (see Metabolism and Bioavailability).
Food sources
R-lipoic acid occurs naturally in food covalently bound to lysine in proteins (lipoyllysine; see Figure 1). Although lipoic acid is found in a wide variety of foods from plant and animal sources, quantitative information on the lipoic acid or lipoyllysine content of food is limited; published databases are lacking. Animal tissues with high lipoyllysine content (~1-3 μg/g dry wt) include kidney, heart, and liver, while lipoyllysine-rich vegetables include spinach and broccoli (107). Somewhat lower amounts of lipoyllysine (~0.5 μg/g dry wt) have been measured in tomatoes, peas, and Brussels sprouts.
Supplements
Unlike lipoic acid in foods, lipoic acid in supplements is not bound to protein. Moreover, the amounts of lipoic acid available in dietary supplements (50-600 mg) are likely as much as 1,000 times greater than the amounts that could be obtained from the diet. In Germany, lipoic acid is approved for the treatment of diabetic neuropathies and is available by prescription (108). Lipoic acid is available as a dietary supplement without a prescription in the US. Most lipoic acid supplements contain a racemic mixture of R-lipoic acid and S-lipoic acid (sometimes noted d,l-lipoic acid). Supplements that claim to contain only R-lipoic acid are usually more expensive, and information regarding their purity is not publicly available (109). Since taking lipoic acid with a meal decreases its bioavailability, it is generally recommended that lipoic acid be taken 30 min prior to a meal (see also Metabolism and Bioavailability) (8).
Racemic mixture versus R-lipoic acid only
R-lipoic acid is the isomer that is synthesized by plants and animals and functions as a cofactor for mitochondrial enzymes in its protein-bound form (see Biological Activities). Direct comparisons of the bioavailability of the oral racemic mixture and R-lipoic acid supplements have not been published. Following the ingestion of R,S-lipoic acid, peak plasma concentrations of R-lipoic acid were found to be 40%-50% higher than S-lipoic acid, suggesting better absorption of R-lipoic acid. Both isomers were nonetheless rapidly metabolized and eliminated (6, 8, 11). In rats, R-lipoic acid was more effective than S-lipoic acid in enhancing insulin-stimulated glucose transport and metabolism in skeletal muscle (110), and R-lipoic acid was more effective than R,S-lipoic acid and S-lipoic acid in preventing cataracts (111). However, all of the published human studies have used R,S-lipoic acid (racemic mixture). It has been suggested that the presence of S-lipoic acid in the racemic mixture may limit the polymerization of R-lipoic acid and enhance its bioavailability (52). At present, it remains unclear which supplemental form is best to use in clinical trials.
Safety
Adverse effects
In general, high-dose lipoic acid administration has been found to have few serious side effects. Intravenous administration of lipoic acid at doses of 600 mg/day for three weeks (112) and oral lipoic acid at doses as high as 1,800 mg/day for six months (113) and 1,200 mg/day for two years (76) did not result in serious adverse effects when used to treat diabetic peripheral neuropathy. There was no significant difference in the incidence of adverse events and serious adverse events in patients with diabetic neuropathy who took 600 mg/day of lipoic acid for four years compared to those in the placebo group (78). Oral intake of 2,400 mg/day for two weeks was also found to be safe in a pilot study that included participants with multiple sclerosis (95). Two mild anaphylactoid reactions and one severe anaphylactic reaction, including laryngospasm, were reported after intravenous lipoic acid administration (55). The most frequently reported side effects of oral lipoic acid supplementation are allergic reactions affecting the skin, including rashes, hives, and itching. Abdominal pain, nausea, vomiting, diarrhea, and vertigo have also been reported, and one trial found that the incidence of nausea, vomiting, and vertigo was dose-dependent (77). Further, malodorous urine has been noted by people taking 1,200 mg/day of lipoic acid orally (95).
Pregnancy and lactation
A retrospective observational study reported that daily oral supplementation with 600 mg of lipoic acid (racemic mixture) during pregnancy and without interruption from a period spanning between week 10 and week 30 of gestation and until the end of week 37 was not associated with any adverse effect in mothers and their newborns (114). In absence of further evidence, lipoic acid supplementation during pregnancy should only be considered under strict medical supervision. The safety of lipoic acid supplements in lactating women has not been established and should thus be discouraged (115).
Children
A case of intoxication was reported in a 20-month old child (10.5 kg bw) after the accidental ingestion of four 600-mg tablets of lipoic acid (116). The child was admitted to hospital with seizure, acidosis, and unconsciousness. Symptomatic management and rapid elimination of lipoic acid led to a full recovery without sequelae within five days. The non-accidental ingestion of a very high dose of lipoic acid led to multi-organ failure and subsequent death of an adolescent girl (117).
Drug interactions
In theory, because lipoic acid supplementation may improve insulin-mediated glucose utilization (see Diabetes mellitus), there is a potential risk of hypoglycemia in diabetic patients using insulin or oral anti-diabetic agents (118). Consequently, blood glucose concentrations should be monitored closely when lipoic acid supplementation is added to diabetes treatment regimens. Yet, one study in 24 healthy volunteers reported no significant drug interactions with the co-administration of a single oral dose of lipoic acid (600 mg) and the oral anti-diabetic agents, glyburide (also called glybenclamide) or acarbose (Precose/Prandase/Glucobay) (119).
Nutrient interactions
Biotin
The chemical structure of biotin is similar to that of lipoic acid, and there is some evidence that high concentrations of lipoic acid can compete with biotin for transport across cell membranes (120, 121). The administration of high doses of lipoic acid by injection to rats decreased the activity of two biotin-dependent enzymes by about 30%-35% (122), but it is not known whether oral or intravenous lipoic acid supplementation substantially increases the requirement for biotin in humans (123).
Authors and Reviewers
Originally written in 2002 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in July 2003 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2006 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in January 2012 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in October 2018 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in January 2019 by:
Tory M. Hagen, Ph.D.
Principal Investigator, Linus Pauling Institute
Professor, Dept. of Biochemistry and Biophysics
Helen P. Rumbel Professor for Healthy Aging Research
Oregon State University
Copyright 2002-2024 Linus Pauling Institute
References
1. Reed LJ. A trail of research from lipoic acid to alpha-keto acid dehydrogenase complexes. J Biol Chem. 2001;276(42):38329-38336. (PubMed)
2. Carreau JP. Biosynthesis of lipoic acid via unsaturated fatty acids. Methods Enzymol. 1979;62:152-158. (PubMed)
3. Smith AR, Shenvi SV, Widlansky M, Suh JH, Hagen TM. Lipoic acid as a potential therapy for chronic diseases associated with oxidative stress. Curr Med Chem. 2004;11(9):1135-1146. (PubMed)
4. Kramer K, Packer L. R-alpha-lipoic acid. In: Kramer K, Hoppe P, Packer L, eds. Nutraceuticals in Health and Disease Prevention. New York: Marcel Dekker, Inc.; 2001:129-164.
5. Mayr JA, Feichtinger RG, Tort F, Ribes A, Sperl W. Lipoic acid biosynthesis defects. J Inherit Metab Dis. 2014;37(4):553-563. (PubMed)
6. Hermann R, Niebch G, Borbe H, et al. Enantioselective pharmacokinetics and bioavailability of different racemic alpha-lipoic acid formulations in healthy volunteers. Eur J Pharm Sci. 1996;4(3):167-174.
7. Teichert J, Hermann R, Ruus P, Preiss R. Plasma kinetics, metabolism, and urinary excretion of alpha-lipoic acid following oral administration in healthy volunteers. J Clin Pharmacol. 2003;43(11):1257-1267. (PubMed)
8. Gleiter CH, Schug BS, Hermann R, Elze M, Blume HH, Gundert-Remy U. Influence of food intake on the bioavailability of thioctic acid enantiomers. Eur J Clin Pharmacol. 1996;50(6):513-514. (PubMed)
9. Brufani M, Figliola R. (R)-alpha-lipoic acid oral liquid formulation: pharmacokinetic parameters and therapeutic efficacy. Acta Biomed. 2014;85(2):108-115. (PubMed)
10. Maglione E, Marrese C, Migliaro E, et al. Increasing bioavailability of (R)-alpha-lipoic acid to boost antioxidant activity in the treatment of neuropathic pain. Acta Biomed. 2015;86(3):226-233. (PubMed)
11. Breithaupt-Grogler K, Niebch G, Schneider E, et al. Dose-proportionality of oral thioctic acid--coincidence of assessments via pooled plasma and individual data. Eur J Pharm Sci. 1999;8(1):57-65. (PubMed)
12. Evans JL, Heymann CJ, Goldfine ID, Gavin LA. Pharmacokinetics, tolerability, and fructosamine-lowering effect of a novel, controlled-release formulation of alpha-lipoic acid. Endocr Pract. 2002;8(1):29-35. (PubMed)
13. Keith DJ, Butler JA, Bemer B, et al. Age and gender dependent bioavailability of R- and R,S-alpha-lipoic acid: a pilot study. Pharmacol Res. 2012;66(3):199-206. (PubMed)
14. Hiltunen JK, Autio KJ, Schonauer MS, Kursu VA, Dieckmann CL, Kastaniotis AJ. Mitochondrial fatty acid synthesis and respiration. Biochim Biophys Acta. 2010;1797(6-7):1195-1202. (PubMed)
15. Bustamante J, Lodge JK, Marcocci L, Tritschler HJ, Packer L, Rihn BH. Alpha-lipoic acid in liver metabolism and disease. Free Radic Biol Med. 1998;24(6):1023-1039. (PubMed)
16. Jones W, Li X, Qu ZC, Perriott L, Whitesell RR, May JM. Uptake, recycling, and antioxidant actions of alpha-lipoic acid in endothelial cells. Free Radic Biol Med. 2002;33(1):83-93. (PubMed)
17. Kozlov AV, Gille L, Staniek K, Nohl H. Dihydrolipoic acid maintains ubiquinone in the antioxidant active form by two-electron reduction of ubiquinone and one-electron reduction of ubisemiquinone. Arch Biochem Biophys. 1999;363(1):148-154. (PubMed)
18. May JM, Qu ZC, Mendiratta S. Protection and recycling of alpha-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch Biochem Biophys. 1998;349(2):281-289. (PubMed)
19. Upston JM, Terentis AC, Stocker R. Tocopherol-mediated peroxidation of lipoproteins: implications for vitamin E as a potential antiatherogenic supplement. Faseb J. 1999;13(9):977-994. (PubMed)
20. Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12(10):1161-1208. (PubMed)
21. Doraiswamy PM, Finefrock AE. Metals in our minds: therapeutic implications for neurodegenerative disorders. Lancet Neurol. 2004;3(7):431-434. (PubMed)
22. Ou P, Tritschler HJ, Wolff SP. Thioctic (lipoic) acid: a therapeutic metal-chelating antioxidant? Biochem Pharmacol. 1995;50(1):123-126. (PubMed)
23. Suh JH, Zhu BZ, deSzoeke E, Frei B, Hagen TM. Dihydrolipoic acid lowers the redox activity of transition metal ions but does not remove them from the active site of enzymes. Redox Rep. 2004;9(1):57-61. (PubMed)
24. Suh JH, Moreau R, Heath SH, Hagen TM. Dietary supplementation with (R)-alpha-lipoic acid reverses the age-related accumulation of iron and depletion of antioxidants in the rat cerebral cortex. Redox Rep. 2005;10(1):52-60. (PubMed)
25. Yamamoto H, Watanabe T, Mizuno H, et al. The antioxidant effect of DL-alpha-lipoic acid on copper-induced acute hepatitis in Long-Evans Cinnamon (LEC) rats. Free Radic Res. 2001;34(1):69-80. (PubMed)
26. Patrick L. Mercury toxicity and antioxidants: Part 1: role of glutathione and alpha-lipoic acid in the treatment of mercury toxicity. Altern Med Rev. 2002;7(6):456-471. (PubMed)
27. Rooney JP. The role of thiols, dithiols, nutritional factors and interacting ligands in the toxicology of mercury. Toxicology. 2007;234(3):145-156. (PubMed)
28. Hagen TM, Vinarsky V, Wehr CM, Ames BN. (R)-alpha-lipoic acid reverses the age-associated increase in susceptibility of hepatocytes to tert-butylhydroperoxide both in vitro and in vivo. Antioxid Redox Signal. 2000;2(3):473-483. (PubMed)
29. Busse E, Zimmer G, Schopohl B, Kornhuber B. Influence of alpha-lipoic acid on intracellular glutathione in vitro and in vivo. Arzneimittelforschung. 1992;42(6):829-831. (PubMed)
30. Monette JS, Gomez LA, Moreau RF, et al. (R)-alpha-Lipoic acid treatment restores ceramide balance in aging rat cardiac mitochondria. Pharmacol Res. 2011;63(1):23-29. (PubMed)
31. Suh JH, Shenvi SV, Dixon BM, et al. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci U S A. 2004;101(10):3381-3386. (PubMed)
32. Suh JH, Wang H, Liu RM, Liu J, Hagen TM. (R)-alpha-lipoic acid reverses the age-related loss in GSH redox status in post-mitotic tissues: evidence for increased cysteine requirement for GSH synthesis. Arch Biochem Biophys. 2004;423(1):126-135. (PubMed)
33. Zhang J, Zhou X, Wu W, Wang J, Xie H, Wu Z. Regeneration of glutathione by alpha-lipoic acid via Nrf2/ARE signaling pathway alleviates cadmium-induced HepG2 cell toxicity. Environ Toxicol Pharmacol. 2017;51:30-37. (PubMed)
34. Fratantonio D, Speciale A, Molonia MS, et al. Alpha-lipoic acid, but not di-hydrolipoic acid, activates Nrf2 response in primary human umbilical-vein endothelial cells and protects against TNF-alpha induced endothelium dysfunction. Arch Biochem Biophys. 2018;655:18-25. (PubMed)
35. Sena CM, Cipriano MA, Botelho MF, Seica RM. Lipoic acid prevents high-fat diet-induced hepatic steatosis in Goto Kakizaki rats by reducing oxidative stress through Nrf2 activation. Int J Mol Sci. 2018;19(9). (PubMed)
36. Pilar Valdecantos M, Prieto-Hontoria PL, Pardo V, et al. Essential role of Nrf2 in the protective effect of lipoic acid against lipoapoptosis in hepatocytes. Free Radic Biol Med. 2015;84:263-278. (PubMed)
37. Fayez AM, Zakaria S, Moustafa D. Alpha lipoic acid exerts antioxidant effect via Nrf2/HO-1 pathway activation and suppresses hepatic stellate cells activation induced by methotrexate in rats. Biomed Pharmacother. 2018;105:428-433. (PubMed)
38. Lin YC, Lai YS, Chou TC. The protective effect of alpha-lipoic Acid in lipopolysaccharide-induced acute lung injury is mediated by heme oxygenase-1. Evid Based Complement Alternat Med. 2013;2013:590363. (PubMed)
39. Segal AW. The function of the NADPH oxidase of phagocytes and its relationship to other NOXs in plants, invertebrates, and mammals. Int J Biochem Cell Biol. 2008;40(4):604-618. (PubMed)
40. Dong Y, Wang H, Chen Z. Alpha-lipoic acid attenuates cerebral ischemia and reperfusion injury via insulin receptor and PI3K/Akt-dependent inhibition of NADPH oxidase. Int J Endocrinol. 2015;2015:903186. (PubMed)
41. Byun E, Lim JW, Kim JM, Kim H. alpha-Lipoic acid inhibits Helicobacter pylori-induced oncogene expression and hyperproliferation by suppressing the activation of NADPH oxidase in gastric epithelial cells. Mediators Inflamm. 2014;2014:380830. (PubMed)
42. Konrad D. Utilization of the insulin-signaling network in the metabolic actions of alpha-lipoic acid-reduction or oxidation? Antioxid Redox Signal. 2005;7(7-8):1032-1039. (PubMed)
43. Diesel B, Kulhanek-Heinze S, Holtje M, et al. Alpha-lipoic acid as a directly binding activator of the insulin receptor: protection from hepatocyte apoptosis. Biochemistry. 2007;46(8):2146-2155. (PubMed)
44. Estrada DE, Ewart HS, Tsakiridis T, et al. Stimulation of glucose uptake by the natural coenzyme alpha-lipoic acid/thioctic acid: participation of elements of the insulin signaling pathway. Diabetes. 1996;45(12):1798-1804. (PubMed)
45. Yaworsky K, Somwar R, Ramlal T, Tritschler HJ, Klip A. Engagement of the insulin-sensitive pathway in the stimulation of glucose transport by alpha-lipoic acid in 3T3-L1 adipocytes. Diabetologia. 2000;43(3):294-303. (PubMed)
46. Ying Z, Kampfrath T, Sun Q, Parthasarathy S, Rajagopalan S. Evidence that alpha-lipoic acid inhibits NF-kappaB activation independent of its antioxidant function. Inflamm Res. 2011;60(3):219-225. (PubMed)
47. Smith AR, Hagen TM. Vascular endothelial dysfunction in aging: loss of Akt-dependent endothelial nitric oxide synthase phosphorylation and partial restoration by (R)-alpha-lipoic acid. Biochem Soc Trans. 2003;31(Pt 6):1447-1449. (PubMed)
48. Wang Y, Li X, Guo Y, Chan L, Guan X. alpha-Lipoic acid increases energy expenditure by enhancing adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor-gamma coactivator-1alpha signaling in the skeletal muscle of aged mice. Metabolism. 2010;59(7):967-976. (PubMed)
49. Moura FA, de Andrade KQ, dos Santos JC, Goulart MO. Lipoic acid: its antioxidant and anti-inflammatory role and clinical applications. Curr Top Med Chem. 2015;15(5):458-483. (PubMed)
50. Packer L, Cadenas E. Lipoic acid: energy metabolism and redox regulation of transcription and cell signaling. J Clin Biochem Nutr. 2011;48(1):26-32. (PubMed)
51. Rochette L, Ghibu S, Richard C, Zeller M, Cottin Y, Vergely C. Direct and indirect antioxidant properties of alpha-lipoic acid and therapeutic potential. Mol Nutr Food Res. 2013;57(1):114-125. (PubMed)
52. Shay KP, Moreau RF, Smith EJ, Smith AR, Hagen TM. Alpha-lipoic acid as a dietary supplement: molecular mechanisms and therapeutic potential. Biochim Biophys Acta. 2009;1790(10):1149-1160. (PubMed)
53. Mayr JA, Zimmermann FA, Fauth C, et al. Lipoic acid synthetase deficiency causes neonatal-onset epilepsy, defective mitochondrial energy metabolism, and glycine elevation. Am J Hum Genet. 2011;89(6):792-797. (PubMed)
54. Tort F, Ferrer-Cortes X, Thio M, et al. Mutations in the lipoyltransferase LIPT1 gene cause a fatal disease associated with a specific lipoylation defect of the 2-ketoacid dehydrogenase complexes. Hum Mol Genet. 2014;23(7):1907-1915. (PubMed)
55. Ziegler D. Thioctic acid for patients with symptomatic diabetic polyneuropathy: a critical review. Treat Endocrinol. 2004;3(3):173-189. (PubMed)
56. Nathan DM, Davidson MB, DeFronzo RA, et al. Impaired fasting glucose and impaired glucose tolerance: implications for care. Diabetes Care. 2007;30(3):753-759. (PubMed)
57. Jacob S, Henriksen EJ, Schiemann AL, et al. Enhancement of glucose disposal in patients with type 2 diabetes by alpha-lipoic acid. Arzneimittelforschung. 1995;45(8):872-874. (PubMed)
58. Jacob S, Rett K, Henriksen EJ, Haring HU. Thioctic acid--effects on insulin sensitivity and glucose-metabolism. Biofactors. 1999;10(2-3):169-174. (PubMed)
59. de Oliveira AM, Rondo PH, Luzia LA, D'Abronzo FH, Illison VK. The effects of lipoic acid and alpha-tocopherol supplementation on the lipid profile and insulin sensitivity of patients with type 2 diabetes mellitus: a randomized, double-blind, placebo-controlled trial. Diabetes Res Clin Pract. 2011;92(2):253-260. (PubMed)
60. Akbari M, Ostadmohammadi V, Lankarani KB, et al. The effects of alpha-lipoic acid supplementation on glucose control and lipid profiles among patients with metabolic diseases: A systematic review and meta-analysis of randomized controlled trials. Metabolism. 2018;87:56-69. (PubMed)
61. Roberts AC, Porter KE. Cellular and molecular mechanisms of endothelial dysfunction in diabetes. Diab Vasc Dis Res. 2013;10(6):472-482. (PubMed)
62. Heitzer T, Finckh B, Albers S, Krohn K, Kohlschutter A, Meinertz T. Beneficial effects of alpha-lipoic acid and ascorbic acid on endothelium-dependent, nitric oxide-mediated vasodilation in diabetic patients: relation to parameters of oxidative stress. Free Radic Biol Med. 2001;31(1):53-61. (PubMed)
63. Heinisch BB, Francesconi M, Mittermayer F, et al. Alpha-lipoic acid improves vascular endothelial function in patients with type 2 diabetes: a placebo-controlled randomized trial. Eur J Clin Invest. 2010;40(2):148-154. (PubMed)
64. Xiang G, Pu J, Yue L, Hou J, Sun H. alpha-lipoic acid can improve endothelial dysfunction in subjects with impaired fasting glucose. Metabolism. 2011;60(4):480-485. (PubMed)
65. Xiang GD, Sun HL, Zhao LS, Hou J, Yue L, Xu L. The antioxidant alpha-lipoic acid improves endothelial dysfunction induced by acute hyperglycaemia during OGTT in impaired glucose tolerance. Clin Endocrinol (Oxf). 2008;68(5):716-723. (PubMed)
66. Sola S, Mir MQ, Cheema FA, et al. Irbesartan and lipoic acid improve endothelial function and reduce markers of inflammation in the metabolic syndrome: results of the Irbesartan and Lipoic Acid in Endothelial Dysfunction (ISLAND) study. Circulation. 2005;111(3):343-348. (PubMed)
67. National Institute of Diabetes and Digestive and Kidney Diseases. Diabetic Neuropathy. Available at: https://www.niddk.nih.gov/health-information/diabetes/overview/preventing-problems/nerve-damage-diabetic-neuropathies. Accessed 9/23/18.
68. Malik RA, Tesfaye S, Ziegler D. Medical strategies to reduce amputation in patients with type 2 diabetes. Diabet Med. 2013;30(8):893-900. (PubMed)
69. Obrosova IG. Diabetes and the peripheral nerve. Biochim Biophys Acta. 2009;1792(10):931-940. (PubMed)
70. Dy SM, Bennett WL, Sharma R, et al. AHRQ Comparative Effectiveness Reviews. Preventing complications and treating symptoms of diabetic peripheral neuropathy. Rockville (MD): Agency for Healthcare Research and Quality (US); 2017. (PubMed)
71. The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329(14):977-986. (PubMed)
72. UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet. 1998;352(9131):837-853. (PubMed)
73. Callaghan BC, Little AA, Feldman EL, Hughes RA. Enhanced glucose control for preventing and treating diabetic neuropathy. Cochrane Database Syst Rev. 2012(6):Cd007543. (PubMed)
74. Han T, Bai J, Liu W, Hu Y. A systematic review and meta-analysis of alpha-lipoic acid in the treatment of diabetic peripheral neuropathy. Eur J Endocrinol. 2012;167(4):465-471. (PubMed)
75. Ruhnau KJ, Meissner HP, Finn JR, et al. Effects of 3-week oral treatment with the antioxidant thioctic acid (alpha-lipoic acid) in symptomatic diabetic polyneuropathy. Diabet Med. 1999;16(12):1040-1043. (PubMed)
76. Ziegler D, Hanefeld M, Ruhnau KJ, et al. Treatment of symptomatic diabetic polyneuropathy with the antioxidant alpha-lipoic acid: a 7-month multicenter randomized controlled trial (ALADIN III Study). ALADIN III Study Group. Alpha-lipoic acid in diabetic neuropathy. Diabetes Care. 1999;22(8):1296-1301. (PubMed)
77. Ziegler D, Ametov A, Barinov A, et al. Oral treatment with alpha-lipoic acid improves symptomatic diabetic polyneuropathy: the SYDNEY 2 trial. Diabetes Care. 2006;29(11):2365-2370. (PubMed)
78. Ziegler D, Low PA, Litchy WJ, et al. Efficacy and safety of antioxidant treatment with alpha-lipoic acid over 4 years in diabetic polyneuropathy: the NATHAN 1 trial. Diabetes Care. 2011;34(9):2054-2060. (PubMed)
79. Ziegler D, Low PA, Freeman R, Tritschler H, Vinik AI. Predictors of improvement and progression of diabetic polyneuropathy following treatment with alpha-lipoic acid for 4 years in the NATHAN 1 trial. J Diabetes Complications. 2016;30(2):350-356. (PubMed)
80. Balcioglu AS, Muderrisoglu H. Diabetes and cardiac autonomic neuropathy: Clinical manifestations, cardiovascular consequences, diagnosis and treatment. World J Diabetes. 2015;6(1):80-91. (PubMed)
81. Ziegler D, Schatz H, Conrad F, Gries FA, Ulrich H, Reichel G. Effects of treatment with the antioxidant alpha-lipoic acid on cardiac autonomic neuropathy in NIDDM patients. A 4-month randomized controlled multicenter trial (DEKAN Study). Deutsche Kardiale Autonome Neuropathie. Diabetes Care. 1997;20(3):369-373. (PubMed)
82. Nguyen N, Takemoto JK. A case for alpha-lipoic acid as an alternative treatment for diabetic polyneuropathy. J Pharm Pharm Sci. 2018;21(1s):177s-191s. (PubMed)
83. National Institute of Diabetes and Digestive and Kidney Diseases. Diabetic Eye Disease. Available at: https://www.niddk.nih.gov/health-information/diabetes/overview/preventing-problems/diabetic-eye-disease. Accessed 9/24/18.
84. Gebka A, Serkies-Minuth E, Raczynska D. Effect of the administration of alpha-lipoic acid on contrast sensitivity in patients with type 1 and type 2 diabetes. Mediators Inflamm. 2014;2014:131538. (PubMed)
85. National Multiple Sclerosis Society. Definition of Multiple Sclerosis (MS). Available at: https://www.nationalmssociety.org/What-is-MS/Definition-of-MS. Accessed 9/28/18.
86. National Multiple Sclerosis Society. Types of Multiple Sclerosis (MS). Available at: https://www.nationalmssociety.org/What-is-MS/Types-of-MS. Accessed 9/28/18.
87. Marracci GH, Jones RE, McKeon GP, Bourdette DN. Alpha lipoic acid inhibits T cell migration into the spinal cord and suppresses and treats experimental autoimmune encephalomyelitis. J Neuroimmunol. 2002;131(1-2):104-114. (PubMed)
88. Morini M, Roccatagliata L, Dell'Eva R, et al. Alpha-lipoic acid is effective in prevention and treatment of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2004;148(1-2):146-153. (PubMed)
89. Schreibelt G, Musters RJ, Reijerkerk A, et al. Lipoic acid affects cellular migration into the central nervous system and stabilizes blood-brain barrier integrity. J Immunol. 2006;177(4):2630-2637. (PubMed)
90. Salinthone S, Schillace RV, Marracci GH, Bourdette DN, Carr DW. Lipoic acid stimulates cAMP production via the EP2 and EP4 prostanoid receptors and inhibits IFN gamma synthesis and cellular cytotoxicity in NK cells. J Neuroimmunol. 2008;199(1-2):46-55. (PubMed)
91. Schillace RV, Pisenti N, Pattamanuch N, et al. Lipoic acid stimulates cAMP production in T lymphocytes and NK cells. Biochem Biophys Res Commun. 2007;354(1):259-264. (PubMed)
92. George JD, Kim E, Spain R, Bourdette D, Salinthone S. Effects of lipoic acid on migration of human B cells and monocyte-enriched peripheral blood mononuclear cells in relapsing remitting multiple sclerosis. J Neuroimmunol. 2018;315:24-27. (PubMed)
93. Chaudhary P, Marracci GH, Bourdette DN. Lipoic acid inhibits expression of ICAM-1 and VCAM-1 by CNS endothelial cells and T cell migration into the spinal cord in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2006;175(1-2):87-96. (PubMed)
94. Marracci GH, McKeon GP, Marquardt WE, Winter RW, Riscoe MK, Bourdette DN. Alpha lipoic acid inhibits human T-cell migration: implications for multiple sclerosis. J Neurosci Res. 2004;78(3):362-370. (PubMed)
95. Yadav V, Marracci G, Lovera J, et al. Lipoic acid in multiple sclerosis: a pilot study. Mult Scler. 2005;11(2):159-165. (PubMed)
96. Yadav V, Marracci GH, Munar MY, et al. Pharmacokinetic study of lipoic acid in multiple sclerosis: comparing mice and human pharmacokinetic parameters. Mult Scler. 2010;16(4):387-397. (PubMed)
97. Khalili M, Eghtesadi S, Mirshafiey A, et al. Effect of lipoic acid consumption on oxidative stress among multiple sclerosis patients: a randomized controlled clinical trial. Nutr Neurosci. 2014;17(1):16-20. (PubMed)
98. Khalili M, Azimi A, Izadi V, et al. Does lipoic acid consumption affect the cytokine profile in multiple sclerosis patients: a double-blind, placebo-controlled, randomized clinical trial. Neuroimmunomodulation. 2014;21(6):291-296. (PubMed)
99. Khalili M, Soltani M, Moghadam SA, Dehghan P, Azimi A, Abbaszadeh O. Effect of alpha-lipoic acid on asymmetric dimethylarginine and disability in multiple sclerosis patients: A randomized clinical trial. Electron Physician. 2017;9(7):4899-4905. (PubMed)
100. Molz P, Schroder N. Potential therapeutic effects of lipoic acid on memory deficits related to aging and neurodegeneration. Front Pharmacol. 2017;8:849. (PubMed)
101. Hager K, Marahrens A, Kenklies M, Riederer P, Munch G. Alpha-lipoic acid as a new treatment option for Azheimer type dementia. Arch Gerontol Geriatr. 2001;32(3):275-282. (PubMed)
102. Hager K, Kenklies M, McAfoose J, Engel J, Munch G. Alpha-lipoic acid as a new treatment option for Alzheimer's disease--a 48 months follow-up analysis. J Neural Transm Suppl. 2007(72):189-193. (PubMed)
103. Dana Consortium on the Therapy of HIV Dementia and Related Cognitive Disorders. A randomized, double-blind, placebo-controlled trial of deprenyl and thioctic acid in human immunodeficiency virus-associated cognitive impairment. Neurology. 1998;50(3):645-651. (PubMed)
104. Shinto L, Quinn J, Montine T, et al. A randomized placebo-controlled pilot trial of omega-3 fatty acids and alpha lipoic acid in Alzheimer's disease. J Alzheimers Dis. 2014;38(1):111-120. (PubMed)
105. Namazi N, Larijani B, Azadbakht L. Alpha-lipoic acid supplement in obesity treatment: A systematic review and meta-analysis of clinical trials. Clin Nutr. 2018;37(2):419-428. (PubMed)
106. Kucukgoncu S, Zhou E, Lucas KB, Tek C. Alpha-lipoic acid (ALA) as a supplementation for weight loss: results from a meta-analysis of randomized controlled trials. Obes Rev. 2017;18(5):594-601. (PubMed)
107. Lodge JK, Youn HD, Handelman GJ, et al. Natural sources of lipic acid: determination of lipoyllysine released from protease-digested tissues by high performance liquid chromatography incorporating electrochemical detection. J Appl Nutr. 1997;49(1 & 2):3-11.
108. Biewenga GP, Haenen GR, Bast A. The pharmacology of the antioxidant lipoic acid. Gen Pharmacol. 1997;29(3):315-331. (PubMed)
109. ConsumerLab.com. Alpha-Lipoic Acid Supplements Review July 2017. Available at: https://www.consumerlab.com/reviews/Alpha-Lipoic_Acid_Supplements/alphalipoic/. Accessed 9/27/18.
110. Streeper RS, Henriksen EJ, Jacob S, Hokama JY, Fogt DL, Tritschler HJ. Differential effects of lipoic acid stereoisomers on glucose metabolism in insulin-resistant skeletal muscle. Am J Physiol. 1997;273(1 Pt 1):E185-191. (PubMed)
111. Maitra I, Serbinova E, Tritschler HJ, Packer L. Stereospecific effects of R-lipoic acid on buthionine sulfoximine-induced cataract formation in newborn rats. Biochem Biophys Res Commun. 1996;221(2):422-429. (PubMed)
112. Ziegler D, Nowak H, Kempler P, Vargha P, Low PA. Treatment of symptomatic diabetic polyneuropathy with the antioxidant alpha-lipoic acid: a meta-analysis. Diabet Med. 2004;21(2):114-121. (PubMed)
113. Reljanovic M, Reichel G, Rett K, et al. Treatment of diabetic polyneuropathy with the antioxidant thioctic acid (alpha-lipoic acid): a two year multicenter randomized double-blind placebo-controlled trial (ALADIN II). Alpha Lipoic Acid in Diabetic Neuropathy. Free Radic Res. 1999;31(3):171-179. (PubMed)
114. Parente E, Colannino G, Picconi O, Monastra G. Safety of oral alpha-lipoic acid treatment in pregnant women: a retrospective observational study. Eur Rev Med Pharmacol Sci. 2017;21(18):4219-4227. (PubMed)
115. Natural Medicines. Alpha-Lipoic Acid/Safety - Professional Handout. Available at: https://naturalmedicines.therapeuticresearch.com. Accessed 9/26/18.
116. Karaarslan U, Isguder R, Bag O, Kisla M, Agin H, Unal N. Alpha lipoic acid intoxication, treatment and outcome. Clin Toxicol (Phila). 2013;51(6):522. (PubMed)
117. Hadzik B, Grass H, Mayatepek E, Daldrup T, Hoehn T. Fatal non-accidental alpha-lipoic acid intoxication in an adolescent girl. Klin Padiatr. 2014;226(5):292-294. (PubMed)
118. Natural Medicines. Alpha-Lipoic acid/Interactions with Drugs - Professional handout. Available at: https://naturalmedicines.therapeuticresearch.com. Accessed 9/25/18.
119. Gleiter CH, Schreeb KH, Freudenthaler S, et al. Lack of interaction between thioctic acid, glibenclamide and acarbose. Br J Clin Pharmacol. 1999;48(6):819-825. (PubMed)
120. Prasad PD, Wang H, Huang W, et al. Molecular and functional characterization of the intestinal Na+-dependent multivitamin transporter. Arch Biochem Biophys. 1999;366(1):95-106. (PubMed)
121. Balamurugan K, Vaziri ND, Said HM. Biotin uptake by human proximal tubular epithelial cells: cellular and molecular aspects. Am J Physiol Renal Physiol. 2005;288(4):F823-831. (PubMed)
122. Zempleni J, Trusty TA, Mock DM. Lipoic acid reduces the activities of biotin-dependent carboxylases in rat liver. J Nutr. 1997;127(9):1776-1781. (PubMed)
123. Zempleni J, Mock DM. Biotin biochemistry and human requirements. J Nutr Biochem. 1999;10(3):128-138. (PubMed)
Phytochemicals
Phytochemicals can be defined, in the strictest sense, as chemicals produced by plants. However, the term is generally used to describe chemicals from plants that may affect health, but are not essential nutrients. While there is ample evidence to support the health benefits of diets rich in fruit, vegetables, legumes, whole grains, and nuts, evidence that these effects are due to specific nutrients or phytochemicals is limited. Because plant-based foods are complex mixtures of bioactive compounds, information on the potential health effects of individual phytochemicals is linked to information on the health effects of foods that contain those phytochemicals.
Select a phytochemical from the list for more information.
The information on dietary phytochemicals from the Linus Pauling Institute's Micronutrient Information Center is now available in a book titled, An Evidence-based Approach to Phytochemicals and Other Dietary Factors. The book can be purchased from the Linus Pauling Institute or Thieme Medical Publishers.
Carotenoids
Contents
α-Carotene, β-Carotene, β-Cryptoxanthin, Lycopene, Lutein, and Zeaxanthin
Summary
- Carotenoids are yellow, orange, and red pigments synthesized by plants. The most common carotenoids in North American diets are α-carotene, β-carotene, β-cryptoxanthin, lutein, zeaxanthin, and lycopene. (More information)
- Provitamin A carotenoids, α-carotene, β-carotene, and β-cryptoxanthin, can be converted by the body to retinol (vitamin A). In contrast, no vitamin A activity can be derived from lutein, zeaxanthin, and lycopene. (More information)
- Dietary lutein and zeaxanthin are selectively taken up into the macula of the eye, where they absorb up to 90% of blue light and help maintain optimal visual function. (More information)
- At present, it is unclear whether the biological effects of carotenoids in humans are related to their antioxidant activity and/or other non-antioxidant activities. (More information)
- Although the results of observational studies suggest that diets high in carotenoid-rich fruit and vegetables are associated with reduced risks of cardiovascular disease and some cancers, high-dose β-carotene supplements did not reduce the risk of cardiovascular disease or cancer in large randomized controlled trials. (More information)
- Two randomized controlled trials found that high-dose β-carotene supplementation increased the risk of lung cancer in smokers and former asbestos workers. (More information)
- Meta-analyses of observational studies have reported an inverse association between dietary lycopene intake or blood lycopene concentration and risk of developing prostate cancer. To date, most small-scale intervention studies have found little-to-no benefit of lycopene supplements in reducing the incidence or severity of prostate cancer in high-risk patients. (More information)
- Observational studies have suggested that diets rich in lutein and zeaxanthin may help slow the development of age-related macular degeneration (AMD). Randomized controlled trials in subjects with AMD have found that lutein and zeaxanthin supplementation improves visual acuity and slows the progression to advanced AMD in subjects with AMD. (More information)
- Evidence is lacking to suggest a role for lutein and zeaxanthin in the management of other eye conditions, including cataracts, diabetic retinopathy, and retinopathy of maturity. (More information)
- Carotenoids are best absorbed with fat in a meal. Chopping, puréeing, and cooking carotenoid-containing vegetables in oil generally increase the bioavailability of the carotenoids they contain. (More information)
Introduction
Carotenoids are a class of more than 750 naturally occurring pigments synthesized by plants, algae, and photosynthetic bacteria (1). These richly colored molecules are the sources of the yellow, orange, and red colors of many plants. Fruit and vegetables provide most of the 40 to 50 carotenoids found in the human diet. α-Carotene, β-carotene, β-cryptoxanthin, lutein, zeaxanthin, and lycopene are the most common dietary carotenoids (1). α-Carotene, β-carotene and β-cryptoxanthin are provitamin A carotenoids, meaning they can be converted by the body to retinol (Figure 1). Lutein, zeaxanthin, and lycopene are nonprovitamin A carotenoids because they cannot be converted to retinol (Figure 2).
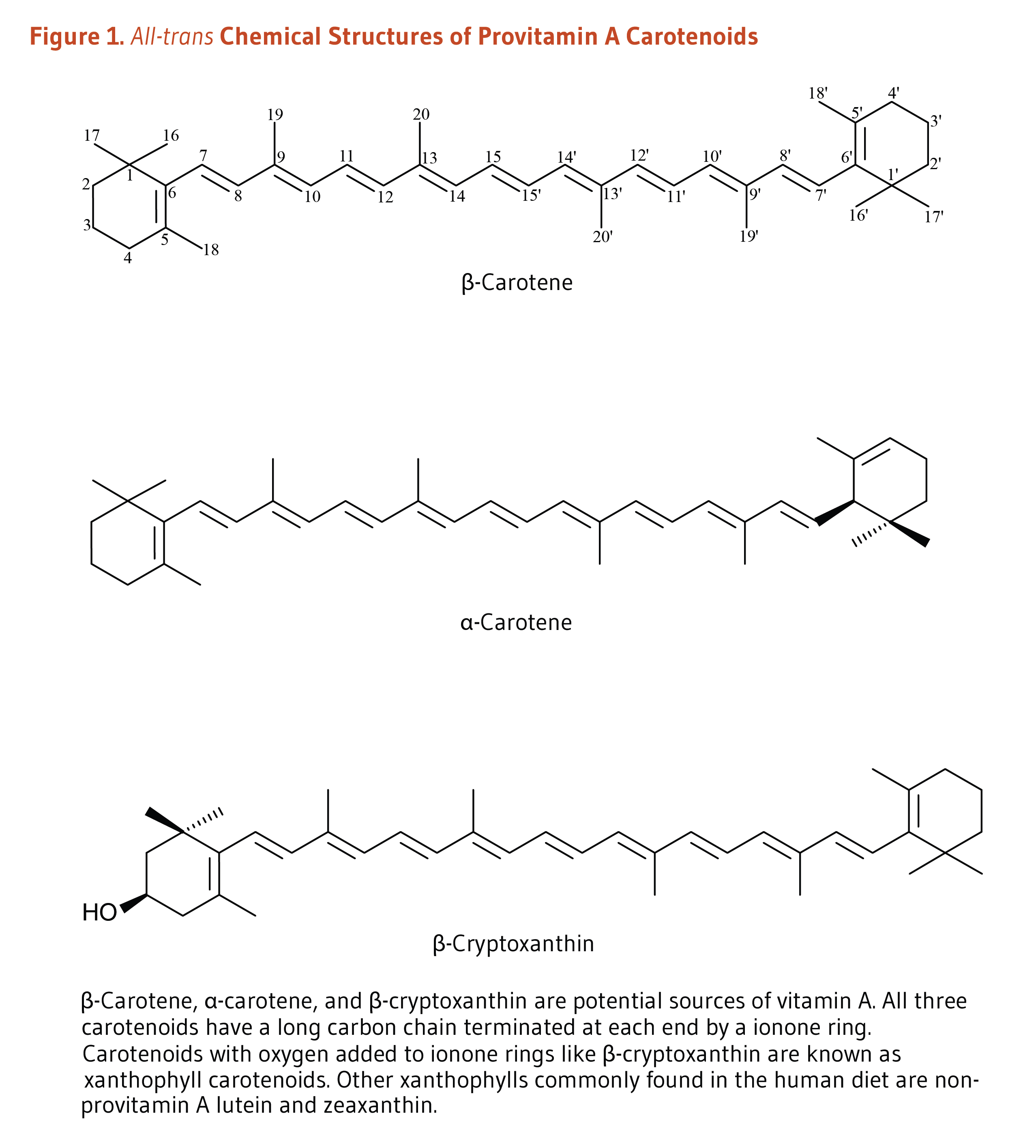
Absorption, Metabolism, and Bioavailability
For dietary carotenoids to be absorbed intestinally, they must be released from the food matrix and incorporated into mixed micelles (mixtures of bile salts and several types of lipids). Food processing and cooking help release carotenoids embedded in their food matrix and increase intestinal absorption (1). Moreover, carotenoid absorption requires the presence of fat in a meal. As little as 3 to 5 g of fat in a meal appears sufficient to ensure carotenoid absorption (2, 3), although the minimum amount of dietary fat required may be different for each carotenoid. The type of fat (e.g., medium-chain vs. long-chain triglycerides), the presence of soluble fiber, and the type and amount of carotenoids (e.g., esterified vs. non-esterified) in the food also appear to influence the rate and extent of carotenoid absorption (reviewed in 4). Because they do not need to be released from the plant matrix, carotenoid supplements (in oil) are more efficiently absorbed than carotenoids in food (3, 5). Although carotenoids were initially thought to be absorbed within the cells that line the intestine (enterocytes) only by passive diffusion, carotenoids are also actively absorbed via the apical membrane transporters, Scavenger Receptor-class B type I (SR-BI), Cluster Determinant 36 (CD36), and Niemann-Pick C1 like intracellular transporter 1 (NPC1L1) (6-8).
Within the enterocytes, provitamin A carotenoids may be cleaved by either β-carotene 15,15’-oxygenase 1 (BCO1) or by β-carotene 9’,10’-oxygenase 2 (BCO2) (Figure 3). BCO1 catalyzes the cleavage of provitamin A carotenoids into retinal, which is further reduced to retinol (vitamin A) or oxidized to retinoic acid (the biologically active form of vitamin A). β-Apocarotenal derived from the cleavage of β-carotene by BCO2 can be cleaved further by BCO1 to produce retinal. Although provitamin A carotenoids can be converted into apocarotenals by BCO2, the activity of this enzyme is higher toward nonprovitamin A carotenoids. Conversely, BCO1 shows limited affinity toward nonprovitamin A carotenoids (1).
Within the enterocytes, uncleaved carotenoids and retinyl esters (derived from retinol) are incorporated into triglyceride-rich lipoproteins called chylomicrons, secreted into lymphatic vessels, and then released into the bloodstream (1). Triglycerides are depleted from circulating chylomicrons through the activity of an enzyme called lipoprotein lipase, resulting in the formation of chylomicron remnants. Chylomicron remnants are taken up by the liver, where carotenoids can be cleaved by BCO1/BCO2 or incorporated into lipoproteins and secreted back into the circulation for delivery to extrahepatic tissues. Of note, more hydrophilic molecules in the enterocytes like retinoic acid and apocarotenals can be transported directly to the liver through the portal blood system.
The conversion of provitamin A carotenoids to retinol is influenced by the vitamin A status of the individual (9). The regulatory mechanism involving the intestine-specific homeobox (ISX) transcription factor can block carotenoid uptake and vitamin A production by inhibiting the expression of SR-BI and BCO1. ISX is under the control of retinoic acid and retinoic acid receptor (RAR)-dependent mechanisms such that, when vitamin A stores are high, ISX is activated and both provitamin A carotenoid absorption and conversion to retinol are inhibited. Conversely, during vitamin A insufficiency, the expression of both SR-BI and BCO1 is no longer repressed by ISX, allowing for provitamin A carotenoid absorption and conversion to retinol (1).
Interindividual variations in blood and tissue concentrations of carotenoids have been attributed to genetic differences among individuals. Specifically, a number of single nucleotide polymorphisms (SNPs) — corresponding to changes of one nucleotide in the sequence of genes — have been identified in genes coding for proteins involved in intestinal uptake, transport, and metabolism of carotenoids (10). Specifically, SNPs within genes coding for SR-BI, CD36, and BCO1 are suspected to affect the expression and/or activity of these proteins and, in turn, individual carotenoid status (10). For more information genetic variants affecting carotenoid status, see the review by Moran et al. (5).
Biological Activities
Provitamin A function
Vitamin A is essential for normal growth and development, immune system function, and vision (see the article on Vitamin A). Currently, the only essential function of carotenoids recognized in humans is that of the provitamin A carotenoids, α-carotene, β-carotene, and β-cryptoxanthin, to serve as a source of vitamin A (11).
Retinol activity equivalents (RAEs)
Provitamin A carotenoids are less easily absorbed than preformed vitamin A and must be converted to retinol and other retinoids by the body (see Figure 3). The efficiency of conversion of provitamin A carotenoids into retinol is highly variable, depending on factors like food matrix, food preparation, and one’s digestive and absorptive capacities (12).
The most recent international standard of measure for vitamin A is retinol activity equivalent (RAE), which represents vitamin A activity as retinol. It has been determined that 2 micrograms (µg) of β-carotene in oil provided as a supplement could be converted by the body to 1 µg of retinol, giving it an RAE ratio of 2:1. However, 12 µg of β-carotene from food are required to provide the body with 1 µg of retinol, giving dietary β-carotene an RAE ratio of 12:1. Other provitamin A carotenoids in food are less easily absorbed than β-carotene, resulting in RAE ratios of 24:1. RAE ratios are shown in Table 1.
Antioxidant activity
In plants, carotenoids have the important antioxidant function of quenching (deactivating) singlet oxygen, an oxidant formed during photosynthesis (13). Test tube studies indicated that lycopene is one of the most effective quenchers of singlet oxygen among carotenoids (14). They also suggested that carotenoids could inhibit the oxidation of fats (i.e., lipid peroxidation) under certain conditions, but their actions in humans appear to be more complex (15). Although important for plants, the relevance of singlet oxygen quenching to human health is less clear (1).
Nrf2-dependent antioxidant pathway
Some evidence suggests that carotenoids and/or their metabolites may upregulate the expression of antioxidant and detoxifying enzymes via the activation of the nuclear factor erythroid 2-related factor 2 (Nrf2)-dependent pathway (reviewed in 16). Briefly, Nrf2 is a transcription factor that is bound to the protein Kelch-like ECH-associated protein 1 (Keap1) in the cytosol. Keap1 responds to oxidative stress signals by freeing Nrf2. Upon release, Nrf2 translocates to the nucleus and binds to the antioxidant response element (ARE) located in the promoter of genes coding for antioxidant/detoxifying enzymes and scavengers. Nrf2/ARE-dependent genes code for numerous mediators of the antioxidant response, including glutamate-cysteine ligase (GCL), glutathione S-transferases (GSTs), thioredoxin, NAD(P)H quinone oxidoreductase 1 (NQO-1), and heme oxygenase 1 (HO-1) (17). One study showed an increase in the level of the major antioxidant glutathione and a protection against TNFα-induced oxidative stress in retinal pigment epithelial cells (RPE) following lycopene-mediated Nrf2 activation and GCL induction (18). Nrf2 activation by lycopene also protected RPE against TNFα-mediated proinflammatory signaling involving nuclear factor-κB (NF-κB) activation and intercellular adhesion molecule-1 (ICAM-1) expression (18). Lycopene was shown to trigger Nrf2-mediated antioxidant pathway in various cell types (19-21). At present, evidence from animal and human studies is very limited (16).
Blue light filtering
The long system of alternating double and single bonds common to all carotenoids allows them to absorb light in the visible range of the spectrum (13). This feature has particular relevance to the eye, where lutein, zeaxanthin, and meso-zeaxanthin (derived from in vivo conversion from lutein) efficiently absorb blue light. Depending on the carotenoid pigment density at the center of the eye’s retina (macula), up to 90% of blue light can be absorbed by these pigments. Reducing the amount of short-wavelength light that reaches the critical visual structures of the eye may protect them from light-induced oxidative damage (22). Because the only source of these plant pigments in the eye is diet, a number of observational and intervention studies have examined the potential of dietary and supplemental lutein and zeaxanthin to protect against age-related eye diseases (see Age-related macular degeneration and Cataracts). Supplemental lutein, alone or with zeaxanthin, was found to improve contrast sensitivity and protect against visual fatigue in young and/or healthy individuals (23-26). Lutein has also been suggested to improve visual function through stimulating neuronal signaling efficiency in the eye (27).
Intercellular communication
Carotenoids can facilitate communication between neighboring cells grown in culture by stimulating the synthesis of connexin proteins (28). Connexins form pores (gap junctions) in cell membranes, allowing cells to communicate through the exchange of small molecules. This type of intercellular communication is important for maintaining cells in a differentiated state and is often lost in cancer cells. Carotenoids facilitate intercellular communication by increasing the expression of the gene encoding a connexin protein, an effect that appears unrelated to the vitamin A or antioxidant activities of various carotenoids (29) and involving a retinoic acid receptor (RAR)-independent mechanism (30).
Immune function
Because vitamin A is essential for normal immune system function, it is difficult to determine whether the effects of provitamin A carotenoids are related to their vitamin A activity or other activities of carotenoids. Although some clinical trials have found that β-carotene supplementation improves several biomarkers of immune function (31-33), increasing intakes of lycopene and lutein — carotenoids without vitamin A activity — have not resulted in similar improvements in biomarkers of immune function (34-36).
Deficiency
Although consumption of provitamin A carotenoids (α-carotene, β-carotene, and β-cryptoxanthin) can prevent vitamin A deficiency (see the article on Vitamin A), no overt deficiency symptoms have been identified in people consuming low-carotenoid diets if they consume adequate vitamin A (11). After reviewing the published scientific research in 2000, the Food and Nutrition Board of the Institute of Medicine concluded that the existing evidence was insufficient to establish a recommended dietary allowance (RDA) or adequate intake (AI) for carotenoids. The Board has set an RDA for vitamin A (see the article on Vitamin A). Recommendations by the National Cancer Institute, American Cancer Society, and American Heart Association to consume a variety of fruit and vegetables daily are aimed, in part, at increasing intakes of carotenoids.
Disease Prevention
Cancer
Lung cancer
In the US, lung cancer is the leading cause of death by cancer among adults, representing about 20% of all cancer-related deaths (37).
Dietary carotenoids: Several large prospective cohort studies, including the Nurses’ Health Study (NHS) and the Health Professionals Follow-up Study (HPFS), have examined potential associations between carotenoid intake and/or blood concentrations and lung cancer (38). In a meta-analysis of eight prospective cohort studies, including NHS and HPFS, the highest versus lowest quantile of total carotenoid intake was significantly associated with a 21% reduced risk of lung cancer. For the individual carotenoids, the risk of lung cancer was estimated to be 20% and 14% lower with the highest versus lowest intakes of β-cryptoxanthin and lycopene, respectively. In contrast, dietary intakes of β-carotene, α-carotene, and lutein/zeaxanthin were not found to be significantly linked to a reduced risk of developing lung cancer (38). In addition, an analysis of the pooled results of 11 nested case-control and four prospective cohort studies found no association between serum concentrations of total carotenoids, β-carotene, α-lycopene, β-cryptoxanthin, and lutein/zeaxanthin and lung cancer. Only high versus low serum lycopene concentrations could be linked to a 29% lower risk of lung cancer (38). Any protective effect of dietary carotenoids against the development of lung cancer is likely small and not statistically significant (38).
Supplemental β-carotene: The effect of β-carotene supplementation on the risk of developing lung cancer has been examined in large randomized, placebo-controlled trials. In Finland, the Α-Tocopherol Βeta-Carotene (ATBC) cancer prevention trial evaluated the effects of 20 mg/day of β-carotene and/or 50 mg/day of α-tocopherol on more than 29,000 male smokers (39), and in the United States, the β-Carotene And Retinol Efficacy Trial (CARET) evaluated the effects of a combination of 30 mg/day of β-carotene and 25,000 IU/day of retinol (preformed vitamin A) in 18,314 men and women who were smokers, former smokers, or had a history of occupational asbestos exposure (40). Unexpectedly, the risk of lung cancer in the groups taking β-carotene supplements was increased by 16% after six years in the ATBC participants and by 28% after four years in the CARET participants. In contrast, the Physicians’ Health Study (PHS) examined the effect of β-carotene supplementation (50 mg every other day) on cancer risk in 22,071 male physicians in the United States, of whom only 11% were current smokers (41). In that lower risk population, β-carotene supplementation for more than 12 years was not associated with an increased risk of lung cancer. In addition, in the Linxian General Population trial conducted in about 29,000 Chinese participants, randomization to 15 mg/day of β-carotene, 30 mg/day of α-tocopherol, and 15 µg/day of selenium, was not found to be associated with lung cancer mortality 10 years after the intervention ended (42). Moreover, five-year follow-up of the Age-Related Eye Disease Study 2 (AREDS2) trial found that β-carotene supplementation nearly doubled the risk of developing lung cancer in former smokers compared to nonsmokers (current smokers did not receive β-carotene supplements) (43). AREDS2 was a multicenter, randomized double-blind, placebo-controlled trial that evaluated the effects of supplementation with antioxidant vitamins and minerals for five years to treat age-related macular degeneration (see below). Finally, a meta-analysis of four randomized controlled trials, including but not limited to trials in high-risk populations like smokers, found β-carotene supplementation (alone or with retinol; 3.7 to 12 years) increased the risk of lung cancer by 20% compared to control (OR, 1.20; 95% CI, 1.01-1.42) (44).
Although the reasons for the increase in lung cancer risk are not yet clear, several mechanisms have been proposed (45). Baseline β-carotene status might be one factor that influences whether with β-carotene supplementation promotes carcinogenesis in the lungs of smokers (46). The US Preventive Services Task Force estimated that the risks of high-dose β-carotene supplementation outweigh any potential benefits for cancer prevention and recommended against supplementation, especially in smokers or other high-risk populations (44, 47).
Prostate cancer
Prostate cancer is the one of the most prevalent cancers among US men, second only to non-melanoma skin cancer (48).
Dietary lycopene: Several early prospective cohort studies suggested that lycopene-rich diets were associated with significant reductions in the risk of prostate cancer, particularly more aggressive forms (49). Several pooled data analyses of observational studies that examining potential links between dietary intakes and/or circulating concentrations of lycopene and risk of prostate cancer have been completed. A 2015 meta-analysis of observational studies found no association of prostate cancer risk with dietary lycopene intakes (10 case-cohort and two prospective cohort studies) but an inverse association with blood lycopene concentrations (two case-control, nine nested case-control, and one cohort studies) (50, 51). Additionally, a meta-analysis of 15 nested case-control studies conducted by the Endogenous Hormones, Nutritional Biomarkers, and Prostate Cancer Collaborative Group showed an inverse association between circulating lycopene concentrations and risk of advanced stage and/or aggressive prostate cancer, while no association was found with risk of non-aggressive or localized disease (52). A 2017 meta-analysis of observational studies found inverse associations between both dietary (6 cohort/case-cohort and 15 case-control studies) and circulating (1 cohort study, 4 case-control studies, and 12 nested case-control studies) lycopene and prostate cancer risk; risk reductions were 12% in both analyses (RR, 0.88; 95% CI, 0.78-0.98) (53). However, this meta-analysis found no associations between dietary lycopene (5 studies) or circulating lycopene (6 studies) and advanced prostate cancer (53).
While there is considerable scientific interest in the potential for lycopene to help prevent prostate cancer, it is not yet clear whether the prostate cancer risk reduction observed in some observational studies is related to lycopene itself, other compounds in tomatoes, or other factors associated with lycopene-rich diets (54). Experimental studies in rodents suggest that lycopene is protective against prostate cancer but not the only protective compound found in tomatoes (reviewed in 5). Of note, the 2014 World Cancer Research Fund International report on Diet, Nutrition, Physical Activity, and Prostate Cancer suggested the need for better designed studies to establish whether consumption of lycopene-containing foods could be linked to a lower risk of prostate cancer (55).
Supplemental lycopene: To date, a few short-term, dietary intervention studies using lycopene in patients with precancerous prostate lesions (high grade prostatic intraepithelial neoplasia; HGPIN) or prostate cancer have been completed. Specifically, two small randomized controlled studies examined the effect of lycopene supplementation for up to six months in men with HGPIN (56, 57). The consumption of 30 to 35 mg/day of supplemental lycopene in the form of tomato extract (56), or together with selenium (55 mg/day) and green tea catechins (600 mg/day) (57), showed no benefit on the rate of progression to prostate cancer at six-month (56, 57) and 37-month follow-ups (57). Earlier small trials in men with HGPIN led to similar conclusions (reviewed in 58). Additionally, a randomized controlled trial in men with localized prostate cancer found that supplementation with 15, 30, or 45 mg of lycopene (until prostatectomy) did not significantly increase plasma lycopene concentration, modify the ratio of steroid hormones in blood, or reduce the concentration of markers of proliferation (i.e., prostate-specific antigen [PSA] and Ki-67) compared to placebo (59). In another trial, 54 patients with metastatic prostate cancer were randomized to orchidectomy alone or orchidectomy plus 4 mg/day of lycopene (60). The proportion of complete clinical response to treatment — assessed by serum PSA and/or bone scan returning to normal — and patient survival rate were found to be significantly higher in patients supplemented with lycopene (60).
Moreover, a meta-analysis of six randomized controlled trials in patients with non-metastatic prostate cancer found that supplemental lycopene (15-30 mg/day for 3 to 24 weeks) had no effect on circulating PSA concentrations (61). Yet, a subgroup analysis revealed a benefit of PSA reduction in patients with higher concentrations at baseline (PSA ≥6.5 µg/L) (61).
Large-scale, controlled clinical trials are needed to further examine the safety and efficacy of long-term use of lycopene supplements for prostate cancer prevention or treatment.
Other types of cancer
In a meta-analysis of 12 prospective cohort studies, no association was found between total and individual carotenoid intake and risk of breast cancer, except with β-carotene for which a 5% reduction in breast cancer risk was estimated for every 5 mg/day increment in consumption (62). In a pooled analysis of 14 nested case-control studies and one follow-up study of a clinical trial, reductions in breast cancer risk were found to be associated with blood concentrations of total carotenoids (-26%), α-carotene (-20%), and lutein (-30%) (62). Another study that recalibrated data for consistency across eight large prospective cohorts before pooled analysis found reduced breast cancer risk to be associated with the highest versus lowest quintile of blood concentrations of total carotenoids (-21%), β-carotene (-17%), and lycopene (-22%) (63). Further analyses found an inverse association between the blood concentrations of β-carotene and α-carotene and risk of estrogen receptor-negative (ER-), but not estrogen receptor-positive (ER+), breast tumors (63). A similar result was reported in a case-control study nested within the multicenter, large, European Prospective Investigation into Cancer and Nutrition (EPIC) study (64). In a nested case-control study of the Nurses’ Health Study (NHS) and NHSII, a 20% reduction in risk of breast cancer was seen in those with the highest total plasma carotenoids (≥142.1 µg/dL) compared to the lowest (<84.6 µg/dL), and this association was strongest in those presumed to be at higher risk of breast cancer (65). Protective associations were observed for higher circulating levels of the carotenoids, α-carotene (-20%), β-carotene (-18%), as well as lutein and zeaxanthin (-17%) (65). In contrast to the abovementioned pooled analysis (63), this study found a protective association between higher circulating carotenoids and ER+, but not ER-, breast tumors (65).
A case-control study nested within the EPIC study found a 31% lower risk of colorectal cancer with the highest versus lowest quartile of β-carotene intake, while no associations were shown with blood concentrations of other carotenoids or total carotenoids (66). The nested case-control study was included in a meta-analysis of 22 observational studies that failed to find associations between carotenoid intakes and colorectal cancer (67). A meta-analysis of 15 observational studies (11 case-control and 4 prospective cohort studies) reported no association between lycopene intake and colorectal cancer (68).
Pooled data also suggested that higher intakes of individual carotenoids, especially β-cryptoxanthin and lycopene, might be associated with a reduced risk of cancers of the mouth, pharynx, and larynx (69), but most of the data come from case-control control studies (70). The results of case-control studies are more likely to be distorted by bias than results of prospective cohort studies.
Examining blood concentrations of each carotenoid in relation to cancer subsites may help overcome limitations associated with dietary data and differences in carotenoid absorption. In a recent dose-response meta-analysis of observational studies, higher blood concentrations of several carotenoids, including α-carotene (3 studies), β-carotene (4 studies), as well as combined lutein and zeaxanthin (3 studies), were linked to a lower risk of developing bladder cancer (71).
Eye diseases
Age-related macular degeneration
In Western countries, degeneration of the macula, the center of the eye’s retina, is a leading cause of blindness among older adults. Long-term blue light exposure and oxidative damage in the outer segments of photoreceptors may lead to drusen and/or pigment abnormalities in the macula, increasing the risk of age-related macular degeneration (AMD) and central blindness.
Dietary lutein and zeaxanthin: The carotenoids found in the retina are lutein and zeaxanthin, which are both of dietary origin, and meso-zeaxanthin, which is derived in vivo conversion from lutein. These three carotenoids are present in high concentrations in the macula (known as macular pigment), where they are efficient absorbers of blue light. They may prevent a substantial amount of the blue light entering the eye from reaching the underlying structures involved in vision and protect against light-induced oxidative damage, which is thought to play a role in the pathology of age-related macular degeneration (reviewed in 22). It is also possible, though not proven, that lutein, zeaxanthin, and meso-zeaxanthin, act directly to neutralize oxidants formed in the retina.
Increasing dietary consumption of lutein and zeaxanthin was shown to raise their serum concentration and macular pigment density (72, 73). Some, but not all, observational studies have provided evidence that higher intakes of lutein and zeaxanthin are associated with lower risk of age-related macular degeneration (AMD) (74). While cross-sectional and retrospective case-control studies found that higher levels of lutein and zeaxanthin in the diet (75-77), blood (78, 79), and retina (80, 81) were associated with a lower incidence of AMD, several prospective cohort studies found no relationship between baseline dietary intakes or serum concentrations of lutein and zeaxanthin and the risk of developing AMD over time (82-85). One report examined the association between the incidence of AMD and calculated dietary intakes and predicted plasma concentrations of lutein and zeaxanthin in older adults (≥50 years) from two large prospective cohorts, the Nurses’ Health Study (NHS; 63,443 women) and the Health Professionals Follow-up Study (HPFS; 38,603 men), followed for 26 years and 24 years, respectively (86). The highest versus lowest quintile of predicted plasma lutein and zeaxanthin scores was associated with a 41% lower risk of advanced AMD, yet no association was found with intermediate AMD. Evidence has suggested that the consumption of about 6 mg/day of lutein and zeaxanthin from fruit and vegetables (compared with less than 2 mg/day) may decrease the risk of advanced AMD (77, 86). In a small population-based prospective study among 609 older adults in France, followed for a median of 7.6 years, found an inverse association between plasma lutein concentration at baseline and advanced AMD (HR, 0.63; 95% CI, 0.41-0.97) (87). No association was observed for circulating zeaxanthin and advanced AMD (87).
Supplemental lutein and zeaxanthin: The first randomized controlled trial in patients with atrophic AMD found that supplementation with 10 mg/day of lutein slightly improved visual acuity after one year compared to a placebo (88). The effects of long-term lutein supplementation on atrophic AMD were further investigated in combination with antioxidant vitamins and minerals in the Age-Related Eye Disease Study 2 (AREDS2), a multicenter, randomized, double-blind, placebo-controlled trial. In AREDS, oral supplementation with β-carotene (15 mg/day), vitamin C (500 mg/day), vitamin E (400 IU/day), zinc (80 mg/day as zinc oxide), and copper (2 mg/day as cupric oxide) for five years reduced the risk of developing advanced AMD by 25% (89). In the AREDS2 study conducted in 4,203 participants at risk for developing late AMD, supplemental lutein (10 mg/day) and zeaxanthin (2 mg/day), in combination with β-carotene, vitamin C, vitamin E, zinc, and copper (the ‘AREDS’ formulation), did not slow the progression to advanced AMD, although subgroup analyses revealed a benefit in those with the lowest dietary intakes of lutein + zeaxanthin (89). A total of 3,036 subjects were further randomized to various combinations of carotenoids; supplementation with lutein and zeaxanthin significantly reduced the risk of progression to late AMD and to neovascular AMD compared to supplementation with β-carotene (90).
Several smaller (n=30-433) randomized controlled trials also suggested that supplementation with xanthophyll carotenoids would be beneficial in the management of AMD (reviewed in 91). A meta-analysis of eight trials that examined the effect of supplemental lutein (6 to 20 mg/day) or/and zeaxanthin (0 to 10 mg/day) in 1,176 AMD subjects for up to 36 months found improvements in visual acuity and contrast sensitivity with increased levels of xanthophyll carotenoids (92). More recently, a randomized, placebo-controlled trial found daily consumption of a buttermilk drink with egg yolks enriched with lutein (1.4 mg), zeaxanthin (0.2 mg), and an omega-3 polyunsaturated fatty acid (DHA; 160 mg) for one year improved visual acuity and macular pigment optical density in subjects with drusen and/or retinal pigment abnormalities (about half of them being classified as having early AMD) (93). In a randomized, placebo-controlled trial in 74 patients with intermediate AMD, daily supplementation with lutein (10 mg/day) and zeaxanthin (2 mg) for two years, along with daily astaxanthin (4 mg), vitamin C (90 mg), vitamin E (30 mg), zinc (22.5 mg), copper (1 mg), and fish oil (containing 185 mg EPA and 140 mg DHA), resulted in a lower incidence of disease progression (2.1% of patients) when compared to placebo (15.4% of patients) (94).
Supplemental β-carotene: The first randomized controlled trial designed to examine the effect of a carotenoid supplement on AMD used β-carotene in combination with vitamin C, vitamin E, and zinc because lutein and zeaxanthin were not commercially available as supplements at the time the trial began (95). Although the combination of antioxidants and zinc lowered the risk of developing advanced macular degeneration in individuals with signs of moderate-to-severe macular degeneration in at least one eye, it is unlikely that the benefit was related to β-carotene since it is not present in the retina. Supplementation of male smokers in Finland with 20 mg/day of β-carotene for six years did not decrease the risk of AMD compared to placebo (96). A placebo-controlled trial in a cohort of 22,071 healthy US men found that β-carotene supplementation (50 mg every other day) had no effect on the incidence of age-related maculopathy — an early stage of AMD (97). Recent systematic reviews of randomized controlled trials have concluded that there is no evidence that β-carotene supplementation prevents or delays the onset of AMD (98, 99).
Other retinopathies
Retinopathy of prematurity
Preterm infants have an immature retinal vascular system that places them at risk of developing retinopathy of prematurity (ROP). Basically, preterm birth halts the normal development of retinal vascular system, which results in a retina that is poorly vascularized and highly susceptible to hyperoxia. In order to meet metabolic demand, the hypoxic retina induces the production of proangiogenic factors like VEGF (vascular endothelial growth factor). These factors stimulate the development of new blood vessels (angiogenesis), causing aberrant vessels sprouting from the retina into the vitreous. It is thought that an imbalance between the production of reactive oxygen species (ROS) and the reduced levels of antioxidants in preterm infants contributes to ROP pathogenesis and causes additional damage to the retina (reviewed in 100).
Supplemental lutein and zeaxanthin: A randomized controlled trial in 62 preterm infants (≤32 weeks of gestational age) failed to observe any benefits regarding ROP incidence and severity with lutein (0.5 mg/kg/day) and zeaxanthin (0.02 mg/kg/day) supplemented from the seventh day post birth until about 10 weeks of age (101). In two other multicenter, placebo-controlled trials in a total of 343 preterm infants, a daily oral dose of 0.14 mg of lutein and 0.006 mg of zeaxanthin administered from the first week after birth did not significantly reduce ROP incidence or the rate of progression from early to more advanced stages of ROP occurrence (102, 103). A fourth multicenter, randomized controlled trial in 203 preterm infants found that administration of a formula containing carotenoids (lutein/zeaxanthin, lycopene, and β-carotene) had no effect on ROP incidence but limited the progression to severe ROP stages in infants with mild ROP compared to a carotenoid-free formula (104). Compared with the control formula, preterm infants free of ROP fed the carotenoid-containing formula had significantly increased plasma carotenoid concentrations, which were correlated with greater rod photoreceptor sensitivity (104). More research is needed to examine whether carotenoid supplementation might promote normal photoreceptor development and prevent ROP in preterm infants.
Diabetic retinopathy
Supplementation with lutein and zeaxanthin has been shown to help preserve retinal integrity in diabetic rodents by reducing oxidative stress and inflammatory mediators (100). A higher ratio of plasma nonprovitamin A carotenoids (lutein, zeaxanthin, and lycopene) to vitamin A carotenoids (α-carotene, β-carotene, β-cryptoxanthin) was associated with a reduced risk of diabetic retinopathy in a cross-sectional analysis in 111 individuals with type 2 diabetes mellitus (105). A small observational study also found lower macular pigment optical density in those with type 2 diabetes (n=17) or mild nonproliferative diabetic retinopathy (n=12) compared to those without either condition (n=14) (106). Data from 1,430 individuals participating in the Atherosclerosis Risk In Communities (ARIC) study found no association between lutein intake and diabetic retinopathy after adjustment for confounding variables (107).
It is not known if supplementation with lutein and zeaxanthin might prevent or help treat diabetic retinopathy; well-designed, placebo-controlled studies would be needed to address these questions.
Cataracts
The primary function of the eye’s lens is to collect and focus light on the retina. Ultraviolet light and oxidants can damage proteins in the lens, causing structural changes that result in the formation of opacities known as cataracts. As people age, cumulative damage to lens proteins often results in cataracts that are large enough to interfere with vision (13). Risk factors other than aging and sunlight exposure include smoking, diabetes mellitus, higher body mass index, and use of estrogen replacement therapy. An estimated 50 million US adults are projected to be affected by age-related cataracts by 2050 (108).
Dietary lutein and zeaxanthin: The observation that lutein and zeaxanthin are the only carotenoids in the human lens has stimulated interest in the potential for increased intakes of lutein and zeaxanthin to prevent or slow the progression of cataracts (109). Large prospective cohort studies have found that men and women with the highest intakes of foods rich in lutein and zeaxanthin, particularly spinach, kale, and broccoli, were 18%-50% less likely to require cataract extraction (110, 111) or develop cataracts (112-114). Moreover, plasma concentrations of lutein and zeaxanthin have been found to be inversely associated to the progression of nuclear cataract. Additional research is required to determine whether these findings are related specifically to lutein and zeaxanthin intake or to other factors associated with diets high in carotenoid-rich foods (74).
Supplemental lutein and zeaxanthin: The Age-related Eye Disease Study 2 (AREDS2) failed to show an effect of supplemental lutein (10 mg/day) and zeaxanthin (2 mg/day; supplementation for a median of 4.7 years) on the risk of developing cataract, on the progression to severe cataract or to cataract surgery, and on visual acuity (115). However, the results might have been confounded by the fact that most participants were better nourished than the general population and/or used multivitamins that have been found to decrease the risk of developing cataract (116). Additional limitations to consider in interpreting the results have been reviewed elsewhere (115). Additional interventions are required to study whether supplemental lutein and zeaxanthin might be helpful in the prevention of cataract.
Supplemental β-carotene: Evidence from observational studies that cataracts were less prevalent in people with high dietary intakes and blood concentrations of carotenoids led to the inclusion of β-carotene supplements in several large randomized controlled trials of antioxidants. The results of those trials have been somewhat conflicting. β-Carotene supplementation (20 mg/day) for more than six years did not affect the prevalence of cataracts or the frequency of cataract surgery in male smokers living in Finland (96). In contrast, a 12-year study of male physicians in the US found that β-carotene supplementation (50 mg every other day) decreased the risk of cataracts in smokers but not in nonsmokers (117). Note that use of β-carotene supplements have been shown to increase the risk of lung cancer in smokers (see Supplemental β-carotene in lung cancer). Three randomized controlled trials examined the effect of an antioxidant combination that included β-carotene, vitamin C, and vitamin E on the progression of cataracts. Two trials found no benefit after supplementation for five years (118) or more than six years (119), but one trial found a small decrease in the progression of cataracts after three years of supplementation (120). Overall, the results of randomized controlled trials suggest that the benefit of β-carotene supplementation in slowing the progression of age-related cataracts does not outweigh the potential risks.
Cardiovascular disease
Dietary carotenoids: Because carotenoids are very soluble in fat and very insoluble in water, they circulate in lipoproteins, along with cholesterol and other fats. Evidence that low-density lipoprotein (LDL) oxidation plays a role in the development of atherosclerosis led scientists to investigate the role of antioxidant compounds like carotenoids in the prevention of cardiovascular disease (121). The thickness of the inner layers of the carotid arteries can be measured noninvasively using ultrasound technology. This measurement of carotid intima-media thickness is considered a reliable marker of atherosclerosis (122). A number of case-control and cross-sectional studies have found higher blood concentrations of carotenoids to be associated with significantly lower measures of carotid artery intima-media thickness (123-128). Additionally, higher plasma carotenoids at baseline have been associated with significant reductions in risk of cardiovascular disease in some prospective cohort studies (129-133) but not in others (134-137).
In a US national survey, National Health And Nutrition Examination Survey (NHANES) 2003-2006, serum total carotenoid concentration was inversely associated with blood concentrations of two cardiovascular risk factors, C-reactive protein (CRP) and total homocysteine (138). HDL-cholesterol concentration was found to be positively associated with α-carotene, β-cryptoxanthin, and lutein/zeaxanthin concentrations, but only the latter was inversely associated with LDL-cholesterol (138). NHANES 2007-2014 also found an inverse association between total dietary carotenoid intake (sum of α-carotene, β-carotene, β-cryptoxanthin, lycopene, lutein, and zeaxanthin) and risk of hypertension (139). Finally, a meta-analysis of observational studies reported lower risks of coronary heart disease (-12%) and stroke (-18%) in individuals in the highest versus lowest tertile of blood lutein concentration (140). In a recent meta-analysis, higher lycopene intake — from diet and/or supplements — was not associated with improvements in blood pressure or concentrations of blood lipids; the included studies were heterogeneous with respect to lycopene delivery (i.e., as a supplement or as food, varying tomato-containing products or extracts) and dose, characteristics of participants (healthy or with disease), and study duration (141).
While the results of several prospective studies indicate that people with higher intakes of carotenoid-rich fruit and vegetables are at lower risk of cardiovascular disease (137, 142-144), it is not yet clear whether this effect is a result of carotenoids or other factors associated with diets high in carotenoid-rich fruit and vegetables.
Supplemental β-carotene: In contrast to the results of observational studies suggesting that high dietary intakes of carotenoid-rich fruit and vegetables may decrease cardiovascular disease risk, four randomized controlled trials found no evidence that β-carotene supplements in doses ranging from 20 to 50 mg/day were effective in preventing cardiovascular disease (39, 41, 145, 146). Based on the results of these randomized controlled trials, the US Preventive Health Services Task Force found good evidence to suggest that β-carotene supplements provided no benefit in the prevention of cardiovascular disease in healthy adults (47). Moreover, a recent meta-analysis of 12 randomized, placebo-controlled trials found that β-carotene supplementation increased mortality related to cardiovascular disease (RR, 1.12; 95% CI, 1.04-1.19) (147). Thus, although diets rich in β-carotene have generally been associated with reduced cardiovascular disease risk in observational studies, there is no evidence that β-carotene supplementation reduces cardiovascular disease risk (148).
Supplemental lycopene: Several randomized controlled trials have examined whether supplementation with lycopene, tomato products, or tomato extracts might benefit cardiovascular health by improving blood pressure, lipid profiles, or function of the vascular endothelium. A recent meta-analysis of eight trials found no effect of supplemental lycopene on systolic or diastolic blood pressure; no benefits were found in healthy subjects or in hypertensive subjects (141). The meta-analysis also showed no effect of lycopene supplementation on total cholesterol (10 trials), LDL cholesterol (11 trials), or HDL (11 trials) cholesterol (141). Moreover, some, but not all (149, 150) short-term trials have indicated supplemental lycopene might improve function of the vascular endothelium in healthy subjects (151, 152); however, large, long-term placebo-controlled studies are needed.
Osteoporosis
An experimental study found that the administration of β-cryptoxanthin to ovariectomized mice limited bone resorption by inhibiting osteoclast differentiation but had no effect on osteoblast-driven bone formation (153). Yet, evidence of a protective role of β-cryptoxanthin — and other individual carotenoids — against bone loss in humans is scarce. In the prospective Framingham Osteoporosis Study, data analysis of 603 participants found no association between β-cryptoxanthin intake and changes in bone mineral density (BMD) over a four-year period (154). In contrast, intakes of total and individual carotenoids (β-carotene, lycopene, and lutein/zeaxanthin) were positively associated with proximal femur BMD in men over a four-year period, while lycopene intake was positively linked to lumbar spine BMD in women (154). The 17-year follow-up of participants in the Framingham Osteoporosis Study (946 participants) showed those in the highest tertile of total carotenoid intake (median intake: 23.7 mg/day) had a 51% lower risk of hip fracture compared to those in the lowest tertile (median intake: 7.3 mg/day) (155). In a much larger prospective study — the Singapore Chinese Health Study — in 63,257 men and women followed for 10 years, the highest versus lowest quartile of total carotenoid intake was associated with a 37% lower risk of hip fracture in men, but no association was observed in women (156). Dietary intakes of α- and β-carotene — but not of β-cryptoxanthin, lycopene, and lutein/zeaxanthin — were inversely associated with hip fracture risk in men (156). Nevertheless, in a small, four-year, Japanese prospective cohort study in 457 adults, the risk of osteoporosis was 93% lower in postmenopausal women in the highest versus lowest tertile of serum β-cryptoxanthin concentration (157). No such association was reported with serum concentrations of other individual carotenoids. More recently, in a cross-sectional analysis of the European Prospective Investigation into Cancer and Nutrition-Norfolk cohort, higher dietary intakes of β-carotene and combined lutein and zeaxanthin were linked to higher bone density at the heel in women (158). No associations of dietary carotenoid intake and heel bone density were found in men, and serum concentration of carotenoids was not linked to heel bone density in either men or women (158).
While a few studies have found a protective association between higher carotenoid intake and osteoporosis or bone fracture, the available studies are observational. Whether carotenoid supplementation may help prevent bone loss and reduce the risk of osteoporosis in older individuals is currently unknown; randomized controlled trials would be needed to address this question. It is important to note that high-dose supplementation with preformed vitamin A (retinol) has been associated with adverse effects on bone health (see the article on Vitamin A).
Cognitive function
Observational studies have suggested that dietary lutein may be of benefit in maintaining cognitive health (159-162), and a cross-sectional study of 4,076 older adults associated higher blood lutein concentrations with improved cognition, including memory and executive function (163). As stated above, among the carotenoids, lutein and its isomer zeaxanthin are the only two that cross the blood-retina barrier to form macular pigment in the eye. Lutein also preferentially accumulates in the brain (164, 165). A few studies have suggested that lutein and zeaxanthin concentrations in the macula correlate with brain lutein and zeaxanthin status and therefore might be used as a biomarker of cognitive health (165-168). Additionally, in the Georgia Centenarian Study, the analysis of cross-sectional data from 47 centenarian decedents showed a positive association between post-mortem measures of brain lutein concentrations and pre-mortem measures of cognitive function (164). Brain lutein concentrations were found to be significantly lower in individuals with mild cognitive impairment compared to those with normal cognitive function (164). In a small, four-month, double-blind, placebo-controlled study in older women (ages, 60 to 80 years) without cognitive impairment, supplementation with lutein (12 mg/day) and zeaxanthin (~0.5 mg/day) significantly improved cognitive test performance (169). Two small trials in younger adults found supplementation with lutein (10 mg/day) and zeaxanthin (2 mg/day) for one year improved some measures of cognitive function, including memory (170, 171). However, the Age-related Eye Disease Study 2 (AREDS2) failed to show an effect of supplemental lutein (10 mg/day) and zeaxanthin (2 mg/day; supplementation for a median of 4.7 years) on the cognitive test scores of 3,741 older participants (mean age, 72.7 years) (172), perhaps because the trial was conducted in a highly educated and well-nourished population.
Sources
Food sources
The most prevalent carotenoids in the human diet are α-carotene, β-carotene, β-cryptoxanthin, lycopene, lutein, and zeaxanthin (11). Most carotenoids in foods are found in the all-trans form (see Figure 1 and Figure 2 above), although cooking may result in the formation of other isomers. The relatively low bioavailability of carotenoids from most foods compared to supplements is partly due to the fact that they are associated with proteins in the plant matrix (173). Chopping, homogenizing, and cooking disrupt the plant matrix, increasing the bioavailability of carotenoids (3). For example, the bioavailability of lycopene from tomatoes is substantially improved by heating tomatoes in oil (174, 175). For information on other factors that affect carotenoid bioavailability, see above and the Moran et al. review (5).
α-Carotene and β-carotene
α-Carotene and β-carotene are provitamin A carotenoids, meaning they can be converted in the body to vitamin A. The vitamin A activity of β-carotene in food is 1⁄12 that of retinol (preformed vitamin A). Thus, it would take 12 µg of β-carotene from food to provide the equivalent of 1 µg (0.001 mg) of retinol. The vitamin A activity of α-carotene from foods is 1⁄24 that of retinol, so it would take 24 µg of α-carotene from food to provide the equivalent of 1 µg of retinol. Orange and yellow vegetables like carrots and winter squash are rich sources of α- and β-carotene. Spinach is also a rich source of β-carotene, although the chlorophyll in spinach leaves hides the yellow-orange pigment. Some foods that are good sources of α-carotene and β-carotene are listed in Table 2 and Table 3 (176).
β-Cryptoxanthin
Like α- and β-carotene, β-cryptoxanthin is a provitamin A carotenoid. The vitamin A activity of β-cryptoxanthin from food was estimated to be 1⁄24 that of retinol, so it would take 24 µg of β-cryptoxanthin from food to provide the equivalent of 1 µg of retinol. Orange and red fruit and vegetables like sweet red peppers and oranges are particularly rich sources of β-cryptoxanthin. Some foods that are good sources of β-cryptoxanthin are listed in Table 4 (176).
Lycopene
Lycopene gives tomatoes, pink grapefruit, watermelon, and guava their red color. It has been estimated that 80% of the lycopene in the US diet comes from tomatoes and tomato products like tomato sauce, tomato paste, and ketchup (catsup) (177). Lycopene is not a provitamin A carotenoid because it cannot be converted to retinol. Some foods that are good sources of lycopene are listed in Table 5 (176).
Lutein and zeaxanthin
Although lutein and zeaxanthin are different compounds, they are both classified as xanthophylls and nonprovitamin A carotenoids (see Figure 2 above). Some methods used to quantify lutein and zeaxanthin in food do not separate the two compounds, so they are typically reported as lutein and zeaxanthin or lutein + zeaxanthin. Both pigments are present in a variety of fruit and vegetables. Dark green leafy vegetables like spinach and kale are particularly rich sources of lutein but poor sources of zeaxanthin (178). Although relatively low in lutein, egg yolks and avocados are highly bioavailable sources of lutein. Good sources of dietary zeaxanthin include yellow corn, corn-based products, bell peppers, and egg yolk (179). Some foods containing lutein and zeaxanthin are listed in Table 6 (176).
For more information on the carotenoid content of certain foods, search USDA's FoodData Central database.
Supplements
Dietary supplements providing purified carotenoids and combinations of carotenoids are commercially available in the US without a prescription. Carotenoids are best absorbed when taken with a meal containing fat.
α-Carotene
Supplements containing a mixture of carotenoids may include α-carotene. As a provitamin A carotenoid, supplemental α-carotene can contribute to fulfill vitamin A requirements. It is not known whether the relative bioavailability of supplemental α-carotene is greater than that of dietary α-carotene.
α-Cryptoxanthin
Supplements containing a mixture of carotenoids may include α-cryptoxanthin. As a provitamin A carotenoid, supplemental α-cryptoxanthin can contribute to fulfill vitamin A requirements. It is not known whether the relative bioavailability of supplemental α-cryptoxanthin is greater than that of dietary α-cryptoxanthin.
β-Carotene
β-Carotene is sold as individual supplements and also found in supplements marketed to promote visual health (180). Commercially available β-carotene supplements usually contain between 1.5 mg and 15 mg of either synthetic β-carotene or natural β-carotene (mainly from the algae Dunaliella salina) per softgel capsule (178). As a provitamin A carotenoid, β-carotene may be used to provide all or part of the vitamin A in multivitamin supplements. The provitamin A activity of β-carotene from supplements is much higher than that of β-carotene from food: it takes only 2 micrograms [µg] (0.002 mg) of β-carotene from supplements to provide 1 µg of retinol (preformed vitamin A) compared to 12 µg of dietary β-carotene.
Of note, the β-carotene content of supplements is often listed in international units (IU) rather than µg: 3,000 µg (3 mg) of supplemental β-carotene provides 5,000 IU of vitamin A.
Lycopene
Lycopene has no provitamin A activity. Synthetic lycopene and lycopene from natural sources, mainly tomatoes, are available as nutritional supplements containing up to 15 mg of lycopene per softgel capsule (178).
Lutein and zeaxanthin
Lutein and zeaxanthin are not provitamin A carotenoids. Lutein and zeaxanthin supplements are available as free carotenoids (non-esterified) or as esters (esterified to fatty acids). Both forms appear to have comparable bioavailability (181). Many commercially available lutein and zeaxanthin supplements have much higher amounts of lutein than zeaxanthin (178). Supplements containing only lutein or zeaxanthin are also available.
Safety
Toxicity
β-Carotene
Although β-carotene can be converted to vitamin A, the conversion of β-carotene to vitamin A decreases when body stores of vitamin A are high (see Absorption, Metabolism, and Bioavailability). This may explain why high doses of β-carotene have never been found to cause vitamin A toxicity. High doses of β-carotene (up to 180 mg/day) have been used to treat erythropoietic protoporphyria, a photosensitivity disorder, without toxic side effects (11).
Lycopene, lutein, and zeaxanthin
No toxicities have been reported (182).
Adverse effects
β-Carotene
Increased lung cancer risk: Two randomized controlled trials in smokers and former asbestos workers found that supplementation with 20 to 30 mg/day of β-carotene for 4 to 6 years was associated with significant 16%-28% increases in the risk of lung cancer compared to placebo (see Supplemental β-carotene in lung cancer). Although the reasons for these findings are not yet clear, the potential risk of lung cancer in smokers and other high-risk groups supplemented with high-dose β-carotene outweigh any possible benefits for chronic disease prevention (47). It should be noted that there is no evidence that β-carotene supplementation may harm nonsmokers.
Carotenodermia: High doses of β-carotene supplements (≥30 mg/day) and the consumption of large amounts of carotene-rich food have resulted in a yellow discoloration of the skin (xanthoderma) known as carotenodermia, also called carotenemia. Carotenodermia is not associated with any underlying health problems and resolves when supplementation with β-carotene is discontinued or dietary carotene intake is reduced.
Lycopene
Lycopenodermia: High intakes of lycopene-rich food or supplements may result in a deep orange discoloration of the skin known as lycopenodermia. Because lycopene is more intensely colored than the carotenes, lycopenodermia may occur at lower doses than carotenodermia (11).
Lutein and zeaxanthin
A risk assessment analysis of 11 human studies concluded that lutein is likely safe at intake levels below 20 mg/day (183). A more recent case report documented "foveal sparkles" (eye crystals) in an older woman with glaucoma taking 20 mg/day of lutein for eight years; the patient also had very high dietary lutein intake, and total daily intake of lutein was not known (184).
β-Cryptoxanthin
A double-blind, placebo controlled trial in 90 young healthy women found that β-cryptoxanthin supplementation up to 6 mg/day for eight weeks was well tolerated (185).
Safety in pregnancy and lactation
β-Carotene
Unlike vitamin A, high doses of β-carotene taken by pregnant women have not been associated with increased risk of birth defects (11). However, the safety of high-dose β-carotene supplements in pregnancy and lactation has not been well studied. Although there is no reason to limit dietary β-carotene intake, pregnant and breast-feeding women should avoid consuming more than 3 mg/day (1,500 µg RAE/day; 5,000 IU/day) of β-carotene from supplements unless they prescribed under medical supervision.
Other carotenoids
The safety of carotenoid supplements other than β-carotene in pregnancy and lactation has not been established, so pregnant and breast-feeding women should obtain carotenoids from food rather than supplements. There is no reason to limit the consumption of carotenoid-rich fruit and vegetables during pregnancy (178).
Drug interactions
The cholesterol-lowering agents, cholestyramine (Questran) and colestipol (Colestid), can reduce absorption of fat-soluble vitamins and carotenoids, as can mineral oil and Orlistat (Xenical), a drug used to treat obesity (178). Colchicine, a drug used to treat gout, can cause intestinal malabsorption. However, long-term use of 1 to 2 mg/day of colchicine did not affect serum β-carotene concentrations in one study (186). Increasing gastric pH through the use of proton-pump inhibitors (Omeprazole, Lansoprazole) may decrease the absorption of a single dose of a β-carotene supplement, but the effect is unlikely to be clinically significant (187).
Antioxidant supplements and statins (3-hydroxy-3-methylglutaryl-coenzyme A [HMG-CoA] reductase inhibitors)
A three-year randomized controlled trial in 160 patients with documented coronary heart disease (CHD) and low serum high density lipoprotein (HDL) concentrations found that a combination of simvastatin (Zocor) and niacin increased HDL2 levels, inhibited the progression of coronary artery stenosis, and decreased the frequency of cardiovascular events, including myocardial infarction and stroke (188). Surprisingly, when an antioxidant combination of 1,000 mg of vitamin C, 536 mg of RRR-α-tocopherol (vitamin E), 100 µg of selenium, and 25 mg of β-carotene daily was taken with the simvastatin-niacin combination, the protective effects were diminished. Since the antioxidants were taken together in this trial, the individual contribution of β-carotene cannot be determined. In contrast, a much larger randomized controlled trial of simvastatin and an antioxidant combination of 297 mg of RRR-α-tocopherol, 250 mg of vitamin C, and 20 mg of β-carotene daily in more than 20,000 men and women with CHD or diabetes mellitus found that the antioxidant combination did not diminish the cardioprotective effects of simvastatin therapy over a five-year period (189). These contradictory findings indicate that further research is needed on potential interactions between antioxidant supplements and cholesterol-lowering agents, such as niacin and statins.
Interactions with food
Fat substitute Olestra
In a controlled feeding study, consumption of 18 g/day of the fat substitute Olestra (sucrose polyester; Olean) resulted in a 27% decrease in serum carotenoid concentrations after three weeks (190). Studies in people before and after the introduction of Olestra-containing snacks to the marketplace found that total serum carotenoid concentrations decreased by 15% in those who reported consuming at least 2 g/day of Olestra (191). One study in adults found that those who consumed more than 4.4 g of Olestra weekly experienced a 9.7% decline in total serum carotenoids compared to those not consuming Olestra (192).
Plant sterol- or stanol-containing foods
Some studies found that the regular use of plant sterol-containing spreads resulted in modest, 10%-20% decreases in the plasma concentrations of some carotenoids, particularly α-carotene, β-carotene, and lycopene (see the article on Phytosterols) (193, 194). However, advising people who use plant sterol- or stanol-containing margarines to consume an extra serving of carotenoid-rich fruit or vegetables daily prevented decreases in plasma carotenoid concentrations (195).
Alcohol
The relationships between alcohol consumption and carotenoid metabolism are not well understood. There is some evidence that regular alcohol consumption inhibits the conversion of β-carotene to retinol (196). Increases in lung cancer risk associated with high-dose β-carotene supplementation in two randomized controlled trials were enhanced in those with higher alcohol intakes (40, 197).
Interactions among carotenoids
The results of metabolic studies suggested that high doses of β-carotene compete with lutein and lycopene for absorption when consumed at the same time (198-200). However, the consumption of high-dose β-carotene supplements did not adversely affect serum carotenoid concentrations in long-term clinical trials (201-204).
Authors and Reviewers
Originally written in 2004 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Updated in December 2005 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in May 2009 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in July 2016 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in September 2023 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in October 2023 by:
Elizabeth J. Johnson, Ph.D., F.A.C.N., F.I.C.S.
Associate Professor,
Friedman School of Nutrition and Science & Policy
Tufts University
Copyright 2004-2024 Linus Pauling Institute
References
1. Wang XD. Carotenoids. In: Ross CA, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed: Lippincott Williams & Wilkins; 2014:427-439.
2. Jalal F, Nesheim MC, Agus Z, Sanjur D, Habicht JP. Serum retinol concentrations in children are affected by food sources of beta-carotene, fat intake, and anthelmintic drug treatment. Am J Clin Nutr. 1998;68(3):623-629. (PubMed)
3. van Het Hof KH, West CE, Weststrate JA, Hautvast JG. Dietary factors that affect the bioavailability of carotenoids. J Nutr. 2000;130(3):503-506. (PubMed)
4. Priyadarshani AM. A review on factors influencing bioaccessibility and bioefficacy of carotenoids. Crit Rev Food Sci Nutr. 2015;57(8):1710-1717. (PubMed)
5. Moran NE, Mohn ES, Hason N, Erdman JW, Jr., Johnson EJ. Intrinsic and extrinsic factors impacting absorption, metabolism, and health effects of dietary carotenoids. Adv Nutr. 2018;9(4):465-492. (PubMed)
6. Reboul E. Absorption of vitamin A and carotenoids by the enterocyte: focus on transport proteins. Nutrients. 2013;5(9):3563-3581. (PubMed)
7. Bohn T, Desmarchelier C, Dragsted LO, et al. Host-related factors explaining interindividual variability of carotenoid bioavailability and tissue concentrations in humans. Mol Nutr Food Res. 2017;61(6):1600685. (PubMed)
8. Reboul E. Mechanisms of carotenoid intestinal absorption: where do we stand? Nutrients. 2019;11(4):838. (PubMed)
9. Tanumihardjo SA, Palacios N, Pixley KV. Provitamin a carotenoid bioavailability:what really matters? Int J Vitam Nutr Res. 2010;80(4-5):336-350. (PubMed)
10. Borel P. Genetic variations involved in interindividual variability in carotenoid status. Mol Nutr Food Res. 2012;56(2):228-240. (PubMed)
11. Food and Nutrition Board, Institute of Medicine. Beta-carotene and other carotenoids. Dietary reference intakes for vitamin C, vitamin E, selenium, and carotenoids. Washington, D.C.: National Academy Press; 2000:325-400. (National Academy Press)
12. Weber D, Grune T. The contribution of beta-carotene to vitamin A supply of humans. Mol Nutr Food Res. 2012;56(2):251-258. (PubMed)
13. Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 3rd ed. New York, NY: Oxford University Press; 1999.
14. Di Mascio P, Kaiser S, Sies H. Lycopene as the most efficient biological carotenoid singlet oxygen quencher. Arch Biochem Biophys. 1989;274(2):532-538. (PubMed)
15. Young AJ, Lowe GM. Antioxidant and prooxidant properties of carotenoids. Arch Biochem Biophys. 2001;385(1):20-27. (PubMed)
16. Kaulmann A, Bohn T. Carotenoids, inflammation, and oxidative stress--implications of cellular signaling pathways and relation to chronic disease prevention. Nutr Res. 2014;34(11):907-929. (PubMed)
17. Ben-Dor A, Steiner M, Gheber L, et al. Carotenoids activate the antioxidant response element transcription system. Mol Cancer Ther. 2005;4(1):177-186. (PubMed)
18. Yang PM, Wu ZZ, Zhang YQ, Wung BS. Lycopene inhibits ICAM-1 expression and NF-kappaB activation by Nrf2-regulated cell redox state in human retinal pigment epithelial cells. Life Sci. 2016;155:94-101. (PubMed)
19. Lian F, Wang XD. Enzymatic metabolites of lycopene induce Nrf2-mediated expression of phase II detoxifying/antioxidant enzymes in human bronchial epithelial cells. Int J Cancer. 2008;123(6):1262-1268. (PubMed)
20. Sung LC, Chao HH, Chen CH, et al. Lycopene inhibits cyclic strain-induced endothelin-1 expression through the suppression of reactive oxygen species generation and induction of heme oxygenase-1 in human umbilical vein endothelial cells. Clin Exp Pharmacol Physiol. 2015;42(6):632-639. (PubMed)
21. Yang CM, Huang SM, Liu CL, Hu ML. Apo-8'-lycopenal induces expression of HO-1 and NQO-1 via the ERK/p38-Nrf2-ARE pathway in human HepG2 cells. J Agric Food Chem. 2012;60(6):1576-1585. (PubMed)
22. Krinsky NI, Landrum JT, Bone RA. Biologic mechanisms of the protective role of lutein and zeaxanthin in the eye. Annu Rev Nutr. 2003;23:171-201. (PubMed)
23. Kvansakul J, Rodriguez-Carmona M, Edgar DF, et al. Supplementation with the carotenoids lutein or zeaxanthin improves human visual performance. Ophthalmic Physiol Opt. 2006;26(4):362-371. (PubMed)
24. Ma L, Lin XM, Zou ZY, Xu XR, Li Y, Xu R. A 12-week lutein supplementation improves visual function in Chinese people with long-term computer display light exposure. Br J Nutr. 2009;102(2):186-190. (PubMed)
25. Stringham JM, Hammond BR. Macular pigment and visual performance under glare conditions. Optom Vis Sci. 2008;85(2):82-88. (PubMed)
26. Yagi A, Fujimoto K, Michihiro K, Goh B, Tsi D, Nagai H. The effect of lutein supplementation on visual fatigue: a psychophysiological analysis. Appl Ergon. 2009;40(6):1047-1054. (PubMed)
27. Stringham JM, Hammond BR, Jr. Dietary lutein and zeaxanthin: possible effects on visual function. Nutr Rev. 2005;63(2):59-64. (PubMed)
28. Bertram JS. Carotenoids and gene regulation. Nutr Rev. 1999;57(6):182-191. (PubMed)
29. Stahl W, Nicolai S, Briviba K, et al. Biological activities of natural and synthetic carotenoids: induction of gap junctional communication and singlet oxygen quenching. Carcinogenesis. 1997;18(1):89-92. (PubMed)
30. Vine AL, Leung YM, Bertram JS. Transcriptional regulation of connexin 43 expression by retinoids and carotenoids: similarities and differences. Mol Carcinog. 2005;43(2):75-85. (PubMed)
31. van Poppel G, Spanhaak S, Ockhuizen T. Effect of beta-carotene on immunological indexes in healthy male smokers. Am J Clin Nutr. 1993;57(3):402-407. (PubMed)
32. Hughes DA, Wright AJ, Finglas PM, et al. The effect of beta-carotene supplementation on the immune function of blood monocytes from healthy male nonsmokers. J Lab Clin Med. 1997;129(3):309-317. (PubMed)
33. Santos MS, Gaziano JM, Leka LS, Beharka AA, Hennekens CH, Meydani SN. Beta-carotene-induced enhancement of natural killer cell activity in elderly men: an investigation of the role of cytokines. Am J Clin Nutr. 1998;68(1):164-170. (PubMed)
34. Hughes DA, Wright AJ, Finglas PM, et al. Effects of lycopene and lutein supplementation on the expression of functionally associated surface molecules on blood monocytes from healthy male nonsmokers. J Infect Dis. 2000;182 Suppl 1:S11-15. (PubMed)
35. Watzl B, Bub A, Blockhaus M, et al. Prolonged tomato juice consumption has no effect on cell-mediated immunity of well-nourished elderly men and women. J Nutr. 2000;130(7):1719-1723. (PubMed)
36. Corridan BM, O'Donoghue M, Hughes DA, Morrissey PA. Low-dose supplementation with lycopene or beta-carotene does not enhance cell-mediated immunity in healthy free-living elderly humans. Eur J Clin Nutr. 2001;55(8):627-635. (PubMed)
37. American Cancer Society. Key Statistics for Lung Cancer. Available at: https://www.cancer.org/cancer/types/lung-cancer/about/key-statistics.html. Accessed 9/27/2023.
38. Gallicchio L, Boyd K, Matanoski G, et al. Carotenoids and the risk of developing lung cancer: a systematic review. Am J Clin Nutr. 2008;88(2):372-383. (PubMed)
39. The Alpha-Tocopherol Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N Engl J Med. 1994;330(15):1029-1035. (PubMed)
40. Omenn GS, Goodman GE, Thornquist MD, et al. Risk factors for lung cancer and for intervention effects in CARET, the Beta-Carotene and Retinol Efficacy Trial. J Natl Cancer Inst. 1996;88(21):1550-1559. (PubMed)
41. Hennekens CH, Buring JE, Manson JE, et al. Lack of effect of long-term supplementation with beta carotene on the incidence of malignant neoplasms and cardiovascular disease. N Engl J Med. 1996;334(18):1145-1149. (PubMed)
42. Kamangar F, Qiao YL, Yu B, et al. Lung cancer chemoprevention: a randomized, double-blind trial in Linxian, China. Cancer Epidemiol Biomarkers Prev. 2006;15(8):1562-1564. (PubMed)
43. Chew EY, Clemons TE, Agron E, et al. Long-term outcomes of adding lutein/zeaxanthin and omega-3 fatty acids to the AREDS supplements on age-related macular degeneration progression: AREDS2 report 28. JAMA Ophthalmol. 2022;140(7):692-698. (PubMed)
44. O'Connor EA, Evans CV, Ivlev I, et al. Vitamin and mineral supplements for the primary prevention of cardiovascular disease and cancer: updated evidence report and systematic review for the US Preventive Services Task Force. JAMA. 2022;327(23):2334-2347. (PubMed)
45. Veeramachaneni S, Wang XD. Carotenoids and lung cancer prevention. Front Biosci (Schol Ed). 2009;1:258-274. (PubMed)
46. Mayne ST, Ferrucci LM, Cartmel B. Lessons learned from randomized clinical trials of micronutrient supplementation for cancer prevention. Annu Rev Nutr. 2012;32:369-390. (PubMed)
47. Moyer VA, US Preventive Services Task Force. Vitamin, mineral, and multivitamin supplements for the primary prevention of cardiovascular disease and cancer: U.S. Preventive services Task Force recommendation statement. Ann Intern Med. 2014;160(8):558-564. (PubMed)
48. Centers for Disease Control and Prevention. Prostate Cancer Statistics. Available at: https://www.cdc.gov/cancer/prostate/statistics/index.htm. Accessed 9/27/2023.
49. Giovannucci E. A review of epidemiologic studies of tomatoes, lycopene, and prostate cancer. Exp Biol Med (Maywood). 2002;227(10):852-859. (PubMed)
50. Wang Y, Cui R, Xiao Y, Fang J, Xu Q. Effect of carotene and lycopene on the risk of prostate cancer: a systematic review and dose-response meta-analysis of observational studies. PLoS One. 2015;10(9):e0137427. (PubMed)
51. Wang Y, Cui R, Xiao Y, Fang J, Xu Q. Correction: effect of carotene and lycopene on the risk of prostate cancer: a systematic review and dose-response meta-analysis of observational studies. PLoS One. 2015;10(10):e0140415. (PubMed)
52. Key TJ, Appleby PN, Travis RC, et al. Carotenoids, retinol, tocopherols, and prostate cancer risk: pooled analysis of 15 studies. Am J Clin Nutr. 2015;102(5):1142-1157. (PubMed)
53. Rowles JL, 3rd, Ranard KM, Smith JW, An R, Erdman JW, Jr. Increased dietary and circulating lycopene are associated with reduced prostate cancer risk: a systematic review and meta-analysis. Prostate Cancer Prostatic Dis. 2017;20(4):361-377. (PubMed)
54. Melendez-Martinez AJ, Mapelli-Brahm P, Benitez-Gonzalez A, Stinco CM. A comprehensive review on the colorless carotenoids phytoene and phytofluene. Arch Biochem Biophys. 2015;572:188-200. (PubMed)
55. World Cancer Research Fund International/American Institute for Cancer Research Continuous Update Project Report. Diet, Nutrition, Physical Activity, and Prostate Cancer 2014.
56. Gann PH, Deaton RJ, Rueter EE, et al. A phase II randomized trial of lycopene-rich tomato extract among men with high-grade prostatic intraepithelial neoplasia. Nutr Cancer. 2015;67(7):1104-1112. (PubMed)
57. Gontero P, Marra G, Soria F, et al. A randomized double-blind placebo controlled phase I-II study on clinical and molecular effects of dietary supplements in men with precancerous prostatic lesions. Chemoprevention or "chemopromotion"? Prostate. 2015;75(11):1177-1186. (PubMed)
58. Holzapfel NP, Holzapfel BM, Champ S, Feldthusen J, Clements J, Hutmacher DW. The potential role of lycopene for the prevention and therapy of prostate cancer: from molecular mechanisms to clinical evidence. Int J Mol Sci. 2013;14(7):14620-14646. (PubMed)
59. Kumar NB, Besterman-Dahan K, Kang L, et al. Results of a randomized clinical trial of the action of several doses of lycopene in localized prostate cancer: administration prior to radical prostatectomy. Clin Med Urol. 2008;1:1-14. (PubMed)
60. Ansari MS, Gupta NP. A comparison of lycopene and orchidectomy vs orchidectomy alone in the management of advanced prostate cancer. BJU Int. 2003;92(4):375-378; discussion 378. (PubMed)
61. Sadeghian M, Asadi M, Rahmani S, Sadeghi N, Hosseini SA, Zare Javid A. Lycopene does not affect prostate-specific antigen in men with non-metastatic prostate cancer: a systematic review and meta-analysis of randomized controlled trials. Nutr Cancer. 2021;73(11-12):2796-2807. (PubMed)
62. Aune D, Chan DS, Vieira AR, et al. Dietary compared with blood concentrations of carotenoids and breast cancer risk: a systematic review and meta-analysis of prospective studies. Am J Clin Nutr. 2012;96(2):356-373. (PubMed)
63. Eliassen AH, Hendrickson SJ, Brinton LA, et al. Circulating carotenoids and risk of breast cancer: pooled analysis of eight prospective studies. J Natl Cancer Inst. 2012;104(24):1905-1916. (PubMed)
64. Bakker MF, Peeters PH, Klaasen VM, et al. Plasma carotenoids, vitamin C, tocopherols, and retinol and the risk of breast cancer in the European Prospective Investigation into Cancer and Nutrition cohort. Am J Clin Nutr. 2016;103(2):454-464. (PubMed)
65. Peng C, Gao C, Lu D, et al. Circulating carotenoids and breast cancer among high-risk individuals. Am J Clin Nutr. 2021;113(3):525-533. (PubMed)
66. Leenders M, Leufkens AM, Siersema PD, et al. Plasma and dietary carotenoids and vitamins A, C and E and risk of colon and rectal cancer in the European Prospective Investigation into Cancer and Nutrition. Int J Cancer. 2014;135(12):2930-2939. (PubMed)
67. Panic N, Nedovic D, Pastorino R, Boccia S, Leoncini E. Carotenoid intake from natural sources and colorectal cancer: a systematic review and meta-analysis of epidemiological studies. Eur J Cancer Prev. 2017;26(1):27-37. (PubMed)
68. Wang X, Yang HH, Liu Y, Zhou Q, Chen ZH. Lycopene consumption and risk of colorectal cancer: a meta-analysis of observational studies. Nutr Cancer. 2016;68(7):1083-1096. (PubMed)
69. Leoncini E, Nedovic D, Panic N, Pastorino R, Edefonti V, Boccia S. Carotenoid intake from natural sources and head and neck cancer: a systematic review and meta-analysis of epidemiological studies. Cancer Epidemiol Biomarkers Prev. 2015;24(7):1003-1011. (PubMed)
70. Brewczynski A, Jablonska B, Kentnowski M, Mrowiec S, Skladowski K, Rutkowski T. The association between carotenoids and head and neck cancer risk. Nutrients. 2021;14(1):88. (PubMed)
71. Wu S, Liu Y, Michalek JE, et al. Carotenoid intake and circulating carotenoids are inversely associated with the risk of bladder cancer: a dose-response meta-analysis. Adv Nutr. 2020;11(3):630-643. (PubMed)
72. Hammond BR, Jr., Johnson EJ, Russell RM, et al. Dietary modification of human macular pigment density. Invest Ophthalmol Vis Sci. 1997;38(9):1795-1801. (PubMed)
73. Mares JA, LaRowe TL, Snodderly DM, et al. Predictors of optical density of lutein and zeaxanthin in retinas of older women in the Carotenoids in Age-Related Eye Disease Study, an ancillary study of the Women's Health Initiative. Am J Clin Nutr. 2006;84(5):1107-1122. (PubMed)
74. Mares-Perlman JA, Millen AE, Ficek TL, Hankinson SE. The body of evidence to support a protective role for lutein and zeaxanthin in delaying chronic disease. Overview. J Nutr. 2002;132(3):518S-524S. (PubMed)
75. Snellen EL, Verbeek AL, Van Den Hoogen GW, Cruysberg JR, Hoyng CB. Neovascular age-related macular degeneration and its relationship to antioxidant intake. Acta Ophthalmol Scand. 2002;80(4):368-371. (PubMed)
76. Mares-Perlman JA, Fisher AI, Klein R, et al. Lutein and zeaxanthin in the diet and serum and their relation to age-related maculopathy in the third national health and nutrition examination survey. Am J Epidemiol. 2001;153(5):424-432. (PubMed)
77. Seddon JM, Ajani UA, Sperduto RD, et al. Dietary carotenoids, vitamins A, C, and E, and advanced age-related macular degeneration. Eye Disease Case-Control Study Group. JAMA. 1994;272(18):1413-1420. (PubMed)
78. Antioxidant status and neovascular age-related macular degeneration. Eye Disease Case-Control Study Group. Arch Ophthalmol. 1993;111(1):104-109. (PubMed)
79. Gale CR, Hall NF, Phillips DI, Martyn CN. Lutein and zeaxanthin status and risk of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2003;44(6):2461-2465. (PubMed)
80. Bone RA, Landrum JT, Mayne ST, Gomez CM, Tibor SE, Twaroska EE. Macular pigment in donor eyes with and without AMD: a case-control study. Invest Ophthalmol Vis Sci. 2001;42(1):235-240. (PubMed)
81. Beatty S, Murray IJ, Henson DB, Carden D, Koh H, Boulton ME. Macular pigment and risk for age-related macular degeneration in subjects from a Northern European population. Invest Ophthalmol Vis Sci. 2001;42(2):439-446. (PubMed)
82. Cho E, Seddon JM, Rosner B, Willett WC, Hankinson SE. Prospective study of intake of fruits, vegetables, vitamins, and carotenoids and risk of age-related maculopathy. Arch Ophthalmol. 2004;122(6):883-892. (PubMed)
83. Flood V, Smith W, Wang JJ, Manzi F, Webb K, Mitchell P. Dietary antioxidant intake and incidence of early age-related maculopathy: the Blue Mountains Eye Study. Ophthalmology. 2002;109(12):2272-2278. (PubMed)
84. Mares-Perlman JA, Klein R, Klein BE, et al. Association of zinc and antioxidant nutrients with age-related maculopathy. Arch Ophthalmol. 1996;114(8):991-997. (PubMed)
85. Mares-Perlman JA, Brady WE, Klein R, et al. Serum antioxidants and age-related macular degeneration in a population-based case-control study. Arch Ophthalmol. 1995;113(12):1518-1523. (PubMed)
86. Wu J, Cho E, Willett WC, Sastry SM, Schaumberg DA. Intakes of lutein, zeaxanthin, and other carotenoids and age-related macular degeneration during 2 decades of prospective follow-up. JAMA Ophthalmol. 2015;133(12):1415-1424. (PubMed)
87. Merle BMJ, Cougnard-Gregoire A, Korobelnik JF, et al. Plasma lutein, a nutritional biomarker for development of advanced age-related macular degeneration: the Alienor study. Nutrients. 2021;13(6):2047. (PubMed)
88. Richer S, Stiles W, Statkute L, et al. Double-masked, placebo-controlled, randomized trial of lutein and antioxidant supplementation in the intervention of atrophic age-related macular degeneration: the Veterans LAST study (Lutein Antioxidant Supplementation Trial). Optometry. 2004;75(4):216-230. (PubMed)
89. Age-Related Eye Disease Study 2 Research Group. Lutein + zeaxanthin and omega-3 fatty acids for age-related macular degeneration: the Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial. JAMA. 2013;309(19):2005-2015. (PubMed)
90. Age-Related Eye Disease Study 2 Research Group, Chew EY, Clemons TE, et al. Secondary analyses of the effects of lutein/zeaxanthin on age-related macular degeneration progression: AREDS2 report No. 3. JAMA Ophthalmol. 2014;132(2):142-149. (PubMed)
91. Scripsema NK, Hu DN, Rosen RB. Lutein, zeaxanthin, and meso-zeaxanthin in the clinical management of eye disease. J Ophthalmol. 2015;2015:865179. (PubMed)
92. Liu R, Wang T, Zhang B, et al. Lutein and zeaxanthin supplementation and association with visual function in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2015;56(1):252-258. (PubMed)
93. van der Made SM, Kelly ER, Kijlstra A, Plat J, Berendschot TT. Increased macular pigment optical density and visual acuity following consumption of a buttermilk drink containing lutein-enriched egg yolks: a randomized, double-blind, placebo-controlled trial. J Ophthalmol. 2016;2016:9035745. (PubMed)
94. Piatti A, Croce A, Mazzacane D, et al. Effect of 2-year nutritional supplementation on progression of age-related macular degeneration. Eur J Ophthalmol. 2020;30(2):376-381. (PubMed)
95. Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119(10):1417-1436. (PubMed)
96. Teikari JM, Laatikainen L, Virtamo J, et al. Six-year supplementation with alpha-tocopherol and beta-carotene and age-related maculopathy. Acta Ophthalmol Scand. 1998;76(2):224-229. (PubMed)
97. Christen WG, Manson JE, Glynn RJ, et al. Beta carotene supplementation and age-related maculopathy in a randomized trial of US physicians. Arch Ophthalmol. 2007;125(3):333-339. (PubMed)
98. Evans J. Antioxidant supplements to prevent or slow down the progression of AMD: a systematic review and meta-analysis. Eye. 2008;22(6):751-760. (PubMed)
99. Evans JR, Henshaw K. Antioxidant vitamin and mineral supplements for preventing age-related macular degeneration. Cochrane Database Syst Rev. 2008(1):CD000253. (PubMed)
100. Gong X, Rubin LP. Role of macular xanthophylls in prevention of common neovascular retinopathies: retinopathy of prematurity and diabetic retinopathy. Arch Biochem Biophys. 2015;572:40-48. (PubMed)
101. Romagnoli C, Giannantonio C, Cota F, et al. A prospective, randomized, double blind study comparing lutein to placebo for reducing occurrence and severity of retinopathy of prematurity. J Matern Fetal Neonatal Med. 2011;24 Suppl 1:147-150. (PubMed)
102. Dani C, Lori I, Favelli F, et al. Lutein and zeaxanthin supplementation in preterm infants to prevent retinopathy of prematurity: a randomized controlled study. J Matern Fetal Neonatal Med. 2012;25(5):523-527. (PubMed)
103. Manzoni P, Guardione R, Bonetti P, et al. Lutein and zeaxanthin supplementation in preterm very low-birth-weight neonates in neonatal intensive care units: a multicenter randomized controlled trial. Am J Perinatol. 2013;30(1):25-32. (PubMed)
104. Rubin LP, Chan GM, Barrett-Reis BM, et al. Effect of carotenoid supplementation on plasma carotenoids, inflammation and visual development in preterm infants. J Perinatol. 2012;32(6):418-424. (PubMed)
105. Brazionis L, Rowley K, Itsiopoulos C, O'Dea K. Plasma carotenoids and diabetic retinopathy. Br J Nutr. 2009;101(2):270-277. (PubMed)
106. Lima VC, Rosen RB, Maia M, et al. Macular pigment optical density measured by dual-wavelength autofluorescence imaging in diabetic and nondiabetic patients: a comparative study. Invest Ophthalmol Vis Sci. 2010;51(11):5840-5845. (PubMed)
107. Sahli MW, Mares JA, Meyers KJ, et al. Dietary intake of lutein and diabetic retinopathy in the Atherosclerosis Risk in Communities Study (ARIC). Ophthalmic Epidemiol. 2016;23(2):99-108. (PubMed)
108. The National Institutes of Heath/National Eye Institute. Cataracts. Available at: https://nei.nih.gov/eyedata/cataract. Accessed 6/9/2016.
109. Yeum KJ, Shang FM, Schalch WM, Russell RM, Taylor A. Fat-soluble nutrient concentrations in different layers of human cataractous lens. Curr Eye Res. 1999;19(6):502-505. (PubMed)
110. Brown L, Rimm EB, Seddon JM, et al. A prospective study of carotenoid intake and risk of cataract extraction in US men. Am J Clin Nutr. 1999;70(4):517-524. (PubMed)
111. Chasan-Taber L, Willett WC, Seddon JM, et al. A prospective study of carotenoid and vitamin A intakes and risk of cataract extraction in US women. Am J Clin Nutr. 1999;70(4):509-516. (PubMed)
112. Lyle BJ, Mares-Perlman JA, Klein BE, Klein R, Greger JL. Antioxidant intake and risk of incident age-related nuclear cataracts in the Beaver Dam Eye Study. Am J Epidemiol. 1999;149(9):801-809. (PubMed)
113. Christen WG, Liu S, Glynn RJ, Gaziano JM, Buring JE. Dietary carotenoids, vitamins C and E, and risk of cataract in women: a prospective study. Arch Ophthalmol. 2008;126(1):102-109. (PubMed)
114. Moeller SM, Voland R, Tinker L, et al. Associations between age-related nuclear cataract and lutein and zeaxanthin in the diet and serum in the carotenoids in the Age-Related Eye Disease Study, an ancillary study of the Women's Health Initiative. Arch Ophthalmol. 2008;126(3):354-364. (PubMed)
115. Age-Related Eye Disease Study 2 Research Group, Chew EY, SanGiovanni JP, et al. Lutein/zeaxanthin for the treatment of age-related cataract: AREDS2 randomized trial report no. 4. JAMA Ophthalmol. 2013;131(7):843-850. (PubMed)
116. Glaser TS, Doss LE, Shih G, et al. The association of dietary lutein plus zeaxanthin and B vitamins with cataracts in the Age-Related Eye Disease Study: AREDS Report No. 37. Ophthalmology. 2015;122(7):1471-1479. (PubMed)
117. Christen WG, Manson JE, Glynn RJ, et al. A randomized trial of beta carotene and age-related cataract in US physicians. Arch Ophthalmol. 2003;121(3):372-378. (PubMed)
118. Gritz DC, Srinivasan M, Smith SD, et al. The Antioxidants in Prevention of Cataracts Study: effects of antioxidant supplements on cataract progression in South India. Br J Ophthalmol. 2006;90(7):847-851. (PubMed)
119. Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E and beta carotene for age-related cataract and vision loss: AREDS report no. 9. Arch Ophthalmol. 2001;119(10):1439-1452. (PubMed)
120. Chylack LT, Jr., Brown NP, Bron A, et al. The Roche European American Cataract Trial (REACT): a randomized clinical trial to investigate the efficacy of an oral antioxidant micronutrient mixture to slow progression of age-related cataract. Ophthalmic Epidemiol. 2002;9(1):49-80. (PubMed)
121. Kritchevsky SB. beta-Carotene, carotenoids and the prevention of coronary heart disease. J Nutr. 1999;129(1):5-8. (PubMed)
122. Bots ML, Grobbee DE. Intima media thickness as a surrogate marker for generalised atherosclerosis. Cardiovasc Drugs Ther. 2002;16(4):341-351. (PubMed)
123. Rissanen TH, Voutilainen S, Nyyssonen K, Salonen R, Kaplan GA, Salonen JT. Serum lycopene concentrations and carotid atherosclerosis: the Kuopio Ischaemic Heart Disease Risk Factor Study. Am J Clin Nutr. 2003;77(1):133-138. (PubMed)
124. Dwyer JH, Paul-Labrador MJ, Fan J, Shircore AM, Merz CN, Dwyer KM. Progression of carotid intima-media thickness and plasma antioxidants: the Los Angeles Atherosclerosis Study. Arterioscler Thromb Vasc Biol. 2004;24(2):313-9. (PubMed)
125. McQuillan BM, Hung J, Beilby JP, Nidorf M, Thompson PL. Antioxidant vitamins and the risk of carotid atherosclerosis. The Perth Carotid Ultrasound Disease Assessment study (CUDAS). J Am Coll Cardiol. 2001;38(7):1788-1794. (PubMed)
126. Rissanen T, Voutilainen S, Nyyssonen K, Salonen R, Salonen JT. Low plasma lycopene concentration is associated with increased intima-media thickness of the carotid artery wall. Arterioscler Thromb Vasc Biol. 2000;20(12):2677-2681. (PubMed)
127. D'Odorico A, Martines D, Kiechl S, et al. High plasma levels of alpha- and beta-carotene are associated with a lower risk of atherosclerosis: results from the Bruneck study. Atherosclerosis. 2000;153(1):231-239. (PubMed)
128. Iribarren C, Folsom AR, Jacobs DR, Jr., Gross MD, Belcher JD, Eckfeldt JH. Association of serum vitamin levels, LDL susceptibility to oxidation, and autoantibodies against MDA-LDL with carotid atherosclerosis. A case-control study. The ARIC Study Investigators. Atherosclerosis Risk in Communities. Arterioscler Thromb Vasc Biol. 1997;17(6):1171-1177. (PubMed)
129. Sesso HD, Buring JE, Norkus EP, Gaziano JM. Plasma lycopene, other carotenoids, and retinol and the risk of cardiovascular disease in women. Am J Clin Nutr. 2004;79(1):47-53. (PubMed)
130. Rissanen TH, Voutilainen S, Nyyssonen K, et al. Low serum lycopene concentration is associated with an excess incidence of acute coronary events and stroke: the Kuopio Ischaemic Heart Disease Risk Factor Study. Br J Nutr. 2001;85(6):749-754. (PubMed)
131. Street DA, Comstock GW, Salkeld RM, Schuep W, Klag MJ. Serum antioxidants and myocardial infarction. Are low levels of carotenoids and alpha-tocopherol risk factors for myocardial infarction? Circulation. 1994;90(3):1154-1161. (PubMed)
132. Ito Y, Kurata M, Suzuki K, Hamajima N, Hishida H, Aoki K. Cardiovascular disease mortality and serum carotenoid levels: a Japanese population-based follow-up study. J Epidemiol. 2006;16(4):154-160. (PubMed)
133. Buijsse B, Feskens EJ, Kwape L, Kok FJ, Kromhout D. Both alpha- and beta-carotene, but not tocopherols and vitamin C, are inversely related to 15-year cardiovascular mortality in Dutch elderly men. J Nutr. 2008;138(2):344-350. (PubMed)
134. Sesso HD, Buring JE, Norkus EP, Gaziano JM. Plasma lycopene, other carotenoids, and retinol and the risk of cardiovascular disease in men. Am J Clin Nutr. 2005;81(5):990-997. (PubMed)
135. Hak AE, Stampfer MJ, Campos H, et al. Plasma carotenoids and tocopherols and risk of myocardial infarction in a low-risk population of US male physicians. Circulation. 2003;108(7):802-807. (PubMed)
136. Evans RW, Shaten BJ, Day BW, Kuller LH. Prospective association between lipid soluble antioxidants and coronary heart disease in men. The Multiple Risk Factor Intervention Trial. Am J Epidemiol. 1998;147(2):180-186. (PubMed)
137. Sahyoun NR, Jacques PF, Russell RM. Carotenoids, vitamins C and E, and mortality in an elderly population. Am J Epidemiol. 1996;144(5):501-511. (PubMed)
138. Wang Y, Chung SJ, McCullough ML, et al. Dietary carotenoids are associated with cardiovascular disease risk biomarkers mediated by serum carotenoid concentrations. J Nutr. 2014;144(7):1067-1074. (PubMed)
139. Li Z, Chen J, Zhang D. Association between dietary carotenoid intakes and hypertension in adults: National Health and Nutrition Examination Survey 2007-2014. J Hypertens. 2019;37(12):2371-2379. (PubMed)
140. Leermakers ET, Darweesh SK, Baena CP, et al. The effects of lutein on cardiometabolic health across the life course: a systematic review and meta-analysis. Am J Clin Nutr. 2016;103(2):481-494. (PubMed)
141. Tierney AC, Rumble CE, Billings LM, George ES. Effect of dietary and supplemental lycopene on cardiovascular risk factors: a systematic review and meta-analysis. Adv Nutr. 2020;11(6):1453-1488. (PubMed)
142. Rimm EB, Stampfer MJ, Ascherio A, Giovannucci E, Colditz GA, Willett WC. Vitamin E consumption and the risk of coronary heart disease in men. N Engl J Med. 1993;328(20):1450-1456. (PubMed)
143. Gaziano JM, Manson JE, Branch LG, Colditz GA, Willett WC, Buring JE. A prospective study of consumption of carotenoids in fruits and vegetables and decreased cardiovascular mortality in the elderly. Ann Epidemiol. 1995;5(4):255-260. (PubMed)
144. Osganian SK, Stampfer MJ, Rimm E, Spiegelman D, Manson JE, Willett WC. Dietary carotenoids and risk of coronary artery disease in women. Am J Clin Nutr. 2003;77(6):1390-1399. (PubMed)
145. Greenberg ER, Baron JA, Karagas MR, et al. Mortality associated with low plasma concentration of beta carotene and the effect of oral supplementation. JAMA. 1996;275(9):699-703. (PubMed)
146. Omenn GS, Goodman GE, Thornquist MD, et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N Engl J Med. 1996;334(18):1150-1155. (PubMed)
147. Yang J, Zhang Y, Na X, Zhao A. beta-Carotene supplementation and risk of cardiovascular disease: a systematic review and meta-analysis of randomized controlled trials. Nutrients. 2022;14(6):1284. (PubMed)
148. Voutilainen S, Nurmi T, Mursu J, Rissanen TH. Carotenoids and cardiovascular health. Am J Clin Nutr. 2006;83(6):1265-1271. (PubMed)
149. Gajendragadkar PR, Hubsch A, Maki-Petaja KM, Serg M, Wilkinson IB, Cheriyan J. Effects of oral lycopene supplementation on vascular function in patients with cardiovascular disease and healthy volunteers: a randomised controlled trial. PLoS One. 2014;9(6):e99070. (PubMed)
150. Stangl V, Kuhn C, Hentschel S, et al. Lack of effects of tomato products on endothelial function in human subjects: results of a randomised, placebo-controlled cross-over study. Br J Nutr. 2011;105(2):263-267. (PubMed)
151. Kim JY, Paik JK, Kim OY, et al. Effects of lycopene supplementation on oxidative stress and markers of endothelial function in healthy men. Atherosclerosis. 2011;215(1):189-195. (PubMed)
152. Xaplanteris P, Vlachopoulos C, Pietri P, et al. Tomato paste supplementation improves endothelial dynamics and reduces plasma total oxidative status in healthy subjects. Nutr Res. 2012;32(5):390-394. (PubMed)
153. Ozaki K, Okamoto M, Fukasawa K, et al. Daily intake of beta-cryptoxanthin prevents bone loss by preferential disturbance of osteoclastic activation in ovariectomized mice. J Pharmacol Sci. 2015;129(1):72-77. (PubMed)
154. Sahni S, Hannan MT, Blumberg J, Cupples LA, Kiel DP, Tucker KL. Inverse association of carotenoid intakes with 4-y change in bone mineral density in elderly men and women: the Framingham Osteoporosis Study. Am J Clin Nutr. 2009;89(1):416-424. (PubMed)
155. Sahni S, Hannan MT, Blumberg J, Cupples LA, Kiel DP, Tucker KL. Protective effect of total carotenoid and lycopene intake on the risk of hip fracture: a 17-year follow-up from the Framingham Osteoporosis Study. J Bone Miner Res. 2009;24(6):1086-1094. (PubMed)
156. Dai Z, Wang R, Ang LW, Low YL, Yuan JM, Koh WP. Protective effects of dietary carotenoids on risk of hip fracture in men: the Singapore Chinese Health Study. J Bone Miner Res. 2014;29(2):408-417. (PubMed)
157. Sugiura M, Nakamura M, Ogawa K, Ikoma Y, Yano M. High serum carotenoids associated with lower risk for bone loss and osteoporosis in post-menopausal Japanese female subjects: prospective cohort study. PLoS One. 2012;7(12):e52643. (PubMed)
158. Hayhoe RPG, Lentjes MAH, Mulligan AA, Luben RN, Khaw KT, Welch AA. Carotenoid dietary intakes and plasma concentrations are associated with heel bone ultrasound attenuation and osteoporotic fracture risk in the European Prospective Investigation into Cancer and Nutrition (EPIC)-Norfolk cohort. Br J Nutr. 2017;117(10):1439-1453. (PubMed)
159. Kang JH, Ascherio A, Grodstein F. Fruit and vegetable consumption and cognitive decline in aging women. Ann Neurol. 2005;57(5):713-720. (PubMed)
160. Morris MC, Evans DA, Tangney CC, Bienias JL, Wilson RS. Associations of vegetable and fruit consumption with age-related cognitive change. Neurology. 2006;67(8):1370-1376. (PubMed)
161. Christensen K, Gleason CE, Mares JA. Dietary carotenoids and cognitive function among US adults, NHANES 2011-2014. Nutr Neurosci. 2020;23(7):554-562. (PubMed)
162. Edwards CG, Walk AM, Thompson SV, et al. Dietary lutein plus zeaxanthin and choline intake is interactively associated with cognitive flexibility in middle-adulthood in adults with overweight and obesity. Nutr Neurosci. 2022;25(7):1437-1452. (PubMed)
163. Feeney J, O'Leary N, Moran R, et al. Plasma lutein and zeaxanthin are associated with better cognitive function across multiple domains in a large population-based sample of older adults: findings from the Irish Longitudinal Study on Aging. J Gerontol A Biol Sci Med Sci. 2017;72(10):1431-1436. (PubMed)
164. Johnson EJ, Vishwanathan R, Johnson MA, et al. Relationship between serum and brain carotenoids, alpha-tocopherol, and retinol concentrations and cognitive performance in the oldest old from the Georgia Centenarian Study. J Aging Res. 2013;2013:951786. (PubMed)
165. Vishwanathan R, Kuchan MJ, Sen S, Johnson EJ. Lutein and preterm infants with decreased concentrations of brain carotenoids. J Pediatr Gastroenterol Nutr. 2014;59(5):659-665. (PubMed)
166. Feeney J, Finucane C, Savva GM, et al. Low macular pigment optical density is associated with lower cognitive performance in a large, population-based sample of older adults. Neurobiol Aging. 2013;34(11):2449-2456. (PubMed)
167. Renzi LM, Dengler MJ, Puente A, Miller LS, Hammond BR, Jr. Relationships between macular pigment optical density and cognitive function in unimpaired and mildly cognitively impaired older adults. Neurobiol Aging. 2014;35(7):1695-1699. (PubMed)
168. Kelly D, Coen RF, Akuffo KO, et al. Cognitive function and its relationship with macular pigment optical density and serum concentrations of its constituent carotenoids. J Alzheimers Dis. 2015;48(1):261-277. (PubMed)
169. Johnson EJ, McDonald K, Caldarella SM, Chung HY, Troen AM, Snodderly DM. Cognitive findings of an exploratory trial of docosahexaenoic acid and lutein supplementation in older women. Nutr Neurosci. 2008;11(2):75-83. (PubMed)
170. Renzi-Hammond LM, Bovier ER, Fletcher LM, et al. Effects of a lutein and zeaxanthin intervention on cognitive function: a randomized, double-masked, placebo-controlled trial of younger healthy adults. Nutrients. 2017;9(11):1246. (PubMed)
171. Power R, Coen RF, Beatty S, et al. Supplemental retinal carotenoids enhance memory in healthy individuals with low levels of macular pigment in a randomized, double-blind, placebo-controlled clinical trial. J Alzheimers Dis. 2018;61(3):947-961. (PubMed)
172. Chew EY, Clemons TE, Agron E, et al. Effect of omega-3 fatty acids, lutein/zeaxanthin, or other nutrient supplementation on cognitive function: the AREDS2 randomized clinical trial. JAMA. 2015;314(8):791-801. (PubMed)
173. Yeum KJ, Russell RM. Carotenoid bioavailability and bioconversion. Annu Rev Nutr. 2002;22:483-504. (PubMed)
174. Gartner C, Stahl W, Sies H. Lycopene is more bioavailable from tomato paste than from fresh tomatoes. Am J Clin Nutr. 1997;66(1):116-122. (PubMed)
175. Stahl W, Sies H. Uptake of lycopene and its geometrical isomers is greater from heat-processed than from unprocessed tomato juice in humans. J Nutr. 1992;122(11):2161-2166. (PubMed)
176. US Department of Agriculture, Agricultural Research Service. FoodData Central. Available at https://fdc.nal.usda.gov/. Accessed 9/28/2023.
177. Clinton SK. Lycopene: chemistry, biology, and implications for human health and disease. Nutr Rev. 1998;56(2 Pt 1):35-51. (PubMed)
178. Hendler SS, Rorvik DM, eds. PDR for Nutritional Supplements. 2nd ed. Thomson Reuters; 2008.
179. Tudor C, Pintea A. A brief overview of dietary zeaxanthin occurrence and bioaccessibility. Molecules. 2020;25(18):4067. (PubMed)
180. Tanvetyanon T, Bepler G. Beta-carotene in multivitamins and the possible risk of lung cancer among smokers versus former smokers: a meta-analysis and evaluation of national brands. Cancer. 2008;113(1):150-157. (PubMed)
181. Bowen PE, Herbst-Espinosa SM, Hussain EA, Stacewicz-Sapuntzakis M. Esterification does not impair lutein bioavailability in humans. J Nutr. 2002;132(12):3668-3673. (PubMed)
182. Solomons NW. Vitamin A and carotenoids. In: Bowman BA, Russell RM, eds. Present Knowledge in Nutrition. 8th ed. Washington, D.C.: ILSI Press; 2001:127-145.
183. Shao A, Hathcock JN. Risk assessment for the carotenoids lutein and lycopene. Regul Toxicol Pharmacol. 2006;45(3):289-298. (PubMed)
184. Choi RY, Chortkoff SC, Gorusupudi A, Bernstein PS. Crystalline maculopathy associated with high-dose lutein supplementation. JAMA Ophthalmol. 2016;134(12):1445-1448. (PubMed)
185. Tan KML, Chee J, Lim KLM, et al. Safety, tolerability, and pharmacokinetics of beta-cryptoxanthin supplementation in healthy women: a double-blind, randomized, placebo-controlled clinical trial. Nutrients. 2023;15(10):2325. (PubMed)
186. Ehrenfeld M, Levy M, Sharon P, Rachmilewitz D, Eliakim M. Gastrointestinal effects of long-term colchicine therapy in patients with recurrent polyserositis (familial mediterranean fever). Dig Dis Sci. 1982;27(8):723-727. (PubMed)
187. Natural-Medicines. Beta carotene/Drug interactions - Professional handout; 2016.
188. Brown BG, Zhao XQ, Chait A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345(22):1583-1592. (PubMed)
189. Collins R, Peto R, Armitage J. The MRC/BHF Heart Protection Study: preliminary results. Int J Clin Pract. 2002;56(1):53-56. (PubMed)
190. Koonsvitsky BP, Berry DA, Jones MB, et al. Olestra affects serum concentrations of alpha-tocopherol and carotenoids but not vitamin D or vitamin K status in free-living subjects. J Nutr. 1997;127(8 Suppl):1636S-1645S. (PubMed)
191. Thornquist MD, Kristal AR, Patterson RE, et al. Olestra consumption does not predict serum concentrations of carotenoids and fat-soluble vitamins in free-living humans: early results from the sentinel site of the olestra post-marketing surveillance study. J Nutr. 2000;130(7):1711-1718. (PubMed)
192. Neuhouser ML, Rock CL, Kristal AR, et al. Olestra is associated with slight reductions in serum carotenoids but does not markedly influence serum fat-soluble vitamin concentrations. Am J Clin Nutr. 2006;83(3):624-631. (PubMed)
193. Katan MB, Grundy SM, Jones P, Law M, Miettinen T, Paoletti R. Efficacy and safety of plant stanols and sterols in the management of blood cholesterol levels. Mayo Clin Proc. 2003;78(8):965-978. (PubMed)
194. Weststrate JA, Meijer GW. Plant sterol-enriched margarines and reduction of plasma total- and LDL-cholesterol concentrations in normocholesterolaemic and mildly hypercholesterolaemic subjects. Eur J Clin Nutr. 1998;52(5):334-343. (PubMed)
195. Noakes M, Clifton P, Ntanios F, Shrapnel W, Record I, McInerney J. An increase in dietary carotenoids when consuming plant sterols or stanols is effective in maintaining plasma carotenoid concentrations. Am J Clin Nutr. 2002;75(1):79-86. (PubMed)
196. Leo MA, Lieber CS. Alcohol, vitamin A, and beta-carotene: adverse interactions, including hepatotoxicity and carcinogenicity. Am J Clin Nutr. 1999;69(6):1071-1085. (PubMed)
197. Albanes D, Heinonen OP, Taylor PR, et al. Alpha-tocopherol and beta-carotene supplements and lung cancer incidence in the alpha-tocopherol, beta-carotene cancer prevention study: effects of base-line characteristics and study compliance. J Natl Cancer Inst. 1996;88(21):1560-1570. (PubMed)
198. van den Berg H. Carotenoid interactions. Nutr Rev. 1999;57(1):1-10. (PubMed)
199. Micozzi MS, Brown ED, Edwards BK, et al. Plasma carotenoid response to chronic intake of selected foods and beta-carotene supplements in men. Am J Clin Nutr. 1992;55(6):1120-1125. (PubMed)
200. Kostic D, White WS, Olson JA. Intestinal absorption, serum clearance, and interactions between lutein and beta-carotene when administered to human adults in separate or combined oral doses. Am J Clin Nutr. 1995;62(3):604-610. (PubMed)
201. Albanes D, Virtamo J, Taylor PR, Rautalahti M, Pietinen P, Heinonen OP. Effects of supplemental beta-carotene, cigarette smoking, and alcohol consumption on serum carotenoids in the Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study. Am J Clin Nutr. 1997;66(2):366-372. (PubMed)
202. Nierenberg DW, Dain BJ, Mott LA, Baron JA, Greenberg ER. Effects of 4 y of oral supplementation with beta-carotene on serum concentrations of retinol, tocopherol, and five carotenoids. Am J Clin Nutr. 1997;66(2):315-319. (PubMed)
203. Wahlqvist ML, Wattanapenpaiboon N, Macrae FA, Lambert JR, MacLennan R, Hsu-Hage BH. Changes in serum carotenoids in subjects with colorectal adenomas after 24 mo of beta-carotene supplementation. Australian Polyp Prevention Project Investigators. Am J Clin Nutr. 1994;60(6):936-943. (PubMed)
Chlorophyll and Metallo-Chlorophyll Derivatives
Contents
Summary
- Chlorophyll a and chlorophyll b are natural, fat-soluble chlorophylls found in plants. (More information)
- Sodium copper chlorophyllin (SCC) is a semi-synthetic mixture of water-soluble sodium copper salts derived from chlorophyll. (More information)
- SCC has been used orally as an internal deodorant and topically in the treatment of slow-healing wounds for more than 50 years without any serious side effects. (More information)
- Chlorophylls and SCC form tight molecular complexes with some chemicals known or suspected to cause cancer, and in doing so, may block carcinogenic effects. Carefully controlled studies have not been undertaken to determine whether a similar mechanism might limit uptake of required nutrients. (More information)
- Supplementation with SCC before meals substantially decreased a urinary biomarker of aflatoxin-induced DNA damage in a Chinese population at high risk of liver cancer due to unavoidable, dietary aflatoxin exposure from moldy grains and legumes. (More information)
- Scientists are hopeful that SCC supplementation will be helpful in decreasing the risk of liver cancer in high-risk populations with unavoidable, dietary aflatoxin exposure. However, it is not yet known whether SCC or natural chlorophylls will be useful in the prevention of cancers in people who are not exposed to significant levels of dietary aflatoxin. (More information)
Introduction
Chlorophyll is the pigment that gives plants and algae their green color. Plants use chlorophyll to trap light needed for photosynthesis (1). The basic structure of chlorophyll is a porphyrin ring similar to that of heme in hemoglobin, although the central atom in chlorophyll is magnesium instead of iron. The long hydrocarbon (phytol) tail attached to the porphyrin ring makes chlorophyll fat-soluble and insoluble in water. Chlorophyll a and chlorophyll b represent about 99% of the chlorophyll species found in edible plants (Figure 1; 2), while some algae and microalgae contain minor quantities of chlorophyll c pigments (e.g., Laminaria ochroleuca, Undaria pinnatifida) (3). Chlorophyll a and b only have a small difference in one of the side chains but an intact phytol tail, while the common characteristic of chlorophyll c isoforms is the absence of a phytol tail. These structural differences cause each type of chlorophyll to absorb light at slightly different wavelengths.
Metallo-chlorophyll derivatives, including chlorophyllins, can be chemically synthesized or produced in industrial food processing; these compounds contain zinc, iron, or copper in place of the central magnesium atom (2). The most studied chlorophyllin, sodium copper chlorophyllin (SCC), is a semi-synthetic mixture of sodium copper salts derived from chlorophyll (4, 5). SCC is often simply called ‘chlorophyllin’ in the older scientific literature, with newer publications specifying whether iron, zinc, copper, or magnesium chlorophyllin were studied. During its synthesis, the magnesium atom at the center of the ring is replaced with copper (or other metals), and the phytol tail is lost. Unlike natural chlorophyll, chlorophyllins (regardless of the metal used) are water-soluble. Although the content of different SCC mixtures may vary, two compounds commonly found in commercial SCC are trisodium copper chlorin e6 and disodium copper chlorin e4 (Figure 2).


