Minerals
Minerals are elements that originate in the Earth and cannot be made by living organisms. Plants obtain minerals from the soil, and most of the minerals in our diets come directly from plants or indirectly from animal sources. Minerals may also be present in the water we drink, but this varies with geographic locale. Minerals from plant sources may also vary from place to place, because soil mineral content varies geographically.
The information from the Linus Pauling Institute's Micronutrient Information Center on vitamins and minerals is now available in a book titled, An Evidence-based Approach to Vitamins and Minerals: Health Benefits and Intake Recommendations. The book can be purchased from the Linus Pauling Institute or Thieme Medical Publishers.
Select a mineral from the list for more information.
Calcium
Contents
Summary
- Calcium is a major constituent of bones and teeth and also plays an essential role as second messenger in cell-signaling pathways. Circulating calcium concentrations are tightly controlled by the parathyroid hormone (PTH) and vitamin D at the expense of the skeleton when dietary calcium intakes are inadequate. (More information)
- The recommended dietary allowance (RDA) for calcium is 1,000 mg/day-1,200 mg/day for adults. (More information)
- The skeleton is a reserve of calcium drawn upon to maintain normal serum calcium in case of inadequate dietary calcium. Thus, calcium sufficiency is required to maximize the attainment of peak bone mass during growth and to limit the progressive demineralization of bones later in life, which leads to osteoporosis, bone fragility, and an increased risk of fractures. (More information)
- High concentrations of calcium and oxalate in the urine are major risk factors for the formation of calcium oxalate stones in the kidneys. Because dietary calcium intake has been inversely associated with stone occurrence, it is thought that adequate calcium consumption may reduce the absorption of dietary oxalate, thus reducing urinary oxalate and kidney stone formation. (More information)
- Data from observational studies and randomized controlled trials support calcium supplementation in reducing the risk of high blood pressure and preeclampsia in pregnant women. The World Health Organization advises that all pregnant women in areas of low calcium intake (i.e., low-income countries with intakes around 300 to 600 mg/day) be given supplemental calcium starting in the 20th week of pregnancy. (More information)
- Prospective cohort studies have reported an association between higher calcium intakes and lower risk of developing colorectal cancer; however, large clinical trials of calcium supplementation are needed. (More information)
- Current available data suggest that adequate calcium intakes may play a role in body weight regulation and have therapeutic benefits in the management of moderate-to-severe premenstrual symptoms. (More information)
- Adequate calcium intake is critical for maintaining a healthy skeleton. Calcium is found in a variety of foods, including dairy products, beans, and vegetables of the kale family. Yet, content and bioavailability vary among foods, and certain drugs are known to adversely affect calcium absorption. (More information)
- Hypercalcemia, a condition of abnormally high concentrations of calcium in blood, is usually due to malignancy or primary hyperparathyroidism. However, the use of large doses of supplemental calcium, together with absorbable alkali, increases the risk of hypercalcemia, especially in postmenopausal women. Often associated with gastrointestinal disturbances, hypercalcemia can be fatal if left untreated. (More information)
- High calcium intakes — either from dairy foods or from supplements — have been associated with increased risks of prostate cancer and cardiovascular events in some, but not all, observational and intervention studies. However, there is currently no evidence of such detrimental effects when people consume a total of 1,000 to 1,200 mg/day of calcium (diet and supplements combined), as recommended by the Food and Nutrition Board of the Institute of Medicine. (More information)
Calcium is the most abundant mineral in the human body. About 99% of the calcium in the body is found in bones and teeth, while the other 1% is found in the blood and soft tissue. Calcium concentrations in the blood and fluid surrounding the cells (extracellular fluid) must be maintained within a narrow concentration range for normal physiological functioning. The physiological functions of calcium are so vital to survival that the body will stimulate bone resorption (demineralization) to maintain normal blood calcium concentrations when calcium intake is inadequate. Thus, adequate intake of calcium is a critical factor in maintaining a healthy skeleton (1).
Function
Structure
Calcium is a major structural element in bones and teeth. The mineral component of bone consists mainly of hydroxyapatite [Ca10(PO4)6(OH)2] crystals, which contain large amounts of calcium, phosphorus, and oxygen. Bone is a dynamic tissue that is remodeled throughout life. Bone cells called osteoclasts begin the process of remodeling by dissolving or resorbing bone. Bone-forming cells called osteoblasts then synthesize new bone to replace the bone that was resorbed. During normal growth, bone formation exceeds bone resorption. Osteoporosis may result when bone resorption chronically exceeds formation (1).
Calcium homeostasis
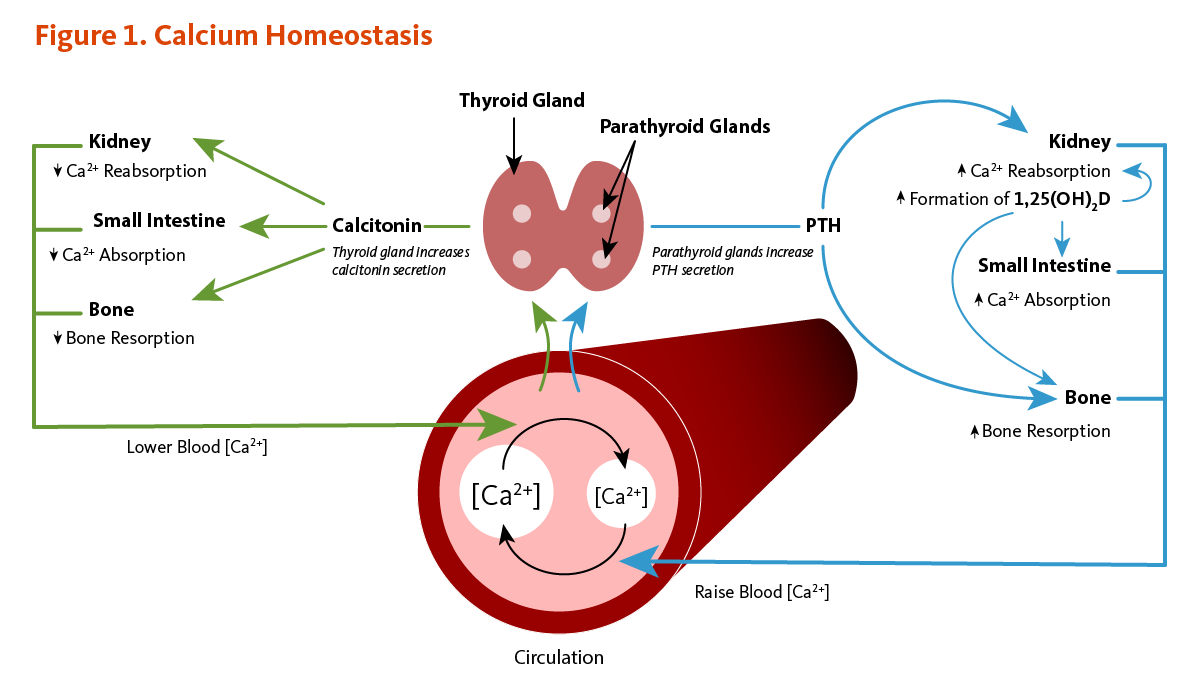
Calcium concentrations in the blood and fluid that surround cells are tightly controlled in order to preserve normal physiological function. A slight drop in blood calcium concentration (e.g., in the case of inadequate calcium intake) is sensed by the parathyroid glands, resulting in their increased secretion of parathyroid hormone (PTH). In the kidneys, PTH stimulates the conversion of vitamin D into its active form (1,25-dihydroxyvitamin D; calcitriol), which rapidly decreases urinary excretion of calcium but increases urinary excretion of phosphorus. Elevations in PTH also stimulates bone resorption, resulting in the release of bone mineral (calcium and phosphate) — actions that also contribute to restoring serum calcium concentrations. Increased circulating 1,25-dihydroxyvitamin D also triggers intestinal absorption of both calcium and phosphorus. Like PTH, 1,25-dihydroxyvitamin D stimulates the release of calcium from bone by activating osteoclasts (bone-resorbing cells). When blood calcium rises to normal levels, the parathyroid glands stop secreting PTH. A slight increase in blood calcium concentration stimulates the production and secretion of the peptide hormone, calcitonin, by the thyroid gland. Calcitonin inhibits PTH secretion, decreases both bone resorption and intestinal calcium absorption, and increases urinary calcium excretion (Figure 1). Finally, acute changes in blood calcium concentrations do not seem to elicit the secretion of the phosphaturic hormone fibroblast growth factor 23 (FGF-23), which is produced by bone-forming cells (osteoblasts/osteocytes) in response to increases in phosphorus intake (see the article on Phosphorus) (2). While this complex system allows for rapid and tight control of blood calcium concentrations, it does so at the expense of the skeleton (1).
Cell signaling
Calcium plays a role in mediating the constriction and relaxation of blood vessels (vasoconstriction and vasodilation), nerve impulse transmission, muscle contraction, and the secretion of hormones like insulin (1). Excitable cells, such as skeletal muscle and nerve cells, contain voltage-dependent calcium channels in their cell membranes that allow for rapid changes in calcium concentrations. For example, when a nerve impulse stimulates a muscle fiber to contract, calcium channels in the cell membrane open to allow calcium ions into the muscle cell. Within the cell, these calcium ions bind to activator proteins, which help release a flood of calcium ions from storage vesicles of the endoplasmic reticulum (ER) inside the cell. The binding of calcium to the protein troponin-c initiates a series of steps that lead to muscle contraction. The binding of calcium to the protein calmodulin activates enzymes that break down muscle glycogen to provide energy for muscle contraction. Upon completion of the action, calcium is pumped outside the cell or into the ER until the next activation (reviewed in 3).
Regulation of protein function
Calcium is necessary to stabilize a number of proteins, including enzymes, optimizing their activities. The binding of calcium ions is required for the activation of the seven "vitamin K-dependent" clotting factors in the coagulation cascade. The term, "coagulation cascade," refers to a series of events, each dependent on the other that stops bleeding through clot formation (see the article on Vitamin K).
Nutrient interactions
Vitamin D
Vitamin D is required for optimal calcium absorption (see Function or the article on Vitamin D). Several other nutrients (and non-nutrients) influence the retention of calcium by the body and may affect calcium nutritional status.
Sodium
Dietary sodium is a major determinant of urinary calcium loss (1). High-sodium intake results in increased loss of calcium in the urine, possibly due to competition between sodium and calcium for reabsorption in the kidneys or by an effect of sodium on parathyroid hormone (PTH) secretion. Every 1-gram (g) increment in sodium (2.5 g of sodium chloride; NaCl salt) excreted by the kidneys has been found to draw about 26.3 milligrams (mg) of calcium into the urine (1). A study conducted in adolescent girls reported that a high-salt diet had a greater effect on urinary sodium and calcium excretion in White compared to Black girls, suggesting differences among ethnic groups (4). In adult women, each extra gram of sodium consumed per day is projected to produce an additional rate of bone loss of 1% per year if all of the calcium loss comes from the skeleton.
A number of cross-sectional and intervention studies have suggested that high-sodium intakes are deleterious to bone health, especially in older women (5). A two-year longitudinal study in postmenopausal women found increased urinary sodium excretion (an indicator of increased sodium intake) to be associated with decreased bone mineral density (BMD) at the hip (6). Another study in 40 postmenopausal women found that adherence to a low-sodium diet (2 g/day) for six months was associated with significant reductions in sodium excretion, calcium excretion, and amino-terminal propeptide of type I collagen, a biomarker of bone resorption. Yet, these associations were only observed in women with elevated baseline urinary sodium excretions (7). Finally, in a randomized, placebo-controlled study in 60 postmenopausal women, potassium citrate supplementation has been found to prevent an increase in calcium excretion induced by the consumption of a high-sodium diet (≥5,000 mg/day of elemental sodium) for four weeks (8).
Protein
Increasing dietary protein intake enhances intestinal calcium absorption, as well as urinary calcium excretion (9). The RDA for protein is 46 grams (g)/day for adult women and 56 g/day for adult men; however, the average intake of protein in the US tends to be higher (about 70 g/day in adult women and over 100 g per day in adult men) (10). It was initially thought that high-protein diets may result in a negative calcium balance (when the sum of urinary and fecal calcium excretion becomes greater than calcium intake) and thus increase bone loss (11). However, most observational studies have reported either no association or positive associations between protein intake and bone mineral density in children, adults, and elderly subjects (reviewed in 12). The overall calcium balance appears to be unchanged by high dietary protein intake in healthy individuals (13), and current evidence suggests that increased protein intakes in those with adequate supplies of protein, calcium, and vitamin D do not adversely affect BMD or fracture risk (14).
Phosphorus
Phosphorus, which is typically found in protein-rich food, tends to increase the excretion of calcium in the urine. Diets with low calcium-to-phosphorus ratios (Ca:P ≤0.5) have been found to increase parathyroid hormone (PTH) secretion and urinary calcium excretion (15, 16). Also, the intestinal absorption and fecal excretion of calcium and phosphorus are influenced by calcium-to-phosphorus ratios of ingested food. Indeed, in the intestinal lumen, calcium salts can bind to phosphorus to form complexes that are excreted in the feces. This forms the basis for using calcium salts as phosphorus binders to lower phosphorus absorption in individuals with kidney insufficiency (17). Increasing phosphorus intakes from cola soft drinks (high in phosphoric acid) and food additives (high in phosphates) may have adverse effects on bone health (18). At present, there is no convincing evidence that the dietary phosphorus levels experienced in the US adversely affect bone health. Yet, the substitution of large quantities of phosphorus-containing soft drinks for milk or other sources of dietary calcium may represent a serious risk to bone health in adolescents and adults (see the article on Phosphorus).
Caffeine
Exposure to caffeine concentrations ≤400 mg/day have led to increased urinary calcium content in two randomized controlled trials (19, 20). However, caffeine intakes of 400 mg/day did not significantly change urinary calcium excretion over 24 hours in premenopausal women when compared to a placebo (21). A systematic review of 14 studies recently concluded that daily intake of ≤400 mg of caffeine was unlikely to interfere with calcium homeostasis, impact negatively bone mineral density, or increase the risks of osteoporosis and fracture in individuals with adequate calcium intakes (22).
Deficiency
A low blood calcium level (hypocalcemia) usually implies abnormal parathyroid function since the skeleton provides a large reserve of calcium for maintaining normal blood levels, especially in the case of low dietary calcium intake. Other causes of abnormally low blood calcium concentrations include chronic kidney failure, vitamin D deficiency, and low blood magnesium levels often observed in cases of severe alcoholism. Magnesium deficiency can impair parathyroid hormone (PTH) secretion by the parathyroid glands and lower the responsiveness of osteoclasts to PTH. Thus, magnesium supplementation is required to correct hypocalcemia in people with low serum magnesium concentrations (see the article on Magnesium). Chronically low calcium intakes in growing individuals may prevent the attainment of optimal peak bone mass. Once peak bone mass is achieved, inadequate calcium intake may contribute to accelerated bone loss and ultimately to the development of osteoporosis (see Disease Prevention) (1).
The Recommended Dietary Allowance (RDA)
Updated recommendations for calcium intake based on the optimization of bone health were released by the Food and Nutrition Board (FNB) of the Institute of Medicine in 2011 (9). The Recommended Dietary Allowance (RDA) for calcium is listed in Table 1 by life stage and gender.
| Life Stage | Age |
Males (mg/day) |
Females (mg/day) |
|---|---|---|---|
| Infants | 0-6 months | 200 (AI) | 200 (AI) |
| Infants | 6-12 months | 260 (AI) | 260 (AI) |
| Children | 1-3 years | 700 | 700 |
| Children | 4-8 years | 1,000 | 1,000 |
| Children | 9-13 years | 1,300 | 1,300 |
| Adolescents | 14-18 years | 1,300 | 1,300 |
| Adults | 19-50 years | 1,000 | 1,000 |
| Adults | 51-70 years | 1,000 | 1,200 |
| Adults | >70 years | 1,200 | 1,200 |
| Pregnancy | 14-18 years | - | 1,300 |
| Pregnancy | 19-50 years | - | 1,000 |
| Breast-feeding | 14-18 years | - | 1,300 |
| Breast-feeding | 19-50 years | - | 1,000 |
Disease Prevention
Osteoporosis
Osteoporosis is a skeletal disorder in which bone mass and strength are compromised, resulting in an increased risk of fracture. Sustaining a hip fracture is one of the most serious consequences of osteoporosis. Nearly one-third of those who sustain osteoporotic hip fractures enter nursing homes within a year following the fracture, and one person in four dies within one year of experiencing an osteoporotic hip fracture (23). Despite being a common diagnosis in postmenopausal women, osteoporosis also affects 4%-6% of men over the age of 50 years (24).
Osteoporosis is a multifactorial disorder, and nutrition is only one factor contributing to its development and progression (25). Other factors that increase the risk of developing osteoporosis include, but are not limited to, increased age, female gender, estrogen deficiency, smoking, high alcohol intake (three or more drinks/day), metabolic disease (e.g., hyperthyroidism), and the use of certain medications (e.g., corticosteroids and anticonvulsants) (26). A predisposition to osteoporotic fracture is related to one's peak bone mass and to the rate of bone loss after peak bone mass has been attained. After adult height has been reached, the skeleton continues to accumulate bone until the third decade of life. Genetic factors exert a strong influence on peak bone mass, but lifestyle factors can also play a significant role. Strategies for reducing the risk of osteoporotic fracture include the attainment of maximal peak bone mass and the reduction of bone loss later in life. A number of lifestyle factors, including diet (especially calcium and protein intake) and physical activity, are amenable to interventions aimed at maximizing peak bone mass and limiting osteoporotic fracture risk (27).
Physical exercise is a lifestyle factor that has been associated with numerous health benefits and is likely to contribute to the prevention of osteoporosis and osteoporotic fracture. There is evidence to suggest that physical activity early in life contributes to the attainment of higher peak bone mass (27). Moreover, lifelong participation in physical activities in the presence of adequate calcium and vitamin D supply (from dietary sources and/or sunlight exposure) may have a modest effect on slowing the rate of bone loss later in life (28). Current National Osteoporosis Foundation guidelines include recommendations of regular muscle-strengthening and weight-bearing exercise to all postmenopausal women and men ages 50 and older (29). Although benefits in reducing bone loss might be limited, muscle-strengthening exercise, including weight training and other resistive exercises (e.g., yoga and Pilates) and weight-bearing exercise (e.g., walking, jogging, and stair climbing), may improve strength, posture, balance, and coordination, thus contributing to reduced risk of falls (29). One compilation of published calcium trials indicated that the beneficial skeletal effect of increased physical activity was achievable only at calcium intakes above 1,000 mg/day in women in late menopause (reviewed in 28).
The progressive loss of bone mineral density (BMD) leading to osteopenia (pre-osteoporosis) and osteoporosis is usually assessed by dual-energy x-ray absorptiometry (DEXA) at the hip and lumbar spine (30). Several randomized, placebo-controlled clinical trials have evaluated the effect of supplemental calcium in the preservation of BMD and the prevention of fracture risk in men and women aged 50 years and older. A meta-analysis of 15 randomized controlled trials, including 1,533 men and women >50 years of age, found that increasing calcium intake from dietary sources (i.e., milk, milk powder, dairy products, or hydroxyapatite preparations) increased BMD by 0.6%-1% at the hip (+0.6%) and total body (+1.0%) after one year and by 0.7%-1.8% at the lumbar spine (+0.7%), femoral neck (+1.8%), total hip (+1.5%), and total body (+0.9%) sites after two years (31). A meta-analysis of 51 randomized controlled trials in 12,257 adults (>50 years) found that BMD at all bone sites (lumbar spine, femoral neck, total hip, forearm) increased by 0.7%-1.4% after one year and 0.8%-1.5% after two years of supplemental calcium, alone or in combination with vitamin D (31). Such modest increases may help limit the average rate of BMD loss after menopause but are unlikely to translate into meaningful fracture risk reductions. A meta-analysis of 20 randomized controlled trials that reported on total fracture risk found an 11% risk reduction associated with supplemental calcium with or without vitamin D (32). However, there was no effect when the analysis was restricted to the largest trials with the lowest risk of bias. Additionally, no reductions were found in risks of hip, vertebral and forearm fractures with calcium supplementation (32). Because estrogen withdrawal significantly impairs intestinal absorption and renal reabsorption of calcium, the level of calcium requirement might depend on whether postmenopausal women receive hormone replacement therapy (28).
The US Preventive Services Task Force conducted a meta-analysis of 11 randomized placebo-controlled trials that included 52,915 older people (of whom 69% were postmenopausal women) and reported that the supplementation of vitamin D (300-1,000 IU/day) and calcium (500-1,200 mg/day) for up to seven years resulted in a 12% reduction in the risk of any new fracture (33). There was no significant effect of vitamin D without calcium (33). A recently updated meta-analysis of randomized, placebo-controlled trials commissioned by the National Osteoporosis Foundation found a 15% reduction in risk of total fracture (8 studies) and a 30% reduction in risk of hip fractures (six studies) with calcium and vitamin D supplementation in older people (34). The National Osteoporosis Foundation advises that adequate intake of calcium (1,000-1,200 mg/day) and vitamin D (800-1,000 IU/day) be included in the diet of all middle-aged men and women (35).
The role and efficacy of vitamin D supplementation in strengthening bone and preventing fracture in older people remain controversial topics. The active form of vitamin D, 1,25-dihydroxyvitamin D, stimulates calcium absorption by promoting the synthesis of calcium-binding proteins in the intestine. While no amount of vitamin D can compensate inadequate total calcium intake, vitamin D insufficiency (defined as circulating concentrations of 25-hydroxyvitamin D below 20 ng/mL [50 nmol/L]) can lead to secondary hyperparathyroidism and an increased risk of osteoporosis (9, 36). Conversely, in postmenopausal women (ages 57-90 years) with adequate total calcium intakes (1,400 IU/day), serum 25-hydroxyvitamin D concentrations ranging from 20 ng/mL to 66 ng/mL had little effect on calcium absorption (only 6% increase over the range) (37). In a randomized, placebo-controlled trial, the supplementation of 1,000 IU/day of vitamin D to postmenopausal women (mean age, 77.2 years) for one year was found to significantly increase circulating 25-hydroxyvitamin D concentrations by 34% from baseline but failed to enhance calcium absorption in the presence of high total calcium intakes (dietary plus supplemental calcium corresponding to an average 2,100 mg/day) (38). This study also reported no significant difference in measures of BMD at the hip and total body between placebo- and vitamin D-treated women. In addition, the pooled analysis of seven randomized controlled trials, including 65,517 older individuals living in the community or in an institution, found that vitamin D (400-800 IU/day) could reduce the risk of any fracture only when combined with calcium (1,000 mg/day) (39). Interestingly, the results of a series of trials included in three recent meta-analyses (33, 40, 41) have suggested that supplemental vitamin D and calcium may have greater benefits in the prevention of fracture in institutionalized, older people who are also at increased risk of vitamin D deficiency and fractures compared to community dwellers (42, 43).
For more information about bone health and osteoporosis, see the article, Micronutrients and Bone Health, and visit the National Osteoporosis Foundation website.
Kidney stones
Approximately 6% of women and 15% of men in industrialized countries will have a kidney stone during their lifetime. Most kidney stones are composed of calcium oxalate or calcium phosphate. Subjects with an abnormally high level of calcium in the urine (hypercalciuria) are at higher risk of developing kidney stones (a process called nephrolithiasis) (44). High urinary oxalate level is another risk factor for calcium oxalate stone formation. Most subjects with a history of kidney stones and/or idiopathic hypercalciuria have increased intestinal calcium absorption (45). Although it was initially recommended to limit dietary calcium intake in these patients, a number of prospective cohort studies have reported associations between lower total dietary calcium intake and increased risk of incident kidney stones (46-48). The prospective analyses of three large cohorts, including a total of 30,762 men and 195,865 women followed for a combined 56 years, have indicated that the risk of kidney stones was significantly lower in individuals in the highest versus lowest quintile of dietary calcium intake from dairy or nondairy sources (49). Additionally, a five-year randomized intervention study that enrolled 120 men with idiopathic hypercalciuria (mean age, 45 years) reported that those assigned to a low-calcium diet (approximately 400 mg/day) had a 51% higher risk of kidney stone recurrence compared to those on a normal-to-high calcium (1,200 mg/day), low animal-protein, low-salt diet (50).
Mechanisms by which increased dietary calcium might reduce the risk of incident kidney stones are not fully understood. An inverse relationship was reported between total calcium intake and intestinal calcium absorption in the recent cross-sectional analysis of a cohort of 5,452 postmenopausal women (45). Moreover, women with higher supplemental calcium intake and lower calcium absorption were less likely to report a history of kidney stones (45). Adequate intake of calcium with food may reduce the absorption of dietary oxalate and lower urinary oxalate through formation of the insoluble calcium oxalate salt (51, 52). A recent small intervention study in 10 non-stone-forming young adults observed that the ingestion of large amounts of oxalate did not increase the risk of calcium oxalate stone occurrence in the presence of recommended level of dietary calcium (53).
However, a randomized, double-blind, placebo-controlled trial in 36,282 postmenopausal women reported that a combination of supplemental calcium (1,000 mg/day) and vitamin D (400 IU/day) was associated with a significantly increased incidence of self-reported kidney stones during a seven-year treatment period. More controlled trials may be necessary to determine whether supplemental calcium affects kidney stone risk (54). However, a systematic review of observational studies and randomized controlled trials that primarily reported on bone-related outcomes failed to find an effect of calcium supplementation on stone incidence (55). A potential kidney stone risk associated with calcium supplementation may likely depend on whether supplemental calcium is co-ingested with oxalate-containing foods or consumed separately. Further research is needed to verify whether osteoporosis treatment drugs (e.g., biphosphonates) rather than calcium supplements might influence the risk of stone occurrence (56).
Current data suggest that diets providing adequate dietary calcium and low levels of animal protein, oxalate, and sodium may benefit the prevention of stone recurrence in subjects with idiopathic hypercalciuria (57-59).
Hypertensive disorders of pregnancy
Pregnancy-induced hypertensive disorders, including gestational hypertension, preeclampsia, and eclampsia, complicate approximately 10% of pregnancies and are a major health risk for pregnant women and their offspring (60). Gestational hypertension is defined as an abnormally high blood pressure that usually develops after the 20th week of pregnancy. Preeclampsia is characterized by poor placental perfusion and a systemic inflammation that may involve several organ systems, including the cardiovascular system, kidneys, liver, and hematological system (61). In addition to gestational hypertension, preeclampsia is associated with the development of severe swelling (edema) and the presence of protein in the urine (proteinuria). Eclampsia is the occurrence of seizures in association with the syndrome of preeclampsia and is a significant cause of maternal and perinatal mortality.
Although cases of preeclampsia are at high risk of developing eclampsia, one-quarter of women with eclampsia do not initially exhibit preeclamptic symptoms. Risk factors for preeclampsia include genetic predisposition, advanced maternal age, first pregnancies, multiple pregnancies (e.g., twins or triplets), obesity, diabetes, and some autoimmune diseases (61). While the pathogenesis of preeclampsia is not entirely understood, nutrition and especially calcium metabolism appear to play a role. Data from epidemiological studies have suggested an inverse relationship between calcium intake during pregnancy and the incidence of preeclampsia (reviewed in 62). Impairment of calcium metabolism when circulating vitamin D concentration is low and/or when dietary calcium intake is inadequate may contribute to the risk of hypertension during pregnancy.
Secondary hyperparathyroidism (high PTH level) due to vitamin D deficiency in young pregnant women has been associated with high maternal blood pressure and increased risk of preeclampsia (63). The risk for elevated PTH concentration was also found to be increased in vitamin D-sufficient women with low-calcium intakes (<480 mg/day) during pregnancy when compared with adequate-to-high calcium intakes (≥1,000 mg/day) (64). In addition, vitamin D deficiency may trigger hypertension through the inappropriate activation of the renin-angiotensin system (see the article on Vitamin D).
Potential beneficial effects of calcium in the prevention of preeclampsia have been investigated in several randomized, placebo-controlled studies. The most recent meta-analysis of 13 trials in 15,730 pregnant women found that calcium supplementation with at least 1,000 mg/day (mostly 1,500-2,000 mg/day) from about 20 weeks of pregnancy (34 weeks of pregnancy at the latest) was associated with significant reductions in the risk of high blood pressure, preeclampsia, and preterm birth (62). Greater risk reductions were reported among pregnant women at high risk of preeclampsia (5 trials; 587 women) or with low dietary calcium intake (8 trials; 10,678 women). Another meta-analysis of nine randomized controlled trials in high-risk women indicated that lower doses of calcium supplementation (≤800 mg/day), alone or with a co-treatment (i.e., vitamin D, linoleic acid, or antioxidants), could also lower the risk of preeclampsia by 62% (65). Yet, based on the systematic review of high-quality randomized controlled trials, which used mostly high-dose calcium supplements, the World Health Organization (WHO) recently recommended that all pregnant women in areas of low-calcium intake (i.e., low-income countries with intakes around 300-600 mg/day) be given 1.5 to 2 g (1,500 to 2,000 mg)/day of elemental calcium from the 20th week of pregnancy (66).
Because excessive calcium supplementation may be harmful (see Safety), further research is required to verify whether calcium supplementation above the current IOM recommendation (1,000 mg/day for pregnant women, ages 19-50 years) would provide greater benefits to women at high risk of preeclampsia. Finally, the lack of effect of supplemental calcium on proteinuria (reported in two trials only) suggested that calcium supplementation from mid-pregnancy might be too late to oppose the genesis of preeclampsia (67, 68). A randomized, double-blind, placebo-controlled study — the WHO Calcium and Pre-eclampsia (CAP) trial — is ongoing to evaluate the effect of calcium supplementation with 500 mg/day, starting before pregnancy and until the 20th week of pregnancy, on the risk of preeclampsia in high-risk women (69, 70).
Colorectal cancer
Colorectal cancer (CRC) is the most common gastrointestinal cancer and the second leading cause of cancer death in the US (71). CRC is caused by a combination of genetic and environmental factors, but the degree to which these two types of factors influence CRC risk in individuals varies widely. In individuals with familial adenomatous polyposis (FAP) or hereditary nonpolyposis colorectal cancer (HNPCC), the cause of CRC is almost entirely genetic, while modifiable lifestyle factors, including dietary habits, tobacco use, and physical activities, greatly influence the risk of sporadic (non-hereditary) CRC.
Prospective cohort studies have consistently reported an inverse association between dairy food consumption and CRC risk. Experimental studies in cell culture and animal models have suggested plausible mechanisms underlying a role for calcium, a major nutrient in dairy products, in preventing CRC (72). In the multicenter European Prospective Investigation into Cancer and Nutrition (EPIC) prospective study of 477,122 individuals, followed for an average of 11 years, 4,513 CRC cases were documented (73). Intakes of milk, cheese, and yogurt, were inversely associated with CRC risk. The highest versus lowest quintile of total dairy intake (≥490 g/day vs. <134 g/day) was associated with a 23% lower risk of CRC. Similarly, CRC risk was 25% lower in those in the top versus bottom quintile of calcium intake from dairy food (≥839 mg/day vs. <308 mg/day). The 16-year follow-up of 41,403 women (ages 26-46 years at inclusion) from the prospective Nurses’ Health Study II (NHS II) documented 2,273 diagnoses of colorectal adenomas (precancerous polyps). The analysis of the prospective cohort found that women with total calcium intake of 1,001-1,250 mg/day had a 76% lower risk of developing advanced adenomas (i.e., adenomas more likely to become malignant) compared to those with intakes equal to and below 500 mg/day (74). In addition, a dose-response analysis using data from eight prospective studies (11,005 CRC cases) estimated that an increase of 300 mg/day in total calcium intake was associated with a 5% reduction in CRC risk (75). Total daily intake of calcium ranged from 333 to 2,229 mg in the examined studies. In addition, the dose-response analysis of six prospective studies (8,839 CRC cases among 920,837 participants) showed 11% lower odds of high-risk adenomas for each 300 mg/day increment in total calcium (75).
However, the meta-analysis of seven randomized, double-blind, placebo-controlled studies found no evidence of an effect of calcium supplementation (≥500 mg/day) for a median period of 45 months on total cancer risk and CRC risk (76). In addition, the re-analysis of the Women’s Health Initiative placebo-controlled trial failed to show a reduction in CRC risk in postmenopausal women supplemented with both vitamin D (400 IU/day) and calcium (1,000 mg/day) for seven years (77). Finally, the results of the meta-analysis of four randomized, placebo-controlled trials have suggested that calcium supplementation (1,200-2,000 mg/day) may reduce the risk of adenoma recurrence by 13% over three to five years in subjects with a history of adenomas (78). At present, it is not clear whether calcium supplementation is beneficial in CRC prevention. Larger trials designed to assess primarily the effect of long-term calcium supplementation on the incidence of adenomas and/or CRC are needed before conclusions can be drawn.
Lead toxicity
Children who are chronically exposed to lead, even in small amounts, are more likely to develop learning disabilities, behavioral problems, and to have low IQs. Deficits in growth and neurological development may occur in the infants of women exposed to lead during pregnancy and lactation. In adults, lead toxicity may result in kidney damage and high blood pressure. Although the use of lead in paint products, gasoline, and food cans has been discontinued in the US, lead toxicity continues to be a significant health problem, especially in children living in urban areas (79).
In 2012, the US Centers for Disease Control and Prevention set the reference value for blood lead concentration at 5 micrograms per deciliter (mg/dL) to identify children at risk (80). Yet, there is no known blood lead concentration below which children are 100% safe. An early study of over 300 children in an urban neighborhood found that 49% of children ages 1 to 8 years had blood lead levels above the threshold of 10 mg/dL, indicating excessive lead exposure. In this study, only 59% of children ages 1 to 3 years and 41% of children ages 4 to 8 years met the recommended levels for calcium intakes (81).
Adequate calcium intake could be protective against lead toxicity in at least two ways. Increased dietary intake of calcium is known to decrease the gastrointestinal absorption of lead. Once lead enters the body it tends to accumulate in the skeleton, where it may remain for more than 20 years. Adequate calcium intake also prevents lead mobilization from the skeleton during bone demineralization. A study of circulating concentrations of lead during pregnancy found that women with inadequate calcium intake during the second half of pregnancy were more likely to have elevated blood lead levels, probably because of increased bone demineralization, leading to the release of accumulated lead into the blood (82). Lead in the blood of a pregnant woman is readily transported across the placenta resulting in fetal lead exposure at a time when the developing nervous system is highly vulnerable. In a randomized, double-blind, placebo-controlled study in 670 pregnant women (≤14 weeks’ gestation) with average dietary calcium intakes of 900 mg/day, daily supplementation of 1,200 mg of calcium throughout the pregnancy period resulted in 8%-14% reductions in maternal blood lead concentrations (83). Similar reductions in maternal lead concentrations in the blood and breast milk of lactating mothers supplemented with calcium were reported in earlier trials (84, 85). In postmenopausal women, factors known to decrease bone demineralization, including estrogen replacement therapy and physical activity, have been inversely associated with blood lead levels (86).
Disease Treatment
Overweight and obesity
High dietary calcium intake, usually associated with dairy product consumption, has been inversely related to body weight and central obesity in a number of cross-sectional studies (reviewed in 87). Cross-sectional baseline data analyses of a number of prospective cohort studies that were not designed and powered to examine the effect of calcium intake or dairy consumption on obesity or body fat have given inconsistent results (87). Yet, a meta-analysis of 18 cross-sectional and prospective studies predicted a reduction in body mass index (a relative measure of body weight; BMI) of 1.1 kg/m2 with an increase in calcium intake from 400 mg/day to 1,200 mg/day (87). In a placebo-controlled intervention study, 32 obese subjects were randomized to energy restriction regimens (500 kCal/day deficit) for 24 weeks with (1) a standard diet providing 400 to 500 mg/day of dietary calcium and a placebo ("low calcium" diet), (2) a standard diet and 800 mg/day of supplemental calcium ("high calcium" diet), or (3) a high-dairy diet providing 1,200 mg/day of dietary calcium and a placebo (88). Energy-restricted diets resulted in significant body weight and fat loss in all three groups. Yet, body weight and fat loss were significantly more reduced with the high-calcium diet compared to the standard diet, and further reductions were measured with the high-dairy diet compared to both high-calcium and low-calcium diets. These results suggested that while calcium intake may play a role in body weight regulation, additional benefits might be attributable to other bioactive components of dairy products, such as proteins, fatty acids, and branched chain amino acids.
Yet, several mechanisms have been proposed to explain the potential impact of calcium on body weight (reviewed in 87). The most-cited mechanism is based on studies in the agouti mouse model showing that low-calcium intakes, through increasing circulating parathyroid hormone (PTH) and vitamin D, could stimulate the accumulation of fat (lipogenesis) in adipocytes (fat cells) (89). Conversely, higher intakes of calcium may reduce fat storage, stimulate the breakdown of lipids (lipolysis), and drive fat oxidation. A recent meta-analysis of randomized controlled trials estimated that high (1,300 mg/day) versus low (488 mg/day) calcium intake for a minimum of seven days increased fat oxidation by 11% (90). However, a double-blind, placebo-controlled, randomized, cross-over trial in 10 low-calcium consuming overweight or obese individuals reported that the supplementation with 800 mg/day of calcium for 5 weeks failed to modify the expression of key factors involved in fat metabolism (91). Moreover, while the model suggests a role for vitamin D in lipogenesis (fat storage), human studies have shown that vitamin D deficiency — rather than sufficiency — is often associated with obesity, and supplemental vitamin D might be effective in lowering body weight when caloric restriction is imposed (92, 93). Another mechanism suggests that high-calcium diets may limit dietary fat absorption in the intestine and increase fecal fat excretion. Indeed, in the gastrointestinal tract, calcium may trap dietary fat into insoluble calcium soaps of fatty acids that are then excreted (94). In addition, despite very limited evidence, it has also been proposed that calcium might be involved in regulating appetite and energy intake (95).
