Vitaminas
El término vitamina proviene de las palabras vital y amina, ya que las vitaminas son necesarias para la vida y originalmente se pensaba que eran aminas. Aunque no todas las vitaminas son aminas, son compuestos orgánicos que los seres humanos necesitan en pequeñas cantidades en la dieta. Un compuesto orgánico se considera una vitamina cuando la falta de ese compuesto en la dieta provoca síntomas evidentes de su deficiencia.
La información sobre vitaminas y minerales del Centro de Información de Micronutrientes del Instituto Linus Pauling se encuentra ahora disponible en un libro titulado Un acercamiento basado en la evidencia a las vitaminas y minerales: Beneficios para la salud y recomendaciones de consumo (An Evidence-based Approach to Vitamins and Minerals: Health Benefits and Intake Recommendations). El libro esta disponible para la venta en Linus Pauling Institute o Thieme Medical Publishers.
Seleccione una vitamina de la lista para más información.
Ácido Pantoténico
Contenido
Resumen
- El ácido pantoténico — también conocido como vitamina B5 — es una vitamina hidrosoluble que a su vez es un precursor en la síntesis de la coenzima A. La coenzima A es esencial para muchas reacciones bioquímicas que sostienen la vida. También, el grupo funcional fosfopanteteinilo de la coenzima A es requerido para la actividad biológica de varias proteínas, incluyendo la proteína portadora de acilo involucrada en la síntesis de ácidos grasos. (Más información)
- El ácido pantoténico es esencial para todas las formas de vida. Se encuentra de forma ubicua en alimentos de origen vegetal y animal, y una deficiencia dietaría es bastante rara. (Más información)
- La Junta de Alimentos y Nutrición del Instituto de Medicina de los EE.UU. estableció una ingesta adecuada (IA) de 5 miligramos (mg)/día para adultos basándose en la ingesta diaria promedio del ácido pantoténico. (Más información)
- Evidencia proveniente de estudios de intervención limitados sugiere que el ácido pantoténico y/o el pantotenol (alcohol análogo) podrían mejorar el proceso de curación de heridas de la piel. Sin embargo, estudios de mayor escala son requeridos. (Más información)
- Se ha demostrado que el tratamiento con altas dosis de pantetina — un derivado del ácido pantoténico — disminuye las concentraciones de colesterol y lípidos del suero. Aunque la terapia con pantetina parece ser bien tolerada, supervisión médica es indispensable. (Más información)
- Alimentos ricos en acido pantoténico incluyen órganos animales (hígado y riñones), pescado, mariscos, productos lácteos, huevos, aguacates, legumbres, champiñones, y camotes. (Más información)
- Poca o ninguna toxicidad ha sido asociada con el ácido pantoténico dietario y suplementario tanto que ningún nivel máximo de ingesta tolerable (NM) ha sido establecido. (Más información)
El ácido pantoténico, también conocido como vitamina B5, es esencial para todas las formas de vida (1). El ácido pantoténico se encuentra a través de todas las ramificaciones de la vida en la forma de coenzima A, una coenzima vital en numerosas reacciones químicas (2).
Función
Síntesis de los cofactores del ácido pantoténico
Coenzima A
El ácido pantoténico es un precursor en la biosíntesis de la coenzima A (CoA) (Figura 1), una coenzima esencial en una variedad de reacciones bioquímicas que soportan la vida (véase abajo). La quinasa ácido pantoténico II (PANKII) cataliza el paso inicial para la fosforilación del ácido pantoténico a ácido 4’-fosfopantoténico. La coenzima A y sus derivados inhiben la síntesis del ácido 4’-fosfopantoténico, pero la inhibición puede ser revertida por la carnitina, requerida para el transporte de ácidos grasos en la mitocondria (3). Las reacciones subsecuentes en esta ruta biosintetica incluyen la síntesis del intermedio 4'-fosfopanteteína, como también el reciclaje de la coenzima A a 4'-fosfopanteteína (Figura 1).
4’-fosfopanteteína
El grupo funcional 4'-fosfopanteteinilo de la coenzima A puede ser transferido a enzimas en las cuales 4'-fosfopanteteína es un cofactor esencial para sus actividades biológicas (véase 4'-fosfopanteteinilación).
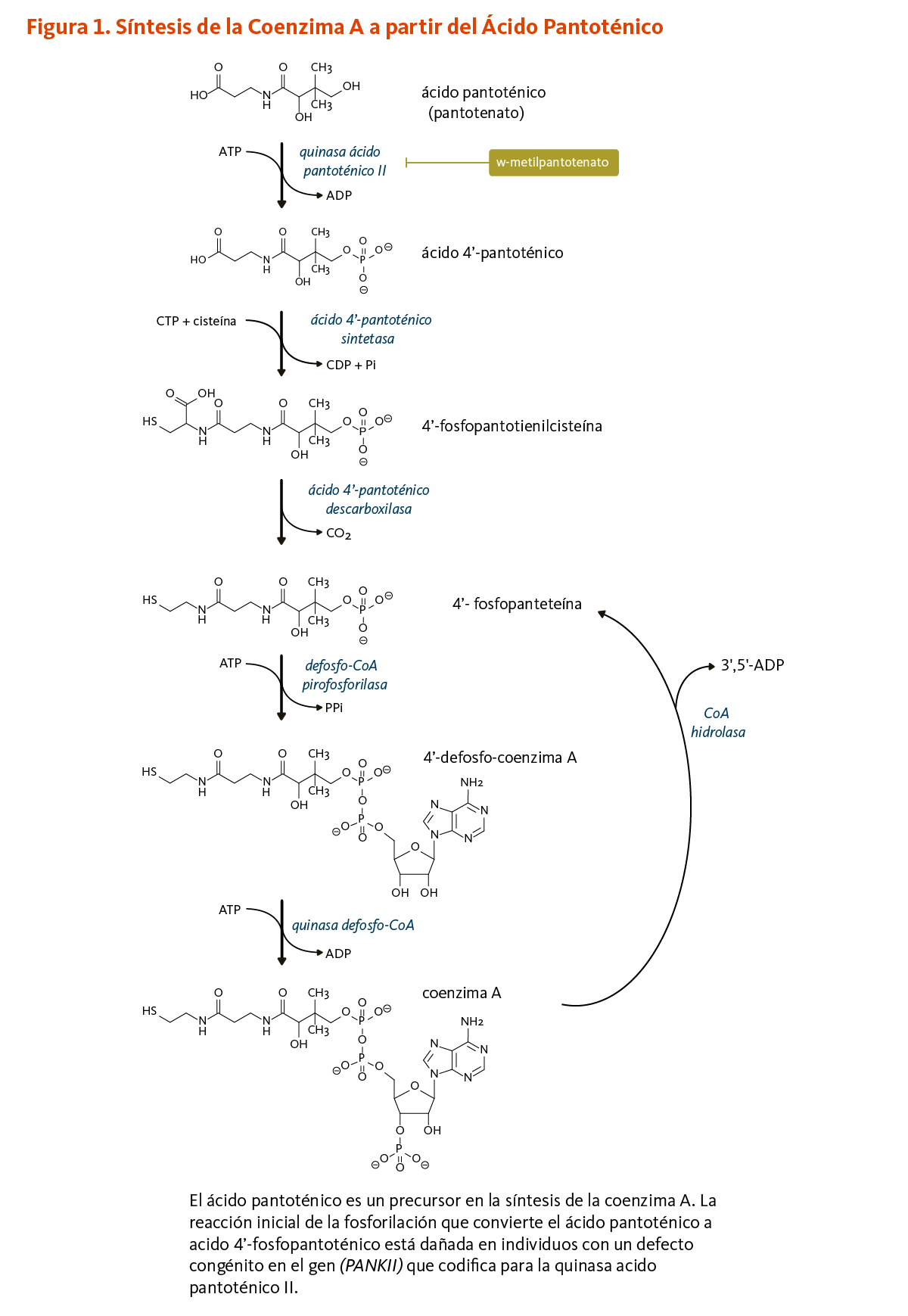
[Figura 1 - Clic para Agrandar]
Función de cofactor y co-sustrato
Coenzima A
La coenzima A reacciona con los grupos acilo, dando lugar a derivados tioéster, como la acetil-CoA, succinil-CoA, malonil-CoA, y 3-hidroxi-3-metilglutaril (HMG)-CoA. La coenzima A y sus derivados de acilo son requeridos para reacciones que generan energía a partir de la degradación de grasas, carbohidratos y proteínas dietarías. Además, la coenzima A en la forma de acetil-CoA y succinil-CoA está involucrada en el ciclo del ácido cítrico, en la síntesis de grasas esenciales, colesterol, hormonas esteroidales, vitamina A y D, el neurotransmisor acetilcolina, y en la ruta de β-oxidación de ácidos grasos. Los derivados de la coenzima A son también requeridos para la síntesis de la hormona melatonina, y para un componente de la hemoglobina llamado hemo. Además, el metabolismo de un cierto número de drogas y toxinas por el hígado requieren de la coenzima A (4).
La coenzima A fue nombrada por su papel en las reacciones de acetilación. La mayoría de las proteínas acetiladas en el cuerpo han sido modificadas por la adición de un grupo acetato que fue donando por el derivado tioéster de la coenzima A, la acetil-CoA. La acetilación de las proteínas altera la carga general de las proteínas, modificando su estructura tridimensional y, alterando potencialmente su función. Por ejemplo, la acetilación es un mecanismo que regula la actividad de hormonas peptídicas, incluyendo aquellas producidas por la glándula pituitaria (5). También, se ha mostrado que la acetilación proteínica, como otras modificaciones postraduccionales, regula la localización subcelular, la función, y el periodo de semidesintegración de muchas moléculas de señalización, factores de transcripción y enzimas. Notablemente, la acetilación de histonas juega un papel en la regulación de la expresión de genes al facilitar la transcripción de (es decir, ARNm síntesis), mientras que las histonas desacetiladas son usualmente asociadas con la compactación de la cromatina y el silenciamiento de genes. Se encontró que la acetilación de histonas resulta en cambios estructurales de la cromatina, que afectan tanto la interacción ADN-proteína como las interacciones proteína-proteína. La diafonía entre marcas de acetilación y otras modificaciones postranscripcionales de las histonas también facilitan el reclutamiento de reguladores transcripcionales para el promotor de los genes que son posteriormente transcritos (revisado en 6).
Finalmente, un cierto número de moléculas de señalización son modificadas por la unión de ácidos grasos de cadena larga donados por la coenzima A. Estas modificaciones son conocidas como acilación de proteínas y tienen papeles centrales en las rutas de señalización celular (4).
4’-fosfopanteteinilación
Complejos multi-enzimáticos específicos, que necesitan llevar a cabo varias reacciones en una manera ordenada, pueden requerir del enlace covalente de un brazo de 4'-fosfopanteteína a un dominio "portador" (o proteína). Este dominio portador mantiene sustratos o intermedios de reacción durante la progresión a través de varias reacciones enzimáticas. En los mamíferos, la transferencia del grupo funcional 4'-fosfopanteteinil proveniente de la coenzima A a un residuo de serina conservado de un dominio portador especifico es catalizado por una única fosfopanteteinil transferasa (7). La 4’-fosfopanteteinilación es necesaria para la conversión de apoenzimas a holoenzimas completamente activas (véase abajo).
Proteína transportadora de grupos acilo
Los lípidos son moléculas grasas esenciales para la función fisiológica normal, y entre otros tipos, incluyen los esfingolípidos (componentes esenciales de la vaina de mielina que mejora la transmisión nerviosa), los fosfolípidos (componentes estructurales importantes de las membranas celulares), y los ácidos grasos. La ácido graso sintasa (FAS) es un complejo multi-enzimático que cataliza la síntesis de ácidos grasos. Dentro del complejo de la FAS, la proteína transportadora de grupos acilo (ACP) requiere de ácido pantoténico en la forma de 4'-fosfopanteteína para su actividad como proteína transportadora (3). Un grupo, como lo es el grupo funcional 4'-fosfopanteteinilo para la ACP, es llamado un grupo prostético, el grupo prostético no está compuesto de aminoácidos y es un cofactor estrechamente unido requerido para la actividad biológica de ciertas proteínas (Figura 2). La acetil-CoA, malonil-CoA, y ACP son todos requeridos para la síntesis de ácidos grasos en el citosol. Durante la síntesis de ácidos grasos, los grupos acilo de la acetil-CoA y malonil-CoA son transferidos al grupo sulfhidrilo (-SH) del grupo funcional 4'-fosfopanteteinilo de la ACP. El grupo prostético es usado como un brazo flexible para transferir la cadena de ácidos grasos creciente a cada uno de los centros enzimáticos del complejo FAS tipo I. En la mitocondria, la 4'-fosfopanteteína también sirve como un grupo prostético para un homólogo de la ACP presente en el complejo FAS tipo II mitocondrial (8).
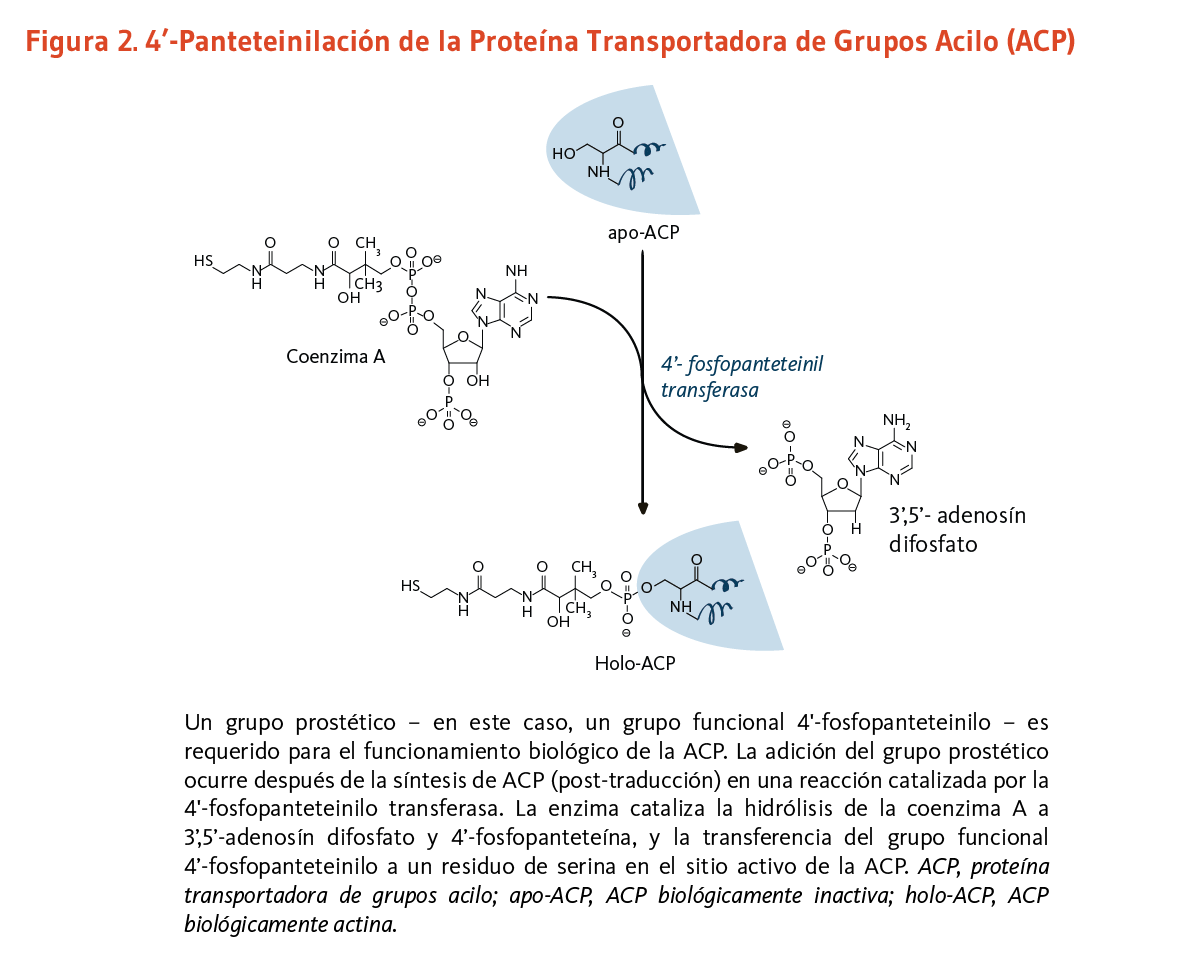
[Figura 2 - Clic para Agrandar]
10-formiltetrahidrofolato deshidrogenasa
La enzima 10-formiltetrahidrofolato deshidrogenasa (FDH) cataliza la conversión de 10-formiltetrahidrofolato a tetrahidrofolato, un cofactor esencial en el metabolismo de ácidos nucleicos y aminoácidos (Figura 3). Similar a la ACP, la FDH requiere de un grupo prostético 4'-fosfopanteteína para su actividad biológica. El grupo prostético actúa como un brazo oscilante para acoplar las actividades de los dos dominios catalíticos de la FDH (9, 10). Un homólogo de la FDH en la mitocondria también requiere de 4’-fosfopanteteinilación para ser biológicamente activo (11).
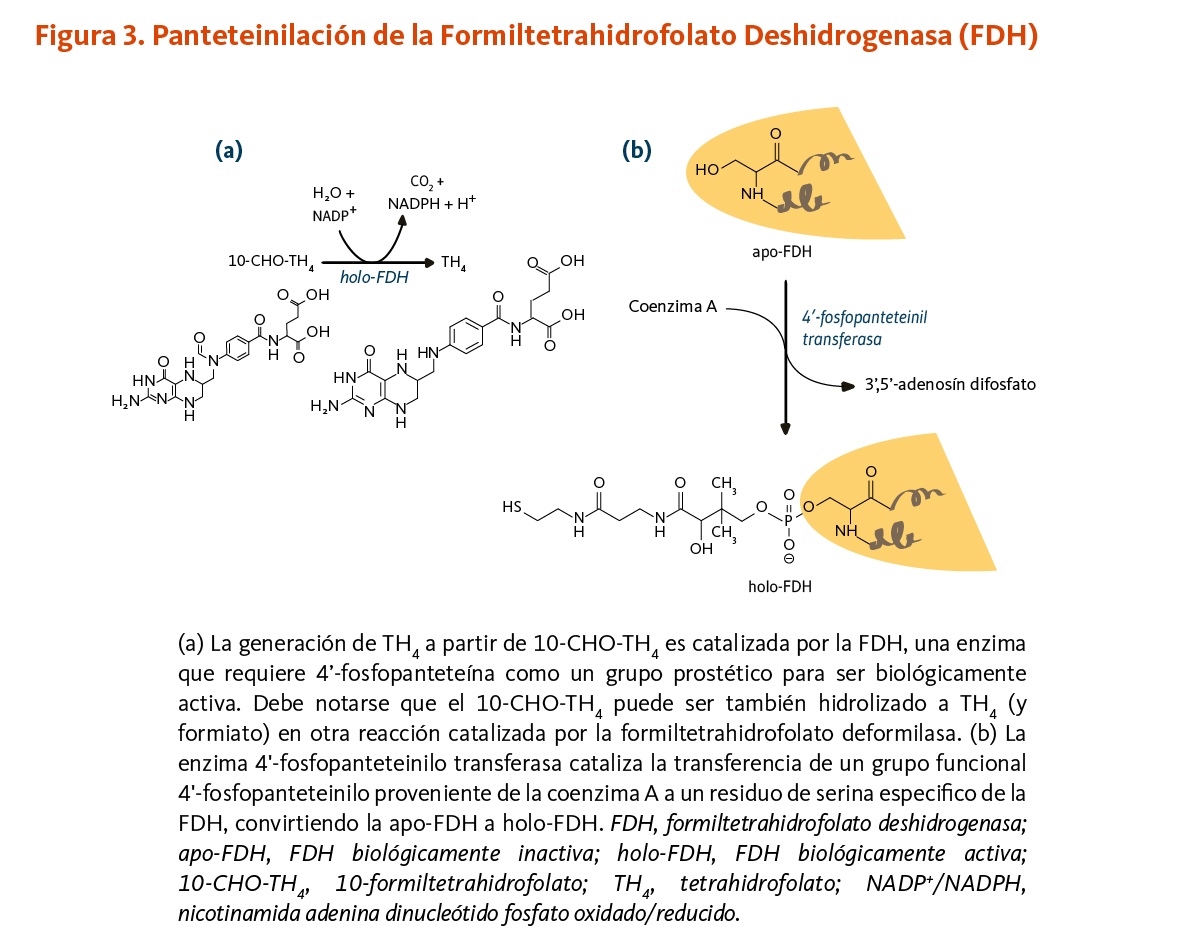
[Figura 3 - Clic para Agrandar]
α-Aminoadipato semialdehído sintasa
La 4’-fosfopanteteinilación es requerida para la actividad biológica de la apoenzima α-aminoadipato semialdehído sintasa (AASS). AASS cataliza las reacciones iniciales en la ruta mitocondrial para la degradación de lisina — un aminoácido esencial para los humanos. AASS está hecha de dos dominios catalíticos. El dominio de la lisina-cetoglutarato reductasa primero cataliza la conversión de lisina a sacaropina. La sacaropina es convertida adicionalmente a α-aminoadipato semialdehído en una reacción catalizada por el dominio de la sacaropina deshidrogenasa (Figura 4).
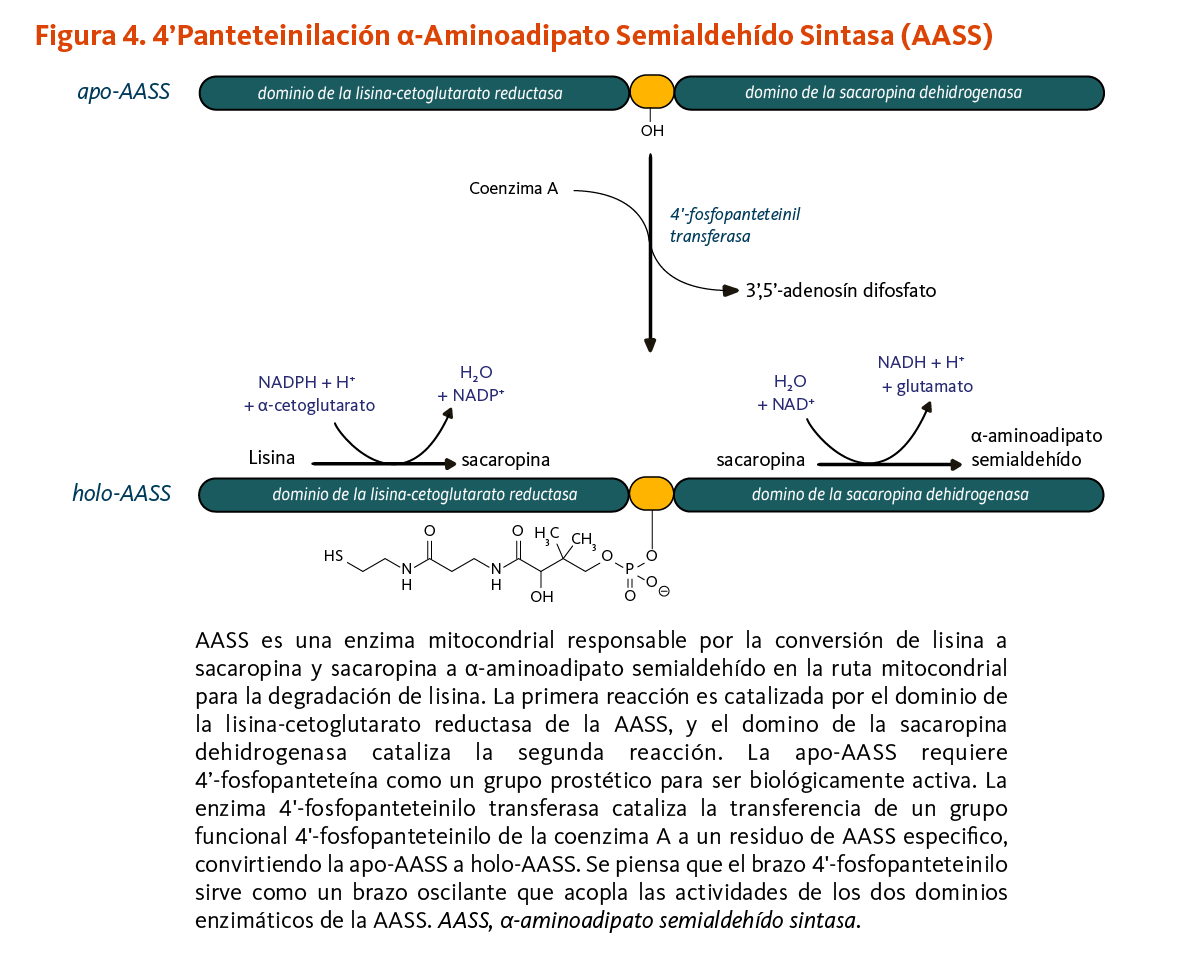
[Figura 4 - Clic para Agrandar]
Deficiencia
Una deficiencia de ácido pantoténico de origen natural en seres humanos es muy rara y sólo ha sido observada en casos de desnutrición severa. Los prisioneros de la Segunda Guerra Mundial en las Filipinas, Birmania, y Japón experimentaron entumecimiento y ardor doloroso y hormigueo en los pies; estos síntomas fueron aliviados específicamente con la suplementación con ácido pantoténico (4). La deficiencia de ácido pantoténico en los humanos ha sido inducida experimentalmente co-administrando un inhibidor de la quinasa del ácido pantoténico (w-metilpantotenato; vease Figura 1 arriba) y una dieta deficiente de ácido pantoténico. Los participantes en este experimento se quejaron de dolor de cabeza, fatiga, insomnio, molestias intestinales, y entumecimiento y cosquilleo en manos y pies (12). En otro estudio, los participantes alimentados con solo una dieta libre de ácido pantoténico no desarrollaron signos clínicos de deficiencia, aunque algunos parecían apáticos y se quejaban de fatiga (13).
El homopantotenato de calcio (o homopantotenato) es un antagonista del ácido pantoténico con efectos colinérgicos (es decir, similar a aquellos del neurotransmisor, acetilcolina). Este compuesto es usado en Japón para mejorar la función mental, especialmente en la enfermedad de Alzheimer. Un efecto secundario raro fue el desarrollo de encefalopatía hepática, una condición de funcionamiento anormal del cerebro producto de una falla en la eliminación de toxinas por parte del hígado. La encefalopatía fue revertida por la suplementación con ácido pantoténico, sugiriendo que se debió a una deficiencia de ácido pantoténico inducida por el homopantotenato (14). Debe notarse, que mutaciones en el gen humano PANKII, el cual codifica para la quinasa ácido pantoténico II (véase Figura 1 arriba), resultan en la síntesis alterada de la 4'-fosfopanteteína y de la coenzima A (véase Función). Este desorden, llamado neurodegeneración asociada a pantotenato quinasa, está caracterizado por alteraciones visuales e intelectuales, distonía, anomalías del habla, dificultades del comportamiento, y desordenes de personalidad (15).
Sin embargo, debido a que el ácido pantoténico es ampliamente distribuido en la naturaleza y la deficiencia es extremadamente rara en humanos, la mayoría de la información con respecto a las consecuencias de la deficiencia ha sido recolectada de investigación experimental en animales (revisada en 3). La deficiencia de ácido pantoténico en ratas desarrollo daño a las glándulas adrenales, mientras que monos desarrollaron anemia debido a la síntesis disminuida de grupos hemo, un componente de la hemoglobina. Perros con deficiencia de ácido pantoténico desarrollaron niveles bajos de glucosa, respiración y frecuencia cardiaca aceleradas y convulsiones. Los pollos desarrollaron irritación de la piel, anomalías en el plumaje y daño en nervios espinales asociado con la degeneración de la vaina de mielina. Los ratones con deficiencia de ácido pantoténico mostraron tolerancia disminuida al ejercicio y almacenaje reducido de glucosa (en la forma de glicógeno) en músculos e hígado. Los ratones también desarrollaron irritación de la piel y canosidad del pelaje, que fueron revertidos por la administración de ácido pantoténico.
La diversidad de los síntomas enfatiza las numerosas funciones del ácido pantoténico en sus formas de coenzima.
La Ingesta Adecuada (IA)
Debido a que existe poca información sobre los requerimientos de ácido pantoténico en humanos, la Junta de Nutrición y Alimentos del Instituto de Medicina estableció un nivel de Ingesta Adecuada (IA) basado en las ingestas dietarías observadas en grupos saludables de la población (Tabla 1) (16).
Tratamiento de Enfermedades
Cicatrización de heridas
Se encontró que la adición de D-pantotenato de calcio y/o pantotenol (Figura 5) al medio de cultivo de fibroblastos cutáneos con una herida artificial aumentó la proliferación y migración celular, acelerando así el proceso de cicatrización in vitro (17, 18). De la misma manera, la deficiencia in vitro de ácido pantoténico indujo la expresión de marcadores de diferenciación en la proliferación de fibroblastos cutáneos e inhibió la proliferación en queratinocitos humanos (19). La aplicación de ungüentos que contienen calcio D-pantotenato o pantotenol — también conocido como D-pantenol o dexpantenol — sobre la piel ha mostrado que acelera el cierre de las heridas de la piel y aumentan la fuerza del tejido cicatricial en animales (3).
Los efectos del dexpantenol en la cicatrización de heridas no están claros. En un estudio controlado con placebo que incluyo 12 voluntarios sanos, la aplicación de un ungüento que contenía dexpantenol (cada 12 horas por 1 a 6 días) en un modelo de cicatrización de herida cutánea fue asociada con una expresión mejorada de los marcadores de proliferación, inflamación, y reparación de tejidos (20). Sin embargo, el estudio fallo en reportar si estos cambios en respuesta al dexpantenol tópico mejoro el proceso de reparación de la herida en comparación al placebo (20). Algunos estudios no han mostrado efectos. Ensayos controlados aleatorios tempranos en pacientes sometidos a cirugía de remoción de tatuajes encontraron que la suplementación diaria con 1 gramo o 3 gramos de vitamina C y 200 mg o 900 mg de ácido pantoténico por 21 días no mejoraron significantemente el proceso de cicatrización (21, 22). A pesar de todo, en un reciente estudio aleatorio, doble ciego, controlado con placebo, el uso de pastillas de dexpantenol (300 mg/día por hasta 14 días post-cirugía) se encontró que acelero la cicatrización de la mucosa después de una amigdalectomía en niños (23).
Colesterol alto
Estudios tempranos sugieren que dosis farmacológicas de pantetina, un derivado del ácido pantoténico, podrían tener un efecto hipocolesterolemiante (24, 25). La pantetina está hecha por dos moléculas de panteteína unidas por un enlace disulfuro (un enlace químico entre dos átomos de azufre) (Figura 5). La pantetina está estructuralmente relacionada a la coenzima A y se encuentra en el grupo prostético que es requerido para la función biológica de la proteína transportadora de acilo, formiltetrahidrofolato dehidrogenasa, y de la α-aminoadipato semialdehído sintasa (véase Función). En un estudio aleatorio, doble ciego, controlado con placebo de 16 semanas, la suplementación diaria con pantetina (600 mg/día por 8 semanas, seguido por 900 mg/día por otras 8 semanas) significantemente mejoró el perfil de los parámetros lípidos en 120 individuos en bajo a moderado riego de enfermedades cardiovasculares (ECV). Después de ajustarse al valor basal, se encontró que la pantetina era significantemente más efectiva que el placebo en la disminución de las concentraciones de colesterol de lipoproteínas de baja densidad (LDL-C) y apolipoproteína B (apoB), como también en la reducción de la proporción de triglicéridos a colesterol de lipoproteínas de alta densidad (TG:HDL-C) (26). Aunque parece ser bien tolerada y potencialmente beneficial en el mejoramiento del metabolismo del colesterol, la pantetina no es una vitamina, y la decisión de usar dosis farmacológicas de pantetina para tratar el colesterol sanguíneo elevado o los triglicéridos deber ser hecha solo en colaboración con proveedores de la salud cualificados que proveen seguimiento adecuado.
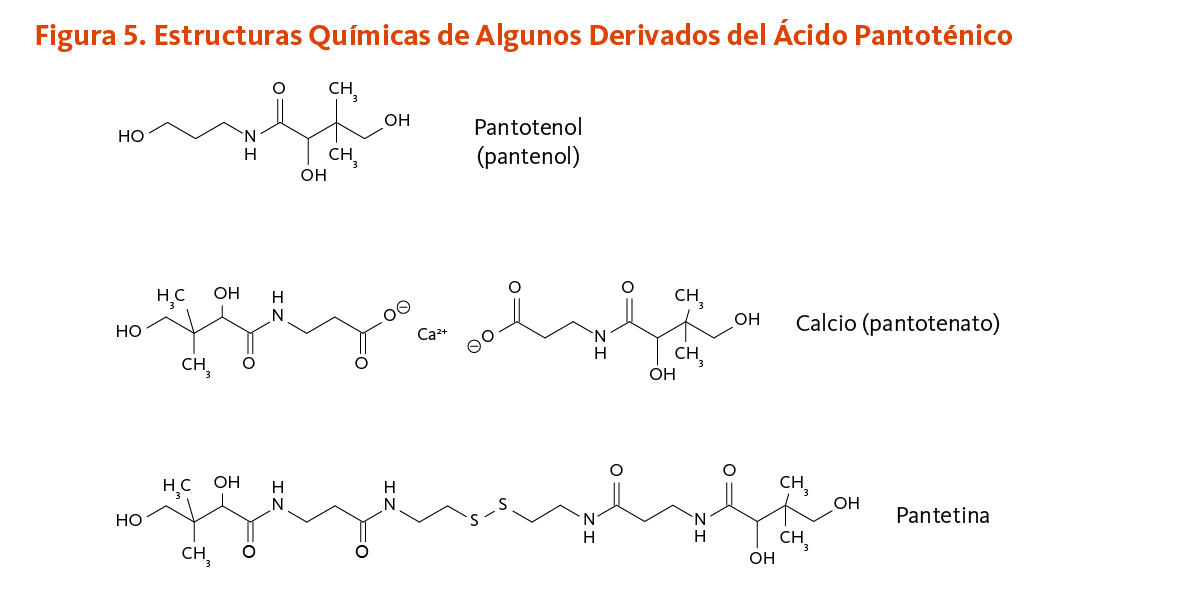
[Figura 5 - Clic para Agrandar]
Encanecimiento del cabello
Ratones deficientes de ácido pantoténico desarrollan irritación cutánea y encanecimiento del pelaje, que es revertido por la administración de ácido pantoténico. No existe evidencia en humanos de que tomando suplementos de ácido pantoténico o usando champú que contengan ácido pantoténico pueda prevenir o restaurar el color del cabello.
Fuentes
Fuentes alimenticias
El ácido pantoténico está disponible en una variedad de alimentos, usualmente como un componente de la coenzima A (CoA) y 4’-fosfopanteteína (véase Figura 1 arriba). Tras ingerirse, la coenzima A dietaría y la fosfopanteteína son hidrolizadas a ácido pantoténico antes de la absorción intestinal (3). El hígado y riñones de animales, pescado, mariscos, carne de cerdo, pollo, yema de huevo, leche, yogurt, legumbres, setas, aguacates, brócoli, y camotes son buenas fuentes de ácido pantoténico. Granos enteros son también buenas fuentes de ácido pantoténico, pero el procesamiento y refinamiento de los granos podría resultar en una pérdida del 35 a 75%. Congelado y enlatado de alimentos resulta en pérdidas similares (16). Encuestas nutricionales nacionales de gran envergadura fallaron en estimar la ingesta de ácido pantoténico, principalmente debido a la escasez de información sobre el contenido de ácido pantoténico de los alimentos (16). Estudios más pequeños estimaron ingestas diarias promedio de ácido pantoténico de entre 4 a 7 mg/día en adultos. La Tabla 2 lista algunas fuentes ricas en ácido pantoténico, junto a su contenido en miligramos (mg). Para mayor información sobre el contenido de nutrientes de los alimentos, revise la base de datos de composición de los alimentos del USDA.
Bacterias intestinales
Las bacterias que normalmente colonizan el colon (intestino grueso) son capaces de sintetizar ácido pantoténico. Un transportador especializado para la ingesta de biotina y ácido pantoténico fue identificado en células cultivadas derivadas del revestimiento del colon, sugiriendo que los humanos pueden ser capaces de absorber el ácido pantoténico y biotina producidos por las bacterias intestinales (27). Sin embargo, el grado al cual la síntesis bacteriana contribuye a la ingesta de ácido pantoténico en humanos es aún desconocido.
Suplementos
Pantotenol y pantotenato
Los suplementos comúnmente contienen pantotenol (pantenol), un alcohol análogo estable del ácido pantoténico, el cual puede ser rápidamente convertido a ácido pantoténico por los humanos. El calcio y el D-pantotenato de sodio, las sales de calcio y sodio del ácido pantoténico, están también disponibles como suplementos.
Pantetina
La pantetina es utilizada como un agente hipocolesterolemiante en Japón y se encuentra disponible en los EE.UU. como un suplemento dietario (29).
Seguridad
Toxicidad
El ácido pantoténico no es conocido por ser toxico en los humanos. El único efecto adverso observado fue diarrea, resultado de ingestas muy altas de 10 a 20 g/día de D-pantotenato de calcio (30). Sin embargo, existe un reporte de caso de derrame pleuropericárdico eosinofílico con riesgo vital en una mujer de edad avanzada, que tomó una combinación de 10 mg/día de biotina y 300 mg/día de ácido pantoténico por dos meses (31). Debido a la falta de reportes de efectos adversos cuándo se estableció la Ingesta Diaria Recomendada (IDR) de ácido pantoténico en 1998, la Junta de Nutrición y Alimentos del Instituto de Medicina no estableció un nivel máximo de ingesta tolerable (NM) para el ácido pantoténico (16). La pantetina es generalmente bien tolerada en dosis de hasta 1,200 mg/día. Sin embargo, los efectos secundarios gastrointestinales, como las náuseas y agruras, han sido reportadas (29). También, las formulaciones tópicas que contienen hasta un 5% de dexpantenol (D-pantenol) han sido usadas de forma segura por hasta un mes. Sin embargo, algunos casos de irritación de la piel, dermatitis de contacto, y eczema han sido reportados con el uso de ungüentos que contienen dexpantenol (32, 33).
Interacción con nutrientes
Altas dosis de ácido pantoténico tienen el potencial de competir con la biotina por la absorción intestinal y celular por el transportador multivitamínico dependiente de sodio humano (hSMVT) (27, 34).
Interacción con fármacos/drogas
Anticonceptivos orales (pastillas anticonceptivas) que contienen estrógeno y progestina pueden incrementar el requerimiento de ácido pantoténico (30). El uso de pantetina en combinación con medicamentos que disminuyen el colesterol llamados estatinas (inhibidores de HMG-CoA reductasa) o con ácido nicotínico (véase el artículo en Niacina) pueden producir efectos aditivos en los lípidos sanguíneos (29).
Recomendación del Instituto Linus Pauling
Más datos son necesarios para definir la cantidad de ácido pantoténico dietario necesario para promover la salud optima o prevenir enfermedades crónicas. El Instituto Linus Pauling apoya la recomendación dada por la Junta de Nutrición y Alimentos de 5 mg/día de ácido pantoténico para adultos. Una dieta variada debería proveer ácido pantoténico suficiente para la mayor parte de la gente. Siguiendo la recomendación del Instituto Linus Pauling de tomar un suplemento multivitamínico-mineral diariamente, que contenga el 100% del Valor Diario (VD) de ácido pantoténico asegurará una ingesta de al menos 5 mg/día.
Adultos mayores (>50 años)
Actualmente existe poca evidencia de que adultos mayores difieren en su ingesta de o en su requerimiento de ácido pantoténico. La mayoría de suplementos multivitamínicos-minerales proveen de por lo menos 5 mg/día de ácido pantoténico.
Autores y Críticos
Escrito en Mayo de 2004 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Abril de 2008 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Abril de 2015 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Julio de 2015 por:
Robert B. Rucker, Ph.D.
Distinguido Profesor Emeritus
Departamento de Nutrición y Escuela de Medicina
Universidad de California, Davis
Traducido al Español en 2017 por:
Silvia Vazquez Lima
Instituto Linus Pauling
Universidad Estatal de Oregon
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Derechos de autoría 2000-2024 Instituto Linus Pauling
Referencias
1. Trumbo PR. Pantothenic acid. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore: Lippincott Williams & Wilkins; 2014:351-357.
2. Martinez DL, Tsuchiya Y, Gout I. Coenzyme A biosynthetic machinery in mammalian cells. Biochem Soc Trans. 2014;42(4):1112-1117. (PubMed)
3. Miller JW, Rucker RB. Pantothenic acid. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Ames: Wiley-Blackwell; 2012:375-390.
4. Bauerly K, Rucker RB. Pantothenic acid. In: Zempleni J, Rucker RB, McCormick DB, Suttie JW, eds. Handbook of vitamins. 4th ed. Boca Raton: CRC Press; 2007:289-314.
5. Takahashi A, Mizusawa K. Posttranslational modifications of proopiomelanocortin in vertebrates and their biological significance. Front Endocrinol (Lausanne). 2013;4:143. (PubMed)
6. Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014;15(8):536-550. (PubMed)
7. Beld J, Sonnenschein EC, Vickery CR, Noel JP, Burkart MD. The phosphopantetheinyl transferases: catalysis of a post-translational modification crucial for life. Nat Prod Rep. 2014;31(1):61-108. (PubMed)
8. Bunkoczi G, Pasta S, Joshi A, et al. Mechanism and substrate recognition of human holo ACP synthase. Chem Biol. 2007;14(11):1243-1253. (PubMed)
9. Donato H, Krupenko NI, Tsybovsky Y, Krupenko SA. 10-formyltetrahydrofolate dehydrogenase requires a 4'-phosphopantetheine prosthetic group for catalysis. J Biol Chem. 2007;282(47):34159-34166. (PubMed)
10. Strickland KC, Hoeferlin LA, Oleinik NV, Krupenko NI, Krupenko SA. Acyl carrier protein-specific 4'-phosphopantetheinyl transferase activates 10-formyltetrahydrofolate dehydrogenase. J Biol Chem. 2010;285(3):1627-1633. (PubMed)
11. Strickland KC, Krupenko NI, Dubard ME, Hu CJ, Tsybovsky Y, Krupenko SA. Enzymatic properties of ALDH1L2, a mitochondrial 10-formyltetrahydrofolate dehydrogenase. Chem Biol Interact. 2011;191(1-3):129-136. (PubMed)
12. Hodges RE, Ohlson MA, Bean WB. Pantothenic acid deficiency in man. J Clin Invest. 1958;37(11):1642-1657. (PubMed)
13. Fry PC, Fox HM, Tao HG. Metabolic response to a pantothenic acid deficient diet in humans. J Nutr Sci Vitaminol (Tokyo). 1976;22(4):339-346. (PubMed)
14. Bender DA. Optimum nutrition: thiamin, biotin and pantothenate. Proc Nutr Soc. 1999;58(2):427-433. (PubMed)
15. Kurian MA, Hayflick SJ. Pantothenate kinase-associated neurodegeneration (PKAN) and PLA2G6-associated neurodegeneration (PLAN): review of two major neurodegeneration with brain iron accumulation (NBIA) phenotypes. Int Rev Neurobiol. 2013;110:49-71. (PubMed)
16. Food and Nutrition Board, Institute of Medicine. Pantothenic acid. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B6, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, D.C.: National Academy Press; 1998:357-373. (National Academy Press)
17. Weimann BI, Hermann D. Studies on wound healing: effects of calcium D-pantothenate on the migration, proliferation and protein synthesis of human dermal fibroblasts in culture. Int J Vitam Nutr Res. 1999;69(2):113-119. (PubMed)
18. Wiederholt T, Heise R, Skazik C, et al. Calcium pantothenate modulates gene expression in proliferating human dermal fibroblasts. Exp Dermatol. 2009;18(11):969-978. (PubMed)
19. Kobayashi D, Kusama M, Onda M, Nakahata N. The effect of pantothenic acid deficiency on keratinocyte proliferation and the synthesis of keratinocyte growth factor and collagen in fibroblasts. J Pharmacol Sci. 2011;115(2):230-234. (PubMed)
20. Heise R, Skazik C, Marquardt Y, et al. Dexpanthenol modulates gene expression in skin wound healing in vivo. Skin Pharmacol Physiol. 2012;25(5):241-248. (PubMed)
21. Vaxman F, Olender S, Lambert A, et al. Effect of pantothenic acid and ascorbic acid supplementation on human skin wound healing process. A double-blind, prospective and randomized trial. Eur Surg Res. 1995;27(3):158-166. (PubMed)
22. Vaxman F, Olender S, Lambert A, Nisand G, Grenier JF. Can the wound healing process be improved by vitamin supplementation? Experimental study on humans. Eur Surg Res. 1996;28(4):306-314. (PubMed)
23. Celebi S, Tepe C, Yelken K, Celik O. Efficacy of dexpanthenol for pediatric post-tonsillectomy pain and wound healing. Ann Otol Rhinol Laryngol. 2013;122(7):464-467. (PubMed)
24. Coronel F, Tornero F, Torrente J, et al. Treatment of hyperlipemia in diabetic patients on dialysis with a physiological substance. Am J Nephrol. 1991;11(1):32-36. (PubMed)
25. Gaddi A, Descovich GC, Noseda G, et al. Controlled evaluation of pantethine, a natural hypolipidemic compound, in patients with different forms of hyperlipoproteinemia. Atherosclerosis. 1984;50(1):73-83. (PubMed)
26. Rumberger JA, Napolitano J, Azumano I, Kamiya T, Evans M. Pantethine, a derivative of vitamin B(5) used as a nutritional supplement, favorably alters low-density lipoprotein cholesterol metabolism in low- to moderate-cardiovascular risk North American subjects: a triple-blinded placebo and diet-controlled investigation. Nutr Res. 2011;31(8):608-615. (PubMed)
27. Said HM, Ortiz A, McCloud E, Dyer D, Moyer MP, Rubin S. Biotin uptake by human colonic epithelial NCM460 cells: a carrier-mediated process shared with pantothenic acid. Am J Physiol. 1998;275(5 Pt 1):C1365-1371. (PubMed)
28. Said HM. Intestinal absorption of water-soluble vitamins in health and disease. Biochem J. 2011;437(3):357-372. (PubMed)
29. Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. 2nd ed. Montvale: Thomson Reuters; 2008.
30. Flodin N. Pharmacology of micronutrients. New York: Alan R. Liss, Inc.; 1988.
31. Debourdeau PM, Djezzar S, Estival JL, Zammit CM, Richard RC, Castot AC. Life-threatening eosinophilic pleuropericardial effusion related to vitamins B5 and H. Ann Pharmacother. 2001;35(4):424-426. (PubMed)
32. Herbst RA, Uter W, Pirker C, Geier J, Frosch PJ. Allergic and non-allergic periorbital dermatitis: patch test results of the Information Network of the Departments of Dermatology during a 5-year period. Contact Dermatitis. 2004;51(1):13-19. (PubMed)
33. Schmuth M, Wimmer MA, Hofer S, et al. Topical corticosteroid therapy for acute radiation dermatitis: a prospective, randomized, double-blind study. Br J Dermatol. 2002;146(6):983-991. (PubMed)
34. Chirapu SR, Rotter CJ, Miller EL, Varma MV, Dow RL, Finn MG. High specificity in response of the sodium-dependent multivitamin transporter to derivatives of pantothenic acid. Curr Top Med Chem. 2013;13(7):837-842. (PubMed)
Biotina
Contenido
Resumen
- La biotina hidrosoluble es un cofactor esencial para las enzimas en metabolismo intermediario y un regulador clave de la expresión de genes. (Más información)
- Ambos la nutrición parenteral desprovista de biotina y el consumo prolongado de la clara de huevo cruda han sido asociados con síntomas de la deficiencia de biotina franca, incluyendo perdida del cabello, dermatitis, y erupción cutánea, ataxia, convulsiones, y otras disfunciones neurológicas. (Más información)
- La deficiencia de biotinidasa es un desorden hereditario que perjudica la absorción y reciclaje de la biotina, resultando en una deficiencia de biotina secundaria. (Más información)
- La ingesta adecuada recomendada (IA) de biotina está establecida en 30 microgramos (μg)/día en los adultos. Los requerimientos de biotina son más propensos a aumentar durante el embarazo y el periodo de lactancia. (Más información)
- Los estudios en animales han mostrado que la suficiencia de biotina es esencial para el desarrollo fetal normal. Si la deficiencia de biotina marginal durante el embarazo aumenta el riesgo de anomalías congénitas en los humanos es un área actualmente de preocupación e investigación. (Más información)
- La biotina es usada en el tratamiento de un desorden hereditario del transporte de tiamina, llamado enfermedad de los ganglios basales sensible a la biotina, y está actualmente siendo probado en ensayos para limitar o revertir las discapacidades funcionales en personas con esclerosis múltiple. (Más información)
- La evidencia definitiva que establece si la suplementación con biotina mejora la homeostasis de la glucosa y lípidos en individuos con diabetes mellitus tipo 2 es actualmente escasa, pero observaciones sugestivas han sido publicadas. (Más información)
- La biotina no puede ser sintetizada por las células mamíferas y debe obtenerse de fuentes exógenas. La biotina se encuentra ampliamente en los alimentos, y buenas fuentes dietarías incluyendo la yema de huevo, hígado, cereales de grano entero, y algunos vegetales. (Más información)
- La terapia con anticonvulsivos (anti-convulsiones) a largo plazo puede incrementar el requerimiento dietario de biotina debido a que los anticonvulsivos pueden interferir con la absorción intestinal y reabsorción renal de la biotina y probablemente también incrementen la degradación de la biotina a metabolitos inactivos. (Más información)
La biotina es una vitamina hidrosoluble que es generalmente clasificada como una vitamina del complejo B. Después de su descubrimiento inicial en 1927, se necesitaron cerca de 40 años de investigación para inequívocamente establecer a la biotina como una vitamina (1). La biotina es requerida por todos los organismos pero puede ser sintetizada por algunas cepas de bacterias, levaduras, hongos, algas, y algunas especies de plantas (2).
Función
Biotinilación
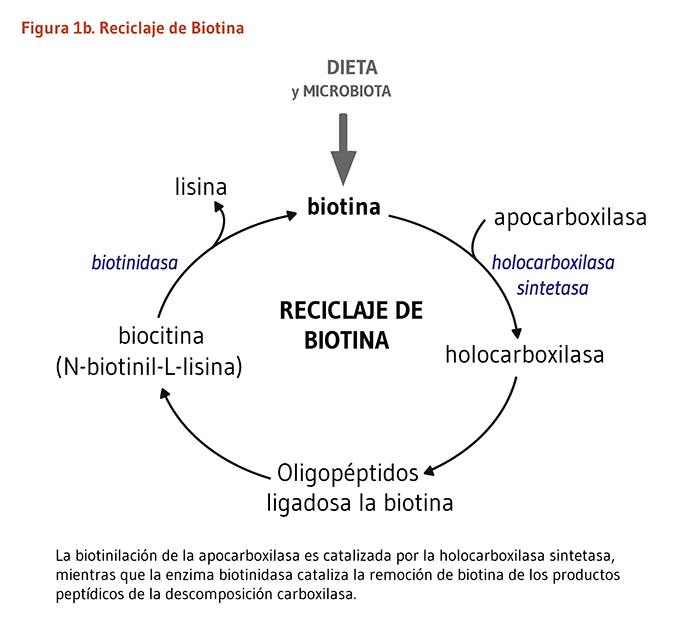
La biotina funciona como un cofactor unido covalentemente requerido para la actividad biológica de las cinco enzimas carboxilasas dependientes de la biotina conocidas de los mamíferos (véase abajo). Tal cofactor no-proteico es conocido como un “grupo prostético.” El enlace covalente de la biotina a la apocarboxilasa (es decir, una carboxilasa catalíticamente inactiva) es catalizado por la enzima, holocarboxilasa sintetasa (HCS). El término “biotinilación” se refiere a la adición covalente de biotina a cualquier molécula, incluyendo apocarboxilasas e histonas. La HCS cataliza la biotinilación postraduccional del grupo épsilon amino de un residuo de lisina en el sitio activo de cada apocarboxilasa, convirtiendo a la apocarboxilasa inactiva en una holocarboxilasa completamente activa (Figura 1a). Particularmente los residuos de lisina dentro del extremo N-terminal de histonas específicas, los cuales ayudan a empaquetar el ADN en el núcleo eucariótico, pueden también ser biotinilados (3). La biotinidasa es la enzima que cataliza la liberación de la biotina proveniente de las histonas biotiniladas y de los productos de péptidos de la degradación de la holocarboxilasa (Figura 1b).

Cofactor de enzimas
Cinco carboxilasas mamíferas que catalizan reacciones metabólicas esenciales:
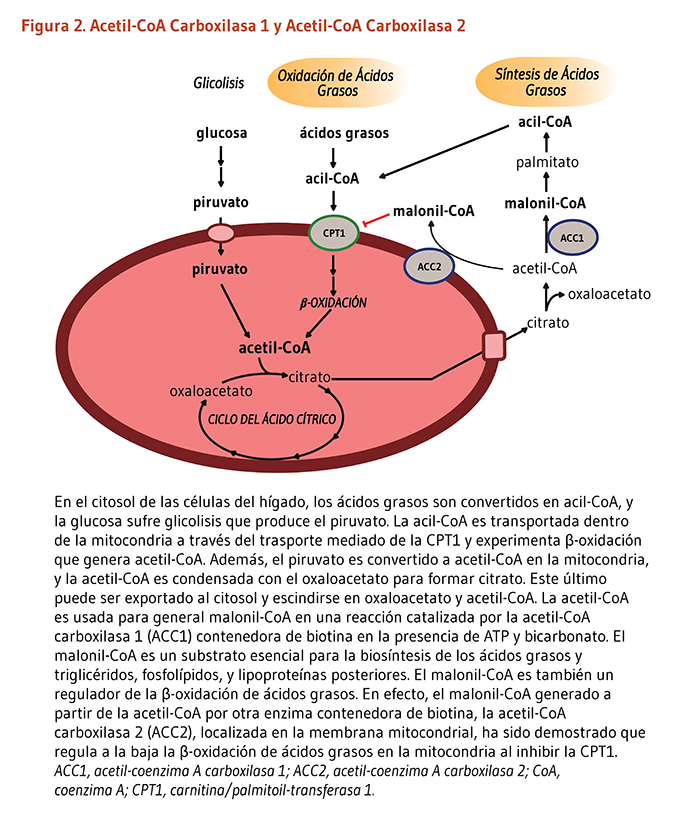
- Ambas la acetil-Coenzima A (CoA) carboxilasa 1 (ACC1) y la acetil-CoA carboxilasa 2 (ACC2) catalizan la conversión de acetil-CoA a malonil-CoA usando bicarbonato y ATP; el malonil CoA generado vía ACC1 es un sustrato limitante para la síntesis de ácidos grasos en el citosol, y el malonil CoA generado vía ACC2 inhibe la CPT1, una enzima de la membrana mitocondrial externa importante en la oxidación de ácidos grasos (Figura 2). La ACC1 se encuentra en todos los tejidos y es particularmente activa en tejidos lipogenicos (es decir, hígado, tejido adiposo blanco, y gandulas mamarias), el corazón, e islotes pancreáticos. La ACC2 es especialmente abundante en el musculo esquelético y corazón (4).
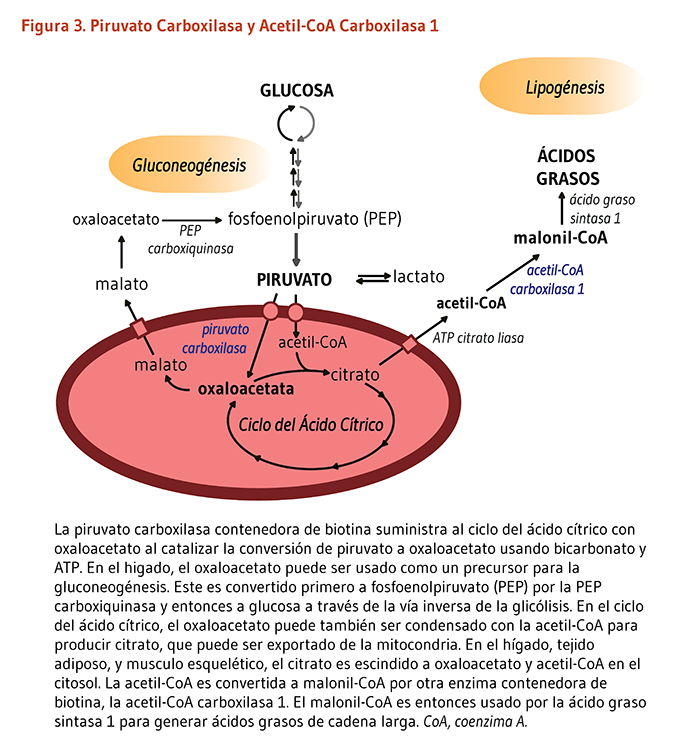
- Piruvato carboxilasa es una enzima critica en la gluconeogénesis — la formación de glucosa proveniente de fuentes distintas de los carbohidratos, como el piruvato, lactato, glicerol, y los aminoácidos glucogénicos. La piruvato carboxilasa cataliza la incorporación dependiente de ATP de bicarbonato en piruvato, produciendo oxaloacetato; de ahí que la piruvato carboxilasa es anaplerótica para el ciclo del ácido cítrico (Figura 3). El oxaloacetato puede entonces ser convertido a fosfoenolpiruvato y eventualmente a glucosa.
- Metilcrotonil-CoA carboxilasa cataliza un paso esencial en el catabolismo de la leucina, un aminoácido esencial de cadena ramificada. Esta enzima que contiene biotina cataliza la producción de 3-metilglutaconil-CoA a partir del metilcrotonil-CoA (Figura 4a).
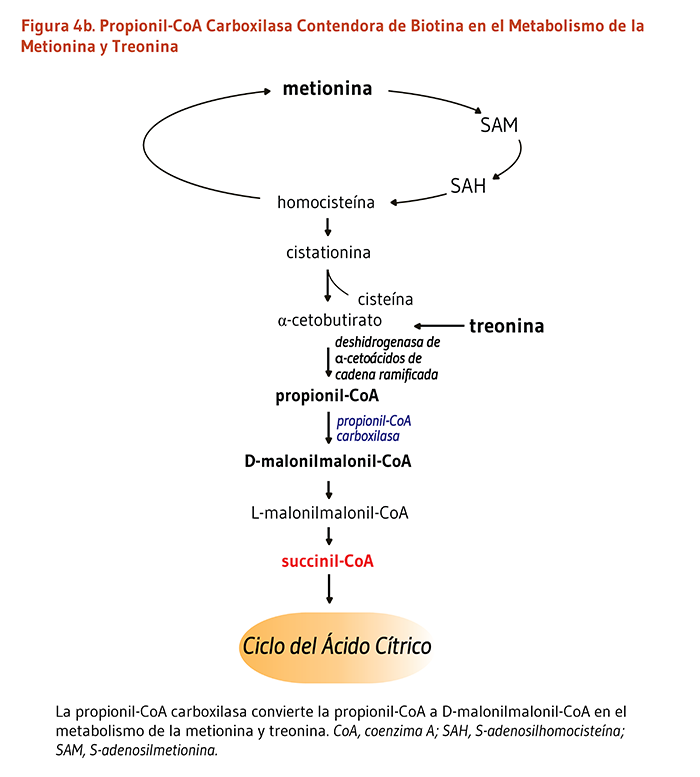
- Propionil-CoA carboxilasa produce D-malonilmalonil-CoA a partir del propionil-CoA, un producto derivado de la β-oxidación de ácidos grasos con un número impar de átomos de carbono (Figura 4a). La conversión de propionil-CoA a D-malonilmalonil-CoA es también requerida en las vías catabólicas de dos aminoácidos de cadena ramificada (isoleucina y valina), de la metionina, la treonina, y la cadena lateral del colesterol (Figura 4a y 4b).

Regulación de la estructura de la cromatina y la expresión de genes
En los núcleos eucariotas, el ADN es empaquetado en estructuras compactas para formar los nucleosomas — unidades fundamentales de la cromatina. Cada nucleosoma está compuesto de 147 pares de bases de ADN envueltos alrededor de ocho histonas (emparejamiento de histonas: H2A, H2B, H3, y H4). Otra histona, llamada H1 ligador, está localizada en la en la superficie exterior de cada nucleosoma y sirve como un ancla para fijar el ADN alrededor del núcleo de la histona. El embalaje compacto de la cromatina debe estar relajado de vez en cuando para permitir procesos biológicos, tales como la replicación y la transcripción del ADN. Modificaciones químicas del ADN y las histonas afectan el plegamiento de la cromatina, incrementando o reduciendo la accesibilidad del ADN a factores involucrados en los procesos previamente mencionados. Juntos con la metilación del ADN, un cierto número de modificaciones químicas dentro la dentro del extremo N-terminal de las histonas del núcleo modifica sus cargas eléctricas y estructuras, de este modo cambiando la conformación de la cromatina y la actividad transcripcional de los genes.
Las diversas modificaciones de las colas de las histonas, incluyendo la acetilación, metilación, fosforilación, ubiquitinación, SUMOilación, ADP-ribosilación, carbonilación, deiminación, hidroxilación, y la biotinilación, tiene diferentes funciones regulatorias. Varios sitios de biotinilación han sido identificados en las histonas H2A, H3, y H4 (5). Entre ellas, la biotinilación de la histona H4 en la lisina (K) 12 (señalado H4K12bio) parece ser enriquecida en la heterocromatina, una cromatina estrechamente condensada asociada con regiones de repetición en los (peri)centrómeros y telómeros, y con elementos transponibles conocidos como repeticiones terminales largas (3). Además, las marcas de biotinilación se co-localizan con marcas de la represión de genes bien conocidas como la lisina 9 metilada en la histona H3 (H3K9me) en la cromatina transcripcionalmente competente (6). Por ejemplo, H4K12bio se puede encontrar en el promotor del gen SLC5A6 que codifica para el transportador de la mediación de la asimilación de biotina en las células, el transportador multivitamínico dependiente del sodio humano (hSMVT). Cuando la biotina es abundante, la HCS puede biotinilar las histonas H4 en el promotor del SLC5A6, el cual detiene la síntesis del hSMVT y reduce la asimilación de biotina. Inversamente, en las células deficientes de biotina, las marcas de biotinilación en el promotor del LC5A6 son removidas de tal manera que se puede producir la expresión de genes, permitiendo la síntesis del hSMVT e incrementando posteriormente la asimilación de biotina (7).
Deficiencia
Aunque la deficiencia de biotina evidente es bastante rara, el requerimiento humano de biotina dietaría ha sido demostrado en tres situaciones diferentes: alimentación intravenosa (parenteral) prolongada sin suplementación de biotina, infantes alimentados con una formula elemental carente de biotina, y el consumo de clara de huevo cruda por un período prolongado (de varias semanas a varios años) (8). La clara de huevo cruda contiene una proteína antimicrobiana conocida como avidina que puede unirse a la biotina y prevenir su absorción. El cocinar la clara de huevo desnaturaliza la avidina, haciéndola susceptible a la digestión y por lo tanto, incapaz de impedir la absorción de la biotina dietaría (5).
Signos y síntomas de la deficiencia de biotina
Los signos de la deficiencia evidente de biotina incluyen la caída de cabello (alopecia) y una erupción escamosa rojiza alrededor de los ojos, nariz, boca, y área genital. Síntomas neurológicos en adultos han incluido depresión, letargo, alucinaciones, y entumecimiento y cosquilleo de las extremidades, ataxia, y convulsiones. Las erupciones faciales características, junto con la distribución inusual de la grasa facial, han sido llamados "rasgos carentes de biotina" por algunos investigadores (1). Los individuos con desordenes hereditarios del metabolismo de la biotina (véase Trastornos metabólicos congénitos) que resultan en la deficiencia de biotina funcional, a menudo tienen hallazgos físicos similares, así como también convulsiones y evidencia de una función alterada del sistema inmune y una susceptibilidad aumentada a infecciones bacterianas y fúngicas (9, 10).
Factores de riesgo de la deficiencia de biotina
Aparte del consumo prolongado de la clara de huevo cruda o el soporte nutricional intravenoso total carente de biotina, otras condiciones pueden incrementar el riesgo de depleción de biotina. El fumar ha sido asociado con un incremento del catabolismo de la biotina (11). Las células que se dividen rápidamente del feto en desarrollo requieren biotina para la síntesis de carboxilasas esenciales y para la biotinilación de las histonas; por consiguiente, el requerimiento de biotina es propenso a incrementar durante el embarazo. Investigación sugiere que un número substancial de mujeres desarrollan una deficiencia marginal o subclínica durante el embarazo normal (véase también Prevención de Enfermedades) (8, 12, 13). Además, ciertos tipos de enfermedades hepáticas pueden disminuir la actividad de la biotinidasa y en teoría incrementar el requerimiento de biotina. Un estudio de 62 niños con enfermedad crónica del hígado y 27 controles sanos encontró que la actividad de la biotinidasa del suero se encontraba anormalmente baja en aquellos con una función severamente deteriorada debido a la cirrosis (14). Sin embargo, este estudio no aporto evidencia de la deficiencia de biotina. Además, los medicamentos anticonvulsivos, usados para prevenir convulsiones en individuos con epilepsia, incrementaron el riesgo de una depleción de biotina (para más información acerca de la biotina y los anticonvulsivos, véase Interacción con drogas/fármacos).
Trastornos metabólicos congénitos
Deficiencia de biotinidasa
Existen varias maneras en que el desorden hereditario, la deficiencia de biotinidasa, conduce a una deficiencia secundaria de biotina. La absorción intestinal es disminuida porque una escases de biotinidasa previene la liberación de biotina proveniente de las proteínas dietarías (15). El reciclaje de nuestra propia biotina unida a las carboxilasas e histonas es también alterado, y la perdida urinaria de biocitina (N-biotinil-lisina) y biotina es incrementada (véase Figura 1 arriba) (5). La deficiencia de biotinidasa uniformemente responde a la biotina suplementaria. La suplementación oral con hasta 5 a 10 miligramos (mg) de biotina diariamente es a veces requerida, aunque dosis más pequeñas son frecuentemente suficientes (revisado en 16).
Deficiencia de holocarboxilasa sintetasa (HCS)
Algunas formas de la deficiencia de HCS responden a la suplementación con dosis farmacológicas de biotina. La deficiencia de HCS resulta en la disminución de la formación de todas las holocarboxilasas a concentraciones sanguíneas fisiológicas de la biotina; así, la suplementación de biotina de alta dosis (10-80 mg de biotina diaria es requerida (10).
La prognosis de estos dos desordenes es usualmente buena si la terapia con biotina es introducida tempranamente (infancia o niñez) y se continua de por vida (10).
Deficiencia del transporte de biotina
Ha habido un reporte de caso de un niño con deficiencia del transporte de biotina que respondió a una suplementación de altas dosis de biotina (17). Debe notarse que la presencia de un transportador multivitamínico dependiente del sodio humano (hSMVT) defectuoso fue descartado como la causa de la deficiencia del transporte de biotina.
Fenilcetonuria (FCU)
Las concentraciones anormalmente elevadas del aminoacido, fenilalanina, en la sangre de individuos afectados con FCU puede inhibir la actividad de la biotinidasa. Schulpis et al. especuló que la dermatitis seborreica asociada con la actividad baja de la biotinidasa en estos pacientes se resolvería a partir del cumplimiento con una dieta especial baja en proteína pero no con la suplementación de biotina (18).
Marcadores del estatus de la biotina
Cuatro medidas de la deficiencia de biotina marginal han sido validados como indicadores del estatus de la biotina: (1) la excreción urinaria reducida de la biotina y algunos de sus catabolitos; (2) la alta excreción urinaria de un ácido orgánico, ácido 3-hidroxiisovalérico, y su derivado, ácido carnitil-3-hidroxiisovalérico, ambos de los cuales se refleja la actividad disminuida de la metilcrotonil-CoA carboxilasa dependiente de la biotina; (3) la actividad reducida de la propionil-CoA carboxilasa en los linfocitos sanguíneos periféricos (5); y (4) los niveles reducidos de holo-metilcrotonil-CoA carboxilasa y holo-propionil-CoA carboxilasa en los linfocitos — los indicadores más fiables del estatus de biotina (19). Estos marcadores han sido solo validados en hombres y mujeres no embarazadas, y podrían no reflejar precisamente el estatus de la biotina en mujeres embarazadas y mujeres que lactan (12).
La Ingesta Adecuada (IA)
La suficiente evidencia científica es escaza para estimar el requerimiento dietario para la biotina; por lo tanto no se ha establecido una Ingesta Diaria Recomendada (IDR) para la biotina. En su lugar, la Junta de Nutrición y Alimentos (JNA) del Instituto de Medicina (IOM) estableció recomendaciones para una Ingesta Adecuada (IA; Tabla 1). La IA para los adultos (30 microgramos [μg]/día) se extrapolo de la IA para infantes exclusivamente alimentados con leche humana y se espera que sobreestime el requerimiento dietario para la biotina en los adultos. Las ingestas dietarías de adultos generalmente saludables se han estimado son de entre 40-60 μg/día de biotina (1). El requerimiento para la biotina en el embarazo puede ser incrementada (20).
| Etapa de la Vida | Edad | Machos (μg/día) | Hembras (μg/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 5 | 5 |
| Infantes | 7-12 meses | 6 | 6 |
| Niños | 1-3 años | 8 | 8 |
| Niños | 4-8 años | 12 | 12 |
| Niños | 9-13 años | 20 | 20 |
| Adolescentes | 14-18 años | 25 | 25 |
| Adultos | 19 años y más | 30 | 30 |
| Embarazo | Todas las edades | - | 30 |
| Período de lactancia | Todas las edades | - | 35 |
Prevención de Enfermedades
Anomalías congénitas
Investigación actual indica que por lo menos un tercio de las mujeres desarrollan deficiencia de biotina marginal durante el embarazo (8), pero no se sabe si esto podría incrementar el riesgo de anomalías congénitas. Estudios basados en la observación pequeños en mujeres embarazadas han reportado una excreción urinaria anormalmente alta de ácido 3-hidroxiisovalérico tanto temprano como tarde en el embarazo, sugiriendo una actividad disminuida de metilcrotonil-CoA carboxilasa dependiente de la biotina (21, 22). En un estudio de intervención, aleatorio, simple ciego en 26 mujeres embarazadas, la suplementación con 300 μg/día de biotina por dos semanas limitó la excreción de ácido 3-hidroxiisovalérico en comparación al placebo, confirmando que la excreción incrementada de ácido 3-hidroxiisovalérico en efecto reflejó la deficiencia de biotina marginal en el embarazo (23). Un estudio de corte transversal pequeño en 22 mujeres embarazadas reportó una incidencia de actividad baja de propionil-CoA carboxilasa linfocitaria (otro marcador de la deficiencia de biotina) mayor al 80% (13). Aunque el nivel de deficiencia de biotina no está asociado con signos manifestados de deficiencia en mujeres embarazadas, tales observaciones son fuentes de preocupación porque se ha demostrado que la deficiencia subclínica de biotina causa paladar hendido e hipoplasia de extremidades en varias especies de animales (revisado en 13). Además, se ha encontrado que la depleción de biotina suprime la expresión de carboxilasas dependientes de la biotina, remueve las marcas de biotina de las histonas, y disminuye la proliferación en células mesenquimales palatales embrionarias humanas en cultivo (24). La actividad alterada de las carboxilasas puede resultar en alteraciones en el metabolismo de los lípidos, el cual ha sido ligado al paladar hendido y anormalidades del esqueleto en animales. Además, la deficiencia de biotina que conduce a la reducción de la biotinilación de histonas en específicos loci del genoma podría posiblemente incrementar la inestabilidad genómica y resultar en anomalías cromosómicas y malformaciones fetales (13).
Mientras que a las mujeres embarazadas se les aconseja consumir ácido fólico suplementario antes de y durante el embarazo para prevenir defectos de tubo neural (véase Folato), sería también prudente asegurar la ingesta adecuada de biotina durante el embarazo. La IA actual para las mujeres embarazadas es de 30 μg/dia de biotina, y toxicidad alguna no ha sido reportada a este nivel de ingesta (véase Seguridad).
Tratamiento de Enfermedades
Enfermedad de los ganglios basales sensible a la biotina
La enfermedad de los ganglios basales sensible a la biotina, también llamada síndrome-2 de disfunción del metabolismo de la tiamina es causada por mutaciones en el gen que codifica para el transportador de tiamina tipo 2 (THTR-2). Las características clínicas aparecen alrededor de los tres a cuatro años de edad e incluyen encefalopatía subaguda (confusión, somnolencia y alteración del nivel de conciencia), ataxia, y convulsiones. Un estudio retrospectivo de 18 individuos afectados provenientes de la misma familia o de la misma tribu en Arabia Saudita fue recientemente conducido. Los datos mostraron que la monoterapia con biotina (5-10 mg/kg/día) eficientemente suprimió las manifestaciones clínicas de la enfermedad, aunque un tercio de los pacientes sufrieron de crisis agudas recurrentes. A menudo asociadas con malos resultados, las crisis agudas no fueron observadas después de que la suplementación con tiamina iniciara (300-400 mg/día) y durante un periodo de seguimiento de cinco años. Un diagnóstico temprano y el tratamiento inmediato con biotina y tiamina llevaron a resultados positivos (25). El mecanismo para el efecto beneficial de la suplementación con biotina aún no ha sido dilucidado.
Esclerosis múltiple
La esclerosis múltiple (EM) es una enfermedad autoinmune caracterizada por daños progresivos a la vaina de mielina que rodea las fibras nerviosas (axones) y perdida neuronal en el cerebro y medula espinal de los individuos afectados. La progresión de las discapacidades neurológicas en los pacientes con EM es a menudo evaluado por la Escala Expandida del Estado de Discapacidad (EDSS) con puntuaciones de entre 1 a 10, partiendo con señales mínimas de la disfunción motora (puntuación de 1) hasta muerte por EM (puntuación de 10). La deficiencia de ATP debido a la disfunción mitocondrial y el estrés oxidativo incrementado pueden ser parcialmente responsables por la degeneración progresiva de las neuronas en las EM (26). Dado su papel en el metabolismo intermediario y en la síntesis de ácidos grasos (requerido para la formación de mielina) (véase Función), se ha postulado que la biotina podría ejercer efectos beneficiosos que limitarían o revertirían alteraciones funcionales asociadas con la EM (26).
Un estudio piloto no aleatorio, no controlado en 23 pacientes con EM progresiva encontró que altas dosis de biotina (100-600 mg/día) se asociaban con mejoras clínicas sostenidas en 5 (de 5) pacientes con perdida visual progresiva y en 16 (de 18) pacientes con parálisis parcial de las extremidades después de un periodo medio de seguimiento de tres meses tras el inicio del tratamiento (27). Además los resultados preliminares del ensayo multicéntrico, aleatorio, controlado con placebo en 154 sujetos con EM progresiva indicó que 13 de 103 pacientes que recibieron aleatoriamente biotina oral diaria (300 mg) por 48 semanas lograron un criterio de valoración funcional compuesto que incluyo una disminución en las puntuaciones de la EDSS. En comparación, ninguno de los 51 pacientes asignados aleatoriamente al grupo de placebo mostró mejoramientos clínicos significativos (28). Dos ensayos en proceso están evaluando el efecto de la suplementación con altas dosis de biotina en el tratamiento de la EM (véase ensayos NCT02220933 y NCT02220244 en www.clinicaltrials.gov).
Diabetes mellitus
Se ha demostrado que la deficiencia de biotina manifiesta afecta la utilización de glucosa en ratones (29) y causa hipoglicemia fatal en pollos. La deficiencia de biotina manifiesta probablemente también causa anormalidades en la regulación de glucosa en humanos (véase Función). Un estudio temprano en humanos reportó concentraciones más bajas de biotina del suero en 43 pacientes con diabetes mellitus tipo 2 en comparación con 64 sujetos de control no diabéticos, como también una relación inversa entre las concentraciones de glucosa en ayunas y biotina sanguíneas (30). En un estudio de intervención aleatorio, controlado con placebo en 28 pacientes con diabetes tipo 2, la suplementación diaria con 9 miligramos (mg) de biotina por un mes resultó en una disminución media del 45% en las concentraciones de glucosa sanguínea en ayunas (30). A pesar de todo, otro estudio pequeño en 10 pacientes con diabetes tipo 2 y 7 controles no diabéticos no encontró efecto alguno de la suplementación con biotina (15 mg/día) por 28 días en las concentraciones de glucosa sanguínea en ayunas en ambos grupos (31). Un estudio doble ciego, controlado con placebo más reciente por el mismo grupo de investigación mostró que el mismo régimen de biotina disminuyó las concentraciones de triglicéridos del plasma en tanto los pacientes diabéticos como los no diabéticos con hipertrigliceridemia (32). En este estudio, la administración de biotina no afectó las concentraciones de glucosa sanguínea en ambos grupos de pacientes. Adicionalmente, unos pocos estudios han demostrado que la co-suplementación con biotina y picolinato de cromo puede ser una terapia adjunta beneficiosa para pacientes con diabetes tipo 2 (33-36). Sin embargo, se ha demostrado que la administración de picolinato de cromo por si sola mejora el control glicémico en sujetos diabéticos (véase el artículo en Cromo) (37).
Como un cofactor de carboxilasas requerido para la síntesis de ácidos grasos, la biotina puede incrementar la utilización de glucosa para la síntesis grasa. Se ha encontrado que la biotina estimula la glucoquinasa, una enzima hepática que incrementa la síntesis de glicógeno, la forma almacenada de la glucosa. La biotina también parece desencadenar la secreción de insulina en el páncreas de ratas y mejorar la homeostasis de la glucosa (38). Sin embargo, se esperaría la actividad reducida de la ACC1 y la ACC2 para reducir la síntesis de ácidos grasos e incrementar la oxidación de ácidos grasos, respectivamente. No es de sorprenderse que actualmente no está claro si las dosis farmacológicas de biotina podrían beneficiar el manejo de la hiperglicemia en pacientes con intolerancia a la glucosa. Por otra parte, queda por ser demostrado si la biotina suplementaria disminuye el riesgo de complicaciones cardiovasculares en pacientes diabéticos al reducir los triglicéridos y el colesterol LDL del suero (32-34).
Uñas quebradizas (onicorrexis)
El descubrimiento de que los suplementos de biotina eran efectivos en el tratamiento de anormalidades de las pezuñas en animales ungulados, sugirió que la biotina podría también ser beneficiosa en el fortalecimiento de uñas quebradizas en humanos (39-41). Tres ensayos no controlados que examinaron los efectos de la suplementación con biotina (2.5 mg/día por varios meses) en mujeres con uñas quebradizas han sido publicados (42-44). En dos de los ensayos, se reportó evidencia subjetiva de mejora clínica en 67%-91% de las participantes disponibles para seguimiento al final del período de tratamiento (42, 43). Un ensayo que utilizó microscopía electrónica de barrido para evaluar la fragilidad de las uñas reportó menos resquebrajamiento de las uñas y un aumento del grosor del 25% de la placa ungueal en pacientes suplementados con biotina por 6 a 15 meses (44). Se encontró también que la suplementación con biotina (5 mg/día) era efectiva en controlar el cabello rebelde y resquebrajamiento de uñas en dos niños pequeños con síndrome del cabello impeinable hereditario (45). Aunque evidencia preliminar sugiere que la suplementación con biotina puede ayudar a fortalecer las uñas frágiles, ensayos controlados con placebo de mayor tamaño son necesarios para evaluar la eficacia de la suplementación con altas dosis de biotina para el tratamiento de uñas quebradizas.
Pérdida del cabello (alopecia)
Se encontró que la administración de biotina revierte la alopecia en niños tratados con el anticonvulsivo, ácido valproico (véase Interacción con drogas/fármacos). Sin embargo, aunque la pérdida de cabello es un síntoma de la deficiencia severa de biotina (véase Deficiencia), no existen estudios científicos publicados que apoyen la aseveración de que los suplementos de altas dosis de biotina son efectivos en la prevención o tratamiento de la pérdida del cabello en hombres y mujeres (46).
Fuentes
Fuentes alimenticias
La biotina se encuentra en muchos alimentos, ya sea como la forma libre que es directamente tomado por los enterocitos o como biotina unida a las proteínas dietarías. La yema de huevo, hígado, y las levaduras son fuentes ricas de biotina. Estimados de las ingestas diarias promedio de biotina provenientes de estudios pequeños oscilaron entre 40 a 60 microgramos (μg) por día en adultos (1). Sin embargo, cuestionarios nutricionales nacionales estadounidenses no han aun sido capaces de estimar la ingesta de biotina debido a la escasez y la falta de fiabilidad de los datos con respecto al contenido de biotina de los alimentos. Tablas de composición de alimentos para la biotina están incompletas de manera que la ingesta diaria no puede ser estimada de forma fiable en los seres humanos. Un estudio por Staggs et al. (47) empleó un método de cromatografía líquida de alta resolución en lugar de bioensayos (48) y reportó un contenido de biotina relativamente diferente para algunos de los alimentos seleccionados. La Tabla 2 lista algunas fuentes alimenticias de biotina, junto con su contenido en μg.
| Alimento | Porción | Biotina (μg) |
|---|---|---|
| Levadura | 1 paquete (7 gramos) | 1.4-14 |
| Pan, trigo entero | 1 rebanada | 0.02-6 |
| Huevo, cocido | 1 grande | 13-25 |
| Queso, cheddar | 1 onza | 0.4-2 |
| Hígado, cocido | 3 onzas* | 27-35 |
| Cerdo, cocido | 3 onzas* | 2-4 |
| Salmón, cooked | 3 ounces* | 4-5 |
| Aguacate | 1 entero | 2-6 |
| Frambuesas | 1 taza | 0.2-2 |
| Coliflor, cruda | 1 taza | 0.2-4 |
| *Una porción de tres onzas de carnes es del tamaño de una baraja de cartas. | ||
Síntesis bacteriana
La mayoría de las bacterias que normalmente colonizan el intestino delgado y el intestino grueso (colon) sintetizan biotina (49). Permanece sin conocerse si la biotina es liberada y absorbida por los humanos en cantidades significativas. La asimilación de biotina libre en las células intestinales a través del transportador multivitamínico dependiente del sodio humano (hSMVT) ha sido identificada en células en cultivo derivadas del revestimiento del intestino delgado y colon (50), sugiriendo que los humanos pueden ser capaces de absorber biotina producida por bacterias entéricas — un fenómeno documentado en porcinos.
Seguridad
Toxicidad
No se sabe que la biotina sea toxica. En las personas sin desordenes del metabolismo de biotina, dosis de hasta 5 mg/día por dos años no fueron asociados con efectos adversos (52). La suplementación oral con biotina ha sido bien tolerada en dosis de hasta 200 mg/día (casi 7,000 veces la IA) en personas con desordenes hereditarios del metabolismo de la biotina (1). Se encontró también que la suplementación diaria con una formulación altamente concentrada de biotina (100-600 mg) fue bien tolerada en individuos con esclerosis múltiple progresiva (27, 28). Sin embargo, hay un reporte de caso de derrame pleuropericárdico eosinofílico que pone en peligro la vida en una mujer de edad avanzada que tomó una combinación de 10 mg/día de biotina y 300 mg/día de ácido pantoténico (vitamina B5) por dos meses (53). Debido a que reportes de eventos adversos fueron escasos cuando las Ingestas Dietéticas de Referencia (IDR) para la biotina fueron establecidas en 1998, el Instituto de Medicina no estableció un nivel máximo de ingesta tolerable (NM) para la biotina (1).
Interacción con nutrientes
Grandes dosis de ácido pantoténico (vitamina B5) tienen el potencial de competir con la biotina por la asimilación intestinal y celular por el transportador multivitamínico dependiente del sodio humano (hSMVT) (54, 55). La biotina también comparte el hSMVT con el ácido α-lipoico (56). Se ha encontrado que las dosis farmacológicas (muy altas) de ácido α-lipoico disminuyen la actividad de carboxilasas dependientes de la biotina en ratas, pero tal efecto no ha sido demostrado en humanos (57).
Interacción con drogas
Individuos en terapia de anticonvulsivos (anti-convulsiones) a largo plazo según se informa, han reducido las concentraciones sanguíneas de biotina así como también incrementaron la excreción urinaria de ácidos orgánicos (p. ej. ácido 3-hidroxiisovalérico) que indican un descenso en la actividad carboxilasa (véase Marcadores del estatus de la biotina) (5). Los mecanismos potenciales de la depleción de la biotina por los anticonvulsivos, primidona (Mysoline), fenitoína (Dilantin), y carbamazepina (Carbatrol, Tegretol), incluyen la inhibición de la absorción intestinal y la reabsorción renal de la biotina, como también un catabolismo incrementado de la biotina (51). El uso del anticonvulsivo ácido valproico en niños ha resultado en la pérdida de cabello revertida por la suplementación con biotina (58-61). El tratamiento a largo plazo con drogas sulfonamidas antibacterianas (sulfa) u otros antibióticos puede disminuir la síntesis bacteriana de biotina. Sin embargo, dado que la medida en la que la síntesis bacteriana contribuye a la ingesta de biotina en los humanos no es conocida, los efectos de las drogas antimicrobianas en el estatus de la biotina nutricional permanecen siendo inciertas (51).
Recomendación del Instituto Linus Pauling
Poco se conoce con respecto a la cantidad de biotina dietaría requerida para promover la salud optima o prevenir enfermedades crónicas. El Instituto Linus Pauling apoya la recomendación hecha por el Instituto de Medicina, la cual es de 30 microgramos (μg) de biotina por día para los adultos. Una dieta variada debería proveer biotina suficiente para la mayoría de las personas. Sin embargo, siguiendo la recomendación del Instituto Linus Pauling de tomar un suplemento multivitamínico-mineral diariamente proveerá generalmente de una ingesta de al menos 30 μg/día de biotina.
Adultos mayores (>50 años)
Actualmente, no existe indicación de que los adultos mayores tengan un requerimiento de biotina aumentado. Si la ingesta dietaría de biotina no es suficiente, un suplemento multivitamínico-mineral diariamente va generalmente a proveer de una ingesta de al menos 30 μg de biotina por día.
Autores y Críticos
Originalmente escrito en 2000 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Junio de 2004 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Agosto de 2008 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Julio de 2015 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Julio de 2015 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Septiembre de 2015 por:
Donald Mock, M.D., Ph.D.
Profesor
Departamentos de Bioquímica y Biología Molecular y Pediatría
Universidad de Arkansas para las Ciencias Médicas
Traducido al Español en 2016 por:
Silvia Vazquez Lima
Instituto Linus Pauling
Universidad Estatal de Oregon
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Última actualización 10/21/15 Derechos de autoría 2000-2024 Instituto Linus Pauling
Referencias
1. Food and Nutrition Board, Institute of Medicine. Biotin. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B6, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, D.C.: National Academy Press; 1998:374-389. (National Academy Press)
2. Mock DM. Biotin. Handbook of vitamins. 4th ed. Boca Raton, FL: CRC Press; 2007:361-383.
3. Zempleni J, Teixeira DC, Kuroishi T, Cordonier EL, Baier S. Biotin requirements for DNA damage prevention. Mutat Res. 2012;733(1-2):58-60. (PubMed)
4. Saggerson D. Malonyl-CoA, a key signaling molecule in mammalian cells. Annu Rev Nutr. 2008;28:253-272. (PubMed)
5. Zempleni J, Wijeratne SSK, Kuroishi T. Biotin. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed.: John Wiley & Sons, Inc.; 2012:359-374.
6. Zempleni J, Li Y, Xue J, Cordonier EL. The role of holocarboxylase synthetase in genome stability is mediated partly by epigenomic synergies between methylation and biotinylation events. Epigenetics. 2011;6(7):892-894. (PubMed)
7. Zempleni J, Gralla M, Camporeale G, Hassan YI. Sodium-dependent multivitamin transporter gene is regulated at the chromatin level by histone biotinylation in human Jurkat lymphoblastoma cells. J Nutr. 2009;139(1):163-166. (PubMed)
8. Mock DM. Biotin. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed.: Lippincott Williams & Wilkins; 2014:390-398.
9. Baumgartner ER, Suormala T. Inherited defects of biotin metabolism. Biofactors. 1999;10(2-3):287-290. (PubMed)
10. Elrefai S, Wolf B. Disorders of biotin metabolism. In: Rosenberg RN, Pascual JM, eds. Rosenberg's Molecular and Genetic basis of Neurological and Psychiatric Disease. 5th ed. United States of America: Elsevier; 2015:531-539.
11. Sealey WM, Teague AM, Stratton SL, Mock DM. Smoking accelerates biotin catabolism in women. Am J Clin Nutr. 2004;80(4):932-935. (PubMed)
12. Perry CA, West AA, Gayle A, et al. Pregnancy and lactation alter biomarkers of biotin metabolism in women consuming a controlled diet. J Nutr. 2014;144(12):1977-1984. (PubMed)
13. Mock DM. Marginal biotin deficiency is common in normal human pregnancy and is highly teratogenic in mice. J Nutr. 2009;139(1):154-157. (PubMed)
14. Pabuccuoglu A, Aydogdu S, Bas M. Serum biotinidase activity in children with chronic liver disease and its clinical significance. J Pediatr Gastroenterol Nutr. 2002;34(1):59-62. (PubMed)
15. Zempleni J, Hassan YI, Wijeratne SS. Biotin and biotinidase deficiency. Expert Rev Endocrinol Metab. 2008;3(6):715-724. (PubMed)
16. Wolf B. Biotinidase deficiency: "if you have to have an inherited metabolic disease, this is the one to have." Genet Med. 2012;14(6):565-575. (PubMed)
17. Mardach R, Zempleni J, Wolf B, et al. Biotin dependency due to a defect in biotin transport. J Clin Invest. 2002;109(12):1617-1623. (PubMed)
18. Schulpis KH, Nyalala JO, Papakonstantinou ED, et al. Biotin recycling impairment in phenylketonuric children with seborrheic dermatitis. Int J Dermatol. 1998;37(12):918-921. (PubMed)
19. Eng WK, Giraud D, Schlegel VL, Wang D, Lee BH, Zempleni J. Identification and assessment of markers of biotin status in healthy adults. Br J Nutr. 2013;110(2):321-329. (PubMed)
20. Mock DM. Adequate intake of biotin in pregnancy: why bother? J Nutr. 2014;144(12):1885-1886. (PubMed)
21. Mock DM, Stadler DD. Conflicting indicators of biotin status from a cross-sectional study of normal pregnancy. J Am Coll Nutr. 1997;16(3):252-257. (PubMed)
22. Mock DM, Stadler DD, Stratton SL, Mock NI. Biotin status assessed longitudinally in pregnant women. J Nutr. 1997;127(5):710-716. (PubMed)
23. Mock DM, Quirk JG, Mock NI. Marginal biotin deficiency during normal pregnancy. Am J Clin Nutr. 2002;75(2):295-299. (PubMed)
24. Takechi R, Taniguchi A, Ebara S, Fukui T, Watanabe T. Biotin deficiency affects the proliferation of human embryonic palatal mesenchymal cells in culture. J Nutr. 2008;138(4):680-684. (PubMed)
25. Alfadhel M, Almuntashri M, Jadah RH, et al. Biotin-responsive basal ganglia disease should be renamed biotin-thiamine-responsive basal ganglia disease: a retrospective review of the clinical, radiological and molecular findings of 18 new cases. Orphanet J Rare Dis. 2013;8:83. (PubMed)
26. Sedel F, Bernard D, Mock DM, Tourbah A. Targeting demyelination and virtual hypoxia with high-dose biotin as a treatment for progressive multiple sclerosis. Neuropharmacology. 2015. (PubMed)
27. Sedel F, Papeix C, Bellanger A, et al. High doses of biotin in chronic progressive multiple sclerosis: a pilot study. Mult Scler Relat Disord. 2015;4(2):159-169. (PubMed)
28. Tourbah A LFC, Edan G, Clanet M, Papeix C, Vukusic S, et al. Effect of MD1003 (high doses of biotin) in progressive multiple sclerosis: results of a pivotal phase III randomized double blind placebo controlled study. Paper presented at: American Association of Neurological Surgeons (AANS) Annual Scientific Meeting 2015; Washington, D.C. Available at: http://www.neurology.org/content/84/14_Supplement/PL2.002. Accessed 9/17/15.
29. Larrieta E, Vega-Monroy ML, Vital P, et al. Effects of biotin deficiency on pancreatic islet morphology, insulin sensitivity and glucose homeostasis. J Nutr Biochem. 2012;23(4):392-399. (PubMed)
30. Maebashi M, Makino Y, Furukawa Y, Ohinata K, Kimura S, Sato T. Therapeutic evaluation of the effect of biotin on hyperglycemia in pateints with non-insulin dependent diabetes mellitus. J Clin Biochem Nutr. 1993;14:211-218.
31. Baez-Saldana A, Zendejas-Ruiz I, Revilla-Monsalve C, et al. Effects of biotin on pyruvate carboxylase, acetyl-CoA carboxylase, propionyl-CoA carboxylase, and markers for glucose and lipid homeostasis in type 2 diabetic patients and nondiabetic subjects. Am J Clin Nutr. 2004;79(2):238-243. (PubMed)
32. Revilla-Monsalve C, Zendejas-Ruiz I, Islas-Andrade S, et al. Biotin supplementation reduces plasma triacylglycerol and VLDL in type 2 diabetic patients and in nondiabetic subjects with hypertriglyceridemia. Biomed Pharmacother. 2006;60(4):182-185. (PubMed)
33. Geohas J, Daly A, Juturu V, Finch M, Komorowski JR. Chromium picolinate and biotin combination reduces atherogenic index of plasma in patients with type 2 diabetes mellitus: a placebo-controlled, double-blinded, randomized clinical trial. Am J Med Sci. 2007;333(3):145-153. (PubMed)
34. Albarracin C, Fuqua B, Geohas J, Juturu V, Finch MR, Komorowski JR. Combination of chromium and biotin improves coronary risk factors in hypercholesterolemic type 2 diabetes mellitus: a placebo-controlled, double-blind randomized clinical trial. J Cardiometab Syndr. 2007;2(2):91-97. (PubMed)
35. Singer GM, Geohas J. The effect of chromium picolinate and biotin supplementation on glycemic control in poorly controlled patients with type 2 diabetes mellitus: a placebo-controlled, double-blinded, randomized trial. Diabetes Technol Ther. 2006;8(6):636-643. (PubMed)
36. Albarracin CA, Fuqua BC, Evans JL, Goldfine ID. Chromium picolinate and biotin combination improves glucose metabolism in treated, uncontrolled overweight to obese patients with type 2 diabetes. Diabetes Metab Res Rev. 2008;24(1):41-51. (PubMed)
37. Suksomboon N, Poolsup N, Yuwanakorn A. Systematic review and meta-analysis of the efficacy and safety of chromium supplementation in diabetes. J Clin Pharm Ther. 2014;39(3):292-306. (PubMed)
38. Lazo de la Vega-Monroy ML, Larrieta E, German MS, Baez-Saldana A, Fernandez-Mejia C. Effects of biotin supplementation in the diet on insulin secretion, islet gene expression, glucose homeostasis and beta-cell proportion. J Nutr Biochem. 2013;24(1):169-177. (PubMed)
39. Randhawa SS, Dua K, Randhawa CS, Randhawa SS, Munshi SK. Effect of biotin supplementation on hoof health and ceramide composition in dairy cattle. Vet Res Commun. 2008;32(8):599-608. (PubMed)
40. Reilly JD, Cottrell DF, Martin RJ, Cuddeford DJ. Effect of supplementary dietary biotin on hoof growth and hoof growth rate in ponies: a controlled trial. Equine Vet J Suppl. 1998(26):51-57. (PubMed)
41. Zenker W, Josseck H, Geyer H. Histological and physical assessment of poor hoof horn quality in Lipizzaner horses and a therapeutic trial with biotin and a placebo. Equine Vet J. 1995;27(3):183-191. (PubMed)
42. Romero-Navarro G, Cabrera-Valladares G, German MS, et al. Biotin regulation of pancreatic glucokinase and insulin in primary cultured rat islets and in biotin-deficient rats. Endocrinology. 1999;140(10):4595-4600. (PubMed)
43. Floersheim GL. [Treatment of brittle fingernails with biotin]. Z Hautkr. 1989;64(1):41-48. (PubMed)
44. Hochman LG, Scher RK, Meyerson MS. Brittle nails: response to daily biotin supplementation. Cutis. 1993;51(4):303-305. (PubMed)
45. Boccaletti V, Zendri E, Giordano G, Gnetti L, De Panfilis G. Familial Uncombable Hair Syndrome: Ultrastructural Hair Study and Response to Biotin. Pediatr Dermatol. 2007;24(3):E14-16. (PubMed)
46. Famenini S, Goh C. Evidence for supplemental treatments in androgenetic alopecia. J Drugs Dermatol. 2014;13(7):809-812. (PubMed)
47. Staggs CG, Sealey WM, McCabe BJ, Teague AM, Mock DM. Determination of the biotin content of select foods using accurate and sensitive HPLC/avidin binding. J Food Compost Anal. 2004;17(6):767-776. (PubMed)
48. Briggs DR, Wahlqvist ML. Food facts: the complete no-fads-plain-facts guide to healthy eating. Victoria, Australia: Penguin Books; 1988.
49. Magnusdottir S, Ravcheev D, de Crecy-Lagard V, Thiele I. Systematic genome assessment of B-vitamin biosynthesis suggests co-operation among gut microbes. Front Genet. 2015;6:148. (PubMed)
50. Said HM. Cell and molecular aspects of human intestinal biotin absorption. J Nutr. 2009;139(1):158-162. (PubMed)
51. Natural-Medicines. Biotin/Drug interactions. http://www.naturaldatabase.com/. 2014 ed.; 2014
52. Koutsikos D, Agroyannis B, Tzanatos-Exarchou H. Biotin for diabetic peripheral neuropathy. Biomed Pharmacother. 1990;44(10):511-514. (PubMed)
53. Debourdeau PM, Djezzar S, Estival JL, Zammit CM, Richard RC, Castot AC. Life-threatening eosinophilic pleuropericardial effusion related to vitamins B5 and H. Ann Pharmacother. 2001;35(4):424-426. (PubMed)
54. Chirapu SR, Rotter CJ, Miller EL, Varma MV, Dow RL, Finn MG. High specificity in response of the sodium-dependent multivitamin transporter to derivatives of pantothenic acid. Curr Top Med Chem. 2013;13(7):837-842. (PubMed)
55. Said HM, Ortiz A, McCloud E, Dyer D, Moyer MP, Rubin S. Biotin uptake by human colonic epithelial NCM460 cells: a carrier-mediated process shared with pantothenic acid. Am J Physiol. 1998;275(5 Pt 1):C1365-1371. (PubMed)
56. Prasad PD, Wang H, Kekuda R, et al. Cloning and functional expression of a cDNA encoding a mammalian sodium-dependent vitamin transporter mediating the uptake of pantothenate, biotin, and lipoate. J Biol Chem. 1998;273(13):7501-7506. (PubMed)
57. Zempleni J, Trusty TA, Mock DM. Lipoic acid reduces the activities of biotin-dependent carboxylases in rat liver. J Nutr. 1997;127(9):1776-1781. (PubMed)
58. Castro-Gago M, Gomez-Lado C, Eiris-Punal J, Diaz-Mayo I, Castineiras-Ramos DE. Serum biotinidase activity in children treated with valproic acid and carbamazepine. J Child Neurol. 2010;25(1):32-35. (PubMed)
59. Castro-Gago M, Perez-Gay L, Gomez-Lado C, Castineiras-Ramos DE, Otero-Martinez S, Rodriguez-Segade S. The influence of valproic acid and carbamazepine treatment on serum biotin and zinc levels and on biotinidase activity. J Child Neurol. 2011;26(12):1522-1524. (PubMed)
60. Schulpis KH, Karikas GA, Tjamouranis J, Regoutas S, Tsakiris S. Low serum biotinidase activity in children with valproic acid monotherapy. Epilepsia. 2001;42(10):1359-1362. (PubMed)
61. Yilmaz Y, Tasdemir HA, Paksu MS. The influence of valproic acid treatment on hair and serum zinc levels and serum biotinidase activity. Eur J Paediatr Neurol. 2009;13(5):439-443. (PubMed)
Folato
Contenido
Resumen
- Folato es un término genérico que hace referencia tanto a los folatos naturales en los alimentos como al ácido fólico, la forma sintética usada en suplementos y alimentos fortificados. El folato es crítico en el metabolismo de los precursores del ácido nucleico y varios aminoácidos, como también en las reacciones de metilación. (Más información)
- Una severa deficiencia de folato o vitamina B12 puede conducir a una anemia megaloblástica, la cual causa fatiga, debilitamiento, y dificultad para respirar. El tratamiento inadecuado de la anemia megaloblástica dependiente de la vitamina B12 con altas dosis de ácido fólico suplementario puede potencialmente retrasar el diagnóstico de la deficiencia de vitamina B12 y así dejar al individuo en un riesgo de desarrollar un daño cerebral irreversible. (Más información)
- El estatus del folato es influenciado por la presencia de variaciones genéticas en el metabolismo del folato, particularmente aquellos encontrados en el gen 5,10-metilentetrahidrofolato reductasa (MTHFR). (Más información)
- Un estatus inadecuado de folato durante el embarazo incrementa el riesgo de anomalías congénitas. La introducción de la fortificación obligatoria con ácido fólico de productos de granos refinados en los EE. UU. en 1998 ha reducido la prevalencia de defectos del tubo neural en recién nacidos. A pesar de todo, el estatus del folato es considerado inadecuado en la mayoría de mujeres a nivel mundial en edad de procrear. Por otra parte, los factores genéticos podrían modificar el riesgo de defectos del tubo neural al incrementar la susceptibilidad a una deficiencia de folato durante el embarazo. Varios estudios están actualmente investigando el papel de la suplementación con ácido fólico en la prevención de anomalías congénitas aparte de los defectos del tubo neural. (Más información)
- La deficiencia de folato y las concentraciones de homocisteína en la sangre están asociadas con un riesgo incrementado de contraer enfermedades cardiovasculares (ECV). Aunque se ha probado que la suplementación con ácido fólico ha sido efectiva en el control de las concentraciones de homocisteína circulante, el efecto de la disminución de la homocisteína en la incidencia de las ECV es todavía debatido. (Más información)
- Un estatus bajo del folato ha sido ligado con un riesgo incrementado de cáncer. Sin embargo, ensayos de intervención con altas dosis de ácido fólico no han mostrado generalmente algún beneficio en la incidencia de cáncer. (Más información)
- Estudios de cohorte prospectivos han reportado una asociación inversa entre en el estatus del folato y el riesgo de cáncer colorrectal especialmente entre hombres. La relación entre el estatus del folato y el riesgo de cáncer es sin embargo complejo y requiere de más investigación. (Más información)
- El folato es esencial para el desarrollo y función del cerebro. Un estatus bajo de folato y/o altas concentraciones de homocisteína se asocian con una disfunción cognitiva en el envejecimiento (desde deterioros leves hasta demencia). Aún se desconoce si las vitaminas B suplementarias, incluyendo el ácido fólico, tendrán beneficios a largo plazo en el mantenimiento de la salud cognitiva. (Más información)
-
Varios trastornos autosómicos recesivos que afectan el transporte y metabolismo del folato pueden ser tratados con altas dosis de ácido folínico un derivado del folato. (Más información)
Los términos de folato y el ácido fólico se usan indistintamente para esta vitamina B soluble en agua, que también es conocida como vitamina B9 o folacina. Los folatos producidos naturalmente existen en muchas formas químicas; los folatos se encuentran en los alimentos, como también en formas metabólicamente activas en cuerpo humano. El ácido fólico es la mayor forma sintética encontrada en alimentos fortificados y suplementos vitamínicos. Otras formas sintéticas incluyen ácido folínico (Figura 1) y ácido levomefólico. El ácido fólico no tiene alguna actividad biológica a menos que sea convertido en folatos (1). En la siguiente discusión, las formas encontradas en alimentos o en el cuerpo son referidos como “folatos,” mientras que la forma encontrada en suplementos o alimentos fortificados es referida como “ácido fólico.”
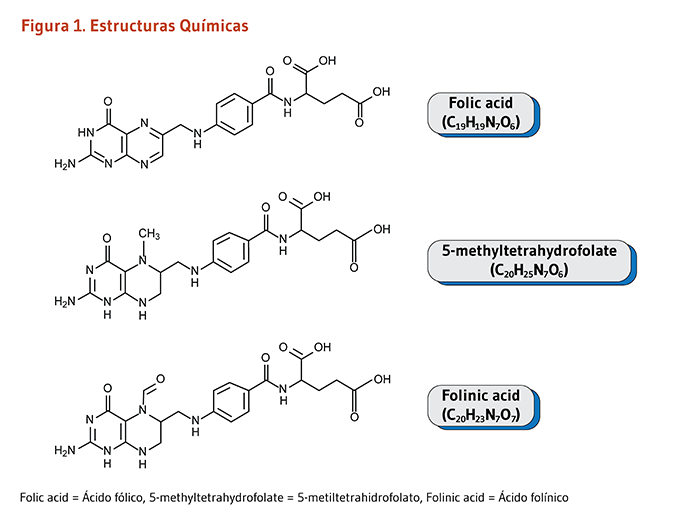
Función
Metabolismo de un carbono
La única función de las coenzimas del folato en el cuerpo es aparentemente mediar la transferencia de unidades de un carbono (2). Las coenzimas del folato actúan como aceptores y dadores de unidades de un carbono en una variedad de reacciones críticas para el metabolismo de los ácidos nucleicos y los aminoácidos (Figura 2) (3).

Metabolismo de los ácidos nucleicos
Las coenzimas folato juegan un papel vital en el metabolismo del ADN a través de dos diferentes vías. (1) La síntesis de ADN desde sus precursores (timidinas y purinas) depende de las coenzimas folato. (2) Para la síntesis de metionina de la homocisteína se requiere de una coenzima folato, y la metionina es necesaria para la síntesis de S-adenosilmetionina (SAM). SAM es un dador de grupos metilo (una unidad de carbono) usado en muchas reacciones biológicas de metilación, incluyendo la metilación de una serie de sitios en el ADN, ARN, proteínas, y fosfolípidos. La metilación del ADN juega un importante papel en el control de la expresión de genes y es crítica durante la diferenciación celular. Aberraciones en la metilación del ADN han sido ligadas al desarrollo de cáncer (véase Cáncer).
Metabolismo de los aminoácidos
Las coenzimas del folato son requeridas para el metabolismo de varios aminoácidos importantes, es decir la metionina, cisteína, serina, glicina, e histidina. La síntesis de la metionina a partir de la homocisteína es catalizada por la metionina sintasa, una enzima que no solo el folato (como 5-metiltetrahidrofolato) pero también la vitamina B12. Por lo tanto, una deficiencia de folato (y/o vitamina B12) puede resultar en una disminución de la síntesis de metionina y una acumulación de homocisteína. Las concentraciones sanguíneas elevadas de homocisteína han sido consideradas por muchos años como un factor de riesgo para algunas enfermedades crónicas, incluyendo las enfermedades cardiovasculares y demencia (véase Prevención de Enfermedades).
Interacción con nutrientes
Vitamina B12 y vitamina B6
El metabolismo de la homocisteína, un intermediario en el metabolismo de los aminoácidos que contienen sulfuro, es un ejemplo de las interrelaciones entre los nutrientes necesarios para una salud y funcionalidad fisiológica óptimas. Los individuos sanos utilizan dos vías diferentes para metabolizar la homocisteína (Figura 3). Una vía (metionina sintasa) sintetiza la metionina de la homocisteína, y es dependiente tanto del folato como la vitamina B12 como cofactores. La otra vía convierte la homocisteína en otro aminoácido, denominado cisteína y requiere de dos enzimas dependientes de la vitamina B6. Por lo tanto, la cantidad de homocisteína en la sangre es regulada por tres vitaminas B: folato, vitamina B12, y vitamina B6 (4). En algunos individuos, la riboflavina (vitamina B2) también se relaciona en la regulación de las concentraciones de homocisteína (véase Riboflavina).
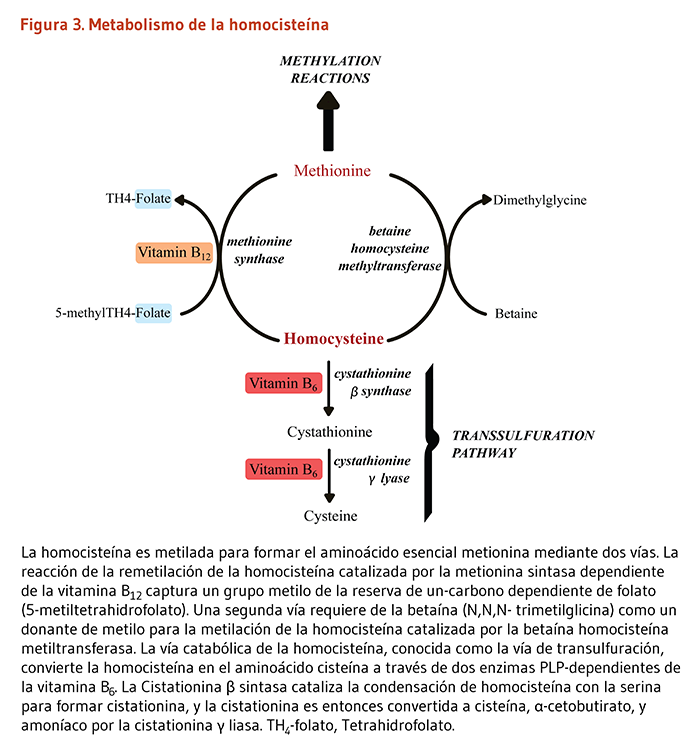
Riboflavina
Aunque es menos reconocida, el folato tiene una importante interacción metabólica con la riboflavina. La riboflavina es un precursor del flavín adenín dinucleótido (FAD), una coenzima necesaria para la actividad de las enzimas del folato encargadas de la metabolización, la 5,10-metilentetrahidrofolato reductasa (MTHFR). El FAD dependiente de la MTHFR en turno cataliza la reacción que genera el 5-metiltetrahidrofolato (ver Figura 2 arriba). Esta forma activa del folato es requerida para la formación de la metionina a partir de la homocisteína. Junto con otras vitaminas B, las ingestas más elevadas de riboflavina han sido asociadas con una disminución de las concentraciones de homocisteína en el plasma (5). Los efectos de la riboflavina en el metabolismo del folato parecen ser mayores en individuos homocigotos del polimorfismo común c.677C>T (genotipo TT) en el gen MTHFR (ver Variaciones genéticas en los requerimientos de folato) (6). Estos individuos (alrededor del 10% de los adultos a nivel mundial) típicamente presentan bajos estatus de folato, junto con elevadas concentraciones de homocisteína, particularmente cuando la ingesta de folato y/o riboflavina es subóptima. Las concentraciones elevadas de homocisteína en estos individuos, sin embargo, es altamente sensible a disminuir con la suplementación de riboflavina, confirmando la importancia de la interacción de la riboflavina y MTHFR (7).
Vitamina C
La vitamina C puede limitar la degradación de coenzimas folato naturales y ácido fólico suplementario en el estómago y así mejorar la biodisponibilidad del folato. Un ensayo cruzado en nueve hombres sanos encontró que la administración oral de ácido 5-metiltetrahidrofólico (343 μg) y vitamina C (289 mg o 974 mg) fue asociada con concentraciones más altas de folato en el suero en comparación con el ácido 5-metiltetrahidrofólico solo (8). Por otra parte, un reciente estudio sugirió que algunas variaciones genéticas del metabolismo del folato podrían influenciar el efecto de la vitamina C en el metabolismo del folato (9).
Biodisponibilidad
El folato dietario existe predominantemente en la forma de poliglutamilo (el cual contiene varios residuos de glutamato) mientras que el ácido fólico – la forma sintética de la vitamina- es un monoglutamato, es decir que contiene solo una fracción del glutamato. Además, folatos naturales son moléculas reducidas, mientras que el ácido fólico es completamente oxidado. Estas diferencias químicas tienen mayores implicaciones para la biodisponibilidad de la vitamina tal como que el ácido fólico es considerablemente más biodisponible que los folatos naturales en los alimentos a niveles de consumo equivalentes.
La absorción intestinal de folatos dietarios es un proceso de dos pasos que involucra la hidrolisis de poliglutamatos del folato a los derivados monoglutamil correspondientes, seguidos por su transporte dentro de las células intestinales. Allí, el ácido fólico es convertido en un folato natural, llamado 5-metiltetrahidrofolato, el cual es la mayor forma de folato circulante en el cuerpo humano (vea Figura 1 arriba).
La biodisponibilidad de folatos de procedencia natural es inherentemente limitada y variable. Existe mucha variabilidad en la facilidad con la que los folatos son liberados de diferentes matrices alimenticias, y la “cola” del poliglutamil es removida (desconjugación) antes de la absorción por las células intestinales. Además, otros componentes de la dieta pueden contribuir a la inestabilidad de los folatos lábiles durante los procesos de digestión. Como resultado, los folatos naturales muestran una biodisponibilidad incompleta en comparación con el ácido fólico. La biodisponibilidad del ácido fólico, en contraste, se asume es un 100% cuando este es ingerido como suplemento, mientras que el ácido fólico en alimentos fortificados se estima que posee alrededor de un 85% de la biodisponibilidad del ácido fólico suplementario.
Nótese que las recomendaciones del folato en los EE.UU. y otros ciertos países están ahora expresadas como Equivalentes de Folato Dietario (EFD), un cálculo que fue ideado para tener en cuenta la extensiva biodisponibilidad del ácido fólico en comparación con los folatos de origen natural (ver La Ingesta Diaria Recomendada).
Transporte
El folato y sus coenzimas requieren de transportadores para atravesar las membranas celulares. Los transportadores del folato incluyen el transportador de folato reducido (RFC), el transportador de folato acoplado a protones (PCFT), y proteínas receptoras del folato, FRα y FRβ. La homeostasis del folato es soportada por la distribución ubicua de los transportadores de folato, aunque la abundancia e importancia varía entre los tejidos (10). El PCFT juega un papel mayor en el transporte del folato intestinal debido a que las mutaciones que afectan el gen que codifica el PCFT causa una malabsorción hereditaria del folato. Además, un PCFT defectuoso conlleva a un deterioro en el transporte de folato al cerebro (ver Tratamiento de Enfermedades). La FRα y el RFC son también críticos para el transporte de folato a través de la barrera hematoencefálica cuando el folato extracelular es bajo o alto, respectivamente. El folato es esencial para el desarrollo adecuado del embrión y el feto. La placenta es conocida por concentrar el folato a la circulación fetal, llevando a concentraciones altas de folato en el feto a comparación a aquellas encontradas en la mujer embarazada. Los tres tipos de receptores han sido asociados con el transporte de folato a través de la placenta durante el embarazo (11).
Deficiencia
Causas
La mayoría de las veces, una deficiencia de folato es causada por una insuficiencia alimentaria; sin embargo, una deficiencia de folato puede también presentarse en un cierto número de otras situaciones. Por ejemplo, el consumo crónico y pesado de alcohol está asociado con una disminución de la absorción del folato (sumada a una ingesta dietaría baja), las cuales pueden conducir a una deficiencia de folato (12). Fumar esta también asociado con un estatus bajo de folato. En un estudio, las concentraciones de folato en la sangre fueron un 15% más bajas en fumadores que en no fumadores (13). Además, un transporte deteriorado del folato hacia el feto ha sido observado en mujeres embarazadas que fumaron o abusaron del alcohol durante el embarazo (14, 15).
El embarazo es el momento cuando él requerimiento de folato se incrementa en gran medida para mantener la demanda de la rápida replicación celular y para el crecimiento del feto, la placenta, y tejido materno. Ciertas condiciones como el cáncer o inflamaciones pueden derivar en un incremento en las tasas de división celular y metabolismo, causando un incremento en la demanda corporal por folato (16). Por otra parte, la deficiencia de folato puede resultar de condiciones de malabsorción, incluyendo enfermedades inflamatorias intestinales (enfermedad de Crohn y colitis ulcerosa) y la enfermedad celiaca (17). Varios medicamentos podrían también contribuir a una deficiencia de folato (véase Interacción con drogas). Finalmente, un cierto número de enfermedades genéticas que afectan la absorción, el transporte, o el metabolismo pueden causar una deficiencia de folato o impedir sus funciones metabólicas (véase Tratamiento de Enfermedades).
Síntomas
Una deficiencia clínica de folato conlleva a una anemia megaloblástica, la cual es reversible con un tratamiento con ácido fólico. Las células de división rápida como aquellas derivadas de la médula ósea son más vulnerables a los efectos de la deficiencia de folato debido a que la síntesis del ADN y la división celular dependen de las coenzimas del folato. Cuando el suministro de folato a las células de división rápida de la médula ósea es inadecuada, la división de glóbulos rojos es reducida, resultando en menos pero más grandes glóbulos rojos. Este tipo de anemia es llamada megaloblástica o anemia macrocítica, ya que hace referencia a glóbulos rojos inmaduros y agrandados. Los neutrófilos, un tipo de leucocito, se vuelven hipersegmentados, un cambio que pude ser observado al analizar microscópicamente una muestra de sangre. Debido a que los glóbulos rojos normales tienen un tiempo de vida en la circulación de aproximadamente cuatro meses, puede tomar meses para que los individuos deficientes de folato desarrollen la anemia megaloblástica característica. La progresión de este tipo de anemia conduce a una disminución de la capacidad de la sangre de transportar oxígeno y en última instancia, podría resultar en síntomas de fatiga, debilidad y dificultad para respirar (1). Es importante señalar que la anemia megaloblástica resultante de una deficiencia de folato es idéntica a la anemia megaloblástica resultante de una deficiencia de vitamina B12, y ensayos clínicos adicionales son requeridos para diagnosticar la verdadera causa de la anemia megaloblástica (véase Toxicidad).
Los individuos en etapas tempranas de una deficiencia de folato pueden no mostrar síntomas obvios, pero las concentraciones de homocisteína podrían incrementar (véase Prevención de Enfermedades). Sin embargo, la concentración de homocisteína circulante no es un indicador específico del estatus del folato, porque una homocisteína elevada puede ser el resultado de una deficiencia de vitamina B12 y de otras vitaminas B, factores del estilo de vida, e insuficiencia renal. Una deficiencia subclínica es comúnmente detectada midiendo las concentraciones de folato en el suero/plasma o en los glóbulos rojos.
La Ingesta Diaria Recomendada (IDR)
Determinación de la IDR
Tradicionalmente, el requerimiento de folato dietario fue definido como la cantidad necesaria para prevenir una deficiencia severa capaz de causar síntomas como la anemia. La IDR más reciente (1998) se basó principalmente en la adecuación de las concentraciones de folato presentes en glóbulos rojos a diferentes niveles de la ingesta de folato a juzgar por la ausencia de indicadores hematológicos anormales. Se ha demostrado que el folato de los glóbulos rojos se correlaciona con los depósitos hepáticos de folato y es usado como un indicador a largo plazo del estatus del folato. El folato en el plasma refleja la reciente ingesta de folato y no es un biomarcador seguro para el estatus del folato. El mantenimiento de niveles normales de homocisteína en la sangre, un indicador del metabolismo de un carbono, fue considerado sólo como un indicador auxiliar de la ingesta adecuada de folato.
Debido a que el embarazo es asociado con un aumento significativo en la división celular y otros procesos metabólicos que requieren coenzimas folato, la IDR para mujeres embarazadas es considerablemente más alta que para las mujeres que no están embarazadas (3). Sin embargo, la prevención de defectos del tubo neural no fue considerada cuando se estableció la IDR para mujeres embarazadas. Más bien la reducción del riesgo de defectos del tubo neural fue considerada una recomendación separada para mujeres capaces de quedar embarazadas (véase Prevención de Enfermedades), debido a que los eventos más importantes en el desarrollo del tubo neural ocurren antes de que muchas mujeres sean conscientes de su embarazo (18).
Equivalentes de Folato Dietario (EFD)
Cuando la Junta de Alimentos y Nutrición del Instituto de Medicina de los EE.UU. estableció la nueva recomendación dietaría de folato, ellos introdujeron una nueva unidad, el Equivalente de Folato Dietario (EFD) (1). El uso del EFD refleja una mayor biodisponibilidad del ácido fólico sintético encontrado en suplementos y alimentos fortificados, en comparación con la de folatos naturales presentes en los alimentos (18).
- 1 microgramo (μg) de folato alimentario aporta 1 μg de EFD
- 1 μg de ácido fólico ingerido con las comidas o como alimento fortificado aporta 1.7 μg de EFD
- 1 μg de ácido fólico (suplemento) ingerido con el estómago vacío aporta 2 μg de EFD
Por ejemplo una porción de alimento que contenga 60 μg de folato aportaría 60 μg de EFD, mientras una porción de tallarines fortificados con 60 μg de ácido fólico aportaría 1.7 x 60 = 102 μg de EFD, debido a una biodisponibilidad más alta de ácido fólico. Un suplemento de 400 μg de ácido fólico ingerido con el estómago vacío aportaría 800 μg de EFD. Debe tenerse en cuenta que los EFDs fueron determinados en estudios con adultos, y no se ha estudiado si el ácido fólico en la fórmula infantil es más biodisponible que los folatos en la leche materna. El uso de EFD para determinar el requerimiento de folato para infantes no sería deseable.
| Ingesta Diaria Recomendada (IDR) para Folato en Equivalentes de Folato Dietario (EFD) | |||
| Etapa de la Vida | Edad | Machos (μg/día) | Hembras (μg/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 65 (IA) | 65 (IA) |
| Infantes | 7-12 meses | 80 (IA) | 80 (IA) |
| Niños | 1-3 años | 150 | 150 |
| Niños | 4-8 años | 200 | 200 |
| Niños | 9-13 años | 300 | 300 |
| Adolescentes | 14-18 años | 400 | 400 |
| Adultos | 19 años y más | 400 | 400 |
| Embarazo | Todas las edades | - | 600 |
| Período de lactancia | Todas las edades | - | 500 |
Variación genética en las requerimientos de folato
Un polimorfismo o variación común en la secuencia del gen para la enzima, 5,10-metilentetrahidrofolato reductasa (MTHFR), conocido como polimorfismo C677T MTHFR, resulta en una enzima termolábil (19). La sustitución de una citosina (C) por una tiamina (T) en el nucleótido 677 en el exón 4 del gen MTHFR lleva a una transición de alanina a valina en el dominio catalítico de la enzima. Dependiendo en la población, un 20% a 53% de los individuos podrían haber heredado una copia T (677C/T genotipo), y de un 3% a un 32% de los individuos podrían haber heredado dos copias T (677T/T genotipo) del gen MTHFR (20). La MTHFR cataliza la reducción de 5,10-5,10-metilentetrahidrofolato (5,10-metilen THF) en 5-metil tretrahidrofolato (5- MeTHF). Este último es la coenzima folato requerida para formar metionina de la homocisteína (ver Figura 2 arriba). La actividad de MTHFR es altamente disminuida en individuos heterocigóticos 677C/T (-30%) y homocigotos 677T/T (-65%) en comparación con aquellos con el genotipo 677C/C (21). La homocigocidad de la mutación (677T/T) está ligada a bajas concentraciones de folato en los eritrocitos y altas concentraciones de homocisteína (22, 23). El mejoramiento del estatus nutricional del folato en mujeres mayores con el alelo T redujo la concentración de homocisteína en el plasma (24). Una pregunta importante sin responder sobre el folato es si la presente IDR es suficiente para compensar por la actividad reducida de la enzima MTHR en individuos con al menos un alelo T, o si aquellos individuos tiene un requerimiento de folato superior que el IDR (25).
Prevención de Enfermedades
Complicaciones del embarazo
Defectos del tubo neural
El crecimiento y desarrollo fetal están caracterizados por una división celular generalizada. Disponer de suficiente folato es crítico para la síntesis del ADN y ARN. Los defectos del tubo neural (DTN) resultan del fracaso del cierre del tubo neural embrionario entre los días 21 y 28 después de la concepción, un tiempo en el que muchas mujeres no se dan cuenta de que están embarazadas (26). Los defectos del tubo neural incluyen varias malformaciones, como lesiones del cerebro (ej. anencefalia, encefalocele) o lesiones de la espina dorsal (espina bífida), los cuales son defectos de nacimiento devastadores, y algunas veces, fatales (27). La prevalencia de DTN en los EE.UU. previa a la fortificación de alimentos con ácido fólico se estimo fue 1 por cada 1,000 embarazos (1). Resultados de ensayos aleatorios han demostrado una reducción del 60% a 100% en casos de DTN cuando las mujeres consumieron suplementos de ácido fólico en adición a una dieta variada durante el período periconcepcional (aproximadamente un mes antes y al menos un mes después de la concepción) (28, 29). Los resultados de estos y otros estudios inspiraron al Servicio de Salud Pública de los EE.UU. a recomendar que todas las mujeres capaces de embarazarse consumieran 400 μg de ácido fólico diariamente para prevenir DTN. Las mujeres con un embarazo afectado previamente fueron también aconsejadas a recibir 4,000 μg de ácido fólico diariamente para así reducir la recurrencia de DTN (30). Estas recomendaciones fueron hechas para todas la mujeres en edad de procrear porque una cantidad folato adecuado debe estar disponible muy temprano en el embarazo, y porque muchos embarazos en los EE.UU. no son planeados (31).
A pesar de la efectividad de la suplementación con ácido fólico en el mejoramiento del estatus del folato, parece que globalmente solo un 30% de mujeres que se embarazan siguen correctamente la recomendación, y existe un poco de preocupación de que mujeres jóvenes de grupos étnicos minoritarios y niveles socioeconómicos más bajos son las menos propensas a seguir la recomendación (32-34). Para disminuir la incidencia de DTN, la legislación implementada por la FDA en 1998 exige la fortificación de todos los productos enriquecidos con granos con 1.4 mg de ácido fólico (véase Fuentes). El nivel de fortificación con ácido fólico requerido en los EE.UU. se estimó inicialmente que aportaba 100 μg de ácido fólico adicional a la dieta de una persona en promedio, aunque probablemente este aporte es aún mayor debido al abuso de ácido fólico por los fabricantes de alimentos (25, 35). La Red Nacional de la Prevención de Defectos del Nacimiento reporto aproximadamente una disminución del 30% en la frecuencia de DTN en los EE.UU. en comparación al periodo de pre-fortificación, y la prevalencia post-fortificación de DTN es 0.69 casos por cada 1,000 nacimientos vivos y muertes fetales (36).
También, un componente genético en la etiología de los DTN es evidenciado por el incremento del riesgo en mujeres con historial familiar de DTN y también por variaciones de riesgo entre etnias (37). Por otra parte, la ocurrencia de defectos del tubo neural puede ser atribuida a específicas interacciones gen-folato. El polimorfismo MTHFR c.677C>T y otras variaciones genéticas pueden incrementar el requerimiento de folato y susceptibilidad a un embarazo afectados con DTN. Previamente a la era de fortificación un estudio de caso y control mostro que ambas concentraciones de eritrocitos y folato del suero fueron significativamente más bajas en mujeres embarazadas con las variantes T/T y C/T en comparación al tipo natural del genotipo C/C (22), sugiriendo un metabolismo inadecuado del folato con genotipos maternos específicos. Un meta-análisis de 25 estudios de casos y controles, incluyendo 2,429 madres de caso y 3,570 madres de control, mostro una asociación positiva entre el polimorfismo maternal MTHFR c.677C>T y DTN (38). Otra variante del MTHFR, un cambio de A a C en la posición 1298, también ha sido asociado con una actividad reducida de MTHFR y un incremento en el riesgo de DTN (39). Los individuos heterocigotos para ambas de estas variantes (677C/T + 1298A/C) mostraron concentraciones más bajas de folato en el plasma y altas concentraciones de homocisteína que en los individuos con 677C/T + 1298A/A (40). Genotipos combinados con la homocigocidad G/G para el transportador de folato reducido (RFC-1) polimorfismo (c.80A>G) podrían contribuir aún más en la ocurrencia de DTN (41). El grado de riesgo de DTN fue también evaluado con polimorfismos MTHFR adicionales (c.116C>T, c.1793G>A) (42) como también con mutaciones que afectan otras enzimas del metabolismo de un carbono, incluyendo la metionina sintasa (MTR c.2756A>G) (43), metionina sintasa reductasa (MTRR c.66A>G) (44), y metilentetrahidrofolato deshidrogenasa (MTHFD1 c.1958G>A) (45). Mientras que el genotipo materno puede impactar el resultado del embarazo, parece que las interacciones de gen a gen entre la madre y el feto es influenciado aún más. El riesgo de DTN se incrementa debido a ciertas combinaciones genéticas, incluyendo interacciones materno (MTHFR c.677C>T)-fetal (MTHFR c.677C>T) y materno (MTRR c.66A>G)-fetal (MTHFR c.677C>T) (43, 44, 46). Finalmente, el estatus de la vitamina B12 ha sido asociado con la modificación del riesgo de DTN en la presencia de polimorfismos específicos en el metabolismo de un carbono (47).
Malformaciones cardiovasculares
Fisuras orofaciales
Otros resultados adversos del embarazo
Un bajo peso al nacer ha sido asociado con un incremento en el riesgo de mortalidad durante el primer año de vida y podría también influenciar los resultados en la salud en la edad adulta (57). Una reciente revisión sistemática y un meta-análisis de ocho ensayos controlados aleatorios encontraron una asociación positiva entre la suplementación de ácido fólico y el peso al nacer; ninguna asociación fue observada con respecto a la duración del periodo de gestación (58). Adicionalmente, un estudio prospectivo de cohorte de 306 adolescentes embarazadas asocio bajas ingestas de folato y el estatus del folato materno durante el tercer trimestre de embarazo con una alta incidencia de nacimientos pequeños para su edad gestacional (peso al nacer <10o percentil) (59). Por otra parte, el genotipo materno c677C>T MTHFR y un incremento en las concentraciones de homocisteína, considerados un indicador de la deficiencia de folato funcional, han sido ligados a bajos pesos al nacer (60).
Las concentraciones elevadas de homocisteína han sido también asociadas con un incremento en la incidencia de abortos espontáneos y otras complicaciones del embarazo, incluyendo preeclampsia y el desprendimiento de la placenta (61). Un exhaustivo estudio retrospectivo mostro que la homocisteína del plasma en mujeres Noruegas estuvo fuertemente relacionado a resultados y complicaciones adversas, incluyendo preeclampsia, parto prematuro, y un peso muy bajo al nacer, en embarazos previos (62). Un reciente meta-analisis de 51 estudios de cohorte prospectivos ligados a la variante c677C>T MTHFR con un riesgo incrementado de preeclampsia en poblaciones caucásicas y asiático orientales, reforzó la noción de que el metabolismo del folato puede jugar un papel en la condición (63). Un ensayo multicéntrico, controlado aleatorio, el Ensayo Clínico de Ácido Fólico (FACT) ha sido iniciado para evaluar si la suplementación diaria de hasta 5.1 mg de ácido fólico durante el embarazo podría prevenir preeclampsia y otro resultado adverso (ej. muerte materna, desprendimiento placentario, parto prematuro) en mujeres de alto riesgo (64). Una ingesta adecuada de folato durante el embarazo protege contra la anemia megaloblástica (65). Un reciente estudio de caso y control encontró una reducción en el riesgo de trastornos del espectro autista con un consumo de ácido fólico diario de 600 μg o más antes y durante el embarazo cuando la madre y el niño poseían el genotipo c677C>T del gen MTHFR (66).
Enfermedades cardiovasculares
Homocisteína y enfermedades cardiovasculares
Los resultados de más de 80 estudios indicaron que incluso moderadas concentraciones de homocisteína en la sangre incrementan el riesgo de enfermedades cardiovasculares (ECV) (4). Posibles predisposiciones a accidentes vasculares han sido también ligadas a deficiencias genéticas en el metabolismo de la homocisteína en ciertas poblaciones (69). El mecanismo por el cual la homocisteína puede incrementar el riesgo de enfermedades vasculares ha sido el tema de una gran cantidad de investigación, pero podría involucrar efectos adversos de la homocisteína en la coagulación sanguínea, vasodilatación arterial, y engrosamiento de las paredes arteriales (70). Aunque un incremento en los niveles de homocisteína en la sangre se ha asociado consistentemente con el riesgo aumentado de enfermedades cardiovasculares, no está claro aún, si al disminuir los niveles de homocisteína se reducirá el riesgo de una enfermedad cardiovascular (véase Folato y homocisteína). Investigaciones han inicialmente predicho que un descenso prolongado en los niveles de homocisteína en el suero de 3 micromoles/litro reduciría el riesgo de ECV por hasta un 25% y sería un objetivo de tratamiento razonable para los individuos con alto riesgo (71, 72). Sin embargo, el análisis de ensayos clínicos recientes de la suplementación con vitaminas B ha mostrado que al disminuir las concentraciones de homocisteína no previno la ocurrencia de un segundo evento cardiovascular en pacientes con ECV existentes (73, 74). Consecuentemente, la Asociación Americana del Corazón recomienda la detección de niveles elevados de homocisteína total sólo en personas de "alto riesgo," por ejemplo, aquellos con historial personal o familiar de enfermedad cardiovascular prematura, malnutrición o síndromes de malabsorción, hipotiroidismo, falla renal, lupus, o personas que toman ciertos medicamentos (ácido nicotínico, teofilina, resinas fijadoras de ácidos biliares, metotrexato, y L-dopa).
Folato y homocisteína
Se ha asociado a las dietas ricas en folato con una disminución del riesgo de ECV, incluyendo la enfermedad coronaria cardíaca, infarto al miocardio (ataque al corazón), y accidente cerebrovascular. Un estudio que dio seguimiento a 1,980 hombres finlandeses por 10 años encontró que aquellos que consumieron más folato dietario tenían un riesgo 55% más bajo de un evento coronario agudo, en comparación con aquellos que consumieron menos folato dietario (75). De las tres vitaminas B que regulan las concentraciones de homocisteína, se ha demostrado que el ácido fólico tiene el mayor efecto en la disminución de los niveles basales de homocisteína en la sangre, cuando no hay una deficiencia de vitamina B12 o vitamina B6 coexistentes (véase Interacciones con nutrientes) (76). Se ha encontrado que un incremento en la ingesta de folato a través de alimentos ricos en folato o suplementos, reduce las concentraciones de homocisteína. Más aún, las concentraciones de homocisteína sanguínea han disminuido desde que la FDA ordenó la fortificación con ácido fólico del suministro de granos en los EE.UU. (25). Un meta-análisis de 25 ensayos controlados aleatorios, incluyendo casi 3,000 sujetos, encontró que la suplementación con 800 μg/día de ácido fólico podría alcanzar un máximo de un 25% en la disminución de las concentraciones de homocisteína en el plasma. En este meta-análisis dosis diarias de 200 μg y 400 μg de ácido fólico fueron asociadas con reducciones del 60% y 90% respectivamente, de homocisteína plasmática (78). Un régimen suplementario de 400 μg de ácido fólico, 2 mg de vitamina B6, y 6 μg de vitamina B12 ha sido ha sido recomendado por la Asociación Americana del Corazón, si una prueba inicial con una dieta rica en folato (véase Fuentes) no es exitosa en reducir adecuadamente las concentraciones de homocisteína (79).
Varios polimorfismos en el metabolismo de un carbono del folato modifican las concentraciones de homocisteína en la sangre (80). En particular el efecto de la variante c.677C>T de MTHFR ha sido examinada en relación a las políticas de fortificación del ácido fólico en todo el mundo. El análisis de ensayos aleatorios, incluyendo 59,995 sujetos sin un historial de ECV, revelo que la diferencia en las concentraciones de homocisteína entre los genotipos T/T y C/C fue mayor en regiones bajas en folato en comparación con regiones con políticas de fortificación de alimentos (3.12 vs. 0.13 micromoles/litro) (81). Aunque la suplementación con ácido fólico disminuye efectivamente las concentraciones de homocisteína, no está claro aún si también disminuye el riesgo de ECV. Un reciente meta-análisis de 19 ensayos clínicos aleatorios, incluyendo 47,921 sujetos con enfermedades cardiovasculares o renales preexistentes, encontró que la disminución de la homocisteína a través de la suplementación con ácido fólico y otra vitamina B fallo en reducir la incidencia de ECV a pesar de las significantes reducciones de las concentraciones de homocisteína en el plasma (74). Otros meta-análisis han confirmado la carencia de causalidad entre la disminución de homocisteína y el riesgo de ECV (80-82), incluyendo el riesgo de un accidente cerebrovascular (83, 84). Por consiguiente, La Asociación Americana del Corazón removió su recomendación del uso de ácido fólico para la prevención de enfermedades cardiovasculares en mujeres de alto riesgo (85). Debe notarse que la mayoría de ensayos preventivos a la fecha han sido realizados en pacientes con ECV con enfermedades avanzadas. La evidencia que apoya un papel beneficial del folato y relacionada con las vitaminas B parece ser más fuerte para la prevención primaria de accidentes cerebrovasculares (86). La introducción de la fortificación obligatoria con ácido fólico ha sido asociada con un decline en mortalidad relacionada a accidentes cerebrovasculares en Norte América, agregando más apoyo a el beneficio potencial del mejoramiento del estatus del folato y/o la disminución de homocisteína en la prevención de accidentes cerebrovasculares (87).
A pesar de la controversia sobre el papel de la disminución de la homocisteína en la prevención de ECV, algunos estudios han investigado el efecto de la suplementación del ácido fólico en el desarrollo de la aterosclerosis, un conocido factor de riesgo en accidentes vasculares. La medición del grosor íntima-media carotídeo (GIMc) es un objetivo subrogado para la aterosclerosis temprana y un pronosticador para eventos cardiovasculares (88). El meta-análisis de 10 ensayos aleatorios que probaron el efecto de la suplementación con ácido fólico mostro una significante reducción del GIMc en sujetos con enfermedades renales crónicas y en aquellos en riesgo de padecer de EC, pero no en participantes saludables (89). La disfunción endotelial es una característica común en la aterosclerosis y enfermedad vascular. Altas dosis de ácido fólico (400-10,000 μg/día) han sido asociadas con mejoramientos de la salud vascular en sujetos sanos y con ECV (90). Aunque recientes ensayos fallaron en demostrar alguna protección de enfermedades cardiovasculares de la suplementación con ácido fólico, una ingesta baja de folato es un conocido factor de riesgo para las enfermedades cardiovasculares y más investigación es necesaria para explorar el papel del folato en el mantenimiento de la salud vascular (91).
Cáncer
Se piensa que el cáncer surge de un daño al ADN por el exceso en la reparación continua de éste y/o de la expresión inadecuada de genes fundamentales. Debido a las importantes funciones que desempeña el folato en la síntesis y metilación del ADN y ARN, es posible que la ingesta inadecuada de folato contribuya a la inestabilidad del genoma y rotura de cromosomas que a menudo caracteriza el desarrollo del cáncer. En particular, la replicación y reparación del ADN son críticas para el mantenimiento del genoma, y la escasez en los nucleótidos causada por la deficiencia de folato podría llevar a una inestabilidad del genoma y mutaciones en el ADN. Una disminución de 5,10-metileno THF puede comprometer la conversión de desoxiuridina monofosfato (dUMP) a desoxitimidina monofosfato (dTMP) por la enzima timidilato sintasa (TS), causando una acumulación de uracilo y una reducción de timina. Esto podría entonces llevar a una incorporación errónea en el ADN durante la replicación o reparación, y causar un daño al ADN, incluyendo mutaciones puntuales y roturas de la cadena (92). Debido a que la 5,10-metileno THF es también el sustrato de la enzima MTHFR, es plausible que una reducción de la actividad de MTHFR con el polimorfismo c.677C>T pudiese incrementar el uso de 5,10-metileno THF para la síntesis de timidilato y prevención de daño al ADN. Sin embargo, la hipótesis podría solo ser validad en una situación de deficiencia de folato (93). Al contrario, se argumentó que la suplementación con ácido fólico podría alimentar la síntesis de ADN, de este modo promoviendo el crecimiento tumoral. Esto es apoyado por la observación de que la TS puede funcionar como un promotor de tumores (oncogén), mientras que una reducción en la actividad de la TS está ligada a un menor riesgo de cáncer (94, 95). Adicionalmente, las moléculas anti-folato que bloquean la vía de la síntesis de timidilato se utilizan con éxito en la terapia contra el cáncer (96). El folato también controla el ciclo de la homocisteína/metionina y el depósito de S-adenosilmetionina (SAM), el donante de metilo para las reacciones de metilación. De esta manera, la deficiencia de folato puede perjudicar la metilación del ADN y proteínas y alterar la expresión de genes involucrados en la reparación del ADN, proliferación y muerte celular. La hipometilación global del ADN, una característica distintiva del cáncer, causa inestabilidad del genoma y ruptura de cromosomas (revisado en 97).
El consumo de por lo menos cinco porciones de frutas y vegetales diariamente ha sido consistentemente asociado con una disminución de la incidencia de cáncer (98). Las frutas y vegetales son excelentes fuentes de folato, las cuales pueden jugar un papel en su efecto anti-cancerígeno. Estudios basados en la observación han encontrado que un estatus disminuido del folato está asociado con canceres de sitio específico. Mientras que la fortificación de alimentos es obligatoria en los EE.UU. (desde 1998; vea Fuentes), preocupaciones acerca del impacto de altas ingestas de ácido fólico en la salud han retrasado la practica en otros países (99). Sin embargo, los meta-análisis más recientes de ensayos de intervención del ácido fólico (dosis suplementarias de entre 500 a 5,000 μg/día durante al menos un año) no mostro algún beneficio especifico o daño con respecto a la incidencia de cáncer total o de sitio especifico (100, 101).
Cáncer colorrectal
Un análisis combinado de 13 estudios de cohorte prospectivos, los cuales dieron seguimiento a un total de 725,134 individuos por un periodo de 7 a 20 años, revelo una modesta asociación inversa entre la ingesta dietaría y total (proveniente de alimentos y suplementos) de folato y el riesgo de cáncer de colon. Específicamente, se estimó un decline del 2% del riesgo de cáncer de colon por cada incremento de 100 μg/día en la ingesta total de folato (102). Un exhaustivo estudio prospectivo estadounidense, el cual dio seguimiento a 525,488 sujetos, de edades entre 50 a 71 años entre 1995 y 2006, correlacionaron el folato dietario, ácido fólico suplementario, e ingestas totales de folato con una disminución del riesgo de cáncer colorrectal (103). Sin embargo, cuando se estratifico por género, no hubo asociación entre la ingesta de folato dietario y el riesgo de cáncer colorrectal en mujeres (103, 104). Una falta de asociación entre el riesgo de cáncer colorrectal e ingestas dietarías, suplementarias, y totales de folato, fue también reportada en otro estudio perspectivo que dio seguimiento a más de 90,000 mujeres postmenopáusicas estadounidenses durante un periodo de 11 años abarcando los periodos de pre y post fortificación (105). Estos datos sugirieron la posible influencia de género sobre la modificación del riesgo de cáncer colorrectal por el folato. En el último estudio, una elevación significante pero transitoria del riesgo fue también observada durante la era de post-fortificación; sin embargo algunos han afirmado que es poco probable que esto sea causado por una ingesta incrementada de folato debido a la fortificación obligatoria (106). Finalmente, un meta-análisis de 18 estudios de casos de controles encontró una ligera reducción en el riesgo de cáncer colorrectal con folato proveniente de alimentos (107). Sin embargo, es importante tener en cuenta que los estudios de caso y control fueron altamente heterogéneos, y que los autores declararon que la fibra dietaría, vitaminas, y el consumo de alcohol pudieran haber alterado sus resultados. Por otra parte, el límite más bajo del cuantil más alto de la ingesta de folato fue altamente variable, oscilando de entre 270 a 1,367 μg/día (107).
Mientras que la mayoría de la investigación epidemiológica muestra un efecto protector del folato en contra del desarrollo del cáncer colorrectal, se ha sugerido que las altas dosis de ácido fólico suplementario en realidad podrían acelerar el crecimiento del tumor en pacientes con cáncer (108). Mientras que un estatus más alto de folato dentro el rango dietario normal es extensamente considerado como protector contra el cáncer, algunos investigadores permanecen preocupados de que la exposición excesiva de altas ingestas de ácido fólico podrían incrementar el crecimiento de neoplasias pre-existentes (108). Varios ensayos clínicos abordaron el efecto de la suplementación de ácido fólico en pacientes con un historial de adenoma colorrectal, con ensayos que encontraron una reducción del riesgo o ningún efecto del ácido fólico suplementario (109-112). Un reciente meta-análisis de tres ensayos controlados aleatorios exhaustivos en sujetos de alto riesgo no demostró algún incremento en la recurrencia de adenoma colorrectal en sujetos suplementados con 500 o 1,000 μg/día de ácido fólico por 24 a 42 meses cuando se comparó con el tratamiento con placebo (113).
Como se sugirió anteriormente, el genotipo MTHFR 677T/T podría prevenir la incorporación errónea de uracilo y proteger la integridad y estabilidad del ADN bajo condiciones de folato bajo. Un meta-análisis de 62 estudios de caso-control y dos de cohorte revelaron que mientras la variante T/T reduce el riesgo de cáncer colorrectal un 12% en comparación a ambos C/T y C/C genotipos, el riesgo se redujo un 30% con altas (348-1,583 μg/día) frente a ingestas bajas totales de folato (264-450 μg/día), independientemente del genotipo (114). Un polimorfismo común (c.2756A>G) en el gen MTR, el cual codifica para la metionina sintasa, fue también examinado en relación con el riesgo de adenoma colorrectal y cáncer. La metionina sintasa cataliza la conversión simultánea de homocisteína y 5-metileno THF en metionina y TFH, respectivamente. El reciente meta-análisis de 27 estudios de caso y control mostro ninguna asociación entre la variante del MTR y el riesgo de cáncer (115).
Cáncer de seno
Varios estudios de cohorte prospectivos y de caso y control han reportado resultados mixtos al investigar si la ingesta de folato afecta el riesgo de cáncer de seno (119). Un meta-análisis de 15 estudios prospectivos y un estudio caso-control anidado no encontró relación alguna con la ingesta de folato dietaría (120). Una ingesta de alcohol moderada ha sido asociada con un incremento en el riesgo de cáncer de seno en mujeres (121). Los resultados de tres estudios prospectivos sugirieron que una ingesta incrementada de folato puede reducir el riesgo de cáncer de seno en mujeres que regularmente consumen alcohol (122-124). De esta manera, una ingesta alta de folato podría estar asociada con una reducción del riesgo solo en mujeres cuyo riesgo de cáncer es incrementado por el consumo de alcohol. Un estudio prospectivo bastante exhaustivo en más de 88,000 enfermeras reporto que una ingesta de ácido fólico no estaba asociada con cáncer de seno en mujeres que consumieron menos de una bebida alcohólica por día. Sin embargo, en mujeres que consumieron por lo menos una bebida alcohólica por día, la ingesta de ácido fólico de al menos 600 μg diariamente resulto en aproximadamente la mitad del riesgo de cáncer de seno en comparación con las mujeres que consumieron 300 μg de ácido fólico diariamente (124). No obstante, si y como el consumo de alcohol incrementa el riesgo de cáncer de seno es aún un tema de discusión (125, 126). Finalmente, recientes meta-análisis que evaluaron la influencia de polimorfismos en el metabolismo de un carbono en el riesgo de cáncer de seno encontraron que variantes específicas en el gen que codifica la timidilato sintasa incrementaron el riesgo de cáncer de seno en ciertas poblaciones étnicas (127, 128).
Cáncer infantil
La incidencia de tumores de Wilm (cáncer de riñón) y ciertos tipos de cánceres cerebrales (neuroblastoma, ganglioneuroblastoma, y ependimoma) en niños ha disminuido desde la fortificación obligatoria del suministro de granos estadounidense en 1998 (129). Sin embargo, las tasas de incidencia se mantuvieron sin cambios entre los periodos de pre y post fortificación para la leucemia- una malignidad predominante en la infancia. A pesar de estudios tempranos que ligaban la suplementación de ácido fólico materna durante el embarazo con la disminución del riesgo de leucemia infantil, investigaciones más recientes han encontrado poca evidencia para apoyar un efecto preventivo del ácido fólico (130). Varios meta-análisis también han encontrado poco a ningún efecto protector con los polimorfismos de MTHFR; sin embargo el meta-análisis más reciente de 22 estudios de caso y control encontró una reducción en el riesgo de leucemia linfoblástica aguda (LLA) con la variante c.677C>T en caucásicos y asiáticos (131).
Enfermedad de Alzheimer y deterioro cognitivo
La enfermedad de Alzheimer es la forma de demencia más común, afectando más de 5 millones de individuos mayores de 65 años de edad en los EE.UU. (132). Se han asociado la deposición de la placa beta-amiloide, la proteína formadora de ovillos Tau, y un incremento de la muerte celular en el cerebro de los pacientes con Alzheimer con el descenso cognitivo y la perdida de la memoria. Un estudio asocio el incremento del consumo de frutas y verduras, las cuales son fuentes abundantes de folato con un riesgo reducido de desarrollar demencia y Alzheimer en mujeres (133). A través de su papel en la síntesis de ácidos nucleicos y la provisión de donantes de metilo para las reacciones de metilación, el folato es crítico para el funcionamiento y desarrollo normal del cerebro, no solamente durante el embarazo y después del nacimiento, sino también posteriormente en la vida (134). En un estudio de corte transversal en mujeres de edad avanzada, los pacientes con Alzheimer tuvieron concentraciones significantemente más altas de homocisteína y concentraciones más bajas de folato en los glóbulos rojos en comparación con individuos saludables. Sin embargo, no hubo diferencia en el nivel de folato en el suero entre los grupos, sugiriendo que el estatus de folato a largo plazo en lugar de la ingesta reciente de folato, podría estar asociado con el riesgo de Alzheimer (135).