Metabolism and Bioavailability
Little is known about the bioavailability and metabolism of chlorophyll in humans, although it is known that chlorophyll undergoes extensive metabolism once consumed. Animal model studies show only about 1%-3% of chlorophyll is absorbed, while the rest is excreted in the feces, primarily as pheopytin and pyropheophytin metabolites, indicating that significant transformation and microbial metabolism occur in the gastrointestinal tract (reviewed in 2). A recent study in eight healthy adults found pheophytin and pheophorbide derivatives in the blood of most subjects following consumption of 1.2 kg boiled spinach, a concentrated source of chlorophyll (6).
Sodium copper chlorophyllin was originally thought to be poorly absorbed because of its lack of apparent toxicity. However, a placebo-controlled clinical trial found that significant amounts of copper chlorin e4 in the serum of people taking chlorophyllin tablets (300 mg/day) (7), indicating that it is indeed absorbed. In vitro studies have found chlorin e4 to have a higher stability than chlorin e6 (2).
More research, however, is needed to understand the bioavailability and metabolism of natural chlorophylls and chlorin compounds in synthetic chlorophyllin.
Biological Activities
Complex formation with other molecules
Chlorophyll and sodium copper chlorophyllin are able to form tight molecular complexes with certain chemicals known or suspected to cause cancer, including polyaromatic hydrocarbons found in tobacco smoke (8), some heterocyclic amines found in cooked meat (9), and aflatoxin-B1 (10). The binding of chlorophyll or SCC to these potential carcinogens may interfere with gastrointestinal absorption of potential carcinogens, reducing the amount that reaches susceptible tissues (11). This has been demonstrated in humans: a cross-over study in three volunteers that used accelerator mass spectrometry to study the pharmacokinetics of an ultra-low dose of aflatoxin-B1 found a 150-mg dose of either SCC or chlorophyll could decrease absorption of aflatoxin-B1 (12).
Antioxidant effects
SCC can neutralize several physically relevant oxidants in vitro (13-16), and limited data from animal studies suggest that SCC supplementation may decrease oxidative damage induced by chemical carcinogens and radiation (17, 18). While chlorophyll and its derivatives have demonstrated antioxidant activity in in vitro assays (15, 19), the relevance of these findings to humans is not clear.
Modification of the metabolism and detoxification of carcinogens
To initiate the development of cancer, some chemicals (procarcinogens) must first be metabolized to active carcinogens that are capable of damaging DNA or other critical molecules in susceptible tissues. Since enzymes in the cytochrome P450 family are required for the activation of some procarcinogens, inhibition of cytochrome P450 enzymes may decrease the risk of some types of chemically induced cancers. In vitro studies indicate that SCC may decrease the activity of cytochrome P450 enzymes (8, 20, 21). Phase II biotransformation enzymes promote the elimination of potentially harmful toxins and carcinogens from the body. Limited data from animal studies indicate that SCC may increase the activity of the phase II enzyme quinone reductase (22).
Therapeutic effects
One in vitro study showed that human colon cancer cells undergo cell cycle arrest after treatment with SCC (23). The mechanism involved inhibition of ribonucleotide reductase activity. Ribonucleotide reductase plays a pivotal role in DNA synthesis and repair and is a target of currently used cancer therapeutic agents, such as hydroxyurea (23). While this may provide a potential avenue for SCC in the clinical setting, sensitizing cancer cells to DNA damaging agents, in vivo studies are needed.
Metal absorption
The porphyrin structure of chlorophyll is analogous to the heme structure found in blood and muscle tissue. Because heme-bound iron has higher bioavailability than nonheme iron (the most common form of iron in plant-based sources, e.g., legumes, spinach), iron uptake from iron chlorophyll is of interest. Toxicity studies in rats suggest iron chlorophyllin is generally safe for mammalian consumption (24). In vitro studies demonstrate that iron chlorophyllin is as good as heme in delivering iron to intestinal cells, and significantly better than the most common supplemental form of iron (i.e., ferrous sulfate) when incorporated into most food matrices (25). However, work in this area is nascent and has not yet been validated in humans. It is also not known if metallo-chlorophyll derivatives of copper or zinc increase absorption of these essential divalent metals.
Disease Prevention
Aflatoxin-associated liver cancer
Aflatoxin-B1 (AFB1), a liver carcinogen produced by certain species of fungus, is found in moldy grains and legumes, including corn, peanuts, and soybeans (4, 11). In hot, humid regions of Africa and Asia with improper grain storage facilities, high levels of dietary AFB1 are associated with increased risk of hepatocellular carcinoma. Moreover, the combination of hepatitis B infection and high dietary AFB1 exposure increases the risk of hepatocellular carcinoma still further. In the liver, AFB1 is metabolized to a carcinogen capable of binding DNA and causing mutations. In animal models of AFB1-induced liver cancer, administration of SCC at the same time as dietary AFB1 exposure significantly reduces AFB1-induced DNA damage in the livers of rainbow trout and rats (26-28) and dose-dependently inhibits the development of liver cancer in trout (29). Likewise, natural chlorophyll has also been found to inhibit AFB1-induced liver cancer in the rat (28). Collectively, this evidence supports a role for SCC and/or chlorophyll itself in limiting cancer initiation. In contrast, data suggest a limited role for SCC in influencing cancer progression. For example, one rat study found that SCC did not protect against aflatoxin-induced liver damage when given after tumor initiation (30).
Because of the long time period between AFB1 exposure and the development of cancer in humans, an intervention trial might require as long as 20 years to determine whether SCC supplementation can reduce the incidence of hepatocellular carcinoma in people exposed to high levels of dietary AFB1. However, a biomarker of AFB1-induced DNA damage (AFB1-N7-guanine) can be measured in the urine, and high urinary levels of AFB1-N7-guanine have been associated with significantly increased risk of developing hepatocellular carcinoma (31). In order to determine whether chlorophyllin could decrease AFB1-induced DNA damage in humans, a randomized, placebo-controlled intervention trial was conducted in 180 adults residing in a region in China where the risk of hepatocellular carcinoma is very high due to unavoidable, dietary AFB1 exposure and a high prevalence of chronic hepatitis B infection (32). Participants took either 100 mg of SCC or a placebo before meals three times daily. After 16 weeks of treatment, urinary levels of AFB1-N7-guanine were 55% lower in those taking SCC than in those taking the placebo, suggesting that SCC supplementation before meals can substantially decrease AFB1-induced DNA damage. Although a reduction in hepatocellular carcinoma has not yet been demonstrated in humans taking SCC, scientists are hopeful that supplementation will provide some protection to high-risk populations with unavoidable, dietary AFB1 exposure (11).
It is not known whether SCC will be useful in the prevention of cancers in people who are not exposed to significant levels of dietary AFB1, as is the case for most people living in the US. Many questions remain to be answered regarding the exact mechanisms of cancer prevention by SCC, the implications for the prevention of other types of cancer, and the potential for natural chlorophylls in the diet to provide cancer protection.
Therapeutic Uses of Chlorophyllin
Internal deodorant
Observations in the 1940s and 1950s that topical SCC had deodorizing effects on foul-smelling wounds led clinicians to administer SCC orally to patients with colostomies and ileostomies in order to control fecal odor (33). While early case reports indicated that SCC doses of 100 to 200 mg/day were effective in reducing fecal odor in ostomy patients (34, 35), a placebo-controlled trial found that 75 mg of oral SCC three times daily was no more effective than placebo in decreasing fecal odor assessed by colostomy patients 36. Several case reports have been published indicating that oral SCC (100-300 mg/day) decreased subjective assessments of urinary and fecal odor in incontinent patients (33, 37).
Trimethylaminuria is a hereditary disorder characterized by the excretion of trimethylamine, a compound with a “fishy” or foul odor. One study in a small number of Japanese patients with trimethylaminuria found that oral SCC (60 mg three times daily) for three weeks significantly decreased urinary trimethylamine concentrations (38).
Wound healing
Research in the 1940s indicated that chlorophyllin slowed the growth of certain anaerobic bacteria in the test tube and accelerated the healing of experimental wounds in animals. These findings led to the use of topical SCC solutions and ointments in the treatment of persistent open wounds in humans (39). During the late 1940s and 1950s, a series of largely uncontrolled studies in patients with slow-healing wounds, such as vascular ulcers and pressure (decubitus) ulcers, reported that the application of topical SCC promoted healing more effectively than other commonly used treatments (40, 41). In the late 1950s, SCC was added to papain and urea-containing ointments used for the chemical debridement of wounds in order to reduce local inflammation, promote healing, and control odor (33). SCC-containing papain/urea ointments are still available in the US by prescription (42). Several studies have reported that such ointments are effective in wound healing (43). A spray formulation of the papain/urea/SCC therapy is also available (44).
Skin conditions
A few small studies have investigated SCC as a topical treatment for various skin conditions. In a pilot study of 10 adults (ages 18-30 years) who had mild-to-moderate acne vulgaris and enlarged facial pores, twice daily application of a 0.1% liposomal SCC gel for three weeks improved a number of clinical parameters of the Global Acne Assessment Scale (i.e., facial oiliness, facial blotchiness, presence and size of facial pores, and number of acne lesions) compared to baseline (45). Additionally, a pilot study in 10 women (ages 40 years or older) with noticeable photodamage and solar lentigines found that twice daily topical application of a gel containing 0.66% SCC complex salts for eight weeks improved various clinical measures, including tactile and visual roughness of facial skin, skin radiance, fine lines, pore size, and overall photodamage (46). A few case reports have also observed some improvement in facial redness and rosacea with application of topical SCC (47).
While the reports from these studies are interesting, placebo-controlled clinical trials are needed to determine whether SCC may have utility in treating various skin conditions.
Sources
Chlorophylls
Chlorophylls are the most abundant pigments in plants, with chlorophyll a being two to four times as prevalent as chlorophyll b (6, 48). Dark-green leafy vegetables like spinach are rich sources of natural chlorophylls. The chlorophyll content of selected vegetables are presented in Table 1 (49).
Food and supplements
Chlorophyll
Green algae like chlorella are often marketed as supplemental sources of chlorophyll. Because natural chlorophyll is not as stable as SCC and is much more expensive, most over-the-counter chlorophyll supplements actually contain sodium copper chlorophyllin.
Sodium copper chlorophyllin
Oral preparations of sodium copper chlorophyllin (also called chlorophyllin copper complex) are available as a dietary supplement and as an over-the-counter drug (Derifil) used to reduce odor from colostomies and ileostomies, or to reduce fecal odor due to incontinence (50). Oral doses of 100 to 300 mg/day in three divided doses have been used to control fecal and urinary odor (see Therapeutic Uses of Chlorophyllin).
In the US, SCC is found in minor quantities in some types of green table olives (51). It is also used as a green color additive in foods like chewing gum (52), as well as in drugs and cosmetics (53).
Zinc chlorophyll derivatives
In US supermarkets, canned green beans thermally processed in a zinc chloride solution to produce zinc chlorophyll derivatives within the green beans themselves are sold under the trademarked name "veri-green" (54). Because zinc chlorophyll derivatives are more robust to heat and acid treatment, they better retain a bright green color as compared to native magnesium-bound chlorophyll (48).
Safety
Natural chlorophylls are not known to be toxic, and no toxic effects have been attributed to chlorophyllin despite more than 50 years of clinical use in humans (11, 33, 39). When taken orally, supplemental chlorophyll or sodium copper chlorophyllin may cause green discoloration of urine or feces, or yellow or black discoloration of the tongue (55). There have also been occasional reports of diarrhea related to oral SCC use. When applied topically to wounds, SCC has been reported to cause mild burning or itching in some cases (56). Oral chlorophyllin may result in false positive results on guaiac card tests for occult blood (57). Since the safety of chlorophyll or chlorophyllin supplements has not been tested in pregnant or lactating women, they should be avoided during pregnancy and lactation.
Authors and Reviewers
Originally written in 2004 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in December 2005 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in June 2009 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in September 2021 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in March 2022 by:
Rachel E. Kopec, Ph.D.
Assistant Professor of Human Nutrition
The Ohio State University
Copyright 2004-2022 Linus Pauling Institute
References
1. Matthews CK, van Holde KE. Biochemistry. 2nd ed. Menlo Park: The Benjamin/Cummings Publishing Company; 1996.
2. Zhong S, Bird A, Kopec RE. The metabolism and potential bioactivity of chlorophyll and metallo-chlorophyll derivatives in the gastrointestinal tract. Mol Nutr Food Res. 2021;65(7):e2000761. (PubMed)
3. Chen K, Rios JJ, Perez-Galvez A, Roca M. Comprehensive chlorophyll composition in the main edible seaweeds. Food Chem. 2017;228:625-633. (PubMed)
4. Sudakin DL. Dietary aflatoxin exposure and chemoprevention of cancer: a clinical review. J Toxicol Clin Toxicol. 2003;41(2):195-204. (PubMed)
5. Dashwood RH. The importance of using pure chemicals in (anti) mutagenicity studies: chlorophyllin as a case in point. Mutat Res. 1997;381(2):283-286. (PubMed)
6. Chao PY, Huang MY, Huang WD, Lin KH, Chen SY, Yang CM. Study of chlorophyll-related compounds from dietary spinach in human blood. Not Bot Horti Agrobo. 2018;46(2):309-316.
7. Egner PA, Stansbury KH, Snyder EP, Rogers ME, Hintz PA, Kensler TW. Identification and characterization of chlorin e(4) ethyl ester in sera of individuals participating in the chlorophyllin chemoprevention trial. Chem Res Toxicol. 2000;13(9):900-906. (PubMed)
8. Tachino N, Guo D, Dashwood WM, Yamane S, Larsen R, Dashwood R. Mechanisms of the in vitro antimutagenic action of chlorophyllin against benzo[a]pyrene: studies of enzyme inhibition, molecular complex formation and degradation of the ultimate carcinogen. Mutat Res. 1994;308(2):191-203. (PubMed)
9. Dashwood R, Yamane S, Larsen R. Study of the forces of stabilizing complexes between chlorophylls and heterocyclic amine mutagens. Environ Mol Mutagen. 1996;27(3):211-218. (PubMed)
10. Breinholt V, Schimerlik M, Dashwood R, Bailey G. Mechanisms of chlorophyllin anticarcinogenesis against aflatoxin B1: complex formation with the carcinogen. Chem Res Toxicol. 1995;8(4):506-514. (PubMed)
11. Egner PA, Munoz A, Kensler TW. Chemoprevention with chlorophyllin in individuals exposed to dietary aflatoxin. Mutat Res. 2003;523-524:209-216. (PubMed)
12. Jubert C, Mata J, Bench G, et al. Effects of chlorophyll and chlorophyllin on low-dose aflatoxin B(1) pharmacokinetics in human volunteers. Cancer Prev Res (Phila). 2009;2(12):1015-1022. (PubMed)
13. Kumar SS, Devasagayam TP, Bhushan B, Verma NC. Scavenging of reactive oxygen species by chlorophyllin: an ESR study. Free Radic Res. 2001;35(5):563-574. (PubMed)
14. Kamat JP, Boloor KK, Devasagayam TP. Chlorophyllin as an effective antioxidant against membrane damage in vitro and ex vivo. Biochim Biophys Acta. 2000;1487(2-3):113-127. (PubMed)
15. Vankova K, Markova I, Jasprova J, et al. Chlorophyll-mediated changes in the redox status of pancreatic cancer cells are associated with its anticancer effects. Oxid Med Cell Longev. 2018;2018:4069167. (PubMed)
16. Domijan AM, Gajski G, Novak Jovanovic I, Geric M, Garaj-Vrhovac V. In vitro genotoxicity of mycotoxins ochratoxin A and fumonisin B(1) could be prevented by sodium copper chlorophyllin--implication to their genotoxic mechanism. Food Chem. 2015;170:455-462. (PubMed)
17. Park KK, Park JH, Jung YJ, Chung WY. Inhibitory effects of chlorophyllin, hemin and tetrakis(4-benzoic acid)porphyrin on oxidative DNA damage and mouse skin inflammation induced by 12-O-tetradecanoylphorbol-13-acetate as a possible anti-tumor promoting mechanism. Mutat Res. 2003;542(1-2):89-97. (PubMed)
18. Kumar SS, Shankar B, Sainis KB. Effect of chlorophyllin against oxidative stress in splenic lymphocytes in vitro and in vivo. Biochim Biophys Acta. 2004;1672(2):100-111. (PubMed)
19. Perez-Galvez A, Viera I, Roca M. Carotenoids and chlorophylls as antioxidants. Antioxidants (Basel). 2020;9(6):505. (PubMed)
20. Yun CH, Jeong HG, Jhoun JW, Guengerich FP. Non-specific inhibition of cytochrome P450 activities by chlorophyllin in human and rat liver microsomes. Carcinogenesis. 1995;16(6):1437-1440. (PubMed)
21. John K, Divi RL, Keshava C, et al. CYP1A1 and CYP1B1 gene expression and DNA adduct formation in normal human mammary epithelial cells exposed to benzo[a]pyrene in the absence or presence of chlorophyllin. Cancer Lett. 2010;292(2):254-260. (PubMed)
22. Dingley KH, Ubick EA, Chiarappa-Zucca ML, et al. Effect of dietary constituents with chemopreventive potential on adduct formation of a low dose of the heterocyclic amines PhIP and IQ and phase II hepatic enzymes. Nutr Cancer. 2003;46(2):212-221. (PubMed)
23. Chimploy K, Diaz GD, Li Q, et al. E2F4 and ribonucleotide reductase mediate S-phase arrest in colon cancer cells treated with chlorophyllin. Int J Cancer. 2009;125(9):2086-2094. (PubMed)
24. Toyoda T, Cho YM, Mizuta Y, Akagi J, Nishikawa A, Ogawa K. A 13-week subchronic toxicity study of sodium iron chlorophyllin in F344 rats. J Toxicol Sci. 2014;39(1):109-119. (PubMed)
25. Miret S, Tascioglu S, van der Burg M, Frenken L, Klaffke W. In vitro bioavailability of iron from the heme analogue sodium iron chlorophyllin. J Agric Food Chem. 2010;58(2):1327-1332. (PubMed)
26. Dashwood RH, Breinholt V, Bailey GS. Chemopreventive properties of chlorophyllin: inhibition of aflatoxin B1 (AFB1)-DNA binding in vivo and anti-mutagenic activity against AFB1 and two heterocyclic amines in the Salmonella mutagenicity assay. Carcinogenesis. 1991;12(5):939-942. (PubMed)
27. Kensler TW, Groopman JD, Roebuck BD. Use of aflatoxin adducts as intermediate endpoints to assess the efficacy of chemopreventive interventions in animals and man. Mutat Res. 1998;402(1-2):165-172. (PubMed)
28. Simonich MT, Egner PA, Roebuck BD, et al. Natural chlorophyll inhibits aflatoxin B1-induced multi-organ carcinogenesis in the rat. Carcinogenesis. 2007;28(6):1294-1302. (PubMed)
29. Breinholt V, Hendricks J, Pereira C, Arbogast D, Bailey G. Dietary chlorophyllin is a potent inhibitor of aflatoxin B1 hepatocarcinogenesis in rainbow trout. Cancer Res. 1995;55(1):57-62. (PubMed)
30. Orner GA, Roebuck BD, Dashwood RH, Bailey GS. Post-initiation chlorophyllin exposure does not modulate aflatoxin-induced foci in the liver and colon of rats. J Carcinog. 2006;5:6. (PubMed)
31. Qian GS, Ross RK, Yu MC, et al. A follow-up study of urinary markers of aflatoxin exposure and liver cancer risk in Shanghai, People's Republic of China. Cancer Epidemiol Biomarkers Prev. 1994;3(1):3-10. (PubMed)
32. Egner PA, Wang JB, Zhu YR, et al. Chlorophyllin intervention reduces aflatoxin-DNA adducts in individuals at high risk for liver cancer. Proc Natl Acad Sci U S A. 2001;98(25):14601-14606. (PubMed)
33. Chernomorsky SA, Segelman AB. Biological activities of chlorophyll derivatives. N J Med. 1988;85(8):669-673. (PubMed)
34. Siegel LH. The control of ileostomy and colostomy odors. Gastroenterology. 1960;38:634-636. (PubMed)
35. Weingarten M, Payson B. Deodorization of colostomies with chlorophyll. Rev Gastroenterol. 1951;18(8):602-604. (PubMed)
36. Christiansen SB, Byel SR, Stromsted H, Stenderup JK, Eickhoff JH. [Can chlorophyll reduce fecal odor in colostomy patients?]. Ugeskr Laeger. 1989;151(27):1753-1754. (PubMed)
37. Young RW, Beregi JS, Jr. Use of chlorophyllin in the care of geriatric patients. J Am Geriatr Soc. 1980;28(1):46-47. (PubMed)
38. Yamazaki H, Fujieda M, Togashi M, et al. Effects of the dietary supplements, activated charcoal and copper chlorophyllin, on urinary excretion of trimethylamine in Japanese trimethylaminuria patients. Life Sci. 2004;74(22):2739-2747. (PubMed)
39. Kephart JC. Chlorophyll derivatives - their chemistry, commercial preparation and uses. Econ Bot. 1955;9:3-38.
40. Bowers WF. Chlorophyll in wound healing and suppurative disease. Am J Surg. 1947;73:37-50. (PubMed)
41. Carpenter EB. Clinical experiences with chlorophyll preparations. Am J Surg. 1949;77(2):167-171. (PubMed)
42. Physicians' Desk Reference. 58th ed. Stamford: Thomson Health Care, Inc.; 2003.
43. Smith RG. Enzymatic debriding agents: an evaluation of the medical literature. Ostomy Wound Manage. 2008;54(8):16-34. (PubMed)
44. Weir D, Farley KL. Relative delivery efficiency and convenience of spray and ointment formulations of papain/urea/chlorophyllin enzymatic wound therapies. J Wound Ostomy Continence Nurs. 2006;33(5):482-490. (PubMed)
45. Stephens TJ, McCook JP, Herndon JH, Jr. Pilot study of topical copper chlorophyllin complex in subjects with facial acne and large pores. J Drugs Dermatol. 2015;14(6):589-592. (PubMed)
46. Sigler ML, Stephens TJ. Assessment of the safety and efficacy of topical copper chlorophyllin in women with photodamaged facial skin. J Drugs Dermatol. 2015;14(4):401-404. (PubMed)
47. Vasily DB. Topical treatment with liposomal sodium copper chlorophyllin complex in subjects with facial redness and erythematotelangiectatic rosacea: case studies. J Drugs Dermatol. 2015;14(10):1157-1159. (PubMed)
48. Hayes M, Ferruzzi MG. Update on the bioavailability and chemopreventative mechanisms of dietary chlorophyll derivatives. Nutr Res. 2020;81:19-37. (PubMed)
49. Bohn T, Walczyk S, Leisibach S, Hurrell RF. Chlorophyll-bound magnesium in commonly consumed vegetables and fruits: relevance to magnesium nutrition. J Food Sci. 2004;69(9):S347-S350.
50. Food and Drug Administration. Code of Federal Regulations: Miscellaneous Internal Drug Products for Over the Counter Use [Web page]. April 1, 2002. Available at: http://www.fda.gov/cder/otcmonographs/Internal_Deodorant/internal_deodorant(357I).html. Accessed 6/9/04.
51. Aparicio-Ruiz R, Riedl KM, Schwartz SJ. Identification and quantification of metallo-chlorophyll complexes in bright green table olives by high-performance liquid chromatography-mass spectrometry quadrupole/time-of-flight. J Agric Food Chem. 2011;59(20):11100-11108. (PubMed)
52. Viera I, Perez-Galvez A, Roca M. Green natural colorants. Molecules. 2019;24(1):154. (PubMed)
53. US Food and Drug Administration. CFR - Code of Federal Regulations Title 21. Available at: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=73.3110. Accessed 9/29/21.
54. Von Elbe JH, Huang AS, Attoe EL, Nank WK. Pigment composition and color of conventional and Veri-Green canned beans. J Agric Food Chem. 1986;34(1):52-54.
55. Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. 2nd ed. Montvale: Physicians' Desk Reference, Inc.; 2008.
56. Smith LW. The present status of topical chlorophyll therapy. N Y State J Med. 1955;55(14):2041-2050. (PubMed)
57. Gogel HK, Tandberg D, Strickland RG. Substances that interfere with guaiac card tests: implications for gastric aspirate testing. Am J Emerg Med. 1989;7(5):474-480. (PubMed)
Curcumin
Contents
Summary
- Curcumin is a biologically active polyphenolic compound found in turmeric, a spice derived from the rhizomes of the plant Curcuma longa Linn. Commonly consumed in Asian countries, turmeric has been used for medicinal purposes for centuries. (More information)
- Mounting evidence from preclinical studies shows that curcumin modulates numerous molecular targets and exerts antioxidant, anti-inflammatory, anticancer, and neuroprotective activities. (More information)
- In humans, curcumin taken orally is poorly absorbed and rapidly metabolized and eliminated. Therefore, the potential of curcumin as a therapeutic agent is limited by its poor bioavailability. (More information)
- Current evidence suggesting that curcumin may help prevent and/or treat colorectal cancer and type 2 diabetes mellitus is very limited. Yet, several clinical trials designed to assess the safety and efficacy of curcumin alone or with first-line treatment in patients with breast, prostate, pancreatic, lung, or colorectal cancer are under way. (More information)
- While a few preliminary trials suggested that curcumin may have anti-inflammatory activities in humans, larger randomized controlled trials are still needed to establish the efficacy of curcumin as an anti-inflammatory agent against rheumatoid arthritis, ulcerative colitis, and radiotherapy-induced dermatitis. (More information)
- There is currently no substantial evidence showing that curcumin may improve cognitive performance in older adults with or without cognitive impairments. Yet, some preclinical studies have found curcumin prevented or reversed certain pathological features of Alzheimer’s disease (AD). A number of clinical trials designed to assess whether curcumin might help prevent or treat AD are under way. (More information)
- Long-term clinical trials are required to confirm whether curcumin could exhibit long-lasting antidepressant effects in patients suffering from major depressive disorder. (More information)
- Oral supplementation with curcumin is generally regarded as safe, especially because of its low bioavailability. However, use of curcumin supplements may affect the efficacy or increase the toxicity of a wide range of drugs when taken concurrently. (More information)
Introduction
Turmeric is a spice derived from the rhizomes of the tropical plant Curcuma longa Linn, which is a member of the ginger family (Zingiberaceae). Rhizomes are horizontal underground stems that send out shoots, as well as roots. The bright yellow-orange color of turmeric comes mainly from fat-soluble, polyphenolic pigments known as curcuminoids. Curcumin, the principal curcuminoid found in turmeric, is generally considered its most active constituent (1). Other curcuminoids found in turmeric include demethoxycurcumin and bisdemethoxycurcumin (Figure 1). In addition to its use as a spice and pigment, turmeric has been used in India for medicinal purposes for centuries (2). More recently, evidence that curcumin may have anti-inflammatory and anticancer activities has renewed scientific interest in its potential to prevent and treat disease.
Metabolism and Bioavailability
Clinical trials in humans indicate that the systemic bioavailability of orally administered curcumin is relatively low (3-5) and that mostly metabolites of curcumin, instead of curcumin itself, are detected in plasma or serum following oral consumption (6, 7). In the intestine and liver, curcumin is readily conjugated to form curcumin glucuronide and curcumin sulfate or, alternately, reduced to tetrahydrocurcumin, hexahydrocurcumin, and octahydrocurcumin (Figure 2) (4). An early clinical trial conducted in Taiwan indicated that serum curcumin concentrations peaked at 0.41 to 1.75 micromoles/liter (μM) one hour after oral doses of 4 to 8 g of curcumin (8). Another clinical trial conducted in the UK found that plasma concentrations of curcumin, curcumin sulfate, and curcumin glucuronide were in the range of 0.01 μM one hour after a 3.6 g oral dose of curcumin (9). Curcumin and its metabolites could not be detected in plasma at doses lower than 3.6 g/day. There is some evidence that orally administered curcumin accumulates in gastrointestinal tissues. For instance, when colorectal cancer patients took 3.6 g/day of curcumin orally for seven days prior to surgery, curcumin was detected in both malignant and normal colorectal tissue (10). In contrast, curcumin was not detected in the liver tissue of patients with liver metastases of colorectal cancer after the same oral dose of curcumin (11), suggesting that oral curcumin administration may not effectively deliver curcumin to tissues outside the gastrointestinal tract.
The safety and efficacy of several curcumin formulations are currently being explored in (pre)clinical settings with the aim of increasing the absorption, bioavailability, and tissue-targeted delivery of curcumin (12-16). Examples of approaches include conjugation to peptide carriers (e.g., to polylactic-co-glycolic acid [PLGA]); complexation with essential oils; coadministration with piperine; and encapsulation into nanoparticles, liposomes, phytosomes, polymeric micelles, and cyclodextrins (reviewed in 17).