To date, there is no consensus regarding the effect of calcium on body weight changes. A meta-analysis of 29 randomized controlled trials in 2,441 participants (median age, 41.4 years) found that calcium supplementation was only associated with body weight and fat loss in short-term studies (<1 year) that used energy-restricted diets (96). Another meta-analysis of 41 randomized controlled trials (4,802 participants) found little-to-no effect of increased calcium intake from supplements or dairy foods for >12 weeks on body weight and body composition (97). Finally, a meta-analysis of 33 randomized controlled trials (4,733 participants) found no overall effect of calcium supplementation (from food or supplements) for >12 weeks on body weight changes. Yet, further subgroup analyses showed weight reductions in children and adolescents (mean, -0.26 kg), in adults (mean, -0.91 kg), and in those with normal BMI (mean, -0.53 kg). Supplemental calcium did not lead to weight loss in postmenopausal women or in overweight/obese individuals (98). At present, additional research is warranted to examine the effect of calcium intake on fat metabolism, as well as its potential benefits in the management of body weight with or without caloric restriction (99).
Premenstrual Syndrome (PMS)
PMS refers to a cluster of symptoms, including but not limited to fatigue, irritability, moodiness/depression, fluid retention, and breast tenderness, that begins sometime after ovulation (mid-cycle) and subsides with the onset of menstruation (the monthly period) (100). A severe form of PMS called premenstrual dysphoric disorder (PMDD) has been described in 3%-8% of women of childbearing age. PMDD interferes with normal functioning, affecting daily activities and relationships (101).
Low dietary calcium intakes have been linked to PMS in early reports, and supplemental calcium has been shown to decrease symptom severity (102). A nested case-control study within the Nurses' Health Study II (NHS II) found that women in the highest quintile of dietary (but not supplemental) calcium intake (median of 1,283 mg/day) had a 30% lower risk of developing PMS compared to those in the lowest quintile (median of 529 mg/day). Similarly, women in the highest versus lowest quintile of skim or low-fat milk intake (≥4 servings/day vs. ≤1 serving/week) had a 46% lower risk of PMS (103). In a randomized, double-blind, placebo-controlled clinical trial of 466 women with moderate-to-severe premenstrual symptoms, supplemental calcium (1,200 mg/day) for three menstrual cycles was associated with a 48% reduction in total symptom scores, compared to a 30% reduction observed in the placebo group (104). Similar positive effects were reported in earlier double-blind, placebo-controlled, cross-over trials that administered 1,000 mg of calcium daily (105, 106). Recent small randomized controlled trials also reported that supplemental calcium (400-500 mg/day) for three weeks to three months reduced severity and/or frequency of symptoms in women with mild-to-moderate PMS (107-110). Currently available data indicate that daily calcium intakes from food and/or supplements may have therapeutic benefits in women diagnosed with PMS or PMDD (111, 112).
Hypertension
The relationship between calcium intake and blood pressure has been investigated extensively over the past decades. A meta-analysis of 23 large observational studies conducted in different populations worldwide found a reduction in systolic blood pressure of 0.34 millimeters of mercury (mm Hg) per 100 mg of calcium consumed daily and a reduction in diastolic blood pressure of 0.15 mm Hg per 100 mg calcium (113). In the DASH (Dietary Approaches to Stop Hypertension) study, 549 people were randomized to one of three diets for eight weeks: (1) a control diet that was low in fruit, vegetables, and dairy products; (2) a diet rich in fruit (~5 servings/day) and vegetables (~3 servings/day); and (3) a combination diet rich in fruit and vegetables, as well as low-fat dairy products (~3 servings/day) (114). The combination diet represented an increase of about 800 mg of calcium/day over the control and fruit/vegetable-rich diets for a total of about 1,200 mg of calcium/day. Overall, the reduction in systolic blood pressure was greater with the combination diet than with the fruit/vegetable diet or the control diet. Among participants diagnosed with hypertension, the combination diet reduced systolic blood pressure by 11.4 mm Hg and diastolic pressure by 5.5 mm Hg more than the control diet, while the reduction for the fruit/vegetable diet was 7.2 mm Hg for systolic and 2.8 mm Hg for diastolic blood pressure compared to the control diet (115). This research suggested that calcium intake at the recommended level (1,000-1,200 mg/day) may be helpful in preventing and treating moderate hypertension (116).
Yet, two large systematic reviews and meta-analyses of randomized controlled trials have examined the effect of calcium supplementation on blood pressure compared to placebo in either normotensive or hypertensive individuals (117, 118). Neither of the analyses reported any significant effect of supplemental calcium on blood pressure in normotensive subjects. A small but significant reduction in systolic blood pressure, but not in diastolic blood pressure, was reported in participants with hypertension. Of note, calcium supplementation in these randomized controlled trials ranged from 400 to 2,200 mg/day, with 1,000 to 1,500 mg/day being the more common dosages. A more recent meta-analysis of 13 randomized controlled studies in 485 individuals with elevated blood pressure found a significant reduction of 2.5 mm Hg in systolic blood pressure but no change in diastolic blood pressure with calcium supplementation (119). The modest effect of calcium on blood pressure needs to be confirmed in larger, high-quality, well-controlled trials before any recommendation is made regarding the management of hypertension. Finally, a review of the literature on the effect of high-calcium intake (dietary and supplemental) in postmenopausal women found either no reduction or mild and transient reductions in blood pressure (120).
More information about the DASH diet is available from the National Institutes of Health (NIH).
Sources
Food sources
Data analysis of the US National Health and Nutrition Examination Surveys (NHANES) 2009-2010 and 2011-2012 found inadequate calcium intakes (defined as intakes below the Estimated Average Requirement [EAR]) in 37.7% of non-supplemented adults (ages, ≥19 years) and 19.6% of adults taking multivitamin/mineral supplements (121). Dairy foods provide 75% of the calcium in the American diet. However, it is typically during the most critical period for peak bone mass development that adolescents tend to replace milk with soft drinks (122). Dairy products represent rich and absorbable sources of calcium, but certain vegetables and grains also provide calcium.
However, the bioavailability of the calcium must be taken into consideration. The calcium content in calcium-rich plants in the kale family (broccoli, bok choy, cabbage, mustard, and turnip greens) is as bioavailable as that in milk; however, other plant-based foods contain components that inhibit the absorption of calcium. Oxalic acid, also known as oxalate, is the most potent inhibitor of calcium absorption and is found at high concentrations in spinach and rhubarb and somewhat lower concentrations in sweet potatoes and dried beans. Phytic acid (phytate) is a less potent inhibitor of calcium absorption than oxalate. Yeast possess an enzyme (phytase) that breaks down phytate in grains during fermentation, lowering the phytate content of breads and other fermented foods. Only concentrated sources of phytate, such as wheat bran or dried beans, substantially reduce calcium absorption (123).
Additional dietary constituents may affect calcium absorption (see Nutrient interactions). Table 2 lists a number of calcium-rich foods, along with their calcium content. For more information on the nutrient content of foods, search USDA's FoodData Central.
Supplements
Most experts recommend obtaining as much calcium as possible from food because calcium in food is accompanied by other important nutrients that assist the body in utilizing calcium. However, calcium supplements may be necessary for those who have difficulty consuming enough calcium from food (124). No multivitamin/mineral tablet contains 100% of the recommended daily value (DV) for calcium because it is too bulky, and the resulting pill would be too large to swallow. The "Supplement Facts" label, required on all supplements marketed in the US, lists the calcium content of the supplement as elemental calcium. Calcium preparations used as supplements include calcium carbonate, calcium citrate, calcium citrate malate, calcium lactate, and calcium gluconate. To determine which calcium preparation is in your supplement, you may have to look at the ingredient list. Calcium carbonate is generally the most economical calcium supplement. To maximize absorption, take no more than 500 mg of elemental calcium at one time. Most calcium supplements should be taken with meals, although calcium citrate and calcium citrate malate can be taken anytime. Calcium citrate is the preferred calcium formulation for individuals who lack stomach acids (achlorhydria) or those treated with drugs that limit stomach acid production (H2 blockers and proton-pump inhibitors) (reviewed in 125).
Lead in calcium supplements
Several decades ago, concern was raised regarding lead concentrations in calcium supplements obtained from natural sources (oyster shell, bone meal, dolomite) (126). In 1993, investigators found measurable quantities of lead in most of the 70 different preparations they tested (127). Since then, manufacturers have reduced the amount of lead in calcium supplements to less than 0.5 micrograms (mg)/1,000 mg of elemental calcium (128). The US Food and Drug Administration (FDA) has developed provisional total tolerable intake levels (PTTI) for lead for specific age and sex groups (129). Because lead is so widespread and long lasting, no one can guarantee entirely lead-free food or supplements. A study found measurable lead in 8 out of 21 supplements, in amounts averaging 1 to 2 mg/1,000 mg of elemental calcium, which is below the tolerable limit of 7.5 mg/1,000 mg of elemental calcium (130). A more recent survey of 324 multivitamin/mineral supplements labeled for use in children or women found that most supplements would result in lead exposure ranging from 1%-4% of the PTTI (131).
Calcium inhibits intestinal absorption of lead, and adequate calcium intake is protective against lead toxicity, so trace amounts of lead in calcium supplementation may pose less of a risk of excessive lead exposure than inadequate calcium consumption. While most calcium sources today are relatively safe, look for supplements approved or certified by independent testing (e.g., US Pharmacopeia, ConsumerLab.com) (125), follow label instructions, and avoid large doses of supplemental calcium (≥1,500 mg/day).
Safety
Toxicity
Malignancy and primary hyperparathyroidism are the most common causes of elevated calcium concentrations in the blood (hypercalcemia) (132). Hypercalcemia has not been associated with the over consumption of calcium occurring naturally in food. Hypercalcemia has been initially reported with the consumption of large quantities of calcium supplements in combination with antacids, particularly in the days when peptic ulcers were treated with large quantities of milk, calcium carbonate (antacid), and sodium bicarbonate (absorbable alkali). This condition is termed calcium-alkali syndrome (formerly known as milk-alkali syndrome) and has been associated with calcium supplement levels from 1.5 to 16.5 g/day for 2 days to 30 years. Since the treatment for peptic ulcers has evolved and because of the widespread use of over-the-counter calcium supplements, the demographic of this syndrome has changed in that those at greater risk are now postmenopausal women, pregnant women, transplant recipients, patients with bulimia, and patients on dialysis, rather than men with peptic ulcers (reviewed in 133). Supplementation with calcium (0.6 g/day-2 g/day for two to five years) has been associated with a higher risk of adverse gastrointestinal events like constipation, cramping, bloating, pain, diarrhea (134). Mild hypercalcemia may be without symptoms or may result in loss of appetite, nausea, vomiting, constipation, abdominal pain, fatigue, frequent urination (polyuria), and hypertension (132). More severe hypercalcemia may result in confusion, delirium, coma, and if not treated, death (1).
In 2011, the Food and Nutrition Board of the Institute of Medicine updated the tolerable upper intake level (UL) for calcium (9). The UL is listed in Table 3 by age group.
Although the risk of forming kidney stones is increased in individuals with abnormally elevated urinary calcium (hypercalciuria), this condition is not usually related to calcium intake, but rather to increased absorption of calcium in the intestine or increased excretion by the kidneys (9). Overall, increased dietary calcium intake has been associated with a decreased risk of kidney stones (see Kidney stones). Concerns have also been raised regarding the risks of prostate cancer and vascular disease with high intakes of calcium.
Do high calcium intakes increase the risk for prostate cancer?
Prostate cancer is the second most common cancer in men worldwide (135). Several observational studies have raised concern that high-dairy intakes are associated with increased risk of prostate cancer (136-138).
The analysis of a prospective cohort study (2,268 men followed for nearly 25 years) conducted in Iceland, a country with a high incidence of prostate cancer, found a positive association between the consumption of milk (at least once daily) during adolescence and developing prostate cancer later in life (139). Another large prospective cohort study in the US followed 21,660 male physicians for 28 years and found that men with daily skim or low-fat milk intake of at least 237 mL (8 oz) had a higher risk of developing prostate cancer compared to occasional consumers (140). The risk of low-grade, early-stage prostate cancer was associated with higher intake of skim milk, and the risk of developing fatal prostate cancer was linked to the regular consumption of whole milk (140). In a cohort of 3,918 male health professionals diagnosed with prostate cancer, 229 men died of prostate cancer and 69 developed metastasized prostate cancer during a median follow-up of 7.6 years (141). The risk of prostate cancer death was found to be increased in men with high (>4 servings/week) versus low (≤3 servings/month) intakes of whole milk. Yet, no increase in risk of prostate cancer-related mortality was associated with consumption of skim and low-fat milk, total milk, low-fat dairy products, full-fat dairy products, or total dairy products (141). A recent meta-analysis of 32 prospective cohort studies found high versus low intakes of total dairy product (15 studies), total milk (15 studies), whole milk (6 studies), low-fat milk (5 studies), cheese (11 studies), and dairy calcium (7 studies) to be associated with modest, yet significant, increases in the risk of developing prostate cancer (142). However, there was no increase in prostate cancer risk with nondairy calcium (4 studies) and calcium from supplements (8 studies). Moreover, high dairy intakes were not linked to fatal prostate cancer (142).
There is some evidence to suggest that milk consumption may result in higher circulating concentrations of insulin-like growth factor-I (IGF-I), a protein known to regulate cell proliferation (143). Circulating IGF-I concentrations have been positively correlated to the risk of developing prostate cancer in a recent meta-analysis of observational studies (144). Milk-borne IGF-I, as well as dairy proteins and calcium, may contribute to increasing circulating IGF-I in milk consumers (143). In the large EPIC study, which examined the consumption of dairy products in relation to cancer in 142,520 men, the risk of prostate cancer was found to be significantly higher in those in the top versusbottom quintile of both protein and calcium intakes from dairy foods (145). Another mechanism underlying the potential relationship between calcium intake and prostate cancer proposed that high levels of dietary calcium may lower circulating concentrations of 1,25-dihydroxyvitamin D, the active form of vitamin D, thereby suppressing vitamin D-mediated cell differentiation (146). However, studies to date have provided little evidence to suggest that vitamin D status can modify the association between dairy calcium and risk of prostate cancer development and progression (147-149).
In a multicenter, double-blind, placebo-controlled trial, 672 healthy men (mean age of 61.8 years) were randomized to daily calcium supplementation (1,200 mg) for four years. While no increase in the risk for prostate cancer has been reported during a 10.3-year follow-up, calcium supplementation resulted in a significant risk reduction in the period spanning from two years after treatment started to two years after treatment ended (150). In a review of the literature published in 2009, the US Agency for Healthcare Research and Quality indicated that not all epidemiological studies found an association between calcium intake and prostate cancer (151). The review reported that 6 out of 11 observational studies failed to find statistically significant positive associations between prostate cancer and calcium intake. Yet, in five studies, daily intakes of 921 to 2,000 mg of calcium were found to be associated with an increased risk of developing prostate cancer when compared to intakes ranging from 455 to 1,000 mg/day (151). Inconsistencies among studies suggest complex interactions between the risk factors for prostate cancer, as well as reflect the difficulties of assessing the effect of calcium intake in free-living individuals. For example, the fact that individuals with higher dairy and/or calcium intakes were found to be more likely to be engaged in healthy lifestyles or more likely to seek medical attention can mitigate the statistical significance of an association with prostate cancer risk (152). Until the relationship between calcium and prostate cancer is clarified, it is reasonable for men to consume a total of 1,000 to 1,200 mg/day of calcium (diet and supplements combined), which is recommended by the Food and Nutrition Board of the Institute of Medicine (see RDA) (9).
Do calcium supplements increase the risk for cardiovascular disease?
Several observational studies and randomized controlled trials have raised concerns regarding the potential adverse effects of calcium supplements on cardiovascular risk. The analysis of data from the Kuopio Osteoporosis Risk Factor and Prevention (OSTPRE) prospective study found that users of calcium supplements amongst 10,555 Finnish women (ages 52-62 years) had a 14% greater risk of developing coronary artery disease compared to non-supplement users during a mean follow-up of 6.75 years (153). The prospective study of 23,980 participants (35-64 years old) of the Heidelberg cohort of the European Prospective Investigation into Cancer and Nutrition cohort (EPIC-Heidelberg) observed that supplemental calcium intake was positively associated with the risk of myocardial infarction (heart attack) but not with the risk of stroke or cardiovascular disease (CVD)-related mortality after a mean follow-up of 11 years (154). Yet, the use of calcium supplements (≥400 mg/day vs. 0 mg/day) was associated with an increased risk of CVD-related mortality in 219,059 men, but not in 169,170 women, included in the National Institute of Health (NIH)-AARP Diet and Health study and followed for a mean period of 12 years. CVD mortality in men was also found to be significantly higher with total (dietary plus supplemental) calcium intakes of 1,500 mg/day and above (155).
In addition, the secondary analyses of two randomized placebo-controlled trials initially designed to assess the effect of calcium on bone health outcomes also suggested an increased risk of CVD in participants daily supplemented with 1,000 mg of calcium for five to seven years (156, 157). In the Auckland Calcium Study of 1,471 healthy postmenopausal women (ages ≥55 years), calcium supplementation resulted in increased risks of myocardial infarction and of a composite cardiovascular endpoint, including myocardial infarction, stroke, or sudden death (156). The analysis of data from 36,282 healthy postmenopausal women randomized to receive a combination of calcium (1,000 mg/day) and vitamin D (400 IU/day) or a placebo in the Women’s Health Initiative/Calcium-Vitamin D supplementation study (WHI/CaD study) initially reported no adverse effect on any cardiovascular endpoints with calcium (and vitamin D) compared to placebo (158). A re-analysis was performed with data from 16,718 women who did not take personal calcium supplements (outside protocol) during the five-year study (157). Although criticized on the approach taken (134, 159), the investigators estimated that women supplemented with calcium and vitamin D had a 16% increased risk of clinical myocardial infarction or stroke and a 21% increased risk of myocardial infarction compared to those who received a placebo (157). However, in another randomized, double-blind, placebo-controlled trial — the Calcium Intake Fracture Outcome (CAIFOS) study — in elderly women (median age, 75.1 years), the supplementation of 1,200 mg/day of calcium for five years was not found to increase the risk of vascular disease or related mortality (160). The WHI/CaD data re-analysis also failed to show an increased risk of mortality due to myocardial infarction or coronary artery disease with calcium therapy (156). Also, after an additional follow-up of 4.5 years at the end of the treatment period in the CAIFOS trial, the investigators reported fewer cases of heart failure-related deaths with supplemental calcium compared to placebo (160). In another randomized, placebo-controlled trial of calcium and/or vitamin D3 (RECORD trial), the evaluation of the effect of 1,000 mg/day of calcium (alone or with 800 IU/day of vitamin D) reported no significant increase in the rate of mortality due to vascular disease in 5,292 participants ages 70 years and older (161). A recent cross-sectional analysis of the Third National Health and Nutrition Examination Survey (NHANES III) evaluated the association between calcium intakes and cardiovascular mortality in 18,714 adults with no history of heart disease. No evidence of an association was observed between dietary calcium intake, supplemental calcium intake, or total calcium intake and cardiovascular mortality in either men or women (162).
A few prospective studies have reported positive correlations between high calcium concentrations in the blood and increased rates of cardiovascular events (163, 164). Because supplemental calcium may have a greater effect than dietary calcium on circulating calcium concentrations (see Toxicity), it has been speculated that the use of calcium supplements might promote vascular calcification — a surrogate marker of the burden of atherosclerosis and a major risk factor for cardiovascular events — by raising calcium serum concentrations. In 1,471 older women from the Auckland Calcium Study and 323 healthy older men from another randomized, placebo-controlled trial of daily calcium supplementation (600 mg or 1,200 mg) for two years, serum calcium concentrations were found to be positively correlated with abdominal aortic calcification or coronary artery calcification (165). However, there was no effect of calcium supplementation on measures of vascular calcification scores in men or women. Data from 1,201 participants of the Framingham Offspring study were also used to assess the relationship between calcium intake and vascular calcification. Again, no association was found between coronary calcium scores and total, dietary, or supplemental calcium intake in men or women (166). Nonetheless, in the Multi-Ethnic Study of Atherosclerosis (MESA), a US multicenter prospective study in 6,814 participants followed for a mean 10 years, the greatest risk of developing coronary artery calcification was found in supplement users with the lowest total calcium intake (~306 mg/day of dietary calcium and ~91 mg/day of supplemental calcium), when compared to supplement users with higher total calcium intakes and nonusers (167). Finally, an assessment of atherosclerotic lesions in the carotid artery wall of 1,103 participants in the CAIFOS trial was also conducted after three years of supplementation (168). When compared with placebo, calcium supplementation showed no effect on carotid artery intimal medial thickness (CIMT) and carotid atherosclerosis. Yet, carotid atherosclerosis (but not CIMT) was significantly reduced in women in the highest versus lowest tertile of total (diet and supplements) calcium intakes (≥1,795 mg/day vs. <1,010 mg/day) (168).
The most recent meta-analysis of 18 randomized clinical trials, including a total of 63,563 postmenopausal women, found no evidence of an increased risk for coronary artery disease and all-cause mortality with calcium (≥500 mg/day) supplementation for at least one year (169). Because these clinical trial data are limited to analyses of secondary endpoints, meta-analyses should be interpreted with caution. There is a need for studies designed to examine the effect of calcium supplements on CVD risk as a primary outcome before definite conclusions can be drawn. Based on an updated review of the literature that included four randomized controlled trials, one nested case-control study, and 26 prospective cohort studies (170), the National Osteoporosis Foundation (NOF) and the American Society for Preventive Cardiology (ASPC) concluded that the use of supplemental calcium for generally healthy individuals was safe from a cardiovascular health standpoint when total calcium intakes did not exceed the UL (171). NOF and ASPC support the use of calcium supplements to correct shortfalls in dietary calcium intake and meet current recommendations (171).
Drug interactions
Taking calcium supplements in combination with thiazide diuretics (e.g., hydrochlorothiazide) increases the risk of developing hypercalcemia due to increased reabsorption of calcium in the kidneys. High doses of supplemental calcium could increase the likelihood of abnormal heart rhythms in people taking digoxin (Lanoxin) for heart failure (172). Calcium, when provided intravenously, may decrease the efficacy of calcium channel blockers (173). However, dietary and oral supplemental calcium do not appear to affect the action of calcium channel blockers (174). Calcium may decrease the absorption of tetracycline, quinolone class antibiotics, bisphosphonates, sotalol (a β-blocker), and levothyroxine; therefore, it is advisable to separate doses of these medications and calcium-rich food or supplements by two hours before calcium or four-to-six hours after calcium (175). Supplemental calcium can decrease the concentration of dolutegravir (Tivicay), elvitegravir (Vitekta), and raltegravir (Isentress), three antiretroviral medications, in blood such that patients are advised to take them two hours before or after calcium supplements (175). Intravenous calcium should not be administrated within 48 hours following intravenous ceftriaxone (rocephine), a cephalosporin antibiotic, since a ceftriaxone-calcium salt precipitate can form in the lungs and kidneys and be a cause of death (175). Use of H2 blockers (e.g., cimetidine) and proton-pump inhibitors (e.g., omeprazole) may decrease the absorption of calcium carbonate and calcium phosphate (reviewed in 176, 177), whereas lithium may increase the risk of hypercalcemia in patients (175). The topical use of calcipotriene, a vitamin D analog, in the treatment of psoriasis places patients at risk of hypercalcemia if they take calcium supplements.
Calcium-nutrient interactions
The presence of calcium decreases iron absorption from nonheme sources (i.e., most supplements and food sources other than meat). However, calcium supplementation up to 12 weeks has not been found to change iron nutritional status, probably due to a compensatory increase in iron absorption (1). Individuals taking iron supplements should take them two hours apart from calcium-rich food or supplements to maximize iron absorption. Although high calcium intakes have not been associated with reduced zinc absorption or zinc nutritional status, an early study in 10 men and women found that 600 mg of calcium consumed with a meal halved the absorption of zinc from that meal (see the article on Zinc) (178). Supplemental calcium (500 mg calcium carbonate) has been found to prevent the absorption of lycopene (a nonprovitamin A carotenoid) from tomato paste in 10 healthy adults randomized into a cross-over study (179).
Linus Pauling Institute Recommendation
The Linus Pauling Institute supports the recommended dietary allowance (RDA) set by the Food and Nutrition Board of the Institute of Medicine. Following these recommendations should provide adequate calcium to promote skeletal health and may also decrease the risks of some chronic diseases.
Children and adolescents (9-18 years)
To promote the attainment of maximal peak bone mass, children and adolescents should consume a total (diet plus supplements) of 1,300 mg/day of calcium.
Adults (women: 19-50 years; men: 19-70 years)
After adult height has been reached, the skeleton continues to accumulate bone until the third decade of life when peak bone mass is attained. To promote the attainment of maximal peak bone mass and to minimize bone loss later in life, adult women (50 years of age and younger) and adult men (70 years of age and younger) should consume a total (diet plus supplements) of 1,000 mg/day of calcium.
Older women (>50 years)
To minimize bone loss, postmenopausal women should consume a total (diet plus supplements) of 1,200 mg/day of calcium. Taking a multivitamin/mineral supplement containing at least 10 μg (400 IU)/day of vitamin D will help to ensure adequate calcium absorption (see the article on Vitamin D).
Older men (>70 years)
To minimize bone loss, older men should consume a total (diet plus supplements) of 1,200 mg/day of calcium. Taking a multivitamin/mineral supplement containing at least 10 μg (400 IU)/day of vitamin D will help to ensure adequate calcium absorption (see the article on Vitamin D).
Pregnant and breast-feeding women
Pregnant and breast-feeding adolescents (<19 years) should consume a total of 1,300 mg/day of calcium, while pregnant and breast-feeding adults (≥19 years) should consume a total of 1,000 mg/day of calcium.
Authors and Reviewers
Originally written in 2001 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2003 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in October 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in August 2014 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in May 2017 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in September 2017 by:
Connie M. Weaver, Ph.D.
Distinguished Professor and Head of Foods and Nutrition
Purdue University
The 2017 update of this article was supported by a grant from Pfizer Inc.
Copyright 2001-2024 Linus Pauling Institute
References
1. Weaver CM. Calcium. In: Erdman JJ, Macdonald I, Zeisel S, eds. Present Knowledge in Nutrition. 10th ed: John Wiley & Sons, Inc.; 2012:434-446.
2. Wesseling-Perry K, Wang H, Elashoff R, Gales B, Juppner H, Salusky IB. Lack of FGF23 response to acute changes in serum calcium and PTH in humans. J Clin Endocrinol Metab. 2014;99(10):E1951-E1956. (PubMed)
3. Clapham DE. Calcium signaling. Cell. 2007;131(6):1047-1058. (PubMed)
4. Wigertz K, Palacios C, Jackman LA, et al. Racial differences in calcium retention in response to dietary salt in adolescent girls. Am J Clin Nutr. 2005;81(4):845-850. (PubMed)
5. Frassetto LA, Morris RC, Jr., Sellmeyer DE, Sebastian A. Adverse effects of sodium chloride on bone in the aging human population resulting from habitual consumption of typical American diets. J Nutr. 2008;138(2):419S-422S. (PubMed)
6. Devine A, Criddle RA, Dick IM, Kerr DA, Prince RL. A longitudinal study of the effect of sodium and calcium intakes on regional bone density in postmenopausal women. Am J Clin Nutr. 1995;62(4):740-745. (PubMed)
7. Carbone LD, Barrow KD, Bush AJ, et al. Effects of a low sodium diet on bone metabolism. J Bone Miner Metab. 2005;23(6):506-513. (PubMed)
8. Sellmeyer DE, Schloetter M, Sebastian A. Potassium citrate prevents increased urine calcium excretion and bone resorption induced by a high sodium chloride diet. J Clin Endocrinol Metab. 2002;87(5):2008-2012. (PubMed)
9. Food and Nutrition Board, Institute of Medicine. Dietary Reference Intakes for Calcium and Vitamin D. Washington, D.C.; 2011. (The National Academies Press)
10. Fulgoni VL, 3rd. Current protein intake in America: analysis of the National Health and Nutrition Examination Survey, 2003-2004. Am J Clin Nutr. 2008;87(5):1554S-1557S. (PubMed)
11. Ince BA, Anderson EJ, Neer RM. Lowering dietary protein to U.S. Recommended dietary allowance levels reduces urinary calcium excretion and bone resorption in young women. J Clin Endocrinol Metab. 2004;89(8):3801-3807. (PubMed)
12. Calvez J, Poupin N, Chesneau C, Lassale C, Tome D. Protein intake, calcium balance and health consequences. Eur J Clin Nutr. 2012;66(3):281-295. (PubMed)
13. Kerstetter JE, O'Brien KO, Caseria DM, Wall DE, Insogna KL. The impact of dietary protein on calcium absorption and kinetic measures of bone turnover in women. J Clin Endocrinol Metab. 2005;90(1):26-31. (PubMed)
14. Darling AL, Millward DJ, Torgerson DJ, Hewitt CE, Lanham-New SA. Dietary protein and bone health: a systematic review and meta-analysis. Am J Clin Nutr. 2009;90(6):1674-1692. (PubMed)
15. Grimm M, Muller A, Hein G, Funfstuck R, Jahreis G. High phosphorus intake only slightly affects serum minerals, urinary pyridinium crosslinks and renal function in young women. Eur J Clin Nutr. 2001;55(3):153-161. (PubMed)
16. Kemi VE, Karkkainen MU, Rita HJ, Laaksonen MM, Outila TA, Lamberg-Allardt CJ. Low calcium:phosphorus ratio in habitual diets affects serum parathyroid hormone concentration and calcium metabolism in healthy women with adequate calcium intake. Br J Nutr. 2010;103(4):561-568. (PubMed)
17. Heaney RP. Phosphorus. In: Erdman JJ, Macdonald I, Zeisel S, eds. Present Knowledge in Nutrition. 10th ed. Ames: John Wiley & Sons, Inc.; 2012:447-458.
18. Calvo MS, Moshfegh AJ, Tucker KL. Assessing the health impact of phosphorus in the food supply: issues and considerations. Adv Nutr. 2014;5(1):104-113. (PubMed)
19. Heaney RP, Rafferty K. Carbonated beverages and urinary calcium excretion. Am J Clin Nutr. 2001;74(3):343-347. (PubMed)
20. Ribeiro-Alves MA, Trugo LC, Donangelo CM. Use of oral contraceptives blunts the calciuric effect of caffeine in young adult women. J Nutr. 2003;133(2):393-398. (PubMed)
21. Barger-Lux MJ, Heaney RP, Stegman MR. Effects of moderate caffeine intake on the calcium economy of premenopausal women. Am J Clin Nutr. 1990;52(4):722-725. (PubMed)
22. Wikoff D, Welsh BT, Henderson R, et al. Systematic review of the potential adverse effects of caffeine consumption in healthy adults, pregnant women, adolescents, and children. Food Chem Toxicol. 2017;Apr 21. pii: S0278-6915(17)30170-9. doi: 10.1016/j.fct. 2017.04.002. [Epub ahead of print]. (PubMed)
23. Haleem S, Lutchman L, Mayahi R, Grice JE, Parker MJ. Mortality following hip fracture: trends and geographical variations over the last 40 years. Injury. 2008;39(10):1157-1163. (PubMed)
24. Kaufman JM, Reginster JY, Boonen S, et al. Treatment of osteoporosis in men. Bone. 2013;53(1):134-144. (PubMed)
25. Heaney RP. Calcium, dairy products and osteoporosis. J Am Coll Nutr. 2000;19(2 Suppl):83S-99S. (PubMed)
26. Crandall CJ, Newberry SJ, Diamant A, et al. Treatment to prevent fractures in men and women with low bone density or osteoporosis: update of a 2007 report. Rockville (MD); 2012. (PubMed)
27. Rizzoli R, Bianchi ML, Garabedian M, McKay HA, Moreno LA. Maximizing bone mineral mass gain during growth for the prevention of fractures in the adolescents and the elderly. Bone. 2010;46(2):294-305. (PubMed)
28. Borer KT. Physical activity in the prevention and amelioration of osteoporosis in women : interaction of mechanical, hormonal and dietary factors. Sports Med. 2005;35(9):779-830. (PubMed)
29. National Osteoporosis Foundation. Clinician's Guide to Prevention and Treatment of Osteoporosis. Washington, D.C. 2014.
30. Levis S, Theodore G. Summary of AHRQ's comparative effectiveness review of treatment to prevent fractures in men and women with low bone density or osteoporosis: update of the 2007 report. J Manag Care Pharm. 2012;18(4 Suppl B):S1-15; discussion S13. (PubMed)
31. Tai V, Leung W, Grey A, Reid IR, Bolland MJ. Calcium intake and bone mineral density: systematic review and meta-analysis. BMJ. 2015;351:h4183. (PubMed)
32. Bolland MJ, Leung W, Tai V, et al. Calcium intake and risk of fracture: systematic review. BMJ. 2015;351:h4580. (PubMed)
33. Chung M, Lee J, Terasawa T, Lau J, Trikalinos TA. Vitamin D with or without calcium supplementation for prevention of cancer and fractures: an updated meta-analysis for the U.S. Preventive Services Task Force. Ann Intern Med. 2011;155(12):827-838. (PubMed)
34. Weaver CM, Alexander DD, Boushey CJ, et al. Calcium plus vitamin D supplementation and risk of fractures: an updated meta-analysis from the National Osteoporosis Foundation. Osteoporos Int. 2016;27(1):367-376. (PubMed)
35. Cosman F, de Beur SJ, LeBoff MS, et al. Clinician's Guide to Prevention and Treatment of Osteoporosis. Osteoporos Int. 2014;25(10):2359-2381. (PubMed)
36. Lips P, van Schoor NM. The effect of vitamin D on bone and osteoporosis. Best Pract Res Clin Endocrinol Metab. 2011;25(4):585-591. (PubMed)
37. Gallagher JC, Yalamanchili V, Smith LM. The effect of vitamin D on calcium absorption in older women. J Clin Endocrinol Metab. 2012;97(10):3550-3556. (PubMed)
38. Zhu K, Bruce D, Austin N, Devine A, Ebeling PR, Prince RL. Randomized controlled trial of the effects of calcium with or without vitamin D on bone structure and bone-related chemistry in elderly women with vitamin D insufficiency. J Bone Miner Res. 2008;23(8):1343-1348. (PubMed)
39. Dipart Group. Patient level pooled analysis of 68 500 patients from seven major vitamin D fracture trials in US and Europe. BMJ. 2010;340:b5463. (PubMed)
40. Avenell A, Mak JC, O'Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev. 2014;4:CD000227. (PubMed)
41. Bischoff-Ferrari HA, Willett WC, Orav EJ, et al. A pooled analysis of vitamin D dose requirements for fracture prevention. N Engl J Med. 2012;367(1):40-49. (PubMed)
42. Aspray TJ, Francis RM. Fracture prevention in care home residents: is vitamin D supplementation enough? Age Ageing. 2006;35(5):455-456. (PubMed)
43. Murad MH, Elamin KB, Abu Elnour NO, et al. Clinical review: The effect of vitamin D on falls: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2011;96(10):2997-3006. (PubMed)
44. Lerolle N, Lantz B, Paillard F, et al. Risk factors for nephrolithiasis in patients with familial idiopathic hypercalciuria. Am J Med. 2002;113(2):99-103. (PubMed)
45. Sorensen MD, Eisner BH, Stone KL, et al. Impact of calcium intake and intestinal calcium absorption on kidney stones in older women: the study of osteoporotic fractures. J Urol. 2012;187(4):1287-1292. (PubMed)
46. Curhan GC, Willett WC, Knight EL, Stampfer MJ. Dietary factors and the risk of incident kidney stones in younger women: Nurses' Health Study II. Arch Intern Med. 2004;164(8):885-891. (PubMed)
47. Curhan GC, Willett WC, Rimm EB, Stampfer MJ. A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med. 1993;328(12):833-838. (PubMed)
48. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol. 2004;15(12):3225-3232. (PubMed)
49. Taylor EN, Curhan GC. Dietary calcium from dairy and nondairy sources, and risk of symptomatic kidney stones. J Urol. 2013;190(4):1255-1259. (PubMed)
50. Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med. 2002;346(2):77-84. (PubMed)
51. Hess B, Jost C, Zipperle L, Takkinen R, Jaeger P. High-calcium intake abolishes hyperoxaluria and reduces urinary crystallization during a 20-fold normal oxalate load in humans. Nephrol Dial Transplant. 1998;13(9):2241-2247. (PubMed)
52. Liebman M, Chai W. Effect of dietary calcium on urinary oxalate excretion after oxalate loads. Am J Clin Nutr. 1997;65(5):1453-1459. (PubMed)
53. Lange JN, Wood KD, Mufarrij PW, et al. The impact of dietary calcium and oxalate ratios on stone risk. Urology. 2012;79(6):1226-1229. (PubMed)
54. Jackson RD, LaCroix AZ, Gass M, et al. Calcium plus vitamin D supplementation and the risk of fractures. N Engl J Med. 2006;354(7):669-683. (PubMed)
55. Heaney RP. Calcium supplementation and incident kidney stone risk: a systematic review. J Am Coll Nutr. 2008;27(5):519-527. (PubMed)
56. Candelas G, Martinez-Lopez JA, Rosario MP, Carmona L, Loza E. Calcium supplementation and kidney stone risk in osteoporosis: a systematic literature review. Clin Exp Rheumatol. 2012;30(6):954-961. (PubMed)
57. Escribano J, Balaguer A, Roque i Figuls M, Feliu A, Ferre N. Dietary interventions for preventing complications in idiopathic hypercalciuria. Cochrane Database Syst Rev. 2014;2:CD006022. (PubMed)
58. Heilberg IP, Goldfarb DS. Optimum nutrition for kidney stone disease. Adv Chronic Kidney Dis. 2013;20(2):165-174. (PubMed)
59. Prezioso D, Strazzullo P, Lotti T, et al. Dietary treatment of urinary risk factors for renal stone formation. A review of CLU Working Group. Arch Ital Urol Androl. 2015;87(2):105-120. (PubMed)
60. Duley L. The global impact of pre-eclampsia and eclampsia. Semin Perinatol. 2009;33(3):130-137. (PubMed)
61. Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet. 2010;376(9741):631-644. (PubMed)
62. Hofmeyr GJ, Lawrie TA, Atallah AN, Duley L, Torloni MR. Calcium supplementation during pregnancy for preventing hypertensive disorders and related problems. Cochrane Database Syst Rev. 2014;6:CD001059. (PubMed)
63. Scholl TO, Chen X, Stein TP. Vitamin D, secondary hyperparathyroidism, and preeclampsia. Am J Clin Nutr. 2013;98(3):787-793. (PubMed)
64. Scholl TO, Chen X, Stein TP. Maternal calcium metabolic stress and fetal growth. Am J Clin Nutr. 2014;99(4):918-925. (PubMed)
65. Hofmeyr GJ, Belizan JM, von Dadelszen P, Calcium, Pre-eclampsia Study G. Low-dose calcium supplementation for preventing pre-eclampsia: a systematic review and commentary. BJOG. 2014;121(8):951-957. (PubMed)
66. World Health Organization. Calcium supplementation in pregnant women; 2013.
67. Hofmeyr GJ, Mlokoti Z, Nikodem VC, et al. Calcium supplementation during pregnancy for preventing hypertensive disorders is not associated with changes in platelet count, urate, and urinary protein: a randomized control trial. Hypertens Pregnancy. 2008;27(3):299-304. (PubMed)
68. Villar J, Abdel-Aleem H, Merialdi M, et al. World Health Organization randomized trial of calcium supplementation among low calcium intake pregnant women. Am J Obstet Gynecol. 2006;194(3):639-649. (PubMed)
69. Hofmeyr GJ, Novikova N, Singata M, et al. Protocol 11PRT/4028: Long term calcium supplementation in women at high risk of pre-eclampsia: a randomised, placebo-controlled trial (PACTR201105000267371). The Lancet; 2011.