Varios investigadores han descrito asociaciones entre el incremento de las concentraciones de homocisteína y el deterioro cognitivo en las personas mayores (136), pero estudios de cohorte prospectivos no han encontrado que altas ingestas de folato estén asociadas con un mejoramiento de la cognición (137, 138). Altas concentraciones de homocisteína fueron encontradas en individuos que sufrían de demencia, incluyendo Alzheimer, y demencia vascular, en comparación a sujetos sanos (139, 140). Aunque las deficiencias de folato, vitamina B12, y vitamina B6 podrían incrementar las concentraciones de homocisteína, una reducción en las concentraciones de vitamina en el suero de pacientes con Alzheimer en comparación a individuos sanos no pudo ser atribuido a una disminución de las ingestas de vitaminas (141). No está claro actualmente si la homocisteína sérica es un factor de riesgo para el desarrollo de la demencia o simplemente está asociada con la disminución cognitiva. En la última década, un cierto número de ensayos clínicos han probado el uso de las vitaminas B para disminuir la homocisteína y prevenir o retrasar el deterioro cognitivo. Un meta-análisis de nueve ensayos controlados aleatorios con placebo de la suplementación con ácido fólico (0.2 a 15 mg/día por una duración media de seis meses) en individuos mayores de 45 años de edad fallo en encontrar un efecto a corto plazo en las funciones cognitivas, incluyendo memoria, velocidad, lenguaje, y funciones ejecutivas (142). Más recientemente, un meta-análisis de 19 ensayos controlados aleatorios con placebo de la suplementación con vitaminas B no encontró alguna diferencia en los parámetros cognitivos entre los grupos de tratamiento y placebo, a pesar de que el tratamiento efectivamente disminuyo las concentraciones de homocisteína (143). Hallazgos inconsistentes a través de los ensayos puede deberse a diferencias en el diseño y metodología (revisado en 144).
No obstante, un ensayo controlado aleatorio con placebo de dos años en 168 sujetos de edad avanzada con deterioro cognitivo leve describió los beneficios de un régimen diario de 800 μg de ácido fólico, 500 μg de vitamina B12 y 20 mg de vitamina B6 (145, 146). La atrofia de regiones cerebrales especificas afectadas por el Alzheimer fue observada en individuos de ambos grupos, y esta atrofia estaba correlacionada con el deterioro cognitivo; sin embargo, el grupo de tratamiento con vitaminas-B experimento una menor perdida de materia gris en comparación a el grupo de placebo (0.5% vs. 3.7%). Un beneficio mayor fue observado en sujetos con concentraciones mayores de homocisteína al inicio del estudio, sugiriendo la importancia de la disminución de homocisteína circulante en la prevención del deterioro cognitivo y la demencia. Aunque alentador, el efecto de la suplementación con vitaminas B necesita ser estudiado más a fondo en ensayos exhaustivos que evalúen resultados a largo plazo, como la incidencia del Alzheimer.
Tratamiento de Enfermedades
Enfermedades metabólicas
El ácido folínico (vea Figura 1 arriba), un derivado del ácido tetrahidrofólico, es usado en el manejo clínico de errores congénitos raros que afectan el transporte y metabolismo del folato (revisado en 147). Tales condiciones son de una herencia autosómica recesiva, lo que significa que solo individuos que reciben dos copias del gen mutado (uno de cada padre) desarrollan la enfermedad.
Malabsorción del folato hereditaria
La malabsorción del folato hereditaria es causada por mutaciones en la codificación del gen SLC46A1 para el trasportador de folato PCFT y típicamente afecta la absorción gastrointestinal del folato y el transporte del folato en el cerebro (148). Los pacientes se presentan con bajas o indetectables concentraciones de folato en el suero y en el fluido cerebroespinal, pancitopenia (baja cantidad de todos los glóbulos rojos), respuestas inmunes deterioradas que incrementan la susceptibilidad a infecciones, y un fallo general para prosperar (149). Síntomas neurológicos incluyendo convulsiones, han sido también observadas (150). Mejoras clínicas han sido registradas tras la suministración parenteral de ácido folínico (151).
Síndrome de la deficiencia de folato cerebral (DFC)
La DFC se caracteriza por bajos niveles de coenzimas de folato en el fluido cerebroespinal a pesar de concentraciones normales de folato en la sangre. El transporte de folato a través de la barrera hematoencefálica está comprometida en la DFC y ha sido ligada ya sea a la presencia de anticuerpos bloqueando el receptor de folato FRα o a mutaciones en el gen FOLR1 que codifica el FRα (152, 153). Anormalidades neurológicas, junto con deficiencias visuales y auditivas, han sido descritas en niños con DFC; el desorden del espectro autista (TEA) está también presente en algunos casos. El ácido folínico (también conocido como leucovorina) puede entrar en el cerebro y normalizar el nivel de coenzimas de folato y se ha mostrado que normaliza las concentraciones de folato y mejora varias interacciones sociales en la DFC, incluyendo el estado de ánimo, comportamiento, y comunicación verbal en niños con (TEA) (152, 154, 155).
Deficiencia de dihidrofolato reductasa (DHFR)
La DHFR es la enzima dependiente de la NADPH que cataliza la reducción del ácido dihidrofólico (DHF) a ácido tetrahidrofólico (THF). La DHFR es también requerida para convertir el ácido fólico a DHF. La deficiencia de DHFR está caracterizada por una anemia megaloblástica y una deficiencia de folato cerebral causando convulsiones intratables y déficits mentales. Aunque el tratamiento con ácido folínico puede aliviar los síntomas de la deficiencia de DHFR, un diagnóstico temprano es esencial para prevenir un daño irreversible al cerebro y mejorar los resultados clínicos (156, 157).
Fuentes
Fuentes alimenticias
Las verduras de hojas verdes (follaje) son fuentes ricas en folato y proporcionan la base para su nombre. Los jugos de frutas cítricas, las legumbres, y los alimentos fortificados son también excelentes fuentes de folato (1); el contenido de folato de cereales fortificados varía en gran medida. La siguiente tabla muestra una serie de alimentos ricos en folato, junto con su contenido de folato en microgramos (μg). Para mayor información del contenido de nutrientes de alimentos específicos, busque en la base de datos de composición de los alimentos del USDA.
| Alimento | Porción | Folato (μg) |
|---|---|---|
| Lentejas (semillas maduras, cocidas, hervidas) | ½ taza | 179 |
| Garbanzos (cocidos, hervidos) | ½ taza | 141 |
| Espárragos (cocidos, hervidos) | ½ taza (~6 varitas) | 134 |
| Espinacas (cocidas, hervidas) | ½ taza | 131 |
| Habas (grandes, semillas maduras, cocidas, hervidas) | ½ taza | 78 |
| Jugo de naranja (recién extraído) | 6 onzas | 56 |
| Espagueti (enriquecido, cocido) | 1 taza | 167* |
| Arroz blanco (enriquecido, cocido) | 1 taza | 153* |
| Pan (enriquecido) | 1 rebana | 84* |
| *Para ayudar a prevenir defectos del tubo neural, la FDA ordenó que se agregara 1.4 miligramos (mg) de ácido fólico por cada kilogramos (kg) de cereal a los productos de grano refinado, los que ya estaban enriquecidos con niacina, tiamina, riboflavina, y hierro desde el 1ro de enero de 1998. La adición de nutrientes en los alimentos con el fin de prevenir una deficiencia nutricional, o restaurar los nutrientes perdidos en el proceso, se conoce como fortificación. La FDA inicialmente estimo que este nivel de fortificación incrementaría la ingesta dietaría 100 μg de ácido fólico al día en promedio. Sin embargo evaluaciones adicionales basadas en estudios basados en la observación sugirieron aumentos del doble que lo predicho por la FDA (35). La prevalencia de bajas concentraciones de folato en ambos suero y eritrocitos es actualmente menos de 1% en la población de los EE.UU., en comparación a 24% y 3.5% respectivamente antes de los periodos de fortificación (158). | ||
Suplementos
La principal forma de folato suplementario es el ácido fólico. Este se encuentra disponible en un sólo ingrediente y en productos combinados como vitaminas del complejo B y multivitamínicos. Dosis iguales o superiores a 1 mg requieren de receta médica (159). Adicionalmente, el ácido folínico, un derivado del ácido tetrahidrofólico, es usado para el manejo de ciertas enfermedades metabólicas (véase Tratamiento de Enfermedades). Ademas, la FDA estadounidense ha aprobado la suplementación de folato en anticonceptivos orales. La adición de levomefolato de calcio (la sal de calcio del meTHF; 451 μg/tableta) a anticonceptivos orales está destinada a elevar el nivel de folato en las mujeres en edad fértil (160). De acuerdo a un estudio nacional estadounidense, solo un 24% de mujeres no embarazadas entre la edad de 15-44 años están cumpliendo con la recomendación actual de 400 μg/día de ácido fólico (161).
Seguridad
Toxicidad
No hay efectos adversos asociados con el consumo en exceso de folato proveniente de alimentos. Las preocupaciones en cuanto a su seguridad se limitan a la ingesta de ácido fólico sintético. Una deficiencia de vitamina B12, aunque con frecuencia sin diagnosticar, podría afectar a un número significativo de personas, especialmente a adultos mayores (véase el artículo separado en Vitamina B12). Uno de los síntomas de la deficiencia de vitamina B12 es la anemia megaloblástica, la cual es indistinguible de la que se asocia con la deficiencia de folato (véase Deficiencia). Grandes dosis de ácido fólico, administradas a un individuo con deficiencia de vitamina B12 no diagnosticada, pueden corregir la anemia megaloblástica sin corregir la deficiencia de vitamina B12 subyacente, dejando al individuo en riesgo de desarrollar un daño neurológico irreversible. Tales casos de progresión neurológica en la deficiencia de vitamina B12, se han visto en su mayoría en dosis de ácido fólico de 5,000 μg (5 mg) o más altas. Con el fin de asegurar la prevención de un daño neurológico irreversible en personas con deficiencia de vitamina B12, La Junta de Nutrición y Alimentos del Instituto de Medicina estadounidense recomienda que todos los adultos limiten su ingesta de ácido fólico (suplementos y fortificación) a 1,000 μg (1 mg diario). La junta también señaló que la deficiencia de vitamina B12, es muy rara en mujeres en edad fértil, haciendo poco probable que un consumo de ácido fólico igual o mayor a 1,000 μg/día, cause problemas (1); sin embargo, la información sobre los efectos de grandes dosis es limitada.
| Nivel de Ingesta Tolerable Máximo (NM) para el Ácido Fólico | |
| Grupo Etario | NM (μg/día) |
|---|---|
| Infantes 0-12 meses | No es posible establecer* |
| Niños 1-3 años | 300 |
| Niños 4-8 años | 400 |
| Niños 9-13 años | 600 |
| Adolescentes 14-18 años | 800 |
| Adultos 19 años o más | 1,000 |
| *La fuente de ingesta debiese ser solo de alimentos y formula. | |
La saturación de la capacidad metabólica de la DHFR por dosis orales de ácido fólico ha sido asociada con la aparición de ácido fólico sin metabolizar en la sangre (162). Alteraciones hematológicas y una cognición más pobre han sido asociadas con la presencia de ácido fólico sin metabolizar en adultos mayores (≥60 años) con una deficiencia de vitamina B12 (163, 164). Un estudio pequeño conducido en mujeres postmenopáusicas también planteo preocupaciones acerca del efecto de la exposición de ácido fólico sin metabolizar en la función inmune (165). En un pequeño ensayo aleatorio abierto en 38 mujeres en edad reproductiva que recibieron 30 semanas de suplementos multivitamínicos diarios, una suplementación diaria con ya sea 1.1 mg o 5mg de ácido fólico resulto en la aparición transitoria de ácido fólico sin metabolizar en la sangre en las primeras 12 semanas de la suplementación (166). Sin embargo las concentraciones de ácido fólico sin metabolizar regresaron a los niveles del inicio del estudio al final del estudio, sugiriendo que los mecanismos de adaptación eventualmente convirtieron el ácido fólico a formas reducidas de folato. No obstante, el uso de levomefolato (5-metil THF) suplementario podría proveer de una alternativa para prevenir los potenciales efectos negativos del ácido fólico sin convertir en adultos mayores.
Interacción con drogas/fármacos
Cuando se toman drogas anti-inflamatorias no esteroidales (AINEs), como la aspirina o el ibuprofeno, en dosis terapéuticas muy grandes (por ejemplo, para tratar artritis severa), éstas podrían interferir con el metabolismo del folato. En comparación, no se ha demostrado que el uso rutinario de AINEs en bajas dosis afecte negativamente el estatus del folato. Se ha demostrado que el anticonvulsivo fenitoina inhibe la absorción intestinal de folato, y varios estudios han asociado un estado de folato disminuido con el uso a largo plazo de los anticonvulsivos fenitoina, fenobarbital, y pirimidona (167). Sin embargo, pocos estudios controlaron las diferencias en la ingesta de folato dietario, entre los usuarios, y los no usuarios de anticonvulsivos. Además, tomar ácido fólico al mismo tiempo que los agentes para reducir colesterol, colestiramina y colestipol, podría disminuir la absorción de ácido fólico (159). El metotrexato es un antagonista del ácido fólico, usado para tratar una serie de enfermedades incluyendo cáncer, artritis reumatoide y psoriasis. Algunos de los efectos secundarios del metotrexato son parecidos a los de una deficiencia de folato severa, y la suplementación con ácido fólico o folínico es usada para reducir la toxicidad del anti-folato. Otras moléculas anti-folato actualmente usadas en la terapia contra el cáncer incluyen aminopterina, pemetrexed, y raltitrexed (96). Además se ha demostrado que una serie de otros medicamentos tienen una actividad anti-folato, incluyendo trimetoprima (un antibiótico), pirimetamina (un antimalarial), triamtereno (un medicamento para la presión sanguínea), y sulfasalazina (un tratamiento para la colitis ulcerosa). Los primeros estudios de anticonceptivos orales (píldoras anticonceptivas), que contenían altas dosis de estrógeno, mostraron efectos adversos sobre los niveles del folato; sin embargo, estos hallazgos no han sido respaldados por estudios más recientes con bajas dosis de anticonceptivos donde la ingesta de folato fue controlada (168).
Recomendación del Instituto Linus Pauling
La evidencia científica disponible demuestra que una ingesta de folato adecuada previene defectos del tubo neural y otros problemas del embarazo, es útil en la disminución del riesgo de algunas formas de cáncer, especialmente en personas genéticamente susceptibles, y que podría reducir el riesgo de enfermedades cardiovasculares. El Instituto Linus Pauling recomienda que los adultos tomen un suplemento multivitamínico-mineral diario, el cual típicamente contiene 400 μg de ácido fólico, el Valor Diario (VD). Incluso con una ingesta de ácido fólico mayor al promedio de alimentos fortificados, es poco probable que la ingesta de ácido fólico diaria de una persona regularmente exceda el nivel de ingesta máxima de 1,000 μg/día, establecida por la Junta de Nutrición y Alimentos (véase Seguridad).
Adultos mayores (>50 años)
La recomendación de 400 μg/día de ácido fólico suplementario, como parte de un suplemento multivitamínico-mineral diario, además de una dieta rica en folato, es especialmente importante en adultos mayores debido a que los niveles de homocisteína sanguínea tienden a aumentar con la edad (véase Prevención de Enfermedades).
Autores y Críticos
Escrito en 2000 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Abril de 2002 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Septiembre de 2007 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Junio de 2014 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Diciembre de 2014 por:
Helene McNulty, Ph.D., R.D.
Profesor de Nutrición Humana y Dietética
Centro para la Alimentación y Salud de Irlanda del Norte (NICHE)
Universidad de Ulster
Coleraine, Reino Unido
Traducido al Español en 2015 por:
Silvia Vazquez Lima
Instituto Linus Pauling
Universidad Estatal de Oregon
La actualización del 2014 de este artículo fue suscrito, por una subvención de Bayer Consumer Care AG, Basel, Suiza.
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Derechos de autoría 2009-2024 Instituto Linus Pauling
Referencias
1. Food and Nutrition Board, Institute of Medicine. Folate. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, D.C.: National Academy Press; 1998:196-305. (National Academy Press)
2. Choi SW, Mason JB. Folate and carcinogenesis: an integrated scheme. J Nutr. 2000;130(2):129-132. (PubMed)
3. Bailey LB, Gregory JF, 3rd. Folate metabolism and requirements. J Nutr. 1999;129(4):779-782. (PubMed)
4. Gerhard GT, Duell PB. Homocysteine and atherosclerosis. Curr Opin Lipidol. 1999;10(5):417-428. (PubMed)
5. Jacques PF, Bostom AG, Wilson PW, Rich S, Rosenberg IH, Selhub J. Determinants of plasma total homocysteine concentration in the Framingham Offspring cohort. Am J Clin Nutr. 2001;73(3):613-621. (PubMed)
6. Jacques PF, Kalmbach R, Bagley PJ, et al. The relationship between riboflavin and plasma total homocysteine in the Framingham Offspring cohort is influenced by folate status and the C677T transition in the methylenetetrahydrofolate reductase gene. J Nutr. 2002;132(2):283-288. (PubMed)
7. McNulty H, Dowey le RC, Strain JJ, et al. Riboflavin lowers homocysteine in individuals homozygous for the MTHFR 677C->T polymorphism. Circulation. 2006;113(1):74-80. (PubMed)
8. Verlinde PH, Oey I, Hendrickx ME, Van Loey AM, Temme EH. L-ascorbic acid improves the serum folate response to an oral dose of [6S]-5-methyltetrahydrofolic acid in healthy men. Eur J Clin Nutr. 2008;62(10):1224-1230. (PubMed)
9. Lucock M, Yates Z, Boyd L, et al. Vitamin C-related nutrient-nutrient and nutrient-gene interactions that modify folate status. Eur J Nutr. 2013;52(2):569-582. (PubMed)
10. Desmoulin SK, Hou Z, Gangjee A, Matherly LH. The human proton-coupled folate transporter: Biology and therapeutic applications to cancer. Cancer Biol Ther. 2012;13(14):1355-1373. (PubMed)
11. Solanky N, Requena Jimenez A, D'Souza SW, Sibley CP, Glazier JD. Expression of folate transporters in human placenta and implications for homocysteine metabolism. Placenta. 2010;31(2):134-143. (PubMed)
12. Halsted CH, Villanueva JA, Devlin AM, Chandler CJ. Metabolic interactions of alcohol and folate. J Nutr. 2002;132(8 Suppl):2367S-2372S. (PubMed)
13. Pfeiffer CM, Sternberg MR, Schleicher RL, Rybak ME. Dietary supplement use and smoking are important correlates of biomarkers of water-soluble vitamin status after adjusting for sociodemographic and lifestyle variables in a representative sample of US adults. J Nutr. 2013;143(6):957S-965S. (PubMed)
14. Stark KD, Pawlosky RJ, Sokol RJ, Hannigan JH, Salem N, Jr. Maternal smoking is associated with decreased 5-methyltetrahydrofolate in cord plasma. Am J Clin Nutr. 2007;85(3):796-802. (PubMed)
15. Hutson JR, Stade B, Lehotay DC, Collier CP, Kapur BM. Folic acid transport to the human fetus is decreased in pregnancies with chronic alcohol exposure. PLoS One. 2012;7(5):e38057. (PubMed)
16. Herbert V. Folic acid. In: Shils M, Olson JA, Shike M, Ross AC, eds. Modern Nutrition in Health and Disease. 9th ed. Baltimore: Lippincott Williams & Wilkins; 1999:433-446.
17. Stabler SP. Clinical folate deficiency. In: Bailey LB, ed. Folate in Health and Disease. 2nd edition ed. Boca Raton, FL: CRC press, Taylor & Francis Group; 2010:409-428.
18. Bailey LB. Dietary reference intakes for folate: the debut of dietary folate equivalents. Nutr Rev. 1998;56(10):294-299. (PubMed)
19. Bailey LB, Gregory JF, 3rd. Polymorphisms of methylenetetrahydrofolate reductase and other enzymes: metabolic significance, risks and impact on folate requirement. J Nutr. 1999;129(5):919-922. (PubMed)
20. Wilcken B, Bamforth F, Li Z, et al. Geographical and ethnic variation of the 677C>T allele of 5,10 methylenetetrahydrofolate reductase (MTHFR): findings from over 7000 newborns from 16 areas world wide. J Med Genet. 2003;40(8):619-625. (PubMed)
21. Guenther BD, Sheppard CA, Tran P, Rozen R, Matthews RG, Ludwig ML. The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomocysteinemia. Nat Struct Biol. 1999;6(4):359-365. (PubMed)
22. Molloy AM, Daly S, Mills JL, et al. Thermolabile variant of 5,10-methylenetetrahydrofolate reductase associated with low red-cell folates: implications for folate intake recommendations. Lancet. 1997;349(9065):1591-1593. (PubMed)
23. Rozen R. Genetic predisposition to hyperhomocysteinemia: deficiency of methylenetetrahydrofolate reductase (MTHFR). Thromb Haemost. 1997;78(1):523-526. (PubMed)
24. Kauwell GP, Wilsky CE, Cerda JJ, et al. Methylenetetrahydrofolate reductase mutation (677C-->T) negatively influences plasma homocysteine response to marginal folate intake in elderly women. Metabolism. 2000;49(11):1440-1443. (PubMed)
25. Shane B. Folic acid, vitamin B-12, and vitamin B-6. In: Stipanuk M, ed. Biochemical and Physiological Aspects of Human Nutrition. Philadelphia: W.B. Saunders Co.; 2000:483-518.
26. Eskes TK. Open or closed? A world of difference: a history of homocysteine research. Nutr Rev. 1998;56(8):236-244. (PubMed)
27. Czeizel AE, Dudas I, Vereczkey A, Banhidy F. Folate deficiency and folic acid supplementation: the prevention of neural-tube defects and congenital heart defects. Nutrients. 2013;5(11):4760-4775. (PubMed)
28. MRC Vitamin Study Research Group. Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet. 1991;338(8760):131-137. (PubMed)
29. Czeizel AE, Dudas I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med. 1992;327(26):1832-1835. (PubMed)
30. Talaulikar VS, Arulkumaran S. Folic acid in obstetric practice: a review. Obstet Gynecol Surv. 2011;66(4):240-247. (PubMed)
31. American College of Obstetricians and Gynecologists (ACOG). Neural tube defects. Washington, DC. 2003. Available at: http://www.guideline.gov/content.aspx?id=3994. Accessed 12/19/14.
32. McNulty B, Pentieva K, Marshall B, et al. Women's compliance with current folic acid recommendations and achievement of optimal vitamin status for preventing neural tube defects. Hum Reprod. 2011;26(6):1530-1536. (PubMed)
33. Nilsen RM, Vollset SE, Gjessing HK, et al. Patterns and predictors of folic acid supplement use among pregnant women: the Norwegian Mother and Child Cohort Study. Am J Clin Nutr. 2006;84(5):1134-1141. (PubMed)
34. Ray JG, Singh G, Burrows RF. Evidence for suboptimal use of periconceptional folic acid supplements globally. BJOG. 2004;111(5):399-408. (PubMed)
35. Quinlivan EP, Gregory JF, 3rd. Effect of food fortification on folic acid intake in the United States. Am J Clin Nutr. 2003;77(1):221-225. (PubMed)
36. National Birth Defects Prevention Network. Neural Tube Defect Ascertainment Project. Available at: http://www.nbdpn.org/ntd_folic_acid_information.php. Accessed 12/16/14.
37. Copp AJ, Stanier P, Greene ND. Neural tube defects: recent advances, unsolved questions, and controversies. Lancet Neurol. 2013;12(8):799-810. (PubMed)
38. Yan L, Zhao L, Long Y, et al. Association of the maternal MTHFR C677T polymorphism with susceptibility to neural tube defects in offsprings: evidence from 25 case-control studies. PLoS One. 2012;7(10):e41689. (PubMed)
39. De Marco P, Calevo MG, Moroni A, et al. Study of MTHFR and MS polymorphisms as risk factors for NTD in the Italian population. J Hum Genet. 2002;47(6):319-324. (PubMed)
40. van der Put NM, Gabreels F, Stevens EM, et al. A second common mutation in the methylenetetrahydrofolate reductase gene: an additional risk factor for neural-tube defects? Am J Hum Genet. 1998;62(5):1044-1051. (PubMed)
41. De Marco P, Calevo MG, Moroni A, et al. Reduced folate carrier polymorphism (80A-->G) and neural tube defects. Eur J Hum Genet. 2003;11(3):245-252. (PubMed)
42. O'Leary VB, Mills JL, Parle-McDermott A, et al. Screening for new MTHFR polymorphisms and NTD risk. Am J Med Genet A. 2005;138A(2):99-106. (PubMed)
43. Christensen B, Arbour L, Tran P, et al. Genetic polymorphisms in methylenetetrahydrofolate reductase and methionine synthase, folate levels in red blood cells, and risk of neural tube defects. Am J Med Genet. 1999;84(2):151-157. (PubMed)
44. Relton CL, Wilding CS, Pearce MS, et al. Gene-gene interaction in folate-related genes and risk of neural tube defects in a UK population. J Med Genet. 2004;41(4):256-260. (PubMed)
45. Brody LC, Conley M, Cox C, et al. A polymorphism, R653Q, in the trifunctional enzyme methylenetetrahydrofolate dehydrogenase/methenyltetrahydrofolate cyclohydrolase/formyltetrahydrofolate synthetase is a maternal genetic risk factor for neural tube defects: report of the Birth Defects Research Group. Am J Hum Genet. 2002;71(5):1207-1215. (PubMed)
46. van der Put NM, van den Heuvel LP, Steegers-Theunissen RP, et al. Decreased methylene tetrahydrofolate reductase activity due to the 677C-->T mutation in families with spina bifida offspring. J Mol Med (Berl). 1996;74(11):691-694. (PubMed)
47. Wilson A, Platt R, Wu Q, et al. A common variant in methionine synthase reductase combined with low cobalamin (vitamin B12) increases risk for spina bifida. Mol Genet Metab. 1999;67(4):317-323. (PubMed)
48. Gilboa SM, Salemi JL, Nembhard WN, Fixler DE, Correa A. Mortality resulting from congenital heart disease among children and adults in the United States, 1999 to 2006. Circulation. 2010;122(22):2254-2263. (PubMed)
49. van Beynum IM, Kapusta L, Bakker MK, den Heijer M, Blom HJ, de Walle HE. Protective effect of periconceptional folic acid supplements on the risk of congenital heart defects: a registry-based case-control study in the northern Netherlands. Eur Heart J. 2010;31(4):464-471. (PubMed)
50. Yin M, Dong L, Zheng J, Zhang H, Liu J, Xu Z. Meta analysis of the association between MTHFR C677T polymorphism and the risk of congenital heart defects. Ann Hum Genet. 2012;76(1):9-16. (PubMed)
51. Wang W, Wang Y, Gong F, Zhu W, Fu S. MTHFR C677T polymorphism and risk of congenital heart defects: evidence from 29 case-control and TDT studies. PLoS One. 2013;8(3):e58041. (PubMed)
52. Badovinac RL, Werler MM, Williams PL, Kelsey KT, Hayes C. Folic acid-containing supplement consumption during pregnancy and risk for oral clefts: a meta-analysis. Birth Defects Res A Clin Mol Teratol. 2007;79(1):8-15. (PubMed)
53. Wilcox AJ, Lie RT, Solvoll K, et al. Folic acid supplements and risk of facial clefts: national population based case-control study. BMJ. 2007;334(7591):464. (PubMed)
54. Boyles AL, Wilcox AJ, Taylor JA, et al. Folate and one-carbon metabolism gene polymorphisms and their associations with oral facial clefts. Am J Med Genet A. 2008;146A(4):440-449. (PubMed)
55. Boyles AL, Wilcox AJ, Taylor JA, et al. Oral facial clefts and gene polymorphisms in metabolism of folate/one-carbon and vitamin A: a pathway-wide association study. Genet Epidemiol. 2009;33(3):247-255. (PubMed)
56. Luo YL, Cheng YL, Ye P, Wang W, Gao XH, Chen Q. Association between MTHFR polymorphisms and orofacial clefts risk: a meta-analysis. Birth Defects Res A Clin Mol Teratol. 2012;94(4):237-244. (PubMed)
57. Wilcox AJ. On the importance--and the unimportance--of birthweight. Int J Epidemiol. 2001;30(6):1233-1241. (PubMed)
58. Fekete K, Berti C, Trovato M, et al. Effect of folate intake on health outcomes in pregnancy: a systematic review and meta-analysis on birth weight, placental weight and length of gestation. Nutr J. 2012;11:75. (PubMed)
59. Baker PN, Wheeler SJ, Sanders TA, et al. A prospective study of micronutrient status in adolescent pregnancy. Am J Clin Nutr. 2009;89(4):1114-1124. (PubMed)
60. Lee HA, Park EA, Cho SJ, et al. Mendelian randomization analysis of the effect of maternal homocysteine during pregnancy, as represented by maternal MTHFR C677T genotype, on birth weight. J Epidemiol. 2013;23(5):371-375. (PubMed)
61. Scholl TO, Johnson WG. Folic acid: influence on the outcome of pregnancy. Am J Clin Nutr. 2000;71(5 Suppl):1295S-1303S. (PubMed)
62. Vollset SE, Refsum H, Irgens LM, et al. Plasma total homocysteine, pregnancy complications, and adverse pregnancy outcomes: the Hordaland Homocysteine study. Am J Clin Nutr. 2000;71(4):962-968. (PubMed)
63. Wang XM, Wu HY, Qiu XJ. Methylenetetrahydrofolate reductase (MTHFR) gene C677T polymorphism and risk of preeclampsia: an updated meta-analysis based on 51 studies. Arch Med Res. 2013;44(3):159-168. (PubMed)
64. Wen SW, Champagne J, Rennicks White R, et al. Effect of folic acid supplementation in pregnancy on preeclampsia: the folic acid clinical trial study. J Pregnancy. 2013;2013:294312. (PubMed)
65. Lassi ZS, Salam RA, Haider BA, Bhutta ZA. Folic acid supplementation during pregnancy for maternal health and pregnancy outcomes. Cochrane Database Syst Rev. 2013;3:CD006896. (PubMed)
66. Schmidt RJ, Tancredi DJ, Ozonoff S, et al. Maternal periconceptional folic acid intake and risk of autism spectrum disorders and developmental delay in the CHARGE (CHildhood Autism Risks from Genetics and Environment) case-control study. Am J Clin Nutr. 2012;96(1):80-89. (PubMed)
67. Crider KS, Cordero AM, Qi YP, Mulinare J, Dowling NF, Berry RJ. Prenatal folic acid and risk of asthma in children: a systematic review and meta-analysis. Am J Clin Nutr. 2013;98(5):1272-1281. (PubMed)
68. Brown SB, Reeves KW, Bertone-Johnson ER. Maternal folate exposure in pregnancy and childhood asthma and allergy: a systematic review. Nutr Rev. 2014;72(1):55-64. (PubMed)
69. Ding R, Lin S, Chen D. The association of cystathionine β synthase (CBS) T833C polymorphism and the risk of stroke: a meta-analysis. J Neurol Sci. 2012;312(1-2):26-30. (PubMed)
70. Seshadri N, Robinson K. Homocysteine, B vitamins, and coronary artery disease. Med Clin North Am. 2000;84(1):215-237. (PubMed)
71. Wald DS, Law M, Morris JK. Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis. BMJ. 2002;325(7374):1202. (PubMed)
72. Homocysteine Studies Collaboration. Homocysteine and risk of ischemic heart disease and stroke: a meta-analysis. JAMA. 2002;288(16):2015-2022. (PubMed)
73. Clarke R, Halsey J, Bennett D, Lewington S. Homocysteine and vascular disease: review of published results of the homocysteine-lowering trials. J Inherit Metab Dis. 2011;34(1):83-91. (PubMed)
74. Huang T, Chen Y, Yang B, Yang J, Wahlqvist ML, Li D. Meta-analysis of B vitamin supplementation on plasma homocysteine, cardiovascular and all-cause mortality. Clin Nutr. 2012;31(4):448-454. (PubMed)
75. Voutilainen S, Rissanen TH, Virtanen J, Lakka TA, Salonen JT. Low dietary folate intake is associated with an excess incidence of acute coronary events: The Kuopio Ischemic Heart Disease Risk Factor Study. Circulation. 2001;103(22):2674-2680. (PubMed)
76. Brattstrom L. Vitamins as homocysteine-lowering agents. J Nutr. 1996;126(4 Suppl):1276S-1280S. (PubMed)
77. Rader JI. Folic acid fortification, folate status and plasma homocysteine. J Nutr. 2002;132(8 Suppl):2466S-2470S. (PubMed)
78. Homocysteine Lowering Trialists' Collaboration. Dose-dependent effects of folic acid on blood concentrations of homocysteine: a meta-analysis of the randomized trials. Am J Clin Nutr. 2005;82(4):806-812. (PubMed)
79. Malinow MR, Bostom AG, Krauss RM. Homocyst(e)ine, diet, and cardiovascular diseases: a statement for healthcare professionals from the Nutrition Committee, American Heart Association. Circulation. 1999;99(1):178-182. (PubMed)
80. van Meurs JB, Pare G, Schwartz SM, et al. Common genetic loci influencing plasma homocysteine concentrations and their effect on risk of coronary artery disease. Am J Clin Nutr. 2013;98(3):668-676. (PubMed)
81. Holmes MV, Newcombe P, Hubacek JA, et al. Effect modification by population dietary folate on the association between MTHFR genotype, homocysteine, and stroke risk: a meta-analysis of genetic studies and randomised trials. Lancet. 2011;378(9791):584-594. (PubMed)
82. Clarke R, Bennett DA, Parish S, et al. Homocysteine and coronary heart disease: meta-analysis of MTHFR case-control studies, avoiding publication bias. PLoS Med. 2012;9(2):e1001177. (PubMed)
83. Ji Y, Tan S, Xu Y, et al. Vitamin B supplementation, homocysteine levels, and the risk of cerebrovascular disease: A meta-analysis. Neurology. 2013;81(15):1298-1307. (PubMed)
84. Zhang C, Chi FL, Xie TH, Zhou YH. Effect of B-vitamin supplementation on stroke: a meta-analysis of randomized controlled trials. PLoS One. 2013;8(11):e81577. (PubMed)
85. Mosca L, Benjamin EJ, Berra K, et al. Effectiveness-based guidelines for the prevention of cardiovascular disease in women--2011 update: a guideline from the American Heart Association. J Am Coll Cardiol. 2011;57(12):1404-1423. (PubMed)
86. Wang X, Qin X, Demirtas H, et al. Efficacy of folic acid supplementation in stroke prevention: a meta-analysis. Lancet. 2007;369(9576):1876-1882. (PubMed)
87. Yang Q, Botto LD, Erickson JD, et al. Improvement in stroke mortality in Canada and the United States, 1990 to 2002. Circulation. 2006;113(10):1335-1343. (PubMed)
88. Lorenz MW, Markus HS, Bots ML, Rosvall M, Sitzer M. Prediction of clinical cardiovascular events with carotid intima-media thickness: a systematic review and meta-analysis. Circulation. 2007;115(4):459-467. (PubMed)
89. Qin X, Xu M, Zhang Y, et al. Effect of folic acid supplementation on the progression of carotid intima-media thickness: a meta-analysis of randomized controlled trials. Atherosclerosis. 2012;222(2):307-313. (PubMed)
90. de Bree A, van Mierlo LA, Draijer R. Folic acid improves vascular reactivity in humans: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2007;86(3):610-617. (PubMed)
91. McNeil CJ, Beattie JH, Gordon MJ, Pirie LP, Duthie SJ. Nutritional B vitamin deficiency disrupts lipid metabolism causing accumulation of proatherogenic lipoproteins in the aorta adventitia of ApoE null mice. Mol Nutr Food Res. 2012;56(7):1122-1130. (PubMed)
92. Blount BC, Mack MM, Wehr CM, et al. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. Proc Natl Acad Sci U S A. 1997;94(7):3290-3295. (PubMed)
93. Narayanan S, McConnell J, Little J, et al. Associations between two common variants C677T and A1298C in the methylenetetrahydrofolate reductase gene and measures of folate metabolism and DNA stability (strand breaks, misincorporated uracil, and DNA methylation status) in human lymphocytes in vivo. Cancer Epidemiol Biomarkers Prev. 2004;13(9):1436-1443. (PubMed)
94. Rahman L, Voeller D, Rahman M, et al. Thymidylate synthase as an oncogene: a novel role for an essential DNA synthesis enzyme. Cancer Cell. 2004;5(4):341-351. (PubMed)
95. Hubner RA, Liu JF, Sellick GS, Logan RF, Houlston RS, Muir KR. Thymidylate synthase polymorphisms, folate and B-vitamin intake, and risk of colorectal adenoma. Br J Cancer. 2007;97(10):1449-1456. (PubMed)
96. Desmoulin SK, Wang L, Polin L, et al. Functional loss of the reduced folate carrier enhances the antitumor activities of novel antifolates with selective uptake by the proton-coupled folate transporter. Mol Pharmacol. 2012;82(4):591-600. (PubMed)
97. Crider KS, Yang TP, Berry RJ, Bailey LB. Folate and DNA methylation: a review of molecular mechanisms and the evidence for folate's role. Adv Nutr. 2012;3(1):21-38. (PubMed)
98. Butrum RR, Clifford CK, Lanza E. NCI dietary guidelines: rationale. Am J Clin Nutr. 1988;48(3 Suppl):888-895. (PubMed)
99. Crider KS, Bailey LB, Berry RJ. Folic acid food fortification-its history, effect, concerns, and future directions. Nutrients. 2011;3(3):370-384. (PubMed)
100. Qin X, Cui Y, Shen L, et al. Folic acid supplementation and cancer risk: A meta-analysis of randomized controlled trials. Int J Cancer. 2013;133(5):1033-1041. (PubMed)
101. Vollset SE, Clarke R, Lewington S, et al. Effects of folic acid supplementation on overall and site-specific cancer incidence during the randomised trials: meta-analyses of data on 50,000 individuals. Lancet. 2013;381(9871):1029-1036. (PubMed)
102. Kim DH, Smith-Warner SA, Spiegelman D, et al. Pooled analyses of 13 prospective cohort studies on folate intake and colon cancer. Cancer Causes Control. 2010;21(11):1919-1930. (PubMed)
103. Gibson TM, Weinstein SJ, Pfeiffer RM, et al. Pre- and postfortification intake of folate and risk of colorectal cancer in a large prospective cohort study in the United States. Am J Clin Nutr. 2011;94(4):1053-1062. (PubMed)
104. Stevens VL, McCullough ML, Sun J, Jacobs EJ, Campbell PT, Gapstur SM. High levels of folate from supplements and fortification are not associated with increased risk of colorectal cancer. Gastroenterology. 2011;141(1):98-105, 105 e101. (PubMed)
105. Zschabitz S, Cheng TY, Neuhouser ML, et al. B vitamin intakes and incidence of colorectal cancer: results from the Women's Health Initiative Observational Study cohort. Am J Clin Nutr. 2013;97(2):332-343. (PubMed)
106. Keum N, Giovannucci EL. Folic acid fortification and colorectal cancer risk. Am J Prev Med. 2014;46(3 Suppl 1):S65-72. (PubMed)
107. Kennedy DA, Stern SJ, Moretti M, et al. Folate intake and the risk of colorectal cancer: a systematic review and meta-analysis. Cancer Epidemiol. 2011;35(1):2-10. (PubMed)
108. Kim YI. Folate: a magic bullet or a double edged sword for colorectal cancer prevention? Gut. 2006;55(10):1387-1389. (PubMed)
109. Paspatis GA, Kalafatis E, Oros L, Xourgias V, Koutsioumpa P, Karamanolis DG. Folate status and adenomatous colonic polyps. A colonoscopically controlled study. Dis Colon Rectum. 1995;38(1):64-67; discussion 67-68. (PubMed)
110. Jaszewski R, Misra S, Tobi M, et al. Folic acid supplementation inhibits recurrence of colorectal adenomas: a randomized chemoprevention trial. World J Gastroenterol. 2008;14(28):4492-4498. (PubMed)
111. Wu K, Platz EA, Willett WC, et al. A randomized trial on folic acid supplementation and risk of recurrent colorectal adenoma. Am J Clin Nutr. 2009;90(6):1623-1631. (PubMed)
112. Logan RF, Grainge MJ, Shepherd VC, Armitage NC, Muir KR. Aspirin and folic acid for the prevention of recurrent colorectal adenomas. Gastroenterology. 2008;134(1):29-38. (PubMed)
113. Figueiredo JC, Mott LA, Giovannucci E, et al. Folic acid and prevention of colorectal adenomas: a combined analysis of randomized clinical trials. Int J Cancer. 2011;129(1):192-203. (PubMed)
114. Kennedy DA, Stern SJ, Matok I, et al. Folate intake, MTHFR polymorphisms, and the risk of colorectal cancer: a systematic review and meta-analysis. J Cancer Epidemiol. 2012;2012:952508. (PubMed)
115. Ding W, Zhou DL, Jiang X, Lu LS. Methionine synthase A2756G polymorphism and risk of colorectal adenoma and cancer: evidence based on 27 studies. PLoS One. 2013;8(4):e60508. (PubMed)
116. Nan H, Lee JE, Rimm EB, Fuchs CS, Giovannucci EL, Cho E. Prospective study of alcohol consumption and the risk of colorectal cancer before and after folic acid fortification in the United States. Ann Epidemiol. 2013;23(9):558-563. (PubMed)
117. Slattery ML, Potter JD, Samowitz W, Schaffer D, Leppert M. Methylenetetrahydrofolate reductase, diet, and risk of colon cancer. Cancer Epidemiol Biomarkers Prev. 1999;8(6):513-518. (PubMed)
118. Ma J, Stampfer MJ, Giovannucci E, et al. Methylenetetrahydrofolate reductase polymorphism, dietary interactions, and risk of colorectal cancer. Cancer Res. 1997;57(6):1098-1102. (PubMed)
119. Larsson SC, Giovannucci E, Wolk A. Folate and risk of breast cancer: a meta-analysis. J Natl Cancer Inst. 2007;99(1):64-76. (PubMed)
120. Liu M, Cui LH, Ma AG, Li N, Piao JM. Lack of effects of dietary folate intake on risk of breast cancer: an updated meta-analysis of prospective studies. Asian Pac J Cancer Prev. 2014;15(5):2323-2328. (PubMed)
121. Brooks PJ, Zakhari S. Moderate alcohol consumption and breast cancer in women: from epidemiology to mechanisms and interventions. Alcohol Clin Exp Res. 2013;37(1):23-30. (PubMed)
122. Rohan TE, Jain MG, Howe GR, Miller AB. Dietary folate consumption and breast cancer risk. J Natl Cancer Inst. 2000;92(3):266-269. (PubMed)
123. Sellers TA, Kushi LH, Cerhan JR, et al. Dietary folate intake, alcohol, and risk of breast cancer in a prospective study of postmenopausal women. Epidemiology. 2001;12(4):420-428. (PubMed)
124. Zhang S, Hunter DJ, Hankinson SE, et al. A prospective study of folate intake and the risk of breast cancer. JAMA. 1999;281(17):1632-1637. (PubMed)
125. Tjonneland A, Christensen J, Olsen A, et al. Alcohol intake and breast cancer risk: the European Prospective Investigation into Cancer and Nutrition (EPIC). Cancer Causes Control. 2007;18(4):361-373. (PubMed)
126. Bassett JK, Baglietto L, Hodge AM, et al. Dietary intake of B vitamins and methionine and breast cancer risk. Cancer Causes Control. 2013;24(8):1555-1563. (PubMed)
127. Wang J, Wang B, Bi J, Di J. The association between two polymorphisms in the TYMS gene and breast cancer risk: a meta-analysis. Breast Cancer Res Treat. 2011;128(1):203-209. (PubMed)
128. Weiner AS, Boyarskikh UA, Voronina EN, et al. Polymorphisms in the folate-metabolizing genes MTR, MTRR, and CBS and breast cancer risk. Cancer Epidemiol. 2012;36(2):e95-e100. (PubMed)
129. Linabery AM, Johnson KJ, Ross JA. Childhood cancer incidence trends in association with US folic acid fortification (1986-2008). Pediatrics. 2012;129(6):1125-1133. (PubMed)
130. Milne E, Royle JA, Miller M, et al. Maternal folate and other vitamin supplementation during pregnancy and risk of acute lymphoblastic leukemia in the offspring. Int J Cancer. 2010;126(11):2690-2699. (PubMed)
131. Yan J, Yin M, Dreyer ZE, et al. A meta-analysis of MTHFR C677T and A1298C polymorphisms and risk of acute lymphoblastic leukemia in children. Pediatr Blood Cancer. 2012;58(4):513-518. (PubMed)
132. Alzheimer's Association. 2013 Alzheimer's Disease Fact and Figures. Alzheimer's & Dementia. 9(2). Available at: http://www.alz.org/downloads/facts_figures_2013.pdf. Accessed 9/9/13.
133. Hughes TF, Andel R, Small BJ, et al. Midlife fruit and vegetable consumption and risk of dementia in later life in Swedish twins. Am J Geriatr Psychiatry. 2010;18(5):413-420. (PubMed)
134. Weir DG, Scott JM. Brain function in the elderly: role of vitamin B12 and folate. Br Med Bull. 1999;55(3):669-682. (PubMed)
135. Faux NG, Ellis KA, Porter L, et al. Homocysteine, vitamin B12, and folic acid levels in Alzheimer's disease, mild cognitive impairment, and healthy elderly: baseline characteristics in subjects of the Australian Imaging Biomarker Lifestyle study. J Alzheimers Dis. 2011;27(4):909-922. (PubMed)
136. Van Dam F, Van Gool WA. Hyperhomocysteinemia and Alzheimer's disease: A systematic review. Arch Gerontol Geriatr. 2009;48(3):425-430. (PubMed)
137. Morris MC, Evans DA, Bienias JL, et al. Dietary folate and vitamin B12 intake and cognitive decline among community-dwelling older persons. Arch Neurol. 2005;62(4):641-645. (PubMed)
138. Morris MC, Evans DA, Schneider JA, Tangney CC, Bienias JL, Aggarwal NT. Dietary folate and vitamins B-12 and B-6 not associated with incident Alzheimer's disease. J Alzheimers Dis. 2006;9(4):435-443. (PubMed)
139. Wald DS, Kasturiratne A, Simmonds M. Serum homocysteine and dementia: meta-analysis of eight cohort studies including 8669 participants. Alzheimers Dement. 2011;7(4):412-417. (PubMed)
140. Ho RC, Cheung MW, Fu E, et al. Is high homocysteine level a risk factor for cognitive decline in elderly? A systematic review, meta-analysis, and meta-regression. Am J Geriatr Psychiatry. 2011;19(7):607-617. (PubMed)
141. Nilforooshan R, Broadbent D, Weaving G, et al. Homocysteine in Alzheimer's disease: role of dietary folate, vitamin B6 and B12. Int J Geriatr Psychiatry. 2011;26(8):876-877. (PubMed)
142. Wald DS, Kasturiratne A, Simmonds M. Effect of folic acid, with or without other B vitamins, on cognitive decline: meta-analysis of randomized trials. Am J Med. 2010;123(6):522-527 e522. (PubMed)
143. Ford AH, Almeida OP. Effect of homocysteine lowering treatment on cognitive function: a systematic review and meta-analysis of randomized controlled trials. J Alzheimers Dis. 2012;29(1):133-149. (PubMed)
144. Nachum-Biala Y, Troen AM. B-vitamins for neuroprotection: narrowing the evidence gap. Biofactors. 2012;38(2):145-150. (PubMed)
145. Smith AD, Smith SM, de Jager CA, et al. Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: a randomized controlled trial. PLoS One. 2010;5(9):e12244. (PubMed)
146. Douaud G, Refsum H, de Jager CA, et al. Preventing Alzheimer's disease-related gray matter atrophy by B-vitamin treatment. Proc Natl Acad Sci U S A. 2013;110(23):9523-9528. (PubMed)
147. Watkins D, Rosenblatt DS. Update and new concepts in vitamin responsive disorders of folate transport and metabolism. J Inherit Metab Dis. 2012;35(4):665-670. (PubMed)
148. Zhao R, Min SH, Qiu A, et al. The spectrum of mutations in the PCFT gene, coding for an intestinal folate transporter, that are the basis for hereditary folate malabsorption. Blood. 2007;110(4):1147-1152. (PubMed)
149. Borzutzky A, Crompton B, Bergmann AK, et al. Reversible severe combined immunodeficiency phenotype secondary to a mutation of the proton-coupled folate transporter. Clin Immunol. 2009;133(3):287-294. (PubMed)
150. Sofer Y, Harel L, Sharkia M, Amir J, Schoenfeld T, Straussberg R. Neurological manifestations of folate transport defect: case report and review of the literature. J Child Neurol. 2007;22(6):783-786. (PubMed)
151. Diop-Bove N, Kronn D, Goldman ID. Hereditary folate malabsorption. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, eds. GeneReviews™ [Internet]. Seattle, WA: University of Washington, Seattle; 2008. (PubMed)
152. Frye RE, Sequeira JM, Quadros EV, James SJ, Rossignol DA. Cerebral folate receptor autoantibodies in autism spectrum disorder. Mol Psychiatry. 2013;18(3):369-381. (PubMed)
153. Grapp M, Just IA, Linnankivi T, et al. Molecular characterization of folate receptor 1 mutations delineates cerebral folate transport deficiency. Brain. 2012;135(Pt 7):2022-2031. (PubMed)
154. Ramaekers VT, Blau N, Sequeira JM, Nassogne MC, Quadros EV. Folate receptor autoimmunity and cerebral folate deficiency in low-functioning autism with neurological deficits. Neuropediatrics. 2007;38(6):276-281. (PubMed)
155. Ramaekers VT, Hausler M, Opladen T, Heimann G, Blau N. Psychomotor retardation, spastic paraplegia, cerebellar ataxia and dyskinesia associated with low 5-methyltetrahydrofolate in cerebrospinal fluid: a novel neurometabolic condition responding to folinic acid substitution. Neuropediatrics. 2002;33(6):301-308. (PubMed)
156. Banka S, Blom HJ, Walter J, et al. Identification and characterization of an inborn error of metabolism caused by dihydrofolate reductase deficiency. Am J Hum Genet. 2011;88(2):216-225. (PubMed)
157. Cario H, Smith DE, Blom H, et al. Dihydrofolate reductase deficiency due to a homozygous DHFR mutation causes megaloblastic anemia and cerebral folate deficiency leading to severe neurologic disease. Am J Hum Genet. 2011;88(2):226-231. (PubMed)
158. Pfeiffer CM, Hughes JP, Lacher DA, et al. Estimation of trends in serum and RBC folate in the US population from pre- to postfortification using assay-adjusted data from the NHANES 1988-2010. J Nutr. 2012;142(5):886-893. (PubMed)
159. Folate. In: Hendler SS, Rorvik, D.R., ed. PDR for Nutritional Supplements. 2nd ed. Montvale: Physicians' Desk Reference Inc.; 2008.
160. Wiesinger H, Eydeler U, Richard F, et al. Bioequivalence evaluation of a folate-supplemented oral contraceptive containing ethinylestradiol/drospirenone/levomefolate calcium versus ethinylestradiol/drospirenone and levomefolate calcium alone. Clin Drug Investig. 2012;32(10):673-684. (PubMed)
161. Tinker SC, Cogswell ME, Devine O, Berry RJ. Folic acid intake among US women aged 15-44 years, National Health and Nutrition Examination Survey, 2003-2006. Am J Prev Med. 2010;38(5):534-542. (PubMed)
162. Kelly P, McPartlin J, Goggins M, Weir DG, Scott JM. Unmetabolized folic acid in serum: acute studies in subjects consuming fortified food and supplements. Am J Clin Nutr. 1997;65(6):1790-1795. (PubMed)
163. Morris MS, Jacques PF, Rosenberg IH, Selhub J. Folate and vitamin B-12 status in relation to anemia, macrocytosis, and cognitive impairment in older Americans in the age of folic acid fortification. Am J Clin Nutr. 2007;85(1):193-200. (PubMed)
164. Morris MS, Jacques PF, Rosenberg IH, Selhub J. Circulating unmetabolized folic acid and 5-methyltetrahydrofolate in relation to anemia, macrocytosis, and cognitive test performance in American seniors. Am J Clin Nutr. 2010;91(6):1733-1744. (PubMed)
165. Troen AM, Mitchell B, Sorensen B, et al. Unmetabolized folic acid in plasma is associated with reduced natural killer cell cytotoxicity among postmenopausal women. J Nutr. 2006;136(1):189-194. (PubMed)
166. Tam C, O'Connor D, Koren G. Circulating unmetabolized folic acid: relationship to folate status and effect of supplementation. Obstet Gynecol Int. 2012;2012:485179. (PubMed)
167. Apeland T, Mansoor MA, Strandjord RE. Antiepileptic drugs as independent predictors of plasma total homocysteine levels. Epilepsy Res. 2001;47(1-2):27-35. (PubMed)
168. Wilson SM, Bivins BN, Russell KA, Bailey LB. Oral contraceptive use: impact on folate, vitamin B(6), and vitamin B(1)(2) status. Nutr Rev. 2011;69(10):572-583. (PubMed)
Niacina
Contenido
Resumen
- Los precursores dietarios del nicotinamida adenina dinucleótido (NAD), incluyendo el ácido nicotínico, la nicotinamida, y la nicotinamida ribósido, son colectivamente referidos como niacina o vitamina B3. El aminoácido esencial triptófano puede también ser convertido en NAD a través de la vía de quinurenina. (Más información)
- El NAD puede ser fosforilado (NADP) y reducido (NADH y NADPH). El NAD funciona en las reacciones de oxidación-reducción (redox) y en las reacciones no-redox. (Más información)
- La pelagra es una enfermedad de deficiencia severa de niacina. Se caracteriza por síntomas que afectan la piel, el sistema digestivo, y el sistema nervioso; la pelagra puede causar la muerte si no es tratada. (Más información)
- Las causas de deficiencia de niacina incluyen una ingesta oral inadecuada, la escasa biodisponibilidad de granos sin nixtamalizar, la absorción defectuosa de triptófano, desordenes metabólicos, y el uso a largo plazo de tratamientos quimioterapéuticos. (Más información)
- Los requerimientos de la ingesta dietaria para la niacina están basados en la excreción urinaria de los metabolitos de niacina. (Más información)
- El NAD es el único sustrato para las enzimas PARP y sirtuinas implicadas en las actividades de reparación del ADN; por lo que NAD es crítico para la estabilidad del genoma. Varios estudios, la mayoría usando modelos in vitro y animales, sugieren un posible papel para la niacina en la prevención del cáncer. En un reciente ensayo de fase III, se encontró que una dosis farmacológica de nicotinamida redujo la tasa de lesiones premalignas de la piel y cánceres no melanoma en sujetos en alto riesgo. (Más información)
- A pesar de los resultados prometedores iniciales, la administración de nicotinamida ha fallado en prevenir o retrasar la aparición de la diabetes mellitus tipo 1 en familiares en alto riesgo de pacientes diabéticos tipo 1. Investigación futura podría explorar el uso de nicotinamida en la terapia combinada y evaluar los activadores de las enzimas dependiente de NAD. (Más información)
- En dosis farmacológicas, el ácido nicotínico mejoró los perfiles lipídicos de pacientes con un historial de enfermedad vascular, sin embargo, falló en reducir eventos cardiovasculares recurrentes o la mortalidad. (Más información)
- El desglose elevado de triptófano en la vía de quinurenina y la deficiencia de niacina han sido reportados en personas VIH positivas. Sin embargo, en la actualidad, es necesario un mejor entendimiento del papel de la vía de quinurenina y otras rutas biosintéticas del NAD durante la infección del VIH para establecer si esta población podría beneficiarse de la suplementación con niacina. (Más información)
- La mayoría de los efectos adversos de la niacina (ácido nicotínico y nicotinamida) han sido reportados con dosis farmacológicas de ácido nicotínico. El nivel máximo de ingesta tolerable (NM) para la niacina está basado en la prevención del enrojecimiento de la piel, el efecto secundario más importante de la niacina. La coadministración con laropiprant — un antagonista del receptor 1 de prostaglandina D2 — ayuda a reducir el enrojecimiento de la piel inducido por el ácido nicotínico. (Más información)
Niacina o vitamina B3 es una vitamina hidrosoluble usada por el cuerpo para formar la coenzima nicotinamida, NAD+. El termino 'niacina' es frecuentemente usado para referirse al acido nicotínico (ácido piridina-3-carboxílico) únicamente, aunque otros vitámeros con un anillo de piridina, incluyendo la nicotinamida (piridina-3-carboxamida) y nicotinamida ribósido, también contribuyen a la formación de NAD+ (1). Ninguno de los vitámeros está relacionado a la nicotina encontrada en el tabaco, aunque sus nombres sean similares. De igual manera la nicotina — pero no el ácido nicotínico — es un agonista de los receptores nicotínicos que responden al neurotransmisor, acetilcolina.
Metabolismo
Esencial para todas las formas de vida, la coenzima nicotinamida NAD+ es sintetizada en el cuerpo a partir de cuatro precursores que son proporcionados en la dieta: el acido nicotínico, la nicotinamida, nicotinamida ribósido, y el triptófano (Figura 1).
La Figura 2 ilustra las distintas rutas biosintéticas que conducen a la producción de NAD+ proveniente de varios precursores dietarios. NAD+ es sintetizada a partir de la nicotinamida y nicotinamida ribósido a través de dos reacciones enzimáticas, mientras que la ruta que produce NAD+ del ácido nicotínico — conocida como la ruta de Preiss-Handler — incluye tres pasos. La vía de quinurenina es la ruta biosintetica mas larga del NAD+: el catabolismo del triptófano a través de la quinurenina produce ácido quinolínico, el cual es entonces convertido a ácido nicotínico mononucleótido, un intermedio en el metabolismo del NAD+. NAD+ es entonces sintetizado a partir del ácido nicotínico mononucleótido en la ruta de Preiss-Handler (2).
Todas las rutas generan mononucleótidos intermediarios — ácido nicotínico mononucleótido o nicotinamida mononucleótido. Enzimas específicas, conocidas como fosforribosiltransferasas, catalizan la adición de un grupo fosforribosa al ácido nicotínico o ácido quinolínico para producir ácido nicotínico mononucleótido o en la nicotinamida para generar nicotinamida mononucleótido. El nicotinamida mononucleótido también es generado por la fosforilación de nicotinamida ribósido, catalizada por quinasas nicotinamida ribosa (NRK). Además, las adenililtransferasas catalizan la adenilación de estos mononucleótidos para formar ya sea ácido nicotínico adenina dinucleótido o NAD+. El ácido nicotínico adenina dinucleótido es luego convertido a NAD+ por la NAD+ sintetasa dependiente de glutamina (NADSYN), la cual usa glutamina con un donante de grupo amida (Figura 2) (2). Debe notarse que se ha reportado que el ácido nicotínico adenina dinucleótido se forma después de la administración de nicotinamida ribósido en alta dosis, sugiriendo que una potencial desamidación podría ocurrir para convertir el NAD+ a ácido nicotínico adenina dinucleótido cuando el depósito de NAD+ es alto (1).
La NAD quinasa cataliza la fosforilación del NAD a NADP usando adenosil trifosfato (ATP) como donador de fosforilo (3). Las propiedades de oxido-reducción (redox) del dinucleótido no son afectadas por la fosforilación tal que las parejas redox NAD+/NADH y NADP+/NADPH muestran potenciales redox similares (4). La oxidación y reducción de la posición C-4 del grupo nicotinamida del NAD y su forma fosforilada son esenciales para las reacciones de transferencia de electrones que soportan funciones metabólicas y bioenergéticas vitales en todas las células (véase Función). Así, NAD y NADP son reciclados una y otra vez entre la forma oxidada (NAD+ y NADP+) y reducida, como se muestra en Figura 3.

[Figura 1 - Clic para Agrandar]

[Figura 2 - Clic para Agrandar]
[Figura 3 - Clic para Agrandar]
Función
NAD como una coenzima en las reacciones de transferencia de electrones
Los organismos vivos derivan la mayoría de su energía de reacciones redox, procesos que involucran la transferencia de electrones. Mas de 400 enzimas requieren las coenzimas de niacina, NAD y NADP, principalmente para aceptar o donar electrones para las reacciones redox (5). NAD y NADP parecen soportar funciones distintas (Figura 4). NAD funciona más frecuentemente en reacciones productoras de energía que involucran la degradación (catabolismo) de carbohidratos, grasas, proteínas, y alcohol. NADP generalmente sirve en las reacciones biosintéticas (anabolismo), como en la síntesis de ácidos grasos, esteroides (por ejemplo, colesterol, ácidos biliares y hormonas esteroides), y como bloques de construcción de otras macromoléculas (4). NADP es también esencial para la regeneración de componentes de la detoxificación y de sistemas antioxidantes (4). Para soportar estas funciones, la célula mantiene NAD en un estado ampliamente oxidado (NAD+) para que sirva como un agente oxidante en las reacciones catabólicas, mientras que NADP se mantiene en gran medida en un estado reducido (NADPH) para donar fácilmente electrones para los procesos celulares reductivos (4, 6).
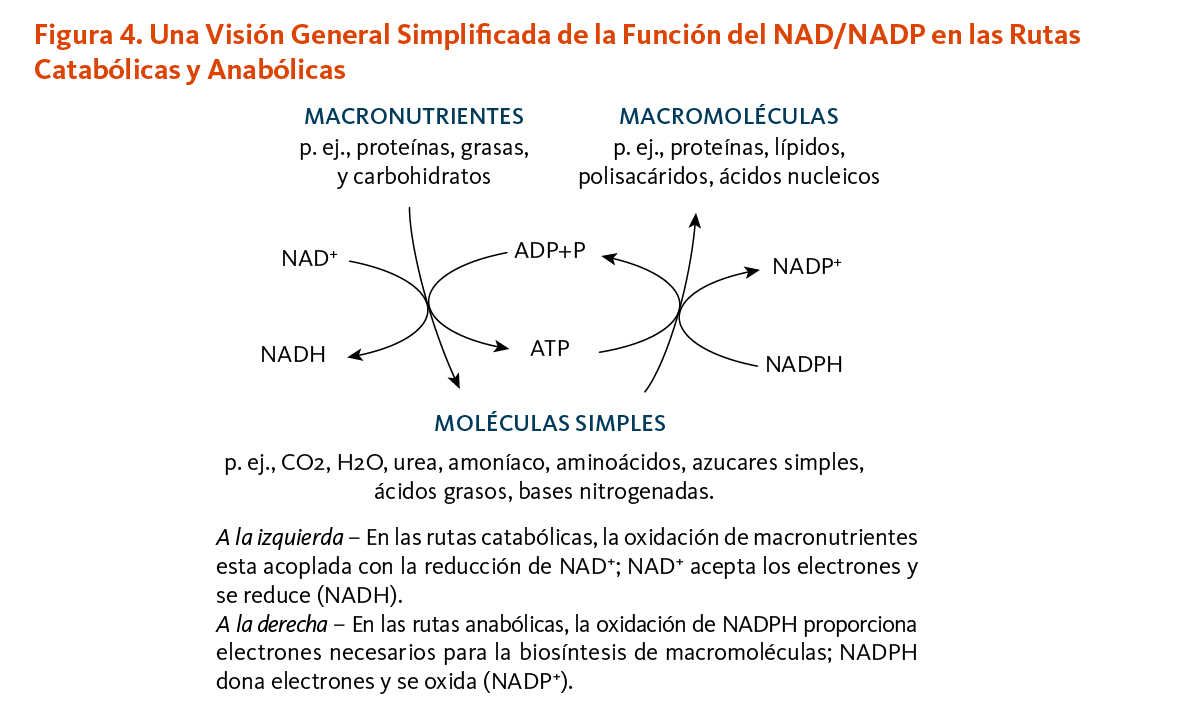
[Figura 4 - Clic para Agrandar]
NAD como un sustrato para las enzimas que consumen NAD
La coenzima de la niacina, NAD, es el sustrato (reactante) de por lo menos cuatros clases de enzimas. Dos clases de enzimas con actividades de mono adenosina difosfato (ADP)- ribosiltransferasa y/o poli (ADP-ribosa) polimerasa catalizan las reacciones de transferencia de ADP-ribosilo. Proteínas parecidas al regulador de información silencioso 2 (Sir2) (sirtuinas) catalizan la eliminación de los grupos acetilo provenientes de proteínas acetiladas, utilizando ADP-ribosa del NAD como un como un aceptor para grupos de acetilo. Finalmente, las ADP-ribosil ciclasas están involucradas en la regulación de la señalización del calcio intracelular.
ADP-ribosilación
Las enzimas con actividades ADP-ribosiltransferasa estaban previamente divididas entre mono ADP-ribosiltransferasas (ART) y poli (ADP-ribosa) polimerasas (PARP). Las ART fueron primeramente descubiertas en ciertas bacterias patogénicas — como aquellas causantes del cólera y difteria — donde median las acciones de toxinas. Estas enzimas transfieren un grupo de residuo ADP-ribosa del NAD a un amino acido especifico de una proteína objetivo, con la creación de una proteína ADP-ribosilada y la liberación de nicotinamida.
Debido a que se ha encontrado que la mayoría de las PARP exhiben solo actividades mono ADP-ribosiltransferasa una nueva nomenclatura fue propuesta para las enzimas que catalizan la ADP-ribosilación: Una familia de mono ADP-ribosiltransferasas con homología con toxinas diftéricas bacterianas fue llamada ARTD, mientras que las enzimas con actividades mono o poli ADP-ribosiltransferasa y relacionadas a las toxinas clostridiales C2 y C3 fueron incluidas en la familia ARTC (7, 8).
ARTC son enzimas extracelulares que catalizan la mono ADP-ribosilación de proteínas secretadas o de membrana involucradas en la inmunidad innata y comunicación celular (2).
ARTD son enzimas intracelulares con actividades mono o poli ADP-ribosiltransferasa. Por lo menos 18 ARTD han sido identificadas. Todas las ARTD poseen un dominio catalítico similar a la toxina diftérica que se une a NAD+. Solo las ARTD 1, 2, 5 y 6 catalizan transferencias poli (ADP-ribosa); las otras tienen actividades mono ADP-ribosiltransferasas. Se demostró que las ARTD están involucradas en la reparación del ADN y respuestas al estrés, señalización celular, regulación de la transcripción, apoptosis, diferenciación celular, mantenimiento de la integridad genómica, y defensa antiviral (revisado en 8).
Desacetilación dependiente de NAD
Siete sirtuinas (SIRT 1-7) han sido identificadas en humanos. Las sirtuinas son una clase de enzimas deacetilasa dependientes de NAD que remueven grupos de acetilo de los restos de lisina acetilados de proteínas diana. Durante el proceso de desacetilación, el grupo acetilo es transferido al grupo ADP-ribosa escindido del NAD produciendo O-acetil-ADP-ribosa. La nicotinamida puede ejercer una inhibición de retroalimentación en la reacción de desacetilación (9). Al igual que la ADP-ribosilación, la acetilación es una modificación postraduccional que afecta la función de las proteínas diana. El interés inicial en las sirtuinas ocurrió después del descubrimiento de que su activación podía imitar la restricción calórica, la cual se ha mostrado incrementa la esperanza de vida en organismos inferiores. Tal papel en los mamíferos es controversial, aunque las sirtuinas son reguladores de detección de energía involucrados en las rutas de señalización que podrían jugar papeles importantes en el retraso de la aparición de enfermedades relacionadas a la edad (p. ej., enfermedades cardiovasculares, cáncer, demencia, artritis). Hasta la fecha, el espectro de sus funciones biológicas incluye el silenciamiento de genes, la reparación del daño al ADN, la regulación del ciclo celular, y la diferenciación celular.
Movilización del calcio
En los humanos CD38 y CD157 pertenecen a una familia de NAD+glicohidrolasas/ADP-ribosil ciclasas. Estas enzimas catalizan la formación de reguladores clave de la señalización por calcio, es decir ADP-ribosa (lineal), ADP-ribosa cíclica, y ácido nicotínico adenina dinucleótido fosfato (Figura 5). El ADP-ribosa cíclica y el ácido nicotínico adenina dinucleótido fosfato trabajan dentro de las células para provocar la liberación de los iones de calcio de los sitios de almacenamiento internos (es decir, el retículo endoplásmico, lisosomas, mitocondrias), mientras que el ADP-ribosa estimula la entrada de calcio extracelular a través de los canales de cationes TRPM2 de la membrana celular (2). Otro agonista de, TRPM2, 2’-desoxi-ADP-ribosa, fue identificado recientemente in vitro. Se encontró que CD38 cataliza la síntesis de 2’-desoxi-ADP-ribosa a partir del nicotinamida mononucleótido y 2’-desoxi-ATP (11). O-acetil-ADP-ribosa generada por la actividad de las sirtuinas también controla la entrada de calcio a través de los canales TRPM2 (6). La transducción de señal mediada por calcio intracelular es regulada por la entrada transitoria de calcio en la célula o la liberación de calcio de almacenamientos intracelulares. La señalización por calcio esta críticamente involucrada en procesos como la neurotransmisión, la liberación de insulina de células β pancreáticas, la contracción muscular, y la activación de linfocitos-T (6).
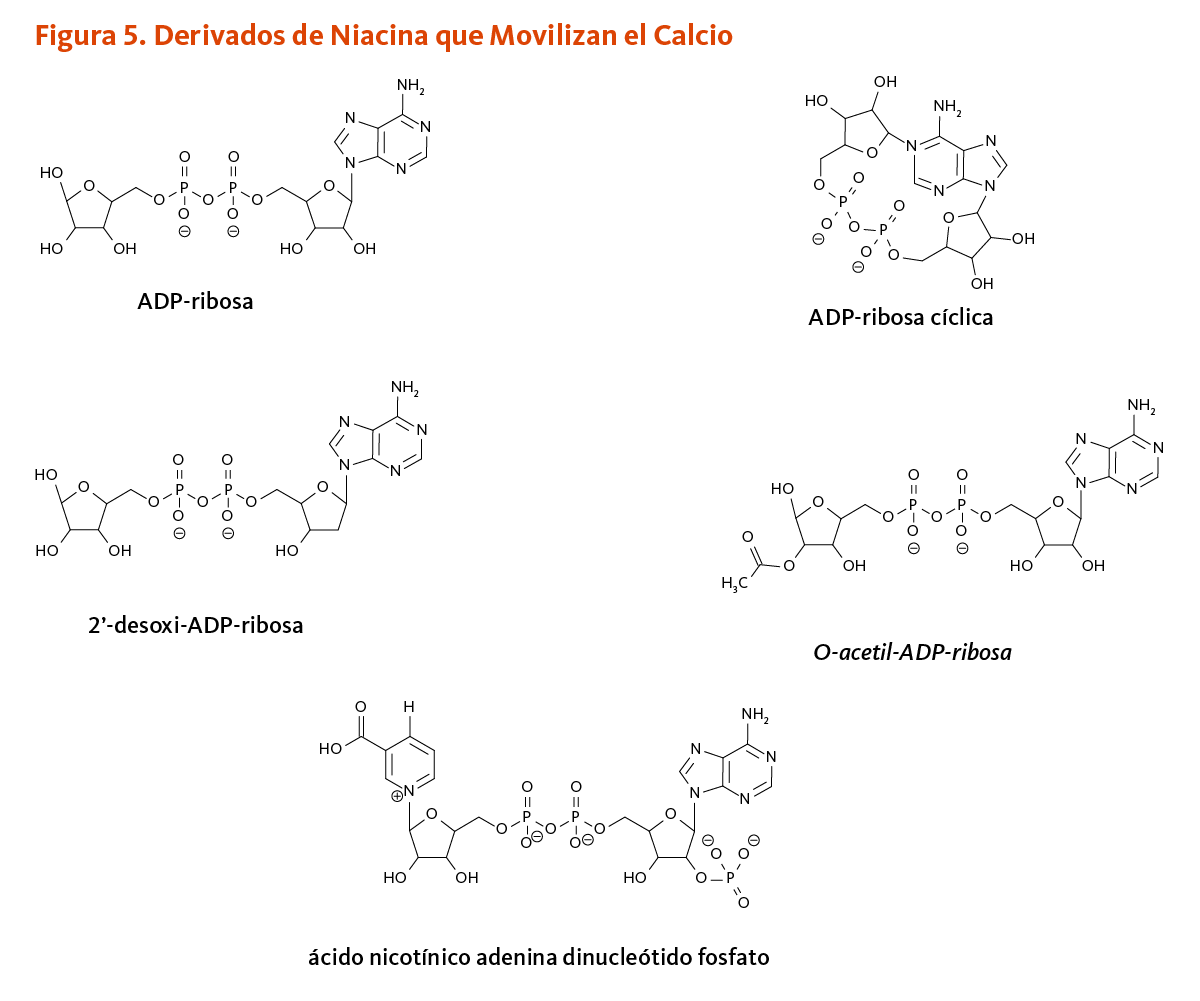
[Figura 5 - Clic para Agrandar]
NAD como un ligando
NAD+ ha sido identificado como un agonista endógeno de receptores de membrana purinérgicos de subclase P2Y. en particular, se encontró que NAD se une al receptor P2Y1 y actúa como un neurotransmisor inhibitorio en las uniones neuromusculares en los músculos lisos viscerales (12). También se encontró que el NAD+ extracelular se comporta como una citoquina proinflamatoria, desencadenando la activación de granulocitos aislados. La unión de NAD+ al receptor P2Y11 en la superficie del granulocito activa una cascada de señalización involucrando la ADP-ribosa cíclica y el aumento del calcio intracelular, eventualmente estimulando la generación de superóxido y la quimiotaxis (13). Observaciones similares fueron hechas con monolitos activados por lipopolisacáridos (14). NAADP+ extracelular y ADP-ribosa podrían también unirse a los receptores P2Y y desencadenar NAADP+ intracelular — y ADP-ribosa-dependiente de la movilización del calcio (véase Movilización del calcio) (15, 16).
Efectos hipolipemiantes con dosis farmacológicas de ácido nicotínico
Por más de medio siglo, las dosis farmacológicas de ácido nicotínico, pero no de nicotinamida, han sido conocidas por reducir el colesterol del suero (véase Tratamiento de Enfermedades) (17). Sin embargo, los mecanismos exactos que subyacen el efecto hipolipemiante del ácido nicotínico siguen siendo especulativos. Dos receptores de membrana acoplados a proteína G, GPR109A y GPR109B, se unen al acido nicotínico con alta y baja afinidad, respectivamente. Estos receptores de ácido nicotínico son principalmente expresados en el tejido adiposo y células inmunes (pero no los linfocitos). También son encontrados en las células epiteliales colónicas y pigmentadas de la retina, queratinocitos, células mamarias, microglía, y posiblemente en niveles bajos en el hígado (18). Así, los efectos modificadores de lípidos del ácido nicotínico son mas propensos a ser mediados por mecanismos independientes de receptores en tejidos principales del metabolismo de los lípidos como el hígado y el musculo esquelético. Datos in vitro tempranos sugirieron que el ácido nicotínico podría perjudicar la secreción de lipoproteínas de muy baja densidad (VLDL) al inhibir la síntesis de triglicéridos y desencadenar la degradación de lipoproteína ApoB en los hepatocitos (19). En otro estudio, el ácido nicotínico afecto la absorción hepática de lipoproteínas ApoAI, reduciendo así la eliminación de lipoproteínas de alta densidad (HDL) de la circulación (revisado en 20). En los adipocitos se encontró que la unión del acido nicotínico al GPR109A inicio una cascada de transducción de señales resultando en reducciones en la producción de ácidos grasos libres mediante la inhibición de la lipasa sensible a las hormonas involucrada en la lipólisis de triglicéridos (21). Sin embargo, observaciones recientes han sugerido que el efecto hipolipemiante del ácido nicotínico no se debe a su actividad anti-lipolítica (22). Ensayos mostraron que los agonistas sintéticos del GPR109A agudamente disminuyeron los ácidos grasos libres, pero fallaron en afectar los lípidos del suero (22). Aparte de su impacto en los HDL y otros lípidos del plasma, el ácido nicotínico ha exhibido actividades anti-ateroscleróticas en monocitos, macrófagos, o células endoteliales vasculares cultivadas, al modular la inflamación y estrés oxidativo y regulando la adhesión, migración, y diferenciación celular (revisado en 18).
Deficiencia
Pelagra
Causas
La última etapa de la deficiencia severa de niacina se conoce como pelagra. Los primeros registros de pelagra ocurrieron luego del cultivo generalizado de maíz en Europa en 1700 (23). La enfermedad es generalmente asociada con las clases sociales más pobres en la cuales el alimento básico principal consistía en cereales como el maíz o el sorgo. La pelagra también fue común en el sur de los Estados Unidos durante los inicios del año 1900, donde el ingreso era bajo y los productos de maíz eran un alimento básico importante (24). Curiosamente, la pelagra no era conocida en México, donde el maíz era también un alimento básico importante y mucha de la población era también pobre. De hecho, si el maíz contiene cantidades considerables de niacina, esta se encuentra presente en una forma ligada que no está nutricionalmente disponible para los seres humanos. La preparación tradicional de tortillas de maíz en México involucra remojar el maíz en una solución de cal (óxido de calcio) antes de cocinarlo. Calentar el maíz en una solución alcalina produce la liberación de la niacina ligada, aumentando así su biodisponibilidad (25). La epidemia de pelagra fue también desconocida por los nativos americanos quienes consumían maíz inmaduro que contiene predominantemente niacina desligada (biodisponible) (24).
La deficiencia de niacina o pelagra pueden resultar de la ingesta dietaría inadecuada de precursores de NAD, incluyendo triptófano. La deficiencia de niacina — frecuentemente asociada con la malnutrición — se observa en la población sin hogar, en individuos que sufren de anorexia nerviosa u obesidad, y en consumidores de dietas altas en maíz y pobres en proteína animal (26-29). Deficiencias de otras vitaminas B y algunos minerales traza pueden agravar la deficiencia de niacina (30, 31). Trastornos de malabsorción que pueden conducir a la pelagra incluyen la enfermedad de Crohn y megaduodeno (32, 33). Pacientes con la enfermedad de Hartnup, un trastorno hereditario que resulta en una absorción defectuosa de triptófano, han desarrollado pelagra (véase Trastornos genéticos sensibles a la niacina). El síndrome carcinoide, una condición de aumento de la secreción de serotonina y otras catecolaminas por tumores carcinoides, puede también resultar en pelagra debido al incremento en la utilización de triptófano para la serotonina en lugar de la síntesis de niacina. Además, el tratamiento prolongado con el fármaco anti-tuberculosis isoniacida ha resultado en deficiencia de niacina (34). Otros agentes farmacéuticos, incluyendo las drogas inmunosupresoras azatioprina (Imuran) y 6- mercaptopurina, el fármaco contra el cáncer 5-fluorouracilo (5-FU, Adrucil), y levodopa/carbidopa (Sinemet; dos fármacos dados a personas con la enfermedad de Parkinson) son conocidos por incrementar la dependencia en la niacina dietaría al interferir con la vía triptófano-quinurenina-niacina (35). Finalmente, otras poblaciones en riesgo de una deficiencia de niacina incluyen pacientes en diálisis, pacientes con cáncer (36, 37), individuos que sufren de alcoholismo crónico (38), y personas con VIH (véase VIH/SIDA abajo). Además, la ingesta crónica de alcohol puede llevar a una deficiencia de niacina severa a través de la reducción de la ingesta de niacina dietaría y al interferir con la conversión de triptófano-a-NAD (30).
Síntomas de la deficiencia
Los síntomas más comunes de la deficiencia de niacina involucran a la piel, al sistema digestivo, y al sistema nervioso. Los síntomas de pelagra son comúnmente referidos como a las tres D: dermatitis sensible al sol, diarrea, y demencia. Una cuarta D, deceso, ocurre si la pelagra no es tratada (5). En la piel, un sarpullido de pigmentación oscura, grueso y escamoso, se desarrolla simétricamente en las áreas expuestas a la luz del sol. De hecho, la palabra "pelagra" deriva de "pelle agra" la expresión italiana para la piel áspera. Los síntomas relacionados al sistema digestivo incluyen inflamación de la boca y lengua ("lengua roja brillante"), vómitos, constipación, dolor abdominal y por último diarrea. Los trastornos gastrointestinales y la diarrea contribuyen a la desnutrición continua de los pacientes. Los síntomas neurológicos incluyen dolor de cabeza, apatía, fatiga, depresión, desorientación, y pérdida de memoria y son más consistentes con delirio que con la demencia descrita históricamente (38). La presentación de la enfermedad varía en apariencia ya que la triada clásica raramente se presenta en su totalidad. La ausencia de la dermatitis, por ejemplo, es conocida como pelagra sinusoidal pelagra.
Tratamiento
Para tratar la pelagra, la Organización Mundial de la Salud (OMS) recomienda administrar nicotinamida para prevenir el enrojecimiento comúnmente causado por el ácido nicotínico (véase Seguridad). Los lineamientos del tratamiento sugieren usar 300 mg/día de nicotinamida oral en dosis divididas, o 100 mg/día administrados parenteralmente en dosis divididas, por tres a cuatro semanas (37, 39). Debido a que los pacientes con pelagra frecuentemente muestran deficiencias vitamínicas adicionales, la administración de una preparación de complejo de vitamina B es aconsejada (39).
La Ingesta Diaria Recomendada (IDR)
Equivalente de niacina (EN)
El termino "equivalente de niacina" (EN) es usado para describir la contribución de la ingesta dietaría de todas las formas de niacina que están disponibles para el cuerpo. En los individuos sanos, menos del 2% del triptófano dietario es convertido a NAD en la vía de quinurenina (40). La síntesis del NAD a partir del triptófano es bastante ineficiente y depende de enzimas que requieren de vitamina B6 y riboflavina como también de un enzima que contiene hemo (hierro). Sin embargo, el triptófano es esencial como precursor para el NAD+. Defectos congénitos en el trasporte del triptófano y el metabolismo resultan en trastornos clínicos severos atribuidos al agotamiento de NAD+ (véase Trastornos genéticos sensibles a la niacina). En promedio, 60 miligramos (mg) de triptófano se considera corresponden a 1 mg de niacina o 1 mg de EN.
Ingestas diarias recomendadas
La ingesta diaria recomendada (IDR) para la niacina esta basada en la prevención de la deficiencia. La pelagra puede ser prevenida con alrededor de 11 mg de EN/día, pero se ha encontrado que, de 12 mg a 16 mg de EN/día, se normaliza la excreción urinaria de metabolitos de niacina (productos de degradación) en adultos jóvenes sanos. Debido a que la pelagra representa una deficiencia severa, la Junta de Nutrición y Alimentos (JNA) del Instituto de Medicina de EE.UU. optó por utilizar la excreción de metabolitos de niacina como un indicador del estado nutricional de la niacina más que como un indicador de síntomas de pelagra (41). Sin embargo, se ha argumentado que el contenido de NAD y NADP celulares pueden ser indicadores más relevantes del estatus nutricional de la niacina (24).
| Etapa de la Vida | Edad | Machos (mg EN*/día) | Hembras (mg EN/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 2 (IA) | 2 (IA) |
| Infantes | 7-12 meses | 4 (IA) | 4 (IA) |
| Niños | 1-3 años | 6 | 6 |
| Niños | 4-8 años | 8 | 8 |
| Niños | 9-13 años | 12 | 12 |
| Adolescentes | 14-18 años | 16 | 14 |
| Adultos | 19 años y más | 16 | 14 |
| Embarazo | Todas las edades | - | 18 |
| Período de lactancia | Todas las edades | - | 17 |
| *EN, equivalentes de niacina: 1 mg de EN = 60 mg de triptófano = 1 mg de niacina | |||
Prevención de Enfermedades
Cáncer
Estudios de cultivos celulares (in vitro) aportan evidencia de que el contenido de NAD influencia mecanismos que mantienen la estabilidad genómica. La pérdida de estabilidad genómica, caracterizada por una alta tasa de daño en el ADN y cromosomas, es un sello distintivo del cáncer (42). La comprensión actual es que se reduce la piscina de NAD durante la deficiencia de niacina y esto afecta la actividad de enzimas que consumen NAD en lugar de funciones redox y metabólicas (43). Entre las reacciones dependientes de NAD, poli ADP-ribosilaciones catalizadas por enzimas PARP (ARTD) son críticas para la respuesta celular al daño del ADN. Después del daño al ADN, las PARP son activadas; se mostró que las subsiguientes poli ADP-ribosilaciones de una serie de señalización y moléculas estructurales por las PARP facilitan la reparación del ADN en roturas de la de cadena del ADN (44). Se ha encontrado que el agotamiento celular de NAD disminuye los niveles de la proteína supresora de tumores, p53, un objetivo para la poli ADP-ribosilación en células de mama, piel y pulmón humanas (45). La expresión de p53 fue también alterada por la deficiencia de niacina en las células de la medula ósea de ratas (46). El deterioro de la reparación del ADN causado por la deficiencia de niacina pudo conducir a la inestabilidad genómica e impulsar el desarrollo de tumores en modelos de ratas (47, 48). Ambas PARP y las sirtuinas han participado recientemente en el mantenimiento de la heterocromatina, un domino cromosómico asociado con la estabilidad del genoma, así como en el silenciamiento génico transcripcional, la integridad de los telómeros, y la segregación cromosómica durante la división celular (49, 50). Ni el contenido celular de NAD ni la ingesta dietaría de precursores de NAD necesarios para optimizar las respuestas de protección luego de un daño al ADN han sido determinados, pero ambos son posiblemente más altos que lo requerido para la prevención de la pelagra.
Medula ósea
Los pacientes con cáncer frecuentemente sufren de supresión de la medula ósea luego de la quimioterapia, dado que la medula ósea es uno de los tejidos mas prolíferos en el cuerpo y por lo tanto un objetivo principal para los agentes quimioterapéuticos. Se encontró que una deficiencia de niacina disminuye los niveles de NAD y poli-ADP-ribosa en la médula ósea y aumenta el riesgo de leucemia en ratas (51). Por el contrario, una dosis farmacológica de ácido nicotínico o nicotinamida fue capaz de aumentar el NAD y poli-ADP-ribosa en la medula ósea y disminuir el desarrollo de la leucemia en ratas (52). Se ha sugerido que la deficiencia de niacina a menudo observada en pacientes con cáncer podría sensibilizar el tejido de la medula ósea al efecto supresor de la quimioterapia. Sin embargo, poco se sabe acerca de los niveles de NAD celular y la prevención del daño al ADN o del cáncer en humanos. Un estudio en dos individuos sanos involucró el aumento de los niveles de NAD en leucocitos sanguíneos a través de la suplementación con 100 mg/día de ácido nicotínico por ocho semanas. En comparación con individuos no suplementados, los individuos con suplementación experimentaron una disminución en la ruptura de hebras del ADN en los linfocitos expuestos a radicales libres en un ensayo de tubo de ensayo (53). Sin embargo, la suplementación con ácido nicotínico de hasta 100 mg/día por 14 semanas en 21 fumadores sanos falló en aportar alguna evidencia de una disminución en el daño genético inducido por el humo del cigarrillo en linfocitos sanguíneos, en comparación con el placebo (38). Más reciente, se utilizó la frecuencia de translocación cromosómica para evaluar daños al ADN en los linfocitos de sangre periférica de 82 pilotos crónicamente expuestos a la radiación ionizante, un conocido carcinógeno humano. En este estudio basado en la observación, la tasa de aberraciones cromosómicas fue significativamente menor en los sujetos con una ingesta más alta de niacina dietaría (28.4 mg/día) en comparación con una más baja (20.5 mg/día) (55). La disponibilidad mas alta de NAD+ en linfocitos de sangre periférica irradiados con rayos X se encontró favorece la reparación del ADN al mejorar la supervivencia, particularmente a través de la deacetilación de p53 mediada por SIRT (56).
Tracto digestivo superior
Generalmente, las relaciones entre los factores dietarios y el cáncer son establecidos primero en los estudios epidemiológicos y seguidos luego por investigación básica del cáncer a nivel celular. En el caso de la niacina, la investigación sobre aspectos bioquímicos y celulares de la reparación del ADN ha estimulado un interés en la relación entre la ingesta de niacina y el riesgo de cáncer en la población humana (57). Un estudio de caso y control de gran magnitud encontró que el consumo aumentado de niacina, junto con nutrientes antioxidantes, se asociaba con una incidencia disminuida de cáncer oral (boca), faríngeo (garganta), y esofágico en el norte de Italia y Suiza. Un aumento de la ingesta de niacina diaria de 6.2 mg se asoció con un descenso de alrededor del 40% de los casos de cáncer de boca y garganta, mientras un aumento de 5.2 mg en la ingesta diaria de niacina se asoció con un descenso similar en los casos de cáncer esofágico (58, 59).
Piel
La deficiencia de niacina puede llevar a una sensibilidad severa a la luz solar en la piel expuesta. Dada la implicación de las enzimas dependientes de NAD en la reparación del ADN, ha habido cierto interés en el efecto de la niacina en la salud de la piel. Experimentos in vitro y en animales han ayudado a obtener información, pero los datos en humanos sobre el estatus de la niacina/NAD y el cáncer de piel son muy limitados. Un estudio reportó que la suplementación con niacina disminuyó el riesgo de canceres en la piel inducidos por luz ultravioleta (UV) en ratones, a pesar del hecho de que los ratones convierten triptófano a NAD más eficientemente que las ratas y los humanos y que por lo tanto no se ven severamente deficientes (60). La hiperproliferación y la diferenciación alterada de células de la piel pueden alterar la integridad de la barrera cutánea y aumentar la ocurrencia de enfermedades de la piel pre-malignas y malignas. Un efecto protector de la niacina fue sugerido a través de la aplicación tópica de miristil nicotinato, un derivado de niacina, que aumentó con éxito la expresión de marcadores de diferenciación epidérmica en sujetos con piel fotodañada (61). La activación de receptores del ácido nicotínico, GPR109A y GPR109B, por dosis farmacológicas de niacina podría estar involucrada en la mejora de la función de la barrera cutánea. Por lo contrario, los defectos de la diferenciación en células cancerígenas cutáneas fueron ligadas a la localización celular anormal de receptores del ácido nicotínico defectuosos (62). La restricción de nicotinamida con el subsecuente agotamiento de NAD celular ha demostrado incrementar el daño del ADN inducido por el estrés oxidativo en un modelo de células precancerosas de la piel, implicando un papel protector de las vías dependientes de NAD en el cáncer (63). La disponibilidad alterada del NAD también afecta la expresión y la actividad de las sirtuinas en las células de la piel humana expuestas a UV. Junto con las PARP, las sirtuinas que consumen NAD podrían jugar un papel importante en la respuesta celular al fotodaño y la homeostasis de la piel (64).
Un análisis agrupado de dos estudios de cohorte prospectivos de gran magnitud de los EE.UU. que dio seguimiento a 41,808 hombres y 72,308 mujeres por hasta 26 años sugirió que la ingesta mas alta de niacina versus la mas baja (proveniente de la dieta y suplementos) podría proteger contra el carcinoma de células escamosas pero no contra el carcinoma de células basales y melanoma (65). Un ensayo aleatorio, doble ciego, fase III, controlado con placebo en 386 sujetos con un historial de cáncer de piel no melanoma recientemente examinó el efecto de la suplementación diaria con nicotinamida (1 g) por 12 meses en la recurrencia del cáncer de piel en intervalos de tres meses sobre un periodo de 18 meses (66). La nicotinamida efectivamente redujo la tasa de queratosis actínica premaligna (-11%), carcinoma de células escamosas (-30%) y carcinoma de células basales (-20%) en comparación al placebo después de 12 meses, sin embargo, esta protección no fue sostenida durante el periodo de seis meses posterior a la suplementación (66). Ensayos de mayor magnitud son necesarios para evaluar si la nicotinamida podría reducir el riesgo de melanomas, los cuales no son tan comunes como otros canceres de piel, pero son más mortales (67).
Diabetes mellitus tipo 1
La diabetes mellitus tipo 1 en niños es causada por la destrucción autoinmune de las células beta secretoras de insulina en el páncreas. Antes del inicio de una diabetes sintomática, anticuerpos específicos, incluyendo autoanticuerpos de las células de los islotes (ACI), pueden ser detectados en la sangre de individuos en alto riesgo (68). En un modelo experimental animal de la diabetes, niveles altos de nicotinamida son administrados para proteger las células β del daño causado por la estreptozocina (69).
Sin embargo, no se ha encontrado que dosis farmacológicas de nicotinamida (de hasta 3 g/día) sean efectivas en el retraso o prevención de la aparición de la diabetes tipo 1 en sujetos en alto riesgo. Un análisis de 10 ensayos, de los cuales cinco fueron controlados con placebo, encontró evidencia de una función de células β mejorada después de un año de tratamiento con nicotinamida, pero el análisis falló en encontrar alguna evidencia clínica de un control glucémico mejorado (70). Un ensayo controlado aleatorio multicéntrico grande de la nicotinamida en familiares ACI positivos (edades, 3-12 años) de pacientes diabéticos tipo 1 también falló en encontrar una diferencia en la incidencia de diabetes tipo 1 después de tres años (70). Un ensayo multicéntrico aleatorio, doble ciego, controlado con placebo de la nicotinamida (un máximo de 3 g/día) fue conducido en 552 familiares ACI positivos de pacientes con diabetes tipo 1. La proporción de familiares que desarrollaron diabetes tipo 1 dentro del periodo de cinco años fue comparable ya fuera si fueron tratados con nicotinamida o placebo (71). La nicotinamida pudo reducir los parámetros relacionados a la inflamación en estos sujetos en alto riesgo, pero fue ineficaz para prevenir el inicio de la enfermedad (72). Mas recientemente, reportes de caso del uso combinado de nicotinamida (25 mg/kg/día) y acetil-L-carnitina (50 mg/kg/día) en niños en riesgo de diabetes tipo 1 mostró resultados prometedores, lo que justifica una mayor investigación (73).
Tratamiento de Enfermedades
Los suplementos con niacina en dosis farmacológicas (es decir, dosis más altas que aquellas necesarias para prevenir la deficiencia) han sido usados en un intento por tratar un cierto rango de condiciones, algunas de las cuales son discutidas a continuación.
Trastornos genéticos sensibles a la niacina
Trastornos congénitos relacionados a la deficiencia de NAD pueden resultar de mutaciones en genes involucrados en la absorción y transporte de varios precursores dietarios del NAD+ o en las distintas rutas metabólicas que conllevan a la producción de NAD+ (véase Metabolismo). Algunos de estos trastornos podrían responder a la suplementación con niacina. Por ejemplo, el transporte alterado de triptófano a las células resulta en la enfermedad de Hartnup, la cual presenta signos de deficiencia grave de niacina (74). La enfermedad de Hartnup se debe a mutaciones en el gen SLCA19, el cual codifica para un transportador de aminoácidos neutros dependiente de sodio principalmente expresado en los riñones e intestinos. El manejo de la enfermedad involucra la suplementación con ácido nicotínico o nicotinamida (75). Mutaciones recesivas en genes que codifican para las enzimas de la vía de quinurenina — es decir quinureninasa y ácido 3-hidroxiantranil 3,4-dioxigenasa — conducen a malformaciones congénitas vertebrales, anales, cardíacas, traqueoesofágicas, renales y de las extremidades (VACTERL) combinadas (76). Se encontró que el agotamiento de NAD+, en lugar de la acumulación de metabolitos intermedios en la vía de quinurenina era responsable de estas malformaciones. La suplementación con niacina a lo largo del embarazo aseguró niveles adecuados de NAD+ y previno anomalías congénitas en ratones con mutaciones en la vía de quinurenina (76). En los humanos, la dosis de precursores de NAD+ necesaria para evitar malformaciones congénitas VACTERL inducidas por deficiencia de NAD falta aun por definirse (77).
La nicotinamida puede también rescatar la depleción de NAD+ secundaria a un error innato ultra raro del metabolismo de la glutamina (78). La glutamina es requerida para la conversión del ácido nicotínico adenina dinucleótido a NAD+ catalizada por la NAD+ sintetasa (Figura 2). De este modo, la deficiencia de glutamina sintetasa hereditaria específicamente afecta la síntesis de NAD+ a partir de los precursores de NAD+, triptófano y ácido nicotínico. Si las deficiencias combinadas de glutamina y NAD+ son responsables por el fenotipo clínico severo de sujetos con deficiencia hereditaria de glutamina sintetasa, es probable que la suplementación con ambos glutamina y nicotinamida proporcionaría algo de alivio (78).
Finalmente, muchos errores innatos del metabolismo resultan de mutaciones genéticas que disminuyen la afinidad de unión del cofactor y, subsecuentemente, la eficacia de la enzima (79). En muchos casos, la administración de altas dosis de las vitaminas que sirven como precursores de cofactores puede restaurar la actividad enzimática — por lo menos parcialmente — y disminuir los signos de las enfermedades genéticas (79). Dado que un gran numero de enzimas requieren NAD, se especula que muchas de las condiciones debidas a enzimas defectuosas podrían ser rescatadas por la suplementación de niacina (5).
Enfermedades cardiovasculares
Factores de riesgo cardiovascular
El ácido nicotínico es un agente hipolipemiante bien conocido: la terapia con ácido nicotínico aumenta notablemente las concentraciones de colesterol de lipoproteínas de alta densidad (HDL), disminuyendo las concentraciones séricas de lipoproteína(a) y desplaza partículas de lipoproteínas de baja densidad (LDL) pequeñas y densas a partículas LDL grandes y boyantes. Todos estos cambios en el perfil sanguíneo lipídico son considerados cardioprotectores. Bajas concentraciones de colesterol HDL son un factor de gran riesgo para la enfermedad coronaria cardiaca (ECC), y un incremento en las concentraciones HDL esta asociado con una reducción de ese riesgo (80). Debido a los efectos secundarios adversos asociados con las altas dosis de ácido nicotínico (véase Seguridad), el ácido nicotínico ha sido utilizado a menudo en combinación con otros medicamentos hipolipemiantes en dosis ligeramente menores (17). En particular, el ácido nicotínico en dosis bajas es frecuentemente coadministrado con inhibidores de la 3-hidroxi-3-metilglutaril-coenzima A reductasa (estatinas), la piedra angular del tratamiento para la hiperlipidemia, un factor de riesgo importante para la ECC. Un estudio controlado con placebo en 39 pacientes que tomaron estatinas (cerivastatina, atorvastatina o simvastatina) encontró que una dosis muy baja de ácido nicotínico (100 mg/día) incremento el colesterol HDL en solo 2.1 mg/dL, y la combinación no tuvo efecto en las concentraciones de colesterol LDL, colesterol total, o triglicéridos (81).
El estudio de la Biología Arterial para la Investigación de los Efectos del Tratamiento de Reducción del Colesterol (ARBITER) 2 — un ensayo doble ciego, controlado con placebo — investigó el efecto incremental de la adición de niacina (1 g/día) a la terapia con estatinas en 167 pacientes con ECC y niveles bajos de HDL en el grosor íntima-media carotídeo (CIMT) (82), un punto final sustituto para el desarrollo de la aterosclerosis. La adición del ácido nicotínico de liberación prolongada a la simvastatina previene el aumento en CIMT en comparación a la monoterapia de simvastatina. Un análisis post-hoc de datos del ARBITER2 mostró que el bloqueo de la progresión de la aterosclerosis se relacionaba con el aumento en las concentraciones de HDL en pacientes con un estado glucémico normal. Sin embargo, en la presencia de factores de riesgo adicionales, como la glucosa en ayunas alterada o la diabetes mellitus, el incremento de las concentraciones de HDL no fue predictivo de reducción del CIMT y retraso aterosclerótico (83). Un ensayo comparativo de eficacia (ARBITER6) también mostro una reducción significativa del CIMT basal con ácido nicotínico de liberación extendida (2 g/día por 14 meses), a diferencia de ezetimibe (un fármaco hipolipemiante) en pacientes que tomaban estatinas (84).
Estudios adicionales han examinado el impacto del acido nicotínico en la dilatación mediada por flujo (DMF) braquial dependiente del endotelio en pacientes en riesgo de ECC o con ECC establecida. La medición de la DMF es frecuentemente usada como un marcador sustituto de la función endotelial; los valores de la DMF están inversamente correlacionados con el riesgo de eventos cardiovasculares futuros (85). Un meta-análisis de siete ensayos controlados aleatorios, que incluyo 441 participantes, mostro un incremento significante del 2% con el acido nicotínico (1-2 g/día) administrado por 12 semanas a un año (86).
Eventos cardiovasculares
Varios ensayos aleatorios, controlados con placebo, multicéntricos han investigado la eficacia y seguridad de la terapia con ácido nicotínico, sola o en combinación con otros agentes hipolipemiantes en los resultados de las enfermedades cardiovasculares (ECV). Específicamente, el Proyecto de Drogas Coronarias (CDP) dio seguimiento a mas de 8,000 hombres con un infarto al corazón previo por seis años (87). En comparación al grupo de placebo, los pacientes que tomaron diariamente 3 g de ácido nicotínico de liberación inmediata una reducción del 10% en el colesterol total, una disminución del 26% en los triglicéridos, una reducción del 27% en infartos al miocardio no letales recurrentes, y una reducción del 26% en eventos cerebrovasculares (accidente cerebrovascular, y ataque isquémico transitorio). Además, un ensayo posterior de nueve años de seguimiento reveló una reducción del 10% en el total de muertes con el tratamiento con ácido nicotínico.
El Estudio del Tratamiento de la Aterosclerosis HDL (HATS), un ensayo controlado aleatorio de tres años en 160 pacientes con ECC documentada y concentraciones bajas de HDL, encontró que una combinación de simvastatina y ácido nicotínico (2-3 g/día) incrementó los niveles HDL, inhibió la progresión de la estenosis (estrechamiento) de la arteria coronaria, y disminuyó la frecuencia de eventos cardiovasculares, incluyendo el infarto al miocardio y accidentes cerebrovasculares, en comparación al placebo (88). Un análisis de subgrupo de pacientes del HATS con síndrome metabólico mostro una reducción en la tasa de eventos clínicos primarios, aunque la glucosa y el metabolismo de la insulina estaban moderadamente alterados por el acido nicotínico (89). Además, una revisión de la seguridad y tolerabilidad al ácido nicotínico entre los sujetos del HATS mostró que el control glucémico en pacientes con diabetes volvió a los valores de pretratamiento luego de ocho meses del manejo de la enfermedad con medicamento y dieta (90). Similarmente, el beneficio cardiovascular de la terapia con acido nicotínico a largo plazo superó el modesto aumento en el riesgo de una nueva aparición de diabetes tipo 2 en pacientes del estudio CDP (91).
En contraste, el ensayo AIM-HIGH (Intervención de la aterotrombosis en el síndrome metabólico con bajo HDL/triglicéridos altos: impacto en los resultados salud global), el cual examino el efecto incremental del ácido nicotínico de liberación extendida (1.5-2 g/día) en 3,414 pacientes que tenían ECV establecidas y dislipidemia aterogénica y eran tratados con simvastatina (+/- ezetimibe), proporcionó resultados decepcionantes. Ciertamente, en estos pacientes que habían alcanzado concentraciones objetivo de colesterol LDL (<70 mg/dL) antes de la aleatorización, el efecto de elevación de HDL del tratamiento con ácido nicotínico falló en reducir el numero de eventos cardiovasculares después de un seguimiento promedio de tres años (92, 93). Mientras que algunas de las limitaciones, como un uso mayor de simvastatina y ezetimibe en el grupo de control, han confundido los resultados, se sugirió también que el colesterol HDL bajo podría ser un marcador del riesgo en lugar de ser un factor de riesgo causal para la predicción de ECV (93). Además, un análisis post-hoc de 505 participantes con enfermedad crónica renal en etapa 3 encontró un incremento en la mortalidad por todas las causas en aquellos aleatorizados para el ácido nicotínico en comparación a aquellos en el grupo de placebo (94).
Aunque el acido nicotínico falló en reducir el numero de eventos cardiovasculares en pacientes con colesterol LDL bajo tratados con simvastatina, estos resultados no pueden extrapolar a pacientes con colesterol LDL más alto al inicio del estudio. Un ensayo multicéntrico, aleatorio, doble ciego, controlado con placebo de mayor magnitud — el ensayo HPS2-THRIVE (Estudio para la protección del corazón 2: tratamiento de HDL para reducir la incidencia de eventos vasculares) — en 25,673 participantes con enfermedad vascular examinó el efecto incremental del ácido nicotínico de liberación extendida (2 g/día) y el laropiprant (un antagonista del receptor-1 de la prostaglandina D2; 40 mg/día) en la incidencia de eventos cardiovasculares. En comparación al placebo, el ácido nicotínico/laropiprant redujo el colesterol LDL en un promedio de 10 mg/dL, disminuyó los triglicéridos 33 mg/dL, e incrementó el colesterol HDL 6 mg/dL después de un periodo medio de seguimiento de 3.9 años. No obstante, el ácido nicotínico/laropiprant mostró no efecto en la incidencia de eventos vasculares mayores y muerte por cualquier causa (95).
Un reciente meta-análisis de 23 ensayos controlados aleatorios — incluyendo los ensayos CDP, AIM-HIGH y HPS2-THRIVE — en 39,195 sujetos con un historial de enfermedad vascular comparó el efecto del acido nicotínico solo o como un agregado a otros agentes hipolipemiantes. Ningún beneficio cardiovascular fue asociado con la terapia con ácido nicotínico: el numero de infartos al miocardio letales y no letales y accidentes cerebrovasculares no disminuyo con la suplementación de acido nicotínico (dosis mediana de 2 g/día por un periodo medio de 11.5 meses) (96).
A pesar de la falta de evidencia para un papel del acido nicotínico en la prevención de ECV (96, 97), el uso de la terapia con ácido nicotínico ha rápidamente incrementado a través de los años en los Estados Unidos (98).
Ataxia de Friedreich
La ataxia de Friedreich, una forma común de ataxia hereditaria es un trastorno recesivo de aparición temprana con características clínicas que incluyen ataxia progresiva, escoliosis, cardiomiopatía, y diabetes mellitus (99). La mayoría de los sujetos afectados tienen expansiones repetidas homocigotas de guanina-adenina-adenina (GAA) en el primer intrón del gen FXN que codifica para la proteína frataxina. Estas repeticiones GAA anormales e inestables desencadenan el silenciamiento genético a través de la formación de heterocromatina, llevando a una expresión de frataxina significantemente reducida (100). La frataxina es una proteína mitocondrial necesaria para la creación de agrupaciones de hierro y sulfuro (ISC). Las subunidades que contienen ISC son especialmente importantes para la cadena respiratoria mitocondrial y para la síntesis de proteínas que contienen hemo (99).
Predominantemente localizada en el núcleo, SIRT1 es una deacetilasa dependiente de NAD+ que promueve el silenciamiento genético a través de la formación de la heterocromatina. Se ha mostrado que la nicotinamida antagoniza la heterocromatización del locus FXN y regula positivamente la expresión de frataxina en células linfoblastoides derivadas de pacientes afectados por la ataxia de Friedreich, posiblemente a través la inhibición de la actividad de SIRT1 (100). En un ensayo piloto, abierto de aumento de dosis en 10 pacientes adultos con ataxia de Friedreich, encontró que dosis solas y repetidas de nicotinamida (2-8 g) por un periodo de hasta ocho semanas eran bien toleradas (101). Dosis repetidas de 3.5 a 6 g de nicotinamida condujeron a incrementos significantes en la concentración de frataxina en glóbulos blancos periféricos (101). Sin embargo, ninguna mejora neurológica fue reportada, sugiriendo que la duración del tratamiento fue muy corta y/o el sistema nervioso de los participantes no respondió a los aumentos de la frataxina (102). Que sepamos, no existe actualmente algún ensayo en progreso diseñado para investigar más allá el efecto de la nicotinamida en pacientes afectados con la ataxia de Friedreich.
VIH/SIDA
El primer paso en la vía de quinurenina es catalizado por la enzima extrahepática, indoleamina 2,3-dioxigenasa (IDO), la cual es responsable por la escisión oxidativa del triptófano. La estimulación crónica de la oxidación del triptófano, mediada por un incremento en la actividad de IDO y/o las ingestas dietarías inadecuadas de niacina, es observada por la infección del virus de inmunodeficiencia humana (VIH), el virus que causa el síndrome de inmunodeficiencia adquirida (SIDA). El interferón-gamma (IFN- γ) es una citoquina producida por células del sistema inmunitario en respuesta a la infección. A través de la estimulación de la enzima IDO, el IFN- γ incrementa la descomposición del triptófano, apoyando de esta manera el descubrimiento de que la concentración de triptófano promedio en la sangre es significantemente menor en pacientes con VIH en comparación con sujetos no infectados (40). Una degradación incrementada del triptófano a través de la vía de quinurenina parece coexistir con la deficiencia intracelular de niacina/NAD en la infección con VIH (103). Un modelo explicativo de estas observaciones paradójicas incrimina al estrés oxidativo inducido por múltiples deficiencias en los pacientes con VIH (103). En particular la activación de enzimas PARP (ARTD) por el daño oxidativo al ADN podría ser responsable por inducir el agotamiento de niacina/NAD (véase Función). La descomposición del triptófano seria entonces una respuesta compensatoria a los niveles de niacina/NAD inadecuados.
Sin embargo, los metabolismos derivados de la oxidación del triptófano en la vía de quinurenina regulan subgrupos de linfocitos T específicos. Como fue mencionado anteriormente, el IFN-γ circulante, pero también los productos virales y bacterianos, pueden activar la IDO durante la infección con VIH. La sobreestimulación de la ruta del triptófano ha sido involucrada en la perdida de la función normal de los linfocitos T, que caracteriza la infección por VIH (104, 105). El aumento en la actividad de la IDO ha sido ligado a la respuesta inmune alterada que contribuye a la persistencia del VIH (104). La terapia antirretroviral (TARV) solo restaura parcialmente la actividad normal de la IDO, sin normalizarla, pero induce la supresión viral y la recuperación de células T CD4 (106). En un modelo de mono para la infección por VIH, un bloqueo parcial y transitorio de la IDO con el inhibidor de IDO 1-metil-triptófano probó ser ineficaz para reducir la carga viral en el plasma y los tejidos intestinales más allá de los niveles logrados por la TARV (107). En la actualidad, un mejor entendimiento del papel de la vía de quinurenina y otras rutas NAD biosintéticas durante la infección por VIH es necesario antes de que se pueda considerar la relevancia y las implicaciones clínicas de la administración de suplementos de niacina en el tratamiento del VIH.
No obstante, las dosis farmacológicas de ácido nicotínico han mostrado ser bien toleradas en los pacientes con VIH con hiperlipidemia (108). Los perfiles de lípidos anormales observados en pacientes han sido atribuidos a la infección por VIH y al tratamiento antirretroviral altamente activo (TARAA) (109). Además, la resistencia a la insulina ha sido detectada junto con dislipidemia en pacientes tratados con TARV (110). Las enfermedades cardiovasculares (ECV) son la segunda causa más frecuente de muerte en la población con VIH, y se predice que la tasa de ECV incrementara aún más ya que los pacientes están viviendo más debido a las terapias antirretrovirales exitosas. Con respecto a la población general, la terapia basada en estatinas parece beneficiar los pacientes con VIH en términos de protección aterogénica y reducción del riesgo de ECV, aunque contraindicaciones existen debido a la interacción de drogas con la TARV. Otros tratamientos de primera línea incluyen fibratos hipolipemiantes, los cuales son preferidos al acido nicotínico debido al riesgo incrementado de intolerancia a la glucosa y resistencia a la insulina (111). Sin embargo, un estudio piloto controlado, sin cegar mostró que el ácido nicotínico de liberación extendida (0.5-1.5 g/día por 12 semanas) pudo efectivamente mejorar la función endotelial de la arteria braquial en los sujetos con VIH tratados con TARV con bajo colesterol HDL y sin historial de ECV (112). Además, un tratamiento combinado de fibratos, acido nicotínico de liberación extendida (0.5-2 g/día), y cambios en el estilo de vida (dieta baja en grasa y ejercicio) por 24 semanas fue eficaz en la normalización de los parámetros lipídicos en una cohorte de 191 pacientes tratados con TARV. El riesgo incrementado de disfunción hepática fue detectado en sujetos que recibían ambos fibratos y niacina, pero la sensibilidad a la insulina no fue afectada por el tratamiento con ácido nicotínico administrado solo o cuando fue combinado con fibratos (113). Otro ensayo no controlado, abierto, de 24 semanas en 99 pacientes tratados con TARV encontró que la aleatorización de ácido nicotínico de liberación extendida (0.5-2 g/día) o fenofibratos incrementó el colesterol HDL sanguíneo, pero no redujo los marcadores inflamatorios o mejoro la función endotelial en comparación al valor basal (114).
Esquizofrenia
La esquizofrenia es un trastorno neurológico de etiología poco clara que se diagnostica solo a partir de su presentación clínica. Debido a que los trastornos neurológicos asociados con la pelagra se asemejan a la esquizofrenia aguda, la terapia basada en niacina para la condición fue investigada durante 1950-70 (revisado en 115). El uso adyuvante de nutrientes como la niacina para corregir las deficiencias asociadas con síntomas neurológicos se llama psiquiatría ortomolecular (116). Tal enfoque no se ha incluido en la práctica psiquiátrica; los profesionales se han basado únicamente en fármacos antipsicóticos para eliminar los síntomas clínicos de la esquizofrenia. Sin embargo, avances científicos recientes y nuevas hipótesis en el beneficio de la suplementación de nutrientes en el tratamiento de trastornos psiquiátricos han sugerido la reevaluación de la medicina ortomolecular por la comunidad médica (117, 118).
El enrojecimiento de la piel es uno de los principales efectos secundarios del uso terapéutico del ácido nicotínico y la razón principal de la falta de adherencia al tratamiento (ver Toxicidad). El enrojecimiento es causado por la activación de la fosfolipasa A2, una enzima que estimula la producción de un lípido específico de la familia prostanoide llamado prostaglandina D2. La prostaglandina D2, sintetizada por las células presentadoras de antígenos de la piel y la mucosa (es decir, las células de Langerhans), puede inducir la dilatación de los vasos sanguíneos y desencadenar una respuesta de enrojecimiento. Curiosamente, los pacientes con esquizofrenia tienden a no enrojecer luego del tratamiento con ácido nicotínico. Esta respuesta de enrojecimiento de piel deficiente sugiere una señalización prostanoide en pacientes esquizofrénicos (119, 120). Se ha encontrado una asociación entre la sensibilidad de niacina alterada y un mayor deterioro funcional en pacientes esquizofrénicos (121), que apoya otros hallazgos que sugieren que el metabolismo lipídico alterado podría críticamente alterar el desarrollo del cerebro y contribuir a la enfermedad (122). Curiosamente, las respuestas de enrojecimiento cutáneas deficientes son más prevalentes en familiares de primer grado de personas con esquizofrenia que en la población general, sugiriendo que la sensibilidad reducida a la niacina es un rasgo hereditario dentro de las familias afectadas (123).
Fuentes
Fuentes alimenticias
Buenas fuentes de niacina incluyen la levadura, carne, aves de corral, pescados rojos (p.ej. atún, salmón), los cereales (especialmente los cereales fortificados), legumbres, y semillas. La leche, verduras de hojas verdes, el café y té también aportan una pequeña cantidad de niacina (124). En las plantas, especialmente los granos de cereal maduro como el maíz y el trigo, la niacina podría estar ligada a moléculas de azúcar en la forma de glucósidos, lo que disminuye significativamente su biodisponibilidad (25).
En los Estados Unidos, la ingesta dietaría promedio de niacina es de alrededor de 30 mg/día para hombres adultos jóvenes y de 20 mg/día para las mujeres adultas jóvenes. En una muestra de adultos mayores de 60 años, se encontró que hombres y mujeres tenían una ingesta dietaría promedio de 21 mg/día y 17 mg/día, respectivamente (41). Algunos alimentos con cantidades sustanciales de niacina son listados en la Tabla 2, junto con su contenido de niacina en miligramos (mg). Las tablas de composición de los alimentos generalmente muestran el contenido de niacina sin incluir los equivalentes de niacina (EN) del triptófano o algún ajuste para la biodisponibilidad de niacina. Para mayor información en el contenido de nutrientes de alimentos específicos, revise la base de datos de composición de los alimentos del USDA; la información incluida en la Tabla 2 proviene de esta base de datos (125).
Suplementos
Los suplementos de niacina están disponibles como nicotinamida o acido nicotínico. La nicotinamida es la forma de la niacina típicamente usada en suplementos nutricionales y en la fortificación de alimentos. El ácido nicotínico esta disponible sin receta y con una prescripción como un agente reductor del colesterol (126). El ácido nicotínico para su uso anti-hiperlipidémico en tres formulaciones: acido nicotínico de liberación inmediata (cristalino) (tiempo de absorción, 1-2 horas), acido nicotínico de liberación extendida (tiempo de absorción, 8-12 horas), y acido nicotínico de liberación sostenida (tiempo de absorción, >12 horas) (127). En la dosis farmacológica requerida para los efectos de reducción del colesterol, el uso de ácido nicotínico debe ser abordado como si este fuese un fármaco (véase Seguridad). Las personas solo deben tomar una terapia de reducción del colesterol con ácido nicotínico bajo la supervisión de un proveedor del cuidado de la salud calificado con el fin de minimizar los efectos potencialmente adversos y maximizar los beneficios terapéuticos.
Seguridad
Toxicidad
A la niacina de los alimentos no se le conoce por causar efectos adversos. Aunque un estudio notó efectos adversos luego del consumo de bagels con 60 veces más la cantidad normal de la fortificación con niacina, la mayoría de los efectos han sido reportados con preparaciones farmacológicas de niacina (41).
Acido nicotínico
Los efectos secundarios comunes de ácido nicotínico incluyen enrojecimiento, prurito (picazón severa de la piel), erupciones cutáneas y trastornos gastrointestinales, como las náuseas y vómitos (97). También se han informado episodios transitorios de presión arterial baja (hipotensión) y dolor de cabeza. La hepatotoxicidad (daño celular hepático), que incluye enzimas hepáticas elevadas e ictericia, ha sido observada en ingestas tan bajas de hasta 750 mg/día de ácido nicotínico (128). Aunque la hepatitis ha sido observada con dosis de ácido nicotínico de liberación extendida de tan poco como 500 mg/día por dos meses, casi todos los reportes de hepatitis severa han sido asociados con dosis de 3 a 9 g/día usados para tratar el colesterol alto por meses o años (41). No esta claro si el ácido nicotínico de liberación inmediata (cristalino) es menos toxico para el hígado que las formas de liberación extendida (41). Sin embargo, el ácido nicotínico de liberación inmediata es frecuentemente usado en dosis más altas que las formas de liberación extendida, y la toxicidad severa hepática ha ocurrido en individuos que sustituyeron el ácido nicotínico de liberación extendida por acido nicotínico de liberación inmediata en dosis equivalentes (126). Se ha observado que grades dosis de ácido nicotínico alteran la tolerancia a la glucosa probablemente debido a una disminución en la sensibilidad a la insulina. La tolerancia a la glucosa alterada en individuos susceptibles (prediabéticos) podría resultar en concentraciones elevadas de glucosa en la sangre y diabetes mellitus tipo 2. Un análisis del ensayo HPS2-THRIVE (véase Enfermedades cardiovasculares), usando datos de 17,374 participantes sin diabetes tipo 2 al inicio del estudio, encontró una proporción significantemente mas alta de casos recientemente diagnosticados entre aquellos aleatorizados para el ácido nicotínico/laropiprant que para el placebo (5.7% versus 4.3%) sobre un periodo de 3.9 años (95). De igual manera, la aleatorización para el ácido nicotínico/laropiprant significantemente incremento el riesgo de disturbios serios en el control para la diabetes (llevando a la hospitalización) en comparación al placebo entre 8,299 participantes con diabetes al inicio del estudio (95). Las concentraciones sanguíneas elevadas de ácido úrico, ocasionalmente resultando en ataques de gota en los individuos susceptibles, han sido también observados con la terapia con ácido nicotínico de alta dosis (126). La niacina en dosis de 1.5 a 5 g/día ha resultado en unos pocos reportes de caso de visión borrosa y otros problemas oculares, los cuales han sido generalmente reversibles luego de la discontinuación (41). Personas con una función hepática anormal o un historial de enfermedad hepática, diabetes, enfermedad de úlcera péptica activa, gota, arritmias cardiacas, enfermedad intestinal inflamatoria, migrañas, o alcoholismo pueden ser mas susceptibles a los efectos adversos de la ingesta de niacina en exceso que la población en general (41).
Nicotinamida
La nicotinamida es generalmente mejor tolerada que el acido nicotínico; generalmente no causa enrojecimiento (126). Sin embargo, náuseas, vómitos, y signos de toxicidad hepática (enzimas hepáticas elevadas, ictericia) han sido observados en dosis muy altas (≥10 g/día) (126).
Nicotinamida ribósido
Un estudio en 12 sujetos saludables encontró que la nicotinamida ribósido en tres dosis únicas (100 mg, 300 mg, y 1,000 mg) incremento el NAD+ sanguíneo sin peligro. Dos de los participantes auto-reportaron enrojecimiento de la piel después de tomar la dosis de 300 mg, y dos otros reportaron una sensación de calor luego de la ingesta de 1,000 mg de nicotinamida ribósido (1). En un ensayo aleatorio, controlado con placebo reciente en 120 adultos saludables (edades, 60-80 años), la suplementación diaria con nicotinamida ribósido (250 mg o 500 mg) y pteroestilbeno (un activador de SIRT; 50 mg o 100 mg) por ocho semanas mostro un perfil favorable de efectos secundarios, sin evidencia de una incidencia mayor de efectos adversos en comparación al placebo (129). Más recientemente, un ensayo aleatorio, controlado con placebo en 40 hombres obesos (edades, 40-70 años) encontró que la suplementación diaria con nicotinamida ribósido (2,000 mg/día dividida en dos dosis diarias) durante 12 semanas fue asociada con reportes de solo efectos secundarios, incluyendo sudoración excesiva, prurito y síntomas gastrointestinales leves como la hinchazón (139).
El nivel máximo de ingesta tolerable (NM)
El enrojecimiento de la piel principalmente en la cara, brazos, y pecho, es un efecto secundario común del ácido nicotínico y puede ocurrir inicialmente con dosis tan bajas como de 30 mg/día. Aunque el enrojecimiento causado por la nicotinamida es raro, la Junta de Nutrición y Alimentos estableció el nivel máximo de ingesta tolerable (NM) para la niacina (ácido nicotínico y nicotinamida) en 35 mg/día para los adultos para evitar el enrojecimiento como un efecto adverso (41). El análisis de datos de la Encuesta de Evaluación Nacional de Salud y Nutrición Estadounidense (NHANES) 2003-2006 encontró que el 15.8% de los niños y adolescentes (edades 2-18 años) y el 8.5% de los adultos (≥19 años) tuvieron ingestas usuales de niacina totales que excedían el NM (130). El NM se aplica a toda la población en general y no esta destinado a ser aplicado en individuos que están siendo tratados con un nutriente bajo supervisión médica (p. ej., acido nicotínico en altas dosis para las concentraciones elevadas de colesterol sanguíneo).
| Grupo Etario | NM (mg/día) |
|---|---|
| Infantes 0-12 meses | Imposible de determinar* |
| Niños 1-3 años | 10 |
| Niños 4-8 años | 15 |
| Niños 9-13 años | 20 |
| Adolescentes 14-18 años | 30 |
| Adultos 19 años y más | 35 |
| *La fuente de la ingesta debiera ser sólo de alimentos y fórmula. | |
Interacción con drogas/fármacos
La ocurrencia de rabdomiolisis se incrementa en los pacientes tratados con estatinas (inhibidores de la HMG-CoA reductasa). La rabdomiolisis es una condición relativamente poco común en la cual las células musculares se degradan, liberando enzimas y electrolitos a la sangre, y algunas veces resultando en falla renal (131). La coadministración de ácido nicotínico con una estatina parece incrementar el riesgo de rabdomiolisis (132). Un nuevo fármaco, laropiprant, bloquea los receptores prostanoides y reduce el enrojecimiento inducido por el ácido nicotínico (133). Un ensayo aleatorio, controlado con placebo fue designado para identificar efectos adversos posibles de la combinación de niacina/laropiprant en más de 25,000 pacientes tratados con simvastatina (134). Cuando se agregó a la terapia con estatinas, la combinación de niacina/laropiprant aumentó el riesgo de miopatía y rabdomiolisis, particularmente en sujetos asiáticos. Es posible que la combinación de niacina/laropirant reduzca aún más la baja tolerabilidad del tratamiento con estatinas observadas en ciertas poblaciones (135).
En el estudio aleatorio controlado HATS de tres años, la terapia concurrente con antioxidantes (1,000 mg/día de vitamina C, 800 UI/día de RRR-α-tocoferol, 100 µg/día de selenio, y 25 mg/día de β-caroteno) disminuyó los efectos protectores de la combinación de simvastatina-acido nicotínico (136). Aunque el mecanismo para estos efectos es desconocido, el beneficio de la terapia antioxidante concurrente en pacientes que toman agentes hipolipemiantes ha sido cuestionado (137).
Los efectos adversos de grandes dosis de ácido nicotínico pueden ser exacerbados por el uso concomitante de ciertos medicamentos. El riesgo de miopatía puede aumentar aún más en aquellos que toman ácido nicotínico y secuestrantes de ácidos biliares (p. ej., colestiramina, colestipol) o la droga antilipidémica, gemfibrozil (Lopid), y el riesgo de hepatotoxicidad observado con el ácido nicotínico podría ser incrementado por fármacos como el paracetamol, la amiodarona (Cordarone) o la carbamazepina (Tegretol) (35). Además, grandes dosis de ácido nicotínico pueden reducir la excreción de ácido úrico, oponiéndose de este modo a la acción de los agentes uricosúricos como el probenecid (Probalan) (35).
Varios otros medicamentos pueden interactuar con la terapia con niacina o con la absorción y metabolismo de la vitamina (126). El estrógeno y los anticonceptivos que contienen estrógeno incrementan la eficacia de la síntesis de niacina a partir del triptófano, resultando en un requerimiento dietario disminuido para la niacina (138).
Se ha reportado que la administración a largo plazo de agentes de la quimioterapia causa síntomas de pelagra; por lo tanto, la suplementación con niacina puede ser necesaria (véase Causas de la pelagra).
Recomendación del Instituto Linus Pauling
La ingesta optima de niacina para la promoción de la salud y prevención de enfermedades crónicas no es aun conocida. La IDR (16 mg EN/día para hombres y 14 mg EN/día para mujeres) es obtenible fácilmente consumiendo una dieta variada y debería prevenir una deficiencia en la mayoría de las personas. Siguiendo la recomendación del Instituto Linus Pauling de tomar a diario un suplemento multivitamínico/mineral que contenga el 100% del Valor Diario (VD) para niacina, aportará diariamente al menos 20 mg de niacina.
Autores y Críticos
Escrito en 2002 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Agosto 2002 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Junio 2007 por:
Victoria J. Drake, Ph.D
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Julio 2013 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Diciembre de 2017 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Marzo de 2018 por:
Mirella Meyer-Ficca, Ph.D.
Profesor Asistente de Investigación
Universidad del Estado de Utah
Traducido al Español en 2018 por:
Silvia Vazquez Lima
Instituto Linus Pauling
Universidad Estatal de Oregon
La actualización y traducciones al español de este artículo en el 2017 fue respaldada por una subvención de ChromaDex, Inc.
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Derechos de autoría 2000-2024 Instituto Linus Pauling
Referencias
1. Trammell SA, Schmidt MS, Weidemann BJ, et al. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun. 2016;7:12948. (PubMed)
2. Nikiforov A, Kulikova V, Ziegler M. The human NAD metabolome: functions, metabolism and compartmentalization. Crit Rev Biochem Mol Biol. 2015;50(4):284-297. (PubMed)
3. Kawai S, Murata K. Structure and function of NAD kinase and NADP phosphatase: key enzymes that regulate the intracellular balance of NAD(H) and NADP(H). Biosci Biotechnol Biochem. 2008;72(4):919-930. (PubMed)
4. Agledal L, Niere M, Ziegler M. The phosphate makes a difference: cellular functions of NADP. Redox Rep. 2010;15(1):2-10. (PubMed)
5. Penberthy WT, Kirkland JB. Niacin. In: Erdman JW, MacDonald I, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Ames: International Life Sciences Institute; 2012:293-306.
6. Kirkland JB. Niacin. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore: Lippincott Williams & Wilkins; 2014:331-340.
7. Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci. 2010;35(4):208-219. (PubMed)
8. Liu C, Yu X. ADP-ribosyltransferases and poly ADP-ribosylation. Curr Protein Pept Sci. 2015;16(6):491-501. (PubMed)
9. Hwang ES, Song SB. Nicotinamide is an inhibitor of SIRT1 in vitro, but can be a stimulator in cells. Cell Mol Life Sci. 2017;74(18):3347-3362. (PubMed)
10. Morris BJ. Seven sirtuins for seven deadly diseases of aging. Free Radic Biol Med. 2013;56:133-171. (PubMed)
11. Fliegert R, Bauche A, Wolf Perez AM, et al. 2'-Deoxyadenosine 5'-diphosphoribose is an endogenous TRPM2 superagonist. Nat Chem Biol. 2017;13(9):1036-1044. (PubMed)
12. Mutafova-Yambolieva VN, Hwang SJ, Hao X, et al. Beta-nicotinamide adenine dinucleotide is an inhibitory neurotransmitter in visceral smooth muscle. Proc Natl Acad Sci U S A. 2007;104(41):16359-16364. (PubMed)
13. Moreschi I, Bruzzone S, Nicholas RA, et al. Extracellular NAD+ is an agonist of the human P2Y11 purinergic receptor in human granulocytes. J Biol Chem. 2006;281(42):31419-31429. (PubMed)
14. Klein C, Grahnert A, Abdelrahman A, Muller CE, Hauschildt S. Extracellular NAD(+) induces a rise in [Ca(2+)](i) in activated human monocytes via engagement of P2Y(1) and P2Y(11) receptors. Cell Calcium. 2009;46(4):263-272. (PubMed)
15. Moreschi I, Bruzzone S, Bodrato N, et al. NAADP+ is an agonist of the human P2Y11 purinergic receptor. Cell Calcium. 2008;43(4):344-355. (PubMed)
16. Huang C, Hu J, Subedi KP, et al. Extracellular adenosine diphosphate ribose mobilizes intracellular Ca2+ via purinergic-dependent Ca2+ pathways in rat pulmonary artery smooth muscle cells. Cell Physiol Biochem. 2015;37(5):2043-2059. (PubMed)
17. Knopp RH. Drug treatment of lipid disorders. N Engl J Med. 1999;341(7):498-511. (PubMed)
18. Graff EC, Fang H, Wanders D, Judd RL. Anti-inflammatory effects of the hydroxycarboxylic acid receptor 2. Metabolism. 2016;65(2):102-113. (PubMed)
19. Jin FY, Kamanna VS, Kashyap ML. Niacin accelerates intracellular ApoB degradation by inhibiting triacylglycerol synthesis in human hepatoblastoma (HepG2) cells. Arterioscler Thromb Vasc Biol. 1999;19(4):1051-1059. (PubMed)
20. Kamanna VS, Ganji SH, Kashyap ML. Recent advances in niacin and lipid metabolism. Curr Opin Lipidol. 2013;24(3):239-245. (PubMed)
21. Carlson LA. Studies on the effect of nicotinic acid on catecholamine stimulated lipolysis in adipose tissue in vitro. Acta Med Scand. 1963;173:719-722. (PubMed)
22. Lauring B, Taggart AK, Tata JR, et al. Niacin lipid efficacy is independent of both the niacin receptor GPR109A and free fatty acid suppression. Sci Transl Med. 2012;4(148):148ra115. (PubMed)
23. Brody T. Nutritional Biochemistry. 2nd ed. San Diego: Academic Press; 1999.
24. Kirkland JB. Niacin. In: Zempleni J, Suttie JW, Gregory III JF, Stover PJ, eds. Handbook of Vitamins. 5th ed. Boca Raton: CRC Press; 2013:149-190.
25. Gregory JF, 3rd. Nutritional properties and significance of vitamin glycosides. Annu Rev Nutr. 1998;18:277-296. (PubMed)
26. Dawson B, Favaloro EJ, Taylor J, Aggarwal A. Unrecognized pellagra masquerading as odynophagia. Intern Med J. 2006;36(7):472-474. (PubMed)
27. Jagielska G, Tomaszewicz-Libudzic EC, Brzozowska A. Pellagra: a rare complication of anorexia nervosa. Eur Child Adolesc Psychiatry. 2007;16(7):417-420. (PubMed)
28. Kertesz SG. Pellagra in 2 homeless men. Mayo Clin Proc. 2001;76(3):315-318. (PubMed)
29. Prakash R, Gandotra S, Singh LK, Das B, Lakra A. Rapid resolution of delusional parasitosis in pellagra with niacin augmentation therapy. Gen Hosp Psychiatry. 2008;30(6):581-584. (PubMed)
30. Badawy AA. Pellagra and alcoholism: a biochemical perspective. Alcohol Alcohol. 2014;49(3):238-250. (PubMed)
31. Majewski M, Kozlowska A, Thoene M, Lepiarczyk E, Grzegorzewski WJ. Overview of the role of vitamins and minerals on the kynurenine pathway in health and disease. J Physiol Pharmacol. 2016;67(1):3-19. (PubMed)
32. Rosmaninho A, Sanches M, Fernandes IC, et al. Letter: Pellagra as the initial presentation of Crohn disease. Dermatol Online J. 2012;18(4):12. (PubMed)
33. Zaraa I, Belghith I, El Euch D, et al. A case of pellagra associated with megaduodenum in a young woman. Nutr Clin Pract. 2013;28(2):218-222. (PubMed)
34. Bilgili SG, Karadag AS, Calka O, Altun F. Isoniazid-induced pellagra. Cutan Ocul Toxicol. 2011;30(4):317-319. (PubMed)
35. Natural Medicines. Professional Monograph - Niacin/Interactions with drugs. Available at: https://naturalmedicines.therapeuticresearch.com/. Accessed 8/2/17.
36. Dreizen S, McCredie KB, Keating MJ, Andersson BS. Nutritional deficiencies in patients receiving cancer chemotherapy. Postgrad Med. 1990;87(1):163-167, 170. (PubMed)
37. Nogueira A, Duarte AF, Magina S, Azevedo F. Pellagra associated with esophageal carcinoma and alcoholism. Dermatol Online J. 2009;15(5):8. (PubMed)
38. Oldham MA, Ivkovic A. Pellagrous encephalopathy presenting as alcohol withdrawal delirium: a case series and literature review. Addict Sci Clin Pract. 2012;7(1):12. (PubMed)
39. World Health Organization, United Nations High Commissions for Refugees. Pellagra and its prevention and control in major emergencies. World Health Organization. 2000. Available at: http://www.who.int/nutrition/publications/emergencies/WHO_NHD_00.10/en/. Accessed 6/20/13.
40. Murray MF. Tryptophan depletion and HIV infection: a metabolic link to pathogenesis. Lancet Infect Dis. 2003;3(10):644-652. (PubMed)
41. Food and Nutrition Board, Institute of Medicine. Niacin. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B-6, Vitamin B-12, Pantothenic Acid, Biotin, and Choline. Washington, D.C.: The National Academies Press; 1998:123-149. (The National Academies Press)
42. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11(3):220-228. (PubMed)
43. Kirkland JB. Niacin requirements for genomic stability. Mutat Res. 2012;733(1-2):14-20. (PubMed)
44. Burkle A. Poly(ADP-ribose). The most elaborate metabolite of NAD+. FEBS J. 2005;272(18):4576-4589. (PubMed)
45. Jacobson EL, Shieh WM, Huang AC. Mapping the role of NAD metabolism in prevention and treatment of carcinogenesis. Mol Cell Biochem. 1999;193(1-2):69-74. (PubMed)
46. Spronck JC, Nickerson JL, Kirkland JB. Niacin deficiency alters p53 expression and impairs etoposide-induced cell cycle arrest and apoptosis in rat bone marrow cells. Nutr Cancer. 2007;57(1):88-99. (PubMed)
47. Spronck JC, Kirkland JB. Niacin deficiency increases spontaneous and etoposide-induced chromosomal instability in rat bone marrow cells in vivo. Mutat Res. 2002;508(1-2):83-97. (PubMed)
48. Kostecki LM, Thomas M, Linford G, et al. Niacin deficiency delays DNA excision repair and increases spontaneous and nitrosourea-induced chromosomal instability in rat bone marrow. Mutat Res. 2007;625(1-2):50-61. (PubMed)
49. Dantzer F, Santoro R. The expanding role of PARPs in the establishment and maintenance of heterochromatin. FEBS J. 2013;280(15):3508-3518. (PubMed)
50. El Ramy R, Magroun N, Messadecq N, et al. Functional interplay between Parp-1 and SirT1 in genome integrity and chromatin-based processes. Cell Mol Life Sci. 2009;66(19):3219-3234. (PubMed)
51. Boyonoski AC, Spronck JC, Gallacher LM, et al. Niacin deficiency decreases bone marrow poly(ADP-ribose) and the latency of ethylnitrosourea-induced carcinogenesis in rats. J Nutr. 2002;132(1):108-114. (PubMed)
52. Boyonoski AC, Spronck JC, Jacobs RM, Shah GM, Poirier GG, Kirkland JB. Pharmacological intakes of niacin increase bone marrow poly(ADP-ribose) and the latency of ethylnitrosourea-induced carcinogenesis in rats. J Nutr. 2002;132(1):115-120. (PubMed)
53. Weitberg AB. Effect of nicotinic acid supplementation in vivo on oxygen radical-induced genetic damage in human lymphocytes. Mutat Res. 1989;216(4):197-201. (PubMed)
54. Hageman GJ, Stierum RH, van Herwijnen MH, van der Veer MS, Kleinjans JC. Nicotinic acid supplementation: effects on niacin status, cytogenetic damage, and poly(ADP-ribosylation) in lymphocytes of smokers. Nutr Cancer. 1998;32(2):113-120. (PubMed)
55. Yong LC, Petersen MR. High dietary niacin intake is associated with decreased chromosome translocation frequency in airline pilots. Br J Nutr. 2011;105(4):496-505. (PubMed)
56. Weidele K, Beneke S, Burkle A. The NAD+ precursor nicotinic acid improves genomic integrity in human peripheral blood mononuclear cells after X-irradiation. DNA Repair (Amst). 2017;52:12-23. (PubMed)
57. Jacobson EL. Niacin deficiency and cancer in women. J Am Coll Nutr. 1993;12(4):412-416. (PubMed)
58. Negri E, Franceschi S, Bosetti C, et al. Selected micronutrients and oral and pharyngeal cancer. Int J Cancer. 2000;86(1):122-127. (PubMed)
59. Franceschi S, Bidoli E, Negri E, et al. Role of macronutrients, vitamins and minerals in the aetiology of squamous-cell carcinoma of the oesophagus. Int J Cancer. 2000;86(5):626-631. (PubMed)
60. Gensler HL, Williams T, Huang AC, Jacobson EL. Oral niacin prevents photocarcinogenesis and photoimmunosuppression in mice. Nutr Cancer. 1999;34(1):36-41. (PubMed)
61. Jacobson EL, Kim H, Kim M, et al. A topical lipophilic niacin derivative increases NAD, epidermal differentiation and barrier function in photodamaged skin. Exp Dermatol. 2007;16(6):490-499. (PubMed)
62. Bermudez Y, Benavente CA, Meyer RG, Coyle WR, Jacobson MK, Jacobson EL. Nicotinic acid receptor abnormalities in human skin cancer: implications for a role in epidermal differentiation. PLoS One. 2011;6(5):e20487. (PubMed)
63. Benavente CA, Jacobson EL. Niacin restriction upregulates NADPH oxidase and reactive oxygen species (ROS) in human keratinocytes. Free Radic Biol Med. 2008;44(4):527-537. (PubMed)
64. Benavente CA, Schnell SA, Jacobson EL. Effects of niacin restriction on sirtuin and PARP responses to photodamage in human skin. PLoS One. 2012;7(7):e42276. (PubMed)
65. Park SM, Li T, Wu S, et al. Niacin intake and risk of skin cancer in US women and men. Int J Cancer. 2017;140(9):2023-2031. (PubMed)
66. Chen AC, Martin AJ, Choy B, et al. A phase 3 randomized trial of nicotinamide for skin-cancer chemoprevention. N Engl J Med. 2015;373(17):1618-1626. (PubMed)
67. Minocha R, Damian DL, Halliday GM. Melanoma and nonmelanoma skin cancer chemoprevention: A role for nicotinamide? Photodermatol Photoimmunol Photomed. 2018;34(1):5-12. (PubMed)
68. Orban T, Sosenko JM, Cuthbertson D, et al. Pancreatic islet autoantibodies as predictors of type 1 diabetes in the Diabetes Prevention Trial-Type 1. Diabetes Care. 2009;32(12):2269-2274. (PubMed)
69. Szkudelski T. Streptozotocin-nicotinamide-induced diabetes in the rat. Characteristics of the experimental model. Exp Biol Med (Maywood). 2012;237(5):481-490. (PubMed)
70. Lampeter EF, Klinghammer A, Scherbaum WA, et al. The Deutsche Nicotinamide Intervention Study: an attempt to prevent type 1 diabetes. DENIS Group. Diabetes. 1998;47(6):980-984. (PubMed)
71. Gale EA, Bingley PJ, Emmett CL, Collier T, European Nicotinamide Diabetes Intervention Trial Group. European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet. 2004;363(9413):925-931. (PubMed)
72. Hedman M, Ludvigsson J, Faresjo MK. Nicotinamide reduces high secretion of IFN-gamma in high-risk relatives even though it does not prevent type 1 diabetes. J Interferon Cytokine Res. 2006;26(4):207-213. (PubMed)
73. Fernandez IC, Del Carmen Camberos M, Passicot GA, Martucci LC, Cresto JC. Children at risk of diabetes type 1. Treatment with acetyl-L-carnitine plus nicotinamide - Case reports. J Pediatr Endocrinol Metab. 2013;26(3-4):347-355. (PubMed)
74. Patel AB, Prabhu AS. Hartnup disease. Indian J Dermatol. 2008;53(1):31-32. (PubMed)
75. Oakley A, Wallace J. Hartnup disease presenting in an adult. Clin Exp Dermatol. 1994;19(5):407-408. (PubMed)
76. Shi H, Enriquez A, Rapadas M, et al. NAD deficiency, congenital malformations, and niacin supplementation. N Engl J Med. 2017;377(6):544-552. (PubMed)
77. Vander Heiden MG. Metabolism and congenital malformations - NAD's effects on development. N Engl J Med. 2017;377(6):509-511. (PubMed)
78. Hu L, Ibrahim K, Stucki M, et al. Secondary NAD+ deficiency in the inherited defect of glutamine synthetase. J Inherit Metab Dis. 2015;38(6):1075-1083. (PubMed)
79. Ames BN, Elson-Schwab I, Silver EA. High-dose vitamin therapy stimulates variant enzymes with decreased coenzyme binding affinity (increased K(m)): relevance to genetic disease and polymorphisms. Am J Clin Nutr. 2002;75(4):616-658. (PubMed)
80. Bays HE, Shah A, Lin J, Sisk CM, Dong Q, Maccubbin D. Consistency of extended-release niacin/laropiprant effects on Lp(a), ApoB, non-HDL-C, Apo A1, and ApoB/ApoA1 ratio across patient subgroups. Am J Cardiovasc Drugs. 2012;12(3):197-206. (PubMed)
81. Wink J, Giacoppe G, King J. Effect of very-low-dose niacin on high-density lipoprotein in patients undergoing long-term statin therapy. Am Heart J. 2002;143(3):514-518. (PubMed)
82. Taylor AJ, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation. 2004;110(23):3512-3517. (PubMed)
83. Taylor AJ, Zhu D, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Relationship between glycemic status and progression of carotid intima-media thickness during treatment with combined statin and extended-release niacin in ARBITER 2. Vasc Health Risk Manag. 2007;3(1):159-164. (PubMed)
84. Villines TC, Stanek EJ, Devine PJ, et al. The ARBITER 6-HALTS Trial (Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol 6-HDL and LDL Treatment Strategies in Atherosclerosis): final results and the impact of medication adherence, dose, and treatment duration. J Am Coll Cardiol. 2010;55(24):2721-2726. (PubMed)
85. Ras RT, Streppel MT, Draijer R, Zock PL. Flow-mediated dilation and cardiovascular risk prediction: a systematic review with meta-analysis. Int J Cardiol. 2013;168(1):344-351. (PubMed)
86. Sahebkar A. Effect of niacin on endothelial function: a systematic review and meta-analysis of randomized controlled trials. Vasc Med. 2014;19(1):54-66. (PubMed)
87. Canner PL, Berge KG, Wenger NK, et al. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol. 1986;8(6):1245-1255. (PubMed)
88. Brown BG, Zhao XQ, Chait A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345(22):1583-1592. (PubMed)
89. Vittone F, Chait A, Morse JS, Fish B, Brown BG, Zhao XQ. Niacin plus simvastatin reduces coronary stenosis progression among patients with metabolic syndrome despite a modest increase in insulin resistance: a subgroup analysis of the HDL-Atherosclerosis Treatment Study (HATS). J Clin Lipidol. 2007;1(3):203-210. (PubMed)
90. Zhao XQ, Morse JS, Dowdy AA, et al. Safety and tolerability of simvastatin plus niacin in patients with coronary artery disease and low high-density lipoprotein cholesterol (The HDL Atherosclerosis Treatment Study). Am J Cardiol. 2004;93(3):307-312. (PubMed)
91. Sazonov V, Maccubbin D, Sisk CM, Canner PL. Effects of niacin on the incidence of new onset diabetes and cardiovascular events in patients with normoglycaemia and impaired fasting glucose. Int J Clin Pract. 2013;67(4):297-302. (PubMed)
92. Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255-2267. (PubMed)
93. Michos ED, Sibley CT, Baer JT, Blaha MJ, Blumenthal RS. Niacin and statin combination therapy for atherosclerosis regression and prevention of cardiovascular disease events: reconciling the AIM-HIGH (Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides: Impact on Global Health Outcomes) trial with previous surrogate endpoint trials. J Am Coll Cardiol. 2012;59(23):2058-2064. (PubMed)
94. Kalil RS, Wang JH, de Boer IH, et al. Effect of extended-release niacin on cardiovascular events and kidney function in chronic kidney disease: a post hoc analysis of the AIM-HIGH trial. Kidney Int. 2015;87(6):1250-1257. (PubMed)
95. Landray MJ, Haynes R, Hopewell JC, et al. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371(3):203-212. (PubMed)
96. Schandelmaier S, Briel M, Saccilotto R, et al. Niacin for primary and secondary prevention of cardiovascular events. Cochrane Database Syst Rev. 2017;6:Cd009744. (PubMed)
97. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 Pt B):2889-2934. (PubMed)
98. Jackevicius CA, Tu JV, Ko DT, de Leon N, Krumholz HM. Use of niacin in the United States and Canada. JAMA Intern Med. 2013;173(14):1379-1381. (PubMed)
99. Burk K. Friedreich ataxia: current status and future prospects. Cerebellum Ataxias. 2017;4:4. (PubMed)
100. Chan PK, Torres R, Yandim C, et al. Heterochromatinization induced by GAA-repeat hyperexpansion in Friedreich's ataxia can be reduced upon HDAC inhibition by vitamin B3. Hum Mol Genet. 2013;22(13):2662-2675. (PubMed)
101. Libri V, Yandim C, Athanasopoulos S, et al. Epigenetic and neurological effects and safety of high-dose nicotinamide in patients with Friedreich's ataxia: an exploratory, open-label, dose-escalation study. Lancet. 2014;384(9942):504-513. (PubMed)
102. Lynch DR, Fischbeck KH. Nicotinamide in Friedreich's ataxia: useful or not? Lancet. 2014;384(9942):474-475. (PubMed)
103. Taylor EW. The oxidative stress-induced niacin sink (OSINS) model for HIV pathogenesis. Toxicology. 2010;278(1):124-130. (PubMed)
104. Favre D, Mold J, Hunt PW, et al. Tryptophan catabolism by indoleamine 2,3-dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci Transl Med. 2010;2(32):32ra36. (PubMed)
105. Jenabian MA, Patel M, Kema I, et al. Distinct tryptophan catabolism and Th17/Treg balance in HIV progressors and elite controllers. PLoS One. 2013;8(10):e78146. (PubMed)
106. Chen J, Shao J, Cai R, et al. Anti-retroviral therapy decreases but does not normalize indoleamine 2,3-dioxygenase activity in HIV-infected patients. PLoS One. 2014;9(7):e100446. (PubMed)
107. Dunham RM, Gordon SN, Vaccari M, et al. Preclinical evaluation of HIV eradication strategies in the simian immunodeficiency virus-infected rhesus macaque: a pilot study testing inhibition of indoleamine 2,3-dioxygenase. AIDS Res Hum Retroviruses. 2013;29(2):207-214. (PubMed)
108. Souza SA, Chow DC, Walsh EJ, Ford S, 3rd, Shikuma C. Pilot study on the safety and tolerability of extended release niacin for HIV-infected patients with hypertriglyceridemia. Hawaii Med J. 2010;69(5):122-125. (PubMed)
109. Dube MP, Lipshultz SE, Fichtenbaum CJ, et al. Effects of HIV infection and antiretroviral therapy on the heart and vasculature. Circulation. 2008;118(2):e36-40. (PubMed)
110. Carr A, Samaras K, Burton S, et al. A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS. 1998;12(7):F51-58. (PubMed)
111. Giannarelli C, Klein RS, Badimon JJ. Cardiovascular implications of HIV-induced dyslipidemia. Atherosclerosis. 2011;219(2):384-389. (PubMed)
112. Chow DC, Stein JH, Seto TB, et al. Short-term effects of extended-release niacin on endothelial function in HIV-infected patients on stable antiretroviral therapy. AIDS. 2010;24(7):1019-1023. (PubMed)
113. Balasubramanyam A, Coraza I, Smith EO, et al. Combination of niacin and fenofibrate with lifestyle changes improves dyslipidemia and hypoadiponectinemia in HIV patients on antiretroviral therapy: results of "heart positive," a randomized, controlled trial. J Clin Endocrinol Metab. 2011;96(7):2236-2247. (PubMed)
114. Dube MP, Komarow L, Fichtenbaum CJ, et al. Extended-release niacin versus fenofibrate in HIV-infected participants with low high-density lipoprotein cholesterol: effects on endothelial function, lipoproteins, and inflammation. Clin Infect Dis. 2015;61(5):840-849. (PubMed)
115. Hoffer LJ. Vitamin therapy in schizophrenia. Isr J Psychiatry Relat Sci. 2008;45(1):3-10. (PubMed)
116. Pauling L. Orthomolecular psychiatry. Varying the concentrations of substances normally present in the human body may control mental disease. Science. 1968;160(3825):265-271. (PubMed)
117. Seybolt SE. Is it time to reassess alpha lipoic acid and niacinamide therapy in schizophrenia? Med Hypotheses. 2010;75(6):572-575. (PubMed)
118. Zell M, Grundmann O. An orthomolecular approach to the prevention and treatment of psychiatric disorders. Adv Mind Body Med. 2012;26(2):14-28. (PubMed)
119. Yao JK, Dougherty GG, Jr., Gautier CH, et al. Prevalence and specificity of the abnormal niacin response: a potential endophenotype marker in schizophrenia. Schizophr Bull. 2016;42(2):369-376. (PubMed)
120. Sun L, Yang X, Jiang J, et al. Identification of the niacin-blunted subgroup of schizophrenia patients from mood disorders and healthy individuals in Chinese population. Schizophr Bull. 2017; doi: 10.1093/schbul/sbx150. [Epub ahead of print]. (PubMed)
121. Messamore E. Niacin subsensitivity is associated with functional impairment in schizophrenia. Schizophr Res. 2012;137(1-3):180-184. (PubMed)
122. Horrobin DF. The membrane phospholipid hypothesis as a biochemical basis for the neurodevelopmental concept of schizophrenia. Schizophr Res. 1998;30(3):193-208. (PubMed)
123. Messamore E. The niacin response biomarker as a schizophrenia endophenotype: A status update. Prostaglandins Leukot Essent Fatty Acids. 2017; pii: S0952-3278(16)30249-6. doi: 10.1016/j.plefa.2017.06.014. [Epub ahead of print]. (PubMed)
124. Jacob R, Swenseid M. Niacin. In: Ziegler E, Filer L, eds. Present Knowledge in Nutrition. 7th ed. Washington D.C.: ILSI Press; 1996:185-190.