Biological Activities
Antioxidant activity
Curcumin is an effective scavenger of reactive oxygen species (ROS) and reactive nitrogen species in the test tube (18, 19). However, it is not clear whether curcumin acts as a direct antioxidant in vivo. Due to its limited oral bioavailability in humans (see Metabolism and Bioavailability), plasma and tissue curcumin concentrations are likely to be much lower than those of other fat-soluble antioxidants like α-tocopherol (vitamin E). Yet, curcumin taken orally may reach sufficient concentrations in the gastrointestinal tract and protect the intestinal mucosa against oxidative DNA damage (11). In addition to a potentially direct antioxidant activity, curcumin can induce the expression of phase II antioxidant enzymes, including glutamate-cysteine ligase (GCL), the rate-limiting enzyme in glutathione synthesis. Glutathione (GSH) is an important intracellular antioxidant that plays a critical role in cellular adaptation to stress (20). Curcumin was found to upregulate the expression of GCL through the activation of different signaling pathways (21). In particular, curcumin increases the expression of GCL and other detoxifying enzymes via the activation of the nuclear factor E2-related factor 2 (Nrf2)-dependent pathway.
Nrf2-dependent antioxidant pathway
Briefly, Nrf2 is a transcription factor that is bound to the protein Kelch-like ECH-associated protein 1 (Keap1) in the cytosol. Keap1 responds to oxidative stress signals by freeing Nrf2. Upon release, Nrf2 translocates to the nucleus and binds to the antioxidant response element (ARE) located in the promoter of genes coding for antioxidant/detoxifying enzymes and scavengers. Nrf2/ARE-dependent genes code for numerous mediators of the antioxidant response, including GCL, glutathione S-transferases (GSTs), thioredoxin, NAD(P)H quinone oxidoreductase 1 (NQO-1), and heme oxygenase 1 (HO-1) (22). Nrf2-dependent upregulation of HO-1 in curcumin-treated renal tubular epithelial cells challenged with high glucose concentrations was shown to prevent phenotype changes resembling fibrosis and known to occur at an early stage of diabetic renal injury (23). Curcumin also inhibited the progression of fibrosis in liver and lung in animal models of chronic inflammatory diseases (24, 25). Curcumin mitigated the effect of chronic ethanol intake on mouse liver, partly by upregulating Nrf2 target genes coding for NQO-1, HO-1, glutathione peroxidase (GSH-Px), and superoxide dismutase (SOD) (26). Curcumin treatment also counteracted oxidative damage induced by heavy ion irradiation by upregulating Nrf2 downstream genes for GCL, HO-1, NQO-1, and SOD in the brain of rats (27). Additional studies have demonstrated the ability of curcumin to reduce oxidative stress in different experimental settings via the induction of Nrf2/ARE pathway (reviewed in 22).
Anti-inflammatory activity
Curcumin has been shown to inhibit mediators of the inflammatory response, including cytokines, chemokines, adhesion molecules, growth factors, and enzymes like cyclooxygenase (COX), lipoxygenase (LOX), and inducible nitric oxide synthase (iNOS). Nuclear factor-kappa B (NF-κB) is a transcription factor that binds DNA and induces the transcription of the COX-2 gene, other pro-inflammatory genes, and genes involved in cell proliferation, adhesion, survival, and differentiation. The anti-inflammatory effects of curcumin result from its ability to inhibit the NF-κB pathway, as well as other pro-inflammatory pathways like the mitogen-activated protein kinase (MAPK)- and the Janus kinase (JAK)/Signal transducer and activator of transcription (STAT)-dependent signaling pathways (28). Inhibition of dextran sulfate sodium (DSS)-induced colitis by curcumin in mice has been associated with a downregulation of the expression of p38-MAPK and pro-inflammatory cytokine TNF-α and a reduction of myeloperoxidase (MPO) activity, a marker of neutrophil infiltration in intestinal mucosa (29). Curcumin has also been shown to improve colitis by preventing STAT3 activation and STAT3-dependent induction of cell proliferation in mouse colon (30). Moreover, curcumin was shown to attenuate the immune response triggered by collagen injections in a mouse model of rheumatoid arthritis, partly by blocking the proliferation of T lymphocytes in mouse splenocytes (31). In addition, curcumin has been found to reduce the secretion of TNF-α and IL-1β and the production of COX-2-induced prostaglandin G2. In one study, curcumin inhibited the secretion of matrix metalloproteins (MMPs) — responsible for the degradation of the synovial joints — in human fibroblast-like synoviocytes (31) and in human articular chondrocytes (32). Curcumin has also been found to alleviate neuro-inflammation in a mouse model of traumatic brain injury, reducing macrophage and microglial activation and increasing neuronal survival (33).
Anticancer activity
Effects on biotransformation enzymes
Some compounds are not carcinogenic until they are metabolized in the body by phase I biotransformation enzymes, such as enzymes of the cytochrome P450 (CYP) family (34). Primarily based on evidence from rodent studies, it is thought that curcumin may inhibit procarcinogen bioactivation and help prevent cancer by inhibiting the activity of multiple CYP enzymes in humans (35-37). Curcumin may also increase the activity of phase II detoxification enzymes, such as GSTs and quinone reductase (QR) (see also Nrf2-dependent antioxidant pathway) (35, 38, 39). However, it is important to note that the effect of curcumin on biotransformation enzymes may vary depending on the route of administration, the dose, and the animal model. In addition, curcumin intakes ranging from 0.45 to 3.6 g/day for up to four months did not increase leukocyte GST activity in humans (9).
Inhibition of proliferation and induction of apoptosis
Following DNA damage, the cell cycle can be transiently arrested to allow for DNA repair or for activation of pathways leading to programmed cell death (apoptosis) if the damage is irreparable (40). Defective cell-cycle regulation may result in the propagation of mutations that contribute to the development of cancer. Unlike normal cells, cancer cells proliferate rapidly and are unable to respond to cell death signals that initiate apoptosis. Curcumin has been found to induce cell-cycle arrest and apoptosis by regulating a variety of cell-signaling pathways (3, 41-45). For example, the inhibition of cell proliferation by curcumin has been associated with the Nrf2-dependent downregulation of DNA repair-specific flap endonuclease 1 (Fen1) in breast cancer cells in culture (46). Curcumin has been shown to induce p53-dependent or -independent apoptosis depending on the cancer cell type (47). In a panel of cancer cell lines, p53-independent apoptosis induced by curcumin was mediated by the rapid increase of ROS and the activation of MAPK and c-jun kinase (JNK) signaling cascades (48). Inhibition of NF-κB signaling by curcumin also suppresses proliferation and induces apoptosis in cancer cells (47).
Inhibition of tumor invasion and angiogenesis
Malignant and aggressive forms of cancer can invade surrounding tissues and spread to distant tissues once cancer cells have acquired the ability to leave the primary site (reduced cell-to-cell adhesion and loss of polarity), migrate, and disseminate. Epithelial-mesenchymal transition (EMT) is the process by which epithelial cells acquire the ability to migrate and invade through downregulating proteins like E-cadherin and γ-catenin and expressing mesenchymal markers like MMPs, N-cadherin, and vimentin. In breast cancer cells, curcumin prevented EMT-associated morphological changes induced by lipopolysaccharide (LPS) while upregulating E-cadherin and downregulating vimentin. It was further shown that curcumin inhibited NF-κB/Snail signaling involved in LPS-induced EMT (49). In another study, curcumin increased the expression of the small non-coding RNA miR181b, which then downregulated proinflammatory cytokines, CXCL1 and CXCL2, as well as MMPs, thereby reducing the metastatic potential of breast cancer cells. Curcumin inhibited IL-6-induced proliferation, migration, and invasiveness of human small cell lung cancer (SCLC) cells by reducing JAK/STAT3 phosphorylation (i.e., activation) and downstream genes coding for cyclin B1, survivin, Bcl-XL, MMPs, intercellular adhesion molecule 1 (ICAM-1), and vascular endothelial growth factor (VEGF) (30).
Curcumin was found to exert its anticancer activities in many different types of cancer cells by regulating a variety of signaling pathways (reviewed in 2, 47).
Neuroprotective activity
In Alzheimer’s disease (AD), a peptide called β-amyloid (Aβ peptide) aggregates into oligomers and fibrils and forms deposits known as amyloid (or senile) plaques outside neurons in the hippocampus and cerebral cortex of patients. Another feature of AD is the accumulation of intracellular neurofibrillary tangles formed by phosphorylated Tau protein (50). Abnormal microglial activation, oxidative stress, and neuronal death are also associated with the progression of the disease. Curcumin has been found to inhibit Aβ fibril formation and extension and to destabilize preformed fibrils in vitro (51-53). Metal chelation by curcumin might interfere with metal ion (Cu2+/Zn2+)-induced Aβ aggregation. Curcumin might also affect the trafficking of Aβ peptide precursor (APP) and the generation of Aβ peptides from APP (54, 55). Abnormally activated microglia and hypertrophic astrocytes around amyloid plaques in AD brains release cytotoxic molecules, such as proinflammatory cytokines and ROS, which enhance Aβ formation and deposition and further damage neurons. Curcumin was found to reduce the inflammatory response triggered by Aβ peptide-induced microglial activation and increase neuronal cell survival (56). When injected into the carotid artery of a transgenic mouse model of AD, curcumin was found to cross the blood-brain barrier, bind to amyloid plaques, and block the formation of Aβ oligomers and fibrils (53). In other animal models of AD, dietary curcumin decreased biomarkers of inflammation and oxidative damage, increased Aβ peptide clearance by macrophages, dismantled amyloid plaques in the brain, stimulated neuronal cell growth in the hippocampus, and improved Aβ-induced memory deficits (reviewed in 57).
Note: It is important to keep in mind that some of the biological activities discussed above were observed in cultured cells and animal models exposed to curcumin at concentrations unlikely to be achieved in cells of humans consuming curcumin orally (see Metabolism and Bioavailability).
Disease Prevention
Cancer
Oral curcumin administration has been found to inhibit the development of chemically-induced cancer in animal models of oral (58, 59), stomach (60, 61), liver (62), and colon (63-65) cancer. ApcMin/+ mice have a mutation in the Apc (adenomatous polyposis coli) gene similar to that in humans with familial adenomatous polyposis, a genetic condition characterized by the development of numerous colorectal adenomas (polyps) and a high risk for colorectal cancer. Oral curcumin administration has been found to inhibit the development of intestinal adenomas in ApcMin/+ mice (66, 67). Despite promising results in animal studies, there is presently little evidence that high intakes of curcumin or turmeric are associated with decreased cancer risk in humans. A 30-day phase II clinical trial in 40 smokers with at least eight rectal aberrant crypt foci (ACF; precancerous lesions) found that the number of ACF was significantly lower with a daily supplementation with 4 g/day of curcumin compared to 2 g/day (68). Several controlled clinical trials in humans designed to evaluate the effect of oral curcumin supplementation on precancerous colorectal lesions, such as adenomas, are under way (69).
Type 2 diabetes mellitus
Oxidative stress and inflammation have been implicated in the pathogenesis of type 2 diabetes mellitus and related vascular complications. A large body of preclinical evidence suggests that the antioxidant, anti-inflammatory, and glucose-lowering activities of curcumin and its analogs may be useful in the prevention and/or treatment of type 2 diabetes (70). In a nine-month, randomized, double-blind, placebo-controlled study in 237 subjects with impaired glucose tolerance (pre-diabetes), no progression to overt diabetes was reported with a daily ingestion of a mixture of curcuminoids (0.5 g), while 16.4% of placebo-treated participants developed diabetes (71). In addition, curcumin supplementation was shown to reduce insulin resistance and improve measures of pancreatic β-cell function and glucose tolerance. In an eight-week, randomized, placebo-controlled study in 67 individuals with type 2 diabetes, oral curcumin (a mixture of all three major curcuminoids; 0.6 g/day) failed to significantly lower the level of glycated hemoglobin A1c (HbA1c; a measure of glycemic control), plasma fasting glucose, total serum cholesterol, LDL-cholesterol, and serum triglycerides (72). Yet, supplemental curcumin was found to be as effective as lipid-lowering drug atorvastatin (10 mg/day) in reducing circulating markers of oxidative stress (malondialdehyde) and inflammation (endothelin-1, TNFα, IL-6) and in improving endothelial function. Another randomized controlled trial also reported that oral curcumin supplementation (1.5 g/day) for six months improved endothelial function, insulin sensitivity, and metabolic markers associated with atherogenesis (plasma triglycerides, visceral fat, total body fat) in participants with type 2 diabetes (73). Finally, in a two-month randomized, double-blind, placebo-controlled study in 40 individuals with type 2 diabetic nephropathy (kidney disease), daily curcumin ingestion (66.3 mg) significantly reduced urinary concentrations of proteins and inflammation markers (TGF-β, IL-8), suggesting that curcumin might be helpful with slowing the progression of kidney damage and preventing kidney failure (74). Larger trials are needed to assess whether curcumin could be useful in the prevention or management of type 2 diabetes and vascular complications.
Disease Treatment
Cancer
The ability of curcumin to regulate a variety of signaling pathways involved in cell growth, apoptosis, invasion, metastasis, and angiogenesis in preclinical studies elicited scientific interest in its potential as an anticancer agent in tumor therapy (75). To date, most of the controlled clinical trials of curcumin supplementation in cancer patients have been phase I trials, which are aimed at determining feasibility, tolerability, safety, and providing early evidence of efficacy (76). A phase I clinical trial in patients with advanced colorectal cancer found that doses up to 3.6 g/day for four months were well tolerated, although the systemic bioavailability of oral curcumin was low (77). When colorectal cancer patients with liver metastases took 3.6 g/day of curcumin orally for seven days, trace levels of curcumin metabolites were measured in liver tissue, but curcumin itself was not detected (11). In contrast, curcumin was measurable in normal and malignant colorectal tissue after patients with advanced colorectal cancer took 3.6 g/day of curcumin orally for seven days (10). In a pilot trial in patients awaiting gastrointestinal endoscopy or colorectal cancer resection, the administration of a mixture of three major curcuminoids (2.35 g/day for 14 days) resulted in detectable amounts of curcumin in colonic mucosa (mean concentration, 48.4 μg/g of tissue), demethoxycurcumin (7.1 μg/g), and bisdemethoxycurcumin (0.7 μg/g) (78).
While these findings suggested that oral curcumin may likely be more effective as a therapeutic agent in cancers of the gastrointestinal tract than of other tissues, some phase I/II trials have also examined whether supplemental curcumin may confer additional benefits to conventional drugs against different types of cancer. Combining curcumin with anticancer drugs like gemcitabine in pancreatic cancer (79, 80), docetaxel in breast cancer (81), and imatinib in chronic myeloid leukemia (82) may be safe and well tolerated. A recent single-arm, phase II trial combining three cycles of docetaxel/prednisone and curcumin (6 g/day) was carried out in 26 patients with castration-resistant prostate cancer (83). The level of prostate-specific antigen (PSA) was decreased in most patients and was normalized in 36% of them, and the co-administration of curcumin with drugs showed no toxicity beyond adverse effects already related to docetaxel monotherapy. Many registered phase I/II clinical trials designed to investigate the effectiveness of curcumin alone or with first-line treatment in patients with breast, prostate, pancreatic, lung, or colorectal cancer are under way (69).
Inflammatory diseases
Although curcumin has been demonstrated to have anti-inflammatory and antioxidant activities in cell culture and animal studies, few randomized controlled trials have examined the efficacy of curcumin in the treatment of inflammatory conditions. A placebo-controlled trial in 40 men who had surgery to repair an inguinal hernia or hydrocele found that oral curcumin supplementation (1.2 g/day) for five days was more effective than placebo in reducing post-surgical edema, tenderness and pain, and was comparable to phenylbutazone therapy (300 mg/day) (84).
Rheumatoid arthritis
A preliminary intervention trial that compared curcumin with a nonsteroidal anti-inflammatory drug (NSAID) in 18 patients with rheumatoid arthritis (RA) found that improvements in morning stiffness, walking time, and joint swelling after two weeks of curcumin supplementation (1.2 g/day) were comparable to those experienced after two weeks of phenylbutazone (NSAID) therapy (300 mg/day) (85). In a more recent randomized, open-label study in 45 RA patients, supplementation with a mixture of all three major curcuminoids (0.5 g/day for eight weeks) was found to be as effective as diclofenac (NSAID; 50 mg/day) in reducing measures of disease activity, tenderness, and swelling joints (86). Larger randomized controlled trials are needed to determine whether oral curcumin supplementation is effective in the treatment of RA.
Radiation dermatitis
Radiation-induced skin inflammation occurs in most patients receiving radiation therapy for sarcoma, lung, breast, or head and neck cancer. One randomized, double-blind, placebo-controlled trial in 30 women prescribed radiation therapy for breast carcinoma in situ reported a reduction of radiation-induced dermatitis severity and moist desquamation with a supplemental curcuminoid mixture (6 g/day for four to seven weeks). Curcumin failed to reduce skin redness and radiation-induced pain at the site of treatment (87).
Ulcerative colitis
Ulcerative colitis (UC) is a long-term condition characterized by diffuse and superficial inflammation of the colonic mucosa. Disease activity may fluctuate between periods of remission and periods of relapse. Preliminary evidence suggests that curcumin might be useful as an add-on therapy to control disease activity. One multicenter, randomized, double-blind, placebo-controlled study has examined the efficacy of curcumin enema (2 g/day) in the prevention of relapse in 82 patients with quiescent UC (88). Six-month treatment with curcumin significantly reduced measures of disease activity and severity and resulted in a lower relapse rate than with placebo in subjects on standard-of-care medication (sulfasalazine or mesalamine); yet, there was no difference in the proportion of patients who experienced relapse six months after curcumin was discontinued (88). In another randomized controlled trial in active UC patients treated with mesalamine, the percentage of patients in clinical remission was significantly higher after a one-month treatment with oral curcumin (3 g/day) than with placebo (89). Larger trials are needed to ensure that curcumin can be safely used with conventional UC treatments and to further support its potential therapeutic benefits for relapsing-remitting UC.
Oral health
Emerging evidence suggests that curcumin has anti-inflammatory and antimicrobial properties that could be beneficial in the treatment of certain diseases of the oral cavity. For example, the topical application of a curcumin gel was found to reduce gingival bleeding and periodontal bacteria after conventional periodontal therapy (scaling and root planing) (90-92). A mouthwash containing curcumin was also found to be as effective as chlorhexidine in reducing inflammation in individuals who underwent periodontal therapy for gingivitis (93).
Oral submucous fibrosis
Any part of the oral cavity may be affected by oral submucous fibrosis (OSMF), a currently incurable condition especially prevalent in Southeast Asia and India. OSMF is characterized by the formation of excess fibrous tissue (fibrosis) that leads to stiffness of the mucosa and restricted mouth opening. A few recent intervention studies showed that curcumin could improve some symptoms, such as burning sensations and reduced mouth opening (reviewed in 94). In an open-label intervention study in 40 OSMF patients randomized to receive either the conventional treatment (weekly intra-lesional injections of steroids) or daily oral administration of a Curcuma longa Linn extract (600 mg/day) for three months, the burning sensation significantly improved in the curcumin-treated group, while tongue protrusion was reduced with conventional therapy. No differences between the two treatment groups were seen with respect to mouth opening (95). A six-month follow-up of the effect of oral curcumin (2 g/day) in OSMF patients treated for three months found that curcumin outperformed steroid ointment in its ability to increase maximum mouth opening and to reduce self-reported burning sensation (96). Further studies should assess the appropriate dose of curcumin to achieve the greatest benefits and determine whether curcumin can enhance the effect of standard-of-care treatment in limiting OSMF disease progression.
Cognitive decline and Alzheimer’s disease
Alzheimer’s disease (AD) is a form of dementia characterized by extracellular deposition of β-amyloid plaques, intracellular formation of neurofibrillary tangles, and neuronal loss, eventually leading to brain atrophy and cognitive impairment in affected individuals (57). When injected into the carotid artery, curcumin was found to cross the blood-brain barrier in an animal model of AD (53), though it is not known whether curcumin taken orally can reach the blood-brain barrier at sufficient concentrations and impede cognitive decline in humans. As a result of promising findings in animal models (see Neuroprotective activity), a few recent clinical trials have examined the effect of oral curcumin supplementation on cognition in healthy older adults and AD patients (57). A randomized, double-blind, placebo-controlled trial in 60 healthy older adults (mean age, 68.5 years) investigated whether acute (80 mg) or chronic (80 mg/day for 4 weeks) oral intake of curcumin could improve their ability to cope with the mental stress and change in mood usually associated with undergoing a battery of cognitive tests (97). A significant reduction in mental fatigue and higher levels of calmness and contentedness following cognitive test sessions were observed in individuals who consumed curcumin (either acutely or chronically) compared to the placebo group. Additionally, the results of cognitive ability tests suggested that curcumin treatment had limited benefits on cognitive function, as shown by better scores in measures of sustained attention and working memory compared to placebo (97).
The results of a six-month trial in 27 patients with AD found that oral supplementation with up to 4 g/day of curcumin — containing all three major curcuminoids — was safe (6). Yet, measures of cognitive performance (using the Mini Mental State Examination [MMSE] scoring scale) and levels of F2-isoprostanes (oxidative stress markers) and antioxidants in blood were not found to be significantly different between curcumin- and placebo-treated subjects at the end of the intervention period. In another six-month, randomized, double-blind, placebo-controlled study of subjects with mild-to-moderate AD, curcumin failed to improve cognitive test scores and to reduce blood and cerebrospinal fluid (CSF) concentrations of β-amyloid peptide, CSF concentrations of total and phosphorylated Tau protein, and CSF concentrations of F2-isoprostanes (98).
Despite the lack of encouraging results from completed trials, several randomized controlled studies are under way to determine whether supplemental curcumin has the ability to reverse or prevent cognitive deficits in both healthy and cognitively impaired individuals (57).
Major depressive disorder
Major depressive disorder (MDD) is a neuropsychiatric disorder associated with abnormal neurotransmission; it is primarily treated with drugs that improve the bioavailability of neurotransmitters like serotonin, noradrenaline, and dopamine in the brain (99). Characteristics of MDD also include alterations in the hypothalamus-pituitary-adrenal axis, increased neuroinflammation, defective neurogenesis, and neuronal death.
A few clinical studies have examined the effect of curcumin alone or with conventional antidepressant drugs in MDD patients. A recent meta-analysis of six randomized controlled trials found that supplementation with curcumin significantly reduced depression symptoms (100). However, in one of the studies included in this meta-analysis — a double-blind, controlled study in 56 adults diagnosed with MDD — curcumin treatment (~880 mg/day of curcuminoids) for eight weeks was no more effective than placebo in reducing self-reported depression- and anxiety-related symptoms (101). Significant improvements in the severity and frequency of specific depression-related symptoms only occurred after four weeks of treatment, suggesting that a longer treatment period might be needed to uncover the antidepressant effects of curcumin (100, 101). In another randomized, placebo-controlled trial, supplemental curcumin (330 mg/day) for five weeks failed to relieve depressive symptoms in patients treated with conventional antidepressants (102). In contrast, in a six-week, randomized, single-blinded, placebo-controlled study in 60 MDD patients, supplemental curcumin (~880 mg/day of curcuminoids) alone yielded a similar response rate to the antidepressant, fluoxetine (a serotonin reuptake inhibitor [Prozac]; 20 mg/day) in terms of depressive symptoms; no additional effect was observed when both curcumin and fluoxetine treatments were combined (103). Moreover, in a randomized controlled study in 100 participants taking escitalopram (a serotonin reuptake inhibitor [Lexapro]; 5 to 15 mg/week), supplemental curcumin (1,000 mg/day) for six weeks increased the antidepressant effect of the medication (104). Curcumin also induced a reduction in plasma concentrations of inflammatory markers and an increase in plasma concentrations of brain-derived neurotrophic factor compared to placebo (antidepressant drug alone) (104).
Larger clinical trials are needed to address the long-term effect of curcumin in subjects with major depression.
Premenstrual syndrome
Premenstrual syndrome (PMS) refers to a range of emotional (e.g., irritability, anxiety), behavioral (e.g., fatigue, insomnia), and physical symptoms (e.g., breast tenderness, headache) occurring prior to the monthly menstrual period in up to 90% of premenopausal women. In a recent randomized, double-blind, placebo-controlled trial in 70 women with PMS, the daily supplementation with 0.2 g of curcumin for 10 days during three consecutive menstrual cycles significantly reduced overall PMS severity, as assessed by a composite measure of all emotional, behavioral, and physical symptoms (105). Additional trials are necessary to evaluate the efficacy of curcumin in the management of PMS.
Sources
Food sources
Turmeric is the dried ground rhizome of Curcuma longa Linn (106). It is used as a spice in Indian, Southeast Asian, and Middle Eastern cuisines. Curcuminoids comprise about 2%-9% of turmeric (107). Curcumin is the most abundant curcuminoid in turmeric, providing about 75% of the total curcuminoids, while demethoxycurcumin and bisdemethoxycurcumin generally represent 10%-20% and less than 5% of the total curcuminoids, respectively (108). Curry powder contains turmeric along with other spices, but the amount of curcumin in curry powders is variable and often relatively low (109). Curcumin extracts are also used as food-coloring agents (110).
Supplements
Commercial curcumin is usually a mixture of curcumin, demethoxycurcumin, and bisdemethoxycurcumin (see Figure 1 above). Curcuminoid extracts are available as dietary supplements without a prescription in the US. The labels of a number of these extracts state that they are standardized to contain 95% curcuminoids, although such claims are not strictly regulated by the US Food and Drug Administration (FDA). Some curcumin preparations also contain piperine, which may increase the bioavailability of curcumin by inhibiting its metabolism (108). However, piperine may also affect the metabolism of drugs (see Drug interactions). Optimal doses of curcumin for cancer chemoprevention or therapeutic uses have not been established. It is unclear whether doses less than 3.6 g/day are biologically active in humans (see Metabolism and Bioavailability). Curcuminoid-containing supplements taken on an empty stomach may cause gastritis and peptic ulcer disease (108).
Safety
Adverse effects
In the United States, turmeric is generally recognized as safe (GRAS) by the FDA as a food additive (110). An increase in gallbladder contractions was observed in 12 healthy people supplemented with single doses of 20 to 40 mg of curcumin (111, 112). Yet, serious adverse effects have not been reported in humans taking high doses of curcumin. A dose escalation trial in 24 adults found that single oral dosages up to 12 g were safe, and adverse effects, including diarrhea, headache, rash, yellow stool, were not related to dose (7). In a phase I trial in Taiwan, curcumin supplementation up to 8 g/day for three months was reported to be well tolerated in patients with precancerous conditions or noninvasive cancer (8). Another clinical trial in the UK found that curcumin supplementation ranging from 0.45 to 3.6 g/day for four months was generally well tolerated by people with advanced colorectal cancer, although two participants experienced diarrhea and another reported nausea (9). Increases in serum alkaline phosphatase and lactate dehydrogenase were also observed in several participants, but it was not clear whether these increases were related to curcumin supplementation or cancer progression (3). In an open-label phase II trial, curcumin treatment (8 g/day) in combination with the anticancer drug gemcitabine was associated with severe abdominal pain in 7 out of 17 patients with advanced pancreatic cancer, leading to the treatment being discontinued in five patients while curcumin dosage was reduced to 4 g/day in two patients (79).
Pregnancy and lactation
Although there is no evidence that dietary consumption of turmeric as a spice adversely affects pregnancy or lactation, the safety of curcumin supplements in pregnancy and lactation has not been established.
Drug interactions
Curcumin has been found to inhibit platelet aggregation in vitro (113, 114), suggesting a potential for curcumin supplementation to increase the risk of bleeding in people taking anticoagulant or antiplatelet medications, such as aspirin, clopidogrel (Plavix), dalteparin (Fragmin), enoxaparin (Lovenox), heparin, ticlopidine (Ticlid), and warfarin (Coumadin). In cultured breast cancer cells, curcumin inhibited apoptosis induced by the chemotherapeutic agents, camptothecin, mechlorethamine, and doxorubicin at concentrations of 1 to 10 μM (115). In an animal model of breast cancer, dietary curcumin inhibited cyclophosphamide-induced tumor regression. Yet, it is not known whether oral curcumin administration will result in breast tissue concentrations that are high enough to inhibit cancer chemotherapeutic agents in humans (11). Curcuminoids may interfere with the activity of efflux drug transporters of the ATP-binding cassette (ABC) family, including P-glycoprotein, multidrug resistance protein (MRP), and breast cancer-resistant protein (BCRP), which function as ATP-dependent efflux pumps that actively regulate the excretion of a number of drugs limiting their systemic bioavailability (116, 117). Curcumin was also found to affect the activity of phase I biotransformation enzymes like cytochrome P450 (CYP) 3A4 (CYP3A4) (118), which catalyzes the metabolism of about one-half of all marketed drugs in the US (119). In healthy Japanese volunteers, curcumin (2 g) was found to increase plasma sulfasalazine concentration following the administration of a therapeutic dose (2 g) of the anti-rheumatic drug sulfasalazine (Salazopyrin, Azulfidine) (120).
Some curcumin supplements also contain piperine to increase the bioavailability of curcumin. Piperine may also interfere with efflux drug transporters and phase I cytochrome P450 enzymes and increase the bioavailability and slow the elimination of a number of drugs, including phenytoin (Dilantin), propranolol (Inderal), theophylline, and carbamazepine (Tegretol) (121-123).
Authors and Reviewers
Originally written in 2005 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in January 2009 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in February 2016 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in March 2016 by:
Lynne Howells, Ph.D.
Research Fellow
Experimental Cancer Medicine Centre Lab Quality Manager
University of Leicester
Copyright 2005-2024 Linus Pauling Institute
References
1. Gupta SC, Kismali G, Aggarwal BB. Curcumin, a component of turmeric: from farm to pharmacy. Biofactors. 2013;39(1):2-13. (PubMed)
2. Bandyopadhyay D. Farmer to pharmacist: curcumin as an anti-invasive and antimetastatic agent for the treatment of cancer. Front Chem. 2014;2:113. (PubMed)
3. Sharma RA, Gescher AJ, Steward WP. Curcumin: The story so far. Eur J Cancer. 2005;41(13):1955-1968. (PubMed)
4. Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: problems and promises. Mol Pharm. 2007;4(6):807-818. (PubMed)
5. Maheshwari RK, Singh AK, Gaddipati J, Srimal RC. Multiple biological activities of curcumin: a short review. Life Sci. 2006;78(18):2081-2087. (PubMed)
6. Baum L, Lam CW, Cheung SK, et al. Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J Clin Psychopharmacol. 2008;28(1):110-113. (PubMed)
7. Lao CD, Ruffin MTt, Normolle D, et al. Dose escalation of a curcuminoid formulation. BMC Complement Altern Med. 2006;6:10. (PubMed)
8. Cheng AL, Hsu CH, Lin JK, et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res. 2001;21(4B):2895-2900. (PubMed)
9. Sharma RA, Euden SA, Platton SL, et al. Phase I clinical trial of oral curcumin: biomarkers of systemic activity and compliance. Clin Cancer Res. 2004;10(20):6847-6854. (PubMed)
10. Garcea G, Berry DP, Jones DJ, et al. Consumption of the putative chemopreventive agent curcumin by cancer patients: assessment of curcumin levels in the colorectum and their pharmacodynamic consequences. Cancer Epidemiol Biomarkers Prev. 2005;14(1):120-125. (PubMed)
11. Garcea G, Jones DJ, Singh R, et al. Detection of curcumin and its metabolites in hepatic tissue and portal blood of patients following oral administration. Br J Cancer. 2004;90(5):1011-1015. (PubMed)
12. Aggarwal ML, Chacko KM, Kuruvilla BT. Systematic and comprehensive investigation of the toxicity of curcuminoidessential oil complex: A bioavailable turmeric formulation. Mol Med Rep. 2016;13(1):592-604. (PubMed)
13. Jager R, Lowery RP, Calvanese AV, Joy JM, Purpura M, Wilson JM. Comparative absorption of curcumin formulations. Nutr J. 2014;13:11. (PubMed)
14. Kanai M, Imaizumi A, Otsuka Y, et al. Dose-escalation and pharmacokinetic study of nanoparticle curcumin, a potential anticancer agent with improved bioavailability, in healthy human volunteers. Cancer Chemother Pharmacol. 2012;69(1):65-70. (PubMed)
15. Mendonca LM, Machado Cda S, Teixeira CC, Freitas LA, Bianchi ML, Antunes LM. Comparative study of curcumin and curcumin formulated in a solid dispersion: Evaluation of their antigenotoxic effects. Genet Mol Biol. 2015;38(4):490-498. (PubMed)
16. Shakeri A, Sahebkar A. Optimized curcumin formulations for the treatment of Alzheimer's disease: A patent evaluation. J Neurosci Res. 2016;94(2):111-113. (PubMed)
17. Prasad S, Tyagi AK, Aggarwal BB. Recent developments in delivery, bioavailability, absorption and metabolism of curcumin: the golden pigment from golden spice. Cancer Res Treat. 2014;46(1):2-18. (PubMed)
18. Sreejayan, Rao MN. Nitric oxide scavenging by curcuminoids. J Pharm Pharmacol. 1997;49(1):105-107. (PubMed)
19. Sreejayan N, Rao MN. Free radical scavenging activity of curcuminoids. Arzneimittelforschung. 1996;46(2):169-171. (PubMed)
20. Dickinson DA, Levonen AL, Moellering DR, et al. Human glutamate cysteine ligase gene regulation through the electrophile response element. Free Radic Biol Med. 2004;37(8):1152-1159. (PubMed)
21. Dickinson DA, Iles KE, Zhang H, Blank V, Forman HJ. Curcumin alters EpRE and AP-1 binding complexes and elevates glutamate-cysteine ligase gene expression. FASEB J. 2003;17(3):473-475. (PubMed)
22. Scapagnini G, Vasto S, Abraham NG, Caruso C, Zella D, Fabio G. Modulation of Nrf2/ARE pathway by food polyphenols: a nutritional neuroprotective strategy for cognitive and neurodegenerative disorders. Mol Neurobiol. 2011;44(2):192-201. (PubMed)
23. Zhang X, Liang D, Guo L, et al. Curcumin protects renal tubular epithelial cells from high glucose-induced epithelial-to-mesenchymal transition through Nrf2-mediated upregulation of heme oxygenase-1. Mol Med Rep. 2015;12(1):1347-1355. (PubMed)
24. Suzuki M, Betsuyaku T, Ito Y, et al. Curcumin attenuates elastase- and cigarette smoke-induced pulmonary emphysema in mice. Am J Physiol Lung Cell Mol Physiol. 2009;296(4):L614-623. (PubMed)
25. Yao QY, Xu BL, Wang JY, Liu HC, Zhang SC, Tu CT. Inhibition by curcumin of multiple sites of the transforming growth factor-β1 signalling pathway ameliorates the progression of liver fibrosis induced by carbon tetrachloride in rats. BMC Complement Altern Med. 2012;12:156. (PubMed)
26. Xiong ZE, Dong WG, Wang BY, Tong QY, Li ZY. Curcumin attenuates chronic ethanol-induced liver injury by inhibition of oxidative stress via mitogen-activated protein kinase/nuclear factor E2-related factor 2 pathway in mice. Pharmacogn Mag. 2015;11(44):707-715. (PubMed)
27. Xie Y, Zhao QY, Li HY, Zhou X, Liu Y, Zhang H. Curcumin ameliorates cognitive deficits heavy ion irradiation-induced learning and memory deficits through enhancing of Nrf2 antioxidant signaling pathways. Pharmacol Biochem Behav. 2014;126:181-186. (PubMed)
28. Ghosh S, Banerjee S, Sil PC. The beneficial role of curcumin on inflammation, diabetes and neurodegenerative disease: A recent update. Food Chem Toxicol. 2015;83:111-124. (PubMed)
29. Li CP, Li JH, He SY, Chen O, Shi L. Effect of curcumin on p38MAPK expression in DSS-induced murine ulcerative colitis. Genet Mol Res. 2015;14(2):3450-3458. (PubMed)
30. Yang JY, Zhong X, Yum HW, et al. Curcumin inhibits STAT3 signaling in the colon of dextran sulfate sodium-treated mice. J Cancer Prev. 2013;18(2):186-191. (PubMed)
31. Moon DO, Kim MO, Choi YH, Park YM, Kim GY. Curcumin attenuates inflammatory response in IL-1β-induced human synovial fibroblasts and collagen-induced arthritis in mouse model. Int Immunopharmacol. 2010;10(5):605-610. (PubMed)
32. Shakibaei M, John T, Schulze-Tanzil G, Lehmann I, Mobasheri A. Suppression of NF-κB activation by curcumin leads to inhibition of expression of cyclo-oxygenase-2 and matrix metalloproteinase-9 in human articular chondrocytes: Implications for the treatment of osteoarthritis. Biochem Pharmacol. 2007;73(9):1434-1445. (PubMed)
33. Zhu HT, Bian C, Yuan JC, et al. Curcumin attenuates acute inflammatory injury by inhibiting the TLR4/MyD88/NF-κB signaling pathway in experimental traumatic brain injury. J Neuroinflammation. 2014;11:59. (PubMed)
34. Baird WM, Hooven LA, Mahadevan B. Carcinogenic polycyclic aromatic hydrocarbon-DNA adducts and mechanism of action. Environ Mol Mutagen. 2005;45(2-3):106-114. (PubMed)
35. Sehgal A, Kumar M, Jain M, Dhawan DK. Modulatory effects of curcumin in conjunction with piperine on benzo(a)pyrene-mediated DNA adducts and biotransformation enzymes. Nutr Cancer. 2013;65(6):885-890. (PubMed)
36. Thapliyal R, Maru GB. Inhibition of cytochrome P450 isozymes by curcumins in vitro and in vivo. Food Chem Toxicol. 2001;39(6):541-547. (PubMed)
37. Volak LP, Ghirmai S, Cashman JR, Court MH. Curcuminoids inhibit multiple human cytochromes P450, UDP-glucuronosyltransferase, and sulfotransferase enzymes, whereas piperine is a relatively selective CYP3A4 inhibitor. Drug Metab Dispos. 2008;36(8):1594-1605. (PubMed)
38. Das L, Vinayak M. Long term effect of curcumin in restoration of tumour suppressor p53 and phase-II antioxidant enzymes via activation of Nrf2 signalling and modulation of inflammation in prevention of cancer. PLoS One. 2015;10(4):e0124000. (PubMed)
39. Iqbal M, Sharma SD, Okazaki Y, Fujisawa M, Okada S. Dietary supplementation of curcumin enhances antioxidant and phase II metabolizing enzymes in ddY male mice: possible role in protection against chemical carcinogenesis and toxicity. Pharmacol Toxicol. 2003;92(1):33-38. (PubMed)
40. Stewart ZA, Westfall MD, Pietenpol JA. Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol Sci. 2003;24(3):139-145. (PubMed)
41. Duvoix A, Blasius R, Delhalle S, et al. Chemopreventive and therapeutic effects of curcumin. Cancer Lett. 2005;223(2):181-190. (PubMed)
42. Surh YJ, Chun KS. Cancer chemopreventive effects of curcumin. Adv Exp Med Biol. 2007;595:149-172. (PubMed)
43. Singh S, Khar A. Biological effects of curcumin and its role in cancer chemoprevention and therapy. Anticancer Agents Med Chem. 2006;6(3):259-270. (PubMed)
44. Kuttan G, Kumar KB, Guruvayoorappan C, Kuttan R. Antitumor, anti-invasion, and antimetastatic effects of curcumin. Adv Exp Med Biol. 2007;595:173-184. (PubMed)
45. Kunnumakkara AB, Anand P, Aggarwal BB. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett. 2008;269(2):199-225. (PubMed)
46. Chen B, Zhang Y, Wang Y, Rao J, Jiang X, Xu Z. Curcumin inhibits proliferation of breast cancer cells through Nrf2-mediated down-regulation of Fen1 expression. J Steroid Biochem Mol Biol. 2014;143:11-18. (PubMed)
47. Zhou H, Beevers CS, Huang S. The targets of curcumin. Curr Drug Targets. 2011;12(3):332-347. (PubMed)
48. Han X, Xu B, Beevers CS, et al. Curcumin inhibits protein phosphatases 2A and 5, leading to activation of mitogen-activated protein kinases and death in tumor cells. Carcinogenesis. 2012;33(4):868-875. (PubMed)
49. Huang T, Chen Z, Fang L. Curcumin inhibits LPS-induced EMT through downregulation of NF-κB-Snail signaling in breast cancer cells. Oncol Rep. 2013;29(1):117-124. (PubMed)
50. Prvulovic D, Hampel H. Amyloid beta (Aβ) and phospho-tau (p-τ) as diagnostic biomarkers in Alzheimer's disease. Clin Chem Lab Med. 2011;49(3):367-374. (PubMed)
51. Ono K, Hasegawa K, Naiki H, Yamada M. Curcumin has potent anti-amyloidogenic effects for Alzheimer's β-amyloid fibrils in vitro. J Neurosci Res. 2004;75(6):742-750. (PubMed)
52. Reinke AA, Gestwicki JE. Structure-activity relationships of amyloid β-aggregation inhibitors based on curcumin: influence of linker length and flexibility. Chem Biol Drug Des. 2007;70(3):206-215. (PubMed)
53. Yang F, Lim GP, Begum AN, et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280(7):5892-5901. (PubMed)
54. Lin R, Chen X, Li W, Han Y, Liu P, Pi R. Exposure to metal ions regulates mRNA levels of APP and BACE1 in PC12 cells: blockage by curcumin. Neurosci Lett. 2008;440(3):344-347. (PubMed)
55. Zhang C, Browne A, Child D, Tanzi RE. Curcumin decreases amyloid-β peptide levels by attenuating the maturation of amyloid-β precursor protein. J Biol Chem. 2010;285(37):28472-28480. (PubMed)
56. Shi X, Zheng Z, Li J, et al. Curcumin inhibits Aβ-induced microglial inflammatory responses in vitro: Involvement of ERK1/2 and p38 signaling pathways. Neurosci Lett. 2015;594:105-110. (PubMed)
57. Goozee KG, Shah TM, Sohrabi HR, et al. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer's disease. Br J Nutr. 2015:1-17. (PubMed)
58. Krishnaswamy K, Goud VK, Sesikeran B, Mukundan MA, Krishna TP. Retardation of experimental tumorigenesis and reduction in DNA adducts by turmeric and curcumin. Nutr Cancer. 1998;30(2):163-166. (PubMed)
59. Li N, Chen X, Liao J, et al. Inhibition of 7,12-dimethylbenz[a]anthracene (DMBA)-induced oral carcinogenesis in hamsters by tea and curcumin. Carcinogenesis. 2002;23(8):1307-1313. (PubMed)
60. Ikezaki S, Nishikawa A, Furukawa F, et al. Chemopreventive effects of curcumin on glandular stomach carcinogenesis induced by N-methyl-N'-nitro-N-nitrosoguanidine and sodium chloride in rats. Anticancer Res. 2001;21(5):3407-3411. (PubMed)
61. Huang MT, Lou YR, Ma W, Newmark HL, Reuhl KR, Conney AH. Inhibitory effects of dietary curcumin on forestomach, duodenal, and colon carcinogenesis in mice. Cancer Res. 1994;54(22):5841-5847. (PubMed)
62. Chuang SE, Kuo ML, Hsu CH, et al. Curcumin-containing diet inhibits diethylnitrosamine-induced murine hepatocarcinogenesis. Carcinogenesis. 2000;21(2):331-335. (PubMed)
63. Pereira MA, Grubbs CJ, Barnes LH, et al. Effects of the phytochemicals, curcumin and quercetin, upon azoxymethane-induced colon cancer and 7,12-dimethylbenz[a]anthracene-induced mammary cancer in rats. Carcinogenesis. 1996;17(6):1305-1311. (PubMed)
64. Rao CV, Rivenson A, Simi B, Reddy BS. Chemoprevention of colon carcinogenesis by dietary curcumin, a naturally occurring plant phenolic compound. Cancer Res. 1995;55(2):259-266. (PubMed)
65. Kawamori T, Lubet R, Steele VE, et al. Chemopreventive effect of curcumin, a naturally occurring anti-inflammatory agent, during the promotion/progression stages of colon cancer. Cancer Res. 1999;59(3):597-601. (PubMed)
66. Mahmoud NN, Carothers AM, Grunberger D, et al. Plant phenolics decrease intestinal tumors in an animal model of familial adenomatous polyposis. Carcinogenesis. 2000;21(5):921-927. (PubMed)
67. Perkins S, Verschoyle RD, Hill K, et al. Chemopreventive efficacy and pharmacokinetics of curcumin in the min/+ mouse, a model of familial adenomatous polyposis. Cancer Epidemiol Biomarkers Prev. 2002;11(6):535-540. (PubMed)
68. Carroll RE, Benya RV, Turgeon DK, et al. Phase IIa clinical trial of curcumin for the prevention of colorectal neoplasia. Cancer Prev Res (Phila). 2011;4(3):354-364. (PubMed)
69. National Institutes of Health. Clinical Trials.gov [Website]. Available at: http://clinicaltrials.gov/. Accessed 1/27/16.
70. Rivera-Mancia S, Lozada-Garcia MC, Pedraza-Chaverri J. Experimental evidence for curcumin and its analogs for management of diabetes mellitus and its associated complications. Eur J Pharmacol. 2015;756:30-37. (PubMed)
71. Chuengsamarn S, Rattanamongkolgul S, Luechapudiporn R, Phisalaphong C, Jirawatnotai S. Curcumin extract for prevention of type 2 diabetes. Diabetes Care. 2012;35(11):2121-2127. (PubMed)
72. Usharani P, Mateen AA, Naidu MU, Raju YS, Chandra N. Effect of NCB-02, atorvastatin and placebo on endothelial function, oxidative stress and inflammatory markers in patients with type 2 diabetes mellitus: a randomized, parallel-group, placebo-controlled, 8-week study. Drugs R D. 2008;9(4):243-250. (PubMed)
73. Chuengsamarn S, Rattanamongkolgul S, Phonrat B, Tungtrongchitr R, Jirawatnotai S. Reduction of atherogenic risk in patients with type 2 diabetes by curcuminoid extract: a randomized controlled trial. J Nutr Biochem. 2014;25(2):144-150. (PubMed)
74. Khajehdehi P, Pakfetrat M, Javidnia K, et al. Oral supplementation of turmeric attenuates proteinuria, transforming growth factor-β and interleukin-8 levels in patients with overt type 2 diabetic nephropathy: a randomized, double-blind and placebo-controlled study. Scand J Urol Nephrol. 2011;45(5):365-370. (PubMed)
75. Schaffer M, Schaffer PM, Zidan J, Bar Sela G. Curcuma as a functional food in the control of cancer and inflammation. Curr Opin Clin Nutr Metab Care. 2011;14(6):588-597. (PubMed)
76. National Institutes of Health. An Introduction to Clinical Trials. Available at: https://clinicaltrials.gov/ct2/about-studies/learn. Accessed 2/8/16.
77. Mall M, Kunzelmann K. Correction of the CF defect by curcumin: hypes and disappointments. Bioessays. 2005;27(1):9-13. (PubMed)
78. Irving GR, Howells LM, Sale S, et al. Prolonged biologically active colonic tissue levels of curcumin achieved after oral administration — a clinical pilot study including assessment of patient acceptability. Cancer Prev Res (Phila). 2013;6(2):119-128. (PubMed)
79. Epelbaum R, Schaffer M, Vizel B, Badmaev V, Bar-Sela G. Curcumin and gemcitabine in patients with advanced pancreatic cancer. Nutr Cancer. 2010;62(8):1137-1141. (PubMed)
80. Kanai M, Yoshimura K, Asada M, et al. A phase I/II study of gemcitabine-based chemotherapy plus curcumin for patients with gemcitabine-resistant pancreatic cancer. Cancer Chemother Pharmacol. 2011;68(1):157-164. (PubMed)
81. Bayet-Robert M, Kwiatkowski F, Leheurteur M, et al. Phase I dose escalation trial of docetaxel plus curcumin in patients with advanced and metastatic breast cancer. Cancer Biol Ther. 2010;9(1):8-14. (PubMed)
82. Ghalaut VS, Sangwan L, Dahiya K, Ghalaut PS, Dhankhar R, Saharan R. Effect of imatinib therapy with and without turmeric powder on nitric oxide levels in chronic myeloid leukemia. J Oncol Pharm Pract. 2012;18(2):186-190. (PubMed)
83. Mahammedi H, Planchat E, Pouget M, et al. The new combination docetaxel, prednisone and curcumin in patients with castration-resistant prostate cancer: a pilot phase II study. Oncology. 2016;90(2):69-78. (PubMed)
84. Satoskar RR, Shah SJ, Shenoy SG. Evaluation of anti-inflammatory property of curcumin (diferuloyl methane) in patients with postoperative inflammation. Int J Clin Pharmacol Ther Toxicol. 1986;24(12):651-654. (PubMed)
85. Deodhar SD, Sethi R, Srimal RC. Preliminary study on antirheumatic activity of curcumin (diferuloyl methane). Indian J Med Res. 1980;71:632-634.
86. Chandran B, Goel A. A randomized, pilot study to assess the efficacy and safety of curcumin in patients with active rheumatoid arthritis. Phytother Res. 2012;26(11):1719-1725. (PubMed)
87. Ryan JL, Heckler CE, Ling M, et al. Curcumin for radiation dermatitis: a randomized, double-blind, placebo-controlled clinical trial of thirty breast cancer patients. Radiat Res. 2013;180(1):34-43. (PubMed)
88. Hanai H, Iida T, Takeuchi K, et al. Curcumin maintenance therapy for ulcerative colitis: randomized, multicenter, double-blind, placebo-controlled trial. Clin Gastroenterol Hepatol. 2006;4(12):1502-1506. (PubMed)
89. Lang A, Salomon N, Wu JC, et al. Curcumin in combination with mesalamine induces remission in patients with mild-to-moderate ulcerative colitis in a randomized controlled trial. Clin Gastroenterol Hepatol. 2015;13(8):1444-1449 e1441. (PubMed)
90. Anuradha BR, Bai YD, Sailaja S, Sudhakar J, Priyanka M, Deepika V. Evaluation of anti-inflammatory effects of curcumin gel as an adjunct to scaling and root planing: A Clinical Study. J Int Oral Health. 2015;7(7):90-93. (PubMed)
91. Nagasri M, Madhulatha M, Musalaiah SV, Kumar PA, Krishna CH, Kumar PM. Efficacy of curcumin as an adjunct to scaling and root planning in chronic periodontitis patients: A clinical and microbiological study. J Pharm Bioallied Sci. 2015;7(Suppl 2):S554-558. (PubMed)
92. Sreedhar A, Sarkar I, Rajan P, et al. Comparative evaluation of the efficacy of curcumin gel with and without photo activation as an adjunct to scaling and root planing in the treatment of chronic periodontitis: A split mouth clinical and microbiological study. J Nat Sci Biol Med. 2015;6(Suppl 1):S102-109. (PubMed)
93. Muglikar S, Patil KC, Shivswami S, Hegde R. Efficacy of curcumin in the treatment of chronic gingivitis: a pilot study. Oral Health Prev Dent. 2013;11(1):81-86. (PubMed)
94. Alok A, Singh ID, Singh S, Kishore M, Jha PC. Curcumin — pharmacological actions and its role in oral submucous fibrosis: a review. J Clin Diagn Res. 2015;9(10):ZE01-03. (PubMed)
95. Yadav M, Aravinda K, Saxena VS, et al. Comparison of curcumin with intralesional steroid injections in Oral Submucous Fibrosis - A randomized, open-label interventional study. J Oral Biol Craniofac Res.2014;4(3):169-173. (PubMed)
96. Hazarey VK, Sakrikar AR, Ganvir SM. Efficacy of curcumin in the treatment for oral submucous fibrosis — a randomized clinical trial. J Oral Maxillofac Pathol. 2015;19(2):145-152. (PubMed)
97. Cox KH, Pipingas A, Scholey AB. Investigation of the effects of solid lipid curcumin on cognition and mood in a healthy older population. J Psychopharmacol. 2015;29(5):642-651. (PubMed)
98. Ringman JM, Frautschy SA, Teng E, et al. Oral curcumin for Alzheimer's disease: tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimers Res Ther. 2012;4(5):43. (PubMed)
99. Davidson JR. Major depressive disorder treatment guidelines in America and Europe. J Clin Psychiatry. 2010;71 Suppl E1:e04. (PubMed)
100. Al-Karawi D, Al Mamoori DA, Tayyar Y. The role of curcumin administration in patients with major depressive disorder: mini meta-analysis of clinical trials. Phytother Res. 2016;30(2):175-183. (PubMed)
101. Lopresti AL, Maes M, Maker GL, Hood SD, Drummond PD. Curcumin for the treatment of major depression: a randomised, double-blind, placebo controlled study. J Affect Disord. 2014;167:368-375. (PubMed)
102. Bergman J, Miodownik C, Bersudsky Y, et al. Curcumin as an add-on to antidepressive treatment: a randomized, double-blind, placebo-controlled, pilot clinical study. Clin Neuropharmacol. 2013;36(3):73-77. (PubMed)
103. Sanmukhani J, Satodia V, Trivedi J, et al. Efficacy and safety of curcumin in major depressive disorder: a randomized controlled trial. Phytother Res. 2014;28(4):579-585. (PubMed)
104. Yu JJ, Pei LB, Zhang Y, Wen ZY, Yang JL. Chronic supplementation of curcumin enhances the efficacy of antidepressants in major depressive disorder: a randomized, double-blind, placebo-controlled pilot study. J Clin Psychopharmacol. 2015;35(4):406-410. (PubMed)
105. Fanaei H, Khayat S, Kasaeian A, Javadimehr M. Effect of curcumin on serum brain-derived neurotrophic factor levels in women with premenstrual syndrome: a randomized, double-blind, placebo-controlled trial. Neuropeptides. 2015. Nov 11. pii: S0143-4179(15)00118-3. doi: 10.1016/j.npep.2015.11.003. [Epub ahead of print]. (PubMed)
106. Prasad S, Gupta SC, Tyagi AK, Aggarwal BB. Curcumin, a component of golden spice: from bedside to bench and back. Biotechnol Adv. 2014;32(6):1053-1064. (PubMed)
107. Lechtenberg M, Quandt B, Nahrstedt A. Quantitative determination of curcuminoids in Curcuma rhizomes and rapid differentiation of Curcuma domestica Val. and Curcuma xanthorrhiza Roxb. by capillary electrophoresis. Phytochem Anal. 2004;15(3):152-158. (PubMed)
108. Hendler SS, Rorvik DM. PDR for Nutritional Supplements. 2nd ed: Thomson Reuters; 2008.
109. Heath DD, Khwaja F, Rock CL. Curcumin content of turmeric and curry powders. FASEB J. 2004;18(4):A125-A125. (PubMed)
110. US Food and Drug Administration. Food Additive Status List: GRN number 460. Aug 23, 2013. Available at: http://www.accessdata.fda.gov/scripts/fdcc/?set=GRASNotices. Accessed 1/25/16.
111. Rasyid A, Lelo A. The effect of curcumin and placebo on human gall-bladder function: an ultrasound study. Aliment Pharmacol Ther. 1999;13(2):245-249. (PubMed)
112. Rasyid A, Rahman AR, Jaalam K, Lelo A. Effect of different curcumin dosages on human gall bladder. Asia Pac J Clin Nutr. 2002;11(4):314-318. (PubMed)
113. Shah BH, Nawaz Z, Pertani SA, et al. Inhibitory effect of curcumin, a food spice from turmeric, on platelet-activating factor- and arachidonic acid-mediated platelet aggregation through inhibition of thromboxane formation and Ca2+ signaling. Biochem Pharmacol. 1999;58(7):1167-1172. (PubMed)
114. Srivastava KC, Bordia A, Verma SK. Curcumin, a major component of food spice turmeric (Curcuma longa) inhibits aggregation and alters eicosanoid metabolism in human blood platelets. Prostaglandins Leukot Essent Fatty Acids. 1995;52(4):223-227. (PubMed)
115. Somasundaram S, Edmund NA, Moore DT, Small GW, Shi YY, Orlowski RZ. Dietary curcumin inhibits chemotherapy-induced apoptosis in models of human breast cancer. Cancer Res. 2002;62(13):3868-3875. (PubMed)
116. Chearwae W, Shukla S, Limtrakul P, Ambudkar SV. Modulation of the function of the multidrug resistance-linked ATP-binding cassette transporter ABCG2 by the cancer chemopreventive agent curcumin. Mol Cancer Ther. 2006;5(8):1995-2006. (PubMed)
117. Chearwae W, Wu CP, Chu HY, Lee TR, Ambudkar SV, Limtrakul P. Curcuminoids purified from turmeric powder modulate the function of human multidrug resistance protein 1 (ABCC1). Cancer Chemother Pharmacol. 2006;57(3):376-388. (PubMed)
118. Hsieh YW, Huang CY, Yang SY, et al. Oral intake of curcumin markedly activated CYP 3A4: in vivo and ex-vivo studies. Sci Rep. 2014;4:6587. (PubMed)
119. Koe XF, Tengku Muhammad TS, Chong AS, Wahab HA, Tan ML. Cytochrome P450 induction properties of food and herbal-derived compounds using a novel multiplex RT-qPCR in vitro assay, a drug-food interaction prediction tool. Food Sci Nutr. 2014;2(5):500-520. (PubMed)
120. Kusuhara H, Furuie H, Inano A, et al. Pharmacokinetic interaction study of sulphasalazine in healthy subjects and the impact of curcumin as an in vivo inhibitor of BCRP. Br J Pharmacol. 2012;166(6):1793-1803. (PubMed)
121. Bano G, Raina RK, Zutshi U, Bedi KL, Johri RK, Sharma SC. Effect of piperine on bioavailability and pharmacokinetics of propranolol and theophylline in healthy volunteers. Eur J Clin Pharmacol. 1991;41(6):615-617. (PubMed)
122. Pattanaik S, Hota D, Prabhakar S, Kharbanda P, Pandhi P. Pharmacokinetic interaction of single dose of piperine with steady-state carbamazepine in epilepsy patients. Phytother Res. 2009;23(9):1281-1286. (PubMed)
123. Velpandian T, Jasuja R, Bhardwaj RK, Jaiswal J, Gupta SK. Piperine in food: interference in the pharmacokinetics of phenytoin. Eur J Drug Metab Pharmacokinet. 2001;26(4):241-247. (PubMed)
Fiber
You should be automatically redirected to the current page, if not click here.
Flavonoids
Contents
Summary
- Flavonoids are a large family of polyphenolic plant compounds. Six major subclasses of flavonoids, namely anthocyanidins, flavan-3-ols, flavonols, flavanones, flavones, and isoflavones, flavonols are the most widespread in the human diet. (More information)
- Dietary flavonoids are naturally occurring in fruit, vegetables, chocolate, and beverages like wine and tea. There has been much interest in the potential health benefits of flavonoids associated with fruit- and vegetable-rich diets. (More information)
- The physicochemical properties of flavonoids influence their metabolic fate, i.e., their digestion, absorption, and biotransformation. The bioavailability of these polyphenols in vivo is a major determinant in their ability to exert biological activities relevant to human health. (More information)
- Many of the biological effects of flavonoids appear to be related to their ability to modulate a number of cell-signaling cascades. Flavonoids have been shown to exhibit antiinflammatory, antithrombogenic, antidiabetic, anticancer, and neuroprotective activities through different mechanisms of action in vitro and in animal models. (More information)
- Accumulating evidence from randomized controlled trials suggests that consumption of flavan-3-ols and anthocyanidins can be beneficial for metabolic and cardiovascular health. (More information)
- The results of small-scale randomized controlled trials suggest that consumption of flavonoid-rich food and beverages containing anthocyanins or flavan-3-ols may improve vascular endothelial function. As yet, it is not known whether these acute improvements result in long-term reductions in risk of cardiovascular disease. (More information)
- Promising findings in randomized controlled studies indicate that supplementation with flavan-3-ols or anthocyanidins may improve glycemic control in subjects at-risk or diagnosed with type 2 diabetes mellitus. (More information)
- Despite promising results in animal studies, only a limited number of observational studies have reported potential cancer preventive effects of flavonoids in humans. Higher intakes of soy isoflavones may be associated with reduced risks of breast cancer in postmenopausal women and prostate cancer in men. (More information)
- Evidence suggesting that some flavonoids or flavonoid-rich foods may enhance cognitive function is currently limited, and it is not yet known whether their consumption could lower the risk of cognitive impairments and dementia in humans. (More information)
- High intakes of dietary flavonoids are generally regarded as safe, especially because of their low bioavailability. However, flavonoid supplements may affect the action of anticoagulants and increase the toxicity of a wide range of drugs when taken concurrently. (More information)
Introduction
Flavonoids are a large family of over 5,000 hydroxylated polyphenolic compounds that carry out important functions in plants, including attracting pollinating insects; combating environmental stresses, such as microbial infection; and regulating cell growth (1). Their bioavailability and biological activities in humans appear to be strongly influenced by their chemical nature. Since the 1990s, there has been a growing interest in dietary flavonoids due to their likely contribution to the health benefits of fruit- and vegetable-rich diets. This article reviews some of the scientific evidence regarding the role of dietary flavonoids in health promotion and disease prevention in humans; it is not meant to be a comprehensive review on every health topic studied.
Flavonoid Subclasses
Flavonoids are classified into 12 major subclasses based on chemical structures, six of which, namely anthocyanidins, flavan-3-ols, flavonols, flavones, flavanones, and isoflavones (Table 1 and Figures 1-9) are of dietary significance. Glycosylated flavonols (bound to at least one sugar molecule) are the most widely distributed flavonoids in the diet (2, 3).
| Flavonoid Subclass | Dietary Flavonoids (aglycones) | Some Common Food Sources (see also Sources) |
|---|---|---|
| Anthocyanidins* | Cyanidin, Delphinidin, Malvidin, Pelargonidin, Peonidin, Petunidin | Red, blue, and purple berries; red and purple grapes; red wine |
|
|
Monomers (Catechins): (+)-Catechin, (-)-Epicatechin, (-)-Epigallocatechin, (+)-Gallocatechin; and their gallate derivatives |
Teas (particularly white, green, and oolong), cocoa-based products, grapes, berries, apples |
|
Dimers and Polymers: Proanthocyanidins# |
Apples, berries, cocoa-based products, red grapes, red wine |
|
| Theaflavins, Thearubigins |
Black tea |
|
| Flavonols | Isorhamnetin, Kaempferol, Myricetin, Quercetin |
Onions, scallions, kale, broccoli, apples, berries, teas |
| Flavones | Apigenin, Luteolin, Baicalein, Chrysin |
Parsley, thyme, celery, hot peppers |
| Flavanones | Eriodictyol, Hesperetin, Naringenin |
Citrus fruit and juices, e.g., oranges, grapefruits, lemons |
| Isoflavones |
Daidzein, Genistein, Glycitein, Biochanin A, Formononetin |
Soybeans, soy foods, legumes |
|
*Anthocyanidins with one or more sugar moieties (anthocyanidin glycosides) are called anthocyanins. #Proanthocyanidin oligomers formed from (+)-catechin and (-)-epicatechin subunits are called procyanidins. |
||
For more detailed information on the health effects of isoflavones, a subclass of flavonoids with estrogenic activity, see the article on Soy Isoflavones.
For more information on the health benefits of foods that are rich in flavonoids, see the articles on Fruit and Vegetables, Legumes, and Tea.

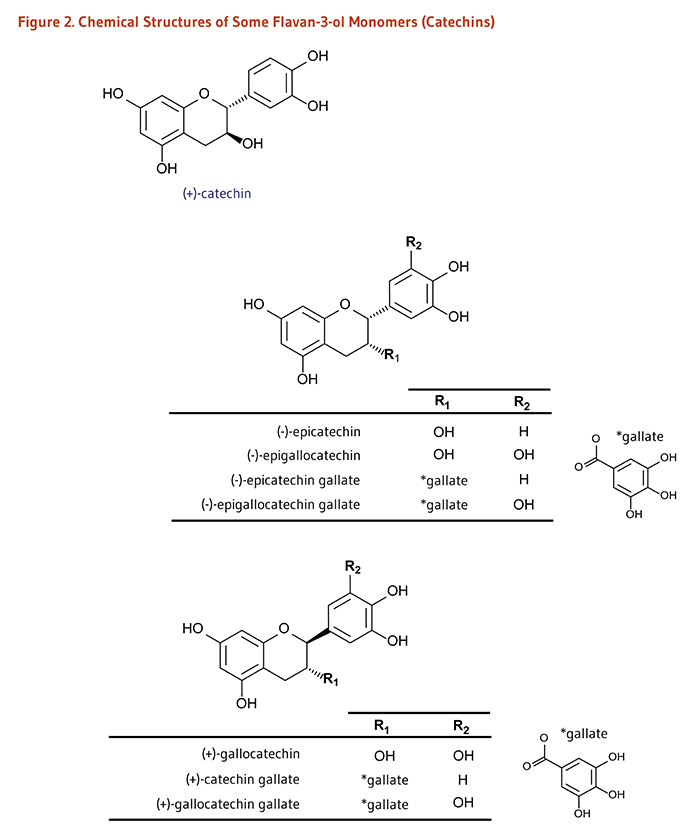

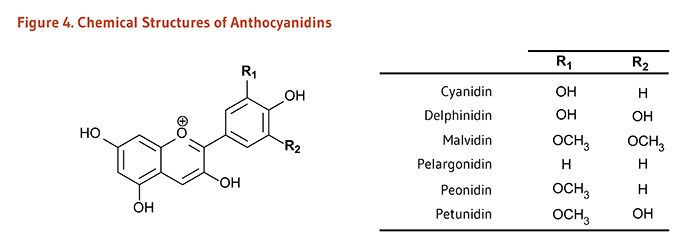
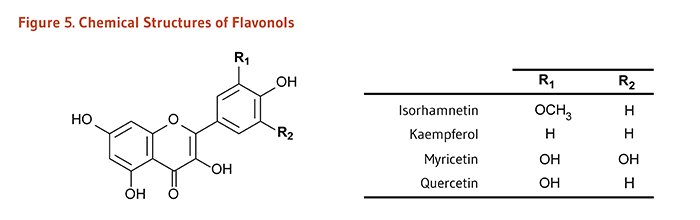
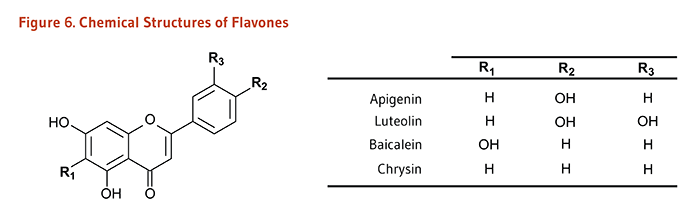
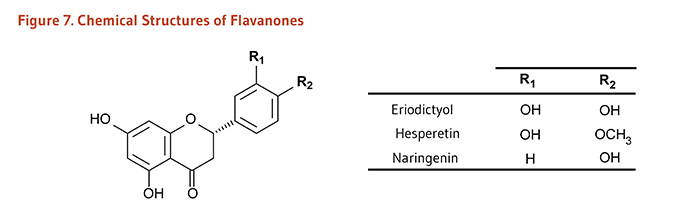
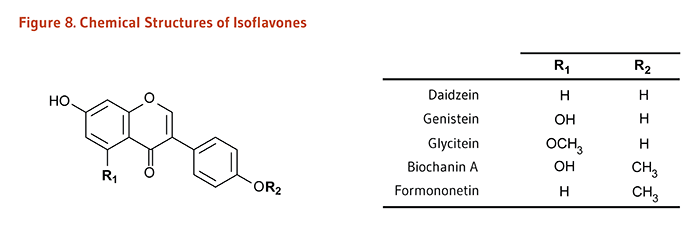
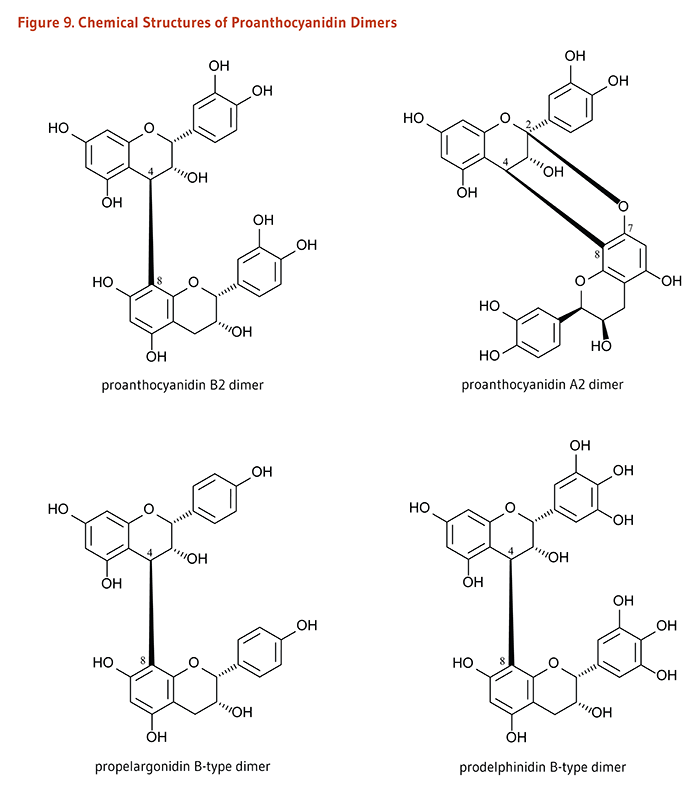
Metabolism and Bioavailability
The amount of flavonoids present in ingested food has little importance unless dietary flavonoids are absorbed and become available to target tissues within the body. During and after intestinal absorption, flavonoids are rapidly and extensively metabolized in intestinal and liver cells such that they are likely to appear as metabolites (e.g., phase II metabolites) in the bloodstream and urine (4). Additionally, the biological activities of flavonoid metabolites are likely to be different from those of their parent compounds (5). Some of the factors influencing the metabolic fate and bioavailability of dietary flavonoids are mentioned below.
Chemical structure of flavonoids
Most flavonoids occur in edible plants and foods as β-glycosides, i.e., bound to one or more sugar molecules (6). Exceptions include flavan-3-ols (catechins and proanthocyanidins) and fermented soy-based products that are exposed to microbial β-glucosidases, which catalyze the release of sugar molecules from glycosylated isoflavones (7). Even after food processing and cooking, most flavonoid glycosides reach the small intestine intact. Only flavonoid aglycones (not bound to a sugar molecule) and a few flavonoid glucosides (bound to glucose) are easily absorbed in the small intestine (8). Glycosylated flavonoids might be able to penetrate the mucus layer of the intestine and be deglycosylated on the cell surface before absorption. Those that cannot be deglycosylated in the small intestine may be hydrolyzed by bacterial enzymes in the colon (7). Nevertheless, colonic bacteria might remove sugar moieties and rapidly degrade aglycone flavonoids, thus limiting their absorption in the colon (9).
In contrast to monomeric flavan-3-ols (catechins), the polymeric nature of proanthocyanidins likely prevents their intestinal absorption. Flavan-3-ol monomers and procyanidins are transformed by the intestinal microbiota to 5-(hydroxyphenyl)-γ-valerolactones which appear in the circulatory system and are excreted in urine as sulfate and glucuronide metabolites (Figure 10). Valerolactones may be further degraded by the colonic microbiota to smaller phenolic acids and aromatic compounds. The colonic microbiota also metabolize the gallate esters of flavonoids, generating gallate, which is further catabolized to pyrogallol. Microbe-derived flavonoid metabolites are readily absorbed into the circulatory system and excreted in both free forms and as phase II metabolites in urine (9).