70. Hofmeyr GJ, Seuc AH, Betran AP, et al. The effect of calcium supplementation on blood pressure in non-pregnant women with previous pre-eclampsia: An exploratory, randomized placebo controlled study. Pregnancy Hypertens. 2015;5(4):273-279. (PubMed)
71. US Centers for Disease Control and Prevention. Colorectal Cancer Statistics. Available at: http://www.cdc.gov/cancer/colorectal/statistics/. Accessed 5/29/17.
72. Whitfield JF. Calcium, calcium-sensing receptor and colon cancer. Cancer Lett. 2009;275(1):9-16. (PubMed)
73. Murphy N, Norat T, Ferrari P, et al. Consumption of dairy products and colorectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC). PLoS One. 2013;8(9):e72715. (PubMed)
74. Massa J, Cho E, Orav EJ, Willett WC, Wu K, Giovannucci EL. Total calcium intake and colorectal adenoma in young women. Cancer Causes Control. 2014;25(4):451-460. (PubMed)
75. Keum N, Lee DH, Greenwood DC, Zhang X, Giovannucci EL. Calcium intake and colorectal adenoma risk: dose-response meta-analysis of prospective observational studies. Int J Cancer. 2015;136(7):1680-1687. (PubMed)
76. Bristow SM, Bolland MJ, MacLennan GS, et al. Calcium supplements and cancer risk: a meta-analysis of randomised controlled trials. Br J Nutr. 2013;110(8):1384-1393. (PubMed)
77. Bolland MJ, Grey A, Gamble GD, Reid IR. Calcium and vitamin D supplements and health outcomes: a reanalysis of the Women's Health Initiative (WHI) limited-access data set. Am J Clin Nutr. 2011;94(4):1144-1149. (PubMed)
78. Bonovas S, Fiorino G, Lytras T, Malesci A, Danese S. Calcium supplementation for the prevention of colorectal adenomas: A systematic review and meta-analysis of randomized controlled trials. World J Gastroenterol. 2016;22(18):4594-4603. (PubMed)
79. Mielke HW, Gonzales C, Powell E, Mielke PW. Evolving from reactive to proactive medicine: community lead (Pb) and clinical disparities in pre- and post-Katrina New Orleans. Int J Environ Res Public Health. 2014;11(7):7482-7491. (PubMed)
80. Centers for Disease Control and Prevention. New blood lead level information. Available at: http://www.cdc.gov/nceh/lead/acclpp/blood_lead_levels.htm, 15 August 2014.
81. Bruening K, Kemp FW, Simone N, Holding Y, Louria DB, Bogden JD. Dietary calcium intakes of urban children at risk of lead poisoning. Environ Health Perspect. 1999;107(6):431-435. (PubMed)
82. Hertz-Picciotto I, Schramm M, Watt-Morse M, Chantala K, Anderson J, Osterloh J. Patterns and determinants of blood lead during pregnancy. Am J Epidemiol. 2000;152(9):829-837. (PubMed)
83. Ettinger AS, Lamadrid-Figueroa H, Tellez-Rojo MM, et al. Effect of calcium supplementation on blood lead levels in pregnancy: a randomized placebo-controlled trial. Environ Health Perspect. 2009;117(1):26-31. (PubMed)
84. Ettinger AS, Tellez-Rojo MM, Amarasiriwardena C, et al. Influence of maternal bone lead burden and calcium intake on levels of lead in breast milk over the course of lactation. Am J Epidemiol. 2006;163(1):48-56. (PubMed)
85. Hernandez-Avila M, Gonzalez-Cossio T, Hernandez-Avila JE, et al. Dietary calcium supplements to lower blood lead levels in lactating women: a randomized placebo-controlled trial. Epidemiology. 2003;14(2):206-212. (PubMed)
86. Muldoon SB, Cauley JA, Kuller LH, Scott J, Rohay J. Lifestyle and sociodemographic factors as determinants of blood lead levels in elderly women. Am J Epidemiol. 1994;139(6):599-608. (PubMed)
87. Dougkas A, Reynolds CK, Givens ID, Elwood PC, Minihane AM. Associations between dairy consumption and body weight: a review of the evidence and underlying mechanisms. Nutr Res Rev. 2011;24(1):72-95. (PubMed)
88. Zemel MB, Thompson W, Milstead A, Morris K, Campbell P. Calcium and dairy acceleration of weight and fat loss during energy restriction in obese adults. Obes Res. 2004;12(4):582-590. (PubMed)
89. Zemel MB, Shi H, Greer B, Dirienzo D, Zemel PC. Regulation of adiposity by dietary calcium. Faseb J. 2000;14(9):1132-1138. (PubMed)
90. Gonzalez JT, Rumbold PL, Stevenson EJ. Effect of calcium intake on fat oxidation in adults: a meta-analysis of randomized, controlled trials. Obes Rev. 2012;13(10):848-857. (PubMed)
91. Bortolotti M, Rudelle S, Schneiter P, et al. Dairy calcium supplementation in overweight or obese persons: its effect on markers of fat metabolism. Am J Clin Nutr. 2008;88(4):877-885. (PubMed)
92. Gallagher JC, Yalamanchili V, Smith LM. The effect of vitamin D supplementation on serum 25(OH)D in thin and obese women. J Steroid Biochem Mol Biol. 2013;136:195-200. (PubMed)
93. Pathak K, Soares MJ, Calton EK, Zhao Y, Hallett J. Vitamin D supplementation and body weight status: a systematic review and meta-analysis of randomized controlled trials. Obes Rev. 2014;15(6):528-537. (PubMed)
94. Christensen R, Lorenzen JK, Svith CR, et al. Effect of calcium from dairy and dietary supplements on faecal fat excretion: a meta-analysis of randomized controlled trials. Obes Rev. 2009;10(4):475-486. (PubMed)
95. Tordoff MG. Calcium: taste, intake, and appetite. Physiol Rev. 2001;81(4):1567-1597. (PubMed)
96. Chen M, Pan A, Malik VS, Hu FB. Effects of dairy intake on body weight and fat: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2012;96(4):735-747. (PubMed)
97. Booth AO, Huggins CE, Wattanapenpaiboon N, Nowson CA. Effect of increasing dietary calcium through supplements and dairy food on body weight and body composition: a meta-analysis of randomised controlled trials. Br J Nutr. 2015;114(7):1013-1025. (PubMed)
98. Li P, Fan C, Lu Y, Qi K. Effects of calcium supplementation on body weight: a meta-analysis. Am J Clin Nutr. 2016;104(5):1263-1273. (PubMed)
99. Soares MJ, Pathak K, Calton EK. Calcium and vitamin D in the regulation of energy balance: where do we stand? Int J Mol Sci. 2014;15(3):4938-4945. (PubMed)
100. Freeman EW. Premenstrual syndrome and premenstrual dysphoric disorder: definitions and diagnosis. Psychoneuroendocrinology. 2003;28 Suppl 3:25-37. (PubMed)
101. Pearlstein T, Steiner M. Premenstrual dysphoric disorder: burden of illness and treatment update. J Psychiatry Neurosci. 2008;33(4):291-301. (PubMed)
102. Bendich A. The potential for dietary supplements to reduce premenstrual syndrome (PMS) symptoms. J Am Coll Nutr. 2000;19(1):3-12. (PubMed)
103. Bertone-Johnson ER, Hankinson SE, Bendich A, Johnson SR, Willett WC, Manson JE. Calcium and vitamin D intake and risk of incident premenstrual syndrome. Arch Intern Med. 2005;165(11):1246-1252. (PubMed)
104. Thys-Jacobs S, Starkey P, Bernstein D, Tian J. Calcium carbonate and the premenstrual syndrome: effects on premenstrual and menstrual symptoms. Premenstrual Syndrome Study Group. Am J Obstet Gynecol. 1998;179(2):444-452. (PubMed)
105. Thys-Jacobs S, Ceccarelli S, Bierman A, Weisman H, Cohen MA, Alvir J. Calcium supplementation in premenstrual syndrome: a randomized crossover trial. J Gen Intern Med. 1989;4(3):183-189. (PubMed)
106. Alvir JM, Thys-Jacobs S. Premenstrual and menstrual symptom clusters and response to calcium treatment. Psychopharmacol Bull. 1991;27(2):145-148. (PubMed)
107. Bharati M. Comparing the Effects of Yoga & Oral Calcium Administration in Alleviating Symptoms of Premenstrual Syndrome in Medical Undergraduates. J Caring Sci. 2016;5(3):179-185. (PubMed)
108. Masoumi SZ, Ataollahi M, Oshvandi K. Effect of combined use of calcium and vitamin B6 on premenstrual syndrome symptoms: a randomized clinical trial. J Caring Sci. 2016;5(1):67-73. (PubMed)
109. Shehata NA. Calcium versus oral contraceptive pills containing drospirenone for the treatment of mild to moderate premenstrual syndrome: a double blind randomized placebo controlled trial. Eur J Obstet Gynecol Reprod Biol. 2016;198:100-104. (PubMed)
110. Shobeiri F, Araste FE, Ebrahimi R, Jenabi E, Nazari M. Effect of calcium on premenstrual syndrome: A double-blind randomized clinical trial. Obstet Gynecol Sci. 2017;60(1):100-105. (PubMed)
111. Nevatte T, O'Brien PM, Backstrom T, et al. ISPMD consensus on the management of premenstrual disorders. Arch Womens Ment Health. 2013;16(4):279-291. (PubMed)
112. Whelan AM, Jurgens TM, Naylor H. Herbs, vitamins and minerals in the treatment of premenstrual syndrome: a systematic review. Can J Clin Pharmacol. 2009;16(3):e407-429. (PubMed)
113. Cappuccio FP, Elliott P, Allender PS, Pryer J, Follman DA, Cutler JA. Epidemiologic association between dietary calcium intake and blood pressure: a meta-analysis of published data. Am J Epidemiol. 1995;142(9):935-945. (PubMed)
114. Appel LJ, Moore TJ, Obarzanek E, et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N Engl J Med. 1997;336(16):1117-1124. (PubMed)
115. Conlin PR, Chow D, Miller ER, 3rd, et al. The effect of dietary patterns on blood pressure control in hypertensive patients: results from the Dietary Approaches to Stop Hypertension (DASH) trial. Am J Hypertens. 2000;13(9):949-955. (PubMed)
116. Miller GD, DiRienzo DD, Reusser ME, McCarron DA. Benefits of dairy product consumption on blood pressure in humans: a summary of the biomedical literature. J Am Coll Nutr. 2000;19(2 Suppl):147S-164S. (PubMed)
117. Allender PS, Cutler JA, Follmann D, Cappuccio FP, Pryer J, Elliott P. Dietary calcium and blood pressure: a meta-analysis of randomized clinical trials. Ann Intern Med. 1996;124(9):825-831. (PubMed)
118. Bucher HC, Cook RJ, Guyatt GH, et al. Effects of dietary calcium supplementation on blood pressure. A meta-analysis of randomized controlled trials. JAMA. 1996;275(13):1016-1022. (PubMed)
119. Dickinson HO, Nicolson DJ, Cook JV, et al. Calcium supplementation for the management of primary hypertension in adults. Cochrane Database Syst Rev. 2006(2):CD004639. (PubMed)
120. Challoumas D, Cobbold C, Dimitrakakis G. Effects of calcium intake on the cardiovascular system in postmenopausal women. Atherosclerosis. 2013;231(1):1-7. (PubMed)
121. Blumberg JB, Frei BB, Fulgoni VL, Weaver CM, Zeisel SH. Impact of frequency of multi-vitamin/multi-mineral supplement intake on nutritional adequacy and nutrient deficiencies in US adults. Nutrients. 2017;9(8). (PubMed)
122. Kit BK, Fakhouri TH, Park S, Nielsen SJ, Ogden CL. Trends in sugar-sweetened beverage consumption among youth and adults in the United States: 1999-2010. Am J Clin Nutr. 2013;98(1):180-188. (PubMed)
123. Zhu K, Prince RL. Calcium and bone. Clin Biochem. 2012;45(12):936-942. (PubMed)
124. Bailey RL, Dodd KW, Goldman JA, et al. Estimation of total usual calcium and vitamin D intakes in the United States. J Nutr. 2010;140(4):817-822. (PubMed)
125. Straub DA. Calcium supplementation in clinical practice: a review of forms, doses, and indications. Nutr Clin Pract. 2007;22(3):286-296. (PubMed)
126. Roberts HJ. Potential toxicity due to dolomite and bonemeal. South Med J. 1983;76(5):556-559. (PubMed)
127. Bourgoin BP, Evans DR, Cornett JR, Lingard SM, Quattrone AJ. Lead content in 70 brands of dietary calcium supplements. Am J Public Health. 1993;83(8):1155-1160. (PubMed)
128. Scelfo GM, Flegal AR. Lead in calcium supplements. Environ Health Perspect. 2000;108(4):309-313. (PubMed)
129. Carrington CD, Bolger PM. An assessment of the hazards of lead in food. Regul Toxicol Pharmacol. 1992;16(3):265-272. (PubMed)
130. Ross EA, Szabo NJ, Tebbett IR. Lead content of calcium supplements. Jama. 2000;284(11):1425-1429. (PubMed)
131. Mindak WR, Cheng J, Canas BJ, Bolger PM. Lead in women's and children's vitamins. J Agric Food Chem. 2008;56(16):6892-6896. (PubMed)
132. Moe SM. Disorders involving calcium, phosphorus, and magnesium. Prim Care. 2008;35(2):215-237, v-vi. (PubMed)
133. Patel AM, Goldfarb S. Got calcium? Welcome to the calcium-alkali syndrome. J Am Soc Nephrol. 2010;21(9):1440-1443. (PubMed)
134. Lewis JR, Zhu K, Prince RL. Adverse events from calcium supplementation: relationship to errors in myocardial infarction self-reporting in randomized controlled trials of calcium supplementation. J Bone Miner Res. 2012;27(3):719-722. (PubMed)
135. World Cancer Research Fund International. Cancer facts and figures - worldwide data. 2012. Available at: http://www.wcrf.org/int/cancer-facts-figures/worldwide-data. Accessed 4/29/17.
136. Gonzalez CA, Riboli E. Diet and cancer prevention: Contributions from the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Eur J Cancer. 2010;46(14):2555-2562. (PubMed)
137. Kurahashi N, Inoue M, Iwasaki M, Sasazuki S, Tsugane AS, Japan Public Health Center-Based Prospective Study G. Dairy product, saturated fatty acid, and calcium intake and prostate cancer in a prospective cohort of Japanese men. Cancer Epidemiol Biomarkers Prev. 2008;17(4):930-937. (PubMed)
138. Raimondi S, Mabrouk JB, Shatenstein B, Maisonneuve P, Ghadirian P. Diet and prostate cancer risk with specific focus on dairy products and dietary calcium: a case-control study. Prostate. 2010;70(10):1054-1065. (PubMed)
139. Torfadottir JE, Steingrimsdottir L, Mucci L, et al. Milk intake in early life and risk of advanced prostate cancer. Am J Epidemiol. 2012;175(2):144-153. (PubMed)
140. Song Y, Chavarro JE, Cao Y, et al. Whole milk intake is associated with prostate cancer-specific mortality among U.S. male physicians. J Nutr. 2013;143(2):189-196. (PubMed)
141. Pettersson A, Kasperzyk JL, Kenfield SA, et al. Milk and dairy consumption among men with prostate cancer and risk of metastases and prostate cancer death. Cancer Epidemiol Biomarkers Prev. 2012;21(3):428-436. (PubMed)
142. Aune D, Navarro Rosenblatt DA, Chan DS, et al. Dairy products, calcium, and prostate cancer risk: a systematic review and meta-analysis of cohort studies. Am J Clin Nutr. 2015;101(1):87-117. (PubMed)
143. Qin LQ, He K, Xu JY. Milk consumption and circulating insulin-like growth factor-I level: a systematic literature review. Int J Food Sci Nutr. 2009;60 Suppl 7:330-340. (PubMed)
144. Rowlands MA, Gunnell D, Harris R, Vatten LJ, Holly JM, Martin RM. Circulating insulin-like growth factor peptides and prostate cancer risk: a systematic review and meta-analysis. Int J Cancer. 2009;124(10):2416-2429. (PubMed)
145. Allen NE, Key TJ, Appleby PN, et al. Animal foods, protein, calcium and prostate cancer risk: the European Prospective Investigation into Cancer and Nutrition. Br J Cancer. 2008;98(9):1574-1581. (PubMed)
146. Moreno J, Krishnan AV, Peehl DM, Feldman D. Mechanisms of vitamin D-mediated growth inhibition in prostate cancer cells: inhibition of the prostaglandin pathway. Anticancer Res. 2006;26(4A):2525-2530. (PubMed)
147. Brandstedt J, Almquist M, Manjer J, Malm J. Vitamin D, PTH, and calcium in relation to survival following prostate cancer. Cancer Causes Control. 2016;27(5):669-677. (PubMed)
148. Brandstedt J, Almquist M, Ulmert D, Manjer J, Malm J. Vitamin D, PTH, and calcium and tumor aggressiveness in prostate cancer: a prospective nested case-control study. Cancer Causes Control. 2016;27(1):69-80. (PubMed)
149. Rowland GW, Schwartz GG, John EM, Ingles SA. Protective effects of low calcium intake and low calcium absorption vitamin D receptor genotype in the California Collaborative Prostate Cancer Study. Cancer Epidemiol Biomarkers Prev. 2013;22(1):16-24. (PubMed)
150. Baron JA, Beach M, Wallace K, et al. Risk of prostate cancer in a randomized clinical trial of calcium supplementation. Cancer Epidemiol Biomarkers Prev. 2005;14(3):586-589. (PubMed)
151. Chung M, Balk EM, Brendel M, et al. Vitamin D and calcium: a systematic review of health outcomes. Evid Rep Technol Assess (Full Rep). 2009(183):1-420. (PubMed)
152. Huncharek M, Muscat J, Kupelnick B. Dairy products, dietary calcium and vitamin D intake as risk factors for prostate cancer: a meta-analysis of 26,769 cases from 45 observational studies. Nutr Cancer. 2008;60(4):421-441. (PubMed)
153. Pentti K, Tuppurainen MT, Honkanen R, et al. Use of calcium supplements and the risk of coronary heart disease in 52-62-year-old women: The Kuopio Osteoporosis Risk Factor and Prevention Study. Maturitas. 2009;63(1):73-78. (PubMed)
154. Li K, Kaaks R, Linseisen J, Rohrmann S. Associations of dietary calcium intake and calcium supplementation with myocardial infarction and stroke risk and overall cardiovascular mortality in the Heidelberg cohort of the European Prospective Investigation into Cancer and Nutrition study (EPIC-Heidelberg). Heart. 2012;98(12):920-925. (PubMed)
155. Xiao Q, Murphy RA, Houston DK, Harris TB, Chow WH, Park Y. Dietary and supplemental calcium intake and cardiovascular disease mortality: the National Institutes of Health-AARP diet and health study. JAMA Intern Med. 2013;173(8):639-646. (PubMed)
156. Bolland MJ, Barber PA, Doughty RN, et al. Vascular events in healthy older women receiving calcium supplementation: randomised controlled trial. BMJ. 2008;336(7638):262-266. (PubMed)
157. Bolland MJ, Grey A, Avenell A, Gamble GD, Reid IR. Calcium supplements with or without vitamin D and risk of cardiovascular events: reanalysis of the Women's Health Initiative limited access dataset and meta-analysis. BMJ. 2011;342:d2040. (PubMed)
158. Hsia J, Heiss G, Ren H, et al. Calcium/vitamin D supplementation and cardiovascular events. Circulation. 2007;115(7):846-854. (PubMed)
159. Abrahamsen B, Sahota O. Do calcium plus vitamin D supplements increase cardiovascular risk? BMJ. 2011;342:d2080. (PubMed)
160. Lewis JR, Calver J, Zhu K, Flicker L, Prince RL. Calcium supplementation and the risks of atherosclerotic vascular disease in older women: results of a 5-year RCT and a 4.5-year follow-up. J Bone Miner Res. 2011;26(1):35-41. (PubMed)
161. Avenell A, MacLennan GS, Jenkinson DJ, et al. Long-term follow-up for mortality and cancer in a randomized placebo-controlled trial of vitamin D(3) and/or calcium (RECORD trial). J Clin Endocrinol Metab. 2012;97(2):614-622. (PubMed)
162. Van Hemelrijck M, Michaelsson K, Linseisen J, Rohrmann S. Calcium intake and serum concentration in relation to risk of cardiovascular death in NHANES III. PLoS One. 2013;8(4):e61037. (PubMed)
163. Foley RN, Collins AJ, Ishani A, Kalra PA. Calcium-phosphate levels and cardiovascular disease in community-dwelling adults: the Atherosclerosis Risk in Communities (ARIC) Study. Am Heart J. 2008;156(3):556-563. (PubMed)
164. Lutsey PL, Alonso A, Michos ED, et al. Serum magnesium, phosphorus, and calcium are associated with risk of incident heart failure: the Atherosclerosis Risk in Communities (ARIC) Study. Am J Clin Nutr. 2014;100(3):756-764. (PubMed)
165. Wang TK, Bolland MJ, van Pelt NC, et al. Relationships between vascular calcification, calcium metabolism, bone density, and fractures. J Bone Miner Res. 2010;25(12):2777-2785. (PubMed)
166. Samelson EJ, Booth SL, Fox CS, et al. Calcium intake is not associated with increased coronary artery calcification: the Framingham Study. Am J Clin Nutr. 2012;96(6):1274-1280. (PubMed)
167. Anderson JJ, Kruszka B, Delaney JA, et al. Calcium intake from diet and supplements and the risk of coronary artery calcification and its progression among older adults: 10-year follow-up of the Multi-Ethnic Study of Atherosclerosis (MESA). J Am Heart Assoc. 2016;5(10). (PubMed)
168. Lewis JR, Zhu K, Thompson PL, Prince RL. The effects of 3 years of calcium supplementation on common carotid artery intimal medial thickness and carotid atherosclerosis in older women: an ancillary study of the CAIFOS randomized controlled trial. J Bone Miner Res. 2014;29(3):534-541. (PubMed)
169. Lewis JR, Radavelli-Bagatini S, Rejnmark L, et al. The effects of calcium supplementation on verified coronary heart disease hospitalization and death in postmenopausal women: a collaborative meta-analysis of randomized controlled trials. J Bone Miner Res. 2015;30(1):165-175. (PubMed)
170. Chung M, Tang AM, Fu Z, Wang DD, Newberry SJ. Calcium intake and cardiovascular disease risk: an updated systematic review and meta-analysis. Ann Intern Med. 2016;165(12):856-866. (PubMed)
171. Kopecky SL, Bauer DC, Gulati M, et al. Lack of evidence linking calcium with or without vtamin D supplementation to cardiovascular disease in generally healthy adults: a clinical guideline from the National Osteoporosis Foundation and the American Society for Preventive Cardiology. Ann Intern Med. 2016;165(12):867-868. (PubMed)
172. Vella A, Gerber TC, Hayes DL, Reeder GS. Digoxin, hypercalcaemia, and cardiac conduction. Postgrad Med J. 1999;75(887):554-556. (PubMed)
173. Moser LR, Smythe MA, Tisdale JE. The use of calcium salts in the prevention and management of verapamil-induced hypotension. Ann Pharmacother. 2000;34(5):622-629. (PubMed)
174. Bania TC, Blaufeux B, Hughes S, Almond GL, Homel P. Calcium and digoxin vs. calcium alone for severe verapamil toxicity. Acad Emerg Med. 2000;7(10):1089-1096. (PubMed)
175. Natural Medicines. Calcium - Professional handout/Drug interactions. Available at: https://naturalmedicines.therapeuticresearch.com. Accessed 5/31/17.
176. Ito T, Jensen RT. Association of long-term proton pump inhibitor therapy with bone fractures and effects on absorption of calcium, vitamin B12, iron, and magnesium. Curr Gastroenterol Rep. 2010;12(6):448-457. (PubMed)
177. Wright MJ, Proctor DD, Insogna KL, Kerstetter JE. Proton pump-inhibiting drugs, calcium homeostasis, and bone health. Nutr Rev. 2008;66(2):103-108. (PubMed)
178. Wood RJ, Zheng JJ. High dietary calcium intakes reduce zinc absorption and balance in humans. Am J Clin Nutr. 1997;65(6):1803-1809. (PubMed)
179. Borel P, Desmarchelier C, Dumont U, et al. Dietary calcium impairs tomato lycopene bioavailability in healthy humans. Br J Nutr. 2016;116(12):2091-2096. (PubMed)
Chromium
Contents
Summary
- Chromium (Cr0) is a ubiquitous trace metal. The predominant chromium form in the body is trivalent chromium (Cr3+), which may potentiate insulin function when provided at pharmacological doses. (More information)
- The nutritional essentiality of chromium has been questioned, as a dietary deficiency has not been described. In 2001, the US Institute of Medicine set the adequate intake (AI) of chromium at 20-35 μg/day for adults. However, in 2014 the European Food Safety Authority concluded that requirements for chromium could not be established. (More information)
- Results of randomized controlled trials of chromium supplementation in the prevention or treatment of impaired glucose tolerance and type 2 diabetes mellitus have been mixed, with few finding benefits that may be clinically meaningful. (More information)
- Evidence that supplemental chromium can help treat metabolic syndrome or polycystic ovary syndrome is largely lacking. (More information)
- A well-balanced diet that includes fruit, vegetables, meat, fish, and grains should easily cover dietary needs of chromium. (More information)
-
Few adverse events have been reported with chromium supplementation. (More information)
Chromium was first discovered in 1797. The most stable oxidation state of chromium in biological systems is trivalent chromium (Cr3+), which forms relatively inert complexes with proteins and nucleic acids (1). The essentiality of trivalent chromium is questioned, and its proposed function in the body remains poorly understood. In fact, in 2014 the European Food Safety Authority concluded that a dietary requirement — or even an Adequate Intake — cannot be set for trivalent chromium as no conclusive evidence exists that chromium is essential at any dietary intake (2). Trivalent chromium appears to have health effects only at pharmacological doses (reviewed in 3), and a dietary deficiency of the mineral has not been observed.
Another common and stable form of chromium in the environment is hexavalent chromium (Cr6+). Hexavalent chromium is derived from trivalent chromium by heating at alkaline pH and is used as a source of chromium for industrial purposes. Hexavalent chromium is highly toxic and is classified as a human carcinogen when inhaled (4). In the acidic environment of the stomach, hexavalent chromium can be readily reduced to trivalent chromium by reducing substances present in food, which limits the ingestion of hexavalent chromium (5-7).
Function
Trivalent chromium has been proposed to be the cofactor for a biologically active molecule that could enhance the effects of insulin on target tissues. Insulin is secreted by specialized cells in the pancreas in response to increased blood glucose concentration, such as after a meal. Insulin binds to insulin receptors on the surface of cells, activating the receptors and stimulating glucose uptake by cells. Through its interaction with insulin receptors, insulin provides cells with glucose for energy and helps maintain blood glucose within a narrow range of concentrations. In addition to its effects on carbohydrate (glucose) metabolism, insulin also influences the metabolism of fat and protein (8). Together, a decreased response to insulin or decreased insulin sensitivity in peripheral tissues (adipose tissue, muscle, and liver) and a progressive defect in insulin secretion may result in impaired glucose tolerance, frequently leading to overt type 2 diabetes mellitus. The body initially increases the secretion of insulin by specialized pancreatic cells to overcome the decrease in insulin sensitivity. However, the pancreas eventually fails to produce enough insulin to maintain normal blood glucose concentrations. Individuals with type 2 diabetes are at increased risk for cardiovascular disease (9).
Possible mechanism of action
The precise composition and structure of the biologically active form of chromium is not known. One model postulates that trivalent chromium might be the cofactor of a low-molecular-weight chromium-binding substance known as LMWCr or chromodulin (10). Chromodulin has been shown to play a role in the transport of chromium from the tissues to the bloodstream for ultimate elimination in the urine (11). When chromium is consumed at high levels, such as from dietary supplements, levels of chromodulin in tissues rise. At these high levels, chromodulin is proposed to enhance the cascade of signaling events induced by the binding of insulin to extracellular α-subunit of the insulin receptor (IR) (12). Upon insulin binding, the tyrosine kinase domain of the intracellular β-subunit of the IR becomes activated and causes the phosphorylation of tyrosine residues in the β-subunit itself. Subsequently, IR activation triggers a series of rapid phosphorylation reactions that activate many downstream effectors, eventually resulting in an increase in glucose uptake and storage (10). While this model is supported by in vitro studies, this mode of action for chromodulin has not been confirmed to date by in vivo studies.
Some, but not all, studies conducted in cell-based and animal models of insulin resistance and diabetes mellitus have found that chromium inhibits the activity of protein tyrosine phosphatase-1B (PTP-1B) and other negative regulators of insulin signaling, suggesting that chromium might improve insulin sensitivity under insulin-resistant conditions (reviewed in 10). A study in diabetic mice also suggested that chromium may reduce insulin clearance and enhance insulin signaling by inhibiting the proteolysis (degradation) of insulin and some downstream effectors (13). Additional mechanisms that may underlie the effect of chromium on insulin sensitivity, such as the reduction of markers of oxidative stress and inflammation known to contribute to insulin resistance, are under investigation (reviewed in 10, 14).
Nutrient interactions
Iron
Chromium competes for one of the binding sites on the iron transport protein, transferrin. However, supplementation of older men with 925 μg/day of chromium for 12 weeks did not significantly affect measures of iron nutritional status (15). A study of younger men found an insignificant decrease in transferrin saturation with iron after supplementation of 200 μg/day of chromium for eight weeks, but no long-term studies have addressed this issue (16). In a 12-week, randomized controlled trial, supplementation with chromium picolinate (200 μg/day) did not affect iron nutritional status in premenopausal women when compared to picolinic acid or placebo (17). Iron overload in hereditary hemochromatosis may interfere with chromium transport by competing for transferrin binding. It has been hypothesized that decreased chromium transport might contribute to the pathogenesis of diabetes mellitus in patients with hereditary hemochromatosis (5).
Vitamin C
Chromium uptake is enhanced in animals when given at the same time as vitamin C (7). In a study of three women, administration of 100 mg of vitamin C together with 1 mg of chromium resulted in higher plasma levels of chromium than 1 mg of chromium without vitamin C (5).
Carbohydrates
Compared to diets rich in complex carbohydrates (e.g., whole grains), diets high in simple sugars (e.g., sucrose) result in increased urinary chromium excretion in adults. This effect may be related to increased insulin secretion in response to the consumption of simple sugars compared to complex carbohydrates (5).
Deficiency
Dietary chromium deficiency has not been observed in humans. Potential cases of chromium deficiency were thought to have been observed in a few patients on long-term intravenous feeding (parenteral nutrition) who did not receive supplemental chromium in their intravenous solutions. The subjects developed abnormal glucose utilization and increased insulin requirements that responded to chromium supplementation (18). However, because intravenous solutions provide chromium at doses well above dietary levels, it has been suggested that chromium might produce biological effects only at pharmacological doses (3, 14). However, the differences in symptoms among subjects, the range of doses of chromium utilized over varying windows of time, and the different reported outcomes are now realized to make interpretation of these results difficult, even in terms of any potential pharmacological effects from the chromium supplementation. Because chromium appeared to enhance the action of insulin and chromium deficiency has been proposed to result in impaired glucose tolerance, chromium insufficiency has been hypothesized to be a contributing factor to the development of type 2 diabetes mellitus (5, 19). However, evidence for this is ambiguous at best.
Urinary chromium loss was reportedly increased by endurance exercise in male runners, suggesting that chromium needs may be greater in individuals who exercise regularly (20). In one study, weightlifting (resistive exercise) was found to increase urinary excretion of chromium in older men. However, chromium absorption also increased, leading to little or no net loss of chromium as a result of resistive exercise (21).
The absence of animal models for chromium deficiency makes it difficult to study possible biochemical, physiological, and functional abnormalities associated with inadequate intakes of chromium (if chromium is nutritionally essential) (2).
The Adequate Intake (AI)
Because there was not enough information to set an estimated average requirement (EAR), the Food and Nutrition Board (FNB) of the US Institute of Medicine (now the National Academy of Medicine) established an adequate intake (AI) based on the chromium content in healthy diets (Table 1; 5).
The case of trivalent chromium essentiality has been questioned in both animals and humans in the last decades, and in 2014 the European Food Safety Authority's Panel on Dietetic Products, Nutrition, and Allergies — which provides dietary guidelines for the EU community — concluded that requirements for chromium could not be established (2). The FNB, which sets dietary intake recommendations for the United States and Canada, is not currently reconsidering the AI level for chromium (22).
Disease Prevention
Impaired glucose tolerance and type 2 diabetes mellitus
Early controlled studies in subjects with impaired glucose tolerance reported that chromium supplementation improved some measure of glucose utilization or had beneficial effects on blood lipid profiles (23). Impaired glucose tolerance refers to a prediabetic state and is currently defined by the presence of impaired fasting glucose (fasting plasma glucose concentration of 100-125 mg/dL) and impaired glucose tolerance status (plasma glucose concentration of 140-199 mg/dL during a two-hour challenge test with a 75-g oral glucose load) (24). Impaired glucose tolerance is associated with modest increases in risk of cardiovascular disease, as well as other traditional microvascular complications of diabetes mellitus (25). Current estimates suggest that up to 70% of individuals with impaired glucose tolerance eventually develop type 2 diabetes (26).
In a randomized, double-blind, placebo-controlled study in 56 subjects at risk of developing type 2 diabetes, six months of daily chromium picolinate supplementation (500 μg or 1,000 μg) had no effect on glucose and insulin concentrations, insulin sensitivity, and blood lipid profiles (27). Another randomized, placebo-controlled trial in 31 individuals without diabetes reported a great variability in serum and urinary chromium concentrations in response to a daily supplementation with 1,000 μg of chromium picolinate for 16 weeks. Also, in the chromium-supplemented group, participants with higher vs. lower serum chromium concentrations (>3.1 μg/L vs. ≤3.1 μg/L) exhibited a decline in insulin sensitivity that could not be explained by expression changes in the genes involved in insulin signaling (28). Additionally, a meta-analysis of nine randomized clinical trials published between 1992 and 2010 reported that chromium at doses of 200-1,000 μg/day for 8-16 weeks had no effect on fasting glucose concentrations in 309 individuals without diabetes (29).
Cardiovascular disease
Impaired glucose tolerance and type 2 diabetes mellitus are associated with adverse changes in lipid profiles and increased risk of cardiovascular disease. Studies examining the effects of chromium supplementation on lipid profiles have given inconsistent results. While some studies have observed reductions in serum total cholesterol, LDL-cholesterol, and triglyceride levels or increases in HDL-cholesterol levels, others have observed no effect. Such mixed responses of lipid and lipoprotein levels to chromium supplementation may reflect differences in chromium nutritional status. It is possible that only individuals with insufficient dietary intake of chromium will experience beneficial effects on lipid profiles after chromium supplementation (6, 7, 30).
Moreover, a recent meta-analysis of 10 randomized controlled trials, mostly in patients with type 2 diabetes mellitus or metabolic syndrome, found chromium supplementation had no effect on either systolic or diastolic blood pressure (31). Yet, another meta-analysis of randomized controlled trials found chromium supplementation decreased circulating levels of two proinflammatory biomarkers associated with increased cardiovascular risk, hs-CRP and TNF-α, but not blood levels of IL-6 (32).