125. US Department of Agriculture. USDA National Nutrient Database for Standard Reference, Release 25. 2012. https://ndb.nal.usda.gov/ndb/. Accessed 7/30/17.
126. Hendler SS, Rorvik DR. PDR for Nutritional Supplements. 2nd ed. Montvale: Thomson Reuters; 2008.
127. Minto C, Vecchio MG, Lamprecht M, Gregori D. Definition of a tolerable upper intake level of niacin: a systematic review and meta-analysis of the dose-dependent effects of nicotinamide and nicotinic acid supplementation. Nutr Rev. 2017;75(6):471-490. (PubMed)
128. MacKay D, Hathcock J, Guarneri E. Niacin: chemical forms, bioavailability, and health effects. Nutr Rev. 2012;70(6):357-366. (PubMed)
129. Dellinger RW, Santos SR, Morris M, et al. Repeat dose NRPT (nicotinamide riboside and pterostilbene) increases NAD(+) levels in humans safely and sustainably: a randomized, double-blind, placebo-controlled study. NPJ Aging Mech Dis. 2017;3:17. (PubMed)
130. Fulgoni VL, 3rd, Keast DR, Bailey RL, Dwyer J. Foods, fortificants, and supplements: Where do Americans get their nutrients? J Nutr. 2011;141(10):1847-1854. (PubMed)
131. Kar S, Chockalingam A. Statin-associated rhabdomyolysis with acute renal failure complicated by intradialytic NSTEMI: a review of lipid management considerations. Am J Ther. 2013;20(1):57-60. (PubMed)
132. Cziraky MJ, Willey VJ, McKenney JM, et al. Risk of hospitalized rhabdomyolysis associated with lipid-lowering drugs in a real-world clinical setting. J Clin Lipidol. 2013;7(2):102-108. (PubMed)
133. Maccubbin DL, Chen F, Anderson JW, et al. Effectiveness and safety of laropiprant on niacin-induced flushing. Am J Cardiol. 2012;110(6):817-822. (PubMed)
134. Hps Thrive Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J. 2013;34(17):1279-1291. (PubMed)
135. Lewey J, Shrank WH, Bowry AD, Kilabuk E, Brennan TA, Choudhry NK. Gender and racial disparities in adherence to statin therapy: a meta-analysis. Am Heart J. 2013;165(5):665-678, 678 e661. (PubMed)
136. Cheung MC, Zhao XQ, Chait A, Albers JJ, Brown BG. Antioxidant supplements block the response of HDL to simvastatin-niacin therapy in patients with coronary artery disease and low HDL. Arterioscler Thromb Vasc Biol. 2001;21(8):1320-1326. (PubMed)
137. Brown BG, Cheung MC, Lee AC, Zhao XQ, Chait A. Antioxidant vitamins and lipid therapy: end of a long romance? Arterioscler Thromb Vasc Biol. 2002;22(10):1535-1546. (PubMed)
138. Rios-Avila L, Coats B, Chi YY, et al. Metabolite profile analysis reveals association of vitamin B-6 with metabolites related to one-carbon metabolism and tryptophan catabolism but not with biomarkers of inflammation in oral contraceptive users and reveals the effects of oral contraceptives on these processes. J Nutr. 2015;145(1):87-95. (PubMed)
139. Dollerup OL, Christensen B,Svart M, et al. A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: safety, insulin-sensitivity, and lipid-mobilizing effects. Am J Clin Nutr. 2018;108:343-353. (PubMed)
Riboflavina
Contenido
Resumen
- La riboflavina es el precursor de las coenzimas, flavín adenín dinucleótido (FAD) y flavín mononucleótido (FMN). Estas actúan como transportadores de electrones en un cierto número de reacciones de oxidación-reducción (redox) involucradas en la producción de energía y en numerosas rutas metabólicas. (Más información)
- La deficiencia de riboflavina puede afectar múltiples rutas en el metabolismo de la vitamina B6, folato, niacina, y del hierro. (Más información)
- La deficiencia de riboflavina ha sido ligada a la preeclampsia en mujeres embarazadas. Esta condición puede progresar a eclampsia y causar hemorragias severas y la muerte. El riesgo de preeclampsia ha sido recientemente asociado con la presencia de una variante genética (C677T) en el gene de la metilentetrahidrofolato reductasa (MTHFR). Este gene codifica para la enzima MTHFR, la cual es dependiente de FAD. (Más información)
- El estrés oxidativo puede causar opacificación del cristalino del ojo, conduciendo a las cataratas en individuos mayores. Mientras los resultados de algunos estudios basados en la observación son prometedores, estudios de intervención son necesarios para evaluar el beneficio potencial de la riboflavina en la prevención de cataratas. (Más información)
- La riboflavina (en la forma de FAD) es requerida como cofactor para la enzima clave que metaboliza el folato, MTHFR. Un estatus bajo del estatus de la riboflavina puede interferir con el metabolismo del folato, particularmente en individuos homocigotos para la variante del gen MTHFR C677T; estos individuos exhiben un riesgo mayor de enfermedades cardiovasculares (ECV). Evidencia emergente proveniente de ensayos de intervención apoya un papel protector de la riboflavina contra la hipertensión en individuos con el genotipo MTHFR 677TT. (Más información)
- Se ha evaluado a la riboflavina como un agente profiláctico en estudios con niños y adultos que padecen de migraña. La suplementación con riboflavina ha mostrado disminuir la frecuencia y severidad de los ataques de dolor de cabeza en adultos pero no en niños. (Más información)
- Reportes de caso han mostrado que los pacientes con trastornos autosómicos recesivos del metabolismo de riboflavina podrían beneficiarse de la suplementación con riboflavina. (Más información)
-
Se ha evaluado a la riboflavina como una terapia adyuvante potencial en el cáncer y ciertos desordenes oculares. (Más información)
La riboflavina es una vitamina B hidrosoluble, también conocida como vitamina B2. En el cuerpo, la riboflavina es principalmente encontrada como un componente integral de las coenzimas, flavín adenín dinucleótido (FAD) y flavín mononucleótido (FMN) (1). Las coenzimas derivadas de la riboflavina se denominan flavocoenzimas, y enzimas que usan una flavocoenzima son llamadas flavoproteínas (2).
Función
Reacciones de oxidación-reducción (redox)
Los organismos vivos derivan la mayoría de su energía de las reacciones redox, las cuales son procesos que involucran la transferencia de electrones. Las flavocoenzimas participan en las reacciones redox en numerosas rutas metabólicas (3). Son críticas para el metabolismo de los carbohidratos, lípidos, y proteínas. El FAD es parte de la cadena de transporte (respiratoria) de electrones, la cual es fundamental para la producción de energía. En conjunción con el citocromo P-450, las flavocoenzimas también participan en el metabolismo de drogas y toxinas (4).
Funciones antioxidantes
La glutatión reductasa es una enzima dependiente de FAD que participa en el ciclo redox del glutatión. El ciclo redox del glutatión juega un papel fundamental en la protección de organismos de especies reactivas del oxígeno, como los hidroperóxidos. La glutatión reductasa (GR) requiere de FAD para regenerar dos moléculas de glutatión reducido a partir del glutatión oxidado. La deficiencia de riboflavina ha sido asociada con un incremento del estrés oxidativo (4). La medición de la actividad de la GR en los glóbulos rojos es comúnmente usada para evaluar el estatus nutricional de la riboflavina (5). El ensayo del coeficiente de activación de la glutatión reductasa eritrocitaria (EGRAC) evalúa el estatus de la riboflavina al medir la actividad de la GR antes y después de la reactivación in vitro con su grupo prostético FAD; el EGRAC es calculado como la proporción de FAD-estimulado con respecto a la actividad enzimática no estimulada e indica el grado de saturación con riboflavina del tejido. De esta manera el EGRAC es una medida fundamental del estatus de riboflavina y ha mostrado ser efectiva en reflejar el estatus del biomarcador de una deficiencia severa a un estatus normal (6).
Las glutatión peroxidasas, enzimas que contienen selenio, necesitan de dos moléculas de glutatión reducido para degradar hidroperóxidos. Las GPx están involucradas en el ciclo de oxidación-reducción (redox) del glutatión (Figura 1).
La xantina oxidasa, otra enzima dependiente de FAD, cataliza la oxidación de hipoxantina y xantina a ácido úrico. El ácido úrico es uno de los antioxidantes hidrosolubles más efectivos en la sangre. Por lo tanto, la deficiencia de riboflavina puede derivar en una actividad disminuida de la xantina oxidasa, reduciendo los niveles sanguíneos de ácido úrico (7).
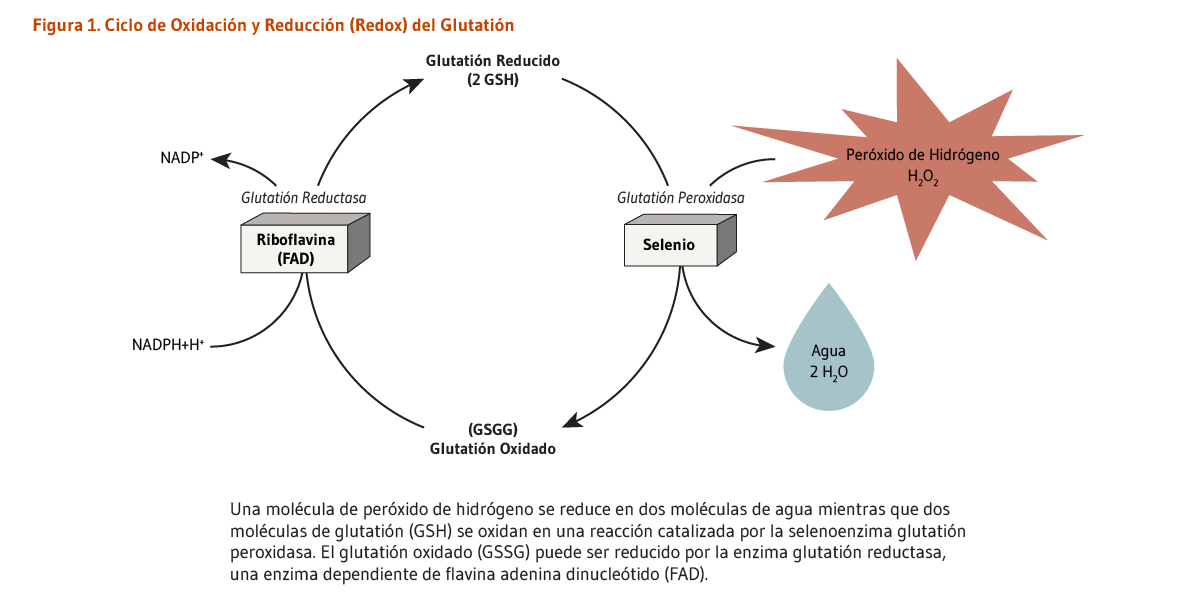
[Figura 1 - Clic para Agrandar]
Interacción con nutrientes
Vitaminas del complejo B
Las flavoproteínas están involucradas en el metabolismo de varias otras vitaminas: (vitamina B6, niacina, y folato). Por lo tanto, una deficiencia severa de riboflavina podría afectar muchos sistemas de enzimas. La conversión de la mayoría de vitamina B6 naturalmente disponible a su forma de coenzima, piridoxal 5’-fosfato (PLP), requiere de la enzima dependiente de FMN, piridoxina 5’-fosfato oxidasa (PPO) (8). Por lo menos dos estudios en adultos mayores han documentado interacciones significantes entre los indicadores de vitamina B6 y el estatus nutricional de la riboflavina (9, 10). La síntesis de coenzimas que contienen niacina, NAD y NADP, a partir del aminoácido triptófano, requiere de la enzima dependiente de FAD, quinurenina mono-oxigenasa. Una deficiencia severa de riboflavina puede disminuir la conversión de triptófano a NAD y NADP, incrementando el riesgo de una deficiencia de niacina (3). La 5,10-metilentetrahidrofolato reductasa (MTHFR) es una enzima dependiente de FAD que juega un papel importante en el mantenimiento de una coenzima folato específica, requerida para formar metionina a partir de homocisteína (Figura 2). Junto con otras vitaminas del complejo B, se ha asociado a las ingestas elevadas de riboflavina con niveles disminuidos de homocisteína en el plasma (11). Niveles incrementados de riboflavina en el plasma han sido también asociados con niveles disminuidos de homocisteína del plasma, principalmente en individuos homocigotos para el polimorfismo C677T en el gen de la MTHFR y en individuos con una baja ingesta de folato (12). Tales resultados muestran que el riesgo de enfermedades crónicas podría ser influenciado por interacciones complejas entre factores genéticos y dietarios (véase Enfermedades cardiovasculares y Cáncer).

[Figura 2 - Clic para Agrandar]
Hierro
La deficiencia de riboflavina altera el metabolismo del hierro. Aunque el mecanismo no está aún claro, investigación en animales sugiere que la deficiencia de riboflavina podría perjudicar la absorción del hierro, incrementar la pérdida intestinal de hierro y/o afectar la utilización del hierro para la síntesis de hemoglobina (Hb) (13). En humanos, se ha encontrado que al mejorar el estatus nutricional de la riboflavina se aumentan los niveles de Hb circulante (14). La corrección de una deficiencia de riboflavina en individuos que son deficientes de ambos riboflavina y hierro mejora la respuesta de individuos con anemia ferropénica a la terapia con hierro (15). La anemia durante el embarazo, un problema de salud pública alrededor del mundo, es responsable por una considerable morbilidad y mortalidad perinatal (16, 17). El manejo de la anemia materna incluye la suplementación con hierro solo o hierro en combinación con ácido fólico (18), y se ha considerado que la suplementación con riboflavina podría mejorar la suplementación con hierro-ácido fólico. Ensayos de intervención, aleatorios, doble ciegos conducidos en mujeres embarazadas con anemia en el Sureste Asiático mostraron que una combinación de ácido fólico, hierro, vitamina A, y riboflavina mejoró los niveles de Hb y disminuyó la prevalencia de anemia en comparación a la suplementación sola con hierro-ácido fólico (19, 20).
Deficiencia
Ariboflavonosis es el término médico para una deficiencia clínica de riboflavina. Una deficiencia de riboflavina es raramente encontrada de manera aislada; ocurre con frecuencia en combinación con deficiencia de otras vitaminas hidrosolubles. Los síntomas de una deficiencia de riboflavina incluyen dolor de garganta, enrojecimiento e hinchazón de la mucosa de la boca y garganta, grietas o llagas en el exterior de los labios (quelosis) y en las comisuras de la boca (estomatitis angular), inflamación y enrojecimiento de la lengua (lengua magenta), y una inflamación cutánea húmeda y escamosa (dermatitis seborreica). Otros síntomas podrían involucrar la formación de vasos sanguíneos en la capa transparente del ojo (vascularización de la córnea) y conteo disminuido de glóbulos rojos en el cual los glóbulos rojos existentes contienen niveles normales de hemoglobina y son de tamaño normal (anemia normocítica normocrómica) (1, 3). Una deficiencia severa de riboflavina podría resultar en la conversión disminuida de vitamina B6 a su forma de coenzima (PLP) y una conversión disminuida de triptófano a niacina (véase Interacciones con nutrientes).
La preeclampsia es definida como la presencia de presión sanguínea elevada, proteínas en la orina y edema (hinchazón significativa) durante el embarazo. Cerca del 5% de las mujeres con preeclampsia progresan a eclampsia, una causa importante de muerte materna y fetal. La eclampsia es caracterizada por convulsiones, además de presión sanguínea alta y un riesgo aumentado de hemorragia (sangramiento severo) (21). Un estudio en 154 mujeres embarazadas en riesgo incrementado de preeclampsia encontró que aquellas que estaban deficientes de riboflavina eran 4.7 veces más propensas a desarrollar preeclampsia que aquellas que tenían un estatus nutricional adecuado de riboflavina (22). La causa de la preeclampsia-eclampsia es desconocida. Niveles intracelulares de flavocoenzimas disminuidos podrían causar disfunción mitocondrial, aumento del estrés oxidativo e interferencia con la liberación de óxido nítrico y, por lo tanto, con la dilatación de vasos sanguíneos — todos estos cambios han sido asociados con la preeclampsia (22).
Un reciente meta-análisis de 51 estudios encontró que el polimorfismo C677T del gen de la metilentetrahidrofolato reductasa (MTHFR) estaba asociado con la preeclamsia en las poblaciones caucásicas y del Este de Asia (23). Sin embargo un meta-análisis temprano reportó que el genotipo 677TT MTHFR acarreaba un riesgo significantemente mayor de preeclampsia entre mujeres asiáticas únicamente, mientras que en mujeres caucásicas el incremento del riesgo no fue significante (24). Tal heterogeneidad entre estudios podría sugerir que el efecto del genotipo 677TT MTHFR podría ser modulado por la riboflavina y otros factores dietarios relevantes que podrían variar considerablemente entre las diferentes poblaciones. La reducción en la actividad de la flavoproteína de la MTHFR observada en sujetos con la variante genética C677T conduce a un incremento leve en las concentraciones de homocisteína del plasma; los niveles incrementados de homocisteína han sido asociados con la preeclampsia (25). Un ensayo pequeño doble ciego, aleatorio, controlado con placebo en 450 mujeres embarazadas en alto riesgo de preeclampsia encontró que la suplementación con 15 mg de riboflavina diariamente no impidió la condición (26). Sin embargo, estudios son necesarios para evaluar el beneficio potencial de la suplementación con riboflavina en la reducción de complicaciones perinatales específicamente en mujeres preeclámpticas con el genotipo C677T.
Factores de riesgo para la deficiencia de riboflavina
Los alcohólicos están en un riesgo incrementado de deficiencia de riboflavina debido a una ingesta disminuida, una absorción disminuida y a la utilización deteriorada de la riboflavina. Curiosamente, los niveles elevados de homocisteína asociados con la deficiencia de riboflavina rápidamente declinaron durante la abstinencia de alcohol (27). Adicionalmente, las personas anoréxicas rara vez consumen riboflavina adecuada, e individuos con intolerancia a la lactosa podrían no consumir leche u otros productos lácteos que son buenas fuentes de riboflavina. La conversión de riboflavina a FAD y FMN es alterada en el hipotiroidismo e insuficiencia adrenal (3, 4). Además, las personas que son muy activas físicamente (atletas, trabajadores) pudiesen tener un requerimiento de riboflavina levemente incrementado. Sin embargo, generalmente no se ha encontrado que la suplementación con riboflavina incremente el desempeño o la tolerancia al ejercicio (28).
Aunque la deficiencia clínica de riboflavina (es decir, incluyendo signos tales como la estomatitis angular, queilosis, y glositis) es rara en el mundo desarrollado, existe evidencia que sugiere que el estatus subóptimo de la riboflavina (determinado por el biomarcador funcional EGRAC) podría ser un problema generalizado que afecta a muchas poblaciones de otro modo sanas dentro del mundo desarrollado. Por ejemplo, se reportó que una proporción alta de la población británica adulta poseía un estatus de riboflavina pobre como se determinó de los datos de la encuesta nacional usando EGRAC (29). Las ingestas dietarías sin embargo fueron generalmente favorables al ser comparadas con los valores recomendados, excepto en mujeres jóvenes que tuvieron ingestas bajas. La gran proporción de la población británica con valores anormales de EGRAC, a pesar de ingestas dietarías aparentemente adecuadas, requiere de investigación adicional.
La Ingesta Diaria Recomendada (IDR)
La IDR para la riboflavina, revisada en 1998, está basada en la prevención de su deficiencia (Tabla 1). En humanos los signos clínicos de la deficiencia aparecen a ingestas de menos de 0.5-0.6 miligramos (mg)/día, y la excreción urinaria de riboflavina es vista a niveles de ingesta de aproximadamente 1 mg/día (1).
| Etapa de la Vida | Edad | Machos (mg/día) | Hembras (mg/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 0.3 (IA) | 0.3 (IA) |
| Infantes | 7-12 meses | 0.4 (IA) | 0.4 (IA) |
| Niños | 1-3 años | 0.5 | 0.5 |
| Niños | 4-8 años | 0.6 | 0.6 |
| Niños | 9-13 años | 0.9 | 0.9 |
| Adolescentes | 14-18 años | 1.3 | 1.0 |
| Adultos | 19 años y más | 1.3 | 1.1 |
| Embarazo | Todas las edades | - | 1.4 |
| Período de lactancia | Todas las edades | - | 1.6 |
Prevención de Enfermedades
Cataratas
Las cataratas seniles son la causa principal de discapacidad visual en los EE.UU. y otros países desarrollados. La investigación se ha enfocado en el papel de los antioxidantes nutricionales debido a la evidencia de que el daño oxidativo foto-inducido de las proteínas del cristalino podría conducir al desarrollo de cataratas relacionadas con la edad. Un estudio de caso y control encontró un riesgo significativamente disminuido de cataratas seniles (33 a 51%) en hombres y mujeres en el quintil más alto de la ingesta de riboflavina (mediana de 1.6 a 2.2 mg/día) en comparación a aquellos en el quintil más bajo (mediana de 0.08 mg/día tanto en hombres como en mujeres) (30). Otro estudio de caso y control reportó que los individuos en el quintil más elevado del estatus nutricional de la riboflavina, medido por la actividad de la glutatión reductasa de los glóbulos rojos, tenían aproximadamente la mitad de la incidencia de cataratas seniles que aquellos en el quintil más bajo del estatus de riboflavina, aunque estos resultados no fueron estadísticamente significativos (31). Un estudio de corte transversal de 2,900 hombres y mujeres australianos, de 49 años de edad y mayores, encontró que aquellos en el quintil más alto de la ingesta de riboflavina eran un 50% menos propensos a tener cataratas que aquellos en el quintil más bajo (32). Un estudio prospectivo de más de 50,000 mujeres no observo alguna diferencia entre las tasas de extracción de cataratas entre mujeres en el quintil más alto de la ingesta de riboflavina (mediana de 1.5 mg/día) y mujeres en el quintil más bajo (mediana de 1.2 mg/día) (33). Sin embargo, el rango entre el quintil más alto y el más bajo era pequeño, y los niveles de ingesta medianos para ambos quintiles estaban por sobre la actual IDR para la riboflavina. Un estudio en 480 mujeres encontró que ingestas recomendada más altas de riboflavina se asociaban inversamente con cambios en la opacificación de los cristalinos en un período de cinco años (34). Aunque estos estudios basados en la observación proporcionan apoyo al papel de la riboflavina en la prevención de las cataratas, es necesario realizar ensayos de intervención aleatorios, controlados con placebo que incluyan un biomacador de respuesta (como la EGRAC) para confirmar esta relación.
Enfermedades cardiovasculares
Por muchos años, los niveles elevados de homocisteína en el plasma han sido considerados como un factor de riesgo para las enfermedades cardiovasculares (ECV), aunque esto recientemente se ha convertido de alguna manera en algo controversial (35). La homocisteína del plasma responde a los efectos reductores de las intervenciones con folato y vitaminas B relacionadas metabólicamente, incluyendo la riboflavina. La riboflavina actúa como un cofactor para la MTHFR y es por ello necesaria para generar 5-metiltetrahidrofolato requerido en la remetilación de la homocisteína a metionina (véase Figura 2 arriba). Estas vitaminas B, sin embargo, podrían tener papeles en la prevención de ECV que son independientes de sus efectos sobre la homocisteína.
Estudios genéticos proporcionan evidencia convincente para apoyar un vínculo entre el estatus sub-óptimo de vitamina B y el riesgo de ECV. Un meta-análisis de tales estudios mostró que los individuos que son homocigotos para la variante del gen MTHFR C677T tuvieron un riesgo significantemente mayor de ECV (por un 14% a 21%) en comparación a aquellos sin este polimorfismo, pero hubo una gran cantidad de variación geográfica en el incremento del riesgo de ECV (36), sugiriendo fuertemente que los factores dietarios pueden modular el riesgo de la enfermedad relacionado a este factor genético.
La acumulación de pruebas vincula este polimorfismo común del folato con la hipertensión (definida como una presión sanguínea de 140/90 mm Hg o mayor), un factor de riesgo principal para las ECV, particularmente los accidentes cerebrovasculares. La evidencia emergente de que la presión sanguínea en pacientes homocigotos para este polimorfismo es altamente sensible a dosis bajas de riboflavina (véase Tratamiento de Enfermedades) aumenta la posibilidad de que el mejoramiento del estatus de la riboflavina tendrá un papel importante en la prevención de la hipertensión. Esto en turno podría potencialmente reducir el riesgo de accidentes cerebrovasculares específicamente en individuos con el genotipo relevante. Notablemente, la frecuencia reportada del genotipo MTHFR C677T es de un 10% en todo el mundo, oscilando entre 4%-26% en Europa, 20% en el norte de China, y tan alto como un 32% en México (37).
Cáncer
La flavoproteína, metilentetrahidrofolato reductasa (MTHFR), juega un papel fundamental en el metabolismo de la homocisteína mediada por el folato. La MTHFR convierte el 5,10-metilentetrahidrofolato a 5-metiltetrahidrofolato, el cual es necesario para le re-metilación de la homocisteína a metionina (véase Figura 2 arriba). La conversión de la homocisteína a metionina es de importancia para la desintoxicación de la homocisteína y para la producción de S-adenosilmetionina (SAM), el donante de metilo para la metilación del ADN y las histonas. La deficiencia de folato y elevadas concentraciones de homocisteína podrían incrementar el riesgo de cáncer (véase el artículo en Folato). Los cambios aberrantes en la metilación, son también conocidos por alterar la estructura y función del ADN y las histonas durante el desarrollo del cáncer (38). Ya que la MTHFR controla la desintoxicación de la homocisteína y el suministro de grupos de metilo para la síntesis de SAM, una reducción en su actividad puede afectar el metabolismo de la homocisteína y alterar los procesos de metilación celular. La sustitución de una citosina por una timina en la posición 677 (c.677C>T) en el gen de la MTHFR es un polimorfismo que afecta la unión de FAD y conduce a una mayor propensión de la MTHFR a perder su coenzima flavina (39). Individuos homocigotos para esta mutación (es decir, genotipo MTHFR 677TT) exhiben una actividad de la MTHFR reducida, y evidencia demuestra que tales individuos están un riesgo incrementado de cáncer en varios sitios (40-42); sin embargo, la naturaleza de la asociación entre este polimorfismo común y el riesgo de cáncer permanece incierto.
Como se mencionó previamente (véase Vitaminas del complejo B), la ingesta de riboflavina es un determinante de la concentración de homocisteína. Esto sugiere que el estatus de la riboflavina puede influenciar la actividad de la MTHFR y el metabolismo del folato, afectando potencialmente así el riesgo de cáncer (42). En un estudio aleatorio, doble ciego, controlado con placebo, a 93 sujetos con pólipos colorrectales y 86 sujetos saludable se les dio o un placebo, ácido fólico (400 o 1,200 mcg/día), o ácido fólico (400 mcg/día) más riboflavina (5 mg/día) por 45 días. Estas intervenciones significantemente mejoraron el estatus del folato y la riboflavina en los individuos suplementados con vitaminas en comparación con aquellos que tomaron el placebo. Curiosamente, la riboflavina mejoró el efecto de 400 mcg de ácido fólico en la 5-metil tetrahidrafolato (5-MeTH4) circulante específicamente en los pacientes con pólipos con la variante genética C677T (43). Esto sugiere que la riboflavina pudiese mejorar la respuesta a la suplementación con ácido fólico en individuos con una actividad de la MTHFR reducida. Adicionalmente, un estudio de cohorte prospectivo de 88,045 mujeres postmenopáusicas encontró que la ingesta total (dietaría más suplementaria) de riboflavina estaba inversamente correlacionada con el riesgo de cáncer colorrectal cuando se compararon los cuartiles más altos (>3.97 mg) y los más bajo (<1.80 mg) de la ingesta diaria (44); la ingesta en el grupo de referencia estaba muy por arriba de la IDR de 1.1 mg/día. Los sujetos en este estudio no fueron pre-cribados para identificar aquellos con el genotipo C677T, y la asociación entre el polimorfismo C677T y el cáncer colorrectal no es claro, con reportes sugiriendo una reducción en el riesgo con el alelo T (45).
Asociaciones entre la ingesta de riboflavina y el riesgo de cáncer han sido evaluadas en otros tipos de cáncer. Un estudio de intervención de siete años evaluó el uso de sal fortificada con riboflavina en 22,093 individuos en un alto riesgo de cáncer de esófago en China. El estatus de la riboflavina y la patología esofágica (porcentaje de tejidos normales, displásicos y cancerosos) mejoró en el grupo de intervención en comparación al grupo de control, pero la menor incidencia de cáncer de esófago encontrada en el grupo de intervención no fue estadísticamente significante (46). Adicionalmente, el Estudio de Cohorte Colaborativo de Melbourne, el cual dio seguimiento a 41,514 hombres y mujeres sobre un periodo de 15 años, encontró asociaciones inversas débiles entre la ingesta de riboflavina y el cáncer de pulmón (47) y el cáncer de seno (48) y no asociación alguna con el cáncer de próstata (49). Sin embargo, un seguimiento de 10 años de un ensayo de intervención en pacientes en alto riesgo de cáncer gástrico (estomago) encontró que la suplementación dietaría con minerales y vitaminas, incluyendo riboflavina (3.2 mg/día) y niacina (40 mg/día), por cinco años falló en disminuir la incidencia o la tasa de mortalidad del cáncer gástrico (50).
Tratamiento de Enfermedades
Migrañas
Cierta evidencia indica que un metabolismo mitocondrial del oxígeno deteriorado en el cerebro podría jugar un papel en la patología de las migrañas. Debido a que la riboflavina es el precursor de dos flavocoenzimas (FAD y FMN) requeridas por las flavoproteínas de la cadena transportadora de electrones mitocondrial, se ha investigado a la riboflavina suplementaria como un tratamiento para la migraña. Un ensayo aleatorio, controlado con placebo examinó el efecto de 400 mg/día de riboflavina al día por tres meses en la prevención de migraña en 54 hombres y mujeres con un historial de migrañas recurrentes (51). La riboflavina fue significativamente mejor que el placebo en la reducción de la frecuencia de los ataques y el número de días con dolores de cabeza, aunque el efecto benéfico fue más pronunciado durante el tercer mes de tratamiento. Otro estudio por los mismos investigadores encontró que el tratamiento tanto con un medicamento β-bloqueador o con riboflavina en dosis elevadas (400 mg/día) por cuatro meses resultó en una mejora clínica, pero cada terapia pareció actuar sobre un mecanismo patológico diferente: los β-bloqueadores en el procesamiento de información cortical anormal y la riboflavina sobre las reservas cerebrales de energía mitocondrial disminuidas (52). Un estudio pequeño en 23 pacientes reportó una reducción en la frecuencia media de ataques de migraña después de la suplementación con 400 mg de riboflavina diariamente por tres meses (53). Sin embargo, un estudio de 3 meses, aleatorio, doble ciego, controlado con placebo que administró una combinación de riboflavina (400 mg/día), magnesio, y matricaria a personas que sufren de migraña reportó ningún beneficio terapéutico más allá del asociado con consumir un placebo que contenga 25 mg/día de riboflavina (54). En comparación con las mediciones basales en este estudio, tanto el grupo que recibió el placebo como el grupo en tratamiento experimentaron ciertos beneficios con respecto al promedio de migrañas, días con migraña, o en el índice de las migrañas (54). Aunque estos resultados son preliminares, los datos procedentes de la mayoría de los estudios hasta la fecha sugieren que la suplementación con riboflavina en adultos podría ser un complemento útil para la terapia farmacológica en la prevención de migrañas.
Dos ensayos aleatorios, doble ciegos, controlados con placebo investigaron el efecto de la suplementación con riboflavina en la frecuencia y severidad de los ataques de dolor de cabeza en niños con migrañas. El primer estudio evaluó la riboflavina en 200 mg/día por 12 semanas en 48 niños de entre 5 a 15 años de edad (55). El segundo estudio fue un ensayo cruzado con la mitad de los 42 niños, de entre 6 a 13 años de edad, recibiendo 50 mg/día de riboflavina por 16 semanas luego placebo (100 mg/día caroteno) por 16 semanas con un periodo de lavado de cuatro semanas de por medio, mientras que a la otra mitad se les dio primero placebo luego riboflavina (56). Ambos estudios no mostraron diferencias en la frecuencia, duración, o intensidad de migrañas entre tratamientos. La efectividad de la suplementación con riboflavina en niños con migraña no es aun establecida, e investigación futura debe primero determinar la dosis más apropiada de riboflavina para esta población.
Trastornos metabólicos
Evidencia creciente indica que pacientes con trastornos autosómicos recesivos causados por enzimas dependientes de FAD defectuosas podrían beneficiarse de la suplementación con riboflavina.
Deficiencia múltiple de acil CoA-deshidrogenasa (MADD)
MADD, también conocida como aciduria glutárica tipo II (o acidemia), es un trastorno del metabolismo de ácidos grasos caracterizado por la acumulación de acil-carnitias de cadena corta, mediana, y larga en varios tejidos. Formas neonatales y de aparición tardía de la MADD muestran un amplio espectro de severidad clínica y presentaciones variables, incluyendo encefalopatía episódica, vómitos periódicos, hepatopatía y rabdomiólisis (57). La MADD es causada por mutaciones en genes que perjudican la actividad de enzimas involucradas en la transferencia de electrones a partir de la acil-Coenzima A (acil-CoA) a la Coenzima Q10 ubiquinona dentro de la mitocondria (Figura 3). ETFA, ETFB, y ETFDH codifican para las dos subunidades de la flavoproteína de transferencia de electrones (ETF-A y -B), y para la ETF deshidrogenasa/ubiquinona oxidorreductasa (ETFDH/ETFQO), respectivamente.
Deficiencias en estas enzimas (ETF o ETFDH) conducen a una disminución en la FAD oxidada, la cual se torna no disponible para las reacciones de deshidrogenación dependientes de FAD, incluyendo el primer paso en la β-oxidación — un principal proceso catabólico de los ácidos grasos que tiene lugar en la mitocondria. Un defecto en la β-oxidación de ácidos grasos causa acumulación de lípidos en el musculo esquelético, llevando a una miopatía de almacenamiento de lípidos caracterizada por dolor y debilidad muscular e intolerancia al ejercicio. Junto con una dieta baja en grasas y alta en carbohidratos, la suplementación con riboflavina ha llevado a mejoramientos clínicos significantes en pacientes con mutaciones de la ETFDH. El tipo específico de mutación en ETF/ETFDH contribuye a la edad de inicio, gravedad y capacidad de respuesta al tratamiento con riboflavina (58). Adicionalmente, el reciente reporte de un hombre de 20 años de edad con una MADD sensible a la riboflavina falló en encontrar mutaciones en los genes de la ETF y ETFDH, sugiriendo que otros sitios de mutación no deben ser excluidos (57). Finalmente, deficiencias secundarias en la cadena respiratoria son observadas en MADD y parecen responder favorablemente a la suplementación con riboflavina (58, 59).

[Figura 3 - Clic para Agrandar]
Deficiencia de acil CoA-deshidrogenasa 9 (ACAD9)
Mutaciones recesivas en el gen de la ACAD9 que codifica para la acil CoA deshidrogenasa dependiente de FAD fueron encontradas en pacientes con deficiencia del complejo I mitocondrial, un trastorno de la cadena respiratoria (60). La deficiencia de ACAD9 no ha sido ligada a defectos de la β-oxidación de ácidos grasos, sugiriendo en cambio un papel en el ensamblaje del complejo I para la ACAD9 (61). El complejo I transporta electrones de la NADH a la Coenzima Q10 en la cadena transportadora de electrones. La fosforilación oxidativa defectuosa (síntesis de ATP por la cadena respiratoria) debido a la deficiencia del complejo I ha sido ligada a una amplia variedad de manifestaciones clínicas desde muerte neonatal hasta la aparición tardía de enfermedades neurodegenerativas. Los síntomas de la deficiencia de complejo I debido a mutaciones en ACAD9 incluyen debilidad muscular, intolerancia al ejercicio, acidosis láctica, encefalopatía, y cardiomiopatía. La suplementación con riboflavina (100-300 mg/día) incremento la actividad del complejo I en pacientes con formas clínicas de la deficiencia de ACAD9 que aparecieron en la infancia. Mejoras en la resistencia muscular y tolerancia al ejercicio han sido también asociadas con la suplementación de riboflavina (62-64).
Trastornos asociados con el transporte defectuoso de la riboflavina
Los genes SLC52A1, SLC52A3, y SLC52A2 codifican para los transportadores de riboflavina humanos hRTF1, hRTF2, y hRTF3, respectivamente. Mutaciones en estos genes han sido ligados con el síndrome de Brown-Vialetto-Van Laere (BVVL), un desorden neurodegenerativo raro caracterizado por su aparición de edades variable. El síndrome incluye parálisis bulbar con hipotonía y debilidad facial, sordera neurosensorial, e insuficiencia respiratoria. La falta de pérdida auditiva en la descripción clínica de los síntomas del BVVL es conocida como síndrome de Fazio-Londe (FL). Las características transitorias clínicas y bioquímicas de MADD fueron descritas en un recién nacido de una madre con deficiencia de riboflavina; esta leve deficiencia, causada por una mutación en el hRTF1, fue corregida rápidamente por la suplementación con riboflavina (65, 66). Una reciente revisión de la literatura, la cual analizó reportes de 74 pacientes afectados por el síndrome BVVL/FL, encontró que 8 de los 13 pacientes a los que se les dio riboflavina suplementaria (dosis promedio de 10 mg/kg/día) experimentaron mejoras clínicas, y los individuos tratados tuvieron una tasa de supervivencia del 100% (67). La riboflavina también restauró los niveles normales de flavina y acilcarnitina en pacientes que presentaban perfiles anormales.
Trimetilaminuria sensible a la riboflavina
La trimetilaminuria primaria es causada por la oxidación defectuosa de la trimetilamina por una flavoproteina del hígado llamada flavina monooxigenasa 3 (FMO3). Individuos con deficiencia de FMO3 tienen niveles incrementados de trimetilamina en la orina, sudor, y aliento (68). Esta condición socialmente angustiante es conocida como "síndrome de olor a pescado" debido al olor a pescado y naturaleza volátil de la trimetilamina. Mutaciones del gen FMO3 son usualmente asociadas con trimetilaminuria leve o intermitente; la condición es a veces limitada a periodos perimenstruales en sujetos femeninos o al consumo de alimentos ricos en trimetilamina. El manejo clínico de esta condición incluye restricciones dietarías de trimetilamina y sus precursores, como los alimentos ricos en colina y los mariscos, como también los vegetales crucíferos que contienen ambos precursores de la trimetilamina y antagonistas de la FMO3 (69). El uso de suplementos de riboflavina fue recientemente reportado en un paciente femenino de 17 años de edad afectado por homocistinuria no responsiva a la piridoxina (70). La enfermedad fue inicialmente tratada con betaína (un derivado de la colina), la cual causó olor corporal secundario a la deficiencia de FMO3. La suplementación con riboflavina (200 mg/día) redujo la excreción de trimetilamina y el olor corporal relacionado al tratamiento con betaína. Estos datos sugieren que la riboflavina podría ayudar a maximizar la actividad residual de la enzima FMO3 en pacientes con trimetilaminuria primaria.
Hipertensión
Aunque la etiología de la hipertensión no es clara, el polimorfismo C677T del gen de la metilentetrahidrofolato reductasa (MTHFR) es el principal determinante de las concentraciones de homocisteína y ha sido relacionado a una presión sanguínea elevada (un marcador de hipertensión) (71) y un riesgo incrementado de enfermedad coronaria cardíaca y accidente vascular (72-74). Dado que esta variante genética lleva a una actividad de la MTHFR disminuida, individuos con el genotipo 677TT podrían beneficiarse de la suplementación con riboflavina. En un ensayo inicial aleatorio, doble ciego, controlado con placebo en 77 sujetos sanos que habían sido pre-cribados para el genotipo de la MTHFR, la suplementación con riboflavina (1.6 mg/día por 12 semanas) disminuyó los niveles de homocisteína en el grupo con el genotipo 677TT MTHFR, pero en no en los grupos 677CC y 677CT de la misma edad que exhibieron niveles normales al inicio del estudio (75). Dos ensayos aleatorios, doble ciegos, controlados con placebo subsecuentes investigaron la posibilidad de la modulación de la hipertensión por la riboflavina en pacientes con enfermedades cardiovasculares (ECV) prematuras (pre-cribados para el polimorfismo MTHFR 677C→T) (76, 77). Los resultados mostraron una disminución significante de la presión sanguínea solo en pacientes con el genotipo 677TT suplementados con riboflavina (1.6 mg/día por 16 semanas) en comparación al placebo, tanto en la examinación inicial (69) como cuando la misma cohorte de pacientes en alto riesgo de ECV fue investigada nuevamente cuatro años después del ensayo original (70). Otro estudio investigó el efecto de la riboflavina en 88 pacientes hipertensos (pero sin ECV presentes) con el genotipo 677TT de la MTHFR, la mayoría de los cuales habían sido tratados con terapia antihipertensiva. Al inicio del estudio, el 60% de los participantes habían fallado en lograr el objetivo de los niveles de presión sanguínea (≤140/90 mm Hg), a pesar de haber tomado tres o más medicamentos antihipertensivos. La reducción en la presión sanguínea después de la suplementación con riboflavina (1.6 mg/día por 16 semanas) en estos pacientes sugirió que el exceso del riesgo de hipertensión ligado a esta variación genética pudo ser superado al optimizar el estatus de la riboflavina (78).
Cáncer
Los agentes anticancerosos frecuentemente muestran varios efectos secundarios que pueden forzar a los pacientes a limitar la dosis o a descontinuar el tratamiento. El efecto antioxidante de la coadministración de riboflavina (10 mg/día), niacina (50 mg/día), y coenzima Q10 (100 mg/día) fue evaluado en 78 pacientes postmenopáusicos con cáncer de seno tratados con tamoxifeno por 90 días. Esta suplementación previno efectivamente el estrés oxidativo asociado con el tratamiento con tamoxifeno (79). La riboflavina puede también actuar como un fotosensibilizador, y esta propiedad pudiese ser de valor en la terapia fotodinámica del cáncer. Un modelo de ratón fue usado para evaluar el efecto de la riboflavina en combinación con cisplatino, uno de los agentes anticancerosos más efectivos. Bajo exposición a la luz, la administración de riboflavina redujo el daño al ADN inducido por el cisplatino en el hígado y los riñones (80). Estos resultados son prometedores, pero estudios en humanos son necesarios para examinar si la riboflavina podría ser un adjunto para la quimioterapia.
Trastornos de la córnea
La ectasia corneal es una condición del ojo caracterizada por irregularidades de la córnea que afectan la visión. La reticulación corneal — un nuevo procedimiento usado por profesionales para limitar la progresión del daño corneal — involucra el uso de riboflavina en conjunción con irradiación de luz ultravioleta. La reticulación corneal modifica las propiedades de la córnea y fortalece su arquitectura (81).
Fuentes
Fuentes alimenticias
La mayoría de los alimentos derivados de plantas y animales contienen al menos pequeñas cantidades de riboflavina. En los EE.UU. se ha enriquecido la harina de trigo y el pan con riboflavina (y también con tiamina, niacina, y hierro) desde 1943. Datos provenientes de encuestas dietarías masivas indican que la ingesta promedio de riboflavina en hombres es de alrededor de 2 mg/día, y en mujeres es alrededor de 1.5 mg/día; ambas ingestas están muy por encima de la IDR. Los niveles de ingesta fueron similares para una población de hombres y mujeres de la tercera edad (1). La riboflavina es fácilmente destruida luego de su exposición a la luz. Por ello, hasta un 50% de la riboflavina de la leche contenida en una botella de vidrio transparente puede destruirse luego de dos horas de exposición a luz solar intensa (7). Algunos alimentos con cantidades substanciales de riboflavina están listados en la Tabla 2, junto con su contenido de riboflavina en miligramos (mg). Para mayor información sobre el contenido de nutrientes de los alimentos, busque la base de datos de composición de los alimentos del USDA.
Suplementos
Las formas más comunes de riboflavina disponible en los suplementos son riboflavina y riboflavina-5’-monofosfato. La riboflavina se encuentra con frecuencia en preparaciones multivitamínicas y de vitaminas del complejo B (26).
Seguridad
Toxicidad
Se desconocen efectos tóxicos o adversos debido a ingestas elevadas de riboflavina en humanos. Estudios en células de cultivo señalan que un exceso de riboflavina podría aumentar el riesgo de ruptura de hebras de ADN en presencia de cromio (IV), un conocido carcinógeno (27). Esto podría ser motivo de preocupación para trabajadores expuestos al cromo, pero no hay información disponible de estudios en humanos. Se ha encontrado que la terapia con dosis elevadas de riboflavina intensifica el color de la orina a un amarillo fuerte (flavinuria), pero este es un efecto secundario inocuo. La Junta de Nutrición y Alimentos no estableció un nivel máximo de ingesta tolerable (NM) cuando se revisó la IDR en 1998 (1).
Interacción con drogas
Varios estudios preliminares señalaron que las mujeres tomando anticonceptivos orales (AO) en dosis altas habían disminuido su estado nutricional de riboflavina. Sin embargo, cuando los investigadores controlaron la ingesta de riboflavina alimentaria, no encontraron diferencias entre las usuarias de AO y las no usuarias (1). Los derivados de la fenotiazina (como el medicamento antipsicótico clorpromazina y los antidepresivos tricíclicos) inhiben la incorporación de riboflavina en el FAD y el FMN, así como lo hace la quinacrina, un medicamento contra la malaria, y la adriamicina, un agente quimioterapéutico contra el cáncer (4). La utilización a largo plazo del anticonvulsivo fenobarbital podría incrementar la destrucción de riboflavina por parte de las enzimas hepáticas, aumentando el riesgo de una deficiencia (3).
Recomendación del Instituto Linus Pauling
La IDR de riboflavina (1.3 mg/día para hombres y 1.1 mg/día para mujeres), que debiera prevenir una deficiencia en la mayoría de las personas, se alcanza fácilmente consumiendo una dieta variada. Consumir una dieta variada debería proporcionar 1.5 mg a 2 mg de riboflavina al día. Siguiendo la recomendación del Instituto Linus Pauling de tomar un suplemento multivitamínico/multimineral que contenga el 100% del Valor Diario (VD) asegurará una ingesta de al menos 1.7 mg de riboflavina al día.
Adultos mayores (>50 años)
Algunos expertos en nutrición y envejecimiento consideraron que la IDR (1.3 mg/día para hombres y 1.1 mg/día para mujeres) dejaba un pequeño margen de error en personas sobre los 50 años de edad (28, 29). Un estudio reciente en personas que viven de manera independiente entre 65 y 90 años de edad reveló que casi el 25% consumía menos riboflavina que la ingesta recomendada, y que un 10% presentaba evidencia bioquímica de una deficiencia (30). Adicionalmente, estudios epidemiológicos de prevalencia de cataratas señalaron que la ingesta de riboflavina de 1.6 a 2.2 mg/día podría reducir el riesgo de sufrir de cataratas relacionadas con la edad. En individuos donde sus dietas podrían no proporcionar riboflavina suficiente, especialmente en aquellos sobre 50 años, deberían considerar consumir un suplemento multivitamínico/multimineral, el cual aporta generalmente, al menos 1.7 mg de riboflavina al día.
Autores y Críticos
Escrito en 2000 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Septiembre de 2002 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Junio de 2007 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Julio de 2013 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Diciembre de 2013 por:
Helene McNulty, Ph.D., R.D.
Profesor de Nutrición Humana y Dietética
Centro para la Alimentación y la Salud de Irlanda del Norte (NICHE)
Universidad de Ulster
Coleraine, Reino Unido
Revisado en Diciembre de 2013 por:
Adrian McCann, Ph.D.
Asociado en Investigación
Centro para la Alimentación y la Salud de Irlanda del Norte (NICHE)
Universidad de Ulster
Coleraine, Reino Unido
Traducido al Español en 2016 por:
Silvia Vazquez Lima
Instituto Linus Pauling
Universidad Estatal de Oregon
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Derechos de autoría 2000-2024 Instituto Linus Pauling
Referencias
1. Food and Nutrition Board, Institute of Medicine. Riboflavin. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B6, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington D.C.: National Academy Press; 1998:87-122. (National Academy Press)
2. Brody T. Nutritional Biochemistry. 2nd ed. San Diego: Academic Press; 1999.
3. McCormick DB. Riboflavin. In: Shils M, Olson JA, Shike M, Ross AC, eds. Modern Nutrition in Health and Disease. 9th ed. Baltimore: Williams & Wilkins; 1999:391-399.
4. Powers HJ. Current knowledge concerning optimum nutritional status of riboflavin, niacin and pyridoxine. Proc Nutr Soc. 1999;58(2):435-440. (PubMed)
5. Rivlin RS. Riboflavin. In: Ziegler EE, Filer LJ, eds. Present Knowledge in Nutrition. 7th ed. Washington D.C.: ILSI Press; 1996:167-173.
6. Hoey L, McNulty H, Strain JJ. Studies of biomarker responses to intervention with riboflavin: a systematic review. Am J Clin Nutr. 2009;89(6):1960S-1980S. (PubMed)
7. Bohles H. Antioxidative vitamins in prematurely and maturely born infants. Int J Vitam Nutr Res. 1997;67(5):321-328. (PubMed)
8. McCormick DB. Two interconnected B vitamins: riboflavin and pyridoxine. Physiol Rev. 1989;69(4):1170-1198. (PubMed)
9. Madigan SM, Tracey F, McNulty H, et al. Riboflavin and vitamin B-6 intakes and status and biochemical response to riboflavin supplementation in free-living elderly people. Am J Clin Nutr. 1998;68(2):389-395. (PubMed)
10. Lowik MR, van den Berg H, Kistemaker C, Brants HA, Brussaard JH. Interrelationships between riboflavin and vitamin B6 among elderly people (Dutch Nutrition Surveillance System). Int J Vitam Nutr Res. 1994;64(3):198-203. (PubMed)
11. Jacques PF, Bostom AG, Wilson PW, Rich S, Rosenberg IH, Selhub J. Determinants of plasma total homocysteine concentration in the Framingham Offspring cohort. Am J Clin Nutr. 2001;73(3):613-621. (PubMed)
12. Jacques PF, Kalmbach R, Bagley PJ, et al. The relationship between riboflavin and plasma total homocysteine in the Framingham Offspring cohort is influenced by folate status and the C677T transition in the methylenetetrahydrofolate reductase gene. J Nutr. 2002;132(2):283-288. (PubMed)
13. Powers HJ, Weaver LT, Austin S, Beresford JK. A proposed intestinal mechanism for the effect of riboflavin deficiency on iron loss in the rat. Br J Nutr. 1993;69(2):553-561. (PubMed)
14. Powers HJ, Hill MH, Mushtaq S, Dainty JR, Majsak-Newman G, Williams EA. Correcting a marginal riboflavin deficiency improves hematologic status in young women in the United Kingdom (RIBOFEM). Am J Clin Nutr. 2011;93(6):1274-1284. (PubMed)
15. Powers HJ. Riboflavin-iron interactions with particular emphasis on the gastrointestinal tract. Proc Nutr Soc. 1995;54(2):509-517. (PubMed)
16. Kalaivani K. Prevalence & consequences of anaemia in pregnancy. Indian J Med Res. 2009;130(5):627-633. (PubMed)
17. Worldwide prevalence of anaemia 1993-2005: WHO global database on anaemia. de Benoist B, McLean E, Egli I, Cogswell M, eds. 2008; World Health Organization Press. Available at: http://www.who.int/nutrition/publications/micronutrients/anaemia_iron_deficiency/9789241596657/en/. Accessed 7/22/13.
18. Pena-Rosas JP, Viteri FE. Effects of routine oral iron supplementation with or without folic acid for women during pregnancy. Cochrane Database Syst Rev. 2006(3):CD004736. (PubMed)
19. Suprapto B, Widardo, Suhanantyo. Effect of low-dosage vitamin A and riboflavin on iron-folate supplementation in anaemic pregnant women. Asia Pac J Clin Nutr. 2002;11(4):263-267. (PubMed)
20. Ma AG, Schouten EG, Zhang FZ, et al. Retinol and riboflavin supplementation decreases the prevalence of anemia in Chinese pregnant women taking iron and folic Acid supplements. J Nutr. 2008;138(10):1946-1950. (PubMed)
21. Crombleholme WR. Obstetrics. In: Tierney LM, McPhee SJ, Papadakis MA, eds. Current Medical Treatment and Diagnosis. 37th ed. Stamford: Appleton and Lange; 1998:731-734.
22. Wacker J, Fruhauf J, Schulz M, Chiwora FM, Volz J, Becker K. Riboflavin deficiency and preeclampsia. Obstet Gynecol. 2000;96(1):38-44. (PubMed)
23. Wang XM, Wu HY, Qiu XJ. Methylenetetrahydrofolate reductase (MTHFR) gene C677T polymorphism and risk of preeclampsia: an updated meta-analysis based on 51 studies. Arch Med Res. 2013;44(3):159-168. (PubMed)
24. Xia XP, Chang WW, Cao YX. Meta-analysis of the methylenetetrahydrofolate reductase C677T polymorphism and susceptibility to pre-eclampsia. Hypertens Res. 2012;35(12):1129-1134. (PubMed)
25. Braekke K, Ueland PM, Harsem NK, Karlsen A, Blomhoff R, Staff AC. Homocysteine, cysteine, and related metabolites in maternal and fetal plasma in preeclampsia. Pediatr Res. 2007;62(3):319-324. (PubMed)
26. Neugebauer J, Zanre Y, Wacker J. Riboflavin supplementation and preeclampsia. Int J Gynaecol Obstet. 2006;93(2):136-137. (PubMed)
27. Heese P, Linnebank M, Semmler A, et al. Alterations of homocysteine serum levels during alcohol withdrawal are influenced by folate and riboflavin: results from the German Investigation on Neurobiology in Alcoholism (GINA). Alcohol Alcohol. 2012;47(5):497-500. (PubMed)
28. Soares MJ, Satyanarayana K, Bamji MS, Jacob CM, Ramana YV, Rao SS. The effect of exercise on the riboflavin status of adult men. Br J Nutr. 1993;69(2):541-551. (PubMed)
29. Ruston D, Hoare J, Henderson L, et al. The National Diet & Nutrition Survey: adults aged 19 to 64 years: Nutritional status (anthropometry and blood analytes), blood pressure and physical activity. Volume 4. London: The Stationary Office; 2004.
30. Mares-Perlman JA, Brady WE, Klein BE, et al. Diet and nuclear lens opacities. Am J Epidemiol. 1995;141(4):322-334. (PubMed)
31. Leske MC, Wu SY, Hyman L, et al. Biochemical factors in the lens opacities. Case-control study. The Lens Opacities Case-Control Study Group. Arch Ophthalmol. 1995;113(9):1113-1119. (PubMed)
32. Cumming RG, Mitchell P, Smith W. Diet and cataract: the Blue Mountains Eye Study. Ophthalmology. 2000;107(3):450-456. (PubMed)
33. Hankinson SE, Stampfer MJ, Seddon JM, et al. Nutrient intake and cataract extraction in women: a prospective study. BMJ. 1992;305(6849):335-339. (PubMed)
34. Jacques PF, Taylor A, Moeller S, et al. Long-term nutrient intake and 5-year change in nuclear lens opacities. Arch Ophthalmol. 2005;123(4):517-526. (PubMed)
35. McNulty H, Strain JJ, Pentieva K, Ward M. C(1) metabolism and CVD outcomes in older adults. Proc Nutr Soc. 2012;71(2):213-221. (PubMed)
36. Holmes MV, Newcombe P, Hubacek JA, et al. Effect modification by population dietary folate on the association between MTHFR genotype, homocysteine, and stroke risk: a meta-analysis of genetic studies and randomised trials. Lancet. 2011;378(9791):584-594. (PubMed)
37. Wilcken B, Bamforth F, Li Z, et al. Geographical and ethnic variation of the 677C>T allele of 5,10 methylenetetrahydrofolate reductase (MTHFR): findings from over 7000 newborns from 16 areas world wide. J Med Genet. 2003;40(8):619-625. (PubMed)
38. McGlynn AP, Wasson GR, O'Reilly SL, et al. Low colonocyte folate is associated with uracil misincorporation and global DNA hypomethylation in human colorectum. J Nutr. 2013;143(1):27-33. (PubMed)
39. Guenther BD, Sheppard CA, Tran P, Rozen R, Matthews RG, Ludwig ML. The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomocysteinemia. Nat Struct Biol. 1999;6(4):359-365. (PubMed)
40. Yin G, Ming H, Zheng X, Xuan Y, Liang J, Jin X. Methylenetetrahydrofolate reductase C677T gene polymorphism and colorectal cancer risk: A case-control study. Oncol Lett. 2012;4(2):365-369. (PubMed)
41. Gao S, Ding LH, Wang JW, Li CB, Wang ZY. Diet folate, DNA methylation and polymorphisms in methylenetetrahydrofolate reductase in association with the susceptibility to gastric cancer. Asian Pac J Cancer Prev. 2013;14(1):299-302. (PubMed)
42. Wen YY, Yang SJ, Zhang JX, Chen XY. Methylenetetrahydrofolate reductase genetic polymorphisms and esophageal squamous cell carcinoma susceptibility: a meta-analysis of case-control studies. Asian Pac J Cancer Prev. 2013;14(1):21-25. (PubMed)
43. Powers HJ, Hill MH, Welfare M, et al. Responses of biomarkers of folate and riboflavin status to folate and riboflavin supplementation in healthy and colorectal polyp patients (the FAB2 Study). Cancer Epidemiol Biomarkers Prev. 2007;16(10):2128-2135. (PubMed)
44. Zschabitz S, Cheng TY, Neuhouser ML, et al. B vitamin intakes and incidence of colorectal cancer: results from the Women's Health Initiative Observational Study cohort. Am J Clin Nutr. 2013;97(2):332-343. (PubMed)
45. Kennedy DA, Stern SJ, Matok I, et al. Folate Intake, MTHFR Polymorphisms, and the Risk of Colorectal Cancer: A Systematic Review and Meta-Analysis. J Cancer Epidemiol. 2012:952508. doi: 10.1155/2012/952508. (PubMed)
46. He Y, Ye L, Shan B, Song G, Meng F, Wang S. Effect of riboflavin-fortified salt nutrition intervention on esophageal squamous cell carcinoma in a high incidence area, China. Asian Pac J Cancer Prev. 2009;10(4):619-622. (PubMed)
47. Bassett JK, Hodge AM, English DR, et al. Dietary intake of B vitamins and methionine and risk of lung cancer. Eur J Clin Nutr. 2012;66(2):182-187. (PubMed)
48. Bassett JK, Baglietto L, Hodge AM, et al. Dietary intake of B vitamins and methionine and breast cancer risk. Cancer Causes Control. 2013;24(8):1555-1563. (PubMed)
49. Bassett JK, Severi G, Hodge AM, et al. Dietary intake of B vitamins and methionine and prostate cancer incidence and mortality. Cancer Causes Control. 2012;23(6):855-863. (PubMed)
50. Qiao YL, Dawsey SM, Kamangar F, et al. Total and cancer mortality after supplementation with vitamins and minerals: follow-up of the Linxian General Population Nutrition Intervention Trial. J Natl Cancer Inst. 2009;101(7):507-518. (PubMed)
51. Schoenen J, Jacquy J, Lenaerts M. Effectiveness of high-dose riboflavin in migraine prophylaxis. A randomized controlled trial. Neurology. 1998;50(2):466-470. (PubMed)
52. Sandor PS, Afra J, Ambrosini A, Schoenen J. Prophylactic treatment of migraine with β-blockers and riboflavin: differential effects on the intensity dependence of auditory evoked cortical potentials. Headache. 2000;40(1):30-35. (PubMed)
53. Boehnke C, Reuter U, Flach U, Schuh-Hofer S, Einhaupl KM, Arnold G. High-dose riboflavin treatment is efficacious in migraine prophylaxis: an open study in a tertiary care centre. Eur J Neurol. 2004;11(7):475-477. (PubMed)
54. Maizels M, Blumenfeld A, Burchette R. A combination of riboflavin, magnesium, and feverfew for migraine prophylaxis: a randomized trial. Headache. 2004;44(9):885-890. (PubMed)
55. MacLennan SC, Wade FM, Forrest KM, Ratanayake PD, Fagan E, Antony J. High-dose riboflavin for migraine prophylaxis in children: a double-blind, randomized, placebo-controlled trial. J Child Neurol. 2008;23(11):1300-1304. (PubMed)
56. Bruijn J, Duivenvoorden H, Passchier J, Locher H, Dijkstra N, Arts WF. Medium-dose riboflavin as a prophylactic agent in children with migraine: a preliminary placebo-controlled, randomised, double-blind, cross-over trial. Cephalalgia. 2010;30(12):1426-1434. (PubMed)
57. Cotelli MS, Vielmi V, Rimoldi M, et al. Riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency with unknown genetic defect. Neurol Sci. 2012;33(6):1383-1387. (PubMed)
58. Olsen RK, Olpin SE, Andresen BS, et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain. 2007;130(Pt 8):2045-2054. (PubMed)
59. Liang WC, Ohkuma A, Hayashi YK, et al. ETFDH mutations, CoQ10 levels, and respiratory chain activities in patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Neuromuscul Disord. 2009;19(3):212-216. (PubMed)
60. Haack TB, Danhauser K, Haberberger B, et al. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat Genet. 2010;42(12):1131-1134. (PubMed)
61. Nouws J, Nijtmans L, Houten SM, et al. Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 2010;12(3):283-294. (PubMed)
62. Scholte HR, Busch HF, Bakker HD, Bogaard JM, Luyt-Houwen IE, Kuyt LP. Riboflavin-responsive complex I deficiency. Biochim Biophys Acta. 1995;1271(1):75-83. (PubMed)
63. Gerards M, van den Bosch BJ, Danhauser K, et al. Riboflavin-responsive oxidative phosphorylation complex I deficiency caused by defective ACAD9: new function for an old gene. Brain. 2011;134(Pt 1):210-219. (PubMed)
64. Garone C, Donati MA, Sacchini M, et al. Mitochondrial Encephalomyopathy Due to a Novel Mutation in ACAD9. JAMA Neurol. 2013:1-3. (PubMed)
65. Chiong MA, Sim KG, Carpenter K, et al. Transient multiple acyl-CoA dehydrogenation deficiency in a newborn female caused by maternal riboflavin deficiency. Mol Genet Metab. 2007;92(1-2):109-114. (PubMed)
66. Ho G, Yonezawa A, Masuda S, et al. Maternal riboflavin deficiency, resulting in transient neonatal-onset glutaric aciduria Type 2, is caused by a microdeletion in the riboflavin transporter gene GPR172B. Hum Mutat. 2011;32(1):E1976-1984. (PubMed)
67. Bosch AM, Stroek K, Abeling NG, Waterham HR, Ijlst L, Wanders RJ. The Brown-Vialetto-Van Laere and Fazio Londe syndrome revisited: natural history, genetics, treatment and future perspectives. Orphanet J Rare Dis. 2012;7:83. doi: 10.1186/1750-1172-7-83. (PubMed)
68. Mackay RJ, McEntyre CJ, Henderson C, Lever M, George PM. Trimethylaminuria: causes and diagnosis of a socially distressing condition. Clin Biochem Rev. 2011;32(1):33-43. (PubMed)
69. Phillips IR, Shephard EA. Trimethylaminuria. 2007 Oct 8 [Updated 2011 Apr 19]. In: Pagon RA, Adam MP, Bird TD, et al., editors. GeneReviews™ [Internet]. Seattle: University of Washington, Seattle; 1993-2013. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1103/.
70. Manning NJ, Allen EK, Kirk RJ, Sharrard MJ, Smith EJ. Riboflavin-responsive trimethylaminuria in a patient with homocystinuria on betaine therapy. JIMD Rep. 2012;5:71-75. (PubMed)
71. Niu WQ, You YG, Qi Y. Strong association of methylenetetrahydrofolate reductase gene C677T polymorphism with hypertension and hypertension-in-pregnancy in Chinese: a meta-analysis. J Hum Hypertens. 2012;26(4):259-267. (PubMed)
72. Almawi WY, Khan A, Al-Othman SS, Bakhiet M. Case-control Study of methylenetetrahydrofolate reductase mutations and hyperhomocysteinemia and risk of stroke. J Stroke Cerebrovasc Dis. 2009;18(5):407-408. (PubMed)
73. Heux S, Morin F, Lea RA, Ovcaric M, Tajouri L, Griffiths LR. The methylentetrahydrofolate reductase gene variant (C677T) as a risk factor for essential hypertension in Caucasians. Hypertens Res. 2004;27(9):663-667. (PubMed)
74. Klerk M, Verhoef P, Clarke R, et al. MTHFR 677C→T polymorphism and risk of coronary heart disease: a meta-analysis. JAMA. 2002;288(16):2023-2031. (PubMed)
75. McNulty H, Dowey le RC, Strain JJ, et al. Riboflavin lowers homocysteine in individuals homozygous for the MTHFR 677C→T polymorphism. Circulation. 2006;113(1):74-80. (PubMed)
76. Horigan G, McNulty H, Ward M, Strain JJ, Purvis J, Scott JM. Riboflavin lowers blood pressure in cardiovascular disease patients homozygous for the 677C→T polymorphism in MTHFR. J Hypertens. 2010;28(3):478-486. (PubMed)
77. Wilson CP, Ward M, McNulty H, et al. Riboflavin offers a targeted strategy for managing hypertension in patients with the MTHFR 677TT genotype: a 4-y follow-up. Am J Clin Nutr. 2012;95(3):766-772. (PubMed)
78. Wilson CP, McNulty H, Ward M, et al. Blood pressure in treated hypertensive individuals with the MTHFR 677TT genotype is responsive to intervention with riboflavin: findings of a targeted randomized trial. Hypertension. 2013;61(6):1302-1308. (PubMed)
79. Yuvaraj S, Premkumar VG, Vijayasarathy K, Gangadaran SG, Sachdanandam P. Augmented antioxidant status in Tamoxifen treated postmenopausal women with breast cancer on co-administration with Coenzyme Q10, Niacin and Riboflavin. Cancer Chemother Pharmacol. 2008;61(6):933-941. (PubMed)
80. Hassan I, Chibber S, Khan AA, Naseem I. Riboflavin ameliorates cisplatin induced toxicities under photoillumination. PLoS One. 2012;7(5):e36273. (PubMed)
81. Raiskup F, Spoerl E. Corneal crosslinking with riboflavin and ultraviolet A. I. Principles. Ocul Surf. 2013;11(2):65-74. (PubMed)
82. Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. Montvale: Medical Economics Company, Inc; 2001.
83. Sugiyama M. Role of physiological antioxidants in chromium(VI)-induced cellular injury. Free Radic Biol Med. 1992;12(5):397-407. (PubMed)
84. Subramanian VS, Subramanya SB, Ghosal A, Said HM. Chronic alcohol feeding inhibits physiological and molecular parameters of intestinal and renal riboflavin transport. Am J Physiol Cell Physiol. 2013; 305(5):C539-C546. (PubMed)
85. Russell RM, Suter PM. Vitamin requirements of elderly people: an update. Am J Clin Nutr. 1993;58(1):4-14. (PubMed)
86. Blumberg J. Nutritional needs of seniors. J Am Coll Nutr. 1997;16(6):517-523. (PubMed)
87. Lopez-Sobaler AM, Ortega RM, Quintas ME, et al. The influence of vitamin B2 intake on the activation coefficient of erythrocyte glutation reductase in the elderly. J Nutr Health Aging. 2002;6(1):60-62. (PubMed)
88. Gariballa S, Ullegaddi R. Riboflavin status in acute ischaemic stroke. Eur J Clin Nutr. 2007;61(10):1237-1240. (PubMed)
89. Yazdanpanah N, Uitterlinden AG, Zillikens MC, et al. Low dietary riboflavin but not folate predicts increased fracture risk in postmenopausal women homozygous for the MTHFR 677 T allele. J Bone Miner Res. 2008;23(1):86-94. (PubMed)
Tiamina
Contenido
Resumen
- Pirofosfato de tiamina (TPP), la forma activa de la tiamina, está envuelto en varias funciones enzimáticas asociadas con el metabolismo carbohidratos, aminoácidos de cadena ramificada, y los ácidos grasos. (Más información)
- La deficiencia severa de tiamina provoca beriberi, una enfermedad que afecta múltiples sistemas orgánicos, incluyendo los sistemas nerviosos central y periférico. (Más información)
- Encefalopatía de Wernicke se refiere a un desorden neurológica agudo secundario a la deficiencia de tiamina. El síndrome de Wernicke-Korsakoff resulta en alteraciones persistentes en la formación de la memoria, junto con los síntomas relacionados con la encefalopatía. (Más información)
- La deficiencia de tiamina puede resultar de una baja ingesta dietética, inadecuada provisión de nutrición parental, la reducción de la absorción gastrointestinal, aumento de las necesidades metabólicas, o pérdida excesiva de tiamina. El consumo crónico de alcohol es la causa principal de la deficiencia de tiamina en los países industrializados. (Más información)
- La alteración del metabolismo de la glucosa se ha asociado con la disminución de las concentraciones plasmáticas de tiamina en pacientes diabéticos. Corrección de la deficiencia de tiamina puede reducir el riesgo de complicaciones vasculares en los sujetos diabéticos. (Más información)
- La deficiencia de tiamina y la disminución de actividad enzimática dependiente de tiamina están asociadas con la enfermedad de Alzheimer. Aunque la complementación de tiamina invierte notablemente el deterioro cognitivo en modelos de animales de deficiencia de tiamina, el efecto de la suplementación de tiamina en pacientes con la enfermedad de Alzheimer es aun no conocida. (Más información)
- La excreción inducida por diuréticos puede aumentar el riesgo de deficiencia de tiamina y la gravedad en personas con insuficiencia cardiaca congestiva. Se necesitan más estudios para evaluar la inclusión de la suplementación de la tiamina en el tratamiento de la enfermedad. (Más información)
La tiamina es una vitamina hidrosoluble del complejo B, tambien conocida como vitamina B1 o aneurina (1). Aislada y caracterizada en la década de 1930, la tiamina fue uno de los primeros compuestos orgánicos en ser reconocido como una vitamina (2). La tiamina se encuentra en el cuerpo humano como tiamina libre y como varias formas fosforilada: tiamina monofosfato (TMP), tiamina trifosfato (TTP), y tiamina pirofosfato (TPP), conocida también como tiamina difosfato.
Función
Función como coenzima
La síntesis de TPP de tiamina libre requiere magnesio, trifosfato de adenosina (ATP) y la enzima tiamina pyrophosphokinase. TPP se requiere como una coenzima para cuatro complexos enzimáticos de varios componentes asociados con el metabolismo de carbohidratos y cadenas ramificadas de aminoácidos.
Piruvato deshidrogenasa, α-cetoglutarato deshidrogenasa, y deshidrogenasa de α-cetoácidos de cadena ramificada, cada una comprende un complejo enzimático diferente encontrados al interior de organelos celulares llamados mitocondrias. Ellos catalizan la descarboxilación de piruvato, a-cetoglutarato, y aminoácidos de cadena ramificada para formar acetil-coenzima A, succinil-coenzima A, y derivados de aminoácidos de cadena ramificada, respectivamente. Todos estos productos juegan un papel crítico en la producción de energía de los alimentos a través de su conexión con el ciclo del ácido cítrico (Kreb) (2). Aminoacidos de cadena ramificada (BCAA) incluyen leucina, isoleucina, y valina, son eventualmente degradados en acetil-CoA y sucinil-CoA como combustible para el ciclo de ácido cítrico. El catabolismo de las tres BCAAs también contribuye a las producción de colesterol y dona nitrógeno para la síntesis de los neurotransmisores, glutamato y acido γ-aminobutírico (GABA) (3). Además de la coenzima de la tiamina (TTP), cada complejo deshidrogenasa necesita de una coenzima que contenga niacina (NAD), una enzima que contenga riboflavina (FAD), y ácido lipoico.
La transcetolasa cataliza reacciones fundamentales en otra vía metabólica que ocurre en el citosol conocida como la vía de las pentosas fosfato. Uno de los intermediarios más importantes de esta vía es la ribosa-5-fosfato, un azúcar fosforilado de 5 carbonos, necesario para la síntesis de ribonucleótidos tales como, ATP y guanosina trifosfato (GTP). Nucleicos son los bloques de construcción de acidos nucleicos, ADN y ARN. La via del fosfato de pentosa también suministra diversas vías anabólicas, incluyendo ácidos grasos, con la coenzima que contiene niacina (1, 4). Debido a que la transcetolasa disminuye con rapidez ante una deficiencia de tiamina, y a diferencia de la mayoría de enzimas dependientes de tiamina, está presente en las células rojas, se ha utilizado la medición de su actividad en eritrocitos para evaluar el estado nutricional de la tiamina (2).
Deficiencia
El beriberi, la enfermedad causada por una deficiencia severa de tiamina, fue descrita en la literatura china ya en el 2600 A.C. La deficiencia de tiamina afecta el cardiovascular, muscular, gastrointestinal, y el sistema nervioso central y periférico (2). El beriberi dividido entre seco, húmedo cerebral, o gastrointestinal dependiendo de los sistemas afectados por la deficiencia severa de tiamina (1, 5).
Beriberi seco
La característica principal del beriberi seco (paralítico o nervioso) es la neuropatía periférica. Al inicio del transcurso de la neuropatía, podría tener lugar el "síndrome pies ardientes." Otros síntomas incluyen reflejos anormales (exagerados) así como también sensación disminuida y debilidad en brazos y piernas. También se han descrito dolores musculares, sensibilidad y dificultad al levantarse de una posición agachada (6).
Beriberi húmedo
Además de los síntomas neurológicos, el beriberi húmedo (cardiaco) se caracteriza por manifestaciones cardiovasculares de la deficiencia de tiamina, que incluyen ritmo cardiaco acelerado, agrandamiento del corazón, hinchazón severa (edema), dificultad para respirar, y en última instancia insuficiencia cardiaca congestiva. La literatura Japonesa describe la forma aguda fulminante del beriberi húmedo como "shoshin" (7).
Beriberi cerebral
El beriberi cerebral podría derivar en encefalopatía de Wernicke y psicosis de Korsakoff, especialmente en personas que abusan del alcohol. El diagnóstico de la encefalopatía de Wernicke se basa en una triada de señales, que incluye movimientos anormales del ojo, alteraciones de la postura y la marcha, la ataxia y deterioros cognitivos. Si no se trata el daño neurológico irreversible puede causar manifestaciones clínicas conocidas como la psicosis de Korsakoff. Este síndrome también conocido como demencia de Korsakoff, amnesia de Korsakoff, o síndrome amnésico confabulatoria implica un confuso estado apático y un trastorno profundo de la memoria, con amnesia severa y perdida de la memoria reciente y activa.
La deficiencia de tiamina que afecta al sistema nervioso central se define como la enfermedad de Wernicke cuando no se presenta el estado amnésico, y síndrome de Wernicke-Korsakoff (SWK) cuando se presentan los síntomas amnésicos junto a los trastornos del movimiento del ojo y la marcha. Las manifestaciones neurológicas más raras pueden incluir convulsiones (8). La mayoría de los que sufren de SWK son alcohólicos, aunque este síndrome se ha observado en otros trastornos de malnutrición grave, incluyendo cáncer al estómago y SIDA. Generalmente la administración de tiamina intravenosa a pacientes con SWK resulta en un rápido mejoramiento de los síntomas oculares, pero las mejoras en la coordinación motora y la memoria podrían ser menores, dependiendo de cuánto tiempo han estado presentes los síntomas. Evidencia reciente de activación aumentada de células inmunes y producción incrementada de radicales libres en las áreas del cerebro que son dañadas selectivamente, sugiere que el stress oxidativo juega un papel importante en la patología neurológica de la deficiencia de tiamina (9).
Beriberi gastrointestinal
TPP es crítico para las reacciones metabólicas que utilizan glucosa en la glicolisis y el ciclo ácido cítrico (Figura 1). Una disminución en la actividad de enzimas dependientes de tiamina limita la conversión de piruvato a acetil-CoA y la utilización del ciclo acido critico, lo que lleva a la acumulación de piruvato y lactato. Acidosis láctico, una condición resultante de la acumulación de lactato, se asocia con nauseas, vomito, y dolor abdominal severo en un síndrome descrito como beriberi gastrointestinal (5).
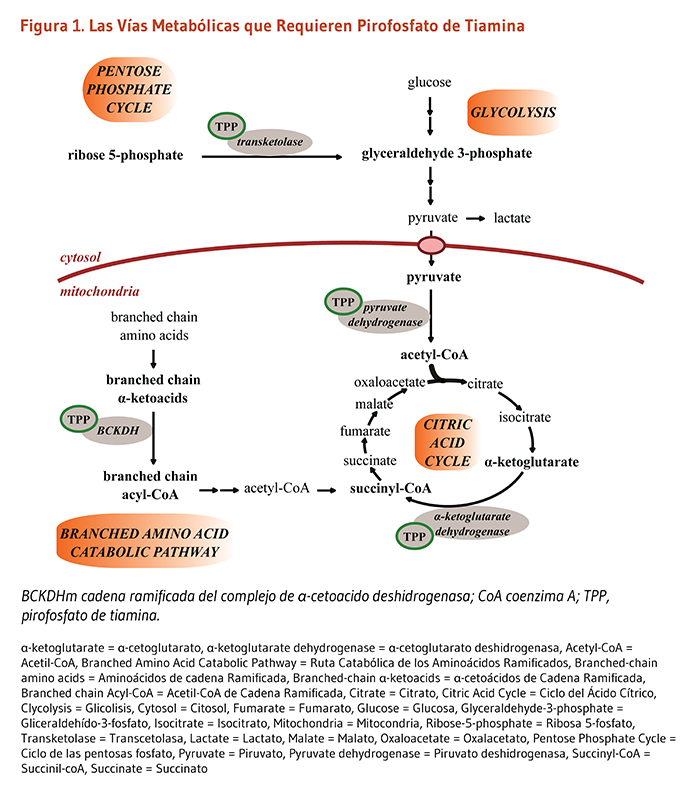
Causas de la deficiencia de tiamina
La deficiencia de tiamina podría derivar de una ingesta de tiamina insuficiente, necesidad aumentada de tiamina, pérdida excesiva de tiamina desde el cuerpo, consumos de factores anti-tiamina en los alimentos o una combinación de estos factores.
Ingesta insuficiente
El consumo insuficiente de tiamina es la causa principal de la deficiencia de tiamina en países subdesarrollados (2). La deficiencia de tiamina es común en poblaciones de bajos ingresos cuya dieta es alta en carbohidratos y baja en tiamina (por ejemplo, arroz blanqueado y pulido). Los infantes amamantados por madres con deficiencia de tiamina son propensos a desarrollar beriberi infantil. El alcoholismo, que se asocia con baja ingesta de tiamina, entre otros nutrientes, es la causa principal de la deficiencia de tiamina en países industrializados. Algunos de los condiciones no alcohólicas asociadas con WKS incluye anorexia, la cirugía bariatrica (cirugía para adelgazar) neoplasias gastrointestinales, y síndromes de mala absorción (10-13). Casos de encefalopia de Wernicke también se han relacionado a la falta de nutrición parental de suplementos vitamínicos (14, 15).
Requerimientos aumentados
Las condiciones que derivan en un requerimiento aumentado de tiamina incluyen esfuerzo físico intenso, fiebre, embarazo, amamantamiento y crecimiento durante la adolescencia. Tales condiciones colocan a los individuos con ingesta marginal de tiamina en riesgo de desarrollar una deficiencia de tiamina sintomática. Pacientes con Malaria en el Sur de Asia, se descubrió que pacientes con malaria sufrían con más frecuencia de deficiencia severa de tiamina que individuos no infectados (16, 17). La infección con malaria conduce a un gran aumento en la demanda metabólica por glucosa. Debido a que la tiamina es requerida por las enzimas involucradas en el metabolismo de la glucosa, el stress inducido por una infección con malaria podría exacerbar una deficiencia de tiamina en individuos predispuestos. También se encontró en personas infectadas con VIH, aún si no han desarrollado SIDA, que se encontraban en un riesgo mayor de sufrir una deficiencia de tiamina (18). Además, el abuso crónico del alcohol perjudica la absorción intestinal y la utilización de la tiamina (1); por lo tanto, los alcohólicos tienen un requerimiento de tiamina aumentado. La deficiencia de tiamina se observada también como una complicación del síndrome de realimentación. La introducción de carbohidratos en individuos severamente hambreados conduce a un aumento de demanda por tiamina en glucolisis y el ciclo ácido cítrico que precipita la deficiencia de tiamina (19).
Pérdida excesiva
La pérdida excesiva de tiamina podría precipitar una deficiencia de tiamina. Mediante el aumento del flujo urinario, los diuréticos podrían evitar la reabsorción de tiamina en los riñones y aumentar su excreción en la orina (20, 21). El riesgo de la deficiencia de tiamina puede aumentar en pacientes tratados con diuréticos con una ingesta marginal de tiamina (22) y en individuos que reciben terapias de diuréticos a largo plazo (23). Los individuos con falla renal que necesitan de hemodiálisis pierden tiamina a un ritmo aumentado y están en riesgo de una deficiencia de tiamina (24). Los alcohólicos que mantienen una ingesta de fluidos altos y un ritmo de flujo urinario altos podrían también experimentar una pérdida incrementada de tiamina, exacerbando los efectos de una baja ingesta de tiamina (25).
Factores Anti-Tiamínicos (FAT)
La presencia de factores anti-tiamínicos (FAT) en los alimentos contribuye también al riesgo de una deficiencia de tiamina. Ciertas plantas contienen FAT, los que reaccionan con la tiamina formando un producto oxidado inactivo. Beber grandes cantidades de té y café (incluso descafeinado), así como mascar hojas de té y nueces de betel, se han asociado con disminución de tiamina en seres humanos debido a la presencia de FAT (26, 27). FAT incluye micotoxinas (índole) y tiaminases que degradan la tiamina de los alimentos. Las personas que habitualmente comen ciertos pescados de agua dulce crudos, mariscos y helechos crudos están en un riesgo más alto de sufrir una deficiencia de tiamina debido a que estos alimentos contienen tiaminasa, la que normalmente se inactiva por el calor al cocinarse (1). En Nigeria se ha asociado a un grave síndrome neurológico (ataxia estacional) con la deficiencia de tiamina provocada por una tiaminasa en los gusanos de seda Africanos, una comida rica en proteínas tradicional para algunos Nigerianos (28).
La Ingesta Diaria Recomendada (IDR)
La IDR de tiamina, revisada en 1998por la Junta Directiva de Alimentación y Nutrición del Instituto de Medicinase basó en la prevención de su deficiencia en individuos generalmente sanos (29).
| Etapa de la Vida | Edad | Machos (mg/día) | Hembras (mg/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 0.2 (IA) | 0.2 (IA) |
| Infantes | 7-12 meses | 0.3 (IA) | 0.3 (IA) |
| Niños | 1-3 años | 0.5 | 0.5 |
| Niños | 4-8 años | 0.6 | 0.6 |
| Niños | 9-13 años | 0.9 | 0.9 |
| Adolescentes | 14-18 años | 1.2 | 1.0 |
| Adultos | 19 años y más | 1.2 | 1.1 |
| Embarazo | Todas las edades | - | 1.4 |
| Período de lactancia | Todas las edades | - | 1.4 |
Prevención de Enfermedades
Cataratas
Un estudio transversal de 2,900 hombres y mujeres Australianos, de 49 años de edad o mayores, encontró que aquellos en el quintil más alto de ingesta de tiamina eran un 40% menos propensos a tener cataratas nucleares que aquellos en el quintil más bajo (30). Además, un estudio reciente en 480 mujeres Estadounidenses encontró que ingesta recomendada más altas de tiamina se asociaban inversamente con cambios en la opacificación de las lentes (cristalinos) en un periodo de cinco años (31). Sin embargo, las asociaciones de este estudio transversal aún deben ser aclaradas por estudios de causalidad.
Diabetes y complicaciones vasculares
Concentración plasmáticas bajas y el aclaramiento renal de tiamina alta se han observado en pacientes diabéticos en comparación con individuos saludables (32) lo que sugiere que individuaos con diabetes mellitus tipo 1 o tipo 2 tienen un mayor riesgo de deficiencia de tiamina. Dos transportadores de tiamina, transportador-1 (THTR-1) y THTR-2 están implicados en la absorción de tiamina por los enterocitos en el intestino delgado y de la re-absorción en los túbulos próximos de los riñones. Un estudio reciente sugiere que la hiperglicemia en pacientes diabéticos podría afectar la re-absorción de tiamina por la disminución de la expresión de transportadores de tiamina en los riñones (33). Por lo contrario, la deficiencia de tiamina parece dañar la función endocrinal normal del páncreas y exacerbar la hiperglicemia. Los primeros estudios mostraron que la síntesis y la secreción de la insulina fueron alteradas en las células endocrinas pancreáticas de ratas con deficiencia de tiamina (34, 35). En los seres humanos, la deficiencia de tiamina causada por mutaciones recesivas en el gen que codifica para THTR-1 conduce a diabetes mellitus en el síndrome de anemia megaloblastica sensible a tiamina.
En un estudio piloto aleatorizado, de doble ciego, los suplementos de dosis alta de tiamina (300 mg/día) fueron repartidos por seis semanas a individuos hyperglicemicos (ya intolerantes a la glucosa o con diagnostico reciente de diabetes tipo 2). La complementación de tiamina impidió cualquier aumento adicional de la glucosa en ayunas y niveles de insulina en comparación con el tratamiento con placebo, pero no redujo la hiperglicemia (36). Sin embargo, un estudio sugerio que la complementación de tiamina puede mejorar los niveles de glucosa en diabetes tipo 2 en las primeras etapas de la enfermedad (es decir, pre-diabéticos o principios de diabetes) (37).
Hiperglicemia crónica en individuos con diabetes mellitus contribuye a la patogénesis enfermedades micros vasculares. Daños vasculares relacionados con la diabetes pueden afectar el corazón (cardiomiopatía), riñones (nefropatía), retina (retinopatía) y el sistema nervioso periférico (neuropatía). En individuos con diabetes, la hiperglicemia altera la función de la medula ósea derivada de las células progenitoras endoteliales (CPE) que son críticas para el crecimiento de los vasos sanguíneos (38). Curiosamente, una ingesta diaria alta de tiamina en la dieta se correlaciono con mayor circulación de CPE y con una mejor salud del endotelio vascular en 88 individuos con diabetes tipo 2 (39). Inversamente asociado se ha encontrado entre las concentraciones plasmáticas de tiamina en pacientes con diabetes y la presencia de la molecula-1 de adhesión vascular soluble (sVCAM-1), un marcado de disfunción vascular (32, 40). Marcadores anteriores de nefropatía diabetes incluye la presencia de suero de albumina en la orina, conocida como micro-albuminuria. La administración de tiamina o benfotiamina (un derivado de tiamina) previno el desarrollo de complicaciones renales en ratas diabéticas químicamente inducidas (41). Un estudio aleatorizado, de doble ciego conducido a cabo en 40 pacientes tipo 2 con micro-albuminuria encontró que los complementos con altas dosis de tiamina (300 mg/día) disminuyen la excreción de albumina en orina en comparación con placebo durante un periodo de tres meses (40). Dado que el tratamiento de tiamina ha mostrado comprometedores resultados en células cultivadas y en modelos animales (42-44), los efectos de tiamina y derivados de las complicaciones vasculares deben ser examinados en pacientes con diabetes.
Tratamiento de Enfermedades
Enfermedad de Alzheimer
Algunas personas mayores están a un riesgo mayor de desarrollar deficiencia sub-clínica de tiamina secundaria a una dieta deficiente, reducción de la absorción gastrointestinal, y varias condiciones médicas (45, 46). Debido a que la deficiencia de tiamina puede derivar en una forma de demencia (síndrome de Wernicke-Korsakoff), se ha investigado su relación con la enfermedad de Alzheimer y a otras formas de demencia. La EA está caracterizado por la disminución de funciones cognitivas en las personas de edad avanzada, acompañado por características patológicas que incluyen deposiciones de placa β-amiloidea y ovillos formados por la proteína Tau fosforada (47). Utilizando la tomografía por emisión de positrones (TEP), se observó un metabolismo de glucosa reducido en los pacientes con la EA (48). Un estudio grande, multe-céntrico PET usando un radio marcador con analogía de glucosa, 18 F-Fluoro-desoxiglucosa (FDG), correlacionado con la reducción en el consumo FDG (un marcador sustituto para el metabolismo de la glucosa) con el grado de deterior cognitivo en pacientes con la EA. Este estudio que incluye 822 individuos mayores de 55 años de edad que eran cognitivamente normales n=299) se muestra un deterior cognitivo leve (n=405) o tenía la EA leve (=188), demostró que la utilización de glucosa cerebral podría predecir el progresión del deterioro cognitivo leve a EA (49). Curiosamente, un estudio longitudinal de nueve años asocio la presencia de diabetes mellitus en personas de edad avanzada (mayores de 55 años) con un aumento en el riesgo de desarrollo EA (50).
Una reducción en los procesos de tiamina dependiente en el cerebro puede alterar el metabolismo de glucosa en pacientes con EA (51), Un estudio de caso y control en 38 mujeres ancianas encontró que los niveles sanguíneos de tiamina (TPF), y (TMF) eran más bajos en aquellos con demencia tipo Alzheimer (DTA) comparado aquellos en el grupo control (52). Por otra parte, varios investigadores han encontrado evidencia de actividad disminuida de las enzimas dependientes de TPP, a-cetoglutarato deshidrogenasa y transcetolasa, en los cerebros de pacientes que murieron de EA (53). El hallazgo de niveles cerebrales de TPF disminuidos en la presencia de niveles normales de tiamina libre y TMF sugiere modificaciones en la síntesis TPP en lugar de escasas biodisponibilidad de tiamina. Sin embargo no está claro si las actividades de las enzimas que metabolizan TPP (incluyendo tiamina pyrophophokinase) son alteradas en pacientes con EA (54, 55). Administración crónica de tiamina derivad de benfotiamina alivio alteraciones cognitivas y disminuyo el número de placas β-amiloidea en un modelo de ratón de ED sin aumentar los nivels de TMP y TPP en el cerebro. Esto sugiere que los efectos beneficiosos de la benfotiamina en el cerebro fueron probablemente mediados por la estimulación de las vías independientes de TPP (56).
La deficiencia de tiamina ha sido relacionada con el aumento de producción de β-amiloidea en células neurales cultivadas y para la formación de placas en modelos de animales (57, 58). Estas características patológicas de la EA pueden ser reversibles por la complementación de tiamina sugiriendo que la tiamina puede tener un efecto protector en AE. Por otra parte, otros desordenes incluyen las disfunción mitocondrial, y el estrés oxidativo crónico se han relacionada con la deficiencia de tiamina y la patogénesis y la progresión de EA (9, 59). Actualmente, sólo hay evidencia escasa e inconsistente de que los suplementos con tiamina sean beneficiosos en la EA. Un estudio de doble ciego controlado con placebo de 15 pacientes (10 completaron el estudio) no informó de ningún efecto benéfico de 3 gramos de tiamina al día sobre el deterioro cognitivo en un periodo de 12 meses (60). Un reporte preliminar de otro estudio informó de un leve beneficio de 3 a 8 gramos de tiamina por día en DTA, sin embargo no hay información adicional disponible sobre dicho estudio (61). Se reportó un leve efecto benéfico en pacientes con EA luego de 12 semanas de tratamiento con 100 mg/día de un derivado de la tiamina (disulfuro tetrahidrofurfuril tiamina), sin embargo, este estudio no fue controlado con placebo (62). Una revisión sistemática de ensayos aleatorizados, de doble ciego y controlados con placebo de tiamina en pacientes con DTA, no encontró evidencia de que la tiamina fuera un tratamiento útil para los síntomas de la enfermedad de Alzheimer (63).
Insuficiencia Cardiaca Congestiva (ICC)
La deficiencia severa de tiamina (beriberi húmedo) puede derivar en función cardiaca deteriorada y, en última instancia, en insuficiencia cardiaca congestiva (ICC). Aunque las manifestaciones cardiacas del beriberi se observan muy rara vez en países industrializados, la ICC debida a otras causas es común, especialmente entre los ancianos. Los diuréticos utilizados en el tratamiento de la ICC, en particular la furosemida (Lasix), incrementan la excreción de tiamina, conduciendo potencialmente a una deficiencia marginal de tiamina (64). Una serie de estudios han examinado el estado nutricional de la tiamina en pacientes con ICC y la mayoría descubrió una incidencia bastante baja de deficiencia de tiamina, medida a través de ensayos de actividad transcetolasa. Al igual que en la población general, se descubrió que los paciente con ICC más viejos están en un riesgo más alto de sufrir una deficiencia de tiamina que los más jóvenes (65). La fracción de eyección del ventrículo izquierdo (FEVI) es una medida importante de la función cardiaca en ICC, la que puede ser evaluada por medio de un ecocardiograma. Un estudio en 25 pacientes descubrió que el uso de furosemida, en dosis de 80 mg/día o más altas, se asociaba con un 98% de prevalencia de la deficiencia de tiamina (23). En un estudio aleatorizado y de doble ciego, de 30 pacientes con ICC, de los cuales todos habían estado tomando furosemida (80 mg/día) por al menos tres meses, la terapia con tiamina intravenosa (IV) (200 mg/día) por siete días dio como resultado una mejora en la FEVI comparado con la terapia de placebo IV (66). Cuando cada uno de los 30 pacientes con ICC en ese estudio recibió subsecuentemente seis semanas de terapia con tiamina oral (200 mg/día), el promedio de la FEVI mejoró en 22%. Este hallazgo podría ser relevante debido a que se han asociado las mejoras en la FEVI con una mejor supervivencia en pacientes con ICC (67). Sin embargo, las conclusiones de los estudios publicados hasta la fecha son limitadas, debido al pequeño tamaño de los grupos de muestra de estos estudios, a la falta de aleatorización en algunos de ellos, y a la necesidad de ensayos más precisos del estado nutricional de la tiamina. En la actualidad, la necesidad de la suplementación con tiamina en la mantención de la función cardiaca en pacientes con ICC.
Cáncer
Se ha visto deficiencia de tiamina en algunos pacientes con cancer con tumores de crecimiento rápido. La investigación en células de cultivo y en modelos animales señala que las células cancerígenas que se dividen rápidamente tienen un requerimiento de tiamina elevado (68). Todas las células que se dividen rápidamente necesitan ácidos nucleicos a un ritmo aumentado, y algunas células cancerígenas parecen depender fuertemente en la enzima dependiente de TPP, transcetolasa, para proveer la ribosa-5-fosfato necesaria para la síntesis de ácidos nucleicos. Un reciente estudio encontró que los niveles de THTR-1 transcetolasa, y TPP transportadores mitocondriales se aumentos en muestras de tejido humano con cáncer de mama a comparación de tejido normal, sugiriendo, una adaptación en la homeostasis de la tiamina en apoyo del metabolismo del cáncer (69). La suplementación con tiamina es común en pacientes con cáncer para prevenir una deficiencia de tiamina, pero Boros et al. advierten que demasiada tiamina podría de hecho estimular el crecimiento de algunos tumores malignos (70), sugiriendo que la suplementación con tiamina se reserve para aquellos pacientes con cáncer que sean efectivamente deficientes de tiamina. En la actualidad no existe evidencia disponible de estudios en humanos que apoye o refute esta teoría. Sin embargo, sería prudente que las personas con cáncer que estén considerando la suplementación con tiamina lo discutieran con el médico a cargo de su terapia contra el cáncer.
Enfermedades metabólicas
La suplementación de tiamina está incluida en el manejo clínico de las enfermedades dietéticas que afectan el metabolismo de carbohidratos y aminoácidos de cadena ramificada (BCAAs).
Deficiencia del complejo piruvato deshidrogenasa de tiamina sensible
Las mutaciones en PDHC previenen la oxidación eficiente de carbohidratos en individuos afectados. Deficiencia de PDHC es caracterizada comúnmente por acidosis láctica, la degeneración neurológica y muscular, y muerte en la infancia. Los pacientes que responden al tratamiento de la tiamina (de pocos mg/día a dosis superiores a 1,000 mg/día) presentan deficiencia de PDHC debido a la disminución de la afinidad de PDHC de TPP (71, 72). Aunque la complementación de vitamina puede reducir la acumulación de lactato y mejorar las características clínicas en pacientes que respondan a la tiamina, esto no constituye una cura (73).
Enfermedad de Orina de Jarabe de Arce
Errores innatos de BCAA metabolismo conducen a la tiamina sensible ketoaciduria de cadena ramificada, también conocida como enfermedad de orina olor a jarabe de Arce. Las alteraciones en la BCAA vía catabólica resultado de la disfunción por la acumulación de BCAAs y sus derivados, ketoacidos de cadena ramificada. El enfoque terapéutico incluye una dienta con una contenido reducido de BCAA y tiamina (10-1,000 mg/día) se complementa a los pacientes con mutaciones en la subunidad E2 del complejo BCKDH (74). En individuos sensibles a la tiamina, la complementación ha demostrado ser eficaz para corregir el fenotipo sin recurrir a la BCAA dieta restringida.
Anemia megaloblastica sensible a la tiamina
Mutaciones en THTR-1 que deterioran la absorción de la tiamina intestinal y causan deficiencia de tiamina se ha encontrado en pacientes con anemia megaloblastica sensibles a la tiamina. Este síndrome es caracterizado por anemia megaloblastica, diabetes mellitus, y la sordera. Una revisión de 30 casos reporto neurológicos adicionales, visual, y deterioro cardiaco (75). Dosis orales de tiamina (hasta 300 mg/día) mantiene la salud y corrige la hiperglicemia en niños pre-púberes. Sin embargo, después de la pubertad, a un deterioro en la función del páncreas resulta en requisito de la insulina junto con tiamina para controlar la hiperglicemia. Un estudio también reporto que el tratamiento de una niña de cuatro meses de edad, con 100 mg/dia de tiamina no evito la pérdida de audición a los 20 meses de edad (76).
Enfermedad de los ganglios basales de biotina sensible
Enfermedad de los ganglios basales de biotina sensible, también nombrada síndrome-2 de disfunción de tiamina, es causada por mutaciones en el código genético para THTR-2. Las características clínicas aparecen alrededor de tres y cuatro años de edad e incluye encefalopatía subaguda (confusión, somnolencia, alteración del nivel de conciencia) ataxia y convulsiones. Un estudio retrospectivo de 18 individuos afectados de la misma familia o de la misma tribu en Arabia Saudita fue conducido reciente. Los datos mostrados que la monoterapia de biotina (5-10 mg/kg/día) abolió efectivamente las manifestaciones clínicas de la enfermedad, aunque un tercio de las pacientes sufrieron de crisis agudas recurrentes. A menudo se asocia con malos resultados, crisis agudas no fueron observados después de que la complementación de tiamina comenzó 300-400 mg/día) y durante un periodo de cinco años. El tratamiento precoz y tratamiento inmediato con biotina y tiamina conducen a resultados positivos (77).
Fuentes
Los seres humanos obtienen tiamina a partir de fuentes dietéticas y de la microflora del colon, aunque la contribución de este último no es claro con respecto de la necesidad de tiamina del cuerpo (78).
Fuentes alimenticias
Una dieta variada debería aportar a la mayoría de las personas la tiamina suficiente para prevenir su deficiencia. En los EE.UU. la ingesta recomendada promedio de tiamina para hombres adultos jóvenes es cercana a los 2 mg/día, y 1.2 mg/día para mujeres adultas jóvenes. Una encuesta en personas sobre los 60 años de edad se encontró con una ingesta recomendada promedio de tiamina de 1.4 mg/día para hombres y 1.1 mg/día para mujeres (29). Sin embargo, tanto la institucionalización como la pobreza aumentan la probabilidad de una ingesta de tiamina insuficiente en los adultos mayores (79). Los cereales de grano entero, las legumbres (por ejemplo, porotos y lentejas), los frutos secos, el cerdo magro, y la levadura son fuentes ricas en tiamina (1). Debido a que la mayoría de la tiamina se pierde durante la producción de la harina blanca y del arroz blanqueado y pulido, el arroz blanco y los alimentos hechos a base harina blanca (por ejemplo, pan y tallarines) se fortifican con tiamina en mucho países occidentales. La siguiente tabla muestra una serie de alimentos ricos en tiamina junto a su contenido en miligramos (mg). Para mayor información sobre el contenido de nutrientes de los alimentos, consulte la base de datos de composición de los alimentos del USDA (80).
| Alimento | Porción | Tiamina (mg) |
|---|---|---|
| Lentejas (cocidas, hervidas) | ½ taza | 0.17 |
| Arvejas (cocidas, hervidas y escurridas) | ½ taza | 0.21 |
| Arroz integral grano largo (cocido) | 1 taza | 0.19 |
| Arroz blanco grano largo, enriquecido (cocido) | 1 taza | 0.26 |
| Arroz blanco grano largo, no enriquecido (cocido) | 1 taza | 0.04 |
| Pan de trigo entero | 1 rebanada | 0.10 |
| Pan blanco, enriquecido | 1 rebanada | 0.23 |
| Cereal de desayuno fortificado | 1 taza | 0.31 |
| Cereal de desayuno de germen de trigo (tostados claro) | 1 taza | 1.88 |
| Cerdo, magro (lomo, filete, cocido, rostisado) | 3 onzas* | 0.81 |
| Pacanas | 1 onza | 0.19 |
| Espinaca (cocida, hervidas, escurridas) | ½ taza | 0.09 |
| Naranja | 1 fruta | 0.11 |
| Melón calameño (cantalupo) | ½ fruta | 0.11 |
| Leche | 1 taza | 0.10 |
| Huevo (cocido) | 1 grande | 0.03 |
| *Una porción de tres onzas de carnes es del tamaño de una baraja de cartas | ||
Suplementos
La tiamina se encuentra disponible en suplementos nutricionales, y, para la fortificación, como clorhidrato de tiamina y nitrato de tiamina (81).
Seguridad
Toxicidad
La Junta de Nutrición y Alimentos no estableció un nivel máximo de ingesta de ingesta tolerable (NM) para la tiamina debido a que no existen efectos tóxicos bien establecidos producto del excesivo consumo de tiamina a través de los alimentos o a través de la suplementación oral a largo plazo (hasta 200 mg/día). Se ha visto un número pequeño de reacciones anafilácticas con riesgo vital con grandes dosis intravenosas de tiamina (29).
Interacción con drogas
Se han reportado niveles sanguíneos de tiamina reducidos en individuos con trastornos convulsivos (epilepsia) tomando el medicamento anticonvulsivo, fenitoina, por periodos largos de tiempo (82). El 5-Fluorouracil, un medicamento utilizado en la terapia contra el cáncer, inhibe la fosforilación de la tiamina a TPP (83). Los diuréticos, especialmente la furosemida (Lasix), podría incrementar el riesgo de una deficiencia de tiamina en personas con ingestas marginales de tiamina debido a una excreción urinaria de tiamina aumentada (21). Más aún, se asocia al abuso crónico del alcohol con deficiencia de tiamina debido a una baja ingesta recomendada, absorción y utilización deterioradas, y excreción incrementada de esta vitamina (1). La alimentación crónica de alcohol a ratas mostro una disminución en la absorción active de tiamina relacionada con la inhibición de los transportadores de membranas de tiamina THTR-1 en el epitelio intestinal (84). El consume de alcohol en ratas también disminuyo los nivels de THTR-1 y THTH-2 en las células epiteliales renales, limitando de este modo la reabsorción por los riñones (85).
Recomendación del Instituto Linus Pauling
El Instituto Linus Pauling apoya la recomendación de la Junta de Nutrición y Alimentos de 1.2 mg de tiamina al día para hombres y 1.1 mg/día para mujeres. Una dieta variada debería aportar tiamina suficiente a la mayoría de las personas. Siguiendo la recomendación del Instituto Linus Pauling de tomar diariamente un suplemento multivitamínico/mineral que contenga el 100% del Valor Diario (VD), se asegurará una ingesta de al menos 1.5 mg de tiamina al día.
Adultos mayores (>50 años)
Actualmente no existe evidencia de que el requerimiento de tiamina sea mayor en adultos mayores, pero algunos estudios han encontrado que la ingesta recomendada insuficiente y la insuficiencia de tiamina son más comunes en poblaciones ancianas (79). Por lo tanto, sería prudente para los adultos mayores tomar un suplemento multivitamínico/mineral, el que aportará, generalmente, al menos 1.5 mg/día de tiamina.
Autores y Críticos
Escrito en 2000 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Septiembre de 2002 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Junio de 2007 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Junio de 2013 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Junio de 2013 por:
Christopher Bates, D.Phil.
Científico Senior Honor
Formerly Head of Micronutrient Status Research
MRC Human Nutrition Research
Elsie Widdowson Laboratorio
Cambridge, UK
Traducido al Español en 2013 por:
Guillermo Sandoval, Facultad de Odontologia, Universidad de Chile;
Revisado y editado en Diciembre 2012 por:
Andrew F.G. Quest, Ph.D. y Lisette Leyton, Ph.D.,
Profesores Titulares del Instituto de Ciencias Biomédicas,
Facultad de Medicina, Universidad de Chile,
en el marco del proyecto Anillo #ACT1111, grupo NEMESIS.
La traducción de el MIC del Inglés al Español fue asegurado, en parte, por una subvención de Bayer Consumer Care AG, Basel, Switzerland.
Derechos de autoría 2000-2024 Instituto Linus Pauling
Referencias
1. Tanphaichitr V. Thiamin. In: Shils M, Olson JA, Shike M, Ross AC, eds. Modern Nutrition in Health and Disease. 9th ed. Baltimore: Williams & Wilkins; 1999:381-389.
2. Rindi G. Thiamin. In: Ziegler EE, Filer LJ, eds. Present Knowledge in Nutrition. 7th ed. Washington D.C.: ILSI Press; 1996:160-166.
3. Hutson SM, Sweatt AJ, Lanoue KF. Branched-chain [corrected] amino acid metabolism: implications for establishing safe intakes. J Nutr. 2005;135(6 Suppl):1557S-1564S. (PubMed)
4. Brody T. Nutritional Biochemistry. 2nd ed. San Diego: Academic Press; 1999.
5. Donnino M. Gastrointestinal beriberi: a previously unrecognized syndrome. Ann Intern Med. 2004;141(11):898-899. (PubMed)
6. McDowell L. Thiamin. In: Vitamins in Animal and Human Nutrition. 2nd ed. Ames: Iowa State University Press; 2000:265-310. Available at: http://www.ucv.ve/fileadmin/user_upload/facultad_agronomia/Producion_
Animal/Vitamins_in_Animal_and_Human_Nutrition.pdf
7. Yamasaki H, Tada H, Kawano S, Aonuma K. Reversible pulmonary hypertension, lactic acidosis, and rapidly evolving multiple organ failure as manifestations of shoshin beriberi. Circ J. 2010;74(9):1983-1985. (PubMed)
8. Doss A, Mahad D, Romanowski CA. Wernicke encephalopathy: unusual findings in nonalcoholic patients. J Comput Assist Tomogr. 2003;27(2):235-240. (PubMed)
9. Hazell AS, Faim S, Wertheimer G, Silva VR, Marques CS. The impact of oxidative stress in thiamine deficiency: a multifactorial targeting issue. Neurochem Int. 2013;62(5):796-802. (PubMed)
10. Saad L, Silva LF, Banzato CE, Dantas CR, Garcia C, Jr. Anorexia nervosa and Wernicke-Korsakoff syndrome: a case report. J Med Case Rep. 2010;4:217. (PubMed)
11. Becker DA, Balcer LJ, Galetta SL. The Neurological Complications of Nutritional Deficiency following Bariatric Surgery. J Obes. 2012;2012:608534. (PubMed)
12. Jung ES, Kwon O, Lee SH, et al. Wernicke's encephalopathy in advanced gastric cancer. Cancer Res Treat. 2010;42(2):77-81. (PubMed)
13. Greenspon J, Perrone EE, Alaish SM. Shoshin beriberi mimicking central line sepsis in a child with short bowel syndrome. World journal of pediatrics : World J Pediatr. 2010;6(4):366-368. (PubMed)
14. Sequeira Lopes da Silva JT, Almaraz Velarde R, Olgado Ferrero F, et al. Wernicke's encephalopathy induced by total parental nutrition. Nutr Hosp. 2010;25(6):1034-1036. (PubMed)
15. Francini-Pesenti F, Brocadello F, Manara R, Santelli L, Laroni A, Caregaro L. Wernicke's syndrome during parenteral feeding: not an unusual complication. Nutrition. 2009;25(2):142-146. (PubMed)
16. Krishna S, Taylor AM, Supanaranond W, et al. Thiamine deficiency and malaria in adults from southeast Asia. Lancet. 1999;353(9152):546-549. (PubMed)
17. Mayxay M, Taylor AM, Khanthavong M, et al. Thiamin deficiency and uncomplicated falciparum malaria in Laos. Trop Med Int Health. 2007;12(3):363-369. (PubMed)
18. Muri RM, Von Overbeck J, Furrer J, Ballmer PE. Thiamin deficiency in HIV-positive patients: evaluation by erythrocyte transketolase activity and thiamin pyrophosphate effect. Clin Nutr. 1999;18(6):375-378. (PubMed)
19. Stanga Z, Brunner A, Leuenberger M, et al. Nutrition in clinical practice-the refeeding syndrome: illustrative cases and guidelines for prevention and treatment. Eur J Clin Nutr. 2008;62(6):687-694. (PubMed)
20. Suter PM, Haller J, Hany A, Vetter W. Diuretic use: a risk for subclinical thiamine deficiency in elderly patients. J Nutr Health Aging. 2000;4(2):69-71. (PubMed)
21. Rieck J, Halkin H, Almog S, et al. Urinary loss of thiamine is increased by low doses of furosemide in healthy volunteers. J Lab Clin Med. 1999;134(3):238-243. (PubMed)
22. Sica DA. Loop diuretic therapy, thiamine balance, and heart failure. Congestive heart failure 2007;13(4):244-247. (PubMed)
23. Zenuk C, Healey J, Donnelly J, Vaillancourt R, Almalki Y, Smith S. Thiamine deficiency in congestive heart failure patients receiving long term furosemide therapy. Can J Clin Pharmacol. 2003;10(4):184-8. (PubMed)
24. Hung SC, Hung SH, Tarng DC, Yang WC, Chen TW, Huang TP. Thiamine deficiency and unexplained encephalopathy in hemodialysis and peritoneal dialysis patients. Am J Kidney Dis. 2001;38(5):941-947. (PubMed)
25. Wilcox CS. Do diuretics cause thiamine deficiency? J Lab Clin Med. 1999;134(3):192-193.
26. Vimokesant SL, Hilker DM, Nakornchai S, Rungruangsak K, Dhanamitta S. Effects of betel nut and fermented fish on the thiamin status of northeastern Thais. Am J Clin Nutr. 1975;28(12):1458-1463. (PubMed)
27. Ventura A, Mafe MC, Bourguet M, Tornero C. Wernicke's encephalopathy secondary to hyperthyroidism and ingestion of thiaminase-rich products. Neurologia. 2013;28(4):257-259. (PubMed)
28. Nishimune T, Watanabe Y, Okazaki H, Akai H. Thiamin is decomposed due to Anaphe spp. entomophagy in seasonal ataxia patients in Nigeria. J Nutr. 2000;130(6):1625-1628. (PubMed)
29. Food and Nutrition Board, Institute of Medicine. Thiamin. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B6, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington D.C.: National Academy Press; 1998:58-86. (National Academy Press)
30. Cumming RG, Mitchell P, Smith W. Diet and cataract: the Blue Mountains Eye Study. Ophthalmology. 2000;107(3):450-456. (PubMed)
31. Jacques PF, Taylor A, Moeller S, et al. Long-term nutrient intake and 5-year change in nuclear lens opacities. Arch Ophthalmol. 2005;123(4):517-526. (PubMed)
32. Thornalley PJ, Babaei-Jadidi R, Al Ali H, et al. High prevalence of low plasma thiamine concentration in diabetes linked to a marker of vascular disease. Diabetologia. 2007;50(10):2164-2170. (PubMed)
33. Larkin JR, Zhang F, Godfrey L, et al. Glucose-induced down regulation of thiamine transporters in the kidney proximal tubular epithelium produces thiamine insufficiency in diabetes. PLoS One. 2012;7:e53175. (PubMed)
34. Rathanaswami P, Sundaresan R. Effects of thiamine deficiency on the biosynthesis of insulin in rats. Biochem Int. 1991;24(6):1057-1062. (PubMed)
35. Rathanaswami P, Pourany A, Sundaresan R. Effects of thiamine deficiency on the secretion of insulin and the metabolism of glucose in isolated rat pancreatic islets. Biochem Int. 1991;25(3):577-583. (PubMed)
36. Alaei Shahmiri F, Soares MJ, Zhao Y, Sherriff J. High-dose thiamine supplementation improves glucose tolerance in hyperglycemic individuals: a randomized, double-blind cross-over trial. Eur J Clin Nutr. 2013. May 29 [Epub ahead of print] (PubMed)
37. Gonzalez-Ortiz M, Martinez-Abundis E, Robles-Cervantes JA, Ramirez-Ramirez V, Ramos-Zavala MG. Effect of thiamine administration on metabolic profile, cytokines and inflammatory markers in drug-naive patients with type 2 diabetes. Eur J Clin Nutr. 2011;50(2):145-149. (PubMed)
38. Tepper OM, Galiano RD, Capla JM, et al. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106(22):2781-2786. (PubMed)
39. Wong CY, Qiuwaxi J, Chen H, et al. Daily intake of thiamine correlates with the circulating level of endothelial progenitor cells and the endothelial function in patients with type II diabetes. Mol Nutr Food Res. 2008;52(12):1421-1427. (PubMed)
40. Rabbani N, Alam SS, Riaz S, et al. High-dose thiamine therapy for patients with type 2 diabetes and microalbuminuria: a randomised, double-blind placebo-controlled pilot study. Diabetologia. 2009;52(2):208-212. (PubMed)
41. Babaei-Jadidi R, Karachalias N, Ahmed N, Battah S, Thornalley PJ. Prevention of incipient diabetic nephropathy by high-dose thiamine and benfotiamine. Diabetes. 2003;52(8):2110-2120. (PubMed)
42. Hammes HP, Du X, Edelstein D, et al. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nature Med. 2003;9(3):294-299. (PubMed)
43. Varkonyi T, Kempler P. Diabetic neuropathy: new strategies for treatment. Diabetes Obes Metab. 2008;10(2):99-108. (PubMed)
44. Kohda Y, Shirakawa H, Yamane K, et al. Prevention of incipient diabetic cardiomyopathy by high-dose thiamine. J Toxicol Sci. 2008;33(4):459-472. (PubMed)
45. Lee DC, Chu J, Satz W, Silbergleit R. Low plasma thiamine levels in elder patients admitted through the emergency department. Acad Emerg Med. 2000;7(10):1156-1159. (PubMed)
46. Ito Y, Yamanaka K, Susaki H, Igata A. A cross-investigation between thiamin deficiency and the physical condition of elderly people who require nursing care. J Nutr Sci Vitaminol. 2012;58(3):210-216. (PubMed)
47. Prvulovic D, Hampel H. Amyloid beta(A-beta) and phospho-tau (p-tau) as diagnostic biomarkers in Alzheimer's disease. Clin Chem Lab Med. 2011;49:367-374. (PubMed)
48. Kish SJ. Brain energy metabolizing enzymes in Alzheimer's disease: alpha-ketoglutarate dehydrogenase complex and cytochrome oxidase. Ann N Y Acad Sci. 1997;826:218-228. (PubMed)
49. Langbaum JB, Chen K, Lee W, et al. Categorical and correlational analyses of baseline fluorodeoxyglucose positron emission tomography images from the Alzheimer's Disease Neuroimaging Initiative (ADNI). NeuroImage. 2009;45(4):1107-1116. (PubMed)
50. Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 2004;61(5):661-666. (PubMed)
51. Gibson GE, Hirsch JA, Cirio RT, Jordan BD, Fonzetti P, Elder J. Abnormal thiamine-dependent processes in Alzheimer's Disease. Lessons from diabetes. Mol Cell Neurosci. 2013;55:17-25. (PubMed)
52. Glaso M, Nordbo G, Diep L, Bohmer T. Reduced concentrations of several vitamins in normal weight patients with late-onset dementia of the Alzheimer type without vascular disease. J Nutr Health Aging. 2004;8(5):407-413. (PubMed)
53. Bender DA. Optimum nutrition: thiamin, biotin and pantothenate. Proc Nutr Soc. 1999;58(2):427-433. (PubMed)
54. Mastrogiacoma F, Bettendorff L, Grisar T, Kish SJ. Brain thiamine, its phosphate esters, and its metabolizing enzymes in Alzheimer's disease. Ann Neurol. 1996;39(5):585-591. (PubMed)
55. Heroux M, Raghavendra Rao VL, Lavoie J, Richardson JS, Butterworth RF. Alterations of thiamine phosphorylation and of thiamine-dependent enzymes in Alzheimer's disease. Metab Brain Dis. 1996;11(1):81-88. (PubMed)
56. Pan X, Gong N, Zhao J, et al. Powerful beneficial effects of benfotiamine on cognitive impairment and beta-amyloid deposition in amyloid precursor protein/presenilin-1 transgenic mice. Brain. 2010;133(Pt 5):1342-1351. (PubMed)
57. Karuppagounder SS, Xu H, Shi Q, et al. Thiamine deficiency induces oxidative stress and exacerbates the plaque pathology in Alzheimer's mouse model. Neurobiol Aging. 2009;30(10):1587-1600. (PubMed)
58. Zhang Q, Yang G, Li W, et al. Thiamine deficiency increases beta-secretase activity and accumulation of beta-amyloid peptides. Neurobiol Aging. 2011;32(1):42-53. (PubMed)
59. Dumont M, Beal MF. Neuroprotective strategies involving ROS in Alzheimer disease. Free Rad Res Med. 2011;51(5):1014-1026. (PubMed)
60. Nolan KA, Black RS, Sheu KF, Langberg J, Blass JP. A trial of thiamine in Alzheimer's disease. Arch Neurology. 1991;48(1):81-83. (PubMed)
61. Meador K, Loring D, Nichols M, et al. Preliminary findings of high-dose thiamine in dementia of Alzheimer's type. J Geriatr Psychiatry Neurol. 1993;6(4):222-229. (PubMed)
62. Mimori Y, Katsuoka H, Nakamura S. Thiamine therapy in Alzheimer's disease. Metab Brain Dis. 1996;11(1):89-94. (PubMed)
63. Rodriguez-Martin JL, Qizilbash N, Lopez-Arrieta JM. Thiamine for Alzheimer's disease (Cochrane Review). Cochrane Database Syst Rev. 2001;2:CD001498. (PubMed)
64. Hanninen SA, Darling PB, Sole MJ, Barr A, Keith ME. The prevalence of thiamin deficiency in hospitalized patients with congestive heart failure. J Am Coll Cardiol. 2006;47(2):354-361. (PubMed)
65. Wilkinson TJ, Hanger HC, George PM, Sainsbury R. Is thiamine deficiency in elderly people related to age or co-morbidity? Age Ageing. 2000;29(2):111-116. (PubMed)
66. Shimon I, Almog S, Vered Z, et al. Improved left ventricular function after thiamine supplementation in patients with congestive heart failure receiving long-term furosemide therapy. Am J Med. 1995;98(5):485-490. (PubMed)
67. Leslie D, Gheorghiade M. Is there a role for thiamine supplementation in the management of heart failure? Am Heart J. 1996;131(6):1248-1250.
68. Comin-Anduix B, Boren J, Martinez S, et al. The effect of thiamine supplementation on tumour proliferation. A metabolic control analysis study. Eur J Biochem. 2001;268(15):4177-4182. (PubMed)
69. Zastre JA, Hanberry BS, Sweet RL, et al. Up-regulation of vitamin B1 homeostasis genes in breast cancer. J Nutr Biochem. 2013. May 1 [Epub ahead of print] (PubMed)
70. Boros LG, Brandes JL, Lee WN, et al. Thiamine supplementation to cancer patients: a double edged sword. Anticancer Res. 1998;18(1B):595-602. (PubMed)
71. Naito E, Ito M, Yokota I, Saijo T, Ogawa Y, Kuroda Y. Diagnosis and molecular analysis of three male patients with thiamine-responsive pyruvate dehydrogenase complex deficiency. J Neurological Sci. 2002;201(1-2):33-37. (PubMed)
72. Patel KP, O'Brien TW, Subramony SH, Shuster J, Stacpoole PW. The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol Genet Metab. 2012;106(3):385-394. (PubMed)
73. Lee EH, Ahn MS, Hwang JS, Ryu KH, Kim SJ, Kim SH. A Korean female patient with thiamine-responsive pyruvate dehydrogenase complex deficiency due to a novel point mutation (Y161C)in the PDHA1 gene. J Korean Med Sci. 2006;21(5):800-804. (PubMed)
74. Chuang DT, Chuang JL, Wynn RM. Lessons from genetic disorders of branched-chain amino acid metabolism. J Nutr. 2006;136(1 Suppl):243S-249S. (PubMed)
75. Shaw-Smith C, Flanagan SE, Patch AM, et al. Recessive SLC19A2 mutations are a cause of neonatal diabetes mellitus in thiamine-responsive megaloblastic anaemia. Pediatr Diabetes. 2012;13(4):314-321. (PubMed)
76. Akin L, Kurtoglu S, Kendirci M, Akin MA, Karakukcu M. Does early treatment prevent deafness in thiamine-responsive megaloblastic anaemia syndrome? J Clin Res Pediatr Endocrinol. 2011;3(1):36-39. (PubMed)
77. Alfadhel M, Almuntashri M, Jadah RH, et al. Biotin-responsive basal ganglia disease should be renamed biotin-thiamine-responsive basal ganglia disease: a retrospective review of the clinical, radiological and molecular findings of 18 new cases. Orphanet J Rare Dis. 2013;8:83. (PubMed)
78. LeBlanc JG, Milani C, de Giori GS, Sesma F, van Sinderen D, Ventura M. Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr Opin Biotechnol. 2013;24(2):160-168. (PubMed)
79. Russell RM, Suter PM. Vitamin requirements of elderly people: an update. Am J Clin Nutr. 1993;58(1):4-14. (PubMed)
80. US Department of Agriculture, Agricultural Research Service. USDA National Nutrient Database for Standard Reference, Release 25. 2012. Available at: http://ndb.nal.usda.gov//. Accessed 6/21/13.
81. Thiamin (vitamin B1). In: Hendler S, Rorvik D, eds. PDR for Nutritional Supplements. 2nd ed. Montvale: Physicians' Desk Reference Inc.; 2008:609-615.
82. Flodin N. Pharmacology of micronutrients. New York: Alan R. Liss, Inc.; 1988.
83. Schumann K. Interactions between drugs and vitamins at advanced age. Int J Vitam Nutr Res. 1999;69(3):173-178. (PubMed)
84. Subramanya SB, Subramanian VS, Said HM. Chronic alcohol consumption and intestinal thiamin absorption: effects on physiological and molecular parameters of the uptake process. Am J Physiol Gastrointest Liver Physiol. 2010;299(1):G23-G31. (PubMed)
85. Subramanian VS, Subramanya SB, Tsukamoto H, Said HM. Effect of chronic alcohol feeding on physiological and molecular parameters of renal thiamin transport. Am J Physiol Renal Physiol. 2010;299(1):F28-F34. (PubMed)
Vitamina A
Contenido
Resumen
- La vitamina A es un término genérico que se refiere a compuestos liposolubles encontrados como vitamina A preformada (retinol) en productos de origen animal y como carotenoides provitamina A en frutas y vegetales. Las tres formas activas de la vitamina A en el cuerpo son el retinol, retinal, y ácido retinoico. (Más información)
- La vitamina A está involucrada en la regulación del crecimiento y especialización (diferenciación) de virtualmente todas las células del cuerpo humano. La vitamina A tiene papeles importantes en el desarrollo embriónico, la formación de órganos durante el desarrollo fetal, funciones inmunes normales, y el desarrollo de los ojos y la visión. (Más información)
- La deficiencia de vitamina A es una causa mayor de ceguera evitable en el mundo. Esta es más prevalente entre niños y mujeres en edad de procrear. La deficiencia de vitamina A esta asociada con una susceptibilidad incrementada a infecciones, así como a desordenes de la tiroides y de la piel. (Más información)
- La ingesta diaria recomendada (IDR) es de 700 microgramos de equivalentes de actividad de retinol (μg EAR)/día para mujeres y 900 μg de EAR/día para hombres. (Más información)
- La profilaxis de la vitamina A parece reducir significantemente la mortalidad infantil en regiones con alto riesgo de deficiencia de vitamina A. Además, una suplementación de altas-dosis de vitamina A es ampliamente recomendada para niños mayores de seis meses de edad cuando son infectados con sarampión mientras están desnutridos, inmunodeficientes, o en riesgo de complicaciones por el sarampión. (Más información)
- El ácido retinoico y sus análogos son usados en dosis farmacológicas en el tratamiento de la leucemia promielocítica aguda y varias enfermedades de la piel. (Más información)
- Fuentes animales ricas en vitamina A preformada incluyen productos lácteos, cereal fortificado, el hígado, y aceites de pescado. Fuentes ricas de carotenoides provitamina A incluyen los vegetales naranjas y verdes, como el camote y las espinacas. (Más información)
- El consumo excesivo de la vitamina A preformada puede ser altamente toxico y es especialmente contraindicado antes de y durante el embarazo debido a que puede resultar en defectos de nacimiento severos. El nivel máximo de ingesta tolerable (NM) para la vitamina A en adultos está establecida en 3,000 μg de EAR/día. El NM no se aplica a la vitamina A derivada de los carotenoides. (Más información)
Vitamina A es un término genérico que abarca un cierto número de compuestos relacionados (Figura 1). El retinol y los retinil ésteres son frecuentemente referidos como vitamina A preformada. El retinol puede ser convertido por el cuerpo a retinal, que puede a su vez ser oxidado a ácido retinoico, la forma de la vitamina A conocida por afectar la transcripción de genes. El retinol, retinal, ácido retinoico, y compuestos relacionados son conocidos como retinoides. El β-caroteno y otros carotenoides alimenticios que pueden ser convertidos por el cuerpo en retinol son referidos como carotenoides provitamina A (véase el artículo en Carotenoides). Cientos de carotenoides diferentes son sintetizados por las plantas, pero solo cerca de un 10% de ellos son capaces de ser convertidos a retinol (1). La siguiente discusión se enfocara principalmente en compuestos de vitamina A preformada y en el ácido retinoico.
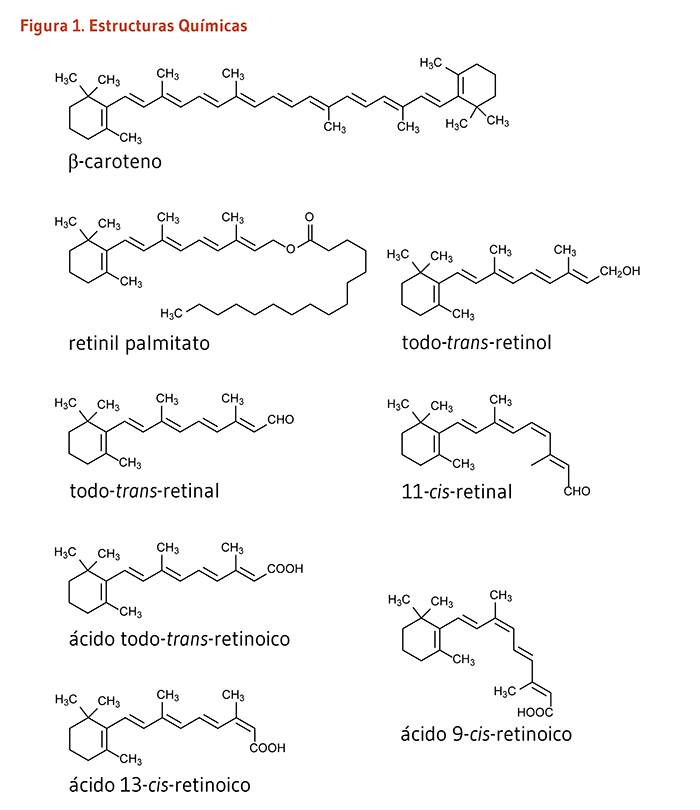
Función
Los compuestos de vitamina A son moléculas liposolubles esenciales predominantemente almacenadas en el hígado en la forma de retinil ésteres (p. ej. retinil palmitato). Cuando es apropiado, los retinil ésteres son hidrolizados para generar todo-trans-retinol, el cual se une a la proteína de unión al retinol (RBP) antes de ser liberado en el torrente sanguíneo. El complejo todo-trans-retinol/RBP circula unido a la proteína, transtiretina, la cual entrega el todo-trans-retinol a los tejidos periféricos (revisado en 2). Se encontró también que la vitamina A como retinil ésteres en quilomicrones tiene un papel apreciable en la entrega de vitamina A a tejidos extrahepáticos, especialmente en los primeros años de vida (3, 4).
Sistema visual y la vista
Localizada en la parte posterior del ojo, la retina contiene dos tipos principales de células receptoras sensibles a la luz — conocidas como células fotorreceptoras de conos y bastones. Los fotones (partículas de luz) que pasan a través del cristalino son detectados por las células fotorreceptoras de la retina y convertidos a impulsos nerviosos (señales eléctricas) para la interpretación por el cerebro. El todo-trans-retinol es transportado a la retina a través de la circulación y acumulado en las células del epitelio pigmentario retinal (EPR) (Figura 2) (5). Aquí, el todo-trans-retinol es esterificado para formar un retinil éster, el cual puede ser almacenado. Cuando es necesario, los retinil esteres son descompuestos (hidrolizados) e isomerizados para formar 11-cis-retinol, el cual puede ser oxidado para formar 11-cis-retinal. El 11-cis retinal puede ser transportado a través del espacio interfotoreceptor hacia la célula fotorreceptora bastón que está especializada para la visión en condiciones de escasa luz y para la detección de movimiento. En las células bastones, el 11-cis-retinal se une a una proteína llamada opsina para formar el pigmento visual, rodopsina (también conocido como púrpura visual). La absorción de un fotón de luz cataliza la isomerización del 11-cis-retinal a todo-trans-retinal que es liberado de la molécula de la opsina. Esta fotoisomerización desencadena una cascada de eventos, que conducen a la generación de un impulso nervioso transmitido por el nervio óptico hacia la corteza visual del cerebro. El todo-trans-retinal es convertido a todo-trans-retinol y transportado a través del espacio intersticial hacia las células del EPR, completando así el ciclo visual.
Un ciclo similar ocurre en las células cono que contienen proteínas opsina rojas, verdes, y azules requeridas para la absorción de fotones provenientes del espectro de luz visible (2). La vitamina A es también esencial para el desarrollo del ojo mamífero (6). Por lo tanto, dado que la vitamina A es requerida para el funcionamiento normal de la retina, la visión en luz tenue, y la visión a color, el retinol inadecuado y la disponibilidad de retinal para la retina resulta en una adaptación a la oscuridad dañada. En los casos más severos de deficiencia de vitamina A, el adelgazamiento y ulceración de la córnea conducen a la ceguera (véase Deficiencia).
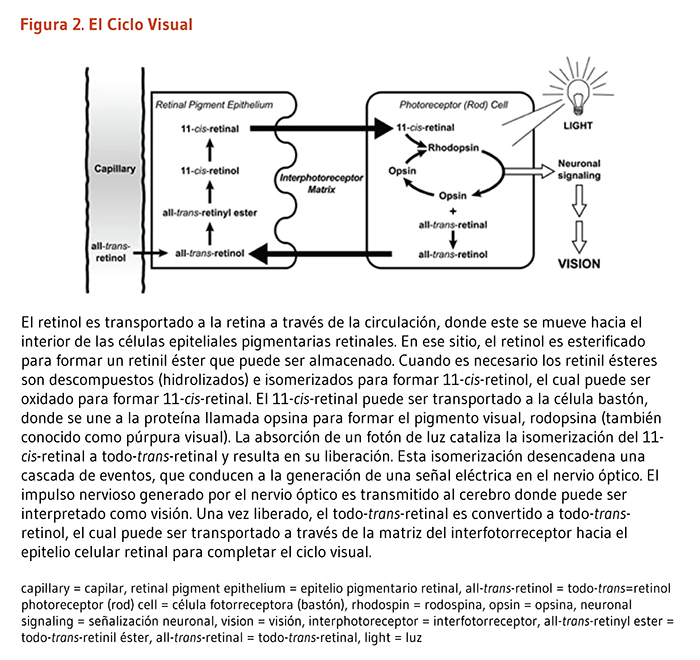
Regulación de la expresión de genes
Capacidad regulatoria del ácido retinoico
En las células, el todo-trans-retinol puede ser tanto almacenado (en la forma de éster retinil) como oxidado a todo-trans-retinal por las alcohol deshidrogenasas. En turno, las retinaldehído deshidrogenasas pueden catalizar la conversión de todo-trans-retinal en dos isómeros del ácido retinoico (AR) biológicamente activos: el ácido todo-trans-retinoico y el ácido 9-cis-retinoico. Los isómeros del AR actúan como hormonas para afectar la expresión de genes y por lo tanto influir en numerosos procesos fisiológicos. El ácido todo-trans-retinoico y el ácido 9-cis-retinoico son transportados hacia el núcleo de la célula unidos a las proteínas celulares de unión al ácido retinoico (CRABP). Dentro del núcleo, los isómeros del AR se unen a proteínas de receptores nucleares específicas que son factores de transcripción dependientes de ligandos. Tanto el ácido todo-trans-retinoico como el ácido 9-cis-retinoico pueden unirse a los receptores de ácido retinoico (RARα, RARβ, y RARγ), mientras que solo el ácido 9-cis-retinoico se use a los receptores X retinoides (RXRα, RXRβ, y RXRβ) (7). Los subtipos RAR y RXR forman tanto complejos de dos de la misma proteína (RAR/RAR y RXR/RXR homodímeros) o complejos de dos proteínas diferentes (RAR/RXR heterodímeros). Los heterodímeros RAR/RXR pueden unirse a la secuencia de ADN regulatoria llamada elemento de respuesta al ácido retinoico (RARE) localizada dentro del promotor de genes retinoide-sensibles. La actividad transcripcional de los heterodímeros RAR/RXR parece estar basada principalmente por la unión del ácido todo-trans-retinoico al RAR.
La activación del RAR por la unión del AR desencadena el reclutamiento de correguladores transcripcionales que apuntan a los promotores, de este modo inhibiendo o permitiendo la transcripción de genes (8). El RXR también forma heterodímeros con otros varios receptores nucleares, incluyendo el receptor de hormona tiroidea (TR), el receptor de vitamina D (RVD), receptores de esteroides, y el receptor activado por el proliferador de peroxisomas (PPAR) (9). De esta manera, la vitamina A pudiese interactuar con la hormona tiroidea, la vitamina D, los esteroides (p. ej., el estrógeno), o los ligandos del PPAR de las rutas de señalización e influenciar la transcripción de una amplia gama de genes.
Existe también evidencia de que el AR/RAR puede afectar la expresión de genes en una manera independiente del RARE. Por ejemplo, se reportó que el RAR pudo interferir con la ruta de señalización TGFβ/Smad a través de la interacción directa del RAR con el factor de transcripción heterodimérico, Smad3/Smad4. En la ausencia del AR, se encontró que el RAR actuó como un coactivador de la transcripción mediada por el Smad3/Smad4, mientras que los agonistas del RAR reprimieron la actividad transcripcional del Smad3/Smad4 (10). En las células del retinoblastoma, el RAR también estuvo involucrado en la activación inducida por el AR de las cascadas de señalización mediadas por las tirosina quinasas conocida como fosfoinositol 3-quinasa (PI3K) y condujo a la diferenciación celular (11, 12). El AR también pareció inducir la diferenciación neuronal mediante la activación de la ruta de señalización ERK1/2 MAP quinasa que fosforila el factor de transcripción, CREB (proteína de unión al elemento de respuesta al AMP cíclico). CREB fosforilado puede subsecuentemente unirse al elemento de respuesta del CREB en el promotor de los genes involucrados en la diferenciación celular (13). También, independientemente del RAR, se encontró que el AR inhibe la fosforilación/activación de las ERK1/2 y la subsecuente expresión mediada por la AP1 de la interleucina-6 en las células sinoviales (14). Por lo tanto el AR puede influenciar la expresión de genes cuyos promotores no contienen RARE.
Mediante la regulación de la expresión de más de 500 genes retinoide-sensibles (incluyendo varios genes involucrados en el metabolismo de la vitamina A en sí), los isómeros de ácido retinoico desempeñan papeles principales en la proliferación y diferenciación (es decir, el compromiso celular para funciones altamente especializadas).
Capacidad regulatoria del retinol
En el ojo y los tejidos como el adiposo blanco y el musculo, el receptor/transportador STRA6 de la membrana plasmática del retinol acepta el retinol proveniente de la RBP extracelular y lo descarga a la proteína de unión al retinol intracelular (CRBP). STRA6 también coopera con la lecitina:retinol aciltransferasa (LRAT), una enzima que cataliza la esterificación y almacenaje de retinol, para mantener un gradiente de concentración interno del retinol (15). Curiosamente, se encontró que la captación de retinol por el STRA6 desencadena la activación de la cascada de señalización mediada por las tirosina quinasas conocidas como quinasas Janus (JAK) y factores de transcripción asociados (STAT). La vía de señalización JAK/STAT regula la expresión de un amplio rango de citoquinas, hormonas, y factores de crecimiento (16). Estudios en animales han reportado que un incremento en la expresión de genes, como el SOCS3 por la vía JAK/STAT, pudo resultar en la inhibición del señalamiento de la insulina. Por lo tanto, ratones obesos faltos de LRAT o STRA6 parecen estar protegidos de la resistencia a la insulina inducida por el retinol/STRA6 (17, 18).
Capacidad regulatoria del retinal
Aparte de su papel como un ligando para la opsina en la cascada visual (véase Sistema visual y la vista), el retinal ha sido específicamente implicado en la regulación de genes importantes para el metabolismo de los lípidos.
En los humanos, dos tipos de tejido adiposo han sido distinguidos basados en sus respectivas funciones: el tejido adiposo blanco (WUT) almacena ácidos grasos como triglicéridos, y el tejido adiposo café (BAT) oxida ácidos grasos para generar calor (termogénesis). En la cadena transportadora de electrones de las células del tejido adiposo café, los procesos de transporte de electrones y la producción de ATP son desacoplados (disociados) para permitir la producción rápida de calor a partir de la oxidación de ácidos grasos (19).
La retinaldehído deshidrogenasa 1 (RALDH1), la cual convierte el retinal a ácido retinoico, es altamente expresada en el WAT pero no en el BAT. La supresión de la expresión de RALDH1 en el WAT puede inducir un fenotipo termogénico parecido al del BAT (20). Durante la diferenciación adipocitaria, se ha encontrado que la estimulación de células con todo-trans retinal activa el gen UCP1 requerido por la termogénesis mientras que inhibe genes que promueven la adipogénesis, como el PPARγ (20). También el retinal parece regular el metabolismo de los lípidos y la adiposidad en la médula ósea mediante la inhibición de la expresión de genes mediada por el heterodímero PPARγ/RXR (21). Además, se encontró que el retinal inhibe la expresión del gen gluconeogénico y la producción de glucosa en el hígado de ratones deficientes en RALDH1 (22).
Inmunidad
La vitamina A fue inicialmente acuñada como "la vitamina anti-infectiva" debido a su importancia en el funcionamiento normal del sistema inmune (23). Las células de la piel y de las mucosas, que recubren las vías respiratorias, el tracto digestivo, y el tracto urinario funcionan como una barrera y forman la primera línea de defensa del cuerpo contra alguna infección. El ácido retinoico (AR) es producido por las células presentadoras de antígeno (CPA), incluyendo macrófagos y células dendríticas, encontrados en estas interfaces mucosas y ganglios linfáticos asociados. El AR parece actuar en las propias células dendríticas para regular su diferenciación, migración y capacidad presentadora de antígenos. Además, la producción del AR por las CPA es requerida para la diferenciación de linfocitos-T CD4 naïve en los linfocitos-T reguladores inducidos (Tregs). Critica para el mantenimiento de la integridad de la mucosa, la diferenciación de Tregs es llevada a cabo por el ácido todo-trans-retinoico a través de la regulación de la expresión de genes mediada por el RARα (véase Regulación de la expresión de genes). También, durante la inflamación, la vía de señalización acido todo-trans-retinoico/RARα promueve la conversión de linfocitos-T CD4 naïve a linfocitos-T efectores — tipo 1 células-T colaboradoras (Th1) — (en lugar de Tregs) e induce la producción de citoquinas proinflamatorias por los linfocitos-T efectores en respuesta a la infección. Existe también evidencia substancial que sugiere que el AR pudiese ayudar a prevenir el desarrollo de la autoinmunidad (revisado en 24).
Desarrollo prenatal y postnatal
Ambos el exceso y la deficiencia de vitamina A son conocidos por causar defectos de nacimiento. La señalización retinoide comienza poco después de la fase temprana del desarrollo embriónico conocido como gastrulación. Durante el desarrollo fetal, el AR es crítico para el desarrollo de órganos, incluyendo el corazón, los ojos, oídos, pulmones, como también otros miembros y órganos viscerales. La vitamina A ha sido implicada en la maduración pulmonar fetal (2). El estatus de la vitamina A es más bajo en recién nacidos prematuros que en infantes nacidos a término (25). Existe algo de evidencia que sugiere que la suplementación con vitamina A puede ayudar a reducir la incidencia de enfermedad pulmonar crónica, y la mortalidad en recién nacidos prematuros (véase Prevención de Enfermedades). La señalización retinoide está también involucrada en la expresión de muchas proteínas de la matriz extracelular (MEC; material que rodea las células), incluyendo colágeno, laminina, y proteoglicanos (26). La deficiencia de vitamina A puede resultar en alteraciones de la composición de la MEC, interrumpiendo de esta manera la morfología y función de órganos (revisado en 26).
Producción de glóbulos rojos (eritropoyesis)
Los glóbulos rojos (eritrocitos), como todas las células sanguíneas, son derivadas de las células madre pluripotentes en la medula ósea. Estudios que involucraron sistemas de cultivo in vitro han sugerido un papel para los retinoides en el compromiso y diferenciación de las células madre al linaje de los glóbulos rojos. Los retinoides podrían también regular la apoptosis (muerte celular programada) de los precursores de los glóbulos rojos (células progenitoras eritropoyéticas) (27). Sin embargo, no ha sido establecido si los retinoides regulan la eritropoyesis in vivo. A pesar de todo, se ha demostrado que la suplementación con vitamina A en individuos deficientes de vitamina A incrementa las concentraciones de hemoglobina. Además, la vitamina A parece facilitar la movilización de hierro de sitios de depósito hacia los de desarrollo de glóbulos rojos para su incorporación a la hemoglobina, el transportador de oxígeno en los glóbulos rojos (27, 28).
Interacción con nutrientes
Zinc
Se piensa que la deficiencia de zinc interfiere con el metabolismo de la vitamina A de varias maneras (29): (1) la deficiencia de zinc resulta en una síntesis de la proteína de unión al retinol (RBP) disminuida, la cual transporta el retinol a través de la circulación hacia los tejidos periféricos y protege el organismo contra la toxicidad potencial del retinol; (2) la deficiencia de zinc resulta en la disminución de la actividad de la enzima que libera retinol de su forma de almacenamiento, retinil palmitato, en el hígado; y (3) el zinc es requerido para la enzima que convierte el retinol en retinal (30). Las consecuencias a la salud de la deficiencia de zinc en el estatus nutricional de la vitamina A en los humanos están aún por definirse (29).
Hierro
La deficiencia de vitamina A frecuentemente coexiste con la deficiencia de hierro y puede exacerbar la anemia por deficiencia de hierro al alterar el metabolismo del hierro (27). La suplementación con vitamina A tiene efectos beneficiales en la anemia por deficiencia de hierro y mejora el estatus nutricional del hierro entre niños y mujeres embarazadas (27, 28). La combinación de vitamina A y hierro suplementarios parece reducir la anemia más efectivamente que la suplementación con hierro o vitamina A solamente (31). Además, estudios en ratas han mostrado que la deficiencia de hierro altera los niveles de vitamina A del plasma y del hígado (32, 33).
Deficiencia
La deficiencia de vitamina A usualmente resulta de las ingestas inadecuadas de vitamina A proveniente de productos animales (como vitamina A preformada) y frutas y vegetales (como carotenoides provitamina A). En países en desarrollo, la deficiencia de vitamina A y trastornos asociados predominantemente afectan niños y mujeres en edad reproductiva. Otros individuos en riesgo de una deficiencia de vitamina A son aquellos con una pobre absorción de lípidos debido a una secreción pancreática o biliar alterada y aquellos con enfermedades inflamatorias intestinales, como la enfermedad de Crohn y la enfermedad celíaca (2). La deficiencia subclínica de vitamina A es frecuentemente definida por concentraciones de retinol en el suero menores de 0.70 mcmol/L (20 μg/dL). En la deficiencia severa de vitamina A, los depósitos de vitamina A del cuerpo se agotan y las concentraciones de retinol del suero caen por debajo de los 0.35 mcmol/L (10 μg/dL). Otros biomarcadores han sido calibrados para evaluar el estatus nutricional de la vitamina A (revisado en 34). Debe notarse que la Organización Mundial de la Salud considera la deficiencia de vitamina A un problema público de salud cuando la prevalencia de retinol del suero bajo (<0.70 mcmol/L) alcanza el 15% o más de una población definida.
Trastornos relacionados a la deficiencia de vitamina A
Enfermedad del ojo y ceguera
Con un estimado de entre 250,000 a 500,000 niños volviéndose ciegos anualmente, la deficiencia de vitamina A constituye la principal causa evitable de ceguera en naciones con bajos y medios ingresos (35). El síntoma más temprano de la deficiencia de vitamina A es la alteración de la adaptación a la oscuridad conocida como ceguera nocturna o nictalopía. La etapa clínica siguiente es la ocurrencia de cambios anormales en la conjuntiva (comisura del ojo), manifestada por la presencia de manchas de Bitot. La deficiencia prolongada o severa de vitamina A eventualmente resulta en una condición llamada xeroftalmia (ojo seco en griego), caracterizada por cambios en las células de la córnea (capa transparente del ojo) que en última instancia resulta en úlceras corneales, cicatrices y ceguera (36). La inmediata administración de 200,000 unidades internacionales (UI) de vitamina A por dos días consecutivos es requerida para prevenir la xeroftalmia cegadora (36).