Interactions with food matrix
The presence of macronutrients in food influences the bioavailability of co-ingested flavonoids (reviewed in 8, 10, 11). The binding affinity and potential (non-) covalent interactions of flavonoids with food proteins, carbohydrates, and fats are directly associated with the physicochemical properties of flavonoids (reviewed in 8). Proteins in milk might reduce the absorption of polyphenols from cocoa or black tea. The presence of milk proteins bound to flavonoids was shown to weaken the flavonoid antioxidant capacity in vitro (12), and milk consumption has been shown to blunt the vascular benefits of tea flavonoids in healthy volunteers (13). Some carbohydrate-rich foods may increase the deglycosylation and absorption of flavonoids by stimulating gastrointestinal motility, mucosal blood flow, and colonic fermentation. Conversely, dietary flavonoids have been shown to interfere with carbohydrate digestion and absorption (see Biological Activities).
Composition of gut microbiota
In the large intestine, gut microbial enzymes transform flavonoids through deglycosylation, ring fission, dehydroxylation, demethylation, etc. into metabolites that can then be absorbed or excreted (9, 14). The diversity and activity of colonic bacteria, which are partly dependent on a person’s dietary habits, will determine which metabolites can be produced from ingested flavonoids (15, 16). The composition of the colonic microbiota can therefore affect the metabolic fate and bioavailability of dietary flavonoids (17).
The detoxification pathway
Flavonoids are recognized as xenobiotics by the body such that they undergo extensive modifications first in the intestinal mucosa and then in the liver.
Phase II enzymes
Depending on their structural characteristics, flavonoids can be rapidly transformed by phase II detoxification enzymes to form methylated, glucuronidated, and/or sulfated metabolites (2). This metabolic pathway increases the solubility of phenolic aglycones and facilitates their excretion in the bile and urine (11). Free (unconjugated) aglycones are generally absent from the bloodstream, with the possible exception of trace levels of catechins (17). Catechol-O-methyltransferase (COMT) is the detoxifying enzyme responsible for the methylation of the hydroxyl groups of flavonoids, producing O-methylated flavonoids. A single nucleotide polymorphism (SNP) in the gene for COMT — known as SNP rs4680 G>A — causes a valine-to-methionine substitution in the sequence of the enzyme. Individuals with the A/A genotype have a form of the enzyme that is three- to four-fold less active than the wild-type variant in G/G genotype carriers (18). It has been suggested that subjects who are less efficient at eliminating green tea flavonoids may be more likely to benefit from their consumption (19).
Efflux transporters
Flavonoid conjugates are excreted via the action of efflux transporters from the ATP-binding cassette (ABC) family, including P-glycoprotein, MRPs (multidrug resistance proteins), or BCRPs (breast cancer-resistant proteins). Depending on their physicochemical properties, some flavonoids may interfere with the activity of ABC transporters (20). This implies that flavonoids can affect their own bioavailability, as well as that of other substrates of these transporters (e.g., pharmacological drugs) (see Drug interactions).
Binding to plasma proteins
Flavonoid bioavailability may be inversely related to their binding affinity to plasma proteins (21). Greater binding affinity to plasma proteins (and thus, possibly, lower flavonoid bioavailability) has been linked to structural characteristics, such as methylation and galloylation. On the contrary, glycosylation reduced binding affinity to plasma proteins, suggesting that aglycones might have a limited bioavailability compared to glycosylated flavonoids. While glucuronidation is thought to facilitate the excretion of flavonoids from the body, glucuronides show little affinity to plasma proteins and might thus be able to diffuse to target tissues where deglucuronidation can take place (8).
Summary
In general, the bioavailability of flavonoids is low due to limited absorption, extensive metabolism, and rapid excretion. Isoflavones are thought to be the most bioavailable of all flavonoid subclasses, while anthocyanins and galloylated catechins are very poorly absorbed (8, 22). Yet, given the wide variability in structures within subclasses, it is difficult to generalize the absorbability and bioavailability of flavonoids based only on their structural classification. In addition, when evaluating the data from flavonoid research in cultured cells, it is important to consider whether the flavonoid concentrations and metabolites used are physiologically relevant (23). In humans, peak plasma concentrations of soy isoflavones and citrus flavanones have not been found to exceed 10 micromoles/liter (μM) after oral consumption. Peak plasma concentrations measured after the consumption of anthocyanins, flavan-3-ols, and flavonols (including those from tea) are generally lower than 1 μM (2). A recent quantitative analysis of 88 polyphenolic metabolites (not limited to flavonoids) identified in human blood and urine found median peak concentrations of 0.9 μM and 3.2 μM after food intake and oral supplementation, respectively (4).
Biological Activities
Direct antioxidant activity
Flavonoids are effective scavengers of free radicals in the test tube (in vitro) (24, 25). However, even with very high flavonoid intakes, plasma and intracellular flavonoid concentrations in humans are likely to be 100 to 1,000 times lower than concentrations of other antioxidants, such as ascorbate (vitamin C), uric acid, and glutathione. Moreover, most circulating flavonoids are actually flavonoid metabolites, some of which have lower antioxidant activity than the parent flavonoid (5). For these reasons, the relative contribution of dietary flavonoids to plasma and tissue antioxidant function in vivo is likely to be very small or negligible (26-28).
Metal chelation
Metal ions, such as iron and copper, can catalyze the production of free radicals. The ability of flavonoids to chelate (bind) metal ions appears to contribute to their antioxidant activity in vitro (29, 30). In living organisms, most iron and copper are bound to proteins, limiting their participation in reactions that produce free radicals. Although the metal-chelating activities of flavonoids may be beneficial in pathological conditions of iron or copper excess, it is not known whether flavonoids or their metabolites function as effective metal chelators in vivo (26).
Effects on cell-signaling pathways
Cells are capable of responding to a variety of different stresses or signals by increasing or decreasing the availability of specific proteins. The complex cascades of events that lead to changes in the expression of specific genes are known as cell-signaling pathways or signal transduction pathways. These pathways regulate numerous cell processes, such as proliferation, differentiation, inflammatory responses, apoptosis (programmed cell death), and survival. Although it was initially hypothesized that the biological effects of flavonoids would be related to their antioxidant activity, available evidence from cell culture experiments suggests that many of the effects of flavonoids, including antiinflammatory, antidiabetic, anticancer, and neuroprotective activities, are related to their ability to modulate cell-signaling pathways (27). Intracellular concentrations of flavonoids required to affect cellular signaling are considerably lower than those required to affect cellular antioxidant capacity. Flavonoid metabolites may retain their ability to interact with cell-signaling proteins even if their antioxidant activity is diminished (31, 32).
Effective signal transduction requires proteins known as kinases that catalyze the phosphorylation of target proteins, which become either activated or inhibited. Cascades involving specific phosphorylations or dephosphorylations of signal transduction proteins ultimately affect the activity of transcription factors — proteins that bind to specific response elements on DNA and promote or prevent the transcription of target genes. Results of numerous studies in cell culture suggest that flavonoids may affect chronic disease by selectively inhibiting kinases (27, 33). Cell growth and proliferation are also regulated by growth factors that initiate cell-signaling cascades by binding to specific receptors in cell membranes. Flavonoids may alter growth factor signaling by inhibiting receptor phosphorylation or blocking receptor binding by growth factors (34).
Each flavonoid subclass contains many types of chemicals with varying biological activities (and potential health benefits) such that the activity of a specific flavonoid cannot easily be generalized. Some examples of major biological activities of flavonoids are highlighted below.
Biological activities related to the prevention of cardiovascular disease
Flavonoids have been shown to (1) reduce inflammation by suppressing the expression of pro-inflammatory mediators (35-37); (2) down-regulate the expression of vascular cell adhesion molecules, which contribute to the recruitment of inflammatory white blood cells from the blood to the arterial wall (38, 39); (3) increase the production of nitric oxide (NO) by endothelial nitric oxide synthase (eNOS), thus improving vascular endothelial function (40); (4) inhibit angiotensin-converting enzyme, thus inducing vascular relaxation (41); (5) inhibit platelet aggregation (42); and (6) oppose smooth muscle cell proliferation and migration occurring during atherogenesis (43).
Biological activities related to the prevention of diabetes
Flavonoids have been found to interfere with the digestion, absorption, and metabolism of carbohydrates (reviewed in 44). Each subclass of flavonoids has also demonstrated anti-diabetic properties, including (1) improving insulin secretion and viability of pancreatic β-cells under glucotoxic or pro-inflammatory conditions, (2) increasing insulin-stimulated glucose uptake by target cells, (3) protecting muscle cells against fatty acid-induced insulin resistance, and (4) reducing hyperglycemia and improving glucose tolerance in animal models of obesity and/or type 2 diabetes mellitus (45).
Biological activities related to the prevention of cancer
Flavonoids have been found to (1) scavenge free radicals that can damage macromolecules, including DNA (46, 47); (2) interfere with biotransformation enzymes and efflux transporters, possibly preventing the activation of procarcinogenic chemicals and promoting their excretion from the body (48, 49); (3) regulate proliferation, DNA repair, or activation of pathways leading to apoptosis (programmed cell death) in case of irreversible DNA damage (50); and (4) inhibit tumor invasion and angiogenesis (51, 52).
Biological activities related to neuroprotection and cognitive function
Flavonoids are thought to (1) promote neurogenesis, synaptic growth, and neuron survival in the learning and memory-related brain regions (e.g., hippocampus) by stimulating the production of neurotrophins like BDNF; (2) protect hippocampal cells and striatal dopaminergic cells from cytotoxic molecules (pro-inflammatory mediators and ROS) released by abnormally activated microglia and hypertrophic astrocytes in neurodegenerative disorders; (3) reduce neuroinflammation by inhibiting the generation of pro-inflammatory cytokines, lipid mediators, and reactive oxygen species by astrocytes and microglial cells; (4) stimulate the production of nitric oxide (NO), which improves endothelial function, increases cerebral blood flow, and protects artery walls against the buildup of atherosclerotic plaques (reviewed in 53, 54).
Disease Prevention
Cardiovascular disease
Several prospective cohort studies conducted in the US and Europe have examined the relationship between some measure of dietary flavonoid intake and cardiovascular disease (CVD) or mortality. A recent meta-analysis of 14 prospective studies published between 1996 and 2012 reported that higher intakes in each flavonoid subclass were significantly associated with a reduced risk of cardiovascular events (55). Top versus bottom quantiles of intake for each of the flavonoid subclasses were associated with an approximate 10% reduction in the risk of CVD. Another meta-analysis of eight prospective studies found a 14% reduced risk of stroke with the highest versus lowest quintile of flavonol intakes (56). However, several serious limitations highlighted in a recent publication by Jacques et al. suggested caution when interpreting these results (57). In particular, most of the prospective studies in these meta-analyses did not include all flavonoid subclasses nor calculate intakes using the latest and more complete versions of the USDA databases for the flavonoid content of foods (58-60). Another major concern is the lack of adjustment regarding the overall quality of the diet. Consumers with higher flavonoid intakes are likely to have a greater consumption of fruit and vegetables and overall healthier diets than those with poor flavonoid intakes. Additionally, none of the studies excluded potential bias due to constituents of flavonoid-rich foods that are known to either lower (e.g., other phytochemicals, vitamins, dietary fiber) or increase (e.g., sodium, saturated fat) the risk of cardiovascular events (discussed in 57).
In the Framingham Offspring Cohort study that followed 2,880 adults for a mean of 14.9 years, consumption of all flavonoid subclasses except flavones and flavanones was inversely associated with CVD (57). Yet, adjusting for confounding factors, including fruit and vegetable intake and overall diet quality, attenuated these relationships such that they were no longer statistically significant. An analysis of a larger prospective study of the EPIC-Norfolk cohort (24,885 participants) that considered confounding by many dietary factors (vitamin C, dietary fiber, fat, saturated fat, potassium, sodium, and alcohol) found no significant association between flavan-3-ol intake and CVD-related or all-cause mortality (61).
A number of large prospective studies and small-scale, randomized controlled trials have investigated the effects of flavonoids on established biomarkers of CVD, including those involved in oxidative stress, inflammation, abnormal blood lipid profile, endothelial dysfunction, and hypertension; some of these studies are highlighted below.
Biochemical markers of cardiovascular disease
In a cross-sectional analysis of the Framingham Offspring Cohort study, the highest versus lowest intake of anthocyanins (≤3.5 mg/day versus ≥23.5 mg/day) was associated with lower concentrations of acute-phase reactant proteins (-100%), pro-inflammatory cytokines (-75%), and markers of oxidative stress (-52%), even after adjustment for confounding variables (62). Interestingly, a food-based analysis revealed that intakes of foods rich in anthocyanins, e.g., apples, red wine, and strawberries, were also inversely associated with an overall inflammation score based on 12 different biomarkers. Higher intakes of polymeric flavan-3-ols (i.e., theaflavins, thearubigins, and proanthocyanidins) were correlated with lower concentrations of pro-inflammatory cytokines and biomarkers of oxidative stress. Intake levels of total flavonoids and flavan-3-ol monomers (i.e., catechins) were inversely associated with concentrations of the biomarkers of oxidative stress. Although tea is a major source of flavan-3-ols, tea consumption was not correlated with the composite inflammation score or any components of this score in this study (62).
Cocoa is another source of flavan-3-ols, in particular (-)-epicatechin and procyanidins, that may provide cardiovascular benefits (63). Indeed, a recent randomized, double-blind, placebo-controlled study in 100 healthy adults (ages, 35-60 years) suggested that short-term benefits of cocoa flavan-3-ol consumption on cardiovascular health, including improvements in lipoprotein profile (i.e., higher HDL-cholesterol and lower total and LDL-cholesterol) and blood pressure, could be extrapolated to predict a 20%-30% reduced 10-year risk of CVD and CVD-related mortality (64).
An increasing number of trials in which participants were fed with berries (65-67) or juices (68) rich in anthocyanins or with purified anthocyanins (69) also reported reduced levels of inflammatory markers and/or improved antioxidant status, decreased LDL-cholesterol, improved insulin sensitivity, and lowered blood pressure (reviewed in 70). In a randomized, double-blind, placebo-controlled study in 150 individuals with hypercholesterolemia, supplementation with a purified anthocyanin mixture (320 mg/day) for 24 weeks reduced circulating markers of inflammation, including C-reactive protein (CRP), interleukin-1β (IL-1β), and soluble vascular adhesion molecule-1 (sVCAM-1) (71). Supplementation of dyslipidemic patients for 12 or 24 weeks with a mixture of 17 anthocyanins improved cholesterol clearance via the HDL-mediated reverse cholesterol transport from extra-hepatic tissues back to the liver and lowered LDL-cholesterol compared to a placebo in two randomized controlled trials (72, 73). However, a 12-week, randomized, double-blind, placebo-controlled study in 52 healthy postmenopausal women found that daily consumption of 500 mg of elderberry anthocyanins (as cyanidin-3-glucoside) had no effect on inflammation markers, markers of vascular health, lipid profile, and glycemia; all of these measures were in normal range of concentrations at baseline (74). Whether exposure to high-dose anthocyanins could lower the risk of CVD in subjects with established CVD risk factors and/or help maintain cardiovascular health in apparently healthy individuals remains to be confirmed.
Endothelial dysfunction
The vascular endothelial cells that line the inner surface of all blood vessels synthesize an enzyme, endothelial nitric oxide synthase (eNOS), whose function is essential to normal vascular physiology. Specifically, eNOS produces nitric oxide (NO), a compound that regulates vascular tone and blood flow by promoting the relaxation (vasodilation) of all types of blood vessels, including arteries (75). NO also regulates vascular homeostasis and protects the integrity of the endothelium by inhibiting vascular inflammation, leukocyte adhesion, platelet adhesion and aggregation, and proliferation of vascular smooth muscle cells (76). In the presence of cardiovascular risk factors (e.g., hypertension, hypercholesterolemia, hyperglycemia), early alterations in the structure and function of the vascular endothelium are associated with the loss of normal NO-mediated endothelium-dependent vasodilation. Endothelial dysfunction results in widespread vasoconstriction and coagulation abnormalities and is considered to be an early step in the development of atherosclerosis. Measures of brachial flow-mediated dilation (FMD), a surrogate marker of endothelial function, have been found to be inversely associated with risk of future cardiovascular events (77).
Preclinical studies have demonstrated the benefits of berry fruits, extracts, or purified anthocyanins on vascular function. Anthocyanin supplementation to diabetic mice was found to improve diabetes-induced vascular dysfunction by promoting NO-mediated endothelium-dependent vasodilation through the upregulation of adipocyte-derived adiponectin (78). Supplementation with purified anthocyanins (320 mg/day for 12 weeks) also increased serum adiponectin concentrations and improved FMD in 58 individuals with type 2 diabetes (78). In a randomized trial of 150 participants with hypercholesterolemia, supplemental anthocyanins increased FMD values by 28.4% compared to 2.2% in the placebo group (79).
Several small-scale, intervention studies have also examined the effect of flavan-3-ol-rich food and beverages, including tea, red wine, purple grape juice, cocoa, and chocolate, on endothelium-dependent vasodilation. A meta-analysis of nine intervention studies in a total of 213 participants estimated that the acute ingestion of 2 to 3 cups of tea (500 mL) — containing about 248 mg of flavonoids in green tea and 415 mg in black tea — significantly increased brachial FMD (see also the article on Tea) (80). Another meta-analysis of 18 randomized controlled studies found that acute (2 h post-ingestion) and chronic (≤18 months) consumption of flavan-3-ol-rich cocoa beverages and chocolate bars significantly increased FMD in participants (81). A small 15-day, cross-over intervention study in hypertensive individuals with endothelial dysfunction found that 100 g/day of flavan-3-ol-rich dark chocolate, but not 90 g/day of flavan-3-ol-free white chocolate, could restore FMD values almost to normal levels (82). Also, using a similar protocol, the authors showed that dark chocolate intake blunted acute endothelial dysfunction-induced by a glucose load challenge in 12 healthy volunteers (83). Other benefits of dark chocolate consumption included reductions in arterial stiffness (measured through pulse wave analysis) and serum concentrations of markers of oxidative stress and vasoconstriction (8-isoprostaglandin F2α and endothelin-1). A randomized controlled trial in overweight and obese participants also reported that the daily consumption of a high-flavan-3-ol cocoa drink (902 mg/day of flavan-3-ols), but not that of a cocoa drink low in flavan-3-ols (38 mg/day), resulted in a sustained increase in FMD during the 12-week study (84). A more recent four-week, randomized, double-blind, cross-over, controlled study in healthy overweight or obese adults found that the consumption of 22 g/day of natural cocoa (in the form of dark chocolate bar and cocoa drink; 814 mg/day of flavan-3-ols) increased arterial diameter and blood flow and lowered peripheral arterial stiffness, but there was no change in FMD (85). Another recent clinical trial found improvements in endothelium-dependent vasodilation in response to acute consumption of one bar (40 g) of dark chocolate (containing 10.8 mg of (+)-catechins and 36 mg of (-)-epicatechins) and daily consumption of two bars (80 g) for up to four weeks in 20 individuals with chronic heart failure (86). Oral administration of pure flavan-3-ol (-)-epicatechin to healthy volunteers showed NO-dependent vasodilatory effects similar to those observed following flavan-3-ol-rich cocoa ingestion (87). Administration of (-)-epicatechin also improved acetylcholine-induced endothelial-dependent vasodilation of thoracic aorta rings from rats with salt-induced hypertension (88).
Endothelial nitric oxide production also inhibits the adhesion and aggregation of platelets, one of the first steps in atherosclerosis and blood clot formation (76). A number of clinical trials that examined the potential for high flavonoid intakes to decrease various measures of platelet function outside of the body (ex vivo) have reported mixed results. A recent systematic review of these intervention studies suggested that consumption of flavan-3-ol-rich cocoa and grape seed extract was generally found to improve platelet function by inhibiting platelet adhesion, activation, and aggregation (89). Interestingly, in a cross-over, controlled study, the acute consumption of a flavan-3-ol-rich cocoa beverage (897 mg of total (-)-EC and procyanidins) exhibited additive anti-platelet effects to aspirin (81 mg) in healthy volunteers (90). In contrast, the results of interventions using apigenin-rich soup, quercetin-rich supplements or onion soups, isoflavone-rich soy protein isolates, black tea, wines, berries, or grape juices have given inconsistent results (reviewed in 89).
Hypertension
A meta-analysis of 20 short-term, randomized controlled trials, including a total of 856 mainly healthy participants, found that consumption of flavan-3-ol-rich dark chocolate and cocoa products significantly reduced systolic blood pressure by 2.77 mm Hg and diastolic blood pressure by 2.20 mm Hg. However, heterogeneity across studies was high, and risk of bias was significant (91). A greater blood pressure-reducing effect was observed in a subanalysis of studies using flavan-3-ol-free rather than flavan-3-ol-low control groups (91). Another meta-analysis of 22 trials (highly heterogeneous) found reductions in diastolic blood pressure (-1.60 mm Hg) and mean arterial pressure (-1.64 mm Hg) with chocolate or cocoa intake but no change in systolic blood pressure (81). Additionally, green tea flavan-3-ols have been shown to lower blood pressure especially in (pre-) hypertensive subjects. A pooled analysis of 13 randomized controlled trials in 1,040 subjects found a 2.05 mm Hg reduction in systolic blood pressure and a 1.71 mm Hg reduction in diastolic blood pressure with green tea consumption for at least three weeks (92). The inhibition of angiotensin-converting enzyme (ACE), a key regulator of arterial blood pressure, may partly explain how flavan-3-ol-rich food and beverages might exert blood pressure-lowering effects (93).
Some intervention trials have also examined the effect of the flavonol quercetin on blood pressure in human subjects. In a randomized, double-blind, cross-over, placebo-controlled trial in 96 participants diagnosed with metabolic disorders, supplementation with 150 mg/day of quercetin aglycone significantly reduced systolic blood pressure by 2.6 mm Hg without affecting diastolic blood pressure and other cardiometabolic markers (94). Similar results were found with 730 mg/day of quercetin in hypertensive individuals (95) and with 500 mg/day of quercetin in women with type 2 diabetes mellitus (96). In a recent six-week, cross-over, randomized, double-blind, placebo-controlled trial, daily ingestion of 162 mg of quercetin decreased 24 h-ambulatory blood pressure — but not systolic blood pressure in the resting state — in hypertensive but not in pre-hypertensive participants (97). There was no change in biomarkers of lipid metabolism, inflammation, oxidative stress, or endothelial function, including total, HDL-, LDL-cholesterol, serum CRP, soluble adhesion molecules, plasma oxidized LDL, urinary 8-isoprostaglandin F2α, serum endothelin-1, serum ACE, and plasma endogenous NOS inhibitor.
Additional trials may help establish whether the blood pressure-lowering effect of some flavonoids could be translated into long-term benefits for cardiovascular health.
Type 2 diabetes mellitus
The association between flavonoid consumption and risk for type 2 diabetes mellitus has been examined in a recent European, multicenter, nested case-control study — the "EPIC-InterAct" project — that included 16,835 diabetes-free participants and 12,043 diabetics. In this study, participants in the highest quintile of total flavonoid intake (>608.1 mg/day) had a 10% lower risk of diabetes than those in the lowest quintile (<178.2 mg/day) (98). Specifically, the risk of diabetes was inversely correlated with the intake of flavan-3-ols (monomers and dimers only) and flavonols (98, 99). Recent meta-analyses of randomized controlled trials have examined the possible health effects of green tea flavan-3-ol monomers (catechins) on glucose metabolism and have provided conflicting results. A meta-analysis of seven trials in pre-diabetic and diabetic patients found no effect of green tea or green tea extracts on fasting plasma glucose, fasting serum insulin, or measures of glycemic control (glycated hemoglobin, HbA1c) and insulin sensitivity (HOMA-IR) (100). Conversely, another meta-analysis of 17 trials in pre-diabetic, diabetic, or overweight/obese subjects found that administration of green tea extracts for 4 to 16 weeks improved fasting plasma glucose and HbA1c level (101). The effect on fasting glucose was observed only with high doses of catechins (≥457 mg/day) and when the confounding effect of caffeine was removed. Finally, a third meta-analysis of 25 trials found that ingestion of green tea extracts for at least two weeks could lower fasting blood glucose in both the presence or absence of caffeine (102).
Dark chocolate is another good source of flavan-3-ols such that the effects of cocoa flavan-3-ols have been examined in individuals at-risk or with established type 2 diabetes. In a 15-day, cross-over, randomized controlled study, the daily consumption of 100 g of dark chocolate bars containing 110.9 mg of (-)-EC and 36.1 mg of (+)-C significantly improved measures of pancreatic β-cell function and insulin sensitivity, along with cardiometabolic markers in glucose-intolerant and hypertensive subjects (103). Daily supplementation with flavonoid-enriched chocolate containing 850 mg of flavan-3-ols and 100 mg of isoflavones for one year significantly improved insulin sensitivity and reduced a predicted risk of coronary heart disease (CHD) at 10 years in 93 postmenopausal women treated for type 2 diabetes (104).
The EPIC-InterAct study did not find any association between dietary anthocyanin intake and risk of diabetes (98, 99). Yet, a 10-fold increase in anthocyanin consumption was correlated with a 15% lower risk of diabetes in the pooled analysis of three large US prospective cohorts (120,003 participants) (105). Of note, this pooled analysis also reported a moderately higher risk of diabetes (+6%) in individuals in the highest versus lowest quintiles of flavone and flavanone intakes. Moreover, the consumption of berries, rich in anthocyanins, has been shown to trigger favorable glycemic responses in type 2 diabetics (reviewed in 70). In recent intervention studies, anthocyanins demonstrated beneficial effects on metabolic abnormalities in patients at-risk or diagnosed with diabetes. In an eight-week, randomized, double-blind, placebo-controlled trial in 38 healthy overweight and obese subjects, the consumption of 2 g/day of grape polyphenols rich in proanthocyanidins and anthocyanins prevented increases in oxidative stress and insulin resistance induced by a six-day, high-fructose challenge (106). Another six-week randomized trial in individuals with diabetes showed that daily supplementation with Cornelian cherry (Cornus mas) extracts containing 600 mg of anthocyanins significantly lowered serum levels of HbA1c and triglycerides and increased serum insulin concentrations (107). The administration of 320 mg/day of anthocyanins for 24 weeks also improved serum lipid and lipoprotein profile, decreased markers of oxidative stress and inflammation, elevated antioxidant capacity, and reduced insulin resistance compared to a placebo in patients with diabetes (108). Further, supplemental anthocyanins up-regulated adiponectin expression and improved nitric oxide-mediated endothelium-dependent vasodilation within 12 weeks of treatment (see also Cardiovascular disease) (78).
These promising findings warrant additional randomized controlled trials to confirm preventive and/or therapeutic benefits of (cocoa) flavan-3-ols and anthocyanins in type 2 diabetes.
Cancer
Although various flavonoids have been found to inhibit the development of chemically-induced cancers in animal models of lung (109), oral (110), esophageal (111), gastric (112), colon (113), skin (114), prostate (115, 116), and mammary cancer (117), observational studies do not provide convincing evidence that high intakes of dietary flavonoids are associated with substantial reductions in human cancer risk (reviewed in 118). A meta-analysis of 13 case-control and 10 prospective cohort studies found little-to-no evidence to support a preventive role of dietary flavonoid intake in gastric and colorectal cancer (119). In addition, a recently published analysis of two large prospective studies (the Health Professionals Follow-up Study [HPFS] and the Nurses’ Health Study [NHS]) — using the most up-to-date flavonoid food composition databases — found no association between the risk of colorectal cancer and intakes of each subclass of flavonoids or flavonoid-rich foods (tea, blueberries, oranges) (120). A meta-analysis of 19 case-control studies and 15 cohort studies found that total flavonoid intake and intakes of specific flavonoid subclasses (i.e., flavonols, flavones, flavanones) were inversely correlated with the risk of smoking-sensitive cancers of the aerodigestive tract (mouth, pharynx, larynx, esophagus, and stomach) in smokers but not in nonsmokers (121). The risk of lung cancer was not significantly associated with high flavonoid intakes (121), although an earlier meta-analysis of eight prospective studies (with substantial heterogeneity across them) suggested a protective role of flavonoids against lung cancer in smokers only (122). Further, a prospective analysis of over 45,000 postmenopausal women from the Multiethnic Cohort Study found a reduced risk of endometrial cancer with the highest intakes of total isoflavones, daidzein, and genistein (123). Additionally, limited evidence from observational studies suggests no relationship between total flavonoid intake and ovarian cancer (124-127). To date, there is little evidence that flavonoid-rich diets might protect against various cancers, but larger prospective cohort studies are needed to address the association.
Hormone-dependent cancers
Because isoflavones are phytoestrogens, it is thought that they may interfere with the synthesis and activity of endogenous hormones, eventually influencing hormone-dependent signaling pathways and protecting against breast and prostate cancers (128). A meta-analysis of 14 observational studies that examined breast cancer incidence in 369,934 women found an overall 11% reduced risk of breast cancer with the highest versus lowest intake of soy isoflavones (129). Subgroup analyses revealed a 24% lower risk of cancer in Asian but not in European or US women, and the risk was 22% lower in postmenopausal but not lower in premenopausal women. In addition to the ethnicity and menopausal status, polymorphisms for hormone receptors (130) and phase I biotransformation enzymes (131) have been found to modify the association between isoflavone intake and breast cancer. Another recent meta-analysis of 12 observational studies (six prospective cohort studies, one nested case-control study, and five case-control studies) investigated the chemopreventive effects of flavonoids (except isoflavones) (132). The results suggested that intakes of flavonols and flavones may also be inversely associated with the risk of breast cancer. Further, a pooled analysis of four case-control studies that stratified by menopausal status showed inverse associations between breast cancer and intakes of flavonols, flavones, or flavan-3-ols in postmenopausal women only. Finally, a meta-analysis of four prospective cohort studies found an overall 16% reduced risk of breast cancer recurrence in women with high versus low isoflavone intakes (129).
A meta-analysis of 13 observational studies also suggested an inverse relationship between prostate cancer risk and consumption of soy products, especially tofu (133). Yet, further analyses supported a protective role of soy food based only on case-control studies, which have inherent flaws such that associations may often be overestimated or underestimated. In a recent 12-month, multicenter, randomized, double-blind, placebo-controlled phase II clinical trial in 158 Japanese men (aged ≥50 years) with elevated risk of prostate cancer, oral isoflavone (60 mg/day) resulted in a significant decrease in prostate cancer incidence in participants aged 65 years and older (134). In this study, no changes were reported in sex hormone concentrations in blood, suggesting that isoflavones may reduce prostate cancer incidence without interfering with hormone-dependent pathways.
Additional investigations will be necessary to determine whether supplementation with specific flavonoids could benefit cancer prevention or treatment.
For more information on flavonoid-rich foods and cancer, see articles on Fruit and Vegetables, Legumes, and Tea.
Cognitive function
Inflammation, oxidative stress, and transition metal accumulation appear to play a role in the pathology of several neurodegenerative diseases, including Parkinson’s disease and Alzheimer’s disease (135). Therefore, the various properties of flavonoids, including their role in protecting vascular health, could have beneficial effects on the brain, possibly in the protection against cerebrovascular disorders, cognitive impairments, and subsequent stroke and dementias. Dietary flavonoids and/or their metabolites have been shown to cross the blood-brain barrier (54) and exert preventive effects towards cognitive impairments in animal models of normal and pathological aging (53).
The cross-sectional data analysis of 2,031 participants (ages, 70-74 years) from the Hordaland Health Study in Norway indicated that, when compared to non-consumers, consumers of flavonoid-rich chocolate, tea, and wine had better global cognitive function, assessed by a battery of six cognitive tests (136). The risk of poor performance in all tests was estimated to be 60 to 74% lower in consumers of all three flavonoid-rich foods compared to non-consumers. An early prospective cohort study in 1,367 older French men and women (aged ≥65 years; free from dementia at baseline) found that those with the lowest flavonoid intakes (<11.5 mg/day) had a 50% higher risk of developing dementia over the next five years than those with higher intakes (137). In addition, those with higher dietary flavonoid intakes at baseline experienced significantly less age-related cognitive decline over a 10-year period than those with the lowest flavonoid intakes (138).
The effect of cocoa flavan-3-ols have been investigated in an eight-week, randomized, double-blind trial — the Cognitive, Cocoa, and Aging (CoCoA) study — in 90 individuals (ages, 64-82 years) with mild cognitive impairments (MCI); participants were given dairy-based cocoa drinks with either high (993 mg/day) or low (48 mg/day) levels of flavan-3-ols (139). The daily consumption of the cocoa drink high in flavan-3-ols improved some, but not all, measures of cognitive process speed and flexibility and verbal fluency compared to baseline test scores and scores following low flavan-3-ol drink consumption. A composite test score reflecting overall cognitive performance was found to be significantly greater in those given cocoa drinks high rather than low in flavan-3-ols. The study also reported reductions in cardiovascular risk markers (i.e., systolic and diastolic blood pressure, total and LDL-cholesterol, insulin resistance), and these changes were proposed to partly contribute to ameliorate cognitive performance in those who consumed the flavan-3-ol-rich cocoa drink (139). The data could be replicated in cognitively healthy older people (ages, 61-85 years), suggesting that cocoa flavan-3-ols might enhance some aspects of cognitive function during healthy aging (140). Interestingly, a two-week, randomized, double-blind, controlled study has reported an increase in blood flow velocity in the middle cerebral artery of 21 healthy subjects (mean age, 72 years) following the daily intake of a flavan-3-ol-rich cocoa drink (900 mg/day of flavan-3-ols) (141). Because cerebral blood flow is correlated with cognitive function in humans, these preliminary data suggest that cocoa flavan-3-ol consumption could exert a protective effect against dementia (54).
Yet, in other randomized controlled trials (142-144), the lack of an effect of cocoa flavan-3-ols on blood pressure, cerebral blood flow, mental fatigue, and cognitive performance in healthy young and old adults suggested that benefits may only be seen in very demanding cognitive exercises (145).
Some randomized controlled studies also reported improvements in measures of cognitive function in healthy and cognitively impaired subjects with other flavonoid subclasses, including anthocyanins (146), flavanones (147, 148), and isoflavones (149, 150). Although some flavonoids and flavonoid-rich foods may enhance cognitive function in the aging brain, it is not yet clear whether their consumption could lower the risk of cognitive impairments and dementia in humans.
For more detailed information on flavan-3-ol-rich tea and cognitive function, see the article on Tea.
Sources
Food sources
Recent data analyses of the National Health and Nutrition Examination Survey (NHANES) estimated flavonoid intakes in US adults (aged ≥19 years) average between 200 and 250 mg/day, with 80% being flavan-3-ols, 8% for flavonols, 6% for flavanones, 5% for anthocyanidins, and ≤1% for isoflavones and flavones (151, 152). The main dietary sources of flavonoids include tea, citrus fruit, citrus fruit juices, berries, red wine, apples, and legumes. Individual flavonoid intakes may vary considerably depending on whether tea, red wine, soy products, or fruit and vegetables are commonly consumed (reviewed in 2). Information on the flavonoid content of some flavonoid-rich foods is presented in Tables 2-8. These values should be considered approximate since a number of factors may affect the flavonoid content of foods, including agricultural practices, environmental conditions, ripening, storage, and food processing. For additional information about the flavonoid content of food, the USDA provides databases for the content of selected foods in flavonoids (60) and proanthocyanidins (58). For more information on the isoflavone content of soy foods, see the article on Soy Isoflavones or the USDA database for the isoflavone content of selected foods (59).
Supplements
Anthocyanins
Bilberry, elderberry, black currant, blueberry, red grape, and mixed berry extracts that are rich in anthocyanins are available as dietary supplements without a prescription in the US. The anthocyanin content of these products may vary considerably. Standardized extracts that list the amount of anthocyanins per dose are available.
Flavan-3-ols
Numerous tea extracts are available in the US as dietary supplements and may be labeled as tea catechins or tea polyphenols. Green tea extracts are the most commonly marketed, but black and oolong tea extracts are also available. Green tea extracts generally have higher levels of catechins (flavan-3-ol monomers), while black tea extracts are richer in theaflavins and thearubigins (tea flavan-3-ol dimers and polymers, respectively). Oolong tea extracts fall somewhere in between green and black tea extracts with respect to their flavan-3-ol content. Some tea extracts contain caffeine, while others are decaffeinated. Flavan-3-ol and caffeine content vary considerably among different products, so it is important to check the label or consult the manufacturer to determine the amounts of flavan-3-ols and caffeine that would be consumed daily with each supplement (for more information on tea flavan-3-ols, see the article on Tea).
Flavanones
Citrus bioflavonoid supplements may contain glycosides of hesperetin (hesperidin), naringenin (naringin), and eriodictyol (eriocitrin). Hesperidin is also available in hesperidin-complex supplements, with daily doses from 500 mg to 2 g (153).
Flavones
The peels and tissues of citrus fruit (e.g., oranges, tangerines, and clementines) are rich in polymethoxylated flavones: tangeretin, nobiletin, and sinensetin (2). Although dietary intakes of these naturally occurring flavones are generally low, they are often present in citrus bioflavonoid complex supplements. Several dietary supplements may also contain various amounts of baicalein (aglycone) and/or baicalin (glycoside). Some tea preparations may also include baicalein-7-glucuronide (153).
Flavonols
The flavonol aglycone, quercetin, and its glycoside rutin are available as dietary supplements without a prescription in the US. Other names for rutin include rutoside, quercetin-3-rutinoside, and sophorin (153). Citrus bioflavonoid supplements may also contain quercetin or rutin.
Isoflavones
A 50-mg soy isoflavone supplement usually includes glycosides of the isoflavones: genistein (genistin; 25 mg), daidzein (daidzin; 19 mg), and glycitein (glycitin; about 6 mg). Smaller amounts of daidzein, genistein, and formononetin are also found in biochanin A-containing supplements (derived from red clover) (153).
Safety
Adverse effects
No adverse effects have been associated with high dietary intakes of flavonoids from plant-based food. This lack of adverse effects may be explained by the relatively low bioavailability and rapid metabolism and elimination of most flavonoids.
Quercetin
Oral supplementation with quercetin glycosides at doses ranging between 3 mg/day-1,000 mg/day for up to three months has not resulted in significant adverse effects in clinical studies (reviewed in 154). A randomized, placebo-controlled study in 30 patients with chronic prostatitis reported one case of headache and another of tingling of the extremities associated with supplemental quercetin (1,000 mg/day for one month); both issues resolved after the study ended (155). In a phase I clinical trial in cancer patients unresponsive to standard treatments, administration of quercetin via intravenous infusion resulted in symptoms of nausea, vomiting, sweating, flushing, and dyspnea (difficulty breathing) at doses ≥10.5 mg/kg body weight (~756 mg of quercetin for a 70 kg individual) (156). Higher doses up to 51.3 mg/kg body weight (~3,591 mg of quercetin) were associated with renal (kidney) toxicity, yet without evidence of nephritis, infection, or obstructive uropathy (reviewed in 154).
Cocoa flavan-3-ols
In a recent randomized, double-blind, controlled study in healthy adults, the daily intake of 2 g of cocoa flavan-3-ols for 12 weeks was found to be well tolerated with no adverse side effects (157).
Tea extracts
In clinical trials employing caffeinated green tea extracts, cancer patients who took 6 g/day in three to six divided doses reported mild-to-moderate gastrointestinal side effects, including nausea, vomiting, abdominal pain, and diarrhea (158, 159). Central nervous system symptoms, including agitation, restlessness, insomnia, tremors, dizziness, and confusion, have also been reported. In one case, confusion was severe enough to require hospitalization (158). In a systematic review published in 2008, the US Pharmacopeia (USP) Dietary Supplement Information Expert Committee identified 34 adverse event reports implicating the use of green tea extract products (containing 25%-97% of polyphenols) as the likely cause of liver damage (hepatotoxicity) in humans (160). In a four-week clinical trial that assessed the safety of decaffeinated green tea extracts (800 mg/day of EGCG) in healthy individuals, a few of the participants reported mild nausea, stomach upset, dizziness, or muscle pain (161). In the Minnesota Green Tea Trial (MGTT), 1,075 postmenopausal women were randomized to receive green tea extracts (1,315±116 mg/day of catechins; the equivalent of four 8-ounce mugs of brewed decaffeinated green tea) or a placebo for one year. The total number of adverse events and the number of serious adverse events were not different between the treatment and placebo groups (162). However, the use of green tea extracts was directly associated with abnormally high liver enzyme levels in 7 out of the 12 women who experienced serious adverse events. Also, the incidence of nausea was twice as high in the green tea arm as in the placebo group (162).
Pregnancy and lactation
The safety of flavonoid supplements in pregnancy and lactation has not been established (153).
Drug interactions
Inhibition of ABC drug transporters
ATP-binding cassette (ABC) drug transporters, including P-glycoprotein, multidrug resistance protein (MRP), and breast cancer-resistant protein (BCRP), function as ATP-dependent efflux pumps that actively regulate the excretion of a number of drugs limiting their systemic bioavailability (8). ABC transporters are found throughout the body, yet they are especially important in organs with a barrier function like the intestines, the blood-brain barrier, blood-testis barrier, and the placenta, as well as in liver and kidneys (163). There is some evidence that the consumption of grapefruit juice inhibits the activity of P-glycoprotein (164). Genistein, biochanin A, quercetin, naringenin, hesperetin, green tea flavan-3-ol (-)-CG, (-)-ECG, and (-)-EGCG, and others have been found to inhibit the efflux activity of P-glycoprotein in cultured cells and in animal models (163). Thus, very high or supplemental intakes of these flavonoids could potentially increase the toxicity of drugs that are substrates of P-glycoprotein, e.g., digoxin, antihypertensive agents, antiarrhythmic agents, chemotherapeutic (anticancer) agents, antifungal agents, HIV protease inhibitors, immunosuppressive agents, H2 receptor antagonists, some antibiotics, and others (reviewed in 165).
Many anthocyanins and anthocyanidins, as well as some flavones (apigenin, chrysin), isoflavones (biochanin A, genistein), flavonols (kaempferol), and flavanones (naringenin), have been identified as inhibitors of BRCP-mediated transport, theoretically affecting drugs like anticancer agents (mitoxantrone, topotecan, thyrosine kinase inhibitors), antibiotics (fluoroquinolones), β-blockers (prazosin), and antiarthritics (sulfasalazine). Finally, flavonols (quercetin, kaempferol, myricetin), flavanones (naringenin), flavones (apigenin, robinetin), and isoflavones (genistein) have been reported to inhibit MRP, potentially affecting MRP-mediated transport of many anticancer drugs, e.g., vincristin, etoposide, cisplatin, irinotecan, methotrexate, camptothecin, anthracyclines, vinca alkaloids (reviewed in 163).
Anticoagulant and antiplatelet drugs
High intakes of flavonoids from purple grape juice (500 mL/day) and dark chocolate (235 mg/day of flavan-3-ols) have been found to inhibit platelet aggregation in ex vivo assays (166-169). Theoretically, high intakes of flavonoids (e.g., from supplements) could increase the risk of bleeding when taken with anticoagulant drugs, such as warfarin (Coumadin), heparin, dalteparin (Fragmin), enoxaparin (Lovenox), and antiplatelet drugs, such as clopidogrel (Plavix), dipyridamole (Persantine), non-steroidal anti-inflammatory drugs (NSAIDs: diclofenac, ibuprofen, naproxen), aspirin, and others (170).
Inhibition of CYP 3A4 by flavonoid-rich grapefruit
Cytochrome P450 (CYP) enzymes are phase I biotransformation enzymes involved in the metabolism of a broad range of compounds, from endogenous molecules to therapeutic agents. The most abundant CYP isoform in the liver and intestines is cytochrome P450 3A4 (CYP3A4); the CYP3A family catalyzes the metabolism of about one-half of all marketed drugs in the US and Canada (171). One grapefruit or as little as 200 mL (7 fluid ounces) of grapefruit juice have been found to irreversibly inhibit intestinal CYP3A4 (164). The most potent inhibitors of CYP3A4 in grapefruit are thought to be furanocoumarins, particularly dihydroxybergamottin, rather than flavonoids. All forms of the fruit — freshly squeezed juice, frozen concentrate, or whole fruit — can potentially affect the activity of CYP3A4. Some varieties of other citrus fruit (Seville oranges, limes, and pomelos) that contain furanocoumarins can also interfere with CYP3A4 activity.
Specifically, the inhibition of intestinal CYP3A4 by grapefruit consumption is known or predicted to increase the bioavailability and the risk of toxicity of more than 85 drugs. Because drugs with very low bioavailability are more likely to be toxic when CYP3A4 activity is inhibited, they are associated with a higher risk of overdose with grapefruit compared to drugs with high bioavailability. Some of the drugs with low bioavailability include, but are not limited to, anticancer drugs (everolimus); anti-infective agents halofantrine, maraviroc); statins (atorvastatin, lovastatin, and simvastatin); cardioactive drugs (amiodarone, clopidogrel, dronedarone, eplenorone, ticagrelor); HIV protease inhibitors (saquinavir), immunosuppressants (cyclosporine, sirolimus, tacrolimus, everolimus); antihistamines (terfenadine); gastrointestinal agents (domperidone); central nervous system agents (buspirone, dextromethorphan, oral ketamine, lurasidone, quetiapine, selective serotonin reuptake inhibitors [sertraline]); and urinary tract agents (darifenacin) (reviewed in 171). Because of the potential for adverse drug interactions, some clinicians recommend that people taking medications with low bioavailability (i.e., undergoing extensive metabolism by CYP3A4) avoid consuming grapefruit and grapefruit juice altogether during the treatment period (171).
Nutrient interactions
Iron
Flavonoids can bind nonheme iron, inhibiting its intestinal absorption (172, 173). Nonheme iron is the principal form of iron in plant foods, dairy products, and iron supplements. The consumption of one cup of tea or cocoa with a meal has been found to decrease the absorption of nonheme iron in that meal by about 70% (174, 175). Flavonoids can also inhibit intestinal heme iron absorption (176). Interestingly, ascorbic acid greatly enhances the absorption of iron (see the article on Iron) and is able to counteract the inhibitory effect of flavonoids on nonheme and heme iron absorption (173, 176, 177). To maximize iron absorption from a meal or iron supplements, flavonoid-rich food and beverages and flavonoid supplements should not be consumed at the same time.
Authors and Reviewers
Originally written in 2005 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in June 2008 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in November 2015 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in February 2016 by:
Alan Crozier, Ph.D.
Professor, Department of Nutrition
University of California, Davis
Copyright 2005-2024 Linus Pauling Institute
References
1. Kumar S, Pandey AK. Chemistry and biological activities of flavonoids: an overview. ScientificWorldJournal. 2013; 2013:162750. (PubMed)
2. Manach C, Scalbert A, Morand C, Remesy C, Jimenez L. Polyphenols: food sources and bioavailability. Am J Clin Nutr. 2004;79(5):727-747. (PubMed)
3. Xiao J, Kai G, Yamamoto K, Chen X. Advance in dietary polyphenols as α-glucosidases inhibitors: a review on structure-activity relationship aspect. Crit Rev Food Sci Nutr. 2013;53(8):818-836. (PubMed)
4. Rothwell JA, Urpi-Sarda M, Boto-Ordonez M, et al. Systematic analysis of the polyphenol metabolome using the Phenol-Explorer database. Mol Nutr Food Res. 2016;60(1):203-211. (PubMed)
5. Lotito SB, Zhang WJ, Yang CS, Crozier A, Frei B. Metabolic conversion of dietary flavonoids alters their anti-inflammatory and antioxidant properties. Free Radic Biol Med. 2011;51(2):454-463. (PubMed)
6. Williamson G. Common features in the pathways of absorption and metabolism of flavonoids. In: Meskin MS, R. BW, Davies AJ, Lewis DS, Randolph RK, eds. Phytochemicals: Mechanisms of Action. Boca Raton: CRC Press; 2004:21-33.
7. Nemeth K, Plumb GW, Berrin JG, et al. Deglycosylation by small intestinal epithelial cell β-glucosidases is a critical step in the absorption and metabolism of dietary flavonoid glycosides in humans. Eur J Nutr. 2003;42(1):29-42. (PubMed)
8. Gonzales GB, Smagghe G, Grootaert C, Zotti M, Raes K, Van Camp J. Flavonoid interactions during digestion, absorption, distribution and metabolism: a sequential structure-activity/property relationship-based approach in the study of bioavailability and bioactivity. Drug Metab Rev. 2015;47(2):175-190. (PubMed)
9. Monagas M, Urpi-Sarda M, Sanchez-Patan F, et al. Insights into the metabolism and microbial biotransformation of dietary flavan-3-ols and the bioactivity of their metabolites. Food Funct. 2010;1(3):233-253. (PubMed)
10. Bordenave N, Hamaker BR, Ferruzzi MG. Nature and consequences of non-covalent interactions between flavonoids and macronutrients in foods. Food Funct. 2014;5(1):18-34. (PubMed)
11. Zhang H, Yu D, Sun J, et al. Interaction of plant phenols with food macronutrients: characterisation and nutritional-physiological consequences. Nutr Res Rev. 2014;27(1):1-15. (PubMed)
12. Xiao J, Mao F, Yang F, Zhao Y, Zhang C, Yamamoto K. Interaction of dietary polyphenols with bovine milk proteins: molecular structure-affinity relationship and influencing bioactivity aspects. Mol Nutr Food Res. 2011;55(11):1637-1645. (PubMed)
13. Lorenz M, Jochmann N, von Krosigk A, et al. Addition of milk prevents vascular protective effects of tea. Eur Heart J. 2007;28(2):219-223. (PubMed)
14. Roowi S, Stalmach A, Mullen W, Lean ME, Edwards CA, Crozier A. Green tea flavan-3-ols: colonic degradation and urinary excretion of catabolites by humans. J Agric Food Chem. 2010;58(2):1296-1304. (PubMed)
15. Setchell KD, Brown NM, Lydeking-Olsen E. The clinical importance of the metabolite equol-a clue to the effectiveness of soy and its isoflavones. J Nutr. 2002;132(12):3577-3584. (PubMed)
16. Yuan JP, Wang JH, Liu X. Metabolism of dietary soy isoflavones to equol by human intestinal microflora--implications for health. Mol Nutr Food Res. 2007;51(7):765-781. (PubMed)
17. Marin L, Miguelez EM, Villar CJ, Lombo F. Bioavailability of dietary polyphenols and gut microbiota metabolism: antimicrobial properties. Biomed Res Int. 2015;2015:905215. (PubMed)
18. Inoue-Choi M, Yuan JM, Yang CS, et al. Genetic Association Between the COMT Genotype and Urinary Levels of Tea Polyphenols and Their Metabolites among Daily Green Tea Drinkers. Int J Mol Epidemiol Genet. 2010;1(2):114-123. (PubMed)
19. Wu AH, Tseng CC, Van Den Berg D, Yu MC. Tea intake, COMT genotype, and breast cancer in Asian-American women. Cancer Res. 2003;63(21):7526-7529. (PubMed)
20. Jiang W, Hu M. Mutual interactions between flavonoids and enzymatic and transporter elements responsible for flavonoid disposition via phase II metabolic pathways. RSC Adv. 2012;2(21):7948-7963. (PubMed)
21. Xiao J, Kai G. A review of dietary polyphenol-plasma protein interactions: characterization, influence on the bioactivity, and structure-affinity relationship. Crit Rev Food Sci Nutr. 2012;52(1):85-101. (PubMed)
22. Manach C, Williamson G, Morand C, Scalbert A, Remesy C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am J Clin Nutr. 2005;81(1 Suppl):230S-242S. (PubMed)
23. Kroon PA, Clifford MN, Crozier A, et al. How should we assess the effects of exposure to dietary polyphenols in vitro? Am J Clin Nutr. 2004;80(1):15-21. (PubMed)
24. Heijnen CG, Haenen GR, van Acker FA, van der Vijgh WJ, Bast A. Flavonoids as peroxynitrite scavengers: the role of the hydroxyl groups. Toxicol In Vitro. 2001;15(1):3-6. (PubMed)
25. Chun OK, Kim DO, Lee CY. Superoxide radical scavenging activity of the major polyphenols in fresh plums. J Agric Food Chem. 2003;51(27):8067-8072. (PubMed)
26. Frei B, Higdon JV. Antioxidant activity of tea polyphenols in vivo: evidence from animal studies. J Nutr. 2003;133(10):3275S-3284S. (PubMed)
27. Williams RJ, Spencer JP, Rice-Evans C. Flavonoids: antioxidants or signalling molecules? Free Radic Biol Med. 2004;36(7):838-849. (PubMed)
28. Lotito SB, Frei B. Consumption of flavonoid-rich foods and increased plasma antioxidant capacity in humans: cause, consequence, or epiphenomenon? Free Radic Biol Med. 2006;41(12):1727-1746. (PubMed)
29. Mira L, Fernandez MT, Santos M, Rocha R, Florencio MH, Jennings KR. Interactions of flavonoids with iron and copper ions: a mechanism for their antioxidant activity. Free Radic Res. 2002;36(11):1199-1208. (PubMed)
30. Cheng IF, Breen K. On the ability of four flavonoids, baicilein, luteolin, naringenin, and quercetin, to suppress the Fenton reaction of the iron-ATP complex. Biometals. 2000;13(1):77-83. (PubMed)
31. Spencer JP, Rice-Evans C, Williams RJ. Modulation of pro-survival Akt/protein kinase B and ERK1/2 signaling cascades by quercetin and its in vivo metabolites underlie their action on neuronal viability. J Biol Chem. 2003;278(37):34783-34793. (PubMed)
32. Spencer JP, Schroeter H, Crossthwaithe AJ, Kuhnle G, Williams RJ, Rice-Evans C. Contrasting influences of glucuronidation and O-methylation of epicatechin on hydrogen peroxide-induced cell death in neurons and fibroblasts. Free Radic Biol Med. 2001;31(9):1139-1146. (PubMed)
33. Hou Z, Lambert JD, Chin KV, Yang CS. Effects of tea polyphenols on signal transduction pathways related to cancer chemoprevention. Mutat Res. 2004;555(1-2):3-19. (PubMed)
34. Lambert JD, Yang CS. Mechanisms of cancer prevention by tea constituents. J Nutr. 2003;133(10):3262S-3267S. (PubMed)
35. Espley RV, Butts CA, Laing WA, et al. Dietary flavonoids from modified apple reduce inflammation markers and modulate gut microbiota in mice. J Nutr. 2014;144(2):146-154. (PubMed)
36. Kim MC, Kim SJ, Kim DS, et al. Vanillic acid inhibits inflammatory mediators by suppressing NF-kappaB in lipopolysaccharide-stimulated mouse peritoneal macrophages. Immunopharmacol Immunotoxicol. 2011;33(3):525-532. (PubMed)
37. Lee SG, Kim B, Yang Y, et al. Berry anthocyanins suppress the expression and secretion of proinflammatory mediators in macrophages by inhibiting nuclear translocation of NF-kappaB independent of NRF2-mediated mechanism. J Nutr Biochem. 2014;25(4):404-411. (PubMed)
38. Mauray A, Felgines C, Morand C, Mazur A, Scalbert A, Milenkovic D. Bilberry anthocyanin-rich extract alters expression of genes related to atherosclerosis development in aorta of apo E-deficient mice. Nutr Metab Cardiovasc Dis. 2012;22(1):72-80. (PubMed)
39. Wang D, Wei X, Yan X, Jin T, Ling W. Protocatechuic acid, a metabolite of anthocyanins, inhibits monocyte adhesion and reduces atherosclerosis in apolipoprotein E-deficient mice. J Agric Food Chem. 2010;58(24):12722-12728. (PubMed)
40. Edirisinghe I, Banaszewski K, Cappozzo J, McCarthy D, Burton-Freeman BM. Effect of black currant anthocyanins on the activation of endothelial nitric oxide synthase (eNOS) in vitro in human endothelial cells. J Agric Food Chem. 2011;59(16):8616-8624. (PubMed)
41. Hidalgo M, Martin-Santamaria S, Recio I, et al. Potential anti-inflammatory, anti-adhesive, anti/estrogenic, and angiotensin-converting enzyme inhibitory activities of anthocyanins and their gut metabolites. Genes Nutr. 2012;7(2):295-306. (PubMed)
42. Chen XQ, Wang XB, Guan RF, et al. Blood anticoagulation and antiplatelet activity of green tea (-)-epigallocatechin (EGC) in mice. Food Funct. 2013;4(10):1521-1525. (PubMed)
43. Ahmad A, Khan RM, Alkharfy KM. Effects of selected bioactive natural products on the vascular endothelium. J Cardiovasc Pharmacol. 2013;62(2):111-121. (PubMed)
44. Hanhineva K, Torronen R, Bondia-Pons I, et al. Impact of dietary polyphenols on carbohydrate metabolism. Int J Mol Sci. 2010;11(4):1365-1402. (PubMed)
45. Babu PV, Liu D, Gilbert ER. Recent advances in understanding the anti-diabetic actions of dietary flavonoids. J Nutr Biochem. 2013;24(11):1777-1789. (PubMed)
46. Delgado ME, Haza AI, Arranz N, Garcia A, Morales P. Dietary polyphenols protect against N-nitrosamines and benzo(a)pyrene-induced DNA damage (strand breaks and oxidized purines/pyrimidines) in HepG2 human hepatoma cells. Eur J Nutr. 2008;47(8):479-490. (PubMed)
47. Erba D, Casiraghi MC, Martinez-Conesa C, Goi G, Massaccesi L. Isoflavone supplementation reduces DNA oxidative damage and increases O-β-N-acetyl-D-glucosaminidase activity in healthy women. Nutr Res. 2012;32(4):233-240. (PubMed)
48. Moon YJ, Wang X, Morris ME. Dietary flavonoids: effects on xenobiotic and carcinogen metabolism. Toxicol In Vitro. 2006;20(2):187-210. (PubMed)
49. Schwarz D, Kisselev P, Roots I. CYP1A1 genotype-selective inhibition of benzo[a]pyrene activation by quercetin. Eur J Cancer. 2005;41(1):151-158. (PubMed)
50. Suh Y, Afaq F, Johnson JJ, Mukhtar H. A plant flavonoid fisetin induces apoptosis in colon cancer cells by inhibition of COX2 and Wnt/EGFR/NF-kappaB-signaling pathways. Carcinogenesis. 2009;30(2):300-307. (PubMed)
51. Ravishankar D, Watson KA, Boateng SY, Green RJ, Greco F, Osborn HM. Exploring quercetin and luteolin derivatives as antiangiogenic agents. Eur J Med Chem. 2015;97:259-274. (PubMed)
52. Santos BL, Oliveira MN, Coelho PC, et al. Flavonoids suppress human glioblastoma cell growth by inhibiting cell metabolism, migration, and by regulating extracellular matrix proteins and metalloproteinases expression. Chem Biol Interact. 2015;242:123-138. (PubMed)
53. Sokolov AN, Pavlova MA, Klosterhalfen S, Enck P. Chocolate and the brain: neurobiological impact of cocoa flavanols on cognition and behavior. Neurosci Biobehav Rev. 2013;37(10 Pt 2):2445-2453. (PubMed)
54. Vauzour D, Vafeiadou K, Rodriguez-Mateos A, Rendeiro C, Spencer JP. The neuroprotective potential of flavonoids: a multiplicity of effects. Genes Nutr. 2008;3(3-4):115-126. (PubMed)
55. Wang X, Ouyang YY, Liu J, Zhao G. Flavonoid intake and risk of CVD: a systematic review and meta-analysis of prospective cohort studies. Br J Nutr. 2014;111(1):1-11. (PubMed)
56. Wang ZM, Zhao D, Nie ZL, et al. Flavonol intake and stroke risk: a meta-analysis of cohort studies. Nutrition. 2014;30(5):518-523. (PubMed)
57. Jacques PF, Cassidy A, Rogers G, Peterson JJ, Dwyer JT. Dietary flavonoid intakes and CVD incidence in the Framingham Offspring Cohort. Br J Nutr. 2015;114(9):1496-1503. (PubMed)
58. US Department of Agriculture. USDA Database for the Proanthocyanidin Content of Selected Foods. August, 2004. Available at: http://www.ars.usda.gov/SP2UserFiles/Place/80400525/Data/PA/PA.pdf. Accessed 8/25/15.
59. US Department of Agriculture. USDA Database for the Isoflavone Content of Selected Foods, release 2.0. September 2008. Available at: http://www.ars.usda.gov/SP2UserFiles/Place/80400525/Data/isoflav/Isoflav_R2.pdf. Accessed 8/25/15.
60. US Department of Agriculture. USDA Database for the Flavonoid Content of Selected Foods, release 3.1. May 2014. Available at: http://www.ars.usda.gov/SP2UserFiles/Place/80400525/Data/Flav/Flav_R03-1.pdf. Accessed 8/25/15.
61. Vogiatzoglou A, Mulligan AA, Bhaniani A, et al. Associations between flavan-3-ol intake and CVD risk in the Norfolk cohort of the European Prospective Investigation into Cancer (EPIC-Norfolk). Free Radic Biol Med. 2015;84:1-10. (PubMed)
62. Cassidy A, Rogers G, Peterson JJ, Dwyer JT, Lin H, Jacques PF. Higher dietary anthocyanin and flavonol intakes are associated with anti-inflammatory effects in a population of US adults. Am J Clin Nutr. 2015;102(1):172-181. (PubMed)
63. Grassi D, Desideri G, Ferri C. Protective effects of dark chocolate on endothelial function and diabetes. Curr Opin Clin Nutr Metab Care. 2013;16(6):662-668. (PubMed)
64. Sansone R, Rodriguez-Mateos A, Heuel J, et al. Cocoa flavanol intake improves endothelial function and Framingham Risk Score in healthy men and women: a randomised, controlled, double-masked trial: the Flaviola Health Study. Br J Nutr. 2015;114(8):1246-1255. (PubMed)
65. Basu A, Fu DX, Wilkinson M, et al. Strawberries decrease atherosclerotic markers in subjects with metabolic syndrome. Nutr Res. 2010;30(7):462-469. (PubMed)
66. Kelley DS, Rasooly R, Jacob RA, Kader AA, Mackey BE. Consumption of Bing sweet cherries lowers circulating concentrations of inflammation markers in healthy men and women. J Nutr. 2006;136(4):981-986. (PubMed)
67. Moazen S, Amani R, Homayouni Rad A, Shahbazian H, Ahmadi K, Taha Jalali M. Effects of freeze-dried strawberry supplementation on metabolic biomarkers of atherosclerosis in subjects with type 2 diabetes: a randomized double-blind controlled trial. Ann Nutr Metab. 2013;63(3):256-264. (PubMed)
68. Edirisinghe I, Banaszewski K, Cappozzo J, et al. Strawberry anthocyanin and its association with postprandial inflammation and insulin. Br J Nutr. 2011;106(6):913-922. (PubMed)
69. Karlsen A, Retterstol L, Laake P, et al. Anthocyanins inhibit nuclear factor-kappaB activation in monocytes and reduce plasma concentrations of pro-inflammatory mediators in healthy adults. J Nutr. 2007;137(8):1951-1954. (PubMed)
70. Basu A, Lyons TJ. Strawberries, blueberries, and cranberries in the metabolic syndrome: clinical perspectives. J Agric Food Chem. 2012;60(23):5687-5692. (PubMed)
71. Zhu Y, Ling W, Guo H, et al. Anti-inflammatory effect of purified dietary anthocyanin in adults with hypercholesterolemia: a randomized controlled trial. Nutr Metab Cardiovasc Dis. 2013;23(9):843-849. (PubMed)
72. Qin Y, Xia M, Ma J, et al. Anthocyanin supplementation improves serum LDL- and HDL-cholesterol concentrations associated with the inhibition of cholesteryl ester transfer protein in dyslipidemic subjects. Am J Clin Nutr. 2009;90(3):485-492. (PubMed)
73. Zhu Y, Huang X, Zhang Y, et al. Anthocyanin supplementation improves HDL-associated paraoxonase 1 activity and enhances cholesterol efflux capacity in subjects with hypercholesterolemia. J Clin Endocrinol Metab. 2014;99(2):561-569. (PubMed)
74. Curtis PJ, Kroon PA, Hollands WJ, et al. Cardiovascular disease risk biomarkers and liver and kidney function are not altered in postmenopausal women after ingesting an elderberry extract rich in anthocyanins for 12 weeks. J Nutr. 2009;139(12):2266-2271. (PubMed)
75. Grassi D, Desideri G, Di Giosia P, et al. Tea, flavonoids, and cardiovascular health: endothelial protection. Am J Clin Nutr. 2013;98(6 Suppl):1660S-1666S. (PubMed)
76. Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33(7):829-837, 837a-837d. (PubMed)
77. Ras RT, Streppel MT, Draijer R, Zock PL. Flow-mediated dilation and cardiovascular risk prediction: a systematic review with meta-analysis. Int J Cardiol. 2013;168(1):344-351. (PubMed)
78. Liu Y, Li D, Zhang Y, Sun R, Xia M. Anthocyanin increases adiponectin secretion and protects against diabetes-related endothelial dysfunction. Am J Physiol Endocrinol Metab. 2014;306(8):E975-988. (PubMed)
79. Zhu Y, Xia M, Yang Y, et al. Purified anthocyanin supplementation improves endothelial function via NO-cGMP activation in hypercholesterolemic individuals. Clin Chem. 2011;57(11):1524-1533. (PubMed)
80. Ras RT, Zock PL, Draijer R. Tea consumption enhances endothelial-dependent vasodilation; a meta-analysis. PLoS One. 2011;6(3):e16974. (PubMed)
81. Hooper L, Kay C, Abdelhamid A, et al. Effects of chocolate, cocoa, and flavan-3-ols on cardiovascular health: a systematic review and meta-analysis of randomized trials. Am J Clin Nutr. 2012;95(3):740-751. (PubMed)
82. Grassi D, Necozione S, Lippi C, et al. Cocoa reduces blood pressure and insulin resistance and improves endothelium-dependent vasodilation in hypertensives. Hypertension. 2005;46(2):398-405. (PubMed)
83. Grassi D, Desideri G, Necozione S, et al. Protective effects of flavanol-rich dark chocolate on endothelial function and wave reflection during acute hyperglycemia. Hypertension. 2012;60(3):827-832. (PubMed)
84. Davison K, Coates AM, Buckley JD, Howe PR. Effect of cocoa flavanols and exercise on cardiometabolic risk factors in overweight and obese subjects. Int J Obes (Lond). 2008;32(8):1289-1296. (PubMed)
85. West SG, McIntyre MD, Piotrowski MJ, et al. Effects of dark chocolate and cocoa consumption on endothelial function and arterial stiffness in overweight adults. Br J Nutr. 2014;111(4):653-661. (PubMed)
86. Flammer AJ, Sudano I, Wolfrum M, et al. Cardiovascular effects of flavanol-rich chocolate in patients with heart failure. Eur Heart J. 2012;33(17):2172-2180. (PubMed)
87. Schroeter H, Heiss C, Balzer J, et al. (-)-Epicatechin mediates beneficial effects of flavanol-rich cocoa on vascular function in humans. Proc Natl Acad Sci U S A. 2006;103(4):1024-1029. (PubMed)
88. Gomez-Guzman M, Jimenez R, Sanchez M, et al. Epicatechin lowers blood pressure, restores endothelial function, and decreases oxidative stress and endothelin-1 and NADPH oxidase activity in DOCA-salt hypertension. Free Radic Biol Med. 2012;52(1):70-79. (PubMed)
89. Bachmair EM, Ostertag LM, Zhang X, de Roos B. Dietary manipulation of platelet function. Pharmacol Ther. 2014;144(2):97-113. (PubMed)
90. Pearson DA, Paglieroni TG, Rein D, et al. The effects of flavanol-rich cocoa and aspirin on ex vivo platelet function. Thromb Res. 2002;106(4-5):191-197. (PubMed)
91. Ried K, Sullivan TR, Fakler P, Frank OR, Stocks NP. Effect of cocoa on blood pressure. Cochrane Database Syst Rev. 2012;8:CD008893. (PubMed)
92. Khalesi S, Sun J, Buys N, Jamshidi A, Nikbakht-Nasrabadi E, Khosravi-Boroujeni H. Green tea catechins and blood pressure: a systematic review and meta-analysis of randomised controlled trials. Eur J Nutr. 2014;53(6):1299-1311. (PubMed)
93. Guerrero L, Castillo J, Quinones M, et al. Inhibition of angiotensin-converting enzyme activity by flavonoids: structure-activity relationship studies. PLoS One. 2012;7(11):e49493. (PubMed)
94. Egert S, Bosy-Westphal A, Seiberl J, et al. Quercetin reduces systolic blood pressure and plasma oxidised low-density lipoprotein concentrations in overweight subjects with a high-cardiovascular disease risk phenotype: a double-blinded, placebo-controlled cross-over study. Br J Nutr. 2009;102(7):1065-1074. (PubMed)
95. Edwards RL, Lyon T, Litwin SE, Rabovsky A, Symons JD, Jalili T. Quercetin reduces blood pressure in hypertensive subjects. J Nutr. 2007;137(11):2405-2411. (PubMed)
96. Zahedi M, Ghiasvand R, Feizi A, Asgari G, Darvish L. Does Quercetin Improve Cardiovascular Risk factors and Inflammatory Biomarkers in Women with Type 2 Diabetes: A Double-blind Randomized Controlled Clinical Trial. Int J Prev Med. 2013;4(7):777-785. (PubMed)
97. Brull V, Burak C, Stoffel-Wagner B, et al. Effects of a quercetin-rich onion skin extract on 24 h ambulatory blood pressure and endothelial function in overweight-to-obese patients with (pre-)hypertension: a randomised double-blinded placebo-controlled cross-over trial. Br J Nutr. 2015;114(8):1263-1277. (PubMed)
98. Zamora-Ros R, Forouhi NG, Sharp SJ, et al. The association between dietary flavonoid and lignan intakes and incident type 2 diabetes in European populations: the EPIC-InterAct study. Diabetes Care. 2013;36(12):3961-3970. (PubMed)
99. Zamora-Ros R, Forouhi NG, Sharp SJ, et al. Dietary intakes of individual flavanols and flavonols are inversely associated with incident type 2 diabetes in European populations. J Nutr. 2014;144(3):335-343. (PubMed)
100. Wang X, Tian J, Jiang J, et al. Effects of green tea or green tea extract on insulin sensitivity and glycaemic control in populations at risk of type 2 diabetes mellitus: a systematic review and meta-analysis of randomised controlled trials. J Hum Nutr Diet. 2014;27(5):501-512. (PubMed)
101. Liu K, Zhou R, Wang B, et al. Effect of green tea on glucose control and insulin sensitivity: a meta-analysis of 17 randomized controlled trials. Am J Clin Nutr. 2013;98(2):340-348. (PubMed)
102. Zheng XX, Xu YL, Li SH, Hui R, Wu YJ, Huang XH. Effects of green tea catechins with or without caffeine on glycemic control in adults: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2013;97(4):750-762. (PubMed)
103. Grassi D, Desideri G, Necozione S, et al. Blood pressure is reduced and insulin sensitivity increased in glucose-intolerant, hypertensive subjects after 15 days of consuming high-polyphenol dark chocolate. J Nutr. 2008;138(9):1671-1676. (PubMed)
104. Curtis PJ, Sampson M, Potter J, Dhatariya K, Kroon PA, Cassidy A. Chronic ingestion of flavan-3-ols and isoflavones improves insulin sensitivity and lipoprotein status and attenuates estimated 10-year CVD risk in medicated postmenopausal women with type 2 diabetes: a 1-year, double-blind, randomized, controlled trial. Diabetes Care. 2012;35(2):226-232. (PubMed)
105. Wedick NM, Pan A, Cassidy A, et al. Dietary flavonoid intakes and risk of type 2 diabetes in US men and women. Am J Clin Nutr. 2012;95(4):925-933. (PubMed)
106. Hokayem M, Blond E, Vidal H, et al. Grape polyphenols prevent fructose-induced oxidative stress and insulin resistance in first-degree relatives of type 2 diabetic patients. Diabetes Care. 2013;36(6):1454-1461. (PubMed)
107. Soltani R, Gorji A, Asgary S, Sarrafzadegan N, Siavash M. Evaluation of the Effects of Cornus mas L. Fruit Extract on Glycemic Control and Insulin Level in Type 2 Diabetic Adult Patients: A Randomized Double-Blind Placebo-Controlled Clinical Trial. Evid Based Complement Alternat Med. 2015;2015:740954. (PubMed)
108. Li D, Zhang Y, Liu Y, Sun R, Xia M. Purified anthocyanin supplementation reduces dyslipidemia, enhances antioxidant capacity, and prevents insulin resistance in diabetic patients. J Nutr. 2015;145(4):742-748. (PubMed)
109. Yang CS, Yang GY, Landau JM, Kim S, Liao J. Tea and tea polyphenols inhibit cell hyperproliferation, lung tumorigenesis, and tumor progression. Exp Lung Res. 1998;24(4):629-639. (PubMed)
110. Balasubramanian S, Govindasamy S. Inhibitory effect of dietary flavonol quercetin on 7,12-dimethylbenz[a]anthracene-induced hamster buccal pouch carcinogenesis. Carcinogenesis. 1996;17(4):877-879. (PubMed)
111. Li ZG, Shimada Y, Sato F, et al. Inhibitory effects of epigallocatechin-3-gallate on N-nitrosomethylbenzylamine-induced esophageal tumorigenesis in F344 rats. Int J Oncol. 2002;21(6):1275-1283. (PubMed)
112. Yamane T, Nakatani H, Kikuoka N, et al. Inhibitory effects and toxicity of green tea polyphenols for gastrointestinal carcinogenesis. Cancer. 1996;77(8 Suppl):1662-1667. (PubMed)
113. Guo JY, Li X, Browning JD, Jr., et al. Dietary soy isoflavones and estrone protect ovariectomized ERαKO and wild-type mice from carcinogen-induced colon cancer. J Nutr. 2004;134(1):179-182. (PubMed)
114. Huang MT, Xie JG, Wang ZY, et al. Effects of tea, decaffeinated tea, and caffeine on UVB light-induced complete carcinogenesis in SKH-1 mice: demonstration of caffeine as a biologically important constituent of tea. Cancer Res. 1997;57(13):2623-2629. (PubMed)
115. Gupta S, Hastak K, Ahmad N, Lewin JS, Mukhtar H. Inhibition of prostate carcinogenesis in TRAMP mice by oral infusion of green tea polyphenols. Proc Natl Acad Sci U S A. 2001;98(18):10350-10355. (PubMed)
116. Haddad AQ, Venkateswaran V, Viswanathan L, Teahan SJ, Fleshner NE, Klotz LH. Novel antiproliferative flavonoids induce cell cycle arrest in human prostate cancer cell lines. Prostate Cancer Prostatic Dis. 2006;9(1):68-76. (PubMed)
117. Yamagishi M, Natsume M, Osakabe N, et al. Effects of cacao liquor proanthocyanidins on PhIP-induced mutagenesis in vitro, and in vivo mammary and pancreatic tumorigenesis in female Sprague-Dawley rats. Cancer Lett. 2002;185(2):123-130. (PubMed)
118. Romagnolo DF, Selmin OI. Flavonoids and cancer prevention: a review of the evidence. J Nutr Gerontol Geriatr. 2012;31(3):206-238. (PubMed)
119. Woo HD, Kim J. Dietary flavonoid intake and risk of stomach and colorectal cancer. World J Gastroenterol. 2013;19(7):1011-1019. (PubMed)
120. Nimptsch K, Zhang X, Cassidy A, et al. Habitual intake of flavonoid subclasses and risk of colorectal cancer in 2 large prospective cohorts. Am J Clin Nutr. 2016;103(1):184-191. (PubMed)
121. Woo HD, Kim J. Dietary flavonoid intake and smoking-related cancer risk: a meta-analysis. PLoS One. 2013;8(9):e75604. (PubMed)
122. Tang NP, Zhou B, Wang B, Yu RB, Ma J. Flavonoids intake and risk of lung cancer: a meta-analysis. Jpn J Clin Oncol. 2009;39(6):352-359. (PubMed)
123. Ollberding NJ, Lim U, Wilkens LR, et al. Legume, soy, tofu, and isoflavone intake and endometrial cancer risk in postmenopausal women in the multiethnic cohort study. J Natl Cancer Inst. 2012;104(1):67-76. (PubMed)
124. Bandera EV, King M, Chandran U, Paddock LE, Rodriguez-Rodriguez L, Olson SH. Phytoestrogen consumption from foods and supplements and epithelial ovarian cancer risk: a population-based case control study. BMC Womens Health. 2011;11:40. (PubMed)
125. Cassidy A, Huang T, Rice MS, Rimm EB, Tworoger SS. Intake of dietary flavonoids and risk of epithelial ovarian cancer. Am J Clin Nutr. 2014;100(5):1344-1351. (PubMed)
126. Gates MA, Vitonis AF, Tworoger SS, et al. Flavonoid intake and ovarian cancer risk in a population-based case-control study. Int J Cancer. 2009;124(8):1918-1925. (PubMed)
127. Rossi M, Negri E, Lagiou P, et al. Flavonoids and ovarian cancer risk: A case-control study in Italy. Int J Cancer. 2008;123(4):895-898. (PubMed)
128. Ko KP. Isoflavones: chemistry, analysis, functions and effects on health and cancer. Asian Pac J Cancer Prev. 2014;15(17):7001-7010. (PubMed)
129. Dong JY, Qin LQ. Soy isoflavones consumption and risk of breast cancer incidence or recurrence: a meta-analysis of prospective studies. Breast Cancer Res Treat. 2011;125(2):315-323. (PubMed)
130. Iwasaki M, Hamada GS, Nishimoto IN, et al. Isoflavone, polymorphisms in estrogen receptor genes and breast cancer risk in case-control studies in Japanese, Japanese Brazilians and non-Japanese Brazilians. Cancer Sci. 2009;100(5):927-933. (PubMed)
131. Wang Q, Li H, Tao P, et al. Soy isoflavones, CYP1A1, CYP1B1, and COMT polymorphisms, and breast cancer: a case-control study in southwestern China. DNA Cell Biol. 2011;30(8):585-595. (PubMed)
132. Hui C, Qi X, Qianyong Z, Xiaoli P, Jundong Z, Mantian M. Flavonoids, flavonoid subclasses and breast cancer risk: a meta-analysis of epidemiologic studies. PLoS One. 2013;8(1):e54318. (PubMed)
133. Hwang YW, Kim SY, Jee SH, Kim YN, Nam CM. Soy food consumption and risk of prostate cancer: a meta-analysis of observational studies. Nutr Cancer. 2009;61(5):598-606. (PubMed)
134. Miyanaga N, Akaza H, Hinotsu S, et al. Prostate cancer chemoprevention study: an investigative randomized control study using purified isoflavones in men with rising prostate-specific antigen. Cancer Sci. 2012;103(1):125-130. (PubMed)
135. Ramassamy C. Emerging role of polyphenolic compounds in the treatment of neurodegenerative diseases: a review of their intracellular targets. Eur J Pharmacol. 2006;545(1):51-64. (PubMed)
136. Nurk E, Refsum H, Drevon CA, et al. Intake of flavonoid-rich wine, tea, and chocolate by elderly men and women is associated with better cognitive test performance. J Nutr. 2009;139(1):120-127. (PubMed)
137. Commenges D, Scotet V, Renaud S, Jacqmin-Gadda H, Barberger-Gateau P, Dartigues JF. Intake of flavonoids and risk of dementia. Eur J Epidemiol. 2000;16(4):357-363. (PubMed)
138. Letenneur L, Proust-Lima C, Le Gouge A, Dartigues JF, Barberger-Gateau P. Flavonoid intake and cognitive decline over a 10-year period. Am J Epidemiol. 2007;165(12):1364-1371. (PubMed)
139. Desideri G, Kwik-Uribe C, Grassi D, et al. Benefits in cognitive function, blood pressure, and insulin resistance through cocoa flavanol consumption in elderly subjects with mild cognitive impairment: the Cocoa, Cognition, and Aging (CoCoA) study. Hypertension. 2012;60(3):794-801. (PubMed)
140. Mastroiacovo D, Kwik-Uribe C, Grassi D, et al. Cocoa flavanol consumption improves cognitive function, blood pressure control, and metabolic profile in elderly subjects: the Cocoa, Cognition, and Aging (CoCoA) Study--a randomized controlled trial. Am J Clin Nutr. 2015;101(3):538-548. (PubMed)
141. Sorond FA, Lipsitz LA, Hollenberg NK, Fisher ND. Cerebral blood flow response to flavanol-rich cocoa in healthy elderly humans. Neuropsychiatr Dis Treat. 2008;4(2):433-440. (PubMed)
142. Crews WD, Jr., Harrison DW, Wright JW. A double-blind, placebo-controlled, randomized trial of the effects of dark chocolate and cocoa on variables associated with neuropsychological functioning and cardiovascular health: clinical findings from a sample of healthy, cognitively intact older adults. Am J Clin Nutr. 2008;87(4):872-880. (PubMed)
143. Massee LA, Ried K, Pase M, et al. The acute and sub-chronic effects of cocoa flavanols on mood, cognitive and cardiovascular health in young healthy adults: a randomized, controlled trial. Front Pharmacol. 2015;6:93. (PubMed)
144. Pase MP, Scholey AB, Pipingas A, et al. Cocoa polyphenols enhance positive mood states but not cognitive performance: a randomized, placebo-controlled trial. J Psychopharmacol. 2013;27(5):451-458. (PubMed)
145. Scholey AB, French SJ, Morris PJ, Kennedy DO, Milne AL, Haskell CF. Consumption of cocoa flavanols results in acute improvements in mood and cognitive performance during sustained mental effort. J Psychopharmacol. 2010;24(10):1505-1514. (PubMed)
146. Kent K, Charlton K, Roodenrys S, et al. Consumption of anthocyanin-rich cherry juice for 12 weeks improves memory and cognition in older adults with mild-to-moderate dementia. Eur J Nutr. 2015; Oct 19. [Epub ahead of print]. (PubMed)
147. Alharbi MH, Lamport DJ, Dodd GF, et al. Flavonoid-rich orange juice is associated with acute improvements in cognitive function in healthy middle-aged males. Eur J Nutr. 2015; Aug 18. [Epub ahead of print]. (PubMed)
148. Kean RJ, Lamport DJ, Dodd GF, et al. Chronic consumption of flavanone-rich orange juice is associated with cognitive benefits: an 8-wk, randomized, double-blind, placebo-controlled trial in healthy older adults. Am J Clin Nutr. 2015;101(3):506-514. (PubMed)
149. Casini ML, Marelli G, Papaleo E, Ferrari A, D'Ambrosio F, Unfer V. Psychological assessment of the effects of treatment with phytoestrogens on postmenopausal women: a randomized, double-blind, crossover, placebo-controlled study. Fertil Steril. 2006;85(4):972-978. (PubMed)
150. Kritz-Silverstein D, Von Muhlen D, Barrett-Connor E, Bressel MA. Isoflavones and cognitive function in older women: the SOy and Postmenopausal Health In Aging (SOPHIA) Study. Menopause. 2003;10(3):196-202. (PubMed)
151. Kim K, Vance TM, Chun OK. Estimated intake and major food sources of flavonoids among US adults: changes between 1999-2002 and 2007-2010 in NHANES. Eur J Nutr. 2015; May 31. [Epub ahead of print]. (PubMed)
152. Sebastian RS, Wilkinson Enns C, Goldman JD, et al. A New Database Facilitates Characterization of Flavonoid Intake, Sources, and Positive Associations with Diet Quality among US Adults. J Nutr. 2015;145(6):1239-1248. (PubMed)
153. Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. 2nd ed: Thomson Reuters; 2008.
154. Harwood M, Danielewska-Nikiel B, Borzelleca JF, Flamm GW, Williams GM, Lines TC. A critical review of the data related to the safety of quercetin and lack of evidence of in vivo toxicity, including lack of genotoxic/carcinogenic properties. Food Chem Toxicol. 2007;45(11):2179-2205. (PubMed)
155. Shoskes DA, Zeitlin SI, Shahed A, Rajfer J. Quercetin in men with category III chronic prostatitis: a preliminary prospective, double-blind, placebo-controlled trial. Urology. 1999;54(6):960-963. (PubMed)
156. Ferry DR, Smith A, Malkhandi J, et al. Phase I clinical trial of the flavonoid quercetin: pharmacokinetics and evidence for in vivo tyrosine kinase inhibition. Clin Cancer Res. 1996;2(4):659-668. (PubMed)
157. Ottaviani JI, Balz M, Kimball J, et al. Safety and efficacy of cocoa flavanol intake in healthy adults: a randomized, controlled, double-masked trial. Am J Clin Nutr. 2015;102(6):1425-1435. (PubMed)
158. Jatoi A, Ellison N, Burch PA, et al. A phase II trial of green tea in the treatment of patients with androgen independent metastatic prostate carcinoma. Cancer. 2003;97(6):1442-1446. (PubMed)
159. Pisters KM, Newman RA, Coldman B, et al. Phase I trial of oral green tea extract in adult patients with solid tumors. J Clin Oncol. 2001;19(6):1830-1838. (PubMed)
160. Sarma DN, Barrett ML, Chavez ML, et al. Safety of green tea extracts : a systematic review by the US Pharmacopeia. Drug Saf. 2008;31(6):469-484. (PubMed)
161. Chow HH, Cai Y, Hakim IA, et al. Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon E in healthy individuals. Clin Cancer Res. 2003;9(9):3312-3319. (PubMed)
162. Dostal AM, Samavat H, Bedell S, et al. The safety of green tea extract supplementation in postmenopausal women at risk for breast cancer: results of the Minnesota Green Tea Trial. Food Chem Toxicol. 2015;83:26-35. (PubMed)
163. Li Y, Paxton JW. The effects of flavonoids on the ABC transporters: consequences for the pharmacokinetics of substrate drugs. Expert Opin Drug Metab Toxicol. 2013;9(3):267-285. (PubMed)
164. Bailey DG, Dresser GK. Interactions between grapefruit juice and cardiovascular drugs. Am J Cardiovasc Drugs. 2004;4(5):281-297. (PubMed)
165. Marzolini C, Paus E, Buclin T, Kim RB. Polymorphisms in human MDR1 (P-glycoprotein): recent advances and clinical relevance. Clin Pharmacol Ther. 2004;75(1):13-33. (PubMed)
166. Freedman JE, Parker C, 3rd, Li L, et al. Select flavonoids and whole juice from purple grapes inhibit platelet function and enhance nitric oxide release. Circulation. 2001;103(23):2792-2798. (PubMed)
167. Keevil JG, Osman HE, Reed JD, Folts JD. Grape juice, but not orange juice or grapefruit juice, inhibits human platelet aggregation. J Nutr. 2000;130(1):53-56. (PubMed)
168. Polagruto JA, Schramm DD, Wang-Polagruto JF, Lee L, Keen CL. Effects of flavonoid-rich beverages on prostacyclin synthesis in humans and human aortic endothelial cells: association with ex vivo platelet function. J Med Food. 2003;6(4):301-308. (PubMed)
169. Murphy KJ, Chronopoulos AK, Singh I, et al. Dietary flavanols and procyanidin oligomers from cocoa (Theobroma cacao) inhibit platelet function. Am J Clin Nutr. 2003;77(6):1466-1473. (PubMed)
170. Natural Medicines. Hesperidin Professional Monograph; 2015.
171. Bailey DG, Dresser G, Arnold JM. Grapefruit-medication interactions: forbidden fruit or avoidable consequences? CMAJ. 2013;185(4):309-316. (PubMed)
172. Kim EY, Ham SK, Shigenaga MK, Han O. Bioactive dietary polyphenolic compounds reduce nonheme iron transport across human intestinal cell monolayers. J Nutr. 2008;138(9):1647-1651. (PubMed)
173. Thankachan P, Walczyk T, Muthayya S, Kurpad AV, Hurrell RF. Iron absorption in young Indian women: the interaction of iron status with the influence of tea and ascorbic acid. Am J Clin Nutr. 2008;87(4):881-886. (PubMed)
174. Hurrell RF, Reddy M, Cook JD. Inhibition of non-haem iron absorption in man by polyphenolic-containing beverages. Br J Nutr. 1999;81(4):289-295. (PubMed)
175. Zijp IM, Korver O, Tijburg LB. Effect of tea and other dietary factors on iron absorption. Crit Rev Food Sci Nutr. 2000;40(5):371-398. (PubMed)
176. Ma Q, Kim EY, Lindsay EA, Han O. Bioactive dietary polyphenols inhibit heme iron absorption in a dose-dependent manner in human intestinal Caco-2 cells. J Food Sci. 2011;76(5):H143-150. (PubMed)
177. Kim EY, Ham SK, Bradke D, Ma Q, Han O. Ascorbic acid offsets the inhibitory effect of bioactive dietary polyphenolic compounds on transepithelial iron transport in Caco-2 intestinal cells. J Nutr. 2011;141(5):828-834. (PubMed)
Garlic
You should be automatically redirected to the current page, if not click here.
Indole-3-Carbinol
Contents
Summary
- Indole-3-carbinol (I3C) is derived from the breakdown of glucobrassicin, a compound found in cruciferous vegetables. (More information)
- In the stomach, I3C molecules undergo acid-catalyzed condensation that generates a number of biologically active I3C oligomers, such as 3,3'-diindolylmethane (DIM) and 5,11-dihydroindolo-[3,2-b]carbazole (ICZ). (More information)
- I3C and DIM have been found to modulate the expression and activity of biotransformation enzymes that are involved in the metabolism and elimination of many biologically active compounds, including steroid hormones, drugs, carcinogens, and toxins. (More information)
- Preclinical studies suggested that anti-estrogenic activities of I3C and DIM might help reduce the risk of hormone-dependent cancers. Although supplementation with I3C and DIM could alter urinary estrogen metabolite profiles in women, the effects of I3C and DIM on breast cancer risk are not known. (More information)
- Preclinical studies showed that I3C and I3C oligomers could affect multiple signaling pathways that are dysregulated in cancer cells, such as those controlling cell proliferation, apoptosis, migration, invasion, and angiogenesis. (More information)
- Limited evidence from preliminary trials suggested that I3C supplementation may help treat conditions related to human papilloma virus (HPV) infection, such as cervical/vulvar intraepithelial neoplasias and recurrent respiratory papillomatosis. However, randomized controlled trials are needed to determine whether I3C supplementation is beneficial. (More information)
- The timing of I3C exposure in animal models of chemically-induced cancers seems to determine whether I3C inhibits or promotes the development of tumors. Some experts have cautioned against the widespread use of I3C and DIM supplements for cancer prevention in humans until their potential risks and benefits are better understood. (More information)
Introduction
Some observational studies have reported significant associations between high intakes of cruciferous vegetables and lower risk of several types of cancer (1). Cruciferous vegetables differ from other classes of vegetables in that they are rich sources of sulfur-containing compounds known as glucosinolates (for detailed information, see the article on Cruciferous Vegetables) (2). The potential health benefits of consuming cruciferous vegetables are attributed to compounds derived from the enzymatic hydrolysis (breakdown) of glucosinolates. Among these compounds is indole-3-carbinol (I3C), a compound derived from the degradation of an indole glucosinolate commonly known as glucobrassicin (Figure 1).

Metabolism and Bioavailability
A number of commonly consumed cruciferous vegetables, including broccoli, Brussels sprouts, and cabbage, are good sources of glucobrassicin — the glucosinolate precursor of I3C (see Food sources).
Myrosinase (β-thioglucosidase), an enzyme that catalyzes the hydrolysis of glucosinolates, is physically separated from glucosinolates in intact plant cells (3). When raw cruciferous vegetables are chopped or chewed, plant cells are damaged such that glucobrassicin is exposed to myrosinase. The hydrolysis of glucobrassicin initially produces a glucose molecule and the unstable aglycone, thiohydroximate-O-sulfonate. The spontaneous release of a sulfate ion results in the formation of another unstable intermediate form, 3-indolylmethylisothiocyanate (4). This compound easily splits into thiocyanate ion and I3C (Figure 1). In the acidic environment of the stomach, I3C molecules can combine with each other to form a complex mixture of polycyclic aromatic compounds, known collectively as acid condensation products (Figure 2) (5). Some of the most prominent acid condensation products include 3,3'-diindolylmethane (DIM), 5,11-dihydroindolo-[3,2-b]carbazole (ICZ), and a cyclic triindole (CT) (Figure 2). The biological activities of individual acid condensation products may differ from those of I3C (see Biological Activities).
When cruciferous vegetables are cooked, plant myrosinase is inactivated thus the hydrolysis of glucosinolates is prevented. Intact glucosinolates then transit to the colon and are metabolized by human intestinal bacteria. The generation of I3C from glucobrassicin may still occur to a lesser degree in the large intestine, due to the myrosinase activity of colonic bacteria (4). Thus, when cruciferous vegetables are cooked, I3C can still form in the colon, but I3C-derived acid condensation products are less likely to form in the more alkaline environment of the intestine.
No I3C could be detected in plasma following oral administration of single doses of I3C, ranging from 200 to 1,200 mg, to healthy women at high risk for breast cancer (6). However, DIM was detected and peaked in plasma around two hours after I3C ingestion, at concentrations from <100 nanograms per milliliter (ng/mL) with oral doses of 400 to 600 mg of I3C up to 500 ng/mL-600 ng/mL with oral doses of 1,000 to 1,200 mg of I3C. All DIM disappeared from the blood within 24 hours (6).
Formulation strategies, such as the encapsulation of I3C and DIM into nanoparticles or liposomes (7-9), are being developed with the aim of increasing the bioavailability and evaluating the safety and efficacy of these compounds in humans.
![Figure 2. Condensation Derivatives of Indole-3-Carbinol: 3,3'-diindolylmethane (DIM), 5,6,11,12,17,18-hexahydrocyclononal [1,2-b:4,5-b':7,8-b"]triindole (CT), and 5,11-dihydroindolo-[3,2-b]carbazole (ICZ)](/sites/lpi.oregonstate.edu/files/indole-3-carbinol-figure-2-1200px.png)
Biological Activities
Effects on biotransformation enzymes
Biotransformation enzymes play major roles in the metabolism and elimination of many biologically active compounds, including physiologic regulators (e.g., estrogens), drugs, and environmental chemicals (xenobiotics; e.g., carcinogens, toxins). In general, phase I metabolizing enzymes, including the cytochrome P450 (CYP) family, catalyze reactions that increase the reactivity of hydrophobic (fat-soluble) compounds, which prepares them for reactions catalyzed by phase II detoxifying enzymes. Reactions catalyzed by phase II enzymes usually increase water solubility and promote the elimination of these compounds (10).
Aryl hydrocarbon receptor (AhR) pathway
I3C and some I3C condensation products can bind to a protein in the cytoplasm of cells called the aryl hydrocarbon receptor (AhR) (Figure 3) (11-13). In fact, ICZ is one of the most potent ligands for the AhR known with an affinity approaching that of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). I3C acid condensation products, as well as indoles and their acid condensation products formed from tryptophan metabolism, appear to be important endogenous ligands for the AhR (13). Binding allows AhR to enter the nucleus where it forms a complex with the AhR nuclear translocator (Arnt) protein. This AhR/Arnt complex binds to specific DNA sequences, known as xenobiotic response elements (XRE), in the regulatory regions (promoters) of target genes, especially those involved in xenobiotic metabolism (14). The promoters of genes coding for a number of CYP enzymes and several phase II enzymes contain XREs. Microarray gene expression profiling of I3C- or DIM-treated human prostate cancer cells showed that both compounds upregulated the phase I enzyme, CYP1A1, and the phase II enzymes, glutathione S-transferase theta-1 (GST q1) and aldo-keto reductase (15). Another study in human prostate cancer cells demonstrated that the removal of AhR abolished I3C- or DIM-induced CYP1A1 mRNA expression (16). The expression of CYP1A1 and CYP1A2 was also upregulated in human primary liver cells challenged with DIM (17). Further, I3C and DIM have been found to interfere with CYP activities involved in estrogen metabolism (see Anti-estrogenic activities).
Increasing the activity of biotransformation enzymes is generally considered a beneficial effect because the elimination of potential carcinogens or toxins is enhanced. However, there is a potential for adverse effects because some procarcinogens require biotransformation by phase I enzymes to become active carcinogens (18).
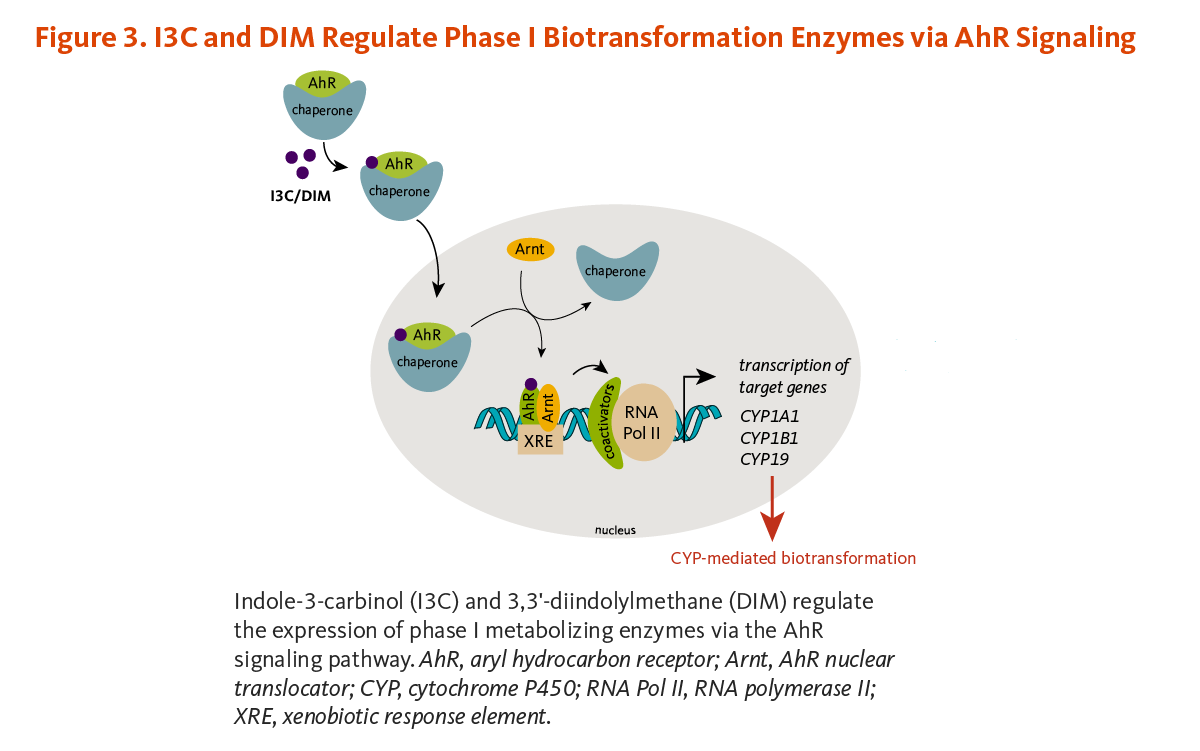
Nuclear factor E2-related factor 2-dependent pathway
I3C and DIM have been shown to induce the expression of phase II detoxifying and antioxidant enzymes via the activation of the nuclear factor E2-related factor 2 (Nrf2)-dependent pathway.
Briefly, Nrf2 is a transcription factor that is bound to the protein Kelch-like ECH-associated protein 1 (Keap1) in the cytosol (Figure 4). Keap1 responds to oxidative stress signals or chemical inducers by freeing Nrf2. Upon release, Nrf2 translocates to the nucleus and binds to the antioxidant response element (ARE) located in the promoters of genes coding for antioxidant/detoxifying enzymes and scavengers. Nrf2/ARE-dependent genes code for numerous mediators of the antioxidant response, including glutathione S-transferases (GSTs), thioredoxin, NAD(P)H quinone oxidoreductase 1 (NQO-1), and heme oxygenase 1 (HO-1) (19). DIM induced Nrf2/ARE-dependent upregulation of HO-1, and I3C stimulated NQO-1 and GST (µ2 isoform) expression in liver cancer cells (20). In addition, the transcription of Nrf2 coding gene, which was abnormally repressed through promoter DNA hypermethylation, was enhanced in mouse prostate cancer cells treated with DIM. DIM subsequently restored the expression of the Nrf2-target genes, NQO1 and GSTµ1 (21). DIM also reversed Nrf2 gene silencing in transgenic mouse prostate cancer tissues, inducing Nrf2 expression, and subsequently, NQO1 expression (Figure 4). This was accompanied by the suppression of proliferation and the induction of apoptosis in prostate cancer tissues (21). Similar observations have been reported with I3C (22).

Anti-estrogenic activities
Endogenous estrogens are steroid hormones synthesized by humans and other mammals.
Inhibition of estrogen synthesis
In breast tissue, CYP19 (aromatase) catalyzes the final steps in the conversion of androgens (testosterone or androstenedione) to estrogens (17β-estradiol or estrone, respectively). Both I3C and DIM have been found to downregulate the expression of CYP19 in non-tumorigenic and tumorigenic estrogen-responsive (ER+) breast cells, whereas CYP19 expression was increased in I3C/DIM-treated tumorigenic estrogen-independent (ER-) breast cells (23).
Inhibition of estrogen metabolic activation
Prolonged exposure to estrogens is thought to play a role in cancer development through CYP-mediated generation of estrogen reactive metabolites that can damage DNA (24, 25).
Phase I metabolizing enzymes, CYP1A1, CYP1A2, and CYP1B1, have been involved in the oxidative metabolism of estrogens. 17β-estradiol can be converted to 2-hydroxyestradiol (2HE2) and 4-hydroxyestradiol (4HE2) by CYP1A1/2 and CYP1B1, respectively. 2HE2 and 4HE2 are further metabolized to 2- and 4-metoxymetabolites by the phase II enzyme, catechol-O-methyltransferase (COMT) (25). 2HE2 is a noncarcinogenic agent with weaker estrogenic potential than 17β-estradiol, while 4-HE2 can be converted to free radicals that can form DNA adducts and promote carcinogenesis (26, 27). In different breast cancer cell lines, I3C and DIM have been shown to upregulate the expression of CYP1A1, CYP1A2, and CYP1B1 at the transcript (mRNA) level but not at the protein level (28).
Additionally, the endogenous estrogens 17β-estradiol and estrone can be irreversibly metabolized to 16a-hydroxyestrone (16HE1) (29). In contrast to 2-hydroxyestrone (2HE1), 16HE1 is highly estrogenic and has been found to stimulate the proliferation of several estrogen-sensitive cancer cell lines (30, 31). It has been hypothesized that shifting the metabolism of 17β-estradiol toward 2HE1, and away from 16HE1, could decrease the risk of estrogen-sensitive cancers, such as breast cancer (32). In controlled clinical trials, oral supplementation with I3C or DIM has consistently increased urinary 2HE1 concentrations or urinary 2HE1:16HE1 ratios in women (33-39). However, large case-control and prospective cohort studies have failed to find significant associations between urinary 2HE1:16HE1 ratios and risk of breast and endometrial cancer (40-43).
Inhibition of estrogen signaling
Endogenous estrogens, including 17β-estradiol, exert their estrogenic effects by binding to specific nuclear receptors called estrogen receptors (ERs). Within the nucleus, estrogen-activated ERs can bind to specific DNA sequences, known as estrogen response elements (EREs), in the promoters of estrogen-responsive genes. ERE-bound estrogen-ER complexes act as transcription factors by recruiting coactivator proteins and chromatin remodeling factors to promoters, thereby triggering the transcription of target genes (44). There are two major ER subtypes, ERα and ERβ, coded by two separate genes ESR1 and ESR2, respectively. ERα is the main driver of the proliferative effect of estrogens, while the expression of ERβ has been inversely associated with mammary gland tumorigenesis (45). Elevated ERα levels promote cellular proliferation in the breast and uterus, possibly increasing the risk of developing estrogen-sensitive cancers (46).
Inhibition of estrogen-dependent cell proliferation
In estrogen-sensitive human breast cancer cells challenged with 17β-estradiol, I3C has been found to inhibit the transcription of estrogen-responsive genes without binding to either ERβ or ERα (47, 48). In fact, the binding of I3C to AhR was shown to trigger the proteasome-dependent degradation of ERα (49). I3C-induced loss of ERα resulted in the downregulation of ERα-responsive gene products like the transcription factor GATA3. Since GATA3 regulates the transcription of the ERα coding gene ESR1, I3C prevented the synthesis of new ERα transcripts and proteins, eventually abolishing the ERα signaling pathway. The disruption of the GATA3/ERα cross-regulatory loop by I3C ultimately halted ERα-dependent cell proliferation (49). Acid condensation products of I3C that bind and activate AhR may also inhibit the transcription of estrogen-responsive genes by competing for co-activators or increasing ERα degradation (14, 50). I3C treatment also affected the expression of other ERα-responsive genes, including those coding for insulin-like growth factor-1 receptor (IGFR1) and insulin receptor substrate-1 (IRS-1), involved in cell proliferation and deregulated in breast cancer (Figure 5) (51).