Health claims
Increases muscle mass
Claims that chromium supplementation increases lean body mass and decreases body fat are based on the relationship between chromium and insulin action (see Function). In addition to regulating glucose metabolism, insulin is known to affect fat and protein metabolism (8). At least 12 placebo-controlled studies have compared the effect of chromium supplementation (172-1,000 μg/day of chromium as chromium picolinate) with or without an exercise program on lean body mass and measures of body fat (reviewed in 33). In general, the studies that used the most sensitive and accurate methods of measuring body fat and lean mass (dual energy x-ray absorbtiometry or DEXA and hydrodensitometry or underwater weighing) did not find a beneficial effect of chromium supplementation on body composition (6, 30). In the US, the claim that chromium picolinate increases lean body mass is not allowed on supplement labels because it is not substantiated by the available research (34, 35).
Promotes weight loss
Controlled studies of chromium supplementation have demonstrated little if any beneficial effect on weight or fat loss, and claims of weight loss in humans appear to be exaggerated. In 1996, the US Federal Trade Commission (FTC) ruled that there was no scientific basis for claims that chromium picolinate could promote weight loss and fat loss in humans (36). A 2013 meta-analysis of 11 randomized, double-blind, placebo-controlled trials in 866 overweight or obese subjects found a significant 0.50-kilogram (1.10-pound) reduction in body weight with supplemental chromium (most exclusively in the form of chromium picolinate) at doses between 137 μg/day and 1,000 μg/day for 8 to 24 weeks (37). However, such a small change did not reach a clinically significant weight loss of ≥5% of the initial body weight (38). A 2019 meta-analysis of 19 clinical trials found similar results: chromium supplementation at doses between 20 μg/day and 1,000 μg/day for 4 to 24 weeks decreased body weight in overweight or obese subjects by only 0.75 kg, a clinically insignificant amount (39). Some reports have suggested that supplemental chromium may reduce food craving and intake in overweight or obese women (40, 41). Yet, current available data remain insufficient to support the use of chromium supplements as a weight-loss strategy (42). In the US, the claim that chromium picolinate promotes weight loss is not allowed on supplement labels because it is unsubstantiated (34, 35).
Reduces insulin resistance
Despite stating that scientific evidence is severely limited, the US FDA allows a single claim on supplement labels: "One small study suggests that chromium picolinate may reduce the risk of insulin resistance, and therefore possibly may reduce the risk of type 2 diabetes. FDA concludes, however, that the existence of such a relationship between chromium picolinate and either insulin resistance or type 2 diabetes is highly uncertain." (35, 43).
Disease Treatment
Type 2 diabetes mellitus
Type 2 diabetes mellitus is characterized by chronic hyperglycemia (elevated blood glucose concentration) and insulin resistance. Because resistance to insulin is usually associated with a compensatory rise in insulin secretion, circulating insulin concentrations in people with type 2 diabetes may be higher than in healthy individuals. Yet, the resistance of peripheral tissues (especially liver and skeletal muscle) to insulin also implies that the physiological effects of insulin are reduced.
Since cell culture and rodent models of diabetes have implicated chromium in the regulation of insulin sensitivity and blood glucose levels, the relationship between chromium nutritional status and type 2 diabetes mellitus has generated considerable scientific interest. Early reports observed that individuals with overt type 2 diabetes for over two years had higher rates of urinary chromium loss than healthy individuals (44). Small, well-designed studies of chromium supplementation in individuals with type 2 diabetes showed no improvement in blood glucose control, although they provided some evidence of reduced insulin concentrations and improved blood lipid profiles (45). In 1997, the results of a placebo-controlled trial conducted in China indicated that chromium supplementation might be beneficial in the treatment of type 2 diabetes (46). One hundred and eighty participants were randomized to receive either a placebo or chromium supplements in the form of chromium picolinate at either 200 μg/day or 1,000 μg/day. After four months of treatment, fasting blood glucose concentrations were found to be 15% to 19% lower in those who took 1,000 μg/day of chromium compared to those who took the placebo. Yet, blood glucose concentrations in those taking 200 μg/day of chromium did not differ significantly from those who took placebo. Chromium picolinate at either 200 μg/day or 1,000 μg/day was also associated with reduced insulin concentrations compared to placebo. The level of glycated hemoglobin A1c (HbA1c), a measure of glycemic control over the past four months, was also significantly reduced in both chromium-supplemented groups. However, a number of limitations made it difficult to extrapolate the results to the US population (47). Besides, the study was excluded from meta-analyses of randomized controlled trials due to insufficient data quality (29, 48).
In a recent systematic review and meta-analysis of randomized controlled trials in patients with type 2 diabetes, chromium supplementation (50-1,000 μg/day for 4 to 25 weeks) reduced concentrations of fasting plasma glucose (23 trials) and insulin (14 trials), and improved HbA1c values (18 trials) (49). These effects were not dose dependent (49). Yet, other meta-analyses in patients with type 2 diabetes did not find significant benefits of chromium supplementation on fasting glucose and insulin (50) or HbA1C (51). Moreover, a systematic review on 20 randomized controlled trials found that only a handful of chromium supplementation studies resulted in glycemic changes that are considered clinically meaningful, i.e., consistent with treatment goals of a 7.2-mmol/dL decrease in fasting glucose, a decrease of 0.5% in HbA1c, or reaching ≤7% HbA1c (52).
Gestational diabetes
Few studies have examined the effects of chromium supplementation on gestational diabetes mellitus, a condition that is estimated to affect 5.8% to 9.2% of pregnant women in the US (53). The occurrence of gestational diabetes during pregnancy is associated with insufficient insulin secretion and glucose intolerance of variable severity (54). Peripheral insulin resistance usually increases in the second or third trimester of pregnancy. Because elevated maternal blood glucose concentrations can have adverse effects on the developing fetus, women with gestational diabetes are at increased risk of pregnancy complications (55). After delivery, impaired glucose tolerance generally reverts to normal glucose tolerance. However, nearly one-third of women who have had gestational diabetes develop postpartum glucose intolerance (prediabetes or type 2 diabetes) (56, 57). The question also arises as to whether low chromium levels might be an effect rather than a contributing factor in gestational diabetes.
An observational study in pregnant women did not find serum chromium levels to be associated with measures of glucose tolerance or insulin resistance in late pregnancy (58). However, it is not known whether measures of serum chromium levels truly reflect tissue chromium levels and chromium status during pregnancy. A more recent prospective study following 425 pregnant women also failed to find a correlation between serum chromium concentrations and incidence of gestational diabetes (59). A cross-sectional study of 90 pregnant women in southern India found that those with gestational diabetes had significantly lower serum chromium concentrations compared to gestational diabetes-free women. Given the above mixed results, it should be noted that different methods were used to measure circulating chromium concentrations in each study. Also, it is possible that nutritional chromium status may vary among ethnically distinct populations so that studies including pregnant women from different ethnic backgrounds and/or geographical areas would give different results.
In addition, there is currently insufficient evidence to evaluate the effect of supplemental chromium on gestational diabetes. Women with gestational diabetes whose diets were supplemented with 4 μg of chromium per kilogram of body weight daily as chromium picolinate for eight weeks had decreased fasting blood glucose and insulin concentrations compared to those who took a placebo. Yet, insulin therapy rather than chromium picolinate was required to normalize severely elevated blood glucose levels (6, 60).
Metabolic syndrome
Metabolic syndrome is a combination of medical conditions, including hypertension, dyslipidemia, central obesity, and insulin resistance, that places one at increased risk for cardiovascular disease and type 2 diabetes mellitus (see the page: Metabolic Syndrome). A prospective cohort study that followed 3,648 young US adults for 23 years found an inverse association between toenail chromium concentration measured at baseline and risk of developing metabolic syndrome (61). An inverse association between plasma chromium concentration and metabolic syndrome was also observed in a case-control study in 4,282 Chinese adults (62). However, low toenail and plasma chromium may be the result of metabolic syndrome, rather than suggesting a cause or contributing factor.
Only a few randomized controlled trials have examined whether chromium supplementation might benefit patients with metabolic syndrome. In a randomized, double-blind, placebo-controlled trial in 65 patients with metabolic syndrome, 300 μg/day of supplemental chromium (from chromium-enriched yeast) for 24 weeks had no effect on measured parameters of glucose, insulin, and lipid metabolism (63). While large-scale trials would be needed, there is presently no evidence that chromium can help treat metabolic syndrome.
Polycystic ovary syndrome
Polycystic ovary syndrome (PCOS) is a common endocrine disorder affecting women of childbearing age, with an estimated worldwide prevalence of 21% (64). The disorder has a multifactorial etiology and is characterized by menstrual irregularities, polycystic ovaries, and infertility. Similar to metabolic syndrome, women with PCOS often have various metabolic abnormalities, including dyslipidemia, obesity (especially abdominal obesity), impaired glucose tolerance, insulin resistance, and are at increased risk for developing type 2 diabetes mellitus (65).
Several randomized controlled trials have evaluated whether chromium supplementation might help treat PCOS, but the results are conflicting. A 2017 systematic review and meta-analysis of seven clinical trials found that chromium picolinate supplementation (of either 200 μg/day or 1,000 ug/day) for 8 to 24 weeks decreased BMI (3 studies), lowered free testosterone concentration (5 studies), and decreased fasting serum insulin concentration (5 studies) but had no effect on concentrations of fasting blood glucose (4 studies), total testosterone (3 studies), luteinizing hormone (3 studies), or follicle-stimulating hormone (2 studies) (66). However, other recent systematic reviews and meta-analyses have concluded that chromium supplementation in women with PCOS does not result in significant or clinically meaningful health benefits, including with respect to glucose and insulin metabolism (67-69).
Sources
Food sources
The amount of chromium in food is variable and has been measured accurately in relatively few foods. Presently, there is no large database for the chromium content of food. Whole-grain products, high-bran cereals, green beans, broccoli, nuts, and egg yolk are good sources of chromium. Processed meats may also be high in chromium, depending on the processing equipment and method (70). Foods high in simple sugars, such as sucrose and fructose, are usually low in chromium and may actually promote chromium excretion (6, 71). Estimated average chromium intakes in the US range from 23 μg/day-29 μg/day for adult women and 39-54 μg/day for adult men (5). The chromium content of some foods is listed in Table 2 and expressed in micrograms (μg) (72). Since chromium content varies significantly between different batches of the same food, the information provided in the table should serve only as a guide to the chromium content of food.
Supplements
Trivalent chromium is available as a supplement in several forms, including chromium chloride, chromium nicotinate, chromium picolinate, and high-chromium yeast. These are available as stand-alone supplements or in combination products, including multivitamin/mineral supplements. Doses typically range from 100 to 300 μg of elemental chromium in single-nutrient supplements and from 10 to 180 μg in multivitamin/mineral supplements (73).
Much of the research on impaired glucose tolerance and type 2 diabetes mellitus uses chromium picolinate as the source of chromium, although investigations suggest that its bioavailability may not be greater than that of dietary chromium (74). Some concerns have been raised over the long-term safety of chromium picolinate supplementation (see Safety).
Safety
Toxicity
Hexavalent chromium (chromium VI; Cr6+) is a recognized carcinogen, with inhalation causing lung, nasal, and sinus cancers (75). Exposure to hexavalent chromium in dust has been associated with an increased incidence of lung cancer and is known to cause inflammation of the skin (dermatitis).
In contrast, there is little evidence that trivalent chromium (chromium III; Cr3+) is toxic to humans. The toxicity from oral intakes is considered to be low because ingested chromium is poorly absorbed, and most absorbed chromium is rapidly excreted in the urine (76). Because no adverse effects have been convincingly associated with excess intake of trivalent chromium from food or supplements, the Food and Nutrition Board (FNB) of the Institute of Medicine (now the National Academy of Medicine) did not set a tolerable upper intake level (UL) for chromium. Yet, despite limited evidence for adverse effects, the FNB acknowledged the possibility of a negative impact of high oral intakes of supplemental trivalent chromium on health and advised caution (5).
Chromium picolinate
Most of the concerns regarding the long-term safety of trivalent chromium supplementation arise from several studies in cell culture, suggesting trivalent chromium, especially in the form of chromium picolinate, may increase DNA damage (77-80). A study in 10 women taking 400 μg/day of chromium as chromium picolinate found no evidence of increased oxidative damage to DNA as measured by antibodies to an oxidized DNA base (81).
Many studies have demonstrated the safety of daily doses of up to 1,000 μg of chromium for several months (46, 82). However, there have been a few isolated reports of serious adverse reactions to chromium picolinate. Kidney failure was reported five months after a six-week course of 600 μg/day of chromium in the form of chromium picolinate (83), while kidney failure and impaired liver function were reported after the use of 1,200 to 2,400 μg/day of chromium in the form of chromium picolinate over a period of four to five months (84). Additionally, a 24-year old healthy male reportedly developed reversible, acute renal failure after taking chromium picolinate-containing supplements for two weeks (85). Individuals with pre-existing kidney or liver disease may be at increased risk of adverse effects and should limit supplemental chromium intake (5).
Drug interactions
Little is known about drug interactions with chromium in humans. Large doses of calcium carbonate or magnesium hydroxide-containing antacids decreased chromium absorption in rats. In contrast, non-steroidal anti-inflammatory drugs, aspirin and indomethacin, can increase chromium absorption in rats (7).
Linus Pauling Institute Recommendation
The lack of any known indicators of chromium nutritional status in humans makes it difficult to determine the level of chromium intake most likely to promote optimum health, if such exists. Following the Linus Pauling Institute recommendation to take a multivitamin/mineral supplement containing 100% of the daily values (DV) of most nutrients may provide 10 to 180 μg/day of chromium, which is generally considered safe.
Older adults (>50 years)
Although the requirement for chromium is not known to be higher for older adults, one study found that chromium concentrations in hair, sweat, and urine decreased with age (86). Following the Linus Pauling Institute recommendation to take a multivitamin/mineral supplement containing 100% of the daily values (DV) of most nutrients should provide sufficient chromium for older adults.
Because impaired glucose tolerance, type 2 diabetes mellitus, and metabolic syndrome are associated with serious health problems, individuals with any of these conditions should seek medical advice if considering the use of high-dose chromium supplements.
Authors and Reviewers
Originally written in 2001 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2003 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in June 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in October 2014 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in November 2022 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in January 2024 by:
John B. Vincent, Ph.D.
Professor, Department of Chemistry
The University of Alabama
Tuscaloosa, Alabama
Copyright 2001-2023 Linus Pauling Institute
References
1. Vaidyanathan VG, Asthana Y, Nair BU. Importance of ligand structure in DNA/protein binding, mutagenicity, excision repair and nutritional aspects of chromium(III) complexes. Dalton Trans. 2013;42(7):2337-2346. (PubMed)
2. Agostoni C, Canani RB, Fairweather-Tait S, et al. Scientific Opinion on Dietary Reference Values for chromium. EFSA J. 2014;12(10):3845.
3. Vincent JB. New evidence against chromium as an essential trace element. J Nutr. 2017;147(12):2212-2219. (PubMed)
4. United States Environmental Protection Agency. Chromium compounds; 2000.
5. Food and Nutrition Board, Institute of Medicine. Chromium. Dietary reference intakes for vitamin A, vitamin K, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. Washington D.C.: National Academy Press; 2001:197-223. (National Academy Press)
6. Lukaski HC. Chromium as a supplement. Annu Rev Nutr. 1999;19:279-302. (PubMed)
7. Stoecker B. Chromium. In: Shils M, Shike M, Ross A, Caballero B, Cousins R, eds. Modern Nutrition in Health and Disease. Philadelphia: Lippincott, Williams & Wilkins; 2006:332-337.
8. Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799-806. (PubMed)
9. Martin-Timon I, Sevillano-Collantes C, Segura-Galindo A, Del Canizo-Gomez FJ. Type 2 diabetes and cardiovascular disease: Have all risk factors the same strength? World J Diabetes. 2014;5(4):444-470. (PubMed)
10. Hua Y, Clark S, Ren J, Sreejayan N. Molecular mechanisms of chromium in alleviating insulin resistance. J Nutr Biochem. 2012;23(4):313-319. (PubMed)
11. Edwards KC, Gannon MW, Frantom PA, Vincent JB. Low-molecular-weight chromium-binding substance (LMWCr) may bind and carry Cr(III) from the endosome. J Inorg Biochem. 2021;223:111555. (PubMed)
12. Nair S. Metabolic effects of chromium — Potential molecular mechanisms. In: Vincent JB, ed. The Nutritional Biochemistry of Chromium (III). Second edition; Elsevier; 2019:175-191.
13. Wang ZQ, Yu Y, Zhang XH, Komorowski J. Chromium-insulin reduces insulin clearance and enhances insulin signaling by suppressing hepatic insulin-degrading enzyme and proteasome protein expression in KKAy mice. Front Endocrinol (Lausanne). 2014;5:99. (PubMed)
14. Vincent JB. Chromium: Is It essential, pharmacologically relevant, or toxic? In: Sigel A, Sigel H, Sigel R, eds. Interrelations between Essential Metal Ions and Human Diseases: Springer Science+Business Media Dordrecht; 2013:171-198.
15. Campbell WW, Beard JL, Joseph LJ, Davey SL, Evans WJ. Chromium picolinate supplementation and resistive training by older men: effects on iron-status and hematologic indexes. Am J Clin Nutr. 1997;66(4):944-949. (PubMed)
16. Lukaski HC, Bolonchuk WW, Siders WA, Milne DB. Chromium supplementation and resistance training: effects on body composition, strength, and trace element status of men. Am J Clin Nutr. 1996;63(6):954-965. (PubMed)
17. Lukaski HC, Siders WA, Penland JG. Chromium picolinate supplementation in women: effects on body weight, composition, and iron status. Nutrition. 2007;23(3):187-195. (PubMed)
18. Rech M, To L, Tovbin A, Smoot T, Mlynarek M. Heavy metal in the intensive care unit: a review of current literature on trace element supplementation in critically ill patients. Nutr Clin Pract. 2014;29(1):78-89. (PubMed)
19. Jeejeebhoy KN. The role of chromium in nutrition and therapeutics and as a potential toxin. Nutr Rev. 1999;57(11):329-335. (PubMed)
20. Lukaski HC. Magnesium, zinc, and chromium nutriture and physical activity. Am J Clin Nutr. 2000;72(2 Suppl):585S-593S. (PubMed)
21. Rubin MA, Miller JP, Ryan AS, et al. Acute and chronic resistive exercise increase urinary chromium excretion in men as measured with an enriched chromium stable isotope. J Nutr. 1998;128(1):73-78. (PubMed)
22. US Department of Health and Human Services. Nutrient Assessment for DRI Review. 8/24/2021. Available at: https://health.gov/our-work/nutrition-physical-activity/dietary-guidelines/dietary-reference-intakes-dris/nutrient-assessment-dri-review. Accessed 12/9/2022.
23. Mertz W. Chromium in human nutrition: a review. J Nutr. 1993;123(4):626-633. (PubMed)
24. Rao SS, Disraeli P, McGregor T. Impaired glucose tolerance and impaired fasting glucose. Am Fam Physician. 2004;69(8):1961-1968. (PubMed)
25. Singleton JR, Smith AG, Russell JW, Feldman EL. Microvascular complications of impaired glucose tolerance. Diabetes. 2003;52(12):2867-2873. (PubMed)
26. Tabak AG, Herder C, Rathmann W, Brunner EJ, Kivimaki M. Prediabetes: a high-risk state for diabetes development. Lancet. 2012;379(9833):2279-2290. (PubMed)
27. Ali A, Ma Y, Reynolds J, Wise JP, Sr., Inzucchi SE, Katz DL. Chromium effects on glucose tolerance and insulin sensitivity in persons at risk for diabetes mellitus. Endocr Pract. 2011;17(1):16-25. (PubMed)
28. Masharani U, Gjerde C, McCoy S, et al. Chromium supplementation in non-obese non-diabetic subjects is associated with a decline in insulin sensitivity. BMC Endocr Disord. 2012;12:31. (PubMed)
29. Bailey CH. Improved meta-analytic methods show no effect of chromium supplements on fasting glucose. Biol Trace Elem Res. 2014;157(1):1-8. (PubMed)
30. Kobla HV, Volpe SL. Chromium, exercise, and body composition. Crit Rev Food Sci Nutr. 2000;40(4):291-308. (PubMed)
31. Ghanbari M, Amini MR, Djafarian K, Shab-Bidar S. The effects of chromium supplementation on blood pressure: a systematic review and meta-analysis of randomized clinical trials. Eur J Clin Nutr. 2022;76(3):340-349. (PubMed)
32. Zhang X, Cui L, Chen B, et al. Effect of chromium supplementation on hs-CRP, TNF-alpha and IL-6 as risk factor for cardiovascular diseases: A meta-analysis of randomized-controlled trials. Complement Ther Clin Pract. 2021;42:101291. (PubMed)
33. Vincent JB. The potential value and toxicity of chromium picolinate as a nutritional supplement, weight loss agent and muscle development agent. Sports Med. 2003;33(3):213-230. (PubMed)
34. Federal Trade Commission. United States of American Before Federal Trade Commission. Available at: https://www.ftc.gov/sites/default/files/documents/cases/1997/07/nutrit2.htm. Accessed 12/8/2022.
35. US Food & Drug Administration. Qualified health claims: letters of denial. Available at: https://www.fda.gov/food/food-labeling-nutrition/qualified-health-claims-letters-denial. Accessed 12/8/2022.
36. Federal Trade Commission. Companies advertising popular dietary supplement "chromium picolinate" can't substantiate weight loss and health benefit claims, says FTC; 1996. Available at: https://www.ftc.gov/news-events/news/press-releases/1996/11/companies-advertising-popular-dietary-supplement-chromium-picolinate-cant-substantiate-weight-loss. Accessed 9/23/2014.
37. Onakpoya I, Posadzki P, Ernst E. Chromium supplementation in overweight and obesity: a systematic review and meta-analysis of randomized clinical trials. Obes Rev. 2013;14(6):496-507. (PubMed)
38. Wing RR, Pinto AM, Crane MM, Kumar R, Weinberg BM, Gorin AA. A statewide intervention reduces BMI in adults: Shape Up Rhode Island results. Obesity (Silver Spring). 2009;17(5):991-995. (PubMed)
39. Tsang C, Taghizadeh M, Aghabagheri E, Asemi Z, Jafarnejad S. A meta-analysis of the effect of chromium supplementation on anthropometric indices of subjects with overweight or obesity. Clin Obes. 2019;9(4):e12313. (PubMed)
40. Anton SD, Morrison CD, Cefalu WT, et al. Effects of chromium picolinate on food intake and satiety. Diabetes Technol Ther. 2008;10(5):405-412. (PubMed)
41. Brownley KA, Von Holle A, Hamer RM, La Via M, Bulik CM. A double-blind, randomized pilot trial of chromium picolinate for binge eating disorder: results of the Binge Eating and Chromium (BEACh) study. J Psychosom Res. 2013;75(1):36-42. (PubMed)
42. Tian H, Guo X, Wang X, et al. Chromium picolinate supplementation for overweight or obese adults. Cochrane Database Syst Rev. 2013;11:CD010063. (PubMed)
43. US Department of Health and Human Services. Qualified Health Claims: Letter of Enforcement Discretion - Chromium Picolinate and Insulin Resistance(Docket No. 2004Q-0144). http://wayback.archive-it.org/7993/20171114183739/https:/www.fda.gov/Food/IngredientsPackagingLabeling/LabelingNutrition/ucm073017.htm. Accessed 11/14/2017.
44. Morris BW, MacNeil S, Hardisty CA, Heller S, Burgin C, Gray TA. Chromium homeostasis in patients with type II (NIDDM) diabetes. J Trace Elem Med Biol. 1999;13(1-2):57-61. (PubMed)
45. Hellerstein MK. Is chromium supplementation effective in managing type II diabetes? Nutr Rev. 1998;56(10):302-306. (PubMed)
46. Anderson RA, Cheng N, Bryden NA, et al. Elevated intakes of supplemental chromium improve glucose and insulin variables in individuals with type 2 diabetes. Diabetes. 1997;46(11):1786-1791. (PubMed)
47. Althuis MD, Jordan NE, Ludington EA, Wittes JT. Glucose and insulin responses to dietary chromium supplements: a meta-analysis. Am J Clin Nutr. 2002;76(1):148-155. (PubMed)
48. Abdollahi M, Farshchi A, Nikfar S, Seyedifar M. Effect of chromium on glucose and lipid profiles in patients with type 2 diabetes; a meta-analysis review of randomized trials. J Pharm Pharm Sci. 2013;16(1):99-114. (PubMed)
49. Asbaghi O, Fatemeh N, Mahnaz RK, et al. Effects of chromium supplementation on glycemic control in patients with type 2 diabetes: a systematic review and meta-analysis of randomized controlled trials. Pharmacol Res. 2020;161:105098. (PubMed)
50. Zhao F, Pan D, Wang N, et al. Effect of chromium supplementation on blood glucose and lipid levels in patients with type 2 diabetes mellitus: a systematic review and meta-analysis. Biol Trace Elem Res. 2022;200(2):516-525. (PubMed)
51. Yin RV, Phung OJ. Effect of chromium supplementation on glycated hemoglobin and fasting plasma glucose in patients with diabetes mellitus. Nutr J. 2015;14:14. (PubMed)
52. Costello RB, Dwyer JT, Bailey RL. Chromium supplements for glycemic control in type 2 diabetes: limited evidence of effectiveness. Nutr Rev. 2016;74(7):455-468. (PubMed)
53. US Preventive Services Task Force, Davidson KW, Barry MJ, et al. Screening for gestational diabetes: US Preventive Services Task Force recommendation statement. JAMA. 2021;326(6):531-538. (PubMed)
54. American Diabetes Association. Gestational diabetes mellitus. Diabetes Care. 2004;27 Suppl 1:S88-90. (PubMed)
55. Mitanchez D. Foetal and neonatal complications in gestational diabetes: perinatal mortality, congenital malformations, macrosomia, shoulder dystocia, birth injuries, neonatal complications. Diabetes Metab. 2010;36(6 Pt 2):617-627. (PubMed)
56. Bellamy L, Casas JP, Hingorani AD, Williams D. Type 2 diabetes mellitus after gestational diabetes: a systematic review and meta-analysis. Lancet. 2009;373(9677):1773-1779. (PubMed)
57. Retnakaran R, Qi Y, Sermer M, Connelly PW, Hanley AJ, Zinman B. Glucose intolerance in pregnancy and future risk of pre-diabetes or diabetes. Diabetes Care. 2008;31(10):2026-2031. (PubMed)
58. Gunton JE, Hams G, Hitchman R, McElduff A. Serum chromium does not predict glucose tolerance in late pregnancy. Am J Clin Nutr. 2001;73(1):99-104. (PubMed)
59. Woods SE, Ghodsi V, Engel A, Miller J, James S. Serum chromium and gestational diabetes. J Am Board Fam Med. 2008;21(2):153-157. (PubMed)
60. Jovanovic-Peterson L, Peterson CM. Vitamin and mineral deficiencies which may predispose to glucose intolerance of pregnancy. J Am Coll Nutr. 1996;15(1):14-20. (PubMed)
61. Bai J, Xun P, Morris S, Jacobs DR, Jr., Liu K, He K. Chromium exposure and incidence of metabolic syndrome among American young adults over a 23-year follow-up: the CARDIA Trace Element Study. Sci Rep. 2015;5:15606. (PubMed)
62. Chen S, Zhou L, Guo Q, et al. Association of plasma chromium with metabolic syndrome among Chinese adults: a case-control study. Nutr J. 2020;19(1):107. (PubMed)
63. Nussbaumerova B, Rosolova H, Krizek M, et al. Chromium supplementation reduces resting heart rate in patients with metabolic syndrome and impaired glucose tolerance. Biol Trace Elem Res. 2018;183(2):192-199. (PubMed)
64. Deswal R, Narwal V, Dang A, Pundir CS. The prevalence of polycystic ovary syndrome: a brief systematic review. J Hum Reprod Sci. 2020;13(4):261-271. (PubMed)
65. Lentscher JA, Decherney AH. Clinical presentation and diagnosis of polycystic ovarian syndrome. Clin Obstet Gynecol. 2021;64(1):3-11. (PubMed)
66. Fazelian S, Rouhani MH, Bank SS, Amani R. Chromium supplementation and polycystic ovary syndrome: A systematic review and meta-analysis. J Trace Elem Med Biol. 2017;42:92-96. (PubMed)
67. Tang XL, Sun Z, Gong L. Chromium supplementation in women with polycystic ovary syndrome: Systematic review and meta-analysis. J Obstet Gynaecol Res. 2018;44(1):134-143. (PubMed)
68. Heshmati J, Omani-Samani R, Vesali S, et al. The effects of supplementation with chromium on insulin resistance indices in women with polycystic ovarian syndrome: a systematic review and meta-analysis of randomized clinical trials. Horm Metab Res. 2018;50(3):193-200. (PubMed)
69. Maleki V, Izadi A, Farsad-Naeimi A, Alizadeh M. Chromium supplementation does not improve weight loss or metabolic and hormonal variables in patients with polycystic ovary syndrome: A systematic review. Nutr Res. 2018;56:1-10. (PubMed)
70. Kumpulainen JT. Chromium content of foods and diets. Biol Trace Elem Res. 1992;32:9-18. (PubMed)
71. Kozlovsky AS, Moser PB, Reiser S, Anderson RA. Effects of diets high in simple sugars on urinary chromium losses. Metabolism. 1986;35(6):515-518. (PubMed)
72. Anderson RA, Bryden NA, Polansky MM. Dietary chromium intake. Freely chosen diets, institutional diet, and individual foods. Biol Trace Elem Res. 1992;32:117-121. (PubMed)
73. US Department of Health and Human Services, National Institutes of Health, Office of Dietary Supplements. Dietary Supplement Label Database (DSLD). [Internet]. Available at: Available from: https://dsld.od.nih.gov/. Accessed 3/18/2024.
74. Laschinsky N, Kottwitz K, Freund B, Dresow B, Fischer R, Nielsen P. Bioavailability of chromium(III)-supplements in rats and humans. Biometals. 2012;25(5):1051-1060. (PubMed)
75. Loomis D, Guha N, Hall AL, Straif K. Identifying occupational carcinogens: an update from the IARC Monographs. Occup Environ Med. 2018;75(8):593-603. (PubMed)
76. Nielsen FH. Manganese, molybdenum, boron, chromium, and other trace elements. In: Erdman JJ, Macdonald I, Zelssel S, eds. Present Knowledge of Nutrition: John Wiley & Sons, Inc.; 2012.
77. Blasiak J, Kowalik J. A comparison of the in vitro genotoxicity of tri- and hexavalent chromium. Mutat Res. 2000;469(1):135-145. (PubMed)
78. Speetjens JK, Collins RA, Vincent JB, Woski SA. The nutritional supplement chromium(III) tris(picolinate) cleaves DNA. Chem Res Toxicol. 1999;12(6):483-487. (PubMed)
79. Stearns DM, Wise JP, Sr., Patierno SR, Wetterhahn KE. Chromium(III) picolinate produces chromosome damage in Chinese hamster ovary cells. FASEB J. 1995;9(15):1643-1648. (PubMed)
80. Jiang L, Vincent JB, Bailey MM. [Cr(3)O(O(2)CCH(2)CH(3))(6)(H(2)O)(3)]NO(3).H(2)O (Cr3) toxicity potential in bacterial and mammalian cells. Biol Trace Elem Res. 2018;183(2):342-350. (PubMed)
81. Kato I, Vogelman JH, Dilman V, et al. Effect of supplementation with chromium picolinate on antibody titers to 5-hydroxymethyl uracil. Eur J Epidemiol. 1998;14(6):621-626. (PubMed)
82. Hathcock JN. Vitamins and minerals: efficacy and safety. Am J Clin Nutr. 1997;66(2):427-437. (PubMed)
83. Wasser WG, Feldman NS, D'Agati VD. Chronic renal failure after ingestion of over-the-counter chromium picolinate. Ann Intern Med. 1997;126(5):410. (PubMed)
84. Cerulli J, Grabe DW, Gauthier I, Malone M, McGoldrick MD. Chromium picolinate toxicity. Ann Pharmacother. 1998;32(4):428-431. (PubMed)
85. Wani S, Weskamp C, Marple J, Spry L. Acute tubular necrosis associated with chromium picolinate-containing dietary supplement. Ann Pharmacother. 2006;40(3):563-566. (PubMed)
86. Davies S, McLaren Howard J, Hunnisett A, Howard M. Age-related decreases in chromium levels in 51,665 hair, sweat, and serum samples from 40,872 patients--implications for the prevention of cardiovascular disease and type II diabetes mellitus. Metabolism. 1997;46(5):469-473. (PubMed)
Copper
Contents
Summary
- Copper is an essential cofactor for oxidase enzymes that catalyze oxidation-reduction reactions in various metabolic pathways. These copper-dependent enzymes, or cuproenzymes, participate in, for example, energy (ATP) production, iron metabolism, connective tissue formation, and neurotransmission. (More information)
- Dietary copper insufficiency in humans has been infrequently described; however, copper depletion may occur due to intestinal defects, high supplemental zinc intake, or in genetic conditions such as Menkes disease. Intestinal copper absorption is severely impaired in Menkes disease, leading to systemic copper deficiency. Symptoms of low body copper include anemia, bone and connective tissue abnormalities, and neurological dysfunction. (More information)
- Assessing copper status in humans is challenging, since no definitive biomarkers exist for detecting moderate, or subclinical, copper deficiency. The development of more precise and sensitive biomarkers of copper nutritional status is thus a critical area for future research. (More information)
- Copper imbalance in humans increases risks of bone demineralization and osteoporosis, fatty liver disease, liver disease mortality, and cardiovascular and neurodegenerative diseases. In certain pathological states, dysregulation of copper homeostasis may not be a primary outcome but could rather be secondary to some aspect of disease pathogenesis. (More information)
- Accurately assessing dietary copper intake is difficult since the copper content of many foods has not been firmly established. Organ meats, shellfish, nuts, seeds, wheat-bran cereals, and whole-grain products are, however, recognized as good sources of dietary copper. (More information)
- Copper toxicity is rare, being most frequently associated with Wilson disease, a rare inborn error of metabolism that causes copper overload initially in the liver and then subsequently in other tissues, particularly the brain. Toxic effects of copper overload in Wilson disease include disruption of lipid metabolism, as well as damage to mitochondria. Toxic copper accumulation is also observed in Indian Childhood Cirrhosis and Endemic Tyrolean Infantile Cirrhosis (or Idiopathic Copper Toxicosis). No causal genetic defects have been linked to these latter disorders, although increased susceptibility to excess copper has been proposed. (More information)
Copper (Cu) is an essential trace element for humans and other mammals. In biological systems, copper readily shifts between the cuprous (Cu1+) and cupric (Cu2+) forms. The redox properties of copper underlie its important role in oxidation-reduction reactions and in scavenging free radicals (1). Although Hippocrates is said to have prescribed copper compounds to treat diseases as early as 400 B.C. (2), scientists are still uncovering new information regarding the functions of copper in the human body (3).
Function
Copper is critical for the function of several essential enzymes known as cuproenzymes, which are integral parts of various metabolic pathways (4, 5). Physiologic functions of these copper-dependent enzymes, and the biochemical pathways in which they function (6, 7), are outlined below.
Energy production
The copper-dependent enzyme cytochrome c oxidase (CCO) plays a critical role in cellular energy production in mitochondria by catalyzing the reduction of molecular oxygen (O2) to water (H2O), thereby generating an electrical gradient that is required for ATP production (8). Redox-active copper contained within the CCO enzyme complex is required for the electron transfer reactions that are critical for its function.
Connective tissue formation
Another cuproenzyme, lysyl oxidase (LOX), is required for the cross-linking of collagen and elastin fibers, which is essential for the formation of strong and flexible connective tissue. LOX function is critical for bone formation and maintenance of connective tissue in the heart and blood vessels (2).
Iron metabolism
Multi-copper oxidases (MCOs) are copper-dependent ferroxidases that function in iron homeostasis. MCOs oxidize ferrous iron (Fe2+) to the ferric (Fe3+) form, which enables binding to transferrin (the main iron carrier) in the blood, thus allowing iron transport to sites of utilization (e.g., the bone marrow). The MCOs include: (1) ceruloplasmin (CP), which contains 60%-95% of plasma copper; (2) a membrane-bound form of CP (GPI-CP), expressed in brain and other tissues; and (3) the membrane-bound ferroxidases hephaestin (HEPH) and zyklopen, which function in the intestine and placenta, respectively (9, 10). CP knockout (Cp-/-) mice accumulate excess hepatic iron but have normal copper status (11, 12). Similarly, humans with aceruloplasminemia, who lack functional CP, display iron overload in liver, brain, and retina but have no observable defects in copper homeostasis (13). Moreover, absorption of dietary iron and iron mobilization from storage sites (e.g., liver) are impaired in copper deficiency, when CP and HEPH activity is reduced, further supporting a role for the MCOs in iron metabolism (14).
Central nervous system
Several physiological processes within the brain and nervous system, including neurotransmitter synthesis and formation and maintenance of myelin, depend upon catalysis mediated by cuproenzymes. Dopamine β-hydroxylase, for example, catalyzes the conversion of dopamine to the neurotransmitter norepinephrine (15). Also, CCO is required for the biosynthesis of phospholipids, which are critical structural components of the myelin sheath (2).
Melanin biosynthesis
The cuproenzyme tyrosinase (TYR) is required for the biosynthesis of melanin in melanocytes, which is critical for normal pigmentation of hair, skin, and eyes (2). Low TYR activity most likely explains the achromotrichia seen in copper-deficient laboratory and agricultural animals, and the depigmentation noted in severely copper-depleted patients with Menkes disease.
Antioxidation
Superoxide dismutase (SOD) functions as an antioxidant by catalyzing the conversion of reactive oxygen species, such as the superoxide anion (O2-) and the hydroxyl radical (•OH), to hydrogen peroxide (H2O2), which is subsequently reduced to water by other antioxidant systems (16). Two forms of SOD contain copper: copper/zinc SOD (SOD1), which is expressed in most cells, including red blood cells; and extracellular SOD (EcSOD), which is highly expressed in the lungs and found at lower levels in plasma (2). Also, as outlined above, ceruloplasmin has antioxidant properties relating to iron metabolism. The ferroxidase activity of ceruloplasmin may prevent ferrous iron (Fe2+) from participating in harmful free-radical-generating reactions via Fenton chemistry (16).