Existe un estimado de 19.1 millones de mujeres embarazadas alrededor del mundo (especialmente en África Sub-sahariana, Sudeste de Asia, y América Central) con deficiencia de vitamina A y más de la mitad son afectadas por la ceguera nocturna (37). La prevalencia de la deficiencia de vitamina A y la ceguera nocturna es especialmente alta durante el tercer trimestre del embarazo debido al crecimiento fetal acelerado. También, aproximadamente 190 millones de niños en edad preescolar tienen concentración bajas de retinol en el suero (<0.70 mcmol/L), con 5.2 millones sufriendo de ceguera nocturna. Además, la mitad de los niños afectados por la xeroftalmia cegadora inducida por la deficiencia severa de vitamina A se estima mueren dentro del lapso de un año de volverse ciegos (37). La Organización Mundial de la Salud (OMS) y el Fondo de las Naciones Unidas para la Infancia (Unicef) promueven la suplementación con vitamina A como una intervención de salud pública para reducir la mortalidad infantil en áreas y poblaciones donde la deficiencia de vitamina A es prevalente (38-40).
Susceptibilidad a enfermedades infecciosas
Las enfermedades infecciosas han sido asociadas con el agotamiento de las reservas hepáticas de vitamina A (ya limitadas en sujetos deficientes de vitamina A), las concentraciones de retinol del suero reducidas, y un incremento en la perdida de vitamina A en la orina (37). Se encontró que la infección con el virus del sarampión precipita el daño conjuntivo y corneal, llevando a la ceguera en niños con un estatus pobre de vitamina A (41). Inversamente, la deficiencia de vitamina A puede ser considerada una enfermedad de inmunodeficiencia nutricionalmente adquirida (42). Incluso niños que están levemente deficientes de vitamina A tienen una incidencia más alta de complicaciones respiratorias y diarrea, como también una tasa mayor de mortalidad por infección de sarampión en comparación con niños que consumen la suficiente vitamina A (43). Debido a que la suplementación con vitamina A puede disminuir tanto la severidad como la incidencia de complicaciones por el sarampión en países en desarrollo (véase Prevención de Enfermedades), la OMS recomienda que los niños de por lo menos un años de edad reciban 200,000 UI de vitamina A (60 mg de EAR) por dos días consecutivos en adición al tratamiento estándar cuando son infectados con el virus de sarampión y viven en áreas de deficiencia de vitamina A (44).
Un reciente estudio de cohorte prospectivo, conducido en 2,774 niños colombianos (edades de entre 5-12 años) con un seguimiento medio de 128 días, también reporto una relación inversa entre las concentraciones de retinol del plasma y las tasas de diarrea con vómito y tos con fiebre, este último siendo un fuerte predictor de la infección por influenza (gripe) (45). Una revisión de cinco estudios aleatorios controlados con placebo que incluyeron 7,528 mujeres VIH positivas embarazadas o lactando no encontró algún beneficio substancial de la suplementación con vitamina A en la reducción de la transmisión del VIH de madre a hijo (46). Un estudio basado en la observación temprano encontró que las mujeres infectadas con VIH que eran deficientes de vitamina A fueron de tres a cuatro veces más propensas de transmitir el VIH a sus infantes (47). Sin embargo, ningún ensayo hasta la fecha ha proporcionado alguna información sobre los efectos adversos potenciales de la suplementación con vitamina A en la transmisión del VIH de madre a hijo (48).
Disfunción tiroidea
En el norte y oeste de África, la deficiencia de vitamina A y la deficiencia de yodo inducida por el bocio pueden coexistir hasta en un 50% de los niños. La respuesta a la profilaxis de yodo en las poblaciones deficientes de yodo parece depender de varios factores nutricionales, incluyendo el estatus de la vitamina A (49, 50). Se encontró que la deficiencia de vitamina A en modelos de animales interfiere con el eje hipófisis-tiroides al (1) incrementar la síntesis y secreción de la hormona estimulante de la tiroides (TSH) por la glándula pituitaria, (2) incrementar el tamaño de la glándula tiroides, (3) reducir la captación de yodo por la glándula tiroides y afectar la síntesis y la yodación de la tiroglobulina, y (4) incrementar las concentraciones circulantes de las hormonas tiroideas (revisado en 51). Un estudio de corte transversal de 138 niños con deficiencias actuales de vitamina A y yodo encontró que la severidad de la deficiencia de vitamina A estaba asociada con un riesgo mayor de bocio y concentraciones mayores de TSH y hormonas tiroideas circulantes (50). Estos niños recibieron sal enriquecida con yodo con vitamina A (200,000 UI al inicio del estudio y 5 meses) o placebo en un ensayo aleatorio, doble ciego, de 10 meses. Esta suplementación con vitamina A significantemente disminuyo la concentración de la TSH y el volumen tiroideo en comparación al placebo (50). En otro ensayo, la suplementación con vitamina A en niños deficientes de yodo no tuvo efecto adicional al yodo en el estatus de la tiroides en comparación al placebo, pero la suplementación con vitamina A sola (sin yodo) redujo el volumen de la glándula tiroides, como también las concentraciones de TSH y tiroglobulina (52).
Otros trastornos
La frinoderma o hiperqueratosis folicular es una condición de la piel caracterizada por una producción excesiva de queratina en los folículos pilosos. Las lesiones aparecen primero en las extremidades, hombros, y glúteos y pudiese extenderse sobre el cuerpo entero en los casos más severos (53). Mientras que la deficiencia de vitamina A pudiese contribuir a la ocurrencia de la frinoderma, la condición ha sido fuertemente asociada con múltiples deficiencias nutricionales y es considerada un signo de malnutrición general. Un raro caso de esofagitis (inflamación del esófago) ha sido recientemente atribuido a la hiperqueratosis secundaria a la deficiencia de vitamina A (54).
También, la deficiencia de vitamina A afecta la movilización de hierro, irrumpe la síntesis de hemoglobina, y precipita la anemia por deficiencia de hierro que es solo aliviada con la suplementación de ambos vitamina A y hierro (véase Interacción con nutrientes) (27).
La Ingesta Diaria Recomendada (IDR)
Equivalentes de Actividad de Retinol (EAR)
La vitamina A puede ser obtenida de los alimentos como vitamina A preformada en productos animales o como carotenoides provitamina A en frutas y vegetales (véase Fuentes alimenticias). A pesar de todo, mientras la vitamina A preformada es efectivamente absorbida, almacenada, e hidrolizada para formar retinol, los carotenoides provitamina A como el β-caroteno son menos fácilmente digeridos y absorbidos, y deben ser convertidos a retinol y otros retinoides por el cuerpo después de la captación en el intestino delgado. La eficacia de la conversión de carotenos provitamina A a retinol es altamente variable, dependiendo de factores como la matriz alimentaria, la preparación de los alimentos, y nuestras propias capacidades digestivas y de absorción (55).
El estándar internacional más reciente de medición de la vitamina A es los equivalentes de actividad de retinol (EAR), los cuales representan la actividad de la vitamina A como retinol. Se ha determinado que 2 microgramos (μg) de β-caroteno en aceite proporcionado como un suplemento podría ser convertido por el cuerpo a 1 μg de retinol dándole una proporción de EAR de 2:1. Sin embargo, 12 μg de β-caroteno proveniente de los alimentos son requeridos para proveer al cuerpo con 1 μg de retinol, dándole al β-caroteno dietario una proporción de EAR de 12:1. Otros carotenoides provitamina A en los alimentos son menos fáciles de absorber que el β-caroteno, resultando en proporciones de EAR de 24:1. Las proporciones de EAR se muestran en la Tabla 1 (56).
Determinación de la IDR
La IDR para la vitamina A fue revisada por la Junta de Nutrición y Alimentos (JNA) del Instituto de Medicina (IOM) de los EE.UU. en el 2001. La IDR está basada en el Requerimiento Estimado Promedio (REP), el cual está definido como el requerimiento biológico para el 50% de la población. La IDR es la ingesta recomendada necesaria por casi toda la población para asegurar depósitos hepáticos adecuados de la vitamina A en el cuerpo (20 μg/g por cuatro meses si la persona consume una dieta deficiente de vitamina A) para soportar la función reproductiva normal, función inmune, expresión de genes, y la visión (para cálculos detallados, vea 56). La Tabla 2 lista los valores de la IDR en microgramos (μg) de Equivalentes de Actividad de Retinol (EAR) por día.
| Etapa de la Vida | Edad | Machos (μg/día) | Hembras (μg/día) |
|---|---|---|---|
| Infantes (IA) | 0-6 meses | 400 | 400 |
| Infantes (IA) | 7-12 meses | 500 | 500 |
| Niños | 1-3 años | 300 | 300 |
| Niños | 4-8 años | 400 | 400 |
| Niños | 9-13 años | 600 | 600 |
| Adolescentes | 14-18 años | 900 | 700 |
| Adultos | 19 años y más | 900 | 700 |
| Embarazo | 18 años y menos | - | 750 |
| Embarazo | 19 años y más | - | 770 |
| Período de lactancia | 18 años y menos | - | 1,200 |
| Período de lactancia | 19 años y más | - | 1,300 |
Prevención de Enfermedades
Displasia broncopulmonar en infantes prematuros
Los infantes prematuros nacen con depósitos inadecuados de vitamina A en el cuerpo, poniéndolos en riesgo de desarrollar enfermedades de los ojos y de los tractos respiratorios y gastrointestinales. Alrededor de un tercio de los infantes prematuros nacidos entre las 22 y 28 semanas de gestación desarrollan displasia broncopulmonar (DBP), una enfermedad pulmonar crónica que puede ser fatal o resultar en morbilidades para toda la vida en los sobrevivientes. Unos cuantos estudios controlados aleatorios han investigado el efecto de la suplementación postnatal en la incidencia de DBP y el riesgo de mortalidad en infantes con pesos muy bajos al nacer (≤1,500 g) requiriendo soporte respiratorio (57-59). En el ensayo de mayor escala, multicéntrico, aleatorio, controlado con placebo que incluyó 807 recién nacidos prematuros con pesos extremadamente bajos al nacer (PEB; ≤1,000 g), la administración intramuscular de 5,000 UI de vitamina A tres veces a la semana por cuatro semanas significantemente, aunque modestamente, disminuyó el riesgo de DBP o muerte a las 36 semanas de edad postmenstrual (edad gestacional más la edad cronológica) (58). Mientras que la suplementación con vitamina A fue incluida en varios programas neonatales después de este ensayo (60), una escases nacional del suministro de vitamina A que ha afectado las unidades de cuidado intensivo neonatales estadounidenses desde el 2010 ha llevado a una significante reducción en el uso de la suplementación con vitamina A en los recién nacidos prematuros (401-1,000 g al nacer) con insuficiencia respiratoria (61, 62). Sin embargo, un análisis retrospectivo de datos a nivel nacional de los EE.UU. de 6,210 infantes prematuros nacidos entre el 2010 y 2012 encontró que una reducción en la profilaxis de la vitamina A de 27.2% a 2.1% durante el mismo periodo de tiempo no tuvo algún impacto significante en la incidencia de DBP o muerte antes del alta hospitalaria (62).
En otro estudio retrospectivo, se encontró que el uso no aleatorio de la suplementación con vitamina A con óxido nítrico inhalado (iNO) resulta en una incidencia menor de DBP (pero no de mortalidad) en comparación a la terapia con solo iNO en recién nacidos prematuros con un peso al nacer de 750-999 g (63). Puntuaciones del índice de desarrollo neurológico al año de edad fueron también mejorados en el grupo de vitamina A de los recién nacidos que pesaban 500-749 g al nacer. Sin embargo, se aconseja precaución con la interpretación de los resultados, especialmente porque el ensayo no fue diseñado para evaluar el efecto de la vitamina A. En Alemania, un estudio aleatorio, multicéntrico, de gran tamaño — el ensayo NeoVitaA — está en curso para explorar el efecto de dosis-altas de vitamina A oral (5,000 UI/kg/día) por 28 días en la incidencia de DBP y mortalidad a las 36 semanas de edad postmenstrual (64).
Mientras que las dosis altas de vitamina A durante el comienzo del embarazo pueden causar defectos de nacimiento (véase Seguridad), la suplementación con vitamina A hacia el final del embarazo puede mejorar el estatus de vitamina A materno y fetal (65). Aunque unos cuantos ensayos controlados aleatorios han fallado en mostrar un efecto en la mortalidad materna y neonatal (66), más investigación es requerida para evaluar si la suplementación con vitamina A durante el embarazo reduce la incidencia de DBP en infantes.
Morbilidad y mortalidad infantil
Un meta-análisis reciente de ensayos controlados aleatorios que evaluaron el efecto preventivo de la vitamina A en la mortalidad infantil indicó que la suplementación con vitamina A (200,000 UI cada 4 o 6 meses) disminuyo la mortalidad por todas las causas en un 25% (13 estudios) y la mortalidad especifica por diarrea en un 30% (7 estudios) en niños de entre 6 a 59 meses de edad. Sin embargo, la administración de vitamina A en este grupo de edad no tuvo efecto preventivo en las tasas de mortalidad específica por neumonía (7 estudios), mortalidad especifica por sarampión (5 estudios), o mortalidad especifica por meningitis (3 estudios). Además, no se encontró reducción en el riesgo de mortalidad por enfermedad especifica en neonatos (0 a 28 días de edad) e infantes de 1 a 6 meses de edad suplementados con vitamina A (67). Otro meta-análisis de ensayos controlados aleatorios no encontró evidencia de una reducción en el riesgo de mortalidad durante la infancia cuando las madres lactantes (7 estudios) o infantes de menos de seis meses de edad (9 estudios) fueron suplementados con vitamina A (68).
Actualmente la política de la OMS recomienda la suplementación con vitamina A en los contactos de vacunación de rutina en niños después de los seis meses de edad que viven en regiones en alto riesgo de una deficiencia de vitamina A. La suplementación con altas dosis de vitamina A — 100,000 UI (30 mg EAR) para infantes de 6 a 11 meses de edad y 200,000 UI (60 mg EAR) para niños de 12 a 59 meses de edad — se piensa que proporciona una protección adecuada por hasta seis meses (38). Un reciente ensayo controlado con placebo en Guinea-Bisáu, en el cual 7,587 niños (6 a 23 meses de edad) aleatoriamente recibieron suplementación con vitamina A en una vacunación de contacto, evaluó la coadministración de la vitamina A y vacunas en la mortalidad infantil (69). El estudio encontró que la suplementación con vitamina A no tuvo efecto en las tasas de mortalidad en general, aunque un seguimiento de seis meses de infantes a los cuales se les dio ambas vacunas de sarampión y DTP (difteria-tétanos-tosferina) mostro una reducción significante de la mortalidad en niñas, pero no en niños (69). Aunque la suplementación neonatal con vitamina A no es actualmente recomendada, un ensayo que evaluó el beneficio de la vacunación temprana contra el sarampión — a los 4.5 en lugar de a los usuales 9 meses de edad — no encontró reducción en las tasas de mortalidad cuando los niños habían recibido suplementación neonatal de vitamina A (70). El análisis agrupado reciente de ensayos previos de la suplementación con vitamina A (VITA I-III) en Guinea-Bisáu confirmó que la suplementación con vitamina A podría interferir con las vacunas. Específicamente, en comparación al placebo, la suplementación neonatal con vitamina A se asoció con un incremento significante en las tasas de mortalidad en niños (pero no en niñas) cuando los infantes habían recibido vacunas contra el virus del sarampión a los 4.5 meses de edad en lugar de a los usuales 9 meses de edad (71). El momento de las intervenciones de vitamina A necesita ser examinado más a fondo en relación con el momento de la vacunación con el fin de maximizar sus beneficios.
Complicaciones de la infección por sarampión
Un meta-análisis temprano de siete ensayos controlados aleatorios que examinaron específicamente el papel de la suplementación con vitamina A en 2,069 niños con sarampión no encontró una reducción en general del riesgo de mortalidad (72). Sin embargo, el análisis agrupado de cuatro estudios que reporto la distribución de la edad de participantes encontró un riesgo 83% menor de mortalidad con dos dosis de 200,000 UI de vitamina A en niños menores de dos años. Además, el análisis agrupado de tres estudios indicó una reducción del 67% del riesgo de mortalidad debido a la neumonía (72). Similar a las pautas de la OMS y Unicef la Academia Americana de Pediatría recomienda la suplementación con vitamina A para niños mayores de seis meses de edad cuando estos son infectados con sarampión mientras se encuentran desnutridos, inmunodeficientes, o en riesgo de complicaciones por el sarampión o trastornos de la deficiencia de vitamina A (73). Aunque la infección por sarampión ha sido asociada con la deficiencia de vitamina A y la ceguera, no existe evidencia actualmente que sugiera que la suplementación con vitamina A reduce el riesgo de ceguera en niños infectados con sarampión (74).
Cáncer
Estudios en células de cultivo y modelos animales han documentado la capacidad de los retinoides naturales y sintéticos de reducir significativamente la carcinogénesis en la piel, seno, colon, próstata, y otros lugares (2). Sin embargo, los resultados de los estudios humanos que examinan la relación entre el consumo de vitamina A preformada y el cáncer actualmente no sugieren que el consumo de vitamina A en ingestas mayores a la IDR benefician la prevención del cáncer (2).
Cáncer de pulmón
Los resultados del Ensayo de la Eficacia del β-Caroteno y Retinol (CARET) han sugerido que la suplementación con altas dosis de vitamina A preformada y β-caroteno deberían ser evitados en personas con alto riesgo de cáncer pulmonar (75). En el estudio CARET, alrededor de 9,000 personas (fumadores y personas con exposición al asbesto) fueron asignadas a un régimen diario de 25,000 UI (7,500 μg EAR) de retinil palmitato y 30 μg de β-caroteno, mientras que a un número similar de personas se les asigno un placebo. Después de cuatro años de seguimiento, la incidencia de cáncer pulmonar fue 28% más alta en el grupo suplementado en comparación al grupo del placebo; sin embargo, la incidencia no fue diferente seis años después de que la intervención finalizara (76). Una posible explicación para el incremento de cáncer de pulmón es que el entorno oxidativo del pulmón, creado por fumar o por la exposición al asbesto, pudo dar lugar a productos de escisión de carotenoides inusuales que podrían promover la carcinogénesis (77). Curiosamente, un estudio de caso y control que incluyo 749 casos de cáncer pulmonar y 679 controles del ensayo CARET encontró una asociación significante entre la reducción del riesgo de cáncer pulmonar y las ingestas altas de vitamina D (≥400 UI/día) en individuos que recibieron suplementos activos del CARET o en aquellos con ingestas de vitamina A iguales o mayores de 1,500 μg EAR/día (78). Además, un reciente meta-análisis de cuatro ensayos controlados aleatorios, incluyendo un total de 202,924 participantes en riesgo bajo de cáncer pulmonar, indicó que la suplementación con retinol y/o β-caroteno no tuvo efecto significante en la incidencia de cáncer de pulmón (79). Actualmente, parece poco probable que la ingesta incrementada de vitamina A preformada (p. ej., retinol) podría disminuir el riesgo de cáncer de pulmón.
Tratamiento de Enfermedades
Los retinoides pueden ser usados en dosis farmacológicas para tratar varias condiciones, incluyendo, leucemia promielocítica aguda, retinitis pigmentosa, y varias enfermedades cutáneas. Es importante destacar que el tratamiento con dosis altas de retinoides naturales o sintéticos anula los mecanismos de control propios del cuerpo; por lo tanto, la terapia con retinoides está asociada con potenciales efectos secundarios y toxicidades. Adicionalmente, se ha encontrado que todos los compuestos retinoides causan defectos de nacimiento. Así, las mujeres que tienen la posibilidad de quedar embarazadas deberían evitar el tratamiento con estos medicamentos. Los retinoides tienden a ser de muy larga duración: se han reportado que efectos secundarios y defectos de nacimiento ocurren meses después de descontinuar la terapia con retinoides (2). Los retinoides discutidos a continuación son medicamentos sujetos a prescripción y no deberían ser utilizados sin supervisión médica.
Leucemia promielocítica aguda
La diferenciación normal de las células madre mieloides en la médula ósea da lugar a las plaquetas, glóbulos rojos, y glóbulos blancos (también llamados leucocitos) que son importantes para la respuesta inmune. La diferenciación alterada de las células mieloides puede resultar en la proliferación de glóbulos blancos inmaduros, dando lugar a leucemia. Translocaciones cromosómicas reciprocas que involucran el gen de la leucemia promielocítica (PML) y el gen que codifica para el receptor del ácido retinoico α (RARα) llevan a un tipo específico de leucemia llamado leucemia promielocítica aguda (LPA). La fusión proteína PML/RAR α reprime la transcripción al unirse al RARE en el promotor de los genes retinoide-sensibles involucrados en la diferenciación celular hematopoyética. La represión de genes por la PML/RARα se lograr mediante el reclutamiento de varios modificadores de la cromatina, incluyendo histona deacetilasas (HDAC) y ADN metiltransferasas (ADNMTasas). Contrario al receptor de tipo silvestre del RARα, la PML/RARα parece ser insensible a concentraciones de ácido retinoico (AR) tal que solo tratamientos con altas dosis de ácido todo-trans-retinoico pueden restaurar la diferenciación normal y conducir a mejoras significantes y remisión completa en algunos pacientes con LPA (80).
Más información sobre los programas de tratamiento para la LPA pueden ser encontrados en el sitio web del Instituto Nacional del Cáncer.
Enfermedades de la piel
Se han utilizado retinoides naturales y sintéticos como agentes farmacológicos para tratar trastornos de la piel. La acitretina es un retinoide sintético que ha sido probado útil en la combinación de tratamientos para la psoriasis (81). La tretinoina tópica (ácido todo-trans-retinoico) y la isotretinoina oral (ácido 13-cis-retinoico) se han utilizado exitosamente para tratar el acné común de leve-a-severo (82, 83). Los retinoides exhiben propiedades antiinflamatorias y regulan la proliferación y diferenciación de las células epiteliales cutáneas, así como también la producción de sebo. El uso de dosis farmacológicas de retinoides (especialmente la isotretinoina oral) por mujeres embarazadas causa defectos de nacimiento y está por lo tanto contraindicado antes de y durante el embarazo (véase Seguridad en el embarazo).
Para más información sobre el uso de retinoides en el manejo del acné, véase el artículo en Vitamina A y la Salud de la Piel.
Retinitis pigmentosa
La retinitis pigmentosa (RP) afecta aproximadamente 1.5 millones de personas alrededor del mundo y es la causa principal de ceguera hereditaria. La RP describe un amplio espectro de trastornos genéticos que resultan en la pérdida progresiva de células fotorreceptoras (bastones y conos) en la retina del ojo (84). Mientras que al menos 45 loci han sido asociados con la RP, las mutaciones en el gen de la rodopsina (RHO), gen de la usherina (USH2A), y en el gen regulador de GTPase de RP (RPGR) representan alrededor del 30% de todos los casos de RP (85).
Los síntomas tempranos de la RP incluyen adaptación a la oscuridad dañada y ceguera nocturna, seguidos por la pérdida progresiva de la visión periférica y central al pasar del tiempo (85). Los resultados de solo un ensayo controlado aleatorio en 601 pacientes con formas comunes de RP indicó que la suplementación con 15,000 UI/día de retinil palmitato (4,500 μg de EAR) desacelero significantemente la perdida de la función de la retina sobre un periodo de cuatro a seis años (86). En contraste, la suplementación con 400 UI/día de vitamina E (dl-α-tocoferol) modestamente pero significantemente incremento la perdida de la función de la retina, sugiriendo que los pacientes con formas comunes de RP pudiesen beneficiarse de la suplementación con vitamina A a largo plazo pero deberían evitar una suplementación con vitamina E en altas dosis. Con un seguimiento de hasta 12 años en estos pacientes no se reveló ningún signo de toxicidad en el hígado como resultado de la ingesta en exceso de vitamina A (87). Debido a que ni los niños menores de 18 años o los adultos afectados por las formas menos comunes de RP fueron incluidos en este ensayo, ninguna recomendación formal sobre la vitamina A y E podría ser hecha (85). La suplementación con vitamina A en altas dosis para desacelerar el curso de la RP requiere supervisión médica y debe ser descontinuada si existe la posibilidad de embarazo (véase Seguridad).
Fuentes
Fuentes alimenticias
El retinol libre no se encuentra generalmente en los alimentos. Los retinil ésteres (incluyendo el retinil palmitato) son las formas de almacenamiento del retinol en animales y de esta manera los precursores principales de retinol en los alimentos provenientes de animales. Las plantas contienen carotenoides, algunos de los cuales son precursores para la vitamina A (p. ej., α-caroteno, β-caroteno y β-criptoxantina). Los vegetales amarillos y naranjas contienen cantidades significantes de carotenoides. Los vegetales verdes también contienen carotenoides, aunque los pigmentos amarillo-a-rojo son enmascarados por el color verde de la clorofila (1). La tabla a continuación lista un cierto número de fuentes alimenticias buenas de vitamina A, incluyendo frutas y vegetales, junto con su contenido de vitamina A. La actividad de retinol es indicada en microgramos de equivalentes de actividad de retinol (μg de EAR). Para más información sobre esta unidad de medición, vea la sección sobre EAR. Además, use la base de datos de la composición de alimentos de la USDA para revisar los alimentos por su contenido de varios carotenoides sin la actividad de la vitamina A, como el licopeno, luteína y zeaxantina.
Unidades Internacionales de vitamina A (UI)
La vitamina A esta actualmente listada en el etiquetado de alimentos y suplementos en unidades internacionales. La base de datos de la USDA también proporciona el contenido de vitamina A de fuentes alimenticias usando la unidad internacional de vitamina A (UI). Sin embargo, contrario a los EAR, el número de UI de vitamina A no refleja la biodisponibilidad de la vitamina A de diferentes fuentes alimenticias. Las tasas de conversión entre las UI y μg de EAR son establecidas de la siguiente manera:
- 1 UI de retinol es equivalente a 0.3 μg de EAR
- 1 UI de β-caroteno suplementario es equivalente a 0.15 μg de EAR
- 1 UI de β-caroteno dietario es equivalente a 0.05 μg de EAR
- 1 UI de α-caroteno o β-criptoxantina es equivalente a 0.025 μg de EAR
Así, en la Tabla 3, el número de UI de vitamina A en los alimentos que contienen carotenoides (números en itálicas) puede ser obtenido multiplicando los EAR por aproximadamente 20.
| Alimento | Porción | Vitamina A Preformada (Retinol), μg | Vitamina A, μg de EAR | Vitamina A, IU |
|---|---|---|---|---|
| Hígado de res, cocido | 1 rebanda (68 g) | 6,421* | 6,421* | 21,566* |
| Aceite de hígado de bacalao | 1 cuchara de pequeña | 1,350 | 1,350 | 4,500 |
| Cereal de desayuno fortificado (avena) | 1 porción (1 oz) | 216 | 216 | 721 |
| Huevo | 1 grande | 80 | 80 | 270 |
| Mantequilla | 1 cucharada grande | 95 | 95 | 355 |
| Leche entera | 1 taza (8 oz fluidas) | 110 | 110 | 395 |
| Leche semi-descremada (2%) (adicionada con vitamina A) | 1 taza (8 oz fluidas) | 134 | 134 | 464 |
| Leche descremada (adicionada con vitamina A) | 1 taza (8 oz fluidas) | 149 | 149 | 500 |
| Camote (enlatado, hecho puré) | ½ taza | 0 | 555 | 11,091 |
| Camote (asado) | ½ taza | 0 | 961 | 19,218 |
| Calabaza (enlatada) | ½ taza | 0 | 953 | 19,065 |
| Zanahoria (cruda, en trozos) | ½ taza | 0 | 534 | 10,692 |
| Melón (cantalupo) | ½ melón mediano | 0 | 466 | 9,334 |
| Mango | 1 fruta | 0 | 181 | 3,636 |
| Espinacas (cocidas) | ½ taza | 0 | 472 | 9,433 |
| Brócoli (cocido) | ½ taza | 0 | 60 | 1,207 |
| Col rizada (cocida) | ½ taza | 0 | 443 | 8,854 |
| Coles (cocidos) | ½ taza | 0 | 361 | 7,220 |
| Calabaza, butternut (cocida) | ½ taza | 0 | 572 | 11,434 |
| *Por arriba del nivel máximo de ingesta tolerable (NM) de 3,000 μg de EAR (10,000 UI)/día | ||||
Suplementos
Las formas principales de vitamina A preformada en los suplementos son el retinil palmitato y retinil acetato. El β-caroteno también es una fuente común de vitamina A en los suplementos, y muchos de ellos proveen una combinación de retinol y β-caroteno (88). Si un porcentaje del total del contenido de vitamina A de un suplemento proviene del β-caroteno, esta información es incluida en la Información Nutricional etiquetada bajo vitamina A. Varios suplementos multivitamínicos disponibles en los EE.UU. proporcionan hasta 5,000 UI de vitamina A preformada, correspondiendo a 1,500 μg de EAR, lo cual es substancialmente más que la actual IDR para la vitamina A. Esto se debe al hecho de que los Valores Diarios (VD) utilizados por la Administración de Alimentos y Drogas de los EE.UU. (FDA) para el etiquetado de suplementos se basaron en la IDR establecida en 1968 en vez de la IDR más reciente, y típicamente los suplementos multivitamínicos aportan el 100% de los VD para la mayoría de los nutrientes. Debido a que las ingestas de retinol de 5,000 UI/día (1,500 μg de EAR) podrían asociarse con un riesgo incrementado de osteoporosis en adultos mayores (véase Seguridad), algunas compañías han reducido el contenido de retinol en sus suplementos multivitamínicos a 2,500 UI (750 μg de EAR).
Seguridad
Toxicidad
La condición causada por la toxicidad de la vitamina A se denomina hipervitaminosis A. Ésta es causada por el sobreconsumo de vitamina A preformada, no de carotenoides. La vitamina A preformada es rápidamente absorbida y lentamente eliminada del cuerpo. Por lo tanto, la toxicidad de la vitamina A preformada podría resultar agudamente debido a una exposición a dosis altas en un periodo de tiempo corto, o crónicamente de una ingesta mucho más baja (2). La toxicidad aguda por vitamina A es relativamente rara, y los síntomas incluyen nauseas, dolor de cabeza, fatiga, pérdida de apetito, mareos, piel seca, descamación y edema cerebral. Los signos de toxicidad crónica incluyen piel seca y pruriginosa, descamación, anorexia, pérdida de peso, dolor de cabeza, edema cerebral, agrandamiento del hígado, agrandamiento del bazo, anemia, y dolor de huesos y articulaciones. También en infantes, los síntomas de toxicidad de vitamina A incluyen fontanelas prominentes. Casos severos de hipervitaminosis A podrían resultar en daño hepático, hemorragia y coma. Generalmente, los signos de toxicidad están asociados con el consumo a largo plazo de vitamina A en excesos de 10 veces la IDR (8,000 a 10,000 EAR/día o 25,000 a 33,000 UI/día). Sin embargo, más investigación es necesaria para determinar si la toxicidad subclínica de vitamina A es un motivo de preocupación en ciertas poblaciones (89). Existe evidencia de que algunas poblaciones podrían ser más susceptibles a la toxicidad en dosis más bajas, incluyendo a los ancianos, bebedores de alcohol crónicos, y a algunas personas con predisposición genética al colesterol alto (90). En enero de 2001, la Junta de Nutrición y Alimentos del Instituto de Medicina de los EE.UU. estableció el nivel máximo de ingesta tolerable (NM) de vitamina A para adultos en 3,000 μg de EAR (10,000 UI)/día de vitamina A preformada (56).
Seguridad en el embarazo
Aunque el desarrollo fetal normal requiere una ingesta de vitamina A suficiente, el consumo en exceso de vitamina A preformada (como el retinol) durante el comienzo del embarazo es conocido por causar defectos de nacimiento. No se ha observado ningún aumento del riesgo de defectos de nacimiento asociados con la vitamina A en dosis de vitamina A preformada de suplementos por debajo de los 3,000 μg de EAR/día (10,000 UI/día) (56). Debe notarse que en el 2011, la Organización Mundial de la Salud (OMS) recomendó la suplementación con vitamina A (de hasta 3,000 μg de EAR/día o 7,500 μg EAR/semana) durante el embarazo en áreas con una alta prevalencia de deficiencia de vitamina A para la prevención de la ceguera (91). En países industrializados, las mujeres potencialmente o ya embarazadas debiesen monitorear sus ingestas de vitamina A provenientes de alimentos fortificados y alimentos naturalmente altos en vitamina A preformada (p. ej., el hígado) y evitar tomar suplementos multivitamínicos que contengan más de 1,500 μg de EAR (5,000 UI) de vitamina A. No existe evidencia de que el consumo de vitamina A del β-caroteno podría incrementar el riesgo de defectos de nacimiento. Se sabe que la isotretinoina, derivado sintético del retinol, causa serios defectos de nacimiento y no deberían tomarse durante el embarazo, o si existe la posibilidad de quedar embarazada (82). La tretinoina (ácido todo-trans-retinoico), otro derivado del retinol, se prescribe como una preparación tópica que se aplica sobre la piel. Aunque la absorción percutánea de tretinoina tópica es mínima, no se recomienda su uso durante el embarazo (92).
¿Las ingestas elevadas de vitamina A incrementan el riesgo de osteoporosis?
Los resultados de algunos estudios prospectivos han sugerido que las ingestas en exceso a largo plazo de vitamina A preformada de 1,500 μg de EAR/día (equivalente a 5,000 UI/día de vitamina A como retinol) se asociaron con una densidad mineral ósea (DMO) disminuida y con un riesgo incrementado de fractura osteoporótica en adultos mayores (93-95). Sin embargo, otros investigadores fallaron en observar tales efectos perjudiciales en la DMO y/o riesgo de fractura (96-98). El reciente meta-análisis de cuatro estudios prospectivos incluyendo casi 183,000 participantes mayores de 40 años de edad, encontró que los quintiles más altos frente a los más bajos de la ingesta de retinol (vitamina A preformada) incrementaron significativamente el riesgo de fractura de cadera (99). Solo las ingestas en exceso de retinol, no de β-caroteno, fueron asociadas con efectos adversos a la salud ósea. Además, el análisis agrupado de cuatro estudios basados en la observación también índico una relación en forma de U entre el retinol circulante y el riesgo de fractura de cadera, sugiriendo que tanto las concentraciones elevadas como las reducidas de retinol en la sangre se asociaron con un riesgo incrementado de fractura de cadera (99).
Hasta la fecha, datos experimentales han sugerido que la vitamina A (como ácido todo-trans-retinoico) puede afectar el desarrollo de las células de remodelación ósea y estimula la degradación de la matriz ósea (reabsorción) (revisado en 100). La vitamina A puede también interferir con la habilidad de la vitamina D para mantener el balance del calcio (101). En el estudio prospectivo de gran tamaño Women’s Health Initiative (WHI) se encontró que el quintil más alto frente al más bajo de la ingesta de retinol (≥1,426 μg/día vs. <474 μg/día) estaba significantemente asociado con un riesgo incrementado de fractura solo en mujeres con las ingestas más bajas de vitamina D (≤440 UI/día) (102).
Hasta que los suplementos y alimentos fortificados estén reformulados para reflejar la IDR actual de la vitamina A, es aconsejable para los individuos mayores el consumir suplementos multivitamínicos que contengan no más de 2,500 UI (750 μg) de vitamina A preformada (usualmente etiquetada acetato de vitamina A o palmitato de vitamina A) y no más de 2,500 UI de vitamina A adicional como β-caroteno.
Interacción con drogas/fármacos
El consumo crónico de alcohol resulta en el agotamiento de los depósitos hepáticos de vitamina A, y podría contribuir al daño hepático inducido por alcohol (cirrosis) (103). Sin embargo, la toxicidad hepática de la vitamina A preformada (retinol) es realzada por el consumo crónico de alcohol, disminuyendo así la ventana terapéutica para la suplementación con vitamina A en alcohólicos (103). Los anticonceptivos orales que contienen estrógeno y progestina incrementan la síntesis de la proteína de unión a retinol (RBP) por el hígado, aumentando la exportación del complejo todo-trans-retinol/RBP en la circulación. No se sabe si esto incrementa el requerimiento dietario de vitamina A. También, el uso de medicamentos que disminuyen el colesterol (como colestiramina y colestipol), como también el orlistat, aceite mineral, y el substituto de grasas, olestra, el cual interfiere con la absorción de grasas, podrían afectar la absorción de vitaminas liposolubles, incluyendo la vitamina A (88). Además, la ingesta de grandes dosis de vitamina A podrían disminuir la absorción de vitamina K. Los retinoides o los retinoides análogos, incluyendo acitretina, ácido todo-trans-retinoico, bexaroteno, etretinato, e isotretinoina no debieran ser usados en combinación con suplementos de vitamina A de un solo nutriente, debido a que podrían incrementar el riesgo de toxicidad de la vitamina A (88).
Recomendación del Instituto Linus Pauling
La IDR para la vitamina A (700 μg de EAR/día para las mujeres y 900 μg de EAR/día para los hombres) es suficiente para mantener una expresión de genes, función inmune, y visión normales. Sin embargo, seguir la recomendación del Instituto Linus Pauling de tomar diariamente un suplemento multivitamínico/mineral podría aportar hasta 5,000 UI (1,500 μg de EAR)/día de vitamina A como retinol, la cantidad que ha sido asociada con efectos adversos sobre la salud ósea en adultos mayores. Por esta razón, recomendamos tomar un suplemento multivitamínico/mineral que no aporte más de 2,500 UI (750 μg) de vitamina A preformada (usualmente etiquetada como acetato de vitamina A o palmitato de vitamina A) y no más de 2,500 UI de vitamina A adicional como β-caroteno. Los suplementos de vitamina A de alta potencia no deberían ser utilizados sin supervisión médica debido al riesgo de toxicidad.
Adultos mayores (>50 años)
Actualmente, existe poco evidencia de que el requerimiento de vitamina A en adultos mayores difiere de aquel de los adultos más jóvenes. Adicionalmente, la toxicidad por vitamina A puede ocurrir con dosis más bajas en los adultos mayores que en los adultos más jóvenes. Además, datos de estudios basados en la observación sugieren una asociación inversa entre las ingestas de vitamina A preformada en exceso de 1,500 μg de EAR (5,000 UI)/día y el riesgo de fractura de cadera en personas mayores (véase Seguridad). Sin embargo al seguir la recomendación del Instituto Linus Pauling de tomar diariamente un suplemento multivitamínico/mineral podría proveer hasta 5,000 UI/día de retinol, la cantidad que ha sido asociada con efectos adversos sobre la salud ósea en adultos mayores. Por esta razón, recomendamos tomar un suplemento multivitamínico/mineral que aporte (750 μg) de vitamina A preformada (usualmente etiquetada como acetato de vitamina A o palmitato de vitamina A) y no más de 2,500 UI de vitamina A adicional como β-caroteno. Con respecto a todos los grupos de edad, los suplementos de vitamina A de alta potencia no deberían utilizarse sin supervisión médica debido al riesgo de toxicidad.
Autores y Críticos
Originalmente escrito en 2000 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Diciembre de 2003 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Noviembre de 2007 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Enero de 2015:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Febrero de 2015 por:
A. Catharine Ross, Ph.D.
Profesor de Nutrición
Dorothy Foehr Huck Catedrático
Departamento de Ciencias Nutricionales
Universidad del Estado de Pennsylvania
Revisado en Marzo de 2015 por:
Libo Tan, Ph.D.
Profesor Adjunto
Departamento de Nutrición Humana
Universidad de Alabama
Traducido al Español en 2016 por:
Silvia Vazquez Lima
Instituto Linus Pauling
Universidad Estatal de Oregon
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Derechos de autoría 2000-2024 Instituto Linus Pauling
Referencias
1. Groff JL. Advanced Nutrition and Human Metabolism. 2nd ed. St. Paul: West Publishing; 1995.
2. Ross AC. Vitamin A. In: Ross A, Caballero B, Cousins R, Tucker K, Ziegler T, eds. Modern Nutrition in Health and Disease. 11th ed: Lippincott Williams & Wilkins; 2014:260-277.
3. Tan L, Green MH, Ross AC. Vitamin A Kinetics in Neonatal Rats vs. Adult Rats: Comparisons from Model-Based Compartmental Analysis. J Nutr. 2014;145(3):403-410. (PubMed)
4. Tan L, Wray AE, Green MH, Ross AC. Compartmental modeling of whole-body vitamin A kinetics in unsupplemented and vitamin A-retinoic acid-supplemented neonatal rats. J Lipid Res. 2014;55(8):1738-1749. (PubMed)
5. Zhong M, Kawaguchi R, Ter-Stepanian M, Kassai M, Sun H. Vitamin A transport and the transmembrane pore in the cell-surface receptor for plasma retinol binding protein. PLoS One. 2013;8(11):e73838. (PubMed)
6. See AW, Clagett-Dame M. The temporal requirement for vitamin A in the developing eye: mechanism of action in optic fissure closure and new roles for the vitamin in regulating cell proliferation and adhesion in the embryonic retina. Dev Biol. 2009;325(1):94-105. (PubMed)
7. Theodosiou M, Laudet V, Schubert M. From carrot to clinic: an overview of the retinoic acid signaling pathway. Cell Mol Life Sci. 2010;67(9):1423-1445. (PubMed)
8. Lefebvre P, Martin PJ, Flajollet S, Dedieu S, Billaut X, Lefebvre B. Transcriptional activities of retinoic acid receptors. Vitam Horm. 2005;70:199-264. (PubMed)
9. Amann PM, Eichmuller SB, Schmidt J, Bazhin AV. Regulation of gene expression by retinoids. Curr Med Chem. 2011;18(9):1405-1412. (PubMed)
10. Pendaries V, Verrecchia F, Michel S, Mauviel A. Retinoic acid receptors interfere with the TGF-beta/Smad signaling pathway in a ligand-specific manner. Oncogene. 2003;22(50):8212-8220. (PubMed)
11. Masia S, Alvarez S, de Lera AR, Barettino D. Rapid, nongenomic actions of retinoic acid on phosphatidylinositol-3-kinase signaling pathway mediated by the retinoic acid receptor. Mol Endocrinol. 2007;21(10):2391-2402. (PubMed)
12. Qiao J, Paul P, Lee S, et al. PI3K/AKT and ERK regulate retinoic acid-induced neuroblastoma cellular differentiation. Biochem Biophys Res Commun. 2012;424(3):421-426. (PubMed)
13. Canon E, Cosgaya JM, Scsucova S, Aranda A. Rapid effects of retinoic acid on CREB and ERK phosphorylation in neuronal cells. Mol Biol Cell. 2004;15(12):5583-5592. (PubMed)
14. Kirchmeyer M, Koufany M, Sebillaud S, Netter P, Jouzeau JY, Bianchi A. All-trans retinoic acid suppresses interleukin-6 expression in interleukin-1-stimulated synovial fibroblasts by inhibition of ERK1/2 pathway independently of RAR activation. Arthritis Res Ther. 2008;10(6):R141. (PubMed)
15. Amengual J, Golczak M, Palczewski K, von Lintig J. Lecithin:retinol acyltransferase is critical for cellular uptake of vitamin A from serum retinol-binding protein. J Biol Chem. 2012;287(29):24216-24227. (PubMed)
16. Noy N. Signaling by retinol and its serum binding protein. Prostaglandins Leukot Essent Fatty Acids. 2014;93:3-7. (PubMed)
17. Berry DC, Jacobs H, Marwarha G, et al. The STRA6 receptor is essential for retinol-binding protein-induced insulin resistance but not for maintaining vitamin A homeostasis in tissues other than the eye. J Biol Chem. 2013;288(34):24528-24539. (PubMed)
18. Marwarha G, Berry DC, Croniger CM, Noy N. The retinol esterifying enzyme LRAT supports cell signaling by retinol-binding protein and its receptor STRA6. FASEB J. 2014;28(1):26-34. (PubMed)
19. Farmer SR. Molecular determinants of brown adipocyte formation and function. Genes Dev. 2008;22(10):1269-1275. (PubMed)
20. Kiefer FW, Vernochet C, O'Brien P, et al. Retinaldehyde dehydrogenase 1 regulates a thermogenic program in white adipose tissue. Nat Med. 2012;18(6):918-925. (PubMed)
21. Nallamshetty S, Le PT, Wang H, et al. Retinaldehyde dehydrogenase 1 deficiency inhibits PPARgamma-mediated bone loss and marrow adiposity. Bone. 2014;67:281-291. (PubMed)
22. Kiefer FW, Orasanu G, Nallamshetty S, et al. Retinaldehyde dehydrogenase 1 coordinates hepatic gluconeogenesis and lipid metabolism. Endocrinology. 2012;153(7):3089-3099. (PubMed)
23. Green HN, Mellanby E. Vitamin A as an anti-infective agent. Br Med J. 1928;2(3537):691-696. (PubMed)
24. Raverdeau M, Mills KH. Modulation of T cell and innate immune responses by retinoic Acid. J Immunol. 2014;192(7):2953-2958. (PubMed)
25. Spears K, Cheney C, Zerzan J. Low plasma retinol concentrations increase the risk of developing bronchopulmonary dysplasia and long-term respiratory disability in very-low-birth-weight infants. Am J Clin Nutr. 2004;80(6):1589-1594. (PubMed)
26. Barber T, Esteban-Pretel G, Marin MP, Timoneda J. Vitamin A Deficiency and Alterations in the Extracellular Matrix. Nutrients. 2014;6(11):4984-5017. (PubMed)
27. Semba RD, Bloem MW. The anemia of vitamin A deficiency: epidemiology and pathogenesis. Eur J Clin Nutr. 2002;56(4):271-281. (PubMed)
28. Allen LH. Iron supplements: scientific issues concerning efficacy and implications for research and programs. J Nutr. 2002;132(4 Suppl):813S-819S. (PubMed)
29. Christian P, West KP, Jr. Interactions between zinc and vitamin A: an update. Am J Clin Nutr. 1998;68(2 Suppl):435S-441S. (PubMed)
30. Auld DS, Bergman T. Medium- and short-chain dehydrogenase/reductase gene and protein families : The role of zinc for alcohol dehydrogenase structure and function. Cell Mol Life Sci. 2008;65(24):3961-3970. (PubMed)
31. Suharno D, West CE, Muhilal, Karyadi D, Hautvast JG. Supplementation with vitamin A and iron for nutritional anaemia in pregnant women in West Java, Indonesia. Lancet. 1993;342(8883):1325-1328. (PubMed)
32. Jang JT, Green JB, Beard JL, Green MH. Kinetic analysis shows that iron deficiency decreases liver vitamin A mobilization in rats. J Nutr. 2000;130(5):1291-1296. (PubMed)
33. Rosales FJ, Jang JT, Pinero DJ, Erikson KM, Beard JL, Ross AC. Iron deficiency in young rats alters the distribution of vitamin A between plasma and liver and between hepatic retinol and retinyl esters. J Nutr. 1999;129(6):1223-1228. (PubMed)
34. Tanumihardjo SA. Vitamin A: biomarkers of nutrition for development. Am J Clin Nutr. 2011;94(2):658S-665S. (PubMed)
35. Underwood BA, Arthur P. The contribution of vitamin A to public health. Faseb J. 1996;10(9):1040-1048. (PubMed)
36. Solomons NW. Vitamin A. In: Erdman JJ, Macdonald I, Zeisel S, eds. Present Knowledge in Nutrition. 10th ed: John Wiley & Sons, Ltd.; 2012:149-184.
37. Sherwin JC, Reacher MH, Dean WH, Ngondi J. Epidemiology of vitamin A deficiency and xerophthalmia in at-risk populations. Trans R Soc Trop Med Hyg. 2012;106(4):205-214. (PubMed)
38. World Health Organization. Guideline - Vitamin A supplementation for infants and children 6-59 months of age - Guideline. Geneva 2011.
39. World Health Organization. Guideline - Neonatal vitamin A supplementation Geneva 2011.
40. World Health Organization. Guideline - Vitamin A supplementation for infants 1–5 months of age - Guideline. Geneva 2011.
41. Gilbert C, Awan H. Blindness in children. BMJ. 2003;327(7418):760-761. (PubMed)
42. Semba RD. Vitamin A and human immunodeficiency virus infection. Proc Nutr Soc. 1997;56(1B):459-469. (PubMed)
43. Field CJ, Johnson IR, Schley PD. Nutrients and their role in host resistance to infection. J Leukoc Biol. 2002;71(1):16-32. (PubMed)
44. WHO, UNICEF, IVACG Task Force. Vitamin A supplements: a guide to their use in the treatment and prevention of vitamin A deficiency and xerophthalmia. Geneva: World Health Organization; 1997.
45. Thornton KA, Mora-Plazas M, Marin C, Villamor E. Vitamin A deficiency is associated with gastrointestinal and respiratory morbidity in school-age children. J Nutr. 2014;144(4):496-503. (PubMed)
46. Wiysonge CS, Shey M, Kongnyuy EJ, Sterne JA, Brocklehurst P. Vitamin A supplementation for reducing the risk of mother-to-child transmission of HIV infection. Cochrane Database Syst Rev. 2011(1):CD003648. (PubMed)
47. Semba RD, Miotti PG, Chiphangwi JD, et al. Maternal vitamin A deficiency and mother-to-child transmission of HIV-1. Lancet. 1994;343(8913):1593-1597. (PubMed)
48. World Health Organization. Guideline - Vitamin A supplementation in pregnancy for reducing the risk of mother-to-child transmission of HIV. Geneva 2011.
49. Zimmermann MB, Adou P, Torresani T, Zeder C, Hurrell RF. Effect of oral iodized oil on thyroid size and thyroid hormone metabolism in children with concurrent selenium and iodine deficiency. Eur J Clin Nutr. 2000;54(3):209-213. (PubMed)
50. Zimmermann MB, Wegmuller R, Zeder C, Chaouki N, Torresani T. The effects of vitamin A deficiency and vitamin A supplementation on thyroid function in goitrous children. J Clin Endocrinol Metab. 2004;89(11):5441-5447. (PubMed)
51. Zimmermann MB. Interactions of vitamin A and iodine deficiencies: effects on the pituitary-thyroid axis. Int J Vitam Nutr Res. 2007;77(3):236-240. (PubMed)
52. Zimmermann MB, Jooste PL, Mabapa NS, et al. Vitamin A supplementation in iodine-deficient African children decreases thyrotropin stimulation of the thyroid and reduces the goiter rate. Am J Clin Nutr. 2007;86(4):1040-1044. (PubMed)
53. Maronn M, Allen DM, Esterly NB. Phrynoderma: a manifestation of vitamin A deficiency?... The rest of the story. Pediatr Dermatol. 2005;22(1):60-63. (PubMed)
54. Herring W, Nowicki MJ, Jones JK. An uncommon cause of esophagitis. Answer to the clinical challenges and images in GI question: image 1: esophageal hyperkeratosis secondary to vitamin A deficiency. Gastroenterology. 2010;139(2):e6-7. (PubMed)
55. Weber D, Grune T. The contribution of beta-carotene to vitamin A supply of humans. Mol Nutr Food Res. 2012;56(2):251-258. (PubMed)
56. Food and Nutrition Board, Institute of Medicine. Vitamin A. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Washington, D.C.: National Academy Press; 2001:65-126. (National Academy Press)
57. Ravishankar C, Nafday S, Green RS, et al. A trial of vitamin A therapy to facilitate ductal closure in premature infants. J Pediatr. 2003;143(5):644-648. (PubMed)
58. Tyson JE, Wright LL, Oh W, et al. Vitamin A supplementation for extremely-low-birth-weight infants. National Institute of Child Health and Human Development Neonatal Research Network. N Engl J Med. 1999;340(25):1962-1968. (PubMed)
59. Wardle SP, Hughes A, Chen S, Shaw NJ. Randomised controlled trial of oral vitamin A supplementation in preterm infants to prevent chronic lung disease. Arch Dis Child Fetal Neonatal Ed. 2001;84(1):F9-F13. (PubMed)
60. Ambalavanan N, Kennedy K, Tyson J, Carlo WA. Survey of vitamin A supplementation for extremely-low-birth-weight infants: is clinical practice consistent with the evidence? J Pediatr. 2004;145(3):304-307. (PubMed)
61. Laughon MM. Vitamin A shortage and risk of bronchopulmonary dysplasia. JAMA Pediatr. 2014;168(11):995-996. (PubMed)
62. Tolia VN, Murthy K, McKinley PS, Bennett MM, Clark RH. The effect of the national shortage of vitamin a on death or chronic lung disease in extremely low-birth-weight infants. JAMA Pediatr. 2014;168(11):1039-1044. (PubMed)
63. Gadhia MM, Cutter GR, Abman SH, Kinsella JP. Effects of early inhaled nitric oxide therapy and vitamin A supplementation on the risk for bronchopulmonary dysplasia in premature newborns with respiratory failure. J Pediatr. 2014;164(4):744-748. (PubMed)
64. Meyer S, Gortner L, NeoVita ATI. Early postnatal additional high-dose oral vitamin A supplementation versus placebo for 28 days for preventing bronchopulmonary dysplasia or death in extremely low birth weight infants. Neonatology. 2014;105(3):182-188. (PubMed)
65. Babu TA, Sharmila V. Vitamin A supplementation in late pregnancy can decrease the incidence of bronchopulmonary dysplasia in newborns. J Matern Fetal Neonatal Med. 2010;23(12):1468-1469. (PubMed)
66. Thorne-Lyman AL, Fawzi WW. Vitamin A and carotenoids during pregnancy and maternal, neonatal and infant health outcomes: a systematic review and meta-analysis. Paediatr Perinat Epidemiol. 2012;26 Suppl 1:36-54. (PubMed)
67. Imdad A, Yakoob MY, Sudfeld C, Haider BA, Black RE, Bhutta ZA. Impact of vitamin A supplementation on infant and childhood mortality. BMC Public Health. 2011;11 Suppl 3:S20. (PubMed)
68. Gogia S, Sachdev HS. Vitamin A supplementation for the prevention of morbidity and mortality in infants six months of age or less. Cochrane Database Syst Rev. 2011(10):CD007480. (PubMed)
69. Fisker AB, Bale C, Rodrigues A, et al. High-dose vitamin A with vaccination after 6 months of age: a randomized trial. Pediatrics. 2014;134(3):e739-748. (PubMed)
70. Aaby P, Martins CL, Garly ML, et al. Non-specific effects of standard measles vaccine at 4.5 and 9 months of age on childhood mortality: randomised controlled trial. BMJ. 2010;341:c6495. (PubMed)
71. Benn CS, Martins CL, Fisker AB, et al. Interaction between neonatal vitamin A supplementation and timing of measles vaccination: a retrospective analysis of three randomized trials from Guinea-Bissau. Vaccine. 2014;32(42):5468-5474. (PubMed)
72. Huiming Y, Chaomin W, Meng M. Vitamin A for treating measles in children. Cochrane Database Syst Rev. 2005(4):CD001479. (PubMed)
73. American Academy of Pediatrics Committee on Infectious Diseases: Vitamin A treatment of measles. Pediatrics. 1993;91(5):1014-1015. (PubMed)
74. Bello S, Meremikwu MM, Ejemot-Nwadiaro RI, Oduwole O. Routine vitamin A supplementation for the prevention of blindness due to measles infection in children. Cochrane Database Syst Rev. 2014;1:CD007719. (PubMed)
75. Omenn GS, Goodman GE, Thornquist MD, et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N Engl J Med. 1996;334(18):1150-1155. (PubMed)
76. Goodman GE, Thornquist MD, Balmes J, et al. The Beta-Carotene and Retinol Efficacy Trial: incidence of lung cancer and cardiovascular disease mortality during 6-year follow-up after stopping beta-carotene and retinol supplements. J Natl Cancer Inst. 2004;96(23):1743-1750. (PubMed)
77. Palozza P, Simone R, Mele MC. Interplay of carotenoids with cigarette smoking: implications in lung cancer. Curr Med Chem. 2008;15(9):844-854. (PubMed)
78. Cheng TY, Goodman GE, Thornquist MD, et al. Estimated intake of vitamin D and its interaction with vitamin A on lung cancer risk among smokers. Int J Cancer. 2014;135(9):2135-2145. (PubMed)
79. Cortes-Jofre M, Rueda JR, Corsini-Munoz G, Fonseca-Cortes C, Caraballoso M, Bonfill Cosp X. Drugs for preventing lung cancer in healthy people. Cochrane Database Syst Rev. 2012;10:CD002141. (PubMed)
80. Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369(2):111-121. (PubMed)
81. Booij MT, Van De Kerkhof PC. Acitretin revisited in the era of biologics. J Dermatolog Treat. 2011;22(2):86-89. (PubMed)
82. Orfanos CE, Zouboulis CC. Oral retinoids in the treatment of seborrhoea and acne. Dermatology. 1998;196(1):140-147. (PubMed)
83. Thielitz A, Gollnick H. Topical retinoids in acne vulgaris: update on efficacy and safety. Am J Clin Dermatol. 2008;9(6):369-381. (PubMed)
84. Vishwanathan R, Johnson EJ. Eye disease. In: Erdman JJ, Macdonald I, Zeisel S, eds. Present Knowledge in Nutrition. 10th ed: John Wiley & Sons, Ltd; 2012:939-981.
85. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795-1809. (PubMed)
86. Berson EL, Rosner B, Sandberg MA, et al. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(6):761-772. (PubMed)
87. Sibulesky L, Hayes KC, Pronczuk A, Weigel-DiFranco C, Rosner B, Berson EL. Safety of <7500 RE (<25000 IU) vitamin A daily in adults with retinitis pigmentosa. Am J Clin Nutr. 1999;69(4):656-663. (PubMed)
88. Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. 2nd edition ed: Thomson Reuters; 2008.
89. Penniston KL, Tanumihardjo SA. The acute and chronic toxic effects of vitamin A. Am J Clin Nutr. 2006;83(2):191-201. (PubMed)
90. Russell RM. The vitamin A spectrum: from deficiency to toxicity. Am J Clin Nutr. 2000;71(4):878-884. (PubMed)
91. World Health Organization. Guideline - Vitamin A supplementation in pregnant women. Geneva 2011.
92. Bozzo P, Chua-Gocheco A, Einarson A. Safety of skin care products during pregnancy. Can Fam Physician. 2011;57(6):665-667. (PubMed)
93. Michaelsson K, Lithell H, Vessby B, Melhus H. Serum retinol levels and the risk of fracture. N Engl J Med. 2003;348(4):287-294. (PubMed)
94. Promislow JH, Goodman-Gruen D, Slymen DJ, Barrett-Connor E. Retinol intake and bone mineral density in the elderly: the Rancho Bernardo Study. J Bone Miner Res. 2002;17(8):1349-1358. (PubMed)
95. Feskanich D, Singh V, Willett WC, Colditz GA. Vitamin A intake and hip fractures among postmenopausal women. JAMA. 2002;287(1):47-54. (PubMed)
96. Rejnmark L, Vestergaard P, Charles P, et al. No effect of vitamin A intake on bone mineral density and fracture risk in perimenopausal women. Osteoporos Int. 2004;15(11):872-880. (PubMed)
97. Sowers MF, Wallace RB. Retinol, supplemental vitamin A and bone status. J Clin Epidemiol. 1990;43(7):693-699. (PubMed)
98. Ballew C, Galuska D, Gillespie C. High serum retinyl esters are not associated with reduced bone mineral density in the Third National Health And Nutrition Examination Survey, 1988-1994. J Bone Miner Res. 2001;16(12):2306-2312. (PubMed)
99. Wu AM, Huang CQ, Lin ZK, et al. The relationship between vitamin a and risk of fracture: meta-analysis of prospective studies. J Bone Miner Res. 2014;29(9):2032-2039. (PubMed)
100. Conaway HH, Henning P, Lerner UH. Vitamin a metabolism, action, and role in skeletal homeostasis. Endocr Rev. 2013;34(6):766-797. (PubMed)
101. Johansson S, Melhus H. Vitamin A antagonizes calcium response to vitamin D in man. J Bone Miner Res. 2001;16(10):1899-1905. (PubMed)
102. Caire-Juvera G, Ritenbaugh C, Wactawski-Wende J, Snetselaar LG, Chen Z. Vitamin A and retinol intakes and the risk of fractures among participants of the Women's Health Initiative Observational Study. Am J Clin Nutr. 2009;89(1):323-330. (PubMed)
103. Lieber CS. Relationships between nutrition, alcohol use, and liver disease. Alcohol Res Health. 2003;27(3):220-231. (PubMed)
Vitamina B6
Contenido
Resumen
- La vitamina B6 y su derivado piridoxal 5’-fosfato (PLP) son esenciales en más de 100 enzimas mayormente involucradas en el metabolismo de las proteínas. (Más información)
- Altos niveles de homocisteína circulante están asociados con un incremento en el riesgo de padecer enfermedades cardiovasculares. Ensayos controlados aleatorios han demostrado que la suplementación con vitaminas B, incluyendo la vitamina B6 podrían efectivamente reducir los niveles de homocisteína. Sin embargo, la disminución de la homocisteína por las vitaminas B ha fallado en disminuir el riesgo de resultados cardiovasculares adversos en individuos de alto riesgo. (Más información)
- Creciente evidencia de estudios clínicos y experimentales sugiere que la inflamación sistémica subyacente en la mayoría de las enfermedades crónicas podría perjudicar el metabolismo de la vitamina B6. (Más información)
- Aunque la suplementación con vitamina B6 y otras vitaminas B no ha sido asociada con un mejoramiento del rendimiento cognitivo o el retraso del deterioro cognitivo en las personas mayores, estudios recientes sugieren que la vitamina B6 podría ayudar a reducir el riesgo de depresión tardía. (Más información)
- Dosis farmacológicas de la vitamina B6 son usadas para tratar convulsiones en errores congénitos raros del metabolismo de la vitamina B6. También, ensayos controlados aleatorios apoyan el uso de la vitamina B6 para tratar nauseas matutinas en mujeres embarazadas y sugieren un posible beneficio en el control del síndrome premenstrual y del síndrome del túnel carpiano. (Más información)
- La vitamina B6 es encontrada en una variedad de alimentos, incluyendo pescado, aves de corral, nueces, legumbres, papas, y bananas. (Más información)
-
Varios medicamentos, incluyendo fármacos anti-tuberculosis, anti-parkinsonianos, fármacos anti-inflamatorios no esteroideos, y anticonceptivos orales, podrían interferir con el metabolismo de la vitamina B6. (Más información)
La vitamina B6 es una vitamina hidrosoluble que se aisló por primera vez en la década de 1930. El termino vitamina B6 se refiere a seis formas comunes conocidas como piridoxal, piridoxina (piridoxol), piridoxamina y sus formas fosforiladas. El derivado éster fosfato, piridoxal 5’-fosfato (PLP), es la forma co-enzimática bioactiva que participa en más de un 4% de todas las reacciones enzimáticas (Figura 1) (1-3).

Función
La vitamina B6 debe ser obtenida de la dieta, debido a que los humanos no pueden sintetizarla. La PLP juega un papel vital en la función de aproximadamente 100 enzimas que catalizan reacciones químicas esenciales en el cuerpo humano (4). Las enzimas dependientes de PLP han sido clasificadas en cinco clases estructurales conocidas como plegamiento tipo I-V (5):
• Plegamiento Tipo I- familia aspartato aminotransferasa
• Plegamiento Tipo II- familia triptófano sintasa
• Plegamiento Tipo III- familia alanina racemasa
• Plegamiento Tipo IV- familia D-aminoácido aminotransferasa
• Plegamiento tipo V- familia glicógeno fosforilasa
Las muchas reacciones biológicas catalizadas por las enzimas dependientes de PLP están involucradas en procesos bilógicos esenciales, tales como la biosíntesis de la hemoglobina y amino ácidos, así como también el metabolismo de los ácidos grasos. Cabe destacar que la PLP también funciona como una coenzima para la glucógeno fosforilasa, una enzima que cataliza la liberación de la glucosa del glicógeno almacenado. Mucha de la PLP en el cuerpo humano se encuentra en el músculo unida a la glucógeno fosforilasa. La PLP es también una coenzima en las reacciones que generan glucosa a partir de aminoácidos, un proceso conocido como gluconeogénesis (6).
Funcionamiento del sistema nervioso
En el cerebro, la enzima L-aminoácido aromático descarboxilasa cataliza la síntesis de dos neurotransmisor principales: la serotonina, a partir del aminoácido triptófano y la dopamina a partir de la L-3,4 dihidroxifenilalanina (L-Dopa). Otros neurotransmisores, incluyendo la glicina, D-serina, glutamato, histamina, y el ácido γ-aminobutírico (GABA), también son sintetizados en reacciones catalizadas por enzimas dependientes de PLP (7).
Función y síntesis de la hemoglobina
Las funciones de la PLP como coenzima del ácido 5-aminolevulínico sintasa el cual está involucrado en la síntesis del grupo hemo, un componente de la hemoglobina que contiene hierro. La hemoglobina se encuentra en los eritrocitos y es fundamental en su capacidad para transportar oxígeno a lo largo del cuerpo. Tanto el piridoxal como la PLP son capaces de unirse a la molécula de hemoglobina e intervenir en su capacidad para captar y liberar el oxígeno. Sin embargo, se desconoce su impacto sobre la entrega normal del oxígeno a los tejidos (6, 8). La deficiencia de vitamina B6 podría perjudicar la síntesis de la hemoglobina y conducir a una anemia microcítica (3).
Metabolismo del triptófano
La deficiencia en otra vitamina B, la niacina, es fácilmente evitada por ingestas dietarías adecuadas. El requerimiento dietario de la niacina y la coenzima de la niacina, nicotinamida adenina dinucleótido (NAD) puede ser también satisfecha, aunque en un grado bastante limitado, debido al catabolismo del amino acido esencial triptófano en la vía triptófano-quinurenina (Figura 2). Reacciones clave en esta vía son la PLP dependiente; en particular, la PLP es el cofactor para la enzima quinureninasa, la cual cataliza la conversión de 3-hidroxiquinurenina a ácido 3-hidroxiantranílico. Una reducción en la disponibilidad de la PLP parece afectar principalmente la actividad de la quinureninasa, limitando la producción de NAD y llevando a altas concentraciones de quinurenina, 3-hidroxiquinurenina y ácido xanturénico en la sangre y orina (Figura 2) (9). De este modo, mientras que la restricción de la vitamina B6 dietaría afecta la síntesis de la NAD a partir del triptófano, niveles adecuados de la PLP ayudan a mantener la formación de la NAD a partir del triptófano (10). El efecto de la insuficiencia de la vitamina B6 en la activación inmune e inflamación podría estar parcialmente relacionada al papel de la PLP en el metabolismo del triptófano-quinurenina (véase Prevención de Enfermedades).
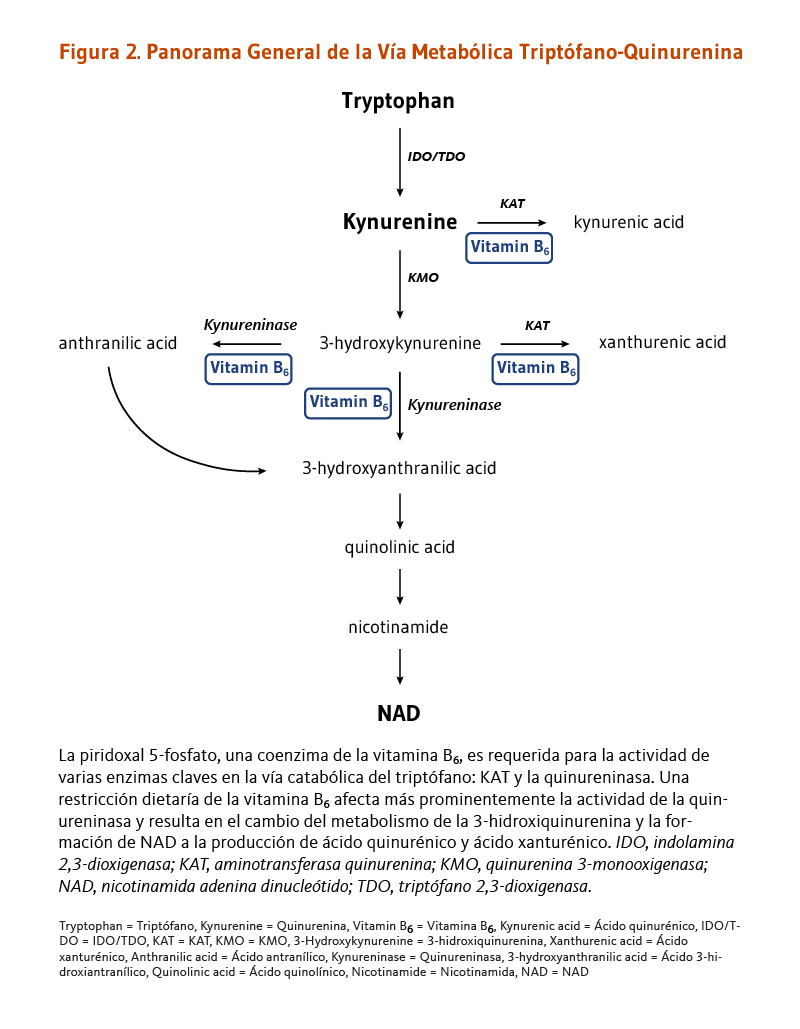
Función hormonal
Las hormonas esteroidales, como el estrógeno y la testosterona, ejercen sus efectos en el cuerpo al unirse a receptores de hormonas esteroidales en el núcleo de células diana. Los receptores nucleares se unen a secuencias reguladoras específicas en el ADN alterando la transcripción de genes diana. Estudios experimentales han descubierto un mecanismo por el cual la PLP podría afectar la actividad de los receptores esteroidales y disminuir sus efectos en la expresión de genes. Se encontró que la PLP pudo interactuar con la RIP140/NRIP1 un represor de los receptores nucleares conocido por su papel en la biología reproductiva (11). A pesar de todo, investigación adicional es necesaria para confirmar que esta interacción puede mejorar la actividad represiva de RIP140/NRIP1 en la expresión de genes mediada por receptores esteroidales. Si la actividad de los receptores esteroidales para el estrógeno, progesterona, testosterona, u otras hormonas esteroidales puede ser inhibida por la PLP, es posible que el estatus de la vitamina B6 influencie el riesgo de desarrollar enfermedades impulsadas por las hormonas esteroides, como el cáncer de próstata o seno (6).
Síntesis de ácidos nucleicos
La síntesis de ácidos nucleicos a partir de precursores de timidina y purinas depende de coenzimas de folato. La ruta biosintética de novo del timidilato (dTMP) involucra tres enzimas: la dihidrofolato reductasa (DHFR), la serina hidroximetiltransferasa (SHMT) y la timidilato sintasa (TS) (Figura 3). La PLP sirve como coenzima para la SHMT, la cual cataliza las conversiones simultaneas de serina a glicina y de tetrahidrofolato (THF) a 5,10-metileno THF. Esta última molécula es el donante de un carbono para la generación del dTMP a partir del dUMP por la TS.
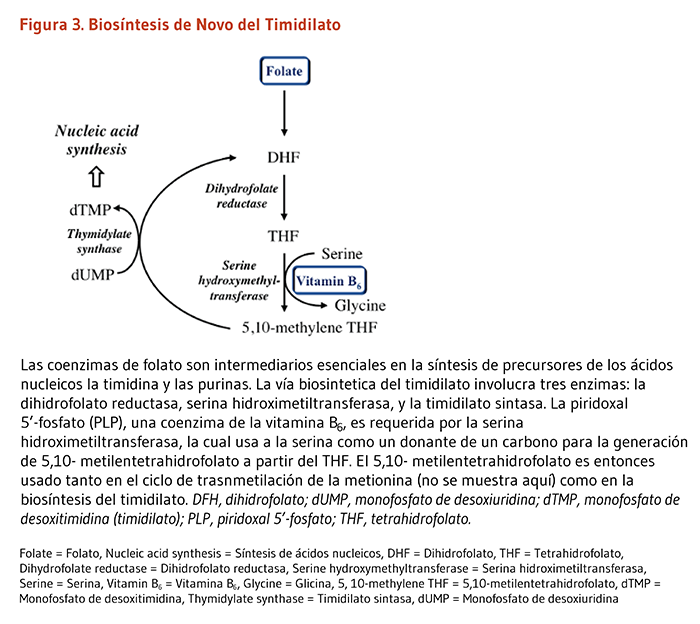
Deficiencia
Una severa deficiencia de vitamina B6 es poco común. Se piensa que alcohólicos se encuentran en un mayor riesgo de una deficiencia debido a ingestas dietarías bajas y a una alteración del metabolismo de la vitamina. A principios de 1950 se observaron convulsiones en infantes como resultado de una deficiencia severa de vitamina B6 provocada por un error en la fabricación de la fórmula infantil (7). También han sido reportados patrones de electroencefalogramas (EEG) alterados en adultos con una deficiencia de vitamina B6. Otros síntomas neurológicos observados en la deficiencia severa de vitamina B6 incluyen irritabilidad, depresión y confusión; síntomas adicionales incluyen inflamación de la lengua, dolor o úlceras en la boca y úlceras en la piel de las comisuras de la boca (12).
La Ingesta Diaria Recomendada (IDR)
Debido a que la vitamina B6 está involucrada en muchos aspectos del metabolismo, especialmente en vías metabólicas del amino ácido, la ingesta de proteína de un individuo es probable que influya en el requerimiento de vitamina B6 (13). A diferencia de las recomendaciones previas, la Junta de Nutrición y Alimentos (JNA) del Instituto de Medicina no expresó la IDR de la vitamina B6 más reciente en términos de la ingesta de proteínas, aunque la relación se consideró en el establecimiento de la IDR (14). La IDR actual fue revisada por la Junta de Nutrición y Alimentos (JNA) en 1998 y se muestra en la Tabla 1.
| Etapa de la Vida | Edad | Machos (mg/día) | Hembras (mg/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 0.1 (IA) | 0.1 (IA) |
| Infantes | 7-12 meses | 0.3 (IA) | 0.3 (IA) |
| Niños | 1-3 años | 0.5 | 0.5 |
| Niños | 4-8 años | 0.6 | 0.6 |
| Niños | 9-13 años | 1.0 | 1.0 |
| Adolescentes | 14-18 años | 1.3 | 1.2 |
| Adultos | 19-50 años | 1.3 | 1.3 |
| Adultos | 51 años y más | 1.7 | 1.5 |
| Embarazo | Todas las edades | - | 1.9 |
| Período de lactancia | Todas las edades | - | 2.0 |
Prevención de Enfermedades
Disfunción inmune
Varias reacciones enzimáticas en la vía triptófano-quinurenina dependen de la coenzima de la vitamina B6, piridoxal-5'-fosfato (PLP) (véase Figura 2 arriba) (véase Metabolismo del triptófano). Esta vía es conocida por estar activada durante las respuestas inmunes pro-inflamatorias y juega un papel crítico en la tolerancia inmune del feto durante el embarazo (15). Intermediarios claves en la vía triptófano-quinurenina están involucrados en la regulación de las respuestas inmunes. Se ha encontrados que varios derivados del triptófano inducen la muerte (apoptosis) o bloquean la proliferación de ciertos tipos de células inmunitarias, como los linfocitos (en particular los linfocitos T colaboradores 1). Estos también pueden inhibir la producción de citoquinas pro-inflamatorias (revisado en 15). Existe evidencia que sugiere que una ingesta adecuada de vitamina B6 es importante para una función óptima del sistema inmunitario, especialmente en individuos mayores (16, 17). A pesar de todo, la inflamación crónica que desencadena la degradación del triptófano y subyace en muchas enfermedades (ej. enfermedades cardiovasculares y canceres) podría precipitar la perdida de la PLP e incrementar los requerimientos de la vitamina B6. Investigación adicional es necesaria para evaluar si las ingestas elevadas de vitamina B6 más que la actual IDR podría prevenir y/o invertir las deficiencias del sistema inmunológico (véase también Vitamina B6 y la inflamación).
Enfermedades cardiovasculares
El uso de suplementos multivitamínicos (incluyendo la vitamina B6) ha sido asociado con un riesgo 24% menor de padecer una enfermedad coronaria cardíaca (ECC) incidental en un estudio prospectivo exhaustivo en 80,082 mujeres de la cohorte del Estudio de Salud de Enfermeras estadounidense (18). Usando cuestionarios de frecuencia de alimentos, los autores observaron que las mujeres en el quintil más alto de la ingesta de vitamina B6 proveniente tanto de alimentos como suplementos (mediana, 4.6 mg/día) tuvieron un riesgo 34% menor de ECC en comparación con aquellas en el quintil más bajo (mediana, 1.1 mg/día). La ECC se caracteriza por la estenosis (estrechamiento) anormal de las arterias coronarias (generalmente debido a la aterosclerosis), la cual puede resultar en un infarto al miocardio (ataque al corazón) potencialmente fatal. Más recientemente, un estudio prospectivo que dio seguimiento a una cohorte japonesa de más de 40,000 individuos de edad media por 11.5 años reporto un riesgo 48% menor de un infarto al miocardio en aquellos en el quintil más alto (promedio, 1.6 mg/día) en comparación con aquellos en el quintil más bajo (promedio 1.3 mg/día) de las ingestas de vitamina B6 en usuarios no suplementados (19).
Estudios basados en la observación tempranos han también demostrado una asociación entre niveles subóptimos de piridoxal-5'-fosfato (PLP) en el plasma, niveles elevados de homocisteína en la sangre, y un riesgo incrementado de enfermedades cardiovasculares (20-22). Investigación más reciente ha confirmado que un estatus bajo de PLP en el plasma es un factor de riesgo para ECC. En un estudio de caso y control, el cual incluyo 184 participantes con ECC y 516 controles sanos, niveles bajos de PLP en el plasma (<30 nanomoles/litro) fueron asociados con casi el doble del riesgo de padecer ECC cuando se comparó con altos niveles de PLP (≥30 nanomoles/litro) (23). En un estudio caso-control anidado basado en la cohorte del Estudio de Salud de Enfermeras e incluyendo 144 casos de infarto al miocardio (de las cuales 21 fueron fatales), las mujeres con niveles de PLP en la sangre en el cuartil más alto (≥70 nanomoles/litro) tuvieron un 79% menos riesgo de padecer un infarto al miocardio en comparación con aquellas en el cuartil más bajo (<27.9 nanomoles/litro) (24).
La vitamina B6 y la homocisteína
Incluso niveles moderadamente elevados de homocisteína en la sangre han sido asociados con un incremento en el riesgo de enfermedades cardiovasculares (ECV), incluyendo insuficiencia cardiaca, ECC, infarto al miocardio, y accidentes cerebrovasculares (25). Durante la digestión de proteínas, los aminoácidos, incluyendo la metionina, son liberados. La metionina es un aminoácido esencial y precursor de la S-adenosilmetionina (SAM), el donante de metilo universal para la mayoría de las reacciones de metilación, incluyendo la metilación del ADN, ARN, proteínas y fosfolípidos (Figura 4). La homocisteína es un intermediario en el metabolismo de la metionina. Los individuos sanos utilizan dos vías diferentes para regenerar la metionina de la homocisteína en el ciclo de la remetilacion de la metionina (Figura 5). Una vía se basa en la metionina sintasa dependiente de la vitamina B12 y el donante de metilo, el 5-metil tetrahidrofolato (un derivado del folato), para convertir la homocisteína de regreso a metionina. La otra reacción es catalizada por la betaína homocisteína metiltransferasa, la cual utiliza la betaína como fuente de grupos metilo para la formación de la metionina a partir de la homocisteína. Por otra parte, dos enzimas dependientes de la PLP son requeridas para convertir la homocisteína al amino acido cisteína en la vía de transulfuración de la homocisteína: la cistationina β sintasa y la cistationina γ liasa (Figura 5). Así, la cantidad de homocisteína en la sangre podría ser influenciada por el estatus nutricional de por lo menos tres vitaminas B, es decir el folato, la vitamina B12 y la vitamina B6.


Deficiencias en una o en todas estas vitaminas B podrían afectar tanto los procesos de remetilación como de transulfuración y resultar en niveles anormalmente elevados de homocisteína. Un estudio temprano encontró que la suplementación con vitamina B6 pudo disminuir los niveles de homocisteína después de que una dosis oral de metionina fue administrada (es decir una prueba de carga de metionina (26), pero la suplementación con vitamina B6 podría no ser efectiva en la disminución de los niveles de homocisteína en ayunas. En un reciente estudio conducido en nueve voluntarios jóvenes saludables, se encontró que el aumento de la homocisteína durante el periodo postprandrial (después de una comida) era mayor con una deficiencia de vitamina B6 marginal (nivel promedio de PLP en el plasma de 19 nanomoles/litro) en comparación con la suficiencia de vitamina B6 (nivel promedio de PLP en el plasma de 49 nanomoles/litro) (27). Los autores reportaron una mayor tasa de la síntesis de cistationina con la restricción de la vitamina B6, sugiriendo que el catabolismo de la homocisteína en la transulfuración podría ser mantenido o mejorado en respuesta a una reducción marginal en la disponibilidad de PLP. A pesar de todo, la proporción del flujo entre el ciclo de la metionina y la vía de transulfuración pareció favorecer el despeje de la homocisteína más bien por la remetilación que por la transulfuración en seis de nueve participantes (27).
Numerosos ensayos controlados aleatorios, muchos basados en sujetos con hiperhomocisteinemia y disfunción vascular existentes han demostrado que la suplementación con ácido fólico, sola o combinada con la vitamina B6 y la vitamina B12, pudo efectivamente reducir las concentraciones de homocisteína en el plasma en ayunas. En 19 estudios de intervención recientemente incluidos en un meta-análisis, las reducciones en el nivel de homocisteína en la sangre después de una suplementación con vitamina B oscilo entre un 7.6% y un 51.7% en comparación con los niveles al inicio del estudio (28). En contraste, estudios que suplementaron a individuos con solo vitamina B6 han usualmente fallado en mostrar un efecto en los niveles de homocisteína en la sangre en ayunas (29, 30). De las tres vitaminas B suplementarias, el ácido fólico parece ser el determinante principal en la regulación de los niveles de homocisteína en ayunas cuando no hay una deficiencia de vitamina B12 o vitamina B6 (31). A pesar de todo, el efecto de la disminución de la homocisteína en la reducción del riesgo de ECV es debatido. Un reciente meta-análisis de nueve ensayos controlados aleatorios reporto una reducción del 10% en casos de accidentes cerebrovasculares con vitaminas B suplementarias, con mayores beneficios para sujetos de alto riesgo (ej. Aquellos con enfermedad renal) (32). Sin embargo, la mayoría de las revisiones sistemáticas y meta-análisis de estudios de intervención de la vitamina B hasta la fecha han indicado una falta de causalidad entre la disminución de los niveles de homocisteína en ayunas y la prevención de eventos cardiovasculares (28, 33-35). Por otra parte, ensayos de suplementación con vitamina B en sujetos de alto riesgo no han resultado en cabios significativos en el grosor íntima-media carotídeo (CIMT) y la dilatación mediada por flujo (DMF) de la arteria braquial, dos marcadores de la salud vascular usada para evaluar la progresión aterosclerótica (36). Finalmente, en el Ensayo de Intervención de la Vitamina B de Noruega Occidental (WENBIT), un ensayo controlado aleatorio, doble ciego con placebo en 87 sujetos con sospecha de ECC, la suplementación con vitamina B6 (40 mg/día de piridoxina) por un promedio de 10 meses no tuvo efecto en la progresión de la estenosis coronaria, evaluada por la angiografía cuantitativa (37).
Se ha sugerido que la terapia antiplaquetaria usada en la prevención primaria de ECV podría interferir con el efecto de la disminución de la homocisteína por las vitaminas B en el riesgo de ECV (38). En este contexto, un análisis post-hoc de un subgrupo del ensayo multicéntrico, aleatorio, doble ciego, controlado con placebo VITATOPS (39) propuso que el pequeño beneficio de la reducción de la homocisteína por las vitaminas B podría ser cancelada en pacientes tratados con fármacos antiplaquetarios (40). A pesar de todo, el beneficio de la suplementación con vitamina B en la prevención primaria (es decir, en usuarios no antiplaquetarios) falta por ser establecida.
La vitamina B6 y la inflamación
Un creciente cuerpo de evidencia actualmente sugiere que un estatus bajo de vitamina B6 pudiese incrementar el riesgo de enfermedades cardiovasculares a través de mecanismos independientes de la disminución de homocisteína (41-43). Marcadores de la activación inmune e inflamación han sido asociados con la hiperhomocisteinemia (niveles de homocisteina >15 micromoles/litro) en individuos con enfermedades coronarias cardíacas (ECC) (44). De hecho, la inflamación está involucrada en las primeras etapas de la aterosclerosis en las cuales los lípidos se depositan en las plaquetas (conocidos como ateromas) dentro de las paredes arteriales y aumentan el riesgo de ECC (45). En un estudio de caso y control que incluyo 267 pacientes con ECC y 475 controles saludables, las concentraciones de PLP en el plasma fueron inversamente correlacionadas con los niveles de dos marcadores de la inflamación sistémica, la proteína C reactiva (PCR) y el fibrinógeno (46). A pesar de todo, el estudio sugirió que niveles subóptimos de PLP (<36.3 nanomoles/litro) podrían contribuir a un riesgo incrementado de ECC independientemente de la inflamación ya que el riesgo se mantuvo sin cambios después del ajuste de los marcadores de la inflamación (razón de momios (RM) sin ajustar: 1.71 vs. RM multivariada ajustada: 1.73). Además, el análisis de los marcadores de inflamación en 2,686 participantes del US National Health and Nutrition Examination Survey (NHANES) del 2003-2004 indico que las concentraciones de la PCR del suero fueron inversamente relacionadas a las ingestas totales de la vitamina B6 (tanto de alimentos como suplementos). Específicamente, el riesgo de tener niveles de PCR en el suero de más de 10 mg/L (correspondientes a una alta actividad inflamatoria) fue 57% mayor en individuos con ingestas de vitamina B6 menores de 2 mg/día en comparación con aquellos con ingestas iguales a o mayores de 5 mg/día (41). Además, la prevalencia de un estatus inadecuado de vitamina B6 (niveles de PLP en el plasma <20 nanomoles/litro) con ingestas menores de 5 mg/día fue sistemáticamente mayor en individuos con concentraciones altas de PCR en el suero frente a las bajas (>10 mg/L vs. ≤3 mg/L), sugiriendo que la inflamación podría perjudicar el metabolismo de la vitamina B6. Estas observaciones fueron confirmadas en el estudio de otra cohorte (Framingham Offspring Study) en el cual el estatus de la vitamina B6 fue ligado a un puntaje inflamatorio general basado en los niveles de 13 marcadores de inflamación (incluyendo PCR, fibrinógeno, factor de necrosis tumoral α, e Interleucina-6) (42). Específicamente, los niveles de la PLP en el plasma fueron 24% menor en individuos en el tercil más alto frente a aquellos en el tercil más bajo del puntaje inflamatorio. Por otra parte, la correlación inversa entre los niveles de PLP y los puntajes inflamatorios permanecieron significativos independientemente de la ingesta de vitamina B6, cuestionando nuevamente la naturaleza de esta relación. Curiosamente, un reciente análisis de datos recolectados del estudio WENBIT demostró que la inflamación sistémica estuvo asociada con un incremento en la degradación de piridoxal (PL) a ácido 4-piridóxico (AP), apoyando el uso de la proporción PA:(PL+PLP) como un marcador de ambos el estatus de la vitamina B6 y la inflamación sistémica (47). Finalmente, mientras que la inflamación podría contribuir a disminuir el estatus de la vitamina B6, evidencia actual falla en apoyar un papel para la vitamina B6 en el control de la inflamación en pacientes con enfermedades cardiovasculares (48, 49).
Deterioro cognitivo y la enfermedad de Alzheimer
Algunos estudios basados en la observación han sido ligados al deterioro cognitivo y la enfermedad de Alzheimer en las personas de edad avanzada con estatus inadecuados de folato, vitamina B12 y vitamina B6 (50). A pesar de todo, la relación entre las vitaminas B y la salud cognitiva en el envejecimiento es complicada por tanto la prevalencia de la hiperhomocisteinemia como por las señales de inflamación sistémica en personas de edad avanzada (51). Por un lado, debido a que la inflamación puede perjudicar el metabolismo de la vitamina B6, niveles bajos de la PLP en el suero pueden bien ser causados por los procesos relacionados al envejecimiento en lugar de ser causados por la malnutrición. Por el otro lado, la homocisteína elevada del suero puede posiblemente ser un factor de riesgo para el declive cognitivo en personas de edad avanzada, aunque el tema aún está bajo debate. Específicamente, el meta-análisis de 19 ensayos aleatorios controlados con placebo de la suplementación de vitamina B fallo en reportar alguna diferencia en varios parámetros de la función cognitiva entre los grupos de tratamiento y placebo, a pesar de que el tratamiento efectivamente disminuyo los niveles de homocisteína (52). En un estudio aleatorio, doble ciego, controlado con placebo reciente de 2,695 sobrevivientes de accidentes cerebrovasculares con o sin alteraciones cognitivas, una suplementación diaria con 2 mg de ácido fólico, 0.5 mg de vitamina B12 y 25 mg de vitamina B6 por 3.4 años resulto en reducciones significativas en los niveles medios de homocisteína (por un 28% y 43% en pacientes con alteraciones cognitivas y sin ellas respectivamente) en comparación con el placebo. A pesar de todo, la intervención de la vitamina B no tuvo efecto en tanto la incidencia de deterioros cognitivos recién diagnosticados como en parámetros del rendimiento cognitivo cuando se compara con el placebo (53). En contraste, otro ensayo controlado con placebo reciente encontró que un régimen diario de vitamina B que llevo a una disminución significativa de la homocisteína en individuos de edad avanzada de alto riesgo pudo limitar la atrofia progresiva de la materia gris de regiones del cerebro asociadas con el proceso del Alzheimer (54). A pesar de todo, los autores atribuyeron los cambios en los niveles de homocisteína principalmente a la vitamina B12. Debido a los descubrimientos mixtos, en la actualidad no está claro si la suplementación con vitaminas B podría atenuar el deterioro cognitivo en las personas mayores. Evidencia es necesaria para determinar si las deficiencias de vitamina B marginales, las cuales son relativamente comunes en las personas mayores, podrían incluso contribuir a deterioros de la función cognitiva asociados con la edad, o si ambas resultan de procesos asociados con el envejecimiento y/o enfermedad.
Depresión
La depresión tardía o en personas de edad avanzada es un trastorno común que a veces ocurre después de enfermedades agudas, como fractura de cadera o accidentes cerebrovasculares (55, 56). La coexistencia de síntomas de depresión y un estatus bajo de la vitamina B6 (niveles de PLP en el plasma ≤20 nanomoles/litro) ha sido reportada en algunos estudios de corte transversal (57, 58). En un estudio prospectivo de 3,503 personas viviendo libremente de 65 años y mayores del Chicago Health and Aging Project, ingestas totales de vitamina B6 (pero ingestas no dietarías únicamente) fueron inversamente correlacionadas con la incidencia de síntomas depresivos durante un periodo medio de seguimiento de 7.2 años (59). En un ensayo aleatorio, doble ciego, controlado con placebo en 563 individuos que sufrieron de un accidente cerebrovascular reciente, una suplementación diaria de 2 mg de ácido fólico, 0.5 mg de vitamina B12, y 25 mg de vitamina B6, redujo a la mitad el riesgo de desarrollar un episodio depresivo mayor durante un periodo de seguimiento medio de 7.1 años (60). Esta reducción del riesgo fue asociada con un nivel 25% inferior de la homocisteína plasmática en pacientes suplementados en comparación a los controles. Evidencia adicional es requerida para evaluar si las vitaminas B podrían ser incluidas en el tratamiento habitual de personas mayores en alto riesgo de depresión.
Cáncer
La inflamación crónica que subyace a la mayoría de canceres podría aumentar la degradación de la vitamina B6 (véase La vitamina B6 y la inflamación). Además, el requerimiento de PLP en el ciclo de la metionina, el catabolismo de la homocisteína, y la síntesis de timidilato que disminuyen el estatus de la vitamina B6 puede contribuir al comienzo y/o progresión de tumores. La revisión sistemática de nueve estudios prospectivos encontró tanto asociaciones inversas como positivas entre las ingestas de vitamina B6 y el riesgo de cáncer colorrectal (CCR) (61). Evidencia inconsistente con respecto a la relación entre las ingestas de vitamina B6 y el cáncer de seno fue también reportada recientemente en un meta-análisis (62). A pesar de todo, un estudio prospectivo que dio seguimiento a aproximadamente 500,000 adultos mayores por nueve años observo que el riesgo de cáncer esofágico y gástrico fue menor en participantes en el quintil más alto que aquellos en el quintil más bajo de las ingestas totales de vitamina B6 (valores medios, 2.7 mg/día vs. 1.4 mg/día) (63). Adicionalmente, un meta-análisis de cuatro estudios de caso y control anidados reporto una reducción del 48% en el riesgo de CCR en el cuartil más elevado frente al cuartil más bajo del nivel de PLP de la sangre (61). Otro meta-análisis de cinco estudios de caso y control anidados encontró que los niveles más altos frente a los más bajos de PLP en el suero estuvieron asociados con un riesgo 29% menor de cáncer de seno en mujeres posmenopáusicas, pero no premenopáusicas (62).
Muy pocos ensayos aleatorios controlados con placebo que investigan la naturaleza de la asociación entre vitaminas B y el riesgo de cáncer se han enfocado en la vitamina B6. Dos estudios tempranos conducidos en sujetos con una enfermedad coronaria cardiaca fallaron en observar algún beneficio de la vitamina B6 suplementaria (40 mg/día) en el riesgo y mortalidad del CCR (revisado en 64). Un estudio aleatorio, doble ciego, controlado con placebo conducido en 1,470 mujeres con un alto riesgo cardiovascular mostro que la suplementación diaria con 2.5 mg de ácido fólico, 1 mg de vitamina B12, y 50 mg de vitamina B6 por un periodo de tratamiento promedio de 7.3 años no tuvo efecto en el riesgo de desarrollar adenoma colorrectal cuando se comparó con el placebo (65).
Cálculos renales
Un estudio prospectivo de gran magnitud examino la relación entre la ingesta de vitamina B6 y la ocurrencia de cálculos renales sintomáticos en mujeres. A un grupo de más de 85,000 mujeres sin un historial previo de cálculos renales se les dio seguimiento por sobre 14 años, y aquellas que consumieron 40 mg o más de vitamina B6 diariamente tuvieron solo dos tercios del riesgo de desarrollar cálculos renales en comparación con aquellas que consumieron 3 mg o menos (66). Sin embargo, en un grupo de ms de 45,000 hombres con un seguimiento de 14 años, no se encontró alguna asociación entre la ingesta de vitamina B6 y la ocurrencia de cálculos renales (67). Datos experimentales limitados han sugerido que la suplementación con altas dosis de piridoxamina puede ayudar a disminuir la formación de cálculos renales de oxalato de calcio y reducir los niveles de oxalato urinario, un determinante importante de la formación de cálculos renales de oxalato de calcio (68, 69). Actualmente, la relación entre la ingesta de vitamina B6 y el riesgo de desarrollar cálculos renales requiere de estudios adicionales antes de que alguna recomendación puede ser hecha.
Tratamiento de Enfermedades
Los suplementos de vitamina B6 en dosis farmacológicas (es decir, en dosis mucho más grandes que las necesarias para prevenir una deficiencia) han sido utilizados en un intento de tratar una amplia variedad de condiciones, algunas de las cuales se comentan a continuación.
Enfermedades metabólicas
Algunos trastornos metabólicos congénitos raros, incluyendo la epilepsia dependiente de piridoxina (EDP) y la deficiencia de fosfato de piridoxamina oxidasa (PNPO), son la causa de la aparición temprana de encefalopatías epilépticas las cuales se encontró son sensibles a dosis farmacológicas de vitamina B6. En individuos afectados por EDP y la deficiencia de PNPO, la biodisponibilidad de la PLP es limitada, y el tratamiento con piridoxina y/o PLP han sido usados para aliviar o abolir las convulsiones epilépticas que caracterizan estas condiciones (70, 71). La terapia con piridoxina, en conjunto con la restricción de proteína dietaría, es también usada en el manejo de la homocistinuria sensible a la vitamina B6 causada por la deficiencia de la enzima dependiente de la PLP, la cistationina β sintasa (72).
Malestar matutino
Las náuseas y vómitos en el embarazo (NVE), frecuentemente referidos como malestar matutino, pueden afectar hasta un 85% de las mujeres durante el inicio del embarazo y usualmente dura entre 12 a 16 semanas (73). La vitamina B6 ha sido usada desde 1940 para tratar las náuseas durante el embarazo. Originalmente la vitamina B6 se incluyó en el medicamento Bendectina, el cual era recetado para el tratamiento del malestar matutino y que posteriormente fue retirado del mercado debido a preocupaciones no probadas que incrementaban el riesgo de defectos de nacimiento. La vitamina B6 por si sola es considerada segura durante el embarazo y ha sido usada en mujeres embarazadas sin ninguna evidencia de daño fetal (74). Los resultados de dos ensayos doble ciegos, controlados con placebo que incluyeron 401 mujeres embarazadas que usaron 25 mg de piridoxina cada ocho horas por tres días (75) o 10 mg de piridoxina cada ocho horas por cinco días (76), sugiriendo que la vitamina B6 puede ser beneficiosa en la reducción de náuseas. Una revisión sistemática reciente de ensayos controlados aleatorios en síntomas del malestar matutino durante el inicio del embarazo encontró que la vitamina B6 suplementaria fue algo eficaz (77). Cabe señalar que las NVE usualmente se resuelven sin ningún tratamiento, por lo que es difícil llevar a cabo ensayos bien controlados. Más recientemente los síntomas del malestar matutino fueron evaluados usando puntuaciones de la Cuantificación Única de Emesis en el Embarazo (PUQE) en un estudio aleatorio, doble ciego, controlado con placebo conducido en 256 mujeres embarazadas (7-14 semanas de gestación) que padecían de NVE (78). La suplementación con piridoxina y el fármaco doxilamina mejoró significativamente los síntomas de NVE, como se evaluó con puntuaciones más bajas de la PUQE en comparación con el placebo. Por otra parte, más de las mujeres suplementadas con piridoxina-doxilamina (48.9%) que aquellas con el tratamiento de placebo (32.8%) pidieron la continuación de su tratamiento al final de la prueba de 15 días. Los Colegios Americanos y Canadienses de Obstetricia y Ginecología han recomendado el uso de la vitamina B6 (hidrocloruro de piridoxina, 10 mg) y el succinato de doxilamina (10 mg) como terapia de primera line para las NVE (73).
Síndrome premenstrual
El síndrome premenstrual SPM hace referencia a un grupo de síntomas incluyendo, aunque no limitado a la fatiga, irritabilidad, mal humor/depresión, la retención de líquidos, y la sensibilidad mamaria, que comienzan poco después de la ovulación (mitad del ciclo) y que disminuyen con el inicio de la menstruación (el periodo menstrual). Una revisión sistemática y meta-análisis de nueve ensayos aleatorios, controlados con placebo sugirieron que la vitamina B6 suplementaria, de hasta 100 mg/día, puede ser de valor para tratar el SPM, incluyendo síntomas del estado de ánimo, sin embargo, sólo se pueden extraer conclusiones limitadas debido a que la mayoría de los estudios eran de baja calidad (79). Otra revisión reciente de 13 estudios controlados aleatorios también enfatizo la necesidad por evidencia concluyente antes de poder hacer recomendaciones (80).
Depresión
La importancia de las enzimas dependientes de la PLP en la síntesis de varios neurotransmisores (vea Funcionamiento del sistema nervioso) ha llevado a investigadores a considerar si la deficiencia de vitamina B6 puede contribuir al comienzo de los síntomas depresivos (véase Prevención de Enfermedades). Existe evidencia limitada que sugiere que la vitamina B6 suplementaria puede tener una eficacia terapéutica en el manejo de la depresión. En un ensayo aleatorio controlado con placebo conducido en 225 pacientes de edad avanzada hospitalizados por una enfermedad aguda, una intervención de seis meses con suplementos minerales/multivitamínicos mejoro el estatus de la vitamina B y disminuyo el número y severidad de los síntomas depresivos al compararse con el placebo (81). Además, mientras la ingesta del suplemento efectivamente redujo los niveles de homocisteína en el plasma en comparación con el placebo, el efecto de la suplementación en los síntomas depresivos al final de la prueba fue mayor en sujetos tratados en el cuartil más bajo de los niveles de homocisteína frente a aquellos en el cuartil más alto (≤10 micromoles/litro vs. ≥16.1 micromoles/litro) (82). A pesar de todo, la etiología de la depresión tardía no es aun clara y evidencia actualmente carece de sugerir si las vitaminas B suplementarias (incluyendo la vitamina B6) podrían aliviar los síntomas depresivos.
Síndrome del túnel carpiano
El síndrome del túnel carpiano causa entumecimiento, dolor y debilidad de la mano y los dedos debido a la compresión del nervio mediano en la muñeca. Esto podría derivar de una lesión de la muñeca por estrés repetitivo o de la hinchazón de tejido blando, lo que algunas veces ocurre con el embarazo o el hipotiroidismo. Estudios tempranos por el mismo investigador sugirieron que la suplementación con 100 a 200 mg/día de vitamina B6 por varios meses podría mejorar los síntomas del síndrome del túnel carpiano en individuos con un estatus bajo de vitamina B6 (83, 84). Además, un estudio transversal en 137 hombres que no tomaban suplementos vitamínicos encontró que la disminución de los niveles de PLP en la sangre se asociaba con un incremento del dolor, hormigueo y despertar nocturno, todos síntomas del síndrome del túnel carpiano (85). Sin embargo, estudios que utilizan mediciones electrofisiológicas de la conducción del nervio mediano han fallado ampliamente en encontrar una asociación entre la deficiencia de vitamina B6 y el síndrome del túnel carpiano (86). Mientras que algunos estudios han notado algún alivio sintomático con la suplementación de la vitamina B6, ensayos doble ciegos controlados con placebo no han encontrado que la vitamina B6 sea generalmente eficaz en el tratamiento del síndrome del túnel carpiano (86). Sin embargo, a pesar de su eficacia equívoca, la suplementación con vitamina B6 es a veces usada en la terapia complementaria en un intento de evitar la cirugía de la mano. Los pacientes que toman altas dosis de vitamina B6 deben ser aconsejados por un médico y monitoreados en caso de síntomas relacionados con la toxicidad de la vitamina B6 (véase Toxicidad) (87).
Fuentes
Fuentes alimenticias
El análisis de los datos recolectados en el US NHANES 2003-2004 ha indicado que las ingestas de vitamina B6 de los alimentos solo promedian alrededor de 1.9 mg/día (88). Sin embargo, a pesar de los valores muy por encima de la actual IDR, las ingestas totales de vitamina B6 (combinando los alimentos y suplementos) por debajo de los 2 mg/día parecieron estar asociados con proporciones relativamente altas de un estatus bajo de vitamina B6 en todos los grupos de edad (véase Suplementos). Muchos alimentos vegetales contienen una forma única de vitamina B6 denominada glucósido de piridoxina; esta forma de vitamina B6 parece ser sólo la mitad de biodisponible que la vitamina B6 de otras fuentes alimenticias o suplementos (7). Se ha encontrado que la vitamina B6 en una dieta variada es biodisponible en aproximadamente un 75% (14). En la mayoría de los casos, incluir en la dieta alimentos ricos en vitamina B6 debería ser suficiente como para satisfacer la IDR actual. Sin embargo, aquellos que siguen una dieta vegetariana estricta podrían necesitar incrementar su ingesta de vitamina B6 consumiendo alimentos fortificados con vitamina B6 o tomando un suplemento. Algunos alimentos que son relativamente ricos en vitamina B6 y su contenido de vitamina B6 en miligramos (mg) se muestran en la Tabla 2. Para más información sobre el contenido de nutrientes de alimentos específicos, revise la base de datos de composición de los alimentos del USDA.
Suplementos
La vitamina B6 está disponible como clorhidrato de piridoxina en suplementos multivitamínicos, de vitaminas del complejo B y en suplementos de vitamina B6 (89). En el NHANES 2003-2004, se reportó un estatus bajo de la vitamina B6 (nivel de PLP en el plasma <20 nanomoles/litro) en un 24% de los usuarios no suplementados y en un 11% en los usuarios suplementados. Además, las ingestas totales de vitamina B6 (proveniente de alimentos y suplementos) menores de 2 mg/día fueron asociadas con altas proporciones de niveles bajos de PLP en el plasma: 16% en hombres de entre 13 a 54 años, 24% en mujeres que menstruaban, y 26% en individuos de 65 años y mayores. Finalmente, se encontró que la prevalencia de niveles bajos de PLP era mayor en individuos que consumían menos de 2 mg/día de vitamina B6 en comparación con ingestas más altas. Por ejemplo, solo el 14% de los hombre y mujeres de 65 y mayores tuvieron valores bajos de PLP con ingestas totales de vitamina B6 de entre 2-2.9 mg/día en comparación con el 26% en aquellos que consumieron menos de 2 mg/día de vitamina B6 (88).
Seguridad
Toxicidad
Debido a que sólo se han documentado efectos adversos provenientes de suplementos de vitamina B6 y nunca de fuentes alimenticias, sólo se discute sobre la seguridad respecto a la forma suplementaria de la vitamina B6 (piridoxina). Aunque la vitamina B6 es una vitamina hidrosoluble y se secreta en la orina, la suplementación a largo plazo con dosis muy altas de piridoxina podría resultar en dolorosos síntomas neurológicos conocidos como neuropatía sensorial. Los síntomas incluyen dolor y entumecimiento de las extremidades y, en casos severos, dificultad para caminar. Típicamente, la neuropatía sensorial se desarrolla con dosis de piridoxina excesivas de 1,000 mg por día. Sin embargo, ha habido algunos reportes de casos de personas que desarrollaron neuropatías sensoriales con dosis de menos de 500 mg al día por un periodo de uno o más meses. A pesar de todo, ninguno de los estudios en los que se realizó un examen neurológico objetivo, reportó evidencia de daño a los nervios sensoriales con ingestas por debajo de los 200 mg de piridoxina diariamente (90). Para prevenir la neuropatía sensorial en virtualmente todos los individuos, la Junta de Nutrición y Alimentos del Instituto de Medicina estableció el nivel máximo de ingesta tolerable (NM) de piridoxina en 100 mg/día para adultos (Tabla 3) (14). Debido a que generalmente los estudios controlados con placebo han fallado en demostrar los beneficios terapéuticos de las dosis elevadas de piridoxina, existe poco fundamento para exceder el NM de 100 mg/día.
Interacción con drogas/fármacos
Ciertos medicamentos interfieren con el metabolismo de la vitamina B6, por lo que algunas personas podrían ser vulnerables a una deficiencia de vitamina B6 si no toman vitamina B6 suplementaria. En el análisis del NHANES 2003-2004, significativamente más usuarias actuales y usuarias pasadas de anticonceptivos orales (AO) de entre mujeres que menstruaban tuvieron niveles bajos de PLP en el plasma en comparación con aquellas que nunca usaron anticonceptivos orales, sugiriendo que el contenido de estrógeno puede interferir con el metabolismo de la vitamina B6 (véase Efectos secundarios de los anticonceptivos orales) (88). Medicamentos antituberculosos (ej. isoniazida y cicloserina), la penicilamina quelante de metales, y fármacos antiparkinsonianos como L-Dopa pueden formar complejos con la vitamina B6 y limitar su biodisponibilidad, de esta manera creando una deficiencia funcional. La biodisponibilidad de la PLP puede ser también reducida por las metilxantinas como la teofilina usada para tratar ciertas condiciones respiratorias (7). El uso a largo plazo de fármacos antinflamatorios no esteroideos (AINE: ej. celecoxib y naproxeno) puede también perjudicar el metabolismo de la vitamina B6 (91). Inversamente, se ha descubierto que altas dosis de vitamina B6 disminuyen la eficiencia de dos anticonvulsivos, el fenobarbital y la fenitoína, y de la L-Dopa (9, 90).
Efectos secundarios de los anticonceptivos orales
Debido a que la vitamina B6 es requerida para el metabolismo del amino ácido triptófano, la prueba de carga de triptófano (un análisis de los metabolitos de triptófano después de una dosis oral de triptófano) ha sido usada como una evaluación funcional del estatus de la vitamina B6. Pruebas de carga de triptófano anormales en mujeres que tomaron altas dosis de anticonceptivos orales (AO) en 1960 y 1970 sugirieron que estas mujeres estaban deficientes de vitamina B6, lo cual condujo a la prescripción de altas dosis de vitamina B6 (100-150 mg/día) en mujeres que toman AO. Sin embargo, la mayoría de los otros índices del estatus de la vitamina B6 fueron normales en mujeres con altas dosis de anticonceptivos orales, y el contenido de estrógeno de los anticonceptivos orales pareció ser más probable responsable de la anormalidad en el metabolismo del triptófano (88). A pesar de todo, más recientemente, el uso de formulaciones de dosis más bajas también se ha asociado con la insuficiencia de vitamina B6 (88, 92). Aunque aún se desconoce si los AO en realidad perjudican el metabolismo de la vitamina B6 o meramente afectan la distribución tisular de la PLP, el uso de AO puede colocar a las mujeres en un riesgo mayor de una deficiencia de vitamina B6 cuando descontinúen los AO y queden embarazadas (93). También es necesario determinar si los usuarios de AO están en un riesgo mayor padecer enfermedades cardiovasculares a pesar de poseer niveles normales de homocisteína. Finalmente, aunque altas dosis de vitamina B6 (piridoxina) no han demostrado algún beneficio en la prevención del riesgo de efectos secundarios de los AO (94), el uso de suplementos de vitamina B6 puede estar justificado en usuarias actuales y pasadas de anticonceptivos orales.
Recomendación del Instituto Linus Pauling
El Instituto Linus Pauling apoya la IDR para la vitamina B6. El Instituto Linus Pauling recomienda que todos los adultos mayores tomen un suplemento multivitamínico/mineral diariamente, el cual usualmente contiene por lo menos 2 mg de vitamina B6. Esta cantidad está ligeramente por encima de la IDR pero 50 veces aún menor que el nivel máximo de ingesta tolerable (NM) establecido por la Junta de Alimentos y Nutrición (véase Seguridad).
Adultos mayores (>50 años)
Estudios metabólicos tempranos han indicado que el requerimiento para la vitamina B6 en adultos mayores es de aproximadamente 2 mg al día (95). A pesar de todo, el análisis de la encuesta de población de EE.UU. (NHANES) 2003-2004 mostro que un estatus adecuado de vitamina B6 y niveles bajos de homocisteína fueron asociados con ingestas totales de vitamina B6 iguales a o por encima de los 3 mg/día en personas de 65 años y mayores (88). El Instituto Linus Pauling recomienda que adultos mayores tomen un suplemento multivitamínico/mineral, el cual provee de al menos 2.0 mg de vitamina B6 diariamente.
Autores y Críticos
Escrito en 2000 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Febrero de 2002 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Noviembre de 2007 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Mayo de 2014 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Junio de 2014 por:
Jesse F. Gregory, Ph.D.
Profesor, Ciencia de los Alimentos y Nutrición Humana
Universidad de Florida
Traducido al Español en 2015 por:
Silvia Vazquez Lima
Instituto Linus Pauling
Universidad Estatal de Oregon
La actualización del artículo del 2014 fue suscrito en parte, por un donación de Bayer Consumer Care AG, Basel, Suiza.
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Derechos de autoría 2000-2024 Instituto Linus Pauling
Referencias
1. Dakshinamurti S, Dakshinamurti K. Vitamin B6. In: Zempleni J, Rucker RB, McCormick DB, Suttie JW, eds. Handbook of Vitamins. 4th ed. New York: CRC Press (Taylor & Fracis Group); 2007:315-359.
2. Galluzzi L, Vacchelli E, Michels J, et al. Effects of vitamin B6 metabolism on oncogenesis, tumor progression and therapeutic responses. Oncogene. 2013;32(42):4995-5004. (PubMed)
3. McCormick DB. Vitamin B6. In: Bowman BA, Russell RM, eds. Present Knowledge in Nutrition. Vol. I. Washington, D.C.: International Life Sciences Institute; 2006:269-277.
4. Da Silva VR, Russell KA, Gregory JF 3rd. Vitamin B6. In: Erdman JW Jr., Macdonald IA, Zeisel SH. Present Knowldege in Nutrition. 10th ed: Wiley-Blackwell; 2012:307-320.
5. Eliot AC, Kirsch JF. Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu Rev Biochem. 2004;73:383-415. (PubMed)
6. Leklem JE. Vitamin B-6. In: Shils M, Olson JA, Shike M, Ross AC, eds. Modern Nutrition in Health and Disease. 9th ed. Baltimore: Williams & Wilkins; 1999:413-422.
7. Clayton PT. B6-responsive disorders: a model of vitamin dependency. J Inherit Metab Dis. 2006;29(2-3):317-326. (PubMed)
8. Schnackerz KD, Benesch RE, Kwong S, Benesch R, Helmreich EJ. Specific receptor sites for pyridoxal 5'-phosphate and pyridoxal 5'-deoxymethylenephosphonate at the α and β NH2-terminal regions of hemoglobin. J Biol Chem. 1983;258(2):872-875. (PubMed)
9. Rios-Avila L, Nijhout HF, Reed MC, Sitren HS, Gregory JF, 3rd. A mathematical model of tryptophan metabolism via the kynurenine pathway provides insights into the effects of vitamin B-6 deficiency, tryptophan loading, and induction of tryptophan 2,3-dioxygenase on tryptophan metabolites. J Nutr. 2013;143(9):1509-1519. (PubMed)
10. Oxenkrug G. Insulin resistance and dysregulation of tryptophan-kynurenine and kynurenine-nicotinamide adenine dinucleotide metabolic pathways. Mol Neurobiol. 2013;48(2):294-301. (PubMed)
11. Huq MD, Tsai NP, Lin YP, Higgins L, Wei LN. Vitamin B6 conjugation to nuclear corepressor RIP140 and its role in gene regulation. Nat Chem Biol. 2007;3(3):161-165. (PubMed)
12. Leklem JE. Vitamin B6. In: Machlin L, ed. Handbook of Vitamins. New York: Marcel Decker Inc; 1991:341-378.
13. Hansen CM, Shultz TD, Kwak HK, Memon HS, Leklem JE. Assessment of vitamin B-6 status in young women consuming a controlled diet containing four levels of vitamin B-6 provides an estimated average requirement and recommended dietary allowance. J Nutr. 2001;131(6):1777-1786. (PubMed)
14. Food and Nutrition Board, Institute of Medicine. Vitamin B6. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B6, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington D.C.: National Academies Press; 1998:150-195. (National Academies Press)
15. Paul L, Ueland PM, Selhub J. Mechanistic perspective on the relationship between pyridoxal 5'-phosphate and inflammation. Nutr Rev. 2013;71(4):239-244. (PubMed)
16. Meydani SN, Ribaya-Mercado JD, Russell RM, Sahyoun N, Morrow FD, Gershoff SN. Vitamin B-6 deficiency impairs interleukin 2 production and lymphocyte proliferation in elderly adults. Am J Clin Nutr. 1991;53(5):1275-1280. (PubMed)
17. Talbott MC, Miller LT, Kerkvliet NI. Pyridoxine supplementation: effect on lymphocyte responses in elderly persons. Am J Clin Nutr. 1987;46(4):659-664. (PubMed)
18. Rimm EB, Willett WC, Hu FB, et al. Folate and vitamin B6 from diet and supplements in relation to risk of coronary heart disease among women. JAMA. 1998;279(5):359-364. (PubMed)
19. Ishihara J, Iso H, Inoue M, et al. Intake of folate, vitamin B6 and vitamin B12 and the risk of CHD: the Japan Public Health Center-Based Prospective Study Cohort I. J Am Coll Nutr. 2008;27(1):127-136. (PubMed)
20. Folsom AR, Nieto FJ, McGovern PG, et al. Prospective study of coronary heart disease incidence in relation to fasting total homocysteine, related genetic polymorphisms, and B vitamins: the Atherosclerosis Risk in Communities (ARIC) study. Circulation. 1998;98(3):204-210. (PubMed)
21. Robinson K, Arheart K, Refsum H, et al. Low circulating folate and vitamin B6 concentrations: risk factors for stroke, peripheral vascular disease, and coronary artery disease. European COMAC Group.Circulation. 1998;97(5):437-443. (PubMed)
22. Robinson K, Mayer EL, Miller DP, et al. Hyperhomocysteinemia and low pyridoxal phosphate. Common and independent reversible risk factors for coronary artery disease. Circulation. 1995;92(10):2825-2830. (PubMed)
23. Lin PT, Cheng CH, Liaw YP, Lee BJ, Lee TW, Huang YC. Low pyridoxal 5'-phosphate is associated with increased risk of coronary artery disease. Nutrition. 2006;22(11-12):1146-1151. (PubMed)
24. Page JH, Ma J, Chiuve SE, et al. Plasma vitamin B(6) and risk of myocardial infarction in women. Circulation. 2009;120(8):649-655. (PubMed)
25. Gerhard GT, Duell PB. Homocysteine and atherosclerosis. Curr Opin Lipidol. 1999;10(5):417-428. (PubMed)
26. Ubbink JB, Vermaak WJ, van der Merwe A, Becker PJ, Delport R, Potgieter HC. Vitamin requirements for the treatment of hyperhomocysteinemia in humans. J Nutr. 1994;124(10):1927-1933. (PubMed)
27. Lamers Y, Coats B, Ralat M, Quinlivan EP, Stacpoole PW, Gregory JF, 3rd. Moderate vitamin B-6 restriction does not alter postprandial methionine cycle rates of remethylation, transmethylation, and total transsulfuration but increases the fractional synthesis rate of cystathionine in healthy young men and women. J Nutr. 2011;141(5):835-842. (PubMed)
28. Huang T, Chen Y, Yang B, Yang J, Wahlqvist ML, Li D. Meta-analysis of B vitamin supplementation on plasma homocysteine, cardiovascular and all-cause mortality. Clin Nutr. 2012;31(4):448-454. (PubMed)
29. Bosy-Westphal A, Holzapfel A, Czech N, Muller MJ. Plasma folate but not vitamin B(12) or homocysteine concentrations are reduced after short-term vitamin B(6) supplementation. Ann Nutr Metab. 2001;45(6):255-258. (PubMed)
30. Lee BJ, Huang MC, Chung LJ, et al. Folic acid and vitamin B12 are more effective than vitamin B6 in lowering fasting plasma homocysteine concentration in patients with coronary artery disease. Eur J Clin Nutr. 2004;58(3):481-487. (PubMed)
31. Bostom AG, Carpenter MA, Kusek JW, et al. Homocysteine-lowering and cardiovascular disease outcomes in kidney transplant recipients: primary results from the Folic Acid for Vascular Outcome Reduction in Transplantation trial. Circulation. 2011;123(16):1763-1770. (PubMed)
32. Qin X, Huo Y, Xie D, Hou F, Xu X, Wang X. Homocysteine-lowering therapy with folic acid is effective in cardiovascular disease prevention in patients with kidney disease: a meta-analysis of randomized controlled trials. Clin Nutr. 2013;32(5):722-727. (PubMed)
33. Clarke R, Halsey J, Bennett D, Lewington S. Homocysteine and vascular disease: review of published results of the homocysteine-lowering trials. J Inherit Metab Dis. 2011;34(1):83-91. (PubMed)
34. Marti-Carvajal AJ, Sola I, Lathyris D, Karakitsiou DE, Simancas-Racines D. Homocysteine-lowering interventions for preventing cardiovascular events. Cochrane Database Syst Rev. 2013;1:CD006612. (PubMed)
35. Zhang C, Chi FL, Xie TH, Zhou YH. Effect of B-vitamin supplementation on stroke: a meta-analysis of randomized controlled trials. PLoS One. 2013;8(11):e81577. (PubMed)
36. Potter K, Hankey GJ, Green DJ, Eikelboom J, Jamrozik K, Arnolda LF. The effect of long-term homocysteine-lowering on carotid intima-media thickness and flow-mediated vasodilation in stroke patients: a randomized controlled trial and meta-analysis. BMC Cardiovasc Disord. 2008;8:24. (PubMed)
37. Loland KH, Bleie O, Blix AJ, et al. Effect of homocysteine-lowering B vitamin treatment on angiographic progression of coronary artery disease: a Western Norway B Vitamin Intervention Trial (WENBIT) substudy. Am J Cardiol. 2010;105(11):1577-1584. (PubMed)
38. Wang X, Qin X, Demirtas H, et al. Efficacy of folic acid supplementation in stroke prevention: a meta-analysis. Lancet. 2007;369(9576):1876-1882. (PubMed)
39. Vitatops Trial Study Group. B vitamins in patients with recent transient ischaemic attack or stroke in the VITAmins TO Prevent Stroke (VITATOPS) trial: a randomised, double-blind, parallel, placebo-controlled trial. Lancet Neurol. 2010;9(9):855-865. (PubMed)
40. Hankey GJ, Eikelboom JW, Yi Q, et al. Antiplatelet therapy and the effects of B vitamins in patients with previous stroke or transient ischaemic attack: a post-hoc subanalysis of VITATOPS, a randomised, placebo-controlled trial. Lancet Neurol. 2012;11(6):512-520. (PubMed)
41. Morris MS, Sakakeeny L, Jacques PF, Picciano MF, Selhub J. Vitamin B-6 intake is inversely related to, and the requirement is affected by, inflammation status. J Nutr. 2010;140(1):103-110. (PubMed)
42. Sakakeeny L, Roubenoff R, Obin M, et al. Plasma pyridoxal-5-phosphate is inversely associated with systemic markers of inflammation in a population of US adults. J Nutr. 2012;142(7):1280-1285. (PubMed)
43. Shen J, Lai CQ, Mattei J, Ordovas JM, Tucker KL. Association of vitamin B-6 status with inflammation, oxidative stress, and chronic inflammatory conditions: the Boston Puerto Rican Health Study. Am J Clin Nutr. 2010;91(2):337-342. (PubMed)
44. Schroecksnadel K, Grammer TB, Boehm BO, Marz W, Fuchs D. Total homocysteine in patients with angiographic coronary artery disease correlates with inflammation markers. Thromb Haemost. 2010;103(5):926-935. (PubMed)
45. Hartman J, Frishman WH. Inflammation and atherosclerosis: a review of the role of interleukin-6 in the development of atherosclerosis and the potential for targeted drug therapy. Cardiol Rev. 2014;22(3):147-151. (PubMed)
46. Friso S, Girelli D, Martinelli N, et al. Low plasma vitamin B-6 concentrations and modulation of coronary artery disease risk. Am J Clin Nutr. 2004;79(6):992-998. (PubMed)
47. Ulvik A, Midttun O, Pedersen ER, Eussen SJ, Nygard O, Ueland PM. Evidence for increased catabolism of vitamin B-6 during systemic inflammation. Am J Clin Nutr. 2014;100(1):250-255. (PubMed)
48. Bleie O, Semb AG, Grundt H, et al. Homocysteine-lowering therapy does not affect inflammatory markers of atherosclerosis in patients with stable coronary artery disease. J Intern Med. 2007;262(2):244-253. (PubMed)
49. Potter K, Lenzo N, Eikelboom JW, Arnolda LF, Beer C, Hankey GJ. Effect of long-term homocysteine reduction with B vitamins on arterial wall inflammation assessed by fluorodeoxyglucose positron emission tomography: a randomised double-blind, placebo-controlled trial. Cerebrovasc Dis. 2009;27(3):259-265. (PubMed)
50. Selhub J, Bagley LC, Miller J, Rosenberg IH. B vitamins, homocysteine, and neurocognitive function in the elderly. Am J Clin Nutr. 2000;71(2):614S-620S. (PubMed)
51. Pawelec G, Goldeck D, Derhovanessian E. Inflammation, ageing and chronic disease. Curr Opin Immunol. 2014;29C:23-28. (PubMed)
52. Ford AH, Almeida OP. Effect of homocysteine lowering treatment on cognitive function: a systematic review and meta-analysis of randomized controlled trials. J Alzheimers Dis. 2012;29(1):133-149. (PubMed)
53. Hankey GJ, Ford AH, Yi Q, et al. Effect of B vitamins and lowering homocysteine on cognitive impairment in patients with previous stroke or transient ischemic attack: a prespecified secondary analysis of a randomized, placebo-controlled trial and meta-analysis. Stroke. 2013;44(8):2232-2239. (PubMed)
54. Douaud G, Refsum H, de Jager CA, et al. Preventing Alzheimer's disease-related gray matter atrophy by B-vitamin treatment. Proc Natl Acad Sci U S A. 2013;110(23):9523-9528. (PubMed)
55. Hackett ML, Yapa C, Parag V, Anderson CS. Frequency of depression after stroke: a systematic review of observational studies. Stroke. 2005;36(6):1330-1340. (PubMed)
56. Lenze EJ, Munin MC, Skidmore ER, et al. Onset of depression in elderly persons after hip fracture: implications for prevention and early intervention of late-life depression. J Am Geriatr Soc. 2007;55(1):81-86. (PubMed)
57. Merete C, Falcon LM, Tucker KL. Vitamin B6 is associated with depressive symptomatology in Massachusetts elders. J Am Coll Nutr. 2008;27(3):421-427. (PubMed)
58. Pan WH, Chang YP, Yeh WT, et al. Co-occurrence of anemia, marginal vitamin B6, and folate status and depressive symptoms in older adults. J Geriatr Psychiatry Neurol. 2012;25(3):170-178. (PubMed)
59. Skarupski KA, Tangney C, Li H, Ouyang B, Evans DA, Morris MC. Longitudinal association of vitamin B-6, folate, and vitamin B-12 with depressive symptoms among older adults over time. Am J Clin Nutr. 2010;92(2):330-335. (PubMed)
60. Almeida OP, Marsh K, Alfonso H, Flicker L, Davis TM, Hankey GJ. B-vitamins reduce the long-term risk of depression after stroke: The VITATOPS-DEP trial. Ann Neurol. 2010;68(4):503-510. (PubMed)
61. Larsson SC, Orsini N, Wolk A. Vitamin B6 and risk of colorectal cancer: a meta-analysis of prospective studies. JAMA. 2010;303(11):1077-1083. (PubMed)
62. Wu W, Kang S, Zhang D. Association of vitamin B6, vitamin B12 and methionine with risk of breast cancer: a dose-response meta-analysis. Br J Cancer. 2013;109(7):1926-1944. (PubMed)
63. Xiao Q, Freedman ND, Ren J, Hollenbeck AR, Abnet CC, Park Y. Intakes of folate, methionine, vitamin B6, and vitamin B12 with risk of esophageal and gastric cancer in a large cohort study. Br J Cancer. 2014;110(5):1328-1333. (PubMed)
64. Zhang XH, Ma J, Smith-Warner SA, Lee JE, Giovannucci E. Vitamin B6 and colorectal cancer: current evidence and future directions. World J Gastroenterol. 2013;19(7):1005-1010. (PubMed)
65. Song Y, Manson JE, Lee IM, et al. Effect of combined folic acid, vitamin B(6), and vitamin B(12) on colorectal adenoma. J Natl Cancer Inst. 2012;104(20):1562-1575. (PubMed)
66. Curhan GC, Willett WC, Speizer FE, Stampfer MJ. Intake of vitamins B6 and C and the risk of kidney stones in women. J Am Soc Nephrol. 1999;10(4):840-845. (PubMed)
67. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol. 2004;15(12):3225-3232. (PubMed)
68. Chetyrkin SV, Kim D, Belmont JM, Scheinman JI, Hudson BG, Voziyan PA. Pyridoxamine lowers kidney crystals in experimental hyperoxaluria: a potential therapy for primary hyperoxaluria. Kidney Int. 2005;67(1):53-60. (PubMed)
69. Scheinman JI, Voziyan PA, Belmont JM, Chetyrkin SV, Kim D, Hudson BG. Pyridoxamine lowers oxalate excretion and kidney crystals in experimental hyperoxaluria: a potential therapy for primary hyperoxaluria. Urol Res. 2005;33(5):368-371. (PubMed)
70. Pearl PL, Gospe SM, Jr. Pyridoxine or pyridoxal-5'-phosphate for neonatal epilepsy: The distinction just got murkier. Neurology. 2014;82(16):1392-1394. (PubMed)
71. Stockler S, Plecko B, Gospe SM, Jr., et al. Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up. Mol Genet Metab. 2011;104(1-2):48-60. (PubMed)
72. Picker JD, Levy HL. Homocystinuria caused by cystathionine β-synthase deficiency. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews®. Seattle, Washington: University of Washington, Seattle 1993-2014. (PubMed)
73. Maltepe C, Koren G. The management of nausea and vomiting of pregnancy and hyperemesis gravidarum--a 2013 update. J Popul Ther Clin Pharmacol. 2013;20(2):e184-192. (PubMed)
74. Magee LA, Mazzotta P, Koren G. Evidence-based view of safety and effectiveness of pharmacologic therapy for nausea and vomiting of pregnancy (NVP). Am J Obstet Gynecol. 2002;186(5 Suppl Understanding):S256-261. (PubMed)
75. Sahakian V, Rouse D, Sipes S, Rose N, Niebyl J. Vitamin B6 is effective therapy for nausea and vomiting of pregnancy: a randomized, double-blind placebo-controlled study. Obstet Gynecol. 1991;78(1):33-36. (PubMed)
76. Vutyavanich T, Wongtra-ngan S, Ruangsri R. Pyridoxine for nausea and vomiting of pregnancy: a randomized, double-blind, placebo-controlled trial. Am J Obstet Gynecol. 1995;173(3 Pt 1):881-884. (PubMed)
77. Matthews A, Haas DM, O'Mathuna DP, Dowswell T, Doyle M. Interventions for nausea and vomiting in early pregnancy. Cochrane Database Syst Rev. 2014;3:CD007575. (PubMed)
78. Koren G, Clark S, Hankins GD, et al. Effectiveness of delayed-release doxylamine and pyridoxine for nausea and vomiting of pregnancy: a randomized placebo controlled trial. Am J Obstet Gynecol. 2010;203(6):571 e571-577. (PubMed)
79. Wyatt KM, Dimmock PW, Jones PW, Shaughn O'Brien PM. Efficacy of vitamin B-6 in the treatment of premenstrual syndrome: systematic review. BMJ. 1999;318(7195):1375-1381. (PubMed)
80. Whelan AM, Jurgens TM, Naylor H. Herbs, vitamins and minerals in the treatment of premenstrual syndrome: a systematic review. Can J Clin Pharmacol. 2009;16(3):e407-429. (PubMed)
81. Gariballa S, Forster S. Effects of dietary supplements on depressive symptoms in older patients: a randomised double-blind placebo-controlled trial. Clin Nutr. 2007;26(5):545-551. (PubMed)
82. Gariballa S. Testing homocysteine-induced neurotransmitter deficiency, and depression of mood hypothesis in clinical practice. Age Ageing. 2011;40(6):702-705. (PubMed)
83. Ellis J, Folkers K, Watanabe T, et al. Clinical results of a cross-over treatment with pyridoxine and placebo of the carpal tunnel syndrome. Am J Clin Nutr. 1979;32(10):2040-2046. (PubMed)
84. Ellis JM, Kishi T, Azuma J, Folkers K. Vitamin B6 deficiency in patients with a clinical syndrome including the carpal tunnel defect. Biochemical and clinical response to therapy with pyridoxine. Res Commun Chem Pathol Pharmacol. 1976;13(4):743-757. (PubMed)
85. Keniston RC, Nathan PA, Leklem JE, Lockwood RS. Vitamin B6, vitamin C, and carpal tunnel syndrome. A cross-sectional study of 441 adults. J Occup Environ Med. 1997;39(10):949-959. (PubMed)
86. Aufiero E, Stitik TP, Foye PM, Chen B. Pyridoxine hydrochloride treatment of carpal tunnel syndrome: a review. Nutr Rev. 2004;62(3):96-104. (PubMed)
87. Ryan-Harshman M, Aldoori W. Carpal tunnel syndrome and vitamin B6. Can Fam Physician. 2007;53(7):1161-1162. (PubMed)
88. Morris MS, Picciano MF, Jacques PF, Selhub J. Plasma pyridoxal 5'-phosphate in the US population: the National Health and Nutrition Examination Survey, 2003-2004. Am J Clin Nutr. 2008;87(5):1446-1454. (PubMed)
89. Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. Montvale: Medical Economics Company, Inc; 2001.
90. Bender DA. Non-nutritional uses of vitamin B6. Br J Nutr. 1999;81(1):7-20 (PubMed)
91. Chang HY, Tang FY, Chen DY, et al. Clinical use of cyclooxygenase inhibitors impairs vitamin B-6 metabolism. Am J Clin Nutr. 2013;98(6):1440-1449. (PubMed)
92. Lussana F, Zighetti ML, Bucciarelli P, Cugno M, Cattaneo M. Blood levels of homocysteine, folate, vitamin B6 and B12 in women using oral contraceptives compared to non-users. Thromb Res. 2003;112(1-2):37-41. (PubMed)
93. Wilson SM, Bivins BN, Russell KA, Bailey LB. Oral contraceptive use: impact on folate, vitamin B(6), and vitamin B(1)(2) status. Nutr Rev. 2011;69(10):572-583. (PubMed)
94. Villegas-Salas E, Ponce de Leon R, Juarez-Perez MA, Grubb GS. Effect of vitamin B6 on the side effects of a low-dose combined oral contraceptive. Contraception. 1997;55(4):245-248. (PubMed)
95. Ribaya-Mercado JD, Russell RM, Sahyoun N, Morrow FD, Gershoff SN. Vitamin B-6 requirements of elderly men and women. J Nutr. 1991;121(7):1062-1074. (PubMed)
Vitamina B12
Contenido
Resumen
- La vitamina B12, o cobalamina juega papeles importantes en el metabolismo del folato y en la síntesis del ciclo del acído cítrico intermedio, succinil-CoA. (Más informacíon)
- La deficiencia de vitamina B12 es comúnmente asociada con una inflamación crónica del estomago, que puede contribuir a un síndrome de malabsorción autoinmune de la vitamina B12 llamado anemia perniciosa y a un síndrome de malabsorción de vitamina B12 unida a los alimentos. Un deterioro de la absorción de la vitamina B12 puede causar una anemia megaloblástica y desordenes neurológicos en sujetos con deficiencia. (Más información)
- Una función normal del sistema digestivo requerida para una absorción de la vitamina B12 esta comúnmente deteriorada en individuos mayores de 60 años de edad, poniéndolos en riesgo de una deficiencia de vitamina B12. (Más información)
- La vitamina B12 y el folato son importantes para el metabolismo de la homocisteína. Niveles elevados de homocisteína en la sangre son un factor de riesgo para enfermedades cardiovasculares (ECV). Aunque la suplementación de vitamina B ha probado un control efectivo en los niveles de homocisteína, datos actuales de ensayos de intervención no han mostrado que al disminuir los niveles de homocisteína un riesgo de padecer ECV disminuya. (Más información)
- La preservación de la integridad del ADN depende del folato y la disponibilidad de la vitamina B12. Un estatus pobre de la vitamina B12 ha sido ligado a un incremento en el riesgo de padecer cáncer de seno en algunos, pero no todos, los estudios observacionales. Existe la necesidad de evaluar si la vitamina B12 suplementaria, junto con el acido fólico, pudieran ayudar a reducir la incidencia de cáncer de seno. (Más información)
- Un nivel bajo de vitamina B12 materno ha sido asociado con un riesgo incrementado de defectos del tubo neural (NTD, por sus siglas en inglés), pero aun se sabe si la suplementación de vitamina B12 podría ayudar a reducir el riesgo de NTD. (Más información)
- La vitamina B12 es esencial para la preservación de la vaina de la mielina alrededor de las neuronas y para la síntesis de neurotransmisores. Mientras que la hiperhomocisteinemia puede incrementar el riesgo de un defecto cognitivo, no está claro si la deficiencia de vitamina B12 contribuye al riesgo de demencia en los ancianos. Aunque la suplementación de vitamina B disminuye los niveles de homocisteína en sujetos de edad mayor, el beneficio a largo plazo es aún desconocido. (Más información)
- Ambos la depresión y la osteoporosis han sido ligadas a una disminución del estado de la vitamina B12 y niveles elevados de homocisteína. (Más información)
- Los productos de origen animal constituyen la fuente primaria de vitamina B12. Individuos de edad avanzada y vegetarianos se les aconseja el uso de alimentos y suplementos fortificados con vitamina B12 para satisfacer sus necesidades. (Más información)
- El uso a largo plazo de ciertos medicamentos, como inhibidores de la secreción del acido estomacal, pueden afectar adversamente la absorción de vitamina B12. (Más información)
La vitamina B12 tiene la estructura química más grande y compleja de todas las vitaminas. Es única entre las vitaminas debido a que contiene un ión metálico, el cobalto. Por esta razón cobalamina es el término usado para referirse a los compuestos que poseen actividad de la vitamina B12. La metilcobalamina y la 5-desoxiadenosilcobalamina son las formas de vitamina B12 utilizadas en el cuerpo humano (1). La forma de cobalamina usada en la mayoría de los suplementos nutricionales y alimentos fortificados, "cianocobalamina," es rápidamente convertida en 5-desoxiadenosil y metilcobalamina en el cuerpo. En los mamíferos, la cobalamina es un cofactor para solo dos enzimas, la metionina sintasa y la L-metilmalonil-CoA mutasa (2).
Función
Cofactor para la metionina sintasa
La metilcobalamina es requerida para el funcionamiento de la enzima folato dependiente, la metionina sintasa. Esta enzima es requerida para la síntesis del aminoácido metionina proveniente de la homocisteína. La metionina a su vez es requerida para la síntesis de S-adenosilmetionina, un donante del grupo metilo utilizado en muchas reacciones de metilación biológica, incluyendo la metilación de una serie de sitios dentro del ADN, ARN y las proteínas (3). La metilación aberrante del ADN y las proteínas, que causa alteraciones en la estructura de la cromatina y la expresión de genes, son una característica común de las células cancerosas. Un funcionamiento inadecuado de la metionina sintasa puede conducir a una acumulación de homocisteína, la cual ha sido asociada con un aumento del riesgo de enfermedades cardiovasculares (Figura 1).
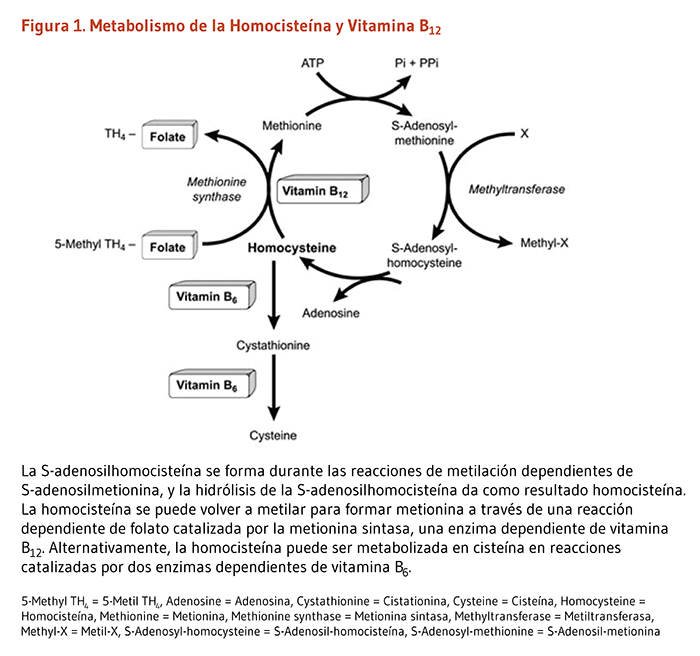
Cofactor para la L-metilmalonil-CoA mutasa
La 5-desoxiadenosilcobalamina es requerida por la enzima que cataliza la conversión de L-metilmalonil-CoA en succinil-CoA, que luego entra en el ciclo del ácido cítrico (Figura 2). El succinil CoA juega un importante papel en la producción de energía de lípidos y proteínas y es también requerido para la síntesis de la hemoglobina, el pigmento en los eritrocitos que transporta el oxígeno (3).
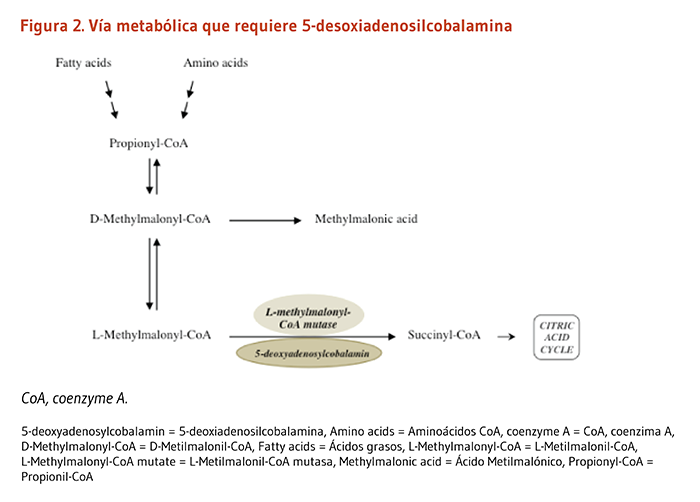
Deficiencia
En adultos sanos, la deficiencia de la vitamina B12 no es común, principalmente porque el suministro total del cuerpo puede exceder 2,500 μg, el desgaste diario es lento, y la ingesta dietaría de solo 2.4 μg/día es suficiente para mantener un nivel adecuado de vitamina B12 (vea IDR debajo) (4). En individuos de mayor edad, la deficiencia de vitamina B12 es más común principalmente debido a una absorción intestinal alterada que puede resultar de marginal a una deficiencia severa de la vitamina B12 en esta población.
Causas de una deficiencia de vitamina B12
La malabsorción intestinal más bien que una inadecuada ingesta dietaría, puede explicar la mayoría de los casos de deficiencia de vitamina B12 (5). La absorción de la vitamina B12 de los alimentos requiere una función normal del estomago, páncreas y el intestino delgado. El acido y las enzimas estomacales liberan la vitamina B12 de los alimentos, permitiéndole unirse a la proteína-R (también conocida como transcobalamina-1 o haptocorrina), encontrada en la saliva y fluidos gástricos. En el ambiente alcalino del intestino delgado, las proteínas-R son degradadas por enzimas pancreáticas, liberando la vitamina B12 para unirse al factor intrínseco (IF), una proteína secretada por células especializadas en el estomago. Los receptores en la superficie del íleon (parte final del intestino delgado) absorben el complejo IF-B12 solo en la presencia de calcio, el cual es suministrado por el páncreas (5). La vitamina B12 puede ser también absorbida por una difusión pasiva, pero este proceso es bastante ineficiente-solo aproximadamente un 1% de la absorción de la dosis de la vitamina B12 es absorbida pasivamente (2). Las frecuentes causas de la deficiencia de vitamina B12 son: (1) una condición autoinmune conocida como anemia perniciosa y (2) malabsorción de la vitamina B12 ligada a los alimentos. Aunque ambas causas han sido asociadas con una enfermedad inflamatoria crónica del estomago conocida como gastritis atrófica.
Gastritis atrófica
La gastritis atrófica se cree afecta del 10% al 30% de las personas mayores de 60 años de edad (6). Esta condición es frecuentemente asociada con la presencia de autoanticuerpos dirigidos hacia las células estomacales (vea Anemia perniciosa) y/o una infección debido a la bacteria Helicobacter pylori (H. pylori) (7). La infección por H. pylori induce a una inflamación crónica del estomago, la cual puede progresar a una enfermedad de ulcera péptica, gastritis atrófica, y/o cáncer gástrico en algunos individuos. Una disminución en la función gástrica en individuos con gastritis atrófica puede resultar en un sobrecrecimiento bacteriano en el intestino delgado y causar una malabsorción de la vitamina B12 ligada a los alimentos. Los niveles de la vitamina B12 en el suero, plasma, y fluidos gástricos son significativamente disminuidos en individuos con una infección por H. pylori y la erradicación de la bacteria ha mostrado que mejora significativamente las concentraciones de vitamina B12 en el suero (8).
Anemia perniciosa
Se ha estimado que la anemia perniciosa se encuentra presente en aproximadamente 2% de los individuos mayores de 60 años de edad (9). Aunque la anemia es con frecuencia un síntoma, la enfermedad es de hecho la etapa final de una inflamación autoinmune del estómago conocida como gastritis atrófica autoinmune, que resulta en la destrucción de las células del estómago por anticuerpos propios (autoanticuerpos). La destrucción progresiva de las células que recubren el estómago provoca una disminución en la secreción del ácido y las enzimas requeridas para liberar la vitamina B12 ligada a los alimentos. Los anticuerpos contra el factor intrínseco (IF) se unen al IF evitando la formación del complejo FI-B12, inhibiendo además la absorción de la vitamina B12. Alrededor del 20% de los familiares de los pacientes con anemia perniciosa también padecen la misma condición, sugiriendo una predisposición genética. También se piensa que la infección por H. pylori podría estar involucrada en la iniciación de la respuesta autoinmune en un subconjunto de individuos (10). Además ha sido reportada la co-ocurrencia de la gastritis atrófica autoinmune con otras condiciones autoinmunes, especialmente la tiroiditis autoinmune y la diabetes mellitus tipo 1 (11, 12).
Generalmente el tratamiento de la anemia perniciosa requiere de inyecciones de vitamina B12 para evitar la absorción intestinal. La suplementación oral en dosis elevadas es otra opción del tratamiento, porque al consumir 1.000 μg (1 mg)/día de vitamina B12 oralmente debería resultar en la absorción de cerca de 10 μg/día (1% de la dosis) por difusión pasiva. De hecho, la terapia oral en dosis elevadas es considerada tan efectiva como una inyección intramuscular (4).
Malabsorción de vitamina B12 ligada a los alimentos
La malabsorción de vitamina B12 ligada a los alimentos se define como un deterioro en la capacidad de absorber la vitamina B12 ligada a los alimentos o a proteínas, individuos con esta condición pueden completamente absorber la forma libre (13). Mientras la condición es la mayor causa de un estado pobre de la vitamina B12 en la población de edad avanzada, es usualmente asociada con la gastritis atrófica, una inflamación crónica de la mucosa del estomago que en última instancia resulta en la pérdida de las glándulas estomacales (atrofia) y el descenso de la producción del acido estomacal (vea Gastritis atrófica arriba). Debido a que el ácido estomacal es requerido para la liberación de la vitamina B12 desde las proteínas en los alimentos, la absorción de la vitamina B12 disminuye. La disminución de la producción de ácido estomacal también provee de un ambiente propicio para el sobrecrecimiento estomacal de bacterias anaeróbicas, las que además interfieren con la absorción de la vitamina B12 (3). Debido a que la vitamina B12 en los suplementos no está unida a las proteínas, y debido a que el factor intrínseco (IF) se encuentra aun disponible, la absorción de la vitamina B12 suplementaria no se reduce como en la anemia perniciosa. Así, individuos con una malabsorción de la vitamina B12 ligada a los alimentos no poseen un requerimiento mayor de vitamina B12; sólo la necesitan en su forma cristalina, la que se encuentra en alimentos fortificados y en suplementos alimenticios.
Otras causas de la deficiencia de vitamina B12
Otras causas de la deficiencia de vitamina B12 incluyen la extracción quirúrgica del estómago o de porciones del intestino delgado donde se localizan los receptores para el complejo FI-B12. Las condiciones que afectan al intestino delgado, como los síndromes de malabsorción (enfermedad celíaca y esprúe tropical), pueden resultar también en una deficiencia de vitamina B12. Debido a que el páncreas provee tanto las enzimas fundamentales como el calcio necesario para la absorción de la vitamina B12, la insuficiencia pancreática puede contribuir a la deficiencia de vitamina B12. Ya que la vitamina B12 sólo se encuentra en alimentos de origen animal, una dieta vegetariana estricta ha resultado en casos de deficiencia de vitamina B12. Más aun, los alcohólicos pueden experimentar una absorción intestinal de vitamina B12 reducida (2), e individuos con síndrome de inmunodeficiencia adquirida (SIDA) parecen estar en un mayor riesgo de deficiencia, posiblemente relacionada a una falla del receptor FI-B12 para captar el complejo FI-B12 (3). Además, el uso a largo plazo de medicamentos antiácidos también se ha implicado en la deficiencia de vitamina B12 (vea Interacción con drogas).
Desordenes hereditarios de la absorción de la vitamina B12
Raros casos de errores innatos del metabolismo de la vitamina B12 se han reportado en la literatura (revisado en 5). El síndrome de Imerslund-Gräsbeck es un síndrome de malabsorción hereditario de la vitamina B12 que causa anemia megaloblástica y desordenes neurológicos de gravedad variable en sujetos afectados. Síntomas clínicos similares son encontrados en individuos con una deficiencia hereditaria del factor intrínseco (FI) (también llamada anemia perniciosa congénita) en los cuales la deficiencia del IF resulta en una deficiente absorción de la vitamina B12. Adicionalmente, mutaciones que afectan el transporte de la vitamina B12 en el cuerpo han sido identificadas (14).
Síntomas de la deficiencia de la vitamina B12
La deficiencia de la vitamina B12 resulta en un deterioro de las actividades de las enzimas que requieren de la vitamina B12. La actividad deteriorada de la metionina sintasa resulta en niveles elevados de homocisteína, mientras que la actividad deteriorada de L-metilmalonil-CoA mutasa resulta en niveles aumentados de un metabolito del metilmalonil-CoA denominado ácido metilmalónico (AMM). Los individuos con una leve deficiencia de vitamina B12 pueden no experimentar síntomas, aunque los niveles sanguíneos de homocisteína y/o AMM podrían estar elevados (15).