Modulation of cell-signaling pathways
I3C and condensation derivatives have been found to affect multiple signaling pathways that are often deregulated in cancer cells. Below are some examples illustrating how I3C, DIM, or ICZ may influence cell proliferation, apoptosis, migration, invasion, angiogenesis, and immunity by targeting specific signaling pathways (23).
Induction of cell cycle arrest and apoptosis
Once a cell divides, it passes through a sequence of stages — collectively known as the cell cycle — before it divides again. Following DNA damage, the cell cycle can be transiently arrested at damage checkpoints, which allows for DNA repair or activation of pathways leading to cell death (apoptosis) if the damage is irreparable (52). Defective cell cycle regulation may result in the propagation of mutations that contribute to the development of cancer. In addition, unlike normal cells, cancerous cells lose their ability to respond to death signals that initiate apoptosis.
I3C-induced downregulation of the phosphatidylinositol 3-kinase (PI3K)/serine-threonine kinase (Akt) cell survival signaling pathway in mice with nasopharyngeal carcinoma resulted in inhibition of cell proliferation and induction of apoptosis (53). Inactivation of the Wnt/β-catenin signaling pathway in DIM-treated colon cancer cells decreased the expression of downstream targets, c-myc and cyclin D1, that promote cell proliferation and survival (54). In prostate cancer cells, I3C opposed the anti-apoptotic effect of epidermal growth factor (EGF) by limiting EGF receptor autophosphorylation (activation) and reducing EGF-induced activation of the PI3K/Akt signaling pathway and expression of the pro-survival target molecules, Bcl-x(L) and BAD (55). In another study, DIM caused apoptosis of prostate cancer cells by stimulating p38 mitogen-activated protein kinase (p38 MAPK)-induced upregulation of tumor suppressor p75NTR (56). The anti-proliferative effect of DIM in cancer cells has also been linked to the inhibition of histone deacetylase (HDAC) activity. Specifically, DIM was found to reverse HDAC-mediated epigenetic silencing of genes coding for key regulators of the cell cycle (57, 58). A recent genome-wide analysis of DNA methylation also showed that DIM could reverse aberrant promoter methylation in prostate cancer cells, at least partly through downregulating the expression of DNA methyltransferases (DNMTs) (59).
Inhibition of cell migration and invasion
The epithelial-to-mesenchymal transition (EMT) describes a process of epithelial cell transformation whereby cells lose their polarity and adhesion properties while gaining migratory and invasive properties through the expression of mesenchymal genes. Inhibition of EMT by I3C and ICZ in breast cancer cells has been associated with upregulation of an epithelial marker, E-cadherin, and downregulation of vimentin, focal adhesion kinase (FAK), and matrix metalloproteins (MMPs) — proteins and enzymes known to promote migration (60). DIM also inhibited migration and invasion of liver cancer cells in vitro and in vivo through inactivating the FAK signaling pathway (61). Moreover, DIM has been shown to reverse methylation-associated dysregulation of genes involved in cell adhesion, chemotaxis, and inflammation that contributes to cancer progression (59). DIM was able to inhibit lung metastasis in mice with liver (61) or mammary tumors (62).
Inhibition of angiogenesis
To fuel their rapid growth, invasive tumors must also develop new capillaries from preexisting blood vessels by a process known as angiogenesis. I3C inhibited lipopolysaccharide (LPS)-induced macrophage activation and secretion of proangiogenic molecules, such as nitric oxide (NO), vascular endothelial growth factor (VEGF), interleukin-6 (IL-6), and MMP-9, and prevented the formation of capillary-like structures from co-cultured human umbilical endothelial cells (63). Similarly, I3C inhibited capillary-like tube formation from phorbol myristate acetate (PMA)-stimulated endothelial cells (64). DIM also blocked PMA-induced angiogenic activities in human umbilical endothelial cells (65).
Regulation of inflammation and cell-mediated immunity
Uncontrolled inflammation has been associated with several chronic diseases, including cancer. In the mouse ear edema model, 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced upregulation of pro-inflammatory mediators, such as cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS), has been found to be mitigated by DIM treatment (66). Nuclear factor-kappa B (NF-κB) is a major transcription factor regulating the expression of many pro-inflammatory genes like those coding for COX-2 and iNOS. Specifically, DIM inhibited TPA-induced activation of kinases (inhibitor of kappa B kinase [IκK] and extracellular signal-regulated kinase [ERK]) that control the transcriptional activity of NF-κB (66). In addition, recent animal studies showed that I3C and/or DIM could modulate cell-mediated immune response in experimental autoimmune encephalomyelitis (67), staphylococcal enterotoxin-induced lung inflammation (68), and delayed-type hypersensitivity (69). Specifically, I3C and/or DIM differentially regulated T-cell subpopulations via the activation or suppression of microRNA-dependent pathways controlling cell cycle progression and apoptosis.
Transplacental cancer prevention
The inclusion of I3C in the maternal diet was found to protect the offspring from lymphoma and lung tumors induced by dibenzo[a]pyrene, a polycyclic aromatic hydrocarbon (70, 71). Polycyclic aromatic hydrocarbons are chemical pollutants formed during incomplete combustion of organic substances, such as coal, oil, wood, and tobacco (72).
However, the physiological relevance of cell culture and animal studies to human health is unclear since little or no I3C is available to tissues after oral administration (see Metabolism and Bioavailability) (6).
Disease Prevention
Cancer
Some observational studies provide some support for the hypothesis that higher intakes of cruciferous vegetables are associated with lower risk for some types of cancer (see the article on Cruciferous Vegetables) (1). Cruciferous vegetables are relatively good sources of nutrients that may have protective effects against cancer, including vitamin C, folate, selenium, carotenoids, and fiber. In addition, glucosinolates can be hydrolyzed to a variety of potentially protective isothiocyanates, in addition to indole-3-carbinol (see the article on Isothiocyanates). Consequently, evidence for an inverse association between cruciferous vegetable intake and cancer risk provides relatively little information about the specific effects of indole-3-carbinol on cancer risk.
At present, the effects of I3C or DIM supplementation on cancer risk in humans are not known.
Disease Treatment
Human papilloma virus infection-related diseases
Cervical intraepithelial neoplasia
Infection with certain strains of human papilloma virus (HPV) is an important risk factor for cervical cancer (73). Transgenic mice that express cancer-promoting HPV genes develop cervical cancer with chronic 17β-estradiol administration. In this model, feeding I3C markedly reduced the number of mice that developed cervical cancer (74). A small placebo-controlled trial in women examined the effect of oral I3C supplementation on the progression of precancerous cervical lesions classified as cervical intraepithelial neoplasia (CIN) 2 or CIN 3 (75). After 12 weeks, four out of the eight women who took 200 mg/day had complete regression of CIN, and four out of the nine who took 400 mg/day had complete regression; none of the 10 women who took a placebo had complete regression. HPV was present in 7 out of the 10 women in the placebo group, seven out of eight women in the 200 mg I3C group, and eight out of nine women in the 400 mg I3C group (75). However compared to placebo, oral supplementation with DIM (2 mg/kg/day) for 12 weeks in 64 women with CIN 2 or CIN 3 lesions failed to improve clinical parameters during a one-year follow-up period (76). In another six-month randomized, double-blind controlled trial, DIM supplementation (150 mg/day) failed to promote HPV clearance and prevent CIN progression in 551 women with low-grade cell abnormalities in cervical smears (77).
Although oral supplementation of I3C or DIM appears relatively safe and well tolerated, results obtained with I3C are only preliminary, and interventions with DIM failed to show preventative or therapeutic efficacy in women with precancerous lesions of the cervix. The intravaginal administration of DIM in the form of suppositories may prove to be a more effective approach to reverse CIN in women (78).
Vulvar intraepithelial neoplasia
HPV infection has also been associated with vulvar intraepithelial neoplasia (VIN), a precancerous condition that may progress to vulval cancer (79). A small randomized trial in 12 women with VIN found that supplementation with 200 mg/day or 400 mg/day of I3C for six months improved overall symptoms, as well as lesion size and appearance (80). Additional trials are necessary to determine whether I3C might be an effective treatment for VIN.
Recurrent respiratory papillomatosis
Recurrent respiratory papillomatosis (RRP) is a rare disease of children and adults, characterized by generally benign growths (papillomas) in the respiratory tract, which are caused by HPV infection (81). These papillomas occur most commonly on or around the vocal cords in the larynx (voice box), but they may also affect the trachea, bronchi, and lungs. The most common treatment for RRP is surgical removal of the papillomas. Since papillomas often recur, adjunct treatments may be used to help prevent or reduce recurrences (82). In immune-compromised mice transplanted with HPV-infected laryngeal tissue, only 25% of the mice fed I3C developed laryngeal papillomas compared to 100% of the control mice (83). In a small observational study of RRP patients, increased ratios of urinary 2HE1:16HE1 resulting from increased cruciferous vegetable consumption were associated with less severe RRP (84). In an uncontrolled pilot study, the effect of daily I3C supplementation (400 mg/day for adults and 10 mg/kg daily for children) on papilloma recurrence has been examined in RRP patients (85). Over a five-year follow-up period, 11 of the original 49 patients experienced no recurrence, 10 experienced a reduction in the rate of recurrence, 12 experienced no improvement, and 12 were lost to follow-up (86). I3C given for 6 months to 3 years to five children with an aggressive form of the disease halted the growth of the papillomas in three children after two years of treatment (87). In some patients, I3C may be an effective adjunct treatment to reduce the growth or recurrence of respiratory papillomas.
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune disorder characterized by chronic inflammation that may result in damage to the joints, skin, kidneys, heart, lungs, blood vessels, or brain (88). Estrogen is thought to play a role in the pathology of SLE because the disorder is much more common in women than men, and its onset is most common during the reproductive years when endogenous estrogen levels are highest (89). The potential for I3C supplementation to shift endogenous estrogen metabolism toward the less estrogenic metabolite 2HE1, and away from the highly estrogenic metabolite 16HE1 (see Anti-estrogenic activities), led to interest in its use in SLE (35). In an animal model of SLE, I3C feeding decreased the severity of kidney disease and prolonged survival (90). A small uncontrolled trial of I3C supplementation (375 mg/day) in female SLE patients found that I3C supplementation increased urinary 2HE1:16HE1 ratios, but the trial found no significant change in SLE symptoms after three months (90). Controlled clinical trials are needed to determine whether I3C supplementation could have beneficial effects in SLE patients.
Sources
Food sources
Glucobrassicin, the glucosinolate precursor of I3C, is found in a number of cruciferous vegetables, including broccoli, Brussels sprouts, cabbage, cauliflower, collard greens, kale, kohlrabi, mustard greens, radish, rutabaga, and turnip (91, 92). Although glucosinolates are present in relatively high concentrations in cruciferous vegetables, glucobrassicin makes up only about 8%-12% of the total glucosinolates (93). Total glucosinolate contents of selected cruciferous vegetables are presented in Table 1. However, the amount of total glucosinolates and the amount of indole-3-carbinol formed from glucobrassicin in food is variable and depends, in part, on the processing and preparation of foods (for more detailed information, see the article on Cruciferous Vegetables).
| Food (raw) | Serving | Total Glucosinolates (mg) |
|---|---|---|
| Brussels sprouts | ½ cup |
104
|
| Garden cress | ½ cup |
98
|
| Mustard greens | ½ cup, chopped |
79
|
| Kale | 1 cup, chopped |
67
|
| Turnip | ½ cup, cubes |
60
|
| Cabbage, savoy | ½ cup, chopped |
35
|
| Watercress | 1 cup, chopped |
32
|
| Kohlrabi | ½ cup, chopped |
31
|
| Cabbage, red | ½ cup, chopped |
29
|
| Broccoli | ½ cup, chopped |
27
|
| Horseradish | 1 tablespoon (15 g) |
24
|
| Cauliflower | ½ cup, chopped |
22
|
| Bok choi (pak choi) | ½ cup, chopped |
19
|
Supplements
Indole-3-Carbinol (I3C)
I3C is available without a prescription as a dietary supplement, alone or in combination products. Dosage ranges between 200 mg/day and 800 mg/day (95). I3C supplementation increased urinary 2HE1 concentrations in adults at doses of 300 to 400 mg/day (39). I3C doses of 200 mg/day or 400 mg/day improved the regression of cervical intraepithelial neoplasia (CIN) in a preliminary clinical trial (75). I3C in doses up to 400 mg/day has been used to treat recurrent respiratory papillomatosis (see Disease Treatment) (85, 86).
3,3'-Diindolylmethane (DIM)
DIM is available without a prescription as a dietary supplement, alone or in combination products. In a small clinical trial, DIM supplementation at a dose of 108 mg/day for 30 days increased urinary 2HE1 excretion in postmenopausal women with a history of breast cancer (34).
Safety
Adverse effects
Slight increases in the serum concentrations of the liver enzyme, alanine aminotransferase (ALT) were observed in two women who took unspecified doses of I3C supplements for four weeks (39). One person reported a skin rash while taking 375 mg/day of I3C (35). High doses of I3C (800 mg/day) have been associated with symptoms of disequilibrium and tremor, which resolved when the dose was decreased (85). In a phase I study in women at high risk for breast cancer, 5 out of 20 participants had gastrointestinal symptoms with single doses ≥600 mg, although others had no adverse effects with single doses up to 1,200 mg (6). No adverse effects were reported with daily consumption of 400 mg of I3C for four weeks (6).
In some animal models, I3C supplementation was found to enhance carcinogen-induced cancer development when given chronically after the carcinogen (96-99). When administered before or at the same time as the carcinogen, oral I3C inhibited tumorigenesis in animal models of cancers of the mammary gland (100, 101), uterus (102), stomach (103), colon (104, 105), lung (106), and liver (107, 108). Although the long-term effects of I3C supplementation on cancer risk in humans are not known, the contradictory results of animal studies have led several experts to caution against the widespread use of I3C and DIM supplements in humans until their potential risks and benefits are better understood (99, 109, 110).
Pregnancy and lactation
The safety of I3C or DIM supplements during pregnancy or lactation has not been established (95).
Drug interactions
No drug interactions with I3C or DIM supplementation in humans have been reported. However, preliminary evidence that I3C and DIM can increase the activity of CYP1A2 (111, 112) suggests the potential for I3C or DIM supplementation to decrease serum concentrations of medications metabolized by CYP1A2 (113). Both I3C and DIM modestly increase the activity of CYP3A4 in rats when administered chronically (114). This observation raises the potential for adverse drug interactions in humans since CYP3A4 is involved in the metabolism of approximately 60% of therapeutic drugs.
The acidic environment of the stomach allows I3C molecules to condense and generate a number of biologically active I3C oligomers (Figure 2). Drugs that block the production of stomach acids, like antacids, Histamine2 (H2) receptor antagonists, and proton-pump inhibitors, would likely prevent the generation of DIM and ICZ. However, it is not known whether these drugs limit the biological activities attributed to I3C and its derivatives (95).
Authors and Reviewers
Originally written in 2005 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in December 2008 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in January 2017 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in July 2017 by:
David E. Williams, Ph.D.
Principal Investigator, and Helen P. Rumbel Professor for Cancer Prevention
Linus Pauling Institute
Professor, Department of Environmental and Molecular Toxicology
Oregon State University
Copyright 2005-2024 Linus Pauling Institute
References
1. Traka M. Chapter nine-health benefits of glucosinolates. Advances in Botanical Research. 2016;80:247-279.
2. Ishida M, Hara M, Fukino N, Kakizaki T, Morimitsu Y. Glucosinolate metabolism, functionality and breeding for the improvement of Brassicaceae vegetables. Breed Sci. 2014;64(1):48-59. (PubMed)
3. Holst B, Williamson G. A critical review of the bioavailability of glucosinolates and related compounds. Nat Prod Rep. 2004;21(3):425-447. (PubMed)
4. Barba FJ, Nikmaram N, Roohinejad S, Khelfa A, Zhu Z, Koubaa M. Bioavailability of glucosinolates and their breakdown products: impact of processing. Front Nutr. 2016;3:24. (PubMed)
5. Wang SQ, Cheng LS, Liu Y, Wang JY, Jiang W. Indole-3-carbinol (I3C) and its major derivatives: their pharmacokinetics and important roles in hepatic protection. Curr Drug Metab. 2016;17(4):401-409. (PubMed)
6. Reed GA, Arneson DW, Putnam WC, et al. Single-dose and multiple-dose administration of indole-3-carbinol to women: pharmacokinetics based on 3,3'-diindolylmethane. Cancer Epidemiol Biomarkers Prev. 2006;15(12):2477-2481. (PubMed)
7. Anderton MJ, Manson MM, Verschoyle R, et al. Physiological modeling of formulated and crystalline 3,3'-diindolylmethane pharmacokinetics following oral administration in mice. Drug Metab Dispos. 2004;32(6):632-638. (PubMed)
8. Luo Y, Wang TT, Teng Z, Chen P, Sun J, Wang Q. Encapsulation of indole-3-carbinol and 3,3'-diindolylmethane in zein/carboxymethyl chitosan nanoparticles with controlled release property and improved stability. Food Chem. 2013;139(1-4):224-230. (PubMed)
9. Song JM, Kirtane AR, Upadhyaya P, et al. Intranasal delivery of liposomal indole-3-carbinol improves its pulmonary bioavailability. Int J Pharm. 2014;477(1-2):96-101. (PubMed)
10. Lampe JW, Peterson S. Brassica, biotransformation and cancer risk: genetic polymorphisms alter the preventive effects of cruciferous vegetables. J Nutr. 2002;132(10):2991-2994. (PubMed)
11. Bjeldanes LF, Kim JY, Grose KR, Bartholomew JC, Bradfield CA. Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proc Natl Acad Sci U S A. 1991;88(21):9543-9547. (PubMed)
12. Bonnesen C, Eggleston IM, Hayes JD. Dietary indoles and isothiocyanates that are generated from cruciferous vegetables can both stimulate apoptosis and confer protection against DNA damage in human colon cell lines. Cancer Res. 2001;61(16):6120-6130. (PubMed)
13. Hubbard TD, Murray IA, Perdew GH. Indole and tryptophan metabolism: endogenous and dietary routes to Ah receptor activation. Drug Metab Dispos. 2015;43(10):1522-1535. (PubMed)
14. Safe S. Molecular biology of the Ah receptor and its role in carcinogenesis. Toxicol Lett. 2001;120(1-3):1-7. (PubMed)
15. Li Y, Li X, Sarkar FH. Gene expression profiles of I3C- and DIM-treated PC3 human prostate cancer cells determined by cDNA microarray analysis. J Nutr. 2003;133(4):1011-1019. (PubMed)
16. Wang TT, Schoene NW, Milner JA, Kim YS. Broccoli-derived phytochemicals indole-3-carbinol and 3,3'-diindolylmethane exerts concentration-dependent pleiotropic effects on prostate cancer cells: comparison with other cancer preventive phytochemicals. Mol Carcinog. 2012;51(3):244-256. (PubMed)
17. Gross-Steinmeyer K, Stapleton PL, Liu F, et al. Phytochemical-induced changes in gene expression of carcinogen-metabolizing enzymes in cultured human primary hepatocytes. Xenobiotica. 2004;34(7):619-632. (PubMed)
18. Baird WM, Hooven LA, Mahadevan B. Carcinogenic polycyclic aromatic hydrocarbon-DNA adducts and mechanism of action. Environ Mol Mutagen. 2005;45(2-3):106-114. (PubMed)
19. Watson GW, Beaver LM, Williams DE, Dashwood RH, Ho E. Phytochemicals from cruciferous vegetables, epigenetics, and prostate cancer prevention. AAPS J. 2013;15(4):951-961. (PubMed)
20. Saw CL, Cintron M, Wu TY, et al. Pharmacodynamics of dietary phytochemical indoles I3C and DIM: Induction of Nrf2-mediated phase II drug metabolizing and antioxidant genes and synergism with isothiocyanates. Biopharm Drug Dispos. 2011;32(5):289-300. (PubMed)
21. Wu TY, Khor TO, Su ZY, et al. Epigenetic modifications of Nrf2 by 3,3'-diindolylmethane in vitro in TRAMP C1 cell line and in vivo TRAMP prostate tumors. AAPS J. 2013;15(3):864-874. (PubMed)
22. Wu TY, Saw CL, Khor TO, Pung D, Boyanapalli SS, Kong AN. In vivo pharmacodynamics of indole-3-carbinol in the inhibition of prostate cancer in transgenic adenocarcinoma of mouse prostate (TRAMP) mice: involvement of Nrf2 and cell cycle/apoptosis signaling pathways. Mol Carcinog. 2012;51(10):761-770. (PubMed)
23. Licznerska BE, Szaefer H, Murias M, Bartoszek A, Baer-Dubowska W. Modulation of CYP19 expression by cabbage juices and their active components: indole-3-carbinol and 3,3'-diindolylmethene in human breast epithelial cell lines. Eur J Nutr. 2013;52(5):1483-1492. (PubMed)
24. Belous AR, Hachey DL, Dawling S, Roodi N, Parl FF. Cytochrome P450 1B1-mediated estrogen metabolism results in estrogen-deoxyribonucleoside adduct formation. Cancer Res. 2007;67(2):812-817. (PubMed)
25. Jefcoate CR, Liehr JG, Santen RJ, et al. Tissue-specific synthesis and oxidative metabolism of estrogens. J Natl Cancer Inst Monogr. 2000(27):95-112. (PubMed)
26. Kwon YJ, Baek HS, Ye DJ, Shin S, Kim D, Chun YJ. CYP1B1 enhances cell proliferation and metastasis through induction of EMT and activation of Wnt/beta-catenin signaling via Sp1 upregulation. PLoS One. 2016;11(3):e0151598. (PubMed)
27. Park SA, Lee MH, Na HK, Surh YJ. 4-Hydroxyestradiol induces mammary epithelial cell transformation through Nrf2-mediated heme oxygenase-1 overexpression. Oncotarget. 2016;8(1):164-178. (PubMed)
28. Szaefer H, Licznerska B, Krajka-Kuzniak V, Bartoszek A, Baer-Dubowska W. Modulation of CYP1A1, CYP1A2 and CYP1B1 expression by cabbage juices and indoles in human breast cell lines. Nutr Cancer. 2012;64(6):879-888. (PubMed)
29. Ziegler RG, Fuhrman BJ, Moore SC, Matthews CE. Epidemiologic studies of estrogen metabolism and breast cancer. Steroids. 2015;99(Pt A):67-75. (PubMed)
30. Telang NT, Suto A, Wong GY, Osborne MP, Bradlow HL. Induction by estrogen metabolite 16 alpha-hydroxyestrone of genotoxic damage and aberrant proliferation in mouse mammary epithelial cells. J Natl Cancer Inst. 1992;84(8):634-638. (PubMed)
31. Yuan F, Chen DZ, Liu K, Sepkovic DW, Bradlow HL, Auborn K. Anti-estrogenic activities of indole-3-carbinol in cervical cells: implication for prevention of cervical cancer. Anticancer Res. 1999;19(3A):1673-1680. (PubMed)
32. Bradlow HL, Telang NT, Sepkovic DW, Osborne MP. 2-Hydroxyestrone: the 'good' estrogen. J Endocrinol. 1996;150 Suppl:S259-265. (PubMed)
33. Bradlow HL, Michnovicz JJ, Halper M, Miller DG, Wong GY, Osborne MP. Long-term responses of women to indole-3-carbinol or a high fiber diet. Cancer Epidemiol Biomarkers Prev. 1994;3(7):591-595. (PubMed)
34. Dalessandri KM, Firestone GL, Fitch MD, Bradlow HL, Bjeldanes LF. Pilot study: effect of 3,3'-diindolylmethane supplements on urinary hormone metabolites in postmenopausal women with a history of early-stage breast cancer. Nutr Cancer. 2004;50(2):161-167. (PubMed)
35. McAlindon TE, Gulin J, Chen T, Klug T, Lahita R, Nuite M. Indole-3-carbinol in women with SLE: effect on estrogen metabolism and disease activity. Lupus. 2001;10(11):779-783. (PubMed)
36. Michnovicz JJ. Increased estrogen 2-hydroxylation in obese women using oral indole-3-carbinol. Int J Obes Relat Metab Disord. 1998;22(3):227-229. (PubMed)
37. Michnovicz JJ, Adlercreutz H, Bradlow HL. Changes in levels of urinary estrogen metabolites after oral indole-3-carbinol treatment in humans. J Natl Cancer Inst. 1997;89(10):718-723. (PubMed)
38. Reed GA, Peterson KS, Smith HJ, et al. A phase I study of indole-3-carbinol in women: tolerability and effects. Cancer Epidemiol Biomarkers Prev. 2005;14(8):1953-1960. (PubMed)
39. Wong GY, Bradlow L, Sepkovic D, Mehl S, Mailman J, Osborne MP. Dose-ranging study of indole-3-carbinol for breast cancer prevention. J Cell Biochem Suppl. 1997;28-29:111-116. (PubMed)
40. Arslan AA, Shore RE, Afanasyeva Y, Koenig KL, Toniolo P, Zeleniuch-Jacquotte A. Circulating estrogen metabolites and risk for breast cancer in premenopausal women. Cancer Epidemiol Biomarkers Prev. 2009;18(8):2273-2279. (PubMed)
41. Eliassen AH, Missmer SA, Tworoger SS, Hankinson SE. Circulating 2-hydroxy- and 16alpha-hydroxy estrone levels and risk of breast cancer among postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2008;17(8):2029-2035. (PubMed)
42. Modugno F, Kip KE, Cochrane B, et al. Obesity, hormone therapy, estrogen metabolism and risk of postmenopausal breast cancer. Int J Cancer. 2006;118(5):1292-1301. (PubMed)
43. Zeleniuch-Jacquotte A, Shore RE, Afanasyeva Y, et al. Postmenopausal circulating levels of 2- and 16alpha-hydroxyestrone and risk of endometrial cancer. Br J Cancer. 2011;105(9):1458-1464. (PubMed)
44. Jordan VC, Gapstur S, Morrow M. Selective estrogen receptor modulation and reduction in risk of breast cancer, osteoporosis, and coronary heart disease. J Natl Cancer Inst. 2001;93(19):1449-1457. (PubMed)
45. Omoto Y, Iwase H. Clinical significance of estrogen receptor beta in breast and prostate cancer from biological aspects. Cancer Sci. 2015;106(4):337-343. (PubMed)
46. Liehr JG. Is estradiol a genotoxic mutagenic carcinogen? Endocr Rev. 2000;21(1):40-54. (PubMed)
47. Ashok BT, Chen Y, Liu X, Bradlow HL, Mittelman A, Tiwari RK. Abrogation of estrogen-mediated cellular and biochemical effects by indole-3-carbinol. Nutr Cancer. 2001;41(1-2):180-187. (PubMed)
48. Meng Q, Yuan F, Goldberg ID, Rosen EM, Auborn K, Fan S. Indole-3-carbinol is a negative regulator of estrogen receptor-alpha signaling in human tumor cells. J Nutr. 2000;130(12):2927-2931. (PubMed)
49. Marconett CN, Sundar SN, Poindexter KM, Stueve TR, Bjeldanes LF, Firestone GL. Indole-3-carbinol triggers aryl hydrocarbon receptor-dependent estrogen receptor (ER)alpha protein degradation in breast cancer cells disrupting an ERalpha-GATA3 transcriptional cross-regulatory loop. Mol Biol Cell. 2010;21(7):1166-1177. (PubMed)
50. Chen I, McDougal A, Wang F, Safe S. Aryl hydrocarbon receptor-mediated antiestrogenic and antitumorigenic activity of diindolylmethane. Carcinogenesis. 1998;19(9):1631-1639. (PubMed)
51. Marconett CN, Singhal AK, Sundar SN, Firestone GL. Indole-3-carbinol disrupts estrogen receptor-alpha dependent expression of insulin-like growth factor-1 receptor and insulin receptor substrate-1 and proliferation of human breast cancer cells. Mol Cell Endocrinol. 2012;363(1-2):74-84. (PubMed)
52. Stewart ZA, Westfall MD, Pietenpol JA. Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol Sci. 2003;24(3):139-145. (PubMed)
53. Mao CG, Tao ZZ, Chen Z, Chen C, Chen SM, Wan LJ. Indole-3-carbinol inhibits nasopharyngeal carcinoma cell growth in vivo and in vitro through inhibition of the PI3K/Akt pathway. Exp Ther Med. 2014;8(1):207-212. (PubMed)
54. Leem SH, Li XJ, Park MH, Park BH, Kim SM. Genome-wide transcriptome analysis reveals inactivation of Wnt/beta-catenin by 3,3'-diindolylmethane inhibiting proliferation of colon cancer cells. Int J Oncol. 2015;47(3):918-926. (PubMed)
55. Chinni SR, Li Y, Upadhyay S, Koppolu PK, Sarkar FH. Indole-3-carbinol (I3C) induced cell growth inhibition, G1 cell cycle arrest and apoptosis in prostate cancer cells. Oncogene. 2001;20(23):2927-2936. (PubMed)
56. Khwaja FS, Wynne S, Posey I, Djakiew D. 3,3'-diindolylmethane induction of p75NTR-dependent cell death via the p38 mitogen-activated protein kinase pathway in prostate cancer cells. Cancer Prev Res. 2009;2(6):566-571. (PubMed)
57. Beaver LM, Yu TW, Sokolowski EI, Williams DE, Dashwood RH, Ho E. 3,3'-Diindolylmethane, but not indole-3-carbinol, inhibits histone deacetylase activity in prostate cancer cells. Toxicol Appl Pharmacol. 2012;263(3):345-351. (PubMed)
58. Li Y, Li X, Guo B. Chemopreventive agent 3,3'-diindolylmethane selectively induces proteasomal degradation of class I histone deacetylases. Cancer Res. 2010;70(2):646-654. (PubMed)
59. Wong CP, Hsu A, Buchanan A, et al. Effects of sulforaphane and 3,3'-diindolylmethane on genome-wide promoter methylation in normal prostate epithelial cells and prostate cancer cells. PLoS One. 2014;9(1):e86787. (PubMed)
60. Ho JN, Jun W, Choue R, Lee J. I3C and ICZ inhibit migration by suppressing the EMT process and FAK expression in breast cancer cells. Mol Med Rep. 2013;7(2):384-388. (PubMed)
61. Li WX, Chen LP, Sun MY, Li JT, Liu HZ, Zhu W. 3'3-Diindolylmethane inhibits migration, invasion and metastasis of hepatocellular carcinoma by suppressing FAK signaling. Oncotarget. 2015;6(27):23776-23792. (PubMed)
62. Kim EJ, Shin M, Park H, et al. Oral administration of 3,3'-diindolylmethane inhibits lung metastasis of 4T1 murine mammary carcinoma cells in BALB/c mice. J Nutr. 2009;139(12):2373-2379. (PubMed)
63. Wang ML, Shih CK, Chang HP, Chen YH. Antiangiogenic activity of indole-3-carbinol in endothelial cells stimulated with activated macrophages. Food Chem. 2012;134(2):811-820. (PubMed)
64. Wu HT, Lin SH, Chen YH. Inhibition of cell proliferation and in vitro markers of angiogenesis by indole-3-carbinol, a major indole metabolite present in cruciferous vegetables. J Agric Food Chem. 2005;53(13):5164-5169. (PubMed)
65. Kunimasa K, Kobayashi T, Kaji K, Ohta T. Antiangiogenic effects of indole-3-carbinol and 3,3'-diindolylmethane are associated with their differential regulation of ERK1/2 and Akt in tube-forming HUVEC. J Nutr. 2010;140(1):1-6. (PubMed)
66. Kim EJ, Park H, Kim J, Park JH. 3,3'-diindolylmethane suppresses 12-O-tetradecanoylphorbol-13-acetate-induced inflammation and tumor promotion in mouse skin via the downregulation of inflammatory mediators. Mol Carcinog. 2010;49(7):672-683. (PubMed)
67. Rouse M, Rao R, Nagarkatti M, Nagarkatti PS. 3,3'-diindolylmethane ameliorates experimental autoimmune encephalomyelitis by promoting cell cycle arrest and apoptosis in activated T cells through microRNA signaling pathways. J Pharmacol Exp Ther. 2014;350(2):341-352. (PubMed)
68. Elliott DM, Nagarkatti M, Nagarkatti PS. 3,3-Diindolylmethane ameliorates Staphylococcal enterotoxin B-induced acute lung injury through alterations in the expression of microRNA that target apoptosis and cell-cycle arrest in activated T cells. J Pharmacol Exp Ther. 2016;357(1):177-187. (PubMed)
69. Singh NP, Singh UP, Rouse M, et al. Dietary indoles suppress delayed-type hypersensitivity by inducing a switch from proinflammatory Th17 Cells to anti-inflammatory regulatory T cells through regulation of microRNA. J Immunol. 2016;196(3):1108-1122. (PubMed)
70. Shorey LE, Madeen EP, Atwell LL, et al. Differential modulation of dibenzo[def,p]chrysene transplacental carcinogenesis: maternal diets rich in indole-3-carbinol versus sulforaphane. Toxicol Appl Pharmacol. 2013;270(1):60-69. (PubMed)
71. Yu Z, Mahadevan B, Lohr CV, et al. Indole-3-carbinol in the maternal diet provides chemoprotection for the fetus against transplacental carcinogenesis by the polycyclic aromatic hydrocarbon dibenzo[a,l]pyrene. Carcinogenesis. 2006;27(10):2116-2123. (PubMed)
72. ATSDR. Toxicological profile for polycyclic aromatic hydrocarbons (PAHs). Agency for Toxic Substances and Disease Registry, U.S. Department of Health and Human Services. Atlanta, GA; August 1995.
73. Rambout L, Hopkins L, Hutton B, Fergusson D. Prophylactic vaccination against human papillomavirus infection and disease in women: a systematic review of randomized controlled trials. CMAJ. 2007;177(5):469-479. (PubMed)
74. Jin L, Qi M, Chen DZ, et al. Indole-3-carbinol prevents cervical cancer in human papilloma virus type 16 (HPV16) transgenic mice. Cancer Res. 1999;59(16):3991-3997. (PubMed)
75. Bell MC, Crowley-Nowick P, Bradlow HL, et al. Placebo-controlled trial of indole-3-carbinol in the treatment of CIN. Gynecol Oncol. 2000;78(2):123-129. (PubMed)
76. Del Priore G, Gudipudi DK, Montemarano N, Restivo AM, Malanowska-Stega J, Arslan AA. Oral diindolylmethane (DIM): pilot evaluation of a nonsurgical treatment for cervical dysplasia. Gynecol Oncol. 2010;116(3):464-467. (PubMed)
77. Castanon A, Tristram A, Mesher D, et al. Effect of diindolylmethane supplementation on low-grade cervical cytological abnormalities: double-blind, randomised, controlled trial. Br J Cancer. 2012;106(1):45-52. (PubMed)
78. Ashrafian L, Sukhikh G, Kiselev V, et al. Double-blind randomized placebo-controlled multicenter clinical trial (phase IIa) on diindolylmethane's efficacy and safety in the treatment of CIN: implications for cervical cancer prevention. EPMA J. 2015;6:25. (PubMed)
79. Pepas L, Kaushik S, Nordin A, Bryant A, Lawrie TA. Medical interventions for high-grade vulval intraepithelial neoplasia. Cochrane Database Syst Rev. 2015(8):Cd007924. (PubMed)
80. Naik R, Nixon S, Lopes A, Godfrey K, Hatem MH, Monaghan JM. A randomized phase II trial of indole-3-carbinol in the treatment of vulvar intraepithelial neoplasia. Int J Gynecol Cancer. 2006;16(2):786-790. (PubMed)
81. Recurrent Respiratory Papillomatosis Foundation. What is recurrent respiratory papillomatosis? Recurrent Respiratory Papillomatosis Foundation [Web page]. Available at: https://rrpf.org/what-is-rrp/. Accessed 1/4/22.
82. Auborn KJ. Therapy for recurrent respiratory papillomatosis. Antivir Ther. 2002;7(1):1-9. (PubMed)
83. Newfield L, Goldsmith A, Bradlow HL, Auborn K. Estrogen metabolism and human papillomavirus-induced tumors of the larynx: chemo-prophylaxis with indole-3-carbinol. Anticancer Res. 1993;13(2):337-341. (PubMed)
84. Auborn K, Abramson A, Bradlow HL, Sepkovic D, Mullooly V. Estrogen metabolism and laryngeal papillomatosis: a pilot study on dietary prevention. Anticancer Res. 1998;18(6B):4569-4573. (PubMed)
85. Rosen CA, Woodson GE, Thompson JW, Hengesteg AP, Bradlow HL. Preliminary results of the use of indole-3-carbinol for recurrent respiratory papillomatosis. Otolaryngol Head Neck Surg. 1998;118(6):810-815. (PubMed)
86. Rosen CA, Bryson PC. Indole-3-carbinol for recurrent respiratory papillomatosis: long-term results. J Voice. 2004;18(2):248-253. (PubMed)
87. Boltezar IH, Bahar MS, Zargi M, Gale N, Maticic M, Poljak M. Adjuvant therapy for laryngeal papillomatosis. Acta Dermatovenerol Alp Pannonica Adriat. 2011;20(3):175-180. (PubMed)
88. Handout on health: systemic lupus erythematosus. National Institute of Arthritis and Musculoskeletal and Skin Diseases [Web page]. June 2016. Available at: https://www.niams.nih.gov/health_info/lupus/. Accessed 1/21/17.
89. McMurray RW, May W. Sex hormones and systemic lupus erythematosus: review and meta-analysis. Arthritis Rheum. 2003;48(8):2100-2110. (PubMed)
90. Auborn KJ, Qi M, Yan XJ, et al. Lifespan is prolonged in autoimmune-prone (NZB/NZW) F1 mice fed a diet supplemented with indole-3-carbinol. J Nutr. 2003;133(11):3610-3613. (PubMed)
91. Carlson DG, Kwolek WF, Williams PH. Glucosinolates in crucifer vegetables: broccoli, Brussels sprouts, cauliflower, collards, kale, mustard greens, and kohlrabi. J Amer Soc Hort Sci. 1987;112(1):173-178.
92. Fenwick GR, Heaney RK, Mullin WJ. Glucosinolates and their breakdown products in food and food plants. Crit Rev Food Sci Nutr. 1983;18(2):123-201. (PubMed)
93. Kushad MM, Brown AF, Kurilich AC, et al. Variation of glucosinolates in vegetable crops of Brassica oleracea. J Agric Food Chem. 1999;47(4):1541-1548. (PubMed)
94. McNaughton SA, Marks GC. Development of a food composition database for the estimation of dietary intakes of glucosinolates, the biologically active constituents of cruciferous vegetables. Br J Nutr. 2003;90(3):687-697. (PubMed)
95. Hendler SS, Rorvik DM. PDR for Nutritional Supplements. 2nd ed: Thomson Reuters; 2008.
96. Kim DJ, Han BS, Ahn B, et al. Enhancement by indole-3-carbinol of liver and thyroid gland neoplastic development in a rat medium-term multiorgan carcinogenesis model. Carcinogenesis. 1997;18(2):377-381. (PubMed)
97. Yoshida M, Katashima S, Ando J, et al. Dietary indole-3-carbinol promotes endometrial adenocarcinoma development in rats initiated with N-ethyl-N'-nitro-N-nitrosoguanidine, with induction of cytochrome P450s in the liver and consequent modulation of estrogen metabolism. Carcinogenesis. 2004;25(11):2257-2264. (PubMed)
98. Pence BC, Buddingh F, Yang SP. Multiple dietary factors in the enhancement of dimethylhydrazine carcinogenesis: main effect of indole-3-carbinol. J Natl Cancer Inst. 1986;77(1):269-276. (PubMed)
99. Stoner G, Casto B, Ralston S, Roebuck B, Pereira C, Bailey G. Development of a multi-organ rat model for evaluating chemopreventive agents: efficacy of indole-3-carbinol. Carcinogenesis. 2002;23(2):265-272. (PubMed)
100. Grubbs CJ, Steele VE, Casebolt T, et al. Chemoprevention of chemically-induced mammary carcinogenesis by indole-3-carbinol. Anticancer Res. 1995;15(3):709-716. (PubMed)
101. Bradlow HL, Michnovicz J, Telang NT, Osborne MP. Effects of dietary indole-3-carbinol on estradiol metabolism and spontaneous mammary tumors in mice. Carcinogenesis. 1991;12(9):1571-1574. (PubMed)
102. Kojima T, Tanaka T, Mori H. Chemoprevention of spontaneous endometrial cancer in female Donryu rats by dietary indole-3-carbinol. Cancer Res. 1994;54(6):1446-1449. (PubMed)
103. Wattenberg LW, Loub WD. Inhibition of polycyclic aromatic hydrocarbon-induced neoplasia by naturally occurring indoles. Cancer Res. 1978;38(5):1410-1413. (PubMed)
104. Wargovich MJ, Chen CD, Jimenez A, et al. Aberrant crypts as a biomarker for colon cancer: evaluation of potential chemopreventive agents in the rat. Cancer Epidemiol Biomarkers Prev. 1996;5(5):355-360. (PubMed)
105. Guo D, Schut HA, Davis CD, Snyderwine EG, Bailey GS, Dashwood RH. Protection by chlorophyllin and indole-3-carbinol against 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)-induced DNA adducts and colonic aberrant crypts in the F344 rat. Carcinogenesis. 1995;16(12):2931-2937. (PubMed)
106. Morse MA, LaGreca SD, Amin SG, Chung FL. Effects of indole-3-carbinol on lung tumorigenesis and DNA methylation induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and on the metabolism and disposition of NNK in A/J mice. Cancer Res. 1990;50(9):2613-2617. (PubMed)
107. Dashwood RH, Arbogast DN, Fong AT, Hendricks JD, Bailey GS. Mechanisms of anti-carcinogenesis by indole-3-carbinol: detailed in vivo DNA binding dose-response studies after dietary administration with aflatoxin B1. Carcinogenesis. 1988;9(3):427-432. (PubMed)
108. Oganesian A, Hendricks JD, Williams DE. Long term dietary indole-3-carbinol inhibits diethylnitrosamine-initiated hepatocarcinogenesis in the infant mouse model. Cancer Lett. 1997;118(1):87-94. (PubMed)
109. Dashwood RH. Indole-3-carbinol: anticarcinogen or tumor promoter in brassica vegetables? Chem Biol Interact. 1998;110(1-2):1-5. (PubMed)
110. Lee BM, Park KK. Beneficial and adverse effects of chemopreventive agents. Mutat Res. 2003;523-524:265-278. (PubMed)
111. He YH, Friesen MD, Ruch RJ, Schut HA. Indole-3-carbinol as a chemopreventive agent in 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) carcinogenesis: inhibition of PhIP-DNA adduct formation, acceleration of PhIP metabolism, and induction of cytochrome P450 in female F344 rats. Food Chem Toxicol. 2000;38(1):15-23. (PubMed)
112. Lake BG, Tredger JM, Renwick AB, Barton PT, Price RJ. 3,3'-Diindolylmethane induces CYP1A2 in cultured precision-cut human liver slices. Xenobiotica. 1998;28(8):803-811. (PubMed)
113. Natural Medicines. Professional monograph: Indole-3-carbinol/Interactions with drugs; 2016.
114. Leibelt DA, Hedstrom OR, Fischer KA, Pereira CB, Williams DE. Evaluation of chronic dietary exposure to indole-3-carbinol and absorption-enhanced 3,3'-diindolylmethane in Sprague-Dawley rats. Toxicol Sci. 2003;74(1):10-21. (PubMed)
Isothiocyanates
Contents
Summary
- Isothiocyanates are derived from the hydrolysis of glucosinolates — sulfur-containing compounds found in cruciferous vegetables. (More information)
- Each glucosinolate forms a different isothiocyanate when hydrolyzed. For example, broccoli is a good source of glucoraphanin, the glucosinolate precursor of sulforaphane, and sinigrin, the glucosinolate precursor of allyl isothiocyanate. (More information)
- Absorbed isothiocyanates are rapidly conjugated to glutathione in the liver, and then sequentially metabolized in the mercapturic acid pathway, before being excreted in the urine. (More information)
- Isothiocyanates may modulate the expression and activity of biotransformation enzymes that are involved in the metabolism and elimination of xenobiotics (e.g., carcinogens) from the body. In cultured cells and animal models, isothiocyanates also exhibited antioxidant and anti-inflammatory activities and interfered with numerous cancer-related targets and pathways. (More information)
- Although high intakes of cruciferous vegetables have been associated with a lower risk for cancer, there is insufficient evidence that exposure to isothiocyanates through cruciferous vegetable consumption decreases cancer risk. (More information)
- Glucosinolates are present in relatively high concentrations in cruciferous vegetables, but the amounts of isothiocyanates formed from glucosinolates in foods are variable and depend partly on food processing and preparation. (More information)
Introduction
Cruciferous vegetables, such as broccoli, cabbage, and kale, are rich sources of sulfur-containing compounds called glucosinolates (see the article on Cruciferous Vegetables). Isothiocyanates are biologically active hydrolysis (breakdown) products of glucosinolates. Cruciferous vegetables contain a variety of glucosinolates, each of which forms a different isothiocyanate when hydrolyzed (Figure 1) (1). For example, broccoli is a good source of glucoraphanin, the glucosinolate precursor of sulforaphane, and sinigrin, the glucosinolate precursor of allyl isothiocyanate (AITC) (see Food sources) (2). Watercress is a rich source of gluconasturtiin, the precursor of phenethyl isothiocyanate (PEITC), while garden cress is rich in glucotropaeolin, the precursor of benzyl isothiocyanate (BITC) (see Food sources). At present, scientists are interested in the cancer-preventive activities of vegetables that are rich in glucosinolates (see the article on Cruciferous Vegetables), as well as individual isothiocyanates (3).
Metabolism and Bioavailability
Metabolism
The hydrolysis of glucosinolates, which is catalyzed by a class of enzymes called myrosinases (β-thioglucosidases), leads to the formation of breakdown compounds, such as thiocyanates, isothiocyanates, indoles, oxazolidine-2-thiones (e.g., goitrin), epithionitrile, and nitrile (see the article on Cruciferous Vegetables). In intact plant cells, myrosinase is physically separated from glucosinolates. Yet, when plant cells are damaged, myrosinase is released and comes in contact with glucosinolates, catalyzing their conversion into highly reactive metabolites that impart a pungent aroma and spicy (some say bitter) taste. Likewise, when raw cruciferous vegetables are chopped during the food preparation process, glucosinolates are rapidly hydrolyzed by myrosinase, generating metabolites that are then absorbed in the proximal intestine. In contrast, cooking cruciferous vegetables before consumption inactivates myrosinase, thus preventing the breakdown of glucosinolates. However, lightly cooking (i.e., light steam for <5 minutes) will preserve some of the myrosinase and allow for isothiocyanate conversion. A small fraction of intact glucosinolates may be absorbed in the small intestine, but a large proportion reaches the colon (4). In the colon, myrosinase produced by the microbiota can catalyze the generation of a wide range of metabolites from glucosinolates, depending on the pH and the presence of cofactors (4, 5).
The hydrolysis of glucosinolates at neutral pH results in the formation of unique isothiocyanates (Figure 1). For example, sinigrin, glucoraphanin, glucotropaeolin, and gluconasturtiin are the glucosinolate precursors of AITC, sulforaphane, BITC, and PEITC, respectively (Figure 1). Once absorbed, glucosinolate-derived isothiocyanates (like sulforaphane) are promptly conjugated to glutathione by a class of phase II detoxification enzymes known as glutathione S-transferases (GSTs) in the liver, and then sequentially metabolized in the mercapturic acid pathway (Figure 2). This mechanism is meant to increase the solubility of isothiocyanates, thereby promoting a rapid excretion in the urine. Using sulforaphane as the model isothiocyanate, it has indeed been established that its metabolites — sulforaphane-glutathione, sulforaphane-cysteine-glycine, sulforaphane-cysteine, and sulforaphane N-acetylcysteine — collectively known as dithiocarbamates (Figure 2), are ultimately excreted in the urine (4).


Bioavailability
The composition and content of glucosinolates in cruciferous vegetables are relatively stable but depend on the genus and species and can vary with plant growing and post-harvest storage conditions and culinary processing (6, 7). Since most cruciferous vegetables are cooked prior to eating, bacterial myrosinase in the gut, rather than plant myrosinase, is responsible for the initial step in glucosinolate degradation (Figure 2). In a feeding study involving 45 healthy subjects, the mean conversion rate of glucosinolates (of which 85% was glucoraphanin) to dithiocarbamates over a 24-hour period was estimated to be around 12% with wide variations among participants (range, 1.1 to 40.7%) (6). In contrast, 70%-75% of ingested isothiocyanates were found to be metabolized to dithiocarbamates. Therefore, following the ingestion of cooked cruciferous vegetables, the conversion of glucosinolates into isothiocyanates by gut bacteria appears to be a limiting step in the generation of dithiocarbamates (6). However, differences in individuals’ capacity to metabolize glucosinolates have not been linked to differences in gut microbiota composition (8).
Biological Activities
Antioxidant activity
Many isothiocyanates, particularly sulforaphane, have been shown to induce the expression of antioxidant enzymes via the activation of the nuclear factor E2-related factor 2 (Nrf2)-dependent pathway (9, 10). Briefly, Nrf2 is a transcription factor that is bound to the protein Kelch-like ECH-associated protein 1 (Keap1) in the cytosol (Figure 3). Keap1 responds to oxidative stress signals or chemical inducers by freeing Nrf2. Isothiocyanates can react with sulfhydryl residues of Keap1, causing the release of Nrf2. Nrf2 can then translocate to the nucleus and bind to the antioxidant response element (ARE) located in the promoters of genes coding for antioxidant/detoxifying enzymes and scavengers. Nrf2/ARE-dependent genes code for several mediators of the antioxidant response, including glutathione S-transferases (GSTs), thioredoxin, NAD(P)H quinone oxidoreductase 1 (NQO-1), and heme oxygenase 1 (HO-1) (11).
In numerous animal models, sulforaphane (often administered ip, iv, or sc, rather than po) was shown to exert protective effects on many tissues and organs by activating the Nrf2/ARE-dependent pathway (12). For example, sulforaphane reduced contrast agent-induced kidney damage in rats by increasing Nrf2 nuclear translocation and upregulating the expression of HO-1 and NQO-1 (13). Upregulation of the Nrf2 pathway by sulforaphane also attenuated oxidative damage-induced vascular endothelial cell injury in a mouse model of type 2 diabetes mellitus (14). In a rat model of hepatic ischemia reperfusion injury — whereby cellular damage is caused by the restoration of oxygen delivery to a hypoxic liver — pre-treatment with sulforaphane limited the reduction in glutathione (GSH) and the antioxidant enzymes, superoxide dismutase (SOD) and GSH peroxidase (GPx). Sulforaphane also upregulated the expression of Nrf2, NQO-1, and HO-1, and decreased ischemic death and apoptosis of liver cells (15).
Human studies are limited. In a placebo-controlled study, oral sulforaphane (in the form of broccoli sprout homogenate) increased the expression of NQO-1 and HO-1 in the upper airway within two hours of ingestion (16). Yet, in a recent trial in patients with chronic obstructive pulmonary disease (COPD), oral administration of sulforaphane for four weeks failed to induce the expression of Nrf2, NQO-1, and HO-1 in alveolar macrophages, bronchial epithelial cells, or peripheral blood mononuclear cells (17).
![Figure 3. Isothiocyanates Target Nrf2 and NF-kappaB Pathways. Isothiocyanates inhibit NF-kappaB-mediated inflammation and increase the expression of phase II detoxifying/antioxidant enzymes via the Nrf2 signaling pathway. [A] Isothiocyanates induce the nuclear translocation of Nrf2 and increase the expression of Nrf2 target genes coding for phase II enzymes and antioxidant enzymes. [B] Isothiocyanates may prevent (1) the phosphorylation of NF-kappaB inhibitor, IkappaB; (2) th nuclear translocation of NF-kappaB; and (3) the transcriptional activity of NF-kappa B.](/sites/lpi.oregonstate.edu/files/isothiocyanates-figure-3-1200px.png)
Anti-inflammatory activity
The therapeutic potential of sulforaphane has also been linked to its capacity to target pro-inflammatory pathways. Sulforaphane was found to attenuate pancreatic injury in a mouse model of acute pancreatitis by stimulating Nrf2-induced antioxidant enzymes (18). Concomitantly, sulforaphane significantly reduced the nuclear translocation of the pro-inflammatory transcription factor nuclear factor (NF)-κB in pancreatic acinar cells, downregulating the expression of NF-κB target genes that code for pro-inflammatory mediators, such as tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1β), and IL-6 (Figure 3) (18). Through inhibiting the NF-κB pathway, sulforaphane also targets other mediators of the inflammatory response, including the enzymes cyclooxygenase-2 (COX-2), prostaglandin E (PGE) synthase, and inducible nitric oxide synthase (iNOS). Sulforaphane exhibited anti-inflammatory effects in the lungs of mice with lipopolysaccharide (LPS)-induced acute respiratory distress syndrome (ARDS) by downregulating the expression of NF-κB, IL-6, TNF-α and COX-2, as well as decreasing production of nitric oxide (NO) and PGE2 (19). Other isothiocyanates have been shown to prevent the degradation of the NF-κB inhibitor, IκB, the nuclear translocation of NF-κB, and/or the transcriptional activity of NF-κB in vitro or in cultured cells (Figure 3) (20), which all can lead to a decrease in inflammatory responses.
The modulation of Nrf2 and NF-κB signaling pathways by isothiocyanates is especially relevant to the prevention of cancer because both oxidative stress and inflammation are significant contributors in the development and progression of cancer.
Anticancer activity
Biotransformation enzymes play important roles in the metabolism and elimination of a variety of chemicals, including drugs, toxins, and carcinogens. In general, phase I metabolizing enzymes catalyze reactions that increase the reactivity of hydrophobic (fat-soluble) compounds, preparing them for reactions catalyzed by phase II biotransformation enzymes. Reactions catalyzed by phase II enzymes generally increase water solubility and promote the elimination of the compound from the body (21).
Inhibition of phase I biotransformation enzymes
Isothiocyanates have been found to modulate the activity of phase I biotransformation enzymes, especially those of the cytochrome P450 (CYP) family. Using a primary rat hepatocyte-based model, both aliphatic (e.g., sulforaphane, AITC) and aromatic (e.g., BITC, PEITC) isothiocyanates (at 20-40 μM) have been found to downregulate CYP3A2 mRNA expression, as well as the activity of benzyloxyquinoline debenzylase, a marker of CYP3As (22). Aromatic isothiocyanates were also able to upregulate CYP1A1 and CYP1A2 mRNA expression and the activity of ethoxyresorufin-O-deethylase (EROD), a marker of CYP1A1/2 activities (22). In this model, sulforaphane inhibited EROD activity, yet failed to affect CYP1A1/2 mRNA expression (22). Using human liver microsomes, it was also recently reported that sulforaphane metabolites (0-200 μM) had little-to-no effect on the activities of CYP1A2, CYP2B6, CYP2D6, and CYP3A4 (23). The ability of PEITC to alter the expression and activity of CYP enzymes has been generally associated with a protective effect against (pro)carcinogen-induced tumor development in animal experiments (reviewed in 24). Increasing the activity of biotransformation enzymes may be beneficial if the elimination of potential carcinogens or toxins is enhanced. Yet, some procarcinogens require phase I enzymes in order to become active carcinogens capable of binding DNA and forming cancer-causing DNA adducts. Inhibition of specific CYP enzymes involved in carcinogen activation has been found to prevent the development of cancer in animal models (3).
Induction of phase II detoxifying enzymes
Many isothiocyanates are potent inducers of phase II detoxifying enzymes, including GSTs, UDP-glucuronosyl transferases (UGTs), NQO1, and glutamate cysteine ligase (GCL), that protect cells from DNA damage by carcinogens and reactive oxygen species (ROS) (25). The genes for these and other phase II enzymes contain AREs and are therefore under the control of Nrf2 (see Antioxidant activity). Limited data from clinical trials suggest that glucosinolate-rich foods can increase phase II enzyme activity in humans. When smokers consumed 170 g/day (6 oz/day) of watercress, urinary excretion of glucuronidated nicotine metabolites increased significantly, suggesting UGT activity increased (26). Brussels sprouts are rich in a number of glucosinolates, including precursors of AITC and sulforaphane. Consumption of 300 g/day (11 oz/day) of Brussels sprouts for one week significantly increased plasma and intestinal GST concentrations in nonsmoking men (27, 28).
Induction of cell cycle arrest and apoptosis
After a cell divides, it passes through a sequence of stages — collectively known as the cell cycle — before dividing again. Following DNA damage, the cell cycle can be transiently arrested to allow for DNA repair or activation of pathways leading to programmed cell death (apoptosis) when the damage cannot be repaired (29). Defective cell cycle regulation and pro-survival mechanisms may result in the propagation of mutations that contribute to the development of cancer. Isothiocyanates have been found to modulate the expression of the cell cycle regulators, cyclins and cyclin-dependent kinases (CDK), as well as trigger apoptosis in a number of cancer cell lines (20). In a mouse model of colorectal cancer, oral administration of PEITC reduced both the number and size of polyps; these changes were associated with activation of the CDK inhibitor, p21, inhibition of various cyclins (A, D1, and E), and induction of apoptosis (30). In a transgenic prostate adenocarcinoma mouse model, BITC limited the progress of prostatic intraepithelial neoplasia (PIN) to a well-differentiated carcinoma (31). This was related to a decreased expression of Ki67 (a marker of cell proliferation) and a downregulation of cyclin A, cyclin D1, and CDK2, which regulate cell cycle progression (31).
Inhibition of cell migration and invasion
The epithelial-to-mesenchymal transition (EMT) describes a process of epithelial cell transformation whereby cells lose their polarity and adhesion properties while gaining migratory and invasive properties through the expression of mesenchymal genes. Inhibition of the EMT by sulforaphane in thyroid cancer cells has been associated with upregulation of an epithelial marker, E-cadherin, and downregulation of a transcription factor (SNAI2), a filament protein (vimentin), and various enzymes (matrix metalloprotein [MMP]-2 and MMP-9) known to contribute to EMT and promote migration (32). In a xenograft mouse model of breast cancer, BITC inhibited high fat diet-driven promotion of breast tumor growth, as well as lung and liver metastasis (33). This study suggested that BITC might prevent the infiltration of macrophages in the tumor environment (33). In another model of breast tumor metastasis, PEITC inhibited the migration of tumor cells to the brain after injection into the heart of mice, limiting the growth of metastatic brain tumors (34).
Inhibition of angiogenesis
To fuel their rapid growth, invasive tumors must also develop new capillaries from preexisting blood vessels by a process known as angiogenesis. Isothiocyanates have been shown to prevent the formation of capillary-like structures from human umbilical endothelial cells (reviewed in 35). Isothiocyanates likely inhibit the expression and function of hypoxia inducible factors (HIFs) that control angiogenesis, as reported in endothelial cells and malignant cell lines (35).
Epigenetic regulation of gene expression
In the nucleus of a cell, DNA is coiled around proteins called histones, thereby forming the chromatin. The N-terminal tails of histones are targets for multiple modifications, including phosphorylation, methylation, acetylation, ubiquitination, poly ADP ribosylation, and sumoylation. Histone modification patterns have differential effects on chromatin structure, and, in synergy with DNA methylation, are implicated in the regulating expression of the genome (36). Within gene regulatory regions, the acetylation of lysine residues of histone tails has been correlated with activation of transcription. Conversely, the deacetylation of histones by histone deacetylases (HDAC) restricts access of transcription factors to the DNA and suppresses transcription. Because abnormal epigenetic marks disrupt the expression of specific tumor suppressor genes in cancer cells, compounds that re-induce their transcription, like those inhibiting HDACs, can potentially promote differentiation and apoptosis in transformed (precancerous) cells (37).
Isothiocyanates have been found to inhibit HDAC expression and/or activity in cultured cancer cells (38-43). Moreover, in vivo evidence for HDAC inhibition by sulforaphane came from a mouse model using prostate cancer xenografts (44). In humans, HDAC activity was reduced in blood cells following ingestion of 68 g (one cup) of sulforaphane-rich broccoli sprouts (44). Isothiocyanates may also affect microRNA-mediated gene silencing. In bladder cancer cells, E-cadherin induction by sulforaphane was partly due to the upregulation of miR-200c expression resulting in the miR-200c-dependent suppression of ZEB-1, a transcriptional repressor of E-cadherin (45). PEITC inhibited androgen receptor (AR) transcriptional activity in prostate cancer cells by repressing miR-141 expression and miR-141-mediated downregulation of small heterodimer partner (shp), a repressor of AR (46).
Antibacterial activity
Bacterial infection with Helicobacter pylori is associated with a marked increase in the risk of peptic ulcer disease and gastric cancer (47). In the test tube and in tissue culture, purified sulforaphane inhibited the growth and killed multiple strains of H. pylori, including antibiotic resistant strains (48). In an animal model of H. pylori infection, sulforaphane administration for five days eradicated H. pylori from 8 out of 11 xenografts of human gastric tissue implanted in immune-compromised mice (49). In another H. pylori-infected mouse model, a functional Nrf2 pathway was found to be required for the reduction of gastric inflammation and infection in mice fed broccoli sprouts (50). In a small clinical trial, consumption of up to 56 g/day (2 oz/day) of glucoraphanin-rich broccoli sprouts for one week was associated with H. pylori eradication in only three out of nine gastritis patients (51). In another trial, daily consumption of 70 g/day (~2-3 servings/day) of glucoraphanin-rich broccoli sprouts for two months significantly reduced markers of inflammation and infection in H. pylori–infected volunteers compared to those who consumed alfalfa sprouts (50). However, the extent to which glucoraphanin was converted to sulforaphane in broccoli sprout-fed participants was not measured.
Disease Prevention
Cancer
Isothiocyanates are thought to play a prominent role in the potential anticancer and cardiovascular benefits associated with cruciferous vegetable consumption (52, 53). Genetic variations in the sequence of genes coding for GSTs may affect the activity of GSTs. Such variations have been identified in humans. Specifically, null variants of the GSTM1 and GSTT1 alleles contain large deletions, and individuals who inherit two copies of the GSTM1-null or GSTT1-null alleles cannot produce the corresponding GST enzymes (54). It has been proposed that a reduced GST activity in these individuals would slow the rate of excretion of isothiocyanates, thereby increasing tissue exposure to isothiocyanates after cruciferous vegetable consumption (55). In addition, GSTs are involved in "detoxifying" potentially harmful substances like carcinogens, suggesting that individuals with reduced GST activity might also be more susceptible to cancer (56-58). Further, induction of the expression and activity of GSTs and other phase II detoxification/antioxidant enzymes by isothiocyanates is an important defense mechanism against oxidative stress and damage associated with the development of diseases like cancer and cardiovascular disease (11). The ability of glucoraphanin-derived sulforaphane to reduce oxidative stress in different settings is linked to activation of the nuclear factor E2-related factor 2 (Nrf2)-dependent pathway (see Biological Activities). Yet, whether potential protection conferred by isothiocyanates via the Nrf2-dependent pathway is diminished in individuals carrying GST null variants is currently unknown. Some, but not all, observational studies have suggested that GST genotypes could influence the associations between cruciferous vegetable consumption and risk of disease (59).
Naturally occurring isothiocyanates and their metabolites have been found to inhibit the development of chemically-induced cancers of the lung, liver, esophagus, stomach, small intestine, colon, and breast in a variety of animal models (20). Although observational studies provide some evidence that higher intakes of cruciferous vegetables are associated with decreased cancer risk in humans (59), it is difficult to determine whether such protective effects are related to isothiocyanates or other factors associated with cruciferous vegetable consumption (see the article on Cruciferous Vegetables). Clinical evidence of a protective effect of isothiocyanates in humans is scarce. For example, in a recent randomized, cross-over intervention, administration of PEITC (40 mg/day for five days) caused a modest, yet significant, 7.7% reduction in the metabolic activation of the tobacco-specific lung carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, in cigarette smokers (60). Another randomized controlled trial in men with biochemically relapsing cancer after radical prostatectomy suggested that prostate-specific antigen (PSA) values tended to increase less in those given daily oral sulforaphane (4.4 or 26.6 mg/day) for six months compared to those receiving the placebo (61). In a recent double-blind, randomized, placebo-controlled trial in women with abnormal mammograms, two-to-eight week consumption of about 250 mg/day of broccoli seed extract (~220 mg of glucoraphanin/day) before surgery failed to affect the expression of markers of proliferation and gene expression, including ki-67, p21, HDACs, and acetylated histones, in breast tissues collected after surgery (62).
Sources
Food sources
Cruciferous vegetables
Cruciferous vegetables, such as bok choi, broccoli, Brussels sprouts, cabbage, cauliflower, horseradish, kale, kohlrabi, mustard, radish, rutabaga, turnip, and watercress, are rich sources of glucosinolate precursors of isothiocyanates (63). Unlike some other phytochemicals, glucosinolates are present in relatively high concentrations in commonly consumed portions of cruciferous vegetables. For example one-half cup of raw broccoli might provide more than 25 mg of total glucosinolates. The glucosinolate content of selected cruciferous vegetables is presented in Table 1 (64). Note that while the composition and content of glucosinolates in cruciferous vegetables are relatively stable, they depend on the genus and species and can vary greatly with plant growing and post-harvest storage conditions, as well as culinary processing.
Table 2 lists vegetables that are relatively good sources of some of the isothiocyanates that are currently being studied for their potential anticancer properties (65).
Amounts of isothiocyanates formed from glucosinolates in foods are variable and depend partly on food processing and preparation (see the article on Cruciferous Vegetables). In a recent study that examined total isothiocyanate content in 73 samples from nine types of raw cruciferous vegetables commonly consumed in the US (namely broccoli, cabbage, cauliflower, Brussels sprout, kale, collard green, mustard green, and turnip greens), an average yield of 16.2 µmol/100 g wet weight was reported, with a 41-fold difference of isothiocyanate yield across the vegetables. The lowest mean level of isothiocyanate yield was found with raw cauliflower (1.5 µmol/100 g), while raw mustard greens had the highest yield (61.3 µmol/100g) (66).
Broccoli sprouts
The amount of glucoraphanin, the precursor of sulforaphane, in broccoli seeds remains more or less constant as those seeds germinate and grow into mature plants. Thus, three-day old broccoli sprouts are concentrated sources of glucoraphanin, which contain 10 to 100 times more glucoraphanin by weight than mature broccoli plants (67). Broccoli sprouts that are certified to contain at least 73 mg of glucoraphanin (also called sulforaphane glucosinolate) per 1-oz serving are available in some health food and grocery stores.
Supplements
Dietary supplements containing extracts of broccoli sprouts, broccoli, and other cruciferous vegetables are available without a prescription. Some products are standardized to contain a minimum amount of glucosinolates and/or sulforaphane. However, the bioavailability of isothiocyanates was found to be much lower with the consumption of broccoli supplements devoid of myrosinase than with the consumption of fresh broccoli sprouts. Peak concentrations of sulforaphane metabolites were found to be eight- and five-times greater in plasma and urine, respectively, following fresh broccoli versus supplement consumption (68). Interestingly, total HDAC activity in peripheral blood mononuclear cells (PBMC) of broccoli sprout consumers was reported to be significantly lower than in PBMC of subjects who consumed the supplement (see Biological Activities) (69).
Safety
Adverse effects
No serious adverse effects of isothiocyanates in humans have been reported. The majority of animal studies have found that isothiocyanates inhibited the development of cancer when given prior to the chemical carcinogen (pre-initiation). However, very high intakes of PEITC or BITC (25 to 250 times higher than average human dietary isothiocyanate intakes) have been found to promote bladder cancer in rats when given after cancer initiation by a chemical carcinogen (70). The relevance of these findings to human urinary bladder cancer is not clear, since at least one prospective cohort study found cruciferous vegetable consumption to be inversely associated with the risk of bladder cancer in men (71). Other potential toxic effects reported in rodents have not been corroborated by observations in humans (20).
Pregnancy and lactation
Although high dietary intakes of glucosinolates from cruciferous vegetables are not known to have adverse effects during pregnancy or lactation, there is no information on the safety of purified isothiocyanates or supplements containing high doses of glucosinolates and/or isothiocyanates during pregnancy or lactation in humans.
Drug interactions
Isothiocyanates are not known to interact with any drugs or medications. However, the potential for isothiocyanates to inhibit various isoforms of the cytochrome P450 (CYP) family of enzymes raises the potential for interactions with drugs that are CYP substrates (see Biological Activities). Isothiocyanates may sensitize cancer cells to anticancer drugs and/or increase drug cytotoxicity, as shown in in vitro and animal models. Yet, these potential benefits of isothiocyanates in cancer therapy have not been explored in clinical trials (72).
Authors and Reviewers
Originally written in 2005 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in November 2008 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in March 2017 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in April 2017 by:
Emily Ho, Ph.D.
Principal Investigator, Linus Pauling Institute
Professor, College of Public Health and Human Sciences
Endowed Director, Moore Family Center for Whole Grain Foods,
Nutrition and Preventive Health
Oregon State University
Copyright 2005-2024 Linus Pauling Institute
References
1. Fahey JW, Zalcmann AT, Talalay P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry. 2001;56(1):5-51. (PubMed)
2. Zhang Y. Cancer-preventive isothiocyanates: measurement of human exposure and mechanism of action. Mutat Res. 2004;555(1-2):173-190. (PubMed)
3. Hecht SS. Chemoprevention by Isothiocyanates. In: Kelloff GJ, Hawk ET, Sigman CC, eds. Promising Cancer Chemopreventive Agents, Volume 1: Cancer Chemopreventive Agents Totowa, NJ: Humana Press; 2004:21-35.
4. Barba FJ, Nikmaram N, Roohinejad S, Khelfa A, Zhu Z, Koubaa M. Bioavailability of Glucosinolates and Their Breakdown Products: Impact of Processing. Front Nutr. 2016;3:24. (PubMed)
5. Luang-In V, Albaser AA, Nueno-Palop C, Bennett MH, Narbad A, Rossiter JT. Glucosinolate and Desulfo-glucosinolate Metabolism by a Selection of Human Gut Bacteria. Curr Microbiol. 2016;73(3):442-451. (PubMed)
6. Fahey JW, Wehage SL, Holtzclaw WD, et al. Protection of humans by plant glucosinolates: efficiency of conversion of glucosinolates to isothiocyanates by the gastrointestinal microflora. Cancer Prev Res (Phila). 2012;5(4):603-611. (PubMed)
7. Verkerk R, Schreiner M, Krumbein A, et al. Glucosinolates in Brassica vegetables: the influence of the food supply chain on intake, bioavailability and human health. Mol Nutr Food Res. 2009;53 Suppl 2:S219. (PubMed)
8. Li F, Hullar MA, Beresford SA, Lampe JW. Variation of glucoraphanin metabolism in vivo and ex vivo by human gut bacteria. Br J Nutr. 2011;106(3):408-416. (PubMed)
9. Hu R, Xu C, Shen G, et al. Identification of Nrf2-regulated genes induced by chemopreventive isothiocyanate PEITC by oligonucleotide microarray. Life Sci. 2006;79(20):1944-1955. (PubMed)
10. Wagner AE, Boesch-Saadatmandi C, Dose J, Schultheiss G, Rimbach G. Anti-inflammatory potential of allyl-isothiocyanate--role of Nrf2, NF-(kappa) B and microRNA-155. J Cell Mol Med. 2012;16(4):836-843. (PubMed)
11. Bryan HK, Olayanju A, Goldring CE, Park BK. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem Pharmacol. 2013;85(6):705-717. (PubMed)
12. Guerrero-Beltran CE, Calderon-Oliver M, Pedraza-Chaverri J, Chirino YI. Protective effect of sulforaphane against oxidative stress: recent advances. Exp Toxicol Pathol. 2012;64(5):503-508. (PubMed)
13. Zhao Z, Liao G, Zhou Q, Lv D, Holthfer H, Zou H. Sulforaphane attenuates contrast-induced nephropathy in rats via Nrf2/HO-1 pathway. Oxid Med Cell Longev. 2016;2016:9825623. (PubMed)
14. Wang Y, Zhang Z, Sun W, et al. Sulforaphane attenuation of type 2 diabetes-induced aortic damage was associated with the upregulation of Nrf2 expression and function. Oxid Med Cell Longev. 2014;2014:123963. (PubMed)
15. Chi X, Zhang R, Shen N, et al. Sulforaphane reduces apoptosis and oncosis along with protecting liver injury-induced ischemic reperfusion by activating the Nrf2/ARE pathway. Hepatol Int. 2015;9(2):321-329. (PubMed)
16. Riedl MA, Saxon A, Diaz-Sanchez D. Oral sulforaphane increases Phase II antioxidant enzymes in the human upper airway. Clin Immunol. 2009;130(3):244-251. (PubMed)
17. Wise RA, Holbrook JT, Criner G, et al. Lack of effect of oral sulforaphane administration on Nrf2 expression in COPD: a randomized, double-blind, placebo controlled trial. PLoS One. 2016;11(11):e0163716. (PubMed)
18. Dong Z, Shang H, Chen YQ, Pan LL, Bhatia M, Sun J. Sulforaphane protects pancreatic acinar cell injury by modulating Nrf2-mediated oxidative stress and NLRP3 inflammatory pathway. Oxid Med Cell Longev. 2016;2016:7864150. (PubMed)
19. Qi T, Xu F, Yan X, Li S, Li H. Sulforaphane exerts anti-inflammatory effects against lipopolysaccharide-induced acute lung injury in mice through the Nrf2/ARE pathway. Int J Mol Med. 2016;37(1):182-188. (PubMed)
20. Kumar G, Tuli HS, Mittal S, Shandilya JK, Tiwari A, Sandhu SS. Isothiocyanates: a class of bioactive metabolites with chemopreventive potential. Tumour Biol. 2015;36(6):4005-4016. (PubMed)
21. Lampe JW, Peterson S. Brassica, biotransformation and cancer risk: genetic polymorphisms alter the preventive effects of cruciferous vegetables. J Nutr. 2002;132(10):2991-2994. (PubMed)
22. La Marca M, Beffy P, Della Croce C, et al. Structural influence of isothiocyanates on expression of cytochrome P450, phase II enzymes, and activation of Nrf2 in primary rat hepatocytes. Food Chem Toxicol. 2012;50(8):2822-2830. (PubMed)
23. Vanduchova A, Tomankova V, Anzenbacher P, Anzenbacherova E. Influence of Sulforaphane Metabolites on Activities of Human Drug-Metabolizing Cytochrome P450 and Determination of Sulforaphane in Human Liver Cells. J Med Food. 2016;19(12):1141-1146. (PubMed)
24. Ioannides C, Konsue N. A principal mechanism for the cancer chemopreventive activity of phenethyl isothiocyanate is modulation of carcinogen metabolism. Drug Metab Rev. 2015;47(3):356-373. (PubMed)
25. Kensler TW, Talalay P. Inducers of enzymes that protect against carcinogens and oxidants: drug- and food-based approaches with dithiolethiones and sulforaphane. In: Kelloff GJ, Hawk ET, Sigman CC, eds. Promising Cancer Chemopreventive Agents, Volume 1: Cancer Chemopreventive Agents Totowa, NJ: Humana Press; 2004:3-20.
26. Hecht SS, Carmella SG, Murphy SE. Effects of watercress consumption on urinary metabolites of nicotine in smokers. Cancer Epidemiol Biomarkers Prev. 1999;8(10):907-913. (PubMed)
27. Nijhoff WA, Grubben MJ, Nagengast FM, et al. Effects of consumption of Brussels sprouts on intestinal and lymphocytic glutathione S-transferases in humans. Carcinogenesis. 1995;16(9):2125-2128. (PubMed)
28. Nijhoff WA, Mulder TP, Verhagen H, van Poppel G, Peters WH. Effects of consumption of brussels sprouts on plasma and urinary glutathione S-transferase class-alpha and -pi in humans. Carcinogenesis. 1995;16(4):955-957. (PubMed)
29. Stewart ZA, Westfall MD, Pietenpol JA. Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol Sci. 2003;24(3):139-145. (PubMed)
30. Khor TO, Cheung WK, Prawan A, Reddy BS, Kong AN. Chemoprevention of familial adenomatous polyposis in Apc(Min/+) mice by phenethyl isothiocyanate (PEITC). Mol Carcinog. 2008;47(5):321-325. (PubMed)
31. Cho HJ, Lim do Y, Kwon GT, et al. Benzyl isothiocyanate inhibits prostate cancer development in the transgenic adenocarcinoma mouse prostate (TRAMP) model, which is associated with the induction of cell cycle G1 arrest. Int J Mol Sci. 2016;17(2):264. (PubMed)
32. Wang L, Tian Z, Yang Q, et al. Sulforaphane inhibits thyroid cancer cell growth and invasiveness through the reactive oxygen species-dependent pathway. Oncotarget. 2015;6(28):25917-25931. (PubMed)
33. Kim M, Cho HJ, Kwon GT, et al. Benzyl isothiocyanate suppresses high-fat diet-stimulated mammary tumor progression via the alteration of tumor microenvironments in obesity-resistant BALB/c mice. Mol Carcinog. 2015;54(1):72-82. (PubMed)
34. Gupta P, Adkins C, Lockman P, Srivastava SK. Metastasis of breast tumor cells to brain is suppressed by phenethyl isothiocyanate in a novel in vivo metastasis model. PLoS One. 2013;8(6):e67278. (PubMed)
35. Cavell BE, Syed Alwi SS, Donlevy A, Packham G. Anti-angiogenic effects of dietary isothiocyanates: mechanisms of action and implications for human health. Biochem Pharmacol. 2011;81(3):327-336. (PubMed)
36. Delage B, Dashwood RH. Targeting the epigenome with dietary agents. Dietary Modulation of Cell Signaling Pathways: CRC Press; 2008.
37. Marks PA, Richon VM, Miller T, Kelly WK. Histone deacetylase inhibitors. Adv Cancer Res. 2004;91:137-168. (PubMed)
38. Abbaoui B, Telu KH, Lucas CR, et al. The impact of cruciferous vegetable isothiocyanates on histone acetylation and histone phosphorylation in bladder cancer. J Proteomics. 2017;156:94-103. (PubMed)
39. Batra S, Sahu RP, Kandala PK, Srivastava SK. Benzyl isothiocyanate-mediated inhibition of histone deacetylase leads to NF-kappaB turnoff in human pancreatic carcinoma cells. Mol Cancer Ther. 2010;9(6):1596-1608. (PubMed)
40. Clarke JD, Hsu A, Yu Z, Dashwood RH, Ho E. Differential effects of sulforaphane on histone deacetylases, cell cycle arrest and apoptosis in normal prostate cells versus hyperplastic and cancerous prostate cells. Mol Nutr Food Res. 2011;55(7):999-1009. (PubMed)
41. Pledgie-Tracy A, Sobolewski MD, Davidson NE. Sulforaphane induces cell type-specific apoptosis in human breast cancer cell lines. Mol Cancer Ther. 2007;6(3):1013-1021. (PubMed)
42. Rajendran P, Delage B, Dashwood WM, et al. Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: competing actions of 14-3-3 and Pin1 in HDAC3/SMRT corepressor complex dissociation/reassembly. Mol Cancer. 2011;10:68. (PubMed)
43. Rajendran P, Kidane AI, Yu TW, et al. HDAC turnover, CtIP acetylation and dysregulated DNA damage signaling in colon cancer cells treated with sulforaphane and related dietary isothiocyanates. Epigenetics. 2013;8(6):612-623. (PubMed)
44. Myzak MC, Tong P, Dashwood WM, Dashwood RH, Ho E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp Biol Med (Maywood). 2007;232(2):227-234. (PubMed)
45. Shan Y, Zhang L, Bao Y, et al. Epithelial-mesenchymal transition, a novel target of sulforaphane via COX-2/MMP2, 9/Snail, ZEB1 and miR-200c/ZEB1 pathways in human bladder cancer cells. J Nutr Biochem. 2013;24(6):1062-1069. (PubMed)
46. Xiao J, Gong AY, Eischeid AN, et al. miR-141 modulates androgen receptor transcriptional activity in human prostate cancer cells through targeting the small heterodimer partner protein. Prostate. 2012;72(14):1514-1522. (PubMed)
47. US National Cancer Institute. Helicobacter pylori and cancer. Available at: https://www.cancer.gov/about-cancer/causes-prevention/risk/infectious-agents/h-pylori-fact-sheet. Accessed 2/28/17.
48. Fahey JW, Haristoy X, Dolan PM, et al. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc Natl Acad Sci U S A. 2002;99(11):7610-7615. (PubMed)
49. Haristoy X, Angioi-Duprez K, Duprez A, Lozniewski A. Efficacy of sulforaphane in eradicating Helicobacter pylori in human gastric xenografts implanted in nude mice. Antimicrob Agents Chemother. 2003;47(12):3982-3984. (PubMed)
50. Yanaka A, Fahey JW, Fukumoto A, et al. Dietary sulforaphane-rich broccoli sprouts reduce colonization and attenuate gastritis in Helicobacter pylori-infected mice and humans. Cancer Prev Res (Phila). 2009;2(4):353-360. (PubMed)
51. Galan MV, Kishan AA, Silverman AL. Oral broccoli sprouts for the treatment of Helicobacter pylori infection: a preliminary report. Dig Dis Sci. 2004;49(7-8):1088-1090. (PubMed)
52. Bai Y, Wang X, Zhao S, Ma C, Cui J, Zheng Y. Sulforaphane protects against cardiovascular disease via Nrf2 activation. Oxid Med Cell Longev. 2015;2015:407580. (PubMed)
53. Higdon JV, Delage B, Williams DE, Dashwood RH. Cruciferous vegetables and human cancer risk: epidemiologic evidence and mechanistic basis. Pharmacol Res. 2007;55(3):224-236. (PubMed)
54. Coles BF, Kadlubar FF. Detoxification of electrophilic compounds by glutathione S-transferase catalysis: determinants of individual response to chemical carcinogens and chemotherapeutic drugs? Biofactors. 2003;17(1-4):115-130. (PubMed)
55. Seow A, Shi CY, Chung FL, et al. Urinary total isothiocyanate (ITC) in a population-based sample of middle-aged and older Chinese in Singapore: relationship with dietary total ITC and glutathione S-transferase M1/T1/P1 genotypes. Cancer Epidemiol Biomarkers Prev. 1998;7(9):775-781. (PubMed)
56. Economopoulos KP, Choussein S, Vlahos NF, Sergentanis TN. GSTM1 polymorphism, GSTT1 polymorphism, and cervical cancer risk: a meta-analysis. Int J Gynecol Cancer. 2010;20(9):1576-1580. (PubMed)
57. Egner PA, Chen JG, Zarth AT, et al. Rapid and sustainable detoxication of airborne pollutants by broccoli sprout beverage: results of a randomized clinical trial in China. Cancer Prev Res (Phila). 2014;7(8):813-823. (PubMed)
58. Yuan JM, Murphy SE, Stepanov I, et al. 2-Phenethyl Isothiocyanate, Glutathione S-transferase M1 and T1 Polymorphisms, and Detoxification of Volatile Organic Carcinogens and Toxicants in Tobacco Smoke. Cancer Prev Res (Phila). 2016;9(7):598-606. (PubMed)
59. Traka MH. Chapter 9: Health benefits of glucosinolates. Advances in Botanical Research. 2016;80:247-279.
60. Yuan JM, Stepanov I, Murphy SE, et al. Clinical trial of 2-phenethyl isothiocyanate as an inhibitor of metabolic activation of a tobacco-specific lung carcinogen in cigarette smokers. Cancer Prev Res (Phila). 2016;9(5):396-405. (PubMed)
61. Cipolla BG, Mandron E, Lefort JM, et al. Effect of sulforaphane in men with biochemical recurrence after radical prostatectomy. Cancer Prev Res (Phila). 2015;8(8):712-719. (PubMed)
62. Atwell LL, Zhang Z, Mori M, et al. Sulforaphane bioavailability and chemopreventive activity in women scheduled for breast biopsy. Cancer Prev Res (Phila). 2015;8(12):1184-1191. (PubMed)
63. International Agency for Research on Cancer. Cruciferous vegetables, isothiocyanates and indoles. Cruciferous vegetables. France: IARC; 2004:1-12.
64. McNaughton SA, Marks GC. Development of a food composition database for the estimation of dietary intakes of glucosinolates, the biologically active constituents of cruciferous vegetables. Br J Nutr. 2003;90(3):687-697. (PubMed)
65. Ishida M, Hara M, Fukino N, Kakizaki T, Morimitsu Y. Glucosinolate metabolism, functionality and breeding for the improvement of Brassicaceae vegetables. Breed Sci. 2014;64(1):48-59. (PubMed)
66. Tang L, Paonessa JD, Zhang Y, Ambrosone CB, McCann SE. Total isothiocyanate yield from raw cruciferous vegetables commonly consumed in the United States. J Funct Foods. 2013;5(4):1996-2001. (PubMed)
67. Fahey JW, Zhang Y, Talalay P. Broccoli sprouts: an exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc Natl Acad Sci U S A. 1997;94(19):10367-10372. (PubMed)
68. Clarke JD, Hsu A, Riedl K, et al. Bioavailability and inter-conversion of sulforaphane and erucin in human subjects consuming broccoli sprouts or broccoli supplement in a cross-over study design. Pharmacol Res. 2011;64(5):456-463. (PubMed)
69. Clarke JD, Riedl K, Bella D, Schwartz SJ, Stevens JF, Ho E. Comparison of isothiocyanate metabolite levels and histone deacetylase activity in human subjects consuming broccoli sprouts or broccoli supplement. J Agric Food Chem. 2011;59(20):10955-10963. (PubMed)
70. Okazaki K, Umemura T, Imazawa T, Nishikawa A, Masegi T, Hirose M. Enhancement of urinary bladder carcinogenesis by combined treatment with benzyl isothiocyanate and N-butyl-N-(4-hydroxybutyl)nitrosamine in rats after initiation. Cancer Sci. 2003;94(11):948-952. (PubMed)
71. Michaud DS, Spiegelman D, Clinton SK, Rimm EB, Willett WC, Giovannucci EL. Fruit and vegetable intake and incidence of bladder cancer in a male prospective cohort. J Natl Cancer Inst. 1999;91(7):605-613. (PubMed)
72. Minarini A, Milelli A, Fimognari C, Simoni E, Turrini E, Tumiatti V. Exploring the effects of isothiocyanates on chemotherapeutic drugs. Expert Opin Drug Metab Toxicol. 2014;10(1):25-38. (PubMed)
Lignans
Contents
Summary
- Lignans are polyphenolic compounds found in plants. (More information)
- Lignan precursors are found in a wide variety of plant-based foods, including seeds, whole grains, legumes, fruit, and vegetables. (More information)
- Flaxseeds are the richest dietary source of lignan precursors. (More information)
- When consumed, lignan precursors may be converted to the enterolignans, enterodiol and enterolactone, by bacteria that normally colonize the human intestine. (More information)
- Enterodiol and enterolactone have weak estrogenic activity but may also exert biological effects through non-estrogenic mechanisms. (More information)
- Lignan-rich foods are part of a healthy diet, but the roles of lignans in the prevention of hormone-associated cancers, osteoporosis, and cardiovascular disease are not yet clear. (More information)
Introduction
The enterolignans, enterodiol and enterolactone (Figure 1), are formed by the action of intestinal bacteria on lignan precursors found in plants (1). Because enterodiol and enterolactone can mimic some of the effects of estrogens, their plant-derived precursors are classified as phytoestrogens. Lignan precursors that have been identified in the human diet include pinoresinol, lariciresinol, secoisolariciresinol, matairesinol, and others (Figure 2). Secoisolariciresinol and matairesinol were among the first lignan precursors identified in the human diet and are therefore the most extensively studied. Lignan precursors are found in a wide variety of foods, including flaxseeds, sesame seeds, legumes, whole grains, fruit, and vegetables. While most research on phytoestrogen-rich diets has focused on soy isoflavones, lignans are the principal source of dietary phytoestrogens in the typical Western diet (2, 3).


Metabolism and Bioavailability
When plant lignans are ingested, they can be metabolized by intestinal bacteria to the enterolignans, enterodiol and enterolactone, in the intestinal lumen and then absorbed into the bloodstream (4). Enterodiol can also be converted to enterolactone by intestinal bacteria. Thus, enterolactone levels measured in blood and urine reflect the activity of intestinal bacteria in addition to dietary intake of plant lignans. Not surprisingly, antibiotic use has been associated with lower serum enterolactone concentrations (5).
Because data on the lignan content of foods are limited, blood and urinary enterolactone levels are sometimes used as markers of dietary lignan intake. A pharmacokinetic study that measured plasma and urinary levels of enterodiol and enterolactone after a single dose (0.9 mg/kg of body weight) of secoisolariciresinol, the principal lignan in flaxseed, found that at least 40% was available to the body as enterodiol and enterolactone (6). Plasma enterodiol concentrations peaked at 73 nanomoles/liter (nmol/L) an average of 15 hours after ingestion of secoisolariciresinol, and plasma enterolactone concentrations peaked at 56 nmol/L an average of 20 hours after ingestion. Thus, substantial amounts of ingested plant lignans are available to humans in the form of enterodiol and enterolactone.
Considerable variation among individuals in urinary and serum enterodiol:enterolactone ratios has been observed in flaxseed feeding studies, suggesting that some individuals convert most enterodiol to enterolactone, while others convert relatively little (1). Individual differences in the metabolism of lignans, likely due to differing composition and activities of gut microbes, can influence the biological activities and health effects of these compounds (7). Several other factors, including antibiotic use, age, BMI, and smoking, may also help explain the variation of circulating enterolignan concentrations among individuals (7); these and other potential confounding factors should be controlled for in observational studies.
Biological Activities
Estrogenic and anti-estrogenic activities
Estrogens are signaling molecules (i.e., hormones) that exert their effects by binding to estrogen receptors within cells (Figure 3). The estrogen-receptor complex interacts with DNA to change the expression of estrogen-responsive genes. Estrogen receptors are present in numerous tissues other than those associated with reproduction, including bone, liver, heart, and brain (8). Although phytoestrogens can also bind to estrogen receptors, their estrogenic activity is much weaker than endogenous estrogens, and they may actually block or antagonize the effects of estrogen in some tissues (9). Scientists are interested in the tissue-selective activities of phytoestrogens because anti-estrogenic effects in reproductive tissue could help reduce the risk of hormone-associated cancers (breast, uterine, ovarian, and prostate cancers), while estrogenic effects in bone could help maintain bone mineral density. The enterolignans, enterodiol and enterolactone, are known to have weak estrogenic activity. At present, the extent to which enterolignans exert weak estrogenic and/or anti-estrogenic effects in humans is not well understood.