Regulation of gene expression
Copper-related gene expression pathways seem to be mainly regulated at a post-translation level, in some cases via protein trafficking-related mechanisms that respond to intracellular copper levels (17). Cytosolic copper may also influence mRNA expression levels of specific genes, in a dose-dependent manner (18-20), implicating possible transcriptional regulation. For example, intracellular copper may alter the redox state of cells and thus induce oxidative stress, which can activate signal transduction pathways that increase the expression of genes encoding proteins involved in the detoxification of reactive oxygen species (21).
Nutrient interactions
Iron
Adequate copper nutritional status is necessary for normal iron metabolism and red blood cell production and function. Copper depletion results in an iron deficiency-like anemia, and iron accumulates in the livers of copper-deficient animals. The development of anemia during copper deficiency may be linked to low CP activity, impaired iron release from stores in the liver, and reduced iron delivery to the erythroid marrow, thus leading to iron-restricted erythropoiesis (see Iron metabolism) (2). However, this may not be the whole story, as was recently suggested by longtime copper researcher, Dr. Joseph R. Prohaska (Univ. of Minnesota, Duluth) (22). Copper depletion also reduces CP activity in humans, leading to hepatic iron overload and thus increasing risk for oxidative damage and liver cirrhosis (14). Oral copper supplementation restored normal serum CP levels and plasma ferroxidase activity, and corrected iron metabolism defects in a copper-deficient subject (23). Moreover, infants fed a formula with a high-iron content absorbed less copper than infants fed a low-iron formulation, suggesting that high iron intakes may interfere with copper absorption in infants (24). This observation was also confirmed in rats and mice, where high dietary iron caused copper depletion, thus increasing the copper nutritional requirement (25, 26).
Zinc
Excess intake of supplemental zinc, at doses of 50 mg/day or more for extended periods of time, may result in copper depletion. The mechanism may relate to increased synthesis of metallothionein (MT), an intracellular zinc- and copper-binding protein. MT has a stronger affinity for copper than zinc, so high levels of MT induced by excess zinc may trap copper within enterocytes thus limiting its bioavailability. This postulate, however, was called into question by studies done in MT-deficient mice, in which high enteral zinc still decreased copper absorption, suggesting that high zinc may block a copper transporter (27). Conversely, elevated copper intakes have not been found to affect zinc nutritional status (2, 24). Moreover, zinc supplementation at 10 mg/day for eight weeks restored normal plasma copper/zinc ratios in 65 subjects on long-term hemodialysis who initially exhibited low serum zinc and high copper. Whether improving zinc and copper status of hemodialysis patients can impact clinical outcomes, however, needs to be assessed (28).
Fructose
Evidence of copper-fructose interactions comes mainly from studies using experimental animals. High-fructose diets exacerbated copper deficiency in male rats, but not in pigs whose gastrointestinal system is anatomically and functionally more like humans. Also, very high levels of dietary fructose (20% of total calories) did not result in copper depletion in humans, suggesting that fructose intake does not result in copper depletion at levels relevant to normal diets (2, 24). However, high fructose consumption and low copper availability may be risk factors for functional copper deficiency in patients with non-alcoholic fatty liver disease (29).
Vitamin C
Although vitamin C supplements have produced copper deficiency in guinea pigs (30), the effect of vitamin C supplementation on copper nutritional status in humans is less clear. Two small studies in healthy, young adult men indicated that ceruloplasmin oxidase activity may be impaired by relatively high doses of supplemental vitamin C. In one study, vitamin C intakes of 1,500 mg/day for two months resulted in a significant decline in CP oxidase activity (31). In the other study, supplements of 605 mg/day of vitamin C for three weeks resulted in decreased CP oxidase activity, although copper absorption did not decline (32). Neither of these studies found vitamin C supplementation to adversely affect copper nutritional status.
Deficiency
Clinically evident, or frank, dietary copper deficiency is relatively uncommon. Serum copper and CP levels may fall to 30% of normal in cases of severe copper deficiency. Hypocupremia is also observed in genetic disorders of copper metabolism, including Wilson disease (WD) and aceruloplasminemia; however, neither disorder has been linked to low dietary copper intake. One of the most common clinical signs of copper deficiency is an anemia that is unresponsive to iron therapy but is corrected by copper supplementation. It was hypothesized that this anemia could result from defective iron mobilization due to decreased CP activity, yet individuals with inherited aceruloplasminemia do not always develop overt anemia (33). Moreover, in copper-deficient swine, intestinal iron absorption is impaired but iron distribution among tissues/organs is normal (34-36). Low serum iron from reduced absorption is an unlikely cause of this anemia since intravenous provision of iron did not correct it. An alternative postulate is that copper-deficiency anemia is caused principally by impaired hemoglobin production and red blood cell proliferation, and a shortened erythrocyte lifespan. These physiological processes are thus likely to require copper. Copper deficiency may also lead to neutropenia, which can increase susceptibility to infection. Copper depletion studies demonstrated that low copper might affect erythroid and myeloid cell lineages, supporting a role for copper in the regulation of blood cell proliferation and maturation (37, 38). More research is clearly needed to further define the mechanisms underlying copper deficiency-induced anemia and neutropenia (4, 39). Furthermore, osteoporosis and other abnormalities of bone development have been described in copper-deficient, low-birth-weight infants and young children. Less common features of copper deficiency may include impaired growth, depigmentation, and development of neurological pathologies (2, 8).
Biomarkers of copper status
Currently, there is not a sensitive and specific biomarker to detect copper inadequacy in humans (5, 40-42). Blood copper (43) and ceruloplasmin concentrations are reduced in severe copper deficiency (3, 6). However, both of these parameters are also influenced by pregnancy, inflammation, and infection (5), thus limiting the usefulness of these assays to estimate body copper status. Experimental work has recently identified other copper-related biomarkers, including erythrocyte copper Cu/Zn superoxide dismutase (SOD1) and copper chaperone for superoxide dismutase (44-46), but further experimental validation is required, including clinical testing in humans.
Individuals at risk of deficiency
Bovine milk is relatively low in copper, and cases of copper deficiency have been reported in infants fed only cow's milk formula (47). Other individuals at elevated risk for copper deficiency include premature and low-birth-weight infants; individuals with severe burns (48) or prolonged diarrhea, which accelerate copper losses; infants and children recovering from malnutrition; and individuals with malabsorption syndromes, including celiac disease, Crohn’s disease, and short bowel syndrome, possibly due to surgical removal of large portions of the intestine (48-50). Also, gastric bypass surgery for morbid obesity significantly increases the risk for copper depletion (51-53). Individuals receiving intravenous total parenteral nutrition lacking copper or those on certain restricted diets may also require supplementation with copper (and other trace elements) (2, 8). Moreover, copper deficiency in infants with cholestasis has been linked to long-term parenteral nutrition lacking copper (54). Case reports indicated that cystic fibrosis patients may also be at increased risk of copper insufficiency (55, 56). And finally, excessive zinc intake has been associated with secondary copper deficiency in individuals taking zinc supplements or using zinc-enriched dental creams (57-59).
Acquired copper deficiency
Copper deficiency is atypical in the general population; however, it was recently suggested that copper deficiency may be more widespread than currently recognized, and that (covert) pathophysiological links exist between low copper nutritional status and Alzheimer’s disease, ischemic heart disease (60), myelodysplastic syndrome, and postmenopausal osteoporosis (61). Part of the rationale for such an assertion relates to the difficulty in clinically evaluating the copper status of humans, so moderate to even severe copper deficiency is unlikely to be detected in individuals with no notable risk factors (62). Firmly establishing links between low copper status and increased risk for these, and possibly other, chronic diseases awaits further epidemiological and clinical testing. Furthermore, a neurological syndrome associated with copper deficiency was recently described in adults (63). The symptoms included central nervous system demyelination, polyneuropathy, myelopathy, and inflammation of the optic nerve. The etiology is unknown and risk factors have not been established (see Individuals at risk of deficiency). Case reports suggested that copper malabsorption underlies this disorder, but mutations in the gene encoding the main intestinal copper exporter, ATP7A, were not detected in affected patients (see Inherited copper deficiency) (64). Oral copper supplementation (2 mg/day) normalized serum copper and CP concentrations, and stabilized individuals afflicted with the disease and significantly improved their quality of life. Optimal duration and dose of copper administration has not, however, been experimentally evaluated (63).
Inherited copper deficiency
ATPase copper transporting alpha, or ATP7A, is a dual function Cu1+-transporting ATPase that is expressed in most cell types, except for, notably, hepatocytes. ATP7A transports copper from the cytosol into the trans-Golgi (TGN), where cuproenzymes are synthesized within the secretory pathway, and exports copper from cells. Mutations in ATP7A, which underlie Menkes disease (MD), critically block copper export from enterocytes and vascular endothelial cells (65). Impaired absorption of dietary copper results in systemic copper deficiency in MD. Decreased cuproenzyme expression/activity is caused by low intracellular copper, and defective copper transport into the TGN. Copper accumulation in microvascular endothelial cells in the blood-brain barrier reduces copper transport into the brain, leading to low brain copper and reduced cuproenzyme activity in neurons. Other mutations in ATP7A have been linked to a less severe neurological copper deficiency disorder called occipital horn syndrome (OHS). The clinical features of MD include intractable seizures, connective tissue disorders, subdural hemorrhage, and hair abnormalities (so called "kinky hair"). OHS patients exhibit muscular hypotonia and connective tissue abnormalities, including formation of exostoses on occipital bones. Subcutaneous injections of copper-histidine improve copper-related metabolic functions in MD and OHS patients. Copper entry into the brain, however, remains limited (reviewed in 66). Furthermore, gene therapy approaches have been recently validated in a pre-clinical mouse model of MD, with long-term goals of using such treatments in humans with the disease (67, 68). Recently, another inherited copper-deficiency disorder was described in identical twin male infants, who were homozygous for a novel missense variant in the gene encoding copper transporter 1 (CTR1) (69). This genetic aberration caused a distinctive autosomal recessive syndrome of infantile seizures and neurodegeneration, consistent with profound central nervous system copper deficiency. Disease pathology was most likely caused by defective intestinal copper transport which resulted in severe systemic copper deficiency. This outcome is supported by experimental laboratory studies which demonstrated that intestine-specific ablation (deletion) of CTR1 significantly impaired absorption of dietary copper in mice (70).
Copper Excess
Inherited copper overload
Patients with Wilson disease (WD) may have increased risk for copper toxicosis even at copper intakes in the normal range. WD is an autosomal recessive disorder that is typified by defective copper distribution and storage (71). The disease is caused by mutations in the ATP7B gene, which encodes a copper-transporting ATPase that is highly expressed in liver and brain. Dysfunctional ATP7B disrupts copper flux in these organs. A recent review provides a nice summary of this devastating human disease (72). WD prevalence is ~1:30,000 individuals globally (73), although much higher prevalence rates have been reported. It was suggested that differences in the penetrance of disease-causing genetic variants explain the apparent discrepancy between epidemiological and genetic prevalence studies of WD (74).
In WD, redox-active copper accumulates in liver, brain, and cornea due to impaired ATP7B-mediated copper excretion, which increases oxidative stress leading to eventual tissue and organ damage. Untreated WD patients are likely to develop liver damage, cancer, and eventual hepatic failure, and severe hemolytic crisis. Elevated brain copper content can lead to neurological damage, and copper accumulation in the eye in so-called Kayser-Fleisher rings can result in abnormal eye movements. Blood concentrations of ceruloplasmin are characteristically low in WD patients, since hepatic ATP7B is required for its biosynthesis, and urinary copper losses may be enhanced. Early intervention can prevent development of some of the most severe pathophysiological outcomes. Treatments for WD include zinc supplementation, which attenuates enteral copper absorption, and/or copper chelation therapy with penicillamine or trientine (75).
Other genetic copper-overload disorders
Additional pathologies associated with liver copper loading include Indian childhood cirrhosis (ICC) and idiopathic copper toxicosis (ICT). In ICC, notable liver copper loading and progressive liver failure are observed (76). In contrast to Wilson disease when ceruloplasmin is low, in ICC, ceruloplasmin is normal or elevated. ICC is most likely caused by inadvertent excess copper ingestion, possibly from the use of copper-lined, food/beverage storage containers in a genetically susceptible individual. It seems likely that the unknown genetic defect in ICC relates to the efficiency of excretion of excess copper in the bile, but this has not been definitively established. Also, about one-third of ICC patients have α-1-antitrypsin deficiency, which calls into question a primary role for copper in disease outcomes (77). ICT is another hepatic copper overload disorder that predominantly afflicts infants and children. ICT displays autosomal recessive inheritance, and an unidentified genetic aberration results in defective copper metabolism leading to an increased susceptibility to excess copper. Affected individuals are at increased risk for copper-related, hepatic toxicosis due to inadvertent consumption of excess copper. However, the source of extra copper remains unidentified in many ICT patients, perhaps suggesting a more complex disease pathogenesis (78).
The Recommended Dietary Allowance (RDA)
A variety of bioindicators were used to establish the RDA for copper, including plasma copper concentration, serum ceruloplasmin activity, superoxide dismutase activity in red blood cells, and platelet copper concentration (24). However, whether these are accurate and sensitive biomarkers of copper nutritional status uncertain (40). Also, estimates of copper concentrations in various foods and water sources may not be accurate and reliable (40, 62). The RDA for copper reflects the results of depletion-repletion studies and is based on the prevention of deficiency (Table 1). For infants up to one year of age, an adequate intake (AI) was established due to the lack of experimental evidence to set a requirement.
| Life Stage | Age Range | Males (μg/day) | Females (μg/day) |
|---|---|---|---|
| Infants | 0-6 months | 200 (AI) | 200 (AI) |
| Infants | 7-12 months | 220 (AI) | 220 (AI) |
| Children | 1-3 years | 340 | 340 |
| Children | 4-8 years | 440 | 440 |
| Children | 9-13 years | 700 | 700 |
| Adolescents | 14-18 years | 890 | 890 |
| Adults | ≥19 years | 900 | 900 |
| Pregnancy | all ages | - | 1,000 |
| Breast-feeding | all ages | - | 1,300 |
Disease Prevention
Cardiovascular disease
Severe copper deficiency results in cardiomyopathy in some animal species (79); however, this pathology differs from the atherosclerotic cardiovascular disease that is prevalent in humans (24). Outcomes of cardiovascular disease (CVD)-related clinical studies in humans are inconsistent, possibly since the copper status of participants is uncertain given the lack of reliable biomarkers of copper nutritional status. Ionic copper is a pro-oxidant, and it can oxidize low-density lipoprotein (LDL) in the test tube. CP can also stimulate LDL oxidation in the laboratory setting (80). As such, some researchers have proposed that excess copper could increase the risk for developing atherosclerosis by promoting the oxidation of LDL in vivo. However, there is scant experimental evidence to support this possibility. Moreover, superoxide dismutase and ceruloplasmin have known antioxidant properties, leading some experts to propose that copper deficiency, rather than copper excess, increases the risk for cardiomyopathy (81, 82). Outcomes of observational and intervention studies relating copper nutritional status to relative risk for CVD are summarized below.
Observational studies
Observational studies have linked elevated serum copper levels to an increased risk for developing CVD. For example, a prospective cohort study examined serum copper levels in more than 4,500 men and women 30 years of age and older in the United States (83). During the subsequent 16 years, 151 participants died from coronary heart disease (CHD). After adjusting for other risk factors, those with serum copper levels in the two highest quartiles had a significantly greater risk of dying from CHD. Case-control studies conducted in Europe also had similar outcomes. For example, a case-cohort study of 2,087 adults in Germany reported an association between higher serum copper concentrations and increased risk of incident CVD, including myocardial infarction and stroke (84). Another study in 60 patients with chronic heart failure or ischemic heart disease reported that serum copper was a predictor of short-term outcomes (85). Higher serum copper was also linked to an increased risk of heart failure in a prospective cohort study of 1,866 middle-aged and older men in Finland (86). Another prospective cohort study in 4,035 middle-aged men in France reported that high serum copper levels were significantly related to a 50% increase in all-cause mortality; however, serum copper was not significantly associated with CVD mortality in this study (87). Serum copper was also elevated in patients with rheumatic heart disease (88). In sum, these studies may indicate that high serum copper reflects elevated body copper content, which increases oxidative stress and accelerates tissue/organ damage and disease development. Importantly, however, most copper in the serum is contained within CP, up to 90% depending upon the species, with the remaining, smaller proportion of serum copper bound to albumin or α2-macroglobulin (89, 90). Serum CP is an acute-phase reactant protein, with levels increasing by up to 50% as a result of trauma or infection and during chronic inflammatory states. Changes in circulating CP are associated with proportional changes in serum copper levels, independent of body copper status. Therefore, elevated serum copper in CHD patients may simply reflect increased CP production due to the inflammation that typifies atherosclerosis. Collectively, these observations raise concerns about linking elevated serum copper to increased tissue copper content and chronic disease development in humans (91).
In contrast to the observational findings discussed above linking high serum copper levels to heart disease, two autopsy studies found copper levels in cardiac muscle were actually lower in patients who died of CHD than in those who died of other causes (92). Additionally, the copper content of white blood cells has been positively correlated with the degree of patency of coronary arteries in CHD patients (93, 94). Further, patients with a history of myocardial infarction (MI) had lower concentrations of copper-dependent, extracellular superoxide dismutase than those without a history of MI (95). Thus, due to the lack of specific, reliable biomarkers of copper nutritional status, it is not clear whether copper is related to cardiovascular disease. It is also important to note that altered copper metabolism may be symptomatic of a cardiovascular condition, rather than a factor that primarily influences its development.
Studies examining dietary intake of copper are scarce. In a prospective cohort study in Japan, which included 58,646 participants followed for a median of 19 years, dietary copper intake — measured by a food frequency questionnaire — was not associated with CHD mortality (96). Yet, this study associated higher copper intakes with an increased risk of mortality from stroke and other cardiovascular diseases (96).
Notably, it was suggested that elevated plasma copper concentrations could be linked to high circulating homocysteine levels in individuals with cardiovascular disease (97-99). Increased blood homocysteine may precipitate development of arterial wall lesions and increase risk of CVD (100); however, this matter is currently open to debate (101). In animal models, copper-homocysteine interactions were linked to impaired vascular endothelial function (102, 103). Copper restriction in experimental animals decreased homocysteine levels and reduced incidence of atherogenic lesions (104, 105), but it is not known whether copper imbalance contributes to a possible atherogenic effect of homocysteine in humans (106).
Intervention studies
Small studies in adults deprived of dietary copper documented adverse changes in blood cholesterol, including increased total and LDL cholesterol concentrations and decreased HDL cholesterol concentrations (107), but these outcomes were not confirmed in other studies (108). For example, in one recent study, copper supplementation of 2 to 3 mg/day for 4 to 8 weeks did not result in clinically significant changes in blood cholesterol levels (81, 109, 110). Additionally, 8 mg/day of copper for six months had no effect on blood cholesterol levels (111). Interpretation of these outcomes is, however, challenging since the copper status of participants was presumably not well defined. Additional research failed to link increased copper intake to elevated oxidative stress. In a multi-center, placebo-controlled clinical trial, copper supplementation of 3 or 6 mg/day for six weeks did not result in increased susceptibility of LDL to oxidation by copper (112). Moreover, supplementation with 3 or 6 mg/day of copper did not increase oxidative damage to red blood cells (113). Collectively, these studies indicated that copper intakes several times above the RDA do not increase oxidative stress, at least as measured by these assays in these populations.
Summary: Copper and cardiovascular disease
Although free copper and CP can promote LDL oxidation in the laboratory, there is little evidence that high dietary copper increases oxidative stress in the human body. Increased serum copper levels have been associated with increased CVD risk, as outlined above, but the significance of these findings is unclear due to the complex association among serum copper, CP, and inflammatory mediators. Clarification of the relationships of copper intake, copper nutritional status, CP levels, and CVD risk thus requires further research.
Immune system function
Copper is known to play several important roles in the development and maintenance of immune system function, including innate and adaptive immunity (reviewed in 114). Neutropenia is a clinical sign of copper deficiency in humans. Adverse effects of insufficient copper on immune system function appear most pronounced in infants. For example, infants with Menkes disease, a genetic copper-deficiency disorder, suffer from frequent and severe infections (115, 116). Moreover, in a study of 11 malnourished infants with evidence of copper deficiency, the ability of white blood cells to engulf pathogens increased significantly after one month of copper supplementation (117). Moreover, 11 men on a low-copper diet (0.66 mg/day of copper for 24 days and 0.38 mg/day for another 40 days) showed an impaired monocyte proliferative response in an ex vivo immune challenge assay (118). Mechanistic studies also support a role for copper in the innate immune response to bacterial and viral infections (reviewed in 119, 120). Severe copper deficiency thus has adverse effects on immune system function; however, whether marginal copper insufficiency impairs immunity in humans has not been established.
Osteoporosis
Progressive decrease of bone mineral density (BMD) is commonly observed in the elderly, frequently leading to development of osteopenia (pre-osteoporosis) and osteoporosis. Women are more often affected by osteoporosis than men, (e.g., prevalence ratio is 5:1 in non-Hispanic whites) (121), primarily due to the postmenopausal reduction in the production of estrogen, which is essential for maintaining strength of muscle, bone, and connective tissue (122). Osteoporosis is associated with an increased risk of falls, bone fracture, and mortality in individuals over 65 years of age (123).
Osteoporosis has been reported in infants with severe copper deficiency (124, 125), but how copper depletion affects bone and connective tissue health in adults is less certain. One recent investigation documented bone resorption (breakdown) in 11 healthy adult males consuming marginal copper for six weeks (0.7 mg/day) (126). Also, supplementation of 3 to 6 mg/day of copper for six weeks had no effect on biochemical markers of bone resorption or bone formation in two studies of healthy adult men and women (127, 128). An effect of copper deficiency on bone integrity seems likely, since a copper-dependent enzyme, lysyl oxidase (LOX), is required for the maturation (cross-linking) of collagen, a key element in the organic matrix of bone. In individuals with marginal copper intake and less efficient copper absorption, such as the elderly, it seems plausible that LOX activity is decreased, possibly increasing risk for bone and connective tissue effects.
Observational studies
Collectively, research examining the role of copper nutritional status in age-related osteoporosis is limited. An early study found that serum copper levels in 46 elderly patients with hip fractures were significantly lower than those of age-matched controls (129). Another study, however, found no differences in serum copper levels among postmenopausal women with normal BMD (N=40), osteopenia (N=40), or osteoporosis (N=40) (130). A cross-sectional study showed that blood copper concentrations were lower than the normal reference range in postmenopausal women with osteopenia (N=28) and osteoporosis (N=23) (131). In another cross-sectional study in 728 postmenopausal women, 491 of which had confirmed osteoporosis, lower serum copper concentrations were associated with osteoporosis in the younger women (ages 40-59 years) but not the older women (ages 60-80 years) (132). Furthermore, in a national survey in the US, including 8,224 adults (compiling data from NHANES 2007-2010, 2013-2014, and 2017-2018), higher daily copper intakes (from diet and supplements) were associated with higher BMD at the femur and lumbar spine and a lower risk of osteoporosis (133).
Intervention studies
Limited studies of copper supplementation and bone health outcomes have been undertaken. A small study in perimenopausal women, who consumed ~1 mg of dietary copper daily, reported decreased loss of BMD from the lumbar spine after copper supplementation of 3 mg/day for two years (134). Additionally, a two-year, double-blind, placebo-controlled trial in 59 postmenopausal women found that daily intake of supplemental calcium plus trace minerals, including 2.5 mg of copper, resulted in maintenance of spinal BMD. Supplemental calcium or trace minerals, alone, were not as effective at preventing loss of bone density (135). Another randomized, double-blind, placebo-controlled study in 224 healthy, postmenopausal women ages 51 to 80 years, found daily supplementation with 600 mg of calcium, 2 mg of copper, and 12 mg of zinc for two years decreased whole-body BMD compared to supplemental calcium alone. Another trial showed that BMD was reduced in subjects with dietary copper intakes below the RDA (0.9 mg/day), but copper supplementation did not prevent the progressive loss of BMD as well as a calcium regimen alone (136). Finally, several studies have suggested that tooth loss might be related to defects in the maintenance of BMD (137, 138). When compared with 20 healthy-matched controls, 50 patients (mean age, 47.5 years) with low spinal BMD and advanced tooth wear were found to have significantly lower copper content in tooth enamel. However, despite evidence of bone demineralization, serum copper levels in this population were similar to those of the healthy group (139). In sum, additional research is required to draw meaningful conclusions regarding the effects of marginal copper depletion and copper supplementation on bone metabolism and risk for developing age-related osteoporosis.
Neurodegenerative diseases
Alzheimer's disease
Cognitive deterioration in individuals with Alzheimer’s disease (AD) is linked to the presence of β-amyloid plaques and abnormal Tau protein-forming aggregates. The possibility that copper imbalance is involved in the onset of AD is under investigation. A recent meta-analysis of case-control studies described higher blood concentrations of copper in AD patients (N=2,929) as compared to healthy subjects (N=3,547), from a total of 46 studies reviewed (140). Also, ‘free’ serum copper (i.e., not bound to CP) was higher in AD patients (N=1,595) than in healthy control subjects (N=2,399), representing 18 total studies. These observations were confirmed in another recent review of published studies (141). An additional meta-analysis of 12 case-control studies revealed AD patients had lower copper concentrations in various brain regions compared to healthy controls (142), further exemplifying dysregulation of copper homeostasis in Alzheimer’s disease.
Among the many hypotheses supporting a role for copper in AD onset or progression includes copper involvement in the formation of senile plaques through hypermetallation of the β-amyloid peptides, possibly leading to zinc depletion, enhanced oxidative stress, and brain damage (143-145). Recent research has also identified polymorphisms in the ATP7B gene that may be associated with the risk of developing copper imbalance and AD (140, 142). ATP7B is a copper-transporting ATPase expressed in the liver and brain. Impairment of ATP7B function causes Wilson disease, which is typified by elevated ‘free’ copper level in blood and copper accumulation in liver and brain.
Additional research is required to determine whether genetic variation could influence individual susceptibility to environmental exposure of high copper levels. Copper administered in drinking water was associated with development of enhanced pathological features in animal models of AD (146, 147). One study in a rabbit model reported that combining a high-cholesterol diet and copper (0.12 mg/L in drinking water) impaired cognition (146). A prospective cohort study in 3,718 elderly participants in the Chicago Health and Aging Project, followed for 5.5 years, evaluated the impact of fat and copper intakes using food frequency questionnaires on cognitive function. For individuals with high intakes of saturated and trans fat, cognitive decline was greater for those in the highest quintile of total copper intake compared to the lowest quintile (median intake of 2.75 vs. 0.88 mg copper per day) (148).
Although dysfunctional copper metabolism is suggested as a risk factor for AD, it could also be symptomatic of the disease, rather than causative. Moreover, it is still unclear whether copper supplementation or restriction could delay the progression of AD. A small, double-blind, placebo-controlled trial in 68 individuals with mild AD found that supplementation of 8 mg/day of copper for one year delayed the decrease of the β-amyloid peptide Aβ42 in cerebrospinal fluid; a decrease in Aβ42 has been linked to cognitive deterioration (149). This delay, however, was not associated with improved cognitive performance (150). Relating to the use of zinc supplementation to block copper absorption in Wilson disease, slow-release zinc acetate administration (150 mg/day for six months) in a randomized, placebo-controlled study of 60 patients with mild-to-moderate AD resulted in a decrease in serum ‘free’ copper and stabilization of cognition deficits (143). A specific role for copper was not, however, determined in these notable outcomes. In summary, additional human studies are needed to clarify the role of copper in AD prevention, development, and progression.
Parkinson's disease
Neurologically presenting Wilson disease and inherited aceruloplasminemia are characterized by copper accumulation in the brain and development of neurological symptoms, including dystonia and cognitive impairment, that resemble Parkinson’s disease (PD) (151). Disruption of copper homeostasis has been documented in PD (152). Copper depletion occurs in brain regions with loss of neurons in PD patients (153, 154). Moreover, some studies have documented lower serum copper and/or CP concentrations in PD patients compared to healthy controls (155-157). Dietary copper intake did not, however, relate to the risk of developing PD in two small case-control studies (158, 159). As in AD, further research is required to elucidate whether copper imbalance contributes to the pathogenesis of PD.
Nonalcoholic fatty liver disease
Similar to findings from animal models (160), human studies have documented low circulating copper (161, 162) or low hepatic copper content (161, 163, 164) in adults and children with nonalcoholic fatty liver disease. An inverse association between hepatic copper content and liver disease severity has also been observed (163, 165). However, it is not known whether low dietary intake of copper might be a contributor to disease pathogenesis or whether dysregulated copper metabolism is only a manifestation of liver disease.
Sources
Food sources
Copper is found in a wide variety of foods and is most plentiful in organ meats, shellfish, nuts, and seeds. Wheat-bran cereals and whole-grain products are also good sources of copper. According to national surveys (NHANES), the mean dietary intake of copper in the US is 1.1 mg/day for adult women and 1.3 mg/day for adult men (166), levels that exceed the established RDA for copper for adults of 900 µg/day (see Table 1). The estimated copper content of some foods that are relatively rich in copper is listed in Table 2. For more information on the nutrient content of foods, search USDA's FoodData Central.
Supplements
Identification of those at risk for copper depletion is challenging since sensitive and specific, copper nutrition-related bioindicators have yet to be identified. Nonetheless, a range of copper supplements are available for purchase, including cupric oxide, copper gluconate, copper sulfate, and copper amino acid chelates (167). The relative bioavailability of these different chemical forms of copper, however, has not yet been established in humans (168). Copper supplements may contain a few µg up to 15 mg of elemental copper, which exceeds the UL for copper by 1.5 fold (169). Moreover, copper is typically included in multivitamin/mineral supplements (169) and added to fortified breakfast cereals. Supplementation of adults with 10 mg/day of cupric gluconate for 12 weeks did not cause copper toxicity (170). Importantly, however, higher copper intakes could be detrimental in some at-risk (unknown) individuals. Copper supplementation of infants should be approached with caution since homeostatic regulators of copper absorption and excretion are not fully developed, thus increasing the potential for copper toxicosis. From a clinical perspective, copper overload most frequently presents in biliary atresia, biliary cirrhosis and in WD patients, and as such, individuals suffering from these conditions should avoid taking supplemental copper.
Safety
Toxicity
Copper toxicity is rare in the general population. Acute copper poisoning has occurred by storing beverages in copper-containing containers, as well as from contaminated water supplies (171). Guideline values for copper in drinking water have been set by the US Environmental Protection Agency (1.3 mg/liter) and by the World Health Organization (2 mg/liter) (172). Symptoms of acute copper toxicity include abdominal pain, nausea, vomiting, and diarrhea; such symptoms help prevent additional ingestion and absorption of copper. More serious signs of acute copper toxicity include severe liver damage, kidney failure, coma, and death.
Of more concern from a nutritional standpoint is the possibility of liver damage resulting from long-term exposure to lower doses of copper. In generally healthy individuals, daily doses of up to 10,000 μg (10 mg) have not resulted in liver damage. The US Food and Nutrition Board has thus set the tolerable upper intake level (UL) in adults at 10 mg/day of copper from food and supplements combined (Table 3) (24). It should be noted that individuals with genetic disorders affecting copper metabolism (e.g., Wilson disease, Indian childhood cirrhosis, and idiopathic copper toxicosis) may be at risk for adverse effects of chronic copper toxicity at significantly lower intake levels. There is some concern that the UL of 10 mg/day might be too high. For example, one study in adult men who consumed 7.8 mg/day of copper for 147 days showed that they loaded excess copper during that time, and some indices of immune function and antioxidant status suggested that these functions were adversely affected by the high intakes of copper (173, 174). However, another study did not report any adverse effects in individuals supplemented with 8 mg/day of copper for six months (150).
Drug interactions
Relatively little is known about the interaction of copper with drugs. Penicillamine is used to bind copper and enhance its elimination in Wilson disease, a genetic disorder resulting in hepatic copper overload. Because penicillamine dramatically increases the urinary excretion of copper, individuals taking the medication for reasons other than copper overload may have an increased dietary copper requirement. Additionally, antacids may interfere with copper absorption when used in very high amounts (2). Also, the anti-tuberculosis drug ethambutol may chelate copper in mitochondria and reduce cytochrome c oxidase activity specifically in optic nerve axons, possibly contributing to optic neuropathy which is a documented side-effect of this drug (175).
Linus Pauling Institute Recommendation
The RDA for copper (900 μg/day for adults) is sufficient to prevent deficiency, but the lack of clear biomarkers of copper nutritional status in humans makes it difficult to determine the level of copper intake most likely to promote optimum health or prevent chronic disease. A varied diet should provide enough copper for most people. For those who are concerned that their diet may not provide adequate copper, a multivitamin/mineral supplement will generally provide at least the RDA for copper.
Older adults (>50 years)
Because aging has not been associated with significant changes in the requirement for copper, our recommendation for older adults is the same as that for adults 50 and younger (176).
Authors and Reviewers
Originally written in 2001 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2003 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in January 2008 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in December 2013 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in October 2022 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed and updated in July 2024 by:
James F. Collins, Ph.D.
Food Science & Human Nutrition department
University of Florida
Reviewed in July 2023 by:
Jason Burkhead, Ph.D.
Biological Sciences department
University of Alaska, Anchorage
Copyright 2001-2024 Linus Pauling Institute
References
1. Linder MC, Hazegh-Azam M. Copper biochemistry and molecular biology. Am J Clin Nutr. 1996;63(5):797S-811S. (PubMed)
2. Turnlund JR. Copper. In: Shils ME, Shike M, Ross A, Caballero B, Cousins RA, eds. Modern Nutrition in Health and Disease. 10th ed. Baltimore: Lipincott Williams & Wilkins; 2006:289-299.
3. Prohaska JR. Copper. In: Erdman JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Ames: Wiley-Blackwell; 2012:540-553.
4. Prohaska JR. Impact of copper limitation on expression and function of multicopper oxidases (ferroxidases). Adv Nutr. 2011;2(2):89-95. (PubMed)
5. Collins JF. Copper nutrition and biochemistry and human (patho)physiology. Adv Food Nutr Res. 2021;96:311-364. (PubMed)
6. Collins JF. Copper. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Philadelphia: Wolter Kluwer; Lippincott Williams & Wilkins; 2014:206-216.
7. Collins JF. Copper. In: Marriott BP, Birt DF, Stalling VA, Yates AA, eds. Present Knowledge in Nutrition. 11th ed: Academic Press; 2020:409-427.
8. Uauy R, Olivares M, Gonzalez M. Essentiality of copper in humans. Am J Clin Nutr. 1998;67(5 Suppl):952S-959S. (PubMed)
9. Vashchenko G, MacGillivray RT. Multi-copper oxidases and human iron metabolism. Nutrients. 2013;5(7):2289-2313. (PubMed)
10. Vasilyev VB. Looking for a partner: ceruloplasmin in protein-protein interactions. Biometals. 2019;32(2):195-210. (PubMed)
11. Meyer LA, Durley AP, Prohaska JR, Harris ZL. Copper transport and metabolism are normal in aceruloplasminemic mice. J Biol Chem. 2001;276(39):36857-36861. (PubMed)
12. Harris ZL, Durley AP, Man TK, Gitlin JD. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc Natl Acad Sci U S A. 1999;96(19):10812-10817. (PubMed)
13. Kono S. Aceruloplasminemia. Curr Drug Targets. 2012;13(9):1190-1199. (PubMed)
14. Thackeray EW, Sanderson SO, Fox JC, Kumar N. Hepatic iron overload or cirrhosis may occur in acquired copper deficiency and is likely mediated by hypoceruloplasminemia. J Clin Gastroenterol. 2011;45(2):153-158. (PubMed)
15. Harris E. Copper. In: O'Dell B, Sunde R, eds. Handbook of Nutritionally Essential Minerals. New York: Marcel Dekker, Inc.; 1997:231-273.