Anemia megaloblástica
La actividad disminuida de la metionina sintasa en la deficiencia de vitamina B12 inhibe la regeneración del tetrahidrofolato (THF) y atrapa el folato en una forma que no es utilizable por el cuerpo (Figura 3), resultando en síntomas de deficiencia de folato incluso en presencia de niveles de folato adecuados. De esta manera, tanto en la deficiencia de folato como en la de vitamina B12, el folato no está disponible para participar en la síntesis de ADN. Este deterioro de la síntesis de ADN afecta a las células de división rápida de la médula ósea más temprano que a otras células, resultando en la producción de eritrocitos grandes, inmaduros, y con poca hemoglobina. La anemia resultante es conocida como anemia megaloblástica y es el síntoma por el cual se nombró a la enfermedad anemia perniciosa (3). La suplementación con ácido fólico proveerá suficiente folato utilizable como para restaurar la formación normal de glóbulos rojos. Sin embargo, si la causa es la deficiencia de vitamina B12, esta persistirá a pesar de la resolución de la anemia. Así, la anemia megaloblástica no debería tratarse con ácido fólico hasta que la causa subyacente haya sido determinada (16).
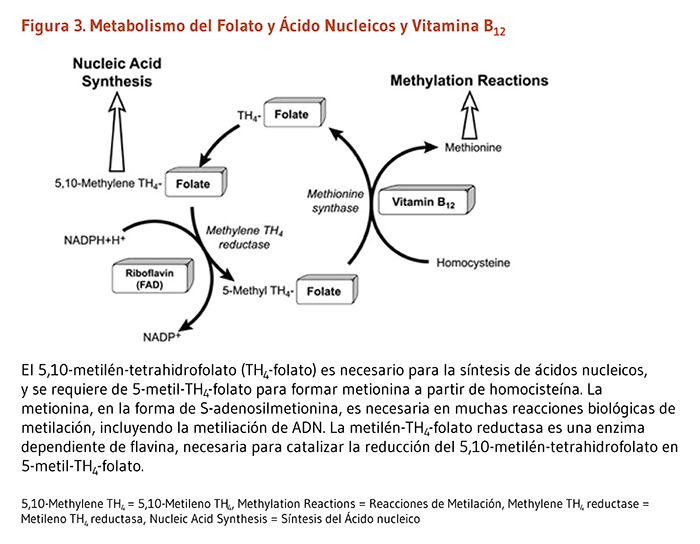
Síntomas neurológicos
Los síntomas neurológicos de la deficiencia de vitamina B12 incluyen entumecimiento y cosquilleo de las manos y, más comúnmente, en los pies; dificultad para caminar, pérdida de memoria, desorientación y demencia con o sin cambios de humor. Aunque, la progresión de las complicaciones neurológicas generalmente es gradual, dichos síntomas no son reversibles con el tratamiento de la deficiencia de vitamina B12, especialmente si han estado presentes por mucho tiempo. Las complicaciones neurológicas no siempre están asociadas con la anemia megaloblástica y son el único síntoma clínico de la deficiencia de vitamina B12 en alrededor de un 25% de los casos (17). Aunque se sabe que la deficiencia de vitamina B12 daña la vaina de mielina que cubre los nervios craneales, espinales y nervios periféricos, los procesos bioquímicos que conducen al daño neurológico en la deficiencia de vitamina B12 aun no son bien comprendidos (18).
Síntomas gastrointestinales
El dolor en la lengua, la pérdida de apetito y la constipación también han sido asociados a la deficiencia de vitamina B12. El origen de estos síntomas no está claro, pero puede estar relacionado con la inflamación del estómago subyacente a algunos casos de deficiencia de vitamina B12, y a la progresiva destrucción del revestimiento estomacal (17).
La Ingesta Diaria Recomendada (IDR)
La IDR para la vitamina B12 fue revisada por la Junta de Nutrición y Alimentos (JNA) del Instituto de Medicina de los Estados Unidos en 1998. Debido al incremento del riesgo de una malabsorción de vitamina B12 ligada a los alimentos en adultos mayores, la JNA recomendó que los adultos sobre los 50 años de edad obtengan la mayor parte de la IDR de alimentos fortificados o de suplementos que contengan vitamina B12 (17).
| Etapa de la Vida | Edad | Machos (μg/día) | Hembras (μg/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 0.4 (IA) | 0.4 (IA) |
| Infantes | 7-12 meses | 0.5 (IA) | 0.5 (IA) |
| Niños | 1-3 años | 0.9 | 0.9 |
| Niños | 4-8 años | 1.2 | 1.2 |
| Niños | 9-13 años | 1.8 | 1.8 |
| Adolescentes | 14-18 años | 2.4 | 2.4 |
| Adultos | 19-50 años | 2.4 | 2.4 |
| Adultos | 51 años y más | 2.4* | 2.4* |
| Embarazo | Todas las edades | - | 2.6 |
| Período de lactancia | Todas las edades | - | 2.8 |
| *La ingesta de vitamina B12 debiera provenir de suplementos o de alimentos fortificados debido al incremento de la malabsorción ligada a los alimentos relacionada con la edad. | |||
Prevención de Enfermedades
Enfermedades cardiovasculares
Como se menciona arriba, la gastritis atrófica crónica y la infección por H. pylori pueden causar una deficiencia de vitamina B12 secundaria a los trastornos de malabsorción (vea Causas de la deficiencia de la vitamina B12). Sin embargo la ocurrencia de la infección por H. pylori y la gastritis atrófica crónica no modificaron la incidencia de 5 años de accidentes cardiovasculares (Accidente cerebrovascular y ataque al corazón) o mortalidad en un exhaustivo estudio de cohorte de aproximadamente 10,000 hombres y mujeres mayores de 50 años de edad (19). Sin embargo, el estado de la vitamina B12 no fue evaluado en este estudio, a pesar de la alta prevalencia de deficiencia de vitamina B12 en personas mayores.
Homocisteína y enfermedades cardiovasculares
Los estudios epidemiológicos indicaron que incluso niveles de homocisteína elevados moderadamente en la sangres incrementan el riesgo de padecer enfermedades cardiovasculares (ECV) (20), aunque el mecanismo por el cual la homocisteína puede incrementar el riesgo de ECV sigue siendo objeto de una gran cantidad de investigación (21). La cantidad de homocisteína en la sangre es regulada por al menos tres vitaminas: folato, vitamina B6, y vitamina B12 (vea Figura 1 arriba). El análisis temprano de los resultados de 12 pruebas aleatorias controladas en la reducción de la homocisteína mostro que la suplementación con ácido fólico (0.5-5 mg/día) tuvo el mayor efecto reductor sobre los niveles de homocisteína sanguínea (reducción del 25%); la co-suplementación con ácido fólico y vitamina B12 (500 μg/día) proporcionó una reducción adicional del 7% (reducción del 32%) en las concentraciones de homocisteína sanguínea (22). Los resultados de un ensayo de suplementación secuencial en 53 hombres y mujeres indicaron que, luego de la suplementación con ácido fólico, la vitamina B12 se convirtió en el determinante principal de los niveles de homocisteína en el plasma (23). Se piensa que la elevación de los niveles de homocisteína podría ser en parte debido a la deficiencia de vitamina B12 en individuos mayores de 60 años de edad. Dos estudios encontraron que los niveles sanguíneos de ácido metilmalónico (AMM) estaban aumentados en más del 60% de los individuos de edad avanzada con niveles de homocisteína elevados. En la ausencia de una insuficiencia renal, un nivel elevado de AMM en conjunto con niveles de homocisteína elevados, indican tanto una deficiencia de vitamina B12 como una deficiencia combinada de vitamina B12 y folato (24). De tal manera, parece importante evaluar el estado de la vitamina B12, como también la función renal, en adultos de edad avanzada con elevados niveles de homocisteína antes de iniciar una terapia de disminución de la homocisteína. Para más información acerca de la homocisteína y ECV, vea el artículo en Folato.
Estudios de intervención
Aunque un incremento en la ingesta de ácido fólico y vitamina B12 es efectivo en la disminución de los niveles de homocisteína, la intervención combinada de estas vitaminas B no disminuyeron el riesgo de ECV. Ciertamente, varias pruebas aleatorias controladas con placebo han sido conducidas a determinar si la disminución de la homocisteína a través de la suplementación con acido fólico, vitamina B12, y vitamina B6 reduce la incidencia de ECV. Un meta-análisis de la información de 11 ensayos que incluyeron 45,000 participantes en riesgo de ECV, mostro que la suplementación de vitamina B no tuvo algún efecto significante en el riesgo de infarto agudo de miocardio (ataque al corazón) o accidente cerebrovascular como tampoco modifico el riesgo de mortalidad por todas las causas (25). Otros meta-análisis que incluyeron pacientes con enfermedad renal crónica han confirmado la deficiencia del efecto de la disminución de la homocisteína en el riesgo de un infarto agudo al miocardio y muerte. Sin embargo, el riesgo de un accidente cerebrovascular fue significantemente reducido en un 7%-12% con la suplementación de vitamina B (26, 27). Otro meta-análisis de 12 ensayos clínicos que midieron la vasodilatación mediada por flujo (VMF un marcador sustituto de la salud vascular) en respuesta a la reducción de la homocisteína revelo que la suplementación de vitamina B estuvo acompañada por un mejoramiento en la VMF a corto plazo <8 semanas) pero no en estudios a largo plazo conducidos en sujetos con enfermedades vasculares preexistentes (28). A pesar de todo, algunos de los estudios incluidos en estos meta-análisis no usaron vitamina B12, y la administración de folato por si sola ha mostrado un papel protector en la función vascular y el riesgo de accidente cerebrovascular (29). Además, la alta prevalencia de los desordenes de malabsorción y la deficiencia de vitamina B12 en individuos de edad avanzada podrían justificar el uso de dosis más altas de vitamina B12 que aquellas usadas en estos ensayos (30); en casos de malabsorción, solo una terapia de altas dosis orales o inyecciones intramusculares pueden superar la deficiencia de vitamina B12 (4).
Cáncer
El folato es requerido para la síntesis del ADN, y existe evidencia que de que la disponibilidad disminuida de folato resulta en hebras de ADN que son más susceptibles a dañarse. La deficiencia de vitamina B12 atrapa el folato en una forma que es inutilizable para el cuerpo en la síntesis del ADN. Tanto la deficiencia de vitamina B12 como la deficiencia de folato derivan en una capacidad disminuida para las reacciones de metilación (vea Figura 3 arriba). De esta manera, la deficiencia de vitamina B12 puede conducir a un a una taza de daño elevada del ADN y a una metilación alterada del ADN, los cuales son factores importantes del riesgo de padecer cáncer. Una serie de estudios en adultos jóvenes y adultos mayores señaló que los niveles incrementados de homocisteína y los niveles disminuidos de vitamina B12 en la sangre fueron asociados con un biomarcador de la ruptura de cromosomas en leucocitos (revisado en 31). En un estudio doble ciego controlado con placebo, el mismo marcador de ruptura de cromosomas fue minimizado en adultos jóvenes que fueron suplementados con 700 μg de ácido fólico y 7 μg de vitamina B12 diariamente en el cereal, por dos meses (32).
Cáncer de seno
Un estudio de caso y control comparó los niveles prediagnósticos del folato, vitamina B6, y vitamina B12 en el suero en 195 mujeres, posteriormente diagnosticadas con cáncer de mama y de 195 mujeres de edades similares que fueron diagnosticadas sin cáncer de mama. Entre las mujeres postmenopáusicas, la asociación entre los niveles sanguíneos de vitamina B12 y el cáncer de mama mostró un efecto umbral. El riesgo de cáncer de mama fue más del doble en mujeres con los niveles de vitamina B12 en el suero en el quintil más bajo comparado con el de las mujeres en los cuatro quintiles más altos (33). Sin embargo el meta-análisis de este estudio en conjunto con tres estudios de caso y control adicionales no encontraron protección asociada con la comparación entre los niveles altos y bajos de la vitamina B12 en el suero (34). Un estudio de caso y control en mujeres mexicanas (475 casos y 1.391 controles) reportó que el riesgo de cáncer de mama para las mujeres en el cuartil más alto de la ingesta de vitamina B12 (7.3-7.7 μg/día) fue un 68% más bajo que el de las mujeres en el cuartil más bajo (2.6 μg/día). La estratificación de la información reveló que la asociación inversa entre la ingesta de vitamina B12 dietaría y el riesgo de cáncer de mama era más fuerte en mujeres postmenopáusicas en comparación con mujeres premenopáusicas, aunque ambas asociaciones fueron estadísticamente significativas. Más aun, entre las mujeres postmenopáusicas, la aparente protección conferida por el folato fue únicamente observada en mujeres en los más altos cuartiles de la ingesta de vitamina B12 (35). Sin embargo estudios de cohorte prospectivos y estudios de casos y controles recientes han reportado una débil o ninguna reducción del riesgo con las ingestas de vitamina B12 en diferentes poblaciones incluyendo mujeres hispanas, afroamericanas, y euroamericanas (36, 37). Un meta-análisis de siete estudios de casos y controles y siete estudios de cohorte prospectivos concluyeron que el riesgo de cáncer de seno no se modifico por altas versus bajas ingestas de vitamina B12 (34). No hubo asociación conjunta entre las ingestas de folato y vitamina B12 y el cáncer de seno. Actualmente, hay poca evidencia para sugerir una relación entre el estatus de la vitamina B12 y el cáncer de mama. En adición, resultados de estudios observacionales no están consistentemente en apoyo a una asociación entre altas ingestas de folato dietario y una reducción en el riesgo de cáncer de seno (vea el artículo en Folato). No existe evidencia para evaluar el efecto de la suplementación de folato y vitamina B12 de ensayos clínicos, aleatorios bien controlados, mientras se consideran varios factores que modifican el riesgo de cáncer de mama, como el estado de la menopausia, etnicidad, y la ingesta de alcohol.
Defectos del tubo neural
Los defectos del tubo neural (DTN) pueden resultar en anencefalia o espina bífida, los cuales son malformaciones congénitas mayormente fatales del sistema nervioso central. Los defectos surgen a partir del fracaso del tubo neural embrionario al cerrar, que ocurre entre el 21. º y el 28. º días después de la concepción, el tiempo cuando muchas mujeres son inconscientes de su embarazo (38). Pruebas aleatorias controladas han demostrado una reducción del 60% al100% en casos de DTN cuando las mujeres consumieron suplementos de acido fólico en adición a una dieta variada durante el anterior y el mes posterior después de la concepción. El incremento de la evidencia indica que el efecto reductor de la homocisteína del acido fólico juega un papel crítico en la reducción del riesgo de DTN (39). La homocisteína puede acumularse en la sangre cuando el folato y/o la vitamina B12 son inadecuados para el funcionamiento eficaz de la enzima metionina sintasa. Niveles de vitamina B12 disminuidos y concentraciones elevadas de homocisteína han sido encontrados en la sangre y el líquido amniótico en mujeres embarazadas en alto riesgo de DTN (40). El reciente meta-análisis de 12 estudios de casos y controles que incluyo 567 madres con un actual o anterior embarazo afectado por DTN y 1566 madres que no fueron afectadas, mostro que un bajo nivel de vitamina B12 materno estuvo asociado con un riesgo incrementado del riesgo de DTN (41). Sin embargo aun no se ha evaluado si la suplementación de vitamina B12 puede ser beneficial en la prevención de DTN (42).
Deterioro cognitivo, demencia, y enfermedad de Alzheimer
La ocurrencia de la deficiencia de vitamina B12 prevalece en la población de edad avanzada y ha sido frecuentemente asociada con la enfermedad de Alzheimer (revisado en 43). Un estudio encontró niveles más bajos de vitamina B12 en el líquido cefalorraquídeo en pacientes con Alzheimer que en pacientes con otros tipos de demencia, aunque los niveles sanguíneos de vitamina B12 no difirieron (44). La razón para la asociación de un bajo estatus de vitamina B12 con la enfermedad de Alzheimer aun no está clara. La deficiencia de vitamina B12, al igual que la deficiencia de folato, puede conducir a un decline de la síntesis de metionina y S-adenosil metionina (SAM), de este modo afecta adversamente las reacciones de metilación. Las reacciones de metilación son esenciales para el metabolismo de los componentes de la vaina de mielina de las células nerviosas así como de los neurotransmisores (18). Otras implicaciones metabólicas de la deficiencia de vitamina B12 incluyen la acumulación de la homocisteína y el ácido metilmalónico, que podría contribuir a las características neuropatológicas de la demencia (43).
Estudios observacionales
Una gran mayoría de estudios transversales y de cohorte prospectivos han asociado elevadas concentraciones de homocisteína con mediciones de resultados cognitivos pobres y un riesgo incrementados de demencia, incluyendo la enfermedad de Alzheimer (revisado en 45). Un estudio de caso y control en 164 pacientes con demencia del tipo de Alzheimer incluyo 76 casos en los que se confirmó el diagnóstico de la enfermedad de Alzheimer al examinarse las células cerebrales después de su muerte. Comparados con los 108 sujetos de control sin evidencia de demencia, los sujetos con demencia del tipo Alzheimer y con enfermedad de Alzheimer confirmada tuvieron niveles sanguíneos de homocisteína más altos y niveles sanguíneos de folato y vitamina B12 más bajos. Las mediciones del estado nutricional general señaló que la asociación de los niveles de homocisteína aumentados y un estado de vitamina B12 disminuido junto a la enfermedad de Alzheimer no se debió a una malnutrición relacionada a la demencia (46). En una muestra de 1,092 hombres y mujeres sin demencia con un seguimiento por un promedio de 10 años, aquellos con niveles más altos de homocisteína en el plasma al inicio del estudio tuvieron un riesgo significativamente más alto de desarrollar la enfermedad de Alzheimer y otros tipos de demencia. Específicamente, aquellos con niveles de homocisteína plasmática mayores a 14 μmol/L tuvieron un riesgo casi doble de desarrollar la enfermedad de Alzheimer (47). Un estudio en 650 hombres y mujeres ancianos reportó que el riesgo de niveles elevados de homocisteína plasmática fue significativamente más elevado en aquellos con resultados más bajos de la función cognitiva (48). Un estudio prospectivo en 816 hombres y mujeres ancianos reportó que aquellos con hiperhomocisteinemia (niveles de homocisteína >15 μmol/L) tuvieron un riesgo significativamente más alto de desarrollar enfermedad de Alzheimer o demencia. Aunque niveles elevados de homocisteína podrían se parcialmente debido a un estatus pobre de vitamina B12, este último no estaba relacionada con el riesgo de padecer la enfermedad de Alzheimer o demencia en este estudio (49).
Una reciente revisión sistemática de 35 estudios de cohorte prospectivo evaluaron la asociación ente el estatus de la vitamina B12 y el deterioro cognitivo en individuos mayores con o sin demencia al inicio del estudio no apoyo una relación entre las concentraciones de vitamina B12 en el suero y el decline cognitivo, demencia o enfermedad de Alzheimer (50). No obstante, estudios que utilizaron biomarcadores más sensibles del estatus de la vitamina B12, incluyendo mediciones de la holo-transcobalamina (holo-TC; un transportador de la vitamina B12), y el ácido metilmalónico mostraron resultados más consistentes y una tendencia hacia las asociaciones entre un estado pobre de la vitamina B12 y un decline cognitivo más rápido y el riesgo de la enfermedad de Alzheimer (51-55). Además, no se puede excluir que la co-ocurrencia de posibles factores de confusión como niveles elevados de la homocisteína y un estado pobre del folato podría mitigar la verdadera contribución del estatus de la vitamina B12 en el funcionamiento cognitivo (45).
Estudios de intervención
Una suplementación de altas dosis de vitamina B ha probado su efectividad al tratar la hiperhomocisteinemia en individuos de edad avanzada con o sin un deterioro cognitivo. Sin embargo, ensayos de la disminución de la homocisteína han producido resultados equívocos respecto a la prevención del deterioro cognitivo en estas población. Una revisión sistemática y un meta-análisis de 18 pruebas aleatorias controladas con placebo que examinaron el efecto de la suplementación de vitamina B no encontraron que la disminución en el nivel de homocisteína previno o retraso el decline cognitivo entre los sujetos de mayor edad (56). Un ensayo clínico aleatorio doble ciego controlado con placebo en 900 individuos de mayor edad en un riesgo alto de deterioro cognitivo encontró que la suplementación diaria de 400 μg de ácido fólico y 100 μg de vitamina B12 por dos años mejoro significativamente las medidas de memoria inmediata y retardada y alentó el aumento de las concentraciones de homocisteína en el plasma (57). Sin embargo sujetos suplementados no tuvieron alguna reducción en las concentraciones de homocisteína comparadas con las del inicio del estudio, ni tampoco se desempeñaron mejor en el procesamiento de pruebas de velocidad en comparación con el placebo. Otro estudio aleatorio controlado con placebo de un lapso de 2 años en adultos ancianos reporto que un régimen diario de 800 μg de ácido fólico, 500 μg de vitamina B12 y 20 mg de vitamina B6, redujeron significantemente la tasa de atrofia cerebral comparado con el tratamiento con placebo (0.5% vs. 3.7%). Curiosamente, un beneficio mayor fue observado en aquellos con altas en comparación con bajas concentraciones de homocisteína al inicio del estudio, sugiriendo la importancia de la disminución de los niveles de homocisteína en la prevención de atrofia muscular y el decline cognitivo (58, 59). Los autores atribuyeron los cambios en los niveles de homocisteína ante todo a la vitamina B12 (59). Finalmente el ensayo clínico, aleatorio, doble ciego, controlado con placebo más reciente en aproximadamente 2,500 individuos que sufrieron un accidente cerebrovascular mostro que la normalización de las concentraciones de homocisteína debido a la suplementación de vitamina B (2 mg de ácido fólico, 500 μg de vitamina B12, y 25 mg de vitamina B6) no mejoro el rendimiento cognitivo o disminuyo la incidencia de un decline cognitivo en comparación con el placebo (60). Actualmente, hay una necesidad de ensayos más amplios para evaluar el efecto de la suplementación de vitamina B en los resultados a largo plazo, como lo es la incidencia de la enfermedad de Alzheimer.
Depresión
Estudios observacionales han encontrado que hasta el 30% de los pacientes hospitalizados por depresión son deficientes en vitamina B12 (61). Un estudio transversal de 700 mujeres físicamente discapacitadas que viven en comunidad sobre los 65 años de edad, encontró que las mujeres deficientes de vitamina B12 eran dos veces más propensas a estar severamente deprimidas que las mujeres sin deficiencia (62). Un estudio de base poblacional en 3,884 hombres y mujeres ancianos con trastornos depresivos encontró que aquellos con una deficiencia de vitamina B12 eran casi 70% más propensos a sufrir de depresión que aquellos con un estatus normal de vitamina B12 (63). Las causas de la relación entre la deficiencia de vitamina B12 y la depresión no son claras, pero podrían involucrar una escazes de S-adenosilmetionina (SAM). La SAM es un donante del grupo metilo en numerosas reacciones de metilación en el cerebro, incluyendo aquellos involucrados en el metabolismo de los neurotransmisores cuya deficiencia ha sido relacionada con la depresión (64). Una deficiencia severa de B12 en un ratón modelo mostro alteraciones dramáticas en la metilación al nivel del ADN en el cerebro, que podrían conducir a alteraciones neurológicas (65). Esta hipótesis se sostiene en varios estudios que han demostrado que la suplementación con SAM mejora los síntomas depresivos (66-69).
Un nivel incrementado de la homocisteína es otro biomarcador no específico de la deficiencia de vitamina B12 que ha sido ligado a síntomas depresivos en los ancianos (70). Sin embargo, en un estudio transversal reciente conducido en 1,677 individuos mayores, niveles altos de vitamina B12 en el plasma, pero ningún cambio en las concentraciones de homocisteína fueron correlacionados con una menor prevalencia de síntomas depresivos (71). Algunos pocos estudios han examinado la relación del estatus de la vitamina B12, los niveles de homocisteína y el desarrollo de la depresión con el tiempo. En un estudio de intervención aleatorio controlado con placebo en un poco mas de 900 participantes mayores que experimentaron malestar psicológico, una suplementación con ácido fólico (400 μg) y vitamina B12 (100 μg) por dos años no redujo la ocurrencia de síntomas de depresión a pesar de la mejora significativa de los niveles de folato, vitamina B12 y homocisteína en la sangre a comparación con el placebo (72). Sin embargo, en un estudio a largo plazo, aleatorio, doble ciego, controlado con placebo entre enfermos con accidentes cerebrovasculares en un riesgo alto de depresión, diariamente suplementados con 2 mg de acido fólico, 25 mg de vitamina B6 y 500 μg de vitamina B12 redujeron significativamente el riesgo de mayores episodios depresivos durante un periodo de seguimiento de siete años comparado con el placebo (73). Aunque aun no puede ser determinado si la deficiencia de vitamina B12 juega un papel causal en la depresión, podría ser beneficial cubrir la deficiencia de vitamina B12 en individuos de edad avanzada como parte de una evaluación médica para la depresión.
Osteoporosis
Niveles altos de homocisteína podrían afectar la remodelación ósea al incrementar la resorción ósea (desglose), disminuyendo la formación ósea, y reduciendo el flujo sanguíneo óseo. Otro mecanismo propuesto involucra la unión de la homocisteína a la matriz de colágeno del hueso, la cual puede modificar las propiedades del colágeno y reducir la resistencia ósea (revisado en 74). Las alteraciones de las propiedades biomecánicas del hueso pueden contribuir a la osteoporosis e incrementar el riesgo de fracturas en los ancianos. Debido a que la vitamina B12 es un determinante del metabolismo de la homocisteína, se sugirió que le riesgo de fracturas osteoporóticas en sujetos de mayor edad podrían ser mas ocurrentes por una deficiencia de vitamina B12. Un meta-ánalisis de cuatro estudios observacionales que dieron seguimiento de 3 a 16 años a un total de 7,475 individuos de mayor edad, encontró una débil asociación entre una elevación en la vitamina B12 de 50 picomoles/L en la sangre y una reducción en el riesgo de fracturas (75). Un ensayo aleatorio controlado con placebo en 559 individuos ancianos con bajos niveles de folato y vitamina B12 en el suero y un riesgo incrementado del riesgo de fracturas evaluó la suplementación combinada de muy altas dosis de acido fólico (5 mg/día) y vitamina B12 (1.5 mg/día). El estudio de dos años encontró que la suplementación mejoro el estatus de la vitamina B, disminuyo las concentraciones de homocisteína, y redujo el riesgo de fracturas totales en comparación con el placebo (76). Sin embargo un estudio multicentrico en 5,485 sujetos con enfermedades cardiovasculares o diabetes mellitus mostro que la suplementación diaria de acido fólico (2.5 mg), vitamina B12 (1mg) y vitamina B6 (50 mg) disminuyo las concentraciones de homocisteína pero no tuvo efecto en el riesgo de fracturas en comparación con el placebo (77). Otro ensayo aleatorio, doble ciego pequeño en 93 individuos con un bajo estado de vitamina D no encontró beneficios adicionales de la suplementación de vitamina B (50 mg/día de vitamina B6, 0.5 mg/día de acido fólico, y 0.5 mg/día de vitamina B12) en marcadores de la salud ósea en un periodo de tiempo de un año después de asociado con la suplementación de vitamina D y calcio. A pesar de todo, la corta escala del estudio no permitió una conclusión sobre si la disminución de la homocisteína a través de la suplementación de vitamina B podría tener beneficios a largo plazo en la resistencia ósea y el riesgo de fracturas (78). Un exhaustivo estudio de intervención conducido en personas de mayor edad sin condiciones preexistentes está evaluando el efecto de la suplementación de vitamina B en marcadores de la salud ósea e incidencias de fracturas; este ensayo podría aclarar si las vitaminas B podría tener un efecto protector en la salud ósea en la población anciana (79).
Fuentes
Fuentes alimenticias
Sólo las bacterias pueden sintetizar la vitamina B12 (80). La vitamina B12 está presente en productos animales como la carne, las aves de corral, el pescado (incluyendo mariscos), y en menor medida, en productos lácteos y huevos (1). La leche fresca pasteurizada contiene 0.9 μg por vaso y es una fuente importante de vitamina B12 para algunos vegetarianos (17). Aquellos vegetarianos estrictos que no comen ningún producto animal (veganos) necesitan vitamina B12 suplementaria para satisfacer sus requerimientos. Análisis recientes revelaron que algunos alimentos vegetales, como ciertos frijoles fermentados y verduras y algas y hongos comestibles, contienen sustanciales cantidades de vitamina B12 bioactiva (81). Junto con alimentos y suplementos fortificados con vitamina B12, estos alimentos pueden contribuir, aunque modestamente, a prevenir una deficiencia de vitamina B12 en individuos que consumen dietas vegetarianas. También, las personas mayores de 50 años deberían obtener su vitamina B12 de suplementos o alimentos fortificados (ej., Cereal fortificado) debido a un incremento en la probabilidad de una malabsorción de vitamina B12 ligada a los alimentos al incrementarse la edad.
La mayoría de las personas no tiene problemas para alcanzar la IDR de 2.4 μg/día de vitamina B12 en la comida. De acuerdo a una encuesta nacional en los Estados Unidos, la ingesta promedio de vitamina B12 es de alrededor de 5.4 μg/día para hombres adultos, y 3.4 μg/día para mujeres adultas. Los adultos mayores de 60 años tuvieron una ingesta promedio de 4.8 μg/día (42). Sin embargo, el consumo de cualquier tipo de dieta vegetariana dramáticamente incrementa la prevalencia de la deficiencia de vitamina B12 en individuos de todas las edades (82). Algunos alimentos con cantidades substanciales de vitamina B12 son listados en la tabla a continuación al igual con su contenido de vitamina B12 en microgramos (μg). Para más información en el contenido nutrimental de alimentos específicos busque en la base de datos de composición de los alimentos del USDA.
| Alimento | Porción | Vitamina B12 (μg) |
|---|---|---|
| Almejas (al vapor) | 3 onzas | 84.1 |
| Mejillones (al vapor) | 3 onzas | 20.4 |
| Caballa (Atlantico, cocido, seco) | 3 onzas | 16.1 |
| Cangrejo (Rey de Alaska, al vapor) | 3 onzas | 9.8 |
| Carne de res (cocida, asada) | 3 onzas | 6.9 |
| Salmon (cocido, al horno) | 3 onzas | 2.4 |
| Pez piedra (cocido, al horno) | 3 onzas | 1.0 |
| Leche (descremada) | 8 onzas | 0.9 |
| Pavo (cocinado, rostizado) | 3 onzas | 0.8 |
| Brie (queso) | 1 onza | 0.5 |
| Huevo (escalfado) | 1 grande | 0.4 |
| Pollo (carne ligera, cocido, rostizado) | 3 onzas | 0.3 |
| *Una porción de tres onzas de carnes es del tamaño de una baraja de cartas. | ||
Suplementos
La cianocobalamina es la forma principal de vitamina B12 utilizada en suplementos orales, pero la metilcobalamina también está disponible como un suplemento. La cianocobalamina está disponible por prescripción médica en una forma inyectable y como un gel nasal para el tratamiento de la anemia perniciosa. Las preparaciones de venta libre que contienen cianocobalamina incluyen multivitamínicos, suplementos de vitaminas del complejo B, y suplementos de vitamina B12 (83).
Seguridad
Toxicidad
Efectos tóxicos o adversos no han sido asociados con grandes ingestas de vitamina B12 proveniente de alimentos o de suplementos en personas sanas. Se han utilizado dosis orales de hasta 2 mg (2,000 μg) diariamente o de 1 mg al mes por inyección intramuscular (IM) para tratar la anemia perniciosa sin efectos secundarios significativos (84). Cuando se administran oralmente altas dosis de vitamina B12, sólo un pequeño porcentaje puede ser absorbido, lo que podría explicar la baja toxicidad (4). Debido a la baja toxicidad de la vitamina B12, ningún nivel máximo de ingesta tolerable (NM) ha sido establecido por la Junta de Nutrición y Alimentos Estadounidense (17).
Interacción con drogas/fármacos
Una serie de drogas reducen la absorción de la vitamina B12. Los inhibidores de la bomba de protones (por ejemplo, omeprazol y lansoprazol), usados en la terapia del síndrome de Zollinger-Ellison y en la enfermedad de reflujo gastroesofágico (ERGE), reducen notablemente la secreción de ácido estomacal requerida para la liberación de la vitamina B12 de los alimentos, aunque no de la de los suplementos. Se ha encontrado que la utilización a largo plazo de inhibidores de la bomba de protones disminuye los niveles sanguíneos de vitamina B12. Sin embargo, la deficiencia de vitamina B12 generalmente no se desarrolla hasta por lo menos después de tres años de terapia continua (85, 86). Otra clase de inhibidores del acido gástrico conocidos como antagonistas de histamina2 receptor-(H2) (ej. cimetidina, famotidina, y ranitidina), usados frecuentemente al tratar la enfermedad de ulcera péptica, han sido también descubierto que disminuyen la absorción de vitamina B12 de los alimentos. No está claro si el uso a largo plazo los antagonistas de los receptores-H2¬ podrían causar una evidente deficiencia de vitamina B12 (87, 88). Los individuos que toman fármacos que inhiben la secreción de acido gástrico deberían considerar tomar vitamina B12 en forma de suplementos porque el acido gástrico no es requerido para su absorción. Otras drogas que se han encontrado que inhiben la absorción de vitamina B12 de los alimentos incluyen colestiramina (una resina que se une a los ácidos biliares utilizada en el tratamiento del colesterol elevado), cloramfenicol y neomicin (antibióticos), y colchicina (medicamento en el tratamiento de la gota) La metformina, un medicamento para individuos con diabetes tipo 2, se encontró que disminuye la absorción de vitamina B12 a través de la ligadura del calcio libre necesario para la absorción del complejo FI-B12 (89). Sin embargo, el significado clínico de esta no está claro (90). No se conoce si la suplementación con calcio puede revertir la malabsorcion de vitamina B12; por ello, la suplementación con calcio no está actualmente prescrita para la prevención o tratamiento de la deficiencia de vitamina B12 inducida por la metformina (91). Reportes previos de que megadosis de vitamina C destruyan la vitamina B12 no han sido apoyados (92) y podrían haber sido un artefacto del ensayo utilizado para medir los niveles de vitamina B12 (17).
El óxido nitroso, un anestésico comúnmente usado, oxida e inactiva la vitamina B12, inhibiendo así ambas enzimas dependientes de vitamina B12 y puede producir muchas de las manifestaciones clínicas de la deficiencia de vitamina B12, tales como la anemia megaloblástica o la neuropatía. Debido a que el óxido nitroso se utiliza comúnmente para la cirugía en ancianos, algunos expertos consideran que la deficiencia de vitamina B12 debe descartarse antes de su uso (6, 15).
Grandes dosis de ácido fólico administradas a un individuo con una deficiencia de vitamina B12 no diagnosticada podría corregir la anemia megaloblástica sin corregir la deficiencia de vitamina B12 subyacente, dejando a la persona en riesgo de sufrir daño neurológico irreversible (17). Por esta razón la Junta de Nutrición y Alimentos del Instituto de Medicina Estadounidense aconseja que todos los adultos limiten su ingesta de ácido fólico (suplementos y fortificación) a 1,000 μg (1 mg) diarios.
Recomendación del Instituto Linus Pauling
Una dieta variada debería proporcionar suficiente vitamina B12 como para prevenir una deficiencia en la mayoría de los individuos de 50 años de edad o menos. Los vegetarianos estrictos y mujeres que planean embarazarse debiesen tomar un suplemento multivitaminico diariamente o comer cereal fortificado, los cuales asegurarían una ingesta de 6 a 30 μg de vitamina B12 en una forma que es fácilmente absorbida. Los suplementos de dosis más altas de vitamina B12 son recomendados a pacientes que toman medicamentos que interfieren con su absorción (vea Interacción con drogas).
Adultos mayores (>50 años)
Debido a que la malabsorción y la deficiencia de vitamina B12 son más comunes en adultos mayores, el instituto Linus Pauling recomienda que los adultos mayores de 50 años tomen entre 100 a 400 μg/día de vitamina B12 suplementaria.
Autores y Críticos
Escrito en Marzo de 2003 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Enero de 2014 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Abril de 2014 por:
Joshua W. Miller, Ph.D.
Profesor y Presidente, Departamento de Ciencias Nutricionales
Rutgers, La Universidad del Estado de New Jersey
Traducido al Español en 2014 por:
Silvia Vazquez Lima
Instituto Linus Pauling
Universidad Estatal de Oregon
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Derechos de autoría 2000-2024 Instituto Linus Pauling
Referencias
1. Brody T. Nutritional Biochemistry. 2nd ed. San Diego: Academic Press; 1999.
2. Carmel R. Cobalamin (Vitamin B-12). In: Shils ME, Shike M, Ross AC, Caballero B, Cousins RJ, eds. Modern Nutrition in Health and Disease. Philadelphia: Lippincott Williams & Wilkins; 2006:482-497.
3. Shane B. Folic acid, vitamin B-12, and vitamin B-6. In: Stipanuk M, ed. Biochemical and Physiological Aspects of Human Nutrition. Philadelphia: W.B. Saunders Co.; 2000:483-518.
4. Carmel R. How I treat cobalamin (vitamin B12) deficiency. Blood. 2008;112(6):2214-2221. (PubMed)
5. Kozyraki R, Cases O. Vitamin B12 absorption: mammalian physiology and acquired and inherited disorders. Biochimie. 2013;95(5):1002-1007. (PubMed)
6. Baik HW, Russell RM. Vitamin B12 deficiency in the elderly. Annu Rev Nutr. 1999;19:357-377. (PubMed)
7. Neumann WL, Coss E, Rugge M, Genta RM. Autoimmune atrophic gastritis--pathogenesis, pathology and management. Nat Rev Gastroenterol Hepatol. 2013;10(9):529-541. (PubMed)
8. Lahner E, Persechino S, Annibale B. Micronutrients (Other than iron) and Helicobacter pylori infection: a systematic review. Helicobacter. 2012;17(1):1-15. (PubMed)
9. Carmel R. Megaloblastic anemias. Curr Opin Hematol. 1994;1(2):107-112. (PubMed)
10. Banka S, Ryan K, Thomson W, Newman WG. Pernicious anemia - genetic insights. Autoimmun Rev. 2011;10(8):455-459. (PubMed)
11. Lam-Tse WK, Batstra MR, Koeleman BP, et al. The association between autoimmune thyroiditis, autoimmune gastritis and type 1 diabetes. Pediatr Endocrinol Rev. 2003;1(1):22-37. (PubMed)
12. Checchi S, Montanaro A, Ciuoli C, et al. Prevalence of parietal cell antibodies in a large cohort of patients with autoimmune thyroiditis. Thyroid. 2010;20(12):1385-1389. (PubMed)
13. Ho C, Kauwell GP, Bailey LB. Practitioners' guide to meeting the vitamin B-12 recommended dietary allowance for people aged 51 years and older. J Am Diet Assoc. 1999;99(6):725-727. (PubMed)
14. Watkins D, Rosenblatt DS. Lessons in biology from patients with inborn errors of vitamin B12 metabolism. Biochimie. 2013;95(5):1019-1022. (PubMed)
15. Weir DG, Scott JM. Vitamin B12 "Cobalamin". In: Shils M, ed. Nutrition in Health and Disease. 9th ed. Baltimore: Williams & Wilkins; 1999:447-458.
16. Herbert V. Vitamin B-12. In: Ziegler EE, Filer LJ, eds. Present Knowledge in Nutrition. 7th ed. Washington D.C.: ILSI Press; 1996:191-205.
17. Food and Nutrition Board, Institute of Medicine. Vitamin B12. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B6, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington D.C.: National Academy Press; 1998:306-356. (National Academy Press)
18. Scalabrino G. The multi-faceted basis of vitamin B12 (cobalamin) neurotrophism in adult central nervous system: Lessons learned from its deficiency. Prog Neurobiol. 2009;88(3):203-220. (PubMed)
19. Schottker B, Adamu MA, Weck MN, Muller H, Brenner H. Helicobacter pylori infection, chronic atrophic gastritis and major cardiovascular events: a population-based cohort study. Atherosclerosis. 2012;220(2):569-574. (PubMed)
20. Gerhard GT, Duell PB. Homocysteine and atherosclerosis. Curr Opin Lipidol. 1999;10(5):417-428. (PubMed)
21. Homocysteine Lowering Trialists' Collaboration. Lowering blood homocysteine with folic acid based supplements: meta-analysis of randomised trials. Homocysteine Lowering Trialists' Collaboration. BMJ. 1998;316(7135):894-898. (PubMed)
22. Lowering blood homocysteine with folic acid based supplements: meta-analysis of randomised trials. Homocysteine Lowering Trialists' Collaboration. Bmj. 1998;316(7135):894-898. (PubMed)
23. Quinlivan EP, McPartlin J, McNulty H, et al. Importance of both folic acid and vitamin B12 in reduction of risk of vascular disease. Lancet. 2002;359(9302):227-228. (PubMed)
24. Stabler SP, Lindenbaum J, Allen RH. Vitamin B-12 deficiency in the elderly: current dilemmas. Am J Clin Nutr. 1997;66(4):741-749. (PubMed)
25. Marti-Carvajal AJ, Sola I, Lathyris D, Karakitsiou DE, Simancas-Racines D. Homocysteine-lowering interventions for preventing cardiovascular events. Cochrane Database Syst Rev. 2013;1:CD006612. (PubMed)
26. Huang T, Chen Y, Yang B, Yang J, Wahlqvist ML, Li D. Meta-analysis of B vitamin supplementation on plasma homocysteine, cardiovascular and all-cause mortality. Clin Nutr. 2012;31(4):448-454. (PubMed)
27. Ji Y, Tan S, Xu Y, et al. Vitamin B supplementation, homocysteine levels, and the risk of cerebrovascular disease: A meta-analysis. Neurology. 2013;81(15):1298-1307. (PubMed)
28. Potter K, Hankey GJ, Green DJ, Eikelboom J, Jamrozik K, Arnolda LF. The effect of long-term homocysteine-lowering on carotid intima-media thickness and flow-mediated vasodilation in stroke patients: a randomized controlled trial and meta-analysis. BMC Cardiovasc Disord. 2008;8:24. (PubMed)
29. Qin X, Xu M, Zhang Y, et al. Effect of folic acid supplementation on the progression of carotid intima-media thickness: a meta-analysis of randomized controlled trials. Atherosclerosis. 2012;222(2):307-313. (PubMed)
30. Spence JD. B vitamin therapy for homocysteine: renal function and vitamin B12 determine cardiovascular outcomes. Clin Chem Lab Med. 2013;51(3):633-637. (PubMed)
31. Fenech M. Folate (vitamin B9) and vitamin B12 and their function in the maintenance of nuclear and mitochondrial genome integrity. Mutat Res. 2012;733(1-2):21-33. (PubMed)
32. Fenech M. Micronucleus frequency in human lymphocytes is related to plasma vitamin B12 and homocysteine. Mutat Res. 1999;428(1-2):299-304. (PubMed)
33. Wu K, Helzlsouer KJ, Comstock GW, Hoffman SC, Nadeau MR, Selhub J. A prospective study on folate, B12, and pyridoxal 5'-phosphate (B6) and breast cancer. Cancer Epidemiol Biomarkers Prev. 1999;8(3):209-217. (PubMed)
34. Wu W, Kang S, Zhang D. Association of vitamin B6, vitamin B12 and methionine with risk of breast cancer: a dose-response meta-analysis. Br J Cancer. 2013;109(7):1926-1944. (PubMed)
35. Lajous M, Lazcano-Ponce E, Hernandez-Avila M, Willett W, Romieu I. Folate, vitamin B(6), and vitamin B(12) intake and the risk of breast cancer among Mexican women. Cancer Epidemiol Biomarkers Prev. 2006;15(3):443-448. (PubMed)
36. Yang D, Baumgartner RN, Slattery ML, et al. Dietary intake of folate, B-vitamins and methionine and breast cancer risk among Hispanic and non-Hispanic white women. PLoS One. 2013;8(2):e54495. (PubMed)
37. Bassett JK, Baglietto L, Hodge AM, et al. Dietary intake of B vitamins and methionine and breast cancer risk. Cancer Causes Control. 2013;24(8):1555-1563. (PubMed)
38. Eskes TK. Open or closed? A world of difference: a history of homocysteine research. Nutr Rev. 1998;56(8):236-244. (PubMed)
39. Mills JL, Scott JM, Kirke PN, et al. Homocysteine and neural tube defects. J Nutr. 1996;126(3):756S-760S. (PubMed)
40. Imbard A, Benoist JF, Blom HJ. Neural tube defects, folic acid and methylation. Int J Environ Res Public Health. 2013;10(9):4352-4389. (PubMed)
41. Wang ZP, Shang XX, Zhao ZT. Low maternal vitamin B(12) is a risk factor for neural tube defects: a meta-analysis. J Matern Fetal Neonatal Med. 2012;25(4):389-394. (PubMed)
42. Dror DK, Allen LH. Interventions with vitamins B6, B12 and C in pregnancy. Paediatr Perinat Epidemiol. 2012;26 Suppl 1:55-74. (PubMed)
43. McCaddon A. Vitamin B12 in neurology and ageing; clinical and genetic aspects. Biochimie. 2013;95(5):1066-1076. (PubMed)
44. Nourhashemi F, Gillette-Guyonnet S, Andrieu S, et al. Alzheimer disease: protective factors. Am J Clin Nutr. 2000;71(2):643S-649S. (PubMed)
45. Smith AD. The worldwide challenge of the dementias: a role for B vitamins and homocysteine? Food Nutr Bull. 2008;29(2 Suppl):S143-172. (PubMed)
46. Clarke R, Smith AD, Jobst KA, Refsum H, Sutton L, Ueland PM. Folate, vitamin B12, and serum total homocysteine levels in confirmed Alzheimer disease. Arch Neurol. 1998;55(11):1449-1455. (PubMed)
47. Seshadri S, Beiser A, Selhub J, et al. Plasma homocysteine as a risk factor for dementia and Alzheimer's disease. N Engl J Med. 2002;346(7):476-483. (PubMed)
48. Ravaglia G, Forti P, Maioli F, et al. Homocysteine and cognitive function in healthy elderly community dwellers in Italy. Am J Clin Nutr. 2003;77(3):668-673. (PubMed)
49. Ravaglia G, Forti P, Maioli F, et al. Homocysteine and folate as risk factors for dementia and Alzheimer disease. Am J Clin Nutr. 2005;82(3):636-643. (PubMed)
50. O'Leary F, Allman-Farinelli M, Samman S. Vitamin B(1)(2) status, cognitive decline and dementia: a systematic review of prospective cohort studies. Br J Nutr. 2012;108(11):1948-1961. (PubMed)
51. Clarke R, Birks J, Nexo E, et al. Low vitamin B-12 status and risk of cognitive decline in older adults. Am J Clin Nutr. 2007;86(5):1384-1391. (PubMed)
52. Tangney CC, Tang Y, Evans DA, Morris MC. Biochemical indicators of vitamin B12 and folate insufficiency and cognitive decline. Neurology. 2009;72(4):361-367. (PubMed)
53. Kivipelto M, Annerbo S, Hultdin J, et al. Homocysteine and holo-transcobalamin and the risk of dementia and Alzheimers disease: a prospective study. Eur J Neurol. 2009;16(7):808-813. (PubMed)
54. Hooshmand B, Solomon A, Kareholt I, et al. Homocysteine and holotranscobalamin and the risk of Alzheimer disease: a longitudinal study. Neurology. 2010;75(16):1408-1414. (PubMed)
55. Hooshmand B, Solomon A, Kareholt I, et al. Associations between serum homocysteine, holotranscobalamin, folate and cognition in the elderly: a longitudinal study. J Intern Med. 2012;271(2):204-212. (PubMed)
56. Ford AH, Almeida OP. Effect of homocysteine lowering treatment on cognitive function: a systematic review and meta-analysis of randomized controlled trials. J Alzheimers Dis. 2012;29(1):133-149. (PubMed)
57. Walker JG, Batterham PJ, Mackinnon AJ, et al. Oral folic acid and vitamin B-12 supplementation to prevent cognitive decline in community-dwelling older adults with depressive symptoms--the Beyond Ageing Project: a randomized controlled trial. Am J Clin Nutr. 2012;95(1):194-203. (PubMed)
58. Smith AD, Smith SM, de Jager CA, et al. Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: a randomized controlled trial. PLoS One. 2010;5(9):e12244. (PubMed)
59. Douaud G, Refsum H, de Jager CA, et al. Preventing Alzheimer's disease-related gray matter atrophy by B-vitamin treatment. Proc Natl Acad Sci U S A. 2013;110(23):9523-9528. (PubMed)
60. Hankey GJ, Ford AH, Yi Q, et al. Effect of B vitamins and lowering homocysteine on cognitive impairment in patients with previous stroke or transient ischemic attack: a prespecified secondary analysis of a randomized, placebo-controlled trial and meta-analysis. Stroke. 2013;44(8):2232-2239. (PubMed)
61. Hutto BR. Folate and cobalamin in psychiatric illness. Compr Psychiatry. 1997;38(6):305-314. (PubMed)
62. Penninx BW, Guralnik JM, Ferrucci L, Fried LP, Allen RH, Stabler SP. Vitamin B(12) deficiency and depression in physically disabled older women: epidemiologic evidence from the Women's Health and Aging Study. Am J Psychiatry. 2000;157(5):715-721. (PubMed)
63. Tiemeier H, van Tuijl HR, Hofman A, Meijer J, Kiliaan AJ, Breteler MM. Vitamin B12, folate, and homocysteine in depression: the Rotterdam Study. Am J Psychiatry. 2002;159(12):2099-2101. (PubMed)
64. Mischoulon D, Fava M. Role of S-adenosyl-L-methionine in the treatment of depression: a review of the evidence. Am J Clin Nutr. 2002;76(5):1158S-1161S. (PubMed)
65. Fernandez-Roig S, Lai SC, Murphy MM, Fernandez-Ballart J, Quadros EV. Vitamin B12 deficiency in the brain leads to DNA hypomethylation in the TCblR/CD320 knockout mouse. Nutr Metab (Lond). 2012;9:41. (PubMed)
66. Bressa GM. S-adenosyl-l-methionine (SAMe) as antidepressant: meta-analysis of clinical studies. Acta Neurol Scand Suppl. 1994;154:7-14. (PubMed)
67. Bell KM, Plon L, Bunney WE, Jr., Potkin SG. S-adenosylmethionine treatment of depression: a controlled clinical trial. Am J Psychiatry. 1988;145(9):1110-1114. (PubMed)
68. Delle Chiaie R, Pancheri P, Scapicchio P. Efficacy and tolerability of oral and intramuscular S-adenosyl-L-methionine 1,4-butanedisulfonate (SAMe) in the treatment of major depression: comparison with imipramine in 2 multicenter studies. Am J Clin Nutr. 2002;76(5):1172S-1176S. (PubMed)
69. Williams AL, Girard C, Jui D, Sabina A, Katz DL. S-adenosylmethionine (SAMe) as treatment for depression: a systematic review. Clin Invest Med. 2005;28(3):132-139. (PubMed)
70. Almeida OP, McCaul K, Hankey GJ, Norman P, Jamrozik K, Flicker L. Homocysteine and depression in later life. Arch Gen Psychiatry. 2008;65(11):1286-1294. (PubMed)
71. Moorthy D, Peter I, Scott TM, et al. Status of vitamins B-12 and B-6 but not of folate, homocysteine, and the methylenetetrahydrofolate reductase C677T polymorphism are associated with impaired cognition and depression in adults. J Nutr. 2012;142(8):1554-1560. (PubMed)
72. Walker JG, Mackinnon AJ, Batterham P, et al. Mental health literacy, folic acid and vitamin B12, and physical activity for the prevention of depression in older adults: randomised controlled trial. Br J Psychiatry. 2010;197(1):45-54. (PubMed)
73. Almeida OP, Marsh K, Alfonso H, Flicker L, Davis TM, Hankey GJ. B-vitamins reduce the long-term risk of depression after stroke: The VITATOPS-DEP trial. Ann Neurol. 2010;68(4):503-510. (PubMed)
74. Vacek TP, Kalani A, Voor MJ, Tyagi SC, Tyagi N. The role of homocysteine in bone remodeling. Clin Chem Lab Med. 2013;51(3):579-590. (PubMed)
75. van Wijngaarden JP, Doets EL, Szczecinska A, et al. Vitamin B12, folate, homocysteine, and bone health in adults and elderly people: a systematic review with meta-analyses. J Nutr Metab. 2013;2013:486186. (PubMed)
76. Sato Y, Honda Y, Iwamoto J, Kanoko T, Satoh K. Effect of folate and mecobalamin on hip fractures in patients with stroke: a randomized controlled trial. JAMA. 2005;293(9):1082-1088. (PubMed)
77. Sawka AM, Ray JG, Yi Q, Josse RG, Lonn E. Randomized clinical trial of homocysteine level lowering therapy and fractures. Arch Intern Med. 2007;167(19):2136-2139. (PubMed)
78. Herrmann W, Kirsch SH, Kruse V, et al. One year B and D vitamins supplementation improves metabolic bone markers. Clin Chem Lab Med. 2013;51(3):639-647. (PubMed)
79. van Wijngaarden JP, Dhonukshe-Rutten RA, van Schoor NM, et al. Rationale and design of the B-PROOF study, a randomized controlled trial on the effect of supplemental intake of vitamin B12 and folic acid on fracture incidence. BMC Geriatr. 2011;11:80. (PubMed)
80. LeBlanc JG, Milani C, de Giori GS, Sesma F, van Sinderen D, Ventura M. Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr Opin Biotechnol. 2013;24(2):160-168. (PubMed)
81. Watanabe F, Yabuta Y, Tanioka Y, Bito T. Biologically active vitamin B12 compounds in foods for preventing deficiency among vegetarians and elderly subjects. J Agric Food Chem. 2013;61(28):6769-6775. (PubMed)
82. Pawlak R, Parrott SJ, Raj S, Cullum-Dugan D, Lucus D. How prevalent is vitamin B(12) deficiency among vegetarians? Nutr Rev. 2013;71(2):110-117. (PubMed)
83. Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. Montvale: Medical Economics Company, Inc; 2001.
84. Kuzminski AM, Del Giacco EJ, Allen RH, Stabler SP, Lindenbaum J. Effective treatment of cobalamin deficiency with oral cobalamin. Blood. 1998;92(4):1191-1198. (PubMed)
85. Dharmarajan TS, Kanagala MR, Murakonda P, Lebelt AS, Norkus EP. Do acid-lowering agents affect vitamin B12 status in older adults? J Am Med Dir Assoc. 2008;9(3):162-167. (PubMed)
86. Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6(4):443-451. (PubMed)
87. Valuck RJ, Ruscin JM. A case-control study on adverse effects: H2 blocker or proton pump inhibitor use and risk of vitamin B12 deficiency in older adults. J Clin Epidemiol. 2004;57(4):422-428. (PubMed)
88. Termanini B, Gibril F, Sutliff VE, Yu F, Venzon DJ, Jensen RT. Effect of long-term gastric acid suppressive therapy on serum vitamin B12 levels in patients with Zollinger-Ellison syndrome. Am J Med. 1998;104(5):422-430. (PubMed)
89. Bauman WA, Shaw S, Jayatilleke E, Spungen AM, Herbert V. Increased intake of calcium reverses vitamin B12 malabsorption induced by metformin. Diabetes Care. 2000;23(9):1227-1231. (PubMed)
90. Obeid R. Metformin causing vitamin B12 deficiency: a guilty verdict without sufficient evidence. Diabetes Care. 2014;37(2):e22-23. (PubMed)
91. Mazokopakis EE, Starakis IK. Recommendations for diagnosis and management of metformin-induced vitamin B12 (Cbl) deficiency. Diabetes Res Clin Pract. 2012;97(3):359-367. (PubMed)
92. Simon JA, Hudes ES. Relation of serum ascorbic acid to serum vitamin B12, serum ferritin, and kidney stones in US adults. Arch Intern Med. 1999;159(6):619-624. (PubMed)
Vitamina C
Contenido
Resumen
- La vitamina C, también conocida como ácido ascórbico-L, es una vitamina soluble en agua. A diferencia de la mayoría de los mamíferos y otros animales, los humanos no tienen la capacidad de sintetizar la vitamina C y deben obtenerla de la dieta. (Más información)
- La vitamina C es un cofactor esencial en numerosas reacciones enzimáticas, p. ej., en la biosíntesis de colágeno, carnitina y neuropéptidos, y en la regulación de la expresión de genes. También es un potente antioxidante. (Más información)
- Los estudios de cohorte prospectivos indicaron que un mayor estado de vitamina C, evaluado por medio de la medición de la vitamina circulante, se asocia con riesgos menores de hipertensión, enfermedad coronaria y accidente cerebrovascular. (Más información)
- Existe cierta evidencia que sugiere que la vitamina C podría ser un complemento útil a la práctica médica convencional para reducir la lesión miocárdica y la arritmia después de un procedimiento cardíaco o cirugía en pacientes con enfermedad cardiovascular. (Más información)
- No hay datos suficientes para sugerir una relación entre el estado de la vitamina C y el riesgo de desarrollar un tipo determinado de cáncer. La mayoría de los estudios observacionales que examinaron la ingesta de vitamina C con relación a la incidencia de cáncer no encontraron alguna asociación. Ensayos controlados aleatorios no reportaron ningún efecto por parte de la suplementación de vitamina C en el riesgo de cáncer. (Más información)
- La evidencia actual de la eficacia de la vitamina C intravenosa en pacientes con cáncer se limita a los estudios observacionales, las intervenciones no controladas, y los reportes de casos. Existe la necesidad de ensayos clínicos de fase II grandes y de más larga duración que evalúen la eficacia de la vitamina C en la progresión del cáncer y la supervivencia general. (Más información)
- En general, el uso regular de suplementos de vitamina C acorta la duración del resfriado común, pero no reduce el riesgo de enfermarse. Tomar suplementos una vez que los síntomas del resfriado ya han comenzado no tiene beneficios comprobados. (Más información)
- Los suplementos de vitamina C están disponibles en muchas formas, pero hay poca evidencia científica de que una forma sea mejor absorbida o más efectiva que otra. (Más información)
- No hay evidencia científica de que grandes cantidades de vitamina C (hasta 10 gramos [g]/día en adultos) ejerzan efectos adversos o tóxicos. Se recomienda un nivel de ingesta inferior a 2 g (2,000 mg)/día para evitar que algunos adultos experimenten diarrea y trastornos gastrointestinales. (Más información)
- La vitamina C suplementaria aumenta las concentraciones de oxalato en la orina, pero aún no se sabe si un aumento en el oxalato en la orina aumenta el riesgo de cálculos renales. Las personas predispuestas a la formación de cálculos renales pueden considerar evitar la suplementación con vitamina C en dosis altas (≥1 g/día). (Más información)
Función
La vitamina C (ácido ascórbico-L) es un potente agente reductor, lo que significa que dona electrones fácilmente a las moléculas receptoras (Figura 1). Con relación a este potencial de oxidación-reducción (redox), dos funciones principales de la vitamina C son como un antioxidante y como un cofactor de enzima (1).
La vitamina C es el principal antioxidante no enzimático, soluble en agua, en el plasma y los tejidos. Incluso en pequeñas cantidades, la vitamina C puede proteger a las moléculas indispensables para el cuerpo, como las proteínas, los lípidos (grasas), los carbohidratos y los ácidos nucleicos (ADN y ARN), del daño causado por los radicales libres y las especies reactivas de oxígeno (ERO) que se generan durante el metabolismo normal, por las células inmunitarias activas y por la exposición a toxinas y contaminantes (p. ej., ciertos medicamentos de quimioterapia y humo de cigarrillo). La vitamina C también participa en el reciclado de la reacción redox de otros antioxidantes importantes; por ejemplo, la vitamina C regenera la vitamina E de su forma oxidada (véase el artículo sobre la Vitamina E).
El papel de la vitamina C como un cofactor también está relacionado con su potencial redox. Al mantener metales unidos a enzimas en sus formas reducidas, la vitamina C asiste funciones mixtas de oxidasas en la síntesis de varias biomoléculas críticas (1). Estas enzimas son monooxigenasas o dioxigenasas (véase la Tabla 1). Los síntomas de la deficiencia de vitamina C, tales como la cicatrización deficiente de las heridas y el letargo, probablemente se deben al deterioro de estas reacciones enzimáticas dependientes de la vitamina C que conduce a la síntesis insuficiente de colágeno, carnitina, y catecolaminas (véase Deficiencia). Además, varias dioxigenasas involucradas en la regulación de la expresión de genes y el mantenimiento de la integridad del genoma requieren vitamina C como un cofactor. De hecho, la investigación ha descubierto recientemente el papel crucial que desempeñan las enzimas, como las TET dioxigenasas y las demetilasas de histonas con dominio de Jumonji, en el destino de las células y los tejidos (Tabla 1). Estas enzimas contribuyen a la regulación epigenética de la expresión de genes al catalizar reacciones involucradas en la desmetilación del ADN y las histonas.
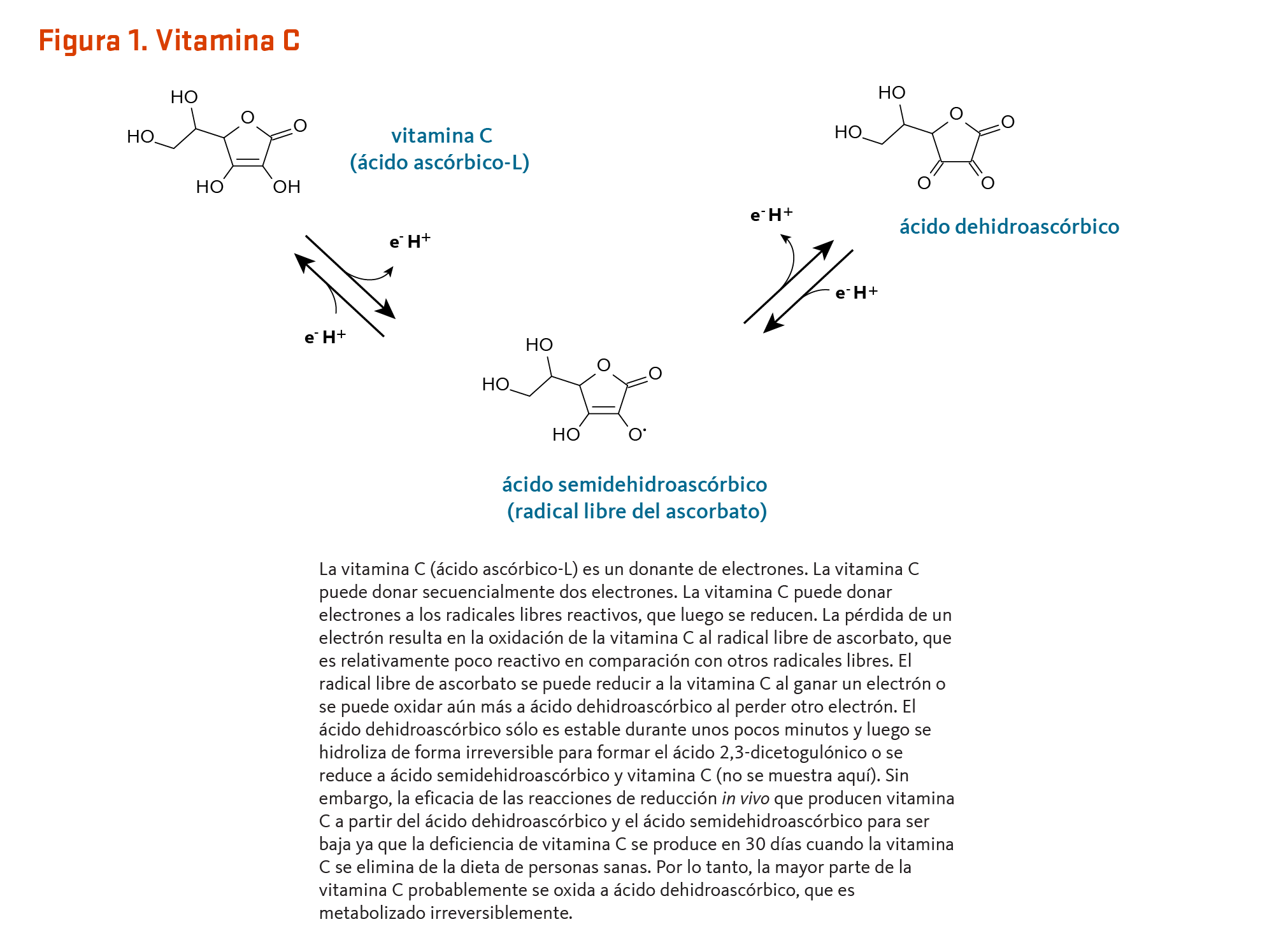
[Figura 1 - Clic para Agrandar]
| Enzimas* | Funciones |
|---|---|
| Monooxigenasas | |
| Dopamina β-monooxigenasa | Biosíntesis de Norepinefrina (Noradrenalina) |
| Peptildiglicano α-amidante monooxigenasa | Amidación de hormonas peptídicas |
| Dioxigenasas | |
| Isoenzimas 3 prolil 4-hidroxilasa | Hidroxilación del colágeno |
|
Isoenzimas 3 prolil 3-hidroxilasa
|
Hidroxilación del colágeno |
| Isoenzimas 3 lisil hidroxilasa | Hidroxilación del colágeno |
| Isoenzimas del 4 factor inducible por hipoxia (HIF) | Hidroxilación de HIF |
| Hidroxilasa tri-metil-lisina | Biosíntesis de carnitina |
| Hidroxilasa γ-butirobetaína | Biosíntesis de carnitina |
| 4-Hidroxifenilpiruvato-dioxigenasa | Metabolismo de tirosina |
| Familia de dioxigenasas de translocación diez-once (TET) | Desmetilación del ADN |
| Demetilasas de histonas con dominio de Jumonji | Desmetilación de histonas |
| *Las monooxigenasas catalizan la hidroxilación de un sustrato, mientras que las dioxigenasas catalizan una reacción que acopla la hidroxilación de un sustrato específico con la conversión (descarboxilación) de α-cetoglutarato a succinato. | |
La capacidad de la vitamina C para influir en el estado de metilación del ADN y en las histonas en las células de los mamíferos apoya el papel de la vitamina en la salud y enfermedad más allá de lo que se entendía previamente, en particular salvaguardando la integridad del genoma (3, 4).
Papel en la inmunidad
La vitamina C afecta a varios componentes del sistema inmunológico humano in vitro; por ejemplo, se ha demostrado que la vitamina C estimula ambas producción (5-9) y función (10, 11) de leucocitos (glóbulos blancos), especialmente neutrófilos, linfocitos, y fagocitos. Medidas específicas de funciones estimuladas por la vitamina C incluyen motilidad celular (10), quimiotaxis (10, 11), y fagocitosis (11). Los neutrófilos, fagocitos mononucleares, y linfocitos acumulan vitamina C en altas concentraciones, las cuales pueden proteger estos tipos de células del daño oxidativo (12-14). En respuesta a microorganismo invasores, leucocitos fagocíticos liberan toxinas no específicas, como radicales superóxidos, ácido hipocloroso ("cloro"), y peroxinitrito; estas especies reactivas de oxígeno matan patógenos y, en el proceso, pueden dañarse los leucocitos también (15). Se ha demostrado que la vitamina C, a través de su funciones antioxidantes, protege los leucocitos de un daño oxidativo auto-infligido (14). Los leucocitos fagocíticos también producen y liberan citoquinas, incluyendo interferones, los cuales tienen una actividad antiviral (16). Se ha mostrado que la vitamina C aumenta la producción de interferones in vitro (17). Otros estudios han informado que la vitamina C mejora la capacidad de destrucción quimiotáctica y microbiana de los neutrófilos y estimula la proliferación y diferenciación de linfocitos B y T (revisado en 18).
El público en general piensa que la vitamina C estimula la función inmunológica, pero los estudios en humanos publicados hasta la fecha son conflictivos. Es probable que los resultados dispares se deban a problemas en el diseño del estudio, a menudo relacionados con una falta de comprensión de la farmacocinética y requisitos de la vitamina C (19, 20).
Finalmente, la vitamina C aumenta la biodisponibilidad del hierro de los alimentos al mejorar la absorción intestinal del hierro no hemo (véase el artículo sobre Hierro) (21).
Biodisponibilidad
Experimentos farmacocinéticos de eliminación y reposición demostraron que la concentración de vitamina C en el plasma está fuertemente controlada por tres mecanismos primarios: absorción intestinal, transporte en el tejido y reabsorción renal (22). En respuesta al incremento de dosis orales de vitamina C, la concentración de vitamina C del plasma se eleva abruptamente en ingestas entre 30 y 100 mg/día. La concentración de ascorbato en el plasma alcanzan un estado estable en concentraciones entre 60 y 80 micromoles/L (µmol/L). Esto se observa normalmente en dosis de 200 a 400 mg/día en adultos jóvenes sanos , con cierto grado de variación individual (23, 24).
Un cien por ciento de la eficacia en la absorción es observada cuando hay una ingesta de vitamina C en dosis separadas de 200 mg a la vez. Las dosis más altas (>500 mg) resultan en que se absorba la vitamina C fraccionadamente menos a medida que aumenta la dosis. Una vez que la concentración de vitamina C en el plasma alcanzan niveles de saturación, vitamina C adicional es excretada en gran parte por la orina. Notablemente, la administración de vitamina C vía intravenosa sobrepasa el control de absorción en el intestino de modo que se pueden alcanzar concentraciones alta muy altas de vitamina C en el plasma; dentro de unas pocas horas, la excreción renal restaura la vitamina C a las concentraciones basales del plasma (véase Tratamiento del Cáncer) (25).
Mientras que la concentración de vitamina C en el plasma refleja la ingesta dietética reciente, se cree que la vitamina C en los leucocitos (glóbulos blancos) es un indicador de las reservas corporales. Sin embargo, la concentración de vitamina en los leucocitos no refleja con precisión la vitamina C en varios tejidos y puede subestimar específicamente la captación de vitamina C en el músculo esquelético (26). No obstante, las concentraciones de vitamina C en el plasma ≥50 µmol/L son suficientes para saturar el tejido muscular en vitamina C.
También hay ciertas evidencias limitadas que sugieren que las personas que portan ciertos polimorfismos en los genes implicados en el transporte de la vitamina C y los mecanismos de desintoxicación pueden tener menores concentraciones de vitamina C en el plasma incluso con altas ingestas de vitamina C (véase también Complicaciones vasculares de la diabetes mellitus) (revisado en 27).
Debido a la farmacocinética y estrecha regulación de la vitamina C en el plasma, la suplementación con vitamina C tendrá efectos variables en vitamina C repleto (concentraciones de plasma cerca a la saturación) versus sub-óptima (concentraciones de plasma <50 µmol/L), ligeramente deficiente (concentraciones de plasma <23 µmol/L), o severamente deficientes (concentraciones de plasma <11 µmol/L) en individuos (28). Estudios científicos investigando la eficacia de la vitamina C para prevenir o tratar enfermedades necesitan evaluar el estatus de la línea de base de la vitamina C antes de embarcarse en una intervención o análisis estadístico (22, 29-31).
Para una discusión más detallada sobre la biodisponibilidad de diferentes formas de vitamina C, véase el artículo separado, La Biodisponibilidad de Diferentes Formas de Vitamina C.
Deficiencia
Una deficiencia severa de vitamina C ha sido conocida por muchos siglos como una enfermedad potencialmente fatal, el escorbuto. A finales de 1700, la marina Británica estaba consciente que el escorbuto podía curarse comiendo naranjas o limones, aunque la vitamina C no sería aislado hasta principios de 1930. Los síntomas del escorbuto incluyen hemorragia subcutánea, deficiencia en la cicatrización de heridas y fácil aparición de moretones, caída de cabello y dientes, y dolor e hinchazón en las articulaciones. Tales síntomas parecen estar relacionados con el debilitamiento de los vasos sanguíneos, tejido conectivo y óseo, los cuales contienen colágeno. Los síntomas iniciales del escorbuto, como fatiga, pueden ser el resultado de la disminución de los niveles de carnitina, la cual se necesita para derivar energía de la grasa, o de la disminución de la síntesis de catecolamina norepinefrina (véase Función). El escorbuto es raro en países desarrollados porque este puede ser prevenido con un mínimo de 10 mg de vitamina C diariamente (32). Sin embargo, han ocurrido casos en niños y ancianos con dietas muy restringidas (33, 34).
La Ingesta Diaria Recomendada (IDR)
La ingesta diaria recomendada (IDR) para la vitamina C está basado en la cantidad de vitamina C ingerida, necesaria para mantener la concentración de vitamina C en los neutrófilos con una mínima excreción urinaria de vitamina C y se propone que provee suficiente protección antioxidante (Tabla 2) (35). La ingesta recomendada para los fumadores es 35 mg/día más alta que para los no fumadores, porque los fumadores están sujetos a un incremento de estrés oxidativo por las toxinas en el humo de cigarrillo y generalmente tienen concentraciones más bajas de vitamina C (36).
| Etapa de la Vida | Edad | Machos (mg/día) | Hembras (mg/día) |
|---|---|---|---|
| Infantes | 0-6 meses | 40 (IA) | 40 (IA) |
| Infantes | 7-12 meses | 50 (IA) | 50 (IA) |
| Niños | 1-3 años | 15 | 15 |
| Niños | 4-8 años | 25 | 25 |
| Niños | 9-13 años | 45 | 45 |
| Adolescentes | 14-18 años | 75 | 65 |
| Adultos | 19 años y más | 90 | 75 |
| Fumadores | 19 años y más | 125 | 110 |
| Embarazo | 18 años y menos | - | 80 |
| Embarazo | 19 años y más | - | 85 |
| Período de lactancia | 18 años y menos | - | 115 |
| Período de lactancia | 19 años y más | - | 120 |
Prevención de Enfermedad
La cantidad de vitamina C requerida para ayudar a prevenir enfermedades crónicas es superior a la cantidad requerida para prevenir el escorbuto. La información con respecto a la vitamina C y la prevención de enfermedades crónicas está basada en ambos estudios cohorte prospectivos observacionales y ensayos controlados aleatorios (29, 37). Estudios de cohorte prospectivos pueden examinar la incidencia de una enfermedad específica con relación a la ingesta de vitamina C o el estado corporal en una cohorte de participantes que son seguidos a lo largo del tiempo. En contraste, los ensayos son estudios de intervención que pueden establecer una relación causal entre una exposición y un resultado, p. ej., al evaluar el efecto de la suplementación con vitamina C en la incidencia de enfermedad crónica en participantes aleatoriamente asignados para recibir ya sea vitamina C o placebo por una determinada duración de tiempo.
Enfermedad cardiovascular
Disfunción endotelial
La disfunción endotelial se considera como un paso temprano en el desarrollo de la aterosclerosis. Las alteraciones en la estructura y función del endotelio vascular que recubre la superficie interna de todos los vasos sanguíneos están asociados con la pérdida de la vasodilatación dependiente del endotelio mediada por el óxido nítrico normal. la disfunción endotelial resulta en la vasoconstricción generalizada y anormalidades en la coagulación. La medición de la dilatación mediada por flujo (DMF) de la arteria braquial es frecuentemente usada como un marcador funcional de la función endotelial; los valores de la DMF están inversamente correlacionados con el riesgo de eventos cardiovasculares futuros (38). Un meta-análisis del 2014 de 44 ensayos controlados aleatorios en sujetos con o sin enfermedades crónicas resumió el efecto de la vitamina C suplementaria en la función endotelial midiendola DMF (19 estudios), evaluando el flujo sanguíneo del antebrazo (20 estudios), o mediante análisis de onda de pulso (5 ensayos) (39). Se encontró que la suplementación a corto plazo con vitamina C reduce la función endotelial en sujetos con falla cardíaca, aterosclerosis o diabetes mellitus, pero no tuvo efecto en aquellos con hipertensión. La vitamina C también limitó la función endotelial que se indujo experimentalmente en voluntarios sanos (39). Se observó una mejora en la función endotelial con dosis diarias de vitamina C por encima de 500 mg (39).
Hipertensión
La hipertensión es un factor de riesgo importante para las enfermedades cardiovascular, incluyendo la enfermedad coronaria, el accidente cerebrovascular y la fibrilación auricular. Un análisis que combinó datos de tres cohortes, grandes, independientes, prospectivas, (1) Estudio de Salud de Enfermeras I (NHS1, por sus siglas en inglés; 88,540 mujeres, edad media de 49 años); (2) Estudio de Salud de Enfermeras 2 (NHS2; 97,315 mujeres, edad media de 36 años); y (3) Estudio de Seguimiento de Profesionales de la Salud (HPFS, por sus siglas en inglés; 37,375 hombres, edad media de 52 años), no encontró asociación entre el nivel de ingesta de vitamina C y el riesgo de desarrollar hipertensión (40). Por otra parte, cuando se midió la concentración de vitamina C en el plasma, los estudios transversales han indicado consistentemente una relación inversa entre la concentración de vitamina C en el plasma y la presión arterial tanto en hombres como en mujeres (41-43). Un seguimiento de 15 años de aproximadamente 2,500 participantes en el estudio de Desarrollo de Arterias Coronarias en Adulto Jóvenes (CARDIA, por sus siglas en inglés) encontró que una mayor cantidad de vitamina C en plasma y una puntuación de calidad dietética más alta se asociaron de forma independiente con un menor riesgo de desarrollar hipertensión (44).
Curiosamente, no hubo relación entre la puntuación de la dieta y el riesgo de hipertensión en aquellos con la vitamina C en plasma más baja, y la vitamina C en plasma se asoció positivamente en las personas con puntuaciones bajas en la dieta (44).
Un meta-análisis de 29 ensayos controlados aleatorios pequeños de corta duración (duración media, 8 semanas) en 1,407 participantes (10 a 120 sujetos por ensayo; incluidos sujetos normotensos e hipertensos) encontró que la suplementación con 60 a 4,000 mg de vitamina C (dosis media, 500 mg) redujo la presión arterial sistólica en 3.84 mm Hg y la presión arterial diastólica en 1.48 mm Hg (45). Se necesitan ensayos a largo plazo de buena calidad para examinar si el efecto antihipertensivo de la vitamina C se mantiene a lo largo del tiempo y eventualmente resulte en un riesgo reducido de eventos cardiovasculares.
Es importante que las personas con presión arterial significativamente elevada no se basen únicamente en los suplementos de vitamina C para reducir su hipertensión. En su lugar, deben buscar o continuar el tratamiento con medicamentos antihipertensivos y hacer cambios en la dieta y el estilo de vida, en consulta con su proveedor del cuidado de la salud.
Riesgo de enfermedad cardiovascular
La enfermedad coronaria (EC) está caracterizada por la acumulación de placa dentro de las arterias que suministran sangre al corazón (aterosclerosis). Después de años de acumulación y daño a las arterias coronarias, la EC puede culminar en un infarto del miocardio o ataque al corazón. Muchos estudios de cohorte prospectivos han examinado la relación entre la ingesta de vitamina C por la dieta y suplementos y el riesgo de padecer enfermedad coronaria, de los cuales los resultados han sido agrupados y analizados en dos análisis diferentes (46, 47). En el 2004, un análisis combinado de nueve estudios de cohorte prospectivos encontró que la ingesta de vitamina suplementaria (≥400 mg/día en un promedio de 10 años), pero no la ingesta dietética de vitamina C, estaba inversamente asociada con riesgo de EC (46). Al contrario, un meta-análisis del 2008 de 14 estudios de cohorte concluyeron que la ingesta dietética, pero no suplementaria, de vitamina C estuvo inversamente relacionada con el riesgo de EC (47). El estudio prospectivo grande más reciente de cohorte encontró una asociación inversa entre la ingesta dietética de vitamina C y la mortalidad de EC en mujeres japonesas, pero no en los hombres (48). A pesar de la variable asociación dependiente de la fuente, estos análisis indican una asociación inversa generalizada entre las altas ingestas de vitamina C y el riesgo de EC.
Las limitaciones inherentes a la metodología de evaluación dietaría, tales como sesgo de recuerdo, errores de medición, y confusión residual, pueden contar como algunas de las asociaciones inconsistentes entre la ingesta de vitamina C y el riesgo de padecer la EC. Para superar tales limitaciones, algunos estudios prospectivos midieron las concentraciones de vitamina C en plasma y suero como un índice más confiable de la ingesta de vitamina C y un biomarcador del estatus de vitamina C en el cuerpo.
El Estudio Prospectivo Europeo sobre Dieta y Cáncer (EPIC), un estudio de cohorte prospectivo de Norfolk, investigó la relación entre el estatus de la vitamina C e incidentes de insuficiencia cardíaca en adultos sanos (9,187 hombres y 11,112 mujeres, entre 58.1+/-9.2 years) (49). Después de un promedio seguido de 12.8 años, la vitamina C del plasma estuvo inversamente asociada con incidentes de casos de insuficiencia cardíaca. Específicamente, la vitamina C del plasma oscilaba entre aproximadamente 23 to 70 µmol/L en los hombres y 33 to 82 µmol/L en las mujeres; a través de este rango, cada 20 µmol/L de incremento de la vitamina C en el plasma se asoció con una reducción en 9% del riesgo de insuficiencia cardíaca. Auto-informes del consumo de frutas y verduras; evaluado por cuestionarios de frecuencia de alimentos — no se asoció con un riesgo menor de insuficiencia cardíaca congestiva (49). Esto resalta el hecho de que las limitaciones asociadas con los métodos de evaluación como los cuestionarios de frecuencia de alimentos, se pueden superar mediante el uso de biomarcadores de la ingesta de nutrientes (50, 51).
Una revisión de 2017 de ocho ensayos controlados aleatorios publicados encontró resultados inconsistentes de siete ensayos que informaron sobre el efecto de la suplementación con vitamina C en el colesterol sérico y triglicéridos, estableció factores de riesgo de enfermedad cardiovascular (52). Sólo un ensayo grande en más de 14,000 hombres mayores que participó en el Estudio de la Salud de Médicos II (PHS II, por sus siglas en inglés) reportó sobre los resultados cardiovasculares. El PHS II encontró que la suplementación con vitamina C (500 mg/día) durante un promedio de ocho años no tuvo un efecto significativo en mayores eventos cardiovasculares, infarto total al miocardio, o mortalidad cardiovascular (53). Notablemente, este estudio tuvo varias limitaciones (54), incluyendo la medición del nivel de vitamina C y el reclutamiento de una población adecuadamente nutrida.
Se necesitan estudios de mejor calidad para examinar el efecto de la vitamina C en los puntos finales cardiovasculares con riesgo elevado de enfermedad cardiovascular.
Accidente cerebrovascular
Un evento cerebrovascular, o accidente cerebrovascular, se puede clasificar como hemorrágico o isquémico. El accidente cerebrovascular hemorrágico ocurre cuando un vaso sanguíneo debilitado se rompe y sangra en el tejido cerebral circundante. El accidente cerebrovascular isquémico ocurre cuando una obstrucción dentro de un vaso sanguíneo bloquea el flujo de sangre al cerebro. La mayoría (~80%) de los eventos cerebrovasculares en países de altos ingresos son de naturaleza isquémica y se asocian a la aterosclerosis como una condición subyacente (55, 56).
Con respecto a la vitamina C y la enfermedad cerebrovascular, un estudio de cohorte prospectivo que siguió a más de 2,000 residentes de una comunidad rural japonesa durante 20 años encontró que el riesgo de accidente cerebrovascular en aquellos con las concentraciones de suero de vitamina C más altas fue 29% más bajo que en aquellos con las concentraciones de suero de vitamina C más bajas (57). De manera similar, el Estudio Prospectivo Europeo sobre Dieta y Cáncer (EPIC)-Norfolk, un estudio de cohorte prospectivo de 10 años en 20,649 adultos, encontró que los individuos con concentraciones de vitamina C en el plasma en el cuartil superior (25%) tuvieron un 42% menos riesgo de accidente cerebrovascular en comparación con aquellos en el cuartil más bajo (≥66 µmol/L vs. <41 µmol/L) (58). Tanto en la población japonesa (57) como en el Estudio Prospectivo Europeo sobre Dieta y Cáncer (EPIC)-Norfolk (58), las concentraciones de vitamina C en la sangre estuvieron altamente correlacionadas con la ingesta de frutas y verduras. Por lo tanto, como en muchos estudios sobre la ingesta de vitamina C y el riesgo de enfermedad crónica, es difícil separar los efectos de la vitamina C de los efectos de otros componentes de las frutas y verduras. Por ejemplo, se sabe que el potasio — que se encuentra en niveles altos en guineos, papas y otras frutas y verduras — es importante en la regulación de la presión arterial, y la presión arterial elevada es un factor de riesgo importante para el accidente cerebrovascular (véase el artículo sobre Potasio). Un meta-análisis del 2013 de 17 estudios de cohorte prospectivos reportó un riesgo 19% menor de accidente cerebrovascular con las ingestas dietéticas de vitamina C más altas versus las más bajas y un riesgo 38% menor con las concentraciones de vitamina C circulantes más altas versus las más bajas (59).
Un ensayo, aleatorio, doble ciego, controlado con placebo en más de 14,000 hombres mayores que participaron en el Estudio de la Salud de Médicos II (PHS II) encontró que la supementación con vitamina C (500 mg/día) por un promedio de ocho años no tuvo un efecto significativo sobre la incidencia o la mortalidad por cualquier tipo de accidente cerebrovascular (53). Otros ensayos tampoco demostraron evidencia de algún efecto de la vitamina C en el riesgo de accidente cerebrovascular. Un meta-análisis de 10 ensayos que examinaron vitaminas antioxidantes, de las cuales cinco incluyeron vitamina C, no encontró asociación entre ninguna vitamina antioxidante (vitamina C, vitamina E, o β-caroteno), administrada por sí sola o en combinación, y el riesgo de accidente cerebrovascular (60).
Cáncer
En general, los estudios de cohorte prospectivos observacionales no reportaron ninguna asociación o alguna asociación inversa modesta entre la ingesta de vitamina C y el riesgo de desarrollo de cierto tipo de cáncer (37, 61-63). A continuación, se proporcionan detalles adicionales para aquellos subtipos de cáncer con información científica sustancial obtenida de estudios de cohorte prospectivos. Los ensayos aleatorios, doble ciego, controlados con placebo que han examinado el efecto de la suplementación con vitamina C (por sí sola o en combinación con otros nutrientes antioxidantes) sobre la incidencia de cáncer o la mortalidad no han mostrado ningún efecto (64).
Cáncer de seno
Dos grandes estudios prospectivos encontraron que la ingesta dietética de vitamina C se asocia inversamente con la incidencia de cáncer de seno en ciertos subgrupos. En el Estudio de Salud de Enfermeras, las mujeres premenopáusicas con antecedentes familiares de cáncer de seno que consumieron un promedio de 205 mg/día de vitamina C de los alimentos tenían un riesgo 63% menor de cáncer de seno que aquellas que consumieron un promedio de 70 mg/día (65). En la Cohorte de Mamografía Sueca, las mujeres con sobrepeso que consumían un promedio de 110 mg/día de vitamina C tenían un riesgo 39% menor de cáncer de seno comparado con las mujeres con sobrepeso que consumían un promedio de 31 mg/día (66). Estudios de cohorte prospectivos más recientes no reportaron ninguna asociación entre la ingesta de de vitamina C dietética o suplementaria y el cáncer de seno (67-69).
Cáncer de estómago
Varios estudios observacionales han encontrado que el aumento en la ingesta de vitamina C dietética está asociado con un menor riesgo de cáncer gástrico (estómago), y los experimentos de laboratorio indican que la vitamina C inhibe la formación de compuestos cancerígenos N-nitroso en el estómago (70-72). Un estudio caso-control anidado en el Estudio Prospectivo Europeo sobre Dieta y Cáncer (EPIC) encontró un riesgo 45% menor de incidencia de cáncer gástrico en individuos en el cuartil más alto (≥51 µmol/L) versus el más bajo (<29 µmol/L) de la concentración de vitamina C en el plasma; no se observó alguna asociación entre la ingesta dietética de vitamina C y el cáncer gástrico (73).
Se sabe que la infección con la bacteria Helicobacter pylori (H. pylori) aumenta el riesgo de cáncer de estómago y se asocia con un menor contenido de vitamina C en las secreciones de estómago (74, 75). Aunque dos estudios de intervención no mostraron una reducción en la incidencia de cáncer con la suplementación con vitamina C (35), algunas investigaciones sugieren que la suplementación con vitamina C puede ser una adición útil a la terapia standard de erradicación de H. pylori para reducir el riesgo de cáncer gástrico (76). Debido a que la vitamina C puede inactivar la ureasa (una enzima que facilita la supervivencia de H. pylori y la colonización de la mucosa gástrica en pH bajo) in vitro, la vitamina C puede ser más efectiva como un agente profiláctico en las personas sin aclorhidria (77, 78).
Cáncer de colon
Al agrupar los datos de 13 estudios de cohorte prospectivos con 676,141 participantes, se determinó que la ingesta dietética de vitamina C no se asoció con el cáncer de colon, mientras que la ingesta total de vitamina C (es decir, de alimentos y suplementos) se asoció con un riesgo 19% menor de cáncer de colon (79). Cada uno de los estudios de cohorte usó cuestionarios de frecuencia de alimentos autoadministrados al inicio de estudio para evaluar la ingesta de vitamina C. Aunque el análisis se ajustó a varios factores de riesgo conocidos y de estilo de vida, los autores notaron que otros comportamientos saludables y/o la ingesta de folato pueden haber confundido la asociación.
Linfoma no Hodgkin
Un estudio prospectivo, basado en la población, el Iowa Women’s Health Study, recolectó datos base en la dieta y suplementos usados en 35,159 mujeres (entre 55-69 años) y evaluó el riesgo de desarrollar linfoma no Hodgkin (NHL) después de 19 años de seguimiento (80). En general, una asociación inversa entre la ingesta de frutas y verduras y el riesgo de padecer linfoma no Hodgkin fue observado. Adicionalmente, una ingesta dietética, pero no suplementaria, y otros nutrientes antioxidantes (carotenoides, proantocianidinas, y manganeso) estuvo inversamente asociada con el riesgo de linfoma no Hodgkin. Otro estudio prospectivo, grande, multicéntrico — la Iniciativa de Salud de las Mujeres — que siguió a 154,363 mujeres posmenopáusicas durante 10 años encontró que la ingesta de vitamina C suplementaria y dietética se asoció inversamente con el linfoma difuso de células B, un subtipo de linfoma no Hodgkin (81).
Otros tipos de cáncer de lugares específicos
El Estudio de la Salud de Médicos II fue un ensayo aleatorizado, controlado con placebo, que examinó el efecto de la vitamina E (400 IU/día), la vitamina C (500 mg/día), y un suplemento multivitamínico sobre el riesgo de cáncer en 14,641 de médicos varones de mediana edad por más de 10.3 años (7.6 años de tratamiento activo más 2.8 años de seguimiento después del tratamiento) (82). La suplementación con vitamina C no tuvo ningún efecto sobre el riesgo general de cáncer ni sobre el riesgo de cáncer de próstata, vejiga o páncreas; hubo una reducción marginal de cáncer colorrectal con la vitamina C en comparación con el placebo (82).
Diabetes mellitus tipo 2
En los Institutos Nacionales de Salud (NIH) — el estudio de Dieta y Salud de la Asociación Americana de Personas Jubiladas (AARP) que incluyó 232,007 participantes, el uso de suplementos de vitamina C durante al menos siete veces a la semana se asoció con un riesgo 9% menor de desarrollar diabetes mellitus tipo 2 comparado con el uso sin suplemento (83). En una cohorte de 21,831 adultos seguidos durante 12 años en el estudio EPIC-Norfolk, se encontró que altos niveles de vitamina C en el plasma estaba fuertemente asociada con un riesgo reducido de diabetes (84). Además, varios estudios transversales reportaron asociaciones inversas entre las concentraciones de vitamina C circulante y marcadores de resistencia a la insulina o intolerancia a la glucosa, como la concentración de hemoglobina glicosilada (HbA1c) (50, 85, 86). Sin embargo, los estudios controlados aleatorios a corto plazo han encontrado efectos de la suplementación con vitamina C en la glucosa en ayunas, insulina en ayunas y concentraciones de HbA1c en individuos sanos (87). No se sabe si la vitamina C suplementaria podría mejorar los marcadores de control glicémico en sujetos con riesgo de diabetes.
Resultados adversos del embarazo
Un meta-análisis del 2015 de 29 ensayos aleatorios controlados encontró que la administración de vitamina C durante el embarazo, por sí sola o en combinación con algunos otros suplementos, no logró reducir el riesgo de muerte fetal, muerte perinatal, restricción del crecimiento intrauterino, parto prematuro, rotura prematura de membranas, y preeclampsia (88) . No obstante, la suplementación con vitamina C llevó a un riesgo 36% menor de desprendimiento de placenta y a un aumento de significativo de la edad gestacional en el nacimiento (88). Otro meta-análisis de 50 ensayos controlados aleatorios en 276,820 mujeres no encontró ningún efecto de la vitamina C, por sí sola o combinada con vitamina E o multivitaminas, cuando se suplementa durante el embarazo (comenzando antes de las 20 semanas de gestación), en los riesgos de pérdida fetal general, aborto espontáneo, y malformación congénita (89).
Fumar cigarrillos durante el embarazo causa restricción del crecimiento intrauterino y partos prematuros, entre otras complicaciones del embarazo (90, 91), y es la causa principal de enfermedad respiratoria infantil (92). Por algunas razones aún no claras, fumar se ha asociado con un menor riesgo de preeclampsia durante el embarazo (93). Un análisis secundario de un ensayo multicéntrico, aleatorio, doble ciego, controlado con placebo en casi 10,000 mujeres embarazadas no encontró reducción en el riesgo de preeclampsia con suplementos de vitamina C (1,000 mg/día) y vitamina E (400 IU/día), independientemente del estado de fumador de las mujeres durante el embarazo. Sin embargo, la suplementación con antioxidantes resultó en riesgos reducidos de desprendimiento de la placenta y el parto prematuro en las mujeres que fumaron durante el embarazo pero no en las no-fumadoras (94). Otro ensayo piloto multicéntrico encontró una mejor función pulmonar durante la primera semana de vida y un menor riesgo de sibilancia durante un año de edad en bebés cuyas madres fumadoras fueron asignadas aleatoriamente para recibir vitamina C (500 mg/día) en vez de un placebo durante el embarazo (95). El estudio de La Vitamina C para Reducir los Efectos de Fumar durante el Embarazo sobre la Función Pulmonar de los Infantes [VCSIP] es un ensayo en curso diseñado para confirmar estas observaciones preliminares utilizando mediciones más precisas de la función pulmonar en una muestra más grande de mujeres asignadas al azar para recibir suplementos de vitamina C o placebo (96).
Enfermedad de Alzheimer
En los EE. UU., la enfermedad de Alzheimer (EA) es la forma más común de demencia, que afecta a 5.5 millones de personas de 65 años y más (97). Estrés oxidativo, neuroinflamación, deposición de placa β-amiloide, marañas formadoras de proteína Tau y muerte de células neuronales en el cerebro de los sujetos afectados por la EA se han asociado con el deterioro cognitivo y pérdida de memoria. Se encontró que las concentraciones más bajas de vitamina C en el fluido cerebroespinal y la matriz extracelular cerebral de un modelo de ratón de EA aumentan el estrés oxidativo y aceleran la deposición de amiloide y la progresión de la enfermedad (98). En otro modelo de ratón de EA que carecía de la capacidad de sintetizar vitamina C, la suplementación con una dosis alta versus baja de vitamina C redujo la deposición de amiloide en la corteza y el hipocampo y limitó los impedimentos de la barrera hematoencefálica y la disfunción mitocondrial (99).
La mayoría de los grandes estudios basados en la población que examinan la relación entre la ingesta de vitamina C o su suplementación con la incidencia de EA han reportado resultados nulos (100). En contraste, los estudios observacionales reportaron concentraciones de vitamina C en el plasma menores en pacientes con EA en comparación con sujetos cognitivamente sanos (101) y se encontró mejor función cognitiva o menor riesgo de deterioro cognitivo con una mayor cantidad de vitamina C en plasma (100).
Pocos estudios han medido la concentración de vitamina C en el fluido cerebroespinal, que refleja mejor el estatus de la vitamina C en el cerebro. La vitamina C se concentra en el cerebro a través de una combinación de transporte activo al tejido cerebral y la retención a través de la barrera hematoencefálica (100). Aunque la vitamina C del fluido cerebroespinal se mantiene en concentraciones muchas veces más altas que la vitamina C en plasma, la función precisa de la vitamina C en la función cognitiva y la etiología de la EA aún no se comprende completamente (102). En un pequeño estudio longitudinal de biomarcadores en 32 individuos con EA probable, una mayor proporción de vitamina C en el fluido cerebroespinal a plasma en la base se asoció con una tasa más lenta de deterioro cognitivo al año de seguimiento (103). La integridad de la barrera hematoencefálica dañada puede afectar la capacidad del cerebro para retener la vitamina C y por lo tanto mantener una alta proporción de vitamina C entre el fluido cerebroespinal y el plasma. La importancia de la proporción de vitamina C en el fluido cerebroespinal a plasma en la progresión de la EA requiere más estudio.
El efecto de la suplementación con vitamina C, en combinación con otros antioxidantes, en los biomarcadores de fluido cerebroespinal y la función cognitiva se ha examinado sólo en unos pocos ensayos con pacientes con EA. En un pequeño (n=23), ensayo abierto, la suplementación combinada con vitamina C (1,000 mg/día) y vitamina E (400 UI/día) a pacientes con EA que tomaron un inhibidor de colinesterasa aumentó significativamente los niveles de antioxidantes y redujo la oxidación de lipoproteínas en el fluido cerebroespinal después de un año, pero no tuvo efecto en el curso clínico de EA comparado con los controles (104). Se obtuvo un resultado similar en un ensayo controlado aleatorio, doble ciego, en el que se combinó la suplementación con vitamina C (500 mg/día), vitamina E (800 UI/día), y el ácido α-lipoico (900 mg/día) durante 16 semanas redujo la oxidación de lipoproteínas en el fluido cerebroespinal, pero no obtuvo beneficios clínicos en individuos con EA leve a moderada (n=78) (105). En este último ensayo, se observó un mayor declive en puntuaciones del Mini Examen del Estado Mental (MEEM) fe observado en el grupo suplementado, sin embargo, la importancia de esta observación permanece inconclusa. Un tercer ensayo controlado con placebo en adultos mayores con deterioro cognitivo leve (edades, 60-75 años) encontró que la suplementación de vitamina C por un año (400 mg/día) y vitamina E (300 mg/día) mejoró la capacidad sanguínea antioxidante, pero sin efecto en los puntajes MEEM (106).
En este momento, parece prudente evitar la deficiencia o insuficiencia de vitamina C, en lugar de la suplementación en individuos repletos, para promover un envejecimiento cerebral saludable (101).
Cataratas
El cristalino del ojo enfoca la luz, produciendo una clara detallada imagen en la retina, una capa de tejido en la pared posterior del globo ocular. Cambios al cristalina relacionados con la edad (engrosamiento, pérdida de flexibilidad) y daño oxidativo contribuyen a la formación de cataratas, p. ej., nubosidad u opacidad en el cristalino que interfiere con un claro enfoque de las imágenes en la retina.
En los seres humanos, la concentración de vitamina es aproximadamente 15 a 20 veces mayor en el humor acuoso — fluido que llena las cámaras anterior y posterior del ojo) que en el plasma, lo cual sugiere que la vitamina C puede estar desempeñando un papel importante en el ojo (107). Disminución de las concentraciones de vitamina C en el cristalino del ojo se han asociado con una mayor severidad de las cataratas (108). Un meta-análisis de estudios observacionales encontró un riesgo reducido de catarata relacionado con la edad con mayores ingestas de vitamina C en estudios caso-control y con mayores concentraciones de vitamina C circulante en estudios transversales. Sin embargo, no se encontraron asociaciones de este tipo en los análisis agrupados de los estudios de cohorte prospectivos (109). De hecho, dos estudios de cohorte prospectivos en hombres suecos (110) y mujeres (111) reportaron que una dosis alta de suplementos con un solo nutriente de vitamina C se asociaron con un mayor riesgo de cataratas, especialmente en aquellos en terapia con corticosteroides.
Una revisión de 2012 de nueve ensayos controlados aleatorios no encontró un efecto sustancial por parte del β-caroteno, la vitamina C, y la vitamina E, administrados individualmente o en combinación durante 2.1 a 12 años, en el riesgo de cataratas o cirugía de cataratas (112). Aunque los ensayos actualmente no respaldan el uso de suplementos con vitamina C de dosis altas en la prevención de cataratas, se observa una asociación inversa consistente entre la ingesta diaria alta de frutas y/o verduras (>5 porciones/día) y el riesgo de catarata (113).
Gota
La gota, una condición que aflige a más del 4% de adultos estadounidenses (114), está caracterizada por concentraciones anormalmente altas de ácido úrico (urato) en la sangre (115). Cristales de urato pueden formarse en las articulaciones, resultando en inflamación y dolor, como también en los riñones y el tracto urinario, resultando en cálculos renales. La tendencia de exhibir elevados niveles de ácido úrico en la sangre y la enfermedad de la gota es a menudo heredada; sin embargo, una modificación en la dieta o el estilo de vida pueden ser provechosos en ambos tratamiento y prevención de la gota (116). En un estudio observacional que incluyó 1,387 hombres, altas ingestas de vitamina C fueron asociadas con concentraciones séricas más bajas de ácido úrico (117). En un estudio transversal realizado en 4,576 afroamericanos, las probabilidades de tener hiperuricemia se asociaron con ingestas dietéticas altas en fructosa, bajas en vitamina C, o con altas proporciones de fructosa a vitamina C (118). Un estudio prospectivo que siguió a una cohorte de 46,994 hombres durante 20 años encontró que la ingesta diaria total de vitamina C se asoció inversamente con la incidencia de gota, mientras que mayores ingestas se asociaron con mayores reducciones de riesgo (119). Los resultados de este estudio también indicaron que la vitamina C suplementaria puede ser útil en la prevención de la gota (119).
Un meta-análisis del 2011 de 13 ensayos controlados aleatorios en individuos sanos con altas concentraciones séricas de ácido úrico reveló que la suplementación con vitamina C (una dosis media de 500 mg/día durante una duración media de 30 días) redujo moderadamente las concentraciones séricas de ácido úrico en 0.35 mg/dL comparado con placebo (120). Esta reducción se encuentra dentro del rango de variabilidad del ensayo y es poco probable que sea clínicamente significativa (121). Un ensayo abierto, de ocho semanas, controlado, aleatorizó a 40 sujetos con gota para recibir ya sea alopurinol (estándar de cuidado), vitamina C, o ambos tratamientos (122). El efecto de la vitamina C, solo o con alopurinol, disminuyendo el ácido úrico en suero fue moderado y mucho menor que el alopurinol solo. El ensayo no examinó el efecto de la vitamina C en otros resultados asociados con la gota (122).
A pesar de que los estudios observacionales sugirieron que la vitamina C suplementaria puede ser útil para prevenir la gota incidente y recurrente, los estudios de intervención realizados hasta el momento no lo han demostrado. Además, actualmente hay poca evidencia para apoyar el papel de la vitamina C en el tratamiento de los pacientes con gota (123).
Mortalidad
Dos grandes estudio de cohorte prospectivos evaluaron la relación entre la ingesta de vitamina C dietética y suplementaria y la mortalidad. En el Vitamins and Lifestyle Study, 55,543 hombres y mujeres (entre 50-76 años) fueron cuestionados como línea base sobre el uso de suplementos dietarios durante los 10 años previos (124). Después de cinco años de seguimiento, el uso de la vitamina C suplementaria se asoció con una ligera disminución del riesgo de mortalidad total, aunque ninguna asociación fue encontrada con enfermedad cardiovascular — o una mortalidad específica de cáncer. En el segundo estudio de cohorte prospectiva, el Diet, Cancer, and Health Study, 55,543 adultos daneses (entre 50-64 años) fueron cuestionados como línea de base acerca de su estilo de vida, dieta, y el uso de suplementos durante los 12 meses previos (125). No se encontró ninguna asociación entre la ingesta de vitamina C dietética o suplementaria y la mortalidad después de aproximadamente 14 años de seguimiento. En contraste, un meta-análisis del 2014 de 10 estudios de cohorte prospectivos en 17,696 mujeres con cáncer de seno encontró un menor riesgo de mortalidad total y específica del cáncer de seno con una mayor ingesta de vitamina C suplementaria y dietética (126). Un meta-análisis del 2012 de de 29 ensayos no encontró ningún efecto de la vitamina C oral, administrada por sí sola o en combinación con otros antioxidantes, en la mortalidad por todas las causas (127).
En paralelo a estos estudios de evaluación dietética, se observó una fuerte asociación inversa entre la vitamina C en el plasma y la mortalidad por todas las causas, la enfermedad cardiovascular y la enfermedad cardíaca isquémica (y el cáncer en hombres solamente) en el estudio multicéntrico prospectivo de cohorte EPIC-Norfolk (128). Después de aproximadamente cuatro años de seguimiento en 19,496 hombres y mujeres (edades 45-79 años), se observó una relación dosis-respuesta tal que cada aumento de 20 µmol/L en la vitamina C en el plasma se asoció con una reducción de riesgo estimada del 20% en mortalidad por todas las causas. De manera similar, las concentraciones más altas de vitamina C en suero se asociaron con menores riesgos de mortalidad por cáncer y por todas las causas en 16,008 adultos de la Encuesta de Evaluación Nacional de Salud y Nutrición (NHANES) III (1994-1998) (129).
Tratamiento de Enfermedad
Enfermedad cardiovascular
Complicaciones de procedimientos cardíacos y cirugías
Lesión miocárdica periprocedimental: La angioplastia coronaria (también llamada angioplastia coronaria transluminal percutánea) es un procedimiento no quirúrgico para el tratamiento de la cardiopatía coronaria obstructiva (CHD, en inglés), incluyendo la angina de pecho inestable, el infarto al miocardio agudo y la CHD de múltiples vasos. La angioplastia consiste en insertar temporalmente e inflar un pequeño globo en la arteria obstruida para ayudar a restablecer el flujo de sangre al corazón. La lesión miocárdica periprocedimental que ocurre en hasta un tercio de los pacientes que se someten a una angioplastia sin complicaciones aumenta el riesgo de morbilidad y mortalidad en seguimiento.
Un ensayo aleatorizado, controlado con placebo, ha examinado el efecto de la vitamina C intravenosa administrada a pacientes con angina estable que se someten a angioplastia coronaria electiva (130). La administración de una infusión de vitamina C de 1 gramo (g) una hora antes de la angioplastia redujo las concentraciones de marcadores de estrés oxidativo y mejoró la perfusión circulatoria en comparación con el placebo (130). Otro ensayo aleatorizó a 532 pacientes para recibir una infusión de 3g de vitamina C o un placebo (solución salina) dentro de las seis horas previas a angioplastia coronaria (131). El tratamiento con vitamina C redujo sustancialmente la incidencia de lesión miocárdica periprocedimental, según se evaluó mediante una reducción en las concentraciones de dos marcadores de lesión miocárdica, a saber, creatina quinasa y troponina-I (131). Un ensayo controlado aleatorio evaluó el efecto de la administración de vitamina C y vitamina E sobre el daño por reperfusión en pacientes que sufrieron un infarto al miocardio agudo y se sometieron a una angioplastia coronaria (ver más abajo) (132).
Lesión por reperfusión miocárdica: La lesión por reperfusión se refiere al daño tisular que se produce en el momento de la restauración del flujo sanguíneo (reperfusión) después de una isquemia transitoria. El músculo cardíaco puede volverse privado de oxígeno (isquémico) como resultado de un infarto al miocardio o con pinzamiento aórtico durante la cirugía de injerto de derivación arterial coronaria (IDAC). El aumento de la generación de especies reactivas de oxígeno (EOR) cuando se restaura el suministro de oxígeno del músculo cardíaco podría ser un contribuyente importante al daño del miocardio que ocurre en la reperfusión (133). La lesión de reperfusión miocárdica conlleva complicaciones, tales como arritmias por reperfusión (véase Fibrilación auricular) y miocardio aturdido.
La vitamina C se agota durante y después de la cirugía cardíaca (134) y esto puede deberse a la extinción de las EOR, a la regeneración de otros antioxidantes, y/o una síntesis masiva de catecolaminas (dopamina, epinefrina, norepinefrina) (135). Dos ensayos aleatorios controlados realizados en la década de los 90 informaron una disminución en el estrés oxidativo inducido por la reperfusión y la lesión miocárdica con la administración de vitamina C intravenosa (136) u oral (137) antes de la cirugía IDAC (revisado en 135). Se ha diseñado un ensayo más reciente aleatorizado, doble ciego, controlado con placebo para examinar el efecto de la administración de vitamina C y vitamina E sobre el daño por isquemia-reperfusión en 99 pacientes con infarto al miocardio agudo sometidos a angioplastia coronaria (132). La infusión de vitamina C (ascorbato de sodio: 3.20 mmol/min durante 1 hora, luego 0.96 mmol/min durante 2 horas) antes de la reperfusión, seguido de una suplementación oral con vitamina C (1 g/día) y vitamina E (400 IU/día) durante 84 días impidió efectivamente una reducción en la capacidad antioxidante en reperfusión y durante las siguientes seis a ocho horas. el protocolo también limitó la disfunción microvascular (es decir, mejoró la perfusión de la microcirculación) y mejoró la fracción de eyección del ventrículo izquierdo al alta (en el día 84) (138, 139). Sin embargo, no se observaron diferencias en el tamaño del infarto entre el tratamiento con vitaminas antioxidantes y el placebo (138).
Fibrilación auricular: La fibrilación auricular es el tipo más común de arritmia cardíaca. También es una complicación común de la cirugía post-cardíaca, que conlleva un mayor riesgo de morbilidad cardiovascular (p. ej., insuficiencia cardíaca, accidente cerebrovascular) y mortalidad. Tres meta-análisis de estudios de cohorte prospectivos y ensayos aleatorios controlados han reportado una reducción general del riesgo de fibrilación auricular postoperatoria después de la administración de vitamina C principalmente por vía oral (140-142). En la mayoría de los ensayos, los participantes recibieron 2 g de vitamina C antes de someterse a una cirugía de reemplazo de válvula o ICAD y 1 a 2 g/día durante cinco días después de la cirugía. Aunque sólo una minoría de los ensayos administraron vitamina C por vía intravenosa, esta vía de administración pareció ser más efectiva para reducir el riesgo de fibrilación auricular — probablemente debido al alcance de concentraciones plasmáticas más altas (140). Es importante señalar que un análisis de subgrupos en uno de los meta-análisis mostró una reducción de la fibrilación auricular postoperatoria con vitamina C en ensayos no basados en los EE. UU. (10 ensayos) pero ningún efecto de la vitamina C en ensayos basados en los EE. UU. (5 ensayos) (140).
Lesión cerebral por isquemia-reperfusión
Un pequeño ensayo controlado aleatorio realizado en 60 pacientes con accidente cerebrovascular isquémico mostró que la administración intravenosa de vitamina C (500 mg/día por 10 días, iniciado el día 1 después del accidente cerebrovascular) no tuvo efecto en los marcadores séricos de estrés oxidativo o resultados neurológicos en comparación con el placebo (143).
Complicaciones vasculares de la diabetes mellitus
La enfermedad cardiovascular (ECV) es la principal causa de muerte en personas con diabetes mellitus. El papel del aumento del estrés oxidativo en la ocurrencia de complicaciones vasculares en sujetos con diabetes ha llevado a la hipótesis de que una mayor ingesta de nutrientes antioxidantes podría ayudar a disminuir el riesgo de ECV en sujetos diabéticos (144). Un meta-análisis de 2018 de ensayos controlados aleatorios investigando el efecto de los suplementos de vitaminas antioxidantes en pacientes con diabetes tipo 2 encontró que la mayor parte de las mejoras en los marcadores de estrés oxidativo y el control de la glucosa en la sangre podrían atribuirse a la vitamina E (145). Otro meta-análisis de los ensayos no encontró efectos de las vitaminas E y C, solas o en combinación, sobre las mediciones de la función de las células β y la resistencia a la insulina (146). Sin embargo, la mayoría de los estudios fueron pequeños y de corta duración y, por lo tanto, no evaluaron las consecuencias del uso a largo plazo de vitaminas antioxidantes en el riesgo de complicaciones vasculares en pacientes diabéticos. Un ensayo aleatorio controlado con placebo de 12 meses en 456 participantes con diabetes tipo 2 tratados con metformina examinó el efecto de la vitamina C (500 mg/día) o del ácido acetilsalicílico (aspirina; 100 mg/día) sobre los factores de riesgo de complicaciones relacionadas a las diabetes como las ECV (147). Tanto la vitamina C como la aspirina redujeron la glucosa en la sangre en ayunas y las concentraciones de HbA1c y mejoraron el perfil de lípidos en la sangre en pacientes tratados con metformina. En comparación con el placebo, se encontró que ambos tratamientos tenían más probabilidades de limitar los factores de riesgo que contribuyen a las complicaciones relacionadas con la diabetes, así como a reducir el riesgo de futuros eventos cardiovasculares durante un período de 10 años (estimado utilizando la puntuación de riesgo de Framingham) (147).
Es de destacar que es posible que las diferencias genéticas entre los pacientes diabéticos influyan en el efecto de la suplementación de la vitamina C en el riesgo cardiovascular. En particular, un alelo específico del gen de la haptoglobina (Hp), a saber, Hp2, parece estar asociado con un mayor riesgo de complicaciones vasculares diabéticas. Los portadores de dos copias del alelo Hp2 (Hp2-2) expresan una proteína Hp que tiene una menor eficacia para unir y eliminar la hemoglobina pro-oxidante libre (Hb) del plasma, en comparación con las proteínas Hp codificadas por los genotipos Hp1-1 y Hp1-2. Cuando se volvieron a analizar los resultados del ensayo Women’s Antioxidant Vitamin Estrogen (WAVE) basado en el genotipo Hp, la terapia antioxidante (1,000 mg/día de vitamina C + 800 UI/día de vitamina E) se asoció con una mejora de la aterosclerosis coronaria en mujeres diabéticas con el genotipo Hp1-1 pero con un empeoramiento de la aterosclerosis en aquellas portadoras del genotipo Hp2-2 (148). Los resultados de otro estudio realizado por los mismos investigadores sugirieron que la vitamina C no pudo prevenir la oxidación de colesterol de lipoproteínas de alta densidad (HDL) por complejos in vitro de Hb-Hp2-2 glicados ni la restauración de la función dañada de HDL en ratones diabéticos portadores del genotipo Hp2-2 (149).
Sepsis
La sepsis y el shock séptico — definidos como presión arterial baja inducida por sepsis persistente — están asociadas con tasas de mortalidad elevadas en pacientes en estado crítico (150, 151). Debido a que las respuestas inflamatorias sistémicas implican un estrés oxidativo excesivo, se ha sugerido que el suministro de nutrientes antioxidantes como la vitamina C puede mejorar el resultado de pacientes en estado crítico en unidades de cuidado intensivo. Además, la hipovitaminosis C es común en pacientes en estado crítico, especialmente en aquellos con shock séptico, y persiste a pesar de la terapia nutricional enteral/parenteral la cual proporciona las cantidades recomendadas de vitamina C (152). Es probable que los requerimientos de vitamina C aumenten en esta población debido a la respuesta hipermetabólica impulsada por la reacción inflamatoria sistémica (152, 153). Se encontró que la administración intravenosa de 50 mg o 200 mg de vitamina C por kg por día durante 96 horas a pacientes con sepsis ingresados en la unidad de cuidado intensivo corrige la deficiencia de vitamina C. La vitamina C también evitó el aumento de las puntuaciones de la Evaluación de Fallo Orgánico Secuencial (SOFA) y del Sistema de Clasificación de Severidad de Enfermedad (APACHE) II — que se utiliza para evaluar la gravedad de la enfermedad y el riesgo de mortalidad — observado en pacientes tratados con placebo (154). La infusión de vitamina C también disminuyó la concentración de marcadores de inflamación y lesión endotelial en pacientes en comparación con placebo (154). En otro ensayo controlado, aleatorio, doble ciego, en 28 pacientes en estado crítico con shock séptico, la infusión de 25 mg de vitamina C por kg cada seis horas por 72 horas limitó significativamente el requerimiento del vasopresor norepinefrina — disminuyendo tanto la dosis como la duración del tratamiento — y mejoró dramáticamente la tasa de supervivencia de 28 días (155). Se han reportado resultados similares en pacientes sépticos que recibieron vitamina C por vía intravenosa (1.5 g/6 h), hidrocortisona (50 mg/6 h), y tiamina (200 mg/12 h) hasta el alta hospitalaria. En comparación con el estándar de atención, este cóctel de intervención redujo en más de la mitad la duración media del uso de vasopresores (18.3 h versus 54.9 h) y redujo las probabilidades de mortalidad en casi un 90% (156). Aunque la administración intravenosa de vitamina C parece ser segura y adecuadamente tolerada, existe un riesgo no despreciable de nefropatía por oxalato (una causa poco frecuente de insuficiencia renal) en estos pacientes en estado críticos (157).
Cáncer
Ruta de administración
En estudios realizados en los años 70 y 80 por Linus Pauling, Ewan Cameron y sus colegas, sugirieron que grandes dosis de vitamina C (10 g/día inyectados por vía intravenosa durante 10 días seguidos de al menos 10 g/día por vía oral indefinidamente) fueron útiles para aumentar el tiempo de supervivencia y la mejora de la calidad de vida de los pacientes con cáncer terminal (158). La controversia sobre la eficacia de la vitamina C en el tratamiento del cáncer se produjo, lo que llevó al reconocimiento de que la ruta de administración de vitamina C es crítica (22, 159). En comparación con la vitamina C administrada por vía oral, la vitamina C intravenosa puede resultar en concentraciones de vitamina C en el plasma de 30 a 70 veces más altas (25). Las concentraciones plasmáticas más altas logradas a través de la administración intravenosa de vitamina C son comparables a las que son tóxicas para las células cancerosas en cultivo. El mecanismo anticancerígeno de la acción intravenosa de la vitamina C está bajo investigación. Este puede implicar la producción de altos niveles de peróxido hidrógeno, selectivamente tóxico para las células cancerosas (22, 160-162), o la desactivación del factor inducible por hipoxia, un factor de transcripción de supervivencia que protege las células cancerosas de varias formas de estrés (159, 163, 164). Es probable que la vitamina C también desempeñe un papel en el mantenimiento de la integridad del genoma y en la protección contra la transformación celular a través de la regulación del ADN y de las enzimas de desmetilación de histonas (véase Función) (165).
Seguridad
La evidencia actual de ensayos clínicos controlados indica que la vitamina C intravenosa es generalmente segura y bien tolerada en pacientes con cáncer. Es de destacar que debido a que la administración intravenosa de 80 g de vitamina C precipitó la anemia hemolítica en dos sujetos con deficiencia de glucosa-6-fosfato deshidrogenasa, los pacientes que van a recibir una infusión de vitamina C en dosis altas se examinan sistemáticamente por este trastorno genético (166). Cuatro ensayos clínicos de fase I en pacientes con cáncer avanzado encontraron que la administración intravenosa de vitamina C en dosis de hasta 1.5 g/kg de peso corporal (equivalente a aproximadamente 100 g/día para un peso promedio [70 kg] por persona) y 70 a 80 g/m2 fue bien tolerado y seguro en pacientes pre-examinados (167-170). Unos pocos estudios observacionales en pacientes con cáncer sometidos a quimioterapia y/o radioterapia informaron que el tratamiento complementario con vitamina C intravenosa se asoció con una reducción de los efectos secundarios asociados con el tratamiento y una mejor calidad de vida (171). Un estudio de fase I en nueve pacientes con cáncer pancreático metastático demostraron que se podían alcanzar concentraciones milimolares de vitamina C en plasma de forma segura cuando se administraba junto con los medicamentos para la quimioterapia contra el cáncer, la gemcitabina y el erlotinib (168).
Sensibilidad a la vitamina C
Los ensayos retrospectivos de formación de colonias in vitro revelaron que las células leucémicas de los pacientes mostraron una sensibilidad variable al tratamiento con vitamina C: las células leucémicas de siete de los nueve pacientes que experimentaron un beneficio clínico significativo fueron sensibles a la vitamina C in vitro (es decir, "respondedores"); las células leucémicas de los seis pacientes restantes no eran sensibles a la vitamina C (es decir, "no-respondedores"). Por lo tanto, los ensayos de sensibilidad a la vitamina C in vitro pueden proporcionar un valor predictivo para la respuesta clínica al tratamiento con vitamina C intravenosa. Los mecanismos que subyacen a la sensibilidad diferencial a la vitamina C están bajo investigación. Los experimentos in vitro realizados con 11 diferentes líneas de células cancerosas demostraron que la sensibilidad a la vitamina C se correlacionó con la expresión de la catalasa, una enzima involucrada en la descomposición del peróxido de hidrógeno (172). Aproximadamente la mitad de las líneas celulares analizadas eran resistentes a la citotoxicidad de la vitamina C, una respuesta asociada con altos niveles de actividad de catalasa.
La sensibilidad a la vitamina C también puede determinarse por la expresión del transportador-2 de la vitamina C dependiente del sodio (SVCT-2), que transporta la vitamina C a las células (173). Los niveles más altos de SVCT-2 se asociaron con una mayor sensibilidad a la vitamina C en nueve niveles diferentes de líneas de células cancerosas de cáncer de seno, pero se expresó débilmente en tejidos normales. Finalmente, las mutaciones en los genes que codifican para las desmetilasas TET dependientes de la vitamina C, mutaciones que son comunes en las células cancerosas, también pueden contribuir a la resistencia al tratamiento con vitamina C (165).
Eficacia
La evidencia actual de la eficacia de la vitamina C intravenosa en pacientes con cáncer se limita a los estudios observacionales, las intervenciones no controladas, y los informes de casos (174, 175). Se necesitan ensayos clínicos de fase II más grandes y de mayor duración que evalúen la eficacia de la vitamina C intravenosa en la progresión de enfermedad y supervivencia en general (176).
El trabajo de Linus Pauling estimuló el interés público en el uso de dosis superiores a 1 g/día de vitamina C para prevenir el resfriado común (177). En los últimos 40 años, numerosos ensayos controlados con placebo han examinado el efecto de la suplementación con vitamina C en la prevención y tratamiento de resfriados. Un meta-análisis de 2013 de 53 ensayos controlados con placebo evaluó el efecto de la suplementación con vitamina C en la incidencia, duración, o gravedad del resfriado común cuando se toma como un suplemento diario continuo (43 ensayos) o como terapia para el inicio de los síntomas del resfriado (10 ensayos) (178). Con respecto a la incidencia de resfriados, se observó una diferencia entre dos grupos de participantes. La suplementación regular con vitamina C (0.25 a 2 g/día) no redujo la incidencia de resfriados en la población general (23 ensayos); sin embargo, en pacientes con estrés físico intenso (p. ej., corredores de maratón, esquiadores, o soldados en condiciones subárticas), la suplementación con vitamina C redujo a la mitad la incidencia de resfriados (5 ensayos). También se observó un beneficio de la suplementación regular con vitamina C en la duración de los resfriados, con un mayor beneficio en niños que en adultos: El efecto combinado de la suplementación con vitamina C fue una reducción del 14% en la duración del resfriado en niños y una reducción del 8% en adultos. Finalmente, no se observó ningún efecto significativo de la suplementación con vitamina C (1-8 g/día) en los ensayos terapéuticos en los que se administró vitamina C después de que ocurrieron los síntomas del resfriado.
Además, una revisión sistemática de 2013 realizada por los mismos investigadores identificó sólo dos pequeños ensayos aleatorios, doble ciego, controlados con placebo que examinaron el efecto de la vitamina C en la incidencia de asma inducida por infección respiratoria (179). Un ensayo encontró que la suplementación con vitamina C (1 g/día) durante 14 semanas redujo el riesgo de ataques de asma precipitados por infección respiratoria. El otro ensayo asignó al azar a sujetos diagnosticados con asma relacionada con la infección para recibir 5 g/día de vitamina C o un placebo durante una semana; se encontró que una proporción menor de participantes presentaba hipersensibilidad bronquial a la histamina — que caracteriza el asma crónica — en el grupo de vitamina C en comparación con el grupo control (revisado en 179). Estas observaciones deben confirmarse en ensayos más grandes y bien diseñados.
Estas observaciones deben confirmarse en ensayos más grandes y bien diseñados.
Asma
Una revisión sistemática de 2013 identificó 11 estudios controlados aleatorios que evaluaron el efecto de la vitamina C en el asma (ocho ensayos) o la broncoconstricción inducida por el ejercicio (tres ensayos) (180). La broncoconstricción inducida por el ejercicio es un estrechamiento transitorio de las vías respiratorias que ocurre después del ejercicio y es indicada por una disminución de ≥10% en Volumen Espiratorio Forzado en 1 segundo (VEF1, o en sus siglas en inglés FEV1). En los tres ensayos que incluyeron un total de 40 participantes con broncoconstricción inducida por el ejercicio, la administración de vitamina C antes del ejercicio (una dosis de 0.5 g en dos días posteriores en un ensayo, una dosis única de 2 g en el segundo ensayo y 1.5 g diariamente durante dos semanas en el tercer ensayo) redujo significativamente la disminución inducida por ejercicio en FEV1. Entre los cinco de los ocho ensayos en sujetos asmáticos que informaron sobre los resultados del FEV1, ninguno encontró una diferencia entre la suplementación con vitamina C y el placebo (180).
Toxicidad del plomo
A pesar de que el uso de pintura con plomo y gasolina con plomo se ha suspendido en los EE. UU., la toxicidad del plomo sigue siendo un problema de salud importante, especialmente en los niños que viven en áreas urbanas. Se ha observado un crecimiento y desarrollo anormales en los bebés de mujeres expuestas al plomo durante el embarazo, mientras que los niños que están expuestos crónicamente al plomo tienen más probabilidades de desarrollar problemas de aprendizaje, problemas de conducta, y tener un CI bajo. En los adultos, la toxicidad del plomo puede causar daño renal, presión arterial alta y anemia.
Varios estudios transversales han informado una asociación inversa entre el estatus de la vitamina C y la concentración de plomo en la sangre. De hecho, en un estudio de 747 hombres mayores, la concentración de plomo en la sangre fue significativamente mayor en aquellos que informaron ingestas totales de vitamina C dietética con un promedio de menos de 109 mg/día en comparación con aquellos con mayores ingestas de vitamina C (181). Un estudio mucho más grande de 19,578 personas, incluyendo 4,214 niños de 6 a 16 años de edad, encontró que concentraciones más altas de vitamina C en suero se asocian con concentraciones de plomo en la sangre significativamente más bajas (182). Una encuesta nacional estadounidense de más de 10,000 adultos encontró que las concentraciones de plomo en la sangre estaban inversamente relacionadas a las concentraciones de vitamina C en suero (183).
Fumar cigarrillos o la exposición de segunda mano al humo del cigarrillo contribuye a aumentar la concentración de plomo en la sangre y a un estado de exposición crónica de bajo nivel al plomo. Un ensayo de intervención en 75 fumadores varones adultos encontró que la suplementación con 1,000 mg/día de vitamina C dio como resultado una concentración de plomo en la sangre significativamente más baja durante un período de tratamiento de cuatro semanas comparación con placebo (184). Una dosis más baja de 200 mg/día no afectó significativamente la concentración de plomo en la sangre, aunque las concentraciones de vitamina C en suero no fueron diferentes de las del grupo que tomó 1,000 mg/día.
El(los) mecanismo(s) por el(los) cual(es) la vitamina C reduce la concentración de plomo en la sangre no se conoce(n), sin embargo, se ha propuesto que la vitamina C podría inhibir la absorción intestinal (184) o mejorar la excreción urinaria de plomo (185).
Fuentes
A diferencia de las plantas y la mayoría de los animales, los humanos han perdido la capacidad de sintetizar la vitamina C de forma endógena y, por lo tanto, tienen un requisito dietético esencial para esta vitamina (véase La Ingesta Diaria Recomendada). Los resultados de 7,277 participantes en la Encuesta Nacional de Salud y Examen Nutricional de los EE. UU. (NHANES) del 2003-2004 indicaron que aproximadamente el 7.1% de los individuos de edades de ≥6 años eran deficientes en vitamina C — basado en concentraciones de vitamina C en suero <11.4 µmol/L (36). El estudio nacional identificó que los fumadores y aquellos con un estatus socioeconómico más bajo tienen un mayor riesgo de deficiencia de vitamina C (36).
Fuentes alimenticias
Como se muestra en la Tabla 3, diferentes frutas y verduras varían en su contenido de vitamina C, pero cinco porciones (equivalentes a 2½ de taza) de una variedad de frutas y verduras deben tener un promedio de entre 150 y 200 mg de vitamina C, especialmente si se consumen frutas ricas en vitamina C. Si desea verificar el contenido de vitamina C, consulte la base de datos de composición de alimentos del USDA.
Suplementos
La vitamina C (ácido ascórbico-L) está disponible en muchas formas, pero hay poca evidencia científica de que una forma sea mejor absorbida o más efectiva que otra. La mayoría de las investigaciones clínicas y experimentales utilizan ácido ascórbico o su sal de sodio, llamado ascorbato de sodio. El ácido ascórbico-L natural y sintético son químicamente idénticos y no se conocen diferencias con respecto a las actividades biológicas o la biodisponibilidad (186).
Ascorbatos minerales
Las sales minerales de la vitamina C se consideran menos ácidas que la vitamina C y, por lo tanto, se consideran "tamponadas." Algunas personas las encuentran menos irritantes para el tracto gastrointestinal que el ácido ascórbico. El ascorbato de sodio y el ascorbato de calcio son las formas más comunes, aunque hay otros ascorbatos minerales disponibles. El ascorbato de sodio proporciona 111 mg de sodio (889 mg de ácido ascórbico) por 1,000 mg de ascorbato de sodio, y el ascorbato de calcio generalmente proporciona de 90 a 110 mg de calcio (890-910 mg de ácido ascórbico) por 1,000 mg de ascorbato de calcio.
Vitamina C con flavonoides
Los flavonoides son una clase de pigmentos vegetales solubles en agua que se encuentran a menudo en las frutas y verduras ricas en vitamina C, especialmente las frutas cítricas y las bayas . Hay poca evidencia de que los flavonoides en la mayoría de las preparaciones comerciales aumenten la biodisponibilidad o la eficacia de la vitamina C (187). Algunos, pero no todos, estudios en modelos animales como las cobayas deficientes en vitamina C o ratas genéticamente escorbúticas encontraron un aumento en la ingesta de vitamina C en circulación periférica y órganos específicos en presencia de flavonoides. Sin embargo, los estudios realizados en seres humanos no encontraron diferencias en la biodisponibilidad de la vitamina C de la fruta entera rica en flavonoides o el jugo de fruta y la vitamina C sintética (revisado en 186).
Vitamina C y metabolitos
Un suplemento, Ester-C®, contiene principalmente ascorbato de calcio e incluye pequeñas cantidades de metabolitos de la vitamina C, ácido deshidroascórbico (ácido ascórbico oxidado), treonato de calcio y cantidades mínimas de xilonato y lixonato. Si bien estos metabolitos pretenden aumentar la biodisponibilidad de la vitamina C, el único estudio publicado en humanos que abordó este problema no encontró diferencias entre el Ester-C® y los comprimidos de vitamina C disponibles comercialmente con respecto a la absorción y excreción urinaria de la vitamina C (187). El Ester-C® no debe confundirse con palmitato de ascorbilo, que también se comercializa como "éster de vitamina C" (véase más abajo).
Palmitato de ascorbilo
El palmitato de ascorbilo es un éster de vitamina C (es decir, ácido ascórbico unido a un ácido graso). En este caso, la vitamina C se esterifica al ácido graso saturado, el ácido palmítico, lo que resulta en una forma soluble en grasa de la vitamina C. Se ha agregado palmitato de ascorbilo a varias cremas para la piel debido al interés en sus propiedades antioxidantes, así como a su importancia en la síntesis de colágeno (188). Aunque el palmitato de ascorbilo también está disponible como un suplemento oral, la mayoría probablemente se hidroliza a ácido ascórbico y ácido palmítico en el tracto digestivo antes de que se absorba (189). El palmitato de ascorbilo se comercializa como "éster de vitamina C," que no debe confundirse con Ester-C® (véase arriba).
Otras formulaciones de vitamina C
Un pequeño ensayo, cruzado, controlado con placebo en 11 hombres mostró que la administración oral de 4 g de vitamina C resultó en una mayor concentración de vitamina C en plasma durante un período de cuatro horas cuando la vitamina C fue encapsulada en liposomas en comparación con la vitamina C no encapsulada (190). Aunque la encapsulación liposomal podría aumentar la biodisponibilidad de la vitamina C, las concentraciones de vitamina C en el plasma fueron mucho más bajas que las logradas con la administración intravenosa de vitamina C (190).
Para una revisión más detallada de la investigación científica sobre la biodisponibilidad de diferentes formas de vitamina C, véase La Disponibilidad de las Diferentes Formas de Vitamina C.
Seguridad
Toxicidad
Se han identificado varios posibles efectos adversos para la salud de dosis muy grandes de vitamina C, principalmente basados en experimentos in vitro o reportes de casos aislados, e incluyen mutaciones genéticas, defectos de nacimiento, cáncer, aterosclerosis, cálculos renales, "escorbuto de rebote," aumento del estrés oxidativo, exceso de absorción de hierro, deficiencia de vitamina B12 y erosión del esmalte dental. Sin embargo, ninguno de estos supuestos efectos adversos para la salud se han confirmado en estudios posteriores, y no existe evidencia científica confiable de que las dosis de vitamina C de hasta 10 g/día en adultos sean tóxicas o perjudiciales para la salud. La preocupación de la formación de cálculos renales con suplementos de vitamina C se discute abajo.
Con la última Ingesta Diaria Recomendada publicada en el 2000, se estableció por primera vez un nivel máximo de ingesta tolerable (NM) para la vitamina C (Tabla 4). Se recomendó un NM de 2 g (2,000 mg) diarios para evitar que los adultos generalmente sanos experimenten diarrea trastornos gastrointestinales (35). Estos síntomas generalmente no son graves, especialmente si se resuelven con la interrupción temporal de la suplementación con vitamina C.
Cálculos renales
Debido a que el oxalato es un metabolito de la vitamina C, existe cierta preocupación de que una ingesta alta de vitamina C podría aumentar el riesgo de cálculos renales de oxalato de calcio. Unos (24, 191, 192), pero no todos los estudios (193-195) han reportado que la vitamina C suplementaria aumenta las concentraciones de oxalato en la orina. Se ha examinado en varios estudios epidemiológicos si un aumento en los niveles de oxalato se traduciría a una elevación en el riesgo de cálculos renales. Dos grandes estudios de cohorte prospectivos, uno siguiendo a 42,251 hombres durante seis años y el otro siguiendo a 85,557 mujeres durante 14 años, reportaron que el consumo de ≥1,500 mg de vitamina C al día no aumentó el riesgo de formación de cálculos renales en comparación con los que consumían <250 mg a diario (196, 197). Por otro lado, otros dos estudios prospectivos grandes informaron que un alto consumo de vitamina C se asoció con un mayor riesgo de formación de cálculos renales en hombres (198, 199). Específicamente, el Estudio de Seguimiento de Profesionales de la Salud recolectó datos en la ingesta dietética y suplementaria de vitamina C cada cuatro años en 45,619 profesionales de la salud masculinos (40-75 años) (198). Después de 14 años de seguimiento, se encontró que los hombres que consumían ≥1,000 mg/día de vitamina C tenían un 41% mayor riesgo de cálculos renales en comparación con los hombres que consumían <90 mg de vitamina C a diario. En el estudio de Cohorte de Hombres Suecos, el uso autoinformado de suplementos de vitamina C con un solo nutriente (tomados siete o más veces por semana) en la base se asoció con un riesgo dos veces mayor de cálculos renales incidentes entre 48,840 hombres (edades 45-79 años) seguidos durante 11 años (199). A pesar de los resultados contradictorios, puede ser prudente que las personas predispuestas a la formación de cálculos renales con oxalato eviten la administración de suplementos de vitamina C en dosis altas.
Interacciones con drogas/fármacos
En general, la evidencia que sugiere que los medicamentos específicos pueden disminuir las concentraciones de vitamina C en la sangre en los seres humanos es limitada. Los bloqueadores de los canales de dihidropiridina del calcio (p. ej., nicardipina, nifedipina) pueden inhibir la captación de vitamina C por las células intestinales in vitro. Sin embargo, no se ha reportado ninguna reducción en las concentraciones de vitamina C en la sangre en humanos con estos medicamentos (200). La aspirina puede afectar el estatus de la vitamina C si se toma con frecuencia (201).
Al contrario, hay reportes de casos sugieren que la vitamina C suplementaria puede disminuir las concentraciones en la sangre de algunos medicamentos, como la flufenazina (el medicamento antipsicótico, Prolixin) y el indinavir (el medicamento antirretroviral, Crixivan) (200). Existe cierta evidencia, aunque controvertida, de que la vitamina C interactúa con medicamentos anticoagulantes como la warfarina (Coumadin). Las dosis grandes de vitamina C pueden bloquear la acción de la warfarina y, por lo tanto, disminuir su eficacia. Las personas que toman anticoagulantes deben limitar su ingesta de vitamina C a <1 g/día y su médico debe controlar su tiempo de protrombina después de la terapia con anticoagulantes (200). Además, la vitamina C puede unir el aluminio en el intestino y aumentar la absorción de compuestos que contienen aluminio (p. ej., antiácidos que contienen aluminio, aglutinantes de fosfato que contienen aluminio). Las personas con insuficiencia renal pueden estar en riesgo de toxicidad por aluminio cuando se toma vitamina C suplementaria al mismo tiempo que estos compuestos (200, 201). Finalmente, la vitamina C suplementaria puede aumentar las concentraciones de estrógeno en la sangre en mujeres que usan anticonceptivos o terapia de reemplazo hormonal (200).
El efecto potencial de los antioxidantes durante la quimioterapia no se conoce bien, pero es probable que sea un problema si un agente quimioterapéutico específico actúa a través de un mecanismo oxidativo, lo cual es poco común (171). No está claro si la vitamina C administrada por vía parenteral podría disminuir o aumentar la eficacia de los fármacos de quimioterapia — en particular, los agentes de alquilación (p. ej., ciclofosfamida, busulfán), antibióticos antitumorales (p. ej., doxorubicina, bleomicina), y trióxido de arsénico. Se recomienda a los pacientes que hablen con su oncólogo antes de usar los suplementos de vitamina C (200, 201).
Debido a que también se ha descubierto que las altas dosis de vitamina C interfieren con la interpretación de ciertas pruebas de laboratorio (p. ej., bilirrubina sérica, creatinina sérica y el análisis de guayaco en heces para detectar la sangre oculta), es importante informar a su proveedor de cuidado de la salud de cualquier reciente uso de suplemento.
Suplementos antioxidantes e inhibidores de la HMG-CoA reductasa (estatinas)
Un ensayo aleatorio controlado de tres años en 160 pacientes con enfermedad coronaria documentada y concentraciones bajas de HDL en la sangre descubrió que una combinación de simvastatina (Zocor) y niacina aumentó la concentración de HDL, inhibió la progresión de estenosis de las arterias coronarias (estrechamiento) y disminuyó la frecuencia de eventos cardiovasculares, tales como el infarto al miocardio y accidente cerebrovascular (202). Sorprendentemente, cuando se tomó una combinación de antioxidantes (1,000 mg de vitamina C, 800 UI de vitamina E, 100 μg de selenio y 25 mg de β-caroteno por día) con la combinación de simvastatina-niacina, se disminuyeron los efectos protectores. Dado que los antioxidantes se tomaron juntos en este ensayo, no se puede determinar la contribución individual de la vitamina C. En contraste, un ensayo mucho más grande en más de 20,000 hombres y mujeres con enfermedad coronaria o diabetes mellitus encontró que la simvastatina y una combinación de antioxidantes (600 mg de vitamina E, 250 mg de vitamina C y 20 mg de β-caroteno por día ) no disminuyeron los efectos cardioprotectores de la terapia con simvastatina durante un período de cinco años (203). Estos hallazgos contradictorios indican que se necesitan más investigaciones sobre las posibles interacciones entre los suplementos antioxidantes y los fármacos reductores de colesterol, tales como los inhibidores de la HMG-CoA reductasa (estatinas).
¿Promueve la vitamina C el daño oxidativo en condiciones fisiológicas?
Se sabe que la vitamina C funciona como un antioxidante altamente efectivo en organismos vivos. Sin embargo, en experimentos de laboratorio, la vitamina C puede interactuar con algunos iones de metal libres y conducir a la generación de radicales libres potencialmente dañinos. Aunque los iones de metal libres generalmente no se encuentran en condiciones fisiológicas, la idea de que altas dosis de vitamina C podrían promover el daño oxidativo in vivo ha recibido mucha atención. Se ha dado publicidad generalizada a algunos estudios que sugieren un efecto pro-oxidante de la vitamina C (204, 205), pero estos estudios resultaron ser defectuosos o sin relevancia fisiológica. Una revisión exhaustiva de la literatura no encontró alguna evidencia científica creíble de que la vitamina C suplementaria promueva el daño oxidativo en condiciones fisiológicas o en humanos (206).
Recomendación del Instituto Linus Pauling
La evidencia combinada de estudios metabólicos, farmacocinéticos y observacionales, y de ensayos aleatorios controlados apoya el consumo de suficiente vitamina C para alcanzar concentraciones plasmáticas de al menos 60 µmol/L. Si bien los adultos jóvenes generalmente sanos pueden alcanzar estas concentraciones plasmáticas con una ingesta diaria de al menos 200 mg/día, algunos individuos pueden tener una capacidad de absorción de vitamina C inferior a lo que se documenta actualmente. Por lo tanto, el Instituto Linus Pauling recomienda una ingesta de vitamina C de 400 mg al día para adultos para garantizar concentraciones tisulares repletas (29) — una cantidad sustancialmente mayor que la Ingesta Diaria Recomendada pero con un riesgo mínimo de efectos secundarios.
Esta recomendación se puede cumplir a través de los alimentos si la dieta incluye al menos varias porciones de frutas y verduras ricas en vitamina C (p. ej., frutas cítricas, kiwi, pimientos; véase Fuentes alimenticias) como parte de la ingesta diaria recomendada de frutas y verduras. La mayoría de los suplementos vitamínicos proporcionan al menos 60 mg de vitamina C.
Adultos mayores (>50 años)
Aún no se sabe con certeza si los adultos mayores tienen mayores requerimientos de vitamina C, sin embargo, se ha encontrado que algunas poblaciones mayores tienen ingestas de vitamina C considerablemente por debajo de la Ingesta Diaria Recomendada de 75 y 90 mg/día para mujeres y hombres, respectivamente (207). Una ingesta de vitamina C de al menos 400 mg al día puede ser particularmente importante para los adultos mayores que tienen un mayor riesgo de enfermedades crónicas relacionadas con la edad. Aún no se han realizado estudios farmacocinéticos en adultos mayores, pero hay cierta evidencia que sugiere que la eficiencia de uno de los mecanismos moleculares para la captación celular de vitamina C disminuye con la edad (208). Debido a que maximizar las concentraciones de la vitamina C en la sangre puede ser importante para proteger contra el daño oxidativo a las células y las moléculas biológicas, una ingesta de vitamina C de al menos 400 mg al día podría beneficiar a los adultos mayores que tienen un mayor riesgo de enfermedades crónicas causadas, en parte, por el daño oxidativo, tales como la enfermedad del corazón, accidente cerebrovascular, ciertos tipos de cáncer, y la catarata.
Autores y Críticos
Escrito originalmente en 2004 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Noviembre 2002 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Septiembre 2003 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Diciembre 2004 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Enero 2006 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Septiembre 2009 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Noviembre 2013 por:
Giana Angelo, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Julio 2018 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Diciembre 2018 por:
Anitra C. Carr, Ph.D.
Profesora Asociada de Investigación
Departamento de Patología y Ciencias Biomédicas
Universidad de Otago
Christchurch, Nueva Zelanda
Revisado en Diciembre 2018 por:
Alexander J. Michels, Ph.D.
Investigador Asociado
Instituto Linus Pauling
Universidad Estatal de Oregon
Traducido al Español en 2019 por:
Natsumi Then Shimazaki
Instituto Linus Pauling
Universidad Estatal de Oregon
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Derechos de autoría 2000-2024 Instituto Linus Pauling
Referencias:
1. Levine M, Padayatty SJ. Vitamin C. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease, 11th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2014:416-426.
2. Englard S, Seifter S. The biochemical functions of ascorbic acid. Annu Rev Nutr. 1986;6:365-406. (PubMed)
3 Camarena V, Wang G. The epigenetic role of vitamin C in health and disease. Cell Mol Life Sci. 2016;73(8):1645-1658. (PubMed)
4 Young JI, Zuchner S, Wang G. Regulation of the epigenome by vitamin C. Annu Rev Nutr. 2015;35:545-564. (PubMed)
5 Jariwalla RJ, Harakeh S. Antiviral and immunomodulatory activities of ascorbic acid. In: Harris JR, ed. Subcellular Biochemistry. Vol. 25. Ascorbic Acid: Biochemistry and Biomedical Cell Biology. New York: Plenum Press; 1996:215-231.
6 Kennes B, Dumont I, Brohee D, Hubert C, Neve P. Effect of vitamin C supplements on cell-mediated immunity in old people. Gerontology. 1983;29(5):305-310. (PubMed)
7 Panush RS, Delafuente JC, Katz P, Johnson J. Modulation of certain immunologic responses by vitamin C. III. Potentiation of in vitro and in vivo lymphocyte responses. Int J Vitam Nutr Res Suppl. 1982;23:35-47. (PubMed)
8 Prinz W, Bortz R, Bregin B, Hersch M. The effect of ascorbic acid supplementation on some parameters of the human immunological defence system. Int J Vitam Nutr Res. 1977;47(3):248-257. (PubMed)
9 Vallance S. Relationships between ascorbic acid and serum proteins of the immune system. Br Med J. 1977;2(6084):437-438. (PubMed)
10. Anderson R, Oosthuizen R, Maritz R, Theron A, Van Rensburg AJ. The effects of increasing weekly doses of ascorbate on certain cellular and humoral immune functions in normal volunteers. Am J Clin Nutr. 1980;33(1):71-76. (PubMed)
11. Levy R, Shriker O, Porath A, Riesenberg K, Schlaeffer F. Vitamin C for the treatment of recurrent furunculosis in patients with impaired neutrophil functions. J Infect Dis. 1996;173(6):1502-1505. (PubMed)
12. Bergsten P, Amitai G, Kehrl J, Dhariwal KR, Klein HG, Levine M. Millimolar concentrations of ascorbic acid in purified human mononuclear leukocytes. Depletion and reaccumulation. J Biol Chem. 1990;265(5):2584-2587. (PubMed)
13. Evans RM, Currie L, Campbell A. The distribution of ascorbic acid between various cellular components of blood, in normal individuals, and its relation to the plasma concentration. Br J Nutr. 1982;47(3):473-482. (PubMed)
14. Jariwalla RJ, Harakeh S. Mechanisms underlying the action of vitamin C in viral and immunodeficiency disease. In: Packer L, Fuchs J, eds. Vitamin C in Health and Disease. New York: Macel Dekker, Inc.; 1997:309-322.
15. Alberts B, Bray D, Lewis J, Raff M. Differentiated cells and the maintenance of tissues. Molecular Biology of the Cell. 3rd ed. New York: Garland Publishing, Inc.; 1994:1139-1193.
16. Pauling L. The immune system. How to Live Longer and Feel Better. 20th Anniversary ed. Corvallis: Oregon State University Press; 2006:105-111.
17. Dahl H, Degre M. The effect of ascorbic acid on production of human interferon and the antiviral activity in vitro. Acta Pathol Microbiol Scand B. 1976;84B(5):280-284. (PubMed)
18. Carr AC, Maggini S. Vitamin C and immune function. Nutrients. 2017;9(11). (PubMed)
19. Lykkesfeldt J, Poulsen HE. Is vitamin C supplementation beneficial? Lessons learned from randomised controlled trials. Br J Nutr. 2010;103(9):1251-1259. (PubMed)
20. Michels AJ, Frei B. Myths, artifacts, and fatal flaws: identifying limitations and opportunities in vitamin C research. Nutrients. 2013;5(12):5161-5192. (PubMed)
21. Johnston CS. Vitamin C. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Ames, Iowa: Wiley-Blackwell; 2012:248-260.