Estrogen receptor-independent activities
Enterolignans have biological activities that are unrelated to their interactions with estrogen receptors. By altering the activity of enzymes involved in estrogen metabolism, lignans may change the biological activity of endogenous estrogens (10). Lignans have also been shown to display antioxidant activity in laboratory studies (11), although the significance in humans is not entirely clear since lignans are rapidly and extensively metabolized. For example, a cross-sectional study found that a biomarker of oxidative damage was inversely associated with serum enterolactone concentrations in men (12), but this association could be due to enterolactone and/or other antioxidants present in lignan-rich foods. Moreover, enterolignans may have anti-inflammatory properties, as well as anti-proliferative and anticancer activities that are independent of estrogen signaling (13-15).
Disease Prevention
Cardiovascular disease
Diets rich in foods containing plant lignans (whole grains, nuts and seeds, legumes, and fruit and vegetables) have been consistently associated with reductions in risk of cardiovascular disease. However, it is likely that numerous nutrients and phytochemicals found in these foods contribute to their cardioprotection.
Higher dietary intakes of two lignans, matairesinol and secoisolariciresinol, were not linked to total cardiovascular disease in 16,162 middle-aged and older women participating in the Dutch PROSPECT European Prospective study Into Cancer and nutrition (EPIC) study (16). A large prospective cohort study conducted within Spain’s PREDIMED trial — a trial evaluating the effects of a Mediterranean diet on cardiovascular disease outcomes — followed 7,172 older adults at high risk of cardiovascular disease for a mean of 4.3 years (17). In this study, the highest quintile of dietary lignan intake (mean intake, 0.94 mg/day), measured by a food frequency questionnaire at baseline, was associated with a 49% lower risk of incident cardiovascular disease compared to the lowest quintile (mean intake, 0.44 mg/day of lignans). The primary dietary source of lignans in this cohort was virgin olive oil (17), which itself is known to be cardioprotective.
Coronary heart disease
In a prospective, nested case-control study in 334 middle-aged Finnish men, followed for an average of 7.7 years, higher serum enterolactone concentrations (a marker of plant lignan intake) were associated with a lower risk of acute coronary events (18). A prospective cohort study of 1,889 Finnish men followed for an average of 12 years, found those with the highest serum enterolactone concentrations were significantly less likely to die from coronary heart disease (CHD) or cardiovascular disease than those with the lowest concentrations (19). However, a study in male smokers did not find strong support for an association between serum enterolactone concentration and CHD (20). Additionally, a nested case-control study in men and women residing in the Netherlands did not find association between plasma concentrations of enterolactone or enterodiol and nonfatal myocardial infarction (236 cases and 283 controls), although the study population was young (ages 20-59 at baseline) and followed for only a mean of 4.5 years (21). A 2017 meta-analysis of three of these studies found no association between blood enterolactone concentration and non-fatal myocardial infarction (22). Moreover, dietary lignan intake was not linked to coronary cardiovascular disease in women participating in the Dutch PROSPECT EPIC study (16).
Clinical trials of lignan supplementation would be needed to determine the effects of lignans on coronary heart disease.
Cardiovascular risk factors
Blood lipids. Flaxseeds are among the richest sources of plant lignans in the human diet, but they are also good sources of other nutrients and phytochemicals with cardioprotective effects, such as omega-3 fatty acids (i.e., α-linolenic acid) and fiber. Supplementation trials have generally used ground or milled flaxseed (i.e., flax meal), which has a higher bioavailability of enterolignans compared to whole flaxseed (23). Five small clinical trials found that adding 30 to 50 g/day of flaxseed to the usual diet for 4 to 12 weeks resulted in modest 8%-20% decreases in low-density lipoprotein (LDL-cholesterol concentration (24-28), but four other trials did not observe significant reductions in LDL-cholesterol after adding 30 to 40 g/day of flaxseed to the diet (29-32). A double-blind, randomized controlled trial in adults, ages 44 to 75 years, found that supplementation with 40 g/day of flaxseed led to significant reductions in LDL-cholesterol after five weeks, but the cholesterol reductions were not statistically significant following 10 weeks’ supplementation (33). Additionally, a one-year clinical trial in postmenopausal women reported that supplementation with 40 g/day of flaxseed did not lower LDL-cholesterol compared to a placebo containing wheat germ (34). Most of these trials were in healthy participants free of cardiovascular disease. In a randomized, double-blind, placebo-controlled trial in 84 patients with peripheral artery disease, 30 g/day of flaxseed for 12 months did not reduce total or LDL-cholesterol compared to placebo, although cholesterol reductions within the flaxseed-supplemented group were evident at 1 month and 6 months compared to baseline but not at 12 months (35).
Any effect of flaxseed supplementation on blood lipids might be attributed to flaxseed constituents other than lignans (i.e., protein, fiber, omega-3 fatty acids, phytochemicals). At least two trials have investigated the effect of supplementation with isolated flaxseed lignan. In a randomized, double-blind, placebo-controlled cross-over trial in 22 healthy postmenopausal women, six-week supplementation with 500 mg/day of secoisolariciresinol diglucoside — derived from flaxseed — had no effect on LDL-cholesterol concentration or other measured blood lipids despite significant increases in serum enterolactone concentration (36). Additionally, a randomized, double-blind, placebo-controlled cross-over trial in 68 patients with type 2 diabetes and mild hypercholesterolemia found no effect of 360 mg/day of secoisolariciresinol diglucoside for 12 weeks on concentration of blood cholesterol or other blood lipids (37).
Large-scale supplementation trials with isolated lignans would be needed to determine whether lignans have cholesterol-lowering effects.
Blood pressure. A 2016 meta-analysis pooled the results of 15 randomized controlled trials, some in healthy participants and some in participants with chronic disease (i.e., type 2 diabetes, metabolic syndrome, peripheral arterial disease) or risk factors of cardiovascular disease. Supplementation with flaxseed was linked to a 2.9 mm Hg reduction in systolic blood pressure and a 2.4 mm Hg reduction in diastolic blood pressure; these blood pressure reductions were greater in trials of longer duration (≥12 weeks vs. <12 weeks; 38). Additionally, data stratification by supplement type revealed a benefit of supplemental flaxseed powder but not of lignan extracts containing 360 to 600 mg/day of secoisolariciresinol diglucoside (38), suggesting the non-lignan constituents of flaxseed may be responsible for any blood pressure-lowering effects.
Hormone-associated cancers
Breast cancer
Overall, there is limited evidence that dietary intake of plant lignans is associated with breast cancer risk; studies on the association have reported conflicting results. Two prospective cohort studies examining plant lignan intake and breast cancer found no association (39, 40). A more recent prospective study reported no association between total lignan intake and breast cancer in premenopausal women (41). In another prospective analysis, the same group of authors found postmenopausal women in the highest quartile of dietary lignan intake had a 17% lower risk of breast cancer compared to women in the lowest quartile, but this protective association was only observed in women with estrogen-positive and progesterone-positive tumors (42). A prospective cohort study of 51,823 postmenopausal Swedish women, followed for an average of 8.3 years, found that women in the highest quartile of lignan intake (≥1,036 mg/day) had a 17% lower risk of invasive breast tumors compared to those in the lowest quartile (lignan intake <712 mg/day) (43). In this study, a strong inverse association of lignan intake and breast cancer risk was observed in women who had used postmenopausal hormones at some point in their life, but no association was evident in those who had never used such hormones (43). A 2009 meta-analysis did not find an overall association between dietary lignan intake and breast cancer, but when the analysis was limited to postmenopausal women, the authors reported a 15% reduction in risk of breast cancer with high lignan intake (44). A similar result was found in a subsequent meta-analysis that included 11 prospective cohort and 10 case-control studies: no association of lignan intake and breast cancer was observed in women overall, but data stratification by menopausal status revealed that the highest lignan intakes were associated with a 14% lower risk of breast cancer among postmenopausal women (13 studies; 45).
Several studies, mainly case-control studies, have examined the relationship between blood or urine concentrations of enterolactone and breast cancer, reporting conflicting results (46-48). Two meta-analyses did not find an association between blood concentrations of enterolactone and breast cancer (44, 45).
At present, it is not clear whether high intakes of plant lignans or high circulating levels of enterolignans offer significant protection against breast cancer. Randomized controlled trials of lignan supplementation would be needed to address this question.
Endometrial and ovarian cancers
Overall, there is limited evidence that dietary lignan intake or circulating enterolignan concentration (a marker of lignan intake) is associated with endometrial cancer or ovarian cancer.
In a case-control study of lignans and endometrial cancer, US women with the highest intakes of plant lignans had the lowest risk of endometrial cancer, but the reduction in risk was statistically significant in postmenopausal women only (49). However, two population-based, case-control studies, one conducted in the US (50) and one in Australia (51), found dietary lignan intake was not linked to endometrial cancer. Moreover, a prospective case-control study in three different countries (US, Sweden, and Italy) did not find an association between circulating enterolactone and endometrial cancer in premenopausal or in postmenopausal women (52). A large case-cohort study among Danish women, ages 50-64 years, also reported no association of plasma enterolactone concentration and endometrial cancer (53).
In an early case-control study among US women ages 40 to 85 years, those with the highest combined intakes of the lignans, secoisolariciresinol and matairesinol, had the lowest risk of ovarian cancer — intakes greater than 708 mg/day of these lignans were associated with a 57% lower risk of ovarian cancer compared with intakes less than 304 mg/day (54). A case-control study in Australia reported no association of total dietary lignans and risk of ovarian cancer but found a significant, inverse association of matairesinol and lariciresinol, individually, with ovarian cancer (51). However, two other studies, a population-based case-control study in the US (55) and a prospective cohort study in Sweden (56) found no relationship between dietary lignan intake and ovarian cancer.
Although some of these studies support the hypothesis that diets rich in plant foods may be helpful in decreasing the risk of hormone-associated cancers, they do not provide strong evidence that lignans in particular are protective against endometrial or ovarian cancer.
Prostate cancer
Several observational studies have examined the association between dietary lignan intake or circulating enterolignan concentration (a marker of lignan intake) and prostate cancer, with most reporting no association. A meta-analysis of three studies (two population-based, case-control studies and one nested case-control study) found lignan intake was not linked to prostate cancer risk (57). Moreover, a meta-analysis of nested case-control studies did not find circulating concentration of enterolactone (total of 2,828 cases and 5,593 controls pooled from five studies) or enterodiol (total of 1,002 cases and 1,197 controls pooled from two studies) to be associated with prostate cancer (58). Yet another meta-analysis found no association between dietary lignan intake (total lignans, or matairesinol or secoisolariciresinol, separately) or circulating enterolactone concentration and prostate cancer risk (59). While adherence to a plant-based diet may be linked to a lower risk of prostate cancer (60), evidence that dietary lignans are protective is lacking.
Osteoporosis
Research on the effects of dietary lignan intake on osteoporosis risk is very limited. In a prospective cohort study of 2,580 postmenopausal women and 4,973 men enrolled in the European Prospective Investigation into Cancer (EPIC) study, dietary intake of matairesinol and secoisolariciresinol was not associated with bone density, when assessed by ultrasound of the heel bone (61). In two much smaller observational studies, urinary enterolactone excretion was used as a marker of dietary lignan intake. One study of 75 postmenopausal Korean women, who were classified as osteoporotic, osteopenic, or normal on the basis of bone mineral density (BMD) measurements, found that urinary enterolactone excretion was positively associated with BMD of the lumbar spine and hip (62). However, a study of 50 postmenopausal Dutch women found that higher levels of urinary enterolactone excretion were associated with higher rates of bone loss (63).
In two separate placebo controlled trials, supplementation of postmenopausal women with 25 to 40 g/day of ground flaxseed for three to four months did not significantly alter biochemical markers of bone formation or bone resorption (loss) (31, 64). In a placebo-controlled trial that included a daily walking intervention in both groups of older adults, supplementation with a flaxseed lignan complex (containing 543 mg/day of secoisolariciresinol) for six months had no effect on bone mineral density measured by DXA (65).
More research is necessary to determine whether high dietary intakes of plant lignans can decrease the risk or severity of osteoporosis.
Type 2 diabetes mellitus
More than 10% of the US population has type 2 diabetes mellitus and another 35% has impaired glucose control (prediabetes) that places them at high risk of developing type 2 diabetes (66). A number of dietary polyphenols found in plant-based foods may affect glucose metabolism and thus aid in the prevention or management of the condition. A few observational studies have examined the association of lignan intake and incidence of diabetes. A prospective cohort study in 6,547 Iranian adults, followed for a mean of 3.0 years, reported an inverse association between dietary lignan intake (measured by food frequency questionnaire) and incidence of type 2 diabetes (67). In particular, this study found the highest versus lowest quartile of lignan intake (median of 9.1 mg/day vs. 1.6 mg/day) to be associated with a 40% lower risk of type 2 diabetes (67). However, no association between lignan intake (highest vs. lowest quintile of intake, median of 2.3 mg/day vs. 0.6 mg/day) and type 2 diabetes was reported in a prospective, case-cohort study conducted in Europe that included more than 15,000 adults (EPIC-InterAct; 68).
Studies that utilize biomarkers of lignan intake, such as urinary concentrations of enterodiol or enterolactone, provide a more accurate estimate of lignan intake compared to self-reported questionnaires (69). A prospective, nested case-control study of two cohorts of US women participating in the Nurses’ Health Study (NHSI with mean age of 66 years and NHSII with mean age of 45 years) found lower concentrations of enterodiol and enterolactone in diabetic case subjects than in controls (70). Upon adjustment for potential confounders, only higher concentrations of urinary enterolactone were associated with a lower risk of developing type 2 diabetes, and this was driven by a significant association in the younger cohort of women (70). A nested case-control study within men and women participating in the Singapore Chinese Health Study (mean age, 59.8 years) reported no association between urinary enterodiol or enterolactone and type 2 diabetes (71).
Because higher lignan intakes may be a marker of a healthy diet in general, randomized controlled trials of lignan supplementation in healthy individuals would inform whether lignans affect glucose homeostasis and risk of developing type 2 diabetes. Interestingly, an eight-week, double-blind, placebo-controlled trial in hypercholesterolemic individuals found that a flaxseed lignan extract containing 600 mg/day of secoisolariciresinol diglucoside decreased fasting glucose concentrations compared to placebo, and the effect was stronger in those with higher baseline glucose concentrations (72). Supplementation with an extract containing 300 mg/day of secoisolariciresinol diglucoside had no effect on fasting glucose concentrations (72).
Mortality
A few studies have examined whether dietary lignan intake is related to all-cause and cause-specific mortality. The European Prospective Investigation into Cancer and Nutrition (EPIC)-Spain prospective cohort study investigated the relationship between lignan intake and all-cause mortality in 40,622 adults (ages 29-70 years) (73). After a mean follow-up of 13.6 years, dietary lignan intake was not associated with all-cause mortality (73). Additionally, dietary lignan intake was not linked to all-cause mortality in a much smaller study that followed 570 older Dutch men for 15 years (74). In this study, an inverse association was observed for intake of a specific lignan, matairesinol, with all-cause mortality and cardiovascular-related mortality, including death from coronary heart disease, although wine consumption modified these associations (74). However, an analysis of a 4.8-year trial that investigated the health effects of a Mediterranean diet in 7,172 older adults at high risk for cardiovascular disease (the PREDIMED trial in Spain) revealed that those in the highest quintile of lignan intake (mean of 0.94 mg/day) had a 40% lower risk of all-cause mortality compared to the lowest quintile (mean of 0.44 mg/day of lignans; 75). A 2017 meta-analysis found no association of lignan intake with all-cause mortality (3 studies mentioned above) or with mortality related to cardiovascular disease (2 studies; 76).
Other studies have assessed whether blood or urinary biomarkers of lignan intake are associated with mortality. In a prospective cohort study of 1,889 healthy, middle-aged Finnish men, followed for a mean of 12.2 years, the highest quartile of serum enterolactone concentration was associated with a 56% lower risk of coronary heart disease-related mortality and a 45% lower risk of cardiovascular disease-related mortality; no association of serum enterlactone and all-cause mortality was found in this study (19). However, serum enterolactone was not associated with coronary death in a case-cohort study in Finnish male smokers (20). In a national cross-sectional survey of US adults (NHANES 1999-2004), those in the highest tertile of urinary total enterolignan concentration had a lower risk of cardiovascular-related and all-cause mortality, and those with the highest urinary enterolactone concentrations had a significantly lower risk of all-cause mortality (77). These measures were not associated with mortality from cancer in this analysis, and urinary enterodiol concentration was not related to any of the mortality endpoints (77). A 2017 meta-analysis that combined results of these studies of lignan biomarkers found that enterolactone was inversely associated with both cardiovascular disease-related mortality and all-cause mortality (22). Most recently, a case-cohort study within the Danish Diet, Cancer and Health cohort found that higher pre-diagnostic plasma concentrations of enterolactone were linked to a lower risk of all-cause and diabetes-specific mortality among adults with type 2 diabetes (78).
Sources
Food sources
Lignans are present in a wide variety of plant foods, including seeds (flax, pumpkin, sunflower, poppy, sesame), whole grains (rye, oats, barley), bran (wheat, oat, rye), beans, fruit (particularly berries), vegetables, and beverages like tea, coffee, and wine (47, 79, 80). Secoisolariciresinol, matairesinol, pinoresinol, and lariciresinol contribute substantially to total dietary lignan intakes, although this varies with dietary pattern (80).
Flaxseed is by far the richest dietary source of plant lignans (81), and lignan bioavailability can be improved by crushing or milling flaxseed (23). Lignans are not associated with the oil fraction of foods, so flaxseed oils do not typically provide lignans unless ground flaxseed has been added to the oil. A variety of factors may affect the lignan content of plants, including geographic location, climate, maturity, and storage conditions. Table 1 provides the total lignan (secoisolariciresinol, matairesinol, pinoresinol, and lariciresinol) content of selected lignan-rich foods (82). The Phenol-Explorer (version 3.6) database lists content of 25 different lignans in various foods.
Surveys have found median total lignan intake to be 0.98 mg/day in the Netherlands (83), 0.85 mg/day in Canada (84), and 0.76 mg/day in Spain (85). Plant lignans are the principal source of phytoestrogens in the diets of people who do not typically consume soy foods. The daily phytoestrogen intake of postmenopausal women in the US was estimated to be less than 1 mg/day, with 80% from lignans and 20% from isoflavones (86).
Supplements
Dietary supplements containing lignans derived from flaxseed are available in the US without a prescription (87); secoisolariciresinol is the primary lignan in such supplements (82).
Safety
Adverse effects
Lignan precursors in food are not known to have any serious adverse effects. Flaxseeds, which are rich in lignan precursors as well as dietary fiber, may increase stool frequency or cause diarrhea in doses of 45 to 50 g/day in adults (24, 88). One small, placebo-controlled study found that 50 mg/day of sesame lignans (1:1 mixture of sesamin and episesamin) for 28 days did not result in any serious adverse effects, although abdominal flatulence was associated with the sesame lignan supplementation (89). The safety of lignan supplements in pregnant or lactating women has not been established; therefore, lignan supplements should be avoided by women who are pregnant, breast-feeding, or trying to conceive.
Authors and Reviewers
Originally written in 2004 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in December 2005 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in January 2010 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in March 2021 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in March 2021 by:
Susan McCann, R.D., Ph.D.
Member and Professor
Roswell Park Comprehensive Cancer Center
Buffalo, New York
Copyright 2004-2024 Linus Pauling Institute
References
1. Lampe JW. Isoflavonoid and lignan phytoestrogens as dietary biomarkers. J Nutr. 2003;133 Suppl 3:956S-964S. (PubMed)
2. de Kleijn MJ, van der Schouw YT, Wilson PW, Grobbee DE, Jacques PF. Dietary intake of phytoestrogens is associated with a favorable metabolic cardiovascular risk profile in postmenopausal U.S. women: the Framingham study. J Nutr. 2002;132(2):276-282. (PubMed)
3. Valsta LM, Kilkkinen A, Mazur W, et al. Phyto-oestrogen database of foods and average intake in Finland. Br J Nutr. 2003;89 Suppl 1:S31-38. (PubMed)
4. Rowland I, Faughnan M, Hoey L, Wahala K, Williamson G, Cassidy A. Bioavailability of phyto-oestrogens. Br J Nutr. 2003;89 Suppl 1:S45-58. (PubMed)
5. Kilkkinen A, Pietinen P, Klaukka T, Virtamo J, Korhonen P, Adlercreutz H. Use of oral antimicrobials decreases serum enterolactone concentration. Am J Epidemiol. 2002;155(5):472-477. (PubMed)
6. Kuijsten A, Arts IC, Vree TB, Hollman PC. Pharmacokinetics of enterolignans in healthy men and women consuming a single dose of secoisolariciresinol diglucoside. J Nutr. 2005;135(4):795-801. (PubMed)
7. Halldin E, Eriksen AK, Brunius C, et al. Factors explaining interpersonal variation in plasma enterolactone concentrations in humans. Mol Nutr Food Res. 2019;63(16):e1801159. (PubMed)
8. National Cancer Institute. Understanding Estrogen Receptors/SERMs. National Cancer Institute. January, 2005. http://www.cancer.gov/cancertopics/understandingcancer/estrogenreceptors. Accessed 1/15/10.
9. Wang LQ. Mammalian phytoestrogens: enterodiol and enterolactone. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;777(1-2):289-309. (PubMed)
10. Brooks JD, Thompson LU. Mammalian lignans and genistein decrease the activities of aromatase and 17beta-hydroxysteroid dehydrogenase in MCF-7 cells. J Steroid Biochem Mol Biol. 2005;94(5):461-467. (PubMed)
11. Moree SS, Rajesha J. Investigation of in vitro and in vivo antioxidant potential of secoisolariciresinol diglucoside. Mol Cell Biochem. 2013;373(1-2):179-187. (PubMed)
12. Vanharanta M, Voutilainen S, Nurmi T, et al. Association between low serum enterolactone and increased plasma F2-isoprostanes, a measure of lipid peroxidation. Atherosclerosis. 2002;160(2):465-469. (PubMed)
13. Sung MK, Lautens M, Thompson LU. Mammalian lignans inhibit the growth of estrogen-independent human colon tumor cells. Anticancer Res. 1998;18(3A):1405-1408. (PubMed)
14. Yeung AWK, Tzvetkov NT, Balacheva AA, et al. Lignans: quantitative analysis of the research literature. Front Pharmacol. 2020;11:37. (PubMed)
15. Zalesak F, Bon DJD, Pospisil J. Lignans and neolignans: Plant secondary metabolites as a reservoir of biologically active substances. Pharmacol Res. 2019;146:104284. (PubMed)
16. van der Schouw YT, Kreijkamp-Kaspers S, Peeters PH, Keinan-Boker L, Rimm EB, Grobbee DE. Prospective study on usual dietary phytoestrogen intake and cardiovascular disease risk in Western women. Circulation. 2005;111(4):465-471. (PubMed)
17. Tresserra-Rimbau A, Rimm EB, Medina-Remon A, et al. Inverse association between habitual polyphenol intake and incidence of cardiovascular events in the PREDIMED study. Nutr Metab Cardiovasc Dis. 2014;24(6):639-647. (PubMed)
18. Vanharanta M, Voutilainen S, Lakka TA, van der Lee M, Adlercreutz H, Salonen JT. Risk of acute coronary events according to serum concentrations of enterolactone: a prospective population-based case-control study. Lancet. 1999;354(9196):2112-2115. (PubMed)
19. Vanharanta M, Voutilainen S, Rissanen TH, Adlercreutz H, Salonen JT. Risk of cardiovascular disease-related and all-cause death according to serum concentrations of enterolactone: Kuopio Ischaemic Heart Disease Risk Factor Study. Arch Intern Med. 2003;163(9):1099-1104. (PubMed)
20. Kilkkinen A, Erlund I, Virtanen MJ, Alfthan G, Ariniemi K, Virtamo J. Serum enterolactone concentration and the risk of coronary heart disease in a case-cohort study of Finnish male smokers. Am J Epidemiol. 2006;163(8):687-693. (PubMed)
21. Kuijsten A, Bueno-de-Mesquita HB, Boer JM, et al. Plasma enterolignans are not associated with nonfatal myocardial infarction risk. Atherosclerosis. 2009;203(1):145-152. (PubMed)
22. Rienks J, Barbaresko J, Nothlings U. Association of polyphenol biomarkers with cardiovascular disease and mortality risk: a systematic review and meta-analysis of observational studies. Nutrients. 2017;9(4):415. (PubMed)
23. Kuijsten A, Arts IC, van't Veer P, Hollman PC. The relative bioavailability of enterolignans in humans is enhanced by milling and crushing of flaxseed. J Nutr. 2005;135(12):2812-2816. (PubMed)
24. Cunnane SC, Hamadeh MJ, Liede AC, Thompson LU, Wolever TM, Jenkins DJ. Nutritional attributes of traditional flaxseed in healthy young adults. Am J Clin Nutr. 1995;61(1):62-68. (PubMed)
25. Arjmandi BH, Khan DA, Jurna S. Whole flaxseed consumption lowers serum LDL-cholesterol and lipoprotein(a) concentrations in postmenopausal women. Nutr Res. 1998;18:1203-1214.
26. Jenkins DJ, Kendall CW, Vidgen E, et al. Health aspects of partially defatted flaxseed, including effects on serum lipids, oxidative measures, and ex vivo androgen and progestin activity: a controlled crossover trial. Am J Clin Nutr. 1999;69(3):395-402. (PubMed)
27. Patade A, Devareddy L, Lucas EA, Korlagunta K, Daggy BP, Arjmandi BH. Flaxseed reduces total and LDL cholesterol concentrations in Native American postmenopausal women. J Womens Health (Larchmt). 2008;17(3):355-366. (PubMed)
28. Saxena S, Katare C. Evaluation of flaxseed formulation as a potential therapeutic agent in mitigation of dyslipidemia. Biomed J. 2014;37(6):386-390. (PubMed)
29. Clark WF, Kortas C, Heidenheim AP, Garland J, Spanner E, Parbtani A. Flaxseed in lupus nephritis: a two-year nonplacebo-controlled crossover study. J Am Coll Nutr. 2001;20(2 Suppl):143-148. (PubMed)
30. Lemay A, Dodin S, Kadri N, Jacques H, Forest JC. Flaxseed dietary supplement versus hormone replacement therapy in hypercholesterolemic menopausal women. Obstet Gynecol. 2002;100(3):495-504. (PubMed)
31. Lucas EA, Wild RD, Hammond LJ, et al. Flaxseed improves lipid profile without altering biomarkers of bone metabolism in postmenopausal women. J Clin Endocrinol Metab. 2002;87(4):1527-1532. (PubMed)
32. Stuglin C, Prasad K. Effect of flaxseed consumption on blood pressure, serum lipids, hemopoietic system and liver and kidney enzymes in healthy humans. J Cardiovasc Pharmacol Ther. 2005;10(1):23-27. (PubMed)
33. Bloedon LT, Balikai S, Chittams J, et al. Flaxseed and cardiovascular risk factors: results from a double blind, randomized, controlled clinical trial. J Am Coll Nutr. 2008;27(1):65-74. (PubMed)
34. Dodin S, Lemay A, Jacques H, Legare F, Forest JC, Masse B. The effects of flaxseed dietary supplement on lipid profile, bone mineral density, and symptoms in menopausal women: a randomized, double-blind, wheat germ placebo-controlled clinical trial. J Clin Endocrinol Metab. 2005;90(3):1390-1397. (PubMed)
35. Edel AL, Rodriguez-Leyva D, Maddaford TG, et al. Dietary flaxseed independently lowers circulating cholesterol and lowers it beyond the effects of cholesterol-lowering medications alone in patients with peripheral artery disease. J Nutr. 2015;145(4):749-757. (PubMed)
36. Hallund J, Tetens I, Bugel S, et al. Daily consumption for six weeks of a lignan complex isolated from flaxseed does not affect endothelial function in healthy postmenopausal women. J Nutr. 2006;136(9):2314-2318. (PubMed)
37. Pan A, Sun J, Chen Y, et al. Effects of a flaxseed-derived lignan supplement in type 2 diabetic patients: a randomized, double-blind, cross-over trial. PLoS One. 2007;2(11):e1148. (PubMed)
38. Ursoniu S, Sahebkar A, Andrica F, et al. Effects of flaxseed supplements on blood pressure: A systematic review and meta-analysis of controlled clinical trial. Clin Nutr. 2016;35(3):615-625. (PubMed)
39. Horn-Ross PL, Hoggatt KJ, West DW, et al. Recent diet and breast cancer risk: the California Teachers Study (USA). Cancer Causes Control. 2002;13(5):407-415. (PubMed)
40. Keinan-Boker L, van Der Schouw YT, Grobbee DE, Peeters PH. Dietary phytoestrogens and breast cancer risk. Am J Clin Nutr. 2004;79(2):282-288. (PubMed)
41. Touillaud MS, Thiebaut AC, Niravong M, Boutron-Ruault MC, Clavel-Chapelon F. No association between dietary phytoestrogens and risk of premenopausal breast cancer in a French cohort study. Cancer Epidemiol Biomarkers Prev. 2006;15(12):2574-2576. (PubMed)
42. Touillaud MS, Thiebaut AC, Fournier A, Niravong M, Boutron-Ruault MC, Clavel-Chapelon F. Dietary lignan intake and postmenopausal breast cancer risk by estrogen and progesterone receptor status. J Natl Cancer Inst. 2007;99(6):475-486. (PubMed)
43. Suzuki R, Rylander-Rudqvist T, Saji S, Bergkvist L, Adlercreutz H, Wolk A. Dietary lignans and postmenopausal breast cancer risk by oestrogen receptor status: a prospective cohort study of Swedish women. Br J Cancer. 2008;98(3):636-640. (PubMed)
44. Velentzis LS, Cantwell MM, Cardwell C, Keshtgar MR, Leathem AJ, Woodside JV. Lignans and breast cancer risk in pre- and post-menopausal women: meta-analyses of observational studies. Br J Cancer. 2009;100(9):1492-1498. (PubMed)
45. Buck K, Zaineddin AK, Vrieling A, Linseisen J, Chang-Claude J. Meta-analyses of lignans and enterolignans in relation to breast cancer risk. Am J Clin Nutr. 2010;92(1):141-153. (PubMed)
46. Velentzis LS, Woodside JV, Cantwell MM, Leathem AJ, Keshtgar MR. Do phytoestrogens reduce the risk of breast cancer and breast cancer recurrence? What clinicians need to know. Eur J Cancer. 2008;44(13):1799-1806. (PubMed)
47. Adlercreutz H. Lignans and human health. Crit Rev Clin Lab Sci. 2007;44(5-6):483-525. (PubMed)
48. Boccardo F, Puntoni M, Guglielmini P, Rubagotti A. Enterolactone as a risk factor for breast cancer: a review of the published evidence. Clin Chim Acta. 2006;365(1-2):58-67. (PubMed)
49. Horn-Ross PL, John EM, Canchola AJ, Stewart SL, Lee MM. Phytoestrogen intake and endometrial cancer risk. J Natl Cancer Inst. 2003;95(15):1158-1164. (PubMed)
50. Bandera EV, Williams MG, Sima C, et al. Phytoestrogen consumption and endometrial cancer risk: a population-based case-control study in New Jersey. Cancer Causes Control. 2009;20(7):1117-1127. (PubMed)
51. Neill AS, Ibiebele TI, Lahmann PH, et al. Dietary phyto-oestrogens and the risk of ovarian and endometrial cancers: findings from two Australian case-control studies. Br J Nutr. 2014;111(8):1430-1440. (PubMed)
52. Zeleniuch-Jacquotte A, Lundin E, Micheli A, et al. Circulating enterolactone and risk of endometrial cancer. Int J Cancer. 2006;119(10):2376-2381. (PubMed)
53. Aarestrup J, Kyro C, Knudsen KE, et al. Plasma enterolactone and incidence of endometrial cancer in a case-cohort study of Danish women. Br J Nutr. 2013;109(12):2269-2275. (PubMed)
54. McCann SE, Freudenheim JL, Marshall JR, Graham S. Risk of human ovarian cancer is related to dietary intake of selected nutrients, phytochemicals and food groups. J Nutr. 2003;133(6):1937-1942. (PubMed)
55. Bandera EV, King M, Chandran U, Paddock LE, Rodriguez-Rodriguez L, Olson SH. Phytoestrogen consumption from foods and supplements and epithelial ovarian cancer risk: a population-based case control study. BMC Womens Health. 2011;11:40. (PubMed)
56. Hedelin M, Lof M, Andersson TM, Adlercreutz H, Weiderpass E. Dietary phytoestrogens and the risk of ovarian cancer in the women's lifestyle and health cohort study. Cancer Epidemiol Biomarkers Prev. 2011;20(2):308-317. (PubMed)
57. He J, Wang S, Zhou M, Yu W, Zhang Y, He X. Phytoestrogens and risk of prostate cancer: a meta-analysis of observational studies. World J Surg Oncol. 2015;13:231. (PubMed)
58. Perez-Cornago A, Appleby PN, Boeing H, et al. Circulating isoflavone and lignan concentrations and prostate cancer risk: a meta-analysis of individual participant data from seven prospective studies including 2,828 cases and 5,593 controls. Int J Cancer. 2018;143(11):2677-2686. (PubMed)
59. Zhang Q, Feng H, Qluwakemi B, et al. Phytoestrogens and risk of prostate cancer: an updated meta-analysis of epidemiologic studies. Int J Food Sci Nutr. 2017;68(1):28-42. (PubMed)
60. Livingstone TL, Beasy G, Mills RD, et al. Plant bioactives and the prevention of prostate cancer: evidence from human studies. Nutrients. 2019;11(9):2245. (PubMed)
61. Kuhnle GG, Ward HA, Vogiatzoglou A, et al. Association between dietary phyto-oestrogens and bone density in men and postmenopausal women. Br J Nutr. 2011;106(7):1063-1069. (PubMed)
62. Kim MK, Chung BC, Yu VY, et al. Relationships of urinary phyto-oestrogen excretion to BMD in postmenopausal women. Clin Endocrinol (Oxf). 2002;56(3):321-328. (PubMed)
63. Kardinaal AF, Morton MS, Bruggemann-Rotgans IE, van Beresteijn EC. Phyto-oestrogen excretion and rate of bone loss in postmenopausal women. Eur J Clin Nutr. 1998;52(11):850-855. (PubMed)
64. Brooks JD, Ward WE, Lewis JE, et al. Supplementation with flaxseed alters estrogen metabolism in postmenopausal women to a greater extent than does supplementation with an equal amount of soy. Am J Clin Nutr. 2004;79(2):318-325. (PubMed)
65. Cornish SM, Chilibeck PD, Paus-Jennsen L, et al. A randomized controlled trial of the effects of flaxseed lignan complex on metabolic syndrome composite score and bone mineral in older adults. Appl Physiol Nutr Metab. 2009;34(2):89-98. (PubMed)
66. Centers for Disease Control and Prevention. National Diabetes Statistics Report, 2020. Atlanta, GA: Centers for Disease Control and Prevention, U.S. Dept of Health and Human Services; 2020. Accessed 3/29/21.
67. Esfandiar Z, Hosseini-Esfahani F, Mirmiran P, Yuzbashian E, Azizi F. The association of dietary polyphenol intake with the risk of type 2 diabetes: Tehran Lipid and Glucose Study. Diabetes Metab Syndr Obes. 2020;13:1643-1652. (PubMed)
68. Zamora-Ros R, Forouhi NG, Sharp SJ, et al. The association between dietary flavonoid and lignan intakes and incident type 2 diabetes in European populations: the EPIC-InterAct study. Diabetes Care. 2013;36(12):3961-3970. (PubMed)
69. Zamora-Ros R, Rabassa M, Llorach R, Gonzalez CA, Andres-Lacueva C. Application of dietary phenolic biomarkers in epidemiology: past, present, and future. J Agric Food Chem. 2012;60(27):6648-6657. (PubMed)
70. Sun Q, Wedick NM, Pan A, et al. Gut microbiota metabolites of dietary lignans and risk of type 2 diabetes: a prospective investigation in two cohorts of U.S. women. Diabetes Care. 2014;37(5):1287-1295. (PubMed)
71. Talaei M, Lee BL, Ong CN, et al. Urine phyto-oestrogen metabolites are not significantly associated with risk of type 2 diabetes: the Singapore Chinese health study. Br J Nutr. 2016;115(9):1607-1615. (PubMed)
72. Zhang W, Wang X, Liu Y, et al. Dietary flaxseed lignan extract lowers plasma cholesterol and glucose concentrations in hypercholesterolaemic subjects. Br J Nutr. 2008;99(6):1301-1309. (PubMed)
73. Zamora-Ros R, Jimenez C, Cleries R, et al. Dietary flavonoid and lignan intake and mortality in a Spanish cohort. Epidemiology. 2013;24(5):726-733. (PubMed)
74. Milder IE, Feskens EJ, Arts IC, Bueno-de-Mesquita HB, Hollman PC, Kromhout D. Intakes of 4 dietary lignans and cause-specific and all-cause mortality in the Zutphen Elderly Study. Am J Clin Nutr. 2006;84(2):400-405. (PubMed)
75. Tresserra-Rimbau A, Rimm EB, Medina-Remon A, et al. Polyphenol intake and mortality risk: a re-analysis of the PREDIMED trial. BMC Med. 2014;12:77. (PubMed)
76. Grosso G, Micek A, Godos J, et al. Dietary flavonoid and lignan intake and mortality in prospective cohort studies: systematic review and dose-response meta-analysis. Am J Epidemiol. 2017;185(12):1304-1316. (PubMed)
77. Reger MK, Zollinger TW, Liu Z, Jones J, Zhang J. Urinary phytoestrogens and cancer, cardiovascular, and all-cause mortality in the continuous National Health and Nutrition Examination Survey. Eur J Nutr. 2016;55(3):1029-1040. (PubMed)
78. Eriksen AK, Kyro C, Norskov NP, et al. Pre-diagnostic plasma enterolactone concentrations are associated with lower mortality among individuals with type 2 diabetes: a case-cohort study in the Danish Diet, Cancer and Health cohort. Diabetologia. 2019;62(6):959-969. (PubMed)
79. Meagher LP, Beecher GR. Assessment of data on the lignan content of foods. J Food Compos Anal. 2000;13(6):935-947.
80. Durazzo A, Lucarini M, Camilli E, et al. Dietary lignans: definition, description and research trends in databases development. Molecules. 2018;23(12):3251. (PubMed)
81. Thompson LU. Experimental studies on lignans and cancer. Baillieres Clin Endocrinol Metab. 1998;12(4):691-705. (PubMed)
82. Milder IE, Arts IC, van de Putte B, Venema DP, Hollman PC. Lignan contents of Dutch plant foods: a database including lariciresinol, pinoresinol, secoisolariciresinol and matairesinol. Br J Nutr. 2005;93(3):393-402. (PubMed)
83. Milder IE, Feskens EJ, Arts IC, Bueno de Mesquita HB, Hollman PC, Kromhout D. Intake of the plant lignans secoisolariciresinol, matairesinol, lariciresinol, and pinoresinol in Dutch men and women. J Nutr. 2005;135(5):1202-1207. (PubMed)
84. Cotterchio M, Boucher BA, Kreiger N, Mills CA, Thompson LU. Dietary phytoestrogen intake--lignans and isoflavones--and breast cancer risk (Canada). Cancer Causes Control. 2008;19(3):259-272. (PubMed)
85. Moreno-Franco B, Garcia-Gonzalez A, Montero-Bravo AM, et al. Dietary alkylresorcinols and lignans in the Spanish diet: development of the alignia database. J Agric Food Chem. 2011;59(18):9827-9834. (PubMed)
86. de Kleijn MJ, van der Schouw YT, Wilson PW, et al. Intake of dietary phytoestrogens is low in postmenopausal women in the United States: the Framingham study(1-4). J Nutr. 2001;131(6):1826-1832. (PubMed)
87. Natural Medicines database. Available at: https://naturalmedicines.therapeuticresearch.com/. Accessed 3/1/21.
88. Clark WF, Parbtani A, Huff MW, et al. Flaxseed: a potential treatment for lupus nephritis. Kidney Int. 1995;48(2):475-480. (PubMed)
89. Tomimori N, Tanaka Y, Kitagawa Y, Fujii W, Sakakibara Y, Shibata H. Pharmacokinetics and safety of the sesame lignans, sesamin and episesamin, in healthy subjects. Biopharm Drug Dispos. 2013;34(8):462-473. (PubMed)
Phytosterols
Contents
Summary
- Plant sterols and plant stanols, known commonly as phytosterols, are plant-derived compounds that are structurally related to cholesterol. (More information)
- Early human diets were likely rich in phytosterols, providing as much as 1 g/day; however, the typical Western diet today is relatively low in phytosterols. (More information)
- Although phytosterols are present in the diet in amounts similar to cholesterol, they are poorly absorbed and blood concentrations tend to be low. After absorption into enterocytes, phytosterols are actively excreted back into the intestinal lumen by the ATP-binding cassette transporter, ABCG5/G8. (More information)
- Phytosterols interfere with the intestinal absorption of dietary cholesterol by displacing cholesterol from micelles; they also facilitate the excretion of biliary cholesterol in the feces. (More information)
- Numerous clinical trials have demonstrated that daily consumption of phytosterols from phytosterol-enriched foods can significantly lower serum low-density lipoprotein (LDL)-cholesterol. An average phytosterol intake of 2 g/day lowers serum LDL-cholesterol by 8%-10%. (More information)
- The effect of long-term use of foods enriched with phytosterols on cardiovascular risk is not known. (More information)
- The results of a few clinical trials suggested that phytosterol supplementation at relatively low doses can improve urinary tract symptoms related to benign prostatic hyperplasia, but further research is needed to confirm these findings. (More information)
- Good food sources of phytosterols include unrefined vegetable oils, whole grains, nuts, seeds, and legumes. (More information)
- Foods and beverages with added phytosterols are now available in many countries throughout the world, and some countries allow health claims on such commercial products. (More information)
- Consumption of phytosterol-enriched foods may have undesirable effects, such as a reduction in plasma carotenoid concentrations. (More information)
Introduction
Throughout much of human evolution, it is likely that large amounts of plant foods were consumed (1). In addition to being rich in fiber and plant protein, the diets of our ancestors were also rich in phytosterols — plant-derived compounds that are structurally very similar to cholesterol (Figure 1). There is increasing evidence to suggest that the reintroduction of plant foods providing phytosterols into the modern diet could improve serum lipid (cholesterol) profiles and help reduce the risk of cardiovascular disease (1).
Cholesterol in human blood and tissues is derived from the diet, as well as from endogenous cholesterol synthesis. In contrast, all phytosterols in human blood and tissues are derived from the diet because humans cannot synthesize phytosterols (2). While cholesterol is the predominant sterol in animals, including humans, a variety of sterols are found in plants (3). Nutritionists recognize two classes of phytosterols:
(1) Plant sterols have a double bond in the sterol ring. The most abundant sterols in plants and the human diet are β-sitosterol, campesterol, and stigmasterol (Figure 2).
(2) Plant stanols lack a double bond in the sterol ring. Stanols, especially sitostanol and campestanol, comprise only about 10% of total dietary phytosterols (Figure 3).