16. Johnson MA, Fischer JG, Kays SE. Is copper an antioxidant nutrient? Crit Rev Food Sci Nutr. 1992;32(1):1-31. (PubMed)
17. van den Berghe PV, Klomp LW. Posttranslational regulation of copper transporters. J Biol Inorg Chem. 2010;15(1):37-46. (PubMed)
18. Armendariz AD, Gonzalez M, Loguinov AV, Vulpe CD. Gene expression profiling in chronic copper overload reveals upregulation of Prnp and App. Physiol Genomics. 2004;20(1):45-54. (PubMed)
19. Armendariz AD, Olivares F, Pulgar R, et al. Gene expression profiling in wild-type and metallothionein mutant fibroblast cell lines. Biol Res. 2006;39(1):125-142. (PubMed)
20. Gonzalez M, Reyes-Jara A, Suazo M, Jo WJ, Vulpe C. Expression of copper-related genes in response to copper load. Am J Clin Nutr. 2008;88(3):830S-834S. (PubMed)
21. Mattie MD, McElwee MK, Freedman JH. Mechanism of copper-activated transcription: activation of AP-1, and the JNK/SAPK and p38 signal transduction pathways. J Mol Biol. 2008;383(5):1008-1018. (PubMed)
22. Prohaska JR. Reflections of a cupromaniac. Metallomics. 2016;8(9):813-815. (PubMed)
23. Videt-Gibou D, Belliard S, Bardou-Jacquet E, et al. Iron excess treatable by copper supplementation in acquired aceruloplasminemia: a new form of secondary human iron overload? Blood. 2009;114(11):2360-2361. (PubMed)
24. Food and Nutrition Board, Institute of Medicine. Copper. Dietary reference intakes for vitamin A, vitamin K, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. Washington D.C.: National Academy Press; 2001:224-257. (National Academy Press)
25. Ha JH, Doguer C, Collins JF. Consumption of a high-iron diet disrupts homeostatic regulation of intestinal copper absorption in adolescent mice. Am J Physiol Gastrointest Liver Physiol. 2017;313(4):G535-G360. (PubMed)
26. Ha JH, Doguer C, Wang X, Flores SR, Collins JF. High-iron consumption impairs growth and causes copper-deficiency anemia in weanling Sprague-Dawley rats. PLoS One. 2016;11(8):e0161033. (PubMed)
27. Reeves PG. Copper metabolism in metallothionein-null mice fed a high-zinc diet. J Nutr Biochem. 1998(10):598-601.
28. Guo CH, Wang CL. Effects of zinc supplementation on plasma copper/zinc ratios, oxidative stress, and immunological status in hemodialysis patients. Int J Med Sci. 2013;10(1):79-89. (PubMed)
29. Song M, Vos MB, McClain CJ. Copper-fructose interactions: A novel mechanism in the pathogenesis of NAFLD. Nutrients. 2018;10(11):1815. (PubMed)
30. Milne DB, Omaye ST. Effect of vitamin C on copper and iron metabolism in the guinea pig. Int J Vitam Nutr Res. 1980;50(3):301-308. (PubMed)
31. Finley EB, Cerklewski FL. Influence of ascorbic acid supplementation on copper status in young adult men. Am J Clin Nutr. 1983;37(4):553-556. (PubMed)
32. Jacob RA, Skala JH, Omaye ST, Turnlund JR. Effect of varying ascorbic acid intakes on copper absorption and ceruloplasmin levels of young men. J Nutr. 1987;117(12):2109-2115. (PubMed)
33. Harris ZL, Klomp LW, Gitlin JD. Aceruloplasminemia: an inherited neurodegenerative disease with impairment of iron homeostasis. Am J Clin Nutr. 1998;67(5 Suppl):972S-977S. (PubMed)
34. Lahey ME, Gubler CJ, Chase MS, Cartwright GE, Wintrobe MM. Studies on copper metabolism. II. Hematologic manifestations of copper deficiency in swine. Blood. 1952;7(11):1053-1074. (PubMed)
35. Wintrobe MM, Cartwright GE, Lahey ME, Gubler CJ. The role of copper in hemopoiesis. Trans Assoc Am Physicians. 1951;64:310-315. (PubMed)
36. Cartwright GE, Gubler CJ, Bush JA, Wintrobe MM. Studies of copper metabolism. XVII. Further observations on the anemia of copper deficiency in swine. Blood. 1956;11(2):143-153. (PubMed)
37. Bustos RI, Jensen EL, Ruiz LM, et al. Copper deficiency alters cell bioenergetics and induces mitochondrial fusion through up-regulation of MFN2 and OPA1 in erythropoietic cells. Biochem Biophys Res Commun. 2013;437(3):426-432. (PubMed)
38. Peled T, Landau E, Prus E, Treves AJ, Nagler A, Fibach E. Cellular copper content modulates differentiation and self-renewal in cultures of cord blood-derived CD34+ cells. Br J Haematol. 2002;116(3):655-661. (PubMed)
39. Lazarchick J. Update on anemia and neutropenia in copper deficiency. Curr Opin Hematol. 2012;19(1):58-60. (PubMed)
40. Bost M, Houdart S, Oberli M, Kalonji E, Huneau JF, Margaritis I. Dietary copper and human health: Current evidence and unresolved issues. J Trace Elem Med Biol. 2016;35:107-115. (PubMed)
41. Harvey LJ, McArdle HJ. Biomarkers of copper status: a brief update. Br J Nutr. 2008;99 Suppl 3:S10-13. (PubMed)
42. Olivares M, Mendez MA, Astudillo PA, Pizarro F. Present situation of biomarkers for copper status. Am J Clin Nutr. 2008;88(3):859S-862S. (PubMed)
43. Harvey LJ, Ashton K, Hooper L, Casgrain A, Fairweather-Tait SJ. Methods of assessment of copper status in humans: a systematic review. Am J Clin Nutr. 2009;89(6):2009S-2024S. (PubMed)
44. Lassi KC, Prohaska JR. Rapid alteration in rat red blood cell copper chaperone for superoxide dismutase after marginal copper deficiency and repletion. Nutr Res. 2011;31(9):698-706. (PubMed)
45. Lassi KC, Prohaska JR. Erythrocyte copper chaperone for superoxide dismutase is increased following marginal copper deficiency in adult and postweanling mice. J Nutr. 2012;142(2):292-297. (PubMed)
46. Dirksen K, Roelen YS, van Wolferen ME, et al. Erythrocyte copper chaperone for superoxide dismutase and superoxide dismutase as biomarkers for hepatic copper concentrations in Labrador retrievers. Vet J. 2016;218:1-6. (PubMed)
47. Shaw JC. Copper deficiency and non-accidental injury. Arch Dis Child. 1988;63(4):448-455. (PubMed)
48. Altarelli M, Ben-Hamouda N, Schneider A, Berger MM. Copper deficiency: causes, manifestations, and treatment. Nutr Clin Pract. 2019;34(4):504-513. (PubMed)
49. Moon N, Aryan M, Westerveld D, Nathoo S, Glover S, Kamel AY. Clinical manifestations of copper deficiency: a case report and review of the literature. Nutr Clin Pract. 2021;36(5):1080-1085. (PubMed)
50. Burkhead JL, Collins JF. Nutrition information brief - copper. Adv Nutr. 2022;13(2):681-683. (PubMed)
51. Griffith DP, Liff DA, Ziegler TR, Esper GJ, Winton EF. Acquired copper deficiency: a potentially serious and preventable complication following gastric bypass surgery. Obesity (Silver Spring). 2009;17(4):827-831. (PubMed)
52. Kirkland Z, Villasmil RJ, Alookaran J, Ward MC, Stone D. Copper deficiency myeloneuropathy following Roux-en-Y gastric bypass in a 72-year-old female. Cureus. 2022;14(5):e25109. (PubMed)
53. Lewis CA, de Jersey S, Seymour M, Hopkins G, Hickman I, Osland E. Iron, vitamin B(12), folate and copper deficiency after bariatric surgery and the impact on anaemia: a systematic review. Obes Surg. 2020;30(11):4542-4591. (PubMed)
54. Blackmer AB, Bailey E. Management of copper deficiency in cholestatic infants: review of the literature and a case series. Nutr Clin Pract. 2013;28(1):75-86. (PubMed)
55. Best K, McCoy K, Gemma S, Disilvestro RA. Copper enzyme activities in cystic fibrosis before and after copper supplementation plus or minus zinc. Metabolism. 2004;53(1):37-41. (PubMed)
56. Seblani MD, McColley SA, Gong S, Bass LM, Badawy SM. Pancytopenia in a child with cystic fibrosis and severe copper deficiency: Insight from bone marrow evaluation. Pediatr Blood Cancer. 2021;68(12):e29276. (PubMed)
57. Rowin J, Lewis SL. Copper deficiency myeloneuropathy and pancytopenia secondary to overuse of zinc supplementation. J Neurol Neurosurg Psychiatry. 2005;76(5):750-751. (PubMed)
58. Nations SP, Boyer PJ, Love LA, et al. Denture cream: an unusual source of excess zinc, leading to hypocupremia and neurologic disease. Neurology. 2008;71(9):639-643. (PubMed)
59. Duncan A, Yacoubian C, Watson N, Morrison I. The risk of copper deficiency in patients prescribed zinc supplements. J Clin Pathol. 2015;68(9):723-725. (PubMed)
60. Klevay LM. IHD from copper deficiency: a unified theory. Nutr Res Rev. 2016;29(2):172-179. (PubMed)
61. Klevay LM. The contemporaneous epidemic of chronic, copper deficiency. J Nutr Sci. 2022;11:e89. (PubMed)
62. Klevay LM. Is the Western diet adequate in copper? J Trace Elem Med Biol. 2011;25(4):204-212. (PubMed)
63. Prodan CI, Bottomley SS, Holland NR, Lind SE. Relapsing hypocupraemic myelopathy requiring high-dose oral copper replacement. J Neurol Neurosurg Psychiatry. 2006;77(9):1092-1093. (PubMed)
64. Kumar N, Gross JB, Jr. Mutation in the ATP7A gene may not be responsible for hypocupraemia in copper deficiency myelopathy. Postgrad Med J. 2006;82(968):416. (PubMed)
65. Tumer Z. An overview and update of ATP7A mutations leading to Menkes disease and occipital horn syndrome. Hum Mutat. 2013;34(3):417-429. (PubMed)
66. Kodama H, Fujisawa C, Bhadhprasit W. Inherited copper transport disorders: biochemical mechanisms, diagnosis, and treatment. Curr Drug Metab. 2012;13(3):237-250. (PubMed)
67. Donsante A, Yi L, Zerfas PM, et al. ATP7A gene addition to the choroid plexus results in long-term rescue of the lethal copper transport defect in a Menkes disease mouse model. Mol Ther. 2011;19(12):2114-2123. (PubMed)
68. Haddad MR, Choi EY, Zerfas PM, et al. Cerebrospinal fluid-directed rAAV9-rsATP7A plus subcutaneous copper histidinate advance survival and outcomes in a Menkes disease mouse model. Mol Ther Methods Clin Dev. 2018;10:165-178. (PubMed)
69. Batzios S, Tal G, DiStasio AT, et al. Newly identified disorder of copper metabolism caused by variants in CTR1, a high-affinity copper transporter. Hum Mol Genet. 2022;31(24):4121-4130. (PubMed)
70. Nose Y, Kim BE, Thiele DJ. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metab. 2006;4(3):235-244. (PubMed)
71. Mak CM, Lam CW. Diagnosis of Wilson's disease: a comprehensive review. Crit Rev Clin Lab Sci. 2008;45(3):263-290. (PubMed)
72. Mulligan C, Bronstein JM. Wilson disease: an overview and approach to management. Neurol Clin. 2020;38(2):417-432. (PubMed)
73. Scheinberg IH, Sternlieb, I. Wilson’s disease. Philadelphia, PA: Saunders; 1984.
74. Wallace DF, Dooley JS. ATP7B variant penetrance explains differences between genetic and clinical prevalence estimates for Wilson disease. Hum Genet. 2020;139(8):1065-1075. (PubMed)
75. LeWitt PA. Penicillamine as a controversial treatment for Wilson's disease. Mov Disord. 1999;14(4):555-556. (PubMed)
76. Washington K. Practical Hepatic Pathology: a Diagnostic Approach. 2nd ed. Philadelphia; 2017.
77. Kishore N, Prasad R. A new concept: pathogenesis of Indian childhood cirrhosis (ICC)--hereditary alpha-I-antitrypsin deficiency. J Trop Pediatr. 1993;39(3):191-192. (PubMed)
78. Coenen ICJ HR. Indian Childhood Cirrhosis and Other Disorders of Copper Handling. 1st ed. London: Academic Press (Elsevier); 2019.
79. Nath R. Copper deficiency and heart disease: molecular basis, recent advances and current concepts. Int J Biochem Cell Biol. 1997;29(11):1245-1254. (PubMed)
80. Fox PL, Mazumder B, Ehrenwald E, Mukhopadhyay CK. Ceruloplasmin and cardiovascular disease. Free Radic Biol Med. 2000;28(12):1735-1744. (PubMed)
81. Jones AA, DiSilvestro RA, Coleman M, Wagner TL. Copper supplementation of adult men: effects on blood copper enzyme activities and indicators of cardiovascular disease risk. Metabolism. 1997;46(12):1380-1383. (PubMed)
82. DiNicolantonio JJ, Mangan D, O'Keefe JH. Copper deficiency may be a leading cause of ischaemic heart disease. Open Heart. 2018;5(2):e000784. (PubMed)
83. Ford ES. Serum copper concentration and coronary heart disease among US adults. Am J Epidemiol. 2000;151(12):1182-1188. (PubMed)
84. Cabral M, Kuxhaus O, Eichelmann F, et al. Trace element profile and incidence of type 2 diabetes, cardiovascular disease and colorectal cancer: results from the EPIC-Potsdam cohort study. Eur J Nutr. 2021;60(6):3267-3278. (PubMed)
85. Malek F, Jiresova E, Dohnalova A, Koprivova H, Spacek R. Serum copper as a marker of inflammation in prediction of short term outcome in high risk patients with chronic heart failure. Int J Cardiol. 2006;113(2):e51-53. (PubMed)
86. Kunutsor SK, Voutilainen A, Kurl S, Laukkanen JA. Serum copper-to-zinc ratio is associated with heart failure and improves risk prediction in middle-aged and older Caucasian men: A prospective study. Nutr Metab Cardiovasc Dis. 2022;32(8):1924-1935. (PubMed)
87. Leone N, Courbon D, Ducimetiere P, Zureik M. Zinc, copper, and magnesium and risks for all-cause, cancer, and cardiovascular mortality. Epidemiology. 2006;17(3):308-314. (PubMed)
88. Kosar F, Sahin I, Acikgoz N, Aksoy Y, Kucukbay Z, Cehreli S. Significance of serum trace element status in patients with rheumatic heart disease: a prospective study. Biol Trace Elem Res. 2005;107(1):1-10. (PubMed)
89. Liu N, Lo LS, Askary SH, et al. Transcuprein is a macroglobulin regulated by copper and iron availability. J Nutr Biochem. 2007;18(9):597-608. (PubMed)
90. Moriya M, Ho YH, Grana A, et al. Copper is taken up efficiently from albumin and alpha2-macroglobulin by cultured human cells by more than one mechanism. Am J Physiol Cell Physiol. 2008;295(3):C708-721. (PubMed)
91. Bertinato J, Zouzoulas A. Considerations in the development of biomarkers of copper status. J AOAC Int. 2009;92(5):1541-1550. (PubMed)
92. Klevay LM. Cardiovascular disease from copper deficiency--a history. J Nutr. 2000;130(2S Suppl):489S-492S. (PubMed)
93. Mielcarz G, Howard AN, Mielcarz B, et al. Leucocyte copper, a marker of copper body status is low in coronary artery disease. J Trace Elem Med Biol. 2001;15(1):31-35. (PubMed)
94. Kinsman GD, Howard AN, Stone DL, Mullins PA. Studies in copper status and atherosclerosis. Biochem Soc Trans. 1990;18(6):1186-1188. (PubMed)
95. Wang XL, Adachi T, Sim AS, Wilcken DE. Plasma extracellular superoxide dismutase levels in an Australian population with coronary artery disease. Arterioscler Thromb Vasc Biol. 1998;18(12):1915-1921. (PubMed)
96. Eshak ES, Iso H, Yamagishi K, Maruyama K, Umesawa M, Tamakoshi A. Associations between copper and zinc intakes from diet and mortality from cardiovascular disease in a large population-based prospective cohort study. J Nutr Biochem. 2018;56:126-132. (PubMed)
97. Mansoor MA, Bergmark C, Haswell SJ, et al. Correlation between plasma total homocysteine and copper in patients with peripheral vascular disease. Clin Chem. 2000;46(3):385-391. (PubMed)
98. Celik C, Bastu E, Abali R, et al. The relationship between copper, homocysteine and early vascular disease in lean women with polycystic ovary syndrome. Gynecol Endocrinol. 2013;29(5):488-491. (PubMed)
99. Gupta M, Meehan-Atrash J, Strongin RM. Identifying a role for the interaction of homocysteine and copper in promoting cardiovascular-related damage. Amino Acids. 2021;53(5):739-744. (PubMed)
100. Gerhard GT, Duell PB. Homocysteine and atherosclerosis. Curr Opin Lipidol. 1999;10(5):417-428. (PubMed)
101. Barter PJ, Rye KA. Homocysteine and cardiovascular disease: is HDL the link? Circ Res. 2006;99(6):565-566. (PubMed)
102. Emsley AM, Jeremy JY, Gomes GN, Angelini GD, Plane F. Investigation of the inhibitory effects of homocysteine and copper on nitric oxide-mediated relaxation of rat isolated aorta. Br J Pharmacol. 1999;126(4):1034-1040. (PubMed)
103. Shukla N, Angelini GD, Jeremy JY. Interactive effects of homocysteine and copper on angiogenesis in porcine isolated saphenous vein. Ann Thorac Surg. 2007;84(1):43-49. (PubMed)
104. Uthus EO, Reeves PG, Saari JT. Copper deficiency decreases plasma homocysteine in rats. J Nutr. 2007;137(6):1370-1374. (PubMed)
105. Wei H, Zhang WJ, McMillen TS, Leboeuf RC, Frei B. Copper chelation by tetrathiomolybdate inhibits vascular inflammation and atherosclerotic lesion development in apolipoprotein E-deficient mice. Atherosclerosis. 2012;223(2):306-313. (PubMed)
106. Tsikas D. Homocysteine and copper ions: is their interaction responsible for cardiovascular-related damage? Amino Acids. 2021;53(8):1297-1298. (PubMed)
107. Klevay LM. Lack of a recommended dietary allowance for copper may be hazardous to your health. J Am Coll Nutr. 1998;17(4):322-326. (PubMed)
108. Milne DB, Nielsen FH. Effects of a diet low in copper on copper-status indicators in postmenopausal women. Am J Clin Nutr. 1996;63(3):358-364. (PubMed)
109. Medeiros DM, Milton A, Brunett E, Stacy L. Copper supplementation effects on indicators of copper status and serum cholesterol in adult males. Biol Trace Elem Res. 1991;30(1):19-35. (PubMed)
110. DiSilvestro RA, Joseph EL, Zhang W, Raimo AE, Kim YM. A randomized trial of copper supplementation effects on blood copper enzyme activities and parameters related to cardiovascular health. Metabolism. 2012;61(9):1242-1246. (PubMed)
111. Rojas-Sobarzo L, Olivares M, Brito A, Suazo M, Araya M, Pizarro F. Copper supplementation at 8 mg neither affects circulating lipids nor liver function in apparently healthy Chilean men. Biol Trace Elem Res. 2013;156(1-3):1-4. (PubMed)
112. Turley E, McKeown A, Bonham MP, et al. Copper supplementation in humans does not affect the susceptibility of low density lipoprotein to in vitro induced oxidation (FOODCUE project). Free Radic Biol Med. 2000;29(11):1129-1134. (PubMed)
113. Rock E, Mazur A, O'Connor J M, Bonham MP, Rayssiguier Y, Strain JJ. The effect of copper supplementation on red blood cell oxidizability and plasma antioxidants in middle-aged healthy volunteers. Free Radic Biol Med. 2000;28(3):324-329. (PubMed)
114. Gombart AF, Pierre A, Maggini S. A review of micronutrients and the immune system-working in harmony to reduce the risk of infection. Nutrients. 2020;12(1):236. (PubMed)
115. Failla ML, Hopkins RG. Is low copper status immunosuppressive? Nutr Rev. 1998;56(1 Pt 2):S59-64. (PubMed)
116. Percival SS. Copper and immunity. Am J Clin Nutr. 1998;67(5 Suppl):1064S-1068S. (PubMed)
117. Heresi G, Castillo-Duran C, Munoz C, Arevalo M, Schlesinger L. Phagocytosis and immunoglobulin levels in hypocupremic children. Nutr Res. 1985;5:1327-1334.
118. Kelley DS, Daudu PA, Taylor PC, Mackey BE, Turnlund JR. Effects of low-copper diets on human immune response. Am J Clin Nutr. 1995;62(2):412-416. (PubMed)
119. Hodgkinson V, Petris MJ. Copper homeostasis at the host-pathogen interface. J Biol Chem. 2012;287(17):13549-13555. (PubMed)
120. Govind V, Bharadwaj S, Sai Ganesh MR, et al. Antiviral properties of copper and its alloys to inactivate covid-19 virus: a review. Biometals. 2021;34(6):1217-1235. (PubMed)
121. Looker AC, Melton LJ, 3rd, Harris TB, Borrud LG, Shepherd JA. Prevalence and trends in low femur bone density among older US adults: NHANES 2005-2006 compared with NHANES III. J Bone Miner Res. 2010;25(1):64-71. (PubMed)
122. Tiidus PM, Lowe DA, Brown M. Estrogen replacement and skeletal muscle: mechanisms and population health. J Appl Physiol. 2013;115(5):569-578. (PubMed)
123. Cauley JA. Public health impact of osteoporosis. J Gerontol A Biol Sci Med Sci. 2013;68(10):1243-51. (PubMed)
124. Kanumakala S, Boneh A, Zacharin M. Pamidronate treatment improves bone mineral density in children with Menkes disease. J Inherit Metab Dis. 2002;25(5):391-398. (PubMed)
125. Marquardt ML, Done SL, Sandrock M, Berdon WE, Feldman KW. Copper deficiency presenting as metabolic bone disease in extremely low birth weight, short-gut infants. Pediatrics. 2012;130(3):e695-698. (PubMed)
126. Baker A, Harvey L, Majask-Newman G, Fairweather-Tait S, Flynn A, Cashman K. Effect of dietary copper intakes on biochemical markers of bone metabolism in healthy adult males. Eur J Clin Nutr. 1999;53(5):408-412. (PubMed)
127. Baker A, Turley E, Bonham MP, et al. No effect of copper supplementation on biochemical markers of bone metabolism in healthy adults. Br J Nutr. 1999;82(4):283-290. (PubMed)
128. Cashman KD, Baker A, Ginty F, et al. No effect of copper supplementation on biochemical markers of bone metabolism in healthy young adult females despite apparently improved copper status. Eur J Clin Nutr. 2001;55(7):525-531. (PubMed)
129. Conlan D, Korula R, Tallentire D. Serum copper levels in elderly patients with femoral-neck fractures. Age Ageing. 1990;19(3):212-214. (PubMed)
130. Mutlu M, Argun M, Kilic E, Saraymen R, Yazar S. Magnesium, zinc and copper status in osteoporotic, osteopenic and normal post-menopausal women. J Int Med Res. 2007;35(5):692-695. (PubMed)
131. Mahdavi-Roshan M, Ebrahimi M, Ebrahimi A. Copper, magnesium, zinc and calcium status in osteopenic and osteoporotic post-menopausal women. Clin Cases Miner Bone Metab. 2015;12(1):18-21. (PubMed)
132. Okyay E, Ertugrul C, Acar B, Sisman AR, Onvural B, Ozaksoy D. Comparative evaluation of serum levels of main minerals and postmenopausal osteoporosis. Maturitas. 2013;76(4):320-325. (PubMed)
133. Fan Y, Ni S, Zhang H. Associations of copper intake with bone mineral density and osteoporosis in adults: data from the National Health and Nutrition Examination Survey. Biol Trace Elem Res. 2022;200(5):2062-2068. (PubMed)
134. Eaton-Evans J, Mellwrath EM, Jackson WE, McCartney H, Strain JJ. Copper supplementation and the maintenance of bone mineral density in middle-aged women. J Trace Elem Exp Med. 1996;9:87-94.
135. Strause L, Saltman P, Smith KT, Bracker M, Andon MB. Spinal bone loss in postmenopausal women supplemented with calcium and trace minerals. J Nutr. 1994;124(7):1060-1064. (PubMed)
136. Nielsen FH, Lukaski HC, Johnson LK, Roughead ZK. Reported zinc, but not copper, intakes influence whole-body bone density, mineral content and T score responses to zinc and copper supplementation in healthy postmenopausal women. Br J Nutr. 2011;106(12):1872-1879. (PubMed)
137. Sidiropoulou-Chatzigiannis S, Kourtidou M, Tsalikis L. The effect of osteoporosis on periodontal status, alveolar bone and orthodontic tooth movement. A literature review. J Int Acad Periodontol. 2007;9(3):77-84. (PubMed)
138. Darcey J, Horner K, Walsh T, Southern H, Marjanovic EJ, Devlin H. Tooth loss and osteoporosis: to assess the association between osteoporosis status and tooth number. Br Dent J. 2013;214(4):E10. (PubMed)
139. Sierpinska T, Konstantynowicz J, Orywal K, Golebiewska M, Szmitkowski M. Copper deficit as a potential pathogenic factor of reduced bone mineral density and severe tooth wear. Osteoporos Int. 2014;25(2):447-54. (PubMed)
140. Squitti R, Ventriglia M, Simonelli I, et al. Copper imbalance in Alzheimer's disease: meta-analysis of serum, plasma, and brain specimens, and replication study evaluating ATP7B gene variants. Biomolecules. 2021;11(7):960. (PubMed)
141. Li DD, Zhang W, Wang ZY, Zhao P. Serum copper, zinc, and iron levels in patients with Alzheimer's disease: a meta-analysis of case-control studies. Front Aging Neurosci. 2017;9:300. (PubMed)
142. Squitti R, Polimanti R. Copper hypothesis in the missing hereditability of sporadic Alzheimer's disease: ATP7B gene as potential harbor of rare variants. J Alzheimers Dis. 2012;29(3):493-501. (PubMed)
143. Brewer GJ. Copper excess, zinc deficiency, and cognition loss in Alzheimer's disease. Biofactors. 2012;38(2):107-113. (PubMed)
144. Squitti R, Polimanti R. Copper phenotype in Alzheimer's disease: dissecting the pathway. Am J Neurodegener Dis. 2013;2(2):46-56. (PubMed)
145. Squitti R, Faller P, Hureau C, Granzotto A, White AR, Kepp KP. Copper imbalance in Alzheimer's disease and its link with the amyloid hypothesis: towards a combined clinical, chemical, and genetic etiology. J Alzheimers Dis. 2021;83(1):23-41. (PubMed)
146. Sparks DL, Schreurs BG. Trace amounts of copper in water induce beta-amyloid plaques and learning deficits in a rabbit model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2003;100(19):11065-11069. (PubMed)
147. Kitazawa M, Cheng D, Laferla FM. Chronic copper exposure exacerbates both amyloid and tau pathology and selectively dysregulates cdk5 in a mouse model of AD. J Neurochem. 2009;108(6):1550-1560. (PubMed)
148. Morris MC, Evans DA, Tangney CC, et al. Dietary copper and high saturated and trans fat intakes associated with cognitive decline. Arch Neurol. 2006;63(8):1085-1088. (PubMed)
149. Kessler H, Pajonk FG, Bach D, et al. Effect of copper intake on CSF parameters in patients with mild Alzheimer's disease: a pilot phase 2 clinical trial. J Neural Transm. 2008;115(12):1651-1659. (PubMed)
150. Kessler H, Bayer TA, Bach D, et al. Intake of copper has no effect on cognition in patients with mild Alzheimer's disease: a pilot phase 2 clinical trial. J Neural Transm. 2008;115(8):1181-1187. (PubMed)
151. Skjorringe T, Moller LB, Moos T. Impairment of interrelated iron- and copper homeostatic mechanisms in brain contributes to the pathogenesis of neurodegenerative disorders. Front Pharmacol. 2012;3:169. (PubMed)
152. Bisaglia M, Bubacco L. Copper ions and Parkinson's disease: why is homeostasis so relevant? Biomolecules. 2020;10(2):195. (PubMed)
153. Akatsu H, Hori A, Yamamoto T, et al. Transition metal abnormalities in progressive dementias. Biometals. 2012;25(2):337-350. (PubMed)
154. Davies KM, Bohic S, Carmona A, et al. Copper pathology in vulnerable brain regions in Parkinson's disease. Neurobiol Aging. 2014;35(4):858-866. (PubMed)
155. Kim MJ, Oh SB, Kim J, et al. Association of metals with the risk and clinical characteristics of Parkinson's disease. Parkinsonism Relat Disord. 2018;55:117-121. (PubMed)
156. Ilyechova EY, Miliukhina IV, Orlov IA, Muruzheva ZM, Puchkova LV, Karpenko MN. A low blood copper concentration is a co-morbidity burden factor in Parkinson's disease development. Neurosci Res. 2018;135:54-62. (PubMed)
157. Younes-Mhenni S, Aissi M, Mokni N, et al. Serum copper, zinc and selenium levels in Tunisian patients with Parkinson's disease. Tunis Med. 2013;91(6):402-405. (PubMed)
158. Miyake Y, Tanaka K, Fukushima W, et al. Dietary intake of metals and risk of Parkinson's disease: a case-control study in Japan. J Neurol Sci. 2011;306(1-2):98-102. (PubMed)
159. Powers KM, Smith-Weller T, Franklin GM, Longstreth WT, Jr., Swanson PD, Checkoway H. Parkinson's disease risks associated with dietary iron, manganese, and other nutrient intakes. Neurology. 2003;60(11):1761-1766. (PubMed)
160. Heffern MC, Park HM, Au-Yeung HY, et al. In vivo bioluminescence imaging reveals copper deficiency in a murine model of nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A. 2016;113(50):14219-14224. (PubMed)
161. Aigner E, Theurl I, Haufe H, et al. Copper availability contributes to iron perturbations in human nonalcoholic fatty liver disease. Gastroenterology. 2008;135(2):680-688. (PubMed)
162. Lan Y, Wu S, Wang Y, et al. Association between blood copper and nonalcoholic fatty liver disease according to sex. Clin Nutr. 2021;40(4):2045-2052. (PubMed)
163. Aigner E, Strasser M, Haufe H, et al. A role for low hepatic copper concentrations in nonalcoholic Fatty liver disease. Am J Gastroenterol. 2010;105(9):1978-1985. (PubMed)
164. Mendoza M, Caltharp S, Song M, et al. Low hepatic tissue copper in pediatric nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr. 2017;65(1):89-92. (PubMed)
165. Stattermayer AF, Traussnigg S, Aigner E, et al. Low hepatic copper content and PNPLA3 polymorphism in non-alcoholic fatty liver disease in patients without metabolic syndrome. J Trace Elem Med Biol. 2017;39:100-107. (PubMed)
166. US Department of Agriculture, Agricultural Research Service. 2022. Nutrient Intakes from Food and Beverages: Mean Amounts Consumed per Individual, by Gender and Age, What We Eat in America, NHANES 2017-March 2020 Prepandemic.
167. Hendler S, Rorvik D, eds. PDR for Nutritional Supplements. Montvale: Medical Economics Company, Inc.; 2001.
168. Rosado JL. Zinc and copper: proposed fortification levels and recommended zinc compounds. J Nutr. 2003;133(9):2985S-2989S. (PubMed)
169. US Department of Health and Human Services, National Institutes of Health, Office of Dietary Supplements. Dietary Supplement Label Database (DSLD). [Internet]. [cited 8/8/2023]. Available from: https://dsld.od.nih.gov.
170. Pratt WB, Omdahl JL, Sorenson JR. Lack of effects of copper gluconate supplementation. Am J Clin Nutr. 1985;42(4):681-682. (PubMed)
171. Bremner I. Manifestations of copper excess. Am J Clin Nutr. 1998;67(5 Suppl):1069S-1073S. (PubMed)
172. Fitzgerald DJ. Safety guidelines for copper in water. Am J Clin Nutr. 1998;67(5 Suppl):1098S-1102S. (PubMed)
173. Turnlund JR, Jacob RA, Keen CL, et al. Long-term high copper intake: effects on indexes of copper status, antioxidant status, and immune function in young men. Am J Clin Nutr. 2004;79(6):1037-1044. (PubMed)
174. Turnlund JR, Keyes WR, Kim SK, Domek JM. Long-term high copper intake: effects on copper absorption, retention, and homeostasis in men. Am J Clin Nutr. 2005;81(4):822-828. (PubMed)
175. Kozak SF, Inderlied CB, Hsu HY, Heller KB, Sadun AA. The role of copper on ethambutol's antimicrobial action and implications for ethambutol-induced optic neuropathy. Diagn Microbiol Infect Dis. 1998;30(2):83-87. (PubMed)
176. Wood RJ, Suter PM, Russell RM. Mineral requirements of elderly people. Am J Clin Nutr. 1995;62(3):493-505. (PubMed)
Fluoride
Contents
Summary
- Fluoride is the ionic form of the naturally occurring fluorine element. The anion increases the structural stability of teeth and bones through interactions with calcium phosphates. (More information)
- The daily intake recommendations for fluoride are based on the safest and most effective intakes to prevent dental caries. (More information)
- The use of fluoridated dental products and adequate intakes of fluoride reduce the occurrence of caries throughout life by promoting tooth mineralization and re-mineralization. Large randomized, placebo-controlled studies are needed to evaluate whether the topical application of fluoridated agents could also prevent dental erosion. (More information)
- Epidemiological and clinical evidence is currently limited to support a role for water fluoridation in the prevention of osteoporosis and bone fracture. (More information)
- Therapeutic trials have found a dose-dependent effect of fluoride on fracture risk in osteoporotic patients. However, the occurrence of numerous side effects warrants additional studies to guarantee that safe and effective doses can be used alone or in combination with current therapies. (More information)
- The major sources of systemic and topical fluoride are drinking water, foods and beverages made with fluoridated water, infant formulas, and fluoride-containing oral care products. Fluoridated salt and milk are currently available outside the US in Europe, Latin America, and Southeast Asia. (More information)
-
While increased exposure to fluoride has led to a decline in dental caries, the prevalence of white speckling or mottling of the permanent teeth, known as dental fluorosis, has increased. Bone tissue homeostasis may also be affected by excess fluoride intake. (More information)
Fluorine occurs naturally as the negatively charged ion, fluoride (F-). Fluoride is considered a trace element because only small amounts are present in the body (about 2.6 grams in adults), and because the daily requirement for maintaining dental health is only a few milligrams a day. About 95% of the total body fluoride is found in bones and teeth (1). Although its role in the prevention of dental caries (tooth decay) is well established, fluoride is not generally considered an essential mineral element because humans do not require it for growth or to sustain life (2). However, if one considers the prevention of chronic disease (dental caries) an important criterion in determining essentiality, then fluoride might well be considered an essential trace element (3).
Function
Fluoride is absorbed in the stomach and small intestine. Once in the bloodstream it rapidly enters mineralized tissue (bones and developing teeth). At usual intake levels, fluoride does not accumulate in soft tissue. The predominant mineral elements in bone are crystals of calcium and phosphate, known as hydroxyapatite crystals. Fluoride's high chemical reactivity and small radius allow it to either displace the larger hydroxyl (-OH) ion in the hydroxyapatite crystal, forming fluoroapatite, or to increase crystal density by entering spaces within the hydroxyapatite crystal. Fluoroapatite hardens tooth enamel and stabilizes bone mineral (4).
Nutrient interactions
Both calcium and magnesium form insoluble complexes with fluoride and are capable of significantly decreasing fluoride absorption when present in the same meal. However, the absorption of fluoride in the form of monofluorophosphate (unlike sodium fluoride) is unaffected by calcium. Also, a diet low in chloride (salt) has been found to increase fluoride retention by reducing urinary excretion of fluoride (1).
Deficiency
In humans, the only clear effect of inadequate fluoride intake is an increased risk of dental caries (tooth decay) for individuals of all ages. Epidemiological investigations of patterns of water consumption and the prevalence of dental caries across various US regions with different water fluoride concentrations led to the development of a recommended optimum range of fluoride concentration of 0.7-1.2 milligrams/liter (mg/L) or parts per million (ppm); the lower concentration was recommended for warmer climates where water consumption is higher, and the higher concentration was recommended for colder climates. In 2015, the US Department of Health and Human Services recommended that all community water systems adjust the fluoride concentration to 0.7 mg/L, as more recent studies did find a relationship between water intake and outdoor air temperature. This recommendation was made in an effort to reduce the risk of dental fluorosis (see below) and in light of the widespread availability of fluoride from other sources, including fluoride-containing oral-care products (5). A number of studies conducted prior to the introduction of fluoride-containing toothpastes demonstrated that the prevalence of dental caries was 40% to 60% lower in communities with optimal water fluoride concentrations than in communities with low water fluoride concentrations (6).
The Adequate Intake (AI)
The Food and Nutrition Board (FNB) of the US Institute of Medicine updated its recommendations for fluoride intake in 1997. Because data were insufficient to establish a Recommended Dietary Allowance (RDA), Adequate Intake (AI) levels were set based on estimated intakes that have been shown to reduce the occurrence of dental caries most effectively without causing the unwanted side effect of tooth enamel mottling known as dental fluorosis (0.05 mg/kg of body weight) (Table 1; 6). See the section below on Safety for a discussion of dental fluorosis.
Disease Prevention
Dental caries (cavities and tooth decay)
Specific cariogenic (cavity-causing) bacteria (mainly Streptococcus mutans and Streptococcus sobrinus) found in dental plaque are capable of metabolizing fermentable carbohydrates (sugars) and converting them to organic acids that can dissolve sensitive tooth enamel. If unchecked, the bacteria may penetrate deeper layers of the tooth and progress into the soft pulp tissue at the center. Untreated caries can lead to severe pain, local infection, tooth loss or extraction, nutritional problems, and serious systemic infections in susceptible individuals (7). Dental caries — both treated and untreated — contribute to diminished overall oral health, which, in turn, may affect systemic health. For example, some observational studies have suggested a link between systemic inflammation in individuals with periodontal (gum) infection and insulin resistance (8), type 2 diabetes mellitus (9), hypertension (10), and coronary heart disease (11). Moreover, the bacterium causing periodontitis, Porphyromonas gingivalis, may be linked to rheumatoid arthritis (12, 13). Poor oral health in general may constitute a risk factor for coronary heart disease (14) and other cardiovascular diseases (15, 16).
Systemic effects of fluoride on teeth
Increased fluoride exposure, most commonly through community water fluoridation, has been found to decrease the incidence of dental caries in children and adults (17, 18). Between 1976 and 1987, clinical studies in several countries demonstrated that the addition of fluoride to community water supplies (0.7-1.2 ppm) reduced caries by 30%-60% in primary (baby) teeth and 15%-35% in permanent teeth (19). A 2015 review and meta-analysis of prospective observational studies found a 35% and 26% reduction in number of decayed, missing, and filled primary and permanent teeth with consumption of fluoridated versus non-fluoridated water, respectively (20). While fluoride’s prevention of dental caries is primarily through topical action, fluoride consumed in water appears to have a systemic effect in children before all teeth have erupted — typically through 12 years of age.