22. Levine M, Padayatty SJ, Espey MG. Vitamin C: a concentration-function approach yields pharmacology and therapeutic discoveries. Adv Nutr. 2011;2(2):78-88. (PubMed)
23. Levine M, Wang Y, Padayatty SJ, Morrow J. A new recommended dietary allowance of vitamin C for healthy young women. Proc Natl Acad Sci U S A. 2001;98(17):9842-9846. (PubMed)
24. Levine M, Conry-Cantilena C, Wang Y, et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci U S A. 1996;93(8):3704-3709. (PubMed)
25. Padayatty SJ, Sun H, Wang Y, et al. Vitamin C pharmacokinetics: implications for oral and intravenous use. Ann Intern Med. 2004;140(7):533-537. (PubMed)
26. Carr AC, Bozonet SM, Pullar JM, Simcock JW, Vissers MC. Human skeletal muscle ascorbate is highly responsive to changes in vitamin C intake and plasma concentrations. Am J Clin Nutr. 2013;97(4):800-807. (PubMed)
27. Michels AJ, Hagen TM, Frei B. Human genetic variation influences vitamin C homeostasis by altering vitamin C transport and antioxidant enzyme function. Annu Rev Nutr. 2013;33:45-70. (PubMed)
28. Carr AC, Pullar JM, Bozonet SM, Vissers MC. Marginal ascorbate status (hypovitaminosis C) results in an attenuated response to vitamin C supplementation. Nutrients. 2016;8(6). (PubMed)
29. Frei B, Birlouez-Aragon I, Lykkesfeldt J. Authors' perspective: What is the optimum intake of vitamin C in humans? Crit Rev Food Sci Nutr. 2012;52(9):815-829. (PubMed)
30. Levine M, Rumsey SC, Daruwala R, Park JB, Wang Y. Criteria and recommendations for vitamin C intake. JAMA. 1999;281(15):1415-1423. (PubMed)
31. Lykkesfeldt J, Christen S, Wallock LM, Chang HH, Jacob RA, Ames BN. Ascorbate is depleted by smoking and repleted by moderate supplementation: a study in male smokers and nonsmokers with matched dietary antioxidant intakes. Am J Clin Nutr. 2000;71(2):530-536. (PubMed)
32. Sauberlich HE. A history of scurvy and vitamin C. In: Packer L, Fuchs J, eds. Vitamin C in health and disease. New York: Marcel Decker, Inc.; 1997:1-24.
33. Stephen R, Utecht T. Scurvy identified in the emergency department: a case report. J Emerg Med. 2001;21(3):235-237. (PubMed)
34. Weinstein M, Babyn P, Zlotkin S. An orange a day keeps the doctor away: scurvy in the year 2000. Pediatrics. 2001;108(3):E55. (PubMed)
35. Food and Nutrition Board, Institute of Medicine. Vitamin C. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids. Washington, D.C.: National Academy Press; 2000:95-185. (National Academy Press)
36. Schleicher RL, Carroll MD, Ford ES, Lacher DA. Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003-2004 National Health and Nutrition Examination Survey (NHANES). Am J Clin Nutr. 2009;90(5):1252-1263. (PubMed)
37. Carr AC, Frei B. Toward a new recommended dietary allowance for vitamin C based on antioxidant and health effects in humans. Am J Clin Nutr. 1999;69(6):1086-1107. (PubMed)
38. Matsuzawa Y, Kwon TG, Lennon RJ, Lerman LO, Lerman A. Prognostic value of flow-mediated vasodilation in brachial artery and fingertip artery for cardiovascular events: a systematic review and meta-analysis. J Am Heart Assoc. 2015;4(11). (PubMed)
39. Ashor AW, Lara J, Mathers JC, Siervo M. Effect of vitamin C on endothelial function in health and disease: a systematic review and meta-analysis of randomised controlled trials. Atherosclerosis. 2014;235(1):9-20. (PubMed)
40. Forman JP, Choi H, Curhan GC. Fructose and vitamin C intake do not influence risk for developing hypertension. J Am Soc Nephrol. 2009;20(4):863-871. (PubMed)
41. Block G, Jensen CD, Norkus EP, Hudes M, Crawford PB. Vitamin C in plasma is inversely related to blood pressure and change in blood pressure during the previous year in young Black and White women. Nutr J. 2008;7:35. (PubMed)
42. Moran JP, Cohen L, Greene JM, et al. Plasma ascorbic acid concentrations relate inversely to blood pressure in human subjects. Am J Clin Nutr. 1993;57(2):213-217. (PubMed)
43. Myint PK, Luben RN, Wareham NJ, Khaw KT. Association between plasma vitamin C concentrations and blood pressure in the European prospective investigation into cancer-Norfolk population-based study. Hypertension. 2011;58(3):372-379. (PubMed)
44. Buijsse B, Jacobs DR, Jr., Steffen LM, Kromhout D, Gross MD. Plasma ascorbic acid, a priori diet quality score, and incident hypertension: a prospective cohort study. PLoS One. 2015;10(12):e0144920. (PubMed)
45. Juraschek SP, Guallar E, Appel LJ, Miller ER, 3rd. Effects of vitamin C supplementation on blood pressure: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2012;95(5):1079-1088. (PubMed)
46 Knekt P, Ritz J, Pereira MA, et al. Antioxidant vitamins and coronary heart disease risk: a pooled analysis of 9 cohorts. Am J Clin Nutr. 2004;80(6):1508-1520. (PubMed)
47. Ye Z, Song H. Antioxidant vitamins intake and the risk of coronary heart disease: meta-analysis of cohort studies. Eur J Cardiovasc Prev Rehabil. 2008;15(1):26-34. (PubMed)
48 Kubota Y, Iso H, Date C, et al. Dietary intakes of antioxidant vitamins and mortality from cardiovascular disease: the Japan Collaborative Cohort Study (JACC) study. Stroke. 2011;42(6):1665-1672. (PubMed)
49. Pfister R, Sharp SJ, Luben R, Wareham NJ, Khaw KT. Plasma vitamin C predicts incident heart failure in men and women in European Prospective Investigation into Cancer and Nutrition-Norfolk prospective study. Am Heart J. 2011;162(2):246-253. (PubMed)
50. Carter P, Gray LJ, Troughton J, Khunti K, Davies MJ. Fruit and vegetable intake and incidence of type 2 diabetes mellitus: systematic review and meta-analysis. BMJ. 2010;341:c4229. (PubMed)
51. Dehghan M, Akhtar-Danesh N, McMillan CR, Thabane L. Is plasma vitamin C an appropriate biomarker of vitamin C intake? A systematic review and meta-analysis. Nutr J. 2007;6:41. (PubMed)
52. Al-Khudairy L, Flowers N, Wheelhouse R, et al. Vitamin C supplementation for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2017;3:Cd011114. (PubMed)
53. Sesso HD, Buring JE, Christen WG, et al. Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians' Health Study II randomized controlled trial. JAMA. 2008;300(18):2123-2133. (PubMed)
54. Roberts LJ, 2nd, Traber MG, Frei B. Vitamins E and C in the prevention of cardiovascular disease and cancer in men. Free Radic Biol Med. 2009;46(11):1558. (PubMed)
55. Hankey GJ. The global and regional burden of stroke. Lancet Glob Health. 2013;1(5):e239-240. (PubMed)
56. Tsivgoulis G, Safouris A, Kim DE, Alexandrov AV. Recent Advances in Primary and Secondary Prevention of Atherosclerotic Stroke. J Stroke. 2018;20(2):145-166. (PubMed)
57. Yokoyama T, Date C, Kokubo Y, Yoshiike N, Matsumura Y, Tanaka H. Serum vitamin C concentration was inversely associated with subsequent 20-year incidence of stroke in a Japanese rural community. The Shibata study. Stroke. 2000;31(10):2287-2294. (PubMed)
58. Myint PK, Luben RN, Welch AA, Bingham SA, Wareham NJ, Khaw KT. Plasma vitamin C concentrations predict risk of incident stroke over 10 y in 20 649 participants of the European Prospective Investigation into Cancer Norfolk prospective population study. Am J Clin Nutr. 2008;87(1):64-69. (PubMed)
59. Chen GC, Lu DB, Pang Z, Liu QF. Vitamin C intake, circulating vitamin C and risk of stroke: a meta-analysis of prospective studies. J Am Heart Assoc. 2013;2(6):e000329. (PubMed)
60. Ye Y, Li J, Yuan Z. Effect of antioxidant vitamin supplementation on cardiovascular outcomes: a meta-analysis of randomized controlled trials. PLoS One. 2013;8(2):e56803. (PubMed)
61. Bertoia M, Albanes D, Mayne ST, Mannisto S, Virtamo J, Wright ME. No association between fruit, vegetables, antioxidant nutrients and risk of renal cell carcinoma. Int J Cancer. 2010;126(6):1504-1512. (PubMed)
62. Heinen MM, Verhage BA, Goldbohm RA, van den Brandt PA. Intake of vegetables, fruits, carotenoids and vitamins C and E and pancreatic cancer risk in The Netherlands Cohort Study. Int J Cancer. 2012;130(1):147-158. (PubMed)
63. Roswall N, Olsen A, Christensen J, Dragsted LO, Overvad K, Tjonneland A. Micronutrient intake and risk of urothelial carcinoma in a prospective Danish cohort. Eur Urol. 2009;56(5):764-770. (PubMed)
64. Goodman M, Bostick RM, Kucuk O, Jones DP. Clinical trials of antioxidants as cancer prevention agents: past, present, and future. Free Radic Biol Med. 2011;51(5):1068-1084. (PubMed)
65. Zhang S, Hunter DJ, Forman MR, et al. Dietary carotenoids and vitamins A, C, and E and risk of breast cancer. J Natl Cancer Inst. 1999;91(6):547-556. (PubMed)
66. Michels KB, Holmberg L, Bergkvist L, Ljung H, Bruce A, Wolk A. Dietary antioxidant vitamins, retinol, and breast cancer incidence in a cohort of Swedish women. Int J Cancer. 2001;91(4):563-567. (PubMed)
67. Hutchinson J, Lentjes MA, Greenwood DC, et al. Vitamin C intake from diary recordings and risk of breast cancer in the UK Dietary Cohort Consortium. Eur J Clin Nutr. 2012;66(5):561-568. (PubMed)
68 Nagel G, Linseisen J, van Gils CH, et al. Dietary beta-carotene, vitamin C and E intake and breast cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC). Breast Cancer Res Treat. 2010;119(3):753-765. (PubMed)
69. Roswall N, Olsen A, Christensen J, Dragsted LO, Overvad K, Tjonneland A. Micronutrient intake and breast cancer characteristics among postmenopausal women. Eur J Cancer Prev. 2010;19(5):360-365. (PubMed)
70. Liu C, Russell RM. Nutrition and gastric cancer risk: an update. Nutr Rev. 2008;66(5):237-249. (PubMed)
71. Mirvish SS, Wallcave L, Eagen M, Shubik P. Ascorbate-nitrite reaction: possible means of blocking the formation of carcinogenic N-nitroso compounds. Science. 1972;177(4043):65-68. (PubMed)
72. Tsugane S, Sasazuki S. Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer. 2007;10(2):75-83. (PubMed)
73 Jenab M, Riboli E, Ferrari P, et al. Plasma and dietary vitamin C levels and risk of gastric cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC-EURGAST). Carcinogenesis. 2006;27(11):2250-2257. (PubMed)
74. Banerjee S, Hawksby C, Miller S, Dahill S, Beattie AD, McColl KE. Effect of Helicobacter pylori and its eradication on gastric juice ascorbic acid. Gut. 1994;35(3):317-322. (PubMed)
75. Zhang ZW, Patchett SE, Perrett D, Katelaris PH, Domizio P, Farthing MJ. The relation between gastric vitamin C concentrations, mucosal histology, and CagA seropositivity in the human stomach. Gut. 1998;43(3):322-326. (PubMed)
76. Chuang CH, Sheu BS, Kao AW, et al. Adjuvant effect of vitamin C on omeprazole-amoxicillin-clarithromycin triple therapy for Helicobacter pylori eradication. Hepatogastroenterology. 2007;54(73):320-324. (PubMed)
77. Krajewska B, Brindell M. Urease activity and L-ascorbic acid. J Enzyme Inhib Med Chem. 2011;26(3):309-318. (PubMed)
78. Pal J, Sanal MG, Gopal GJ. Vitamin-C as anti-Helicobacter pylori agent: More prophylactic than curative- Critical review. Indian J Pharmacol. 2011;43(6):624-627. (PubMed)
79 Park Y, Spiegelman D, Hunter DJ, et al. Intakes of vitamins A, C, and E and use of multiple vitamin supplements and risk of colon cancer: a pooled analysis of prospective cohort studies. Cancer Causes Control. 2010;21(11):1745-1757. (PubMed)
80. Thompson CA, Cerhan JR. Fruit and vegetable intake and survival from non-Hodgkin lymphoma: does an apple a day keep the doctor away? Leuk Lymphoma. 2010;51(6):963-964. (PubMed)
81. Kabat GC, Kim MY, Wactawski-Wende J, Shikany JM, Vitolins MZ, Rohan TE. Intake of antioxidant nutrients and risk of non-Hodgkin's Lymphoma in the Women's Health Initiative. Nutr Cancer. 2012;64(2):245-254. (PubMed)
82. Wang L, Sesso HD, Glynn RJ, et al. Vitamin E and C supplementation and risk of cancer in men: posttrial follow-up in the Physicians' Health Study II randomized trial. Am J Clin Nutr. 2014;100(3):915-923. (PubMed)
83. Song Y, Xu Q, Park Y, Hollenbeck A, Schatzkin A, Chen H. Multivitamins, individual vitamin and mineral supplements, and risk of diabetes among older U.S. adults. Diabetes Care. 2011;34(1):108-114. (PubMed)
84. Harding AH, Wareham NJ, Bingham SA, et al. Plasma vitamin C level, fruit and vegetable consumption, and the risk of new-onset type 2 diabetes mellitus: the European prospective investigation of cancer--Norfolk prospective study. Arch Intern Med. 2008;168(14):1493-1499. (PubMed)
85. Donin AS, Dent JE, Nightingale CM, et al. Fruit, vegetable and vitamin C intakes and plasma vitamin C: cross-sectional associations with insulin resistance and glycaemia in 9-10 year-old children. Diabet Med. 2016;33(3):307-315. (PubMed)
86. Kositsawat J, Freeman VL. Vitamin C and A1c relationship in the National Health and Nutrition Examination Survey (NHANES) 2003-2006. J Am Coll Nutr. 2011;30(6):477-483. (PubMed)
87. Ashor AW, Werner AD, Lara J, Willis ND, Mathers JC, Siervo M. Effects of vitamin C supplementation on glycaemic control: a systematic review and meta-analysis of randomised controlled trials. Eur J Clin Nutr. 2017;71(12):1371-1380. (PubMed)
88. Rumbold A, Ota E, Nagata C, Shahrook S, Crowther CA. Vitamin C supplementation in pregnancy. Cochrane Database Syst Rev. 2015(9):Cd004072. (PubMed)
89. Balogun OO, da Silva Lopes K, Ota E, et al. Vitamin supplementation for preventing miscarriage. Cochrane Database Syst Rev. 2016(5):Cd004073. (PubMed)
90. Hammoud AO, Bujold E, Sorokin Y, Schild C, Krapp M, Baumann P. Smoking in pregnancy revisited: findings from a large population-based study. Am J Obstet Gynecol. 2005;192(6):1856-1862; discussion 1862-1853. (PubMed)
91. Kitsantas P, Christopher KE. Smoking and respiratory conditions in pregnancy: associations with adverse pregnancy outcomes. South Med J. 2013;106(5):310-315. (PubMed)
92. Milner AD, Rao H, Greenough A. The effects of antenatal smoking on lung function and respiratory symptoms in infants and children. Early Hum Dev. 2007;83(11):707-711. (PubMed)
93. Conde-Agudelo A, Althabe F, Belizan JM, Kafury-Goeta AC. Cigarette smoking during pregnancy and risk of preeclampsia: a systematic review. Am J Obstet Gynecol. 1999;181(4):1026-1035. (PubMed)
94. Abramovici A, Gandley RE, Clifton RG, et al. Prenatal vitamin C and E supplementation in smokers is associated with reduced placental abruption and preterm birth: a secondary analysis. Bjog. 2015;122(13):1740-1747. (PubMed)
95. McEvoy CT, Schilling D, Clay N, et al. Vitamin C supplementation for pregnant smoking women and pulmonary function in their newborn infants: a randomized clinical trial. JAMA. 2014;311(20):2074-2082. (PubMed)
96. McEvoy CT, Milner KF, Scherman AJ, et al. Vitamin C to Decrease the Effects of Smoking in Pregnancy on Infant Lung Function (VCSIP): Rationale, design, and methods of a randomized, controlled trial of vitamin C supplementation in pregnancy for the primary prevention of effects of in utero tobacco smoke exposure on infant lung function and respiratory health. Contemp Clin Trials. 2017;58:66-77. (PubMed)
97. Alzheimer's Association. 2018 Alzheimer's Disease Facts and Figures. Available at: https://www.alz.org/alzheimers-dementia/facts-figures. Accessed 6/22/18.
98. Dixit S, Bernardo A, Walker JM, et al. Vitamin C deficiency in the brain impairs cognition, increases amyloid accumulation and deposition, and oxidative stress in APP/PSEN1 and normally aging mice. ACS Chem Neurosci. 2015;6(4):570-581. (PubMed)
99. Kook SY, Lee KM, Kim Y, et al. High-dose of vitamin C supplementation reduces amyloid plaque burden and ameliorates pathological changes in the brain of 5XFAD mice. Cell Death Dis. 2014;5:e1083. (PubMed)
100. Bowman GL. Ascorbic acid, cognitive function, and Alzheimer's disease: a current review and future direction. Biofactors. 2012;38(2):114-122. (PubMed)
101. Harrison J, Rentz DM, McLaughlin T, et al. Cognition in MCI and Alzheimer's disease: baseline data from a longitudinal study of the NTB. Clin Neuropsychol. 2014;28(2):252-268. (PubMed)
102. Hansen SN, Tveden-Nyborg P, Lykkesfeldt J. Does vitamin C deficiency affect cognitive development and function? Nutrients. 2014;6(9):3818-3846. (PubMed)
103. Bowman GL, Dodge H, Frei B, et al. Ascorbic acid and rates of cognitive decline in Alzheimer's disease. J Alzheimers Dis. 2009;16(1):93-98. (PubMed)
104. Arlt S, Muller-Thomsen T, Beisiegel U, Kontush A. Effect of one-year vitamin C- and E-supplementation on cerebrospinal fluid oxidation parameters and clinical course in Alzheimer's disease. Neurochem Res. 2012;37(12):2706-2714. (PubMed)
105. Galasko DR, Peskind E, Clark CM, et al. Antioxidants for Alzheimer disease: a randomized clinical trial with cerebrospinal fluid biomarker measures. Arch Neurol. 2012;69(7):836-841. (PubMed)
106. Naeini AM, Elmadfa I, Djazayery A, et al. The effect of antioxidant vitamins E and C on cognitive performance of the elderly with mild cognitive impairment in Isfahan, Iran: a double-blind, randomized, placebo-controlled trial. Eur J Nutr. 2014;53(5):1255-1262. (PubMed)
107. Reiss GR, Werness PG, Zollman PE, Brubaker RF. Ascorbic acid levels in the aqueous humor of nocturnal and diurnal mammals. Arch Ophthalmol. 1986;104(5):753-755. (PubMed)
108. Tessier F, Moreaux V, Birlouez-Aragon I, Junes P, Mondon H. Decrease in vitamin C concentration in human lenses during cataract progression. Int J Vitam Nutr Res. 1998;68(5):309-315. (PubMed)
109. Wei L, Liang G, Cai C, Lv J. Association of vitamin C with the risk of age-related cataract: a meta-analysis. Acta Ophthalmol. 2016;94(3):e170-176. (PubMed)
110. Zheng Selin J, Rautiainen S, Lindblad BE, Morgenstern R, Wolk A. High-dose supplements of vitamins C and E, low-dose multivitamins, and the risk of age-related cataract: a population-based prospective cohort study of men. Am J Epidemiol. 2013;177(6):548-555. (PubMed)
111. Rautiainen S, Lindblad BE, Morgenstern R, Wolk A. Vitamin C supplements and the risk of age-related cataract: a population-based prospective cohort study in women. Am J Clin Nutr. 2010;91(2):487-493. (PubMed)
112. Mathew MC, Ervin AM, Tao J, Davis RM. Antioxidant vitamin supplementation for preventing and slowing the progression of age-related cataract. Cochrane Database Syst Rev. 2012(6):Cd004567. (PubMed)
113. Pastor-Valero M. Fruit and vegetable intake and vitamins C and E are associated with a reduced prevalence of cataract in a Spanish Mediterranean population. BMC Ophthalmol. 2013;13:52. (PubMed)
114. Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63(10):3136-3141. (PubMed)
115. Saag KG, Choi H. Epidemiology, risk factors, and lifestyle modifications for gout. Arthritis Res Ther. 2006;8 Suppl 1:S2. (PubMed)
116. Choi HK, Curhan G. Gout: epidemiology and lifestyle choices. Curr Opin Rheumatol. 2005;17(3):341-345. (PubMed)
117. Gao X, Curhan G, Forman JP, Ascherio A, Choi HK. Vitamin C intake and serum uric acid concentration in men. J Rheumatol. 2008;35(9):1853-1858. (PubMed)
118. Zheng Z, Harman JL, Coresh J, et al. The dietary fructose: vitamin C intake ratio is associated with hyperuricemia in African-American adults. J Nutr. 2018;148(3):419-426. (PubMed)
119. Choi HK, Gao X, Curhan G. Vitamin C intake and the risk of gout in men: a prospective study. Arch Intern Med. 2009;169(5):502-507. (PubMed)
120. Juraschek SP, Miller ER, 3rd, Gelber AC. Effect of oral vitamin C supplementation on serum uric acid: a meta-analysis of randomized controlled trials. Arthritis Care Res (Hoboken). 2011;63(9):1295-1306. (PubMed)
121. Stamp LK, Zhu X, Dalbeth N, Jordan S, Edwards NL, Taylor W. Serum urate as a soluble biomarker in chronic gout-evidence that serum urate fulfills the OMERACT validation criteria for soluble biomarkers. Semin Arthritis Rheum. 2011;40(6):483-500. (PubMed)
122. Stamp LK, O'Donnell JL, Frampton C, Drake JM, Zhang M, Chapman PT. Clinically insignificant effect of supplemental vitamin C on serum urate in patients with gout: a pilot randomized controlled trial. Arthritis Rheum. 2013;65(6):1636-1642. (PubMed)
123. Andres M, Sivera F, Falzon L, Buchbinder R, Carmona L. Dietary supplements for chronic gout. Cochrane Database Syst Rev. 2014(10):Cd010156. (PubMed)
124. Pocobelli G, Peters U, Kristal AR, White E. Use of supplements of multivitamins, vitamin C, and vitamin E in relation to mortality. Am J Epidemiol. 2009;170(4):472-483. (PubMed)
125. Roswall N, Olsen A, Christensen J, et al. Micronutrient intake in relation to all-cause mortality in a prospective Danish cohort. Food Nutr Res. 2012;56. (PubMed)
126. Harris HR, Orsini N, Wolk A. Vitamin C and survival among women with breast cancer: a meta-analysis. Eur J Cancer. 2014;50(7):1223-1231. (PubMed)
127. Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst Rev. 2012(3):Cd007176. (PubMed)
128. Khaw KT, Bingham S, Welch A, et al. Relation between plasma ascorbic acid and mortality in men and women in EPIC-Norfolk prospective study: a prospective population study. European Prospective Investigation into Cancer and Nutrition. Lancet. 2001;357(9257):657-663. (PubMed)
129. Goyal A, Terry MB, Siegel AB. Serum antioxidant nutrients, vitamin A, and mortality in U.S. Adults. Cancer Epidemiol Biomarkers Prev. 2013;22(12):2202-2211. (PubMed)
130. Basili S, Tanzilli G, Mangieri E, et al. Intravenous ascorbic acid infusion improves myocardial perfusion grade during elective percutaneous coronary intervention: relationship with oxidative stress markers. JACC Cardiovasc Interv. 2010;3(2):221-229. (PubMed)
131. Wang ZJ, Hu WK, Liu YY, et al. The effect of intravenous vitamin C infusion on periprocedural myocardial injury for patients undergoing elective percutaneous coronary intervention. Can J Cardiol. 2014;30(1):96-101. (PubMed)
132. Rodrigo R, Hasson D, Prieto JC, et al. The effectiveness of antioxidant vitamins C and E in reducing myocardial infarct size in patients subjected to percutaneous coronary angioplasty (PREVEC Trial): study protocol for a pilot randomized double-blind controlled trial. Trials. 2014;15:192. (PubMed)
133. Milei J, Forcada P, Fraga CG, et al. Relationship between oxidative stress, lipid peroxidation, and ultrastructural damage in patients with coronary artery disease undergoing cardioplegic arrest/reperfusion. Cardiovasc Res. 2007;73(4):710-719. (PubMed)
134. Lassnigg A, Punz A, Barker R, et al. Influence of intravenous vitamin E supplementation in cardiac surgery on oxidative stress: a double-blinded, randomized, controlled study. Br J Anaesth. 2003;90(2):148-154. (PubMed)
135. Spoelstra-de Man AME, Elbers PWG, Oudemans-van Straaten HM. Making sense of early high-dose intravenous vitamin C in ischemia/reperfusion injury. Crit Care. 2018;22(1):70. (PubMed)
136. Dingchao H, Zhiduan Q, Liye H, Xiaodong F. The protective effects of high-dose ascorbic acid on myocardium against reperfusion injury during and after cardiopulmonary bypass. Thorac Cardiovasc Surg. 1994;42(5):276-278. (PubMed)
137. Sisto T, Paajanen H, Metsa-Ketela T, Harmoinen A, Nordback I, Tarkka M. Pretreatment with antioxidants and allopurinol diminishes cardiac onset events in coronary artery bypass grafting. Ann Thorac Surg. 1995;59(6):1519-1523. (PubMed)
138. Ramos C, Brito R, Gonzalez-Montero J, et al. Effects of a novel ascorbate-based protocol on infarct size and ventricle function in acute myocardial infarction patients undergoing percutaneous coronary angioplasty. Arch Med Sci. 2017;13(3):558-567. (PubMed)
139. Valls N, Gormaz JG, Aguayo R, et al. Amelioration of persistent left ventricular function impairment through increased plasma ascorbate levels following myocardial infarction. Redox Rep. 2016;21(2):75-83. (PubMed)
140. Hemila H, Suonsyrja T. Vitamin C for preventing atrial fibrillation in high risk patients: a systematic review and meta-analysis. BMC Cardiovasc Disord. 2017;17(1):49. (PubMed)
141. Hu X, Yuan L, Wang H, et al. Efficacy and safety of vitamin C for atrial fibrillation after cardiac surgery: A meta-analysis with trial sequential analysis of randomized controlled trials. Int J Surg. 2017;37:58-64. (PubMed)
142. Polymeropoulos E, Bagos P, Papadimitriou M, Rizos I, Patsouris E, Tauoumpoulis I. Vitamin C for the prevention of postoperative atrial fibrillation after cardiac surgery: a meta-analysis. Adv Pharm Bull. 2016;6(2):243-250. (PubMed)
143. Lagowska-Lenard M, Stelmasiak Z, Bartosik-Psujek H. Influence of vitamin C on markers of oxidative stress in the earliest period of ischemic stroke. Pharmacol Rep. 2010;62(4):751-756. (PubMed)
144. Johansen JS, Harris AK, Rychly DJ, Ergul A. Oxidative stress and the use of antioxidants in diabetes: linking basic science to clinical practice. Cardiovasc Diabetol. 2005;4:5. (PubMed)
145. Balbi ME, Tonin FS, Mendes AM, et al. Antioxidant effects of vitamins in type 2 diabetes: a meta-analysis of randomized controlled trials. Diabetol Metab Syndr. 2018;10:18. (PubMed)
146. Khodaeian M, Tabatabaei-Malazy O, Qorbani M, Farzadfar F, Amini P, Larijani B. Effect of vitamins C and E on insulin resistance in diabetes: a meta-analysis study. Eur J Clin Invest. 2015;45(11):1161-1174. (PubMed)
147. Gillani SW, Sulaiman SAS, Abdul MIM, Baig MR. Combined effect of metformin with ascorbic acid versus acetyl salicylic acid on diabetes-related cardiovascular complication; a 12-month single blind multicenter randomized control trial. Cardiovasc Diabetol. 2017;16(1):103. (PubMed)
148. Levy AP, Friedenberg P, Lotan R, et al. The effect of vitamin therapy on the progression of coronary artery atherosclerosis varies by haptoglobin type in postmenopausal women. Diabetes Care. 2004;27(4):925-930. (PubMed)
149. Asleh R, Levy AP. Divergent effects of alpha-tocopherol and vitamin C on the generation of dysfunctional HDL associated with diabetes and the Hp 2-2 genotype. Antioxid Redox Signal. 2010;12(2):209-217. (PubMed)
150. Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31(4):1250-1256. (PubMed)
151. Mann EA, Baun MM, Meininger JC, Wade CE. Comparison of mortality associated with sepsis in the burn, trauma, and general intensive care unit patient: a systematic review of the literature. Shock. 2012;37(1):4-16. (PubMed)
152. Carr AC, Rosengrave PC, Bayer S, Chambers S, Mehrtens J, Shaw GM. Hypovitaminosis C and vitamin C deficiency in critically ill patients despite recommended enteral and parenteral intakes. Crit Care. 2017;21(1):300. (PubMed)
153. Pravda J. Metabolic theory of septic shock. World J Crit Care Med. 2014;3(2):45-54. (PubMed)
154. Fowler AA, 3rd, Syed AA, Knowlson S, et al. Phase I safety trial of intravenous ascorbic acid in patients with severe sepsis. J Transl Med. 2014;12:32. (PubMed)
155. Zabet MH, Mohammadi M, Ramezani M, Khalili H. Effect of high-dose Ascorbic acid on vasopressor's requirement in septic shock. J Res Pharm Pract. 2016;5(2):94-100. (PubMed)
156. Marik PE, Khangoora V, Rivera R, Hooper MH, Catravas J. Hydrocortisone, vitamin C, and thiamine for the treatment of severe sepsis and septic shock: a retrospective before-after study. Chest. 2017;151(6):1229-1238. (PubMed)
157. Spoelstra-de Man AME, Elbers PWG, Oudemans-Van Straaten HM. Vitamin C: should we supplement? Curr Opin Crit Care. 2018;24(4):248-255. (PubMed)
158. Cameron E, Pauling L. Supplemental ascorbate in the supportive treatment of cancer: Prolongation of survival times in terminal human cancer. Proc Natl Acad Sci U S A. 1976;73(10):3685-3689. (PubMed)
159. Ohno S, Ohno Y, Suzuki N, Soma G, Inoue M. High-dose vitamin C (ascorbic acid) therapy in the treatment of patients with advanced cancer. Anticancer Res. 2009;29(3):809-815. (PubMed)
160. Chen Q, Espey MG, Krishna MC, et al. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci U S A. 2005;102(38):13604-13609. (PubMed)
161. Chen Q, Espey MG, Sun AY, et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc Natl Acad Sci U S A. 2007;104(21):8749-8754. (PubMed)
162. Chen Q, Espey MG, Sun AY, et al. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci U S A. 2008;105(32):11105-11109. (PubMed)
163. Gao P, Zhang H, Dinavahi R, et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12(3):230-238. (PubMed)
164. Kuiper C, Molenaar IG, Dachs GU, Currie MJ, Sykes PH, Vissers MC. Low ascorbate levels are associated with increased hypoxia-inducible factor-1 activity and an aggressive tumor phenotype in endometrial cancer. Cancer Res. 2010;70(14):5749-5758. (PubMed)
165. Vissers MCM, Das AB. Potential mechanisms of action for vitamin C in cancer: reviewing the evidence. Front Physiol. 2018;9:809. (PubMed)
166. Carr AC, Cook J. Intravenous vitamin C for cancer therapy - identifying the current gaps in our knowledge. Front Physiol. 2018;9:1182. (PubMed)
167. Hoffer LJ, Levine M, Assouline S, et al. Phase I clinical trial of i.v. ascorbic acid in advanced malignancy. Ann Oncol. 2008;19(11):1969-1974. (PubMed)
168. Monti DA, Mitchell E, Bazzan AJ, et al. Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PLoS One. 2012;7(1):e29794. (PubMed)
169 Riordan HD, Casciari JJ, Gonzalez MJ, et al. A pilot clinical study of continuous intravenous ascorbate in terminal cancer patients. P R Health Sci J. 2005;24(4):269-276. (PubMed)
170. Stephenson CM, Levin RD, Spector T, Lis CG. Phase I clinical trial to evaluate the safety, tolerability, and pharmacokinetics of high-dose intravenous ascorbic acid in patients with advanced cancer. Cancer Chemother Pharmacol. 2013;72(1):139-146. (PubMed)
171. Carr AC, Vissers MC, Cook JS. The effect of intravenous vitamin C on cancer- and chemotherapy-related fatigue and quality of life. Front Oncol. 2014;4:283. (PubMed)
172. Klingelhoeffer C, Kammerer U, Koospal M, et al. Natural resistance to ascorbic acid induced oxidative stress is mainly mediated by catalase activity in human cancer cells and catalase-silencing sensitizes to oxidative stress. BMC Complement Altern Med. 2012;12:61. (PubMed)
173. Hong SW, Lee SH, Moon JH, et al. SVCT-2 in breast cancer acts as an indicator for L-ascorbate treatment. Oncogene. 2013;32(12):1508-1517. (PubMed)
174. Fritz H, Flower G, Weeks L, et al. Intravenous vitamin C and cancer: a systematic review. Integr Cancer Ther. 2014;13(4):280-300. (PubMed)
175. Jacobs C, Hutton B, Ng T, Shorr R, Clemons M. Is there a role for oral or intravenous ascorbate (vitamin C) in treating patients with cancer? A systematic review. Oncologist. 2015;20(2):210-223. (PubMed)
176. Cabanillas F. Vitamin C and cancer: what can we conclude--1,609 patients and 33 years later? P R Health Sci J. 2010;29(3):215-217. (PubMed)
177. Pauling LC. Vitamin C and the Common Cold. San Francisco: W.H. Freeman; 1970.
178. Hemila H, Chalker E. Vitamin C for preventing and treating the common cold. Cochrane Database Syst Rev. 2013(1):Cd000980. (PubMed)
179. Hemila H. Vitamin C and common cold-induced asthma: a systematic review and statistical analysis. Allergy Asthma Clin Immunol. 2013;9(1):46. (PubMed)
180. Milan SJ, Hart A, Wilkinson M. Vitamin C for asthma and exercise-induced bronchoconstriction. Cochrane Database Syst Rev. 2013(10):Cd010391. (PubMed)
181. Cheng Y, Willett WC, Schwartz J, Sparrow D, Weiss S, Hu H. Relation of nutrition to bone lead and blood lead levels in middle-aged to elderly men. The Normative Aging Study. Am J Epidemiol. 1998;147(12):1162-1174. (PubMed)
182. Simon JA, Hudes ES. Relationship of ascorbic acid to blood lead levels. JAMA. 1999;281(24):2289-2293. (PubMed)
183. Lee DH, Lim JS, Song K, Boo Y, Jacobs DR, Jr. Graded associations of blood lead and urinary cadmium concentrations with oxidative-stress-related markers in the U.S. population: results from the third National Health and Nutrition Examination Survey. Environ Health Perspect. 2006;114(3):350-354. (PubMed)
184. Dawson EB, Evans DR, Harris WA, Teter MC, McGanity WJ. The effect of ascorbic acid supplementation on the blood lead levels of smokers. J Am Coll Nutr. 1999;18(2):166-170. (PubMed)
185. Abam E, Okediran BS, Odukoya OO, Adamson I, Ademuyiwa O. Reversal of ionoregulatory disruptions in occupational lead exposure by vitamin C. Environ Toxicol Pharmacol. 2008;26(3):297-304. (PubMed)
186. Carr AC, Vissers MC. Synthetic or food-derived vitamin C--are they equally bioavailable? Nutrients. 2013;5(11):4284-4304. (PubMed)
187. Johnston CS, Luo B. Comparison of the absorption and excretion of three commercially available sources of vitamin C. J Am Diet Assoc. 1994;94(7):779-781. (PubMed)
188. Austria R, Semenzato A, Bettero A. Stability of vitamin C derivatives in solution and topical formulations. J Pharm Biomed Anal. 1997;15(6):795-801. (PubMed)
189. DeRitter E, Cohen N, Rubin SH. Physiological availability of dehydro-L-ascorbic acid and palmitoyl-L-ascorbic acid. Science. 1951;113:628-631. (PubMed)
190. Davis JL, Paris HL, Beals JW, et al. Liposomal-encapsulated ascorbic acid: influence on vitamin C bioavailability and capacity to protect against ischemia-reperfusion injury. Nutr Metab Insights. 2016;9:25-30. (PubMed)
191. Massey LK, Liebman M, Kynast-Gales SA. Ascorbate increases human oxaluria and kidney stone risk. J Nutr. 2005;135(7):1673-1677. (PubMed)
192. Traxer O, Huet B, Poindexter J, Pak CY, Pearle MS. Effect of ascorbic acid consumption on urinary stone risk factors. J Urol. 2003;170(2 Pt 1):397-401. (PubMed)
193. Auer BL, Auer D, Rodgers AL. The effect of ascorbic acid ingestion on the biochemical and physicochemical risk factors associated with calcium oxalate kidney stone formation. Clin Chem Lab Med. 1998;36(3):143-147. (PubMed)
194. Liebman M, Chai WW, Harvey E, Boenisch L. Effect of supplemental ascorbate and orange juice on urinary oxalate. Nutr Res. 1997;17(3):415-425.
195. Wandzilak TR, D'Andre SD, Davis PA, Williams HE. Effect of high dose vitamin C on urinary oxalate levels. J Urol. 1994;151(4):834-837. (PubMed)
196. Curhan GC, Willett WC, Rimm EB, Stampfer MJ. A prospective study of the intake of vitamins C and B6, and the risk of kidney stones in men. J Urol. 1996;155(6):1847-1851. (PubMed)
197. Curhan GC, Willett WC, Speizer FE, Stampfer MJ. Intake of vitamins B6 and C and the risk of kidney stones in women. J Am Soc Nephrol. 1999;10(4):840-845. (PubMed)
198. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol. 2004;15(12):3225-3232. (PubMed)
199. Thomas LD, Elinder CG, Tiselius HG, Wolk A, Akesson A. Ascorbic acid supplements and kidney stone incidence among men: a prospective study. JAMA Intern Med. 2013;173(5):386-388. (PubMed)
200. Natural Medicines. Vitamin C/Professional handout/Interactions with drugs. Available at: https://naturalmedicines.therapeuticresearch.com/. Accessed 7/2/18.
201. Hendler SS, Rorvik DM. PDR for Nutritional Supplements. 2nd ed. Montvale: Thomson Reuters; 2008.
202 Brown BG, Zhao XQ, Chait A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345(22):1583-1592. (PubMed)
203. Collins R, Peto R, Armitage J. The MRC/BHF Heart Protection Study: preliminary results. Int J Clin Pract. 2002;56(1):53-56. (PubMed)
204. Lee SH, Oe T, Blair IA. Vitamin C-induced decomposition of lipid hydroperoxides to endogenous genotoxins. Science. 2001;292(5524):2083-2086. (PubMed)
205. Podmore ID, Griffiths HR, Herbert KE, Mistry N, Mistry P, Lunec J. Vitamin C exhibits pro-oxidant properties. Nature. 1998;392(6676):559. (PubMed)
206. Carr A, Frei B. Does vitamin C act as a pro-oxidant under physiological conditions? Faseb J. 1999;13(9):1007-1024. (PubMed)
207. Brubacher D, Moser U, Jordan P. Vitamin C concentrations in plasma as a function of intake: a meta-analysis. Int J Vitam Nutr Res. 2000;70(5):226-237. (PubMed)
208. Michels AJ, Joisher N, Hagen TM. Age-related decline of sodium-dependent ascorbic acid transport in isolated rat hepatocytes. Arch Biochem Biophys. 2003;410(1):112-120. (PubMed)
Formas Suplementarias
La Biodisponibilidad de las Diferentes Formas de Vitamina C (Ácido Ascórbico)
En un mercado que se extiende rápidamente de suplementos dietarios, es posible encontrar vitamina C en varias formas con diferentes afirmaciones con respecto a su eficacia o biodisponibilidad. La biodisponibilidad se refiere al grado al cual un nutriente (o droga) se encuentra disponible al tejido diana después de que se ha administrado. Nosotros revisamos la parte teórica de los los resultados de investigaciones científicas de la biodisponibilidad de diferentes formas de vitamina C.
Ácido ascórbico natural vs. sintético
El ácido ascórbico-L natural y sintético son químicamente idénticos, y no existen diferencias conocidas en sus actividades biológicas. La posibilidad de que la biodisponibilidad del ácido ascórbico-L de fuentes naturales pueda diferir del ácido ascórbico sintético se investigó en por lo menos dos estudios en seres humanos, y ninguna diferencia clínicamente significante fueron observadas. Un estudio de 12 varones (6 fumadores y 6 no fumadores) encontró que la biodisponibilidad del ácido ascórbico sintético (en polvo y administrado en agua) es ligeramente superior a la del jugo de naranja, basado en los niveles sanguíneos del ácido ascórbico, y sin diferencia basado en el ácido ascórbico en los leucocitos (glóbulos blancos) (1). Un estudio de 68 varones no-fumadores encontró que el ácido ascórbico consumido en brócoli cocido, jugo de naranja, rodajas de naranja, y como tabletas de ácido ascórbico sintético están igualmente biodisponibles, tal como se miden en los niveles de ácido ascórbico en el plasma (2, 3).
Diferentes formas de ácido ascórbico
La absorción gastrointestinal de ácido ascórbico ocurre a través de un activo proceso de transporte, como también a través de una difusión pasiva. En bajas concentraciones de ácido ascórbico gastrointestinal el transporte activo predomina, mientras que en altas concentraciones gastrointestinales el transporte activo se satura, dejando solo una difusión pasiva. En teoría, disminuyendo la velocidad de vaciado del estómago (ej. al tomar ácido ascórbico con comida o al tomar una forma de ácido ascórbico de liberación prolongada) debería incrementar su absorción. Mientras que la biodisponibilidad del ácido ascórbico parece equivalente si está en la forma de polvo, tabletas masticables o tabletas normales, la biodisponibilidad del ácido ascórbico en preparaciones de liberación prolongada es menos exacta.
Un estudio en 3 hombres y una mujer encontró que 1 gramo de ácido ascórbico es igualmente absorbido si proviene de soluciones, tabletas, y tabletas masticables, pero la forma de absorción de una capsula con un cierto tiempo de liberación fue 50% más bajo. La absorción se evaluó midiendo la excreción urinaria del ácido ascórbico después de una dosis intravenosa de ácido ascórbico y entonces comparándola con la excreción urinaria de dosis orales (4).
Un estudio más reciente examino los niveles de ácido ascórbico en el plasma de 59 fumadores varones suplementados durante 2 meses con ya fuese 500 mg/día de ácido ascórbico de liberación prolongada, 500 mg/día de ácido ascórbico simple, o un placebo. Después de dos meses de suplementación ninguna diferencia significativa fue encontrada en los niveles de acido ascórbico del plasma entre el acido ascórbico de liberación prolongada y el acido ascórbico simple en ambos grupos (5). Un segundo ensayo controlado con placebo también evaluó el acido ascórbico simple versus el acido ascórbico de liberación prolongada en 48 fumadores varones (6). Los participantes fueron suplementados con 250 mg de acido ascórbico simple como con 250 mg de acido ascórbico de liberación prolongada, o placebo dos veces al día por cuatro semanas. No se observaron diferencias en el cambio de la concentración de ascorbato en el plasma o el área bajo la curva después de la ingestión de cualquiera de la formulación.
Ascorbatos minerales
Las sales minerales del acido ascórbico (ascorbatos minerales) están "tamponadas" y por lo tanto son menos ácidas. Por lo tanto, estos ascorbatos minerales son recomendados frecuentemente a personas que experimentan problemas gastrointestinales (malestar estomacal, o diarrea) con acido ascórbico plano. Parece haber poca investigación científica que apoye o refute la afirmación de que los ascorbatos minerales son menos irritantes al tracto gastrointestinal. Cuando las sales minerales del acido ascórbico son tomadas, ambos el acido ascórbico y los minerales parecen ser bien absorbidos, por ello es importante considerar la dosis de los minerales que acompañan al acido ascórbico cuando se toman altas dosis de ascorbatos minerales. Para la discusión siguiente, se debe notar que 1 gramo (g)+ 1,000 miligramos (mg) y 1 miligramos (mg)= 1,000 microgramos (μg). Los ascorbatos minerales están disponibles en las siguientes formas:
- Ascorbato de sodio: 1,000 mg de ascorbato de sodio generalmente contiene 111 mg de sodio. Individuos que mantienen una dieta baja en sodio (ej. Presión arterial alta) son generalmente aconsejados a mantener su ingesta dietaría de sodio a un nivel de 2,500 mg/dia o menos. Por lo tanto, mega dosis de vitamina C en la forma de ascorbato de sodio podría significantemente incrementar la ingesta de sodio (vea Cloruro de Sodio).
- Ascorbato de calcio: El ascorbato de calcio generalmente provee de entre 90-110 mg de calcio (890-910 mg de acido ascórbico) por cada 1,000 mg de ascorbato de calcio. En esta forma el calcio parece ser razonablemente bien absorbido. La ingesta dietaría recomendad para personas adultas es de 1,000 a 1,200 mg/día. La ingesta total de calcio no debería exceder la NM, la cual es de 2,500 mg/día para adultos de entre 19-50 años de edad y 2,000 mg/día para adultos mayores de 50 años (vea Calcio).
Los siguientes ascorbatos minerales son más propensos a ser encontrados en combinación con otros ascorbatos minerales como también con otros minerales. Es una buena idea comprobar las etiquetas de suplementos dietarios para la dosis de acido ascórbico así como la de cada mineral. Ingestas dietarías recomendadas y niveles superiores de ingesta máxima (cuando son disponibles) son listados después de los ascorbatos minerales siguientes:
- Ascorbato de potasio: El requerimiento mínimo de potasio se piensa es entre 1.6 y 2.0 g/día. Las frutas y verduras son fuentes ricas de potasio, y una dieta rica en frutas y verduras puede proveer tanto como entre 8 a 11 g/día. Se piensa que la toxicidad aguda y potencialmente fatal del potasio en adultos (hipercalemia) ocurre si hay una ingesta de 18 g/día de potasio. Los individuos que toman diuréticos ahorradores de potasio y aquellos con insuficiencia renal deberían evitar ingestas significantes de ascorbato de potasio. La forma comercial más pura disponible de ascorbato de potasio contiene 0.175 gramos (175 mg) de potasio por gramos de ascorbato (vea Potasio).
- Ascorbato de magnesio: La ingesta diaria recomendada (IDR) para el magnesio es de 400-420 mg/día para hombres adultos y 310-320 mg/día para mujeres adultas. La nivel máximo de ingesta tolerable (NM) para el magnesio proveniente de suplementos no deberían exceder 350 mg/día (ver Magnesio).
- Ascorbato de zinc: El IDR para el zinc es de 11 mg/día para adultos varones y 8 mg/día para mujeres adultas. El nivel máximo de ingesta tolerable (NM) del zinc en adultos no debería exceder 40 mg/día (vea Zinc).
- Ascorbato de molibdeno: La IDR para el molibdeno es de 45 microgramos (μg)/día para hombres y mujeres adultos. El nivel máximo de ingesta tolerable (NM) de molibdeno para adultos no debería exceder 2,000 μg (2mg)/día (ver Molibdeno).
- Ascorbato de cromo: La ingesta diaria recomendada para el cromo es de 30-35 μg/día para adultos varones y 20-25 μg/día para mujeres adultas. El nivel máximo de ingesta tolerable (NM) no ha sido determinado por la Junta de Nutrición y Alimentos de EE.UU. (US Food and Nutrition Board) (vea Cromo).
- Ascorbato de manganeso: La Ingesta diaria recomendada para el manganeso es de 2.3 mg/día para hombres adultos y 1.8 mg/día para mujeres adultas. El nivel máximo de ingesta tolerable (NM) para el manganeso en adultos no debería exceder 11 mg/día. El ascorbato de manganeso se encuentra en algunas preparaciones de glucosamina y sulfato de condroitina, y siguiendo las dosis recomendadas en la etiqueta de dichos suplementos podría resultar en una ingesta diaria que exceda el nivel máximo tolerable de manganeso (vea Manganeso).
La vitamina C con bioflavonoides
Los bioflavonoides o flavonoides son compuestos polifenólicos encontrados en las plantas. Las plantas y verduras ricas en vitamina C, especialmente frutas cítricas, son frecuentemente fuentes de flavonoides también. El efecto de los bioflavonoides en la biodisponibilidad del acido ascórbico ha sido recientemente revisado (7).
Los resultados de 10 estudios clínicos que compararon la absorción de la vitamina C sola o vitamina C contenida en flavonoides en los alimentos no mostraron una apreciable diferencia en la biodisponibilidad del acido ascórbico. Solo un estudio el cual incluyo cinco hombres y tres mujeres encontró que un suplemento de 500-mg de acido ascórbico sintético, dado en un extracto cítrico natural que contenía bioflavonoides, proteínas, y carbohidratos, fue más lentamente absorbida y un 35% mas biodisponible que el acido ascórbico sintético solo, estando basado en los niveles de acido ascórbico en el plasma (8). Los estudios restantes mostraron ningún cambio o niveles de acido ascórbico en el plasma ligeramente más bajas en sujetos que consumieron vitamina C con flavonoides en comparación con los flavonoides por sí solos (7).
Otra evaluación de la biodisponibilidad de la vitamina C se da al medir los niveles de ascorbato urinario a tasas aproximadas de excreción de la vitamina C. Un estudio en seis jóvenes japoneses varones (entre 22-26 años de edad) mostro una significante reducción en la excreción urinaria de acido ascórbico en la presencia de jugo de acerola, una fuente natural tanto de vitamina C como flavonoides (9). Sin embargo, tres estudios separados mostraron que los niveles urinarios de vitamina C se incrementaron después del consumo de kiwi (10), jugo de grosella negra (11), o jugo de naranja (1). En general, el impacto de los flavonoides en la biodisponibilidad de la vitamina C parece ser insignificante; sin embargo, hay una necesidad de estudios cuidadosamente controlados que utilicen extractos de flavonoides específicos (7).
Ascorbato y metabolitos de la vitamina C (Ester-C®)
Ester-C® contiene mayormente ascorbato de calcio, pero también contiene pequeñas cantidades de metabolitos de la vitamina C, ácido dehidroascórbico (ácido ascórbico oxidado), treonato de calcio, y niveles traza de xilófago y lixonato. En la parte teórica, los fabricantes declararon que los metabolitos, especialmente el treonato, incremento la biodisponibilidad de la vitamina C en este producto, y ellos indicaron que habían realizado un estudio en seres humanos que demostró el incremento de la biodisponibilidad de la vitamina C en Ester-C®. Este estudio no ha sido publicado en alguna revista revisada por expertos. Un pequeño estudio publicado de la biodisponibilidad de la vitamina C en ocho mujeres y un hombre no encontró alguna diferencia entre Ester-C® y tabletas de acido ascórbico disponible comercialmente con respecto a la absorción y excreción urinaria de la vitamina C (12). Ester-C® no debería ser confundido con el palmitato de ascorbilo el cual es también comercializado como “éster de vitamina C” (ver a continuación).
Palmitato de ascorbilo
El palmitato de ascorbilo es un antioxidante liposoluble usado para incrementar la vida útil de los aceites vegetales y papas fritas (13). Este es una molécula anfipática es decir un extremo es hidrosoluble y el otro es liposoluble. Esta solubilidad dual le permite incorporarse en las membranas celulares. Se ha encontrado que el palmitato de ascorbilo al ser incorporado en la membrana celular de los glóbulos rojos este los protege del daño oxidativo y protege el α-tocoferol (un antioxidante liposoluble) de la oxidación por radicales libres (14). Sin embargo, los efectos protectores del palmitato de ascorbilo en las membranas celulares han sido solamente demostrados en tubos de ensayo. Al tomar el palmitato de ascorbilo oralmente es probable que no resulte en alguna significativa incorporación en las membranas celulares porque la mayoría de este parece ser hidrolizado (degradado en palmitato y acido ascórbico) en el tracto digestivo humano antes de ser absorbido. El acido ascórbico liberado por la hidrólisis del palmitato de ascorbilo parece estar tan biodisponible como el acido ascórbico solo (15). La presencia del palmitato de ascorbilo en suplementos orales contribuye a el contenido del acido ascórbico del suplemento y probablemente ayude a proteger antioxidantes hidrosolubles en el suplementos. Los papeles de la vitamina C en la promoción de la síntesis de colágeno y como un antioxidante han generado interés en su uso en la piel (vea el artículo, La vitamina C y la Salud de la Piel). El palmitato de ascorbilo es frecuentemente usado en preparaciones tópicas porque es más estable que algunas otras formas acuosas (hidrosolubles) de la vitamina C (16). El palmitato de ascorbilo es también comercializado como un éster de la vitamina C, el cual no debería ser confundido con Ester-C®.
Ácido D-araboascórbico (Acido eritórbico)
El acido eritórbico es un isómero del acido ascórbico. Los isómeros son compuestos que tienen el mismo tipo y numero de átomos, pero diferentes estructuras moleculares. La diferencia en la estructura molecular entre los isómeros puede resultar en diferentes propiedades químicas. El acido eritórbico es usado en los Estados Unidos como un antioxidante alimenticio aditivo y es generalmente reconocido como seguro. Se ha estimado que más de 200 mg de acido eritórbico per cápita es introducido diariamente en el sistema de alimentos del los Estados Unidos. Diferente al acido ascórbico, el acido eritórbico no parece ejercer la actividad de la vitamina C, por ejemplo, no previno el escorbuto en conejillos de india (una de las pocas especies animales distintas de los seres humanos que no sintetizan el ácido ascórbico). Sin embargo, estudios en conejillos de india también indicaron que una ingesta incrementada de acido eritórbico redujo la biodisponibilidad del acido ascórbico hasta un 50%. En contraste, una serie de estudios en mujeres jóvenes encontró que hasta 1,000 mg/día de acido eritórbico por hasta 40 días fue rápidamente eliminado del cuerpo y tuvo poco efecto en la biodisponibilidad del acido ascórbico, indicando que el acido eritórbico no disminuye la biodisponibilidad del acido ascórbico en seres humanos a niveles nutricionalmente relevantes de la ingesta (17).
Otras formulaciones de la vitamina C
PureWay-C® está compuesto de vitamina C y metabolitos lípidos. Dos estudios de cultivos celulares que usaron PureWay-C® han sido publicados por los mismos investigadores (18, 19), pero datos in vivo son necesarios actualmente. Un pequeño estudio en adultos saludables encontró que los niveles plasmáticos de la vitamina C no difirieron cuando una sola dosis oral (1 gramo) de cualquiera PureWay-C® o ácido ascórbico fue administrada (20).
Otra formulación de la vitamina C, la vitamina C encapsulada en liposomas (ej. Lypo-spheric™ de la vitamina C) esta comercialmente disponible ahora. Sin embargo, datos referentes a la biodisponibilidad de la vitamina C encapsulada en liposomas no están actualmente disponibles.
Estudios farmacocinéticos a grande escala son necesarios para determinar como la biodisponibilidad de estas formulaciones de la vitamina C se compara con la del acido ascórbico.
Última actualización 11/27/13 Derechos de autoría 2000-2024 Instituto Linus Pauling
Referencias
1. Pelletier, O. & Keith, M.O. Bioavailability of synthetic and natural ascorbic acid. Journal of the American Dietetic Association. 1974; 64: 271-275
2. Mangels, A.R. et al. The bioavailability to humans of ascorbic acid from oranges, orange juice, and cooked broccoli is similar to that of synthetic ascorbic acid. Journal of Nutrition. 1993; volume 123: pages 1054-1061. (PubMed)
3. Gregory, J.F. Ascorbic acid bioavailability in foods and supplements. Nutrition Reviews. 1993; volume 51: pages 301-309. (PubMed)
4. Yung, S. et al. Ascorbic acid absorption in humans: a comparison among several dosage forms. Journal of Pharmaceutical Sciences. 1982; volume 71: pages 282-285. (PubMed)
5. Nyyssonen, K. et al. Effect of supplementation of smoking men with plain or slow release ascorbic acid on lipoprotein oxidation. European Journal of Clinical Nutrition. 1997; volume 51: pages 154-163. (PubMed)
6. Viscovich M, Lykkesfeldt J, Poulsen HE. Vitamin C pharmacokinetics of plain and slow release formulations in smokers. Clinical nutrition. 2004;23(5):1043-1050. (PubMed)
7. Carr AC, Vissers MC. Synthetic or food-derived vitamin C-are they equally bioavailable? Nutrients. 2013;5(11):4284-4304. (PubMed)
8. Vinson, J.A. & Bose, P. Comparative bioavailability to humans of ascorbic acid alone or in a citrus extract. American Journal of Clinical Nutrition. 1988; volume 48: pages 501-604. (PubMed)
9. Uchida E, Kondo Y, Amano A, et al. Absorption and excretion of ascorbic acid alone and in acerola (Malpighia emarginata) juice: comparison in healthy Japanese subjects. Biol Pharm Bull. 2011;34(11):1744-1747. (PubMed)
10. Carr AC, Bozonet SM, Pullar JM, Simcock JW, Vissers MC. A randomized steady-state bioavailability study of synthetic versus natural (kiwifruit-derived) vitamin C. Nutrients. 2013;5(9):3684-3695. (PubMed)
11. Jones E, Hughes RE. The influence of bioflavonoids on the absorption of vitamin C. IRCS Med Sci. 1984;12:320.
12. Johnston, C.S. & Luo, B. Comparison of the absorption and excretion of three commercially available sources of vitamin C. Journal of the American Dietetic Association. 1994; volume 94: pages 779-781.
13. Cort, W.M. Antioxidant activity of tocopherols, ascorbyl palmitate, and ascorbic acid and their mode of action. Journal of the American Oil Chemists’ Society. 1974; volume 51: pages 321-325.
14. Ross, D. et al. Ascorbate 6-palmitate protects human erythrocytes from oxidative damage. Free Radical Biology and Medicine. 1999; volume 26: pages 81-89. (PubMed)
15. DeRitter, E. et al. Physiologic availability of dehydro-L-ascorbic acid and palmitoyl-L-ascorbic acid. Science. 1951; volume 113: pages 628-631.
16. Austria R. et al. Stability of vitamin C derivatives in solution and in topical formulations. Journal of Pharmacology and Biomedical Analysis. 1997; volume 15: pages 795-801. (PubMed)
17. Sauberlich, H.E. et al. Effects of erythorbic acid on vitamin C metabolism in young women. American Journal of Clinical Nutrition. 1996; volume 64: pages 336-346. (PubMed)
18. Weeks BS, Perez PP. Absorption rates and free radical scavenging values of vitamin C-lipid metabolites in human lymphoblastic cells. Med Sci Monit. 2007;13(10):BR205-210. (PubMed)
19. Weeks BS, Perez PP. A novel vitamin C preparation enhances neurite formation and fibroblast adhesion and reduces xenobiotic-induced T-cell hyperactivation. Med Sci Monit. 2007;13(3):BR51-58. (PubMed)
20. Pancorbo D, Vazquez C, Fletcher MA. Vitamin C-lipid metabolites: uptake and retention and effect on plasma C-reactive protein and oxidized LDL levels in healthy volunteers. Med Sci Monit. 2008;14(11):CR547-551. (PubMed)
Vitamina D
Contenido
Resumen
- La vitamina D puede ser sintetizada en la piel después de exponerse a la luz solar y es entonces metabolizada en el hígado y riñón a la metabólicamente forma activa llamada 1α,25-dihidroxivitamina D. A través de la unión al receptor de la vitamina D (RVD), la 1α,25-dihidroxivitamina D puede regular la expresión de cientos de genes involucrados en funciones del esqueleto y otras funciones biológicas. (Más información)
- La vitamina D es esencial para el mantenimiento de la mineralización ósea a través de la regulación de calcio y homeostasis del fósforo. La vitamina D también exhibe muchos efectos no-esqueléticos, particularmente en los sistemas inmune, endocrino, y cardiovascular. (Más información)
- La vitamina D es importante para el desarrollo y mantenimiento óseo normal. Una deficiencia severa de vitamina D causa raquitismo en niños y osteomalacia en adultos. (Más información)
-
El hiperparatiroidismo secundario debido a la insuficiencia de vitamina D puede incrementar la degradación ósea y precipitar la osteoporosis. Ensayos clínicos aleatorios indican que la suplementación con al menos 800 UI/día de vitamina D puede reducir el riesgo de caídas y fracturas en individuos de edad avanzada. (Más información)
- La vitamina D puede regular la diferenciación celular y crecimiento al unirse al receptor de la vitamina D encontrado en la mayoría de las células del cuerpo. Estudios basados en la observación han reportado asociaciones entre la baja exposición al sol, un estatus pobre de la vitamina D, y el riesgo incrementado de desarrollar cáncer colorrectal y de seno. Ensayos clínicos aleatorios son necesarios para evaluar si la prevención de cáncer podría beneficiarse de la suplementación de vitamina D. (Más información)
- Varios estudios basados en la observación han reportado asociaciones inversas entre el estatus de la vitamina D y la susceptibilidad o severidad de enfermedades autoinmunes, incluyendo la diabetes mellitus tipo 1, esclerosis múltiple, artritis reumatoides, y lupus eritematoso sistémico. (Más información)
- Evidencia actual proveniente de estudios basados en la observación sugiere una relación inversa entre las concentraciones circulantes de vitamina D y el riesgo de diabetes mellitus tipo 2. Aún no se conoce si corrigiendo la deficiencia de vitamina D en individuos con intolerancia a la glucosa puede disminuir el riesgo de la progresión a diabetes tipo 2. (Más información)
- Ensayos clínicos aleatorios están actualmente investigando si la suplementación con vitamina D puede limitar el deterioro cognitivo y progresión de la enfermedad en individuos con enfermedades neurodegenerativas. (Más información)
- La insuficiencia de vitamina D en mujeres embarazadas puede estar asociada con varios efectos adversos para la madre y el recién nacido. La seguridad y beneficios de la suplementación con vitamina D durante el embarazo necesitan ser evaluados en ensayos clínicos. (Más información)
- Estudios observacionales han documentado una asociación entre la deficiencia de vitamina D y una mayor incidencia y gravedad de la enfermedad por coronavirus, COVID-19. (Más información)
- Estudios preliminares han demostrado que la suplementación con vitamina D puede ofrecer mejoras prometedoras en el tratamiento de la dermatitis atópica (eccema) y la enfermedad de Crohn. (Más información)
La vitamina D es una vitamina liposoluble que regula la homeostasis del calcio y es vital para la salud ósea (1). Mientras que esta puede ser también obtenida de fuentes dietéticas o suplementos, la vitamina D3 (colecalciferol) es sintetizada en la piel humana a partir de la conversión del 7-dehidrocolesterol luego de la exposición a la radiación ultravioleta-B (UVB) de la luz solar. La vitamina D2 (ergocalciferol) es una vitamina D análoga fotosintetizada en plantas, hongos, y levaduras; la vitamina D2 es también utilizada en la fortificación de alimentos con vitamina D (2). Cuando la vitamina D3 en la piel es inadecuada debido a insuficiente exposición a la radiación UVB, la ingesta oral de vitamina D es necesaria para satisfacer los requerimientos de vitamina D.
Función
Metabolismo de la vitamina D
El colecalciferol y ergocalciferol son precursores biológicamente inactivos de la vitamina D y deben ser convertidos a formas biológicamente activas en el hígado y riñones (Figura 1). De hecho, luego de la ingesta dietética o síntesis en la epidermis de la piel después de la exposición a la radiación UVB, ambas formas de la vitamina D entran en la circulación y son transportadas al hígado por la proteína de unión a vitamina D (y en menor medida por la albúmina). En los hepatocitos (células del hígado), la vitamina D es hidroxilada para formar 25-hidroxivitamina D (calcidiol; calcifediol). La exposición a la luz solar o la ingesta dietética de vitamina D incrementan las concentraciones en suero de 25-hidroxivitamina D. La 25-hidroxivitamina D constituye la principal forma de vitamina D circulante, y la suma de los niveles de 25-hidroxivitamina D2 y 25-hidroxivitamina D3 en el suero es usada como un indicador del estatus nutricional de la vitamina D (3). La enzima renal 25-hidroxivitamina D-1α-hidroxilasa (también conocida como CYP27B1) eventualmente cataliza una segunda hidroxilación que convierte a la 25-hidroxivitamina D en 1α,25-hidroxivitamina D (calcitriol). La producción de 1α,25-dihidroxivitamina D en los riñones es regulada por varios factores, incluyendo el fósforo, calcio, hormona paratiroidea (PTH), el factor de crecimiento de fibroblastos 23 (FGF-23), y la propia 1α,25-dihidroxivitamina D del suero. Mientras que los riñones son la fuente principal de la actividad de la 1α-hidroxilasa, la producción externo-renal de 1α-dihidroxivitamina D ha sido también demostrada en una variedad de tejidos, incluyendo la piel, glándula paratiroidea, senos, colon, próstata, como también en células del sistema inmunológico y células óseas (2). La mayoría de los efectos fisiológicos de la vitamina D en el cuerpo están relacionados a la actividad de 1α,25-dihidroxivitamina D (4). Varias formas de la vitamina D están listadas en Figura 1.
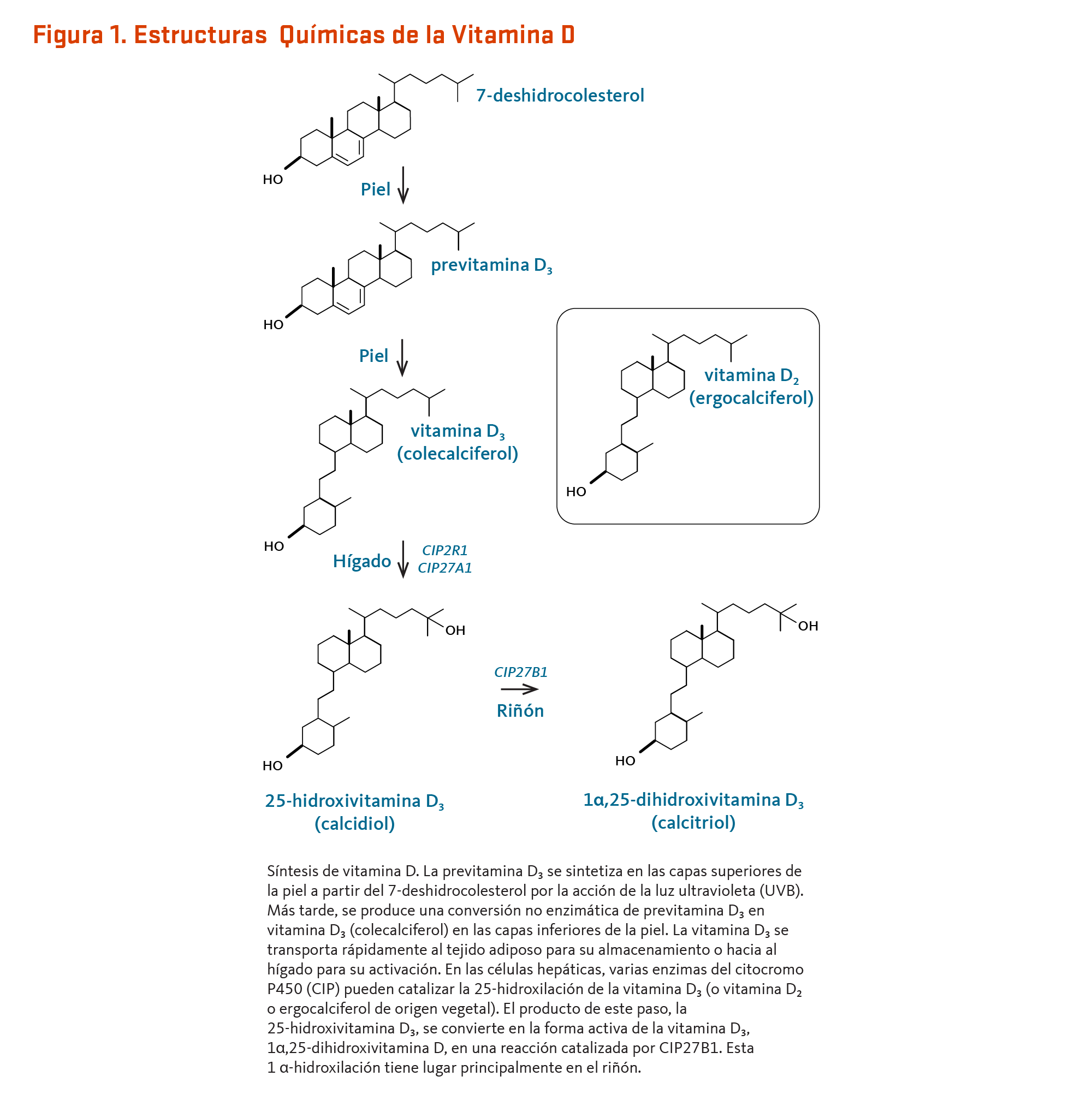
[Figura 1 - Clic para Agrandar]
Mecanismos de acción
La mayoría, sino todas, las acciones de la vitamina D son mediadas a través de un factor de transcripción nuclear conocido como receptor de vitamina D (RVD) (Figura 2) (5). Al entrar al núcleo de una célula, la 1α,25-dihidroxivitamina D se une al RVD y recluta otro receptor nuclear conocido como receptor del ácido retinoico X (RXR). En la presencia de 1α,25-dihidroxivitamina D, el complejo RVD/RXR se une a pequeñas secuencias de ADN conocidas como elementos de respuesta a vitamina D (VDREs) e inicia una cascada de interacciones moleculares que modulan la transcripción de genes específicos. Miles de VDREs han sido identificados a través del genoma, y se piensa que la activación del RVD por la 1α,25-dihidroxivitamina D directamente y/o indirectamente regula de entre 100 a 1,250 genes (6).
![Figura 2. Conversión a la Forma Activa de Vitamina D y Regulación de la Expresión de Genes mediada por RVD. Mecanismos de acción. La 25-hidroxivitamina D (25(OH)D) es la principal forma de vitamina D circulante. La mayoría de las moléculas 25(OH)D y 1α, 25-dihidroxivitamina D (1,25(OH)2D) se transportan unidas a la proteína de unión a la vitamina D y entran a las células a través del complejo megalina/tubulina. En las células renales, la 1α-hidroxilasa (CIP27B1) cataliza la conversión de 25(OH)D en 1,25(OH)2D. La hormona paratiroidea (PTH), el estradiol, y las concentraciones bajas de fósforo ([P]) estimulan esta reacción, mientras que el 1,25(OH)2D, el factor de crecimiento de fibroblastos 23 (FGF-23), y la alta concentración de calcio ([Ca]) la inhibe. El 1,25(OH)2D ingresa a la circulación y se transporta a los tejidos diana extra-renales, donde puede regular la expresión de genes. En el núcleo de las células diana, el 1,25(OH)2D se une al receptor de vitamina D (RVD), que se heterodimeriza con el receptor retinoide X (RXR). RVD-RXR se une a los elementos de respuesta a la vitamina D (VDRE) en el promotor de los genes diana de la vitamina D. 25(OH)D, 25-hidroxivitamina D; 1,25(OH)2D, 1α, 25-dihidroxivitamina D; [Ca], concentración de calcio; CIP2R1, 1α-hidroxilasa; DBP, proteína de unión a la vitamina D, FGF-23, factor de crecimiento de fibroblastos-23; PTH, hormona paratiroidea; [P] concentración de fósforo; ARN Pol II, ARN polimerasa II; RXR, receptor retinoide X; RVD; receptor nuclear de vitamina D; VDRE, elemento de respuesta a la vitamina D.](/sites/lpi.oregonstate.edu/files/vitamina-d-figura-2-2000px.png)
[Figura 2 - Clic para Agrandar]
Balance de calcio
El mantenimiento del calcio en el suero dentro de un rango estrecho es vital para el funcionamiento normal del sistema nervioso, como también para el crecimiento óseo y mantenimiento de la densidad ósea. La vitamina D es esencial para la utilización eficiente del calcio por el cuerpo (1). Las glándulas paratiroideas captan las concentraciones de calcio en el suero y secretan la hormona paratiroidea (PTH) si las concentraciones de calcio disminuyen por debajo de lo normal (Figura 3). Incrementos en la PTH estimulan la actividad de la enzima 25-hidroxivitamina D3-1α-hidroxilasa en el riñón, resultando en una producción incrementada de 1α,25-hidroxivitamina D. La forma activa de vitamina D, 1α,25-hidroxivitamina D, se libera en la circulación y se transporta a los tejidos objetivo. Dentro de las células diana, la 1α,25-dihidroxivitamina se une e induce la activación de RVD, lo que conduce a cambios en la expresión génica que normalizan el calcio sérico al (1) aumentar la absorción intestinal de calcio dietético, (2) aumentar la reabsorción de calcio filtrado por los riñones, y (3) movilizando calcio del hueso cuando no hay suficiente calcio dietético para mantener las concentraciones normales de calcio en suero (7).
![Regulación de la Homeostasis del Calcio y el Fósforo. Homeostasis de calcio y fósforo. Una ligera caída en la concentración de calcio en la sangre ([Ca2 +]) resulta en la secreción de la hormona paratiroidea (PTH) por las glándulas paratiroides. La PTH simula la actividad de CIP27B1 (enzima 25-hidroxivitamina D-1α-hidroxilasa) que cataliza la hidroxilación de 25-hidroxivitamina D en 1α, 25-dihidroxivitamina D. Esta forma activa de vitamina D (i) aumenta la reabsorción de calcio filtrada por los riñones y estimula la excreción de fósforo, (ii) aumenta la absorción intestinal de calcio y fósforo, y (iii) moviliza calcio (y fósforo) del hueso cuando la ingesta de calcio en la dieta es insuficiente. La PTH también estimula la reabsorción ósea que libera calcio y fósforo. La 1α,25-dihidroxivitamina D inhibe su propia producción, así como la síntesis de PTH, a través de bucles de retroalimentación negativa. El FGF-23 es secretado por las células formadoras de hueso (osteoblastos) en respuesta a un aumento en la ingesta de fósforo. El FGF-23 inhibe la síntesis de 1α,25-dihidroxivitamina D y promueve la excreción de fósforo en la orina.<br />
[Ca2+], concentración de calcio; FGF-23, factor de crecimiento de fibroblastos-23; PTH, hormona paratiroidea.<br />](/sites/lpi.oregonstate.edu/files/vitamina-d-figura-3-2000px.png)
[Figura 3 - Clic para Agrandar]
Balance del fósforo
Las regulaciones del calcio y homeostasis del fósforo están estrechamente relacionadas, y las hormonas calciotrópicas de la PTH y la 1α,25-hidroxivitamina D pueden también controlar el fósforo del suero. Específicamente, la 1α,25-hidroxivitamina D incrementa la absorción intestinal de fósforo al estimular la expresión de un cotransportador sodio-fosfato en el intestino delgado. Mientras que la PTH incrementa la excreción urinaria de fósforo al reducir la reabsorción en el riñón, aún no es claro si la 1α,25-dihidroxivitamina D puede directamente regular el transporte renal de fósforo. La hormona fosfatúrica del factor de crecimiento fibroblástico 23 (FGF-23), secretada por los osteoblastos (células formadoras de hueso), limita la producción de la 1,25-hidroxivitamina D al inhibir la 25-hidroxivitamina D-1α-hidroxilasa (Figura 3) (revisado en 8).
Diferenciación celular
Se dice que las células que se dividen rápidamente son células en proliferación. La diferenciación resulta en la especialización de células para funciones específicas. En general, la diferenciación celular lleva a una disminución en la proliferación. Mientras que la proliferación celular es esencial para el crecimiento y cicatrización de heridas, la proliferación incontrolada de células con ciertas mutaciones puede llevar al cáncer. La forma activa de la vitamina D, 1α,25-dihidroxivitamina D inhibe la proliferación y estimula la diferenciación de las células mediante su unión al RVD (1).
Inmunidad
Actuando a través del RVD la 1α,25-dihidroxivitamina D es un potente modulador del sistema inmune. El RVD es expresado por la mayoría de las células del sistema inmune, incluyendo las células T regulatorias y las células presentadoras de antígeno, como las células dendríticas y macrófagos (9). Bajo circunstancias específicas, los monocitos, macrófagos, y células T pueden expresar la enzima 25-dihidroxivitamina D3-1α-hidroxilasa y producir 1α,25-dihidroxivitamina D, la cual actúa localmente para regular la respuesta inmune (10, 11). Existe evidencia científica considerable que la 1α,25-dihidroxivitamina D posee una variedad de efectos en la función del sistema inmune, que podrían mejorar la inmunidad innata e inhibir el desarrollo de la autoinmunidad (12). Inversamente, la deficiencia de vitamina D puede comprometer la integridad del sistema inmunológico y conducir a respuestas inmunes inapropiadas (véase Enfermedades autoinmunes).
Secreción de insulina
El RVD es expresado por las células secretoras de insulina del páncreas, y los resultados de estudios en animales sugieren que la 1α,25-dihidroxivitamina D desempeña un papel en la secreción de insulina bajo condiciones de un incremento de la demanda de insulina (13, 14). Estudios prospectivos y de corte transversal sugieren que niveles insuficientes de vitamina D pueden tener un efecto adverso en la secreción de insulina y tolerancia a la glucosa en la diabetes mellitus tipo 2 (diabetes mellitus no dependiente de insulina) (revisado en 15).
Regulación de la presión sanguínea
El sistema renina-angiotensina juega un papel importante en la regulación de la presión sanguínea (16). La renina es una enzima que cataliza el clivaje (separación) de un péptido pequeño (angiotensina I) de una proteína más grande (angiotensinógeno) producida en el hígado. La enzima convertidora de angiotensina (ECA) cataliza el clivaje de la angiotensina I para formar angiotensina II, un péptido que puede incrementar la presión sanguínea al inducir la constricción de pequeñas arterias y al incrementar la retención de sodio y agua. La tasa de síntesis de angiotensina II es dependiente de la renina (17). Investigación en ratones sin el gen que codifica para el RVD, indica que la 1α,25-dihidroxivitamina D disminuye la expresión del gen que codifica para la renina a través de su interacción con el RVD (18). Ya que la activación inapropiada del sistema renina-angiotensina puede contribuir al desarrollo de hipertensión, alcanzar niveles adecuados de vitamina D podría ser importante para disminuir el riesgo de presión sanguínea alta (véase Hipertensión).
Deficiencia
En la deficiencia de vitamina D, la absorción de calcio no puede ser incrementada lo suficiente para satisfacer las necesidades de calcio del cuerpo (4). En consecuencia, la producción de la PTH por las glándulas paratiroideas es incrementada y calcio es movilizado del esqueleto para mantener niveles normales de calcio en el suero — una condición conocida como hiperparatiroidismo secundario. A pesar de que hace mucho tiempo se sabe que la deficiencia severa de vitamina D tiene consecuencias serias para la salud ósea, investigaciones sugieren que estados menos obvios de deficiencia de vitamina D son comunes, e incrementan el riesgo de osteoporosis y otros problemas de salud (véase Prevención de Enfermedad).
Severa deficiencia de vitamina D
Raquitismo
En infantes y niños la deficiencia severa de vitamina D resulta en el fracaso de la mineralización de los huesos. El proceso de mineralización, que involucra la producción de cristales de fosfato de calcio por las células formadoras de hueso, determina la dureza y resistencia de los huesos. La deficiencia de vitamina D severamente afecta los huesos que crecen rápidamente. Las placas de crecimiento de los huesos continúan alargándose, pero en la ausencia de una mineralización adecuada, los miembros que soportan el peso (brazos y piernas) se arquean. En infantes, el raquitismo podría resultar en un retraso en el cierre de las fontanelas (partes blandas) en el cráneo, y la caja torácica podría deformarse debido a la acción de arrastre del diafragma. En casos severos, bajos niveles de calcio en el suero (hipocalcemia) podrían causar convulsiones. Aunque la fortificación de alimentos ha llevado a la complacencia con respecto a la deficiencia de vitamina D, en ciudades alrededor del mundo aún se reportan casos de raquitismo nutricional (19, 20).
Osteomalacia
Aunque los huesos adultos no están más en crecimiento, estos están en un constante estado de renovación, o "remodelación." En adultos con una deficiencia severa de vitamina D, la matriz de colágeno del hueso se conserva, pero el mineral óseo se pierde progresivamente, resultando en el ablandamiento de huesos (osteomalacia), dolor de huesos, y un incremento en el riesgo de osteoporosis (21).
Debilidad y dolor muscular
La deficiencia de vitamina D causa debilidad y dolor muscular en niños y adultos. El dolor y la debilidad muscular fueron los síntomas prominentes de la deficiencia de vitamina D en un estudio en mujeres musulmanas árabes y danesas viviendo en Dinamarca (22). En un estudio de corte transversal en 150 pacientes consecutivos remitidos a una clínica en Minnesota para la evaluación de dolor musculoesquelético persistente, no especifico, el 93% tuvieron niveles de 25-hidroxivitamina D en el suero iguales o por debajo de los 20 ng/mL, con una concentración media de 12.1 ng/mL, lo cual es un indicativo de la insuficiencia de vitamina D (23). La pérdida de fuerza muscular contribuye en gran medida al incremento del riesgo de caídas y fracturas de hueso, especialmente en personas mayores (24). Además, la insuficiencia de vitamina D a largo plazo puede ser un factor contribuidor a la osteoporosis en los ancianos (véase Osteoporosis).
Factores de riesgo de la deficiencia de vitamina D
Tanto los factores ambientales como las prácticas culturales dan como resultado variaciones en el estatus de la vitamina D:
- Condiciones ambientales: Ubicaciones geográficas, incluyendo latitud y altitudes, y condiciones atmosféricas (p. ej., contaminación del aire, presencia de nubes) influyen en gran medida a la intensidad de la radiación UVB que alcanza el suelo. Cambios estacionales también afectan la calidad y cantidad de los rayos UVB y de este modo la producción de vitamina D en la piel (25-27).
- Estilo de vestimenta cubierta: En un estudio de 2,032 en mujeres del Medio Oriente, que vistieron un velo en la cabeza o cubrieron toda la piel por razones religiosas o culturales, el 96% tuvieron niveles de 25-hidroxivitamina D en el suero por debajo de los 20 ng/mL, y el 60% tuvieron niveles de vitamina D por debajo de los 12 ng/mL (28). El raquitismo y osteomalacia no son comunes en las regiones del Medio Oriente y Norte de África donde niños y mujeres cubren la mayoría de toda su piel cuando salen al exterior (29).
- Medidas de protección solar: Las prácticas de protección solar, incluyendo el limitar la exposición al sol, vestir ropas protectoras y sombreros, y aplicar bloqueador solar, obstaculizan la exposición de la piel a la luz solar y por lo tanto dan lugar a una producción menor de vitamina D3 y metabolitos de la vitamina D circulantes a menos que haya una ingesta oral adecuada. Debe notarse que la aplicación de protector solar (2 mg/cm2) con un factor de protección solar (FPS) de 10 reduce la radiación UVB en un 90% (30).
- Infantes exclusivamente amamantados: Los infantes que son exclusivamente amamantados y no reciben suplementación con vitamina D están en un riesgo alto de deficiencia de vitamina D, particularmente si tienen la piel oscura y/o reciben poca exposición solar (19). La leche humana generalmente provee de entre 10-80 UI de vitamina D por litro (L), que corresponde a 0.2 a 1.5 μg/día (8-60 UI/día) cuando se usa un promedio diario de ingesta de leche de 0.75 L (25 oz) (31). La Academia Americana de Pediatría recomienda que todos los infantes amamantados y parcialmente amamantados les sea dado un suplemento de vitamina D de 400 UI/día (19). La suplementación materna de vitamina D durante el período de lactancia puede contribuir a un mejorado estatus de vitamina D en el infante que está siendo amamantado, especialmente en poblaciones con una prevalencia alta de deficiencia de vitamina D (32). Infantes mayores y niños pequeños que son alimentados exclusivamente con substitutos de la leche (p. ej., fórmulas a base de soya) y alimentos para el destete que no están fortificados con vitamina D están un riesgo de una deficiencia de vitamina D (33).
La eficiencia de la síntesis de vitamina D, absorción, y metabolismo también depende en una variedad de factores biológicos:
- Pigmentación de la piel: Las personas con una complexión oscura sintetizan menos vitamina D al exponerse a la luz solar que aquellos con un color de piel más claro (34). Una encuesta nacional estadounidense reportó niveles promedio de 25-hidroxivitamina D en el suero de 28.1 ng/mL, 21.6 ng/mL, y 16.9 ng/mL en mujeres caucásicas, méxico-americanas, y afroamericanas de ≥20 años de edad, respectivamente (25).
- Variaciones genéticas: Un estudio internacional, multicéntrico, de asociación del genoma completo (en inglés, GWAS) de 15 cohortes, que incluyó a ~ 30,000 participantes de ascendencia europea — conocido como el consorcio SUNLIGHT [Estudio de Determinantes Genéticos Subyacentes de la Vitamina D y Rasgos Altamente Relacionados] — identificó variaciones comunes (llamadas polimorfismos) en genes implicados en la síntesis de colesterol, la hidroxilación y el transporte de vitamina D que influyen en el estado de la vitamina D (35). Si bien los determinantes genéticos del bajo nivel de vitamina D se están identificando en poblaciones de origen europeo (36, 37) y asiático (38, 39), se necesitan estudios de asociación del genoma completo en poblaciones de ascendencia africana.
- Edad avanzada: Las personas de la tercera edad tienen una capacidad reducida para sintetizar vitamina D en la piel cuando se exponen a la radiación UVB y son más propensos a permanecer en interiores y usar bloqueador solar, lo cual previene la síntesis de vitamina D. Se ha estimado que a través de Canadá, los EE.UU., y Europa, la prevalencia de la deficiencia de vitamina D oscila entre 20%-100% en personas de la tercera edad que viven libremente (40). Por otra parte, adultos que se encuentran en instituciones y que no son suplementados con vitamina D están en un riesgo extremadamente alto de una deficiencia de vitamina D (41, 42).
- Enfermedad renal crónica (ERC): La deficiencia de vitamina D en pacientes con insuficiencia renal se debe a una síntesis reducida de 1α,25-dihidroxivitamina D y una mayor pérdida de 25-hidroxivitamina D en la orina (43).
- Síndromes de malabsorción de grasas: La deficiencia de vitamina D es común entre personas con fibrosis quística y con tanto enfermedades hepáticas colestásicas y no colestásicas debido a la disminución de la absorción de la vitamina D dietética y a la alteración de la conversión de vitamina D a 25-hidroxivitamina D (revisado en 44).
- Enfermedad inflamatoria intestinal: Las personas con una enfermedad intestinal inflamatoria como la enfermedad de Crohn parecen estar en un riesgo incrementado de una deficiencia de vitamina D, especialmente aquellas que han tenido resecciones intestinales pequeñas (45).
- Obesidad:
- La obesidad (índice de masa corporal ≥30 kg/m2) incrementa el riesgo de la deficiencia de vitamina D (46). Una vez la vitamina D es sintetizada en la piel o ingerida, esta puede ser secuestrada en los depósitos de grasa corporal, haciéndola menos biodisponible para las personas con una mayor masa corporal grasa. Por otra parte, ensayos de la suplementación de vitamina D han mostrado que las personas obesas alcanzaron niveles mucho más bajos de 25-hidroxivitamina D del suero en comparación a participantes con pesos normales (IMC <25 kg/m2) con dosis orales equivalentes (47).
- Deficiencia de magnesio: Recientes descubrimientos sugieren que altas ingestas de magnesio podrían reducir el riesgo de la insuficiencia de vitamina D. El magnesio regula la actividad de enzimas críticas en el metabolismo de la vitamina D, lo cual explicaría cómo la deficiencia de magnesio afecta negativamente el estatus de la vitamina D (48).
Evaluación del estado nutricional de vitamina D
La creciente toma de conciencia de que la deficiencia de vitamina D tiene serias consecuencias para la salud más allá del raquitismo y la osteomalacia; resalta la necesidad de una evaluación precisa del estatus nutricional de la vitamina D. Actualmente se entiende que la medida de la concentración total de la 25-hidroxivitamina D en el suero (1 ng/mL correspondiente a 2.5 nmol/L) es el mejor indicador para evaluar el estatus de la vitamina D.
Sin embargo, evidencia de alta calidad aún es necesaria para asegurar que los valores de corte actuales son óptimos para definir estados de insuficiencia y deficiencia (40). Además, sólo recientemente, se han realizado esfuerzos para la estandarización de los ensayos de 25-hidroxivitamina D desarrollados comercialmente y en laboratorio, de modo que las directrices se han desarrollado utilizando datos de investigación en gran medida no estandarizados (49). Aunque valores de referencia del laboratorio para los niveles suficientes de vitamina D se habían basado inicialmente en los niveles de 25-hidroxivitamina D del suero provenientes de cohortes de individuos saludables, estudios adicionales han sugerido que los valores de corte basados en la salud que apuntan a la prevención de hiperparatiroidismo secundario y pérdida ósea deberían ser considerablemente más altos. En efecto, mientras que se considera que las concentraciones de 25-hidroxivitamina D del suero menores a 8-10 ng/mL (20-25 nmol/L) indican una deficiencia severa asociada al raquitismo y osteomalacia, varios estudios han observado que los niveles de PTH (50, 51) y de la absorción de calcio (52) no fueron optimizados en relación a los niveles de 25-hidroxivitamina D del suero por debajo de los 32 ng/mL (80 nmol/L).
A pesar de todo, estudios más recientes han fallado en encontrar valores de umbral de las concentraciones de 25-hidroxivitamina D del suero en relación a la supresión de la PTH y la absorción óptima del calcio. Por una parte, el análisis de corte transversal reciente de 312,962 muestras clínicas no encontró evidencia de un umbral para la supresión de la PTH en la curva bien ajustada que muestra la asociación inversa entre las mediciones emparejadas de la PTH del suero y la 25-hidroxivitamina D, incluso con una concentración de 25-hidroxivitamina D más allá de los 70 ng/mL (175 nmol/L) (53). Esto contradijo un reciente análisis de la Encuesta Nacional de Examinación de Salud y Nutrición de los EE.UU. (NHANES 2003-2006) que estimó la supresión máxima de la PTH para las concentraciones de 25-hidroxivitamina D de 40 ng/mL (100 nmol/L) y mayores (54). Además, ambos estudios identificaron evidencia de hiperparatiroidismo leve (PTH sérica >65 pg/mL) en individuos con niveles de 25-hidroxivitamina D del suero mucho más allá de los 20 ng/mL (50 nmol/L), cuestionando el uso de PTH en el suero como un indicador razonable de la insuficiencia de vitamina D (53, 54). Por otra parte, un ensayo aleatorio controlado con placebo reciente en mujeres posmenopáusicas con una insuficiencia de vitamina D (25-hidroxivitamina D sérica <20 ng/mL) suplementadas con dosis diarias de vitamina D3 de entre 400 a 4,800 UI encontró poco cambio (6%) en la absorción de calcio sobre una concentración normal de 25-hidroxivitamina D con un rango de 20 a 66 ng/mL (55).
Los puntos de corte actuales propuestos por el Instituto de Medicina (en inglés, IOM) son los siguientes: como deficiencia valores de 25-hidroxivitamina D del suero ≤12 ng/mL (30 nmol/L); como insuficiencia valores de 25-hidroxivitamina D del suero de 12-19 ng/mL (30-49 nmol/L); y como suficiencia valores de 25-hidroxivitamina D del suero de 20-50 ng/mL (50-125 nmol/L) (56). Las ingestas dietéticas de referencia (REP, IDR) establecidas por el Instituto de Medicina están basadas en el logro de concentraciones de 25-hidroxivitamina D circulante (20-50 ng/mL) que son adecuadas para mantener la salud ósea y una óptima absorción de calcio (57).
Sin embargo, teniendo en cuenta el papel potencial de los niveles circulantes menores de 30 ng/mL en la carga de muchas enfermedades crónicas (6), la Sociedad de Endocrinología Estadounidense ha sugerido definir los valores de corte de la siguiente manera: como deficiencia valores de 25-hidroxivitamina D del suero ≤20 ng/mL (≤ 50 nmol/L); como insuficiencia valores de 25-hidroxivitamina D del suero de 21-29 ng/mL (51-74 nmol/L); como suficiencia valores de 25-hidroxivitamina D del suero de 30-100 ng/mL (75-250 nmol/L) (40). Sin embargo, este rango alternativo deseado es apoyado por algunos estudios basados en la observación, pero no está basado en datos de ensayos controlados aleatorios (véase Prevención de Enfermedad) (47). Con estos últimos valores de corte mencionados, estudios provenientes de todo el mundo han estimado que la hipovitaminosis D está bastante extendida y que niños y adultos de todas las edades están en un riesgo igual de insuficiencia y deficiencia (58). Datos de estudios de suplementación indican que las ingestas de vitamina D de por lo menos 800-1,000 UI/día son requeridas por adultos que viven en latitudes templadas para alcanzar niveles de 25-hidroxivitamina D del suero de por lo menos 30 ng/mL (75 nmol/L) (40).
Finalmente, las concentraciones totales de 25-hidroxivitamina en el suero pueden no siempre adecuadamente reflejar la biodisponibilidad de la vitamina D (59), y evidencia adicional es necesaria para mejorar la determinación del estatus de la vitamina D en diferentes poblaciones étnicas.
La Ingesta Diaria Recomendada (IDR)
En el 2010, la Junta de Nutrición y Alimentos (JNA) del Instituto de Medicina estableció una Ingesta Diaria Recomendada (IDR) basada en la cantidad de vitamina D necesaria para la salud ósea. Mientras que la IDR se incrementó en comparación al nivel de ingesta adecuada (IA) establecida en 1997, los niveles óptimos de las ingestas recomendadas y de 25-hidroxivitamina D del suero que minimizan el hiperparatiroidismo y maximizan la salud ósea en la población general permanecen controversiales (40). La IDR para la vitamina D se muestra en la Tabla 1 por etapa de la vida y género.
| Etapa de la Vida | Edad | Machos | Hembras | ||
|---|---|---|---|---|---|
| μg/día | UI/día | μg/día | UI/día | ||
| Infantes (IA) | 0-6 meses |
10
|
400
|
10
|
400
|
| Infantes (IA) | 6-12 meses |
10
|
400
|
10
|
400
|
| Niños | 1-3 años |
15
|
600
|
15
|
600
|
| Niños | 4-8 años |
15
|
600
|
15
|
600
|
| Niños | 9-13 años |
15
|
600
|
15
|
600
|
| Adolescentes | 14-18 años |
15
|
600
|
15
|
600
|
| Adultos | 19-70 años |
15
|
600
|
15
|
600
|
| Adultos | 71 años y más |
20
|
800
|
20
|
800
|
| Embarazo | Todas las edades |
-
|
-
|
15
|
600
|
| Período de lactancia | Todas las edades |
-
|
-
|
15
|
600
|
Prevención de Enfermedad
Mortalidad
En un análisis de seguimiento de nueve años de la Tercera Encuesta Nacional de Examinación de Salud y Nutrición de los EE.UU. (NHANES III) que incluyó a 15,099 participantes (de los cuales el 77% eran caucásicos), las concentraciones séricas de 25-hidroxivitamina D — estandarizadas según la metodología desarrollada por el Programa de Estandarización de Vitamina D [en inglés, VDSP] — fueron examinados en relación con la mortalidad. El análisis sugirió un aumento en la mortalidad por todas las causas con una disminución de las concentraciones séricas de 25-hidroxivitamina D <16 ng/mL (60). En contraste, el riesgo de mortalidad por todas las causas varió poco para las concentraciones séricas basales de 25-hidroxivitamina D en el rango de 16 a 40 ng/mL (60). Se obtuvieron resultados similares en un meta-análisis de ocho estudios de cohorte prospectivos que consideraron la relación entre las concentraciones estandarizadas de 25-hidroxivitamina D y la mortalidad durante un período de seguimiento medio de 10.5 años. Se encontró que el riesgo de muerte era 19% más alto con concentraciones de 25-hidroxivitamina D entre 12 y 15,99 ng/mL y 56% más alto con concentraciones <12 ng/mL en comparación con el riesgo asociado con concentraciones entre 30 y 39,99 ng/mL (61). Un meta-análisis de 73 estudios de cohorte prospectivos, que incluyó a >800,000 participantes, encontró que el tercil más bajo versus el más alto de las concentraciones séricas de 25-hidroxivitamina D se asoció con un mayor riesgo de mortalidad por todas las causas (+35%), mortalidad por enfermedades cardiovasculares (+35%) y mortalidad por cáncer (+14%) (62). Sin embargo, un análisis de aleatorización mendeliana — que limita el sesgo debido a la confusión y la causalidad inversa (63) — de los datos de tres grandes cohortes danesas de 95,766 adultos encontró una asociación significativa genéticas de bajas concentraciones en plasma de 25-hidroxivitamina D por todas las causas y relacionada con el cáncer, pero no con la mortalidad relacionada con la enfermedad cardiovascular (64). Finalmente, dos metanálisis de ensayos aleatorios controlados han sugerido una moderada reducción en la mortalidad por todas las causas en las personas mayores suplementadas con vitamina D y calcio, pero no sólo con vitamina D (62, 65). Ensayos adicionales controlados con placebo necesitan examinar más a fondo si la suplementación con vitamina D sola o en combinación con calcio podría ayudar a prevenir la muerte prematura en individuos repletos.
Osteoporosis
Estado de vitamina D, osteoporosis y riesgo de fractura
Aunque las causas de la osteoporosis son multifactoriales, la insuficiencia de vitamina D puede ser un factor etiológico importante en los adultos mayores. La osteoporosis afecta un tercio de las mujeres de entre 60-70 años de edad y dos tercios de las mujeres de 80 años y mayores (66). Una encuesta multinacional (18 países diferentes con latitudes en un rango entre los 64 grados norte y 38 grados sur) de más de 2,600 mujeres posmenopáusicas con osteoporosis, reveló que el 31% de los sujetos tenían concentraciones de 25-hidroxivitamina D <20 ng/mL (50 nmol/L) (67). Además, un reciente estudio de caso y control que incluyó 111 pacientes con una fractura de cadera y 73 controles (edad media, 83 años) encontró que bajos niveles séricos tanto de 25-hidroxivitamina D como vitamina K1 en comparación con control se asociaban con un incremento en el riesgo de una fractura de cadera (68). Sin suficiente vitamina D proveniente de la exposición al sol o de la ingesta dietética, la absorción intestinal del calcio puede ser significativamente reducida. Esto incrementa la secreción de PTH por las glándulas paratiroideas; la elevación prolongada de PTH puede resultar en una reabsorción ósea incrementada, la cual en turno puede incrementar el riesgo de fracturas osteoporóticas (69).
Suplementos de vitamina D y densidad mineral ósea
La pérdida progresiva de la densidad mineral ósea (DMO) que conduce a la osteopenia (pre-osteoporosis) y la osteoporosis se observa comúnmente en adultos mayores, especialmente en los ancianos. Los resultados de un meta-análisis de 23 ensayos aleatorios controlados con más de 4,000 participantes (edad media, 59 años) mostraron poca evidencia de un efecto de la suplementación con vitamina D en la DMO en cualquiera de los cinco sitios esqueléticos examinados, incluida la columna lumbar, la fémur cuello, trocánter, antebrazo y cuerpo total. Se informó un aumento significativo en la DMO solo en el cuello femoral (70). Sin embargo, se sugirió que las personas en este grupo de edad tendrían una ingesta adecuada de calcio y, por lo tanto, un metabolismo óseo normal, lo que explica la falta de un efecto de la vitamina D en el fortalecimiento de la masa ósea (71). Por el contrario, en las personas mayores, la suplementación con vitamina D es esencial para corregir y mantener concentraciones adecuadas de 25-hidroxivitamina D en suero y para prevenir el hiperparatiroidismo secundario y la pérdida de DMO (72).
Suplementos de vitamina D y riesgo de fractura
Un estudio de cohorte prospectivo que siguió a más de 72,000 mujeres posmenopáusicas en los EE.UU. durante 18 años encontró que aquellas que consumieron al menos 600 UI/día de vitamina D de la dieta y los suplementos tenían un riesgo 37% menor de fractura de cadera osteoporótica que las mujeres que consumieron menos de 140 UI/día de vitamina D (73). Sin embargo, la suplementación diaria con 400 UI de vitamina D3, en combinación con 1,000 mg de calcio, no redujo significativamente el riesgo de fractura de cadera en comparación con un placebo en 36,282 mujeres posmenopáusicas del ensayo Women's Health Initiative (74), lo que sugiere que puede haber un umbral de ingesta de vitamina D que es necesario para observar reducciones en el riesgo de fractura. Los resultados de un análisis genético de los datos de este ensayo también sugirieron que los efectos beneficiosos de la suplementación con vitamina D y calcio en el riesgo de fractura podrían limitarse a las mujeres con el riesgo genético más bajo de baja DMO (75). Sin embargo, este ensayo ha sido cuestionado por razones que incluyen mala adherencia y el hecho de que a los participantes se les permitió tomar suplementos adicionales de vitamina D y calcio que podrían haber confundido los resultados. Además, el uso de la terapia de reemplazo hormonal no se consideró en el estudio del efecto de la vitamina D y el calcio en la salud esquelética en mujeres posmenopáusicas a pesar de ser un factor de confusión importante en esta población (57, 76).
Otro ensayo, el estudio de la Evaluación Aleatorizada de Calcio o Vitamina D (RECORD), reportó que la vitamina D3 suplementaria en forma oral (800 UI/día) sola, o en combinación con calcio (1,000 mg/día), no previno la ocurrencia de fracturas osteoporóticas en adultos de la tercera edad que ya habían experimentado una fractura osteoporótica de bajo impacto (77). También en este último estudio, un cierto número de limitaciones, incluyendo una pobre adherencia y/o el hecho de que la suplementación con vitamina D no elevó los niveles de 25-hidroxivitamina D del suero a un nivel que protegería contra las fracturas, podrían explicar la carencia de algún efecto (78). A pesar de la alta adherencia al tratamiento, la incidencia de fractura no vertebral fue similar en mujeres posmenopáusicas suplementadas con vitamina D3 (dosis inicial de 200,000 UI seguida de 100,000/mes) o placebo durante más de tres años en el ensayo de Evaluación de Vitamina D (ViDA) (79). Sin embargo, el Equipo de Trabajo de Servicios Preventivos de EE.UU. que realizó el meta-análisis de 11 ensayos aleatorios controlados con placebo, incluidas 52,915 personas mayores (de las cuales el 69% eran mujeres posmenopáusicas), descubrió que la suplementación con vitamina D (300-1,000 UI/día) y calcio (500-1,200 mg/día) por hasta siete años resultaron en una reducción del 12% en el riesgo de cualquier nueva fractura (80). Otro meta-análisis de 11 ensayos aleatorizados, doble ciego, controlados con placebo en el efecto de la suplementación con vitamina D en 31,022 personas (91% mujeres) de 65 años y mayores indicó que aquellos con la mayor ingesta de vitamina D (792-2,000 UI/día) tenían un riesgo 30% menor de fractura de cadera y un riesgo 14% menor de cualquier fractura no espinal (81). Finalmente, un tercer meta-análisis de ensayos que examinó el efecto de la combinación de vitamina D y calcio en la prevención de fracturas en hombres mayores y mujeres posmenopáusicas también concluyó que el riesgo de nuevas fracturas, incluidas las fracturas de cadera, era significativamente reducido en aquellos complementados en comparación con los controles (82). Curiosamente, los tres meta-análisis han encontrado que la prevención de fracturas por la suplementación de vitamina D y calcio se limita a las personas mayores institucionalizadas. De hecho, el riesgo de fractura no se redujo significativamente por la vitamina D en las personas mayores que viven en la comunidad (80-82).
Suplementos de vitamina D y equilibrio postural, fuerza muscular y riesgo de caída
Un meta-análisis de siete estudios observacionales en 840 fallers y 1,330 no fallers encontró concentraciones de 25-hidroxivitamina D en suero significativamente más bajas en fallers que en no fallers (83). Además, otro metaanálisis de cuatro cohortes de tres estudios observacionales reportó una modesta pero significativa asociación inversa entre el estatus de la vitamina C y el riesgo de caída (83). Varios ensayos aleatorios controlados han examinado el impacto de la suplementación con vitamina D en la fuerza muscular, el equilibrio postural o el riesgo de caída en sujetos mayores. Un meta-análisis de estos ensayos encontró evidencia limitada de un efecto de la suplementación con vitamina D sobre la fuerza muscular y la movilidad, basado en un solo tipo de prueba para cada resultado. Sin embargo, en un reciente estudio aleatorizado, doble ciego, controlado con placebo en 160 mujeres posmenopáusicas (edades, 50-65 años) con un estatus subóptimo de vitamina D (concentración media de 25-hidroxivitamina D sérica <20 ng/mL), suplementación con 1,000 UI/día de vitamina D3 mejoró significativamente el estatus de la vitamina D, así como la fuerza muscular de las extremidades superiores e inferiores y los parámetros de equilibrio postural (84, 85). Se encontró que los riesgos de caídas y caídas recurrentes eran de dos a tres veces mayores en mujeres en el grupo de control que en aquellos suplementados con vitamina D3 (85). Por el contrario, otro estudio controlado aleatorio de 12 meses en 200 adultos mayores (de los cuales el 58% tenía una concentración basal de 25-hidroxivitamina D sérica <20 ng/mL) no mostró beneficios con respecto a la función de la extremidad inferior o probabilidades de caer en aquellos suplementados con 2,000 UI/día (+/- 10 µg de calcidiol) en comparación con aquellos que recibieron 800 UI/día (86). El análisis post hoc recientemente publicado del ensayo ViDA no encontró diferencias entre las probabilidades de caer y el número de caídas informadas por 5,108 participantes que viven en la comunidad (edades, 50-84 años) independientemente de si fueron asignados al azar para recibir vitamina D suplementaria (100,000 UI/mes, es decir, ~3.350 UI/día) o un placebo durante una media de 3.4 años (79). a mayoría de los participantes de ViDA tenían concentraciones séricas de 25-hidroxivitamina D ≥20 ng/mL, lo que podría explicar al menos en parte la falta de un efecto de la vitamina D en las caídas (87).
En general, la evidencia actual sugiere que los suplementos de vitamina D3 de 800-1,000 UI/día pueden ser útiles para reducir las caídas y las tasas de fracturas en adultos mayores. Para que la suplementación con vitamina D sea efectiva en la preservación de la salud ósea, se debe consumir calcio dietético adecuado (1,000 a 1,200 mg/día) (véase el artículo sobre Calcio) (88).
Cáncer
Estudios ecológicos primero sugirieron una asociación entre latitudes septentrionales, la deficiencia de vitamina D, y la incidencia de cáncer (89). Desde la década de 1980 varios estudios de cohorte prospectivos han examinado la asociación de la ingesta o estatus de la vitamina D y varios tipos de cáncer. Una reciente revisión sistemática y meta-análisis de 16 estudios prospectivos, incluyendo 137,567 sujetos, reportó una reducción del 11% en la incidencia total de cáncer y una reducción del 17% en la mortalidad por cáncer con cada incremento de 20 ng/mL (50 nmol/L) en las concentraciones de 25-hidroxivitamina D circulante. Sin embargo, un análisis de subgrupos basados en el sexo de ocho estudios encontró una asociación inversa entre la vitamina D circulante y la mortalidad por cáncer en mujeres, pero no en hombres (90). Además, evidencia creciente sugiere que algunas variaciones en el gen que codifica para el receptor de la vitamina D (RVD) podrían influir en el estatus individual de la vitamina D y posteriormente modificar la susceptibilidad a los cánceres de sitio específicos (91) e influir en la supervivencia al cáncer (92). Finalmente, se ha encontrado que muchos tumores malignos expresan el RVD, incluyendo de seno, pulmón, piel (melanoma), colon y hueso (93), sugiriendo que podrían ser susceptibles a los efectos de la vitamina D. Numerosos estudios experimentales han demostrado que las formas biológicamente activas de la vitamina D, como la 1α,25-dihidroxivitamina D y sus análogas, tras la unión al RVD, pueden controlar el destino de la célula al inhibir la proliferación y/o induciendo la diferenciación celular o muerte (apoptosis) de un cierto número de células de tipo cancerosas (94).
Cáncer colorrectal
La distribución geográfica de la mortalidad del cáncer de colon se asemeja a la distribución geográfica histórica del raquitismo (95), aportando evidencia circunstancial de que la exposición reducida a luz solar y un estado nutricional de la vitamina D disminuido podrían estar relacionados con un riesgo aumentado de cáncer de colon. La evidencia de los estudios observacionales ha apoyado en gran medida esta hipótesis. Un meta-análisis reciente de cuatro estudios de cohorte prospectivos, cuatro estudios transversales y siete estudios de caso y control encontró una relación inversa entre la vitamina D circulante y la incidencia de adenoma colorrectal — un tumor benigno que puede transformarse en maligno (96). El análisis identificó una reducción del riesgo del 32% entre los cuantiles superior versus inferior de las concentraciones séricas de 25-hidroxivitamina D (96). Además, existe una fuerte evidencia de los meta-análisis de estudios de cohorte prospectivos para sugerir que mayores ingestas de vitamina D y concentraciones séricas de 25-hidroxivitamina D están asociadas con reducciones en el riesgo de cáncer colorrectal (97-99). El meta-análisis más reciente de cuatro cohortes prospectivas, 17 estudios de caso y control anidados y tres estudios de caso y control encontró un riesgo 38% menor de cáncer colorrectal con cuantiles altos versus bajos de concentraciones circulantes de 25-hidroxivitamina D (100). Un análisis de dosis-respuesta estimó que las concentraciones séricas de 25-hidroxivitamina D de ~20 a 30 ng/mL (en comparación con ≤12 ng mL) se asociaron con un riesgo 17% menor de cáncer colorrectal, y el riesgo fue aún menor (-35%) con una concentración sérica de 55 ng/mL (100). Un análisis anterior de dosis-respuesta basado en cinco estudios de caso y control anidados había estimado que las concentraciones séricas de 25-hidroxivitamina D ≥33 ng/mL (en comparación con ≤12 ng/mL) se asociaron con un riesgo 50% menor de cáncer colorrectal (101).
Sin embargo, en un ensayo aleatorizado, doble ciego, controlado con placebo, de siete años en 36,282 mujeres posmenopáusicas que participaron en el estudio Women's Health Initiative, una combinación de vitamina D3 suplementaria (400 UI/día) y calcio (1,000 mg/día) no disminuyó la incidencia de cáncer colorrectal (102). Otro ensayo aleatorio controlado de suplementos de vitamina D3 (1,000 UI/día), con o sin suplementos de calcio (1,200 mg/día), no encontró reducción en el riesgo de recurrencia de adenoma colorrectal durante un período de tres a cinco años, en comparación con el placebo, después de la extracción inicial de adenoma en los participantes (103). No está claro si estas dosis diarias de vitamina D son demasiado bajas para detectar algún efecto sobre la incidencia de cáncer (101, 104). Se necesitan ensayos clínicos aleatorios adicionales para evaluar si la suplementación con vitamina D podría ayudar a prevenir el cáncer colorrectal. Además, no está claro si las variaciones genéticas (polimorfismos) en la secuencia de genes implicados en el metabolismo y la función de la vitamina D pueden influir en la relación entre el estatus de la vitamina D y el riesgo de adenoma colorrectal o cáncer colorrectal (105-107).
Finalmente, la creciente evidencia sugiere que un estado adecuado de vitamina D puede estar relacionado con una mejor supervivencia de los pacientes con cáncer colorrectal. Un meta-análisis de cinco estudios prospectivos encontró un riesgo 35% menor de mortalidad específica por cáncer colorrectal en pacientes con cáncer con concentraciones séricas de 25-hidroxivitamina D más altas. Un análisis de dosis-respuesta estimó que cada aumento de 8 ng/mL en la concentración de 25-hidroxivitamina D se asoció con una disminución del 10% en la mortalidad por cáncer colorrectal (108).
Cáncer de seno
Aunque la evidencia ecológica sugiere que la mortalidad por cáncer de seno aumenta con el aumento de las latitudes y la disminución de la exposición a la luz solar (109), los datos de observación más recientes proporcionan poco apoyo para una asociación entre el estado nutricional de la vitamina D y el riesgo de cáncer de seno. Un estudio prospectivo temprano en mujeres que participaron en la primera Encuesta Nacional de Examinación de Salud y Nutrición de los EE.UU. (NHANES I) encontró que las mujeres caucásicas con una exposición al sol y una ingesta de vitamina D adecuadas tuvieron un riesgo significativamente reducido de cáncer de mama 20 años después (110). Sin embargo, cuando este estudio se incluyó en un meta-análisis con nueve estudios prospectivos más recientes, no hubo diferencias significativas en el riesgo de desarrollar cáncer de seno entre los niveles más altos y más bajos de ingestas de vitamina D (111). Además, si existe una asociación entre las concentraciones circulantes de vitamina D y el riesgo de cáncer de seno no están claros. Un meta-análisis de 14 estudios observacionales (9,110 casos y 16,244 controles) informó una reducción general del riesgo del 16% cuando se comparó el cuantil más alto de las concentraciones séricas de 25-hidroxivitamina D con el más bajo. Esta asociación inversa fue estadísticamente significativa en mujeres posmenopáusicas pero no en mujeres premenopáusicas (112). Sin embargo, otro meta-análisis que incluyó un conjunto similar de 14 estudios prospectivos (dos estudios fueron diferentes) no encontró una asociación general (111). Además, un meta-análisis de estudios realizados en pacientes en la etapa temprana del cáncer de seno identificaron asociaciones entre un estado inadecuado de vitamina D y un mayor riesgo de recurrencia y muerte (113). La evidencia de los ensayos aleatorios controlados actualmente es demasiado limitada para concluir si la suplementación con vitamina D puede reducir la incidencia de cáncer de seno (revisado en 114).
No obstante, tres meta-análisis han encontrado una asociación inversa entre las concentraciones circulantes de vitamina D y la mortalidad relacionada con el cáncer de seno (111, 115, 116). En un meta-análisis de un estudio retrospectivo y cinco estudios de cohorte prospectivos, las categorías más altas versus más bajas de vitamina sérica; las concentraciones de D se asociaron con una reducción del 33% en la mortalidad; un análisis de dosis-respuesta encontró una reducción del 12% por aumento de 8 ng/mL en la vitamina D en suero (115).
Finalmente, la evidencia actual no sugiere que variaciones genéticas específicas en la codificación genética para el VDR puedan influir en el riesgo de cáncer de seno (117, 118).
Otros tipos de cáncer
Evidencia que asocie el estatus de la vitamina D con otros tipos de cáncer es actualmente limitada. Mientras que la incidencia del cáncer de próstata parece estar inversamente asociada con la disponibilidad de la luz solar, estudios prospectivos de cohorte no han encontrado generalmente relaciones significativas entre los niveles de 25-hidroxivitamina D del suero y el riesgo posterior de desarrollar cáncer de próstata (119, 120). De hecho, algunos estudios han sugerido un mayor riesgo de cáncer de próstata con concentraciones circulantes de vitamina D más altas. Por ejemplo, un estudio anidado de caso y control de hombres (622 casos y 1,451 controles) de Escandinavia encontró una relación en forma de U entre las concentraciones séricas de 25-hidroxivitamina D y el riesgo de cáncer de próstata. En ese estudio, las concentraciones séricas de 25-hidroxivitamina D de 7.6 ng/mL o inferiores, o 32 ng/mL o superiores, se asociaron con un mayor riesgo de cáncer de próstata (121). Un meta-análisis de 17 estudios de caso y control anidado, tres estudios prospectivos de cohorte, y un estudio retrospectivo de cohorte encontraron un riesgo 17% mayor de cáncer de próstata en individuos en las categorías más altas versus más bajas de concentraciones de 25-hidroxivitamina D en sangre (122). Se han destacado, en una publicación reciente, factores de confusión potenciales que podrían explicar la detección de un ligero aumento en los casos de cáncer de próstata en los hombres con altas concentraciones circulantes de vitamina D (123).
Finalmente, meta-análisis recientes de estudios observacionales encontraron una relación inversa entre el estado de la vitamina D y el riesgo de cáncer de pulmón (124, 125) y cáncer de vejiga (126, 127). Sin embargo, en los pocos y a menudo heterogéneos estudios publicados hasta la fecha, las concentraciones séricas de 25-hidroxivitamina D no se asociaron con otros tipos de cáncer, incluido el linfoma no Hodgkin (128), cáncer de ovario (129), cáncer gástrico (130), o cáncer de piel (131).
Enfermedades autoinmunes
La diabetes mellitus dependiente de insulina (diabetes mellitus tipo 1), la esclerosis múltiple (EM), la artritis reumatoide (AR), y el lupus eritematoso sistémico (LES) son ejemplos de enfermedades autoinmunes. Las enfermedades autoinmunes ocurren cuando el cuerpo organiza una respuesta inmune en contra de su propio tejido, en lugar de un patógeno externo. Las enfermedades autoinmunes ocurren cuando el cuerpo monta una respuesta inmune contra su propio tejido, en lugar de un patógeno extraño. En la diabetes mellitus tipo 1, las células β del páncreas productoras de insulina son el objetivo de una respuesta inmune inapropiada. En la EM, los objetivos son las células productoras de mielina del sistema nervioso central, y en la AR, los objetivos son las células productoras de colágeno de las articulaciones (132). El LES se caracteriza por la presencia de un amplio espectro de autoanticuerpos que provocan daños potenciales a múltiples tejidos (133). Las respuestas autoinmunes están mediadas por células inmunes llamadas células T. Se ha encontrado que la forma biológicamente activa de la vitamina D, 1α, 25-dihidroxivitamina D, modula las respuestas de las células T, de modo que las respuestas autoinmunes disminuyen. Los estudios ecológicos han encontrado que la prevalencia de enfermedades autoinmunes (particularmente para MS; 134) aumenta a medida que aumenta la latitud, lo que sugiere que una menor exposición a la radiación UVB y las asociadas disminuciones en la síntesis de vitamina D en la piel pueden desempeñar un papel en la patología de estas enfermedades. Los resultados de varios estudios de cohorte prospectivos también sugieren que un estado adecuado de vitamina D a diferentes edades (incluso in utero, en la primera infancia y durante la adolescencia) podría posiblemente disminuir el riesgo de enfermedades autoinmunes.
Diabetes mellitus tipo 1
Niveles más bajos de vitamina D circulante han sido reportados en pacientes recién diagnosticados con diabetes mellitus tipo 1 en comparación con sujetos no diabéticos del mismo sexo y edad (135, 136). Una mayor prevalencia de una insuficiencia y deficiencia de vitamina D ha sido también observada en niños pre-diabéticos que desarrollaron múltiples autoanticuerpos en los islotes (anticuerpos contra células pancreáticas que secretan insulina) en comparación a niños negativos de autoanticuerpos. Sin embargo, un estudio prospectivo que dio seguimiento al cohorte de niños pre-diabéticos encontró que sus estatus de vitamina D, definidos como insuficiente, deficiente, o suficiente, no se asoció con la tasa de progresión a diabetes tipo 1 después de 5 a 10 años de seguimiento (137). Un estudio de cohorte prospectivo más temprano de niños nacidos en Finlandia durante el año 1966 y con un seguimiento de 30 años encontró que los niños suplementados con vitamina D durante el primer año de vida tuvieron un riesgo 88% menor de desarrollar diabetes tipo 1 en comparación con aquellos que no recibieron una suplementación. Por otra parte, niños con sospecha de haber tenido raquitismo (deficiencia severa de vitamina D) durante el primer año de vida mostraron un riesgo significativamente mayor de desarrollar diabetes tipo 1 (138). Por lo tanto, la suplementación con vitamina D parece proteger contra el inicio de la diabetes tipo 1, y un estatus subóptimo de la vitamina D en la infancia puede tener efectos a largo plazo en las respuestas inmunes más tarde en la vida.
Existen también datos limitados sugiriendo que la insuficiencia de vitamina D materna durante el embarazo puede influir en el riesgo de diabetes tipo 1 en el recién nacido. En un reciente estudio de caso y control, el riesgo de la aparición de diabetes tipo 1 en la niñez era más de dos veces mayor en niños cuyas madres tuvieron niveles de 25-hidroxivitamina D del suero <21.6 ng/mL (54 nmol/L) durante el último trimestre del embarazo en comparación con los niños nacidos de mujeres con 25-hidroxivitamina D en el suero >35.6 ng/mL (89 nmol/L) (139). Otros estudios de caso y control han encontrado que la suplementación con vitamina D durante el embarazo se asoció con un riesgo menor de que sus hijos desarrollarán autoanticuerpos relacionados con la diabetes (140, 141). Sin embargo, un estudio amplio conducido en madres de niños en un riesgo genético incrementado de padecer diabetes no reportó asociación alguna entre la aparición de autoanticuerpos islote y/o aparición de diabetes en sus hijos en el primer año de vida y la ingesta de vitamina D materna durante el embarazo (142). Otro estudio de caso y control falló en observar una relación entre la 25-hidroxivitamina D del suero durante el embarazo temprano y el diagnóstico de diabetes tipo 1 en la descendencia (143). Estudios prospectivos amplios son necesarios para establecer si el estatus de la vitamina D materna durante el embarazo puede influir en el riesgo de diabetes tipo 1 en la descendencia.
Finalmente, la relación de los polimorfismos en genes relacionados con el metabolismo de la vitamina D y la diabetes tipo 1 está actualmente bajo investigación. Se ha propuesto que polimorfismos específicos en los genes, como el CYP27B1 (que codifica para la 25-hidroxivitamina D3-1α-hidroxilasa) y el RVD, pueden ser funcionalmente relevantes para la acción de la vitamina D y podrían de esta manera afectar la susceptibilidad a la enfermedad. En un estudio conducido en 8,517 niños y adolescentes con diabetes tipo 1 y 7,320 sujetos de control, polimorfismos en los genes que involucran la síntesis del colesterol y la hidroxilación de la vitamina D fueron ligados a los niveles de vitamina D circulantes y al estatus diabético (26).
Esclerosis múltiple
Niveles bajos de exposición al sol y una deficiencia de vitamina D parecen estar asociados con el desarrollo de la esclerosis múltiple (EM). Un estatus de la vitamina D pobre podría comprometer la función de células inmunes específicas críticas en la regulación de varias respuestas inmunes y ayudar a desencadenar autoinmunidad en la EM (144). Los determinantes genéticos del estatus bajo de vitamina D se han relacionado recientemente con una mayor susceptibilidad a la EM de inicio en adultos en un análisis aleatorizado mendeliano de datos del Consorcio de Genética de Esclerosis Múltiple (145). Esto se hizo eco de los resultados de varios estudios observacionales que sugirieron una asociación entre la suficiencia de vitamina D y disminución del riesgo de EM. Un estudio retrospectivo de los niveles de radiación UV ambiental y los casos de EM realizados en Australia revelaron que la incidencia de EM en la descendencia estaba inversamente relacionada con la exposición materna a los rayos UV durante el embarazo temprano (146). La exposición al sol fue también usada como un marcador sustituto para la exposición de la vitamina D en un reciente estudio de caso y control que incluyó 1,660 pacientes con EM y 3,050 controles. Los autores encontraron que las actividades al aire libre poco frecuentes y el uso de bloqueador solar durante la infancia temprana y la adolescencia se asociaron con un significante incremento en el riesgo de desarrollar EM más tarde en la vida (147). En un estudio de corte transversal, la exposición al sol y la ingesta de aceite de hígado de bacalao (rico en vitamina D) durante la infancia fueron ligados a un comienzo tardío de los síntomas entre veteranos con EM recidivante (148). Adicionalmente, un estudio de caso y control en el personal militar de los EE.UU. incluyendo 257 casos de EM diagnosticada, encontró que los sujetos caucásicos en el quintil más alto de la 25-hidroxivitamina D del suero (>39.6 ng/mL) tuvieron un riesgo 62% menor de desarrollar EM en comparación al quintil más bajo (<25.3 ng/mL) (149). Además, en dos cohortes amplias de más de 187,000 mujeres estadounidenses con un seguimiento de por lo menos 10 años, el uso de un suplemento de vitamina D (≥400 UI/día) se asoció con una reducción del 41% en el riesgo de desarrollar EM (150). Otro estudio prospectivo no controlado monitoreó la incidencia de recaída en relación con el estatus de la vitamina D en 156 pacientes con EM recurrente-remitente antes y después de recibir vitamina D suplementaria (100,000 UI/mes; 6-42 meses, mediana de 31 meses), en adición al tratamiento inmunomodulador de primera línea (151). Cada aumento de 4 ng/mL en la concentración sérica de 25-hidroxivitamina D se asoció con una disminución del 14,9% en la incidencia de recaída (151). En un estudio multicéntrico realizado en pacientes recién diagnosticados con un síndrome clínicamente aislado (SCA) y tratado con interferón (IFN)-β, el estatus de la vitamina D fue predictivo de la actividad y progresión de la EM. Las concentraciones séricas más altas de 25-hidroxivitamina D (≥20 ng/mL o ≥50 nmol/L) en el primer año después del diagnóstico de SCA predijeron un tiempo más largo para el diagnóstico de EM, menor número de nuevas lesiones, y menores cambios en la lesión y el volumen cerebral durante los siguientes cuatro años de seguimiento (152). Sin embargo, un estudio retrospectivo sugirió que el estatus de la vitamina D en pacientes con EM recurrente-remitente no tenía valor predictivo con respecto al tiempo de conversión a EM progresiva secundaria, que se caracteriza por un empeoramiento de discapacidad (153).
Los ensayos clínicos no han podido demostrar ningún beneficio de la suplementación con vitamina D, sola o en combinación con el tratamiento con IFN-β, con respecto a las tasas de recaída y los síntomas relacionados con la discapacidad en pacientes con EM (154, 155). En otros ensayos, la vitamina D3 suplementaria también falló en demostrar actividades inmunomoduladores (156-159). En un reciente ensayo aleatorizado, controlado con placebo en 53 pacientes tratados con IFN-β con EM recurrente-remitente, la suplementación con vitamina D3 (7,000 UI/día durante cuatro semanas, seguido de 14,000 UI/día hasta la semana 48) mostró poco efecto sobre la proporción de algunos linfocitos T y B reguladores durante el período de estudio de 48 semanas. La vitamina D3 solo pareció ayudar a mantener la proporción de células T CD4+ antiinflamatorias — que disminuyeron en los pacientes que recibieron placebo — pero no pudieron mejorar su reactividad cuando se estimularon con 1,25-dihidroxivitamina D in vitro (157). En otro ensayo, se encontró que la suplementación con vitamina D3 (10,400 UI/día durante tres meses) a pacientes con EM recurrente-remitente reduce la proporción de células T CD4+ productoras de IL-17 proinflamatorias, que se cree que juegan un papel central en el desarrollo de EM (160).
Artritis reumatoide
La deficiencia de vitamina D puede también estar involucrada en la etiología y/o progresión de la artritis reumatoide (AR), aunque la evidencia proviene primeramente de estudios en animales. La ausencia de receptores de la vitamina D (RVD) en ratones genéticamente modificados ha sido ligada a niveles más altos de inflamación y a un incremento en la susceptibilidad a la autoinmunidad (161). Cuando los ratones transgénicos que espontáneamente desarrollan artritis inflamatoria también están deficientes de RVD, estos desarrollan una forma más agresiva de artritis crónica (162). También, polimorfismos específicos en el gen del RVD han sido ligados a un incremento en la susceptibilidad a la AR en ciertas poblaciones, aunque la manera en que estas variantes genéticas incluyen la funcionalidad de la vitamina D no es completamente comprendida (163-165). Los datos actuales, sin embargo, apuntan a un papel de la vitamina D en la modulación del proceso inflamatorio que subyace a muchas enfermedades crónicas, incluyendo la AR. Varios estudios de corte transversal en individuos con niveles moderados a altos de inflamación han reportado tanto ninguna asociación o una asociación inversa entre la 25-dihidroxivitamina D circulante y los marcadores de la inflamación. No obstante, existe una carencia de ensayos de intervención para mostrar si la suplementación con vitamina D podría limitar la inflamación y reducir el riesgo de una enfermedad (incluyendo la AR) en sujetos con niveles altos de inflamación (166).
Actualmente, no está claro si la prevalencia de una deficiencia de vitamina D está ligada a la incidencia de la AR. En un estudio de cohorte amplio de casi 30,000 mujeres posmenopáusicas estadounidenses, los sujetos con las ingestas totales más altas de vitamina D (≥467.7 UI/día) tuvieron un riesgo 33% menor de desarrollar AR después de 11 años de seguimiento que aquellos con las ingestas más bajas (<221.4 UI/día) (167). A pesar de todo, más análisis recientes de dos cohortes amplias de casi 200,000 mujeres estadounidenses con un seguimiento de varias décadas no encontró asociación alguna entre las ingestas dietéticas reportadas de vitamina D (usando cuestionarios de frecuencia de alimentos) durante la adolescencia o adultez y la incidencia de AR más tarde en la vida (168, 169). Además, varios estudios que exploraron la relación entre la vitamina D circulante y la actividad patológica en los pacientes con AR han reportado resultados mixtos (revisado en 170). Sin embargo, dos meta-análisis recientes de estudios observacionales encontraron una relación inversa entre el estado de la vitamina D y la actividad de la enfermedad en pacientes con AR, evaluada mediante el Índice de la Actividad de la Enfermedad 28 (en inglés, DAS28) (171, 172). Finalmente, existe una escasez de estudios que exploren el efecto de la suplementación con vitamina D en la actividad patológica en los sujetos con artritis. Un pequeño estudio aleatorio, doble ciego, controlado con placebo en 22 pacientes con AR falló en demostrar mejoras en la actividad patológica y el nivel de inflamación en sujetos suplementados con calcio (1,500 mg/día) y altas dosis de vitamina D2 (ergocalciferol; un promedio de más de 4,500 UI/día) por un año en comparación al placebo (173). Otro ensayo controlado aleatorio de tres meses en 41 mujeres con AR temprana no encontró beneficios adicionales de la vitamina D3 suplementaria (una dosis en bolo de 300,000 UI) a la atención estándar (metotrexato y glucocorticoides) con respecto al recuento de linfocitos T colaboradores, producción de citocinas, o parámetros clínicos incluida la actividad de la enfermedad (174). La vitamina D suplementaria tampoco logró reducir la tasa de recurrencia de la enfermedad en pacientes con AR incluidos en dos pequeños ensayos aleatorios controlados (175, 176). Dado que estos estudios tienen varias limitaciones, incluidos los pequeños tamaño de la muestra, se justifica la investigación adicional.
Lupus eritematoso sistémico
Más prevalente y severo en poblaciones no caucásicas (hispanos, descendientes de africanos, y asiáticos) (177), el lupus eritematoso sistémico (LES) es una enfermedad autoinmune con manifestaciones clínicas heterogéneas. La enfermedad puede potencialmente afectar la mayoría de los tejidos y órganos, incluyendo la piel (erupciones en la piel y la fotosensibilidad), los riñones (nefritis), y las articulaciones (artritis). Existe evidencia de un papel de la vitamina D en la prevención del LES en modelos animales (178). Curiosamente, un reciente meta-análisis de 11 estudios de caso y control encontró que polimorfismos específicos del RVD estaban ligados al LES particularmente en asiáticos (179). Sin embargo, la relevancia funcional de tales variantes genéticas es desconocida (180). Los análisis de dos estudios de cohorte prospectivos amplios de casi 200,000 mujeres estadounidenses falló en mostrar una asociación entre la ingesta dietética de vitamina D (medida vía cuestionarios de frecuencia de alimentos) durante la adolescencia o adultez y la incidencia de LES más tarde en la vida (168, 169).
A pesar de todo, un estatus subóptimo de la vitamina D es comúnmente observado en sujetos con LES, y esto es parcialmente explicado por la falta de exposición al sol, la cual tiende a agravar los síntomas de la enfermedad (181, 182). Las concentraciones de 25-hidroxivitamina D del suero fueron inversamente correlacionadas con medidas de la actividad patológica en una cohorte de 378 pacientes con LES (183). La corrección de la insuficiencia de vitamina D con altos niveles de vitamina D3 (100,000 UI/semana por un mes de seguimiento por 100,000 UI/mes por seis meses) en 20 sujetos con LES fue ligada a la reducción en los signos de un desequilibrio inmunológico y en los niveles de autoanticuerpos típicamente detectados en LES, sugiriendo un valor terapéutico para la vitamina D en el tratamiento de la enfermedad (184). Otro estudio prospectivo conducido en 52 pacientes deficientes de vitamina D con lupus eritematoso cutáneo (un tipo de lupus con trastornos de la piel sólo) reportó una reducción en la severidad de la enfermedad en el grupo suplementado con vitamina D3 1,400 UI/día inicialmente, seguido por 800 UI/día) y calcio por un año en comparación a los pacientes no tratados (185). La suplementación con vitamina D3 (200 UI/día por un año) fue también capaz de reducir el nivel de citoquinas inflamatorias en un estudio aleatorio controlado con placebo conducido en 267 pacientes con LES (186). En otro ensayo aleatorizado, controlado con placebo, la suplementación con vitamina D3 (50,000 UI/semana durante seis meses) mejoró el índice de actividad de la enfermedad del LES (en inglés, SLEDAI) y los puntajes de la Medida de Actividad del Lupus del Consenso Europeo (en inglés, ECLAM), así como algunas medidas de fatiga en jóvenes adultos con LES de inicio juvenil (187). Sin embargo, en otros dos estudios recientes, la suplementación con vitamina D3 (dosis de bolo semanales/mensuales equivalentes a ~800 a 7,000 UI/día durante 6 a 24 meses) mejoró el estado de la vitamina D en pacientes con LES pero falló en mostrar algún beneficio con respecto a la actividad de la enfermedad (188, 189). Si bien la administración oral de vitamina D a pacientes con LES es bien tolerada, su eficacia sigue siendo cuestionable y merece mayor investigación en ensayos clínicos.
Resumen
Por lo tanto, la evidencia de estudios epidemiológicos en humanos sugiere que, si bien aún no se puede concluir que la suplementación con vitamina D es beneficiosa en la prevención o el tratamiento de enfermedades autoinmunes, es razonable suponer que corregir la insuficiencia de vitamina D y mantener niveles suficientes podría ayudar a disminuir el riesgo de enfermedad (190).
Enfermedad cardiovascular
Hipertensión (presión sanguínea alta)
La hipertensión es un factor de riesgo bien conocido para las enfermedades cardiovasculares (ECV) (191). Los resultados de estudios clínicos y basados en la observación sugieren un papel de la vitamina D en la disminución de la presión sanguínea, el cual podría ser parcialmente explicado por el hecho de que la 1,25-dihidroxivitamina D inhibe la síntesis de renina (véase Función). Así, la deficiencia de vitamina D y la posterior regulación positiva del sistema renina-angiotensina pueden contribuir a la presión sanguínea alta y el riesgo de ECV. Se ha sugerido que los niveles elevados de la PTH pueden incrementar el riesgo de hipertensión y ECV (6). A pesar de todo, en un reciente estudio de cohorte prospectivo de 3,002 individuos (con 59 años de edad al inicio del estudio), la incidencia de hipertensión, la cual afectó al 41% de los participantes durante el período de seguimiento de nueve años, no fue mayor en aquellos con niveles de 25-hidroxivitamina D del suero menores a 20 ng/mL y fue asociada sólo marginalmente con niveles elevados de PTH (192). No obstante, un meta-análisis de siete estudios prospectivos, incluyendo un total de 48,633 con casi 5,000 casos de hipertensión incidente, encontró un riesgo 30% menor de hipertensión en aquellos en el tercil superior vs. aquellos en el tercil más inferior de las concentraciones séricas de 25-hidroxivitamina D. El análisis dosis-respuesta estimó que cada 10 ng/mL de incremento en el nivel de 25-hidroxivitamina D del suero se asoció con un riesgo 12% menor de hipertensión (193). Otro meta-análisis de 14 estudios de corte transversal y 4 prospectivos también reportó una relación inversa entre la 25-hidroxivitamina D y la hipertensión (194).
Disfunción endotelial
La disfunción del endotelio vascular, la cual contribuye a un riesgo incrementado de enfermedad cardiovascular (ECV), es común en pacientes con una enfermedad renal crónica (ERC) (195). En pacientes con ERC, la función endotelial anormal está asociada con valores bajos de la dilatación mediada por flujo (DMF) de la arteria braquial, un marcador sustituto de la salud vascular. En un estudio reciente conducido en sujetos con una leve a moderada ERC, los niveles de 25-hidroxivitamina D del suero fueron positivamente asociados con valores de la DMF, sugiriendo un enlace entre el estatus de la vitamina D sub-óptimo y la disfunción endotelial (196). En un estudio de intervención preliminar, 26 pacientes con una ERC moderada y una insuficiencia/deficiencia de vitamina D fueron suplementados dos veces con 300,000 UI de vitamina D3 (en las semanas 1 y 8) y seguidos por un total de 16 semanas. La suplementación con vitamina D casi duplicó los niveles de 25-hidroxivitamina D del suero y disminuyó los niveles de PTH en un 68.5%; el estatus mejorado de la vitamina D fue acompañado por el incremento de los valores de DMF y la reducción de los niveles de los marcadores de disfunción endotelial (197). Un meta-análisis reciente de 12 pequeños ensayos aleatorios controlados en participantes con alto riesgo de ECV encontró un aumento significativo en la DMF con la suplementación con vitamina D (dosis diarias, 2,500-5,000 UI; dosis semanal, 50,000 UI; dosis mensual, 60,000 UI; dosis única de bolo, 100,000-200,000 UI) durante ocho semanas a seis meses (198).
Eventos cardiovasculares en estudios observacionales y ensayos clínicos
Hasta la fecha, la muchos estudios epidemiológicos que investigan la relación entre la vitamina D y los resultados de las ECV han provisto resultados mixtos (revisado en 199). Estudios recientes de aleatorización mendeliana no encontraron asociación entre las concentraciones séricas de 25-hidroxivitamina D genéticamente bajas y los riesgos de enfermedad coronaria, enfermedad cardíaca isquémica o infarto al miocardio (200, 201), lo que sugiere que las asociaciones informadas en estudios observacionales pueden deberse a confusión o causalidad inversa. En un ensayo de la Evaluación Aleatorizada de Calcio o Vitamina D (RECORD) en 5,292 personas mayores (véase Osteoporosis), la suplementación con 800 UI/día de vitamina D3 (± calcio) redujo el riesgo de primera falla cardíaca, pero no tuvo ningún efecto sobre el riesgo de infarto al miocardio y accidente cerebrovascular en comparación con la suplementación con calcio solo o placebo (202). Los datos sobre el efecto de la suplementación con vitamina D sobre los eventos cardiovasculares se obtuvieron de 21 estudios aleatorios controlados (incluido el ensayo RECORD) en 13,033 participantes (≥60 años) y se combinaron en un meta-análisis (202). No se encontró ningún efecto de la vitamina D (incluidos los análogos de la vitamina D) para eventos cardiovasculares mayores, como fallo cardíaco, infarto al miocardio y accidente cerebrovascular durante los períodos de seguimiento de 1 a 6,2 años (202). Sin embargo, se recomienda precaución al interpretar estos resultados, ya que los ensayos se diseñaron inicialmente para evaluar el efecto de la vitamina D en la salud ósea, y los resultados cardiovasculares no fueron criterios de valoración primarios. Varios ensayos controlados aleatorios que exploran el efecto de la suplementación con vitamina D sobre el riesgo de ECV están actualmente en curso (203), incluyendo dos grandes ensayos, el Ensayo de la Vitamina D y Omega-3 Trial (VITAL) en los EE.UU. (204) y el ensayo D-Health en Australia (205). Recientemente se publicaron los resultados de un ensayo aleatorio controlado, el ensayo de Evaluación de Vitamina D (ViDA) en Nueva Zelanda. El número total de eventos de ECV y el tiempo hasta el primer evento de ECV durante el seguimiento no fue diferente entre los que fueron suplementados con vitamina D3 (dosis inicial de 200,000 UI durante el primer mes seguido de dosis mensuales de 100,000 UI) y aquellos que recibieron un placebo por un mediana 3,3 años (206).
Diabetes mellitus tipo 2
Las personas con un síndrome metabólico están en un riesgo incrementado de contraer diabetes mellitus tipo 2 (diabetes mellitus no dependiente de insulina) y enfermedad cardiovascular (ECV). Un síndrome metabólico se refiere a varios desórdenes metabólicos, incluyendo dislipidemia, hipertensión, resistencia a la insulina y obesidad. Un reciente estudio encontró que la prevalencia de diabetes tipo 2 fue asociada con niveles subóptimos de 25-hidroxivitamina D del suero (<30 ng/mL) en 1,801 pacientes con un síndrome metabólico. Durante un período de seguimiento de ocho años, se reportaron riesgos más bajos de mortalidad por todas las causas (riesgo 72% menor) y de mortalidad por ECV específica (riesgo 64% menor) en individuos con concentraciones séricas de 25-hidroxivitamina D mayores a 30 ng/mL (75 nmol/L) cuando fueron comparados con aquellos con concentraciones por debajo de los 10 ng/mL (25 nmol/L) (207).
En personas saludables, la suficiencia de vitamina D es positivamente correlacionada con una sensibilidad a la insulina y una función de las células β pancreáticas adecuada. De manera contraria, la deficiencia de vitamina D podría afectar la homeostasis de la glucosa y causar una intolerancia a la glucosa y una resistencia a la insulina (208). En un estudio de corte transversal conducido en 12,719 adultos, de los cuales 4,057 tuvieron prediabetes (es decir un riesgo incrementado de desarrollar diabetes tipo 2), la prevalencia de la prediabetes se asoció con concentraciones inferiores de 25-hidroxivitamina D del suero (≤32.4 ng/mL). Los sujetos con las concentraciones más bajas de 25-hidroxivitamina D del suero (≤17.7 ng/mL) fueron más propensos a ser actuales fumadores, obesos, y tener hipertensión (209). La insuficiencia de vitamina D en individuos en alto riesgo puede acelerar la progresión a una diabetes evidente. En un estudio prospectivo de 2,387 hombres y mujeres de mediana edad con un seguimiento de 8 a 10 años, el riesgo de la progresión a diabetes tipo 2 a partir de la prediabetes fue 62% más bajo en mujeres y 60% más bajo en hombres en el cuartil más alto en comparación al más bajo de la vitamina D circulante (>28.4 ng/mL vs. <18.5 ng/mL). Un análisis dosis-respuesta midió una reducción promedio del 23% en el riesgo de la progresión a diabetes tipo 2 por cada incremento de 4 ng/mL (10 nmol/L) en la concentración sérica de 25-hidroxivitamina D (210). Una revisión reciente y un meta-análisis de 18 estudios de cohorte prospectivos, que incluyeron a más de 210,000 participantes seguidos durante un período medio de 10 años, encontraron que las personas en el tercio superior de los niveles de vitamina D (reportados como vitamina D circulante o ingestas dietéticas) tenían menos riesgos de desarrollar diabetes tipo 2 (riesgo 19% menor) y síndrome metabólico (riesgo 14% menor) en comparación con los del tercio inferior (211). En otro meta-análisis de nueve estudios prospectivos, que incluyó a 28,258 personas mayores (edad media, 67.7 años), se encontró que las concentraciones de vitamina D circulantes más bajas versus más altas al inicio del estudio se asociaron con un riesgo 17% mayor de desarrollar diabetes tipo 2 durante una mediana período de seguimiento de 7.3 años (212). Actualmente, la evidencia limitada sugiere que la suplementación con vitamina D puede mejorar la sensibilidad a la insulina en individuos con intolerancia a la glucosa o que manifiestan diabetes tipo 2 (213-216). Existe la necesidad de ensayos clínicos bien diseñados para examinar si mantener niveles adecuados de vitamina D puede prevenir resultados metabólicos adversos en individuos sanos y en riesgo.
Enfermedades neurodegenerativas
El deterioro cognitivo y la enfermedad de Alzheimer
La enfermedad de Alzheimer (EA) es la forma más común de demencia, caracterizada por la presencia de placas β-amiloides extra-neuronales y agregados intra-neuronales de proteínas Tau (conocidos como ovillos neurofibrilares) en el cerebro. Modelos mecanicistas actualmente estudiados en la investigación en animales sugieren que la deficiencia de vitamina D o desórdenes del metabolismo de la vitamina D y/o la interrupción de la vía vitamina D-RVD en las regiones cerebrales de la corteza y el hipocampo pueden estar involucrados en la degeneración de las neuronas y la pérdida de las funciones cognitivas (217). La evidencia experimental que apoya un papel de la vitamina D en la regulación de los canales del calcio, la neuroprotección, y la inmunomodulación en el sistema nervioso central también implica que el estatus bajo de la vitamina D puede preceder o contribuir a la disfunción cognitiva con la edad (218).
Varios estudios observacionales han examinado el deterioro cognitivo y la enfermedad cerebral degenerativa en los ancianos en relación con la ingesta dietética de vitamina D y la concentración sérica de 25-hidroxivitamina D. En un gran estudio de cohorte francés sobre osteoporosis y fracturas de cadera en mujeres posmenopáusicas, las deficiencias en el rendimiento cognitivo global, evaluadas con el Cuestionario Corto Portátil del Estado Mental de Pfeiffer (en inglés, SPMSQ), se asociaron con una menor ingesta de vitamina D dietética (<1,400 UI/semana vs. ≥1,400 UI/semana) en 5,596 mujeres de edad avanzada (edad media, 80.5 años) (219). Un estudio con un seguimiento de siete años de un subgrupo de 498 mujeres indicó que el riesgo de padecer la enfermedad de Alzheimer (pero no otros tipos de demencia) fue 77% menor en aquellas en el quintil más alto vs. aquellas en el quintil más bajo de las ingestas de vitamina D basales (220). Algunos, pero no todos, los estudios observacionales han encontrado una asociación entre las bajas concentraciones séricas de 25-hidroxivitamina D y el deterioro cognitivo leve en adultos mayores (219, 221, 222). El análisis de corte transversal y longitudinal de dos estudios prospectivos, que incluyó 1,604 hombres (223) y 6,257 mujeres (224) de edades a partir de los 65 años, informó una probabilidad 60% mayor de deterioro cognitivo al inicio del estudio y un riesgo 58% mayor de deterioro cognitivo durante un período de seguimiento de cuatro años en mujeres, pero no en hombres, con deficiencia de vitamina D (25-hidroxivitamina D circulante <10 ng/mL vs. ≥30 ng/mL). En el estudio de caso y control anidado, multiétnico, Singapore Kidney Eye Study, que incluyó a 2,273 individuos (edad media, 70.4 años), las concentraciones séricas de 25-hidroxivitamina D se correlacionaron inversamente con los déficits cognitivos que afectan la memoria episódica retrógrada, la memoria semántica y la orientación en el tiempo, según lo evaluado por el Examen Mental Abreviado (en inglés, AMT) (225).
Sin embargo, las revisiones sistemáticas y los meta-análisis de los estudios observacionales han arrojado resultados mixtos sobre la asociación del estado de la vitamina D con el rendimiento cognitivo y la EA (226-230). Además, el análisis reciente de datos de 1,182 hombres seguidos durante 18 años en el Estudio Longitudinal Uppsala de Hombres Adultos (en inglés, ULSAM) no logró encontrar asociaciones de determinantes genéticos de la síntesis de vitamina D, ingestas de vitamina D y concentraciones en el plasma de 25-hidroxivitamina D con riesgos de deficiencias cognitivas, EA, demencia vascular o demencia por todas las causas (231). Por el contrario, otro estudio de aleatorización mendeliano asoció los determinantes genéticos del estatus bajo de vitamina D a un mayor riesgo de EA en el conjunto de datos del Proyecto Internacional del Genoma del Alzheimer (17,008 casos de EA y 37,154 casos saludables) (232).
Sin embargo, la prevalencia de la insuficiencia/deficiencia de vitamina D oscila entre el 70% y 90% en los adultos mayores, y la corrección de niveles bajos de la 25-hidroxivitamina D del suero puede ayudar a mejorar los procesos cognitivos, en particular las funciones ejecutiva (233). En un pequeño estudio controlado no aleatorio en una clínica para pacientes ambulatorios, se evaluó la función cognitiva global al inicio del estudio y después de 16 meses en 20 pacientes suplementados con 800 UI/día (o 100,000 UI/mes) de vitamina D y en 24 sujetos de control. La suplementación de pacientes ambulatorios con vitamina D resultó en la corrección del estatus bajo de vitamina D (la concentración sérica promedio de 25-hidroxivitamina D fue de 16,8 ng/mL al inicio y 30 ng/mL a los 16 meses) y se asoció con una puntuación significativamente mejorada en las pruebas de cognición en comparación con el grupo no suplementado (234). En un ensayo clínico aleatorio controlado con placebo en 32 pacientes con leve a moderada EA que recibieron insulina nasal, altas dosis de la suplementación con vitamina D2 por ocho semanas (hasta 36,000 UI/día) no mejoraron significativamente el rendimiento cognitivo en comparación con dosis bajas (1,000 UI/día) (235). Más investigaciones son necesarias para investigar una relación causal entre la reposición de la vitamina D y los beneficios cognitivos potenciales a largo plazo en adultos mayores. Además, es de gran importancia evaluar si la corrección de la deficiencia de vitamina D en sujetos con deterioro cognitivo pueden mejorar el impacto de la terapia contra la demencia (236).
Enfermedad de Parkinson
La enfermedad de Parkinson (EP) ha sido asociada con una alta prevalencia de insuficiencia de vitamina D entre los pacientes, especialmente aquellos con mayores problemas de movilidad (237). Un estudio de caso y control de 296 pacientes ambulatorios con una edad media de 65 años indicó que el 23% de los sujetos con EP tenían concentraciones séricas de 25-hidroxivitamina D inferiores a 20 ng/mL en comparación con el 16% y el 10% de EA y personas sanas, respectivamente (238). En un estudio de cohorte prospectivo realizado entre 3,173 hombres y mujeres de 50-79 años y libres de EP al inicio del estudio, los individuos en el cuartil más alto de 25-hidroxivitamina D en suero (≥20 ng/mL para mujeres y ≥22.8 ng/mL para hombres) tenían un riesgo 67% menor de EP en comparación con los del cuartil más bajo (≤10 ng/mL para las mujeres y ≤11.2 ng/mL para los hombres) (239). Los meta-análisis que agrupaban datos de estudios observacionales mostraron que la insuficiencia de vitamina D se informaba con mayor probabilidad en sujetos con EP que en controles sanos (240-242).