Definitions
Phytosterols: a collective term for plant-derived sterols and stanols.
Plant sterols or stanols: terms generally applied to plant-derived sterols or stanols; these phytochemicals are added to food or supplements.
Plant sterol or stanol esters: plant sterols or stanols that have been esterified by creating an ester bond between a fatty acid and the sterol or stanol. Esterification occurs in intestinal cells and is also an industrial process. Esterification makes plant sterols and stanols more fat-soluble so they are easily incorporated into fat-containing foods, including margarines and salad dressings. In this article, the weights of plant sterol and stanol esters are expressed as the equivalent weights of free (unesterified) sterols and stanols.
Metabolism and Bioavailability
Absorption and metabolism of dietary cholesterol
Dietary cholesterol must be incorporated into mixed micelles in order to be absorbed by the cells that line the intestine (enterocytes) (4). Mixed micelles are mixtures of bile salts, lipids, and sterols formed in the small intestine after a fat-containing meal is consumed. Transport across the apical membrane of enterocytes is mediated by intestinal cholesterol transporter, Niemann Pick C1-Like 1 (NPC1L1), which is also involved in the uptake of phytosterols (5). Inside the enterocyte, cholesterol is esterified in a reaction catalyzed by intestinal acyl-coenzyme A (CoA) cholesterol acyltransferases (ACATs; also present in the liver) and incorporated into triglyceride-rich lipoproteins known as chylomicrons, which are secreted into the intestinal lymphatics. The thoracic lymphatic duct then collects most of the lymph before draining into the systemic blood circulation (6). As circulating chylomicrons become depleted of triglycerides, they become chylomicron remnants, which are taken up by the liver. In the liver, cholesterol from chylomicron remnants may be repackaged into other lipoproteins for transport throughout the circulation or, alternatively, secreted into bile, which is released into the small intestine.
Absorption and metabolism of dietary phytosterols
Although varied diets typically contain similar amounts of phytosterols and cholesterol, serum phytosterol concentrations are usually several hundred times lower than serum cholesterol concentrations in humans (7). Less than 5% of dietary plant sterols and less than 0.5% of dietary plant stanols are systemically absorbed, in contrast to about 50%-60% of dietary cholesterol (8, 9). Like cholesterol, phytosterols must be incorporated into mixed micelles before they are taken up by enterocytes. Once inside the enterocyte, systemic absorption of phytosterols is inhibited by the activity of an efflux transporter, consisting of a pair of ATP-binding cassette (ABC) proteins known as ABCG5 and ABCG8. ABCG5 and ABCG8 each form one half of a transporter that secretes phytosterols and unesterified cholesterol from the enterocyte into the intestinal lumen. Phytosterols are secreted back into the intestine by ABCG5/G8 transporters at a much greater rate than cholesterol, resulting in much lower intestinal absorption of dietary phytosterols than cholesterol (10).
Within the enterocyte, phytosterols are not as readily esterified as cholesterol, so they are incorporated into chylomicrons at much lower concentrations. Those phytosterols that are incorporated into chylomicrons enter the circulation and are taken up by the liver. Once inside the liver, phytosterols are rapidly secreted into bile by hepatic ABCG5/G8 transporters. Although cholesterol is also secreted into bile, the rate of phytosterol secretion into bile is much greater than cholesterol secretion (11). Thus, the low serum concentrations of phytosterols relative to cholesterol can be explained by decreased intestinal absorption and increased excretion of phytosterols into bile.
Biological Activities
Effects on cholesterol absorption and excretion
It is well established that high intakes of plant sterols or stanols can lower serum total and low-density lipoprotein (LDL)-cholesterol concentrations in humans (see Cardiovascular disease). Different mechanisms appear to underlie the cholesterol-lowering effect of phytosterols (reviewed in 12). In the intestinal lumen, phytosterols displace cholesterol from mixed micelles and reduce cholesterol absorption (13). It is also suggested that phytosterols might interfere with the esterification and incorporation of cholesterol into chylomicrons inside the enterocytes (12). In a placebo-controlled, cross-over trial, the consumption of moderate (0.46 g/day) and high (2.1 g/day) phytosterol-enriched beverages reduced cholesterol absorption by about 10% and 25%, respectively (14). Moderate and high phytosterol intakes also significantly increased the excretion of biliary and dietary cholesterol in the feces by 36% and 74%, respectively (14). Although the mechanisms are currently not clear, phytosterols might facilitate cholesterol efflux from peripheral tissues and macrophages lining vessel walls. Cholesterol is then transported to the liver and incorporated into bile stored in the gallbladder. While plant sterols may promote the hepatobiliary secretion of cholesterol into the intestinal lumen, they are also hypothesized to facilitate the disposal of cholesterol via a nonbiliary route called transintestinal cholesterol efflux (TICE) (12).
Effects on cholesterol metabolism
A decrease in intestinal-derived cholesterol entering the circulation as chylomicrons triggers the endogenous production of cholesterol in order to maintain cholesterol homeostasis (14). Cell surface LDL-receptor expression is also up-regulated to enhance a receptor-mediated uptake of circulating LDL-cholesterol into cells (15). This process results in an increased clearance of circulating LDL from the blood. Within the cells, LDL particles are dismantled in lysosomes and cholesterol becomes available for metabolic needs. Through inhibiting the sterol regulatory element-binding protein (SREBP) pathway, LDL and LDL-derived cholesterol then suppress the transcription of the genes coding for 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase and other enzymes involved in the synthesis of cholesterol and of the LDL-receptor (16). The net result is the maintenance of cellular cholesterol homeostasis within tissues (especially in the liver) and a reduction in serum LDL-cholesterol concentration.
Of note, some individuals with low intestinal cholesterol absorption efficiency (17) and/or high basal cholesterol synthesis rate (18) have been found to be poorly responsive to phytosterol therapy (reviewed in 19).
Other biological activities
Experiments in cell culture and animal models have suggested that phytosterols might have biological activities unrelated to cholesterol lowering. However, their significance in humans is not yet known.
Alterations in cell membrane properties
Cholesterol is an important structural component of mammalian cell membranes (20). Displacement of cholesterol with phytosterols has been found to alter the physical properties of cell membranes in vitro (21), which could potentially affect signal transduction or membrane-bound enzyme activity (22, 23). Limited evidence from an animal model of hemorrhagic stroke suggested that very high intakes of phytosterols could displace cholesterol in red blood cell membranes, resulting in decreased deformability and potentially increased fragility (24, 25). However, daily phytosterol supplementation (1 g/1,000 kcal) for four weeks did not alter red blood cell fragility in humans (26).
Alterations in testosterone metabolism
Limited evidence from animal studies suggests that very high phytosterol intake could alter testosterone metabolism by inhibiting 5-α-reductase, a membrane-bound enzyme that converts testosterone to dihydrotestosterone, a more potent metabolite (27, 28). It is not known whether phytosterol consumption alters testosterone metabolism in humans. No significant changes in free or total serum testosterone concentrations were observed in men who consumed 1.6 g/day of plant sterol esters for one year (29).
Anticancer effects
Phytosterols have been found to inhibit proliferation, induce apoptosis, and reduce invasiveness of cancer cells in culture (reviewed in 30). There is currently little evidence to suggest that phytosterol consumption could substantially contribute to lower the risk of cancer in humans (see Cancer).
Anti-inflammatory effects
Limited data from cell culture and animal studies suggest that phytosterols may attenuate the inflammatory activity of immune cells, including macrophages and neutrophils (31, 32). The result of a recent meta-analysis of 20 randomized controlled trials found that reductions in total cholesterol and LDL-cholesterol concentrations with phytosterol-enriched foods were not associated with changes in plasma concentration of C-reactive protein (CRP), a surrogate marker of chronic low-grade inflammation (33).
Disease Prevention
Cardiovascular disease
Typical diets across different populations have been estimated to provide 150 to 450 mg/day of naturally occurring phytosterols. Nevertheless, the consumption of vegetarian diets and of food products enriched with phytosterols can help achieve much greater intakes of phytosterols (see Food sources). Relatively few studies have considered the effects of naturally occurring dietary phytosterol intakes on serum LDL-cholesterol concentrations, while an abundance of studies have examined the lipid-lowering effect of phytosterol-enriched foods.
Foods enriched with plant sterols or stanols
Lipid-lowering effect: Elevated LDL-cholesterol concentration is a well-established risk factor in the development of atherosclerosis and coronary heart disease (34, 35). Numerous clinical trials have found that daily consumption of foods enriched with free or esterified forms of plant sterols or stanols lowers concentrations of serum total and LDL-cholesterol (36-40). This wealth of evidence has been summarized in several meta-analyses combining the results of randomized controlled trials (41-46). A dose-dependent relationship was reported between total phytosterol intake levels (from less than 1 g/day to 4 g/day) and LDL-cholesterol reduction in a recent meta-analysis of 124 human studies (47). When analyzed separately, plant sterols and stanols showed similar dose-response effects on LDL-cholesterol concentrations for average doses ranging from 0.6 g/day to 3.3 g/day. Average doses of phytosterols between 0.6 and 1.1 g/day were found to significantly lower LDL-cholesterol concentrations by at least 5%, while an average intake of 3.3 g/day resulted in reductions of about 12.4% (47).
Another meta-analysis that analyzed the results of 59 randomized controlled trials suggested that reductions in LDL-cholesterol were greater in those with higher baseline concentrations of LDL-cholesterol (41). Interestingly, a recent meta-analysis of 15 randomized controlled trials investigating the effects of phytosterol-enriched food intake (1.8 to 6 g/day of phytosterols) in patients treated with statins (drugs that inhibit endogenous cholesterol synthesis) found that co-administration of phytosterols and statins significantly reduced total cholesterol and LDL-cholesterol concentrations compared to statin therapy alone (48). The concentrations of HDL-cholesterol and triglycerides were unaffected by the combination of phytosterols and statins compared to statin alone. In subgroup analyses, the effect of combining phytosterols and statins on blood lipid profile was not found to be significantly influenced by lipid baseline values, phytosterol dosage, or study duration (48).
Effect on vascular health: Impairment of vascular endothelial function is considered to be an early step in the development of atherosclerosis and cardiovascular disease (49). A recent 12-week randomized, double-blind, placebo-controlled study in 240 subjects with hypercholesterolemia (serum cholesterol ≥5 mmol/L [≥193 mg/dL]) found no effect of consuming 3 g/day of phytosterols added to low-fat spread on brachial artery flow-mediated dilation (FMD), a surrogate marker of endothelial health (50). Assessment of arterial stiffness — using measures of aortic pulse wave velocity (PWV) and augmentation index (AI) — and blood pressure also showed no difference between supplemented and placebo groups, despite a significant 6.7% reduction in total and LDL-cholesterol. Other trials in individuals with hypercholesterolemia (51, 52) and type 1 diabetes mellitus (53) also failed to find an effect of phytosterol-enriched spread consumption on brachial artery diameter, FMD, and/or arterial stiffness. Nonetheless, the results of a randomized controlled trial in 92 individuals of whom 72% had serum cholesterol ≥5 mmol/L suggested beneficial effects of plant stanol-enriched spread consumption (corresponding to 3 g/day of stanols for six months) on arterial stiffness and endothelial function, as assessed by cardio-ankle vascular index (CAVI) and reactive hyperemia index (RHI) measures, respectively (54). Finally, a 21-month randomized controlled trial used retinal photography to examine the effect of phytosterol-enriched margarine consumption on retinal microcirculation in 43 statin-treated subjects (55). Reductions in LDL-cholesterol concentration by 9.7% and 11.2% with plant sterols (2.5 g/day) and plant stanols (2.5 g/day), respectively, were not accompanied by significant changes in the diameter of retinal arterioles and venules, a proxy measure to assess microvascular health (55). At present, whether phytosterols can improve vascular health in individuals with endothelial dysfunction is unclear. The lack of an effect of phytosterols in most of the abovementioned trials may be due to the inclusion of apparently healthy participants who may have normal endothelial function (50).
Effect on the risk of coronary heart disease: Elevated LDL-cholesterol is an established risk factor for coronary heart disease (CHD) (56). The pooled analysis of 27 randomized controlled trials of statin drug therapy found a 24% decrease in the risk of major coronary events and a 12% decrease in vascular mortality per 1 millimol/L (1 mM) reduction in LDL-cholesterol concentration, irrespective of gender and level of cardiovascular risk (57). Yet, at present, the effect of long-term use of foods enriched with plant sterols or stanols on CHD risk is not known.
The addition of plant sterol- or stanol-enriched foods to a heart-healthy diet that is low in saturated fat and rich in fruit and vegetables, whole grains, and fiber offers the potential for additive effects in CHD risk reduction. For example, following a diet that substituted monounsaturated and polyunsaturated fats for saturated fat resulted in a 9% reduction in serum LDL-cholesterol after 30 days, but the addition of 1.7 g/day of plant sterols to the same diet resulted in a 24% reduction (58). In addition, one-month adherence to a diet providing a portfolio of cholesterol-lowering foods, including plant sterols (1 g/1,000 kcal), soy protein, almonds, and viscous fibers, lowered serum LDL-cholesterol concentrations by an average of 30% — a decrease that was not significantly different from that induced by statin therapy (59).
The National Cholesterol Education Program (NCEP) Adult Treatment Panel III included the use of plant sterol or stanol esters (2 g/day) as a component of maximal dietary therapy for elevated LDL-cholesterol (60). The 2013 report of the American College of Cardiology (ACC) task force advised clinicians to consider the use of phytosterol-enriched foods as dietary adjuncts for high-risk patients with insufficient LDL-cholesterol response to statin therapy (61). However, stepping back from a general recommendation, the ACC and American Heart Association (AHA) did not include phytosterols in their 2013 report on lifestyle management guidelines to reduce cardiovascular risk (62). Likewise, the 2015-2020 Dietary Guidelines for Americans — issued jointly by the US Department of Health and Human Services and the US Department of Agriculture — does not mention phytosterols in the composition of healthy eating patterns (63).
The US Food and Drug Administration (FDA) has authorized the use of health claims on food labels indicating that regular consumption of foods enriched with plant sterol or stanol esters, as part of a diet low in saturated fat and cholesterol, may reduce the risk of heart disease (see Foods enriched with plant sterols and plant stanols) (61, 64). In the EU, disease risk reduction claims for phytosterols are restricted to certain fortified food products and include a number of mandatory statements such as the fact that these products are not intended for people who do not need to control their blood cholesterol level (65).
Dietary phytosterols
Clinical trials finding daily consumption of foods enriched with plant sterols or stanols can significantly lower LDL-cholesterol concentrations do not account for naturally occurring phytosterols in the diet (66). Relatively few studies have considered the effects of dietary phytosterol intakes on serum LDL-cholesterol concentrations. Limited evidence, primarily from cross-sectional studies, suggests that dietary phytosterols may play an important role in decreasing cholesterol absorption. A cross-sectional study in the UK found that dietary phytosterol intakes were inversely related to serum total and LDL-cholesterol concentrations even after adjusting for saturated fat and fiber intake (67). Similarly, an analysis in a Swedish population found that dietary intake of phytosterols was inversely associated with total cholesterol in both men and women and with LDL-cholesterol in women (68). Dietary phytosterol intakes were also found to be inversely associated with LDL-cholesterol concentrations in another cross-sectional study in healthy Spanish participants (69). In single-meal tests, removal of 150 mg of phytosterols from corn oil increased cholesterol absorption by 38% (70), and removal of 328 mg of phytosterols from wheat germ increased cholesterol absorption by 43% (71). Although these findings suggest that moderate intakes of phytosterols could have an important impact on cardiovascular health, the intake of phytosterols (83 to 966 mg/day) from natural sources was not found to be associated with reduced risks of CHD, myocardial infarction, or total cardiovascular disease during the 12.2-year follow-up of 35,597 participants of the European Prospective Investigation into Cancer and Nutrition-The Netherlands (EPIC-NL) (72).
Cancer
Limited data from animal studies suggest that very high intakes of phytosterols, particularly sitosterol, may inhibit the growth of breast and prostate cancer (reviewed in 73). Only a few observational studies have examined associations between dietary phytosterol intakes and cancer risk in humans (30). A series of case-control studies in Uruguay found that dietary phytosterol intakes were lower in people diagnosed with stomach, lung, or breast cancer than in cancer-free control groups (74-76). Case-control studies in the US found that women diagnosed with breast or endometrial (uterine) cancer had lower dietary phytosterol intakes than women who did not have cancer (77, 78). In contrast, another case-control study in the US found that men diagnosed with prostate cancer had higher dietary campesterol intakes than cancer-free men, but total phytosterol consumption was not associated with prostate cancer risk (79). Although higher intakes of plant foods containing phytosterols may be associated with lower cancer risk, it is not clear whether potential anticancer health benefits can be attributed to phytosterols or to other compounds in plant foods (e.g., other phytochemicals, vitamins, minerals, and fiber).
Disease Treatment
Benign prostatic hyperplasia
Benign prostatic hyperplasia (BPH) is the term used to describe a noncancerous enlargement of the prostate. The enlarged prostate may exert pressure on the urethra, resulting in difficulty urinating. Plant extracts that provide a mixture of phytosterols (marketed as β-sitosterol) are often included in herbal therapies for urinary symptoms related to BPH. However, relatively few controlled studies have examined the efficacy of phytosterol supplements in men with symptomatic BPH. In a six-month study of 200 men with symptomatic BPH, 60 mg/day of a β-sitosterol preparation improved symptom scores, increased peak urinary flow, and decreased post-void residual urine volume compared to placebo (80). A follow-up study reported that these improvements were maintained for up to 18 months in the 38 participants who continued β-sitosterol treatment (81). Similarly, in a six-month study of 177 men with symptomatic BPH, 130 mg/day of a different β-sitosterol preparation improved urinary symptom scores, increased peak urinary flow, and decreased post-void residual urine volume compared to placebo (82). A systematic review that combined the results of these and two other controlled clinical trials found that β-sitosterol extracts increased peak urinary flow by an average of 3.9 mL/second and decreased post-void residual volume by an average of 29 mL (83). Although the results of a few clinical trials suggest that relatively low doses of phytosterols can improve lower urinary tract symptoms related to BPH, further research is needed to confirm these findings (84).
Sources
Food
Unlike the typical diet in most developed countries today, the diets of our ancestors were rich in phytosterols, likely providing as much as 1 g/day (1). Present-day dietary phytosterol intakes have been estimated to vary from 150 to 450 mg/day in different populations (85). Vegetarians, particularly vegans, generally have the highest intakes of dietary phytosterols (86). Phytosterols are found in all plant foods, but the highest concentrations are found in unrefined plant oils, including vegetable, nut, and olive oils (3). Nuts, seeds, whole grains, and legumes are also good dietary sources of phytosterols (4). The phytosterol content of selected foods are presented in Table 1. For information on the nutrient content of specific foods, search USDA's FoodData Central.
| Food | Serving | Phytosterols* (mg) |
|---|---|---|
| Soybeans, mature seeds, raw | ½ cup | 149 |
| Peas, green, mature seeds, raw | ½ cup | 133 |
| Sesame oil | 1 tablespoon (14 g) | 118 |
| Kidney beans, mature seeds, raw | ½ cup | 117 |
| Pistachio nuts | 1 ounce (49 kernels) | 61 |
| Safflower oil | 1 tablespoon (14 g) | 60 |
| Lentils, pink or red, mature seeds, raw | ½ cup | 54 |
| Cashew nuts | 1 ounce | 45 |
| Soybeans, green, cooked, boiled | ½ cup | 45 |
| Cottonseed oil | 1 tablespoon (14 g) | 44 |
| Orange, raw | 1 fruit | 34 |
| Macadamia nuts | 1 ounce (10-12 kernels) | 33 |
| Almonds, blanched | 1 ounce | 32 |
| Olive oil | 1 tablespoon (14 g) | 30 |
| Banana, raw | 1 large | 24 |
| Brussels sprouts, raw | 1 cup | 21 |
|
*In the USDA food composition database, the values of phytosterol content of foods are likely to be underestimates since they account only for major sterols (sitosterol, campesterol, and stigmasterol). In addition, the values correspond to the amounts of free and esterified phytosterols in foods, because phytosterol glycosides are not quantified by the current method unless glycosides (sugars) are removed before quantification (87). |
||
Food enriched with plant sterols and plant stanols
Clinical trials that demonstrated a cholesterol-lowering effect have primarily used plant sterol or stanol esters solubilized in fat-containing foods, such as margarine or mayonnaise (44). Additional studies indicate that low-fat or even nonfat foods can effectively deliver plant sterols or stanols if they are adequately solubilized (37, 66). Plant sterols or stanols added to low-fat yogurt (88-91), low-fat milk (92-94), low-fat cheese (95), dark chocolate (96), and orange juice (97, 98) have been reported to lower LDL-cholesterol in randomized controlled trials. A variety of foods containing added plant sterols or stanols, including margarines, mayonnaises, vegetable oils, salad dressings, yogurt, milk, soy milk, orange juice, snack bars, and meats, are available in the US, Europe, Asia, Australia, and New Zealand (37). A 2008 meta-analysis found that phytosterols added to fat spreads, mayonnaise, salad dressings, milk, or yogurt more effectively reduced LDL-cholesterol concentrations compared to phytosterols incorporated into chocolate, orange juice, cheese, meats, and cereal bars (41). In most clinical trials, dividing the daily dose of phytosterols among two or three meals appeared to effectively lower LDL-cholesterol (41). Nevertheless, consumption of the daily dose of plant sterols or stanols with a single meal has also been found to lower LDL-cholesterol in a few clinical trials (89-91, 99, 100).
In the US, FDA-authorized health claims on food labels specify that the daily dietary intake of plant sterol (≥1.3 g/day) or stanol esters (≥3.4 g/day) that has been associated with a reduced risk of heart disease should be consumed in two servings eaten at different times of the day with other foods, as part of a diet low in saturated fat and cholesterol (61, 64). In the EU, food labels must indicate that the beneficial effect of phytosterols is obtained with a daily intake of 1.5 to 3 g of plant sterols/stanols in order to use the following European Food Safety Authority (EFSA)-approved statement: "Plant sterol and stanol esters have been shown to lower blood cholesterol. High cholesterol is a risk factor in the development of coronary heart disease" (65).
Supplements
Available without a prescription in the US, β-sitosterol supplements typically contain a mixture of β-sitosterol with other phytosterols and/or with substances like pumpkin seed oil and saw palmetto extract (101). Doses of 60 to 130 mg/day of β-sitosterol have been found to alleviate the symptoms of benign prostatic hyperplasia in a few clinical trials (see Benign prostatic hyperplasia). Phytosterol and phytostanol supplements should be taken with a meal that contains fat.
Safety
In the US, plant sterols and stanols added to a variety of food products are generally recognized as safe (GRAS) by the FDA (102). Additionally, the Scientific Committee on Foods of the EU concluded that plant sterols and stanols added to various food products are safe for human use (103). However, the Committee recommended that intakes of plant sterols and stanols from food products should not exceed 3 g/day because there is no evidence of health benefits at higher intakes and there might be undesirable effects at high intakes (65).
Adverse effects
Few adverse effects have been associated with regular consumption of plant sterols or stanols for up to one year. People who consumed a plant sterol-enriched spread providing 1.6 g/day did not report any more adverse effects than those consuming a control spread for up to one year (29), and people consuming a plant stanol-enriched spread providing 1.8 to 2.6 g/day for one year did not report any adverse effects (104). Consumption of up to 8.6 g/day of phytosterols in margarine for three to four weeks was well tolerated by healthy men and women and did not adversely affect intestinal bacteria or female hormone levels (105). Although phytosterols are usually well tolerated, nausea, indigestion, diarrhea, and constipation have occasionally been reported (106).
Sitosterolemia
Sitosterolemia, also known as phytosterolemia, is a very rare hereditary disease that results from inheriting a mutation in both copies of the ABCG5 or ABCG8 gene (107). Individuals who are homozygous for a mutation in either transporter protein have dramatically elevated serum phytosterol concentrations due to increased intestinal absorption and decreased biliary excretion of phytosterols. Although serum cholesterol concentrations may be normal or only mildly elevated, individuals with sitosterolemia are at high risk for premature atherosclerosis. Other clinical symptoms include tuberous and tendon xanthomas (i.e., cutaneous lipid depositions), hematological abnormalities, and sometimes joint pain and arthritis.
People with sitosterolemia should avoid foods or supplements with added plant sterols (37). Two studies have examined the effect of plant sterol consumption in heterozygous carriers of sitosterolemia, a more common condition. Consumption of 3 g/day of plant sterols for four weeks by two heterozygous carriers (108) and consumption of 2.2 g/day of plant sterols for 6 to 12 weeks by 12 heterozygous carriers did not result in abnormally elevated serum phytosterols (109). Because atherosclerosis has been reported in subjects with sitosterolemia, phytosterols have been attributed atherogenic effects. However, no relationship between serum concentrations of sitosterol and campesterol and risk of cardiovascular disease has been identified in a recent meta-analysis of 17 observational studies in 11,182 participants (110).
Pregnancy and lactation
Phytosterol-enriched foods and supplements are not recommended for pregnant or breast-feeding women because their safety has not been studied (106). At present, there is no evidence that high dietary intakes of naturally occurring phytosterols, such as those consumed by vegetarian women, adversely affects pregnancy or lactation.
Drug interactions
Statins
There is some evidence showing that statin administration initially reduces plant sterol concentrations in blood. This might be attributed to the reduction of circulating LDL, the major transport lipoprotein of plant sterols, due to enhanced hepatic uptake of LDL. However, statin therapy appears to increase the absorption of plant sterols that can be then transported by the remaining LDL particles (111). Further, the LDL-cholesterol-lowering effect of plant sterols or stanols may be additive to that of statins. The result of a recent meta-analysis of controlled clinical trials suggested that consumption of 2 to 3 g/day of plant sterols or stanols by individuals on statin therapy may lower both total cholesterol and LDL-cholesterol by an additional 0.30 mmol/L (11.6 mg/dL), compared to statin alone (48).
Ezetimibe
Ezetimibe (marketed as Zetia) is another cholesterol-lowering drug that may interfere with the intestinal absorption of phytosterols, thus significantly reducing phytosterol concentration in blood (112).
Nutrient interactions
Fat-soluble vitamins (vitamins A, D, E, and K)
Because plant sterols and stanols decrease cholesterol absorption and serum LDL-cholesterol concentrations, their effects on fat-soluble vitamin status have also been studied in clinical trials. Plasma vitamin A (retinol) concentrations were not affected by consumption of plant sterol esters or plant stanol esters for up to one year (29, 44). Although the majority of studies found no changes in plasma vitamin D (25-hydroxyvitamin D3) concentrations, one placebo-controlled study in individuals consuming 1.6 g/day of sterol esters for one year observed a small (7%) but statistically significant decrease in plasma 25-hydroxyvitamin D3 concentrations (29). There is little evidence that plant sterol or stanol consumption adversely affects vitamin K status. Consumption of 1.6 g/day of sterol esters for six months was associated with a nonsignificant, 14% decrease in plasma vitamin K1 (phylloquinone) concentrations, and the level of carboxylated osteocalcin, a functional indicator of vitamin K status, was unchanged (29). Other studies of shorter duration also found no change in plasma concentrations of phylloquinone (113, 114) or vitamin K-dependent clotting factors with the consumption of plant sterol and stanol esters (115). Consumption of phytosterol-enriched foods has been found to decrease plasma vitamin E (α-tocopherol) concentration in a number of studies (44, 114). However, those decreases generally do not persist when plasma α-tocopherol concentrations are standardized to LDL-cholesterol concentrations, suggesting that observed reductions in plasma α-tocopherol are due in part to reductions in its lipoprotein carrier, LDL.
A recent meta-analysis of intervention studies found no adverse effects of phytosterol-enriched food consumption (average dose of 2.5 g/day) on fat-soluble vitamin status in well-nourished people (116).
Carotenoids
Dietary carotenoids are fat-soluble phytochemicals that circulate in lipoproteins. A recent meta-analysis of randomized controlled studies reported about 5 to 20% reductions in plasma hydrocarbon carotenoids after consumption of plant sterol- or stanol-enriched foods for one month to one year (116). Even when standardized to serum total cholesterol concentrations, decreases in α-carotene, β-carotene, and lycopene may persist, suggesting that phytosterols could inhibit the absorption of these carotenoids. Total cholesterol-standardized concentrations of xanthophyll carotenoids, zeaxanthin and β-cryptoxanthin, but not lutein, were also found to be significantly reduced by 5 to 15% with the consumption of phytosterol-enriched foods (116).
Although it is not clear whether reductions in plasma carotenoid concentrations confer any health risks (see the article on Carotenoids), a few studies showed that increasing intakes of carotenoid-rich fruit and vegetables would prevent phytosterol-induced decreases in plasma concentrations of carotenoids (117). In one randomized controlled study, advice to consume five daily servings of fruit and vegetables, including one serving of carotenoid-rich vegetables, was enough to maintain plasma carotenoid levels in people consuming 2.5 g/day of plant sterol or stanol esters (118).
Authors and Reviewers
Originally written in 2005 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in September 2008 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in November 2016 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in March 2017 by:
Susan B. Racette, Ph.D.
Professor, Program in Physical Therapy
and Department of Medicine
Washington University in St. Louis
Copyright 2005-2024 Linus Pauling Institute
References
1. Jew S, AbuMweis SS, Jones PJ. Evolution of the human diet: linking our ancestral diet to modern functional foods as a means of chronic disease prevention. J Med Food. 2009;12(5):925-934. (PubMed)
2. Sudhop T, Lutjohann D, von Bergmann K. Sterol transporters: targets of natural sterols and new lipid lowering drugs. Pharmacol Ther. 2005;105(3):333-341. (PubMed)
3. Ostlund RE, Jr. Phytosterols in human nutrition. Annu Rev Nutr. 2002;22:533-549. (PubMed)
4. de Jong A, Plat J, Mensink RP. Metabolic effects of plant sterols and stanols (Review). J Nutr Biochem. 2003;14(7):362-369. (PubMed)
5. Davis HR, Zhu LJ, Hoos LM, et al. Niemann-Pick C1 like 1 (NPC1L1) is the intestinal phytosterol and cholesterol transporter and a key modulator of whole-body cholesterol homeostasis. Journal of Biological Chemistry. 2004;279(32):33586-33592. (PubMed)
6. Howles PN. Cholesterol absorption and metabolism. Methods Mol Biol. 2016;1438:177-197. (PubMed)
7. von Bergmann K, Sudhop T, Lutjohann D. Cholesterol and plant sterol absorption: recent insights. Am J Cardiol. 2005;96(1A):10D-14D. (PubMed)
8. Ostlund RE, Jr., McGill JB, Zeng CM, et al. Gastrointestinal absorption and plasma kinetics of soy Delta(5)-phytosterols and phytostanols in humans. Am J Physiol Endocrinol Metab. 2002;282(4):E911-916. (PubMed)
9. Weingartner O, Bohm M, Laufs U. Controversial role of plant sterol esters in the management of hypercholesterolaemia. Eur Heart J. 2009;30(4):404-409. (PubMed)
10. Jones PJ, Rideout T. Lipids, sterols, and their metabolites. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease: Lippincott Williams & Wilkins; 2014:65-87.
11. Sudhop T, Sahin Y, Lindenthal B, et al. Comparison of the hepatic clearances of campesterol, sitosterol, and cholesterol in healthy subjects suggests that efflux transporters controlling intestinal sterol absorption also regulate biliary secretion. Gut. 2002;51(6):860-863. (PubMed)
12. De Smet E, Mensink RP, Plat J. Effects of plant sterols and stanols on intestinal cholesterol metabolism: suggested mechanisms from past to present. Mol Nutr Food Res. 2012;56(7):1058-1072. (PubMed)
13. Nissinen M, Gylling H, Vuoristo M, Miettinen TA. Micellar distribution of cholesterol and phytosterols after duodenal plant stanol ester infusion. Am J Physiol Gastrointest Liver Physiol. 2002;282(6):G1009-1015. (PubMed)
14. Racette SB, Lin X, Lefevre M, et al. Dose effects of dietary phytosterols on cholesterol metabolism: a controlled feeding study. Am J Clin Nutr. 2010;91(1):32-38. (PubMed)
15. Plat J, Mensink RP. Plant stanol and sterol esters in the control of blood cholesterol levels: mechanism and safety aspects. Am J Cardiol. 2005;96(Suppl):15D-22D. (PubMed)
16. Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29(4):431-438. (PubMed)
17. Zhao HL, Houweling AH, Vanstone CA, et al. Genetic variation in ABC G5/G8 and NPC1L1 impact cholesterol response to plant sterols in hypercholesterolemic men. Lipids. 2008;43(12):1155-1164. (PubMed)
18. Rideout TC, Harding SV, Mackay D, Abumweis SS, Jones PJ. High basal fractional cholesterol synthesis is associated with nonresponse of plasma LDL cholesterol to plant sterol therapy. Am J Clin Nutr. 2010;92(1):41-46. (PubMed)
19. Rideout TC, Harding SV, Mackay DS. Metabolic and genetic factors modulating subject specific LDL-C responses to plant sterol therapy. Can J Physiol Pharmacol. 2012;90(5):509-514. (PubMed)
20. Mouritsen OG, Zuckermann MJ. What's so special about cholesterol? Lipids. 2004;39(11):1101-1113. (PubMed)
21. Halling KK, Slotte JP. Membrane properties of plant sterols in phospholipid bilayers as determined by differential scanning calorimetry, resonance energy transfer and detergent-induced solubilization. Biochim Biophys Acta. 2004;1664(2):161-171. (PubMed)
22. Awad AB, Chen YC, Fink CS, Hennessey T. beta-Sitosterol inhibits HT-29 human colon cancer cell growth and alters membrane lipids. Anticancer Res. 1996;16(5A):2797-2804. (PubMed)
23. Leikin AI, Brenner RR. Fatty acid desaturase activities are modulated by phytosterol incorporation in microsomes. Biochim Biophys Acta. 1989;1005(2):187-191. (PubMed)
24. Ratnayake WM, L'Abbe MR, Mueller R, et al. Vegetable oils high in phytosterols make erythrocytes less deformable and shorten the life span of stroke-prone spontaneously hypertensive rats. J Nutr. 2000;130(5):1166-1178. (PubMed)
25. Ratnayake WM, Plouffe L, L'Abbe MR, Trick K, Mueller R, Hayward S. Comparative health effects of margarines fortified with plant sterols and stanols on a rat model for hemorrhagic stroke. Lipids. 2003;38(12):1237-1247. (PubMed)
26. Jones PJ, Raeini-Sarjaz M, Jenkins DJ, et al. Effects of a diet high in plant sterols, vegetable proteins, and viscous fibers (dietary portfolio) on circulating sterol levels and red cell fragility in hypercholesterolemic subjects. Lipids. 2005;40(2):169-174. (PubMed)
27. Awad AB, Hartati MS, Fink CS. Phytosterol feeding induces alteration in testosterone metabolism in rat tissues. J Nutr Biochem. 1998;9(12):712-717.
28. Cabeza M, Bratoeff E, Heuze I, Ramirez E, Sanchez M, Flores E. Effect of beta-sitosterol as inhibitor of 5 alpha-reductase in hamster prostate. Proc West Pharmacol Soc. 2003;46:153-155. (PubMed)
29. Hendriks HF, Brink EJ, Meijer GW, Princen HM, Ntanios FY. Safety of long-term consumption of plant sterol esters-enriched spread. Eur J Clin Nutr. 2003;57(5):681-692. (PubMed)
30. Woyengo TA, Ramprasath VR, Jones PJ. Anticancer effects of phytosterols. Eur J Clin Nutr. 2009;63(7):813-820. (PubMed)
31. Awad AB, Toczek J, Fink CS. Phytosterols decrease prostaglandin release in cultured P388D1/MAB macrophages. Prostaglandins Leukot Essent Fatty Acids. 2004;70(6):511-520. (PubMed)
32. Navarro A, De las Heras B, Villar A. Anti-inflammatory and immunomodulating properties of a sterol fraction from Sideritis foetens Clem. Biol Pharm Bull. 2001;24(5):470-473. (PubMed)
33. Rocha VZ, Ras RT, Gagliardi AC, Mangili LC, Trautwein EA, Santos RD. Effects of phytosterols on markers of inflammation: A systematic review and meta-analysis. Atherosclerosis. 2016;248:76-83. (PubMed)
34. Catapano AL, Graham I, De Backer G, et al. 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias: The Task Force for the Management of Dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur Heart J. 2016;37(39):2999-3058. (PubMed)
35. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 Suppl 2):S1-45. (PubMed)
36. St-Onge MP, Jones PJ. Phytosterols and human lipid metabolism: efficacy, safety, and novel foods. Lipids. 2003;38(4):367-375. (PubMed)
37. Berger A, Jones PJ, Abumweis SS. Plant sterols: factors affecting their efficacy and safety as functional food ingredients. Lipids Health Dis. 2004;3:5. (PubMed)
38. Moruisi KG, Oosthuizen W, Opperman AM. Phytosterols/stanols lower cholesterol concentrations in familial hypercholesterolemic subjects: a systematic review with meta-analysis. J Am Coll Nutr. 2006;25(1):41-48. (PubMed)
39. Ellegard LH, Andersson SW, Normen AL, Andersson HA. Dietary plant sterols and cholesterol metabolism. Nutr Rev. 2007;65(1):39-45. (PubMed)
40. Van Horn L, McCoin M, Kris-Etherton PM, et al. The evidence for dietary prevention and treatment of cardiovascular disease. J Am Diet Assoc. 2008;108(2):287-331. (PubMed)
41. AbuMweis SS, Barake R, Jones P. Plant sterols/stanols as cholesterol lowering agents: A meta-analysis of randomized controlled trials. Food & Nutrition Research. 2008; 52. doi: 10.3402/fnr.v52i0.1811. (PubMed)
42. Chen JT, Wesley R, Shamburek RD, Pucino F, Csako G. Meta-analysis of natural therapies for hyperlipidemia: plant sterols and stanols versus policosanol. Pharmacotherapy. 2005;25(2):171-183. (PubMed)
43. Demonty I, Ras RT, van der Knaap HC, et al. Continuous dose-response relationship of the LDL-cholesterol-lowering effect of phytosterol intake. J Nutr. 2009;139(2):271-284. (PubMed)
44. Katan MB, Grundy SM, Jones P, Law M, Miettinen T, Paoletti R. Efficacy and safety of plant stanols and sterols in the management of blood cholesterol levels. Mayo Clin Proc. 2003;78(8):965-978. (PubMed)
45. Law M. Plant sterol and stanol margarines and health. BMJ. 2000;320(7238):861-864. (PubMed)
46. Musa-Veloso K, Poon TH, Elliot JA, Chung C. A comparison of the LDL-cholesterol lowering efficacy of plant stanols and plant sterols over a continuous dose range: results of a meta-analysis of randomized, placebo-controlled trials. Prostaglandins Leukot Essent Fatty Acids. 2011;85(1):9-28. (PubMed)
47. Ras RT, Geleijnse JM, Trautwein EA. LDL-cholesterol-lowering effect of plant sterols and stanols across different dose ranges: a meta-analysis of randomised controlled studies. Br J Nutr. 2014;112(2):214-219. (PubMed)
48. Han S, Jiao J, Xu J, et al. Effects of plant stanol or sterol-enriched diets on lipid profiles in patients treated with statins: systematic review and meta-analysis. Sci Rep. 2016;6:31337. (PubMed)
49. Landmesser U, Drexler H. The clinical significance of endothelial dysfunction. Curr Opin Cardiol. 2005;20(6):547-551. (PubMed)
50. Ras RT, Fuchs D, Koppenol WP, et al. The effect of a low-fat spread with added plant sterols on vascular function markers: results of the Investigating Vascular Function Effects of Plant Sterols (INVEST) study. Am J Clin Nutr. 2015;101(4):733-741. (PubMed)
51. Hallikainen M, Lyyra-Laitinen T, Laitinen T, et al. Endothelial function in hypercholesterolemic subjects: Effects of plant stanol and sterol esters. Atherosclerosis. 2006;188(2):425-432. (PubMed)
52. Raitakari OT, Salo P, Gylling H, Miettinen TA. Plant stanol ester consumption and arterial elasticity and endothelial function. Br J Nutr. 2008;100(3):603-608. (PubMed)
53. Hallikainen M, Lyyra-Laitinen T, Laitinen T, Moilanen L, Miettinen TA, Gylling H. Effects of plant stanol esters on serum cholesterol concentrations, relative markers of cholesterol metabolism and endothelial function in type 1 diabetes. Atherosclerosis. 2008;199(2):432-439. (PubMed)
54. Gylling H, Halonen J, Lindholm H, et al. The effects of plant stanol ester consumption on arterial stiffness and endothelial function in adults: a randomised controlled clinical trial. BMC Cardiovasc Disord. 2013;13:50. (PubMed)
55. Kelly ER, Plat J, Mensink RP, Berendschot TT. Effects of long term plant sterol and -stanol consumption on the retinal vasculature: a randomized controlled trial in statin users. Atherosclerosis. 2011;214(1):225-230. (PubMed)
56. McCormack T, Dent R, Blagden M. Very low LDL-C levels may safely provide additional clinical cardiovascular benefit: the evidence to date. Int J Clin Pract. 2016;70(11):886-897. (PubMed)
57. Cholesterol Treatment Trialists Collaboration. Fulcher J, O'Connell R, et al. Efficacy and safety of LDL-lowering therapy among men and women: meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet. 2015;385(9976):1397-1405. (PubMed)
58. Jones PJ, Ntanios FY, Raeini-Sarjaz M, Vanstone CA. Cholesterol-lowering efficacy of a sitostanol-containing phytosterol mixture with a prudent diet in hyperlipidemic men. Am J Clin Nutr. 1999;69(6):1144-1150. (PubMed)
59. Jenkins DJ, Kendall CW, Marchie A, et al. Direct comparison of a dietary portfolio of cholesterol-lowering foods with a statin in hypercholesterolemic participants. Am J Clin Nutr. 2005;81(2):380-387. (PubMed)
60. Grundy SM. Stanol esters as a component of maximal dietary therapy in the National Cholesterol Education Program Adult Treatment Panel III Report. Am J Cardiol. 2005;96(1A):47D-50D. (PubMed)
61. Writing C, Lloyd-Jones DM, Morris PB, et al. 2016 ACC expert consensus decision pathway on the role of non-statin therapies for LDL-cholesterol lowering in the management of atherosclerotic cardiovascular disease risk: a report of the American College of Cardiology Task Force on Clinical Expert Consensus Documents. J Am Coll Cardiol. 2016;68(1):92-125. (PubMed)
62. Eckel RH, Jakicic JM, Ard JD, et al. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 Suppl 2):S76-99. (PubMed)
63. US Department of Health and Human Services and US Department of Agriculture. 2015 – 2020 Dietary Guidelines for Americans; 2015. Available at: https://health.gov/dietaryguidelines/2015/guidelines/.
64. Food and Drug Administration. Health claims: plant sterol/stanol esters and risk of coronary heart disease (CHD). U. S. Government Printing Office [Code of Federal Regulations]. April 1, 2002. Available at: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=101.83. Accessed 6/29/05.
65. Shortt C. Authorised EU health claims for phytosterols. In: Sadler M, ed. Foods, Nutrients and Food Ingredients with Authorised EU Health Claims: Elsevier Ltd; 2015:31-47.
66. Ostlund RE, Jr. Phytosterols and cholesterol metabolism. Curr Opin Lipidol. 2004;15(1):37-41. (PubMed)
67. Andersson SW, Skinner J, Ellegard L, et al. Intake of dietary plant sterols is inversely related to serum cholesterol concentration in men and women in the EPIC Norfolk population: a cross-sectional study. Eur J Clin Nutr. 2004;58(10):1378-1385. (PubMed)
68. Klingberg S, Ellegard L, Johansson I, et al. Inverse relation between dietary intake of naturally occurring plant sterols and serum cholesterol in northern Sweden. Am J Clin Nutr. 2008;87(4):993-1001. (PubMed)
69. Sanclemente T, Marques-Lopes I, Fajo-Pascual M, et al. Naturally-occurring phytosterols in the usual diet influence cholesterol metabolism in healthy subjects. Nutr Metab Cardiovasc Dis. 2012;22(10):849-855. (PubMed)
70. Ostlund RE, Jr., Racette SB, Okeke A, Stenson WF. Phytosterols that are naturally present in commercial corn oil significantly reduce cholesterol absorption in humans. Am J Clin Nutr. 2002;75(6):1000-1004. (PubMed)
71. Ostlund RE, Jr., Racette SB, Stenson WF. Inhibition of cholesterol absorption by phytosterol-replete wheat germ compared with phytosterol-depleted wheat germ. Am J Clin Nutr. 2003;77(6):1385-1389. (PubMed)
72. Ras RT, van der Schouw YT, Trautwein EA, et al. Intake of phytosterols from natural sources and risk of cardiovascular disease in the European Prospective Investigation into Cancer and Nutrition-the Netherlands (EPIC-NL) population. Eur J Prev Cardiol. 2015;22(8):1067-1075. (PubMed)
73. Ramprasath VR, Awad AB. Role of phytosterols in cancer prevention and treatment. J AOAC Int. 2015;98(3):735-738. (PubMed)
74. De Stefani E, Boffetta P, Ronco AL, et al. Plant sterols and risk of stomach cancer: a case-control study in Uruguay. Nutr Cancer. 2000;37(2):140-144. (PubMed)
75. Mendilaharsu M, De Stefani E, Deneo-Pellegrini H, Carzoglio J, Ronco A. Phytosterols and risk of lung cancer: a case-control study in Uruguay. Lung Cancer. 1998;21(1):37-45. (PubMed)
76. Ronco A, De Stefani E, Boffetta P, Deneo-Pellegrini H, Mendilaharsu M, Leborgne F. Vegetables, fruits, and related nutrients and risk of breast cancer: a case-control study in Uruguay. Nutr Cancer. 1999;35(2):111-119. (PubMed)
77. McCann SE, Freudenheim JL, Marshall JR, Brasure JR, Swanson MK, Graham S. Diet in the epidemiology of endometrial cancer in western New York (United States). Cancer Causes Control. 2000;11(10):965-974. (PubMed)
78. McCann SE, Freudenheim JL, Marshall JR, Graham S. Risk of human ovarian cancer is related to dietary intake of selected nutrients, phytochemicals and food groups. J Nutr. 2003;133(6):1937-1942. (PubMed)
79. Strom SS, Yamamura Y, Duphorne CM, et al. Phytoestrogen intake and prostate cancer: a case-control study using a new database. Nutr Cancer. 1999;33(1):20-25. (PubMed)
80. Berges RR, Windeler J, Trampisch HJ, Senge T. Randomised, placebo-controlled, double-blind clinical trial of beta-sitosterol in patients with benign prostatic hyperplasia. Beta-sitosterol Study Group. Lancet. 1995;345(8964):1529-1532. (PubMed)
81. Berges RR, Kassen A, Senge T. Treatment of symptomatic benign prostatic hyperplasia with beta-sitosterol: an 18-month follow-up. BJU Int. 2000;85(7):842-846. (PubMed)
82. Klippel KF, Hiltl DM, Schipp B. A multicentric, placebo-controlled, double-blind clinical trial of beta-sitosterol (phytosterol) for the treatment of benign prostatic hyperplasia. German BPH-Phyto Study group. Br J Urol. 1997;80(3):427-432. (PubMed)
83. Wilt TJ, MacDonald R, Ishani A. Beta-sitosterol for the treatment of benign prostatic hyperplasia: a systematic review. BJU Int. 1999;83(9):976-983. (PubMed)
84. Dreikorn K. The role of phytotherapy in treating lower urinary tract symptoms and benign prostatic hyperplasia. World J Urol. 2002;19(6):426-435. (PubMed)
85. Othman RA, Myrie SB, Jones PJ. Non-cholesterol sterols and cholesterol metabolism in sitosterolemia. Atherosclerosis. 2013;231(2):291-299. (PubMed)
86. Nair PP, Turjman N, Kessie G, et al. Diet, nutrition intake, and metabolism in populations at high and low risk for colon cancer. Dietary cholesterol, beta-sitosterol, and stigmasterol. Am J Clin Nutr. 1984;40(4 Suppl):927-930. (PubMed)
87. Racette SB, Spearie CA, Phillips KM, Lin X, Ma L, Ostlund RE, Jr. Phytosterol-deficient and high-phytosterol diets developed for controlled feeding studies. J Am Diet Assoc. 2009;109(12):2043-2051. (PubMed)
88. Mensink RP, Ebbing S, Lindhout M, Plat J, van Heugten MM. Effects of plant stanol esters supplied in low-fat yoghurt on serum lipids and lipoproteins, non-cholesterol sterols and fat soluble antioxidant concentrations. Atherosclerosis. 2002;160(1):205-213. (PubMed)
89. Volpe R, Niittynen L, Korpela R, et al. Effects of yoghurt enriched with plant sterols on serum lipids in patients with moderate hypercholesterolaemia. Br J Nutr. 2001;86(2):233-239. (PubMed)
90. Plana N, Nicolle C, Ferre R, et al. Plant sterol-enriched fermented milk enhances the attainment of LDL-cholesterol goal in hypercholesterolemic subjects. Eur J Nutr. 2008;47(1):32-39. (PubMed)
91. Doornbos AM, Meynen EM, Duchateau GS, van der Knaap HC, Trautwein EA. Intake occasion affects the serum cholesterol lowering of a plant sterol-enriched single-dose yoghurt drink in mildly hypercholesterolaemic subjects. Eur J Clin Nutr. 2006;60(3):325-333. (PubMed)
92. Noakes M, Clifton PM, Doornbos AME, Trautwein EA. Plant sterol ester-enriched milk and yoghurt effectively reduce serum cholesterol in modestly hypercholesterolemic subjects. Eur J Clin Nutr. 2005;44(4):214-222. (PubMed)
93. Thomsen AB, Hansen HB, Christiansen C, Green H, Berger A. Effect of free plant sterols in low-fat milk on serum lipid profile in hypercholesterolemic subjects. Eur J Clin Nutr. 2004;58(6):860-870. (PubMed)
94. Seppo L, Jauhiainen T, Nevala R, Poussa T, Korpela R. Plant stanol esters in low-fat milk products lower serum total and LDL cholesterol. Eur J Nutr. 2007;46(2):111-117. (PubMed)
95. Jauhiainen T, Salo P, Niittynen L, Poussa T, Korpela R. Effects of low-fat hard cheese enriched with plant stanol esters on serum lipids and apolipoprotein B in mildly hypercholesterolaemic subjects. Eur J Clin Nutr. 2006;60(11):1253-1257. (PubMed)
96. Allen RR, Carson L, Kwik-Uribe C, Evans EM, Erdman JW, Jr. Daily consumption of a dark chocolate containing flavanols and added sterol esters affects cardiovascular risk factors in a normotensive population with elevated cholesterol. J Nutr. 2008;138(4):725-731. (PubMed)
97. Devaraj S, Jialal I, Vega-Lopez S. Plant sterol-fortified orange juice effectively lowers cholesterol levels in mildly hypercholesterolemic healthy individuals. Arterioscler Thromb Vasc Biol. 2004;24(3):e25-28. (PubMed)
98. Devaraj S, Autret BC, Jialal I. Reduced-calorie orange juice beverage with plant sterols lowers C-reactive protein concentrations and improves the lipid profile in human volunteers. Am J Clin Nutr. 2006;84(4):756-761. (PubMed)
99. Matvienko OA, Lewis DS, Swanson M, et al. A single daily dose of soybean phytosterols in ground beef decreases serum total cholesterol and LDL cholesterol in young, mildly hypercholesterolemic men. Am J Clin Nutr. 2002;76(1):57-64. (PubMed)
100. Plat J, van Onselen EN, van Heugten MM, Mensink RP. Effects on serum lipids, lipoproteins and fat soluble antioxidant concentrations of consumption frequency of margarines and shortenings enriched with plant stanol esters. Eur J Clin Nutr. 2000;54(9):671-677. (PubMed)
101. Hendler SS, Rorvik DM. Beta-sitosterol. In: Reuters T, ed. PDR for Nutritional Supplements. 2nd ed. Montvale: Physicians' Desk Reference Inc.; 2008:78-80.
102. Food and Drug Administration. GRAS Notice No. GRN 000112. February 4, 2003. http://www.cfsan.fda.gov/~rdb/opa-g112.html. Accessed 7/11/05.
103. Scientific Committee on Food. Opinion on Applications for Approval of a Variety of Plant Sterol-Enriched Foods. March 5, 2003. http://europa.eu.int/comm/food/fs/sc/scf/out174_en.pdf. Accessed 7/11/05.
104. Miettinen TA, Puska P, Gylling H, Vanhanen H, Vartiainen E. Reduction of serum cholesterol with sitostanol-ester margarine in a mildly hypercholesterolemic population. N Engl J Med. 1995;333(20):1308-1312. (PubMed)
105. Ayesh R, Weststrate JA, Drewitt PN, Hepburn PA. Safety evaluation of phytosterol esters. Part 5. Faecal short-chain fatty acid and microflora content, faecal bacterial enzyme activity and serum female sex hormones in healthy normolipidaemic volunteers consuming a controlled diet either with or without a phytosterol ester-enriched margarine. Food Chem Toxicol. 1999;37(12):1127-1138. (PubMed)
106. Hendler SS, Rorvik DM. Phytosterols. In: Reuters T, ed. PDR for Nutritional Supplements. 2nd ed. Montvale: Physicians' Desk Reference Inc.; 2008:500-503.
107. Berge KE. Sitosterolemia: a gateway to new knowledge about cholesterol metabolism. Ann Med. 2003;35(7):502-511. (PubMed)
108. Stalenhoef AF, Hectors M, Demacker PN. Effect of plant sterol-enriched margarine on plasma lipids and sterols in subjects heterozygous for phytosterolaemia. J Intern Med. 2001;249(2):163-166. (PubMed)
109. Kwiterovich PO, Jr., Chen SC, Virgil DG, Schweitzer A, Arnold DR, Kratz LE. Response of obligate heterozygotes for phytosterolemia to a low-fat diet and to a plant sterol ester dietary challenge. J Lipid Res. 2003;44(6):1143-1155. (PubMed)
110. Genser B, Silbernagel G, De Backer G, et al. Plant sterols and cardiovascular disease: a systematic review and meta-analysis. Eur Heart J. 2012;33(4):444-451. (PubMed)
111. Miettinen TA, Gylling H. Effect of statins on noncholesterol sterol levels: implications for use of plant stanols and sterols. Am J Cardiol. 2005;96(1A):40D-46D. (PubMed)
112. Assmann G, Kannenberg F, Ramey DR, Musliner TA, Gutkin SW, Veltri EP. Effects of ezetimibe, simvastatin, atorvastatin, and ezetimibe-statin therapies on non-cholesterol sterols in patients with primary hypercholesterolemia. Curr Med Res Opin. 2008;24(1):249-259. (PubMed)
113. Raeini-Sarjaz M, Ntanios FY, Vanstone CA, Jones PJ. No changes in serum fat-soluble vitamin and carotenoid concentrations with the intake of plant sterol/stanol esters in the context of a controlled diet. Metabolism. 2002;51(5):652-656. (PubMed)
114. Korpela R, Tuomilehto J, Hogstrom P, et al. Safety aspects and cholesterol-lowering efficacy of low fat dairy products containing plant sterols. Eur J Clin Nutr. 2006;60(5):633-642. (PubMed)
115. Plat J, Mensink RP. Vegetable oil based versus wood based stanol ester mixtures: effects on serum lipids and hemostatic factors in non-hypercholesterolemic subjects. Atherosclerosis. 2000;148(1):101-112. (PubMed)
116. Baumgartner S, Ras RT, Trautwein EA, Mensink RP, Plat J. Plasma fat-soluble vitamin and carotenoid concentrations after plant sterol and plant stanol consumption: a meta-analysis of randomized controlled trials. Eur J Nutr. 2017;56(3):909-923. (PubMed)
117. Fardet A, Morise A, Kalonji E, Margaritis I, Mariotti F. Influence of phytosterol and phytostanol food supplementation on plasma liposoluble vitamins and provitamin A carotenoid levels in humans: an updated review of the evidence. Crit Rev Food Sci Nutr. 2015;57(9):1906-1921. (PubMed)
118. Noakes M, Clifton P, Ntanios F, Shrapnel W, Record I, McInerney J. An increase in dietary carotenoids when consuming plant sterols or stanols is effective in maintaining plasma carotenoid concentrations. Am J Clin Nutr. 2002;75(1):79-86. (PubMed)
Resveratrol
Contents
Summary
- Resveratrol is a polyphenolic compound naturally found in peanuts, grapes, red wine, and some berries. (More information)
- When taken orally, resveratrol is well absorbed by humans, but its bioavailability is relatively low because it is rapidly metabolized and eliminated. (More information)
- In preclinical studies, resveratrol has been shown to possess numerous biological activities, which could possibly be applied to the prevention and/or treatment of cancer, cardiovascular disease, and neurodegenerative diseases. (More information)
- Although resveratrol can inhibit the growth of cancer cells in culture and in some animal models, it is not known whether resveratrol can prevent and/or help treat cancer in humans. (More information)
- The presence of resveratrol in red wine was initially thought to be responsible for red wine’s beneficial cardiovascular effects. Two randomized, placebo-controlled trials reported that one-year consumption of a grape supplement containing 8 mg/day of resveratrol improved inflammatory and atherogenic status in subjects at risk for cardiovascular disease, as well as in patients with established coronary heart disease. Yet, there is currently no evidence that the content of resveratrol in red wine confers any additional risk reduction beyond that attributed to the alcohol content and to other wine polyphenols. (More information)
- Resveratrol administration has increased the lifespans of yeast, worms, fruit flies, fish, and mice fed a high-calorie diet, but it is not known whether resveratrol will have similar effects in humans. (More information)
- Experimental animal studies have suggested that resveratrol might be neuroprotective and be beneficial in the prevention and/or treatment of neurodegenerative diseases; however, clinical trials in healthy or cognitively impaired older people are currently very limited. (More information)
- In randomized controlled trials, short-term supplementation with resveratrol significantly improved glucose and lipid metabolic disorders in patients with type 2 diabetes. (More information)
- Long-term, high resveratrol intake might affect the pharmacokinetics of certain drugs (i.e., those metabolized by cytochrome P450 enzymes), potentially reducing their efficacy or increasing their toxicity. (More information)
Introduction
Resveratrol (3,4',5-trihydroxystilbene) belongs to a class of polyphenolic compounds called stilbenes (1). Certain plants produce resveratrol and other stilbenoids in response to stress, injury, fungal infection, or ultraviolet (UV) radiation (2). Resveratrol is a fat-soluble compound that occurs in both trans and cis molecular configurations (Figure 1). Both cis- and trans-resveratrol also occur as glucosides, i.e., bound to a glucose molecule. One major resveratrol derivative is resveratrol-3-O-β-glucoside, also called piceid (Figure 1) (3).
Since the early 1990s, when the presence of resveratrol in red wine was established (4), the scientific community has been exploring the effects of resveratrol on health. Specifically, it was postulated that resveratrol intake via moderate red wine consumption might help explain the fact that French people have a relatively low incidence of coronary heart disease (CHD) in spite of consuming foods high in saturated fat, a phenomenon dubbed the “French Paradox” (see Cardiovascular disease) (5). Since then, reports on the potential for resveratrol to prevent cancer, delay the development of cardiovascular and neurodegenerative diseases, improve glycemic control in type 2 diabetes, and extend lifespan in experimental models have continued to generate scientific interest (see Disease Prevention).
Metabolism and Bioavailability
Initial studies of the pharmacokinetics of trans-resveratrol in humans found only traces of the unmetabolized resveratrol in the plasma upon oral exposure of single trans-resveratrol doses of 5 to 25 mg. Indeed, trans-resveratrol appears to be well absorbed by humans when taken orally, but its bioavailability is relatively low due to its rapid metabolism and elimination (6). Once absorbed, resveratrol is rapidly metabolized by conjugation to glucuronic acid and/or sulfate, forming resveratrol glucuronides, sulfates, and/or sulfoglucuronides. Sulfate conjugates are the major forms of resveratrol metabolites found in plasma and urine in humans (7).
Preliminary studies found that the administration of single oral doses of 25 mg of trans-resveratrol to healthy volunteers resulted in peak blood concentrations of total resveratrol (i.e., trans-resveratrol plus its metabolites) around 60 minutes later, at about 1.8-2 μmoles/liter (μM), depending on whether resveratrol was administered in wine, vegetable juice, or grape juice (8, 9). A study in 40 healthy subjects who received single ascending doses of oral trans-resveratrol (i.e., 0.5 g, 1 g, 2.5 g, and 5 g) showed that plasma concentrations of unmetabolized resveratrol peaked between 0.8 and 1.5 hours after resveratrol administration at levels ranging from 0.3 μM to 2.3 μM (10). Of note, these values were markedly below those used to elicit chemopreventive effects of resveratrol in in vitro experiments (>5 μM). In contrast, following a single oral dose of 5 g of trans-resveratrol, the peak plasma concentrations of certain resveratrol conjugates were found to be about two to eight times higher than those of unmetabolized resveratrol (10). Also, compared to a single dose administration, the repeated intake of 5 g/day of trans-resveratrol for 29 days was found to result in significantly greater peak plasma concentrations of trans-resveratrol and two resveratrol glucuronide conjugates (11). Repeated doses of 1 g/day of trans-resveratrol (a dose less likely to cause side effects; see Safety) could yield maximum plasma concentrations of about 22 μM for resveratrol-3-O-sulfate (the most abundant sulfate conjugate in humans) and about 7-8 μM for typical monoglucuronide conjugates (12).
A few studies have examined the influence of food matrix on resveratrol absorption and/or bioavailability (reviewed in 13). One study has reported that bioavailability of trans-resveratrol from red wine did not differ when the wine was consumed with a meal (low- or high-fat) versus on an empty stomach (14). Yet, in another study, the absorption of supplemental resveratrol was found to be delayed, but not reduced, by the presence of food in the stomach (15). A third study found that the bioavailability of supplemental resveratrol was reduced by the amount of fat in the diet, but not by the co-administration of quercetin (another polyphenol) or alcohol (16).
Information about the bioavailability of resveratrol in humans is important because most of the experimental research conducted to date has been ‘preclinical,’ i.e., in vitro, exposing cells to resveratrol concentrations up to 100 times greater than peak plasma concentrations observed in humans, and in animal models given very high (non dietary) doses of resveratrol (13). While cells that line the digestive tract are exposed to unmetabolized resveratrol, other tissues are likely exposed to resveratrol metabolites. At present, little is known about the biological activity of resveratrol metabolites. Yet, if some tissues are capable of converting resveratrol metabolites back to resveratrol, stable resveratrol conjugates in tissues could serve as a pool in the body from which resveratrol might be regenerated (6, 12).
Biological Activities
The biological significance of resveratrol has been primarily investigated in test tubes and cultured cells, and to a lesser extent, in animal models. Of note, a recent publication by Tomé-Carneiro et al. (13) thoroughly reviewed the most relevant preclinical studies published in the most recent decades. It is important to keep in mind that many of the biological activities discussed below were observed in cells cultured in the presence of resveratrol at higher concentrations than those likely to be achieved in humans consuming resveratrol orally (see Metabolism and Bioavailability).
Direct antioxidant activity
In the test tube, resveratrol effectively scavenges (neutralizes) free radicals and other oxidants (17, 18) and inhibits low-density lipoprotein (LDL) oxidation (19, 20). Resveratrol was found to induce antioxidant enzymes, including superoxide dismutase (SOD), thioredoxin, glutathione peroxidase-1, heme oxygenase-1, and catalase, and/or inhibit reactive oxygen species (ROS) production by nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) (21). However, there is little evidence that resveratrol is an important antioxidant in vivo. Upon oral consumption of resveratrol, circulating and intracellular levels of resveratrol in humans are likely to be much lower than that of other important antioxidants, such as vitamin C, uric acid, vitamin E, and glutathione. Moreover, the antioxidant activity of resveratrol metabolites, which comprise most of the circulating resveratrol, may be lower than that of resveratrol (22).
Estrogenic and anti-estrogenic activities
Endogenous estrogens are steroid hormones synthesized by humans and other mammals; these hormones bind to estrogen receptors within cells. The estrogen-receptor complex interacts with unique sequences in DNA (estrogen response elements; EREs) to modulate the expression of estrogen-responsive genes (23). The chemical structure of resveratrol is very similar to that of the synthetic estrogen agonist, diethylstilbestrol (Figure 2), suggesting that resveratrol might also function as an estrogen agonist, i.e., might bind to estrogen receptors and elicit similar responses to endogenous estrogens. However, in cell culture experiments, resveratrol was found to act either as an estrogen agonist or as an estrogen antagonist depending on such factors as cell type, estrogen receptor isoform (ERα or ERβ), and the presence of endogenous estrogens (23). Most recently, resveratrol was shown to improve endothelial wound healing through an ERα-dependent pathway in an animal model of arterial injury (24).
Biological activities related to cancer prevention
Effects on biotransformation enzymes
Some compounds are not carcinogenic until they have been metabolized in the body by phase I biotransformation enzymes, especially cytochrome P450 enzymes (2). By inhibiting the expression and activity of certain cytochrome P450 enzymes (25, 26), resveratrol might help prevent cancer by limiting the activation of procarcinogens. In contrast, increasing the activity of phase II detoxification enzymes generally promotes the excretion of potentially toxic or carcinogenic chemicals. Resveratrol has been found to increase the expression and activity of NAD(P)H:quinone oxidoreductase-1 (NQO1) in cultured cells (27) and may be a weak inducer of other phase II enzymes (28).
Inhibition of proliferation and induction of apoptosis
Following DNA damage, the cell cycle can be transiently arrested to allow for DNA repair or activation of pathways leading to cell death (apoptosis) if the damage is irreparable (29). Defective cell cycle regulation may result in the propagation of mutations that contribute to the development of cancer. Moreover, unlike normal cells, cancer cells proliferate rapidly and are unable to respond to cell death signals that initiate apoptosis. Resveratrol has been found to induce cell cycle arrest and/or apoptosis (programmed cell death) in a number of cancer cell lines (reviewed in 13).
Inhibition of tumor invasion and angiogenesis
Cancerous cells invade normal tissue aided by enzymes called matrix metalloproteinases. Resveratrol has been found to inhibit the activity of at least one type of matrix metalloproteinase (30, 31). To fuel their rapid growth, invasive tumors must also develop new blood vessels by a process known as angiogenesis. Resveratrol has been found to inhibit angiogenesis in vitro (32-34) and in vivo (35).
Anti-inflammatory effects
Inflammation promotes cellular proliferation and angiogenesis and inhibits apoptosis (36). Resveratrol has been found to inhibit the activity of several inflammatory enzymes in vitro, including cyclooxygenases and lipoxygenases (37, 38). Resveratrol may also inhibit pro-inflammatory transcription factors, such as NFκB or AP-1 (39, 40).
Biological activities related to cardiovascular disease prevention
Inhibition of vascular cell adhesion molecule (VCAM) expression
Atherosclerosis is an inflammatory process in which lipids deposit in plaques (known as atheromas) within arterial walls and increase the risk of myocardial infarction (41). One of the earliest events in the development of atherosclerosis is the recruitment of inflammatory white blood cells from the blood to the arterial wall by vascular cell adhesion molecules (42). Resveratrol has been found to inhibit the expression of adhesion molecules in cultured endothelial cells (43, 44).
Inhibition of vascular smooth muscle cell (VSMC) proliferation
The proliferation of vascular smooth muscle cells (VSMCs) plays an important role in the progression of hypertension, atherosclerosis, and restenosis (when treated arteries become blocked again). Resveratrol has been found to inhibit the proliferation of VSMCs in culture (45-47), as well as in vivo (48). Resveratrol appeared to reduce VSMC proliferation via an ERα-dependent mechanism, hence preventing the narrowing of vessels in a mouse model of arterial injury (48).
Stimulation of endolethelial nitric oxide synthase (eNOS) activity
Endothelial nitric oxide synthase (eNOS) is an enzyme that catalyzes the formation of nitric oxide (NO) by vascular endothelial cells. NO is needed to maintain arterial relaxation (vasodilation), and impaired NO-dependent vasodilation is associated with an increased risk of cardiovascular disease (49). Because physiological concentrations of resveratrol were found to stimulate eNOS activity in cultured endothelial cells (50-52), resveratrol might help maintain or improve endothelial function in vivo (see Cardiovascular disease).
Inhibition of platelet activiation and aggregation
Platelet aggregation is one of the first steps in the formation of a blood clot that can occlude a coronary or cerebral artery, resulting in myocardial infarction or stroke, respectively. Resveratrol has been found to inhibit platelet activation and aggregation in vitro (53-55).
Biological activities related to neurodegenerative disease prevention and treatment
Stimulation of neurogenesis and microvessel formation
Age-related mood alterations and memory deficits result from a decrease in the function of the hippocampus in the elderly. Resveratrol was shown to stimulate neurogenesis and blood vessel formation in the hippocampus of healthy old rats. These structural changes were associated with significant improvements in spatial learning, memory formation, and mood function (56).
Stimulation of β-amyloid peptide clearance
One feature of Alzheimer’s disease (AD) is the accumulation of β-amyloid peptide into senile (amyloid) plaques outside neurons in the hippocampus and cortex of AD patients (57). Senile plaques are toxic to cells, resulting in progressive neuronal dysfunction and death. Resveratrol was found to facilitate the clearance of β-amyloid peptide and promote cell survival in primary neurons in culture and neuronal cell lines (58-60). Resveratrol also reduced senile plaque counts in various brain regions of a transgenic AD mouse model (61).
Inhibition of neuroinflammation
Abnormally activated microglia and hypertrophic astrocytes around the senile plaques in AD brains release cytotoxic molecules, such as proinflammatory mediators and ROS, which enhance the formation and deposition of β-amyloid peptides and further damage neurons (57). Resveratrol was found able to inhibit the inflammatory response triggered by β-amyloid peptide-induced microglial activation in microglial cell lines and in a mouse model of cerebral amyloid deposition (62). A decreased occurrence of microglial activation and astrocyte hypertrophy was also reported in healthy aged rats treated with resveratrol (56).
Reduction of oxidative stress
Mitochondrial dysfunction and oxidative stress are thought to be involved in the etiology and/or progression of several neurodegenerative disorders (63). Resveratrol counteracted oxidative stress and β-amyloid peptide-induced toxicity in cultured neuroblastoma (64). Resistance against oxidative stress-related damage in primary neuronal cells treated with resveratrol has been associated with the induction of heme oxygenase-1 (HO-1), an enzyme that degrades pro-oxidant heme (65). In an experimental model of stroke, resveratrol limited infarct size during ischemia-reperfusion in wild-type mice but not in mice lacking the HO-1 gene (66). Also, resveratrol was able to correct experimentally induced oxidative stress and the associated cognitive dysfunction in rats (67).
Disease Prevention
Cancer
Resveratrol has been found to inhibit the proliferation of a variety of human cancer cell lines, including those from breast, prostate, stomach, colon, pancreatic, and thyroid cancers (2). In animal models, oral administration, topical application, and/or injection of resveratrol inhibited the development of chemically-induced cancer at many sites, including gastrointestinal tract, liver, skin, breast, prostate, and lung (reviewed in 68, 69). The anti-cancer effects of resveratrol in rodent models involved the reduction of cell proliferation, the induction of apoptosis, and the inhibition of angiogenesis, tumor growth, and metastasis (reviewed in 13). Yet, a few animal studies have reported a lack of an effect of oral resveratrol in inhibiting the development of lung cancer induced by carcinogens in cigarette smoke (70, 71), and the study of resveratrol administration on colon cancer has given mixed results (72-74).
At present, it is not known whether resveratrol might be beneficial in the prevention and/or the treatment of cancer in humans. The low bioavailability of resveratrol reported in human studies limits the clinical evaluation of possible systemic health effects of resveratrol in humans (see Metabolism and Bioavailability). Yet, in a pilot study, unmetabolized resveratrol and conjugates have been detected in colorectal tumor tissues from 20 cancer patients following daily oral supplementation with either 4 g or 8 g of resveratrol for 29 days. Resveratrol appeared to be well tolerated and significantly, though modestly, reduced cell proliferation compared to baseline (75). A micronized formulation of resveratrol (named SRT501), which was meant to increase resveratrol delivery to target tissues, was given for 14 days to 6 patients with colorectal cancer and liver metastasis in a small randomized, double-blind, placebo-controlled trial (76). Unmetabolized resveratrol was measurable in the liver of five out of six patients who consumed 5 g of SRT501, and SRT501 administration resulted in an increased detection of the apoptotic marker, cleaved caspase-3, in hepatic tumor tissues. Yet, in an unrandomized and unblinded trial in patients with multiple myeloma, the administration of SRT501 was associated with a number of serious adverse effects, including kidney failure, such that the trial was halted (77). Since kidney failure is a frequent complication in myeloma patients, it is unclear whether kidney failure cases should be solely attributed to the use of SRT501. Nevertheless, there is a need to find safe ways to increase resveratrol bioavailability in humans before exploring its putative benefits in clinical settings (6, 78).
Cardiovascular disease
Red wine polyphenols
Significant reductions in cardiovascular disease risk have been associated with moderate consumption of alcoholic beverages (79). The “French Paradox” — the observation that incidence of coronary heart disease was relatively low in France despite high levels of dietary saturated fat and cigarette smoking — led to the idea that regular consumption of red wine might provide additional protection from cardiovascular disease (80). Red wine contains variable and usually low concentrations of resveratrol (see Sources) and higher concentrations of flavonoids like procyanidins. These polyphenolic compounds have displayed antioxidant, anti-inflammatory, and other potentially anti-atherogenic effects in the test tube and in some animal models of atherosclerosis (81). The results of epidemiological studies addressing this question have been inconsistent. While some large prospective cohort studies found that wine drinkers were at lower risk of cardiovascular disease than beer or liquor drinkers (82-84), others found no difference (85-87). Socioeconomic and lifestyle differences between people who prefer wine and those who prefer beer or liquor may explain part of the additional benefit observed in some studies: people who prefer wine tend to have higher incomes, more education, smoke less, and eat more fruit and vegetables and less saturated fat than those who prefer other alcoholic beverages (87-92).
Although moderate alcohol consumption has been consistently associated with reductions in coronary heart disease risk, it is not yet clear whether red wine polyphenols confer any additional risk reduction. Interestingly, studies that administered alcohol-free red wine to rodents noted improvements in various parameters related to cardiovascular disease (93, 94), and a placebo-controlled human study found that heart disease patients administered red grape polyphenol extract experienced acute improvements in endothelial function (95). Whether increased consumption of polyphenols from red wine provides any additional cardiovascular benefits beyond those associated with red wine’s alcohol content needs to be further examined (see the article on Alcoholic Beverages) (96).
Resveratrol and endothelial function
Endothelial dysfunction is usually associated with the presence of cardiovascular risk factors (e.g., insulin resistance, hypertension, and hypercholesterolemia) and is thought to precede the clinical manifestation of cardiovascular and metabolic disorders. Endothelial dysfunction is characterized by abnormal vasoconstriction, leukocyte adherence to vascular endothelial cells, platelet activation and aggregation, smooth muscle cell proliferation, vascular inflammation, thrombosis (clot formation), impaired coagulation, and atherosclerosis (97).
Experimental studies: Resveratrol has been found to exert a number of protective effects on the cardiovascular system in vitro, including inhibition of both platelet activation and aggregation (53, 98, 99), promotion of vasodilation by enhancing the production of nitric oxide (NO) (52), and control of the production of inflammatory lipid mediators (38, 100, 101). However, the concentrations of resveratrol required to produce these effects are often higher than those measured in human plasma after oral consumption of resveratrol (9). Some animal studies also suggested that high oral doses of resveratrol could decrease the risk of thrombosis and atherosclerosis (102, 103), although one study found increased atherosclerosis in animals fed resveratrol (104). Other protective effects of resveratrol in vivo include the reduction of cardiac hypertrophy and the lowering of blood pressure in various models, as well as the limitation of infarct size in post myocardial infarction rats (reviewed in 13).
Randomized controlled studies: In a six-month, cross-over study, 34 patients with metabolic syndrome were randomized to receive resveratrol (100 mg/day) for three months either immediately at the beginning of the study or three months later. Resveratrol supplementation resulted in improved values of flow-mediated dilation (FMD) of the brachial artery, a surrogate marker of vascular health. Yet, FMD returned to baseline values within three months after discontinuing resveratrol (105). One study limitation was that the resveratrol formulation contained additional compounds (i.e., vitamin D3, quercetin, and rice bran phytate), which may also affect endothelial function. One randomized, placebo-controlled study in healthy overweight or obese volunteers (BMI, 25-34 kg/m2) found that a single dose of trans-resveratrol (30 mg, 90 mg, or 270 mg) improved brachial FMD around 60 minutes after administration (106). In a second study, the same investigators found that FMD improvements were similar whether participants had received a single dose of resveratrol (75 mg) or a daily dose (75 mg/day of resveratrol) for six months (107).
In a few additional studies, resveratrol was shown to improve endothelial function by reducing vascular inflammation and endothelial activation. A randomized, double-blind, placebo-controlled study in 41 healthy subjects found that daily supplementation with resveratrol (400 mg), grapeseed extract (400 mg), and quercetin (100 mg), for one month significantly reduced the expression of interleukin-8 (IL-8) and cell adhesion molecules (ICAM-1 and VCAM-1) in endothelial cells, suggestive of a protective effect against endothelial dysfunction (108). The daily intake of a resveratrol-rich grape supplement was compared to resveratrol-free grape supplement in a year-long, randomized, double blind, placebo-controlled study in 75 individuals at high risk for cardiovascular disease (CVD). Administration of the resveratrol-rich supplement (resveratrol: 8 mg/day for 6 months, then 16 mg/day for another 6 months) significantly improved the profile of circulating inflammatory markers, reducing levels of C-reactive protein (CRP) and Tumor Necrosis Factor-α (TNF-α), as well as the level of thrombogenic factor, Plasminogen Activator Inhibitor-1 (PAI-1) (109). The decreased concentrations of two CVD risk markers, oxidized low-density lipoprotein (oxLDL) and apolipoprotein B (ApoB) after six months further suggested a cardioprotective effect of resveratrol (110). Supplementation of patients with stable coronary heart disease with the same regimen also improved the profile of circulating inflammatory markers and reduced the expression of proinflammatory genes in peripheral blood mononuclear cells (PBMCs) (111). The expression of microRNAs and cytokines specifically involved in atherogenic and pro-inflammatory signals were also found to be downregulated in the PBMCs of supplemented patients (112). Finally, although it is not clear whether hypertension is a cause or an effect of endothelial dysfunction, a recent meta-analysis of randomized controlled trials suggested that high doses of resveratrol (≥150 mg/for at least one month) might help lower systolic blood pressure in individuals at risk for CVD (113).
While preliminary human studies suggest that resveratrol may have beneficial effects on cardiovascular health, there is currently no convincing evidence that these effects can be achieved in the amounts present in one to two glasses of red wine (see Sources). For more information regarding resveratrol and cardiovascular disease, see (114).
Longevity
Caloric restriction is known to extend the lifespan of a number of species, including yeast, worms, flies, fish, rats, and mice (115). In yeast (Saccharomyces cerevisiae), caloric restriction stimulates the activity of an enzyme known as Silent information regulator 2 protein (Sir2) or sirtuin (116). Yeast Sir2 is a nicotinamide adenine dinucleotide (NAD)-dependent deacetylase enzyme that removes the acetyl group from acetylated lysine residues in target proteins (see the article on Niacin).
Providing resveratrol to yeast increased Sir2 activity in the absence of caloric restriction and extended the replicative (but not the chronological) lifespan of yeast by 70% (117). Resveratrol feeding also extended the lifespan of worms (Caenorhabditis elegans) and fruit flies (Drosophila melanogaster) by a similar mechanism (118). Additionally, resveratrol dose-dependently increased the lifespan of a vertebrate fish (Nothobranchius furzeri) (119). Resveratrol was also found to extend the lifespan of mice on a high-calorie diet such that their lifespan was similar to that of mice fed a standard diet (120). Although resveratrol increased the activity of the Sir2 homologous human sirtuin 1 (SIRT1) in the test tube (117), there are no epidemiological data to link resveratrol, SIRT1 activation, and extended human lifespan. Moreover, the supraphysiological concentrations of resveratrol required to increase human SIRT1 activity were considerably higher than concentrations that have been measured in human plasma after oral consumption.
The results of a nine-year prospective cohort study in over 700 older adults (≥65 years) indicated that participants who were alive at the end of the study had baseline concentrations of total urinary resveratrol metabolites (used as a biomarker of resveratrol intake) similar to those who died during the study period (121). Based on a lack of correlation with baseline inflammatory markers, cardiovascular disease and cancer incidence, and all-cause mortality, the authors concluded that higher versus lower quartiles of urinary resveratrol metabolite concentrations did not predict risk of chronic disease or mortality. However, key experts identified several limitations regarding the quality of the research (122, 123). Specifically, the use of single measures of total urinary resveratrol metabolites at baseline has been highlighted as being unlikely to reflect lifetime consumption of wine or exposure to dietary resveratrol (122).
Cognitive decline
In a mouse model of Alzheimer’s disease (AD), caloric restriction has been shown to limit the production and deposition of neurotoxic β-amyloid peptide in the brain (124). Similar to the effect of caloric restriction, resveratrol was found to improve obesity and diabetes-related metabolic deregulations via the activation of metabolic sensors, including SIRT and the AMP-activated protein kinase (AMPK) (125), as well as to promote the AMPK-dependent clearance of β-amyloid peptide in the brain of an AD mouse model (60). Resveratrol has also exhibited additional neuroprotective properties in cultured cells and animal models (see Biological Activities).
Although resveratrol bioavailability to the brain is uncertain (78), a randomized, double-blind, placebo-controlled study has reported an increase in cerebral blood flow in the prefrontal cortex of healthy young subjects (ages, 18-29 years) following a single oral dose of 500 mg of resveratrol. However, resveratrol intake did not improve performance in cognitively demanding tasks undertaken during the post-administration period (126). More recently, the co-administration of resveratrol (200 mg/day) and quercetin (320 mg/day) for 26 weeks in a double-blind, placebo-controlled study significantly improved measures of memory function and enhanced blood glucose control in 46 healthy, overweight older adults (ages, 50-80 years; BMI, 25-30 kg/m2) (127). Additional evidence of the potential of resveratrol to mimic the metabolic benefits of caloric restriction on cognitive health may come from ongoing clinical trials in both healthy older individuals and AD patients (128).
Disease Treatment
Impaired glucose tolerance and type 2 diabetes mellitus
More than one out of three American adults has impaired glucose tolerance (also known as prediabetes), which places them at increased risk of developing type 2 diabetes (129). Impaired glucose tolerance is associated with insulin resistance in skeletal muscle — the major peripheral tissue for insulin-mediated glucose uptake — as well as defective insulin secretion by pancreatic β-cells. Muscle insulin resistance, which is thought to be the earliest stage in the development of type 2 diabetes, is characterized by excess lipid exposure, impaired insulin receptor signaling, impaired glucose uptake, mitochondrial dysfunction, reduced fatty acid oxidation, and increased expression of pro-inflammatory cytokines.
In animal studies, resveratrol has been shown to improve insulin sensitivity, glucose tolerance, and lipid profiles in obese and/or metabolically abnormal animals (reviewed in 130).
In humans, short-term supplementation with resveratrol has been associated with beneficial effects on glucose and lipid metabolism in individuals with type 2 diabetes. In a randomized, double-blind, placebo-controlled study, the effect of oral resveratrol supplementation (1,000 mg/day for 45 days) on the control of glucose metabolism was assessed in 70 subjects with type 2 diabetes (131). Comparison of changes between baseline and end-of-study measures between placebo and intervention groups showed that resveratrol significantly lowered both fasting glucose and fasting insulin concentrations and improved measures of glycemic control (HbA1c level) and insulin sensitivity (HOMA-IR). In addition, the level of HDL-cholesterol was increased while the level of LDL-cholesterol and systolic blood pressure were significantly reduced. No changes were found in measures of diastolic blood pressure, total cholesterol, triglycerides, and markers of liver function (131). Additionally, in a randomized, open-label, and controlled study, the effect of oral resveratrol (250 mg/day) on glycemic control and lipid metabolism was assessed in 62 type 2 diabetics (132). During the three-month study period, changes in biochemical and clinical parameters, including fasting glucose concentration, HbA1c level, systolic and diastolic blood pressure, total cholesterol, and LDL-cholesterol, were significantly improved with resveratrol compared to control (i.e., no resveratrol). Doses as low as 10 mg/day of resveratrol also resulted in lower insulin resistance in a four-week, randomized, placebo-controlled study in 19 male subjects with type 2 diabetes (133).
Obesity (defined as a body mass index [BMI] ≥ 30 kg/m2) is a well-known risk factor for the development of type 2 diabetes. A few clinical studies have evaluated the effects of resveratrol on key metabolic variables in overweight or obese subjects with no overt metabolic dysfunction and found little or no metabolic benefits following resveratrol treatment (134-136). Yet, at present, there is no available evidence to suggest whether overweight or obese individuals with impaired glucose tolerance could benefit from resveratrol supplements and reduce their risk of developing type 2 diabetes (137).
Current data suggest that resveratrol could improve specific metabolic variables in individuals with type 2 diabetes (138, 139), but more research is needed to assess its effect in individuals at risk for diabetes, including obese subjects with impaired glucose tolerance.
Sources
Food sources
Resveratrol is found in grapes, wine, grape juice, peanuts, cocoa, and berries of Vaccinium species, including blueberries, bilberries, and cranberries (140-143). In grapes, resveratrol is found only in the skins (144). The amount of resveratrol in grape skins varies with the grape cultivar, its geographic origin, and exposure to fungal infection (145). The amount of fermentation time a wine spends in contact with grape skins is also an important determinant of its resveratrol content. Because grape skins are removed early during the production process of white and rosé wines, these wines generally contain less resveratrol than red wines (4). Therefore, because of variations between types of wine, vintages, and regions, it is very difficult to provide accurate estimates of resveratrol content in the thousands of wines from worldwide wineries. Yet, it appears that resveratrol content in wine is usually low, highly variable and unpredictable, and resveratrol is only a minor compound in the complete set of grape and wine polyphenols (13).
The predominant form of resveratrol in grapes and grape juice is trans-resveratrol-3-O-β-glucoside (trans-piceid), and wines contain significant amounts of resveratrol aglycones, thought to be the result of sugar cleavage during fermentation (3, 140). Many wines also contain significant amounts of cis-resveratrol (see Figure 1 above), which may be produced during fermentation or released from viniferins (resveratrol polymers) (146). Red wine is a relatively rich source of resveratrol, but other polyphenols are present in red wine at considerably higher concentrations than resveratrol (see the article on Flavonoids) (147). Estimates of resveratrol content of some beverages and foods are listed in Table 1 and Table 2. These values should be considered approximate since the resveratrol content of foods and beverages can vary considerably.
| Variety | Lowest (mg/L) | Highest (mg/L) | Mean (mg/L) | 5-oz Glass (mg) |
|---|---|---|---|---|
| Pinot Noir | 0.2 | 11.9 | 3.6 ± 2.9 | 0.5 |
| Merlot | 0.3 | 14.3 | 2.8 ± 2.6 | 0.4 |
| Zweigelt | 0.6 | 4.7 | 1.9 ± 1.2 | 0.3 |
| Shiraz | 0.2 | 3.2 | 1.8 ± 0.9 | 0.3 |
| Cabernet Sauvignon | - | 9.3 | 1.7 ± 1.7 | 0.2 |
| Red wines (global) | - | 14.3 | 1.9 ± 1.7 | 0.3 |
| Food | Serving | Total Resveratrol (mg) |
|---|---|---|
| Peanuts (raw) | 1 cup (146 g) | 0.01-0.26 |
| Peanuts (boiled) | 1 cup (180 g) | 0.32-1.28 |
| Peanut butter | 1 cup (258 g) | 0.04-0.13 |
| Red grapes | 1 cup (160 g) | 0.24-1.25 |
Supplements
Most resveratrol supplements available in the US contain extracts of the root of Polygonum cuspidatum, also known as Fallopia japonica, Japanese knotweed, or Hu Zhang (150). Red wine extracts and grape extracts (from Vitis vinifera) containing resveratrol and other polyphenols are also available as dietary supplements. Resveratrol supplements may contain anywhere from less than 1 milligram (mg) to 500 mg of resveratrol per tablet or capsule, but it is not known whether there is a safe and effective dosage for chronic disease prevention in humans (also see the section on Safety).
Safety
Adverse effects
In rats, daily oral administration of trans-resveratrol at doses up to 700 mg/kg of body weight for 90 days resulted in no apparent adverse effects (151). Other toxicity studies conducted in animal models estimated that the no-observed-adverse-effect-level (NOAEL) for resveratrol was 200 mg/kg/days and 600 mg/kg/day in rats and dogs, respectively (152). Resveratrol is not known to be toxic or cause significant adverse effects in humans, but there have been only a few controlled clinical trials to date (reviewed in 153). A trial evaluating the safety of oral trans-resveratrol in 10 subjects found that a single dose of 5,000 mg resulted in no serious adverse effects (10). In a follow-up study, mild-to-moderate gastrointestinal side effects, including nausea, abdominal pain, flatulence, and diarrhea, have been reported in participants who consume more than 1,000 mg/day of resveratrol for up to 29 consecutive days (11). Mild diarrhea was also reported in six out of eight individuals who consumed 2,000 mg of resveratrol twice daily for two periods of eight days in an open-label and within subject-control study (16).
Pregnancy and lactation
The safety of resveratrol-containing supplements during pregnancy and lactation has not been established (150). Because there is no known safe amount of alcohol consumption at any stage of pregnancy (154), pregnant women should avoid consuming wine as a source of resveratrol.
Estrogen-sensitive conditions
Until more is known about the estrogenic activity of resveratrol in humans, women with a history of estrogen-sensitive cancers, such as breast, ovarian, and uterine cancers, should avoid resveratrol supplements (see Estrogenic and anti-estrogenic activities) (150).
Drug interactions
Anticoagulant and antiplatelet drugs
Resveratrol has been found to inhibit human platelet aggregation in vitro (53, 155). Theoretically, high intakes of resveratrol (i.e., from supplements) could increase the risk of bruising and bleeding when taken with anticoagulant drugs, such as warfarin (Coumadin) and heparin; antiplatelet drugs, such as clopidogrel (Plavix) and dipyridamole (Persantine); and non-steroidal anti-inflammatory drugs (NSAIDs), including aspirin, ibuprofen, diclofenac, naproxen, and others.
Drugs metabolized by cytochrome P450
Cytochrome P450 (CYP) enzymes are phase I biotransformation enzymes involved in the metabolism of a broad range of compounds, from endogenous molecules to therapeutic agents. The most abundant CYP isoform in the human liver and intestines is cytochrome P450 3A4 (CYP3A4), which catalyzes the metabolism of about half of all marketed drugs in the US (156). Resveratrol has been reported to inhibit CYP3A4 activity in vitro (157, 158) and in healthy volunteers (28). Therefore, high intakes of resveratrol (i.e., from supplements) could potentially reduce the metabolic clearance of drugs that undergo extensive first-pass metabolism by CYP3A4, hence increasing the bioavailability and risk of toxicity of these drugs. Some of the many drugs metabolized by CYP3A4 include HMG-CoA reductase inhibitors (statins), calcium channel antagonists (felodipine, nicardipine, nifedipine, nisoldipine, nitrendipine, nimodipine, and verapamil), anti-arrhythmic agents (amiodarone), HIV protease inhibitors (saquinavir), immunosuppressants (cyclosporine and tacrolimus), antihistamines (terfenadine), benzodiazepines (midazolam and triazolam), and drugs used to treat erectile dysfunction (sildenafil). Of note, a recently completed clinical trial (NCT01173640) examined the potential for single and multiple doses of resveratrol (1,000 mg) to interfere with the metabolism of midazolam in healthy volunteers, and results are soon to be published (153). Other CYP enzymes (e.g., CYP2D6 and CYP2C9) may also be inhibited by resveratrol (reviewed in 159).
Finally, resveratrol was found to be a weak inducer of the expression and activity of CYP1A2, which catalyzes the metabolism of several drugs, including acetaminophen (paracetamol) and the antidepressant drugs, clomipramine and imipramine (28, 156). This suggests that resveratrol may interfere with CYP1A2-mediated drug metabolism by increasing drug clearance, possibly lowering circulating drug concentrations below therapeutic levels.
Authors and Reviewers
Originally written in 2005 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in May 2008 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in May 2015 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in May 2015 by:
Juan Carlos Espín, Ph.D.
Research Professor
Consejo Superior de Investigaciones Científicas (CSIC)
Department of Food Science & Technology
Murcia, Spain
Last updated 6/11/15 Copyright 2005-2024 Linus Pauling Institute
References
1. Soleas GJ, Diamandis EP, Goldberg DM. Resveratrol: a molecule whose time has come? And gone? Clin Biochem. 1997;30(2):91-113. (PubMed)
2. Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y. Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res. 2004;24(5A):2783-2840. (PubMed)
3. Romero-Perez AI, Ibern-Gomez M, Lamuela-Raventos RM, de La Torre-Boronat MC. Piceid, the major resveratrol derivative in grape juices. J Agric Food Chem. 1999;47(4):1533-1536. (PubMed)
4. Siemann EH, Creasey LL. Concentration of the phytoalexin resveratrol in wine. Am J Enol Vitic. 1992;43(1):49-52.
5. Renaud S, de Lorgeril M. Wine, alcohol, platelets, and the French paradox for coronary heart disease. Lancet. 1992;339(8808):1523-1526. (PubMed)
6. Walle T. Bioavailability of resveratrol. Ann N Y Acad Sci. 2011;1215:9-15. (PubMed)
7. Burkon A, Somoza V. Quantification of free and protein-bound trans-resveratrol metabolites and identification of trans-resveratrol-C/O-conjugated diglucuronides - two novel resveratrol metabolites in human plasma. Mol Nutr Food Res. 2008;52(5):549-557. (PubMed)
8. Goldberg DM, Yan J, Soleas GJ. Absorption of three wine-related polyphenols in three different matrices by healthy subjects. Clin Biochem. 2003;36(1):79-87. (PubMed)
9. Walle T, Hsieh F, Delegge MH, Oatis JE, Jr., Walle UK. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab Dispos. 2004;32(12):1377-1382. (PubMed)
10. Boocock DJ, Faust GE, Patel KR, et al. Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol Biomarkers Prev. 2007;16(6):1246-1252. (PubMed)
11. Brown VA, Patel KR, Viskaduraki M, et al. Repeat dose study of the cancer chemopreventive agent resveratrol in healthy volunteers: safety, pharmacokinetics, and effect on the insulin-like growth factor axis. Cancer Res. 2010;70(22):9003-9011. (PubMed)
12. Patel KR, Andreadi C, Britton RG, et al. Sulfate metabolites provide an intracellular pool for resveratrol generation and induce autophagy with senescence. Sci Transl Med. 2013;5(205):205ra133. (PubMed)
13. Tome-Carneiro J, Larrosa M, Gonzalez-Sarrias A, Tomas-Barberan FA, Garcia-Conesa MT, Espin JC. Resveratrol and clinical trials: the crossroad from in vitro studies to human evidence. Curr Pharm Des. 2013;19(34):6064-6093. (PubMed)
14. Vitaglione P, Sforza S, Galaverna G, et al. Bioavailability of trans-resveratrol from red wine in humans. Mol Nutr Food Res. 2005;49(5):495-504. (PubMed)
15. Vaz-da-Silva M, Loureiro AI, Falcao A, et al. Effect of food on the pharmacokinetic profile of trans-resveratrol. Int J Clin Pharmacol Ther. 2008;46(11):564-570. (PubMed)
16. la Porte C, Voduc N, Zhang G, et al. Steady-State pharmacokinetics and tolerability of trans-resveratrol 2000 mg twice daily with food, quercetin and alcohol (ethanol) in healthy human subjects. Clin Pharmacokinet. 2010;49(7):449-454. (PubMed)
17. Leonard SS, Xia C, Jiang BH, et al. Resveratrol scavenges reactive oxygen species and effects radical-induced cellular responses. Biochem Biophys Res Commun. 2003;309(4):1017-1026. (PubMed)
18. Vlachogianni IC, Fragopoulou E, Kostakis IK, Antonopoulou S. In vitro assessment of antioxidant activity of tyrosol, resveratrol and their acetylated derivatives. Food Chem. 2015;177:165-173. (PubMed)
19. Brito P, Almeida LM, Dinis TC. The interaction of resveratrol with ferrylmyoglobin and peroxynitrite; protection against LDL oxidation. Free Radic Res. 2002;36(6):621-631. (PubMed)
20. Frankel EN, Waterhouse AL, Kinsella JE. Inhibition of human LDL oxidation by resveratrol. Lancet. 1993;341(8852):1103-1104. (PubMed)
21. Wang H, Yang YJ, Qian HY, Zhang Q, Xu H, Li JJ. Resveratrol in cardiovascular disease: what is known from current research? Heart Fail Rev. 2012;17(3):437-448. (PubMed)
22. Bradamante S, Barenghi L, Villa A. Cardiovascular protective effects of resveratrol. Cardiovasc Drug Rev. 2004;22(3):169-188. (PubMed)
23. Tangkeangsirisin W, Serrero G. Resveratrol in the chemoprevention and chemotherapy of breast cancer. In: Bagchi D, Preuss HG, eds. Phytopharmaceuticals in Cancer Chemoprevention. Boca Raton: CRC Press; 2005:449-463.
24. Yurdagul A, Jr., Kleinedler JJ, McInnis MC, et al. Resveratrol promotes endothelial cell wound healing under laminar shear stress through an estrogen receptor-alpha-dependent pathway. Am J Physiol Heart Circ Physiol. 2014;306(6):H797-806. (PubMed)
25. Chen ZH, Hurh YJ, Na HK, et al. Resveratrol inhibits TCDD-induced expression of CYP1A1 and CYP1B1 and catechol estrogen-mediated oxidative DNA damage in cultured human mammary epithelial cells. Carcinogenesis. 2004;25(10):2005-2013. (PubMed)
26. Ciolino HP, Yeh GC. Inhibition of aryl hydrocarbon-induced cytochrome P-450 1A1 enzyme activity and CYP1A1 expression by resveratrol. Mol Pharmacol. 1999;56(4):760-767. (PubMed)
27. Hsieh TC, Lu X, Wang Z, Wu JM. Induction of quinone reductase NQO1 by resveratrol in human K562 cells involves the antioxidant response element ARE and is accompanied by nuclear translocation of transcription factor Nrf2. Med Chem. 2006;2(3):275-285. (PubMed)
28. Chow HH, Garland LL, Hsu CH, et al. Resveratrol modulates drug- and carcinogen-metabolizing enzymes in a healthy volunteer study. Cancer Prev Res (Phila). 2010;3(9):1168-1175. (PubMed)
29. Stewart ZA, Westfall MD, Pietenpol JA. Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol Sci. 2003;24(3):139-145. (PubMed)
30. Woo JH, Lim JH, Kim YH, et al. Resveratrol inhibits phorbol myristate acetate-induced matrix metalloproteinase-9 expression by inhibiting JNK and PKC delta signal transduction. Oncogene. 2004;23(10):1845-1853. (PubMed)
31. Yu H, Pan C, Zhao S, Wang Z, Zhang H, Wu W. Resveratrol inhibits tumor necrosis factor-alpha-mediated matrix metalloproteinase-9 expression and invasion of human hepatocellular carcinoma cells. Biomed Pharmacother. 2008;62(6):366-372. (PubMed)
32. Igura K, Ohta T, Kuroda Y, Kaji K. Resveratrol and quercetin inhibit angiogenesis in vitro. Cancer Lett. 2001;171(1):11-16. (PubMed)
33. Lin MT, Yen ML, Lin CY, Kuo ML. Inhibition of vascular endothelial growth factor-induced angiogenesis by resveratrol through interruption of Src-dependent vascular endothelial cadherin tyrosine phosphorylation. Mol Pharmacol. 2003;64(5):1029-1036. (PubMed)
34. Chen Y, Tseng SH. Review. Pro- and anti-angiogenesis effects of resveratrol. In Vivo. 2007;21(2):365-370. (PubMed)
35. Kanavi MR, Darjatmoko S, Wang S, et al. The sustained delivery of resveratrol or a defined grape powder inhibits new blood vessel formation in a mouse model of choroidal neovascularization. Molecules. 2014;19(11):17578-17603. (PubMed)
36. Steele VE, Hawk ET, Viner JL, Lubet RA. Mechanisms and applications of non-steroidal anti-inflammatory drugs in the chemoprevention of cancer. Mutat Res. 2003;523-524:137-144. (PubMed)
37. Donnelly LE, Newton R, Kennedy GE, et al. Anti-inflammatory effects of resveratrol in lung epithelial cells: molecular mechanisms. Am J Physiol Lung Cell Mol Physiol. 2004;287(4):L774-783. (PubMed)
38. Pinto MC, Garcia-Barrado JA, Macias P. Resveratrol is a potent inhibitor of the dioxygenase activity of lipoxygenase. J Agric Food Chem. 1999;47(12):4842-4846. (PubMed)
39. Shankar S, Singh G, Srivastava RK. Chemoprevention by resveratrol: molecular mechanisms and therapeutic potential. Front Biosci. 2007;12:4839-4854. (PubMed)
40. de la Lastra CA, Villegas I. Resveratrol as an anti-inflammatory and anti-aging agent: mechanisms and clinical implications. Mol Nutr Food Res. 2005;49(5):405-430. (PubMed)
41. Hartman J, Frishman WH. Inflammation and atherosclerosis: a review of the role of interleukin-6 in the development of atherosclerosis and the potential for targeted drug therapy. Cardiol Rev. 2014;22(3):147-151. (PubMed)
42. Stocker R, Keaney JF, Jr. Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84(4):1381-1478. (PubMed)
43. Carluccio MA, Siculella L, Ancora MA, et al. Olive oil and red wine antioxidant polyphenols inhibit endothelial activation: antiatherogenic properties of Mediterranean diet phytochemicals. Arterioscler Thromb Vasc Biol. 2003;23(4):622-629. (PubMed)
44. Ferrero ME, Bertelli AE, Fulgenzi A, et al. Activity in vitro of resveratrol on granulocyte and monocyte adhesion to endothelium. Am J Clin Nutr. 1998;68(6):1208-1214. (PubMed)
45. Ekshyyan VP, Hebert VY, Khandelwal A, Dugas TR. Resveratrol inhibits rat aortic vascular smooth muscle cell proliferation via estrogen receptor dependent nitric oxide production. J Cardiovasc Pharmacol. 2007;50(1):83-93. (PubMed)
46. Haider UG, Sorescu D, Griendling KK, Vollmar AM, Dirsch VM. Resveratrol increases serine15-phosphorylated but transcriptionally impaired p53 and induces a reversible DNA replication block in serum-activated vascular smooth muscle cells. Mol Pharmacol. 2003;63(4):925-932. (PubMed)
47. Mnjoyan ZH, Fujise K. Profound negative regulatory effects by resveratrol on vascular smooth muscle cells: a role of p53-p21(WAF1/CIP1) pathway. Biochem Biophys Res Commun. 2003;311(2):546-552. (PubMed)
48. Khandelwal AR, Hebert VY, Dugas TR. Essential role of ER-alpha-dependent NO production in resveratrol-mediated inhibition of restenosis. Am J Physiol Heart Circ Physiol. 2010;299(5):H1451-1458. (PubMed)
49. Duffy SJ, Vita JA. Effects of phenolics on vascular endothelial function. Curr Opin Lipidol. 2003;14(1):21-27. (PubMed)
50. Klinge CM, Blankenship KA, Risinger KE, et al. Resveratrol and estradiol rapidly activate MAPK signaling through estrogen receptors alpha and beta in endothelial cells. J Biol Chem. 2005;280(9):7460-7468. (PubMed)
51. Klinge CM, Wickramasinghe NS, Ivanova MM, Dougherty SM. Resveratrol stimulates nitric oxide production by increasing estrogen receptor alpha-Src-caveolin-1 interaction and phosphorylation in human umbilical vein endothelial cells. FASEB J. 2008;22(7):2185-2197. (PubMed)
52. Takahashi S, Nakashima Y. Repeated and long-term treatment with physiological concentrations of resveratrol promotes NO production in vascular endothelial cells. Br J Nutr. 2012;107(6):774-780. (PubMed)
53. Pace-Asciak CR, Hahn S, Diamandis EP, Soleas G, Goldberg DM. The red wine phenolics trans-resveratrol and quercetin block human platelet aggregation and eicosanoid synthesis: implications for protection against coronary heart disease. Clin Chim Acta. 1995;235(2):207-219. (PubMed)
54. Shen MY, Hsiao G, Liu CL, et al. Inhibitory mechanisms of resveratrol in platelet activation: pivotal roles of p38 MAPK and NO/cyclic GMP. Br J Haematol. 2007;139(3):475-485. (PubMed)
55. Yang YM, Chen JZ, Wang XX, Wang SJ, Hu H, Wang HQ. Resveratrol attenuates thromboxane A2 receptor agonist-induced platelet activation by reducing phospholipase C activity. Eur J Pharmacol. 2008;583(1):148-155. (PubMed)
56. Kodali M, Parihar VK, Hattiangady B, Mishra V, Shuai B, Shetty AK. Resveratrol prevents age-related memory and mood dysfunction with increased hippocampal neurogenesis and microvasculature, and reduced glial activation. Sci Rep. 2015;5:8075. (PubMed)
57. Ma T, Tan MS, Yu JT, Tan L. Resveratrol as a therapeutic agent for Alzheimer's disease. Biomed Res Int. 2014;2014:350516. (PubMed)
58. Chen J, Zhou Y, Mueller-Steiner S, et al. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J Biol Chem. 2005;280(48):40364-40374. (PubMed)
59. Marambaud P, Zhao H, Davies P. Resveratrol promotes clearance of Alzheimer's disease amyloid-beta peptides. J Biol Chem. 2005;280(45):37377-37382. (PubMed)
60. Vingtdeux V, Giliberto L, Zhao H, et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem. 2010;285(12):9100-9113. (PubMed)
61. Karuppagounder SS, Pinto JT, Xu H, Chen HL, Beal MF, Gibson GE. Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer's disease. Neurochem Int. 2009;54(2):111-118. (PubMed)
62. Capiralla H, Vingtdeux V, Zhao H, et al. Resveratrol mitigates lipopolysaccharide- and Abeta-mediated microglial inflammation by inhibiting the TLR4/NF-kappaB/STAT signaling cascade. J Neurochem. 2012;120(3):461-472. (PubMed)
63. Ruszkiewicz J, Albrecht J. Changes in the mitochondrial antioxidant systems in neurodegenerative diseases and acute brain disorders. Neurochem Int. 2015; doi: 10.1016/j.neuint.2014.12.012. [Epub ahead of print]. (PubMed)
64. Albani D, Polito L, Batelli S, et al. The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by alpha-synuclein or amyloid-beta (1-42) peptide. J Neurochem. 2009;110(5):1445-1456. (PubMed)
65. Zhuang H, Kim YS, Koehler RC, Dore S. Potential mechanism by which resveratrol, a red wine constituent, protects neurons. Ann N Y Acad Sci. 2003;993:276-286; discussion 287-278. (PubMed)
66. Sakata Y, Zhuang H, Kwansa H, Koehler RC, Dore S. Resveratrol protects against experimental stroke: putative neuroprotective role of heme oxygenase 1. Exp Neurol. 2010;224(1):325-329. (PubMed)
67. Kumar A, Naidu PS, Seghal N, Padi SS. Neuroprotective effects of resveratrol against intracerebroventricular colchicine-induced cognitive impairment and oxidative stress in rats. Pharmacology. 2007;79(1):17-26. (PubMed)
68. Bishayee A. Cancer prevention and treatment with resveratrol: from rodent studies to clinical trials. Cancer Prev Res (Phila). 2009;2(5):409-418. (PubMed)
69. Bishayee A, Darvesh AS, Politis T, McGory R. Resveratrol and liver disease: from bench to bedside and community. Liver Int. 2010;30(8):1103-1114. (PubMed)
70. Hecht SS, Kenney PM, Wang M, et al. Evaluation of butylated hydroxyanisole, myo-inositol, curcumin, esculetin, resveratrol and lycopene as inhibitors of benzo[a]pyrene plus 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung tumorigenesis in A/J mice. Cancer Lett. 1999;137(2):123-130. (PubMed)
71. Berge G, Ovrebo S, Eilertsen E, Haugen A, Mollerup S. Analysis of resveratrol as a lung cancer chemopreventive agent in A/J mice exposed to benzo[a]pyrene. Br J Cancer. 2004;91(7):1380-1383. (PubMed)
72. Schneider Y, Duranton B, Gosse F, Schleiffer R, Seiler N, Raul F. Resveratrol inhibits intestinal tumorigenesis and modulates host-defense-related gene expression in an animal model of human familial adenomatous polyposis. Nutr Cancer. 2001;39(1):102-107. (PubMed)
73. Sengottuvelan M, Nalini N. Dietary supplementation of resveratrol suppresses colonic tumour incidence in 1,2-dimethylhydrazine-treated rats by modulating biotransforming enzymes and aberrant crypt foci development. Br J Nutr. 2006;96(1):145-153. (PubMed)
74. Ziegler CC, Rainwater L, Whelan J, McEntee MF. Dietary resveratrol does not affect intestinal tumorigenesis in Apc(Min/+) mice. J Nutr. 2004;134(1):5-10. (PubMed)
75. Patel KR, Brown VA, Jones DJ, et al. Clinical pharmacology of resveratrol and its metabolites in colorectal cancer patients. Cancer Res. 2010;70(19):7392-7399. (PubMed)
76. Howells LM, Berry DP, Elliott PJ, et al. Phase I randomized, double-blind pilot study of micronized resveratrol (SRT501) in patients with hepatic metastases--safety, pharmacokinetics, and pharmacodynamics. Cancer Prev Res (Phila). 2011;4(9):1419-1425. (PubMed)
77. Popat R, Plesner T, Davies F, et al. A phase 2 study of SRT501 (resveratrol) with bortezomib for patients with relapsed and or refractory multiple myeloma. Br J Haematol. 2013;160(5):714-717. (PubMed)
78. Smoliga JM, Blanchard O. Enhancing the delivery of resveratrol in humans: if low bioavailability is the problem, what is the solution? Molecules. 2014;19(11):17154-17172. (PubMed)
79. Ronksley PE, Brien SE, Turner BJ, Mukamal KJ, Ghali WA. Association of alcohol consumption with selected cardiovascular disease outcomes: a systematic review and meta-analysis. BMJ. 2011;342:d671. (PubMed)
80. Lippi G, Franchini M, Favaloro EJ, Targher G. Moderate red wine consumption and cardiovascular disease risk: beyond the "French paradox". Semin Thromb Hemost. 2010;36(1):59-70. (PubMed)
81. Salvamani S, Gunasekaran B, Shaharuddin NA, Ahmad SA, Shukor MY. Antiartherosclerotic effects of plant flavonoids. Biomed Res Int. 2014;2014:480258. (PubMed)
82. Gronbaek M, Becker U, Johansen D, et al. Type of alcohol consumed and mortality from all causes, coronary heart disease, and cancer. Ann Intern Med. 2000;133(6):411-419. (PubMed)
83. Klatsky AL, Friedman GD, Armstrong MA, Kipp H. Wine, liquor, beer, and mortality. Am J Epidemiol. 2003;158(6):585-595. (PubMed)
84. Renaud SC, Gueguen R, Siest G, Salamon R. Wine, beer, and mortality in middle-aged men from eastern France. Arch Intern Med. 1999;159(16):1865-1870. (PubMed)
85. Mukamal KJ, Conigrave KM, Mittleman MA, et al. Roles of drinking pattern and type of alcohol consumed in coronary heart disease in men. N Engl J Med. 2003;348(2):109-118. (PubMed)
86. Rimm EB, Klatsky A, Grobbee D, Stampfer MJ. Review of moderate alcohol consumption and reduced risk of coronary heart disease: is the effect due to beer, wine, or spirits. Bmj. 1996;312(7033):731-736. (PubMed)
87. Wannamethee SG, Shaper AG. Type of alcoholic drink and risk of major coronary heart disease events and all-cause mortality. Am J Public Health. 1999;89(5):685-690. (PubMed)
88. Barefoot JC, Gronbaek M, Feaganes JR, McPherson RS, Williams RB, Siegler IC. Alcoholic beverage preference, diet, and health habits in the UNC Alumni Heart Study. Am J Clin Nutr. 2002;76(2):466-472. (PubMed)
89. McCann SE, Sempos C, Freudenheim JL, et al. Alcoholic beverage preference and characteristics of drinkers and nondrinkers in western New York (United States). Nutr Metab Cardiovasc Dis. 2003;13(1):2-11. (PubMed)
90. Mortensen EL, Jensen HH, Sanders SA, Reinisch JM. Better psychological functioning and higher social status may largely explain the apparent health benefits of wine: a study of wine and beer drinking in young Danish adults. Arch Intern Med. 2001;161(15):1844-1848. (PubMed)
91. Johansen D, Friis K, Skovenborg E, Gronbaek M. Food buying habits of people who buy wine or beer: cross sectional study. Bmj. 2006;332(7540):519-522. (PubMed)
92. Ruidavets JB, Bataille V, Dallongeville J, et al. Alcohol intake and diet in France, the prominent role of lifestyle. Eur Heart J. 2004;25(13):1153-1162. (PubMed)
93. Stocker R, O'Halloran RA. Dealcoholized red wine decreases atherosclerosis in apolipoprotein E gene-deficient mice independently of inhibition of lipid peroxidation in the artery wall. Am J Clin Nutr. 2004;79(1):123-130. (PubMed)
94. De Curtis A, Murzilli S, Di Castelnuovo A, et al. Alcohol-free red wine prevents arterial thrombosis in dietary-induced hypercholesterolemic rats: experimental support for the 'French paradox'. J Thromb Haemost. 2005;3(2):346-350. (PubMed)
95. Lekakis J, Rallidis LS, Andreadou I, et al. Polyphenolic compounds from red grapes acutely improve endothelial function in patients with coronary heart disease. Eur J Cardiovasc Prev Rehabil. 2005;12(6):596-600. (PubMed)
96. Karatzi K, Karatzis E, Papamichael C, Lekakis J, Zampelas A. Effects of red wine on endothelial function: postprandial studies vs clinical trials. Nutr Metab Cardiovasc Dis. 2009;19(10):744-750. (PubMed)
97. Grover-Paez F, Zavalza-Gomez AB. Endothelial dysfunction and cardiovascular risk factors. Diabetes Res Clin Pract. 2009;84(1):1-10. (PubMed)
98. Wang Z, Huang Y, Zou J, Cao K, Xu Y, Wu JM. Effects of red wine and wine polyphenol resveratrol on platelet aggregation in vivo and in vitro. Int J Mol Med. 2002;9(1):77-79. (PubMed)
99. Kirk RI, Deitch JA, Wu JM, Lerea KM. Resveratrol decreases early signaling events in washed platelets but has little effect on platalet in whole blood. Blood Cells Mol Dis. 2000;26(2):144-150. (PubMed)
100. Szewczuk LM, Forti L, Stivala LA, Penning TM. Resveratrol is a peroxidase-mediated inactivator of COX-1 but not COX-2: a mechanistic approach to the design of COX-1 selective agents. J Biol Chem. 2004;279(21):22727-22737. (PubMed)
101. Tsai SH, Lin-Shiau SY, Lin JK. Suppression of nitric oxide synthase and the down-regulation of the activation of NFkappaB in macrophages by resveratrol. Br J Pharmacol. 1999;126(3):673-680. (PubMed)
102. Fukao H, Ijiri Y, Miura M, et al. Effect of trans-resveratrol on the thrombogenicity and atherogenicity in apolipoprotein E-deficient and low-density lipoprotein receptor-deficient mice. Blood Coagul Fibrinolysis. 2004;15(6):441-446. (PubMed)
103. Wang Z, Zou J, Huang Y, Cao K, Xu Y, Wu JM. Effect of resveratrol on platelet aggregation in vivo and in vitro. Chin Med J (Engl). 2002;115(3):378-380. (PubMed)
104. Wilson T, Knight TJ, Beitz DC, Lewis DS, Engen RL. Resveratrol promotes atherosclerosis in hypercholesterolemic rabbits. Life Sci. 1996;59(1):PL15-21. (PubMed)
105. Fujitaka K, Otani H, Jo F, et al. Modified resveratrol Longevinex improves endothelial function in adults with metabolic syndrome receiving standard treatment. Nutr Res. 2011;31(11):842-847. (PubMed)
106. Wong RH, Howe PR, Buckley JD, Coates AM, Kunz I, Berry NM. Acute resveratrol supplementation improves flow-mediated dilatation in overweight/obese individuals with mildly elevated blood pressure. Nutr Metab Cardiovasc Dis. 2011;21(11):851-856. (PubMed)
107. Wong RH, Berry NM, Coates AM, et al. Chronic resveratrol consumption improves brachial flow-mediated dilatation in healthy obese adults. J Hypertens. 2013;31(9):1819-1827. (PubMed)
108. Agarwal B, Campen MJ, Channell MM, et al. Resveratrol for primary prevention of atherosclerosis: clinical trial evidence for improved gene expression in vascular endothelium. Int J Cardiol. 2013;166(1):246-248. (PubMed)
109. Tome-Carneiro J, Gonzalvez M, Larrosa M, et al. One-year consumption of a grape nutraceutical containing resveratrol improves the inflammatory and fibrinolytic status of patients in primary prevention of cardiovascular disease. Am J Cardiol. 2012;110(3):356-363. (PubMed)
110. Tome-Carneiro J, Gonzalvez M, Larrosa M, et al. Consumption of a grape extract supplement containing resveratrol decreases oxidized LDL and ApoB in patients undergoing primary prevention of cardiovascular disease: a triple-blind, 6-month follow-up, placebo-controlled, randomized trial. Mol Nutr Food Res. 2012;56(5):810-821. (PubMed)
111. Tome-Carneiro J, Gonzalvez M, Larrosa M, et al. Grape resveratrol increases serum adiponectin and downregulates inflammatory genes in peripheral blood mononuclear cells: a triple-blind, placebo-controlled, one-year clinical trial in patients with stable coronary artery disease. Cardiovasc Drugs Ther. 2013;27(1):37-48. (PubMed)
112. Tome-Carneiro J, Larrosa M, Yanez-Gascon MJ, et al. One-year supplementation with a grape extract containing resveratrol modulates inflammatory-related microRNAs and cytokines expression in peripheral blood mononuclear cells of type 2 diabetes and hypertensive patients with coronary artery disease. Pharmacol Res. 2013;72:69-82. (PubMed)
113. Liu Y, Ma W, Zhang P, He S, Huang D. Effect of resveratrol on blood pressure: A meta-analysis of randomized controlled trials. Clin Nutr. 2015;34(1):27-34. (PubMed)
114. Tome-Carneiro J, Gonzalvez M, Larrosa M, et al. Resveratrol in primary and secondary prevention of cardiovascular disease: a dietary and clinical perspective. Ann N Y Acad Sci. 2013;1290:37-51. (PubMed)
115. Heilbronn LK, Ravussin E. Calorie restriction and aging: review of the literature and implications for studies in humans. Am J Clin Nutr. 2003;78(3):361-369. (PubMed)
116. Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289(5487):2126-2128. (PubMed)
117. Howitz KT, Bitterman KJ, Cohen HY, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425(6954):191-196. (PubMed)
118. Wood JG, Rogina B, Lavu S, et al. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430(7000):686-689. (PubMed)
119. Valenzano DR, Terzibasi E, Genade T, Cattaneo A, Domenici L, Cellerino A. Resveratrol prolongs lifespan and retards the onset of age-related markers in a short-lived vertebrate. Curr Biol. 2006;16(3):296-300. (PubMed)
120. Baur JA, Pearson KJ, Price NL, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444(7117):337-342. (PubMed)
121. Semba RD, Ferrucci L, Bartali B, et al. Resveratrol levels and all-cause mortality in older community-dwelling adults. JAMA Intern Med. 2014;174(7):1077-1084. (PubMed)
122. Brown K, Rufini A, Gescher A. Do not throw out the resveratrol with the bath water. JAMA Intern Med. 2015;175(1):140-141. (PubMed)
123. Glaser JH. Effect of wine consumption on mortality. JAMA Intern Med. 2015;175(4):650. (PubMed)
124. Wang J, Ho L, Qin W, et al. Caloric restriction attenuates beta-amyloid neuropathology in a mouse model of Alzheimer's disease. FASEB J. 2005;19(6):659-661. (PubMed)
125. Aguirre L, Fernandez-Quintela A, Arias N, Portillo MP. Resveratrol: anti-obesity mechanisms of action. Molecules. 2014;19(11):18632-18655. (PubMed)
126. Kennedy DO, Wightman EL, Reay JL, et al. Effects of resveratrol on cerebral blood flow variables and cognitive performance in humans: a double-blind, placebo-controlled, crossover investigation. Am J Clin Nutr. 2010;91(6):1590-1597. (PubMed)
127. Witte AV, Kerti L, Margulies DS, Floel A. Effects of resveratrol on memory performance, hippocampal functional connectivity, and glucose metabolism in healthy older adults. J Neurosci. 2014;34(23):7862-7870. (PubMed)
128. Pasinetti GM, Wang J, Ho L, Zhao W, Dubner L. Roles of resveratrol and other grape-derived polyphenols in Alzheimer's disease prevention and treatment. Biochim Biophys Acta. 2015;1852(6):1202-1208. (PubMed)
129. Centers for Disease Control and Prevention. National Diabetes Statistics Report: Estimates of Diabetes and Its Burden in the United States, 2014. Atlanta, GA: US Department of Health and Human Services. http://www.cdc.gov/diabetes/data/statistics/2014StatisticsReport.html.
130. Szkudelski T, Szkudelska K. Resveratrol and diabetes: from animal to human studies. Biochim Biophys Acta. 2015; 1852(6):1145-1154. (PubMed)
131. Movahed A, Nabipour I, Lieben Louis X, et al. Antihyperglycemic effects of short term resveratrol supplementation in type 2 diabetic patients. Evid Based Complement Alternat Med. 2013;2013:851267. (PubMed)
132. Bhatt JK, Thomas S, Nanjan MJ. Resveratrol supplementation improves glycemic control in type 2 diabetes mellitus. Nutr Res. 2012;32(7):537-541. (PubMed)
133. Brasnyo P, Molnar GA, Mohas M, et al. Resveratrol improves insulin sensitivity, reduces oxidative stress and activates the Akt pathway in type 2 diabetic patients. Br J Nutr. 2011;106(3):383-389. (PubMed)
134. Poulsen MM, Vestergaard PF, Clasen BF, et al. High-dose resveratrol supplementation in obese men: an investigator-initiated, randomized, placebo-controlled clinical trial of substrate metabolism, insulin sensitivity, and body composition. Diabetes. 2013;62(4):1186-1195. (PubMed)
135. Timmers S, Konings E, Bilet L, et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011;14(5):612-622. (PubMed)
136. Yoshino J, Conte C, Fontana L, et al. Resveratrol supplementation does not improve metabolic function in nonobese women with normal glucose tolerance. Cell Metab. 2012;16(5):658-664. (PubMed)
137. Carpene C, Gomez-Zorita S, Deleruyelle S, Carpene MA. Novel strategies for preventing diabetes and obesity complications with natural polyphenols. Curr Med Chem. 2015;22(1):150-164. (PubMed)
138. Hausenblas HA, Schoulda JA, Smoliga JM. Resveratrol treatment as an adjunct to pharmacological management in type 2 diabetes mellitus-systematic review and meta-analysis. Mol Nutr Food Res. 2015;59(1):147-159. (PubMed)
139. Liu K, Zhou R, Wang B, Mi MT. Effect of resveratrol on glucose control and insulin sensitivity: a meta-analysis of 11 randomized controlled trials. Am J Clin Nutr. 2014;99(6):1510-1519. (PubMed)
140. Burns J, Yokota T, Ashihara H, Lean ME, Crozier A. Plant foods and herbal sources of resveratrol. J Agric Food Chem. 2002;50(11):3337-3340. (PubMed)
141. Rimando AM, Kalt W, Magee JB, Dewey J, Ballington JR. Resveratrol, pterostilbene, and piceatannol in vaccinium berries. J Agric Food Chem. 2004;52(15):4713-4719. (PubMed)
142. Sanders TH, McMichael RW, Jr., Hendrix KW. Occurrence of resveratrol in edible peanuts. J Agric Food Chem. 2000;48(4):1243-1246. (PubMed)
143. Hurst WJ, Glinski JA, Miller KB, Apgar J, Davey MH, Stuart DA. Survey of the trans-resveratrol and trans-piceid content of cocoa-containing and chocolate products. J Agric Food Chem. 2008;56(18):8374-8378. (PubMed)
144. Creasey LL, Coffee M. Phytoalexin production potential of grape berries. J Am Soc Hortic Sci. 1988;113(2):230-234.
145. Fremont L. Biological effects of resveratrol. Life Sci. 2000;66(8):663-673. (PubMed)
146. Goldberg DM, Karumanchiri A, Ng E, Yan J, Eleftherios P, Soleas G. Direct gas chromatographic-mass spectrometric method to assay cis-resveratrol in wines: preliminary survey of its concentration in commercial wines. J Agric Food Chem. 1995;43(5):1245-1250.
147. Guerrero RF, Garcia-Parrilla MC, Puertas B, Cantos-Villar E. Wine, resveratrol and health: a review. Nat Prod Commun. 2009;4(5):635-658. (PubMed)
148. Stervbo U, Vang O, Bonnesen C. A review of the content of the putative chemopreventive phytoalexin resveratrol in red wine. Food Chemistry. 2007;101:449-457.
149. Sobolev VS, Cole RJ. trans-resveratrol content in commercial peanuts and peanut products. J Agric Food Chem. 1999;47(4):1435-1439. (PubMed)
150. Hendler SS, Rorvik DR, eds, eds. PDR for Nutritional Supplements. 2nd edition ed: Thomson Reuters; 2008.
151. Williams LD, Burdock GA, Edwards JA, Beck M, Bausch J. Safety studies conducted on high-purity trans-resveratrol in experimental animals. Food Chem Toxicol. 2009;47(9):2170-2182. (PubMed)
152. Johnson WD, Morrissey RL, Usborne AL, et al. Subchronic oral toxicity and cardiovascular safety pharmacology studies of resveratrol, a naturally occurring polyphenol with cancer preventive activity. Food Chem Toxicol. 2011;49(12):3319-3327. (PubMed)
153. Gescher A, Steward WP, Brown K. Resveratrol in the management of human cancer: how strong is the clinical evidence? Ann N Y Acad Sci. 2013;1290:12-20. (PubMed)
154. American Academy of Pediatrics. Committee on Substance Abuse and Committee on Children With Disabilities. Fetal alcohol syndrome and alcohol-related neurodevelopmental disorders. Pediatrics. 2000;106(2 Pt 1):358-361. (PubMed)
155. Bertelli AA, Giovannini L, Giannessi D, et al. Antiplatelet activity of synthetic and natural resveratrol in red wine. Int J Tissue React. 1995;17(1):1-3. (PubMed)
156. Koe XF, Tengku Muhammad TS, Chong AS, Wahab HA, Tan ML. Cytochrome P450 induction properties of food and herbal-derived compounds using a novel multiplex RT-qPCR in vitro assay, a drug-food interaction prediction tool. Food Sci Nutr. 2014;2(5):500-520. (PubMed)
157. Piver B, Berthou F, Dreano Y, Lucas D. Inhibition of CYP3A, CYP1A and CYP2E1 activities by resveratrol and other non volatile red wine components. Toxicol Lett. 2001;125(1-3):83-91. (PubMed)
158. Regev-Shoshani G, Shoseyov O, Kerem Z. Influence of lipophilicity on the interactions of hydroxy stilbenes with cytochrome P450 3A4. Biochem Biophys Res Commun. 2004;323(2):668-673. (PubMed)
159. Detampel P, Beck M, Krahenbuhl S, Huwyler J. Drug interaction potential of resveratrol. Drug Metab Rev. 2012;44(3):253-265. (PubMed)
Soy Isoflavones
Contents
Summary
- Isoflavones are a class of phytoestrogens — plant-derived compounds with estrogenic activity. Soybeans and soy products are the richest sources of isoflavones in the human diet. (More information)
- Some health effects of soy may be dependent on one’s capacity to convert the isoflavone daidzein to equol during digestion. (More information)
- The results of observational studies suggest that higher intakes of soy foods early in life may decrease the risk of breast cancer in adulthood. There is currently little clinical evidence that taking soy isoflavone supplements decreases the risk of incident and recurrent breast cancer. (More information)
- Current evidence from observational studies and small clinical trials is not robust enough to understand whether soy protein/isoflavone supplements may help prevent or inhibit the progression of prostate cancer. (More information)
- To date, randomized controlled trials examining the effect of soy isoflavones on bone mineral density in postmenopausal women have produced mixed results. Potential benefits of soy isoflavones as an alternative to bone-sparing treatments in women undergoing menopause remain to be determined. (More information)
- Current evidence suggests that whole soy components other than isoflavones may have favorable effects on serum lipid profiles. Yet, two recent meta-analyses of randomized controlled trials indicated that isoflavones might exert cardiovascular benefits by improving vascular function in postmenopausal women. (More information)
- Supplementation with isoflavones appeared to be about 40% less efficient than hormone-replacement therapy in attenuating menopausal hot flashes and required more time to reach its maximum effect. Yet, supplements containing primarily the isoflavone genistein have demonstrated consistent alleviation of menopausal hot flashes. (More information)
- Currently available data suggest that breast cancer survivors should not be further discouraged from consuming soy foods in moderation. Moreover, in a pooled analysis of three large prospective cohort studies, soy isoflavone intake ≥10 mg/day was associated with a 25% reduced risk of tumor recurrence in breast cancer survivors. (More information)
- At present, there is no convincing evidence that infants fed soy-based formula are at greater risk for adverse effects than infants fed cow’s milk-based formula. (More information)
Introduction
Isoflavones are polyphenolic compounds that possess both estrogen-agonist and estrogen-antagonist properties (see Biological Activities). For this reason, they are classified as phytoestrogens — plant-derived compounds with estrogenic activity (1). Isoflavones are the major flavonoids found in legumes, particularly soybeans. In soybeans, isoflavones are present as glycosides, i.e., bound to a sugar molecule. Digestion or fermentation of soybeans or soy products results in the release of the sugar molecule from the isoflavone glycoside, leaving an isoflavone aglycone. Soy isoflavone glycosides include genistin, daidzin, and glycitin, while the aglycones are called genistein, daidzein, and glycitein (Figure 1). Unless otherwise indicated, quantities of isoflavones specified in this article refer to aglycones — not glycosides.

Metabolism and Bioavailability
The article on Flavonoids describes some of the factors influencing the absorption, metabolic fate, and bioavailability of flavonoid family members, including isoflavones. Pharmacokinetic studies have indicated that plasma concentrations of daidzein and genistein peaked about six hours after isoflavone intake, preceded by a smaller initial peak one hour post-meal (2, 3). The initial peak reflects isoflavone absorption following the hydrolysis of isoflavone glycosides to aglycones by β-glucosidases in the small intestine, while the second peak corresponds to isoflavone aglycones absorbed after the hydrolysis of glycosides by bacterial β-glucosidases in the colon (2).
The composition of one’s colonic microbiota can influence the metabolic fate and biological effects of isoflavones. Indeed, the extent of at least some of the potential health benefits of soy intake are thought to depend on one’s capacity to convert isoflavones to key metabolites during digestion. Specifically, some colonic bacteria can convert the soy isoflavone daidzein to equol, a metabolite that has greater estrogenic activity than daidzein, and to other metabolites, such as O-desmethylangolensin [O-DMA], that are less estrogenic (Figure 2) (4, 5). Equol appears in plasma about eight hours after isoflavone intake owing to the transit time of daidzein to the colon and its subsequent conversion to equol by the microbiota. Studies measuring urinary equol excretion after soy consumption indicated that equol was produced by about 25%-30% of the adult population in Western countries compared to 50%-60% of adults living in Asian countries and Western adult vegetarians (4, 6). Note that individuals possessing equol-producing bacteria are called "equol producers" as opposed to "equol non-producers."
Although prolonged soy food consumption has not been associated with the ability to produce equol, the type of soy food consumed might influence the composition of microbiota to include equol-producing bacteria (discussed in 4).

Biological Activities
Estrogenic and anti-estrogenic activities
Soy isoflavones are known to have weak estrogenic or hormone-like activity due to their structural similarity with 17-β-estradiol (Figure 3). Estrogens are signaling molecules that exert their effects by binding to estrogen receptors within cells (Figure 3). The estrogen-receptor complex interacts with DNA to change the expression of estrogen-responsive genes. Estrogen receptors (ER) are present in numerous tissues other than those associated with reproduction, including bone, liver, heart, and brain (7). Soy isoflavones can preferentially bind to and transactivate estrogen receptor-β (ER-β) — rather than ER-α — mimicking the effects of estrogen in some tissues and antagonizing (blocking) the effects of estrogen in others (8). Scientists are interested in the tissue-selective activities of phytoestrogens because anti-estrogenic effects in reproductive tissue could help reduce the risk of hormone-associated cancers (breast, uterine, and prostate), while estrogenic effects in other tissues could help maintain bone mineral density and improve blood lipid profiles (see Disease Prevention). The extent to which soy isoflavones exert estrogenic and anti-estrogenic effects in humans is currently the focus of considerable scientific research.