Fluoride is incorporated into the developing enamel of teeth and increases the resistance to caries. Since the caries preventative effect of fluoride is also topical (surface) in children after teeth have erupted and in adults, the optimal protection achieved by fluoridated water likely occurs through both systemic exposure before and after tooth eruption and topical exposure after tooth eruption.
Topical effects of fluoride on teeth
Research has indicated that the primary action of fluoride occurs topically after the teeth erupt into the mouth. Ingested fluoride is secreted in the saliva and contributes to topical protection. When enamel is partially demineralized by organic acids, fluoride in the saliva can enhance the remineralization of enamel through its interactions with calcium and phosphate. Fluoride containing remineralized enamel is more resistant to acid attack and demineralization. In salivary concentrations associated with optimum fluoride intake, fluoride has been found to inhibit bacterial enzymes, resulting in reduced acid production by cariogenic bacteria (7, 17).
The use of topically applied fluoride-containing products, including toothpaste, gel, varnish, and mouth rinse, is thought to have contributed to the substantial decrease in the prevalence of caries over the last decades (21). A 2013 meta-analysis of fluoride interventions in children and adolescents (up to 16 years of age) found that the application of fluoride varnish for at least one year was associated with a 37% reduction in decayed, missing, and filled tooth surfaces in decayed tooth surfaces of primary teeth; the anti-caries effect on the permanent teeth corresponded to a 43% decrease compared to no treatment or placebo (22). A recent (2021) meta-analysis of topical fluoride reviewed 15 clinical trials with 9,541 participants. Specifically, the meta-analysis evaluated 14 trials of fluoride varnish compared to placebo or no fluoride and one trial of fluoride foam, and found that topical fluoride reduced dental caries increment by about one tooth surface over a two-year period (23). The same meta-analysis also found that fluoride varnish significantly prevented any incident dental caries, but only for children who were younger than 2 years of age (23).
The effects of fluoride-containing toothpaste has been more extensive. A 2017 meta-analysis of 96 randomized controlled trials, including more than 65,000 participants, found tooth brushing with a fluoridated toothpaste reduced caries in primary teeth of children and in permanent teeth of adults compared to brushing with non-fluoridated toothpaste (24). In participants overall, use of toothpastes containing 1,000 to 1,500 ppm of fluoride had a caries-preventative effect compared to use of toothpastes with lower fluoride concentrations or with non-fluoridated toothpastes (24). A systematic review of 17 clinical trials found use of fluoridated toothpaste effectively reduced dental caries in primary teeth of children younger than 6 years who were at high risk of developing dental caries (25).
Dental erosion (tooth wear)
The attack of dental hard tissue by acids other than those produced by the bacterial plaque may lead to the loss of tooth enamel, also known as dental erosion. Factors involved in dental erosion include acidic foods and beverages (e.g., carbonated drinks) and acid reflux (26). The protective effect of fluoridated agents against dental erosion has mainly been observed in in vitro studies (reviewed in 26). Nevertheless, a meta-analysis of four small, randomized trials examining the effect of fluoride in toothpaste, varnish, and saliva on dental erosion did not find any overall benefit compared to placebo (27). Larger clinical studies are needed to evaluate whether topical fluoride applications can prevent dental erosion and/or reduce the progression of existing erosive lesions.
Osteoporosis
Although fluoride in pharmacologic doses has been shown to be a potent therapeutic agent for increasing spinal bone mass (see Disease Treatment), there is little evidence that water fluoridation at optimum levels for the prevention of dental caries is helpful in the prevention of osteoporosis. The majority of studies conducted to date have failed to find clinically significant differences in bone mineral density (BMD) or fracture incidence when comparing residents of areas with fluoridated water supplies to residents in areas without fluoridated water supplies (28, 29). However, two studies found that drinking water fluoridation was associated with decreased incidence of hip fracture in the elderly. In addition, one study in Italy found a significantly greater risk of femoral (hip) fractures in men and women residing in an area with low water fluoridation (0.05 ppm) compared to the risk in a similar population whose water supply was naturally fluoridated (1.45 ppm) at higher than optimum levels for prevention of dental caries (30). Another study in Germany found no significant difference in BMD between residents of a community whose water supply had been optimally fluoridated for 30 years (1 ppm) compared with those who resided in a community without fluoridated water. However, this study reported that the incidence of hip fracture in men and women, aged 85 years or older, was significantly lower in the community with fluoridated water compared to the community with non-fluoridated water, despite higher calcium levels in the non-fluoridated water supply (31). Another community-based study in 1,300 women found that elevated serum fluoride concentrations were not related to BMD or to osteoporotic fracture incidence (32). Finally, a nationwide cohort study in Sweden found no association between chronic exposure to fluoridated water and incidence of hip fracture (33). A 2015 meta-analysis, which pooled results of 13 prospective cohort studies and one case-control study, found that fluoride exposure in drinking water was not associated with risk of hip fracture (34).
Moreover, because bone mineral accretion early in life affects risk for osteoporosis in later adulthood, studies have examined the association of fluoride intake during adolescence and bone outcomes. Reports from the Iowa Bone Development Study, an ongoing prospective cohort study, indicate little to no association of fluoride intake during childhood and adolescence with measures of bone microstructure throughout adolescence (35-37) and into early adulthood (38).
Disease Treatment
Osteoporosis
Osteoporosis is characterized by decreased bone mineral density (BMD) and increased bone fragility and susceptibility to fracture. In general, decreased BMD is associated with increased risk of fracture. However, the usual relationship between BMD and fracture risk does not always hold true when very high (pharmacologic) doses of fluoride are used to treat osteoporosis. Most available therapies for osteoporosis (e.g., estrogen, calcitonin, and bisphosphonates) decrease bone loss (resorption), resulting in very small increases in BMD. Pharmacologic doses of fluoride are capable of producing large increases in the BMD of the lumbar spine. Overall, therapeutic trials of fluoride in patients with osteoporosis have not consistently demonstrated significant decreases in the occurrence of vertebral fracture despite dramatic increases in lumbar spine BMD (39). A meta-analysis of 11 controlled studies, including 1,429 participants, found that fluoride treatment resulted in increased BMD at the lumbar spine but was not associated with a lower risk of vertebral fractures (40). This meta-analysis also found that higher concentrations of fluoride were associated with increased risk of non-vertebral fractures after four years of treatment. Early studies using high doses of fluoride (>20 mg/day) may have induced rapid bone mineralization in the absence of adequate calcium and vitamin D, resulting in denser bones that were not mechanically stronger (41, 42). Analysis of bone architecture has also shed some light on the inconsistent effect of fluoride therapy in reducing vertebral fractures. Research has indicated that osteoporosis may be associated with an irreversible change in the architecture of bone known as decreased trabecular connectivity. Normal bone consists of a series of plates interconnected by thick rods. Severely osteoporotic bone has fewer plates, and the rods may be fractured or disconnected (decreased trabecular connectivity) (43). Despite fluoride therapy increasing bone density, it probably cannot reduce bone resorption and restore connectivity in patients with severe bone loss. Thus, fluoride therapy may be less effective in osteoporotic individuals who have already lost substantial trabecular connectivity (39, 44).
On the other hand, randomized controlled trials using lower fluoride doses (≤20 mg/day), intermittent dosage schedules, or slow-release formulations (enteric coated sodium fluoride) have demonstrated a decreased incidence of vertebral and non-vertebral fractures along with increased bone density of the lumbar spine (45). Yet, bone biopsies from postmenopausal, osteoporotic women treated with 20 mg/day of fluoride showed evidence of abnormal bone mineralization despite calcium and vitamin D supplementation (46). Additionally, a randomized, double-blind, placebo-controlled study did not find any increase in lumbar spine BMD in 180 postmenopausal women with osteopenia (early osteoporosis) who were given daily supplements of up to 10 mg/day of fluoride for one year (47). Additional studies are required to assess whether a safe dose of fluoride can be found to maximize bone formation while preventing mineralization defects.
Safety of fluoride therapy for osteoporosis
Serious side effects have been associated with the high doses of fluoride used to treat osteoporosis (45). They include gastrointestinal irritation, joint pain in the lower extremities, and the development of calcium deficiency and stress fractures. The reasons for the occurrence of lower extremity joint pain and stress fractures in patients taking fluoride for osteoporosis remain unclear, but they may be related to rapid increases in bone formation without sufficient calcium to support such an increase (39). Presently, enteric coated sodium fluoride or monofluorophosphate preparations offer a lower side effect profile than the high-dose sodium fluoride used in earlier trials. Additionally, sufficient calcium and vitamin D must be provided to support fluoride-induced bone formation. Although fluoride therapy may be beneficial for the treatment of osteoporosis in appropriately selected and closely monitored individuals, uncertainty about its safety and benefit in reducing fractures has kept the US Food and Drug Administration (FDA) from approving fluoride therapy for osteoporosis (48). Combinations of lower doses of fluoride with antiresorptive agents, such as estrogen or bisphosphonates, may improve therapeutic results while minimizing side effects (49, 50). Yet, randomized studies have shown that the risk of fractures remained unchanged whether treatments include fluoride, antiresorptives, or both (45, 46). Additional studies are warranted to determine whether any treatment combinations could provide substantial therapeutic benefits over monotherapy.
Sources
Water fluoridation
The major source of dietary fluoride in the US diet is drinking water. Controlled addition of fluoride to water is used by communities as a public health measure to adjust fluoride concentration in drinking water to prevent dental caries. Originally, an optimal range of 0.7 to 1.2 milligrams (mg) per liter (corresponding to 0.7-1.2 ppm) was established, which was shown to decrease the incidence of dental caries while minimizing the risk of dental fluorosis and other adverse effects. In 2015, the US Department of Health and Human Services recommended that the optimal concentration in drinking water be set at 0.7 ppm (see Safety) (51). Approximately 73% of the US population receives water with sufficient fluoride for the prevention of dental caries (52). The average fluoride intake for adults living in fluoridated communities ranges from 1.4 to 3.4 mg/day compared to 0.3 to 1 mg/day in non-fluoridated areas (6). Since well water can vary greatly in its fluoride content, people who consume water from wells should have the fluoride content of their water tested by their local water district or health department. Water fluoride testing may also be warranted in households that use home water treatment systems. While water softeners are not thought to change water fluoride levels, reverse osmosis systems, distillation units, and some water filters have been found to remove significant amounts of fluoride from water. However, Brita-type carbon-charcoal filters do not remove fluoride (6, 48).
Bottled water sales have grown exponentially in the US in the last decades, and studies have found that most bottled waters contain sub-optimal levels of fluoride, although there is considerable variation (53). For example, a study of 105 different bottled water products in the Greater Houston metropolitan area found that over 80% had fluoride concentrations of less than 0.4 ppm; only 5% of the tested products had fluoride concentrations within the recommended range (54). Several other studies have reported similar findings, with most bottled waters relatively low in fluoride, but a few in the optimal range or higher (55-57). The FDA-approved claim that "drinking fluoridated water may reduce the risk of tooth decay" is only used by bottlers when the water contains greater than 0.6 ppm of fluoride but no more than 1.0 ppm of fluoride. However, bottlers are not required to provide the fluoride concentration in bottled water unless fluoride was added (58).
Infant formulas
While consumption of fluoride from water presents very little risk of adverse effects in adults except in extreme circumstances (see Safety), consumption of relatively large amounts of water mixed with formula concentrates appears to increase the risk for the development of dental fluorosis in infants (59-61). One study found that, on average, at least half of all fluoride ingested by infants 6 months and younger was from water mixed with formula concentrates (62). The study of 49 commercially available infant formulas in the Chicago area showed that milk-based ready-to-feed, liquid concentrate, and powdered formulas (reconstituted with deionized water) had mean fluoride concentrations of 0.15 ppm, 0.27 ppm, and 0.12 ppm, respectively (63). Fluoride content was significantly higher in soy-based compared to milk-based liquid concentrate formulas (0.50 ppm vs. 0.27 ppm). Using average body weights and total formula intakes during the first year of life, the authors estimated that the risk of exceeding the tolerable upper intake level (UL) for fluoride ingestion was minimal when liquid concentrate and powdered formulas were reconstituted with water containing less than 0.5 ppm of fluoride, but the risk was maximal with 1.0 ppm fluoridated water. Fluoride-free or low-fluoride water labeled as "deionized," "purified," "demineralized," "distilled," or "produced through reverse-osmosis" can be used in order to minimize the risk for mild fluorosis (58). However, infants between 6 and 12 months may not reach the adequate intake of fluoride if they are fed ready-to-feed formulas or formulas reconstituted with water containing less than 0.4 ppm (63).
For additional information on fluoride and infant formulas, see the CDC website.
Food and beverage sources
The fluoride content of most foods is low (less than 0.05 mg/100 grams or 0.5 ppm). Rich sources of fluoride include tea, which concentrates fluoride in its leaves, and marine fish that are consumed with their bones (e.g., sardines). Foods made with mechanically separated (boned) chicken, such as canned meats, hot dogs, and infant foods, also add fluoride to the diet (64). In addition, certain fruit juices, particularly grape juices, often have relatively high fluoride concentrations (65). Foods generally contribute only 0.3-0.6 mg of the daily intake of fluoride. An adult male residing in a community with fluoridated water has an intake range from 1 mg/day-3 mg/day. Intake is less than 1 mg/day in non-fluoridated areas (2). Table 2 provides a range of fluoride content for a few fluoride-rich foods. For more information on the fluoride content of foods and beverages, search the USDA national fluoride database.
Fluoride supplements
Fluoride supplements — available only by prescription in the US — are intended for infants 6 months and older and children up to 16 years of age living in areas with suboptimal water fluoridation for the purpose of bringing their intake to approximately 1 mg/day (6). The American Dental Association Council on Scientific Affairs recommends the prescription of fluoride supplements only to children at high risk of developing dental caries (66). The supplemental fluoride dosage schedule in Table 3 was recommended by the American Dental Association, the American Academy of Pediatric Dentistry, and the American Academy of Pediatrics (66, 67). It requires knowledge of the fluoride concentration of local drinking water, as well as other possible sources of fluoride intake. For more detailed information regarding fluoride and the prevention of dental caries, visit the American Dental Association website.
Toothpaste
Fluoridated toothpastes (1,000-1,100 ppm of fluoride) are very effective in preventing dental caries but also add considerably to fluoride intake of children, especially young children who are more likely to swallow toothpaste. Researchers estimate that children under 6 years of age may ingest an average of 0.3 mg of fluoride from toothpaste with each brushing. Children under the age of 6 years who ingest more than two or three times the recommended fluoride intake are at increased risk of a white speckling or mottling of the permanent teeth, known as dental fluorosis (see Safety section below). A major source of excess fluoride intake in this age group comes from swallowing fluoride-containing toothpaste. To prevent dental fluorosis while providing optimum protection from tooth decay, it is recommended that parents supervise children under 6 years of age while brushing with fluoridated toothpaste. In addition to discouraging the swallowing of toothpaste, children should be supervised during teeth brushing, and young children should be encouraged to use very small amounts of toothpaste — a "smear amount" (a thin layer of toothpaste that covers less than half of the bristle surface of a child-size toothbrush; size equivalent to a grain of rice) for children younger than 3 years, and no more than a pea-sized application of toothpaste for children 3 to 6 years of age (25, 68-70). Interestingly, it has been suggested that the management of the fluorosis risk in young children who ingest fluoridated toothpaste could include the use of toothpaste formulation that reduces gastrointestinal absorption and bioavailability of fluoride (71).
Salt fluoridation
Fluoridation of salt has been implemented in several countries worldwide as an alternative to water fluoridation to promote the ingestion of fluoride and improve oral care. Since the fluoridation of water is extensively practiced in the US, fluoride is not added to salt. Observational studies have shown that the incidence of teeth with caries dramatically decreased in the regions where salt fluoridation programs were developed (reviewed in 72). While concerns around hypertension and the monitoring of population intakes should be addressed, no adverse health effects linked to the fluoridation of salt have been reported (reviewed in 73). According to the World Health Organization (WHO), salt fluoridation and, to a lesser extent, milk fluoridation, are affordable alternatives to improve oral hygiene in areas where access to oral health services is limited and fluoridation of public water is not feasible (74).
Mouth rinses
Mouth rinses that contain fluoride are available over-the-counter in the US. Such products often contain 0.05% sodium fluoride, which translates to about 1 mg of fluoride per 5 mL (one teaspoon) (70). Due to the risk of accidental ingestion, fluoride-containing mouth rinses are not recommended for young children (70).
Professionally applied fluoride products
Professionally applied fluoride products, including fluoride foams, gels, varnish, and silver diamine fluoride, are highly concentrated (9,000 to 44,800 ppm of fluoride) and applied topically to the teeth by dentists, dental hygienists, or other healthcare professionals (75). Because of their high concentrations, they are potential sources of fluoride; however, due to their infrequent use (every 3-6 months) and small amounts used per application, professionally applied fluoride products are not significant sources of fluoride when used as directed (75).
Safety
Adverse effects
Fluoridation of public drinking water in the US was initiated more than 70 years ago. Since then, a number of adverse effects have been attributed to water fluoridation. However, extensive scientific research has uncovered no evidence of increased risks of cancer, heart disease, kidney disease, liver disease, thyroid disease, Alzheimer's disease, birth defects, or Down's syndrome (51, 76, 77).
A number of observational studies, mostly published in Chinese journals, have investigated the association between fluoride content in drinking water and children’s neurodevelopment. Two meta-analyses of observational studies, mainly conducted in China, found lower intelligence quotients (IQs) in children exposed to higher fluoride concentrations in drinking water (78, 79). Serious limitations, including substantial heterogeneity among studies and co-occurrence of neurotoxicants in drinking water, hinder the strength of these findings and their application to US settings. The Academy of Nutrition and Dietetics has estimated that only limited evidence supports an association between fluoride content in water and the IQs of children (58). A prospective study in a New Zealand population-based cohort followed for nearly four decades found no association between fluoride exposure in the context of community water fluoridation programs and IQs measured during childhood and at age 38 years (80). A series of recent studies by a research group in Canada have raised concern over a possible link between higher maternal fluoride exposures during pregnancy and lower IQ (81-83), cognitive delay (84), and attention-deficit/hyperactivity disorder (85) in offspring. However, these studies have been widely criticized for shortcomings in measuring fluoride intake, only reporting significant relationships among subgroups, and being observational and subject to residual confounding. Thus, high-quality prospective studies with more definitive fluoride intake measures and better control of confounders are needed to determine whether fluoride might have neurotoxic effects at usual intake levels.
Acute toxicity
Fluoride is toxic when consumed in excessive amounts, so concentrated fluoride products should be used and stored with caution to prevent the possibility of acute fluoride poisoning, especially in children and other vulnerable individuals. The lowest dose that could trigger adverse symptoms is considered to be 5 mg/kg of body weight, with the lowest potentially fatal dose considered 15 mg/kg of body weight. Nausea, abdominal pain, and vomiting almost always accompany acute fluoride toxicity. Other symptoms like diarrhea, excessive salivation and tearing, sweating, and generalized weakness may also occur (77). In order to prevent acute fluoride poisoning, the American Dental Association has recommended that no more than 120 mg of fluoride (224 mg of sodium fluoride) be dispensed at one time (48). The use of high doses of fluoride to treat osteoporosis has been associated with some adverse effects, which are discussed in the Disease Treatment section above.
Dental fluorosis
Dental fluorosis, also called enamel fluorosis, is a result of excess fluoride intake during the period of tooth formation, with the critical window of susceptibility being the first eight years of life, as this corresponds to the eruption of the permanent teeth (86). Once the tooth enamel has formed, dental fluorosis cannot develop (87). The mildest form of dental fluorosis is detectable only to the trained observer and is characterized by small opaque white flecks or spots on the enamel of the teeth. Moderate dental fluorosis is characterized by mottling and mild staining of the teeth, and severe dental fluorosis results in marked staining and pitting of the teeth. In its moderate to severe forms, dental fluorosis becomes a cosmetic concern when it affects the incisors and canines (front teeth). Dental fluorosis is a dose-dependent condition, with higher fluoride intakes being associated with more pronounced effects on the teeth.
The incidence of mild and moderate dental fluorosis has increased over the past decades, mainly due to increasing fluoride intake from reconstituted infant formula and toothpaste, although inappropriate use of fluoride supplements may also contribute (61). According to a US national survey, the National Health and Nutrition Examination Survey (NHANES) 1999-2004, 23% of people aged 6 to 49 years (n=16,051) had some degree of dental fluorosis (88). National data from NHANES 2011-2012 found that the prevalence of fluorosis among children and adolescents (n=2,283), ages 6 to 19 years, was much higher at 57% (89); however, a quality assessment of these data raised concerns about their validity and suggested such results were not biologically plausible (90). In 1997, the US Food and Nutrition Board (FNB) of the Institute of Medicine set the tolerable upper intake level (UL) for fluoride based on the prevention of moderate enamel fluorosis (Table 4) (6).
The current US EPA maximum allowable level of fluoride in drinking water is 4 mg/L; the EPA also has a non-enforceable standard fluoride level of 2 mg/L to prevent moderate dental fluorosis (91). The EPA recently conducted a six-year review of drinking water standards and concluded that the limit of fluoride in drinking water is not a candidate for regulatory revision at this time (92).
Skeletal fluorosis
Intake of fluoride at excessive levels for long periods of time may lead to changes in bone structure known as skeletal fluorosis. The early stages of skeletal fluorosis are characterized by increased bone mass, detectable by x-ray. If very high fluoride intake persists over many years, joint pain and stiffness may result from the skeletal changes. The most severe form of skeletal fluorosis is known as "crippling skeletal fluorosis," which may result in calcification of ligaments, immobility, muscle wasting, and neurological problems related to spinal cord compression. While skeletal fluorosis is endemic in many world regions with naturally high fluoride concentrations in drinking water, crippling skeletal fluorosis may occur only when fluoride intake exceeds 10 mg/day for at least 10 years (6, 93). Rare cases of skeletal fluorosis in the US have been observed in consumers of large volumes of tea (94-97). Because of the potential risk for skeletal fluorosis, as well as the risks for pitting of tooth enamel and bone fracture, the US EPA set the maximum level of fluoride allowed in drinking water at 4 mg/L (98). The agency also recommended limiting fluoride in drinking water at 2 mg/L to prevent dental fluorosis in children; however, this is only a guideline and not enforceable by law (see the US EPA website).
Drug interactions
Calcium supplements, as well as calcium and aluminum-containing antacids, can decrease the absorption of fluoride. It is best to take these products two hours before or after fluoride supplements (99).
Linus Pauling Institute Recommendation
The safety and public health benefits of optimally fluoridated water for prevention of tooth decay in people of all ages have been well established. The Linus Pauling Institute supports the recommendations of the American Dental Association and the Centers for Disease Control and Prevention, which include optimally fluoridated water and the use of fluoride toothpaste, fluoride mouth rinse, fluoride varnish, and when necessary, fluoride supplementation. Due to the risk of fluorosis, any fluoride supplementation should be prescribed and closely monitored by a dentist or physician.
Authors and Reviewers
Originally written in 2001 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2003 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in September 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in November 2013 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in November 2021 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in December 2021 by:
John J. Warren, D.D.S., M.S.
Professor
Preventive & Community Dentistry
College of Dentistry
The University of Iowa
Copyright 2001-2024 Linus Pauling Institute
References
1. Cerklewski FL. Fluoride bioavailability — nutritional and clinical aspects. Nutr Res. 1997;17:907-929.
2. Nielsen FH. Ultratrace minerals. In: Shils M, Olson JA, Shike M, Ross AC, eds. Modern Nutrition in Health and Disease. 9th ed. Baltimore: Williams & Wilkins; 1999:283-303.
3. Cerklewski FL. Fluoride — essential or just beneficial. Nutrition. 1998;14(5):475-476. (PubMed)
4. Cerklewski FL. Fluorine. In: O'Dell BL, Sunde RA, eds. Handbook of Nutritionally Essential Minerals. New York: Marcel Dekker, Inc.; 1997:583-602.
5. US Department of Health Human Services Federal Panel on Community Water Fluoridation. US Public Health Service Recommendation for Fluoride Concentration in Drinking Water for the Prevention of Dental Caries. Public Health Rep. 2015;130(4):318-331. (PubMed)
6. Food and Nutrition Board, Institute of Medicine. Fluoride. Dietary Reference Intakes: Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington D.C.: National Academy Press; 1997:288-313. (National Academy Press)
7. US Centers for Disease Control and Prevention. Achievements in public health, 1900-1999: fluoridation of drinking water to prevent dental caries. MMWR. 1999;48:933-940.
8. Demmer RT, Squillaro A, Papapanou PN, et al. Periodontal infection, systemic inflammation, and insulin resistance: results from the continuous National Health and Nutrition Examination Survey (NHANES) 1999-2004. Diabetes Care. 2012;35(11):2235-2242. (PubMed)
9. Demmer RT, Jacobs DR, Jr., Desvarieux M. Periodontal disease and incident type 2 diabetes: results from the First National Health and Nutrition Examination Survey and its epidemiologic follow-up study. Diabetes Care. 2008;31(7):1373-1379. (PubMed)
10. Desvarieux M, Demmer RT, Jacobs DR, Jr., et al. Periodontal bacteria and hypertension: the oral infections and vascular disease epidemiology study (INVEST). J Hypertens. 2010;28(7):1413-1421. (PubMed)
11. Bahekar AA, Singh S, Saha S, Molnar J, Arora R. The prevalence and incidence of coronary heart disease is significantly increased in periodontitis: a meta-analysis. Am Heart J. 2007;154(5):830-837. (PubMed)
12. Kriauciunas A, Gleiznys A, Gleiznys D, Januzis G. The influence of Porphyromonas gingivalis bacterium causing periodontal disease on the pathogenesis of rheumatoid arthritis: systematic review of literature. Cureus. 2019;11(5):e4775. (PubMed)
13. Perricone C, Ceccarelli F, Saccucci M, et al. Porphyromonas gingivalis and rheumatoid arthritis. Curr Opin Rheumatol. 2019;31(5):517-524. (PubMed)
14. Batty GD, Jung KJ, Mok Y, et al. Oral health and later coronary heart disease: Cohort study of one million people. Eur J Prev Cardiol. 2018;25(6):598-605. (PubMed)
15. Demmer RT, Desvarieux M. Periodontal infections and cardiovascular disease: the heart of the matter. J Am Dent Assoc. 2006;137 Suppl:14S-20S; quiz 38S. (PubMed)
16. Zoellner H. Dental infection and vascular disease. Semin Thromb Hemost. 2011;37(3):181-192. (PubMed)
17. DePaola DP, Faine MP, Palmer CA. Nutrition in relation to dental medicine. In: Shils M, Olson JA, Shike M, Ross AC, eds. Modern Nutrition in Health and Disease. 9th ed. Baltimore: Williams & Wilkins; 1999:1099-1124.
18. Whelton HP, Spencer AJ, Do LG, Rugg-Gunn AJ. Fluoride revolution and dental caries: evolution of policies for global use. J Dent Res. 2019;98(8):837-846. (PubMed)
19. Newbrun E. Effectiveness of water fluoridation. J Public Health Dent. 1989;49(5 Spec No):279-289. (PubMed)
20. Iheozor-Ejiofor Z, Worthington HV, Walsh T, et al. Water fluoridation for the prevention of dental caries. Cochrane Database Syst Rev. 2015(6):CD010856. (PubMed)
21. Dye BA, Tan S, Smith V, Lewis BG, Barker LK, Thornton-Evans G. Trends in oral health status: United States, 1988-1994 and 1999-2004. National Center for Health Statistics. Vital Health Stat 11(248); 2007.
22. Marinho VC, Worthington HV, Walsh T, Clarkson JE. Fluoride varnishes for preventing dental caries in children and adolescents. Cochrane Database Syst Rev. 2013;7:CD002279. (PubMed)
23. Chou R, Pappas M, Dana T, et al. Screening and interventions to prevent dental caries in children younger than 5 years: updated evidence report and systematic review for the US Preventive Services Task Force. JAMA. 2021;326(21):2179-2192. (PubMed)
24. Walsh T, Worthington HV, Glenny AM, Marinho VC, Jeroncic A. Fluoride toothpastes of different concentrations for preventing dental caries. Cochrane Database Syst Rev. 2019;3:CD007868. (PubMed)
25. Wright JT, Hanson N, Ristic H, Whall CW, Estrich CG, Zentz RR. Fluoride toothpaste efficacy and safety in children younger than 6 years: a systematic review. J Am Dent Assoc. 2014;145(2):182-189. (PubMed)
26. Magalhaes AC, Wiegand A, Rios D, Honorio HM, Buzalaf MA. Insights into preventive measures for dental erosion. J Appl Oral Sci. 2009;17(2):75-86. (PubMed)
27. Zini A, Krivoroutski Y, Vered Y. Primary prevention of dental erosion by calcium and fluoride: a systematic review. Int J Dent Hyg. 2014;12(1):17-24. (PubMed)
28. Krall EA, Dawson-Hughes B. Osteoporosis. In: Shils M, Olson JA, Shike M, Ross AC, eds. Modern Nutrition in Health and Disease. 9th ed. Baltimore: Williams & Wilkins; 1999:1353-1364.
29. Lee N, Kang S, Lee W, Hwang SS. The association between community water fluoridation and bone diseases: a natural experiment in Cheongju, Korea. Int J Environ Res Public Health. 2020;17(24):9170. (PubMed)
30. Fabiani L, Leoni V, Vitali M. Bone-fracture incidence rate in two Italian regions with different fluoride concentration levels in drinking water. J Trace Elem Med Biol. 1999;13(4):232-237. (PubMed)
31. Lehmann R, Wapniarz M, Hofmann B, Pieper B, Haubitz I, Allolio B. Drinking water fluoridation: bone mineral density and hip fracture incidence. Bone. 1998;22(3):273-278. (PubMed)
32. Sowers M, Whitford GM, Clark MK, Jannausch ML. Elevated serum fluoride concentrations in women are not related to fractures and bone mineral density. J Nutr. 2005;135(9):2247-2252. (PubMed)
33. Nasman P, Ekstrand J, Granath F, Ekbom A, Fored CM. Estimated drinking water fluoride exposure and risk of hip fracture: a cohort study. J Dent Res. 2013;92(11):1029-1034. (PubMed)
34. Yin XH, Huang GL, Lin DR, et al. Exposure to fluoride in drinking water and hip fracture risk: a meta-analysis of observational studies. PLoS One. 2015;10(5):e0126488. (PubMed)
35. Levy SM, Eichenberger-Gilmore J, Warren JJ, et al. Associations of fluoride intake with children's bone measures at age 11. Community Dent Oral Epidemiol. 2009;37(5):416-426. (PubMed)
36. Levy SM, Warren JJ, Phipps K, et al. Effects of life-long fluoride intake on bone measures of adolescents: a prospective cohort study. J Dent Res. 2014;93(4):353-359. (PubMed)
37. Oweis RR, Levy SM, Eichenberger-Gilmore JM, et al. Fluoride intake and cortical and trabecular bone characteristics in adolescents at age 17: A prospective cohort study. Community Dent Oral Epidemiol. 2018;46(6):527-534. (PubMed)
38. Saha PK, Oweis RR, Zhang X, et al. Effects of fluoride intake on cortical and trabecular bone microstructure at early adulthood using multi-row detector computed tomography (MDCT). Bone. 2021;146:115882. (PubMed)
39. Cesar Libanati K-H. Fluoride therapy for osteoporosis. In: Marcus R, ed. Osteoporosis. San Diego: Academic Press; 1996:1259-1277.
40. Haguenauer D, Welch V, Shea B, Tugwell P, Adachi JD, Wells G. Fluoride for the treatment of postmenopausal osteoporotic fractures: a meta-analysis. Osteoporos Int. 2000;11(9):727-738. (PubMed)
41. Riggs BL, Hodgson SF, O'Fallon WM, et al. Effect of fluoride treatment on the fracture rate in postmenopausal women with osteoporosis. N Engl J Med. 1990;322(12):802-809. (PubMed)
42. Lundy MW, Stauffer M, Wergedal JE, et al. Histomorphometric analysis of iliac crest bone biopsies in placebo-treated versus fluoride-treated subjects. Osteoporos Int. 1995;5(2):115-129. (PubMed)
43. Fields AJ, Keaveny TM. Trabecular architecture and vertebral fragility in osteoporosis. Curr Osteoporos Rep. 2012;10(2):132-140. (PubMed)
44. Balena R, Kleerekoper M, Foldes JA, et al. Effects of different regimens of sodium fluoride treatment for osteoporosis on the structure, remodeling and mineralization of bone. Osteoporos Int. 1998;8(5):428-435. (PubMed)
45. Vestergaard P, Jorgensen NR, Schwarz P, Mosekilde L. Effects of treatment with fluoride on bone mineral density and fracture risk — a meta-analysis. Osteoporos Int. 2008;19(3):257-268. (PubMed)
46. Reid IR, Cundy T, Grey AB, et al. Addition of monofluorophosphate to estrogen therapy in postmenopausal osteoporosis: a randomized controlled trial. J Clin Endocrinol Metab. 2007;92(7):2446-2452. (PubMed)
47. Grey A, Garg S, Dray M, et al. Low-dose fluoride in postmenopausal women: a randomized controlled trial. J Clin Endocrinol Metab. 2013;98(6):2301-2307. (PubMed)
48. American Dietetic Association. Position of the American Dietetic Association: the impact of fluoride on health. J Am Diet Assoc. 2001;101(1):126-132. (PubMed)
49. Murray TM, Ste-Marie LG. Prevention and management of osteoporosis: consensus statements from the Scientific Advisory Board of the Osteoporosis Society of Canada. 7. Fluoride therapy for osteoporosis. CMAJ. 1996;155(7):949-954. (PubMed)
50. Alexandersen P, Riis BJ, Christiansen C. Monofluorophosphate combined with hormone replacement therapy induces a synergistic effect on bone mass by dissociating bone formation and resorption in postmenopausal women: a randomized study. J Clin Endocrinol Metab. 1999;84(9):3013-3020. (PubMed)
51. US Department of Health and Human Services Federal Panel on Community Water Fluoridation. US Public health service recommendation for fluoride concentration in drinking water for the prevention of dental caries. Public Health Rep. 2015;130(4):318-31. (PubMed)
52. US Centers for Disease Control and Prevention. Community Water Fluoridation. Water Floridation Basics. Available at: https://www.cdc.gov/fluoridation/basics/index.htm. Accessed 11/19/21.
53. Cutrufelli R, Pehrsson P, Haytowitz D, Patterson K, Holden J. USDA National Fluoride Database of Selected Beverages and Foods, Release 2. Nutrient Data Laboratory, Beltsville Human Nutrition Research Center, Agricultural Research Service, U.S. Department of Agriculture; 2005.
54. Quock RL, Chan JT. Fluoride content of bottled water and its implications for the general dentist. Gen Dent. 2009;57(1):29-33. (PubMed)
55. Van Winkle S, Levy SM, Kiritsy MC, Heilman JR, Wefel JS, Marshall T. Water and formula fluoride concentrations: significance for infants fed formula. Pediatr Dent. 1995;17(4):305-310. (PubMed)
56. Tate WH, Chan JT. Fluoride concentrations in bottled and filtered waters. Gen Dent. 1994;42(4):362-366. (PubMed)
57. McGuire S. Fluoride content of bottled water. N Engl J Med. 1989;321(12):836-837. (PubMed)
58. Palmer CA, Gilbert JA, Academy of N, Dietetics. Position of the Academy of Nutrition and Dietetics: the impact of fluoride on health. J Acad Nutr Diet. 2012;112(9):1443-1453. (PubMed)
59. Marshall TA, Levy SM, Warren JJ, Broffitt B, Eichenberger-Gilmore JM, Stumbo PJ. Associations between Intakes of fluoride from beverages during infancy and dental fluorosis of primary teeth. J Am Coll Nutr. 2004;23(2):108-116. (PubMed)
60. Pendrys DG. Risk of enamel fluorosis in nonfluoridated and optimally fluoridated populations: considerations for the dental professional. J Am Dent Assoc. 2000;131(6):746-755. (PubMed)
61. Levy SM, Broffitt B, Marshall TA, Eichenberger-Gilmore JM, Warren JJ. Associations between fluorosis of permanent incisors and fluoride intake from infant formula, other dietary sources and dentifrice during early childhood. J Am Dent Assoc. 2010;141(10):1190-1201. (PubMed)
62. Levy SM, Kohout FJ, Guha-Chowdhury N, Kiritsy MC, Heilman JR, Wefel JS. Infants' fluoride intake from drinking water alone, and from water added to formula, beverages, and food. J Dent Res. 1995;74(7):1399-1407. (PubMed)
63. Siew C, Strock S, Ristic H, et al. Assessing a potential risk factor for enamel fluorosis: a preliminary evaluation of fluoride content in infant formulas. J Am Dent Assoc. 2009;140(10):1228-1236. (PubMed)
64. Fein NJ, Cerklewski FL. Fluoride content of foods made with mechanically separated chicken. J Agric Food Chem. 2001;49(9):4284-4286. (PubMed)
65. Kiritsy MC, Levy SM, Warren JJ, Guha-Chowdhury N, Heilman JR, Marshall T. Assessing fluoride concentrations of juices and juice-flavored drinks. J Am Dent Assoc. 1996;127(7):895-902. (PubMed)
66. Rozier RG, Adair S, Graham F, et al. Evidence-based clinical recommendations on the prescription of dietary fluoride supplements for caries prevention: a report of the American Dental Association Council on Scientific Affairs. J Am Dent Assoc. 2010;141(12):1480-1489. (PubMed)
67. US Centers for Disease Control and Prevention. Recommendations for using fluoride to prevent and control dental caries in the United States. MMWR Recomm Rep. 2001;50(RR-14):1-42.