En un estudio aleatorizado, doble ciego, controlado con placebo, 112 pacientes con EP (edad media, 72 años) en tratamiento estándar con DP fueron suplementados con 1,200 UI/día de vitamina D o un placebo durante 12 meses. La suplementación con vitamina D casi duplicó la concentración sérica de 25-hidroxivitamina D (de una media de 22.5 ng/mL a 41.7 ng/mL) en sujetos suplementados y limitó la progresión de la EP, como lo indica una mayor proporción de pacientes que no mostraron empeoramiento (según lo evaluado en la etapa de Hoehn y Yahr y la United Parkinson Disease Rating Scale parte II) en el grupo suplementado en comparación con el grupo placebo (243). Se desconoce si la insuficiencia de vitamina D tiene un papel en la patogénesis de la enfermedad, pero la reposición de vitamina D puede proveer de beneficios a la salud que van más allá de la prevención y/o el tratamiento de EP. Por ejemplo, la deficiencia de vitamina D puede contribuir al riesgo incrementado de osteoporosis y fracturas óseas en individuos con desórdenes neurológicos, incluyendo la EP y la esclerosis múltiple (244-246). Curiosamente, se encontró que la exposición al sol estaba asociada con un estatus de la vitamina D mejorada, una mayor densidad mineral ósea del segundo hueso metacarpiano, y una menor incidencia de fractura de cadera en un estudio prospectivo conducido en 324 personas de la tercera edad con EP (247).
Resultados adversos del embarazo
Una revisión sistemática y meta-análisis recientes de 31 estudios basados en la observación en el estatus de la vitamina D materno y los resultados del embarazo indicaron que la insuficiencia de vitamina D puede estar asociada con la diabetes mellitus gestacional, preeclampsia, y vaginosis bacteriana en mujeres embarazadas. La vitamina D sérica materna baja durante el embarazo también se relacionó con un mayor riesgo para los lactantes pequeños para la edad gestacional y los lactantes de bajo peso al nacer, pero no para la cesárea (248). Sin embargo, el número de ensayos de intervención actualmente es demasiado limitado para extraer conclusiones sobre si la suplementación con vitamina D durante el embarazo podría reducir la incidencia de los resultados adversos mencionados anteriormente (249).
Diabetes mellitus gestacional
La hiperglicemia anormal debido a la disfunción de células β pancreáticas caracteriza a la aparición de la diabetes mellitus gestacional (DMG) en mujeres embarazadas sin antecedentes de diabetes mellitus tipo 2. Esta condición está asociada con resultados adversos maternos serios, incluyendo preeclampsia, un riesgo alto de un parto por cesárea, y un riesgo de por vida incrementado de desarrollar un síndrome metabólico y diabetes mellitus tipo 2. La DMG puede también contribuir a un riesgo incrementado de macrosomía fetal (peso excesivo al nacer), hipoglicemia neonatal, dificultad respiratoria neonatal, y un riesgo de por vida incrementado de obesidad, intolerancia a la glucosa, diabetes mellitus tipo 2, y enfermedades cardiovasculares en los hijos (revisado en 250).
Un reciente estudio prospectivo conducido en 655 mujeres embarazadas encontró que la concentración media de 25-hidroxivitamina D del suero durante el primer trimestre del embarazo fue significativamente menor en 54 mujeres que desarrollaron una DMG incidente en comparación al resto de la cohorte (23 ng/mL vs. 25.4 ng/mL). Después de varios ajustes para los factores de confusión de estatus de la vitamina D y el riesgo de DMG (incluyendo un historial previo de diabetes tipo 2 y DMG), el estudio encontró que cada disminución de 7.5 ng/mL en la concentración de 25-hidroxivitamina D del suero durante el embarazo temprano fue asociada con un riesgo 48% mayor de desarrollar DMG (251). Las bajas concentraciones séricas de 25-hidroxivitamina D (<29,4 ng/mL) durante el segundo trimestre del embarazo también se asociaron con la incidencia de DMG en un estudio anidado de caso y control de 118 mujeres con DMG y 219 sujetos de control pareados (252). Cinco meta-análisis (248, 253-256), incluyendo estudios observacionales de calidad moderada a alta, también informaron que las concentraciones de vitamina D en suero materno durante el embarazo estaban inversamente relacionadas con el riesgo de desarrollar DMG a pesar de la evidencia de sesgo entre los estudios, como el uso de diferentes métodos para la medición de 25-hidroxivitamina D en suero, mediciones realizadas en diferentes trimestres y diferentes criterios para evaluar la DMG (revisado en 257).
Además, evidencia para el papel de la vitamina D en la regulación de la glucosa durante el embarazo fue recientemente reportada en un ensayo aleatorio, doble ciego, controlado con placebo en 54 mujeres embarazadas diagnosticadas con DMG. La suplementación con 50,000 UI de vitamina D3 dos veces durante un período de seis semanas (en e l día 1 y el día 21) resultó en una concentración significativamente menor de glucosa en plasma en ayunas y de insulina en suero, resistencia a la insulina reducida, y sensibilidad a la insulina mejorada en comparación con el placebo (258). Esto sugiere que la deficiencia de vitamina D puede afectar negativamente la tolerancia a la glucosa durante el embarazo y contribuir a la aparición de DMG. Sin embargo, no se han evaluado los beneficios potenciales de los suplementos de vitamina D en la prevención de la intolerancia a la glucosa y la DMG durante el embarazo. Un ensayo aleatorio controlado multicéntrico (DALI) está en curso en Europa para evaluar los efectos de la vitamina D y las intervenciones en el estilo de vida (alimentación saludable y actividad física) sobre el estado metabólico de las mujeres embarazadas con riesgo de DMG (criterios de inclusión: IMC anterior al embarazo ≥ 29 kg/m2) (259). Hallazgos preliminares sugieren que una alimentación saludable y la actividad física pueden ayudar a reducir el aumento de peso gestacional, en comparación con el estándar de atención; sin embargo, es poco probable que estos cambios en el estilo de vida reduzcan el riesgo de DMG entre las mujeres embarazadas obesas (260). Los resultados con respecto al efecto de la suplementación con vitamina D en el estudio DALI aún no se han publicado.
Resultados de salud en la descendencia
Durante el embarazo, el aumento de la absorción intestinal de calcio y la movilización de calcio desde el esqueleto permite la acumulación de calcio dentro del esqueleto fetal. Sin embargo, los estudios basados en la observación que examinaron la relación entre el estado materno de vitamina D y las medidas del crecimiento óseo fetal no han proporcionado resultados consistentes (261, 262). Además, los datos recientes del Estudio de Osteoporosis con Vitamina D Materna (MAVIDOS) sugirieron que no hay diferencia en la densidad mineral ósea (DMO) corporal total de los recién nacidos de madres aleatorizadas a la suplementación diaria con vitamina D3 (1,000 UI) o placebo a partir de <17 semanas gestación hasta el parto (263). Además, el riesgo de fractura en niños daneses de 10 a 18 años fue similar, independientemente de si sus madres estuvieron expuestas a vitamina D adicional por fortificación durante el embarazo (264). Si bien la suplementación materna con vitamina D durante el embarazo previene efectivamente el riesgo de deficiencia de vitamina D en el recién nacido (265), hay poca evidencia de que el estado neonatal de vitamina D influya en el riesgo de fractura más tarde durante la infancia (266).
Algunos estudios observacionales han aportado pruebas bastante débiles en apoyo de una relación entre la suficiencia materna de vitamina D durante el embarazo y la incidencia de afecciones respiratorias y alergias en los niños (267). Un ensayo aleatorio controlado encontró que la suplementación de 108 mujeres embarazadas en el tercer trimestre (en la semana 27 de gestación hasta el parto) con 800 UI/día o una dosis en bolo de 200,000 UI de vitamina D3 no disminuyó el riesgo de sibilancias, rinitis alérgica, diagnóstico de alergia a los alimentos, infecciones del tracto respiratorio menores, o eccema en la descendencia a los tres años de edad en comparación con placebo (N=50) (268). Un ensayo aleatorio controlado doble ciego más reciente encontró que la suplementación con vitamina D3 de 295 mujeres embarazadas danesas, desde las semanas 22 a 26 de gestación hasta el parto, con 2,800 UI/día (70 μg/día) — comparado con 400 UI/día (10 μg/día) de vitamina D3 (es decir, la recomendación actual en Dinamarca) (N=286) — redujo el riesgo de episodios problemáticos de síntomas pulmonares en un 17% en la descendencia durante los primeros tres años de vida (269). Sin embargo, no se informaron diferencias con respecto al riesgo de sibilancias persistentes, asma, sensibilización alérgica, infecciones del tracto respiratorio o eccema entre los grupos de tratamiento y control (269). En un ensayo similar, aleatorizado, doble ciego y controlado- el Vitamin D Antenatal Asthma Reduction Trial — realizado en 777 mujeres embarazadas de EE.UU. con antecedentes de asma, rinitis alérgica o eccema (o cuya pareja tenía ese historial), suplementación con 2,400 UI/día (60 μg/día) o 400 UI/día (10 μg/día) no dieron lugar a diferencias en el riesgo de desarrollar asma o sibilancias recurrentes en sus hijos a la edad de tres años (270). A pesar de la falta de importancia informada en estudios individuales, el análisis agrupado de los tres ensayos encontró una reducción del 19% en el riesgo de sibilancias recurrentes en niños cuyas madres recibieron suplementos de vitamina D en dosis altas versus bajas durante el embarazo (271). En una cohorte de 378 parejas de madres e hijos, las altas concentraciones séricas de 25-hidroxivitamina D medidas en la semana 34 del embarazo se asociaron con un aumento de la alergia alimentaria del niño durante los primeros dos años de vida, lo que garantiza una evaluación cuidadosa de la seguridad de suplementos de vitamina D durante el embarazo (272).
Dado que la insuficiencia de vitamina D se ha relacionado con la autoinmunidad (ver Enfermedades autoinmunes), también se ha propuesto que el mal estatus de la vitamina D materna durante el embarazo puede contribuir a un mayor riesgo de diabetes autoinmune (diabetes mellitus tipo 1 dependiente de insulina) en la descendencia. Sin embargo, los resultados de un estudio de 3,723 niños con alto riesgo genético de diabetes tipo 1 y seguidos durante un período medio de 4.3 años encontró que la ingesta materna de vitamina D (de alimentos y/o suplementos) durante el tercer trimestre del embarazo (evaluada a través de cuestionarios de frecuencia de alimentos) no se asoció con autoinmunidad avanzada de células β o diabetes clínica (142). En un estudio anidado de caso y control, no hubo diferencia en la concentración sérica media de 25-hidroxivitamina D durante el primer trimestre del embarazo entre 343 madres de niños con diabetes tipo 1 y 343 madres control (143). Un estudio de seguimiento sugirió que los polimorfismos específicos del VDR materno, en lugar del estatus de la vitamina D, pueden estar relacionados con una mayor susceptibilidad a desarrollar diabetes tipo 1 en niños (273). Otro estudio anidado de caso y control (119 madres de niños con diabetes tipo 1 y 129 madres control) encontró una asociación inversa entre la concentración materna de la proteína de unión a vitamina D — pero no la concentración de 25-hidroxivitamina D — durante el tercer trimestre del embarazo y el riesgo de diabetes tipo 1 en niños (274). En la actualidad, no existe una causalidad establecida entre el estatus materno de vitamina D durante el embarazo y el riesgo de enfermedad autoinmune en la descendencia.
Infecciones respiratorias agudas
Más de 200 virus son responsables de causar infecciones familiares del tracto respiratorio superior (TRS), conocidas como el resfriado común, resultando en síntomas de congestión y secreción nasal, tos, dolor de garganta, y estornudos (275). El análisis de datos de corte transversal de 18,883 participantes (de 12 años de edad y mayores) de la Tercera Encuesta Nacional de Examinación de Salud y Nutrición de los EE.UU. (NHANES III) reportó una relación inversa entre las concentraciones séricas de 25-hidroxivitamina D o y una reciente (auto-reportada) infección del TRS (ITRS). En comparación a los niveles de vitamina D circulantes iguales a o por encima de los 30 ng/mL, el riesgo de una ITRS fue 24% mayor en los individuos con concentraciones de entre 10 a 29 ng/mL y 36% mayor en aquellos con niveles por debajo de los 10 ng/mL (276). Un análisis de subgrupo indicó que concentraciones séricas bajas de 25-hidroxivitamina D en sujetos con asma y una enfermedad pulmonar obstructiva crónica (EPOC) fueron ligados a una mayor susceptibilidad a una ITRS al compararse con las personas sin una enfermedad pulmonar.
En un ensayo aleatorio, doble ciego, controlado con placebo conducido en 322 adultos saludables (edades ≥18 years) dosis mensuales de vitamina D3 (200,000 UI por los primeros 2 meses y 100,000 UI por los 16 meses siguientes) aumentaron significativamente la concentración sérica promedio de 25-hidroxivitamina D del suero (de 29 ng/mL a 48 ng/mL) en el grupo de intervención pero no disminuyó la ocurrencia de ITRS en comparación con placebo (277). Además, en un ensayo clínico más grande, multicéntrico, de cuatro brazos en 2,259 sujetos (de entre 45-75 años de edad) con un historial de adenoma colorrectal, la suplementación diaria de vitamina D3 de 1,000 UI no redujo el número o la duración de los episodios de ITRS durante el invierno o el resto del año, incluso entre los participantes con las concentraciones séricas más bajas de 25-hidroxivitamina D basales (278). Además, el análisis post-hoc de los datos de un ensayo aleatorio controlado con placebo en 644 individuos (60-84 años de edad) encontró que la suplementación mensual con 30,000 UI o 60,000 UI de vitamina D3 por un período máximo de un año no disminuyó significativamente la tasa de prescripciones de antibióticos para las infecciones bacterianas de las vías respiratorias. El análisis estratificado, sin embargo, encontró que las dosis de 60,000 UI/mes disminuyeron el riesgo del uso de antibióticos en un 47% en participantes de 70 años de edad y mayores (279). Además, al compararse al placebo, la suplementación con vitamina D3 (con 2,000 UI/día) en mujeres embarazadas por 3 meses hasta el nacimiento seguida por la suplementación de sus infantes (800 UI/día) a partir de su nacimiento hasta la edad de 6 meses significativamente disminuyó el número de infección respiratorias agudas después del período de intervención, en niños de 6-18 meses de edad (280). Curiosamente, a pesar de la heterogeneidad significativa entre los ensayos, el análisis agrupado de estos datos con el de 21 ensayos adicionales sugirió una reducción general del 12% en la incidencia de ITRS con vitamina D3 administrada como dosis en bolo (cada semana, mes, o cada tres meses), dosis diarias, o una combinación de bolo y dosis diarias (281). Los análisis de subgrupos revelaron una reducción del 42% en el riesgo de ITRS con la suplementación con vitamina D3 en sujetos con concentraciones séricas basales de 25-hidroxivitamina D <10 ng/mL, mientras que no hubo efecto protector de la vitamina D3 en aquellos con concentraciones ≥10 ng/mL. Además, se encontró que las grandes dosis de bolo (≥30,000 UI) no eran efectivas en comparación con las dosis diarias o semanales, de modo que, en los análisis de subgrupos que excluían las dosis de bolo, los regímenes diarios y semanales de vitamina D3 parecían proteger contra la ITRS, independientemente del estatus basal de la vitamina D. Finalmente, el efecto de la suplementación con vitamina D3 en ITRS no parecía variar con la edad, el IMC, la presencia de asma o EPOC, y el estado de vacunación contra la gripe (281).
Los resultados de un gran ensayo clínico en Nueva Zelanda, el Estudio de Evaluación de Vitamina D (ViDA), confirmaron la ineficacia de grandes dosis de bolo de vitamina D para reducir el riesgo de infecciones respiratorias agudas (282). En este ensayo doble ciego controlado con placebo en 5,110 adultos mayores (de 50-84 años al inicio del estudio), la suplementación con una dosis inicial de 200,000 UI de vitamina D3 seguida de 100,000 UI/mes de vitamina D3 durante una mediana de 3.3 años no tuvo ningún efecto sobre la incidencia de (autoinformado) infecciones respiratorias agudas (HR, 1.01; IC del 95%, 0.94-1.07). Tras la estratificación de los datos por estatus de vitamina D, se observó un resultado nulo similar (HR, 1.08; IC del 95%, 0.95-1.23) en los participantes del estudio con concentraciones de vitamina D en la sangre ≤20 ng/ml (≤50 nmol/L; 282).
El recientemente completado VITAL (Ensayo de la Vitamina D y Omega-3) puede proporcionar evidencia adicional de un efecto de la dosificación diaria (2,000 UI/día de vitamina D) sobre el riesgo de infecciones de las vías respiratorias en adultos mayores, aunque la prevención de enfermedades infecciosas es un resultado secundario en el ensayo (204, 283, 284).
En un ensayo en niños en edad preescolar, la suplementación con vitamina D con 2,000 UI/día durante al menos cuatro meses no disminuyó la incidencia (285) o la gravedad (286) de infecciones del tracto respiratorio en invierno en comparación con la suplementación con 400 UI/día durante al menos cuatro meses. La suplementación con 2,000 UI/día dio lugar a concentraciones séricas medias de 25-hidroxivitamina D significativamente más altas en comparación con la dosis más baja (48.7 ng/mL vs. 36.8 ng/mL), pero la dosis de 400 UI puede haber sido suficiente para prevenir las infecciones de tracto respiratorio en estos niños pequeños (285). En un ensayo controlado con placebo en 1,300 niños y adolescentes sanos en zonas rurales de Vietnam (concentraciones basales de 25-hidroxivitamina D de 26 ng/mL para el grupo control y de intervención), la suplementación con 14,000 UI/semana de vitamina D durante ocho meses redujo significativamente la incidencia de infecciones respiratorias virales no relacionadas con la influenza en un 24%, pero no la incidencia de la infección por influenza A o B (287).
Enfermedad por coronavirus, COVID-19
La enfermedad por coronavirus, COVID-19, es causada por una infección con el severo síndrome respiratorio agudo coronavirus-2 (SARS-CoV-2). Este virus se originó en Wuhan, China a fines de 2019 y se extendió rápidamente por todo el mundo provocando una pandemia global. Síntomas similares a los de la gripe, incluyendo tos, fiebre, fatiga y dificultad para respirar, así como varios otros síntomas diversos caracterizan a COVID-19 (consulte los sitios web de la CDC y la OMS). Los efectos de la enfermedad varían ampliamente, y los más severos resultan en neumonía, dificultad respiratoria, y muerte. Algunas personas infectadas con SARS-CoV-2, sin embargo, son asintomáticas, pero pueden transmitir el virus a otras (288). Además, las personas infectadas con SARS-CoV-2 pueden propagar la enfermedad antes de que experimenten síntomas (es decir, transmisión presintomática) (289), lo que le da importancia al público medidas de salud como lavarse las manos, usar mascarillas, distanciamiento social, y pruebas y rastreo de contactos para reducir la pandemia.
Varios estudios observacionales han examinado la correlación del estado de la vitamina D y la incidencia de COVID-19, y la mayoría — pero no todos (290-292) — han encontrado una deficiencia o insuficiencia de vitamina D asociada con un mayor riesgo de infección por SARS-CoV-2. En un estudio de cohorte retrospectivo de más de 191,000 residentes de los EE.UU. a los que se les administró una prueba de SARS-CoV-2 durante un período de tres meses en la primavera de 2020, la positividad del SARS-CoV-2 se asoció fuertemente con una concentración sérica más baja de 25-hidroxivitamina D, medida a un solo punto en los 12 meses previos a la prueba viral (293). En este estudio, cada incremento de 1 ng/mL de 25-hidroxivitamina D en suero se vinculó con un 1.6% menos de riesgo de positividad para SARS-CoV-2, siendo el riesgo más bajo a concentraciones séricas de 55 ng/mL y superiores (293). En un estudio poblacional retrospectivo entre 7,807 personas en Israel, incluidos 782 casos positivos de SARS-CoV-2, las concentraciones en el plasma de 25-hidroxivitamina D por debajo de 30 ng/mL se asociaron con un 50% más de riesgo de infección por SARS-CoV-2 en comparación con concentraciones más altas de vitamina D (294). La evaluación del estatus de vitamina D en este estudio se realizó antes de la prueba viral, pero faltan detalles sobre el período de tiempo. La relación temporal de la medida del estatus de la vitamina D y la prueba de diagnóstico de COVID también es una preocupación en un análisis de un gran conjunto de datos del Biobanco del Reino Unido, que encontró que el estatus de la vitamina D no estaba asociado con la infección por SARS-CoV-2: el estudio utilizó evaluaciones de estatus de vitamina D realizadas de 10 a 14 años antes de la prueba de diagnóstico COVID-19 (291). De acuerdo con los hallazgos de la mayoría de los estudios disponibles, un pequeño estudio de cohorte retrospectivo (295) y cinco estudios de caso y control (296-300) informaron una asociación inversa del estatus de vitamina D y la infección por SARS-CoV-2. Un meta-análisis de 2021 de 10 estudios observacionales, que incluyeron 361,934 participantes, informó una asociación entre la insuficiencia o deficiencia de vitamina D y un mayor riesgo de COVID-19 (OR, 1.43; IC del 95%: 1.00-2.05), aunque la heterogeneidad entre los estudios fue alta (301). Además, en un análisis de datos del Biobanco del Reino Unido, se descubrió que el uso regular de suplementos de vitamina D estaba asociado con un riesgo 34% menor de infección por SARS-CoV-2 en comparación con los no consumidores de suplementos de vitamina D (292).
Adicionalmente, la mayor parte de la investigación observacional disponible sugiere una asociación entre el estatus bajo de vitamina D y la gravedad de COVID-19, con estudios que encuentran que la deficiencia de vitamina D está relacionada con un mayor riesgo de gravedad de la enfermedad, medido por la necesidad de hospitalización, ingreso a la unidad de cuidados intensivos (302) etapa de la neumonía (en hombres pero no en mujeres; 303), necesidad de ventilación no invasiva (304), necesidad de ventilación mecánica invasiva (305), o una combinación de estos y otros indicadores (297). Otro estudio encontró que el estatus insuficiente de vitamina D, definido como concentraciones de 25-hidroxivitamina D en plasma por debajo de 30 ng/mL, se asoció con una mayor probabilidad de hospitalización relacionada con COVID, pero la asociación no alcanzó significación estadística (p=0,061) excepto en un sub-análisis de personas mayores de 50 años (OR, 2.71; IC 95%: 1.55-4.78; p<0,001) (294). En contraste con la mayor parte de la evidencia hasta la fecha, un estudio de caso y control encontró que la deficiencia de vitamina D no estaba relacionada con la gravedad de COVID-19 (298). Además, algunos estudios han analizado la relación del estatus de vitamina D y la mortalidad por COVID-19, y tres encontraron que la deficiencia de vitamina D se asocia con un mayor riesgo de muerte (303, 305, 306) y dos no encontraron asociación (304, 307), aunque uno de los estudios que no encontró asociación utilizó mediciones del estatus de la vitamina D más de 10 años antes de la prueba de diagnóstico COVID-19 (307).
Dado que existen varios factores de riesgo conocidos de enfermedad grave por COVID-19, incluida la edad avanzada, la obesidad y la diabetes tipo 2 o enfermedad cardiovascular preexistente (308), es importante que los estudios observacionales controlen posibles factores de confusión. Dos estudios cuasiexperimentales informaron que la suplementación en bolo con vitamina D3 (ya sea 50,000 UI por mes o 80,000-100,000 UI cada dos o tres meses) antes o durante la infección por SARS-CoV-2 se asoció con una enfermedad menos grave y una mejor supervivencia en pacientes ancianos frágiles con COVID-19 (309, 310). Actualmente se están llevando a cabo varios ensayos controlados aleatorios de la suplementación con vitamina D en la prevención y el tratamiento de COVID-19; los resultados de estos ensayos informarán sobre la causalidad de la asociación. Sin embargo, los datos actualmente disponibles indican que la mejora del estatus de la vitamina D a través de la suplementación representa un factor de riesgo modificable para COVID-19.
Tratamiento de Enfermedad
Dermatitis atópica
La dermatitis atópica o eccema es particularmente prevalente en países industrializados, afectando de entre el 10%-20% de los niños y de entre el 1%-3% de los adultos. La dermatitis atópica es un desorden inflamatorio crónico de la piel caracterizada por áreas secas y con prurito (picazón) de la piel en sujetos afectados. La inflamación cutánea local y la disfunción inmune pueden dañar la barrera epidérmica e incrementar la susceptibilidad a las infecciones cutáneas y reacciones atópicas en los individuos afectados. La enfermedad es usualmente asociada con otras enfermedades atópicas, incluyendo alergias a los alimentos, el asma, y la rinitis alérgica (311).
Aunque la etiología de la enfermedad no es completamente explicada, la deficiencia de vitamina D puede contribuir a la aparición y/o severidad de la enfermedad (312). Recientemente, utilizando conjuntos de datos a gran escala de personas caucásicas de ascendencia europea, incluido los consorcios del recurso de UK Biobank (313) y el SUNLIGHT (35), el asma de GABRIEL (314), y eccema EAGLE (315), un estudio de aleatorización mendeliano no encontró asociación entre las concentraciones séricas de 25-hidroxivitamina D genéticamente bajas y los riesgos de dermatitis atópica, asma, o altas concentraciones séricas de inmunoglobulina (Ig)-E (316).
Varios estudios aleatorios controlados han examinado si la vitamina D podría ser una herramienta adjunta eficaz en el manejo de la enfermedad, posiblemente mediante la regulación de las reacciones inflamatorias locales y la estimulación de las actividades antimicrobianas en la piel. Además, el efecto beneficioso de la fototerapia observado en casos específicos de dermatitis atópica puede estar parcialmente mediado por la acción de la vitamina D (311). En un pequeño estudio aleatorizado, doble ciego, controlado con placebo en 45 pacientes con dermatitis atópica y bajo nivel de vitamina D (70% de los sujetos tenían concentraciones séricas de 25-hidroxivitamina D <20 ng/mL), administración diaria de 1,600 UI de administración oral de vitamina D3, sola o junto con 600 UI/día de vitamina E, durante un período de 60 días redujo significativamente el alcance y la intensidad del eccema, según lo evaluado por el puntaje SCORAD (SCORing Atopic Dermatitis) (317). La vitamina D3 (1,600 UI/día durante 60 días) también mejoró el estatus de la vitamina D y redujo la gravedad de la enfermedad en 53 pacientes con dermatitis atópica en otro pequeño ensayo aleatorizado (318). Más recientemente, la vitamina D3 (1,000 UI/día durante un mes) mejoró la gravedad de la dermatitis atópica relacionada con el invierno en niños mongoles, como lo demuestran los cambios en el Eczema Area and Severity Index (EASI) y los puntajes de la Investigator’s Global Assessment (IGA) (319). Un meta-análisis de cuatro ensayos pequeños (incluidos los citados anteriormente) confirmó que la vitamina D suplementaria puede conducir a mejoras clínicas medibles en las personas afectadas (320). Se necesitan ensayos más grandes para fortalecer estos hallazgos preliminares y determinar el régimen de suplementación más apropiado y efectivo. Es de notarse que el tratamiento tópico de la psoriasis con análogos de la vitamina D ha sido aprobado por la Administración de Drogas y Alimentos de los EE.UU. (FDA) y puede ser efectivo en el manejo de otros desórdenes cutáneos (321).
Enfermedades inflamatorias intestinales
Se cree que varios factores ambientales y genéticos mal definidos contribuyen al desarrollo de la respuesta inmune inapropiada de la microbiota intestinal que causa la colitis ulcerosa (CU) y la enfermedad de Crohn (EC). Mientras que los polimorfismos específicos del RVD pueden estar ligados a un incremento en la susceptibilidad de desarrollar CU y EC (322), se encontró que las ingestas altas de vitamina D y niveles circulantes pronosticados estaban asociados con una reducción en la incidencia de CU y EC en una cohorte amplia de 72,719 mujeres (323). Un meta-análisis de seis estudios observacionales encontró una asociación inversa entre el estatus de la vitamina D y la gravedad de la EC (324). Tres estudios han investigado si la vitamina D3 podría beneficiar a los pacientes con EC, posiblemente a través de la reducción de la inflamación intestinal. En un estudio multicéntrico, doble ciego, controlado con placebo, la tasa de recaída en pacientes con EC en remisión después de un año de tratamiento fue significativamente menor en aquellos suplementados diariamente con 1,200 UI de vitamina D3 y 1,200 mg de calcio en comparación con aquellos que recibieron calcio por sí solo (13% vs. a 29%) (325). En un segundo estudio piloto, dosis diarias incrementadas de vitamina D3 partiendo de 1,000 UI hasta 5,000 UI, fueron administradas sobre un período de 24 semanas en 18 pacientes con EC a fin de lograr niveles circulantes de 25-hidroxivitamina D >40 ng/mL. Aunque la mitad de los pacientes fallaron en lograr los 40 ng/mL, la concentración media de 25-hidroxivitamina D fue elevado a 45 ng/mL (a partir de un nivel medio basal de 16 ng/mL), y el mejoramiento general del estatus de la vitamina D fue asociado con una disminución significante de la severidad de la enfermedad según la evaluación de los puntajes vía Actividad de la Enfermedad de Crohn (CDAI) (326). En un ensayo de tres meses, aleatorizado, doble ciego, controlado con placebo, en 27 pacientes con EC en remisión, la suplementación diaria con vitamina D3 (2,000 UI) mejoró el estatus de la vitamina D pero no tuvo un efecto significativo sobre la permeabilidad intestinal ('intestino permeable') o medidas de inflamación y actividad de la enfermedad (327). Sin embargo, el estudio sugirió que lograr concentraciones séricas de 25-hidroxivitamina D ≥30 ng/mL podría ayudar a disminuir la inflamación intestinal y mejorar la calidad de vida de los pacientes. Se necesitan estudios adicionales para confirmar la eficacia terapéutica de la vitamina D en las enfermedades inflamatorias intestinales.
Enfermedad cardiovascular
El análisis prospectivo de 41,504 registros médicos electrónicos del estudio Intermountain Heart Collaborative encontró que sólo un tercio de los pacientes tuvieron concentraciones séricas adecuadas de 25-hidroxivitamina D (>30 ng/mL); la insuficiencia de vitamina D (concentraciones séricas de 25-hidroxivitamina D ≤30 ng/mL) fue asociada con un incremento en la prevalencia e incidencia de muchas condiciones cardiovasculares, incluyendo la hipertensión, enfermedad coronaria cardíaca, insuficiencia cardíaca, y accidentes cerebrovasculares (328). El estatus subóptimo de la vitamina D ha sido también ligado a la rigidez arterial y a la disfunción endotelial vascular — fuertes determinantes de la hipertensión incidente y resultados cardiovasculares adversos (329).
Hipertensión
Varios estudios de intervención han evaluado el efecto de la suplementación con vitamina D en la presión sanguínea alta. Un ensayo clínico controlado temprano de 18 hombres y mujeres con hipertensión leve sin tratar; viviendo en Los Países Bajos encontró que la exposición a la radiación UVB tres veces a la semana por seis semanas durante el invierno incrementó las concentraciones séricas de 25-hidroxivitamina D en un 162%, disminuyó los niveles de PTH en un 15%, y disminuyó las medidas de la presión sanguínea diastólica y sistólica ambulatoria de 24 horas en un promedio de 6 mm Hg (330). Un reciente meta-análisis de 16 ensayos controlados aleatorios que involucraron 1,879 participantes, tanto saludables como con condiciones cardiometabólicas preexistentes (incluyendo la hipertensión), no encontró una reducción significante en las presiones sistólica y diastólica sanguíneas con la suplementación de vitamina D (800-8,571 UI/día por cinco semanas a un año). Sin embargo, un análisis de subgrupo de seis ensayos encontró una reducción significativa de 1.31 mm Hg en la presión sanguínea diastólica en individuos con condiciones preexistentes. Si bien se pueden esperar mejoras en la presión sanguínea en casos de insuficiencia/deficiencia de vitamina D, los autores notaron que el estatus subóptimo de vitamina D en los participantes no se observó exclusivamente en aquellos con condiciones cardiometabólicas (331). Por el contrario, en dos estudios de intervención recientes — el ensayo de hipertensión con vitamina D de Estiria (332) y el ensayo de la terapia con Vitamina D en individuos con alto riesgo de hipertensión [DAYLIGHT] (333) — los sujetos con (pre)hipertensión suplementada con vitamina D3 (400 a 4,000 UI/día durante dos a seis meses) no mostraron evidencia de disminución de la presión sanguínea, independientemente de sus estatus de vitamina D basales (insuficientes o adecuados, de acuerdo con los límites actuales de la OIM).
Las condiciones que disminuyen la síntesis de vitamina D en la piel, tales como tener la piel de color oscuro, vivir en latitudes templadas, y el envejecimiento, están asociados con el incremento de la prevalencia de la hipertensión (334), sugiriendo que la vitamina D puede disminuir los niveles de la presión sanguínea en grupos selectos de individuos. En el meta-análisis previamente mencionado, un ensayo de cuatro brazos, doble ciego, controlado con placebo fue conducido en 283 afroamericanos que al azar recibieron suplementos de vitamina D3 diarios de 1,000 UI, 2,000 UI, o 4,000 UI por un período de tres meses. La presión sanguínea sistólica se redujo unos 0.66 mm Hg con 1,000 UI/día, 3.4 mm Hg con 2,000 UI/día y en unos 4 mm Hg con 4,000 UI/día mientras que incrementó 1.7 mm Hg en el grupo de placebo cuando se comparó con los valores basales. Una reducción significante del 0.2 mm Hg en la presión sanguínea sistólica fue detectada por cada incremento de 1 ng/mL en la concentración de 25-hidroxivitamina D. Sin embargo, no hubo diferencia estadística en el cambio de tres meses en la presión sanguínea entre la vitamina D3 y el placebo (335). Otro estudio aleatorizado, controlado con placebo en 150 participantes de edad avanzada (edad media, 77 años) mostró que la suplementación con 100,000 UI de vitamina D3 cada tres meses durante un año no disminuyó significativamente la presión arterial en comparación con el placebo (336).
Se necesita más investigación para determinar si la suplementación con vitamina D es útil en la prevención o el manejo de la hipertensión.
Insuficiencia cardiaca congestiva
La insuficiencia cardíaca congestiva (también llamada insuficiencia cardíaca) es caracterizada por un aumento del ritmo cardíaco y la posterior hipertrofia del ventrículo izquierdo del corazón. La insuficiencia cardiaca está asociada con una fracción de eyección ventricular izquierda (FEVI) reducida, evaluada con un ecocardiograma. Los inhibidores de la enzima convertidora de angiotensina (ECA) (véase Regulación de la presión sanguínea) son actualmente usados como terapia de primera elección en los pacientes con insuficiencia cardíaca. Curiosamente, en un estudio de corte transversal reciente en pacientes saludables sometidos a una angiografía coronaria, las concentraciones séricas insuficientes de 25-hidroxivitamina D de <30 ng/mL fueron asociadas con tasas de flujo coronario más pobres (337). El estatus subóptimo de vitamina D también se ha relacionado con un peor pronóstico en pacientes con insuficiencia cardíaca (338). En los últimos años, varios estudios de intervención han examinado el efecto de la suplementación con vitamina D en personas con insuficiencia cardíaca. En un estudio aleatorio, doble ciego controlado con placebo de 12 semanas, la suplementación diaria con 1,200 UI de vitamina D3 en niños con una insuficiencia cardíaca congestiva crónica llevó a un incremento significativo del estatus de la vitamina D acompañado por una mejora del rendimiento muscular del corazón (FEVI incrementada) como también por niveles más bajos de PTH y citoquinas pro-inflamatorias (339). En otro ensayo aleatorio, doble ciego, controlado con placebo en 64 pacientes de la tercera edad con insuficiencia cardíaca, los participantes que recibieron 800 mg/día de calcio y 50,000 UI/semana de vitamina D3 no desempeñaron significativamente mejor en las tareas de rendimiento físico (usadas como proxy para evaluar la capacidad aeróbica y la fortaleza músculo esquelética) en comparación con aquellos suplementados con calcio solo (340). Un meta-análisis reciente de siete pequeños ensayos controlados aleatorizados con placebo en 573 sujetos con insuficiencia cardíaca mostró que la suplementación con vitamina D (de 1,000 UI/día a 50,000 UI/semana) durante seis semanas a nueve meses podría reducir las concentraciones séricas de PTH, tumor necrosis factor α (TNF-α), y proteína C reactiva (PCR). Sin embargo, no hubo diferencias significativas en la FEVI, la concentración de interleucina-10 (IL-10) circulante, y la concentración de renina entre los pacientes tratados con vitamina D y los que recibieron un placebo (341). Finalmente, en el ensayo EVITA (Efecto de la vitamina D sobre la mortalidad por todas las causas) en pacientes con insuficiencia cardíaca en etapa terminal y estatus inadecuado de la vitamina D (valores basales de 25-hidroxivitamina D en suero, 8.6-19.7 ng/mL), suplementación con 4,000 UI/día durante tres años no redujo el riesgo de mortalidad en comparación con el placebo (342).
Fuentes
Luz solar
La radiación solar ultravioleta-B (UVB; longitudes de onda de 290 a 315 nanómetros) estimula la producción de vitamina D3 en la epidermis de la piel (343). La exposición a la luz solar puede proveer a la mayoría de las personas de su requerimiento entero de vitamina D. Los niños y adultos jóvenes que pasan un tiempo corto en exteriores, dos o tres veces a la semana, generalmente sintetizan toda la vitamina D que necesitan para prevenir una deficiencia. Un estudio reportó que las concentraciones séricas de vitamina D luego de la exposición a una dosis eritemal mínima de luz solar estimulada (la cantidad de luz requerida para causar un leve enrojecimiento de la piel) en todo el cuerpo fue equivalente a ingerir aproximadamente de 10,000 a 25,000 UI de vitamina D (344). Las personas con un color de piel oscuro sintetizan notablemente menos vitamina D con la exposición a luz solar que aquellas con piel de color más claro (34). Adicionalmente, los adultos mayores tienen una capacidad reducida para sintetizar vitamina D a partir de la exposición a la luz solar y frecuentemente utilizan bloqueador solar o ropa protectora con el fin de prevenir el cáncer a la piel y el daño solar. La aplicación de bloqueador solar con un FPS de 10 reduce la producción de vitamina D en un 90% (30). En latitudes cercanas a los 40 grados norte y 40 grados sur (Boston está a 42 grados norte), hay radiación UVB insuficiente disponible para la síntesis de vitamina D desde noviembre hasta principios de marzo. Diez grados más al norte o al sur (Edmonton, Canadá) el "invierno de vitamina D" se extiende desde mediados de octubre a mediados de marzo. Se ha estimado que unos 15 minutos de exposición solar diaria en manos, brazos, y cara, alrededor de las 12 pm a través de todo el año en latitudes de 25 grados (Miami, FL) y durante la primavera, verano, y otoño, en latitudes de 42 grados (Boston, MA), pueden proveer a un individuo de piel clara con 1,000 UI de vitamina D (345).
Fuentes alimenticias
La vitamina D se encuentra de forma natural en solo algunos pocos alimentos como pescados grasos (macarela, salmón, sardinas), aceites de hígado de pescado, huevos de gallinas que han sido alimentadas con vitamina D, y hongos expuestos a la luz solar o radiación UVB. En los EE.UU., la leche y la fórmula infantil son fortificadas con vitamina D para que contenga 400 UI (10 μg) por cuarto. Sin embargo, otros productos lácteos, como el queso y el yogurt, no siempre son fortificados con vitamina D. Algunos cereales, panes, y jugos de frutas, pueden también ser fortificados con vitamina D. Realizar estimados precisos de la ingesta recomendada promedio de vitamina D resulta difícil debido a la alta variabilidad del contenido de vitamina D de los alimentos fortificados (346). El contenido de vitamina D de algunos productos ricos en vitamina D están listados en la Tabla 2 en ambos unidades internacionales (UI) y microgramos (μg). Para obtener más información sobre el contenido de nutrientes de alimentos específicos, revise la base de datos de composición de alimentos de la USDA. El metabolito 25-hidroxivitamina D también está presente en niveles bajos en ciertos alimentos, como carnes, productos lácteos, y huevos (347, 348).
Suplementos
La mayoría de los suplementos de vitamina D disponibles sin una prescripción contienen colecalciferol (vitamina D3). Los suplementos multivitamínicos generalmente contienen 400 UI-1,000 UI (10 μg-25 μg) de vitamina D2 o vitamina D3. Los suplementos de vitamina D de un solo ingrediente pueden aportar entre 400 a 5,000 UI de vitamina D3, pero 400 UI es la dosis más comúnmente disponible (66). Varios suplementos de calcio también pueden proporcionar vitamina D. Un meta-análisis de ensayos aleatorios controlados sugirió que las dosis en bolo de vitamina D2 (ergocalciferol) pueden no ser siempre tan efectivas como la vitamina D3 para elevar las concentraciones séricas de 25-hidroxivitamina D, pero no hay diferencia en eficacia se encontró con la suplementación diaria con vitamina D2 o vitamina D3 (349). Sin embargo, un ensayo aleatorizado, doble ciego, controlado con placebo de 25 semanas, encontró que la suplementación diaria con 1,000 UI de vitamina D3 iniciada al final del verano es más eficaz que la vitamina D2 para mantener las concentraciones de 25-hidroxivitamina D en verano durante los meses de otoño e invierno (350).
Existe un interés creciente en el uso de la forma hidroxilada de colecalciferol, 25-hidroxivitamina D3 (calcidiol; calcifediol), como suplemento. Este metabolito de la vitamina D es sintetizado por el hígado de humanos y animales a partir del colecalciferol producido en la piel o consumido en la dieta, y está presente en algunos alimentos (por ejemplo, carnes, leche, huevos) en niveles bajos (347, 348). En general, los estudios clínicos han encontrado que el calcifediol es de dos a cinco veces más potente para aumentar las concentraciones de 25-hidroxivitamina D en la sangre en comparación con la suplementación con dosis equivalentes de colecalciferol (351-359). Por lo tanto, la suplementación con calcifediol puede representar un medio para mejorar el estatus de la vitamina D de manera rápida y constante con dosis más bajas que el colecalciferol y puede beneficiar a aquellos individuos con condiciones que disminuyen la absorción intestinal del colecalciferol (359). Sin embargo, la forma de 25-hidroxivitamina D3 no está disponible actualmente como suplemento de venta libre en los Estados Unidos.
Seguridad
Toxicidad
La toxicidad por vitamina D (hipervitaminosis D) no ha sido observada como resultado de la exposición solar. La razón es que la exposición solar excesiva genera un cierto número de fotoproductos biológicamente inertes provenientes del 7-dehidrocolesterol y el colecalciferol (3). La toxicidad por vitamina D induce una concentración sérica anormalmente elevada de calcio (hipercalcemia), los que podrían resultar en pérdida de hueso, cálculos renales, y la calcificación de órganos como el corazón y riñones si no es tratada por un largo período de tiempo. Se ha observado hipercalcemia posterior a dosis diarias superiores a 50,000 UI de vitamina D (360). En general, la investigación sugiere que la toxicidad por vitamina D es muy poco probable en personas saludables con niveles de ingesta menores a 10,000 UI/día (361-363). Sin embargo la Junta de Nutrición y Alimentos del Instituto de Medicina estableció conservadoramente un nivel máximo de ingesta tolerable (NM) de 4,000 UI/día (100 μg/día) para todos los adultos (Tabla 3). Ciertas condiciones médicas pueden incrementar el riesgo de hipercalcemia en respuesta a la vitamina D, incluyendo el hiperparatiroidismo primario, la sarcoidosis, tuberculosis, y linfoma (361). Las personas con estas condiciones podrían desarrollar hipercalcemia en respuesta a cualquier aumento en la nutrición de vitamina D y deben consultar a un profesional de la salud calificado con relación a cualquier aumento en la ingesta de vitamina D.
Interacciones con drogas/fármacos
Los siguientes medicamentos no deben tomarse al mismo tiempo que la vitamina D porque pueden disminuir la absorción intestinal de vitamina D: colestiramina (Questran), colestipol (Colestid), orlistat (Xenical), y aceite mineral (364, 365). Los siguientes medicamentos incrementan el metabolismo de la vitamina D y podrían disminuir las concentraciones séricas de 25-hidroxivitamina D: fenitoína (Dilantin), fosfenitoína (Cerebyx), fenobarbital (Luminal), carbamazepina (Tegretol), y rifampicina (Rimactane) (6). La cimetidina, un bloqueador H2 que suprime la secreción de ácido del estómago, inhibe la hidroxilación de la vitamina D en el hígado (366). El tratamiento del reflujo ácido, la enfermedad por reflujo gastroesofágico (ERGE), o las úlceras con inhibidores de la bomba de protones (omeprazol, lansoprazol) pueden interferir con la absorción de calcio y aumentar el riesgo de fracturas, por lo que se recomienda a los pacientes que tomen suplementos de calcio y vitamina D (367). El medicamento antimicótico oral, ketoconazol, inhibe la enzima 25-hidroxivitamina D3-1α-hidroxilasa y se ha encontrado que reduce las concentraciones séricas de 1α, 25-hidroxivitamina D en hombres sanos (368). La Sociedad de Endocrinología también recomienda monitorear el estatus de la vitamina D de pacientes tratados con glucocorticoides y drogas para el tratamiento del VIH ya que estos medicamentos incrementan el catabolismo de la 25-hidroxivitamina D (40). El uso de algunos agentes citostáticos (inhibidores del crecimiento celular) puede también incrementar la degradación de la 25-hidroxivitamina D y 1α,25-hidroxivitamina D en pacientes con cáncer bajo quimioterapia (6). La inducción de hipercalcemia por niveles tóxicos de vitamina D puede precipitar la arritmia cardíaca en pacientes tratados con digoxina (Lanoxin) (366). La hipercalcemia también puede reducir la efectividad del verapamilo (Calan) y el diltiazem (Cardizem) en la fibrilación auricular (366).
Recomendación del Instituto Linus Pauling
El Instituto Linus Pauling recomienda que adultos generalmente sanos tomen 2,000 UI (50 μg) de vitamina D suplementaria diariamente. La mayoría de los multivitamínicos contienen 400 UI (10 μg) de vitamina D, y los suplementos de vitamina D de un solo ingrediente se encuentran disponibles para la suplementación adicional. La exposición al sol, la dieta, el color de piel, y el índice de masa corporal (IMC) tienen un impacto variable y sustancial sobre los niveles corporales de vitamina D. Para ajustarse a las diferencias individuales y asegurar un estatus adecuado de vitamina D corporal, el Instituto Linus Pauling recomienda apuntar hacia una concentración sérica de 25-hidroxivitamina D de por lo menos 30 ng/mL (75 nmol/L). Estudios basados en la observación sugieren que concentraciones séricas de 25-hidroxivitamina D de entre 30 ng/mL y 60 ng/mL están asociadas con riesgos menores de resultados de la salud adversos, incluyendo cánceres y enfermedades autoinmunes.
La Academia Estadounidense de Pediatría actualmente sugiere que todos los bebés, niños, y adolescentes, reciban 400 UI de vitamina D suplementaria diariamente (19). De acuerdo con las recomendaciones de la Sociedad de Endocrinología (40), el Instituto Linus Pauling recomienda ingestas diarias de 400 a 1,000 UI (10 a 25 μg) de vitamina D en infantes y 600 a 1,000 UI (15 a 25 μg) de vitamina D en niños y adolescentes. Dado el contenido promedio de vitamina D de la leche materna, la fórmula infantil, y las dietas de niños y adolescentes, puede ser necesaria la suplementación para cumplir con estas recomendaciones.
Adultos mayores (>50 años)
La suplementación diaria con 2,000 UI (50 μg) de vitamina D es especialmente importante en adultos mayores, debido a que el envejecimiento es asociado con una capacidad reducida de sintetizar vitamina D en la piel luego de su exposición al sol.
Autores y Críticos
Originalmente escrito en Marzo de 2004 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Julio de 2014 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Julio de 2017 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Octubre de 2017 por:
Adrian F. Gombart
Investigador Principal, Instituto Linus Pauling
Profesor Asociado, Departamento de Bioquímica y Biofísica
Universidad Estatal de Oregon
Traducido al Español en 2019 por:
Natsumi Then Shimazaki
Instituto Linus Pauling
Universidad Estatal de Oregon
La actualización de este artículo en el 2017 fue respaldada por una subvención de Pfizer Inc.
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Última actualización 5/6/21 Derechos de autoría 2000-2023 Instituto Linus Pauling
Referencias
1. Holick MF. Vitamin D: importance in the prevention of cancers, type 1 diabetes, heart disease, and osteoporosis. Am J Clin Nutr. 2004;79(3):362-371. (PubMed)
2. Bikle DD. Vitamin D metabolism, mechanism of action, and clinical applications. Chem Biol. 2014;21(3):319-329. (PubMed)
3. Volmer DA, Mendes LR, Stokes CS. Analysis of vitamin D metabolic markers by mass spectrometry: Current techniques, limitations of the "gold standard" method, and anticipated future directions. Mass Spectrom Rev. 2015;34(1):2-23. (PubMed)
4. Holick MF. Vitamin D: A millenium perspective. J Cell Biochem. 2003;88(2):296-307. (PubMed)
5. Sutton AL, MacDonald PN. Vitamin D: more than a "bone-a-fide" hormone. Mol Endocrinol. 2003;17(5):777-791. (PubMed)
6. Grober U, Spitz J, Reichrath J, Kisters K, Holick MF. Vitamin D: Update 2013: From rickets prophylaxis to general preventive healthcare. Dermatoendocrinol. 2013;5(3):331-347. (PubMed)
7. Lieben L, Carmeliet G. The delicate balance between vitamin D, calcium and bone homeostasis: lessons learned from intestinal- and osteocyte-specific VDR null mice. J Steroid Biochem Mol Biol. 2013;136:102-106. (PubMed)
8. Fukumoto S. Phosphate metabolism and vitamin D. Bonekey Rep. 2014;3:497. (PubMed)
9. Lin R, White JH. The pleiotropic actions of vitamin D. Bioessays. 2004;26(1):21-28. (PubMed)
10. Edfeldt K, Liu PT, Chun R, et al. T-cell cytokines differentially control human monocyte antimicrobial responses by regulating vitamin D metabolism. Proc Natl Acad Sci U S A. 2010;107(52):22593-22598. (PubMed)
11. Smolders J, Thewissen M, Damoiseaux J. Control of T cell activation by vitamin D. Nat Immunol. 2011;12(1):3; author reply 3-4. (PubMed)
12. Aranow C. Vitamin D and the immune system. J Investig Med. 2011;59(6):881-886. (PubMed)
13. Zeitz U, Weber K, Soegiarto DW, Wolf E, Balling R, Erben RG. Impaired insulin secretory capacity in mice lacking a functional vitamin D receptor. Faseb J. 2003;17(3):509-511. (PubMed)
14. Bourlon PM, Billaudel B, Faure-Dussert A. Influence of vitamin D3 deficiency and 1,25 dihydroxyvitamin D3 on de novo insulin biosynthesis in the islets of the rat endocrine pancreas. J Endocrinol. 1999;160(1):87-95. (PubMed)
15. Heer M, Egert S. Nutrients other than carbohydrates: their effects on glucose homeostasis in humans. Diabetes Metab Res Rev. 2015;31(1):14-35. (PubMed)
16. Sheng H-W. Sodium, chloride, and potassium. In: Stipanuk M, ed. Biochemical and Physiological Aspects of Human Nutrition. Philadelphia: W.B. Saunders Company; 2000:686-710.
17. Sigmund CD. Regulation of renin expression and blood pressure by vitamin D(3). J Clin Invest. 2002;110(2):155-156. (PubMed)
18. Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110(2):229-238. (PubMed)
19. Wagner CL, Greer FR, American Academy of Pediatrics Section on B, American Academy of Pediatrics Committee on N. Prevention of rickets and vitamin D deficiency in infants, children, and adolescents. Pediatrics. 2008;122(5):1142-1152. (PubMed)
20. Goldacre M, Hall N, Yeates DG. Hospitalisation for children with rickets in England: a historical perspective. Lancet. 2014;383(9917):597-598. (PubMed)
21. Jones AN, Hansen KE. Recognizing the musculoskeletal manifestations of vitamin D deficiency. J Musculoskelet Med. 2009;26(10):389-396. (PubMed)
22. Bringhurst FR, Demay MB, Kronenberg HM. Mineral Metabolism. In: Larson PR, Kronenberg HM, Melmed S, Polonsky KS, eds. Williams Textbook of Endocrinology. 10th ed. Philadelphia: Saunders Book Company; 2003:1317-1320.
23. Plotnikoff GA, Quigley JM. Prevalence of severe hypovitaminosis D in patients with persistent, nonspecific musculoskeletal pain. Mayo Clin Proc. 2003;78(12):1463-1470. (PubMed)
24. Deandrea S, Lucenteforte E, Bravi F, Foschi R, La Vecchia C, Negri E. Risk factors for falls in community-dwelling older people: a systematic review and meta-analysis. Epidemiology. 2010;21(5):658-668. (PubMed)
25. Al-Khalidi B, Kimball SM, Rotondi MA, Ardern CI. Standardized serum 25-hydroxyvitamin D concentrations are inversely associated with cardiometabolic disease in US adults: a cross-sectional analysis of NHANES, 2001-2010. Nutr J. 2017;16(1):16. (PubMed)
26. Cooper JD, Smyth DJ, Walker NM, et al. Inherited variation in vitamin D genes is associated with predisposition to autoimmune disease type 1 diabetes. Diabetes. 2011;60(5):1624-1631. (PubMed)
27. Webb AR, Kline L, Holick MF. Influence of season and latitude on the cutaneous synthesis of vitamin D3: exposure to winter sunlight in Boston and Edmonton will not promote vitamin D3 synthesis in human skin. J Clin Endocrinol Metab. 1988;67(2):373-378. (PubMed)
28. Nichols EK, Khatib IM, Aburto NJ, et al. Vitamin D status and determinants of deficiency among non-pregnant Jordanian women of reproductive age. Eur J Clin Nutr. 2012;66(6):751-756. (PubMed)
29. Bassil D, Rahme M, Hoteit M, Fuleihan Gel H. Hypovitaminosis D in the Middle East and North Africa: prevalence, risk factors and impact on outcomes. Dermatoendocrinol. 2013;5(2):274-298. (PubMed)
30. Balk SJ, Council on Environmental H, Section on D. Ultraviolet radiation: a hazard to children and adolescents. Pediatrics. 2011;127(3):e791-817. (PubMed)
31. Dawodu A, Tsang RC. Maternal vitamin D status: effect on milk vitamin D content and vitamin D status of breastfeeding infants. Adv Nutr. 2012;3(3):353-361. (PubMed)
32. Thiele DK, Senti JL, Anderson CM. Maternal vitamin D supplementation to meet the needs of the breastfed infant: a systematic review. J Hum Lact. 2013;29(2):163-170. (PubMed)
33. Wharton B, Bishop N. Rickets. Lancet. 2003;362(9393):1389-1400. (PubMed)
34. Chen TC, Chimeh F, Lu Z, et al. Factors that influence the cutaneous synthesis and dietary sources of vitamin D. Arch Biochem Biophys. 2007;460(2):213-217. (PubMed)
35. Wang TJ, Zhang F, Richards JB, et al. Common genetic determinants of vitamin D insufficiency: a genome-wide association study. Lancet. 2010;376(9736):180-188. (PubMed)
36. Ahn J, Yu K, Stolzenberg-Solomon R, et al. Genome-wide association study of circulating vitamin D levels. Hum Mol Genet. 2010;19(13):2739-2745. (PubMed)
37. Wang W, Ingles SA, Torres-Mejia G, et al. Genetic variants and non-genetic factors predict circulating vitamin D levels in Hispanic and non-Hispanic White women: the Breast Cancer Health Disparities Study. Int J Mol Epidemiol Genet. 2014;5(1):31-46. (PubMed)
38. Elkum N, Alkayal F, Noronha F, et al. Vitamin D insufficiency in Arabs and South Asians positively associates with polymorphisms in GC and CYP2R1 genes. PLoS One. 2014;9(11):e113102. (PubMed)
39. Zhang Y, Yang S, Liu Y, Ren L. Relationship between polymorphisms in vitamin D metabolism-related genes and the risk of rickets in Han Chinese children. BMC Med Genet. 2013;14:101. (PubMed)
40. Holick MF, Binkley NC, Bischoff-Ferrari HA, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96(7):1911-1930. (PubMed)
41. Harris SS, Soteriades E, Coolidge JA, Mudgal S, Dawson-Hughes B. Vitamin D insufficiency and hyperparathyroidism in a low income, multiracial, elderly population. J Clin Endocrinol Metab. 2000;85(11):4125-4130. (PubMed)
42. Allain TJ, Dhesi J. Hypovitaminosis D in older adults. Gerontology. 2003;49(5):273-278. (PubMed)
43. Doorenbos CR, van den Born J, Navis G, de Borst MH. Possible renoprotection by vitamin D in chronic renal disease: beyond mineral metabolism. Nat Rev Nephrol. 2009;5(12):691-700. (PubMed)
44. Pappa HM, Bern E, Kamin D, Grand RJ. Vitamin D status in gastrointestinal and liver disease. Curr Opin Gastroenterol. 2008;24(2):176-183. (PubMed)
45. Jahnsen J, Falch JA, Mowinckel P, Aadland E. Vitamin D status, parathyroid hormone and bone mineral density in patients with inflammatory bowel disease. Scand J Gastroenterol. 2002;37(2):192-199. (PubMed)
46. Arunabh S, Pollack S, Yeh J, Aloia JF. Body fat content and 25-hydroxyvitamin D levels in healthy women. J Clin Endocrinol Metab. 2003;88(1):157-161. (PubMed)
47. Gallagher JC, Yalamanchili V, Smith LM. The effect of vitamin D supplementation on serum 25(OH)D in thin and obese women. J Steroid Biochem Mol Biol. 2013;136:195-200. (PubMed)
48. Deng X, Song Y, Manson JE, et al. Magnesium, vitamin D status and mortality: results from US National Health and Nutrition Examination Survey (NHANES) 2001 to 2006 and NHANES III. BMC Med. 2013;11:187. (PubMed)
49. Sempos CT, Durazo-Arvizu RA, Binkley N, Jones J, Merkel JM, Carter GD. Developing vitamin D dietary guidelines and the lack of 25-hydroxyvitamin D assay standardization: The ever-present past. J Steroid Biochem Mol Biol. 2016;164:115-119. (PubMed)
50. Chapuy MC, Preziosi P, Maamer M, et al. Prevalence of vitamin D insufficiency in an adult normal population. Osteoporos Int. 1997;7(5):439-443. (PubMed)
51. Thomas MK, Lloyd-Jones DM, Thadhani RI, et al. Hypovitaminosis D in medical inpatients. N Engl J Med. 1998;338(12):777-783. (PubMed)
52. Heaney RP, Dowell MS, Hale CA, Bendich A. Calcium absorption varies within the referencia range for serum 25-hydroxyvitamin D. J Am Coll Nutr. 2003;22(2):142-146. (PubMed)
53. Valcour A, Blocki F, Hawkins DM, Rao SD. Effects of age and serum 25-OH-vitamin D on serum parathyroid hormone levels. J Clin Endocrinol Metab. 2012;97(11):3989-3995. (PubMed)
54. Ginde AA, Wolfe P, Camargo CA, Jr., Schwartz RS. Defining vitamin D status by secondary hyperparathyroidism in the US population. J Endocrinol Invest. 2012;35(1):42-48. (PubMed)
55. Gallagher JC, Yalamanchili V, Smith LM. The effect of vitamin D on calcium absorption in older women. J Clin Endocrinol Metab. 2012;97(10):3550-3556. (PubMed)
56. Looker AC, Johnson CL, Lacher DA, Pfeiffer CM, Schleicher RL, Sempos CT. Vitamin D status: United States, 2001-2006. NCHS Data Brief. 2011(59):1-8. (PubMed)
57. Food and Nutrition Board, Institute of Medicine. Dietary referencia Intakes for Calcium and Vitamin D. Washington, D.C.: The National Academies Press; 2011. (The National Academies Press)
58. Mithal A, Wahl DA, Bonjour JP, et al. Global vitamin D status and determinants of hypovitaminosis D. Osteoporos Int. 2009;20(11):1807-1820. (PubMed)
59. Powe CE, Evans MK, Wenger J, et al. Vitamin D-binding protein and vitamin D status of black Americans and white Americans. N Engl J Med. 2013;369(21):1991-2000. (PubMed)
60. Durazo-Arvizu RA, Dawson-Hughes B, Kramer H, et al. The reverse J-shaped association between serum total 25-hydroxyvitamin D concentration and all-cause mortality: the impact of assay standardization. Am J Epidemiol. 2017;185(8):720-726. (PubMed)
61. Gaksch M, Jorde R, Grimnes G, et al. Vitamin D and mortality: Individual participant data meta-analysis of standardized 25-hydroxyvitamin D in 26916 individuals from a European consortium. PLoS One. 2017;12(2):e0170791. (PubMed)
62. Chowdhury R, Kunutsor S, Vitezova A, et al. Vitamin D and risk of cause specific death: systematic review and meta-analysis of observational cohort and randomised intervention studies. BMJ. 2014;348:g1903. (PubMed)
63. Gupta V, Walia GK, Sachdeva MP. 'Mendelian randomization': an approach for exploring causal relations in epidemiology. Public Health. 2017;145:113-119. (PubMed)
64. Afzal S, Brondum-Jacobsen P, Bojesen SE, Nordestgaard BG. Genetically low vitamin D concentrations and increased mortality: Mendelian randomisation analysis in three large cohorts. BMJ. 2014;349:g6330. (PubMed)
65. Bjelakovic G, Gluud LL, Nikolova D, et al. Vitamin D supplementation for prevention of mortality in adults. Cochrane Database Syst Rev. 2014(1):Cd007470. (PubMed)
66. Wacker M, Holick MF. Vitamin D - effects on skeletal and extraskeletal health and the need for supplementation. Nutrients. 2013;5(1):111-148. (PubMed)
67. Lips P, Hosking D, Lippuner K, et al. The prevalence of vitamin D inadequacy amongst women with osteoporosis: an international epidemiological investigation. J Intern Med. 2006;260(3):245-254. (PubMed)
68. Torbergsen AC, Watne LO, Wyller TB, et al. Vitamin K1 and 25(OH)D are independently and synergistically associated with a risk for hip fracture in an elderly population: A case control study. Clin Nutr. 2015;34(1):101-106. (PubMed)
69. Lips P, van Schoor NM. The effect of vitamin D on bone and osteoporosis. Best Pract Res Clin Endocrinol Metab. 2011;25(4):585-591. (PubMed)
70. Reid IR, Bolland MJ, Grey A. Effects of vitamin D supplements on bone mineral density: a systematic review and meta-analysis. Lancet. 2014;383(9912):146-155. (PubMed)
71. Rosen CJ. Vitamin D supplementation: bones of contention. Lancet. 2014;383(9912):108-110. (PubMed)
72. Mocanu V, Vieth R. Three-year follow-up of serum 25-hydroxyvitamin D, parathyroid hormone, and bone mineral density in nursing home residents who had received 12 months of daily bread fortification with 125 mug of vitamin D3. Nutr J. 2013;12:137. (PubMed)
73. Feskanich D, Willett WC, Colditz GA. Calcium, vitamin D, milk consumption, and hip fractures: a prospective study among postmenopausal women. Am J Clin Nutr. 2003;77(2):504-511. (PubMed)
74. Jackson RD, LaCroix AZ, Gass M, et al. Calcium plus vitamin D supplementation and the risk of fractures. N Engl J Med. 2006;354(7):669-683. (PubMed)
75. Wang Y, Wactawski-Wende J, Sucheston-Campbell LE, et al. The influence of genetic susceptibility and calcium plus vitamin D supplementation on fracture risk. Am J Clin Nutr. 2017;105(4):970-979. (PubMed)
76. Gurney EP, Nachtigall MJ, Nachtigall LE, Naftolin F. The Women's Health Initiative trial and related studies: 10 years later: a clinician's view. J Steroid Biochem Mol Biol. 2014;142:4-11. (PubMed)
77. Grant AM, Avenell A, Campbell MK, et al. Oral vitamin D3 and calcium for secondary prevention of low-trauma fractures in elderly people (Randomised Evaluation of Calcium Or vitamin D, RECORD): a randomised placebo-controlled trial. Lancet. 2005;365(9471):1621-1628. (PubMed)
78. Bischoff-Ferrari HA, Giovannucci E, Willett WC, Dietrich T, Dawson-Hughes B. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84(1):18-28. (PubMed)
79. Khaw KT, Stewart AW, Waayer D, et al. Effect of monthly high-dose vitamin D supplementation on falls and non-vertebral fractures: secondary and post-hoc outcomes from the randomised, double-blind, placebo-controlled ViDA trial. Lancet Diabetes Endocrinol. 2017;5(6):438-447. (PubMed)
80. Chung M, Lee J, Terasawa T, Lau J, Trikalinos TA. Vitamin D with or without calcium supplementation for prevention of cancer and fractures: an updated meta-analysis for the US Preventive Services Task Force. Ann Intern Med. 2011;155(12):827-838. (PubMed)
81. Bischoff-Ferrari HA, Willett WC, Orav EJ, et al. A pooled analysis of vitamin D dose requirements for fracture prevention. N Engl J Med. 2012;367(1):40-49. (PubMed)
82. Avenell A, Mak JC, O'Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev. 2014;4:CD000227. (PubMed)
83. Annweiler C, Beauchet O. Questioning vitamin D status of elderly fallers and nonfallers: a meta-analysis to address a 'forgotten step'. J Intern Med. 2015;277(1):16-44. (PubMed)
84. Cangussu LM, Nahas-Neto J, Orsatti CL, Bueloni-Dias FN, Nahas EA. Effect of vitamin D supplementation alone on muscle function in postmenopausal women: a randomized, double-blind, placebo-controlled clinical trial. Osteoporos Int. 2015;26(10):2413-2421. (PubMed)
85. Cangussu LM, Nahas-Neto J, Orsatti CL, et al. Effect of isolated vitamin D supplementation on the rate of falls and postural balance in postmenopausal women fallers: a randomized, double-blind, placebo-controlled trial. Menopause. 2016;23(3):267-274. (PubMed)
86. Bischoff-Ferrari HA, Dawson-Hughes B, Orav EJ, et al. Monthly high-dose vitamin D treatment for the prevention of functional decline: a randomized clinical trial. JAMA Intern Med. 2016;176(2):175-183. (PubMed)
87. Murad MH, Elamin KB, Abu Elnour NO, et al. Clinical review: The effect of vitamin D on falls: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2011;96(10):2997-3006. (PubMed)
88. Boonen S, Lips P, Bouillon R, Bischoff-Ferrari HA, Vanderschueren D, Haentjens P. Need for additional calcium to reduce the risk of hip fracture with vitamin d supplementation: evidence from a comparative metaanalysis of randomized controlled trials. J Clin Endocrinol Metab. 2007;92(4):1415-1423. (PubMed)
89. Grant WB. Update on evidence that support a role of solar ultraviolet-B irradiance in reducing cancer risk. Anticancer Agents Med Chem. 2013;13(1):140-146. (PubMed)
90. Yin L, Ordonez-Mena JM, Chen T, Schottker B, Arndt V, Brenner H. Circulating 25-hydroxyvitamin D serum concentration and total cancer incidence and mortality: a systematic review and meta-analysis. Prev Med. 2013;57(6):753-764. (PubMed)
91. Gandini S, Gnagnarella P, Serrano D, Pasquali E, Raimondi S. Vitamin D receptor polymorphisms and cancer. Adv Exp Med Biol. 2014;810:69-105. (PubMed)
92. Vaughan-Shaw PG, O'Sullivan F, Farrington SM, et al. The impact of vitamin D pathway genetic variation and circulating 25-hydroxyvitamin D on cancer outcome: systematic review and meta-analysis. Br J Cancer. 2017;116(8):1092-1110. (PubMed)
93. Gombart AF, Luong QT, Koeffler HP. Vitamin D compounds: activity against microbes and cancer. Anticancer Res. 2006;26(4A):2531-2542. (PubMed)
94. Thorne J, Campbell MJ. The vitamin D receptor in cancer. Proc Nutr Soc. 2008;67(2):115-127. (PubMed)
95. Garland CF, Garland FC, Gorham ED. Calcium and vitamin D. Their potential roles in colon and breast cancer prevention. Ann N Y Acad Sci. 1999;889:107-119. (PubMed)
96. Choi YJ, Kim YH, Cho CH, Kim SH, Lee JE. Circulating levels of vitamin D and colorectal adenoma: A case-control study and a meta-analysis. World J Gastroenterol. 2015;21(29):8868-8877. (PubMed)
97. Gandini S, Boniol M, Haukka J, et al. Meta-analysis of observational studies of serum 25-hydroxyvitamin D levels and colorectal, breast and prostate cancer and colorectal adenoma. Int J Cancer. 2011;128(6):1414-1424. (PubMed)
98. Ma Y, Zhang P, Wang F, Yang J, Liu Z, Qin H. Association between vitamin D and risk of colorectal cancer: a systematic review of prospective studies. J Clin Oncol. 2011;29(28):3775-3782. (PubMed)
99. Touvier M, Chan DS, Lau R, et al. Meta-analyses of vitamin D intake, 25-hydroxyvitamin D status, vitamin D receptor polymorphisms, and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev. 2011;20(5):1003-1016. (PubMed)
100. Ekmekcioglu C, Haluza D, Kundi M. 25-Hydroxyvitamin D status and risk for colorectal cancer and type 2 diabetes mellitus: a systematic review and meta-analysis of epidemiological studies. Int J Environ Res Public Health. 2017;14(2). (PubMed)
101. Gorham ED, Garland CF, Garland FC, et al. Optimal vitamin D status for colorectal cancer prevention: a quantitative meta analysis. Am J Prev Med. 2007;32(3):210-216. (PubMed)
102. Cauley JA, Chlebowski RT, Wactawski-Wende J, et al. Calcium plus vitamin D supplementation and health outcomes five years after active intervention ended: the Women's Health Initiative. J Womens Health (Larchmt). 2013;22(11):915-929. (PubMed)
103. Baron JA, Barry EL, Mott LA, et al. A trial of calcium and vitamin D for the prevention of colorectal adenomas. N Engl J Med. 2015;373(16):1519-1530. (PubMed)
104. Holick MF. Calcium plus vitamin D and the risk of colorectal cancer. N Engl J Med. 2006;354(21):2287-2288; author reply 2287-2288. (PubMed)
105. Barry EL, Peacock JL, Rees JR, et al. Vitamin D receptor genotype, vitamin D3 supplementation, and risk of colorectal adenomas: a randomized clinical trial. JAMA Oncol. 2017;3(5):628-635. (PubMed)
106. Hiraki LT, Joshi AD, Ng K, et al. Joint effects of colorectal cancer susceptibility loci, circulating 25-hydroxyvitamin D and risk of colorectal cancer. PLoS One. 2014;9(3):e92212. (PubMed)
107. Vidigal VM, Silva TD, de Oliveira J, Pimenta CAM, Felipe AV, Forones NM. Genetic polymorphisms of vitamin D receptor (VDR), CYP27B1 and CYP24A1 genes and the risk of colorectal cancer. Int J Biol Markers. 2017;32(2):e224-e230. (PubMed)
108. Maalmi H, Ordonez-Mena JM, Schottker B, Brenner H. Serum 25-hydroxyvitamin D levels and survival in colorectal and breast cancer patients: Systematic review and meta-analysis of prospective cohort studies. 2014;50(8):1510-1521. (PubMed)
109. Mohr SB, Garland CF, Gorham ED, Grant WB, Garland FC. Relationship between low ultraviolet B irradiance and higher breast cancer risk in 107 countries. Breast J. 2008;14(3):255-260. (PubMed)
110. John EM, Schwartz GG, Dreon DM, Koo J. Vitamin D and breast cancer risk: the NHANES I Epidemiologic follow-up study, 1971-1975 to 1992. National Health and Nutrition Examination Survey. Cancer Epidemiol Biomarkers Prev. 1999;8(5):399-406. (PubMed)
111. Kim Y, Je Y. Vitamin D intake, blood 25(OH)D levels, and breast cancer risk or mortality: a meta-analysis. Br J Cancer. 2014;110(11):2772-2784. (PubMed)
112. Wang D, Velez de-la-Paz OI, Zhai JX, Liu DW. Serum 25-hydroxyvitamin D and breast cancer risk: a meta-analysis of prospective studies. Tumour Biol. 2013;34(6):3509-3517. (PubMed)
113. Rose AA, Elser C, Ennis M, Goodwin PJ. Blood levels of vitamin D and early stage breast cancer prognosis: a systematic review and meta-analysis. Breast Cancer Res Treat. 2013;141(3):331-339. (PubMed)
114. Sperati F, Vici P, Maugeri-Sacca M, et al. Vitamin D supplementation and breast cancer prevention: a systematic review and meta-analysis of randomized clinical trials. PLoS One. 2013;8(7):e69269. (PubMed)
115. Hu K, Callen DF, Li J, Zheng H. Circulating vitamin D and overall survival in breast cancer patients: a dose-response meta-analysis of cohort studies. Integr Cancer Ther. 2017:1534735417712007. (PubMed)
116. Mohr SB, Gorham ED, Kim J, Hofflich H, Garland CF. Meta-analysis of vitamin D sufficiency for improving survival of patients with breast cancer. Anticancer Res. 2014;34(3):1163-1166. (PubMed)
117. Lu D, Jing L, Zhang S. Vitamin D receptor polymorphism and breast cancer risk: a meta-analysis. Medicine (Baltimore). 2016;95(18):e3535. (PubMed)
118. Mun MJ, Kim TH, Hwang JY, Jang WC. Vitamin D receptor gene polymorphisms and the risk for female reproductive cancers: A meta-analysis. Maturitas. 2015;81(2):256-265. (PubMed)
119. Gilbert R, Martin RM, Beynon R, et al. Associations of circulating and dietary vitamin D with prostate cancer risk: a systematic review and dose-response meta-analysis. Cancer Causes Control. 2011;22(3):319-340. (PubMed)
120. van der Rhee H, Coebergh JW, de Vries E. Is prevention of cancer by sun exposure more than just the effect of vitamin D? A systematic review of epidemiological studies. Eur J Cancer. 2013;49(6):1422-1436. (PubMed)
121. Tuohimaa P, Tenkanen L, Ahonen M, et al. Both high and low levels of blood vitamin D are associated with a higher prostate cancer risk: a longitudinal, nested case-control study in the Nordic countries. Int J Cancer. 2004;108(1):104-108. (PubMed)
122. Xu Y, Shao X, Yao Y, et al. Positive association between circulating 25-hydroxyvitamin D levels and prostate cancer risk: new findings from an updated meta-analysis. J Cancer Res Clin Oncol. 2014;140(9):1465-1477. (PubMed)
123. Grant WB, Karras SN, Bischoff-Ferrari HA, et al. Do studies reporting 'U'-shaped serum 25-hydroxyvitamin D-health outcome relationships reflect adverse effects? Dermatoendocrinol. 2016;8(1):e1187349. (PubMed)
124. Wu X, Cheng J, Yang K. Vitamin D-related gene polymorphisms, plasma 25-hydroxy-vitamin D, cigarette smoke and non-small cell lung cancer (NSCLC) Risk. Int J Mol Sci. 2016;17(10). (PubMed)
125. Zhang L, Wang S, Che X, Li X. Vitamin D and lung cancer risk: a comprehensive review and meta-analysis. Cell Physiol Biochem. 2015;36(1):299-305. (PubMed)
126. Liao Y, Huang JL, Qiu MX, Ma ZW. Impact of serum vitamin D level on risk of bladder cancer: a systemic review and meta-analysis. Tumour Biol. 2015;36(3):1567-1572. (PubMed)
127. Zhang H, Zhang H, Wen X, Zhang Y, Wei X, Liu T. Vitamin D deficiency and increased risk of bladder carcinoma: a meta-analysis. Cell Physiol Biochem. 2015;37(5):1686-1692. (PubMed)
128. Lu D, Chen J, Jin J. Vitamin D status and risk of non-Hodgkin lymphoma: a meta-analysis. Cancer Causes Control. 2014;25(11):1553-1563. (PubMed)
129. Prescott J, Bertrand KA, Poole EM, Rosner BA, Tworoger SS. Surrogates of long-term vitamin d exposure and ovarian cancer risk in two prospective cohort studies. Cancers (Basel). 2013;5(4):1577-1600. (PubMed)
130. Khayatzadeh S, Feizi A, Saneei P, Esmaillzadeh A. Vitamin D intake, serum Vitamin D levels, and risk of gastric cancer: A systematic review and meta-analysis. J Res Med Sci. 2015;20(8):790-796. (PubMed)
131. Gandini S, Raimondi S, Gnagnarella P, Dore JF, Maisonneuve P, Testori A. Vitamin D and skin cancer: a meta-analysis. Eur J Cancer. 2009;45(4):634-641. (PubMed)
132. Deluca HF, Cantorna MT. Vitamin D: its role and uses in immunology. Faseb J. 2001;15(14):2579-2585. (PubMed)
133. Agmon-Levin N, Mosca M, Petri M, Shoenfeld Y. Systemic lupus erythematosus one disease or many? Autoimmun Rev. 2012;11(8):593-595. (PubMed)
134. Goodin DS. The epidemiology of multiple sclerosis: insights to disease pathogenesis. Handb Clin Neurol. 2014;122:231-266. (PubMed)
135. Littorin B, Blom P, Scholin A, et al. Lower levels of plasma 25-hydroxyvitamin D among young adults at diagnosis of autoimmune type 1 diabetes compared with control subjects: results from the nationwide Diabetes Incidence Study in Sweden (DISS). Diabetologia. 2006;49(12):2847-2852. (PubMed)
136. Pozzilli P, Manfrini S, Crino A, et al. Low levels of 25-hydroxyvitamin D3 and 1,25-dihydroxyvitamin D3 in patients with newly diagnosed type 1 diabetes. Horm Metab Res. 2005;37(11):680-683. (PubMed)
137. Raab J, Giannopoulou EZ, Schneider S, et al. Prevalence of vitamin D deficiency in pre-type 1 diabetes and its association with disease progression. Diabetologia. 2014;57(5):902-908. (PubMed)
138. Hypponen E, Laara E, Reunanen A, Jarvelin MR, Virtanen SM. Intake of vitamin D and risk of type 1 diabetes: a birth-cohort study. Lancet. 2001;358(9292):1500-1503. (PubMed)
139. Sorensen IM, Joner G, Jenum PA, Eskild A, Torjesen PA, Stene LC. Maternal serum levels of 25-hydroxy-vitamin D during pregnancy and risk of type 1 diabetes in the offspring. Diabetes. 2012;61(1):175-178. (PubMed)
140. Brekke HK, Ludvigsson J. Vitamin D supplementation and diabetes-related autoimmunity in the ABIS study. Pediatr Diabetes. 2007;8(1):11-14. (PubMed)
141. Fronczak CM, Baron AE, Chase HP, et al. In utero dietary exposures and risk of islet autoimmunity in children. Diabetes Care. 2003;26(12):3237-3242. (PubMed)
142. Marjamaki L, Niinisto S, Kenward MG, et al. Maternal intake of vitamin D during pregnancy and risk of advanced beta cell autoimmunity and type 1 diabetes in offspring. Diabetologia. 2010;53(8):1599-1607. (PubMed)
143. Miettinen ME, Reinert L, Kinnunen L, et al. Serum 25-hydroxyvitamin D level during early pregnancy and type 1 diabetes risk in the offspring. Diabetologia. 2012;55(5):1291-1294. (PubMed)
144. Smolders J, Thewissen M, Peelen E, et al. Vitamin D status is positively correlated with regulatory T cell function in patients with multiple sclerosis. PLoS One. 2009;4(8):e6635. (PubMed)
145. Mokry LE, Ross S, Ahmad OS, et al. Vitamin D and risk of multiple sclerosis: a Mendelian randomization study. PLoS Med. 2015;12(8):e1001866. (PubMed)
146. Staples J, Ponsonby AL, Lim L. Low maternal exposure to ultraviolet radiation in pregnancy, month of birth, and risk of multiple sclerosis in offspring: longitudinal analysis. BMJ. 2010;340:c1640. (PubMed)
147. Bjornevik K, Riise T, Casetta I, et al. Sun exposure and multiple sclerosis risk in Norway and Italy: The EnvIMS study. Mult Scler. 2014; 20(8):1042-1049. (PubMed)
148. McDowell TY, Amr S, Culpepper WJ, et al. Sun exposure, vitamin D and age at disease onset in relapsing multiple sclerosis. Neuroepidemiology. 2011;36(1):39-45. (PubMed)
149. Munger KL, Levin LI, Hollis BW, Howard NS, Ascherio A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA. 2006;296(23):2832-2838. (PubMed)
150. Munger KL, Zhang SM, O'Reilly E, et al. Vitamin D intake and incidence of multiple sclerosis. Neurology. 2004;62(1):60-65. (PubMed)
151. Pierrot-Deseilligny C, Rivaud-Pechoux S, Clerson P, de Paz R, Souberbielle JC. Relationship between 25-OH-D serum level and relapse rate in multiple sclerosis patients before and after vitamin D supplementation. Ther Adv Neurol Disord. 2012;5(4):187-198. (PubMed)
152. Ascherio A, Munger KL, White R, et al. Vitamin d as an early predictor of multiple sclerosis activity and progression. JAMA Neurol. 2014;71(3):306-314. (PubMed)
153. Muris AH, Smolders J, Rolf L, et al. Vitamin D status does not affect disability progression of patients with multiple sclerosis over three year follow-up. PLoS One. 2016;11(6):e0156122. (PubMed)
154. Kampman MT, Steffensen LH, Mellgren SI, Jorgensen L. Effect of vitamin D3 supplementation on relapses, disease progression, and measures of function in persons with multiple sclerosis: exploratory outcomes from a double-blind randomised controlled trial. Mult Scler. 2012;18(8):1144-1151. (PubMed)
155. Soilu-Hanninen M, Aivo J, Lindstrom BM, et al. A randomised, double blind, placebo controlled trial with vitamin D3 as an add on treatment to interferon beta-1b in patients with multiple sclerosis. J Neurol Neurosurg Psychiatry. 2012;83(5):565-571. (PubMed)
156. Mrad MF, El Ayoubi NK, Esmerian MO, Kazan JM, Khoury SJ. Effect of vitamin D replacement on immunological biomarkers in patients with multiple sclerosis. Clin Immunol. 2017;181:9-15. (PubMed)
157. Muris AH, Smolders J, Rolf L, Thewissen M, Hupperts R, Damoiseaux J. Immune regulatory effects of high dose vitamin D3 supplementation in a randomized controlled trial in relapsing remitting multiple sclerosis patients receiving IFNbeta; the SOLARIUM study. J Neuroimmunol. 2016;300:47-56. (PubMed)
158. O'Connell K, Sulaimani J, Basdeo SA, et al. Effects of vitamin D3 in clinically isolated syndrome and healthy control participants: A double-blind randomised controlled trial. Mult Scler J Exp Transl Clin. 2017;3(3):2055217317727296. (PubMed)
159. Rosjo E, Steffensen LH, Jorgensen L, et al. Vitamin D supplementation and systemic inflammation in relapsing-remitting multiple sclerosis. J Neurol. 2015;262(12):2713-2721. (PubMed)
160. Sotirchos ES, Bhargava P, Eckstein C, et al. Safety and immunologic effects of high- vs low-dose cholecalciferol in multiple sclerosis. Neurology. 2016;86(4):382-390. (PubMed)
161. Bruce D, Whitcomb JP, August A, McDowell MA, Cantorna MT. Elevated non-specific immunity and normal Listeria clearance in young and old vitamin D receptor knockout mice. Int Immunol. 2009;21(2):113-122. (PubMed)
162. Zwerina K, Baum W, Axmann R, et al. Vitamin D receptor regulates TNF-mediated arthritis. Ann Rheum Dis. 2011;70(6):1122-1129. (PubMed)
163. Hitchon CA, Sun Y, Robinson DB, et al. Vitamin D receptor polymorphism rs2228570 (Fok1) is associated with rheumatoid arthritis in North American natives. J Rheumatol. 2012;39(9):1792-1797. (PubMed)
164. Lee YH, Bae SC, Choi SJ, Ji JD, Song GG. Associations between vitamin D receptor polymorphisms and susceptibility to rheumatoid arthritis and systemic lupus erythematosus: a meta-analysis. Mol Biol Rep. 2011;38(6):3643-3651. (PubMed)
165. Mosaad YM, Hammad EM, Fawzy Z, et al. Vitamin D receptor gene polymorphism as possible risk factor in rheumatoid arthritis and rheumatoid related osteoporosis. Hum Immunol. 2014;75(5):452-461. (PubMed)
166. Zanetti M, Harris SS, Dawson-Hughes B. Ability of vitamin D to reduce inflammation in adults without acute illness. Nutr Rev. 2014;72(2):95-98. (PubMed)
167. Merlino LA, Curtis J, Mikuls TR, Cerhan JR, Criswell LA, Saag KG. Vitamin D intake is inversely associated with rheumatoid arthritis: results from the Iowa Women's Health Study. Arthritis Rheum. 2004;50(1):72-77. (PubMed)
168. Costenbader KH, Feskanich D, Holmes M, Karlson EW, Benito-Garcia E. Vitamin D intake and risks of systemic lupus erythematosus and rheumatoid arthritis in women. Ann Rheum Dis. 2008;67(4):530-535. (PubMed)
169. Hiraki LT, Munger KL, Costenbader KH, Karlson EW. Dietary intake of vitamin D during adolescence and risk of adult-onset systemic lupus erythematosus and rheumatoid arthritis. Arthritis Care Res (Hoboken). 2012;64(12):1829-1836. (PubMed)
170. Sen D, Ranganathan P. Vitamin D in rheumatoid arthritis: panacea or placebo? Discov Med. 2012;14(78):311-319. (PubMed)
171. Lee YH, Bae SC. Vitamin D level in rheumatoid arthritis and its correlation with the disease activity: a meta-analysis. Clin Exp Rheumatol. 2016;34(5):827-833. (PubMed)
172. Lin J, Liu J, Davies ML, Chen W. Serum vitamin D level and rheumatoid arthritis disease activity: review and meta-analysis. PLoS One. 2016;11(1):e0146351. (PubMed)
173. Hansen KE, Bartels CM, Gangnon RE, Jones AN, Gogineni J. An evaluation of high-dose vitamin D for rheumatoid arthritis. J Clin Rheumatol. 2014;20(2):112-114. (PubMed)
174. Buondonno I, Rovera G, Sassi F, et al. Vitamin D and immunomodulation in early rheumatoid arthritis: A randomized double-blind placebo-controlled study. PLoS One. 2017;12(6):e0178463. (PubMed)
175. Dehghan A, Rahimpour S, Soleymani-Salehabadi H, Owlia MB. Role of vitamin D in flare ups of rheumatoid arthritis. Z Rheumatol. 2014;73(5):461-464. (PubMed)
176. Yang J, Liu L, Zhang Q, Li M, Wang J. Effect of vitamin D on the recurrence rate of rheumatoid arthritis. Exp Ther Med. 2015;10(5):1812-1816. (PubMed)
177. Gonzalez LA, Toloza SM, McGwin G, Jr., Alarcon GS. Ethnicity in systemic lupus erythematosus (SLE): its influence on susceptibility and outcomes. Lupus. 2013;22(12):1214-1224. (PubMed)
178. Hsieh CC, Lin BF. Dietary factors regulate cytokines in murine models of systemic lupus erythematosus. Autoimmun Rev. 2011;11(1):22-27. (PubMed)
179. Mao S, Huang S. Association between vitamin D receptor gene BsmI, FokI, ApaI and TaqI polymorphisms and the risk of systemic lupus erythematosus: a meta-analysis. Rheumatol Int. 2014;34(3):381-388. (PubMed)
180. Monticielo OA, Teixeira Tde M, Chies JA, Brenol JC, Xavier RM. Vitamin D and polymorphisms of VDR gene in patients with systemic lupus erythematosus. Clin Rheumatol. 2012;31(10):1411-1421. (PubMed)
181. Ruiz-Irastorza G, Egurbide MV, Olivares N, Martinez-Berriotxoa A, Aguirre C. Vitamin D deficiency in systemic lupus erythematosus: prevalence, predictors and clinical consequences. Rheumatology (Oxford). 2008;47(6):920-923. (PubMed)
182. Toloza SM, Cole DE, Gladman DD, Ibanez D, Urowitz MB. Vitamin D insufficiency in a large female SLE cohort. Lupus. 2010;19(1):13-19. (PubMed)
183. Amital H, Szekanecz Z, Szucs G, et al. Serum concentrations of 25-OH vitamin D in patients with systemic lupus erythematosus (SLE) are inversely related to disease activity: is it time to routinely supplement patients with SLE with vitamin D? Ann Rheum Dis. 2010;69(6):1155-1157. (PubMed)
184. Terrier B, Derian N, Schoindre Y, et al. Restoration of regulatory and effector T cell balance and B cell homeostasis in systemic lupus erythematosus patients through vitamin D supplementation. Arthritis Res Ther. 2012;14(5):R221. (PubMed)
185. Cutillas-Marco E, Marquina-Vila A, Grant W, Vilata-Corell J, Morales-Suarez-Varela M. Vitamin D and cutaneous lupus erythematosus: effect of vitamin D replacement on disease severity. Lupus. 2014;23(7):615-623. (PubMed)
186. Abou-Raya A, Abou-Raya S, Helmii M. The effect of vitamin D supplementation on inflammatory and hemostatic markers and disease activity in patients with systemic lupus erythematosus: a randomized placebo-controlled trial. J Rheumatol. 2013;40(3):265-272. (PubMed)
187. Lima GL, Paupitz J, Aikawa NE, Takayama L, Bonfa E, Pereira RM. Vitamin D supplementation in adolescents and young adults With juvenile systemic lupus erythematosus for improvement in disease activity and fatigue scores: a randomized, double-blind, placebo-controlled trial. Arthritis Care Res (Hoboken). 2016;68(1):91-98. (PubMed)
188. Andreoli L, Dall'Ara F, Piantoni S, et al. A 24-month prospective study on the efficacy and safety of two different monthly regimens of vitamin D supplementation in pre-menopausal women with systemic lupus erythematosus. Lupus. 2015;24(4-5):499-506. (PubMed)
189. Karimzadeh H, Shirzadi M, Karimifar M. The effect of Vitamin D supplementation in disease activity of systemic lupus erythematosus patients with Vitamin D deficiency: A randomized clinical trial. J Res Med Sci. 2017;22:4. (PubMed)
190. Antico A, Tampoia M, Tozzoli R, Bizzaro N. Can supplementation with vitamin D reduce the risk or modify the course of autoimmune diseases? A systematic review of the literature. Autoimmun Rev. 2012;12(2):127-136. (PubMed)
191. Wang TJ, Pencina MJ, Booth SL, et al. Vitamin D deficiency and risk of cardiovascular disease. Circulation. 2008;117(4):503-511. (PubMed)
192. van Ballegooijen AJ, Kestenbaum B, Sachs MC, et al. Association of 25-Hydroxyvitamin D and parathyroid hormone with incident hypertension: MESA (Multi-Ethnic Study of Atherosclerosis). J Am Coll Cardiol. 2014;63(12):1214-1222. (PubMed)
193. Kunutsor SK, Apekey TA, Steur M. Vitamin D and risk of future hypertension: meta-analysis of 283,537 participants. Eur J Epidemiol. 2013;28(3):205-221. (PubMed)
194. Burgaz A, Orsini N, Larsson SC, Wolk A. Blood 25-hydroxyvitamin D concentration and hypertension: a meta-analysis. J Hypertens. 2011;29(4):636-645. (PubMed)
195. Moody WE, Edwards NC, Madhani M, et al. Endothelial dysfunction and cardiovascular disease in early-stage chronic kidney disease: cause or association? Atherosclerosis. 2012;223(1):86-94. (PubMed)
196. Chitalia N, Recio-Mayoral A, Kaski JC, Banerjee D. Vitamin D deficiency and endothelial dysfunction in non-dialysis chronic kidney disease patients. Atherosclerosis. 2012;220(1):265-268. (PubMed)
197. Chitalia N, Ismail T, Tooth L, et al. Impact of vitamin d supplementation on arterial vasomotion, stiffness and endothelial biomarkers in chronic kidney disease patients. PLoS One. 2014;9(3):e91363. (PubMed)
198. Mazidi M, Karimi E, Rezaie P, Vatanparast H. The impact of vitamin D supplement intake on vascular endothelial function; a systematic review and meta-analysis of randomized controlled trials. Food Nutr Res. 2017;61(1):1273574. (PubMed)
199. Messa P, Curreri M, Regalia A, Alfieri CM. Vitamin D and the cardiovascular system: an overview of the recent literature. Am J Cardiovasc Drugs. 2014;14(1):1-14. (PubMed)
200. Brondum-Jacobsen P, Benn M, Afzal S, Nordestgaard BG. No evidence that genetically reduced 25-hydroxyvitamin D is associated with increased risk of ischaemic heart disease or myocardial infarction: a Mendelian randomization study. Int J Epidemiol. 2015;44(2):651-661. (PubMed)
201. Manousaki D, Mokry LE, Ross S, Goltzman D, Richards JB. Mendelian randomization studies do not support a role for vitamin D in coronary artery disease. Circ Cardiovasc Genet. 2016;9(4):349-356. (PubMed)
202. Ford JA, MacLennan GS, Avenell A, Bolland M, Grey A, Witham M. Cardiovascular disease and vitamin D supplementation: trial analysis, systematic review, and meta-analysis. Am J Clin Nutr. 2014;100(3):746-755. (PubMed)
203. Chin K, Appel LJ, Michos ED. Vitamin D, calcium, and cardiovascular disease: a"D"vantageous or "D"etrimental? An era of uncertainty. Curr Atheroscler Rep. 2017;19(1):5. (PubMed)
204. Pradhan AD, Manson JE. Update on the Vitamin D and OmegA-3 trial (VITAL). J Steroid Biochem Mol Biol. 2016;155(Pt B):252-256. (PubMed)
205. Neale RE, Armstrong BK, Baxter C, et al. The D-Health Trial: A randomized trial of vitamin D for prevention of mortality and cancer. Contemp Clin Trials. 2016;48:83-90. (PubMed)
206. Scragg R, Stewart AW, Waayer D, et al. Effect of monthly high-dose vitamin D supplementation on cardiovascular disease in the vitamin D assessment study: a randomized clinical trial. JAMA Cardiol. 2017;2(6):608-616. (PubMed)
207. Thomas GN, o Hartaigh B, Bosch JA, et al. Vitamin D levels predict all-cause and cardiovascular disease mortality in subjects with the metabolic syndrome: the Ludwigshafen Risk and Cardiovascular Health (LURIC) Study. Diabetes Care. 2012;35(5):1158-1164. (PubMed)
208. Chiu KC, Chu A, Go VL, Saad MF. Hypovitaminosis D is associated with insulin resistance and beta cell dysfunction. Am J Clin Nutr. 2004;79(5):820-825. (PubMed)
209. Shankar A, Sabanayagam C, Kalidindi S. Serum 25-hydroxyvitamin D levels and prediabetes among subjects free of diabetes. Diabetes Care. 2011;34(5):1114-1119. (PubMed)
210. Deleskog A, Hilding A, Brismar K, Hamsten A, Efendic S, Ostenson CG. Low serum 25-hydroxyvitamin D level predicts progression to type 2 diabetes in individuals with prediabetes but not with normal glucose tolerance. Diabetologia. 2012;55(6):1668-1678. (PubMed)
211. Khan H, Kunutsor S, Franco OH, Chowdhury R. Vitamin D, type 2 diabetes and other metabolic outcomes: a systematic review and meta-analysis of prospective studies. Proc Nutr Soc. 2013;72(1):89-97. (PubMed)
212. Lucato P, Solmi M, Maggi S, et al. Low vitamin D levels increase the risk of type 2 diabetes in older adults: A systematic review and meta-analysis. Maturitas. 2017;100:8-15. (PubMed)
213. George PS, Pearson ER, Witham MD. Effect of vitamin D supplementation on glycaemic control and insulin resistance: a systematic review and meta-analysis. Diabet Med. 2012;29(8):e142-150. (PubMed)
214. Gulseth HL, Wium C, Angel K, Eriksen EF, Birkeland KI. Effects of vitamin D supplementation on insulin sensitivity and insulin secretion in subjects with type 2 diabetes and vitamin D deficiency: a randomized controlled trial. Diabetes Care. 2017;40(7):872-878. (PubMed)
215. Lee CJ, Iyer G, Liu Y, et al. The effect of vitamin D supplementation on glucose metabolism in type 2 diabetes mellitus: A systematic review and meta-analysis of intervention studies. J Diabetes Complications. 2017;31(7):1115-1126. (PubMed)
216. Talaei A, Mohamadi M, Adgi Z. The effect of vitamin D on insulin resistance in patients with type 2 diabetes. Diabetol Metab Syndr. 2013;5(1):8. (PubMed)
217. Gezen-Ak D, Yilmazer S, Dursun E. Why vitamin d in Alzheimer's disease? The hypothesis. J Alzheimers Dis. 2014;40(2):257-269. (PubMed)
218. Landel V, Annweiler C, Millet P, Morello M, Feron F. Vitamin D, cognition and Alzheimer's disease: the therapeutic benefit is in the D-tails. J Alzheimers Dis. 2016;53(2):419-444. (PubMed)
219. Annweiler C, Schott AM, Rolland Y, Blain H, Herrmann FR, Beauchet O. Dietary intake of vitamin D and cognition in older women: a large population-based study. Neurology. 2010;75(20):1810-1816. (PubMed)
220. Annweiler C, Rolland Y, Schott AM, et al. Higher vitamin D dietary intake is associated with lower risk of Alzheimer's disease: a 7-year follow-up. J Gerontol A Biol Sci Med Sci. 2012;67(11):1205-1211. (PubMed)
221. Annweiler C, Fantino B, Schott AM, Krolak-Salmon P, Allali G, Beauchet O. Vitamin D insufficiency and mild cognitive impairment: cross-sectional association. Eur J Neurol. 2012;19(7):1023-1029. (PubMed)
222. Hooshmand B, Lokk J, Solomon A, et al. Vitamin D in relation to cognitive impairment, cerebrospinal fluid biomarkers, and brain volumes. J Gerontol A Biol Sci Med Sci. 2014;69(9):1132-1138. (PubMed)
223. Slinin Y, Paudel ML, Taylor BC, et al. 25-Hydroxyvitamin D levels and cognitive performance and decline in elderly men. Neurology. 2010;74(1):33-41. (PubMed)
224. Slinin Y, Paudel M, Taylor BC, et al. Association between serum 25(OH) vitamin D and the risk of cognitive decline in older women. J Gerontol A Biol Sci Med Sci. 2012;67(10):1092-1098. (PubMed)
225. Annweiler C, Milea D, Whitson HE, et al. Vitamin D insufficiency and cognitive impairment in Asians: a multi-ethnic population-based study and meta-analysis. J Intern Med. 2016;280(3):300-311. (PubMed)
226. Annweiler C, Llewellyn DJ, Beauchet O. Low serum vitamin D concentrations in Alzheimer's disease: a systematic review and meta-analysis. J Alzheimers Dis. 2013;33(3):659-674. (PubMed)
227. Balion C, Griffith LE, Strifler L, et al. Vitamin D, cognition, and dementia: a systematic review and meta-analysis. Neurology. 2012;79(13):1397-1405. (PubMed)
228. Lopes da Silva S, Vellas B, Elemans S, et al. Plasma nutrient status of patients with Alzheimer's disease: Systematic review and meta-analysis. Alzheimers Dement. 2014;10(4):485-502. (PubMed)
229. Shen L, Ji HF. Vitamin D deficiency is associated with increased risk of Alzheimer's disease and dementia: evidence from meta-analysis. Nutr J. 2015;14:76. (PubMed)
230. Sommer I, Griebler U, Kien C, et al. Vitamin D deficiency as a risk factor for dementia: a systematic review and meta-analysis. BMC Geriatr. 2017;17(1):16. (PubMed)
231. Olsson E, Byberg L, Karlstrom B, et al. Vitamin D is not associated with incident dementia or cognitive impairment: an 18-y follow-up study in community-living old men. Am J Clin Nutr. 2017;105(4):936-943. (PubMed)
232. Mokry LE, Ross S, Morris JA, Manousaki D, Forgetta V, Richards JB. Genetically decreased vitamin D and risk of Alzheimer disease. Neurology. 2016;87(24):2567-2574. (PubMed)
233. Annweiler C, Montero-Odasso M, Llewellyn DJ, Richard-Devantoy S, Duque G, Beauchet O. Meta-analysis of memory and executive dysfunctions in relation to vitamin D. J Alzheimers Dis. 2013;37(1):147-171. (PubMed)
234. Annweiler C, Fantino B, Gautier J, Beaudenon M, Thiery S, Beauchet O. Cognitive effects of vitamin D supplementation in older outpatients visiting a memory clinic: a pre-post study. J Am Geriatr Soc. 2012;60(4):793-795. (PubMed)
235. Stein MS, Scherer SC, Ladd KS, Harrison LC. A randomized controlled trial of high-dose vitamin D2 followed by intranasal insulin in Alzheimer's disease. J Alzheimers Dis. 2011;26(3):477-484. (PubMed)
236. Annweiler C, Karras SN, Anagnostis P, Beauchet O. Vitamin D supplements: a novel therapeutic approach for Alzheimer patients. Front Pharmacol. 2014;5:6. (PubMed)
237. Sato Y, Kikuyama M, Oizumi K. High prevalence of vitamin D deficiency and reduced bone mass in Parkinson's disease. Neurology. 1997;49(5):1273-1278. (PubMed)
238. Evatt ML, Delong MR, Khazai N, Rosen A, Triche S, Tangpricha V. Prevalence of vitamin D insufficiency in patients with Parkinson disease and Alzheimer disease. Arch Neurol. 2008;65(10):1348-1352. (PubMed)
239. Knekt P, Kilkkinen A, Rissanen H, Marniemi J, Saaksjarvi K, Heliovaara M. Serum vitamin D and the risk of Parkinson disease. Arch Neurol. 2010;67(7):808-811. (PubMed)
240. Lv Z, Qi H, Wang L, et al. Vitamin D status and Parkinson's disease: a systematic review and meta-analysis. Neurol Sci. 2014;35(11):1723-1730. (PubMed)
241. Shen L, Ji HF. Associations between vitamin D status, supplementation, outdoor work and risk of Parkinson's disease: a meta-analysis assessment. Nutrients. 2015;7(6):4817-4827. (PubMed)
242. Zhao Y, Sun Y, Ji HF, Shen L. Vitamin D levels in Alzheimer's and Parkinson's diseases: a meta-analysis. Nutrition. 2013;29(6):828-832. (PubMed)
243. Suzuki M, Yoshioka M, Hashimoto M, et al. Randomized, double-blind, placebo-controlled trial of vitamin D supplementation in Parkinson disease. Am J Clin Nutr. 2013;97(5):1004-1013. (PubMed)
244. Dobson R, Yarnall A, Noyce AJ, Giovannoni G. Bone health in chronic neurological diseases: a focus on multiple sclerosis and parkinsonian syndromes. Pract Neurol. 2013;13(2):70-79. (PubMed)
245. Torsney KM, Noyce AJ, Doherty KM, Bestwick JP, Dobson R, Lees AJ. Bone health in Parkinson's disease: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2014;85(10):1159-66. (PubMed)
246. van den Bos F, Speelman AD, Samson M, Munneke M, Bloem BR, Verhaar HJ. Parkinson's disease and osteoporosis. Age Ageing. 2013;42(2):156-162. (PubMed)
247. Sato Y, Iwamoto J, Honda Y. Amelioration of osteoporosis and hypovitaminosis D by sunlight exposure in Parkinson's disease. Parkinsonism Relat Disord. 2011;17(1):22-26. (PubMed)
248. Aghajafari F, Nagulesapillai T, Ronksley PE, Tough SC, O'Beirne M, Rabi DM. Association between maternal serum 25-hydroxyvitamin D level and pregnancy and neonatal outcomes: systematic review and meta-analysis of observational studies. BMJ. 2013;346:f1169. (PubMed)
249. Perez-Lopez FR, Pasupuleti V, Mezones-Holguin E, et al. Effect of vitamin D supplementation during pregnancy on maternal and neonatal outcomes: a systematic review and meta-analysis of randomized controlled trials. Fertil Steril. 2015;103(5):1278-1288.e1274. (PubMed)
250. Alzaim M, Wood RJ. Vitamin D and gestational diabetes mellitus. Nutr Rev. 2013;71(3):158-167. (PubMed)
251. Lacroix M, Battista MC, Doyon M, et al. Lower vitamin D levels at first trimester are associated with higher risk of developing gestational diabetes mellitus. Acta Diabetol. 2014;51(4):609-616. (PubMed)
252. Parlea L, Bromberg IL, Feig DS, Vieth R, Merman E, Lipscombe LL. Association between serum 25-hydroxyvitamin D in early pregnancy and risk of gestational diabetes mellitus. Diabet Med. 2012;29(7):e25-32. (PubMed)
253. Lu M, Xu Y, Lv L, Zhang M. Association between vitamin D status and the risk of gestational diabetes mellitus: a meta-analysis. Arch Gynecol Obstet. 2016;293(5):959-966. (PubMed)
254. Poel YH, Hummel P, Lips P, Stam F, van der Ploeg T, Simsek S. Vitamin D and gestational diabetes: a systematic review and meta-analysis. Eur J Intern Med. 2012;23(5):465-469. (PubMed)
255. Wei SQ, Qi HP, Luo ZC, Fraser WD. Maternal vitamin D status and adverse pregnancy outcomes: a systematic review and meta-analysis. J Matern Fetal Neonatal Med. 2013;26(9):889-899. (PubMed)
256. Zhang MX, Pan GT, Guo JF, Li BY, Qin LQ, Zhang ZL. Vitamin D deficiency increases the risk of gestational diabetes mellitus: a meta-analysis of observational studies. Nutrients. 2015;7(10):8366-8375. (PubMed)
257. Triunfo S, Lanzone A, Lindqvist PG. Low maternal circulating levels of vitamin D as potential determinant in the development of gestational diabetes mellitus. J Endocrinol Invest. 2017; May 29. doi: 10.1007/s40618-017-0696-9. [Epub ahead of print]. (PubMed)
258. Asemi Z, Hashemi T, Karamali M, Samimi M, Esmaillzadeh A. Effects of vitamin D supplementation on glucose metabolism, lipid concentrations, inflammation, and oxidative stress in gestational diabetes: a double-blind randomized controlled clinical trial. Am J Clin Nutr. 2013;98(6):1425-1432. (PubMed)
259. Jelsma JG, van Poppel MN, Galjaard S, et al. DALI: Vitamin D and lifestyle intervention for gestational diabetes mellitus (GDM) prevention: an European multicentre, randomised trial - study protocol. BMC Pregnancy Childbirth. 2013;13:142. (PubMed)
260. Simmons D, Devlieger R, van Assche A, et al. Effect of Physical Activity and/or Healthy Eating on GDM Risk: The DALI Lifestyle Study. J Clin Endocrinol Metab. 2017;102(3):903-913. (PubMed)
261. Galthen-Sorensen M, Andersen LB, Sperling L, Christesen HT. Maternal 25-hydroxyvitamin D level and fetal bone growth assessed by ultrasound: a systematic review. Ultrasound Obstet Gynecol. 2014;44(6):633-640. (PubMed)
262. Shor DB, Barzel J, Tauber E, Amital H. The effects of maternal vitamin D on neonatal growth parameters. Eur J Pediatr. 2015;174(9):1169-1174. (PubMed)
263. Cooper C, Harvey NC, Bishop NJ, et al. Maternal gestational vitamin D supplementation and offspring bone health (MAVIDOS): a multicentre, double-blind, randomised placebo-controlled trial. Lancet Diabetes Endocrinol. 2016;4(5):393-402. (PubMed)
264. Handel MN, Frederiksen P, Osmond C, Cooper C, Abrahamsen B, Heitmann BL. Prenatal exposure to vitamin D from fortified margarine and risk of fractures in late childhood: period and cohort results from 222 000 subjects in the D-tect observational study. Br J Nutr. 2017;117(6):872-881. (PubMed)
265. Rodda CP, Benson JE, Vincent AJ, Whitehead CL, Polykov A, Vollenhoven B. Maternal vitamin D supplementation during pregnancy prevents vitamin D deficiency in the newborn: an open-label randomized controlled trial. Clin Endocrinol (Oxf). 2015;83(3):363-368. (PubMed)
266. Handel MN, Frederiksen P, Cohen A, Cooper C, Heitmann BL, Abrahamsen B. Neonatal vitamin D status from archived dried blood spots and future risk of fractures in childhood: results from the D-tect study, a population-based case-cohort study. Am J Clin Nutr. 2017;106(1):155-161. (PubMed)
267. Bountouvi E, Douros K, Papadopoulou A. Can getting enough vitamin D during pregnancy reduce the risk of getting asthma in childhood? Front Pediatr. 2017;5:87. (PubMed)
268. Goldring ST, Griffiths CJ, Martineau AR, et al. Prenatal vitamin D supplementation and child respiratory health: a randomised controlled trial. PLoS One. 2013;8(6):e66627. (PubMed)
269. Chawes BL, Bonnelykke K, Stokholm J, et al. Effect of vitamin D3 supplementation during pregnancy on rsk of persistent wheeze in the offspring: a randomized clinical trial. JAMA. 2016;315(4):353-361. (PubMed)
270. Litonjua AA, Carey VJ, Laranjo N, et al. Effect of prenatal supplementation with vitamin D on asthma or eecurrent wheezing in offspring by age 3 years: the VDAART randomized clinical trial. JAMA. 2016;315(4):362-370. (PubMed)
271. Vahdaninia M, Mackenzie H, Helps S, Dean T. Prenatal intake of vitamins and allergic outcomes in the offspring: a systematic review and meta-analysis. J Allergy Clin Immunol Pract. 2017;5(3):771-778.e775. (PubMed)
272. Weisse K, Winkler S, Hirche F, et al. Maternal and newborn vitamin D status and its impact on food allergy development in the German LINA cohort study. Allergy. 2013;68(2):220-228. (PubMed)
273. Miettinen ME, Smart MC, Kinnunen L, et al. Maternal VDR variants rather than 25-hydroxyvitamin D concentration during early pregnancy are associated with type 1 diabetes in the offspring. Diabetologia. 2015;58(10):2278-2283. (PubMed)
274. Sorensen IM, Joner G, Jenum PA, et al. Vitamin D-binding protein and 25-hydroxyvitamin D during pregnancy in mothers whose children later developed type 1 diabetes. Diabetes Metab Res Rev. 2016;32(8):883-890. (PubMed)
275. Makela MJ, Puhakka T, Ruuskanen O, et al. Viruses and bacteria in the etiology of the common cold. J Clin Microbiol. 1998;36(2):539-542. (PubMed)
276. Ginde AA, Mansbach JM, Camargo CA, Jr. Association between serum 25-hydroxyvitamin D level and upper respiratory tract infection in the Third National Health and Nutrition Examination Survey. Arch Intern Med. 2009;169(4):384-390. (PubMed)
277. Murdoch DR, Slow S, Chambers ST, et al. Effect of vitamin D3 supplementation on upper respiratory tract infections in healthy adults: the VIDARIS randomized controlled trial. JAMA. 2012;308(13):1333-1339. (PubMed)
278. Rees JR, Hendricks K, Barry EL, et al. Vitamin D3 supplementation and upper respiratory tract infections in a randomized, controlled trial. Clin Infect Dis. 2013;57(10):1384-1392. (PubMed)
279. Tran B, Armstrong BK, Ebeling PR, et al. Effect of vitamin D supplementation on antibiotic use: a randomized controlled trial. Am J Clin Nutr. 2014;99(1):156-161. (PubMed)
280. Grant CC, Kaur S, Waymouth E, et al. Reduced primary care respiratory infection visits following pregnancy and infancy vitamin D supplementation: a randomised controlled trial. Acta Paediatr. 2015;104(4):396-404. (PubMed)
281. Martineau AR, Jolliffe DA, Hooper RL, et al. Vitamin D supplementation to prevent acute respiratory tract infections: systematic review and meta-analysis of individual participant data. BMJ. 2017;356:i6583. (PubMed)
282. Scragg R, Waayer D, Stewart AW, et al. The Vitamin D Assessment (ViDA) study: design of a randomized controlled trial of vitamin D supplementation for the prevention of cardiovascular disease, acute respiratory infection, falls and non-vertebral fractures. J Steroid Biochem Mol Biol. 2016;164:318-325. (PubMed)
283. Manson JE, Bassuk SS, Buring JE, Group VR. Principal results of the VITamin D and OmegA-3 TriaL (VITAL) and updated meta-analyses of relevant vitamin D trials. J Steroid Biochem Mol Biol. 2020;198:105522. (PubMed)
284. Gold DR, Litonjua AA, Carey VJ, et al. Lung VITAL: Rationale, design, and baseline characteristics of an ancillary study evaluating the effects of vitamin D and/or marine omega-3 fatty acid supplements on acute exacerbations of chronic respiratory disease, asthma control, pneumonia and lung function in adults. Contemp Clin Trials. 2016;47:185-195. (PubMed)
285. Aglipay M, Birken CS, Parkin PC, et al. Effect of high-dose vs standard-dose wintertime vitamin D supplementation on viral upper respiratory tract infections in young healthy children. JAMA. 2017;318(3):245-254. (PubMed)
286. Hueniken K, Aglipay M, Birken CS, et al. Effect of high-dose vitamin D supplementation on upper respiratory tract infection symptom severity in healthy children. Pediatr Infect Dis J. 2019;38(6):564-568. (PubMed)
287. Loeb M, Dang AD, Thiem VD, et al. Effect of Vitamin D supplementation to reduce respiratory infections in children and adolescents in Vietnam: A randomized controlled trial. Influenza Other Respir Viruses. 2019;13(2):176-183. (PubMed)
288. Kronbichler A, Kresse D, Yoon S, Lee KH, Effenberger M, Shin JI. Asymptomatic patients as a source of COVID-19 infections: A systematic review and meta-analysis. Int J Infect Dis. 2020;98:180-186. (PubMed)
289. Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA. 2020;324(8):782-793. (PubMed)
290. Ferrari D, Locatelli M. No significant association between vitamin D and COVID-19. A retrospective study from a northern Italian hospital. Int J Vitam Nutr Res. 2020:1-4. (PubMed)
291. Hastie CE, Mackay DF, Ho F, et al. Vitamin D concentrations and COVID-19 infection in UK Biobank. Diabetes Metab Syndr. 2020;14(4):561-565. (PubMed)
292. Ma H, Zhou T, Heianza Y, Qi L. Habitual use of vitamin D supplements and risk of coronavirus disease 2019 (COVID-19) infection: a prospective study in UK Biobank. Am J Clin Nutr. 2021. [Epub ahead of print] (PubMed)
293. Kaufman HW, Niles JK, Kroll MH, Bi C, Holick MF. SARS-CoV-2 positivity rates associated with circulating 25-hydroxyvitamin D levels. PLoS One. 2020;15(9):e0239252. (PubMed)
294. Merzon E, Tworowski D, Gorohovski A, et al. Low plasma 25(OH) vitamin D level is associated with increased risk of COVID-19 infection: an Israeli population-based study. FEBS J. 2020;287(17):3693-3702. (PubMed)
295. Meltzer DO, Best TJ, Zhang H, Vokes T, Arora V, Solway J. Association of vitamin D status and other clinical characteristics with COVID-19 test results. JAMA Netw Open. 2020;3(9):e2019722. (PubMed)
296. D'Avolio A, Avataneo V, Manca A, et al. 25-Hydroxyvitamin D concentrations are lower in patients with positive PCR for SARS-CoV-2. Nutrients. 2020;12(5):1359. (PubMed)
297. Ye K, Tang F, Liao X, et al. Does serum vitamin D level affect COVID-19 infection and its severity?-a case-control study. J Am Coll Nutr. 2020:1-8. (PubMed)
298. Hernandez JL, Nan D, Fernandez-Ayala M, et al. Vitamin D status in hospitalized patients with SARS-CoV-2 infection. J Clin Endocrinol Metab. 2020. [Epub ahead of print] (PubMed)
299. Im JH, Je YS, Baek J, Chung MH, Kwon HY, Lee JS. Nutritional status of patients with COVID-19. Int J Infect Dis. 2020;100:390-393. (PubMed)
300. Abdollahi A, Kamali Sarvestani H, Rafat Z, et al. The association between the level of serum 25(OH) vitamin D, obesity, and underlying diseases with the risk of developing COVID-19 infection: A case-control study of hospitalized patients in Tehran, Iran. J Med Virol. 2020. [Epub ahead of print] (PubMed)
301. Liu N, Sun J, Wang X, Zhang T, Zhao M, Li H. Low vitamin D status is associated with coronavirus disease 2019 outcomes: a systematic review and meta-analysis. Int J Infect Dis. 2021;104:58-64. (PubMed)
302. Panagiotou G, Tee SA, Ihsan Y, et al. Low serum 25-hydroxyvitamin D (25[OH]D) levels in patients hospitalized with COVID-19 are associated with greater disease severity. Clin Endocrinol (Oxf). 2020;93(4):508-511. (PubMed)
303. De Smet D, De Smet K, Herroelen P, Gryspeerdt S, Martens GA. Serum 25(OH)D level on hospital admission associated with COVID-19 stage and mortality. Am J Clin Pathol. 2020. [Epub ahead of print] (PubMed)
304. Baktash V, Hosack T, Patel N, et al. Vitamin D status and outcomes for hospitalised older patients with COVID-19. Postgrad Med J. 2020. [Epub ahead of print] (PubMed)
305. Radujkovic A, Hippchen T, Tiwari-Heckler S, Dreher S, Boxberger M, Merle U. Vitamin D deficiency and outcome of COVID-19 patients. Nutrients. 2020;12(9): 2757. (PubMed)
306. Carpagnano GE, Di Lecce V, Quaranta VN, et al. Vitamin D deficiency as a predictor of poor prognosis in patients with acute respiratory failure due to COVID-19. J Endocrinol Invest. 2020. [Epub ahead of print] (PubMed)
307. Hastie CE, Pell JP, Sattar N. Vitamin D and COVID-19 infection and mortality in UK Biobank. Eur J Nutr. 2020;60(1):545-548. (PubMed)
308. Centers for Disease Control and Prevention. Assessing Risk Factors for Severe COVID-19 Illness. Available at: https://www.cdc.gov/coronavirus/2019-ncov/covid-data/investigations-discovery/assessing-risk-factors.html. Accessed 1/22/21.
309. Annweiler G, Corvaisier M, Gautier J, et al. Vitamin D supplementation associated to better survival in hospitalized frail elderly COVID-19 patients: the GERIA-COVID quasi-experimental study. Nutrients. 2020;12(11):3377. (PubMed)
310. Annweiler C, Hanotte B, Grandin de l'Eprevier C, Sabatier JM, Lafaie L, Celarier T. Vitamin D and survival in COVID-19 patients: A quasi-experimental study. J Steroid Biochem Mol Biol. 2020;204:105771. (PubMed)
311. Mesquita Kde C, Igreja AC, Costa IM. Atopic dermatitis and vitamin D: facts and controversies. An Bras Dermatol. 2013;88(6):945-953. (PubMed)
312. Lee SA, Hong S, Kim HJ, Lee SH, Yum HY. Correlation between serum vitamin D level and the severity of atopic dermatitis associated with food sensitization. Allergy Asthma Immunol Res. 2013;5(4):207-210. (PubMed)
313. Sudlow C, Gallacher J, Allen N, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12(3):e1001779. (PubMed)
314. Moffatt MF, Gut IG, Demenais F, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363(13):1211-1221. (PubMed)
315. Paternoster L, Zhurov AI, Toma AM, et al. Genome-wide association study of three-dimensional facial morphology identifies a variant in PAX3 associated with nasion position. Am J Hum Genet. 2012;90(3):478-485. (PubMed)
316. Manousaki D, Paternoster L, Standl M, et al. Vitamin D levels and susceptibility to asthma, elevated immunoglobulin E levels, and atopic dermatitis: A Mendelian randomization study. PLoS Med. 2017;14(5):e1002294. (PubMed)
317. Javanbakht MH, Keshavarz SA, Djalali M, et al. Randomized controlled trial using vitamins E and D supplementation in atopic dermatitis. J Dermatolog Treat. 2011;22(3):144-150. (PubMed)
318. Amestejani M, Salehi BS, Vasigh M, et al. Vitamin D supplementation in the treatment of atopic dermatitis: a clinical trial study. J Drugs Dermatol. 2012;11(3):327-330. (PubMed)
319. Camargo CA, Jr., Ganmaa D, Sidbury R, Erdenedelger K, Radnaakhand N, Khandsuren B. Randomized trial of vitamin D supplementation for winter-related atopic dermatitis in children. J Allergy Clin Immunol. 2014;134(4):831-835.e831. (PubMed)
320. Kim G, Bae JH. Vitamin D and atopic dermatitis: A systematic review and meta-analysis. Nutrition. 2016;32(9):913-920. (PubMed)
321. Wat H, Dytoc M. Off-label uses of topical vitamin d in dermatology: a systematic review. J Cutan Med Surg. 2014;18(2):91-108. (PubMed)
322. Xue LN, Xu KQ, Zhang W, Wang Q, Wu J, Wang XY. Associations between vitamin D receptor polymorphisms and susceptibility to ulcerative colitis and Crohn's disease: a meta-analysis. Inflamm Bowel Dis. 2013;19(1):54-60. (PubMed)
323. Ananthakrishnan AN, Khalili H, Higuchi LM, et al. Higher predicted vitamin D status is associated with reduced risk of Crohn's disease. Gastroenterology. 2012;142(3):482-489. (PubMed)
324. Sadeghian M, Saneei P, Siassi F, Esmaillzadeh A. Vitamin D status in relation to Crohn's disease: meta-analysis of observational studies. Nutrition. 2016;32(5):505-514. (PubMed)
325. Jorgensen SP, Agnholt J, Glerup H, et al. Clinical trial: vitamin D3 treatment in Crohn's disease - a randomized double-blind placebo-controlled study. Aliment Pharmacol Ther. 2010;32(3):377-383. (PubMed)
326. Yang L, Weaver V, Smith JP, Bingaman S, Hartman TJ, Cantorna MT. Therapeutic effect of vitamin d supplementation in a pilot study of Crohn's patients. Clin Transl Gastroenterol. 2013;4:e33. (PubMed)
327. Raftery T, Martineau AR, Greiller CL, et al. Effects of vitamin D supplementation on intestinal permeability, cathelicidin and disease markers in Crohn's disease: Results from a randomised double-blind placebo-controlled study. United European Gastroenterol J. 2015;3(3):294-302. (PubMed)
328. Anderson JL, May HT, Horne BD, et al. Relation of vitamin D deficiency to cardiovascular risk factors, disease status, and incident events in a general healthcare population. Am J Cardiol. 2010;106(7):963-968. (PubMed)
329. Al Mheid I, Patel R, Murrow J, et al. Vitamin D status is associated with arterial stiffness and vascular dysfunction in healthy humans. J Am Coll Cardiol. 2011;58(2):186-192. (PubMed)
330. Krause R, Buhring M, Hopfenmuller W, Holick MF, Sharma AM. Ultraviolet B and blood pressure. Lancet. 1998;352(9129):709-710. (PubMed)
331. Kunutsor SK, Burgess S, Munroe PB, Khan H. Vitamin D and high blood pressure: causal association or epiphenomenon? Eur J Epidemiol. 2014;29(1):1-14. (PubMed)
332. Pilz S, Gaksch M, Kienreich K, et al. Effects of vitamin D on blood pressure and cardiovascular risk factors: a randomized controlled trial. Hypertension. 2015;65(6):1195-1201. (PubMed)
333. Arora P, Song Y, Dusek J, et al. Vitamin D therapy in individuals with prehypertension or hypertension: the DAYLIGHT trial. Circulation. 2015;131(3):254-262. (PubMed)
334. Rostand SG. Ultraviolet light may contribute to geographic and racial blood pressure differences. Hypertension. 1997;30(2 Pt 1):150-156. (PubMed)
335. Forman JP, Scott JB, Ng K, et al. Effect of vitamin D supplementation on blood pressure in blacks. Hypertension. 2013;61(4):779-785. (PubMed)
336. Witham MD, Price RJ, Struthers AD, et al. Cholecalciferol treatment to reduce blood pressure in older patients with isolated systolic hypertension: the VitDISH randomized controlled trial. JAMA Intern Med. 2013;173(18):1672-1679. (PubMed)
337. Oz F, Cizgici AY, Oflaz H, et al. Impact of vitamin D insufficiency on the epicardial coronary flow velocity and endothelial function. Coron Artery Dis. 2013;24(5):392-397. (PubMed)
338. Liu LC, Voors AA, van Veldhuisen DJ, et al. Vitamin D status and outcomes in heart failure patients. Eur J Heart Fail. 2011;13(6):619-625. (PubMed)
339. Shedeed SA. Vitamin D supplementation in infants with chronic congestive heart failure. Pediatr Cardiol. 2012;33(5):713-719. (PubMed)
340. Boxer RS, Kenny AM, Schmotzer BJ, Vest M, Fiutem JJ, Pina IL. A randomized controlled trial of high dose vitamin D3 in patients with heart failure. JACC Heart Fail. 2013;1(1):84-90. (PubMed)
341. Jiang WL, Gu HB, Zhang YF, Xia QQ, Qi J, Chen JC. Vitamin D supplementation in the treatment of chronic heart failure: a meta-analysis of randomized controlled trials. Clin Cardiol. 2016;39(1):56-61. (PubMed)
342. Zittermann A, Ernst JB, Prokop S, et al. Effect of vitamin D on all-cause mortality in heart failure (EVITA): a 3-year randomized clinical trial with 4000 IU vitamin D daily. Eur Heart J. 2017;38(29):2279-2286. (PubMed)
343. Norman AW, Henry HH. Vitamin D. In: Bowman BA, Russell RM, eds. Present Knowledge in Nutrition. 9th ed. Washington, D.C.: ILSI Press; 2006:198-210.
344. Holick MF. Vitamin D: the underappreciated D-lightful hormone that is important for skeletal and cellular health. Curr Opin Endocrinol Diabetes. 2002;9:87-98.
345. Terushkin V, Bender A, Psaty EL, Engelsen O, Wang SQ, Halpern AC. Estimated equivalency of vitamin D production from natural sun exposure versus oral vitamin D supplementation across seasons at two US latitudes. J Am Acad Dermatol. 2010;62(6):929 e921-929. (PubMed)
346. Food and Nutrition Board, Institute of Medicine. Vitamin D. Dietary referencia Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington, D.C.: National Academies Press; 1999:250-287. (National Academy Press)
347. Ovesen L, Brot C, Jakobsen J. Food contents and biological activity of 25-hydroxyvitamin D: a vitamin D metabolite to be reckoned with? Ann Nutr Metab. 2003;47(3-4):107-113. (PubMed)
348. Jakobsen J, Christensen T. Natural vitamin D in food: to what degree does 25-hydroxyvitaminn D contribute to the vitamin D activity in food? JMBR Plus. 2020: doi: 10.1002/jbm1004.10453.
349. Tripkovic L, Lambert H, Hart K, et al. Comparison of vitamin D2 and vitamin D3 supplementation in raising serum 25-hydroxyvitamin D status: a systematic review and meta-analysis. Am J Clin Nutr. 2012;95(6):1357-1364. (PubMed)
350. Logan VF, Gray AR, Peddie MC, Harper MJ, Houghton LA. Long-term vitamin D3 supplementation is more effective than vitamin D2 in maintaining serum 25-hydroxyvitamin D status over the winter months. Br J Nutr. 2013;109(6):1082-1088. (PubMed)
351. Barger-Lux MJ, Heaney RP, Dowell S, Chen TC, Holick MF. Vitamin D and its major metabolites: serum levels after graded oral dosing in healthy men. Osteoporos Int. 1998;8(3):222-230. (PubMed)
352. Cashman KD, Seamans KM, Lucey AJ, et al. Relative effectiveness of oral 25-hydroxyvitamin D3 and vitamin D3 in raising wintertime serum 25-hydroxyvitamin D in older adults. Am J Clin Nutr. 2012;95(6):1350-1356. (PubMed)
353. Bischoff-Ferrari HA, Dawson-Hughes B, Stocklin E, et al. Oral supplementation with 25(OH)D3 versus vitamin D3: effects on 25(OH)D levels, lower extremity function, blood pressure, and markers of innate immunity. J Bone Miner Res. 2012;27(1):160-169. (PubMed)
354. Jetter A, Egli A, Dawson-Hughes B, et al. Pharmacokinetics of oral vitamin D(3) and calcifediol. Bone. 2014;59:14-19. (PubMed)
355. Navarro-Valverde C, Sosa-Henriquez M, Alhambra-Exposito MR, Quesada-Gomez JM. Vitamin D3 and calcidiol are not equipotent. J Steroid Biochem Mol Biol. 2016;164:205-208. (PubMed)
356. Shieh A, Ma C, Chun RF, et al. Effects of cholecalciferol vs calcifediol on total and free 25-hydroxyvitamin D and parathyroid hormone. J Clin Endocrinol Metab. 2017;102(4):1133-1140. (PubMed)
357. Vaes AMM, Tieland M, de Regt MF, Wittwer J, van Loon LJC, de Groot L. Dose-response effects of supplementation with calcifediol on serum 25-hydroxyvitamin D status and its metabolites: A randomized controlled trial in older adults. Clin Nutr. 2018;37(3):808-814. (PubMed)
358. Graeff-Armas LA, Bendik I, Kunz I, Schoop R, Hull S, Beck M. Supplemental 25-hydroxycholecalciferol is more effective than cholecalciferol in raising serum 25-hydroxyvitamin D concentrations in older adults. J Nutr. 2020;150(1):73-81. (PubMed)
359. Quesada-Gomez JM, Bouillon R. Is calcifediol better than cholecalciferol for vitamin D supplementation? Osteoporos Int. 2018;29(8):1697-1711. (PubMed)
360. Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266-281. (PubMed)
361. Vieth R. Vitamin D supplementation, 25-hydroxyvitamin D concentrations, and safety. Am J Clin Nutr. 1999;69(5):842-856. (PubMed)
362. Heaney RP, Davies KM, Chen TC, Holick MF, Barger-Lux MJ. Human serum 25-hydroxycholecalciferol response to extended oral dosing with cholecalciferol. Am J Clin Nutr. 2003;77(1):204-210. (PubMed)
363. Vieth R, Chan PC, MacFarlane GD. Efficacy and safety of vitamin D3 intake exceeding the lowest observed adverse effect level. Am J Clin Nutr. 2001;73(2):288-294. (PubMed)
364. Knodel LC, Talbert RL. Adverse effects of hypolipidaemic drugs. Med Toxicol. 1987;2(1):10-32. (PubMed)
365. McDuffie JR, Calis KA, Booth SL, Uwaifo GI, Yanovski JA. Effects of orlistat on fat-soluble vitamins in obese adolescents. Pharmacotherapy. 2002;22(7):814-822. (PubMed)
366. Natural Medicines. Vitamin D. Professional handout/Drug interactions. Available at: https://naturalmedicines.therapeuticresearch.com. Accessed 6/11/17.
367. Panday K, Gona A, Humphrey MB. Medication-induced osteoporosis: screening and treatment strategies. Ther Adv Musculoskelet Dis. 2014;6(5):185-202. (PubMed)
368. Glass AR, Eil C. Ketoconazole-induced reduction in serum 1,25-dihydroxyvitamin D. J Clin Endocrinol Metab. 1986;63(3):766-769. (PubMed)
Vitamina E
Contenido
Resumen
- La vitamina E de origen natural incluye ocho isoformas liposolubles: α-, β-, γ-, y δ-tocoferol y α-, β-, γ-, y δ-tocotrienol. A pesar de ello, el cuerpo preferencialmente usa el α-tocoferol, y solo la suplementación con α-tocoferol puede revertir los síntomas de la deficiencia de vitamina E. (Más información)
- El α-tocoferol funciona como un antioxidante rompe cadenas, previniendo la propagación de radicales libres en membranas y lipoproteínas del plasma. El α-tocoferol es también más propenso a estar involucrado en el fortalecimiento de ciertos aspectos de la inmunidad celular. (Más información)
- La deficiencia de vitamina E puede ser causad por trastornos de malabsorción de grasas o por anormalidades genéticas que afectan el transporte de vitamina E. Los síntomas de una deficiencia severa incluyen ataxia inducida por deficiencia de vitamina E, neuropatía periférica, debilidad muscular, y daño a la retina del ojo. (Más información)
- La ingesta diaria recomendada (IDR) actual es de 15 mg/día de α-tocoferol. Se estima que más del 90% de los adultos americanos no satisfacen el requerimiento estimado promedio (REP) de 12 mg/día de α-tocoferol. (Más información)
- Ensayos aleatorios controlados que investigan la prevención primaria y/o secundaria de las enfermedades crónicas, como las enfermedades cardiovasculares, el cáncer, y las cataratas, actualmente no apoyan un efecto preventivo del α-tocoferol suplementario. (Más información)
- Evidencia clínica limitada sugiere que la suplementación con vitamina E pudiese ser beneficial para el manejo de la degeneración macular relacionada con la edad y enfermedades por hígado graso secundarias a la diabetes mellitus tipo 2. (Más información)
- Se encontró que la suplementación con α-tocoferol retrasa el deterioro cognitivo o perdida de habilidades funcionales en sujetos con deterioro cognitivo en algunos, pero no en todos, los estudios clínicos. (Más información)
- Semillas de plantas, especialmente semillas de girasol, almendras y avellanas, son fuentes ricas en α-tocoferol tanto que muchos aceites vegetales (p. ej., aceite de oliva y aceite de canola) también contienen α-tocoferol. Otras fuentes incluyen tomates, aguacates, espinacas, espárragos, acelgas, y brócoli. (Más información)
- Altas dosis de α-tocoferol suplementario pueden interferir con la cascada de coagulación sanguínea dependiente de la vitamina K e incrementar el riesgo de sangrado en individuos que toman drogas anticoagulantes. Un nivel máximo de ingesta tolerable (NM) para el α-tocoferol en adultos está establecido en 1,000 mg/día y se aplica a todos los posible estereoisómeros del α-tocoferol. (Más información)
El termino vitamina E describe una familia de ocho moléculas liposolubles con actividades antioxidantes: cuatro isoformas del tocoferol (α-, β-, γ-, y δ-tocoferol) y cuatro isoformas del tocotrienol (α-, β-, γ-, y δ-tocotrienol) (Figura 1). Solo una forma, el α-tocoferol, satisface los requerimientos humanos de vitamina E (véase La IDR). En el hígado humano, el α-tocoferol es la forma de la vitamina E que es preferencialmente unida a la proteína de transferencia de α-tocoferol (α-TTP) e incorporada en las lipoproteínas que transportan el α-tocoferol en la sangre para su entrega a los tejidos extrahepáticos. Por lo tanto, es la forma predominante de la vitamina E encontrada en la sangre y los tejidos (1). Además, el α-tocoferol parece ser la forma de la vitamina E con la mayor importancia nutricional, tanto que será el tema principal de la siguiente discusión.
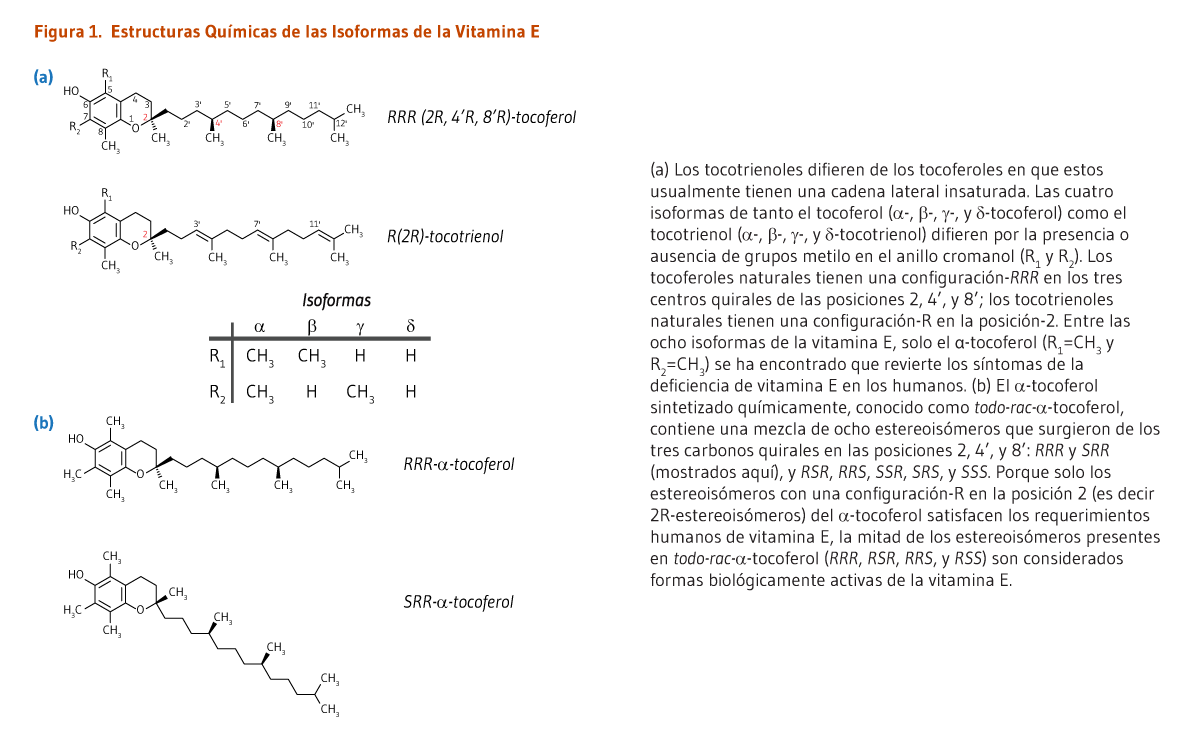
[Figura 1 - Clic para Agrandar]
Función
α-Tocoferol
α-Tocoferol natural versus sintético
El α-tocoferol natural, producido por las plantas encontrado en los alimentos tiene una configuración-RRR en la position-2, 4’, y 8’ de la molécula del α-tocoferol (erróneamente referida como d-α-tocoferol) (véase Figura 1). Sintetizado químicamente el todo-rac-α-tocoferol (todo-racémico-α-tocoferol; incorrectamente etiquetado dl-α-tocoferol) es una mezcla de ocho estereoisómeros del α-tocoferol, la cual se originó a partir de tres carbonos quirales en las posiciones- 2, 4’, y 8’: RRR-, RSR-, RRS-, RSS-, SRR-, SSR-, SRS-, y SSS-α-tocoferol (véase Figura 1). Mientras que todos los estereoisómeros tienen una actividad antioxidante in vitro equivalente, solo las formas en la conformación-R en la posición 2 (nótese 2R) satisfacen los requerimientos de vitamina E en los humanos (2).
Actividad antioxidante
La función principal del α-tocoferol en los humanos es aquella de un antioxidante liposoluble. Las grasas, las cuales son una parte integral de todas las membranas celulares, son vulnerables al daño a través de la peroxidación lipídica por los radicales libres. El α-tocoferol es únicamente adecuado para interceptar los radicales peroxilo y así prevenir una reacción en cadena de la oxidación de los lípidos (Figura 2). Cuando una molécula de α-tocoferol neutraliza un radical libre, esta es oxidada y su capacidad antioxidante se pierde. Otros antioxidantes, como la vitamina C, son capaces de regenerar la capacidad antioxidante del α-tocoferol (Figura 2) (revisado en 1).
Aparte de mantener la integridad de las membranas celulares en todo el cuerpo, el α-tocoferol protege de la oxidación a las grasas en las lipoproteínas de baja densidad (LDL). Las lipoproteínas son partículas compuestas de lípidos y proteínas que transportan grasas a través del torrente sanguíneo. Las LDL transportan específicamente colesterol del hígado a los tejidos del cuerpo. Las LDL oxidadas han sido involucradas en el desarrollo de enfermedades cardiovasculares (3).
Efectos en la inmunidad celular
Otras funciones del α-tocoferol son propensas a ser relacionadas con su capacidad antioxidante (1). Por lo tanto, el α-tocoferol puede proteger las propiedades fisiológicas de las membranas de bicapa lipídica y podría influenciar la actividad de las proteínas y enzimas de la membrana (4). En estudios de cultivo celular, se encontró que el α-tocoferol mejora la formación de una unión adhesiva (conocida como sinapsis inmune) entre los linfocitos T naïve y las células presentadoras de antígeno (CPA), la cual eventualmente estimuló la activación y proliferación de células T (véase Prevención de Enfermedades) (5, 6).

[Figura 2 - Clic para Agrandar]
γ-Tocoferol y tocotrienoles
Las formas de la vitamina E distintas del α-tocoferol también son conocidas por ser potentes antioxidantes. Se piensa que los tocotrienoles y el γ-tocoferol son mejores catadores de radicales peroxilo y de especies reactivas de nitrógeno, respectivamente, que el α-tocoferol (7). A pesar de todo, en el cuerpo, (1) el α-tocoferol es preferencialmente retenido en el hígado por la unión a la proteína de transferencia de α-tocoferol (α-TTP), la cual incorpora α-tocoferol en las lipoproteínas para su entrega en los tejidos extrahepáticos; y (2) las formas de la vitamina E distintas del α-tocoferol son activamente metabolizadas y excretadas. Por lo tanto, mientras el γ-tocoferol es la forma más común de la vitamina E en la dieta americana (8), sus concentraciones en el plasma y tejidos son generalmente más bajas que aquellas del α-tocoferol, y más γ-tocoferol es excretado en la orina que α-tocoferol, sugiriendo que menos γ-tocoferol es necesario para el uso por el cuerpo (1).
Estudios conducidos in vitro y en animales han indicado que el γ-tocoferol y su principal metabolito, γ carboxietil hidroxicroman (γ-CEHC), podrían desempeñar un papel en la protección del cuerpo del daño inducido por radicales libres en varias condiciones de estrés oxidativo e inflamación (revisado en 7). Estudios de intervención limitados (destacados en 7) no han demostrado convincentemente un potencial efecto anti-inflamatorio del γ-tocoferol en humanos. A pesar de todo, en dos estudios aleatorios, controlados con placebo recientes, la suplementación a fumadores con γ-tocoferol potencio beneficios a corto plazo del abandono del hábito de fumar (con o sin terapia de reemplazo de nicotina) en la función endotelial vascular (9, 10).
Numerosos estudios preclínicos han sugerido que los tocotrienoles podrían ser beneficiosos en la prevención de enfermedades crónicas (11). Por lo tanto, los tocotrienoles (especialmente δ-tocotrienol) han mostrado mayores efectos anti-proliferativos y pro-apoptóticos que los tocoferoles en líneas de células malignas (12). Sin embargo, un cierto número de factores, incluyendo dosis, formulación, y tipo de estudio de población, afectan la biodisponibilidad de los tocotrienoles y podrían socavar su eficacia putativa en humanos (13). No existen datos actuales disponibles sobre la eficacia de los tocotrienoles suplementarios en humanos (11).
Interacción con nutrientes
Ácidos grasos dietarios y circulantes
El mecanismo de digestión y absorción de la vitamina E en las células intestinales (enterocitos) no está claro, pero requiere de ácidos biliares y enzimas pancreáticas, y el empaquetado junto con la grasa dietaría en quilomicrones. La eficiencia de la absorción de vitamina E se incrementa con la cantidad de grasa en los alimentos ingeridos, tal que la absorción de vitamina E proveniente de suplementos es más propensa a ser mínima con comidas bajas en grasas (14, 15).
En la circulación, todas las lipoproteínas (es decir, VLDL, LDL, y HDL) están involucradas en el transporte y distribución de α-tocoferol a los tejidos (1). Concentraciones incrementadas de lípidos (colesterol y triglicéridos) en la sangre han sido correlacionadas con concentraciones mayores de α-tocoferol en el suero. Sin embargo, si una alta concentración de lípidos en la sangre está asociada con un recambio más lento de lipoproteínas, entonces la distribución de α-tocoferol a los tejidos podría ser substancialmente alterada (16).
Vitamina C
Unos pocos estudios en humanos que usaron condiciones de estrés oxidativo han demostrado la importancia de la vitamina C (ácido ascórbico) en el reciclaje del α-tocoferol oxidado de regreso a su estado reducido (véase Figura 2). El estrés oxidativo causado por fumar cigarrillos acelera el agotamiento de α-tocoferol del plasma en personas que fuman en comparación con aquellos que no fuman (17). En un ensayo doble ciego, controlado con placebo en 11 fumadores y 13 no fumadores a los cuales se les administro α-tocoferol y γ-tocoferol que fue marcado con deuterio (por lo tanto, rastreable), la suplementación con vitamina C redujo la tasa de perdida de vitamina E en el plasma, muy probablemente por la regeneración de radicales tocoferilo de regreso a formas no oxidadas (18).
Vitamina K
Un estudio en adultos con un estatus de coagulación normal encontró que la suplementación diaria con 1,000 UI (670 mg) de RRR-α-tocoferol por 12 semanas disminuyo la γ-carboxilación de la protrombina, un factor dependiente de la vitamina K en la cascada de coagulación (19). Individuos que toman drogas anticoagulantes como la warfarina y aquellos que están deficientes de vitamina K no deberían tomar suplementos de vitamina E sin supervisión médica debido al riesgo incrementado de sangrado (véase Seguridad) (20).
Deficiencia
Causas
La deficiencia severa de vitamina E raramente ocurre en humanos per ha sido observada como un resultado de la malnutrición (21). La deficiencia severa de vitamina E ha sido asociada con defectos genéticos específicos que afectan el transporte de α-tocoferol por la proteína de transferencia de α-tocoferol (α-TTP) y las lipoproteínas. La deficiencia de vitamina E ha sido también observada en individuos con síndromes de malabsorción de grasas, los cuales perjudican la absorción de grasas dietarías y por consiguiente las vitaminas liposolubles con la vitamina E (véase Interacción con nutrientes) (21).
Síntomas
La deficiencia severa de vitamina E resulta en síntomas neurológicos, incluyendo balance y coordinación deterioradas (ataxia espinocerebelosa), lesión de los nervios sensoriales (neuropatía periférica), debilidad muscular (miopatía) y daño a la retina del ojo (retinopatía). Por esta razón, la gente que desarrolla neuropatía periférica, ataxia, o retinitis pigmentosa (RP) por causas desconocidas debiera ser examinada por deficiencia de vitamina E (21). Los resultados de un ensayo controlado aleatorio en 601 pacientes con formas comunes de RP indicaron que la suplementación diaria con 400 UI de todo-rac-α-tocoferol (180 mg de RRR-α-tocoferol) modestamente pero significantemente incremento la perdida de la función de la retina (22). En contraste, la suplementación diaria con 15,000 UI de vitamina A (4,500 μg de EAR) significantemente alentó la perdida de la función de la retina sobre un periodo de cuatro a seis años, sugiriendo que los pacientes con formas comunes de RP podrían beneficiarse de la suplementación con vitamina A a largo plazo pero deberían evitar altas dosis de vitamina E suplementaria.
Defectos hereditarios en la α-TTP están asociados con un síndrome característico llamando AVED (Ataxia con Deficiencia de Vitamina E). Un reciente estudio de caso reporto que la discapacidad visual en un paciente de edad media con AVED fue causada por tanto la RP como por la aparición temprana de degeneración macular (23). La suplementación con altas dosis de vitamina E (800-1,200 mg/día) es usada para prevenir el deterioro neurológico en sujetos con AVED (21).
Por otra parte, el desarrollo del sistema nervioso parece ser especialmente vulnerable a la deficiencia de vitamina E. Por lo tanto, niños con severa deficiencia de vitamina E al nacer rápidamente experimentan síntomas neurológicos irreversibles si no son tratados con vitamina E. En contraste, individuos que desarrollan desordenes gastrointestinales que afectan la absorción de vitamina E en la adultez podrían no desarrollar síntomas neurológicos por 10-20 años (21). Debe también notarse que los síntomas neurológicos causados por la deficiencia de vitamina E no se han reportado en individuos saludables que consumen dietas bajas en vitamina E.
Deficiencia marginal
Aunque la deficiencia franca de vitamina E es rara, la ingesta marginal de vitamina E es relativamente común. Entre 1988 y 1994, la Encuesta Nacional de Salud y Examen Nutricional III de los EE.UU. (NHANES III) examinó la ingesta recomendada y las concentraciones sanguíneas de α-tocoferol en 16,295 adultos. El estudio reportó que alrededor de un tercio de todos los participantes tuvieron concentraciones sanguíneas de α-tocoferol por debajo de los 20 micromoles/litro (μmol/L) — se eligió este valor de corte debido su asociación con un riesgo incrementado de enfermedades cardiovasculares (24). Datos más recientes de 18,063 participantes en NHANES 2003-2006 indicaron una ingesta dietaría promedio de α-tocoferol de los alimentos (incluyendo fuentes enriquecidas y fortificadas) entre adultos americanos de 7.2 mg/día (25). Esta ingesta se encuentra bien por debajo de la ingesta diaria recomendada actual de 15 mg/día (véase la IDR). En este nivel de ingesta dietaría, más del 93% de los adultos americanos no satisfacen el requerimiento estimado promedio (REP) de 12 mg/día para la vitamina E (25). Además, un reciente estudio de caso y control anidado en mujeres de Bangladesh sugirió que un estatus de vitamina E inadecuado durante el embarazo temprano puede estar asociado con un incremento en el riesgo de aborto (26).