Estrogen receptor-independent activities
Soy isoflavones and their metabolites also have biological activities that are unrelated to their interactions with estrogen receptors (9). By inhibiting the synthesis and activity of certain enzymes involved in estrogen metabolism, soy isoflavones may alter the biological activity of endogenous estrogens and androgens (10-13). Soy isoflavones have also been found to inhibit tyrosine kinases (14), enzymes that play critical roles in the signaling pathways that stimulate cell proliferation. Additionally, isoflavones can act as antioxidants in vitro (15), but the extent to which they contribute to the antioxidant status of humans is not yet clear. Plasma F2-isoprostanes, biomarkers of lipid peroxidation in vivo, were significantly lower after two weeks of daily consumption of soy protein containing 56 mg of isoflavones than after consumption of soy protein providing only 2 mg of isoflavones (16). However, daily supplementation with 50 to 100 mg of isolated soy isoflavones did not significantly alter plasma or urinary F2-isoprostane concentrations (17, 18).
Disease Prevention
Hormone-associated cancers
Since soy isoflavones are structurally similar to endogenous estrogens, it has been suggested that they might help protect against hormone-associated cancers.
Breast cancer
High isoflavone intake from soy foods in Asian countries (average range, 25 to 50 mg/day) has been suggested to contribute to reducing the risk of breast cancer; in contrast, the incidence of breast cancer remains elevated in Europe, North America, and Australia/New Zealand (19) where average isoflavone intakes in non-Asian women are generally less than 2 mg/day (20). Nevertheless, several hereditary and lifestyle factors likely also contribute to this difference (19, 21). In a meta-analysis of one prospective cohort study and seven case-control studies conducted in Asian populations and in Asian Americans, higher versus lower intakes of dietary soy isoflavones (≥20 mg/day vs. ≤5 mg/day) were found to be associated with a 29% reduced risk of breast cancer (22). In observational studies conducted in Western populations, median intake of soy isoflavones was reportedly low (0.3 mg/day) and not associated with a decreased risk of breast cancer (22, 23). Moreover, a lifelong exposure to isoflavones may be needed to lower the risk of developing breast cancer later in life (21). This would explain why moderate versus low intakes of isoflavones (10.8 mg/day vs. 0.23 mg/day; cohort followed for a median of 7.4 years) during adulthood was not associated with a reduced risk of breast cancer in British women enrolled in the European Prospective Investigation into Cancer and Nutrition (EPIC) study (24). A few case-control studies also reported that early soy exposure — during childhood and adolescence — might be associated with a lower risk of breast cancer later in life (25-28). Further, a meta-analysis of four prospective cohort studies suggested that high versus low isoflavone intakes might be associated with a modest reduction in risk of recurrence (RR=0.84, 95% CI: 0.71-0.99) in breast cancer survivors (see Safety for breast cancer survivors) (23).
While lower circulating estrogen concentrations have been linked to a lower risk of breast cancer in postmenopausal women (29), a meta-analysis of randomized controlled trials in this population found no effect of soy isoflavone supplementation on the circulating concentrations of estrogenic hormones, estradiol (21 studies) and estrone (7 studies), and of sex-hormone binding globulin (SHBG; 17 studies) (30). Another meta-analysis of seven randomized controlled trials in 1,287 women found no overall effect of soy isoflavones (40 to 120 mg/day) consumed for six months to three years on mammographic breast density, used as a surrogate marker of breast cancer risk (31). Subgroup analyses showed no effect in postmenopausal women (four studies) but a modest increase in breast density — of unclear clinical significance — in premenopausal women (five studies). Further, in a recent randomized, placebo-controlled trial, soy isoflavone supplementation (50 mg/day for one year) also failed to affect breast density in women (ages, 30 to 75 years) with breast cancer (32).
There is currently little evidence that taking soy isoflavone supplements decreases the risk of incident and recurrent breast cancer.
Endometrial cancer
It is thought that the development of endometrial (uterine) cancer could be related to prolonged exposure to unopposed estrogen, i.e., estrogen not counterbalanced with the hormone progesterone (33). Excess estrogen relative to progesterone may result in endometrial thickening, a potential biomarker of estrogen-induced proliferation and a predictor of endometrial carcinomas (34). Whether high intakes of isoflavones with anti-estrogenic activity in uterine tissue could be associated with a lower risk of endometrial cancer has been examined in a number of observational studies. A recent meta-analysis of two prospective cohort studies and eight case-control studies found the highest versus lowest quantile of isoflavone intake to be associated with a 19% lower risk of endometrial cancer (35). One of the two prospective studies included in the meta-analysis found no association between consumption of total soy foods, legumes, and tofu and risk of endometrial cancer in a cohort of 46,027 multiethnic US women (mean age at cohort entry, 61.6 years) followed for a median 13.6 years (36). Nevertheless, a 34% lower risk of endometrial cancer was found to be associated with the highest versus lowest quintile of total isoflavones (median intakes, 11.23 mg/1,000 kcal/day vs. 0.87 mg/1,000 kcal/day) in this cohort (36). In the second prospective study, no association was observed between the top versus bottom tertile of isoflavone intakes (median intakes, 63.2 mg/day vs. 17.7 mg/day) and the risk of endometrial cancer risk in 49,121 Japanese women (ages, 45 to 74 years) (37). Because the consumption of isoflavones is much lower in non-Asian versus Asian cohorts, comparisons among populations are quite problematic and limit the scope of pooled analyses of observational studies (38).
Finally, a recent meta-analysis of 23 randomized controlled trials found no overall effect of isoflavone supplementation (5 to 154 mg/day) for up to three years on endometrial thickness in postmenopausal women (39). Nevertheless, a subgroup analysis of 10 trials showed that supplementation of postmenopausal women with isoflavone doses >54 mg/day of isoflavones could significantly decrease endometrial thickness (39).
Although limited evidence from case-control studies showed an inverse relationship between consumption of soy foods and endometrial cancer, there is little evidence from intervention trials to suggest that taking soy isoflavone supplements could decrease the risk of endometrial cancer.
Prostate cancer
Incidence rates of prostate cancer are much higher in Northern America, Northern and Western Europe, Australia, and New Zealand compared to Asian countries, such as Japan and China, where isoflavone-rich soybeans are common components of the diet (19). Soy food consumption has been associated with a reduced risk of prostate cancer in recent pooled analyses of observational studies (40, 41). In a study of 19 men with prostate cancer, daily soy supplementation resulted in soy isoflavone concentrations six-fold higher in prostate tissue than in serum (42). The results of cell culture and animal studies have suggested a potential role for soy isoflavones in limiting the progression of prostate cancer (reviewed in 43).
A number of small, short-term randomized controlled interventions have examined the effect of soy foods/isoflavones on biomarkers of prostate cancer risk (44). Compared to supplementation with milk protein, consumption of a diet supplemented with soy protein isolate high in isoflavones (~107 mg/day) limited the rise in androgen receptor density in prostate tissue after six months but did not modify prostatic estrogen receptor-β expression or circulating sex steroid hormone profile in men at high risk of developing prostate cancer (45). In addition, dietary soy protein supplementation had no effect on prostate-specific antigen (PSA) in serum or markers of cell proliferation and apoptosis in premalignant tissue. Yet, supplemental soy protein isolate — regardless of isoflavone content — for six months resulted in a lower cancer incidence compared to milk control (46). In a multicenter, randomized, double-blind, placebo-controlled trial in 158 Japanese men (ages, 50 to 75 years) with negative prostate biopsy but rising serum PSA, supplemental isoflavones (60 mg/day) for one year had no effect on circulating concentrations of PSA and sex steroid hormones, or on the overall incidence of biopsy-detectable prostate cancer. However, prostate cancer incidence was found to be significantly lower with isoflavones compared to placebo in the subset of men aged 65 years and older (47).
Some trials found that supplementation with soy products, soy dietary proteins, or soy isoflavones could reduce or slow down the rising of serum PSA concentration in men with localized prostate cancer prior to therapy (48-50), as well as in those with PSA biochemical recurrence following radiotherapy and/or prostatectomy (51-53). However, other trials failed to show an effect of soy food/isoflavones on serum PSA in prostate cancer patients prior to (54, 55) or after therapy (56-58). Clinical studies have also failed to show a protective effect of soy isoflavones on circulating concentrations of sex hormones (testosterone, dihydrotestosterone, and estradiol) and sex hormone-binding globulin (SHBG) in patients with prostate cancer (44).
Pooled analyses of current data are hindered by the heterogeneity in soy/soy isoflavone preparations and dosage regimens in short-term interventions (mostly ≤6 months) in small sample-size trials. Therefore, despite a good safety profile for supplemental soy isoflavones and soy proteins in prostate cancer patients, larger randomized controlled trials with longer periods of intervention are required to assess whether soy isoflavones could influence the development and/or progression of prostate cancer.
More information about soy foods and prostate cancer risk may be found in the article on Legumes.
Osteoporosis
The decline in estrogen production that accompanies menopause places middle-aged women at risk of osteopenia and osteoporosis. The measurement of bone mineral density (BMD) loss by dual-energy X-ray absorptiometry is generally used in the diagnosis of osteoporosis (59). Whether the estrogenic properties of soy isoflavones might play any role in preserving bone health and preventing bone loss is unclear. To date, the results of observational and intervention studies examining the potential protection of soy isoflavones against BMD loss have been inconsistent. A recent review by Zheng et al. (60) discussed some potential factors relative to study design (e.g., intervention duration, isoflavone dosages) and target populations (e.g., ethnic and genetic differences, hormonal status) that could explain the conflicting study results.
For instance, the results of short-term (≤6 months) clinical trials assessing the effects of increased soy intake on biochemical markers of bone formation and bone resorption are inconsistent. Some controlled trials in postmenopausal women have found that increasing intakes of soy foods, soy protein, or soy isoflavones improved markers of bone resorption and formation (61-64) or attenuated bone loss (64, 65), but other trials have found no significant benefit of increasing soy intakes (66-69). Trials of longer duration also showed conflicting results. A meta-analysis of 10 randomized controlled trials (trial duration, 12 to 24 months) concluded that a mean dose of 87 mg/day of soy isoflavones resulted in no significant changes in lumbar spine, total hip, or femoral neck BMD of postmenopausal women (70).
Mixed results also emerged from studies conducted in different ethnic populations. Compared to Caucasian women, the incidence of hip fractures tend to be lower among Asian women who are habitual soy food consumers (71, 72), suggesting that long-term soy food consumption might protect against bone loss or osteoporotic fracture (73, 74). Moreover, pooled analyses combining intervention trials in Caucasian and Asian postmenopausal women reported significant increases in BMD with supplemental soy isoflavones (75-77). In contrast, a meta-analysis of 12 placebo-controlled trials in Caucasian postmenopausal women found no effect of soy isoflavone supplementation (dose range, 52 to 120 mg/day) for six months to three years on lumbar spine BMD (78).
The rate of bone loss is not linear during the 10-year period surrounding the final menstrual period and is especially increased during the menopausal transition, starting about one year before to two years after the final menstrual period. Surprisingly, the recent US Study of Women’s Health Across the Nation (SWAN) that followed Black, White, Japanese, and Chinese women reported that Japanese women with the highest versus lowest tertile of dietary isoflavone intakes (median intakes, 36.3 mg/day versus 4 mg/day) had an increased rate of BMD loss during menopause transition (79). In other ethnic groups, no such associations could be found between habitual dietary isoflavone intakes and the rate of bone loss before, after, or during menopause transition (79). Most meta-analyses of randomized controlled trials to date have reported a weak, positive effect of supplemental soy isoflavones on BMD in postmenopausal women (70, 75-77). Yet, it remains to be elucidated whether supplementation with soy isoflavones might be of any benefit to perimenopausal/early postmenopausal women before the acute loss of estrogen or to older postmenopausal women with established osteoporosis (60).
Finally, some authors have proposed that the effect of soy isoflavones on bone health may be dependent on whether or not the individual produces the daidzein metabolite, equol (see Metabolism and Bioavailability) (80-84). However, a recent randomized controlled trial found that supplementation with soy isoflavones increased calcium retention capacity in postmenopausal women regardless of their equol-producing capacity (85).
At present, the clinical benefits of soy isoflavones as an alternative to bone-sparing treatments in women undergoing menopause remain to be determined.
Cardiovascular disease
To date, large prospective cohort studies, mainly in Asian populations, investigating whether habitual soy food/isoflavone consumption is related to the incidence of cardiovascular disease (CVD), including coronary heart disease (CHD), ischemic stroke, and myocardial infarction, have found mixed results. In the Japan Public Health Center-based Study (mean follow-up, 13.5 years), consumption of soy foods was associated with a reduced risk of stroke in Japanese women (ages, 40 to 59 years) — but not in men. In this cohort, the highest versus lowest quintile of soy isoflavone intakes was found to be associated with a 65% lower risk of ischemic stroke and a 63% lower risk of myocardial infarction in women (86). In addition, an early data analysis of 64,915 Chinese women (ages 40 to 70 years) enrolled in the Shanghai Women’s Health Study (SWHS) found an inverse relationship between soy food intake and risk of incident CHD during a 2.5-year follow-up period (87). However, a higher soy protein intake was associated with a higher risk of incident CHD in 55,474 Chinese men (ages, 40 to 74 years) from the Shanghai Men’s Health Study (SMHS; mean follow-up of 5.4 years) (88). Moreover, a recent report of the SWHS cohort followed for 10 years reported a 24% higher risk of ischemic stroke with the highest versus lowest quintile of isoflavone intakes (mean intakes of 59.4 mg/d versus 8.6 mg/day) (89). Nevertheless, nested case-control studies within both Shanghai prospective studies found no correlations between soy food intake and incident CVD events when measures of urinary isoflavonoids were used as a more objective estimate of isoflavone exposure compared to dietary assessment with food-frequency questionnaires (89, 90). Further, no significant associations were found between long-term soy food, soy protein, and soy isoflavone consumption and CHD-, stroke-, and total CVD-related mortality in a 14.7-year follow-up of 60,298 participants of the Singapore Chinese Health Study (91).
Low consumption of soy foods in Western cohorts makes it more difficult to analyze possible longitudinal associations between isoflavone intake and CVD incidence or mortality in these populations (92-95).
Cardiometabolic risk factors
A number of intervention studies have examined soy intake in relation to several cardiometabolic risk factors. A recent meta-analysis of randomized controlled trials concluded that intake of either soy products (i.e., whole soybeans, soy milk, nuts, oil, and flour), soy protein isolate, or soy isoflavones for one month to one year could significantly improve serum lipid profiles in healthy and hypercholesterolemic individuals by lowering circulating triglycerides, total cholesterol, and LDL-cholesterol, and by increasing HDL-cholesterol (96). Further analyses suggested that soy protein without isoflavones was more effective at lowering total and LDL-cholesterol than soy protein containing isoflavones, and the consumption of soy isoflavones alone (as supplements or extracts) showed no significant effects on serum lipid profiles (96). In addition, a meta-analysis of 18 randomized controlled studies indicated that neither soy foods nor soy isoflavones could lower blood homocysteine concentrations, a known risk factor for CVD, in high-risk middle-aged and older adults (97). Another meta-analysis of 14 randomized controlled studies reported a reduction in circulating C-reactive protein (CRP) — an inflammation marker associated with increased cardiovascular risk — following soy isoflavone intake (from soy foods or isoflavone extracts) in postmenopausal women with elevated baseline CRP concentrations (>2.2 mg/L) (98). In a recent six-month placebo-controlled intervention study in 253 postmenopausal equol-producing women with prehypertension, supplementation with whole soy — but not daidzein — improved lipid profiles and lowered the concentrations of CRP (99). Whether potential cardiovascular benefits of soy isoflavone intake depend on individuals’ capacity to produce isoflavone metabolites (like equol) needs to be more closely examined.
Current evidence suggests that whole soy components other than isoflavones may have favorable effects on cardiometabolic risk factors.
Vascular function
The preservation of normal arterial function plays an important role in cardiovascular disease prevention. The ability of all types of blood vessels, including arteries, to dilate in response to nitric oxide (NO) produced by the endothelial cells that line their inner surface is compromised in people at high risk for cardiovascular disease (100). In the presence of cardiovascular risk factors (e.g., hypertension, hypercholesterolemia, hyperglycemia), impaired endothelial function results in widespread vasodilation and coagulation abnormalities and is considered to be an early step in the development of atherosclerosis. Measures of brachial flow-mediated dilation (FMD), a surrogate marker of endothelial function, have been found to be inversely associated with risk of future cardiovascular events (101). A meta-analysis of nine small randomized, placebo-controlled trials found that supplementation with soy isoflavones (50 to 99 mg/day; isolated or from isoflavone-containing soy protein) for a median eight-week period significantly increased brachial FMD, especially in postmenopausal women with low FMD levels (102). A more inclusive meta-analysis of 17 trials in either healthy individuals or in individuals with hyperlipidemia showed an increase of FMD with the intake of isolated isoflavones but not of isoflavones-containing soy protein (103).
Arterial stiffness or impaired arterial distensibility, another marker of vascular damage and an indicator of cardiovascular disease risk, is generally assessed using measures of aortic pulse-wave velocity (PWV) (104). Some, but not all, placebo-controlled clinical trials have suggested that supplementation with isoflavone-containing soy protein or isoflavone extracts might significantly decrease arterial stiffness (105-107). A recent randomized, double-blind, placebo-controlled study found that carotid-femoral PWV could be significantly reduced 24 hours after a single oral intake of 80 mg of soy isoflavones but only in participants able to produce equol (3). Long-term interventions are needed to evaluate the clinical relevance of such a result.
Finally, whether whole soy or isoflavones reduce the burden of subclinical atherosclerosis (99, 108) and lower blood pressure (109) in individuals at high CVD risk requires further examination.
Cognitive decline
Scientific research on the effect of soy isoflavones on cognitive function has been recently reviewed by Soni et al. (110). An observational study — the Honolulu-Asia Asian Study — that examined the relationship between soy intake and cognitive function found that Hawaiian men who reported consuming tofu (non-fermented soy product) at least twice weekly during midlife were more likely to have poor cognitive test scores 20 to 25 years later than those who reported consuming tofu less than twice a week (111). In an Indonesian study of elderly men and women, consumption of tofu was associated with worse memory, while consumption of tempeh (fermented soy) was associated with improved memory (112). In the multicenter, prospective, SWAN phytoestrogen ancillary study, the highest versus lowest tertile of isoflavone intakes was found to be associated with better scores in the processing speed test but worse scores in the verbal memory test in late perimenopausal and postmenopausal Asian women (79).
The results of several randomized controlled trials have been mixed. In a review of 12 trials, only half reported an improvement in cognitive function with soy isoflavone supplementation (110). Postmenopausal women given soy extracts, providing 60 mg/day of soy isoflavones for 6 to 12 weeks, performed better on cognitive tests of picture recall (short-term memory), learning rule reversals (mental flexibility), and a planning task compared to women given a placebo (113, 114). In a longer trial, postmenopausal women given supplements that provided 110 mg/day of soy isoflavones for six months performed better on a test of verbal fluency than women given placebos (115). In a cross-over trial lasting six months, women receiving 60 mg/day of soy isoflavones experienced significant improvements in cognitive performance and overall mood compared to when the women were given a placebo (116). However, in larger placebo-controlled trials, 80 mg/day of isoflavones for six months (117) or 99 mg/day of isoflavones for one year (118) did not affect performance on a battery of cognitive function tests, including tests for memory, attention, verbal fluency, motor control, and dementia in postmenopausal women. In addition, in the 30-month Women’s Isoflavone Soy Health trial in 313 postmenopausal women, daily supplementation with 91 mg of soy isoflavones significantly improved visual memory but failed to improve other aspects of cognition or global cognition (119). Nevertheless, a recent meta-analysis of 10 randomized controlled trials found a significant improvement in the pooled summary of cognitive function tests in healthy postmenopausal women supplemented with 60 to 160 mg/day of soy isoflavones for 6 to 30 months (120).
There has been little investigation regarding the potential effects of soy isoflavones in individuals with cognitive impairments (121).
Disease Treatment
Menopausal symptoms
Menopause-related vasomotor symptoms, including hot flashes (flushes) and night sweats, affect over 75% of middle-aged US women (122). Concern over potential adverse effects of hormone replacement therapy (123, 124) has led to an increased interest in the use of phytoestrogen supplements in the management of menopausal symptoms (125).
To date, the effects of increasing soy isoflavone intake on the frequency and severity of menopausal symptoms have been examined in over 60 randomized controlled trials of small sample size (126). The results of these trials have been mixed, as reflected by the conclusions of several systematic reviews and meta-analyses published in the last decade (125, 127-134). However, a systematic Cochrane review published in 2013 concluded that supplements containing primarily genistein (four studies; 30 to 60 mg/day for 12 weeks to one year) — but not dietary soy (13 studies), soy isoflavone extracts (12 studies), or red clover extracts (nine studies) — significantly reduced the frequency of hot flashes (131). This result was consistent with previous analyses reporting alleviation of hot flashes in higher- rather than lower-genistein supplementation trials (126, 133, 134). Nevertheless, in a meta-analysis by Taku et al. (133), supplemental soy isoflavone extract (30 to 80 mg/day for six weeks to one year) was found to result in a net 17.4% reduction in hot flash frequency in 13 placebo-controlled trials (1,196 women). In addition, the meta-analysis of nine trials (988 women) showed a 30.5% reduction in hot flash severity with soy isoflavone extracts (30 to 135 mg/day for 12 weeks to one year) (133). Moreover, the observation that longer trials showed a greater efficacy of soy isoflavones was confirmed in a recent model-based meta-analysis by Li et al. (135). In this analysis of 16 studies, the authors estimated that supplementation with soy isoflavones required 48 weeks of treatment, compared to only 12 weeks for estradiol, in order to achieve close to 80% of its maximum effect (135). Besides, the maximum effect of soy isoflavones was found to account for only 57% of the maximum effect of estradiol (135).
Evidence from observational studies suggested that one’s ability to produce equol might contribute to reducing the occurrence or severity of menopausal symptoms in postmenopausal women (see Metabolism and Bioavailability) (136, 137). Several relatively small intervention studies have examined the potential of equol to relieve these symptoms (reviewed in 138). A recent study in Chinese postmenopausal and equol-producing women showed no benefits of daily supplementation with soy flour (40 g) or daidzein (63 mg) for six months on the frequency or severity of menopausal symptoms (139). Yet, an earlier randomized controlled study found that, compared to equol non-producing women, those able to produce equol experienced improvements in menopausal symptoms like hot flashes following soy isoflavone supplementation (135 mg/day) for six months (140). In addition, the 12-week administration of 10 mg/day of equol to equol non-producing Japanese women experiencing three or more daily hot flashes significantly reduced the frequency and severity of hot flashes and neck and shoulder muscle stiffness compared to a placebo (141). In another study, daily supplementation with soy isoflavone (containing 24 mg of daidzein and 22 mg of genistein) or equol (10, 20, or 40 mg) supplements over an eight-week period resulted in equivalent reductions of the frequency of hot flashes in postmenopausal women with five or more daily hot flashes; however, 20 mg/day and 40 mg/day of equol supplements proved to be more effective than soy isoflavone supplements in the subgroup of women experiencing eight or more hot flashes per day (142). Nevertheless, because this study lacked an inert placebo control group, the results should be viewed with caution.
At present, supplements containing sufficient amounts of genistein may help alleviate vasomotor symptoms in women transitioning through menopause (126, 143).
Sources
Food sources
Isoflavones are found in small amounts in a number of legumes, grains, and vegetables, but soybeans are by far the most concentrated source of isoflavones in the human diet (144, 145). Average dietary isoflavone intakes in Japan, China, and other Asian countries range from 25 to 50 mg/day (20). Dietary isoflavone intakes are considerably lower in Western countries. Twenty-four-hour dietary recall data collected from 36,037 individuals in 10 countries (participating in the EPIC study) showed average isoflavone intakes to be lower than 1 mg/day (146). Compared to other European countries, the isoflavone intake was slightly higher in the British general population (2.3 mg/day) and health-conscious cohort (19.4 mg/day) (146).
Traditional Asian foods made from soybeans include tofu, tempeh, miso, and natto. Edamame refers to varieties of soybeans that are harvested and eaten in their green phase. Soy products that are gaining popularity in Western countries include soy-based meat substitutes, soy milk, soy cheese, and soy yogurt. The isoflavone content of a soy protein isolate depends on the method used to isolate it. Soy protein isolates prepared by an ethanol wash process generally lose most of their associated isoflavones, while those prepared by aqueous wash processes tend to retain them (147). Some foods that are rich in soy isoflavones are listed in Table 1, along with their isoflavone content. Because the isoflavone content of soy foods can vary considerably among brands and among different lots of the same brand (147), these values should be viewed only as a guide. Given the potential health implications of diets rich in soy isoflavones, accurate and consistent labeling of the soy isoflavone content of soy foods is needed. Of note, foods of animal origin also contain low levels of isoflavones (and other phytoestrogens), derived from animal feeds and pastures (148). More information on the isoflavone content of foods is available from the USDA Food Composition Database website and the USDA Database for the Isoflavone Content of Selected Foods report (149).
Supplements
Soy isoflavone extracts and supplements are available as dietary supplements without a prescription in the US. These products are not standardized, and the amounts of soy isoflavones they provide may vary considerably. Moreover, quality control may be an issue with some of these products (150). When isoflavone supplements available in the US were tested for their isoflavone content by an independent laboratory, the isoflavone content in the product differed by more than 10% from the amount claimed on the label in approximately 50% of the products tested (151).
Infant formulas
Soy protein-based infant formulas are made from soy protein isolate and contain significant amounts of soy isoflavones (Table 2). In 1997, the total isoflavone content of soy-based infant formulas that were commercially available in the US ranged from 32 to 47 mg/liter (~34 fluid ounces) (152).
Safety
Soy isoflavones have been consumed by humans as part of soy-based diets for many years without any evidence of adverse effects (145). The 75th percentile of dietary isoflavone intake has been reported to be as high as 65 mg/day in some Asian populations (153). Although diets rich in soy or soy-containing products appear safe and potentially beneficial, the long-term safety of very high supplemental doses of soy isoflavones is not yet known. One study in older men and women found that 100 mg/day of soy isoflavones for six months was well tolerated (154). Yet, longer-term studies are needed to evaluate the safety of isoflavones.
Adverse effects
Safety for breast cancer survivors
The safety of high intakes of soy isoflavones and other phytoestrogens for breast cancer survivors is an area of concern among scientists and clinicians. The results of cell culture and animal studies have been conflicting; some preclinical studies showed that soy isoflavones might stimulate the growth of estrogen receptor-positive (ER+) breast cancer cells (155, 156), while others suggested that they might either potentiate (157, 158) or abrogate the anticancer effects of tamoxifen on breast tissue (159, 160).
Very limited data from clinical trials suggested that increased consumption of soy isoflavones (38 to 45 mg/day) may show weak estrogenic effects in human breast tissue (161, 162). However, a study in women with biopsy-confirmed breast cancer found that supplementation with 200 mg/day of soy isoflavones did not increase breast cell proliferation — a marker of breast cancer risk — over the two to six weeks before surgery when compared to a control group that did not take soy isoflavones (163). A few large prospective cohort studies have examined the association between soy isoflavone intake and breast cancer recurrence and survival. In the Shanghai Breast Cancer Survival Study that followed 5,042 female breast cancer survivors for a median of 3.9 years, consumption of isoflavone-rich soy foods was significantly associated with a 29% lower risk of death and a 32% lower risk of cancer recurrence (164). In this study, soy isoflavone intake was associated with a significant 23% reduced risk of cancer recurrence and a nonsignificant 21% reduced risk of death (164). A pooled analysis of data from 9,514 breast cancer survivors from the Shanghai Breast Cancer Survival Study (164), the Life After Cancer Epidemiology study (165), and the Women’s Healthy Eating and Living study (166) found a 25% reduced risk of recurrence with soy isoflavone intakes ≥10 mg/day (compared to intakes of <4 mg/day) (167). A subgroup analysis showed that the inverse association between soy isoflavone intake and recurrence was significant only among women taking the anticancer drug, tamoxifen. No inverse association was reported between soy isoflavone intake and the risks of all-cause and breast cancer-specific mortality (167).
Given the available data, some experts think that women with a history of breast cancer, particularly ER+ breast cancer, should not increase their consumption of phytoestrogens, including soy isoflavones (168). Nevertheless, there is not enough evidence to discourage breast cancer survivors from consuming soy foods in moderation (143, 169).
Safety of soy protein-based infant formulas
Infant formula made from soy protein isolate has been commercially available since the mid-1960s (170). As much as 25% of the infant formula sold in the US is soy protein-based formula (171). The American Academy of Pediatrics (AAP) supports the use of soy protein isolate-based formula for normal growth and development of term infants whose needs are not being met by human milk or cow’s milk-based formulas (171). Soy protein-based formulas are especially indicated for infants with galactosemia and hereditary lactase deficiency, but they have no proven value in the prevention or management of infantile colic and fussiness (171).
Since infants fed soy-based formulas are exposed to relatively high levels of isoflavones (152), which they can absorb and metabolize, concern has been raised regarding potential long-term effects on anthropometric growth, bone health, as well as reproductive, endocrine, and immune functions (152, 172). In addition to the AAP review (171), a recent systematic review and meta-analysis of data published between 1909 and 2013 found no clinical concerns regarding nutritional adequacy, sexual development, thyroid disease, immune function, and neurodevelopment in infants fed soy protein-fed formulas (173). Specifically, this review identified 14 clinical trials comparing infants fed soy-based formula with infants fed human milk or cow’s milk-based formula and found that soy-based formula adequately supported growth and development in the first year of life (173). In addition, the results of three observational studies suggested no adverse effects of soy protein-based formula on the neurodevelopment of children (174-176). Two of these observational studies of low-to-moderate quality also reported associations between soy protein-based formula intake and marginal adverse events, including early menarche (176, 177) and increased duration of menstrual bleeding (176). A recent prospective cohort study (the Beginnings study) examining the effects of early infant feeding on reproductive organ development during childhood found no differences in the volume and structure of the reproductive organs of 101 five-year-old boys and girls who were either breastfed or fed soy protein- or cow’s milk-based formula as infants (178). Finally, no adverse health effects have been associated with the presence of phytates and aluminum in soy protein-based formulas fed to full-term infants (reviewed in 173).
At present, there is no convincing evidence that infants fed soy protein-based formula are at greater risk for adverse effects than infants fed cow’s milk-based formula. Nonetheless, if current evidence shows a safety profile for use of soy protein-based formulas in term infants, they are not designed or recommended for preterm infants (171). Also, recent preliminary findings suggesting potential links between consumption of soy protein-based formulas and adverse effects in autistic children deserve further investigation (179, 180).
Male reproductive health
Claims that soy food/isoflavone consumption can have adverse effects on male reproductive function, including feminization, erectile dysfunction, and infertility, are primarily based on animal studies and case reports (181). Exposure to isoflavones (including at levels above typical Asian dietary intakes) has not been shown to affect either the concentrations of estrogen and testosterone, or the quality of sperm and semen (181, 182). Thorough reviews of the literature found no basis for concern but emphasized the need for long-term, large scale comprehensive human studies (181, 183).
Thyroid function
In cell culture and animal studies, soy isoflavones have been found to inhibit the activity of thyroid peroxidase, an enzyme required for thyroid hormone synthesis (184, 185). However, high intakes of soy isoflavones do not appear to increase the risk of hypothyroidism as long as dietary iodine consumption is adequate (186). Since the addition of iodine to soy-based formulas in the 1960s, there have been no further reports of hypothyroidism in soy formula-fed infants (187). Several clinical trials, mostly in women with sufficient iodine intakes, have not found increased consumption of soy isoflavones to result in clinically significant changes in circulating thyroid hormone concentrations (188-192).
Pregnancy
To date, studies have not examined the effect of an isoflavone-rich diet on fetal development or pregnancy outcomes in humans, and the safety of isoflavone supplements during pregnancy has not been established.
Drug interactions
Fermented soy foods contain highly variable amounts of the biologically active amine, tyramine, which is catabolized in the body by monoamine oxidase enzyme (MAO) and excreted in the urine. The ingestion of very high amount of tyramine may saturate the detoxification system and lead to clinical symptoms of intoxication. Because individuals taking MAO inhibitors (MAOIs; phenelzine, tranylcypromine) are at greater risk of adverse effects, they should avoid consuming fermented soy products (193, 194). Because colonic bacteria play an important role in the metabolism of soy isoflavones, antibiotic therapy could decrease their biological activity (193). Some evidence from animal studies suggested that high intakes of soy isoflavones, particularly genistein, could interfere with the antitumor effects of tamoxifen (Nolvadex) (159). Yet, a recent pooled analysis of three prospective cohort studies found that the risk of recurrence in breast cancer survivors was reduced to a greater extent with soy isoflavone intake in tamoxifen users than in nonusers (see Safety for breast cancer survivors) (167). Nonetheless, until more is known about potential interactions in humans, those taking tamoxifen or other selective estrogen receptor modulators (SERMs) to treat or prevent breast cancer should be cautious and seek medical advice regarding the use of soy protein supplements or isoflavone extracts (193).
High intakes of soy protein may interfere with the efficacy of the anticoagulant medication warfarin. There is one case report of an individual on warfarin who developed subtherapeutic international normalized ratio (INR; prothrombin time) values upon consuming ~16 ounces of soy milk daily for four weeks (195). INR values returned to therapeutic levels two weeks after discontinuing soy milk.
The amount of levothyroxine required for adequate thyroid hormone replacement has been found to increase in infants with congenital hypothyroidism fed soy formula (187, 196). Taking levothyroxine at the same time as a soy protein supplement also increased the levothyroxine dose required for adequate thyroid hormone replacement in an adult with hypothyroidism (197).
Regular consumption of a diet high in soy — rather than supplementation with isoflavone extracts or isoflavone containing isolated soy protein — may help lower fasting glucose concentrations (198). It is unknown whether individuals taking antidiabetic agents might be at risk of hypoglycemia if they follow a soy-based meal replacement plan rather than a diet plan recommended by the American Diabetes Association (199).
Authors and Reviewers
Originally written in 2004 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in January 2006 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in December 2009 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in August 2016 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in October 2016 by:
Alison M. Duncan, Ph.D., R.D.
Professor
Department of Human Health and Nutritional Sciences
University of Guelph
Guelph, Ontario, Canada
Copyright 2004-2024 Linus Pauling Institute
References
1. Lampe JW. Isoflavonoid and lignan phytoestrogens as dietary biomarkers. J Nutr. 2003;133 Suppl 3:956S-964S. (PubMed)
2. Franke AA, Lai JF, Halm BM. Absorption, distribution, metabolism, and excretion of isoflavonoids after soy intake. Arch Biochem Biophys. 2014;559:24-28. (PubMed)
3. Hazim S, Curtis PJ, Schar MY, et al. Acute benefits of the microbial-derived isoflavone metabolite equol on arterial stiffness in men prospectively recruited according to equol producer phenotype: a double-blind randomized controlled trial. Am J Clin Nutr. 2016;103(3):694-702. (PubMed)
4. Setchell KD, Clerici C. Equol: history, chemistry, and formation. J Nutr. 2010;140(7):1355S-1362S. (PubMed)
5. Setchell KD, Clerici C, Lephart ED, et al. S-equol, a potent ligand for estrogen receptor beta, is the exclusive enantiomeric form of the soy isoflavone metabolite produced by human intestinal bacterial flora. Am J Clin Nutr. 2005;81(5):1072-1079. (PubMed)
6. Setchell KD, Cole SJ. Method of defining equol-producer status and its frequency among vegetarians. J Nutr. 2006;136(8):2188-2193. (PubMed)
7. National Cancer Institute. Understanding Estrogen Receptors/SERMs. National Cancer Institute. January 2005. http://www.cancer.gov/cancertopics/understandingcancer/estrogenreceptors. Accessed 7/12/09.
8. Wang LQ. Mammalian phytoestrogens: enterodiol and enterolactone. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;777(1-2):289-309. (PubMed)
9. Barnes S, Boersma B, Patel R, et al. Isoflavonoids and chronic disease: mechanisms of action. Biofactors. 2000;12(1-4):209-215. (PubMed)
10. Holzbeierlein JM, McIntosh J, Thrasher JB. The role of soy phytoestrogens in prostate cancer. Curr Opin Urol. 2005;15(1):17-22. (PubMed)
11. Kao YC, Zhou C, Sherman M, Laughton CA, Chen S. Molecular basis of the inhibition of human aromatase (estrogen synthetase) by flavone and isoflavone phytoestrogens: A site-directed mutagenesis study. Environ Health Perspect. 1998;106(2):85-92. (PubMed)
12. Whitehead SA, Cross JE, Burden C, Lacey M. Acute and chronic effects of genistein, tyrphostin and lavendustin A on steroid synthesis in luteinized human granulosa cells. Hum Reprod. 2002;17(3):589-594. (PubMed)
13. Ye L, Chan MY, Leung LK. The soy isoflavone genistein induces estrogen synthesis in an extragonadal pathway. Mol Cell Endocrinol. 2009;302(1):73-80. (PubMed)
14. Akiyama T, Ishida J, Nakagawa S, et al. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J Biol Chem. 1987;262(12):5592-5595. (PubMed)
15. Ruiz-Larrea MB, Mohan AR, Paganga G, Miller NJ, Bolwell GP, Rice-Evans CA. Antioxidant activity of phytoestrogenic isoflavones. Free Radic Res. 1997;26(1):63-70. (PubMed)
16. Wiseman H, O'Reilly JD, Adlercreutz H, et al. Isoflavone phytoestrogens consumed in soy decrease F(2)-isoprostane concentrations and increase resistance of low-density lipoprotein to oxidation in humans. Am J Clin Nutr. 2000;72(2):395-400. (PubMed)
17. Hodgson JM, Puddey IB, Croft KD, Mori TA, Rivera J, Beilin LJ. Isoflavonoids do not inhibit in vivo lipid peroxidation in subjects with high-normal blood pressure. Atherosclerosis. 1999;145(1):167-172. (PubMed)
18. Djuric Z, Chen G, Doerge DR, Heilbrun LK, Kucuk O. Effect of soy isoflavone supplementation on markers of oxidative stress in men and women. Cancer Lett. 2001;172(1):1-6. (PubMed)
19. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87-108. (PubMed)
20. Messina M, Nagata C, Wu AH. Estimated Asian adult soy protein and isoflavone intakes. Nutr Cancer. 2006;55(1):1-12. (PubMed)
21. Messina M, Hilakivi-Clarke L. Early intake appears to be the key to the proposed protective effects of soy intake against breast cancer. Nutr Cancer. 2009;61(6):792-798. (PubMed)
22. Wu AH, Yu MC, Tseng CC, Pike MC. Epidemiology of soy exposures and breast cancer risk. Br J Cancer. 2008;98(1):9-14. (PubMed)
23. Dong JY, Qin LQ. Soy isoflavones consumption and risk of breast cancer incidence or recurrence: a meta-analysis of prospective studies. Breast Cancer Res Treat. 2011;125(2):315-323. (PubMed)
24. Travis RC, Allen NE, Appleby PN, Spencer EA, Roddam AW, Key TJ. A prospective study of vegetarianism and isoflavone intake in relation to breast cancer risk in British women. Int J Cancer. 2008;122(3):705-710. (PubMed)
25. Korde LA, Wu AH, Fears T, et al. Childhood soy intake and breast cancer risk in Asian American women. Cancer Epidemiol Biomarkers Prev. 2009;18(4):1050-1059. (PubMed)
26. Shu XO, Jin F, Dai Q, et al. Soyfood intake during adolescence and subsequent risk of breast cancer among Chinese women. Cancer Epidemiol Biomarkers Prev. 2001;10(5):483-488. (PubMed)
27. Thanos J, Cotterchio M, Boucher BA, Kreiger N, Thompson LU. Adolescent dietary phytoestrogen intake and breast cancer risk (Canada). Cancer Causes Control. 2006;17(10):1253-1261. (PubMed)
28. Wu AH, Wan P, Hankin J, Tseng CC, Yu MC, Pike MC. Adolescent and adult soy intake and risk of breast cancer in Asian-Americans. Carcinogenesis. 2002;23(9):1491-1496. (PubMed)
29. Eliassen AH, Hankinson SE. Endogenous hormone levels and risk of breast, endometrial and ovarian cancers: prospective studies. Adv Exp Med Biol. 2008;630:148-165. (PubMed)
30. Hooper L, Ryder JJ, Kurzer MS, et al. Effects of soy protein and isoflavones on circulating hormone concentrations in pre- and post-menopausal women: a systematic review and meta-analysis. Hum Reprod Update. 2009;15(4):423-440. (PubMed)
31. Hooper L, Madhavan G, Tice JA, Leinster SJ, Cassidy A. Effects of isoflavones on breast density in pre- and post-menopausal women: a systematic review and meta-analysis of randomized controlled trials. Hum Reprod Update. 2010;16(6):745-760. (PubMed)
32. Wu AH, Spicer D, Garcia A, et al. Double-blind randomized 12-month soy intervention had no effects on breast MRI fibroglandular tissue density or mammographic density. Cancer Prev Res (Phila). 2015;8(10):942-951. (PubMed)
33. Key TJ, Pike MC. The dose-effect relationship between 'unopposed' oestrogens and endometrial mitotic rate: its central role in explaining and predicting endometrial cancer risk. Br J Cancer. 1988;57(2):205-212. (PubMed)
34. Felix AS, Weissfeld JL, Pfeiffer RM, et al. Endometrial thickness and risk of breast and endometrial carcinomas in the prostate, lung, colorectal and ovarian cancer screening trial. Int J Cancer. 2014;134(4):954-960. (PubMed)
35. Zhang GQ, Chen JL, Liu Q, Zhang Y, Zeng H, Zhao Y. Soy intake is associated with lower endometrial cancer risk: a systematic review and meta-analysis of observational studies. Medicine (Baltimore). 2015;94(50):e2281. (PubMed)
36. Ollberding NJ, Lim U, Wilkens LR, et al. Legume, soy, tofu, and isoflavone intake and endometrial cancer risk in postmenopausal women in the multiethnic cohort study. J Natl Cancer Inst. 2012;104(1):67-76. (PubMed)
37. Budhathoki S, Iwasaki M, Sawada N, et al. Soy food and isoflavone intake and endometrial cancer risk: the Japan Public Health Center-based prospective study. BJOG. 2015;122(3):304-311. (PubMed)
38. Wan YL, Crosbie EJ. Soy intake and endometrial cancer risk varies according to study population. BJOG. 2015;122(3):311. (PubMed)
39. Liu J, Yuan F, Gao J, et al. Oral isoflavone supplementation on endometrial thickness: a meta-analysis of randomized placebo-controlled trials. Oncotarget. 2016;7(14):17369-17379. (PubMed)
40. Hwang YW, Kim SY, Jee SH, Kim YN, Nam CM. Soy food consumption and risk of prostate cancer: a meta-analysis of observational studies. Nutr Cancer. 2009;61(5):598-606. (PubMed)
41. Zhang M, Wang K, Chen L, Yin B, Song Y. Is phytoestrogen intake associated with decreased risk of prostate cancer? A systematic review of epidemiological studies based on 17,546 cases. Andrology. 2016;4(4):745-756. (PubMed)
42. Gardner CD, Oelrich B, Liu JP, Feldman D, Franke AA, Brooks JD. Prostatic soy isoflavone concentrations exceed serum levels after dietary supplementation. Prostate. 2009;69(7):719-726. (PubMed)
43. Mahmoud AM, Yang W, Bosland MC. Soy isoflavones and prostate cancer: a review of molecular mechanisms. J Steroid Biochem Mol Biol. 2014;140:116-132. (PubMed)
44. van Die MD, Bone KM, Williams SG, Pirotta MV. Soy and soy isoflavones in prostate cancer: a systematic review and meta-analysis of randomized controlled trials. BJU Int. 2014;113(5b):E119-130. (PubMed)
45. Hamilton-Reeves JM, Rebello SA, Thomas W, Slaton JW, Kurzer MS. Isoflavone-rich soy protein isolate suppresses androgen receptor expression without altering estrogen receptor-beta expression or serum hormonal profiles in men at high risk of prostate cancer. J Nutr. 2007;137(7):1769-1775. (PubMed)
46. Hamilton-Reeves JM, Rebello SA, Thomas W, Kurzer MS, Slaton JW. Effects of soy protein isolate consumption on prostate cancer biomarkers in men with HGPIN, ASAP, and low-grade prostate cancer. Nutr Cancer. 2008;60(1):7-13. (PubMed)
47. Miyanaga N, Akaza H, Hinotsu S, et al. Prostate cancer chemoprevention study: an investigative randomized control study using purified isoflavones in men with rising prostate-specific antigen. Cancer Sci. 2012;103(1):125-130. (PubMed)
48. Dalais FS, Meliala A, Wattanapenpaiboon N, et al. Effects of a diet rich in phytoestrogens on prostate-specific antigen and sex hormones in men diagnosed with prostate cancer. Urology. 2004;64(3):510-515. (PubMed)
49. Hussain M, Banerjee M, Sarkar FH, et al. Soy isoflavones in the treatment of prostate cancer. Nutr Cancer. 2003;47(2):111-117. (PubMed)
50. Lazarevic B, Boezelijn G, Diep LM, et al. Efficacy and safety of short-term genistein intervention in patients with localized prostate cancer prior to radical prostatectomy: a randomized, placebo-controlled, double-blind Phase 2 clinical trial. Nutr Cancer. 2011;63(6):889-898. (PubMed)
51. Grainger EM, Schwartz SJ, Wang S, et al. A combination of tomato and soy products for men with recurring prostate cancer and rising prostate specific antigen. Nutr Cancer. 2008;60(2):145-154. (PubMed)
52. Kwan W, Duncan G, Van Patten C, Liu M, Lim J. A phase II trial of a soy beverage for subjects without clinical disease with rising prostate-specific antigen after radical radiation for prostate cancer. Nutr Cancer. 2010;62(2):198-207. (PubMed)
53. Pendleton JM, Tan WW, Anai S, et al. Phase II trial of isoflavone in prostate-specific antigen recurrent prostate cancer after previous local therapy. BMC Cancer. 2008;8:132. (PubMed)
54. deVere White RW, Tsodikov A, Stapp EC, Soares SE, Fujii H, Hackman RM. Effects of a high dose, aglycone-rich soy extract on prostate-specific antigen and serum isoflavone concentrations in men with localized prostate cancer. Nutr Cancer. 2010;62(8):1036-1043. (PubMed)
55. Kumar NB, Cantor A, Allen K, et al. The specific role of isoflavones in reducing prostate cancer risk. Prostate. 2004;59(2):141-147. (PubMed)
56. Bosland MC, Kato I, Zeleniuch-Jacquotte A, et al. Effect of soy protein isolate supplementation on biochemical recurrence of prostate cancer after radical prostatectomy: a randomized trial. JAMA. 2013;310(2):170-178. (PubMed)
57. deVere White RW, Hackman RM, Soares SE, Beckett LA, Li Y, Sun B. Effects of a genistein-rich extract on PSA levels in men with a history of prostate cancer. Urology. 2004;63(2):259-263. (PubMed)
58. Napora JK, Short RG, Muller DC, et al. High-dose isoflavones do not improve metabolic and inflammatory parameters in androgen-deprived men with prostate cancer. J Androl. 2011;32(1):40-48. (PubMed)
59. Cosman F, de Beur SJ, LeBoff MS, et al. Clinician's guide to prevention and treatment of osteoporosis. Osteoporos Int. 2014;25(10):2359-2381. (PubMed)
60. Zheng X, Lee SK, Chun OK. Soy isoflavones and osteoporotic bone loss: a review with an emphasis on modulation of bone remodeling. J Med Food. 2016;19(1):1-14. (PubMed)
61. Chiechi LM, Secreto G, D'Amore M, et al. Efficacy of a soy rich diet in preventing postmenopausal osteoporosis: the Menfis randomized trial. Maturitas. 2002;42(4):295-300. (PubMed)
62. Scheiber MD, Liu JH, Subbiah MT, Rebar RW, Setchell KD. Dietary inclusion of whole soy foods results in significant reductions in clinical risk factors for osteoporosis and cardiovascular disease in normal postmenopausal women. Menopause. 2001;8(5):384-392. (PubMed)
63. Arjmandi BH, Khalil DA, Smith BJ, et al. Soy protein has a greater effect on bone in postmenopausal women not on hormone replacement therapy, as evidenced by reducing bone resorption and urinary calcium excretion. J Clin Endocrinol Metab. 2003;88(3):1048-1054. (PubMed)
64. Harkness LS, Fiedler K, Sehgal AR, Oravec D, Lerner E. Decreased bone resorption with soy isoflavone supplementation in postmenopausal women. J Womens Health (Larchmt). 2004;13(9):1000-1007. (PubMed)
65. Ye YB, Tang XY, Verbruggen MA, Su YX. Soy isoflavones attenuate bone loss in early postmenopausal Chinese women : a single-blind randomized, placebo-controlled trial. Eur J Nutr. 2006;45(6):327-334. (PubMed)
66. Wangen KE, Duncan AM, Merz-Demlow BE, et al. Effects of soy isoflavones on markers of bone turnover in premenopausal and postmenopausal women. J Clin Endocrinol Metab. 2000;85(9):3043-3048. (PubMed)
67. Alekel DL, Germain AS, Peterson CT, Hanson KB, Stewart JW, Toda T. Isoflavone-rich soy protein isolate attenuates bone loss in the lumbar spine of perimenopausal women. Am J Clin Nutr. 2000;72(3):844-852. (PubMed)
68. Dalais FS, Ebeling PR, Kotsopoulos D, McGrath BP, Teede HJ. The effects of soy protein containing isoflavones on lipids and indices of bone resorption in postmenopausal women. Clin Endocrinol (Oxf). 2003;58(6):704-709. (PubMed)
69. Cheong JM, Martin BR, Jackson GS, et al. Soy isoflavones do not affect bone resorption in postmenopausal women: a dose-response study using a novel approach with 41Ca. J Clin Endocrinol Metab. 2007;92(2):577-582. (PubMed)
70. Liu J, Ho SC, Su YX, Chen WQ, Zhang CX, Chen YM. Effect of long-term intervention of soy isoflavones on bone mineral density in women: a meta-analysis of randomized controlled trials. Bone. 2009;44(5):948-953. (PubMed)
71. Lauderdale DS, Jacobsen SJ, Furner SE, Levy PS, Brody JA, Goldberg J. Hip fracture incidence among elderly Asian-American populations. Am J Epidemiol. 1997;146(6):502-509. (PubMed)
72. Silverman SL, Madison RE. Decreased incidence of hip fracture in Hispanics, Asians, and blacks: California Hospital Discharge Data. Am J Public Health. 1988;78(11):1482-1483. (PubMed)
73. Koh WP, Wu AH, Wang R, et al. Gender-specific associations between soy and risk of hip fracture in the Singapore Chinese Health Study. Am J Epidemiol. 2009;170(7):901-909. (PubMed)
74. Zhang X, Shu XO, Li H, et al. Prospective cohort study of soy food consumption and risk of bone fracture among postmenopausal women. Arch Intern Med. 2005;165(16):1890-1895. (PubMed)
75. Ma DF, Qin LQ, Wang PY, Katoh R. Soy isoflavone intake increases bone mineral density in the spine of menopausal women: meta-analysis of randomized controlled trials. Clin Nutr. 2008;27(1):57-64. (PubMed)
76. Taku K, Melby MK, Takebayashi J, et al. Effect of soy isoflavone extract supplements on bone mineral density in menopausal women: meta-analysis of randomized controlled trials. Asia Pac J Clin Nutr. 2010;19(1):33-42. (PubMed)
77. Wei P, Liu M, Chen Y, Chen DC. Systematic review of soy isoflavone supplements on osteoporosis in women. Asian Pac J Trop Med. 2012;5(3):243-248. (PubMed)
78. Ricci E, Cipriani S, Chiaffarino F, Malvezzi M, Parazzini F. Soy isoflavones and bone mineral density in perimenopausal and postmenopausal Western women: a systematic review and meta-analysis of randomized controlled trials. J Womens Health (Larchmt). 2010;19(9):1609-1617. (PubMed)
79. Greendale GA, Tseng CH, Han W, et al. Dietary isoflavones and bone mineral density during midlife and the menopausal transition: cross-sectional and longitudinal results from the Study of Women's Health Across the Nation Phytoestrogen Study. Menopause. 2015;22(3):279-288. (PubMed)
80. Frankenfeld CL, McTiernan A, Thomas WK, et al. Postmenopausal bone mineral density in relation to soy isoflavone-metabolizing phenotypes. Maturitas. 2006;53(3):315-324. (PubMed)
81. Ishimi Y. Soybean isoflavones in bone health. Forum Nutr. 2009;61:104-116. (PubMed)
82. Kuhnle GG, Ward HA, Vogiatzoglou A, et al. Association between dietary phyto-oestrogens and bone density in men and postmenopausal women. Br J Nutr. 2011;106(7):1063-1069. (PubMed)
83. Vatanparast H, Chilibeck PD. Does the effect of soy phytoestrogens on bone in postmenopausal women depend on the equol-producing phenotype? Nutr Rev. 2007;65(6 Pt 1):294-299. (PubMed)
84. Wu J, Oka J, Ezaki J, et al. Possible role of equol status in the effects of isoflavone on bone and fat mass in postmenopausal Japanese women: a double-blind, randomized, controlled trial. Menopause. 2007;14(5):866-874. (PubMed)
85. Pawlowski JW, Martin BR, McCabe GP, et al. Impact of equol-producing capacity and soy-isoflavone profiles of supplements on bone calcium retention in postmenopausal women: a randomized crossover trial. Am J Clin Nutr. 2015;102(3):695-703. (PubMed)
86. Kokubo Y, Iso H, Ishihara J, et al. Association of dietary intake of soy, beans, and isoflavones with risk of cerebral and myocardial infarctions in Japanese populations: the Japan Public Health Center-based (JPHC) study cohort I. Circulation. 2007;116(22):2553-2562. (PubMed)
87. Zhang X, Shu XO, Gao YT, et al. Soy food consumption is associated with lower risk of coronary heart disease in Chinese women. J Nutr. 2003;133(9):2874-2878. (PubMed)
88. Yu D, Zhang X, Xiang YB, et al. Association of soy food intake with risk and biomarkers of coronary heart disease in Chinese men. Int J Cardiol. 2014;172(2):e285-287. (PubMed)
89. Yu D, Shu XO, Li H, et al. Dietary isoflavones, urinary isoflavonoids, and risk of ischemic stroke in women. Am J Clin Nutr. 2015;102(3):680-686. (PubMed)
90. Zhang X, Gao YT, Yang G, et al. Urinary isoflavonoids and risk of coronary heart disease. Int J Epidemiol. 2012;41(5):1367-1375. (PubMed)
91. Talaei M, Koh WP, van Dam RM, Yuan JM, Pan A. Dietary soy intake is not associated with risk of cardiovascular disease mortality in Singapore Chinese adults. J Nutr. 2014;144(6):921-928. (PubMed)
92. McCullough ML, Peterson JJ, Patel R, Jacques PF, Shah R, Dwyer JT. Flavonoid intake and cardiovascular disease mortality in a prospective cohort of US adults. Am J Clin Nutr. 2012;95(2):454-464. (PubMed)
93. Mink PJ, Scrafford CG, Barraj LM, et al. Flavonoid intake and cardiovascular disease mortality: a prospective study in postmenopausal women. Am J Clin Nutr. 2007;85(3):895-909. (PubMed)
94. van der Schouw YT, Kreijkamp-Kaspers S, Peeters PH, Keinan-Boker L, Rimm EB, Grobbee DE. Prospective study on usual dietary phytoestrogen intake and cardiovascular disease risk in Western women. Circulation. 2005;111(4):465-471. (PubMed)
95. Zamora-Ros R, Jimenez C, Cleries R, et al. Dietary flavonoid and lignan intake and mortality in a Spanish cohort. Epidemiology. 2013;24(5):726-733. (PubMed)
96. Tokede OA, Onabanjo TA, Yansane A, Gaziano JM, Djousse L. Soya products and serum lipids: a meta-analysis of randomised controlled trials. Br J Nutr. 2015;114(6):831-843. (PubMed)
97. Song X, Zeng R, Ni L, Liu C. The effect of soy or isoflavones on homocysteine levels: a meta-analysis of randomised controlled trials. J Hum Nutr Diet. 2016;29(6):797-804. (PubMed)
98. Dong JY, Wang P, He K, Qin LQ. Effect of soy isoflavones on circulating C-reactive protein in postmenopausal women: meta-analysis of randomized controlled trials. Menopause. 2011;18(11):1256-1262. (PubMed)
99. Liu ZM, Ho SC, Chen YM, et al. Whole soy, but not purified daidzein, had a favorable effect on improvement of cardiovascular risks: a 6-month randomized, double-blind, and placebo-controlled trial in equol-producing postmenopausal women. Mol Nutr Food Res. 2014;58(4):709-717. (PubMed)
100. Landmesser U, Hornig B, Drexler H. Endothelial function: a critical determinant in atherosclerosis? Circulation. 2004;109(21 Suppl 1):II27-33. (PubMed)
101. Ras RT, Streppel MT, Draijer R, Zock PL. Flow-mediated dilation and cardiovascular risk prediction: a systematic review with meta-analysis. Int J Cardiol. 2013;168(1):344-351. (PubMed)
102. Li SH, Liu XX, Bai YY, et al. Effect of oral isoflavone supplementation on vascular endothelial function in postmenopausal women: a meta-analysis of randomized placebo-controlled trials. Am J Clin Nutr. 2010;91(2):480-486. (PubMed)
103. Beavers DP, Beavers KM, Miller M, Stamey J, Messina MJ. Exposure to isoflavone-containing soy products and endothelial function: a Bayesian meta-analysis of randomized controlled trials. Nutr Metab Cardiovasc Dis. 2012;22(3):182-191. (PubMed)
104. Bots ML, Dijk JM, Oren A, Grobbee DE. Carotid intima-media thickness, arterial stiffness and risk of cardiovascular disease: current evidence. J Hypertens. 2002;20(12):2317-2325. (PubMed)
105. Nestel PJ, Yamashita T, Sasahara T, et al. Soy isoflavones improve systemic arterial compliance but not plasma lipids in menopausal and perimenopausal women. Arterioscler Thromb Vasc Biol. 1997;17(12):3392-3398. (PubMed)
106. Teede HJ, Dalais FS, Kotsopoulos D, Liang YL, Davis S, McGrath BP. Dietary soy has both beneficial and potentially adverse cardiovascular effects: a placebo-controlled study in men and postmenopausal women. J Clin Endocrinol Metab. 2001;86(7):3053-3060. (PubMed)
107. Teede HJ, Giannopoulos D, Dalais FS, Hodgson J, McGrath BP. Randomised, controlled, cross-over trial of soy protein with isoflavones on blood pressure and arterial function in hypertensive subjects. J Am Coll Nutr. 2006;25(6):533-540. (PubMed)
108. Chan YH, Lau KK, Yiu KH, et al. Isoflavone intake in persons at high risk of cardiovascular events: implications for vascular endothelial function and the carotid atherosclerotic burden. Am J Clin Nutr. 2007;86(4):938-945. (PubMed)
109. Liu XX, Li SH, Chen JZ, et al. Effect of soy isoflavones on blood pressure: a meta-analysis of randomized controlled trials. Nutr Metab Cardiovasc Dis. 2012;22(6):463-470. (PubMed)
110. Soni M, Rahardjo TB, Soekardi R, et al. Phytoestrogens and cognitive function: a review. Maturitas. 2014;77(3):209-220. (PubMed)
111. White LR, Petrovitch H, Ross GW, et al. Brain aging and midlife tofu consumption. J Am Coll Nutr. 2000;19(2):242-255. (PubMed)
112. Hogervorst E, Sadjimim T, Yesufu A, Kreager P, Rahardjo TB. High tofu intake is associated with worse memory in elderly Indonesian men and women. Dement Geriatr Cogn Disord. 2008;26(1):50-57. (PubMed)
113. Duffy R, Wiseman H, File SE. Improved cognitive function in postmenopausal women after 12 weeks of consumption of a soya extract containing isoflavones. Pharmacol Biochem Behav. 2003;75(3):721-729. (PubMed)
114. File SE, Hartley DE, Elsabagh S, Duffy R, Wiseman H. Cognitive improvement after 6 weeks of soy supplements in postmenopausal women is limited to frontal lobe function. Menopause. 2005;12(2):193-201. (PubMed)
115. Kritz-Silverstein D, Von Muhlen D, Barrett-Connor E, Bressel MA. Isoflavones and cognitive function in older women: the SOy and Postmenopausal Health In Aging (SOPHIA) Study. Menopause. 2003;10(3):196-202. (PubMed)
116. Casini ML, Marelli G, Papaleo E, Ferrari A, D'Ambrosio F, Unfer V. Psychological assessment of the effects of treatment with phytoestrogens on postmenopausal women: a randomized, double-blind, crossover, placebo-controlled study. Fertil Steril. 2006;85(4):972-978. (PubMed)
117. Ho SC, Chan AS, Ho YP, et al. Effects of soy isoflavone supplementation on cognitive function in Chinese postmenopausal women: a double-blind, randomized, controlled trial. Menopause. 2007;14(3 Pt 1):489-499. (PubMed)
118. Kreijkamp-Kaspers S, Kok L, Grobbee DE, et al. Effect of soy protein containing isoflavones on cognitive function, bone mineral density, and plasma lipids in postmenopausal women: a randomized controlled trial. JAMA. 2004;292(1):65-74. (PubMed)
119. Henderson VW, St John JA, Hodis HN, et al. Long-term soy isoflavone supplementation and cognition in women: a randomized, controlled trial. Neurology. 2012;78(23):1841-1848. (PubMed)
120. Cheng PF, Chen JJ, Zhou XY, et al. Do soy isoflavones improve cognitive function in postmenopausal women? A meta-analysis. Menopause. 2015;22(2):198-206. (PubMed)
121. Gleason CE, Fischer BL, Dowling NM, et al. Cognitive Effects of Soy Isoflavones in Patients with Alzheimer's Disease. J Alzheimers Dis. 2015;47(4):1009-1019. (PubMed)
122. Nonhormonal management of menopause-associated vasomotor symptoms: 2015 position statement of The North American Menopause Society. Menopause. 2015;22(11):1155-1172; quiz 1173-1154. (PubMed)
123. Farquhar C, Marjoribanks J, Lethaby A, Suckling JA, Lamberts Q. Long term hormone therapy for perimenopausal and postmenopausal women. Cochrane Database Syst Rev. 2009(2):CD004143. (PubMed)
124. Hardie C, Bain C, Walters M. Hormone replacement therapy: the risks and benefits of treatment. J R Coll Physicians Edinb. 2009;39(4):324-326. (PubMed)
125. Nelson HD, Vesco KK, Haney E, et al. Nonhormonal therapies for menopausal hot flashes: systematic review and meta-analysis. JAMA. 2006;295(17):2057-2071. (PubMed)
126. Messina M. Soy foods, isoflavones, and the health of postmenopausal women. Am J Clin Nutr. 2014;100 Suppl 1:423S-430S. (PubMed)
127. Bolanos R, Del Castillo A, Francia J. Soy isoflavones versus placebo in the treatment of climacteric vasomotor symptoms: systematic review and meta-analysis. Menopause. 2010;17(3):660-666. (PubMed)
128. Chen MN, Lin CC, Liu CF. Efficacy of phytoestrogens for menopausal symptoms: a meta-analysis and systematic review. Climacteric. 2015;18(2):260-269. (PubMed)
129. Franco OH, Chowdhury R, Troup J, et al. Use of Plant-Based Therapies and Menopausal Symptoms: A Systematic Review and Meta-analysis. JAMA. 2016;315(23):2554-2563. (PubMed)
130. Howes LG, Howes JB, Knight DC. Isoflavone therapy for menopausal flushes: a systematic review and meta-analysis. Maturitas. 2006;55(3):203-211. (PubMed)
131. Lethaby A, Marjoribanks J, Kronenberg F, Roberts H, Eden J, Brown J. Phytoestrogens for menopausal vasomotor symptoms. Cochrane Database Syst Rev. 2013(12):CD001395. (PubMed)
132. North American Menopause Society. The role of soy isoflavones in menopausal health: report of The North American Menopause Society/Wulf H. Utian Translational Science Symposium in Chicago, IL (October 2010). Menopause. 2011;18(7):732-753. (PubMed)
133. Taku K, Melby MK, Kronenberg F, Kurzer MS, Messina M. Extracted or synthesized soybean isoflavones reduce menopausal hot flash frequency and severity: systematic review and meta-analysis of randomized controlled trials. Menopause. 2012;19(7):776-790. (PubMed)
134. Williamson-Hughes PS, Flickinger BD, Messina MJ, Empie MW. Isoflavone supplements containing predominantly genistein reduce hot flash symptoms: a critical review of published studies. Menopause. 2006;13(5):831-839. (PubMed)
135. Li L, Xu L, Wu J, Dong L, Zhao S, Zheng Q. Comparative efficacy of nonhormonal drugs on menopausal hot flashes. Eur J Clin Pharmacol. 2016;72(9):1051-1058. (PubMed)
136. Melby MK, Lock M, Kaufert P. Culture and symptom reporting at menopause. Hum Reprod Update. 2005;11(5):495-512. (PubMed)
137. Newton KM, Reed SD, Uchiyama S, et al. A cross-sectional study of equol producer status and self-reported vasomotor symptoms. Menopause. 2015;22(5):489-495. (PubMed)
138. Utian WH, Jones M, Setchell KD. S-equol: a potential nonhormonal agent for menopause-related symptom relief. J Womens Health (Larchmt). 2015;24(3):200-208. (PubMed)
139. Liu ZM, Ho SC, Woo J, Chen YM, Wong C. Randomized controlled trial of whole soy and isoflavone daidzein on menopausal symptoms in equol-producing Chinese postmenopausal women. Menopause. 2014;21(6):653-660. (PubMed)
140. Jou HJ, Wu SC, Chang FW, Ling PY, Chu KS, Wu WH. Effect of intestinal production of equol on menopausal symptoms in women treated with soy isoflavones. Int J Gynaecol Obstet. 2008;102(1):44-49. (PubMed)
141. Aso T, Uchiyama S, Matsumura Y, et al. A natural S-equol supplement alleviates hot flushes and other menopausal symptoms in equol nonproducing postmenopausal Japanese women. J Womens Health (Larchmt). 2012;21(1):92-100. (PubMed)
142. Jenks BH, Iwashita S, Nakagawa Y, et al. A pilot study on the effects of S-equol compared to soy isoflavones on menopausal hot flash frequency. J Womens Health (Larchmt). 2012;21(6):674-682. (PubMed)
143. Schmidt M, Arjomand-Wolkart K, Birkhauser MH, et al. Consensus: soy isoflavones as a first-line approach to the treatment of menopausal vasomotor complaints. Gynecol Endocrinol. 2016;32(6):427-430. (PubMed)
144. Fletcher RJ. Food sources of phyto-oestrogens and their precursors in Europe. Br J Nutr. 2003;89 Suppl 1:S39-43. (PubMed)
145. Munro IC, Harwood M, Hlywka JJ, et al. Soy isoflavones: a safety review. Nutr Rev. 2003;61(1):1-33. (PubMed)
146. Zamora-Ros R, Knaze V, Lujan-Barroso L, et al. Dietary intakes and food sources of phytoestrogens in the European Prospective Investigation into Cancer and Nutrition (EPIC) 24-hour dietary recall cohort. Eur J Clin Nutr. 2012;66(8):932-941. (PubMed)
147. Setchell KD, Cole SJ. Variations in isoflavone levels in soy foods and soy protein isolates and issues related to isoflavone databases and food labeling. J Agric Food Chem. 2003;51(14):4146-4155. (PubMed)
148. Kuhnle GG, Dell'Aquila C, Aspinall SM, Runswick SA, Mulligan AA, Bingham SA. Phytoestrogen content of foods of animal origin: dairy products, eggs, meat, fish, and seafood. J Agric Food Chem. 2008;56(21):10099-10104. (PubMed)
149. US Department of Agriculture. USDA Database for the Isoflavone Content of Selected Foods, Release 2. Available at: http://www.ars.usda.gov/News/docs.htm?docid=6382. Accessed 8/9/16.
150. Chua R, Anderson K, Chen J, Hu M. Quality, labeling accuracy, and cost comparison of purified soy isoflavonoid products. J Altern Complement Med. 2004;10(6):1053-1060. (PubMed)
151. Setchell KD, Brown NM, Desai P, et al. Bioavailability of pure isoflavones in healthy humans and analysis of commercial soy isoflavone supplements. J Nutr. 2001;131(4 Suppl):1362S-1375S. (PubMed)
152. Setchell KD, Zimmer-Nechemias L, Cai J, Heubi JE. Isoflavone content of infant formulas and the metabolic fate of these phytoestrogens in early life. Am J Clin Nutr. 1998;68(6 Suppl):1453S-1461S. (PubMed)
153. Chen Z, Zheng W, Custer LJ, et al. Usual dietary consumption of soy foods and its correlation with the excretion rate of isoflavonoids in overnight urine samples among Chinese women in Shanghai. Nutr Cancer. 1999;33(1):82-87. (PubMed)
154. Gleason CE, Carlsson CM, Barnet JH, et al. A preliminary study of the safety, feasibility and cognitive efficacy of soy isoflavone supplements in older men and women. Age Ageing. 2009;38(1):86-93. (PubMed)
155. Allred CD, Allred KF, Ju YH, Virant SM, Helferich WG. Soy diets containing varying amounts of genistein stimulate growth of estrogen-dependent (MCF-7) tumors in a dose-dependent manner. Cancer Res. 2001;61(13):5045-5050. (PubMed)
156. Ju YH, Allred CD, Allred KF, Karko KL, Doerge DR, Helferich WG. Physiological concentrations of dietary genistein dose-dependently stimulate growth of estrogen-dependent human breast cancer (MCF-7) tumors implanted in athymic nude mice. J Nutr. 2001;131(11):2957-2962. (PubMed)
157. Constantinou AI, Lantvit D, Hawthorne M, Xu X, van Breemen RB, Pezzuto JM. Chemopreventive effects of soy protein and purified soy isoflavones on DMBA-induced mammary tumors in female Sprague-Dawley rats. Nutr Cancer. 2001;41(1-2):75-81. (PubMed)
158. Tanos V, Brzezinski A, Drize O, Strauss N, Peretz T. Synergistic inhibitory effects of genistein and tamoxifen on human dysplastic and malignant epithelial breast cells in vitro. Eur J Obstet Gynecol Reprod Biol. 2002;102(2):188-194. (PubMed)
159. Ju YH, Doerge DR, Allred KF, Allred CD, Helferich WG. Dietary genistein negates the inhibitory effect of tamoxifen on growth of estrogen-dependent human breast cancer (MCF-7) cells implanted in athymic mice. Cancer Res. 2002;62(9):2474-2477. (PubMed)
160. Liu B, Edgerton S, Yang X, et al. Low-dose dietary phytoestrogen abrogates tamoxifen-associated mammary tumor prevention. Cancer Res. 2005;65(3):879-886. (PubMed)
161. Petrakis NL, Barnes S, King EB, et al. Stimulatory influence of soy protein isolate on breast secretion in pre- and postmenopausal women. Cancer Epidemiol Biomarkers Prev. 1996;5(10):785-794. (PubMed)
162. Hargreaves DF, Potten CS, Harding C, et al. Two-week dietary soy supplementation has an estrogenic effect on normal premenopausal breast. J Clin Endocrinol Metab. 1999;84(11):4017-4024. (PubMed)
163. Sartippour MR, Rao JY, Apple S, et al. A pilot clinical study of short-term isoflavone supplements in breast cancer patients. Nutr Cancer. 2004;49(1):59-65. (PubMed)
164. Shu XO, Zheng Y, Cai H, et al. Soy food intake and breast cancer survival. JAMA. 2009;302(22):2437-2443. (PubMed)
165. Caan B, Sternfeld B, Gunderson E, Coates A, Quesenberry C, Slattery ML. Life After Cancer Epidemiology (LACE) Study: a cohort of early stage breast cancer survivors (United States). Cancer Causes Control. 2005;16(5):545-556. (PubMed)
166. Pierce JP, Faerber S, Wright FA, et al. A randomized trial of the effect of a plant-based dietary pattern on additional breast cancer events and survival: the Women's Healthy Eating and Living (WHEL) Study. Control Clin Trials. 2002;23(6):728-756. (PubMed)
167. Nechuta SJ, Caan BJ, Chen WY, et al. Soy food intake after diagnosis of breast cancer and survival: an in-depth analysis of combined evidence from cohort studies of US and Chinese women. Am J Clin Nutr. 2012;96(1):123-132. (PubMed)
168. Duffy C, Perez K, Partridge A. Implications of phytoestrogen intake for breast cancer. CA Cancer J Clin. 2007;57(5):260-277. (PubMed)
169. Rock CL, Doyle C, Demark-Wahnefried W, et al. Nutrition and physical activity guidelines for cancer survivors. CA Cancer J Clin. 2012;62(4):243-274. (PubMed)
170. American Academy of Pediatrics. Committee on Nutrition. Soy protein-based formulas: recommendations for use in infant feeding. Pediatrics. 1998;101(1 Pt 1):148-153. (PubMed)
171. Bhatia J, Greer F. Use of soy protein-based formulas in infant feeding. Pediatrics. 2008;121(5):1062-1068. (PubMed)
172. Setchell KD, Zimmer-Nechemias L, Cai J, Heubi JE. Exposure of infants to phyto-oestrogens from soy-based infant formula. Lancet. 1997;350(9070):23-27. (PubMed)
173. Vandenplas Y, Castrellon PG, Rivas R, et al. Safety of soya-based infant formulas in children. Br J Nutr. 2014;111(8):1340-1360. (PubMed)
174. Andres A, Cleves MA, Bellando JB, Pivik RT, Casey PH, Badger TM. Developmental status of 1-year-old infants fed breast milk, cow's milk formula, or soy formula. Pediatrics. 2012;129(6):1134-1140. (PubMed)
175. Malloy MH, Berendes H. Does breast-feeding influence intelligence quotients at 9 and 10 years of age? Early Hum Dev. 1998;50(2):209-217. (PubMed)
176. Strom BL, Schinnar R, Ziegler EE, et al. Exposure to soy-based formula in infancy and endocrinological and reproductive outcomes in young adulthood. JAMA. 2001;286(7):807-814. (PubMed)
177. Adgent MA, Daniels JL, Rogan WJ, et al. Early-life soy exposure and age at menarche. Paediatr Perinat Epidemiol. 2012;26(2):163-175. (PubMed)
178. Andres A, Moore MB, Linam LE, Casey PH, Cleves MA, Badger TM. Compared with feeding infants breast milk or cow-milk formula, soy formula feeding does not affect subsequent reproductive organ size at 5 years of age. J Nutr. 2015;145(5):871-875. (PubMed)
179. Westmark CJ. Soy Infant Formula may be Associated with Autistic Behaviors. Autism Open Access. 2013;3. (PubMed)
180. Westmark CJ. Soy infant formula and seizures in children with autism: a retrospective study. PLoS One. 2014;9(3):e80488. (PubMed)
181. Messina M. Soybean isoflavone exposure does not have feminizing effects on men: a critical examination of the clinical evidence. Fertil Steril. 2010;93(7):2095-2104. (PubMed)
182. Hamilton-Reeves JM, Vazquez G, Duval SJ, Phipps WR, Kurzer MS, Messina MJ. Clinical studies show no effects of soy protein or isoflavones on reproductive hormones in men: results of a meta-analysis. Fertil Steril. 2010;94(3):997-1007. (PubMed)
183. Cederroth CR, Zimmermann C, Nef S. Soy, phytoestrogens and their impact on reproductive health. Mol Cell Endocrinol. 2012;355(2):192-200. (PubMed)
184. Divi RL, Chang HC, Doerge DR. Anti-thyroid isoflavones from soybean: isolation, characterization, and mechanisms of action. Biochem Pharmacol. 1997;54(10):1087-1096. (PubMed)
185. Doerge DR, Sheehan DM. Goitrogenic and estrogenic activity of soy isoflavones. Environ Health Perspect. 2002;110 Suppl 3:349-353. (PubMed)
186. Messina M, Redmond G. Effects of soy protein and soybean isoflavones on thyroid function in healthy adults and hypothyroid patients: a review of the relevant literature. Thyroid. 2006;16(3):249-258. (PubMed)
187. Chorazy PA, Himelhoch S, Hopwood NJ, Greger NG, Postellon DC. Persistent hypothyroidism in an infant receiving a soy formula: case report and review of the literature. Pediatrics. 1995;96(1 Pt 1):148-150. (PubMed)
188. Bruce B, Messina M, Spiller GA. Isoflavone supplements do not affect thyroid function in iodine-replete postmenopausal women. J Med Food. 2003;6(4):309-316. (PubMed)
189. Persky VW, Turyk ME, Wang L, et al. Effect of soy protein on endogenous hormones in postmenopausal women. Am J Clin Nutr. 2002;75(1):145-153. (PubMed)
190. Duncan AM, Merz BE, Xu X, Nagel TC, Phipps WR, Kurzer MS. Soy isoflavones exert modest hormonal effects in premenopausal women. J Clin Endocrinol Metab. 1999;84(1):192-197. (PubMed)
191. Duncan AM, Underhill KE, Xu X, Lavalleur J, Phipps WR, Kurzer MS. Modest hormonal effects of soy isoflavones in postmenopausal women. J Clin Endocrinol Metab. 1999;84(10):3479-3484. (PubMed)
192. Dillingham BL, McVeigh BL, Lampe JW, Duncan AM. Soy protein isolates of varied isoflavone content do not influence serum thyroid hormones in healthy young men. Thyroid. 2007;17(2):131-137. (PubMed)
193. Natural Medicines. Soy: interactions with drugs - Professional handout. 2016. Available at: https://naturalmedicines-therapeuticresearch-com.ezproxy.proxy.library.oregonstate.edu/databases/food,-herbs-supplements/professional.aspx?productid=975 - interactionsWithDrugs. Accessed 8/8/16.
194. Toro-Funes N, Bosch-Fuste J, Latorre-Moratalla ML, Veciana-Nogues MT, Vidal-Carou MC. Biologically active amines in fermented and non-fermented commercial soybean products from the Spanish market. Food Chem. 2015;173:1119-1124. (PubMed)
195. Cambria-Kiely JA. Effect of soy milk on warfarin efficacy. Ann Pharmacother. 2002;36(12):1893-1896. (PubMed)
196. Jabbar MA, Larrea J, Shaw RA. Abnormal thyroid function tests in infants with congenital hypothyroidism: the influence of soy-based formula. J Am Coll Nutr. 1997;16(3):280-282. (PubMed)
197. Bell DS, Ovalle F. Use of soy protein supplement and resultant need for increased dose of levothyroxine. Endocr Pract. 2001;7(3):193-194. (PubMed)
198. Liu ZM, Chen YM, Ho SC. Effects of soy intake on glycemic control: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2011;93(5):1092-1101. (PubMed)
199. Li Z, Hong K, Saltsman P, et al. Long-term efficacy of soy-based meal replacements vs an individualized diet plan in obese type II DM patients: relative effects on weight loss, metabolic parameters, and C-reactive protein. Eur J Clin Nutr. 2005;59(3):411-418. (PubMed)
Chlorophyll and Metallo-Chlorophyll Derivatives
Contents
Summary
- Chlorophyll a and chlorophyll b are natural, fat-soluble chlorophylls found in plants. (More information)
- Sodium copper chlorophyllin (SCC) is a semi-synthetic mixture of water-soluble sodium copper salts derived from chlorophyll. (More information)
- SCC has been used orally as an internal deodorant and topically in the treatment of slow-healing wounds for more than 50 years without any serious side effects. (More information)
- Chlorophylls and SCC form tight molecular complexes with some chemicals known or suspected to cause cancer, and in doing so, may block carcinogenic effects. Carefully controlled studies have not been undertaken to determine whether a similar mechanism might limit uptake of required nutrients. (More information)
- Supplementation with SCC before meals substantially decreased a urinary biomarker of aflatoxin-induced DNA damage in a Chinese population at high risk of liver cancer due to unavoidable, dietary aflatoxin exposure from moldy grains and legumes. (More information)
- Scientists are hopeful that SCC supplementation will be helpful in decreasing the risk of liver cancer in high-risk populations with unavoidable, dietary aflatoxin exposure. However, it is not yet known whether SCC or natural chlorophylls will be useful in the prevention of cancers in people who are not exposed to significant levels of dietary aflatoxin. (More information)
Introduction
Chlorophyll is the pigment that gives plants and algae their green color. Plants use chlorophyll to trap light needed for photosynthesis (1). The basic structure of chlorophyll is a porphyrin ring similar to that of heme in hemoglobin, although the central atom in chlorophyll is magnesium instead of iron. The long hydrocarbon (phytol) tail attached to the porphyrin ring makes chlorophyll fat-soluble and insoluble in water. Chlorophyll a and chlorophyll b represent about 99% of the chlorophyll species found in edible plants (Figure 1; 2), while some algae and microalgae contain minor quantities of chlorophyll c pigments (e.g., Laminaria ochroleuca, Undaria pinnatifida) (3). Chlorophyll a and b only have a small difference in one of the side chains but an intact phytol tail, while the common characteristic of chlorophyll c isoforms is the absence of a phytol tail. These structural differences cause each type of chlorophyll to absorb light at slightly different wavelengths.
Metallo-chlorophyll derivatives, including chlorophyllins, can be chemically synthesized or produced in industrial food processing; these compounds contain zinc, iron, or copper in place of the central magnesium atom (2). The most studied chlorophyllin, sodium copper chlorophyllin (SCC), is a semi-synthetic mixture of sodium copper salts derived from chlorophyll (4, 5). SCC is often simply called ‘chlorophyllin’ in the older scientific literature, with newer publications specifying whether iron, zinc, copper, or magnesium chlorophyllin were studied. During its synthesis, the magnesium atom at the center of the ring is replaced with copper (or other metals), and the phytol tail is lost. Unlike natural chlorophyll, chlorophyllins (regardless of the metal used) are water-soluble. Although the content of different SCC mixtures may vary, two compounds commonly found in commercial SCC are trisodium copper chlorin e6 and disodium copper chlorin e4 (Figure 2).
Metabolism and Bioavailability
Little is known about the bioavailability and metabolism of chlorophyll in humans, although it is known that chlorophyll undergoes extensive metabolism once consumed. Animal model studies show only about 1%-3% of chlorophyll is absorbed, while the rest is excreted in the feces, primarily as pheopytin and pyropheophytin metabolites, indicating that significant transformation and microbial metabolism occur in the gastrointestinal tract (reviewed in 2). A recent study in eight healthy adults found pheophytin and pheophorbide derivatives in the blood of most subjects following consumption of 1.2 kg boiled spinach, a concentrated source of chlorophyll (6).
Sodium copper chlorophyllin was originally thought to be poorly absorbed because of its lack of apparent toxicity. However, a placebo-controlled clinical trial found that significant amounts of copper chlorin e4 in the serum of people taking chlorophyllin tablets (300 mg/day) (7), indicating that it is indeed absorbed. In vitro studies have found chlorin e4 to have a higher stability than chlorin e6 (2).
More research, however, is needed to understand the bioavailability and metabolism of natural chlorophylls and chlorin compounds in synthetic chlorophyllin.
Biological Activities
Complex formation with other molecules
Chlorophyll and sodium copper chlorophyllin are able to form tight molecular complexes with certain chemicals known or suspected to cause cancer, including polyaromatic hydrocarbons found in tobacco smoke (8), some heterocyclic amines found in cooked meat (9), and aflatoxin-B1 (10). The binding of chlorophyll or SCC to these potential carcinogens may interfere with gastrointestinal absorption of potential carcinogens, reducing the amount that reaches susceptible tissues (11). This has been demonstrated in humans: a cross-over study in three volunteers that used accelerator mass spectrometry to study the pharmacokinetics of an ultra-low dose of aflatoxin-B1 found a 150-mg dose of either SCC or chlorophyll could decrease absorption of aflatoxin-B1 (12).
Antioxidant effects
SCC can neutralize several physically relevant oxidants in vitro (13-16), and limited data from animal studies suggest that SCC supplementation may decrease oxidative damage induced by chemical carcinogens and radiation (17, 18). While chlorophyll and its derivatives have demonstrated antioxidant activity in in vitro assays (15, 19), the relevance of these findings to humans is not clear.
Modification of the metabolism and detoxification of carcinogens
To initiate the development of cancer, some chemicals (procarcinogens) must first be metabolized to active carcinogens that are capable of damaging DNA or other critical molecules in susceptible tissues. Since enzymes in the cytochrome P450 family are required for the activation of some procarcinogens, inhibition of cytochrome P450 enzymes may decrease the risk of some types of chemically induced cancers. In vitro studies indicate that SCC may decrease the activity of cytochrome P450 enzymes (8, 20, 21). Phase II biotransformation enzymes promote the elimination of potentially harmful toxins and carcinogens from the body. Limited data from animal studies indicate that SCC may increase the activity of the phase II enzyme quinone reductase (22).
Therapeutic effects
One in vitro study showed that human colon cancer cells undergo cell cycle arrest after treatment with SCC (23). The mechanism involved inhibition of ribonucleotide reductase activity. Ribonucleotide reductase plays a pivotal role in DNA synthesis and repair and is a target of currently used cancer therapeutic agents, such as hydroxyurea (23). While this may provide a potential avenue for SCC in the clinical setting, sensitizing cancer cells to DNA damaging agents, in vivo studies are needed.
Metal absorption
The porphyrin structure of chlorophyll is analogous to the heme structure found in blood and muscle tissue. Because heme-bound iron has higher bioavailability than nonheme iron (the most common form of iron in plant-based sources, e.g., legumes, spinach), iron uptake from iron chlorophyll is of interest. Toxicity studies in rats suggest iron chlorophyllin is generally safe for mammalian consumption (24). In vitro studies demonstrate that iron chlorophyllin is as good as heme in delivering iron to intestinal cells, and significantly better than the most common supplemental form of iron (i.e., ferrous sulfate) when incorporated into most food matrices (25). However, work in this area is nascent and has not yet been validated in humans. It is also not known if metallo-chlorophyll derivatives of copper or zinc increase absorption of these essential divalent metals.
Disease Prevention
Aflatoxin-associated liver cancer
Aflatoxin-B1 (AFB1), a liver carcinogen produced by certain species of fungus, is found in moldy grains and legumes, including corn, peanuts, and soybeans (4, 11). In hot, humid regions of Africa and Asia with improper grain storage facilities, high levels of dietary AFB1 are associated with increased risk of hepatocellular carcinoma. Moreover, the combination of hepatitis B infection and high dietary AFB1 exposure increases the risk of hepatocellular carcinoma still further. In the liver, AFB1 is metabolized to a carcinogen capable of binding DNA and causing mutations. In animal models of AFB1-induced liver cancer, administration of SCC at the same time as dietary AFB1 exposure significantly reduces AFB1-induced DNA damage in the livers of rainbow trout and rats (26-28) and dose-dependently inhibits the development of liver cancer in trout (29). Likewise, natural chlorophyll has also been found to inhibit AFB1-induced liver cancer in the rat (28). Collectively, this evidence supports a role for SCC and/or chlorophyll itself in limiting cancer initiation. In contrast, data suggest a limited role for SCC in influencing cancer progression. For example, one rat study found that SCC did not protect against aflatoxin-induced liver damage when given after tumor initiation (30).
Because of the long time period between AFB1 exposure and the development of cancer in humans, an intervention trial might require as long as 20 years to determine whether SCC supplementation can reduce the incidence of hepatocellular carcinoma in people exposed to high levels of dietary AFB1. However, a biomarker of AFB1-induced DNA damage (AFB1-N7-guanine) can be measured in the urine, and high urinary levels of AFB1-N7-guanine have been associated with significantly increased risk of developing hepatocellular carcinoma (31). In order to determine whether chlorophyllin could decrease AFB1-induced DNA damage in humans, a randomized, placebo-controlled intervention trial was conducted in 180 adults residing in a region in China where the risk of hepatocellular carcinoma is very high due to unavoidable, dietary AFB1 exposure and a high prevalence of chronic hepatitis B infection (32). Participants took either 100 mg of SCC or a placebo before meals three times daily. After 16 weeks of treatment, urinary levels of AFB1-N7-guanine were 55% lower in those taking SCC than in those taking the placebo, suggesting that SCC supplementation before meals can substantially decrease AFB1-induced DNA damage. Although a reduction in hepatocellular carcinoma has not yet been demonstrated in humans taking SCC, scientists are hopeful that supplementation will provide some protection to high-risk populations with unavoidable, dietary AFB1 exposure (11).
It is not known whether SCC will be useful in the prevention of cancers in people who are not exposed to significant levels of dietary AFB1, as is the case for most people living in the US. Many questions remain to be answered regarding the exact mechanisms of cancer prevention by SCC, the implications for the prevention of other types of cancer, and the potential for natural chlorophylls in the diet to provide cancer protection.
Therapeutic Uses of Chlorophyllin
Internal deodorant
Observations in the 1940s and 1950s that topical SCC had deodorizing effects on foul-smelling wounds led clinicians to administer SCC orally to patients with colostomies and ileostomies in order to control fecal odor (33). While early case reports indicated that SCC doses of 100 to 200 mg/day were effective in reducing fecal odor in ostomy patients (34, 35), a placebo-controlled trial found that 75 mg of oral SCC three times daily was no more effective than placebo in decreasing fecal odor assessed by colostomy patients (36). Several case reports have been published indicating that oral SCC (100-300 mg/day) decreased subjective assessments of urinary and fecal odor in incontinent patients (33, 37).
Trimethylaminuria is a hereditary disorder characterized by the excretion of trimethylamine, a compound with a “fishy” or foul odor. One study in a small number of Japanese patients with trimethylaminuria found that oral SCC (60 mg three times daily) for three weeks significantly decreased urinary trimethylamine concentrations (38).
Wound healing
Research in the 1940s indicated that chlorophyllin slowed the growth of certain anaerobic bacteria in the test tube and accelerated the healing of experimental wounds in animals. These findings led to the use of topical SCC solutions and ointments in the treatment of persistent open wounds in humans (39). During the late 1940s and 1950s, a series of largely uncontrolled studies in patients with slow-healing wounds, such as vascular ulcers and pressure (decubitus) ulcers, reported that the application of topical SCC promoted healing more effectively than other commonly used treatments (40, 41). In the late 1950s, SCC was added to papain and urea-containing ointments used for the chemical debridement of wounds in order to reduce local inflammation, promote healing, and control odor (33). SCC-containing papain/urea ointments are still available in the US by prescription (42). Several studies have reported that such ointments are effective in wound healing (43). A spray formulation of the papain/urea/SCC therapy is also available (44).
Skin conditions
A few small studies have investigated SCC as a topical treatment for various skin conditions. In a pilot study of 10 adults (ages 18-30 years) who had mild-to-moderate acne vulgaris and enlarged facial pores, twice daily application of a 0.1% liposomal SCC gel for three weeks improved a number of clinical parameters of the Global Acne Assessment Scale (i.e., facial oiliness, facial blotchiness, presence and size of facial pores, and number of acne lesions) compared to baseline (45). Additionally, a pilot study in 10 women (ages 40 years or older) with noticeable photodamage and solar lentigines found that twice daily topical application of a gel containing 0.66% SCC complex salts for eight weeks improved various clinical measures, including tactile and visual roughness of facial skin, skin radiance, fine lines, pore size, and overall photodamage (46). A few case reports have also observed some improvement in facial redness and rosacea with application of topical SCC (47).
While the reports from these studies are interesting, placebo-controlled clinical trials are needed to determine whether SCC may have utility in treating various skin conditions.
Sources
Chlorophylls
Chlorophylls are the most abundant pigments in plants, with chlorophyll a being two to four times as prevalent as chlorophyll b (6, 48). Dark-green leafy vegetables like spinach are rich sources of natural chlorophylls. The chlorophyll content of selected vegetables are presented in Table 1 (49).
Food and supplements
Chlorophyll
Green algae like chlorella are often marketed as supplemental sources of chlorophyll. Because natural chlorophyll is not as stable as SCC and is much more expensive, most over-the-counter chlorophyll supplements actually contain sodium copper chlorophyllin.
Sodium copper chlorophyllin
Oral preparations of sodium copper chlorophyllin (also called chlorophyllin copper complex) are available as a dietary supplement and as an over-the-counter drug (Derifil) used to reduce odor from colostomies and ileostomies, or to reduce fecal odor due to incontinence (50). Oral doses of 100 to 300 mg/day in three divided doses have been used to control fecal and urinary odor (see Therapeutic Uses of Chlorophyllin).
In the US, SCC is found in minor quantities in some types of green table olives (51). It is also approved for use as a green color additive in a limited number of foods like chewing gum (52), as well as in drugs and cosmetics, as detailed in the Code of Federal Regulations 21 (53).
Zinc chlorophyll derivatives
In US supermarkets, canned green beans thermally processed in a zinc chloride solution to produce zinc chlorophyll derivatives within the green beans themselves are sold under the trademarked name "veri-green" (54). Because zinc chlorophyll derivatives are more robust to heat and acid treatment, they better retain a bright green color as compared to native magnesium-bound chlorophyll (48).
Safety
Natural chlorophylls are not known to be toxic, and no toxic effects have been attributed to chlorophyllin despite more than 50 years of clinical use in humans (11, 33, 39). When taken orally, supplemental chlorophyll or sodium copper chlorophyllin may cause green discoloration of urine or feces, or yellow or black discoloration of the tongue (55). There have also been occasional reports of diarrhea related to oral SCC use. When applied topically to wounds, SCC has been reported to cause mild burning or itching in some cases (56). Oral chlorophyllin may result in false positive results on guaiac card tests for occult blood (57). Since the safety of chlorophyll or chlorophyllin supplements has not been tested in pregnant or lactating women, they should be avoided during pregnancy and lactation.
Authors and Reviewers
Originally written in 2004 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in December 2005 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in June 2009 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in September 2021 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in March 2022 by:
Rachel E. Kopec, Ph.D.
Assistant Professor of Human Nutrition
The Ohio State University
Copyright 2004-2024 Linus Pauling Institute
References
1. Matthews CK, van Holde KE. Biochemistry. 2nd ed. Menlo Park: The Benjamin/Cummings Publishing Company; 1996.
2. Zhong S, Bird A, Kopec RE. The metabolism and potential bioactivity of chlorophyll and metallo-chlorophyll derivatives in the gastrointestinal tract. Mol Nutr Food Res. 2021;65(7):e2000761. (PubMed)
3. Chen K, Rios JJ, Perez-Galvez A, Roca M. Comprehensive chlorophyll composition in the main edible seaweeds. Food Chem. 2017;228:625-633. (PubMed)
4. Sudakin DL. Dietary aflatoxin exposure and chemoprevention of cancer: a clinical review. J Toxicol Clin Toxicol. 2003;41(2):195-204. (PubMed)
5. Dashwood RH. The importance of using pure chemicals in (anti) mutagenicity studies: chlorophyllin as a case in point. Mutat Res. 1997;381(2):283-286. (PubMed)
6. Chao PY, Huang MY, Huang WD, Lin KH, Chen SY, Yang CM. Study of chlorophyll-related compounds from dietary spinach in human blood. Not Bot Horti Agrobo. 2018;46(2):309-316.
7. Egner PA, Stansbury KH, Snyder EP, Rogers ME, Hintz PA, Kensler TW. Identification and characterization of chlorin e(4) ethyl ester in sera of individuals participating in the chlorophyllin chemoprevention trial. Chem Res Toxicol. 2000;13(9):900-906. (PubMed)
8. Tachino N, Guo D, Dashwood WM, Yamane S, Larsen R, Dashwood R. Mechanisms of the in vitro antimutagenic action of chlorophyllin against benzo[a]pyrene: studies of enzyme inhibition, molecular complex formation and degradation of the ultimate carcinogen. Mutat Res. 1994;308(2):191-203. (PubMed)
9. Dashwood R, Yamane S, Larsen R. Study of the forces of stabilizing complexes between chlorophylls and heterocyclic amine mutagens. Environ Mol Mutagen. 1996;27(3):211-218. (PubMed)
10. Breinholt V, Schimerlik M, Dashwood R, Bailey G. Mechanisms of chlorophyllin anticarcinogenesis against aflatoxin B1: complex formation with the carcinogen. Chem Res Toxicol. 1995;8(4):506-514. (PubMed)
11. Egner PA, Munoz A, Kensler TW. Chemoprevention with chlorophyllin in individuals exposed to dietary aflatoxin. Mutat Res. 2003;523-524:209-216. (PubMed)
12. Jubert C, Mata J, Bench G, et al. Effects of chlorophyll and chlorophyllin on low-dose aflatoxin B(1) pharmacokinetics in human volunteers. Cancer Prev Res (Phila). 2009;2(12):1015-1022. (PubMed)
13. Kumar SS, Devasagayam TP, Bhushan B, Verma NC. Scavenging of reactive oxygen species by chlorophyllin: an ESR study. Free Radic Res. 2001;35(5):563-574. (PubMed)
14. Kamat JP, Boloor KK, Devasagayam TP. Chlorophyllin as an effective antioxidant against membrane damage in vitro and ex vivo. Biochim Biophys Acta. 2000;1487(2-3):113-127. (PubMed)
15. Vankova K, Markova I, Jasprova J, et al. Chlorophyll-mediated changes in the redox status of pancreatic cancer cells are associated with its anticancer effects. Oxid Med Cell Longev. 2018;2018:4069167. (PubMed)
16. Domijan AM, Gajski G, Novak Jovanovic I, Geric M, Garaj-Vrhovac V. In vitro genotoxicity of mycotoxins ochratoxin A and fumonisin B(1) could be prevented by sodium copper chlorophyllin--implication to their genotoxic mechanism. Food Chem. 2015;170:455-462. (PubMed)
17. Park KK, Park JH, Jung YJ, Chung WY. Inhibitory effects of chlorophyllin, hemin and tetrakis(4-benzoic acid)porphyrin on oxidative DNA damage and mouse skin inflammation induced by 12-O-tetradecanoylphorbol-13-acetate as a possible anti-tumor promoting mechanism. Mutat Res. 2003;542(1-2):89-97. (PubMed)
18. Kumar SS, Shankar B, Sainis KB. Effect of chlorophyllin against oxidative stress in splenic lymphocytes in vitro and in vivo. Biochim Biophys Acta. 2004;1672(2):100-111. (PubMed)
19. Perez-Galvez A, Viera I, Roca M. Carotenoids and chlorophylls as antioxidants. Antioxidants (Basel). 2020;9(6):505. (PubMed)
20. Yun CH, Jeong HG, Jhoun JW, Guengerich FP. Non-specific inhibition of cytochrome P450 activities by chlorophyllin in human and rat liver microsomes. Carcinogenesis. 1995;16(6):1437-1440. (PubMed)
21. John K, Divi RL, Keshava C, et al. CYP1A1 and CYP1B1 gene expression and DNA adduct formation in normal human mammary epithelial cells exposed to benzo[a]pyrene in the absence or presence of chlorophyllin. Cancer Lett. 2010;292(2):254-260. (PubMed)
22. Dingley KH, Ubick EA, Chiarappa-Zucca ML, et al. Effect of dietary constituents with chemopreventive potential on adduct formation of a low dose of the heterocyclic amines PhIP and IQ and phase II hepatic enzymes. Nutr Cancer. 2003;46(2):212-221. (PubMed)
23. Chimploy K, Diaz GD, Li Q, et al. E2F4 and ribonucleotide reductase mediate S-phase arrest in colon cancer cells treated with chlorophyllin. Int J Cancer. 2009;125(9):2086-2094. (PubMed)
24. Toyoda T, Cho YM, Mizuta Y, Akagi J, Nishikawa A, Ogawa K. A 13-week subchronic toxicity study of sodium iron chlorophyllin in F344 rats. J Toxicol Sci. 2014;39(1):109-119. (PubMed)
25. Miret S, Tascioglu S, van der Burg M, Frenken L, Klaffke W. In vitro bioavailability of iron from the heme analogue sodium iron chlorophyllin. J Agric Food Chem. 2010;58(2):1327-1332. (PubMed)
26. Dashwood RH, Breinholt V, Bailey GS. Chemopreventive properties of chlorophyllin: inhibition of aflatoxin B1 (AFB1)-DNA binding in vivo and anti-mutagenic activity against AFB1 and two heterocyclic amines in the Salmonella mutagenicity assay. Carcinogenesis. 1991;12(5):939-942. (PubMed)
27. Kensler TW, Groopman JD, Roebuck BD. Use of aflatoxin adducts as intermediate endpoints to assess the efficacy of chemopreventive interventions in animals and man. Mutat Res. 1998;402(1-2):165-172. (PubMed)
28. Simonich MT, Egner PA, Roebuck BD, et al. Natural chlorophyll inhibits aflatoxin B1-induced multi-organ carcinogenesis in the rat. Carcinogenesis. 2007;28(6):1294-1302. (PubMed)
29. Breinholt V, Hendricks J, Pereira C, Arbogast D, Bailey G. Dietary chlorophyllin is a potent inhibitor of aflatoxin B1 hepatocarcinogenesis in rainbow trout. Cancer Res. 1995;55(1):57-62. (PubMed)
30. Orner GA, Roebuck BD, Dashwood RH, Bailey GS. Post-initiation chlorophyllin exposure does not modulate aflatoxin-induced foci in the liver and colon of rats. J Carcinog. 2006;5:6. (PubMed)
31. Qian GS, Ross RK, Yu MC, et al. A follow-up study of urinary markers of aflatoxin exposure and liver cancer risk in Shanghai, People's Republic of China. Cancer Epidemiol Biomarkers Prev. 1994;3(1):3-10. (PubMed)
32. Egner PA, Wang JB, Zhu YR, et al. Chlorophyllin intervention reduces aflatoxin-DNA adducts in individuals at high risk for liver cancer. Proc Natl Acad Sci U S A. 2001;98(25):14601-14606. (PubMed)
33. Chernomorsky SA, Segelman AB. Biological activities of chlorophyll derivatives. N J Med. 1988;85(8):669-673. (PubMed)
34. Siegel LH. The control of ileostomy and colostomy odors. Gastroenterology. 1960;38:634-636. (PubMed)
35. Weingarten M, Payson B. Deodorization of colostomies with chlorophyll. Rev Gastroenterol. 1951;18(8):602-604. (PubMed)
36. Christiansen SB, Byel SR, Stromsted H, Stenderup JK, Eickhoff JH. [Can chlorophyll reduce fecal odor in colostomy patients?]. Ugeskr Laeger. 1989;151(27):1753-1754. (PubMed)
37. Young RW, Beregi JS, Jr. Use of chlorophyllin in the care of geriatric patients. J Am Geriatr Soc. 1980;28(1):46-47. (PubMed)
38. Yamazaki H, Fujieda M, Togashi M, et al. Effects of the dietary supplements, activated charcoal and copper chlorophyllin, on urinary excretion of trimethylamine in Japanese trimethylaminuria patients. Life Sci. 2004;74(22):2739-2747. (PubMed)
39. Kephart JC. Chlorophyll derivatives - their chemistry, commercial preparation and uses. Econ Bot. 1955;9:3-38.
40. Bowers WF. Chlorophyll in wound healing and suppurative disease. Am J Surg. 1947;73:37-50. (PubMed)
41. Carpenter EB. Clinical experiences with chlorophyll preparations. Am J Surg. 1949;77(2):167-171. (PubMed)
42. Physicians' Desk Reference. 58th ed. Stamford: Thomson Health Care, Inc.; 2003.
43. Smith RG. Enzymatic debriding agents: an evaluation of the medical literature. Ostomy Wound Manage. 2008;54(8):16-34. (PubMed)
44. Weir D, Farley KL. Relative delivery efficiency and convenience of spray and ointment formulations of papain/urea/chlorophyllin enzymatic wound therapies. J Wound Ostomy Continence Nurs. 2006;33(5):482-490. (PubMed)
45. Stephens TJ, McCook JP, Herndon JH, Jr. Pilot study of topical copper chlorophyllin complex in subjects with facial acne and large pores. J Drugs Dermatol. 2015;14(6):589-592. (PubMed)
46. Sigler ML, Stephens TJ. Assessment of the safety and efficacy of topical copper chlorophyllin in women with photodamaged facial skin. J Drugs Dermatol. 2015;14(4):401-404. (PubMed)
47. Vasily DB. Topical treatment with liposomal sodium copper chlorophyllin complex in subjects with facial redness and erythematotelangiectatic rosacea: case studies. J Drugs Dermatol. 2015;14(10):1157-1159. (PubMed)
48. Hayes M, Ferruzzi MG. Update on the bioavailability and chemopreventative mechanisms of dietary chlorophyll derivatives. Nutr Res. 2020;81:19-37. (PubMed)
49. Bohn T, Walczyk S, Leisibach S, Hurrell RF. Chlorophyll-bound magnesium in commonly consumed vegetables and fruits: relevance to magnesium nutrition. J Food Sci. 2004;69(9):S347-S350.
50. Food and Drug Administration. Code of Federal Regulations: Miscellaneous Internal Drug Products for Over the Counter Use [Web page]. April 1, 2002. Available at: http://www.fda.gov/cder/otcmonographs/Internal_Deodorant/internal_deodorant(357I).html. Accessed 6/9/04.
51. Aparicio-Ruiz R, Riedl KM, Schwartz SJ. Identification and quantification of metallo-chlorophyll complexes in bright green table olives by high-performance liquid chromatography-mass spectrometry quadrupole/time-of-flight. J Agric Food Chem. 2011;59(20):11100-11108. (PubMed)
52. Viera I, Perez-Galvez A, Roca M. Green natural colorants. Molecules. 2019;24(1):154. (PubMed)
53. US Food and Drug Administration. CFR - Code of Federal Regulations Title 21. Available at: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=73.3110. Accessed 9/29/21.
54. Von Elbe JH, Huang AS, Attoe EL, Nank WK. Pigment composition and color of conventional and Veri-Green canned beans. J Agric Food Chem. 1986;34(1):52-54.
55. Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. 2nd ed. Montvale: Physicians' Desk Reference, Inc.; 2008.
56. Smith LW. The present status of topical chlorophyll therapy. N Y State J Med. 1955;55(14):2041-2050. (PubMed)
57. Gogel HK, Tandberg D, Strickland RG. Substances that interfere with guaiac card tests: implications for gastric aspirate testing. Am J Emerg Med. 1989;7(5):474-480. (PubMed)