Se piensa que el fumar cigarrillos incrementa la utilización de α-tocoferol tal que los fumadores podrían estar en un riesgo incrementado de deficiencia en comparación con los no fumadores (17). También, el análisis de 19 años de seguimiento del estudio del Alfa-Tocoferol y del Beta-Caroteno para prevención del Cáncer (ATBC) en fumadores mayores de sexo masculino indicó que los participantes en el quintil más elevado frente a aquellos en el nivel más inferior de las concentraciones de α-tocoferol en el suero (>31 μmol/L vs. <23 μmol/L) al inicio del estudio tuvieron riesgos reducidos de mortalidad total y por causa especifica (27).
Se desconoce si la deficiencia margina de vitamina E incrementa el riesgo de enfermedades crónicas (1).
La Ingesta Diaria Recomendada (IDR)
La IDR para la vitamina E fue revisada por última vez por la Junta de Nutrición y Alimentos del Instituto de Medicina de los EE.UU. en el 2000 (Tabla 1) (2). La IDR está basada en gran parte en los resultados de estudios realizados en la década de 1950 en hombres alimentados con dietas deficientes de vitamina E. En un análisis de tubos de ensayo, la vitamina E suprimió la degradación de eritrocitos (conocida como hemólisis) inducida por peróxido de hidrógeno. Debido a que también se ha reportado hemólisis en niños con deficiencia severa de vitamina E, el efecto preventivo de la vitamina E contra la hemolisis inducida por el daño oxidativo se consideró a este análisis in vitro como una prueba clínicamente relevante para evaluar el estado de la vitamina E. Es importante destacar que esto significa que la IDR mas reciente para la vitamina E continúa estando basada en la prevención de síntomas de deficiencia, en vez de la promoción de salud y la prevención de enfermedades crónicas.
Las formas del α-tocoferol que satisfacen las ingestas recomendadas son el RRR-α-tocoferol — la única forma de la vitamina E originada naturalmente — y los tres isómeros sintéticos, RRS-, RSR-, y RSS- α-tocoferol, los cuales se encuentran en suplementos nutricionales y alimentos fortificados.
La Tabla 1 lista la IDR para el α-tocoferol expresado en miligramos (mg) y unidades internacionales (UI).
| Etapa de la Vida | Edad | Machos | Hembras | ||
|---|---|---|---|---|---|
| mg/día | UI/día | mg/día | UI/día | ||
| Infantes (IA) | 0-6 meses |
4
|
6
|
4
|
6
|
| Infantes (IA) | 7-12 meses |
5
|
7.5
|
5
|
7.5
|
| Niños | 1-3 años |
6
|
9
|
6
|
9
|
| Niños | 4-8 años |
7
|
10.5
|
7.5
|
10.5
|
| Niños | 9-13 años |
11
|
16.5
|
11
|
16.5
|
| Adolescentes | 14-18 años |
15
|
22.5
|
15
|
22.5
|
| Adultos | 19 años y más |
15
|
22.5
|
15
|
22.5
|
| Embarazo | Todas las edades |
-
|
-
|
15
|
22.5
|
| Período de lactancia | Todas las edades |
-
|
-
|
19
|
28.5
|
|
*Estas ingestas recomendadas se limitan a las formas 2R- estereoisoméricas del α-tocoferol. |
|||||
Prevención de Enfermedades
Deterioro de la función inmune relacionado a la edad
El decline natural relacionado a la edad de la función inmune está acompañado por un incremento en la susceptibilidad a infecciones, una respuesta más pobre a la inmunización, y riesgos mayores de desarrollar canceres y enfermedades autoinmunes. Se ha demostrado que el α-tocoferol mejora específicamente la respuesta inmune mediada por células T la cual declina con el avance de la edad (revisado en 28). La respuesta deteriorada de las células T ha sido parcialmente asociada con una capacidad reducida de las células T naïve para ser activadas durante la presentación de antígenos y para producir interleucina-2 (IL-2) y proliferar como resultado (6). Sin embargo, muy pocos estudios han abordado la asociación potencial entre el α-tocoferol y la función inmune en humanos (28). En un estudio de intervención de menor escala en adultos mayores (edad promedio, 70 años), la suplementación con 200 mg/día de todo-rac-α-tocoferol (equivalente a 100 mg de RRR-α-tocoferol) por tres meses significantemente mejoró la actividad citotóxica de las células asesinas naturales (NK), la quimiotaxis de neutrófilos, la respuesta fagocítica, y mejoró la proliferación de linfocitos inducida por mitógenos y la producción de interleucina-2 (IL-2) en comparación al inicio del estudio (29). En un ensayo más temprano, la suplementación diaria de adultos mayores sanos (≥65 años de edad) con 200 mg de todo-rac-α-tocoferol por 235 días también mejoró la inmunidad mediada por los linfocitos T — medida con una prueba de piel de reacción de hipersensibilidad retardada (DTH) — e incremento la producción de anticuerpos en respuesta a las vacunas contra la hepatitis B y el tétanos (30).
Dosis menores de α-tocoferol fallaron en mejorar la respuesta de DTH en comparación al placebo en otro estudio en participantes saludables (edades, de 65-80 años) (31). Un ensayo aleatorio, controlado con placebo en 617 residentes de asilos de ancianos (≥65 años de edad) reportó que la suplementación con 200 UI de α-tocoferol sintético (90 mg de RRR-α-tocoferol) por un año significantemente disminuyo el riesgo de contraer infecciones de las vías respiratorias superiores, especialmente el resfriado común, pero no tuvo efecto en las infecciones de las vías respiratorias inferiores (pulmones) (32). Mas investigación es necesaria para examinar si la vitamina E suplementaria podría mejorar la función inmune y reducir el riesgo de infecciones en adultos mayores.
Enfermedades cardiovasculares
Prevención primaria: en adultos sanos
Estudios basados en la observación: Los resultados de varios estudios basados en la observación de gran escala en hombres y mujeres han sugerido una relación inversa entre el consumo de vitamina E y el riesgo de infarto al miocardio o muerte por enfermedades cardiacas. Cada estudio tuvo un diseño prospectivo que midió la ingesta de vitamina E en personas generalmente sanas a las cuales se les dio un seguimiento sobre un periodo de tiempo para determinar la aparición de eventos cardiovasculares y analizar la asociación entre la exposición y el (los) resultado(s). En dos de los estudios, los individuos que consumían más de 7 mg/día de α-tocoferol dietario eran aproximadamente 35% menos propensos a morir de una enfermedad cardiaca que aquellos que consumían menos de 3-5 mg/día de α-tocoferol (33, 34). Otros dos estudios de gran escala observaron un riesgo significativamente reducido de una enfermedad cardiaca solo en mujeres y hombres que consumían al menos 100 UI (67 mg)/día de RRR-α-tocoferol (35, 36).
Estudios de intervención: Un ensayo de intervención, aleatorio controlado con placebo en 39,876 mujeres (≥45 años de edad) participantes del Estudio de Salud de las Mujeres (Women's Health Study [WHS]) encontró que la suplementación con 600 UI (400 mg) de RRR-α-tocoferol cada dos días por 10 años resulto en una reducción del 34% de infarto al miocardio no-fatal y una reducción del 49% en muertes relacionadas a lo cardiovascular pero solo en mujeres con edades de por lo menos 65 años al inicio del estudio (representando 10% de los participantes del estudio) (37). Análisis posteriores de los datos del WHS mostraron que las mujeres en el brazo de la vitamina E del estudio experimentaron una reducción del 21% del riesgo de tromboembolismo venoso (TEV) en comparación al placebo: la reducción fue del 12% en mujeres menores de 55 años de edad, 26% en mujeres de 65 años de edad y mayores, y 44% en mujeres con un historial de TEV. Otro ensayo controlado aleatorio de gran escala — el Physicians’ Health Study II (PHSII) — conducido en hombres de mediana edad saludables no encontró algún efecto significante de 400 UI de α-tocoferol sintético (180 mg de RRR-α-tocoferol), administrado cada dos días por ocho años, en el riesgo de eventos cardiovasculares importantes en la cohorte entera y en los análisis de subgrupos (40). Además, preocupaciones fueron planteadas con respecto al posible efecto dañino de la suplementación con altas dosis de vitamina E en el riesgo de accidentes cerebrovasculares hemorrágicos en esta cohorte (40).
Prevención secundaria: en individuos con o en riesgo de enfermedades cardiovasculares
Factores de riesgo convencionales de las enfermedades cardiovasculares (ECV) incluyen fumar cigarrillos, falta de actividad física, hipertensión, dislipidemia y tener sobrepeso o estar obeso. Se piensa también que otros factores tales como el estrés oxidativo y la inflamación contribuyen al incremento del riesgo de ECV, especialmente en pacientes con condiciones crónicas como la diabetes mellitus tipo 2 y la enfermedad renal crónica. Aunque los ensayos no parecen apoyar ningún beneficio cardiovascular en sujetos sanos de mediana edad y mayores, la suplementación con vitamina E podría ayudar a mejorar la salud cardiovascular y/o disminuir el riesgo de ECV en específico, sujetos en alto riesgo.
Estudios basados en la observación: La presencia de placas ateroscleróticas en las paredes arteriales es una de las marcas distintivas de las enfermedades cardiovasculares. La ruptura de placa que causa la formación de coágulos sanguíneos es la causa usual de infartos al miocardio y cerebrovasculares. El estudio de corte transversal Asymptomatic Carotid Atherosclerosis Disease In Manfredonia (ACADIM) en 640 individuos en riesgo reportó una asociación inversa entre el grosor íntima-media carotídeo (GIMC) — un marcador de aterosclerosis — y las concentraciones circulantes de antioxidantes, incluyendo la vitamina E (41). Sin embargo, otros estudios basados en la observación no encontraron asociación alguna entre las concentraciones de vitamina E plasmáticas y el GIMC (revisado en 42).
Estudios de intervención: Un estudio controlado aleatorio pequeño que evaluó el efecto de drogas/fármacos hipolipemiantes en hombres que previamente habían tenido una cirugía de revascularización coronaria encontró que el uso de por lo menos 100 UI/día (en comparación a menos de 100 UI/día) de α-tocoferol suplementario (45 mg de RRR-α-tocoferol) estaba asociado con una progresión reducida del GIMC sobre un periodo de dos años pero solo entre los participantes en el grupo del placebo del estudio (es decir, aquellos que no recibieron los fármacos hipolipemiantes) (43). Sin embargo, un reciente meta-análisis de siete ensayos de menor escala controlados con placebo encontró poca evidencia de que la suplementación con vitamina E pudiese mejorar la dilatación mediada por flujo (DMF) de la arteria braquial, un marcador de la salud vascular endotelial que es adversamente afectada por los factores de riesgo de las ECV (44). En el Cambridge Heart AntiOxidant Study (CHAOS), un ensayo de intervención aleatorio controlado con placebo en 2,002 pacientes con enfermedades coronarias cardiacas, la suplementación diaria con 400 UI o con 800 UI de α-tocoferol sintético (180 mg o 360 mg de RRR-α-tocoferol) por una mediana de 18 meses redujo dramáticamente la ocurrencia de infartos al miocardio no-fatales en un 77%. Sin embargo, la suplementación con vitamina E no redujo significantemente las muertes totales o relacionadas a lo cardiovascular (45).
Otro estudio pequeño en pacientes con insuficiencia renal — la Prevención Secundaria con Antioxidantes de las enfermedades cardiovasculares en la enfermedad renal terminal (ERT) — encontró que la suplementación con 800 UI (536 mg)/día de RRR-α-tocoferol por un promedio de 1.4 años redujo significantemente el riesgo de infarto al miocardio en comparación al placebo (46). Un estudio controlado aleatorio más reciente sugirió que la suplementación con vitamina E puede beneficiar un subgrupo de pacientes con diabetes mellitus tipo 2. El estudio multicéntrico por Milman et al. (47) fue conducido en 1,434 diabéticos tipo 2 (≥55 años de edad) que llevaban una variante especifica de la proteína haptoglobina (Hp), Hp2-2, la cual tiene una menor eficacia para unir y remover la hemoglobina pro-oxidante libre del plasma, en comparación a las variantes Hp-1 y Hp1-2. La suplementación diaria con 400 UI (268 mg) de RRR-α-tocoferol por 18 meses resulto en un riesgo menor de infarto al miocardio en comparación al placebo (47).
Otros ensayos de intervención de gran escala conducidos en fumadores de cigarrillos (el estudio ATBC-Alpha-Tocopherol, Beta-Carotene cancer prevention (48)), en individuos en riego de ECV (the Heart Outcomes Prevention Evaluation [HOPE]-The Ongoing Outcomes [HOPE-TOO study] (49)), o en pacientes que han sufrido un infarto al miocardio (ensayo GISSI-prevenzione - Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocárdico (50)) fallaron en encontrar reducciones significantes del riesgo de ECV con la suplementación de α-tocoferol. Además, efectos potencialmente dañinos de la suplementación con vitamina E fueron reportados en el riesgo de accidentes cerebrovasculares hemorrágicos en el ensayo ATBC y en el riesgo de insuficiencia cardiaca en los ensayos HOPE y GISSI (véase Seguridad) (48-50).
Cáncer
El daño oxidativo al ADN por los radicales libres puede conducir a mutaciones que pueden contribuir a causar cáncer (51). Debido a su habilidad para neutralizar radicales libres, ha sido sugerido que la vitamina E posee actividad anticancerígena al proteger las células contra el daño oxidativo. A pesar de todo, varios estudios de cohorte prospectivos de gran escala han fallado en encontrar asociaciones significantes entre la ingesta de vitamina E y la incidencia de cáncer pulmonar o de seno (2). Más recientemente, el estudio VITAL (the VITamins And Lifestyle study) prospectivamente evaluó la asociación entre el uso a largo plazo de vitaminas suplementarias (ingesta de 10 años) y el riesgo de cáncer de pulmón en una cohorte de 77,126 hombres y mujeres (52). No se reportaron relaciones entre la ingesta de multivitaminas, vitamina C, vitamina E, o folato y el riesgo de cáncer de pulmón. Sin embargo, el uso de vitamina E suplementaria en fumadores actuales, pero no en exfumadores fue asociado con un incremento del 11% del riesgo de cáncer de pulmón por cada incremento de 100 mg/día, y las ingestas mayores de 215 mg/día fueron específicamente ligadas a un incremento del 29% en el riesgo de cáncer pulmonar de células no pequeñas (52).
Hasta la fecha, la mayoría de los ensayos clínicos han fallado en encontrar algún efecto beneficioso de la suplementación con vitamina E en el riesgo de varios cánceres. Un ensayo aleatorio controlado con placebo en 39,876 mujeres participantes en el Estudio de Salud de las Mujeres (Women's Health Study) encontró que la suplementación con 600 UI (400 mg) de RRR-α-tocoferol cada dos días por 10 años no tuvo efecto alguno en la incidencia de cáncer en general, incidencia de cáncer de tejido especifico, o en las muertes relacionadas con cáncer (37). A pesar de todo, los resultados de unos pocos ensayos controlados aleatorios han sugerido que la suplementación con vitamina E podría afectar el riesgo de cáncer de próstata. El estudio de prevención del cáncer Alfa-Tocopherol Beta-Caroteno (ATBC) fue un ensayo controlado con placebo, aleatorio, doble ciego de cuatro brazos diseñado para investigar el efecto de la suplementación con α-tocoferol en el desarrollo del cáncer pulmonar en 29,133 fumadores masculinos. El estudio encontró una reducción del 32% en la incidencia de cáncer de próstata en participantes a los que se les administro diariamente con suplementos de 50 mg de α-tocoferol sintético (equivalente a 25 mg de RRR-α-tocoferol) únicamente, o en combinación con β-caroteno en comparación con aquellos a los que se les administro β-caroteno únicamente o un placebo (53). Sin embargo, no se encontraron diferencia en la incidencia de cáncer de próstata entre los que recibieron α-tocoferol y los que no lo recibieron durante el periodo de post-intervención de 18 años (54). En el estudio PHS II (Physicians’ Health Study II), el cual dio seguimiento a 14,641 hombres sanos de 50 años de edad y mayores, la suplementación con 400 UI de vitamina E sintética (equivalente a 180 mg de RRR-α-tocoferol) cada dos días por ocho años no tuvo efecto en el riesgo de cáncer de próstata, otros canceres de sitio especifico, o en el cáncer total (55). En el ensayo multicéntrico, aleatorio controlado con placebo SELECT (SELenium and vitamin E Cancer prevention Trial), la suplementación con vitamina E (equivalente a 180 mg/día de RRR-α-tocoferol), únicamente o en combinación con selenio, fue detenida porque no había evidencia de algún beneficio en la prevención del cáncer de próstata en 35,533 hombres sano de 50 años de edad y mayores (56). Después de un seguimiento medio de siete años, se encontró que el riesgo de cáncer de próstata había incrementado significantemente en un 17% en participantes suplementados solo con vitamina E durante el periodo del ensayo — pero no cuando la vitamina E era combinada con selenio — en comparación al placebo (57). Un estudio de casos frente a individuos de una sub-cohorte extraídos del estudio SELECT evaluó el efecto de la suplementación con vitamina E y/o selenio en el riesgo de cáncer de próstata en relación a el estatus de selenio de los participantes al inicio del estudio (58). El selenio suplementario con o sin vitamina E estuvo asociado con un significante incremento en el riesgo de cáncer de próstata avanzado en individuos con un estatus de selenio más alto frente aquellos con uno más bajo. Además, los riesgos de cáncer de próstata total y avanzado fueron significantemente elevados con la suplementación con vitamina E en sujetos con estatus de selenio más bajos frente aquellos con estatus más altos (58). Investigaciones recientes han sugerido que variaciones en la secuencia (polimorfismos) de genes relacionados a la vitamina E y genes que codifican para las enzimas antioxidantes, incluyendo selenoproteínas, podrían modificar el impacto de dosis altas de vitamina E y selenio en el riesgo de cáncer de próstata (59-61).
Cataratas
Las cataratas relacionadas a la edad parecen ser el resultado de la oxidación de proteínas en el cristalino (lente) del ojo; antioxidantes como el α-tocoferol podrían proteger el cristalino contra el daño oxidativo proveniente de especies reactivas de oxígeno. En un reciente estudio de corte transversal, se encontró que las concentraciones de vitamina E eran más bajas en el cristalino y en la sangre de los sujetos con cataratas seniles nucleares, pero no corticales cuando se comparó con un grupo de control de la misma edad (62). Sin embargo, estudios tempranos reportaron altas concentración de vitamina E en el cristalino y sangre de pacientes con cataratas (63, 64).
Los resultados de varios estudios basados en la observación que examinaron la asociación entre el consumo de vitamina E y la incidencia o severidad de las cataratas son también mixtos. Algunos reportaron que la ingesta incrementada de vitamina E protegió contra el desarrollo de las cataratas, mientras que otros no encontraron alguna asociación (65). Por otra parte, un meta-análisis de ocho estudios, incluyendo 15,021 participantes, encontró una reducción del 17% en el riesgo de cataratas seniles en sujetos en el cuantil más alto de la ingesta dietaría de vitamina frente aquellos en el cuantil más bajo (66). Un reciente estudio de cohorte prospectivo de 31,120 hombres suecos con un seguimiento promedio de 8.4 años observó un riesgo más alto de desarrollar cataratas en usuarios ocasionales y regulares de altas dosis (alrededor de 100 mg/día) de suplementos de solo vitamina E, cuando se comparó con usuarios que no consumían suplementos (67). Sin embargo, no se encontró que el uso de vitamina E suplementaria en altas dosis con suplementos adicionales o el uso de suplementos multivitamínicos que contienen vitamina E en bajas dosis estuviera asociado con un riesgo elevado de cataratas. Un meta-análisis basado en datos de más de 350,00 participantes en 10 estudios — incluyendo el estudio citado previamente por Zheng Selin et. Al. (67) — no encontró asociación alguna entre la vitamina E suplementaria y el riesgo de cataratas (66).
En situaciones clínicas se encontró que la suplementación con altas dosis de vitamina E — sola o en adición a otros suplementos — era segura, aunque los beneficios con respecto al riesgo de cataratas o progresión fueron limitados. Un ensayo de intervención temprano encontró que un suplemento diario de 50 mg de α-tocoferol sintético (equivalente s 25 mg de RRR-α-tocoferol) no alteró la incidencia de cirugías de cataratas en fumadores masculinos (68). Un ensayo de intervención, aleatorio, controlado con placebo en 4,629 hombres y mujeres encontró que un suplemento antioxidante diario que contenía 500 mg de vitamina C, 400 UI de acetato de todo-rac-α-tocoferol (equivalente a 180 mg de RRR-α-tocoferol), y 15 mg de β-caroteno no afectó el desarrollo y progresión de las cataratas seniles sobre un periodo de siete años (69). Similarmente, un estudio de cuatro brazos controlado con placebo, aleatorio, de 11,267 hombres provenientes del ensayo SELECT falló en observar una reducción en la incidencia de cataratas con 400 UI/día de acetato de todo-rac-α-tocoferol suplementario (180 mg/día de RRR-α-tocoferol), solo o en combinación con selenio (200 μg/día), durante un seguimiento promedio de 5.6 años (70). La suplementación diaria de antioxidantes con 500 mg de vitamina C, 400 UI (268 mg) de RRR-α-tocoferol, y 15 g de β-caroteno no limitó la progresión de cataratas en un ensayo de intervención de cinco años (71). Otro ensayo aleatorio controlado con placebo de cuatro años reportó que los suplementos que contenían 500 UI/día (335 mg/día) de RRR-α-tocoferol no redujeron la incidencia o progresión de cataratas en adultos mayores (72). Datos actuales disponibles de ensayos clínicos no apoyan un efecto preventivo de la vitamina E en las cataratas.
Tratamiento de Enfermedades
Degeneración macular relacionada con la edad
Un reciente análisis combinado de cuatro ensayos controlados aleatorios en 62,520 sujetos encontró que la vitamina E o β-caroteno suplementarios no redujeron el riesgo de desarrollar degeneración macular relacionada a la edad (AMD), una enfermedad multifactorial que afecta el área central de la retina (73). Sin embargo, una revisión de datos actualmente disponibles sugirió que los suplementos de antioxidantes más zinc podrían reducir la progresión de AMD y la perdida de la visión en individuos afectados (74). La evidencia principal provino de un Estudio de la Enfermedad Ocular Relacionada con la Edad (AREDS). En este ensayo clínico, los participantes con degeneración molecular relacionada a la edad (AMD) marginal a avanzada fueron aleatoriamente seleccionados para recibir (1) placebo; (2) vitaminas antioxidantes (15 mg/día de β-caroteno, 500 mg/día de ácido ascórbico, y 400 UI/día de acetato de todo-rac-α-tocoferol); (3) zinc (80 mg/día) y cobre (2 mg/día); o (4) ambos cobre y zinc y vitaminas antioxidantes (75). Los resultados de cinco años indicaron que el riesgo de desarrollar AMD avanzada fue significantemente reducido en aquellos que tomaron zinc con o sin vitaminas antioxidantes. Las vitaminas antioxidantes solas fallaron en prevenir la progresión de la AMD a avanzada, incluso en individuos en un riesgo más alto. Se concluyó a partir de este estudio que una combinación de vitaminas antioxidantes y minerales podría beneficiar a las personas con AMD intermedia o AMD avanzada en un ojo (74, 76).
Diabetes mellitus tipo 2
El estrés oxidativo contribuye a la progresión de la diabetes mellitus tipo 2 y causa daño a muchos órganos y tejidos, incluyendo el páncreas, cerebro, ojos, nervios periféricos, y riñones. Evidencia proveniente de estudios en animales sugiere que la suplementación con vitamina E podría mitigar el papel del daño oxidativo en la ocurrencia de complicaciones de la diabetes (revisado en 77). En el ensayo ATBC (Alpha-Tocopherol Beta-Carotene) para la prevención del cáncer en fumadores masculinos, la suplementación con 50 mg/día de α-tocoferol sintético (25 mg/día de RRR-α-tocoferol) no tuvo efecto en el riesgo de incidencia de diabetes mellitus tipo 2 durante el seguimiento de post-intervención de 19 años. Así mismo, la ingesta de vitamina E durante el ensayo no produjo diferencia alguna en la incidencia de complicaciones macrovasculares o mortalidad en participantes con diabetes tipo 2 establecida (78). Además, un meta-análisis de 14 ensayos controlados aleatorios heterogéneos, incluyendo 714 individuos con diabetes tipo 2, encontró que la suplementación con vitamina E (200-1,800 UI/día por 6-27 semanas) no tuvo efecto en los marcadores del control glucémico, incluyendo el nivel de hemoglobina glicosilada A1c (HbA1c) y las mediciones de glucosa en ayunas y concentraciones de insulina en ayunas (79). Análisis de subgrupo posteriores indicaron que las dosis más altas de vitamina E (>400 UI/día) suplementada por largos periodos (>12 semanas) redujeron significantemente el nivel de HbA1c y la concentración de insulina en ayunas, sugiriendo que la vitamina E podría posiblemente mejorar la acción de la insulina y eliminación de glucosa en individuos con diabetes tipo 2 (79). Otro meta-análisis reciente de ensayos controlados aleatorios encontró que la función endotelial en pacientes con diabetes tipo 2 con pesos normales y con sobrepeso — pero no obesos — fue significantemente mejorada por la suplementación con vitamina E y/o vitamina C (80). Aunque existe una razón para sospechar que la suplementación con vitamina E puede tener utilidad en el manejo de la diabetes tipo 2, evidencia de beneficios proveniente de ensayos clínicos bien controlados de gran escala es aun escaza.
Enfermedades del hígado graso
La creciente incidencia de enfermedad del hígado graso no alcohólico (NAFLD por sus siglas en inglés) en niños y adultos en países industrializados es mayormente atribuida a la epidemia en curso de obesidad y diabetes mellitus tipo 2. La NAFLD resulta de la acumulación anormal de grasa (esteatosis) en el hígado en la ausencia de un alto consumo de alcohol. Aunque la condición se considera en gran medida benigna, la NAFLD puede progresar a una enfermedad más severa llamada esteatohepatitis no alcohólica (EHNA) con riesgos incrementados de cirrosis, carcinoma hepatocelular (cáncer de hígado), y enfermedades cardiovasculares (81). Se piensa que el estrés oxidativo es uno de los posibles mecanismos responsables por estimular los procesos inflamatorios que pueden conducir a la progresión de NAFLD a EHNA.
No existe un tratamiento actual establecido para la NAFLD y EHNA que no sean intervenciones que fomenten cambios en el estilo de vida y el uso de medicinas para controlar o tratar desordenes metabólicos (82). En el ensayo multicéntrico PIVENS (PIoglitazone versus Vitamin E versus placebo for the treatment of Nonalcoholic Steatohepatitis), 247 sujetos no diabéticos con EHNA fueron aleatoriamente seleccionados para recibir 30 mg/día de pioglitazona (un fármaco sensibilizador de insulina), 800 UI/día (536 mg/día) de RRR-α-tocoferol, o un placebo por 96 semanas (83). Solo la suplementación con vitamina E significantemente incremento en general la tasa de mejoría en las anomalías histológicas que caracterizan la EHNA en las biopsias de hígado (es decir, hinchamiento hepatocelular, esteatosis e inflamación lobular) (84). Ambos tratamientos activos mejoraron algunos marcadores de la función hepática (es decir, alanina aminotransferasa y aspartato aminotransferasa) (84). Por otra parte, resultados provenientes de otro ensayo controlado aleatorio de dos años — llamado TONIC por Treatment Of Nonalcoholic fatty liver disease In Children — en 173 niños (edades, 8-17 años) con NAFLD fallaron en observar alguna reducción significante en las concentraciones sanguíneas de alanina y aspartato aminotransferasas tanto con la vitamina E suplementaria (536 mg/día de RRR-α-tocoferol) como con metformina (un fármaco anti-diabético; 1,000 mg/día) en comparación al placebo (85). Sin embargo, la suplementación con vitamina E significantemente mejoró la puntuación de la actividad de la enfermedad en general — usada para cuantificar la severidad de la enfermedad. Además, un reciente meta-análisis de otros seis ensayos encontró que la vitamina E disminuyo significantemente las concentraciones circulantes de aminotransferasa en pacientes con NAFLD y EHNA, sugiriendo mejorías de la función hepática (86). Finalmente, en un estudio controlado, no-aleatorio, no-ciego de menor escala en 42 niños obesos (edad promedio, 8 años) con NAFLD, recomendaciones de estilo de vida en combinación con 600 mg/día de acetato de RRR-α-tocoferol por seis meses redujeron los marcadores del estrés oxidativo y la disfunción del hígado y mejoró la sensibilidad a la insulina y el perfil de lípidos en la sangre, en comparación al valor basal. Tales cambios en los marcadores del estrés oxidativo, la función hepática, y la utilización de glucosa no fueron reportados en el único grupo de intervención de estilo de vida (87).
Deterioro cognitivo y enfermedad de Alzheimer
Se piensa que la disfunción mitocondrial y el estrés oxidativo contribuyen a la aparición y/o progresión de varias enfermedades neurodegenerativas, especialmente la enfermedad de Alzheimer (EA) (88). La degeneración progresiva de las células neuronales que acompañan el declive de la memoria y otras funciones cognitivas en sujetos con la enfermedad de Alzheimer está asociada con una agregación intracelular de fibrillas Tau, una acumulación extraneuronal de péptidos β-amiloides en las placas seniles, y un desequilibrio de oxidación-reducción (redox) de etiología compleja. En el cerebro de los pacientes con deterioro cognitivo leve (DCL) y aquellos con EA, el nivel de marcadores del daño oxidativo al ADN, proteínas, y los lípidos es incrementado, mientras que la expresión y actividades del glutatión y las enzimas antioxidantes son reducidas (revisado en 88). Además, un reciente meta-análisis reporto que las concentraciones circulantes de vitaminas, incluyendo vitamina A, vitamina C, y vitamina E, fueron significativamente más bajas en pacientes con EA que en individuos cognitivamente saludables (89). Otros estudios han documentado bajas concentraciones de vitamina E en el fluido cerebroespinal de pacientes con deterioro cognitivo (revisado en 90).
Debido a que una reducción en el estrés oxidativo podría ayudar a mantener el estatus cognitivo y/o prevenir el deterioro, los efectos de la suplementación con vitamina E han sido evaluados en unos pocos estudios de intervención. Un estudio temprano controlado con placebo, aleatorio, multicéntrico en individuos con EA de severidad moderada encontró que la suplementación con 2,000 UI/día de todo-rac-α-tocoferol (equivalente a 900 mg/día de RRR-α-tocoferol) por dos años significantemente retrasó el declive cognitivo, ralentizó la progresión de la enfermedad, e incremento la supervivencia media (91). Sin embargo, un ensayo controlado con placebo en 769 pacientes con DCL encontró que la misma dosis de vitamina E no afecto la probabilidad de la progresión de DCL a EA sobre un periodo de tres años (92). En otro ensayo controlado con placebo, doble ciego, una mejora en el rendimiento cognitivo — medido por el sistema de puntuación del Mini Examen del Estado Mental (MEEM) — fue reportada en pacientes con EA que aleatoriamente recibieron 800 UI/día de todo-rac-α-tocoferol (360 mg/día de RRR-α-tocoferol) por seis meses solo cuando el tratamiento efectivamente redujo el estrés oxidativo (evaluado por la medición de glutatión total y marcadores de peroxidación lipídica en la sangre) (93). Por el contrario, un fracaso en la reducción del estrés oxidativo dio como resultado que la vitamina E suplementaria fuera más perjudicial para la función cognitiva de pacientes con EA que el placebo. En el estudio multicéntrico, aleatorio, doble ciego, controlado con placebo más reciente, la vitamina E suplementaria (2,000 UI/día; la forma de la vitamina E no es mencionada en la publicación) por más de dos años significativamente retrasó el declive funcional — determinado por la (in)capacidad para realizar actividades básicas de la vida diaria — y redujo la tasa de mortalidad anual en pacientes como enfermedad de Alzheimer moderada (94). A pesar de todo, la vitamina E fallo en afectar el rendimiento cognitivo medido con puntuaciones del MEEM y otras pruebas de habilidad cognitiva.
Mientras que existe poca evidencia que sugiera que la suplementación a largo plazo de vitamina E provee de algunos beneficios cognitivos en adultos mayores sanos (95), investigación adicional necesita confirmar si la suplementación con vitamina E podría beneficiar el manejo de pacientes con deterioros cognitivos de leve-a-moderados.
Fuentes
Fuentes alimenticias
Las principales fuentes de α-tocoferol en la dieta americana incluyen aceites vegetales (aceite de oliva, girasol, y cártamo), nueces, granos enteros, y vegetales de hoja verde. Las ocho formas de vitamina E (α-, β-, γ-, y δ-tocoferoles y α-, β-, γ-, y δ-tocotrienoles) están presentes naturalmente en los alimentos de origen vegetal, pero en cantidades variables. La Tabla 2 lista el contenido de α-tocoferol y γ-tocoferol (en miligramos) en varias fuentes ricas en vitamina E. Para más información sobre el contenido de vitamina E de los alimentos, revise la base de datos de composición de los alimentos de la USDA.
En los EE.UU., la ingesta promedio de α-tocoferol proveniente de los alimentos (incluyendo fuentes enriquecidas y fortalecidas) para los adultos (≥19 años de edad) es de 7.2 mg/día (25); este nivel está por debajo de la IDR de 15 mg/día de α-tocoferol (véase Tabla 1). Mientras que parece factible para los individuos satisfacer la IDR a partir de solo los alimentos, los americanos tendrían que apartarse de sus prácticas dietarías actuales e incluir ingestas mayores de nueces, semillas, frutos y vegetales sin incrementar la ingesta de grasas por arriba de los niveles recomendados (96).
Suplementos
El RRR-α-tocoferol es la única forma estereoisómera del α-tocoferol encontrada en alimentos no fortificados. Lo mismo no siempre es cierto para los suplementos nutricionales. La vitamina E generalmente contiene de 100 UI a 1,000 UI de α-tocoferol. Los suplementos hechos completamente de fuentes naturales contienen solo RRR-α-tocoferol (también denominado d-α-tocoferol). El RRR-α-tocoferol es la forma más biodisponible de α-tocoferol en el cuerpo. El α-tocoferol sintético, el cual frecuentemente se encuentra en alimentos fortificados y suplementos nutricionales y usualmente se etiqueta como todo-rac-α-tocoferol o dl-α-tocoferol, incluye los ocho posibles isómeros del α-tocoferol (véase Función). Debido a que la mitad de los isómeros presentes como una mezcla en el α-tocoferol sintético no son utilizables por el cuerpo, el α-tocoferol sintético es menos biodisponible y que el α-tocoferol natural (véase Figura 1). Para calcular la cantidad (en miligramos) de α-tocoferol biodisponible presente en un suplemento, los factores de conversión son los siguientes:
-
Suplementos que contienen vitamina E natural (RRR-α-tocoferol):
UI de RRR-α-tocoferol x 0.67 = mg de RRR-α-tocoferol
Ejemplo: 100 UI de vitamina E natural proporciona 67 mg de RRR-α-tocoferol -
Suplementos que contienen vitamina E sintética (todo-rac-α-tocoferol):
UI de todo-rac-α-tocoferol x 0.45 = mg de RRR-α-tocoferol
Ejemplo: 100 UI de vitamina E sintética proporciona 45 mg de RRR-α-tocoferol
Además, los alimentos fortificados con vitamina E frecuentemente contienen α-tocoferol sintético, y las cantidades de vitamina E son proporcionad como un porcentaje del valor diario (VD) de 30 UI (aproximadamente 20 mg de RRR-α-tocoferol)
Ésteres α-tocoferilos
El succinato de α-tocoferilo y acetato de α-tocoferilo son las formas esterificadas de la vitamina E en los suplementos nutricionales. Los ésteres del tocoferol son más resistentes a la oxidación durante el almacenamiento que los tocoferoles no esterificados (1). Cuando se toman de manera oral, el grupo funcional succinato o acetato se remueve del α-tocoferol en el intestino. La biodisponibilidad del α-tocoferol a partir del succinato de α-tocoferilo y del acetato de α-tocoferilo, es equivalente a la del α-tocoferol libre (97). Por lo tanto, los factores de conversión usados para determinar la cantidad de α-tocoferol biodisponible proporcionada por el succinato de α-tocoferilo y acetato de α-tocoferilo son los mismos que aquellos usados para el α-tocoferol (vease arriba) (2). Los estudios de cultivos celulares indicaron que el éster de vitamina E, succinato de α-tocoferilo, pudo inhibir la proliferación e inducir apoptosis en un número de líneas celulares cancerígenas (12). Datos limitados provenientes de modelos en animales del cáncer, encontraron que el succinato de α-tocoferilo administrado a través de inyecciones inhibió el crecimiento del tumor (98). Actualmente no existe evidencia en humanos de que tomar suplementos de succinato de α-tocoferilo de forma oral entregue succinato de α-tocoferilo a los tejidos. Es de destacarse que la investigación actual indaga las nanomedicinas para incrementar la biodisponibilidad del succinato de α-tocoferilo antes de explorar los beneficios putativos en el entorno clínico (98).
El nicotinato de α-tocoferilo es otro éster de α-tocoferol formado a partir del α-tocoferol sintético y el ácido nicotínico (niacina). Mientras que el nicotinato de α-tocoferilo puede ser prescrito como un agente hipolipemiante en Europa y Japón, este es comercializado solo como un suplemento en los EE.UU. (99).
Fosfatos de α-tocoferilo (Ester-E®)
No existe actualmente evidencia publicada de que los suplementos que contienen fosfatos de α-tocoferilo sean más eficientemente absorbidos o que tengan una mayor biodisponibilidad en humanos que los suplementos que contienen α-tocoferol (99).
Otras formas suplementarias
Los suplementos que contienen γ-tocoferol, una mezcla de tocoferoles, o tocotrienoles están también disponibles comercialmente (99). La cantidad de α- y γ-tocoferol en los suplementos con mezclas de tocoferoles varia, por eso es importante leer el etiquetado para determinar la cantidad de cada tocoferol presente en una capsula.
Seguridad
Toxicidad
Se han registrado pocos efectos secundarios en adultos que toman diariamente suplementos de menos de 2,000 mg de α-tocoferol (ya sea vitamina E natural o sintética). Sin embargo, la mayoría de los estudios que evalúan los problemas de seguridad o toxicidad de la suplementación con α-tocoferol han durado de sólo unas pocas semanas hasta unos cuantos meses, y los efectos secundarios asociados con la suplementación a largo plazo con α-tocoferol no han sido estudiados adecuadamente. La posibilidad más preocupante es la probabilidad de una alteración de la coagulación de la sangre, la cual aumenta la probabilidad de hemorragia en algunos individuos. Un meta-análisis de ensayos controlados aleatorios encontró que la suplementación diaria con vitamina E — equivalente a 25 a 536 mg/día de RRR-α-tocoferol — por varios años resultó en una reducción significante del 10% en el riesgo de accidentes cerebrovasculares isquémicos (cinco ensayos, 91,393 participantes) y una tendencia no significante hacia un riesgo incrementado de accidentes cerebrovasculares hemorrágicos (cinco ensayos, 100,748 participantes) (100).
Un nivel máximo de ingesta tolerable (NM) para cualquiera de los suplementos de α-tocoferol (todos los posibles estereoisómeros) ha sido establecido por la Junta de Nutrición y Alimentos del Instituto de Medicina para la prevención del riesgo potencial de hemorragia (Tabla 3). Especialmente, el NM de 1,000 mg/día de α-tocoferol en cualquiera de las formas suplementarias (equivalente a 1,500 UI/día de RRR-α-tocoferol o a 1,100 UI/día de todo-rac-α-tocoferol) correspondiente a la dosis más alta menos propensa en resultar en hemorragia en casi todos los adultos (2). Aunque solo ciertos isómeros de α-tocoferol son retenidos en la circulación, todas las formas son absorbidas y metabolizadas por el hígado. Por lo tanto, la justificación por la cual un NM que se refiere a todos los estereoisómeros de α-tocoferol está basado en el hecho de que cualquier forma de α-tocoferol (natural o sintética) puede ser absorbida y de esta manera ser potencialmente dañina.
Algunos doctores recomiendan descontinuar la suplementación con vitamina E en altas dosis de dos a cuatro semanas antes de una cirugía electiva — incluyendo procedimientos dentales — para disminuir el riesgo de hemorragia (99).
Debido a que la vitamina E dietaría es esencial para prevenir la deficiencia de vitamina E en los recién nacidos, la vitamina E debe suministrarse en soluciones de nutrición parenteral en infantes que no pueden ser alimentos de forma enteral, como los infantes nacidos prematuramente. Por otra parte, los infantes prematuros parecen ser especialmente vulnerables a los efectos adversos de la suplementación con α-tocoferol, y la vitamina E suplementaria debe ser administrada solo bajo supervisión controlada por un pediatra (101).
Finalmente, los resultados de solo un ensayo controlado aleatorio en 601 pacientes con formas comunes de retinitis pigmentosa (RP) indicaron que la suplementación con 400 UI/día de vitamina E sintética (equivalente a 180 mg/día de RRR-α-tocoferol) modestamente pero significantemente aceleró la perdida de la función de la retina en comparación al placebo (22). Pacientes con formas comunes de RP debiesen por lo tanto evitar tomar suplementos con altas dosis de vitamina E si no están deficientes de vitamina E (véase Deficiencia).
¿La suplementación con vitamina E incrementa el riesgo de mortalidad por todas las causas?
Un estudio basado en la observación prospectivo en más de 4,000 participantes del Estudio del Corazón de Framinhgam y del Estudio de los Descendientes de Framinhgam — con o sin enfermedades cardiovasculares preexistentes — no encontró una asociación estadísticamente significante entre la ingesta de suplementos de vitamina E y la mortalidad cardiovascular o por todas las causas después de un periodo de seguimiento de 10 años (102). Sin embargo, además de los reportes del riesgo incrementado de hemorragia e insuficiencia cardiaca con vitamina E suplementaria en varios estudios controlados aleatorios (véase Enfermedades cardiovasculares), un meta-análisis por Miller et al. (103) sugirió un riesgo incrementado de muerte con el uso de altas dosis de vitamina E, aun menores que el NM. Específicamente, este meta-análisis combinó los resultados de 19 ensayos clínicos de la suplementación con vitamina E que se enfocaron principalmente en la prevención secundaria y, como tal, incluyó sujetos con condiciones preexistentes incluyendo enfermedad cardiaca, insuficiencia renal en etapa terminal, y la enfermedad de Alzheimer. El estudio encontró que la suplementación diaria con al menos 400 UI de vitamina E sintética (equivalente a 180 mg de RRR-α-tocoferol) resultó en un incremento del 4% en el riesgo de muerte por cualquier causa en comparación al placebo (103). Sin embargo, el análisis de dosis-respuesta y el ajuste para la ingesta de suplementos vitamínicos y minerales posteriores indicaron que el riesgo de mortalidad por todas las causas fue significantemente incrementado en un 7% solo en dosis de 2,000 UI/día, notablemente más altas que el NM para adultos (1,100 UI/día de tocoferol sintético o 1,500 UI/día de tocoferol natural; véase Tabla 3). Adicionalmente, un meta-análisis más reciente de 46 ensayos aleatorios, incluyendo 171,244 participantes, encontró que la vitamina E suplementaria, sola o en combinación con otros antioxidantes, no altero significantemente el riesgo de mortalidad por todas las causas (104). Actualmente, no existe evidencia convincente de que la suplementación con vitamina E por debajo del NM incremente el riesgo de muerte por enfermedades cardiovasculares o por otras causas, especialmente en sujetos generalmente sanos. A pesar de todo, los individuos con condiciones preexistentes podrían estar en un riesgo incrementado de efectos adversos serios (incluyendo muerte) si uno considera la posibilidad de que dosis altas de vitamina E suplementaria podría interferir con medicamentos, y posiblemente disminuir su eficacia o aumentar su toxicidad (1).
Interacción con nutrientes
Las dosis altas de vitamina E pueden inhibir la actividad carboxilasa dependiente de la vitamina K e interferir con la cascada de coagulación (véase el artículo en Vitamina K) (19). Por ello, el uso de suplementos de vitamina E puede incrementar el riesgo de sangrado en individuos que toman drogas anticoagulantes, como la heparina y el antagonista de la vitamina K, la warfarina (Coumadin); fármacos antiplaquetarios, como el clopidogrel (Plavix), ticlopidina (Ticlid), tirofiban (Aggrastat), y dipiridamol (Aggrenox); y fármacos antiinflamatorios no esteroideos (AINE), incluyendo la aspirina, ibuprofeno, y otros. Además, los individuos que podrían estar deficientes de vitamina K debido a una insuficiencia hepática, aquellos propensos a sangrar (es decir, úlceras pépticas sangrantes), y aquellos con trastornos de la coagulación hereditarios (es decir, hemofilia) o un historial de accidente cerebrovascular hemorrágico, no debiesen tomar suplementos de α-tocoferol sin estricta supervisión médica debido al riesgo incrementado de hemorragia (20, 99). Finalmente, no se puede excluir que la vitamina E potenciaría la actividad antitrombótica de los aceites de pescado suplementarios y productos herbales, como el ajo, la curcumina o Ginkgo biloba (99).
Interacción con drogas/fármacos
Un cierto número de medicamentos que disminuyen el colesterol (como la colestiramina y el colestipol), como también el orlistat, sucralfato, aceite mineral, y el sustituto graso, olestra, los cuales interfieren con la absorción de grasas, podrían en teoría disminuir la absorción de vitaminas liposolubles, incluyendo la vitamina E. Los fármacos anticonvulsivos fenobarbital, fenitoína (Dilantin), o carbamazepina (Tegretol), podrían también disminuir las concentraciones de vitamina E del plasma en individuos con epilepsia (105).
Antioxidantes y estatinas (inhibidores de 3-hidroxi-3-metil-glutaril-CoA reductasa)
Un ensayo controlado aleatorio de 3 años en 160 pacientes con enfermedad coronaria cardiaca (ECC) y bajos niveles de lipoproteínas de alta densidad (HDL), encontró que una combinación de simvastatina (Zocor) y niacina incrementó el nivel de subfracción de HDL2 (considerado el más cardioprotector), inhibió la progresión de estenosis de arteria coronaria (estrechamiento), y disminuyó la frecuencia de eventos cardiovasculares, como infarto al miocardio y accidente cerebrovascular (106). Sorprendentemente, cuando se tomó una combinación de antioxidantes de 1,000 mg de vitamina C, 800 UI (536 mg) de RRR-α-tocoferol, 100 μg de selenio, y 25 mg de β-caroteno diariamente junto con la combinación de simvastatina-niacina, los efectos protectores fueron disminuidos. Sin embargo, un ensayo controlado aleatorio de mayor escala de simvastatina y una combinación de antioxidantes de 600 mg de todo-rac-α-tocoferol (297 mg de RRR-α-tocoferol), 250 mg de vitamina C, y 20 mg de β-caroteno diariamente en más de 20,000 hombres y mujeres con ECC o diabetes, la combinación antioxidante no afectó adversamente los efectos cardioprotectores de la terapia con simvastatina sobre un periodo de cinco años (107). Estos hallazgos contradictorios indican que es necesaria más investigación sobre las potenciales interacciones entre la suplementación antioxidante y los agentes hipocolesterolemiantes como las estatinas.
Recomendación del Instituto Linus Pauling
La Ingesta Diaria Recomendada (IDR) de la vitamina E para los hombres y mueres adultos es de 15 mg (22.5 UI) por día. Cabe destacar, que más del 90% de los individuos de 2 años de edad y mayores en los EE.UU. no alcanza el requerimiento diario de vitamina E sólo de fuentes alimenticias. Por lo tanto, el ILP recomienda que los adultos generalmente sanos (19 años de edad y mayores) tomen un suplemento multivitamínico/mineral (MVM) diario, el cual usualmente contiene 30 UI de vitamina E sintética — equivalente a 13.5 mg de RRR-α-tocoferol y 90% de la IDR.
Adultos mayores (>50 años)
La recomendación del Instituto Linus Pauling de tomar un suplemento multivitamínico/mineral (MVM) diario que contenga vitamina E es también apropiado para los adultos mayores generalmente sanos. Los MVM típicamente contienen 30 UI de vitamina E sintética, cubriendo el 90% de la IDR.
Autores y Críticos
Originalmente escrito en 2000 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Noviembre de 2004 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Junio de 2008 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Junio de 2008 por:
Maret G. Traber, Ph.D.
Profesor de Ciencias de la Nutrición y el Ejercicio
Investigador Principal,
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Mayo de 2015 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Reviewed in Octubre 2015 por:
Maret G. Traber, Ph.D.
Helen P. Rumbel Profesor de Investigación de Micronutrientes, Instituto Linus Pauling
Profesor, Colegio de Salud Publica y Ciencias Humanas
Universidad Estatal de Oregon
Traducido al Español en 2017 por:
Silvia Vazquez Lima
Instituto Linus Pauling
Universidad Estatal de Oregon
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Derechos de autoría 2000-2024 Instituto Linus Pauling
Referencias
1. Traber MG. Vitamin E. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Washington, D.C.: Wiley-Blackwell; 2012:214-229.
2. Food and Nutrition Board, Institute of Medicine. Vitamin E. Dietary reference intakes for vitamin C, vitamin E, selenium, and carotenoids. Washington, D.C.: National Academy Press; 2000:186-283. (National Academy Press)
3. Trpkovic A, Resanovic I, Stanimirovic J, et al. Oxidized low-density lipoprotein as a biomarker of cardiovascular diseases. Crit Rev Clin Lab Sci. 2014:1-16. (PubMed)
4. Davis S, Davis BM, Richens JL, et al. α-Tocopherols modify the membrane dipole potential leading to modulation of ligand binding by P-glycoprotein. J Lipid Res. 2015;56(8):1543-1550. (PubMed)
5. Marko MG, Ahmed T, Bunnell SC, et al. Age-associated decline in effective immune synapse formation of CD4(+) T cells is reversed by vitamin E supplementation. J Immunol. 2007;178(3):1443-1449. (PubMed)
6. Molano A, Meydani SN. Vitamin E, signalosomes and gene expression in T cells. Mol Aspects Med. 2012;33(1):55-62. (PubMed)
7. Jiang Q. Natural forms of vitamin E: metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic Biol Med. 2014;72:76-90. (PubMed)
8. Jiang Q, Christen S, Shigenaga MK, Ames BN. γ-Tocopherol, the major form of vitamin E in the US diet, deserves more attention. Am J Clin Nutr. 2001;74(6):714-722. (PubMed)
9. Mah E, Pei R, Guo Y, et al. γ-Tocopherol-rich supplementation additively improves vascular endothelial function during smoking cessation. Free Radic Biol Med. 2013;65:1291-1299. (PubMed)
10. Mah E, Pei R, Guo Y, et al. Greater γ-tocopherol status during acute smoking abstinence with nicotine replacement therapy improved vascular endothelial function by decreasing 8-iso-15(S)-prostaglandin F2α. Exp Biol Med (Maywood). 2015;240(4):527-533. (PubMed)
11. Ahsan H, Ahad A, Iqbal J, Siddiqui WA. Pharmacological potential of tocotrienols: a review. Nutr Metab (Lond). 2014;11(1):52. (PubMed)
12. Constantinou C, Papas A, Constantinou AI. Vitamin E and cancer: An insight into the anticancer activities of vitamin E isomers and analogs. Int J Cancer. 2008;123(4):739-752. (PubMed)
13. Fu JY, Che HL, Tan DM, Teng KT. Bioavailability of tocotrienols: evidence in human studies. Nutr Metab (Lond). 2014;11(1):5. (PubMed)
14. Bruno RS, Leonard SW, Park SI, Zhao Y, Traber MG. Human vitamin E requirements assessed with the use of apples fortified with deuterium-labeled α-tocopheryl acetate. Am J Clin Nutr. 2006;83(2):299-304. (PubMed)
15. Leonard SW, Good CK, Gugger ET, Traber MG. Vitamin E bioavailability from fortified breakfast cereal is greater than that from encapsulated supplements. Am J Clin Nutr. 2004;79(1):86-92. (PubMed)
16. Traber MG, Leonard SW, Bobe G, et al. α-Tocopherol disappearance rates from plasma depend on lipid concentrations: studies using deuterium-labeled collard greens in younger and older adults. Am J Clin Nutr. 2015;101(4):752-759. (PubMed)
17. Leonard SW, Bruno RS, Ramakrishnan R, Bray T, Traber MG. Cigarette smoking increases human vitamin E requirements as estimated by plasma deuterium-labeled CEHC. Ann N Y Acad Sci. 2004;1031:357-360. (PubMed)
18. Bruno RS, Leonard SW, Atkinson J, et al. Faster plasma vitamin E disappearance in smokers is normalized by vitamin C supplementation. Free Radic Biol Med. 2006;40(4):689-697. (PubMed)
19. Booth SL, Golly I, Sacheck JM, et al. Effect of vitamin E supplementation on vitamin K status in adults with normal coagulation status. Am J Clin Nutr. 2004;80(1):143-148. (PubMed)
20. Pastori D, Carnevale R, Cangemi R, et al. Vitamin E serum levels and bleeding risk in patients receiving oral anticoagulant therapy: a retrospective cohort study. J Am Heart Assoc. 2013;2(6):e000364. (PubMed)
21. Traber MG. Vitamin E. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Philadelphia: Lippincott Williams & Wilkins; 2014:293-304.
22. Berson EL, Rosner B, Sandberg MA, et al. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(6):761-772. (PubMed)
23. Iwasa K, Shima K, Komai K, Nishida Y, Yokota T, Yamada M. Retinitis pigmentosa and macular degeneration in a patient with ataxia with isolated vitamin E deficiency with a novel c.717 del C mutation in the TTPA gene. J Neurol Sci. 2014;345(1-2):228-230. (PubMed)
24. Ford ES, Sowell A. Serum α-tocopherol status in the United States population: findings from the Third National Health and Nutrition Examination Survey. Am J Epidemiol. 1999;150(3):290-300. (PubMed)
25. Fulgoni VL, 3rd, Keast DR, Bailey RL, Dwyer J. Foods, fortificants, and supplements: Where do Americans get their nutrients? J Nutr. 2011;141(10):1847-1854. (PubMed)
26. Shamim AA, Schulze K, Merrill RD, et al. First-trimester plasma tocopherols are associated with risk of miscarriage in rural Bangladesh. Am J Clin Nutr. 2015;101(2):294-301. (PubMed)
27. Wright ME, Lawson KA, Weinstein SJ, et al. Higher baseline serum concentrations of vitamin E are associated with lower total and cause-specific mortality in the Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study. Am J Clin Nutr. 2006;84(5):1200-1207. (PubMed)
28. Wu D, Meydani SN. Age-associated changes in immune function: impact of vitamin E intervention and the underlying mechanisms. Endocr Metab Immune Disord Drug Targets. 2014;14(4):283-289. (PubMed)
29. De la Fuente M, Hernanz A, Guayerbas N, Victor VM, Arnalich F. Vitamin E ingestion improves several immune functions in elderly men and women. Free Radic Res. 2008;42(3):272-280. (PubMed)
30. Meydani SN, Meydani M, Blumberg JB, et al. Vitamin E supplementation and in vivo immune response in healthy elderly subjects. A randomized controlled trial. JAMA. 1997;277(17):1380-1386. (PubMed)
31. Pallast EG, Schouten EG, de Waart FG, et al. Effect of 50- and 100-mg vitamin E supplements on cellular immune function in noninstitutionalized elderly persons. Am J Clin Nutr. 1999;69(6):1273-1281. (PubMed)
32. Meydani SN, Leka LS, Fine BC, et al. Vitamin E and respiratory tract infections in elderly nursing home residents: a randomized controlled trial. JAMA. 2004;292(7):828-836. (PubMed)
33. Knekt P, Reunanen A, Jarvinen R, Seppanen R, Heliovaara M, Aromaa A. Antioxidant vitamin intake and coronary mortality in a longitudinal population study. Am J Epidemiol. 1994;139(12):1180-1189. (PubMed)
34. Kushi LH, Folsom AR, Prineas RJ, Mink PJ, Wu Y, Bostick RM. Dietary antioxidant vitamins and death from coronary heart disease in postmenopausal women. N Engl J Med. 1996;334(18):1156-1162. (PubMed)
35. Rimm EB, Stampfer MJ, Ascherio A, Giovannucci E, Colditz GA, Willett WC. Vitamin E consumption and the risk of coronary heart disease in men. N Engl J Med. 1993;328(20):1450-1456. (PubMed)
36. Stampfer MJ, Hennekens CH, Manson JE, Colditz GA, Rosner B, Willett WC. Vitamin E consumption and the risk of coronary disease in women. N Engl J Med. 1993;328(20):1444-1449. (PubMed)
37. Lee IM, Cook NR, Gaziano JM, et al. Vitamin E in the primary prevention of cardiovascular disease and cancer: the Women's Health Study: a randomized controlled trial. JAMA. 2005;294(1):56-65. (PubMed)
38. Glynn RJ, Ridker PM, Goldhaber SZ, Zee RY, Buring JE. Effects of random allocation to vitamin E supplementation on the occurrence of venous thromboembolism: report from the Women's Health Study. Circulation. 2007;116(13):1497-1503. (PubMed)
39. Violi F, Pignatelli P. Letter by Violi and Pignatelli regarding article, "Effects of random allocation to vitamin E supplementation on the occurrence of venous thromboembolism: report from the Women's Health Study". Circulation. 2008;117(15):e312; author reply e313. (PubMed)
40. Sesso HD, Buring JE, Christen WG, et al. Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians' Health Study II randomized controlled trial. JAMA. 2008;300(18):2123-2133. (PubMed)
41. Riccioni G, D'Orazio N, Palumbo N, et al. Relationship between plasma antioxidant concentrations and carotid intima-media thickness: the Asymptomatic Carotid Atherosclerotic Disease In Manfredonia Study. Eur J Cardiovasc Prev Rehabil. 2009;16(3):351-357. (PubMed)
42. Riccioni G, Bazzano LA. Antioxidant plasma concentration and supplementation in carotid intima media thickness. Expert Rev Cardiovasc Ther. 2008;6(5):723-729. (PubMed)
43. Azen SP, Qian D, Mack WJ, et al. Effect of supplementary antioxidant vitamin intake on carotid arterial wall intima-media thickness in a controlled clinical trial of cholesterol lowering. Circulation. 1996;94(10):2369-2372. (PubMed)
44. Joris PJ, Mensink RP. Effects of supplementation with the fat-soluble vitamins E and D on fasting flow-mediated vasodilation in adults: a meta-analysis of randomized controlled trials. Nutrients. 2015;7(3):1728-1743. (PubMed)
45. Stephens NG, Parsons A, Schofield PM, Kelly F, Cheeseman K, Mitchinson MJ. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Lancet. 1996;347(9004):781-786. (PubMed)
46. Boaz M, Smetana S, Weinstein T, et al. Secondary prevention with antioxidants of cardiovascular disease in endstage renal disease (SPACE): randomised placebo-controlled trial. Lancet. 2000;356(9237):1213-1218. (PubMed)
47. Milman U, Blum S, Shapira C, et al. Vitamin E supplementation reduces cardiovascular events in a subgroup of middle-aged individuals with both type 2 diabetes mellitus and the haptoglobin 2-2 genotype: a prospective double-blinded clinical trial. Arterioscler Thromb Vasc Biol. 2008;28(2):341-347. (PubMed)
48. Leppala JM, Virtamo J, Fogelholm R, et al. Controlled trial of α-tocopherol and β-carotene supplements on stroke incidence and mortality in male smokers. Arterioscler Thromb Vasc Biol. 2000;20(1):230-235. (PubMed)
49. Lonn E, Bosch J, Yusuf S, et al. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA. 2005;293(11):1338-1347. (PubMed)
50. Marchioli R, Levantesi G, Macchia A, et al. Vitamin E increases the risk of developing heart failure after myocardial infarction: Results from the GISSI-Prevenzione trial. J Cardiovasc Med (Hagerstown). 2006;7(5):347-350. (PubMed)
51. Dizdaroglu M. Oxidatively induced DNA damage: mechanisms, repair and disease. Cancer Lett. 2012;327(1-2):26-47. (PubMed)
52. Slatore CG, Littman AJ, Au DH, Satia JA, White E. Long-term use of supplemental multivitamins, vitamin C, vitamin E, and folate does not reduce the risk of lung cancer. Am J Respir Crit Care Med. 2008;177(5):524-530. (PubMed)
53. Heinonen OP, Albanes D, Virtamo J, et al. Prostate cancer and supplementation with α-tocopherol and β-carotene: incidence and mortality in a controlled trial. J Natl Cancer Inst. 1998;90(6):440-446. (PubMed)
54. Virtamo J, Taylor PR, Kontto J, et al. Effects of α-tocopherol and β-carotene supplementation on cancer incidence and mortality: 18-year postintervention follow-up of the Alpha-tocopherol, Beta-carotene Cancer Prevention Study. Int J Cancer. 2014;135(1):178-185. (PubMed)
55. Wang L, Sesso HD, Glynn RJ, et al. Vitamin E and C supplementation and risk of cancer in men: posttrial follow-up in the Physicians' Health Study II randomized trial. Am J Clin Nutr. 2014;100(3):915-923. (PubMed)
56. Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2009;301(1):39-51. (PubMed)
57. Klein EA, Thompson IM, Jr., Tangen CM, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2011;306(14):1549-1556. (PubMed)
58. Kristal AR, Darke AK, Morris JS, et al. Baseline selenium status and effects of selenium and vitamin e supplementation on prostate cancer risk. J Natl Cancer Inst. 2014;106(3):djt456. (PubMed)
59. Cheng TY, Barnett MJ, Kristal AR, et al. Genetic variation in myeloperoxidase modifies the association of serum α-tocopherol with aggressive prostate cancer among current smokers. J Nutr. 2011;141(9):1731-1737. (PubMed)
60. Gerstenberger JP, Bauer SR, Van Blarigan EL, et al. Selenoprotein and antioxidant genes and the risk of high-grade prostate cancer and prostate cancer recurrence. Prostate. 2015;75(1):60-69. (PubMed)
61. Major JM, Yu K, Weinstein SJ, et al. Genetic variants reflecting higher vitamin e status in men are associated with reduced risk of prostate cancer. J Nutr. 2014;144(5):729-733. (PubMed)
62. Katta AV, Katkam RV, Geetha H. Lipid peroxidation and the total antioxidant status in the pathogenesis of age related and diabetic cataracts: a study on the lens and blood. J Clin Diagn Res. 2013;7(6):978-981. (PubMed)
63. Ferrigno L, Aldigeri R, Rosmini F, Sperduto RD, Maraini G, Italian-American Cataract Study G. Associations between plasma levels of vitamins and cataract in the Italian-American Clinical Trial of Nutritional Supplements and Age-Related Cataract (CTNS): CTNS Report #2. Ophthalmic Epidemiol. 2005;12(2):71-80. (PubMed)
64. Krepler K, Schmid R. α-Tocopherol in plasma, red blood cells and lenses with and without cataract. Am J Ophthalmol. 2005;139(2):266-270. (PubMed)
65. West AL, Oren GA, Moroi SE. Evidence for the use of nutritional supplements and herbal medicines in common eye diseases. Am J Ophthalmol. 2006;141(1):157-166. (PubMed)
66. Zhang Y, Jiang W, Xie Z, Wu W, Zhang D. Vitamin E and risk of age-related cataract: a meta-analysis. Public Health Nutr. 2015:1-11. (PubMed)
67. Zheng Selin J, Rautiainen S, Lindblad BE, Morgenstern R, Wolk A. High-dose supplements of vitamins C and E, low-dose multivitamins, and the risk of age-related cataract: a population-based prospective cohort study of men. Am J Epidemiol. 2013;177(6):548-555. (PubMed)
68. Teikari JM, Rautalahti M, Haukka J, et al. Incidence of cataract operations in Finnish male smokers unaffected by α-tocopherol or β-carotene supplements. J Epidemiol Community Health. 1998;52(7):468-472. (PubMed)
69. Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E and β-carotene for age-related cataract and vision loss: AREDS report no. 9. Arch Ophthalmol. 2001;119(10):1439-1452. (PubMed)
70. Christen WG, Glynn RJ, Gaziano JM, et al. Age-related cataract in men in the selenium and vitamin E cancer prevention trial eye endpoints study: a randomized clinical trial. JAMA Ophthalmol. 2015;133(1):17-24. (PubMed)
71. Gritz DC, Srinivasan M, Smith SD, et al. The Antioxidants in Prevention of Cataracts Study: effects of antioxidant supplements on cataract progression in South India. Br J Ophthalmol. 2006;90(7):847-851. (PubMed)
72. McNeil JJ, Robman L, Tikellis G, Sinclair MI, McCarty CA, Taylor HR. Vitamin E supplementation and cataract: randomized controlled trial. Ophthalmology. 2004;111(1):75-84. (PubMed)
73. Evans JR, Lawrenson JG. Antioxidant vitamin and mineral supplements for preventing age-related macular degeneration. Cochrane Database Syst Rev. 2012;6:CD000253. (PubMed)
74. Evans JR, Lawrenson JG. Antioxidant vitamin and mineral supplements for slowing the progression of age-related macular degeneration. Cochrane Database Syst Rev. 2012;11:CD000254. (PubMed)
75. Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, β-carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119(10):1417-1436. (PubMed)
76. Hobbs RP, Bernstein PS. Nutrient Supplementation for Age-related Macular Degeneration, Cataract, and Dry Eye. J Ophthalmic Vis Res. 2014;9(4):487-493. (PubMed)
77. Pazdro R, Burgess JR. The role of vitamin E and oxidative stress in diabetes complications. Mech Ageing Dev. 2010;131(4):276-286. (PubMed)
78. Kataja-Tuomola MK, Kontto JP, Mannisto S, Albanes D, Virtamo JR. Effect of α-tocopherol and β-carotene supplementation on macrovascular complications and total mortality from diabetes: results of the ATBC Study. Ann Med. 2010;42(3):178-186. (PubMed)
79. Xu R, Zhang S, Tao A, Chen G, Zhang M. Influence of vitamin E supplementation on glycaemic control: a meta-analysis of randomised controlled trials. PLoS One. 2014;9(4):e95008. (PubMed)
80. Montero D, Walther G, Stehouwer CD, Houben AJ, Beckman JA, Vinet A. Effect of antioxidant vitamin supplementation on endothelial function in type 2 diabetes mellitus: a systematic review and meta-analysis of randomized controlled trials. Obes Rev. 2014;15(2):107-116. (PubMed)
81. Dyson JK, Anstee QM, McPherson S. Non-alcoholic fatty liver disease: a practical approach to diagnosis and staging. Frontline Gastroenterol. 2014;5(3):211-218. (PubMed)
82. Musso G, Cassader M, Rosina F, Gambino R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): a systematic review and meta-analysis of randomised trials. Diabetologia. 2012;55(4):885-904. (PubMed)
83. Chalasani NP, Sanyal AJ, Kowdley KV, et al. Pioglitazone versus vitamin E versus placebo for the treatment of non-diabetic patients with non-alcoholic steatohepatitis: PIVENS trial design. Contemp Clin Trials. 2009;30(1):88-96. (PubMed)
84. Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362(18):1675-1685. (PubMed)
85. Lavine JE, Schwimmer JB, Van Natta ML, et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA. 2011;305(16):1659-1668. (PubMed)
86. Ji HF. Vitamin E therapy on aminotransferase levels in NAFLD/NASH patients. Nutrition. 2015;31(6):899. (PubMed)
87. D'Adamo E, Marcovecchio ML, Giannini C, et al. Improved oxidative stress and cardio-metabolic status in obese prepubertal children with liver steatosis treated with lifestyle combined with Vitamin E. Free Radic Res. 2013;47(3):146-153. (PubMed)
88. Zhao Y, Zhao B. Oxidative stress and the pathogenesis of Alzheimer's disease. Oxid Med Cell Longev. 2013;2013:316523. (PubMed)
89. Lopes da Silva S, Vellas B, Elemans S, et al. Plasma nutrient status of patients with Alzheimer's disease: Systematic review and meta-analysis. Alzheimers Dement. 2014;10(4):485-502. (PubMed)
90. Kontush K, Schekatolina S. Vitamin E in neurodegenerative disorders: Alzheimer's disease. Ann N Y Acad Sci. 2004;1031:249-262. (PubMed)
91. Sano M, Ernesto C, Thomas RG, et al. A controlled trial of selegiline, α-tocopherol, or both as treatment for Alzheimer's disease. The Alzheimer's Disease Cooperative Study. N Engl J Med. 1997;336(17):1216-1222. (PubMed)
92. Petersen RC, Thomas RG, Grundman M, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005;352(23):2379-2388. (PubMed)
93. Lloret A, Badia MC, Mora NJ, Pallardo FV, Alonso MD, Vina J. Vitamin E paradox in Alzheimer's disease: it does not prevent loss of cognition and may even be detrimental. J Alzheimers Dis. 2009;17(1):143-149. (PubMed)
94. Dysken MW, Sano M, Asthana S, et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA. 2014;311(1):33-44. (PubMed)
95. Kang JH, Cook N, Manson J, Buring JE, Grodstein F. A randomized trial of vitamin E supplementation and cognitive function in women. Arch Intern Med. 2006;166(22):2462-2468. (PubMed)
96. Gao X, Wilde PE, Lichtenstein AH, Bermudez OI, Tucker KL. The maximal amount of dietary α-tocopherol intake in U.S. adults (NHANES 2001-2002). J Nutr. 2006;136(4):1021-1026. (PubMed)
97. Cheeseman KH, Holley AE, Kelly FJ, Wasil M, Hughes L, Burton G. Biokinetics in humans of RRR-α-tocopherol: the free phenol, acetate ester, and succinate ester forms of vitamin E. Free Radic Biol Med. 1995;19(5):591-598. (PubMed)
98. Duhem N, Danhier F, Preat V. Vitamin E-based nanomedicines for anti-cancer drug delivery. J Control Release. 2014;182:33-44. (PubMed)
99. Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. 2nd edition ed: Thomson Reuters; 2008.
100. Schurks M, Glynn RJ, Rist PM, Tzourio C, Kurth T. Effects of vitamin E on stroke subtypes: meta-analysis of randomised controlled trials. BMJ. 2010;341:c5702. (PubMed)
101. Brion LP, Bell EF, Raghuveer TS, Soghier L. What is the appropriate intravenous dose of vitamin E for very-low-birth-weight infants? J Perinatol. 2004;24(4):205-207. (PubMed)
102. Dietrich M, Jacques PF, Pencina MJ, et al. Vitamin E supplement use and the incidence of cardiovascular disease and all-cause mortality in the Framingham Heart Study: Does the underlying health status play a role? Atherosclerosis. 2009;205(2):549-553. (PubMed)
103. Miller ER, 3rd, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med. 2005;142(1):37-46. (PubMed)
104. Bjelakovic G, Nikolova D, Gluud C. Meta-regression analyses, meta-analyses, and trial sequential analyses of the effects of supplementation with β-carotene, vitamin A, and vitamin E singly or in different combinations on all-cause mortality: do we have evidence for lack of harm? PLoS One. 2013;8(9):e74558. (PubMed)
105. https://naturalmedicines.therapeuticresearch.com/. Vitamin E professional monograph. Natural Medicines; 2014 Copyright.
106. Brown BG, Zhao XQ, Chait A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345(22):1583-1592. (PubMed)
107. Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360(9326):23-33. (PubMed)
Vitamina K
Contenido
Resumen
- Las formas de la vitamina K que se originan de manera natural incluyen la filoquinona (vitamina K1) y una familia de moléculas llamadas menaquinonas (MKs o vitamina K2). (Más información)
- Con una capacidad de almacenaje limitada de vitamina K, el cuerpo recicla la vitamina K en el ciclo de oxidación y reducción de la vitamina K a fin de que sea reutilizada varias veces. (Más información)
- La vitamina K es un cofactor esencial para la carboxilación de residuos de ácido glutámico en muchas proteínas dependientes de la vitamina K que están involucradas en la coagulación sanguínea, el metabolismo óseo, la prevención de la mineralización de vasos, y la regulación de varias funciones celulares. (Más información)
- La deficiencia de vitamina K incrementa el riesgo de sangrado excesivo (hemorragia). Una inyección de vitamina K es recomendada para proteger a todos los recién nacidos de sangrados dentro del cráneo potencialmente mortales. (Más información)
- El nivel de ingesta adecuada (IA) para la vitamina K está establecido en 90 μg/día para mujeres y 120 μg/día para hombres. (Más información)
- La deficiencia de vitamina K puede perjudicar la actividad de las proteínas dependientes de vitamina K e incrementar el riesgo de osteoporosis y fracturas. A pesar de todo, estudios basados en la observación han fallado en aislar las ingesta de vitamina K de dietas generalmente saludables, lo cual justifica una interpretación cautelosa de asociaciones positivas entre la ingesta de vitamina K y marcadores de la salud ósea. En general, ensayos de intervención han sido inconclusos con respecto al papel de la vitamina K suplementaria en la fomentación de la reducción de la perdida de hueso en adultos de otro modo repletos de calcio y vitamina D. (Más información).
- La mineralización anormal de los vasos sanguíneos incrementa con la edad y es un factor de riesgo principal para las enfermedades cardiovasculares. La insuficiencia de vitamina K puede inactivar varias proteínas dependientes de la vitamina K que inhiben la formación de precipitados de calcio en los vasos. El efecto de la vitamina K suplementaria en la prevención de la calcificación de vasos y eventos cardiovasculares aún necesita ser evaluado en ensayos controlados aleatorios. (Más información)
- La filoquinona se encuentra en altas concentraciones en vegetales de hojas verdes, y ciertos aceites vegetales, mientras que la mayoría de menaquinonas se encuentran en hígados animales y alimentos fermentados. (Más información)
- Varias drogas/fármacos, incluyendo antagonistas de la vitamina K (p. ej., la warfarina), son conocidos por interferir con la absorción y el metabolismo de la vitamina K. (Más información)
La vitamina K es un vitamina liposoluble. Originalmente identificada por su papel en el proceso de la formación de coágulos sanguíneos (la "K" se deriva de la palabra alemana "koagulation"), la vitamina K es esencial para el funcionamiento de varias proteínas involucrada en los procesos fisiológicos que abarca, pero no se limitan a la regulación de coágulos sanguíneos (coagulación) (1). Las formas de la vitamina K que se originan de forma natural incluyen un cierto número de vitámeros conocidos como vitamina K1 y vitamina K2 (Figura 1). La vitamina K1 o filoquinona es sintetizada por plantas y es la forma predominante en la dieta. La vitamina K2 incluye un rango de formas de la vitamina K colectivamente referidas como menaquinonas. La mayoría de las menaquinonas son sintetizadas por la microbiota intestinal humana y se encuentran en alimentos fermentados y productos de origen animal. Las menaquinonas difieren en la longitud de 1 a 14 repeticiones de unidades de 5 carbonos en la cadena lateral de las moléculas. Estas formas de la vitamina K son designadas como menaquinona-n (MK-n), donde la n representa el número de unidades de 5 carbonos (MK-2 a MK-14) (2, 3). Ampliamente usado en la cría de animales, el compuesto sintético conocido como menadiona (vitamina K3) es una provitamina que necesita ser convertida a menaquinona-4 (MK-4) para ser activa (4).
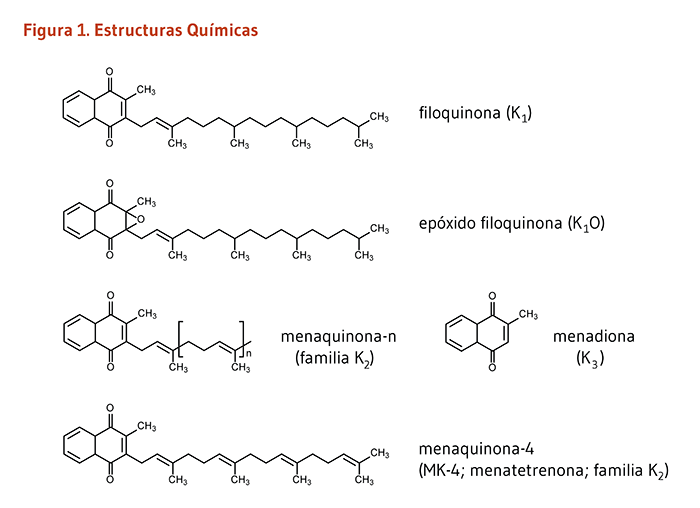
Función
Las funciones de la vitamina K como un cofactor para la enzima, γ-glutamil carboxilasa (GGCX), la cual cataliza la carboxilación del aminoácido ácido glutámico (Glu), a ácido γ-carboxiglutámico (Gla). La γ-carboxilación dependiente de vitamina K que ocurre solo en residuos específicos de ácido glutámico en proteínas dependientes de vitamina K (VKDP) identificadas es fundamental por su habilidad de unir al calcio (5).
Ciclo de oxidación-reducción de la vitamina K
Aunque la vitamina K es una vitamina liposoluble, el cuerpo almacena cantidades muy pequeñas que son rápidamente agotadas sin una ingesta dietaría regular. Talvez debido a su limitada habilidad de almacenar vitamina K, el cuerpo la recicla a través de un proceso llamado ciclo de la vitamina K-epóxido (Figura 2). El ciclo de la vitamina K permite que una pequeña cantidad de vitamina K sea reusada muchas veces en la carboxilación de proteínas disminuyendo así el requerimiento dietario. Brevemente, la vitamina K hidroquinona (una forma reducida) es oxidada a vitamina K epóxido (una forma oxidada). La reacción permite a la γ-glutamil carboxilasa carboxilizar residuos de ácido glutámico selectivos en las proteínas dependientes de la vitamina K. El reciclado de la vitamina K epóxido (forma oxidada) a hidroquinona (forma reducida) es llevada a cabo por dos reacciones que reducen la vitamina K epóxido (KO) a vitamina K quinona y después a vitamina K hidroquinona (KH2; Figura 2). Además, la enzima vitamina K oxidorreductasa (VKOR) cataliza la reducción de la KO a vitamina K quinona y puede estar involucrada — así como otra aun-por-definirse reductasa — en la producción de KH2 proveniente de la vitamina K quinona (6, 7). La droga anticoagulante warfarina actúa con un antagonista de la vitamina K al inhibir la actividad de la VKOR, previniendo por lo tanto el reciclaje de la vitamina K (véase Coagulación).
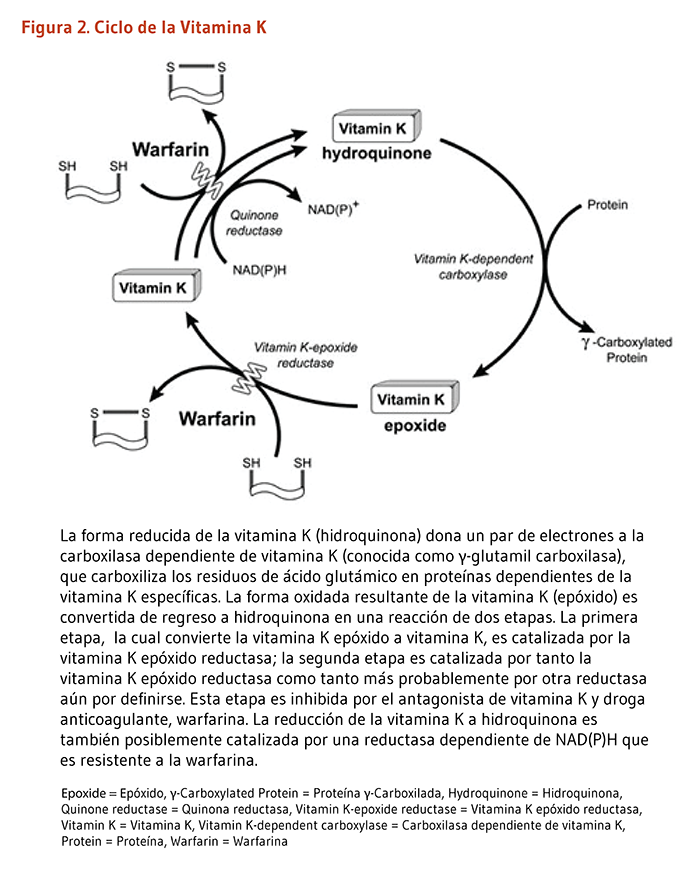
Coagulación
La habilidad para unir iones de calcio (Ca2+) es requerida para la activación de varios factores de la coagulación dependientes de la vitamina K, o proteínas, en la cascada de la coagulación. El termino, cascada de coagulación, se refiere a una serie de eventos, cada uno dependiente del otro, que detienen el sangrado a través de la formación de coágulos. La γ-carboxilación dependiente de vitamina K de residuos específicos de ácido glutámico en tales proteínas hace posible que puedan unir al calcio. Los factores II (protrombina), VII, IX, y X conforman el núcleo de la cascada de la coagulación. La proteína Z parece aumentar la acción de la trombina (forma activada de la protrombina) al promover su asociación con fosfolípidos en las membranas celulares. La proteína C y la proteína S son proteínas anticoagulantes que proveen control y balance en la cascada de la coagulación; la proteína Z también posee una función anticoagulante. Los mecanismos de control de la cascada de la coagulación existen, ya que la coagulación descontrolada puede poner en riesgo la vida como el sangrado descontrolado. Los factores de la coagulación dependientes de vitamina K son sintetizados en el hígado. Como consecuencia, las enfermedades hepáticas graves resultan en bajos niveles sanguíneos de los factores de la coagulación dependientes de la vitamina K y en un riesgo incrementado de sangrado descontrolado (hemorragia) (8).
Terapia oral anticoagulante con antagonistas de la vitamina K
Algunas personas están en un riesgo incrementado de formar coágulos, los cuales podrían bloquear el flujo de la sangre en las arterias del corazón, cerebro o pulmones, resultando en un infarto al miocardio (ataque al corazón), accidentes cerebrovasculares o embolia pulmonar respectivamente. La coagulación anormal no está relacionada con la ingesta excesiva de vitamina K, y no se conoce de alguna toxicidad asociada con la vitamina K1 o la vitamina K2 (véase Toxicidad). Algunos anticoagulantes orales, como la warfarina (Coumadin, Jantoven), inhiben la coagulación a través del antagonismo de la acción de la vitamina K. La warfarina previene el reciclaje de la vitamina K al bloquear la actividad de la VKOR, creando así una deficiencia funcional de vitamina K (véase Figura 2 arriba). La γ-carboxilación inadecuada de proteínas de la coagulación dependientes de la vitamina K interfiere con la cascada de coagulación, lo que inhibe la formación de coágulos. Grandes cantidades de vitamina K dietaría o suplementaria podrían superar el efecto anticoagulante de los antagonistas de la vitamina K, por lo que aquellos pacientes que toman estos medicamentos se les advierte que no consuman cantidades muy grandes o altamente variables de vitamina K (véase Interacción con drogas). Los expertos recomiendan ahora a los pacientes en tratamiento con antagonistas de vitamina K como la warfarina, una ingesta dietaría razonablemente constante que satisfaga las recomendaciones dietarías actuales (90-120 μg/día) (9). Finalmente, debido a la alta variabilidad en la respuesta de pacientes a los antagonistas de la vitamina K, se ha sugerido que la suplementación diaria de dosis bajas de filoquinona puede mejorar la estabilidad de la terapia de anticoagulación. A pesar de todo, varios meta-análisis recientemente destacaron la falta de evidencia suficiente que apoye esta opción para aquellos que toman warfarina (10-12).
Formación esquelética y prevención de calcificación de tejido blando
La γ-carboxilación dependiente de la vitamina K es esencial en varias proteínas relacionadas con los huesos, incluyendo la osteocalcina, el factor de anticoagulación de la proteína S, la proteína glutamato γ-carboxilada (Gla) de la matriz (MGP), la proteína rica en Gla (GRP), y la periostina (originalmente llamada factor osteoblástico específico 2). La osteocalcina (también llamada proteína Gla ósea) es sintetizada por los osteoblastos (células formadoras de hueso); la síntesis de la osteocalcina es regulada por la forma activa de la vitamina D, 1,25-dihidroxivitamina D (calcitriol). La capacidad de unión del calcio de la osteocalcina requiere la γ-carboxilación dependiente de vitamina K de tres residuos de ácido glutámico. Aunque su función en la mineralización ósea no es comprendida del todo, la osteocalcina es requerida para el crecimiento y maduración de los cristales de hidroxiapatita de calcio (véase Osteoporosis) (13).
La proteína S parece desempeñar un papel en la degradación ósea mediada por los osteoclastos. Individuos con una deficiencia hereditaria de la proteína S padecen de complicaciones relacionadas al incremento de coagulación sanguínea, como también de osteonecrosis (14, 15). La proteína S puede unirse y activar los receptores de la familia TAM que están involucrados en fagocitosis. Mutaciones en los receptores TAM pueden resultar en discapacidad visual, espermatogénesis defectuosa, trastornos autoinmunes, y trastornos plaquetarios (16).
La MGP ha sido encontrada en cartílagos, tejido óseo y blando, incluyendo las paredes de vasos sanguinos, donde es secretada por células del músculo liso vascular. La MPG se ha involucrado en la prevención de la calcificación en varios sitios, incluyendo cartílagos, paredes de vasos sanguíneos, fibras elásticas de la piel, o en la malla trabecular en el ojo humano (véase Calcificación vascular) (17). Además, varias VKDP, incluyendo la MGP, han sido asociadas con sitios de calcificación en las arterias, piel, riñones, y ojos en ciertas enfermedades hereditarias, como el pseudoxantoma elástico y la β-talasemia (18, 19).
Las proteínas dependientes de la vitamina K, la GRP y la periostina, son también sintetizadas en el tejido óseo, pero sus papeles en el metabolismo óseo no son claros aun (20, 21). Expresada en la piel humana normal y los tejidos vasculares, la GRP ha sido colocalizada con depósitos minerales anormales en la matriz extracelular en arterias calcificadas y lesiones de la piel calcificadas (22).
Expresada en la mayoría de tejidos conectivos, incluyendo piel y huesos, la periostina fue inicialmente asociada con adhesión y migración celular. Esta VKDP también parece promover la angiogénesis (formación de nuevos vasos sanguíneos) durante la degeneración de la válvula cardiaca y el crecimiento tumoral (23, 24).
Investigación actual sugiere que la actividad reducida de la GGCX y/o la baja biodisponibilidad de la vitamina K puede perjudicar la actividad de las VKPD y contribuir a defectos de la mineralización ósea y a la calcificación anormal del tejido blando (véase Prevención de Enfermedades) (25).
Regulación de funciones celulares
La proteína específica del gen 6 de la detención de crecimiento (Gas6) es un proteína dependiente de la vitamina K identificada en 1993. Ha sido encontrada a lo largo del sistema nervioso, así como en el corazón, pulmones, estómago, riñones, y cartílago. Identificada como un ligando de la familia TAM de receptores de la tirosina quinasa de transmembrana, la Gas6 parece ser un factor de la regulación del crecimiento celular con actividades de señalización celular. La Gas6 ha sido involucrada en funciones celulares diversas, incluyendo fagocitosis, adhesión celular, proliferación celular, y protección contra apoptosis (5). Puede también desempeñar papeles importantes en el desarrollo y envejecimiento del sistema nervioso (revisado en 26). Además, la Gas6 parece regular la señalización plaquetaria y la hemostasia vascular (27). Expresada en la mayoría de tejidos e involucrada en muchas funciones celulares, la Gas6 ha sido también ligada a varias afecciones patológicas, incluyendo la formación de coágulos (trombogénesis), aterosclerosis, inflamación crónica, y crecimiento del cáncer (28-30).
Deficiencia
La deficiencia evidente de vitamina K resulta en una coagulación sanguínea deteriorada, usualmente demostrada por pruebas de laboratorio que miden el tiempo de coagulación. Los síntomas incluyen moretones y sangrado con facilidad que podrían manifestarse como hemorragias nasales, encías sangrantes, sangre en la orina, sangre en las deposiciones, deposiciones negras, o sangramiento menstrual extremadamente profuso. En infantes, la deficiencia de vitamina K puede resultar en hemorragias potencialmente letales dentro del cráneo (hemorragia intracraneal) (8).
Adultos
La deficiencia de vitamina K es poco común en adultos saludables por un cierto número de razones: (1) la vitamina K es ampliamente distribuida en los alimentos (véase Fuentes alimenticias); (2) el ciclo de la vitamina K conserva la vitamina K (véase Ciclo de oxidación-reducción de la vitamina K); y (3) las bacterias que normalmente habitan el intestino grueso sintetizan menaquinonas (vitamina K2), aunque no está claro si cantidades significativas son absorbidas o utilizadas (véase Fuentes alimenticias). Los adultos en riesgo de una deficiencia de vitamina K incluye a aquellos que consumen antagonistas de vitamina K e individuos con enfermedades o daño hepático severo (8). Además, las personas con desordenes de malabsorción de grasas incluyendo enfermedad intestinal inflamatoria y fibrosis quística podrían encontrarse en un mayor riesgo de deficiencia de vitamina K (31-33).
Infantes
Los bebes recién nacidos que son exclusivamente amamantados con leche materna se encuentran en un riesgo incrementado de deficiencia de vitamina K, debido a que la leche humana es relativamente baja en vitamina K en comparación con la leche formulada. En general, los recién nacidos tienen un bajo estatus de vitamina K por las siguientes razones: (1) el transporte de la vitamina K a través de la barrera placentaria es limitada; (2) el almacenamiento de la vitamina K en el hígado es bastante bajo; (3) el ciclo de la vitamina K puede aún no ser totalmente funcional en recién nacidos, especialmente en infantes prematuros; y (4) el contenido de vitamina K de la leche materna es bajo (5). Infantes cuyas madres consumen medicamentos anticonvulsivos para prevenir convulsiones están también en riesgo de una deficiencia de vitamina K. La deficiencia de vitamina K en recién nacidos puede resultar en un trastorno hemorrágico llamado sangrado por deficiencia de vitamina K (SDVK) del recién nacido (revisado en 34). Debido a que el SDVK pone en riesgo la vida y es fácilmente evitado, la Academia Americana de Pediatras y un cierto número de organizaciones internacionales similares recomiendan que una dosis intramuscular de filoquinona (vitamina K1) sea administrada a todos los recién nacidos (35).
Controversias acerca de la administración de vitamina K a recién nacidos
La vitamina K y leucemia infantil: A principios de 1990, se publicaron dos estudios retrospectivos que sugerían una posible asociación entre las inyecciones de filoquinona en recién nacidos y el desarrollo de leucemia infantil y otras formas de cáncer infantil. Sin embargo, dos grandes estudios retrospectivos en los EE.UU. y Suecia los cuales revisaron los registros médicos de 54,000 y 1.3 millones de niños, respectivamente, no encontraron evidencia de una relación entre cánceres infantiles e inyecciones de filoquinona al momento de nacer (36, 37). Además, un análisis agrupado de seis estudios de caso y control, incluyendo a 2,431 niños diagnosticados con cáncer infantil y 6,338 niños libres de cáncer, no encontró evidencia de que las inyecciones de filoquinona en recién nacidos incrementaran el riesgo de leucemia infantil (38). En una declaración de política, la Academia Americana de Pediatría recomendó que se debe continuar la profilaxis de rutina con vitamina K en recién nacidos debido a que la SDVK es potencialmente mortal y los riesgos de cáncer son improbables y no han sido demostrados (35). En los últimos años, doctores han reportado un incremento en casos de aparición tardía de SDVK debido a un una tendencia creciente de omisión o negativa parental del recién nacido a la profilaxis con vitamina K (39).
Dosis menores de vitamina K1 en infantes prematuros: Los resultados de dos estudios de los niveles de vitamina K en infantes prematuros sugiere que la dosis estándar inicial de filoquinona (vitamina K1) para infantes de termino (1.0 mg) podría ser demasiado elevada para infantes prematuros (40, 41). Estos hallazgos han llevado a algunos expertos a sugerir el uso de una dosis inicial de vitamina K1 de 0.3 mg/kg para infantes con pesos al nacer menores a 1,000 g (2 lbs, 3 oz), y una dosis inicial de 0.5 mg de filoquinona probablemente prevendría enfermedades hemorrágicas en recién nacidos (40).
La Ingesta Adecuada (IA)
En enero del 2001, la Junta de Nutrición y Alimentos (JNA) del Instituto de Medicina de los EE.UU. estableció el nivel de ingesta adecuada (IA) de la vitamina K en base a los niveles de consumo de individuos saludables (Tabla 1). La IA para infantes se basó en la ingesta estimada de vitamina K proveniente de la leche materna (42).
Prevención de Enfermedades
Osteoporosis
El descubrimiento de las proteínas dependientes de la vitamina K en el hueso llevo a la investigación del papel de la vitamina K en el mantenimiento de la salud ósea.
La vitamina K y la salud ósea: estudios basados en la observación
Vitamina K1: Estudios basados en la observación han encontrado una relación entre la filoquinona (vitamina K1) y la pérdida ósea relacionada con la edad (osteoporosis). El Estudio de Salud de las Enfermeras (Nurses' Health Study) dio seguimiento a más de 72,000 mujeres por diez años. En un análisis de esta cohorte, las mujeres cuyas ingestas de filoquinona fueron menores a 109 microgramos/día (μg/día) tuvieron un riesgo 30% más alto de fractura de cadera en comparación a aquellas mujeres con ingestas iguales o por encima de los 109 μg/día (43). Otro estudio prospectivo en más de 800 hombres y mujeres de la tercera edad, con un seguimiento en el Estudio del Corazón de Framingham (Framingham Heart Study) por siete años, encontró que los participantes con ingestas de vitamina K dietaría en el cuartil más alto (mediana, 254 μg/día) tuvieron un riesgo 65% más bajo de fractura de cadera que aquellos con ingestas en el cuartil más bajo (mediana, 56 μg/día) (44). Las fracturas osteoporóticas son frecuentemente ligadas a una reducción en la mineralización ósea. A pesar de todo, los investigadores no encontraron alguna asociación entre la ingesta dietaría de filoquinona y la densidad mineral ósea (DMO) en los sujetos del estudio de Framingham (44). Mientras que otros estudios fallaron en observar asociaciones entre la ingesta de filoquinona dietaría y mediciones de la resistencia ósea, DMO, o la incidencia de fractura (45, 46), el estudio de corte trasversal de una cohorte de 3,199 mujeres de edad media encontró que los sujetos en el cuartil más alto de la ingesta de filoquinona dietaría tuvieron una DMO de la cadera y columna lumbar significantemente mayor que aquellos en el cuartil más bajo (162 μg/día vs. 59 μg/día) (47). Además, estudios de corte transversal y de caso y control recientes han reportado asociaciones entre las ingestas altas de filoquinona y la baja incidencia de fractura de cadera (48, 49).
Sin embargo, debido a que vegetales de hoja verde son la fuente dietaría principal de filoquinona y debido a que son usualmente parte de una dieta balanceada, el consumo alto de filoquinona puede ser solo un indicador de hábitos alimenticios saludables que pudiesen, en lugar de la filoquinona en sí, dar cuenta de la totalidad o parte de la asociación descrita en los estudios basados en la observación (50). Los pocos estudios que midieron la filoquinona del plasma generalmente encontraron que los niveles altos circulantes estuvieron asociados con un riesgo menor de fractura (17, 51). Por ejemplo, la incidencia de fracturas vertebrales fue inversamente correlacionada con la DMO y la filoquinona en el plasma en un estudio prospectivo de cuatro años que incluyo 379 mujeres japonesas de edades de entre 30-88 años de edad (51). A pesar de todo, estudios basados en la observación no están diseñados para hacer inferencias de causalidad, y solo ensayos controlados aleatorios pueden evaluar si la filoquinona podría tener efectos benéficos en la salud ósea (véase Estudios de la suplementación con vitamina K y osteoporosis).
Vitamina K2: Existen algunos pocos estudios sobre las asociaciones entre las menaquinonas (vitamina K2) y la salud ósea, talvez debido al número limitado de fuentes dietarías de la menaquinona-4 (MK-4), la forma principal de la vitamina K2 presente en dietas occidentales. La comida japonesa natto, hecha de frijoles de soya cocidos fermentados por la bacteria Bacillus subtilis natto, es rica en MK-7. En un estudio prospectivo que dio seguimiento a 944 mujeres japonesas (edades 20-79 años), la DMO al inicio del estudio fue asociada positivamente con la ingesta de natto en mujeres postmenopáusicas (52). Durante el periodo de seguimiento de tres años, la tasa de perdida de la DMO en el cuello femoral fue significantemente menor en mujeres que consumieron natto (>200 μg/día de MK-7) en comparación con las que no lo consumieron. Ninguna asociación fue encontrada entre la ingesta de natto y la DMO en mujeres postmenopáusicas (52).
La DMO total de la cadera y cuello femoral según los reportes fue también más alta en aproximadamente 2,000 hombres japoneses de 65 años y mayores que regularmente consumieron por lo menos un paquete por día de natto (≥350 μg/día de MK-7) en comparación con aquellos que consumieron menos de un paquete por semana (<50 μg/día de MK-7) (53). A pesar de que incrementar el consumo de natto también maximiza la ingesta de otros compuestos dietarios (p. ej. isoflavonas de soya) que tienen beneficios potenciales para la salud del esqueleto; aún existe la necesidad de encontrar mediciones fiables del estatus de la vitamina K. Hasta la fecha, estudios basados en la observación han fallado inequívocamente en confirmar una asociación entre los niveles de menaquinona circulante (MK-7 y MK-4) y el riesgo de fractura (17, 54).
Biomarcador del estatus de la vitamina K y la salud ósea
Los niveles totales circulantes de la proteína ósea, osteocalcina, han mostrado ser marcadores sensibles de la formación ósea. Varias hormonas y factores de crecimiento, incluyendo la vitamina D pero no la vitamina K, regulan la síntesis de la osteocalcina por los osteoblastos. Sin embargo, la vitamina K es un cofactor esencial para la γ-carboxilación de los tres residuos de ácido glutámico en la osteocalcina. La subcarboxilación de la osteocalcina en el hueso humano y suero ha sido ligada a el estatus pobre de la vitamina K. El grado de γ-carboxilación de la osteocalcina es sensible a intervenciones nutricionales de la vitamina K, y de este modo es usado como un indicador relativo del estatus de la vitamina K (13).
Se encontró que los niveles circulantes de la osteocalcina subcarboxilada (OCsc) eran más altos en mujeres postmenopáusicas que en mujeres premenopáusicas, y marcadamente más altos en mujeres sobre los 70 años. También altas proporciones de OCsc a OC total (OCsc/OC) parecen predecir el riesgo de fractura de cadera en mujeres de edad avanzada (55, 56). Aunque la deficiencia de vitamina K parecería la causa más probable de la proporción de OCsc sanguínea elevada, algunos investigadores también han documentado una relación inversa entre las medidas bioquímicas del estado nutricional de la vitamina D y los niveles de OCsc, así como una disminución significativa de la proporción OCsc/OC a través de la suplementación con vitamina D (57). Se ha sugerido que el incremento de la proporción de OCsc/OC circulante podría reflejar un estatus nutricional pobre en general que incluiría la insuficiencia de vitamina D, la cual explicaría la observaciones anteriormente mencionadas (58). Sin embargo, en varios estudios de intervención aleatorios, controlados con placebo conducidos en niñas jóvenes (58, 59) y mujeres postmenopáusicas (60), la suplementación con vitamina D fallo en disminuir las proporciones de OCsc/OC o mostrar algún efectos adicional en la disminución de OCsc/OC a través de la vitamina K suplementaria.
Estudios de la suplementación con vitamina K y osteoporosis
Suplementación con vitamina K: La revisión sistemática de cinco ensayos clínicos aleatorios que evaluaron el efecto de la suplementación con filoquinona (vitamina K1) en la DMO de la cadera usando dosis de entre 200 μg/día a 5,000 μg/día con duraciones de 12 a 36 meses encontró un beneficio poco prometedor para la salud ósea (17). Aunque la suplementación con filoquinona disminuyo los niveles de OCsc en los cinco estudios, solo un estudio reporto un efecto de la filoquinona suplementaria en la DMO (61). En este estudio, 150 mujeres postmenopáusicas fueron asignadas aleatoriamente para recibir un placebo, minerales (500 mg/día de calcio, 130 mg/día de magnesio, y 10 mg/día de zinc) más vitamina D (320 UI/día), o minerales, vitamina D y filoquinona (1,000 mg/día). El ritmo de la pérdida de DMO en el cuello femoral, pero no en la columna lumbar, fue significativamente menor en sujetos con filoquinona suplementaria en comparación a los otros dos grupos. De esta manera, evidencia de un supuesto beneficio de la filoquinona en la salud ósea en adultos mayores es considerada débil. Ninguno de los estudios fue diseñado para evaluar el efecto de la filoquinona en fracturas relacionadas con la osteoporosis. Investigación adicional podría buscar evaluar si la suplementación con filoquinona podría mejorar la salud del esqueleto en sujetos en alto riesgo de una insuficiencia de vitamina K (es decir, individuos con trastornos de malabsorción, o fibrosis cística).
Suplementación con vitamina K2: Las dosis farmacológicas de manaquinona-4 (MK-4; nombre comercial, menatetrenona) son usadas actualmente en Japón en el tratamiento de la osteoporosis. De acuerdo con la mayoría de los ensayos de intervención la investigación del efecto de una dosis alta de MK-4 en la perdida ósea ha sido conducida en mujeres postmenopáusicas japonesas. Un meta-análisis del 2006 de siete ensayos controlados aleatorios asoció el incremento de la DMO y la incidencia reducida de fractura (62). Todos con la excepción de uno de los siete ensayos individuales emplearon 45 mg de MK-4 diariamente; el otro ensayo uso 15 mg/día (62). Este meta-análisis reporto que la suplementación con MK-4 por más de seis meses significantemente disminuyo el riesgo de fracturas vertebrales en un 60%, fracturas de cadera en un 77%, y fracturas no vertebrales en un 81%. Sin embargo, los resultados de este meta-análisis fueron posteriormente restados de importancia debido al pequeño tamaño de los estudios incluidos y al hecho de que algunos de ellos no fueron controlados con placebo pero en su lugar se usó un tratamiento abierto actual (p. ej., con calcio y vitamina D). Adicionalmente, el análisis no incluyo datos de un estudio no publicado con un tamaño de muestra más amplio que reporto ningún efecto de la MK-4 en el riesgo de fractura y habría alterado la conclusión del meta-análisis (63).
Un estudio controlado sin placebo más reciente asigno aleatoriamente a más de 4,000 mujeres postmenopáusicas japonesas para recibir calcio solo o en combinación con MK-4 (45 mg/día) por tres años. Al final de un seguimiento adicional de un año (cuatro años en total), no hubo diferencias entre los grupos con respecto a la incidencia de fracturas vertebrales, y solo una pequeña reducción fue apreciada en la incidencia de nuevas fracturas clínicas en aquellas que tomaron el tratamiento combinado en comparación con el calcio solo (4.4% vs 3.4%) pero solo en mujeres en alto riesgo de fracturas (64). Resultados dudosos han sido reportados en ensayos adicionales conducidos en Europa y en los EE.UU. Un ensayo de intervención controlado con placebo de tres años en 325 mujeres postmenopáusicas saludables encontró que la MK-4 suplementaria (45 mg/día) por tres años mejoro las medidas de la resistencia ósea en comparación con el placebo (65). Debe notarse, esta dosis de MK-4 usada en la mayoría de los estudios citados es 500 veces más alta que la IA para la vitamina K. Otro ensayo aleatorio, doble ciego, controlado con placebo de un año en 365 mujeres postmenopáusicas americanas con una insuficiencia de vitamina K (osteocalcina subcarboxilada ≥4%) encontró que ni la alta dosis de filoquinona suplementaria (1,000 μg/día) ni la de MK-4 (45 mg/día) tuvieron efecto en los marcadores del suero del recambio óseo o en la DMO (columna lumbar y cadera) cuando se comparó con el placebo (66). En este estudio, todos los sujetos también recibieron diariamente, calcio de etiqueta abierta (630 mg) y vitamina D3 (400 UI).
Aunque unos pocos estudios basados en la observación han sugerido un vínculo entre el consumo de natto (rico en MK-7) y la salud ósea, un reciente estudio aleatorio, doble ciego, controlado con placebo en 334 mujeres postmenopáusicas saludables (1 a 5 años de postmenopausia) no encontró efecto alguno de 360 μg/día de MK-7 (en la forma de capsulas de natto) en la DMO en varios sitios después de un año en comparación con el inicio del estudio (67). Otro ensayo controlado con placebo comparable en 244 mujeres menopaúsicas encontró que la suplementación con 180 μg/día de MK-7 por tres años significantemente limito la perdida ósea en el cuello femoral pero no en otros sitios (68). Actualmente, el papel potencial de las menaquinonas suplementarios sobre la salud ósea aún necesita ser establecido en ensayos de mayor magnitud, aleatorios, y bien controlados.
Antagonistas de la vitamina K en la salud ósea
Ciertos anticoagulantes orales, como la warfarina, son conocidos por ser antagonistas de la vitamina K (véase Coagulación). Pocos estudios han examinado el uso crónico de la warfarina y el riesgo de fractura en mujeres mayores. Un estudio reporto no asociación alguna entre el tratamiento a largo plazo con warfarina y el riesgo de fracturas (69), mientras tanto otro encontró un riesgo significativamente mayor de fracturas vertebrales y de las costillas en los usuarios de warfarina en comparación con los que no la usan (70). Además, un estudio en pacientes de la tercera edad con fibrilación auricular reporto que el tratamiento a largo plazo con warfarina estuvo asociado con un riesgo significativamente mayor de fracturas osteoporóticas en hombres pero no en mujeres (71). Un meta-análisis de resultados de 11 estudios publicados encontró que la terapia oral de anticoagulación estuvo asociada con una reducción muy modesta de la DMO en la muñeca y no cambio alguno en la DMO en la cadera o espina (72). El desarrollo de nuevos anticoagulantes que no bloquean el reciclaje de la vitamina K podrían ofrecer una alternativa más segura para el uso de antagonistas de la vitamina K (73).
Enfermedades cardiovasculares
Una relación inversa entre la ingesta de vitamina K y la mortalidad fue reportada en una encuesta nacional de los EE.UU. (NHANES III) de 3,401 participantes (74). Ingestas de vitamina K adecuadas vs. ingestas inadecuadas (basadas en específico en el sexo de la IA: 90 μg/día para las mujeres y 120 μg/día para los hombres) fueron asociadas con un riesgo 22% menor de mortalidad relacionada a enfermedades cardiovasculares (ECV) y un riesgo 15% menor de mortalidad por todos las causas. El reporte también indio que, mientras más de dos tercios de los individuos con enfermedad renal crónica tuvieron ingestas de vitamina K por debajo de la IA, el riesgo de mortalidad por ECV fue un 41% menor en aquellos con ingestas adecuadas en comparación con ingestas subóptimas (74). Sin embargo, ingestas más altas de vitamina K no fueron asociadas con una mortalidad menor por ECV en un estudio prospectivo que dio seguimiento a 7,216 adultos mayores en riesgo de desarrollar ECV (75). Este estudio asoció altas ingestas de filoquinona, pero no de menaquinonas, con un riesgo menor de mortalidad por todas las causas.
En otro estudio prospectivo reciente, que dio seguimiento a 35,476 hombres y mujeres holandeses saludables durante un periodo medio de 12.1 años, el riesgo de un incidente de un accidente cerebrovascular no fue significativamente asociado con ingestas de filoquinona o menaquinonas (76). Estudios basados en la observación tempranos ofrecen evidencia limitada para una relación inversa entre la ingesta de filoquinona y el riesgo de ECV, a pesar de que a las altas ingestas a veces se les atribuyen como un marcador de hábitos dietarios saludables con un riesgo cardiovascular bajo (revisado en 77). Un cohorte prospectivo de 16,057 mujeres holandesas (edades 49-70 años) con un seguimiento medio de 8.1 años encontró una reducción del 9% del riesgo de enfermedad coronaria cardiaca (ECC) por cada aumento gradual de 10 μg/día en la ingesta de menaquinona (78). En otro estudio holandés temprano que examino 4,807 hombres y mujeres saludables de 55 años y mayores, los participantes en el tercil más alto de la ingesta de menaquinona (>32.7 μg/día) tuvieron un riesgo 41% menor de ECC incidente y un riesgo 26% menor del riesgo de mortalidad por todas la causas que aquellos en el tercil más bajo (<21.6 μg/día) (79). Además, se encontró que la ingesta de menaquinona esta inversamente asociada con la calcificación aortica, un mayor factor de riesgo de ECV (79).
Calcificación vascular
Uno de los sellos distintivos de la enfermedad cardiovascular es la formación de placas ateroscleróticas en las paredes arteriales. La ruptura de placas que causa la formación de coágulos (trombogénesis), es la causa habitual de un infarto al miocardio (ataque al corazón) o de un accidente cerebrovascular. Mientras que la calcificación de las placas ocurre al mismo tiempo que la aterosclerosis progresa, no está claro si la calcificación incrementa la inestabilidad de las placas y podría predecir el riesgo de ruptura y trombogénesis (80). Sin embargo, la calcificación puede ser predictiva de eventos cardiovasculares futuros. Un meta-análisis de 30 estudios de cohorte prospectivos, incluyendo un total de 218,080 participantes, encontró que la presencia de calcificación vascular se asoció con un triple a cuádruple incremento general del riesgo de eventos cardiovasculares y mortalidad (81). Un estudio temprano basado en la población en mujeres postmenopáusicas (edades 60-79 años) observo que las mujeres de menor edad (60-69 años) con calcificaciones aorticas tuvieron menores ingestas de vitamina K que aquellas sin calcificaciones aórticas, sin embargo esto no fue cierto en mujeres de mayor edad (70-79 años) (82). Un estudio de cohorte prospectivo en 807 hombres y mujeres, 39-45 años de edad, no encontró correlación alguna entre la ingesta de filoquinona y la calcificación de arterias coronarias, al medirse de manera no invasiva mediante una tomografía computarizada (83). Además, ni las ingestas de filoquinona o las de menaquinona se asociaron con la calcificación de arterias de los senos en un estudio de corte transversal en 1,689 mujeres de edades de entre 49-70 años (84). Sin embargo, en otro estudio de corte transversal se encontró que el cuartil más elevado frente al más bajo de la ingesta (ingestas medias, 48.5 μg/día vs. 18 μg/día) de menaquinona (MK-4 a MK-10) estaba asociado con una reducción del 20% de la prevalencia de la calcificación arterial coronaria en 564 mujeres postmenopáusicas (85).
Reciente investigación ha descubierto posibles mecanismos por los cuales la vitamina K puede inhibir la mineralización (calcificación) de vasos sanguíneos mientras que promueve la mineralización ósea. Los mecanismos potenciales, aunque no son completamente comprehendidos, implican las proteínas dependientes de la vitamina K, incluyendo la proteína Gla de la matriz (MGP) y la recientemente descrita proteína rica en Gla (GRP). Secretada por varios tipos de células, como las células del musculo liso vascular (CMLV) en las paredes de los vasos arteriales, la MGP parece ser importante para la prevención de la calcificación de tejidos suaves, incluyendo cartílagos, vasculatura, piel, y células de la red trabecular en el ojo (17, 86). En ratones knockout de la MGP, la conversión de CMLV a células similares a las óseas y una calcificación de vasos extensa resulta en una gran ruptura de vasos y muerte prematura. En los seres humanos, el gen defectuoso de la MGP ha sido ligado al síndrome de Keutel, una rara condición hereditaria caracterizada en particular por la calcificación anormal del cartílago y estenosis de la arteria pulmonar (estrechamiento). La prevención de la calcificación por la MGP involucro varios mecanismos, incluyendo la unión a cristales de calcio y la inhibición de las proteínas (proteínas morfogenéticas óseas; PMO) conocidas por promover la formación de hueso ectópico (revisado en 87).
La actividad de la unión del calcio de la MGP es regulada por dos tipos de modificaciones (conocidas como modificaciones postraduccionales ya que toman lugar después de la síntesis de proteínas): la carboxilación dependiente de la vitamina K de hasta 5 residuos de Glu y la fosforilación de residuos de serina. Una variación en la secuencia (polimorfismo) del gene de la MGP que conduce a una transición de treonina a alanina en uno de los cinco dominios de la proteína Gla puede posiblemente prevenir la carboxilación y provocar un cambio en la habilidad de la MGP para unirse al calcio. Este polimorfismo, conocido como MGPThr83Ala ha sido asociado con la progresión de la calcificación de las arterias coronarias durante un periodo medio de 10.6 años en un estudio prospectivo basado en la comunidad que dio seguimiento a 605 mujeres y hombres de edad media (88). Esta asociación fue únicamente observada entre participantes sin una calcificación detectable al inicio del estudio y no en aquellos que tuvieron una calcificación al inicio del estudio (88). Curiosamente, el MGPThr83Ala fue asociado con un alto riesgo de infarto al miocardio y calcificación de la arteria femoral en portadores del genotipo (89).
Además, un estudio menor inicialmente encontró que, mientras la subcarboxilación de la MGP (MGPsc) estuvo ausente en el revestimiento más interno de las arterias carótidas en sujetos saludables, la mayoría de MGP en el revestimiento de la arteria carótida de los pacientes con aterosclerosis estaba subcarboxilada (90). En otro estudio que examino la asociación entre la MGP circulante y eventos cardiovasculares incidentes en 577 hombres y mujeres mayores con un seguimiento medio de 5.6 años, el riesgo de una enfermedad cardiovascular (es decir, enfermedad coronaria cardiaca, enfermedad arterial periférica, y enfermedad cerebrovascular) fue de dos a tres veces mayor en sujetos en el tercil más elevado frente al tercil más bajo de la MGP desfosforilada subcarboxilada (MGPdf-sc) del plasma (91). Los resultados de otro estudio prospectivo sugirieron que la MGPdf-sc circulante podría predecir el riesgo de mortalidad en sujetos con una enfermedad cardiovascular evidente (92). En efecto se encontró que el riesgo de mortalidad por todas las causas y relacionada a lo cardiovascular casi fue duplicado en sujetos con enfermedad coronaria cardiaca o accidentes cerebrovasculares en el cuartil más elevado frente al cuartil más bajo de las concentraciones de la MGPdf-sc (92).
Debido a que el estatus nutricional subóptimo de la vitamina K puede limitar la carboxilación y resultar en MGPsc biológicamente inactiva, se ha especulado que la suplementación con vitamina K podría proteger contra la calcificación vascular. Un ensayo controlado, doble ciego de tres años investigo el efecto potencial de la vitamina K en la progresión de la calcificación coronaria en 401 adultos mayores residentes de la comunidad (edades 60-80 años) sin ECV al inicio del estudio (93). Los participantes fueron aleatoriamente asignados para recibir un multivitamínico diario más calcio y vitamina D con o sin 500 mg de filoquinona. Usando mediciones de la calcificación de la arteria coronaria al inicio del estudio y del seguimiento, se encontró que la suplementación con filoquinona fue capaz de limitar la progresión de la calcificación vascular y reducir la MGPdf-sc del plasma en comparación con el control (93, 94). Aunque la MGPdf-sc circulante fue correlacionada con varios marcadores del estatus de la vitamina K, ninguna asociación con las medidas de la calcificación de la arteria coronaria fueron encontradas (94).
Investigaciones adicionales son necesarias para examinar el papel de otras proteínas dependientes de la vitamina K (p. ej., GRP, periostina, Gas6) en la calcificación de la placa aterosclerótica humana y para evaluar el efecto de la vitamina K en la progresión de la calcificación vascular y el riesgo de ECV.
Antagonistas de la vitamina K y la calcificación vascular
Varios estudios de corte transversal han reportado puntuaciones incrementadas de calcio vascular (una manera de cuantificar la calcificación vascular) en usuarios crónicos de antagonistas de la vitamina K en comparación con aquellos que no los usan (revisado en 95). La terapia con warfarina ha sido también asociada con altas concentraciones circulantes de MGPdf-sc en un estudio prospectivo reciente que examino la calcificación vascular en sujetos con ECV (92). Inhibidores directos recientemente desarrollados de los factores de coagulación que no interfieren con la actividad de las VKDP pueden ser más adecuados que los antagonistas de la vitamina K, especialmente con respecto a la calcificación vascular (95).
Fuentes
Fuentes alimenticias
Encuestas sobre la nutrición en Europa y los EE.UU han mostrado que las ingestas dietarías promedio de vitamina K (todas las formas) varias ampliamente entre individuos y poblaciones, con valores oscilando entre 60 a 200 μg/día (96).
Vitamina K1
La filoquinona (vitamina K1) es la es la principal forma dietaría de la vitamina K en la mayoría de las dietas. Los vegetales de hoja verde y algunos aceites vegetales (de soya, semilla de algodón, canola, y oliva) son los principales contribuyentes de la vitamina K dietaría. Sin embargo, la biodisponibilidad de la filoquinona proveniente de los vegetales de hoja verde es menor que aquella en aceites y suplementos. También, el contenido de filoquinona de los vegetales de hoja verde depende de su contenido de clorofila (pigmento verde), de modo que las hojas exteriores tienen más filoquinona que las hojas interiores. La eficiencia de la absorción intestinal de la filoquinona varía entre las fuentes vegetales y aumenta con la adición de una fuente de grasa a una comida. Finalmente, la hidrogenación de aceites vegetales podría disminuir la absorción y el efecto biológico de la vitamina K dietaría (revisada en 2, 9). Si desea revisar el contenido nutricional de los alimentos, incluyendo el de la filoquinona, busque en la base de datos de composición de los alimentos de la USDA. Una serie de alimentos ricos en filoquinona se muestran en la Tabla 2, junto a su contenido de filoquinona en microgramos (μg).
Vitamina K2
Las menaquinonas (vitamina K2) son principalmente de origen microbiano, y así comúnmente encontrada en alimentos fermentados, como el queso, la cuajada, y el natto (frijoles de soya fermentados). Otra fuente de menaquinonas de cadena larga (MK-7 a MK-13) es los hígados de animales (9). Debido a la disponibilidad limitada de tablas de composición de alimentos para las menaquinonas, su contribución al total de las ingestas de vitamina K es difícil de estimar y tiende a variar entre poblaciones con diferentes prácticas de consumo de alimentos (2). Las bacterias que normalmente colonizan el intestino grueso (colon) pueden sintetizar menaquinonas. Inicialmente se pensó que hasta un 50% del requerimiento podría ser satisfecho por la síntesis bacteriana. Sin embargo, todas las formas de vitamina K son absorbidas en el intestino delgado a través de un mecanismo que requiere de sales biliares, mientras que la mayoría de la producción de menaquinona toma lugar en el colon donde se carece de sales biliares. Investigación actual sugiere que la contribución de la síntesis bacteriana es mucho menos que lo que se pensó previamente, aunque la contribución exacta permanece sin ser clara (97). Entre las menaquinonas, la MK-4 está formada de menadiona (la forma sintética de la vitamina K) encontrada en alimentos para ganado o es convertida en una forma de tejido especifico de la filoquinona dietaría; por lo tanto, es la única menaquinona que no es producida por bacterias (4). Las menaquinonas de cadena más larga son encontradas en productos alimenticios fermentados limitados. El basado de frijoles soja fermentada japonés, el natto, es rico en MK-7 (998 μg/100 g) y también contienen MK-8 (84 μg/100 g). Algunos quesos contienen MK-8 y MK-9 (2).
Suplementos
En los EE.UU., tanto la filoquinona como las menaquinonas están disponible sin prescripción en multivitamínicos y otros suplementos dietarios en dosis que generalmente oscilan entre 25-100 μg por tableta (98). Mientras que la menaquinona-4 (MK-4) es comercializada para el tratamiento de la osteoporosis en Japón, la Administración de Alimentos y Drogas de los Estados Unidos (FDA) actualmente no permite a ninguna de las formas de vitamina K ser usada en la prevención o tratamiento de la osteoporosis.
Seguridad
Toxicidad
Aunque una reacción alérgica es posible, no existe toxicidad conocida asociada con altas dosis (dietaría y suplementaria) de la filoquinona (vitamina K1) o de las formas de menaquinona (vitamina K2) de la vitamina K (42). Lo mismo no es cierto para la menadiona sintética (vitamina K3) y sus derivados. La menadiona puede interferir con la función del glutatión, uno de los antioxidantes naturales del cuerpo, resultando en daño oxidativo a las membranas celulares. La menadiona administrada por inyección ha inducido toxicidad hepática, ictericia, y anemia hemolítica (debido a la ruptura de glóbulos rojos) en infantes; por ello, la menadiona ya no es usada para el tratamiento de la deficiencia de vitamina K (5). Un nivel máximo de ingesta tolerable (NM) para la vitamina K aún no ha sido establecido (42).
Interacción con nutrientes
Se ha encontrado que grandes dosis de vitamina A y vitamina E antagonizan a la vitamina K (8). El exceso de vitamina A parece interferir con la absorción de vitamina K, mientras que la vitamina E podría inhibir la actividad de enzimas carboxilasas dependientes de vitamina K e interferir con la cascada de coagulación (99). Un estudio en adultos con estados de coagulación normales encontró que la suplementación con 1,000 UI de vitamina E por 12 semanas disminuyó la γ-carboxilación de protrombina, una proteína dependiente de vitamina K (100). Individuos que toman drogas anticoagulantes como la warfarina y aquellos que son deficientes de vitamina K no deberían tomar suplementos de vitamina E sin una supervisión médica minuciosa debido al riesgo incrementado de hemorragia (sangrado excesivo) (101).
Interacción con drogas/fármacos
El efecto anticoagulante de los antagonistas de la vitamina K (p. ej., warfarina) podría verse involucrada por una muy alta ingesta recomendada o suplementaria de vitamina K. Por otra parte, los suplementos diarios de filoquinona de hasta 100 μg son considerados seguros para los sujetos que toman warfarina, pero la estabilidad anticoagulante terapéutica puede ser socavada por dosis de MK-7 tan bajas como de 10-20 μg (102). Generalmente se recomienda que los individuos que usan warfarina intenten consumir la IA de vitamina K (90-120 μg), y evitar grandes fluctuaciones en la ingesta de vitamina K que podrían interferir con el ajuste de su dosis de anticoagulante (9). La prescripción de anticoagulantes anti-vitamina K, anticonvulsivos (p. ej. fenitoína), y drogas contra la tuberculosis (p. ej. rifampicina e isoniazida) para mujeres embarazadas o que lactan pueden poner en un riesgo incrementado de deficiencia de vitamina K al recién nacido (103).
El uso prolongado de un amplio espectro de antibióticos, como cefalosporinas y salicilatos, puede interferir con la síntesis de vitamina K por bacterias intestinales y disminuir la absorción de vitamina K. El fármaco amiodarona, usado en el manejo de ciertas arritmias cardiacas (ritmo cardiaco irregular), incluyendo la fibrilación auricular, puede aumentar el efecto anticoagulante de la warfarina y así incrementar el riesgo de hemorragia (104, 105). Además, el uso de medicamentos que disminuyen el colesterol (como la colestiramina y el colestipol), como también el orlistat, el aceite mineral, y el sustituto de las grasas, olestra, que interfiere con la absorción de grasas, puede afectar la absorción de vitaminas liposolubles, incluyendo la vitamina K (106).
Recomendación del Instituto Linus Pauling
No es claro si la IA para la vitamina K es suficiente para optimizar la γ-carboxilación de las proteínas dependientes de vitamina K en el hueso (véase Osteoporosis). Para consumir la cantidad suficiente de vitamina K asociada con un riesgo disminuido de fractura de cadera en el Estudio del Corazón de Framingham (aproximadamente 250 μg/día) (44), un individuo necesitaría comer un poco más de una ½ taza de brócoli picado o una ensalada grande de verduras mixtas cada día. Aunque la ingesta recomendada de vitamina K necesaria para el funcionamiento óptimo de todas las proteínas dependientes de vitamina K aún se desconoce, el Instituto Linus Pauling recomienda tomar diariamente un suplemento multivitamínico-mineral y comer al menos una taza de vegetales de hoja verde. Reemplazar las grasas saturadas de la dieta como la mantequilla y el queso con grasas monosaturadas que se encuentran en el aceite de oliva y de canola, incrementara la ingesta de vitamina K dietética y podría disminuir el riesgo de enfermedades cardiovasculares.
Adultos mayores (>50 años)
Ya que los adultos mayores están un riesgo incrementado de osteoporosis y fractura de cadera, la recomendación anterior de un suplemento multivitamínico/mineral y al menos una taza de vegetales de hoja verde a diario es especialmente relevante.
Autores y Críticos
Originalmente escrito en 2000 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Mayo de 2004 por:
Jane Higdon, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Mayo de 2008 por:
Victoria J. Drake, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Actualizado en Julio de 2014 por:
Barbara Delage, Ph.D.
Instituto Linus Pauling
Universidad Estatal de Oregon
Revisado en Agosto de 2014 por:
Sarah L. Booth, Ph.D.
Directora, Programa de Investigación de la Vitamina K
Jean Mayer USDA Centro de Investigación de Nutrición Humana en el Envejecimiento
Universidad de Tufts
Traducido al Español en 2016 por:
Silvia Vazquez Lima
Instituto Linus Pauling
Universidad Estatal de Oregon
Originalmente traducido al español en 2012 por Guillermo Sandoval y editado por Andrew Quest (Ph.D.) y Lisette Leyton (Ph.D.), todos provenientes de la Universidad de Chile. Estos esfuerzos fueron patrocinados por el projecto Anillo #ACT1111, CONICYT-Chile, programa PIA.
Derechos de autoría 2000-2024 Instituto Linus Pauling
Referencias
1. Brody T. Nutritional Biochemistry. 2nd ed. San Diego: Academic Press; 1999.
2. Booth SL. Vitamin K: food composition and dietary intakes. Food Nutr Res. 2012;56. (PubMed)
3. Kidd PM. Vitamins D and K as pleiotropic nutrients: clinical importance to the skeletal and cardiovascular systems and preliminary evidence for synergy. Altern Med Rev. 2010;15(3):199-222. (PubMed)
4. Nakagawa K, Hirota Y, Sawada N, et al. Identification of UBIAD1 as a novel human menaquinone-4 biosynthetic enzyme. Nature. 2010;468(7320):117-121. (PubMed)
5. Ferland G. Vitamin K. In: Erdman Jr. JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Ames: Wiley-Blackwell; 2012:230-247.
6. Rishavy MA, Hallgren KW, Wilson LA, Usubalieva A, Runge KW, Berkner KL. The vitamin K oxidoreductase is a multimer that efficiently reduces vitamin K epoxide to hydroquinone to allow vitamin K-dependent protein carboxylation. J Biol Chem. 2013;288(44):31556-31566. (PubMed)
7. Tie JK, Jin DY, Straight DL, Stafford DW. Functional study of the vitamin K cycle in mammalian cells. Blood. 2011;117(10):2967-2974. (PubMed)
8. Olson RE. Vitamin K. In: Shils M, Olson JA, Shike M, Ross AC, eds. Modern Nutrition in Health and Disease. 9th ed. Baltimore: Lippincott Williams & Wilkins; 1999:363-380.
9. Holmes MV, Hunt BJ, Shearer MJ. The role of dietary vitamin K in the management of oral vitamin K antagonists. Blood Rev. 2012;26(1):1-14. (PubMed)
10. Kramps M, Flanagan A, Smaldone A. The use of vitamin K supplementation to achieve INR stability: a systematic review and meta-analysis. J Am Assoc Nurse Pract. 2013;25(10):535-544. (PubMed)
11. Lam J, Schulman S, Witt DM, Vandvik PO, Qayyum F, Holbrook AM. Anticoagulation control with daily low-dose vitamin k to reduce clinically adverse outcomes and international normalized ratio variability: a systematic review and meta-analysis. Pharmacotherapy. 2013;33(11):1184-1190. (PubMed)
12. Mahtani KR, Heneghan CJ, Nunan D, Roberts NW. Vitamin K for improved anticoagulation control in patients receiving warfarin. Cochrane Database Syst Rev. 2014;5:CD009917. (PubMed)
13. Gundberg CM, Lian JB, Booth SL. Vitamin K-dependent carboxylation of osteocalcin: friend or foe? Adv Nutr. 2012;3(2):149-157. (PubMed)
14. Pierre-Jacques H, Glueck CJ, Mont MA, Hungerford DS. Familial heterozygous protein-S deficiency in a patient who had multifocal osteonecrosis. A case report. J Bone Joint Surg Am. 1997;79(7):1079-1084. (PubMed)
15. Rawat RS, Mehta Y, Arora D, Trehan N. Asymptomatic type B right atrial thrombus in a case with protein S deficiency. Ann Card Anaesth. 2014;17(3):237-239. (PubMed)
16. van der Meer JH, van der Poll T, van 't Veer C. TAM receptors, Gas6, and protein S: roles in inflammation and hemostasis. Blood. 2014;123(16):2460-2469. (PubMed)
17. Booth SL. Roles for vitamin K beyond coagulation. Annu Rev Nutr. 2009;29:89-110. (PubMed)
18. Boraldi F, Annovi G, Guerra D, et al. Fibroblast protein profile analysis highlights the role of oxidative stress and vitamin K recycling in the pathogenesis of pseudoxanthoma elasticum. Proteomics Clin Appl. 2009;3(9):1084-1098. (PubMed)
19. Boraldi F, Garcia-Fernandez M, Paolinelli-Devincenzi C, et al. Ectopic calcification in β-thalassemia patients is associated with increased oxidative stress and lower MGP carboxylation. Biochim Biophys Acta. 2013;1832(12):2077-2084. (PubMed)
20. Coutu DL, Wu JH, Monette A, Rivard GE, Blostein MD, Galipeau J. Periostin, a member of a novel family of vitamin K-dependent proteins, is expressed by mesenchymal stromal cells. J Biol Chem. 2008;283(26):17991-18001. (PubMed)
21. Viegas CS, Simes DC, Laize V, Williamson MK, Price PA, Cancela ML. Gla-rich protein (GRP), a new vitamin K-dependent protein identified from sturgeon cartilage and highly conserved in vertebrates. J Biol Chem. 2008;283(52):36655-36664. (PubMed)
22. Viegas CS, Cavaco S, Neves PL, et al. Gla-rich protein is a novel vitamin K-dependent protein present in serum that accumulates at sites of pathological calcifications. Am J Pathol. 2009;175(6):2288-2298. (PubMed)
23. Hakuno D, Kimura N, Yoshioka M, et al. Periostin advances atherosclerotic and rheumatic cardiac valve degeneration by inducing angiogenesis and MMP production in humans and rodents. J Clin Invest. 2010;120(7):2292-2306. (PubMed)
24. Kudo Y, Siriwardena BS, Hatano H, Ogawa I, Takata T. Periostin: novel diagnostic and therapeutic target for cancer. Histol Histopathol. 2007;22(10):1167-1174. (PubMed)
25. Vanakker OM, Martin L, Schurgers LJ, et al. Low serum vitamin K in PXE results in defective carboxylation of mineralization inhibitors similar to the GGCX mutations in the PXE-like syndrome. Lab Invest. 2010;90(6):895-905. (PubMed)
26. Ferland G. Vitamin K and the nervous system: an overview of its actions. Adv Nutr. 2012;3(2):204-212. (PubMed)
27. Laurance S, Lemarie CA, Blostein MD. Growth arrest-specific gene 6 (gas6) and vascular hemostasis. Adv Nutr. 2012;3(2):196-203. (PubMed)
28. Robins RS, Lemarie CA, Laurance S, Aghourian MN, Wu J, Blostein MD. Vascular Gas6 contributes to thrombogenesis and promotes tissue factor up-regulation after vessel injury in mice. Blood. 2013;121(4):692-699. (PubMed)
29. Rothlin CV, Leighton JA, Ghosh S. Tyro3, Axl, and Mertk Receptor Signaling in Inflammatory Bowel Disease and Colitis-associated Cancer. Inflamm Bowel Dis. 2014;20(8):1472-1480. (PubMed)
30. Tjwa M, Moons L, Lutgens E. Pleiotropic role of growth arrest-specific gene 6 in atherosclerosis. Curr Opin Lipidol. 2009;20(5):386-392. (PubMed)
31. Jagannath VA, Fedorowicz Z, Thaker V, Chang AB. Vitamin K supplementation for cystic fibrosis. Cochrane Database Syst Rev. 2013;4:CD008482. (PubMed)
32. Nakajima S, Iijima H, Egawa S, et al. Association of vitamin K deficiency with bone metabolism and clinical disease activity in inflammatory bowel disease. Nutrition. 2011;27(10):1023-1028. (PubMed)
33. Nowak JK, Grzybowska-Chlebowczyk U, Landowski P, et al. Prevalence and correlates of vitamin K deficiency in children with inflammatory bowel disease. Sci Rep. 2014;4:4768. (PubMed)
34. Shearer MJ. Vitamin K deficiency bleeding (VKDB) in early infancy. Blood Rev. 2009;23(2):49-59. (PubMed)
35. American Academy of Pediatrics Committee on Fetus and Newborn. Controversies concerning vitamin K and the newborn. Pediatrics. 2003;112(1 Pt 1):191-192. (PubMed)
36. Klebanoff MA, Read JS, Mills JL, Shiono PH. The risk of childhood cancer after neonatal exposure to vitamin K. N Engl J Med. 1993;329(13):905-908. (PubMed)
37. Ekelund H, Finnstrom O, Gunnarskog J, Kallen B, Larsson Y. Administration of vitamin K to newborn infants and childhood cancer. BMJ. 1993;307(6896):89-91. (PubMed)
38. Roman E, Fear NT, Ansell P, et al. Vitamin K and childhood cancer: analysis of individual patient data from six case-control studies. Br J Cancer. 2002;86(1):63-69. (PubMed)
39. Schulte R, Jordan LC, Morad A, Naftel RP, Wellons JC, 3rd, Sidonio R. Rise in late onset vitamin K deficiency bleeding in young infants because of omission or refusal of prophylaxis at birth. Pediatr Neurol. 2014;50(6):564-568. (PubMed)
40. Costakos DT, Greer FR, Love LA, Dahlen LR, Suttie JW. Vitamin K prophylaxis for premature infants: 1 mg versus 0.5 mg. Am J Perinatol. 2003;20(8):485-490. (PubMed)
41. Kumar D, Greer FR, Super DM, Suttie JW, Moore JJ. Vitamin K status of premature infants: implications for current recommendations. Pediatrics. 2001;108(5):1117-1122. (PubMed)
42. Food and Nutrition Board, Institute of Medicine. Vitamin K. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Washington, D.C.: National Academy Press; 2001:162-196. (National Academy Press)
43. Feskanich D, Weber P, Willett WC, Rockett H, Booth SL, Colditz GA. Vitamin K intake and hip fractures in women: a prospective study. Am J Clin Nutr. 1999;69(1):74-79. (PubMed)
44. Booth SL, Tucker KL, Chen H, et al. Dietary vitamin K intakes are associated with hip fracture but not with bone mineral density in elderly men and women. Am J Clin Nutr. 2000;71(5):1201-1208. (PubMed)
45. Rejnmark L, Vestergaard P, Charles P, et al. No effect of vitamin K1 intake on bone mineral density and fracture risk in perimenopausal women. Osteoporos Int. 2006;17(8):1122-1132. (PubMed)
46. McLean RR, Booth SL, Kiel DP, et al. Association of dietary and biochemical measures of vitamin K with quantitative ultrasound of the heel in men and women. Osteoporos Int. 2006;17(4):600-607. (PubMed)
47. Macdonald HM, μguigan FE, Lanham-New SA, Fraser WD, Ralston SH, Reid DM. Vitamin K1 intake is associated with higher bone mineral density and reduced bone resorption in early postmenopausal Scottish women: no evidence of gene-nutrient interaction with apolipoprotein E polymorphisms. Am J Clin Nutr. 2008;87(5):1513-1520. (PubMed)
48. Apalset EM, Gjesdal CG, Eide GE, Tell GS. Intake of vitamin K1 and K2 and risk of hip fractures: The Hordaland Health Study. Bone. 2011;49(5):990-995. (PubMed)
49. Torbergsen AC, Watne LO, Wyller TB, et al. Vitamin K1 and 25(OH)D are independently and synergistically associated with a risk for hip fracture in an elderly population: A case control study. Clin Nutr. 2015;34(1):101-106. (PubMed)
50. Booth SL, Mayer J. Warfarin use and fracture risk. Nutr Rev. 2000;58(1):20-22. (PubMed)
51. Tsugawa N, Shiraki M, Suhara Y, et al. Low plasma phylloquinone concentration is associated with high incidence of vertebral fracture in Japanese women. J Bone Miner Metab. 2008;26(1):79-85. (PubMed)
52. Ikeda Y, Iki M, Morita A, et al. Intake of fermented soybeans, natto, is associated with reduced bone loss in postmenopausal women: Japanese Population-Based Osteoporosis (JPOS) Study. J Nutr. 2006;136(5):1323-1328. (PubMed)
53. Fujita Y, Iki M, Tamaki J, et al. Association between vitamin K intake from fermented soybeans, natto, and bone mineral density in elderly Japanese men: the Fujiwara-kyo Osteoporosis Risk in Men (FORMEN) study. Osteoporos Int. 2012;23(2):705-714. (PubMed)
54. Kaneki M, Hodges SJ, Hosoi T, et al. Japanese fermented soybean food as the major determinant of the large geographic difference in circulating levels of vitamin K2: possible implications for hip-fracture risk. Nutrition. 2001;17(4):315-321. (PubMed)
55. Szulc P, Chapuy MC, Meunier PJ, Delmas PD. Serum undercarboxylated osteocalcin is a marker of the risk of hip fracture in elderly women. J Clin Invest. 1993;91(4):1769-1774. (PubMed)
56. Vergnaud P, Garnero P, Meunier PJ, Breart G, Kamihagi K, Delmas PD. Undercarboxylated osteocalcin measured with a specific immunoassay predicts hip fracture in elderly women: the EPIDOS Study. J Clin Endocrinol Metab. 1997;82(3):719-724. (PubMed)
57. Shearer MJ. The roles of vitamins D and K in bone health and osteoporosis prevention. Proc Nutr Soc. 1997;56(3):915-937. (PubMed)
58. O'Connor E, Molgaard C, Michaelsen KF, Jakobsen J, Cashman KD. Vitamin D-vitamin K interaction: effect of vitamin D supplementation on serum percentage undercarboxylated osteocalcin, a sensitive measure of vitamin K status, in Danish girls. Br J Nutr. 2010;104(8):1091-1095. (PubMed)
59. Kanellakis S, Moschonis G, Tenta R, et al. Changes in parameters of bone metabolism in postmenopausal women following a 12-month intervention period using dairy products enriched with calcium, vitamin D, and phylloquinone (vitamin K(1)) or menaquinone-7 (vitamin K (2)): the Postmenopausal Health Study II. Calcif Tissue Int. 2012;90(4):251-262. (PubMed)
60. Bolton-Smith C, McMurdo ME, Paterson CR, et al. Two-year randomized controlled trial of vitamin K1 (phylloquinone) and vitamin D3 plus calcium on the bone health of older women. J Bone Miner Res. 2007;22(4):509-519. (PubMed)
61. Braam LA, Knapen MH, Geusens P, et al. Vitamin K1 supplementation retards bone loss in postmenopausal women between 50 and 60 years of age. Calcif Tissue Int. 2003;73(1):21-26. (PubMed)
62. Cockayne S, Adamson J, Lanham-New S, Shearer MJ, Gilbody S, Torgerson DJ. Vitamin K and the prevention of fractures: systematic review and meta-analysis of randomized controlled trials. Arch Intern Med. 2006;166(12):1256-1261. (PubMed)
63. Tamura T, Morgan SL, Takimoto H. Vitamin K and the prevention of fractures. Arch Intern Med. 2007;167(1):94; author reply 94-95. (PubMed)
64. Inoue T, Fujita T, Kishimoto H, et al. Randomized controlled study on the prevention of osteoporotic fractures (OF study): a phase IV clinical study of 15-mg menatetrenone capsules. J Bone Miner Metab. 2009;27(1):66-75. (PubMed)
65. Knapen MH, Schurgers LJ, Vermeer C. Vitamin K2 supplementation improves hip bone geometry and bone strength indices in postmenopausal women. Osteoporos Int. 2007;18(7):963-972. (PubMed)
66. Binkley N, Harke J, Krueger D, et al. Vitamin K treatment reduces undercarboxylated osteocalcin but does not alter bone turnover, density, or geometry in healthy postmenopausal North American women. J Bone Miner Res. 2009;24(6):983-991. (PubMed)
67. Emaus N, Gjesdal CG, Almas B, et al. Vitamin K2 supplementation does not influence bone loss in early menopausal women: a randomised double-blind placebo-controlled trial. Osteoporos Int. 2010;21(10):1731-1740. (PubMed)
68. Knapen MH, Drummen NE, Smit E, Vermeer C, Theuwissen E. Three-year low-dose menaquinone-7 supplementation helps decrease bone loss in healthy postmenopausal women. Osteoporos Int. 2013;24(9):2499-2507. (PubMed)
69. Jamal SA, Browner WS, Bauer DC, Cummings SR. Warfarin use and risk for osteoporosis in elderly women. Study of Osteoporotic Fractures Research Group. Ann Intern Med. 1998;128(10):829-832. (PubMed)
70. Caraballo PJ, Heit JA, Atkinson EJ, et al. Long-term use of oral anticoagulants and the risk of fracture. Arch Intern Med. 1999;159(15):1750-1756. (PubMed)
71. Gage BF, Birman-Deych E, Radford MJ, Nilasena DS, Binder EF. Risk of osteoporotic fracture in elderly patients taking warfarin: results from the National Registry of Atrial Fibrillation 2. Arch Intern Med. 2006;166(2):241-246. (PubMed)
72. Caraballo PJ, Gabriel SE, Castro MR, Atkinson EJ, Melton LJ, 3rd. Changes in bone density after exposure to oral anticoagulants: a meta-analysis. Osteoporos Int. 1999;9(5):441-448. (PubMed)
73. Fusaro M, Crepaldi G, Maggi S, et al. Bleeding, vertebral fractures and vascular calcifications in patients treated with warfarin: hope for lower risks with alternative therapies. Curr Vasc Pharmacol. 2011;9(6):763-769. (PubMed)
74. Cheung CL, Sahni S, Cheung BM, Sing CW, Wong IC. Vitamin K intake and mortality in people with chronic kidney disease from NHANES III. Clin Nutr. 2014; pii: S0261-5614(14)00086-7. doi: 10.1016/j.clnu.2014.03.011. [Epub ahead of print] (PubMed)
75. Juanola-Falgarona M, Salas-Salvado J, Martinez-Gonzalez MA, et al. Dietary intake of vitamin K is inversely associated with mortality risk. J Nutr. 2014;144(5):743-750. (PubMed)
76. Vissers LE, Dalmeijer GW, Boer JM, Monique Verschuren WM, van der Schouw YT, Beulens JW. Intake of dietary phylloquinone and menaquinones and risk of stroke. J Am Heart Assoc. 2013;2(6):e000455. (PubMed)
77. Rees K, Guraewal S, Wong YL, et al. Is vitamin K consumption associated with cardio-metabolic disorders? A systematic review. Maturitas. 2010;67(2):121-128. (PubMed)
78. Gast GC, de Roos NM, Sluijs I, et al. A high menaquinone intake reduces the incidence of coronary heart disease. Nutr Metab Cardiovasc Dis. 2009;19(7):504-510. (PubMed)
79. Geleijnse JM, Vermeer C, Grobbee DE, et al. Dietary intake of menaquinone is associated with a reduced risk of coronary heart disease: the Rotterdam Study. J Nutr. 2004;134(11):3100-3105. (PubMed)
80. Otsuka F, Sakakura K, Yahagi K, Joner M, Virmani R. Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler Thromb Vasc Biol. 2014;34(4):724-736. (PubMed)
81. Rennenberg RJ, Kessels AG, Schurgers LJ, van Engelshoven JM, de Leeuw PW, Kroon AA. Vascular calcifications as a marker of increased cardiovascular risk: a meta-analysis. Vasc Health Risk Manag. 2009;5(1):185-197. (PubMed)
82. Jie KS, Bots ML, Vermeer C, Witteman JC, Grobbee DE. Vitamin K intake and osteocalcin levels in women with and without aortic atherosclerosis: a population-based study. Atherosclerosis. 1995;116(1):117-123. (PubMed)
83. Villines TC, Hatzigeorgiou C, Feuerstein IM, O'Malley PG, Taylor AJ. Vitamin K1 intake and coronary calcification. Coron Artery Dis. 2005;16(3):199-203. (PubMed)
84. Maas AH, van der Schouw YT, Beijerinck D, et al. Vitamin K intake and calcifications in breast arteries. Maturitas. 2007;56(3):273-279. (PubMed)
85. Beulens JW, Bots ML, Atsma F, et al. High dietary menaquinone intake is associated with reduced coronary calcification. Atherosclerosis. 2009;203(2):489-493. (PubMed)
86. Borras T, Comes N. Evidence for a calcification process in the trabecular meshwork. Exp Eye Res. 2009;88(4):738-746. (PubMed)
87. Schurgers LJ, Uitto J, Reutelingsperger CP. Vitamin K-dependent carboxylation of matrix Gla-protein: a crucial switch to control ectopic mineralization. Trends Mol Med. 2013;19(4):217-226. (PubMed)
88. Cassidy-Bushrow AE, Bielak LF, Levin AM, et al. Matrix gla protein gene polymorphism is associated with increased coronary artery calcification progression. Arterioscler Thromb Vasc Biol. 2013;33(3):645-651. (PubMed)
89. Herrmann SM, Whatling C, Brand E, et al. Polymorphisms of the human matrix gla protein (MGP) gene, vascular calcification, and myocardial infarction. Arterioscler Thromb Vasc Biol. 2000;20(11):2386-2393. (PubMed)
90. Schurgers LJ, Teunissen KJ, Knapen MH, et al. Novel conformation-specific antibodies against matrix γ-carboxyglutamic acid (Gla) protein: undercarboxylated matrix Gla protein as marker for vascular calcification. Arterioscler Thromb Vasc Biol. 2005;25(8):1629-1633. (PubMed)
91. van den Heuvel EG, van Schoor NM, Lips P, et al. Circulating uncarboxylated matrix Gla protein, a marker of vitamin K status, as a risk factor of cardiovascular disease. Maturitas. 2014;77(2):137-141. (PubMed)
92. Mayer O, Jr., Seidlerova J, Bruthans J, et al. Desphospho-uncarboxylated matrix Gla-protein is associated with mortality risk in patients with chronic stable vascular disease. Atherosclerosis. 2014;235(1):162-168. (PubMed)
93. Shea MK, O'Donnell CJ, Hoffmann U, et al. Vitamin K supplementation and progression of coronary artery calcium in older men and women. Am J Clin Nutr. 2009;89(6):1799-1807. (PubMed)
94. Shea MK, O'Donnell CJ, Vermeer C, et al. Circulating uncarboxylated matrix gla protein is associated with vitamin K nutritional status, but not coronary artery calcium, in older adults. J Nutr. 2011;141(8):1529-1534. (PubMed)
95. Chatrou ML, Winckers K, Hackeng TM, Reutelingsperger CP, Schurgers LJ. Vascular calcification: the price to pay for anticoagulation therapy with vitamin K-antagonists. Blood Rev. 2012;26(4):155-166. (PubMed)
96. Booth SL, Suttie JW. Dietary intake and adequacy of vitamin K. J Nutr. 1998;128(5):785-788. (PubMed)
97. Beulens JW, Booth SL, van den Heuvel EG, Stoecklin E, Baka A, Vermeer C. The role of menaquinones (vitamin K(2)) in human health. Br J Nutr. 2013;110(8):1357-1368. (PubMed)
98. Hendler SS, Rorvik DR. Vitamin K. PDR for Nutritional Supplements. 2nd ed. Montvale: Physicians' Desk Reference Inc.; 2008:708-7123.
99. Traber MG. Vitamin E and K interactions--a 50-year-old problem. Nutr Rev. 2008;66(11):624-629. (PubMed)
100. Booth SL, Golly I, Sacheck JM, et al. Effect of vitamin E supplementation on vitamin K status in adults with normal coagulation status. Am J Clin Nutr. 2004;80(1):143-148. (PubMed)
101. Pastori D, Carnevale R, Cangemi R, et al. Vitamin E serum levels and bleeding risk in patients receiving oral anticoagulant therapy: a retrospective cohort study. J Am Heart Assoc. 2013;2(6):e000364. (PubMed)
102. Shearer MJ, Newman P. Recent trends in the metabolism and cell biology of vitamin K with special reference to vitamin K cycling and MK-4 biosynthesis. J Lipid Res. 2014;55(3):345-362. (PubMed)
103. Thorp JA, Gaston L, Caspers DR, Pal ML. Current concepts and controversies in the use of vitamin K. Drugs. 1995;49(3):376-387. (PubMed)
104. Reiffel JA. An important indirect drug interaction between dronedarone and warfarin that may be extrapolated to other drugs that can alter gastrointestinal function. Am Heart J. 2011;161(2):e5; author reply e7. (PubMed)
105. Shirolkar SC, Fiuzat M, Becker RC. Dronedarone and vitamin K antagonists: a review of drug-drug interactions. Am Heart J. 2010;160(4):577-582. (PubMed)
106. Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. Montvale: Medical Economics Company, Inc.; 2001.