68. Section on Pediatric Dentistry and Oral Health. Preventive oral health intervention for pediatricians. Pediatrics. 2008;122(6):1387-1394. (PubMed)
69. American Dental Association Council on Scientific Affairs. Fluoride toothpaste use for young children. J Am Dent Assoc. 2014;145(2):190-191. (PubMed)
70. Clark MB, Keels MA, Slayton RL, Section On Oral Health. Fluoride use in caries prevention in the primary care setting. Pediatrics. 2020;146(6): e2020034637. (PubMed)
71. Falcao A, Tenuta LM, Cury JA. Fluoride gastrointestinal absorption from Na2FPO3/CaCO3- and NaF/SiO2-based toothpastes. Caries Res. 2013;47(3):226-233. (PubMed)
72. O'Mullane DM, Baez RJ, Jones S, et al. Fluoride and oral health. Community Dent Health. 2016;33(2):69-99. (PubMed)
73. Pollick HF. Salt fluoridation: a review. J Calif Dent Assoc. 2013;41(6):395-397, 400-394. (PubMed)
74. Marthaler TM, Petersen PE. Salt fluoridation — an alternative in automatic prevention of dental caries. Int Dent J. 2005;55(6):351-358. (PubMed)
75. Moss ME, Zero DT. Fluoride and Caries Prevention. In: Mascarenhas AK, Okunseri C, Dye BA, eds. Burt and Eklund’s Dentistry, Dental Practice and the Community. 7th ed. St. Louis: Elsevier; 2021:277-295.
76. Committee on Fluoride in Drinking Water National Research Council. Fluoride in drinking water: a scientific review of EPA’s Standards. Washington D.C.: National Academies Press; 2006. (The National Academies Press)
77. Whitford GM. Acute toxicity of ingested fluoride. Monogr Oral Sci. 2011;22:66-80. (PubMed)
78. Choi AL, Sun G, Zhang Y, Grandjean P. Developmental fluoride neurotoxicity: a systematic review and meta-analysis. Environ Health Perspect. 2012;120(10):1362-1368. (PubMed)
79. Duan Q, Jiao J, Chen X, Wang X. Association between water fluoride and the level of children's intelligence: a dose-response meta-analysis. Public Health. 2018;154:87-97. (PubMed)
80. Broadbent JM, Thomson WM, Ramrakha S, et al. Community Water Fluoridation and Intelligence: Prospective Study in New Zealand. Am J Public Health. 2015;105(1):72-76. (PubMed)
81. Bashash M, Thomas D, Hu H, et al. Prenatal fluoride exposure and cognitive outcomes in children at 4 and 6-12 years of age in Mexico. Environ Health Perspect. 2017;125(9):097017. (PubMed)
82. Green R, Lanphear B, Hornung R, et al. Association between maternal fluoride exposure during pregnancy and IQ scores in offspring in Canada. JAMA Pediatr. 2019;173(10):940-948. (PubMed)
83. Xu K, An N, Huang H, et al. Fluoride exposure and intelligence in school-age children: evidence from different windows of exposure susceptibility. BMC Public Health. 2020;20(1):1657. (PubMed)
84. Valdez Jimenez L, Lopez Guzman OD, Cervantes Flores M, et al. In utero exposure to fluoride and cognitive development delay in infants. Neurotoxicology. 2017;59:65-70. (PubMed)
85. Bashash M, Marchand M, Hu H, et al. Prenatal fluoride exposure and attention deficit hyperactivity disorder (ADHD) symptoms in children at 6-12 years of age in Mexico City. Environ Int. 2018;121(Pt 1):658-666. (PubMed)
86. Buzalaf MAR. Review of fluoride intake and appropriateness of current guidelines. Adv Dent Res. 2018;29(2):157-166. (PubMed)
87. Pollick H. The role of fluoride in the prevention of tooth decay. Pediatr Clin North Am. 2018;65(5):923-940. (PubMed)
88. Beltrán-Aguilar ED, Barker L, Dye BA. Prevalence and severity of dental fluorosis in the United States, 1999-2004. NCHS data brief, no 53. Hyattsville, MD: National Center for Health Statistics; 2010.
89. Neurath C, Limeback H, Osmunson B, Connett M, Kanter V, Wells CR. Dental fluorosis trends in US oral health surveys: 1986 to 2012. JDR Clin Trans Res. 2019;4(4):298-308. (PubMed)
90. National Center for Health Statistics, National Center for Chronic Disease Prevention and Health Promotion. Data quality evaluation of the dental fluorosis clinical assessment data from the National Health and Nutrition Examination Survey, 1999–2004 and 2011–2016. National Center for Health Statistics. Vital Health Stat 2(183); 2019.
91. US Environmental Protection Agency. New fluoride risk assessment and relative source contribution documents; 2011.
92. US Environmental Protection Agency. Six-year review 3 of drinking water standards. Available at: https://www.epa.gov/dwsixyearreview/six-year-review-3-drinking-water-standards. Accessed 11/22/21.
93. Whitford GM. The Metabolism and Toxicity of Fluoride. Basel: S. Karger AG; 1996.
94. Hallanger Johnson JE, Kearns AE, Doran PM, Khoo TK, Wermers RA. Fluoride-related bone disease associated with habitual tea consumption. Mayo Clin Proc. 2007;82(6):719-724. (PubMed)
95. Whyte MP, Totty WG, Lim VT, Whitford GM. Skeletal fluorosis from instant tea. J Bone Miner Res. 2008;23(5):759-769. (PubMed)
96. Izuora K, Twombly JG, Whitford GM, Demertzis J, Pacifici R, Whyte MP. Skeletal fluorosis from brewed tea. J Clin Endocrinol Metab. 2011;96(8):2318-2324. (PubMed)
97. Kakumanu N, Rao SD. Images in clinical medicine. Skeletal fluorosis due to excessive tea drinking. N Engl J Med. 2013;368(12):1140. (PubMed)
98. US Environmental Protection Agency. Fact sheet: Questions and answers on fluoride. January 2011. Available at: https://www.epa.gov/sites/default/files/2015-10/documents/2011_fluoride_questionsanswers.pdf. Accessed 11/22/21.
99. Minerals. Drug Facts and Comparisons. St. Louis: Facts and Comparisons; 2000:27-51.
Iodine
Contents
Summary
- Iodine is a key component of thyroid hormones, which are required throughout life for normal growth, neurological development, and metabolism. (More information)
- Insufficient iodine intake impairs the production of thyroid hormones, leading to a condition called hypothyroidism. Iodine deficiency results in a range of adverse health disorders with varying degrees of severity, from thyroid gland enlargement (goiter) to severe physical and mental retardation known as cretinism. (More information)
- Iodine deficiency-induced hypothyroidism has adverse effects in all stages of development but is most damaging to the developing brain. Maternal iodine deficiency during pregnancy can result in maternal and fetal hypothyroidism, as well as miscarriage, preterm birth, and neurological impairments in offspring. (More information)
- Even in areas with voluntary/mandatory iodization programs and in iodine-replete countries, pregnant women, lactating mothers, and young infants are among the most vulnerable to iodine deficiency due to their special requirements during these life stages. (More information)
- The recommended dietary allowance (RDA) for iodine intake is 150 micrograms (μg)/day in adults, 220 μg/day in pregnant women, and 290 μg/day in breast-feeding women. During pregnancy and lactation, the fetus and infant are entirely reliant on maternal iodine intake for thyroid hormone synthesis. (More information)
- Thyroid accumulation of radioactive iodine (131I) increases the risk of developing thyroid cancer, especially in children. In case of radiation emergencies, current preventive measures include the distribution of pharmacologic doses of potassium iodide that would reduce the risk of significant uptake of 131I by the thyroid gland. (More information)
- Seafood is an excellent source of dietary iodine. Dairy products, grains, eggs, and poultry contribute substantially to dietary iodine intakes in the US. (More information)
- More than 120 countries worldwide have introduced programs of salt fortification with iodine in order to correct iodine deficiency in populations. (More information)
- In iodine-deficient populations, a rapid increase in iodine intake may precipitate iodine-induced hyperthyroidism. The risk of iodine-induced hyperthyroidism is especially high in older people with multi-nodular goiter. (More information)
- In iodine-sufficient adults, long-term iodine intake above the tolerable upper intake level (UL) of 1,100 μg/day may increase the risk of thyroid disorders, including iodine-induced goiter and hypothyroidism. (More information)
Iodine (I), a non-metallic trace element, is required by humans for the synthesis of thyroid hormones. Iodine deficiency is an important health problem throughout much of the world. Most of the Earth's iodine, in the form of the iodide ion (I-), is found in oceans, and iodine content in the soil varies with region. The older an exposed soil surface, the more likely the iodine has been leached away by erosion. Mountainous regions, such as the Himalayas, Atlas, Andes, and Alps; flooded river valleys, such as the Ganges River plain in India; and many inland regions, such as central Asia and Africa, central and eastern Europe, and the Midwestern region of North America are among the most severely iodine-deficient areas in the world (1).
Function
Iodine is an essential component of the thyroid hormones, triiodothyronine (T3) and thyroxine (T4), and is therefore essential for normal thyroid function. To meet the body's demand for thyroid hormones, the thyroid gland traps iodine from the blood and incorporates it into the large (660 kDa) glycoprotein thyroglobulin. The hydrolysis of thyroglobulin by lysosomal enzymes gives rise to thyroid hormones that are stored and released into the circulation when needed. In target tissues, such as the liver and the brain, T4 (the most abundant circulating thyroid hormone) can be converted to T3 by selenium-containing enzymes known as iodothyronine deiodinases (DIOs) (Figure 1; see also Nutrient interactions). T3 is the physiologically active thyroid hormone that can bind to thyroid receptors in the nuclei of cells and regulate gene expression. In this manner, thyroid hormones regulate a number of physiologic processes, including growth, development, metabolism, and reproductive function (2).
The regulation of thyroid function is a complex process that involves the hypothalamus and the pituitary gland. In response to thyrotropin-releasing hormone (TRH) secretion by the hypothalamus, the pituitary gland secretes thyroid-stimulating hormone (TSH), which stimulates iodine trapping, thyroid hormone synthesis, and release of T4 and T3 by the thyroid gland. The presence of adequate circulating T4 and T3 feeds back at the level of both the hypothalamus and pituitary, decreasing TRH and TSH production (Figure 2). When circulating T4 levels decrease, the pituitary gland increases its secretion of TSH, resulting in increased iodine trapping, as well as increased production and release of both T3 and T4. Iodine deficiency results in inadequate production of T4. In response to decreased blood T4 concentrations, the pituitary gland increases its output of TSH. Persistently elevated TSH levels may lead to hypertrophy (enlargement) of the thyroid gland, also known as goiter (see Deficiency) (3).

Deficiency
The thyroid gland of a healthy adult concentrates 70-80% of a total body iodine content of 15-20 mg and utilizes about 80 μg of iodine daily to synthesize thyroid hormones. In contrast, chronic iodine deficiency can result in a dramatic reduction of the iodine content in the thyroid well below 1 mg (1). Iodine deficiency is recognized as the most common cause of preventable brain damage in the world. The spectrum of iodine deficiency disorders (IDD) includes mental retardation, hypothyroidism, goiter, and varying degrees of other growth and developmental abnormalities (4). The World Health Organization (WHO) estimated that over 30% of the world’s population (2 billion people) have insufficient iodine intake as measured by median urinary iodine concentrations below 100 μg/L (5). Moreover, about one-third of school-age children (6-12 years old) worldwide (241 million children in 2011) have insufficient iodine intake (6, 7). Major international efforts have produced dramatic improvements in the correction of iodine deficiency in the 1990s, mainly through the use of iodized salt in iodine-deficient countries (4). Although about 70% of households in the world now have access to iodized salt (8), mild-to-moderate iodine deficiency remains a public health concern in at least 30 countries; there are no iodine excretion data available for 42 other countries, including Israel, Syria, and Sierra Leone (7). For more information on the international effort to eradicate iodine deficiency, visit the websites of the Iodine Global Network (formerly the International Council for the Control of Iodine Deficiency Disorders) and the WHO.
Biomarkers of iodine status
More than 90% of ingested iodine is excreted in the urine within 24-48 hours such that daily iodine intakes in a population can be extrapolated from measures of median spot urinary iodine concentrations (9, 10). According to WHO criteria, population iodine deficiency is defined by median urinary iodine concentrations lower than 150 micrograms (μg)/L for pregnant women and 100 μg/L for all other groups (Table 1). Adequate intakes correspond to median urinary iodine concentrations of 100-199 μg/L in school-age children and 150-249 μg/L in pregnant women (Table 1). While median urinary iodine concentration is a population indicator of recent dietary iodine intake, multiple collections of 24-hour urinary iodine are preferable to estimate intake in individuals (9-11).
| Population Group | Median/Range of Urinary Iodine Concentrations (μg/L) | Iodine Intake |
|---|---|---|
| Children (<2 years) | <100 | Insufficient |
| ≥100 | Adequate | |
| Children (≥6 years), adolescents, and adults* | <100 | Insufficient |
| 100-199 | Adequate | |
| 200-299 | More than adequate | |
| >300 | Excessive | |
| Pregnant women | <150 | Insufficient |
| 150-249 | Adequate | |
| 250-499 | More than adequate | |
| ≥500 | Excessive | |
| Breast-feeding women# | <100 | Insufficient |
| ≥100 | Adequate | |
|
*Excludes pregnant or lactating women. |
||
In many countries, serum TSH concentration is used in the screening for congenital hypothyroidism in newborns. Newborn TSH can be used as an indicator of population iodine status. Yet, in older children and adults, serum TSH is not a sensitive indicator of iodine status as concentrations are usually maintained within a normal range despite frank iodine deficiency (12). Serum thyroglobulin concentration in school-age children is a sensitive marker of iodine status in populations (13). In areas of endemic goiter, changes in thyroid size reflect long-term iodine nutrition (months to years). Assessment of the goiter rate in a population is used to define the severity of iodine deficiency, as well as to monitor the long-term impact of sustained salt iodization programs (4, 10). Finally, serum thyroid hormone concentrations do not adequately reflect iodine nutrition in populations (1).
Iodine deficiency disorders
All the adverse effects of iodine deficiency in animals and humans are collectively termed iodine deficiency disorders (reviewed in 1). Thyroid enlargement, or goiter, is one of the earliest and most visible signs of iodine deficiency. It is a physiologic adaptation of the thyroid gland in response to persistent stimulation by TSH (see Function). In mild iodine deficiency, thyroid enlargement may be enough to maximize the uptake of available iodine and provide the body with sufficient thyroid hormones. Yet, large goiters can obstruct the trachea and esophagus and damage the recurrent laryngeal nerves.
More severe cases of iodine deficiency result in impaired thyroid hormone synthesis known as hypothyroidism. Adequate iodine intake will generally reduce the size of goiters, but the reversibility of the effects of hypothyroidism depends on an individual's life stage. Iodine deficiency-induced hypothyroidism has adverse effects in all stages of development but is most damaging to the developing brain. In addition to regulating many aspects of growth and development, thyroid hormones are important for the migration, proliferation, and differentiation of specific neuronal populations, the overall architecture of the brain’s cortex, the formation of axonal connections, and the myelination of the central nervous system, which occurs both before and shortly after birth (reviewed in 14).
The effects of iodine deficiency at different life stages are discussed below.
Pregnancy and lactation
Daily iodine requirements are significantly increased in pregnant and breast-feeding women because of (1) the increased thyroid hormone production and transfer to the fetus in early pregnancy before the fetal thyroid gland becomes functional, (2) iodine transfer to the fetus during late gestation, (3) increased urinary iodine excretion, and (4) iodine transfer to the infant via breast milk (see also The RDA) (12, 15).
During pregnancy, the size of the thyroid gland is increased by 10% in women residing in iodine-sufficient regions and increased by 20%-40% in those living in iodine-deficient regions (16). Iodine deficiency during pregnancy can result in hypothyroidism in women. Maternal hypothyroidism has been associated with increased risk for preeclampsia, miscarriage, stillbirth, preterm birth, and low-birth-weight infants (reviewed in 16). In addition, severe iodine deficiency during pregnancy may result in congenital hypothyroidism and neurocognitive deficits in the offspring (see Prenatal development) (12).
Iodine-deficient women who are breast-feeding may not be able to provide sufficient iodine to their infants who are particularly vulnerable to the effects of iodine deficiency (see Newborns and infants) (17). A daily prenatal supplement of 150 μg of iodine, as recommended by the American Thyroid Association (ATA) (16), will help to ensure that US pregnant and breast-feeding women consume sufficient iodine during these critical periods. In iodine-deficient areas where iodized salt is not available, the Iodine Global Network (IGN; formerly the International Council for the Control of Iodine Deficiency Disorders), the World Health Organization (WHO), and UNICEF recommend that lactating women receive a single annual dose of 400 mg of iodine (or 250 μg/day) and exclusively breast-feed for at least six months. When breast-feeding is not possible, direct supplementation of the infant (<2 years old) with a single annual dose of 200 mg of iodine (or 90 μg/day) is advised (4). A randomized and placebo-controlled trial recently demonstrated that maternal supplementation (with a single 400-mg dose of iodine) improved the iodine status of breast-fed infants more efficiently than direct infant supplementation (with a single 100 mg-dose of iodine) for a period of at least six months (18). Yet, supplementation of lactating women failed to increase maternal urinary iodine concentrations above 100 μg/L, suggesting that supplemented mothers remained deficient in iodine (18).
Prenatal development
Fetal iodine deficiency is caused by iodine deficiency in the mother (see Pregnancy and lactation). During pregnancy, before the fetal thyroid gland becomes functional at 16-20 weeks’ gestation, maternal thyroxine (T4) crosses the placenta to promote normal embryonic and fetal development. Hence, maternal iodine deficiency and hypothyroidism can result in adverse pregnancy complications, including fetal loss, placental abruption, preeclampsia, preterm delivery, and congenital hypothyroidism in the offspring (16). The effects of maternal hypothyroidism on the offspring depend on the timing and severity of in utero iodine deficiency. A severe form of congenital hypothyroidism may lead to cretinism, a condition associated with irreversible mental retardation. The clinical picture of neurological cretinism in the offspring includes severe mental and physical retardation, deafness, mutism, and motor spasticity.
A myxedematous form of cretinism has been associated with coexisting iodine and selenium deficiency in central Africa (see Nutrient interactions) and is characterized by a less severe degree of mental retardation than in neurological cretinism. Yet, affected individuals exhibit all the features of severe hypothyroidism, including severe growth retardation and delayed sexual maturation (12). Two longitudinal cohort studies (one in the UK and one in Australia) recently observed that even mild-to-moderate iodine deficiency during pregnancy was associated with reduced scores of IQ and various measures of literacy performance in children 8 to 9 years of age (19, 20).
Newborns and infants (up to one year of age)
Infant mortality is higher in areas of severe iodine deficiency than in iodine-replete regions, and several studies have demonstrated an increase in childhood survival upon correction of the iodine deficiency (8, 21, 22). Infancy is a period of rapid brain growth and development. Sufficient thyroid hormone, which depends on adequate iodine intake, is essential for normal brain development. Even in the absence of congenital hypothyroidism, iodine deficiency during infancy may result in abnormal brain development and, consequently, impaired intellectual development (23, 24).
Children and adolescents
Iodine deficiency in children and adolescents is often associated with goiter. The incidence of goiter peaks in adolescence and is more common in girls than boys. School-age children in iodine-deficient areas show poorer school performance, lower IQs, and a higher incidence of learning disabilities than matched groups from iodine-sufficient areas. Three meta-analyses of mainly cross-sectional studies concluded that chronic iodine deficiency was associated with reduced mean IQ scores by 7-13.5 points in participants (primarily children) (25-27). However, these observational studies did not distinguish between iodine deficiency during pregnancy and during childhood, and such observational studies may be confounded by social, economic, and educational factors that influence child development.
Adults
Inadequate iodine intake may also result in goiter and hypothyroidism in adults. Although the effects of hypothyroidism are more subtle in the brains of adults than children, research suggests that hypothyroidism results in poor social and economic achievements due to low educability, apathy, and reduced work productivity (28). Other symptoms of hypothyroidism in adults include fatigue, weight gain, cold intolerance, and constipation.
Finally, because iodine deficiency induces an increase in the iodine trapping capacity of the thyroid, iodine-deficient individuals of all ages are more susceptible to radiation-induced thyroid cancer (see Disease Prevention), as well as to iodine-induced hyperthyroidism after an increase in iodine intakes (see Safety) (2).
Individuals and populations at risk of iodine deficiency
While the risk of iodine deficiency for populations living in iodine-deficient areas without adequate iodine fortification programs is well recognized, concerns have been raised that certain subpopulations in countries considered iodine-sufficient may not consume adequate iodine (7, 29). The greater use of methods assessing iodine status (see Biomarkers of iodine status) has shown that iodine deficiency also occurs in areas where the prevalence of goiter is low, in coastal areas, in highly developed countries, and in regions where iodine deficiency was previously eliminated (4).
The US is currently considered to be iodine-sufficient. Yet, in recent years, dietary intakes of iodine in the US population have decreased. Data from the latest US National Health and Nutrition Examination Survey (NHANES 2009-2010) indicated that the median urinary iodine concentration for the general population was 144 μg/L compared to 164 μg/L reported in previous assessments (NHANES 2005-2006 and 2007-2008) (30, 31). In addition to regional differences across the US, ethnic variations have been found. In all age groups, median urinary iodine concentrations were shown to be lower in African Americans than in Hispanics and Caucasians.
In addition, median urinary iodine concentrations in nonpregnant women of childbearing age and pregnant women indicate that mild iodine deficiency has re-emerged in the US in recent years (31).
Nonpregnant women
Data from US NHANES 2007-2010 indicated that 37.3% of nonpregnant women (ages 15-44 years) had urinary iodine concentrations lower than 100 μg/L, reflecting potentially insufficient iodine intakes (see Biomarkers of iodine status) (31). Only one-fifth of nonpregnant women reported using iodine-containing supplements in an earlier NHANES (2001-2006) (32). Yet, adequate intakes of iodine in women of childbearing age (150 μg/day; see The RDA) are essential for optimum stores of iodine, especially if they are considering pregnancy. Some experts suggested a daily consumption of 250 μg of iodine before conception to ensure adequate thyroid hormone production and iodine supply to the embryo and fetus during pregnancy (see Pregnancy and lactation) (12).
Pregnant women
There are no statistics on the global burden of iodine deficiency in pregnant women, but national and regional data suggest that this group is especially vulnerable. Given the increased iodine requirements during pregnancy, the median urinary iodine concentration should be at least of 150 μg/L (see Biomarkers of iodine status). Pooled data from NHANES 2005-2010 reported that US pregnant women had a median urinary iodine concentration of 129 μg/L, and the lowest median concentration (109 μg/L) was observed during the first trimester of gestation, when the embryo/fetus relies exclusively on maternal thyroid hormones (31).
Breast-feeding women
While data regarding the iodine status of breast-feeding women in the US are limited, dietary intakes that were inadequate during pregnancy are likely to be insufficient in a significant fraction of breast-feeding women (33, 34). A systematic review of the literature recently reported suboptimal dietary iodine intakes in breast-feeding women in some countries with a mandatory fortification program, including Denmark, Australia, and India (35). The American Thyroid Association (ATA) recommends that all North American women who are pregnant or breast-feeding supplement their dietary iodine intake with 150 μg/day of iodine (36).
Breast-fed and weaning infants
The body of a healthy newborn contains only about 300 μg of iodine, which makes newborns extremely vulnerable to iodine deficiency (28), and breast-fed infants are entirely reliant on maternal iodine intakes for thyroid hormone synthesis. Even in areas covered by a salt iodization program, weaning infants are at high risk of iodine deficiency, especially if they are not receiving iodine-containing infant formula (17).
Individuals consuming special diets
Diets that exclude iodized salt, fish, and seaweed have been found to contain very little iodine (9). Individuals consuming branded weight-loss foods may also be at risk of inadequate intakes (37). A small US cross-sectional study in 78 vegetarians and 63 vegans reported median urinary iodine concentrations of 147 μg/L and 78.5 μg/L, respectively, suggesting inadequate iodine intakes among vegans (38). Two cases of goiter and/or hypothyroidism have also been recently reported in children following restrictive diets to control esophageal inflammation (eosinophilic esophagitis) (39) or allergies (40).
Patients requiring parenteral nutrition
Although iodine is usually not added to parenteral nutrition (PN) solutions, topical iodine-containing disinfectants and other adventitious sources provide substantial amounts of iodine to some PN patients such that the occurrence of iodine deficiency is unlikely. Yet, deficiency might occur, especially in preterm infants with limited body stores, if chlorhexidine-based antiseptics replace iodinated antiseptics (28, 41).
Nutrient interactions
Concurrent deficiencies in selenium, iron, or vitamin A may exacerbate the effects of iodine deficiency (reviewed in 42).
Selenium
While iodine is an essential component of thyroid hormones, the selenium-containing iodothyronine deiodinases (DIOs) are enzymes (or selenoenzymes) required for the conversion of T4 to the biologically active thyroid hormone, T3 (see the article on Selenium). DIO1 activity may also be involved in regulating iodine homeostasis (43). In addition, glutathione peroxidases are selenoenzymes that protect the thyroid gland from hydrogen peroxide-induced damage during thyroid hormone synthesis (44). A randomized, placebo-controlled study in 151 pregnant women at risk of developing autoimmune thyroid disease found that selenium supplementation (200 μg/day in the form of selenomethionine) at 12 weeks of gestation until 12 months’ postpartum reduced the risk of thyroid dysfunction and permanent hypothyroidism (45). However, another trial (the Selenium in Pregnancy Intervention Trial) found no benefit of selenium supplementation (60 μg/day from 12-14 weeks of gestation to delivery) over placebo on circulating autoantibody concentrations in pregnant women mildly deficient in iodine (46).
The epidemiology of coexisting iodine and selenium deficiencies in central Africa has been linked to the prevalence of myxedematous cretinism, a severe form of congenital hypothyroidism accompanied by mental and physical retardation. Selenium deficiency may be only one of several undetermined factors that might exacerbate the detrimental effects of iodine deficiency (42). Besides, results from randomized controlled intervention trials have shown that correcting only the selenium deficiency may have a deleterious effect on thyroid hormone metabolism in school-age children with co-existing selenium and iodine deficiency (47, 48). Finally, selenium deficiency in rodents was found to have little impact on DIO activities as it appears that selenium is being supplied in priority for adequate synthesis of DIOs at the expense of other selenoenzymes (44).
Iron
Severe iron-deficiency anemia can impair thyroid metabolism in the following ways: (1) by altering the TSH response of the pituitary gland; (2) by reducing the activity of thyroid peroxidase that catalyzes the iodination of thyroglobulin for the production of thyroid hormones; and (3) in the liver by limiting the conversion of T4 to T3, increasing T3 turnover, and decreasing T3 binding to nuclear receptors (49). It is estimated that goiter and iron-deficiency anemia coexist in up to 25% of school-age children in west and north Africa (42). A randomized controlled study in iron-deficient children with goiter showed a greater reduction in thyroid size following the consumption of iodized salt together with 60 mg/day of iron four times per week compared to placebo (50). Additional interventions have confirmed that correcting iron-deficiency anemia improved the efficacy of iodine supplementation to mitigate thyroid disorders (reviewed in 42, 49).
Vitamin A
In north and west Africa, vitamin A deficiency and iodine deficiency-induced goiter may coexist in up to 50% of children. Vitamin A status, like other nutritional factors, appears to influence the response to iodine prophylaxis in iodine-deficient populations (51). Vitamin A deficiency in animal models was found to interfere with the pituitary-thyroid axis by (1) increasing the synthesis and secretion of thyroid-stimulating hormone (TSH) by the pituitary gland, (2) increasing the size of the thyroid gland, (3) reducing iodine uptake by the thyroid gland and impairing the synthesis and iodination of thyroglobulin, and (4) increasing circulating concentrations of thyroid hormones (reviewed in 52). A cross-sectional study of 138 children with concurrent vitamin A and iodine deficiencies found that the severity of vitamin A deficiency was associated with higher risk of goiter and higher concentrations of circulating TSH and thyroid hormones (51). These children received iodine-enriched salt together with vitamin A (200,000 IU at baseline and at 5 months) or a placebo in a randomized, double-blind, 10-month trial. Vitamin A supplementation significantly decreased TSH concentration and thyroid volume compared to placebo (51). In another trial, vitamin A supplementation alone (without iodine) to iodine-deficient children reduced the volume of the thyroid gland, as well as TSH and thyroglobulin concentrations (53). Yet, supplemental vitamin A had no additional effect on thyroid function/hormone metabolism when children were also given iodized oil.
Goitrogens
Some foods contain substances that interfere with iodine utilization or thyroid hormone production; these substances are called goitrogens. The occurrence of goiter in the Democratic Republic of Congo has been related to the consumption of cassava, which contains linamarin, a compound that is metabolized to thiocyanate and blocks thyroidal uptake of iodine (1). In iodine-deficient populations, tobacco smoking has been associated with an increased risk for goiter (54, 55). Cyanide in tobacco smoke is converted to thiocyanate in the liver, placing smokers with low iodine intake at risk of developing a goiter. Moreover, thiocyanate affects iodine transport into the lactating mammary gland, leading to low iodine concentrations in breast milk and impaired iodine supply to the neonates/infants of smoking mothers (2). Some species of millet, sweet potatoes, beans, and cruciferous vegetables (e.g., cabbage, broccoli, cauliflower, and Brussels sprouts) also contain goitrogens (1). Further, the soybean isoflavones, genistein and daidzein, have been found to inhibit thyroid hormone synthesis (56). Most of these goitrogens are not of clinical importance unless they are consumed in large amounts or there is coexisting iodine deficiency. Industrial pollutants, such as perchlorate (see Safety), resorcinol, and phthalic acid, may also be goitrogenic (1, 57).
The Recommended Dietary Allowance (RDA)
The RDA for iodine was reevaluated by the Food and Nutrition Board (FNB) of the Institute of Medicine (IOM) in 2001 (Table 2). The recommended amounts were calculated using several methods, including the measurement of iodine uptake in the thyroid glands of individuals with normal thyroid function (9). Similar recommendations have been made by several organizations, including the American Thyroid Association (ATA) (16, 58), the World Health Organization (WHO), the Iodine Global Network (IGN; formerly the International Council for the Control of Iodine Deficiency Disorders), and the United Nations Children’s Fund (UNICEF) (4). Of note, the WHO, IGN, and UNICEF recommend daily intakes of 250 μg of iodine for both pregnant and breast-feeding women (4).
| Life Stage | Age | Males (μg/day) | Females (μg/day) |
|---|---|---|---|
| Infants | 0-6 months | 110 (AI) | 110 (AI) |
| Infants | 7-12 months | 130 (AI) | 130 (AI) |
| Children | 1-3 years | 90 | 90 |
| Children | 4-8 years | 90 | 90 |
| Children | 9-13 years | 120 | 120 |
| Adolescents | 14-18 years | 150 | 150 |
| Adults | 19 years and older | 150 | 150 |
| Pregnancy | all ages | - | 220 |
| Breast-feeding | all ages | - | 290 |
Disease Prevention
Radiation-induced thyroid cancer
Radioactive iodine, especially iodine 131 (131I), may be released into the environment as a result of nuclear reactor accidents, such as the 1986 Chernobyl nuclear accident in Ukraine and the 2011 Fukushima Daiichi nuclear accident in Japan. Thyroid accumulation of radioactive iodine increases the risk of developing thyroid cancer, especially in children (59). The increased iodine trapping activity of the thyroid gland in iodine deficiency results in increased thyroid accumulation of radioactive iodine (131I). Thus, iodine-deficient individuals are at increased risk of developing radiation-induced thyroid cancer because they will accumulate greater amounts of radioactive iodine. Potassium iodide administered in pharmacologic doses (up to 130 mg for adults) within 48 hours before or eight hours after radiation exposure from a nuclear reactor accident can significantly reduce thyroid uptake of 131I and decrease the risk of radiation-induced thyroid cancer (60). The prompt and widespread use of potassium iodide prophylaxis in Poland after the 1986 Chernobyl nuclear reactor accident may explain the lack of a significant increase in childhood thyroid cancer compared to fallout areas where potassium iodide prophylaxis was not widely used (61). In the US, the Nuclear Regulatory Commission (NRC) requires that consideration be given to potassium iodide as a protective measure for the general public in the case of a major release of radioactivity from a nuclear power plant (62). See also the US FDA’s Potassium Iodide Information.
Disease Treatment
Fibrocystic breast changes
Fibrocystic breast changes constitute a benign (non-cancerous) condition of the breasts, characterized by lumpiness and discomfort in one or both breasts. Cyst formation and fibrous changes in the appearance of breast tissue occur in at least 50% of premenopausal women and are not usually associated with an increased risk of breast cancer (63). The cause of fibrocystic changes is not known, but variations in hormonal stimulation during menstrual cycles may trigger changes in breast tissue (63).
A few observational studies also suggested an association between benign breast diseases (including but not limited to fibrocystic changes) and thyroid disorders. Recently, a small case-control study (166 cases vs. 72 controls) showed that the frequency of benign breast diseases was greater in women with nodular goiter (54.9%) or Hashimoto thyroiditis (47.4%) than in euthyroid controls (29.2%) (64). Conversely, the prevalence of anti-thyroid autoimmunity and hypothyroidism was found to be significantly higher in women with benign breast diseases compared to controls (65, 66). Interestingly, correcting hypothyroidism with supplemental T4 was found to improve some of the benign breast disease symptoms, including breast pain (mastalgia) and nipple discharge (65).
In estrogen-treated rats, iodine deficiency leads to changes similar to those seen in fibrocystic breasts, while iodine repletion reverses those changes (67). An uncontrolled study of 233 women with fibrocystic changes found that treatment with aqueous molecular iodine (I2) at a dose of 0.08 mg of I2/kg of body weight daily over 6 to 18 months was associated with improvement in pain and other symptoms in over 70% of participants (68). About 10% of the study participants reported side effects that were described by the investigators as minor. A double-blind, placebo-controlled trial of aqueous molecular iodine (0.07-0.09 mg of I2/kg of body weight daily for six months) in 56 women with fibrocystic changes found that 65% of the women taking molecular iodine reported improvement compared to 33% of those taking the placebo (68). A double-blind, placebo-controlled trial in 87 women with documented breast pain reported that molecular iodine (1.5, 3, or 6 mg/day) for six months improved overall pain (69). In this study, 38.5% of the women receiving 1.5 mg/day, 37.9% of those receiving 3 mg/day, and 51.7% of those receiving 6 mg/day reported at least a 50% reduction in self-assessed breast pain compared to 8.3% in the placebo group.
Large-scale, controlled clinical trials are needed to determine the therapeutic value of molecular iodine in fibrocystic breasts. Besides, the doses of iodine used in these studies (1.5 to 6 mg/day for a 60 kg person) are higher than the tolerable upper intake level (UL) recommended by the Food and Nutrition Board of the Institute of Medicine and should only be used under medical supervision (see Safety).
Sources
Food sources
Data from the ongoing US Total Diet Study, which monitors the levels of some contaminants and nutrients in food products, indicates that dietary iodine intakes in adults range between 138 and 268 micrograms (μg)/day. Considerably higher average intakes (304-353 μg/day of iodine) were reported for boys 14 to 16 years of age (70).
Seafood is rich in iodine because marine animals can concentrate the iodine from seawater. Certain types of edible seaweed (e.g., wakame) are also very rich in iodine (71). The iodine content of food that is grown or raised on a particular soil depends on the iodine content of this soil. In the US, dairy products contribute up to 90% of total estimated iodine intakes in infants, at least 70% in children (ages, 2-10 years), 53%-63% in adolescents (ages, 14-16 years), and about 50% in adults (70). In the UK and northern Europe, iodine levels in dairy products tend to be lower in summer when cattle are allowed to graze in pastures with low soil iodine content (9).
Other good sources of dietary iodine include eggs, fruit, grain products, and poultry (70). Processed foods can contribute to iodine intake if iodized salt or food additives, such as calcium iodate and potassium iodate, are added during production. Yet, in the US, virtually no iodized salt is used in the manufacturing of processed food and fast food products, and the food industry is not required to list the iodine content on food packaging (72). Table 3 lists the iodine content of some iodine-rich foods in micrograms (μg). Because the iodine content of foods can vary considerably, these values should be considered approximate (73).
Supplements
Over-the-counter iodine supplements
Potassium iodide is available as a nutritional supplement, typically in combination products, such as multivitamin/mineral supplements. Iodine makes up approximately 77% of the total weight of potassium iodide (56). A multivitamin/mineral supplement that contains 100% of the daily value (DV) for iodine provides 150 μg of iodine. Although most people in the US consume sufficient iodine in their diets (see Sources), an additional 150 μg/day is unlikely to result in excessive iodine intake. The American Thyroid Association (ATA) recommends prenatal supplementation with 150 μg/day of iodine and advises against the ingestion of ≥500 μg/day of iodine from iodine, potassium iodine, and kelp supplements for children and adults, and during pregnancy and lactation (see also Safety) (36, 74).
Iodine fortification programs
The fortification of salt with iodine is a feasible and inexpensive method to eliminate iodine deficiency, and salt iodization programs have been implemented in almost all countries. In North America, salt fortification with iodine is mandated in Canada and some parts of Mexico, but only voluntary in the US such that only 52% of US table salt is iodized and only one-fifth of the total salt consumed in the US is iodized (72, 75). Potassium iodide (KI), cuprous iodide (CuI), and potassium iodate (KIO3) are used to iodize salt. The US Food and Drug Administration (FDA) recommends between 46 and 76 μg of iodine per gram of salt in iodized salt. However, the recent analysis of 88 US iodized food-grade salt samples revealed that the iodine content was below the recommended range in 52% of the samples and above the range in 7% of the samples (76).
In other countries, salt commonly contains 20-50 μg of iodine per gram of salt, depending on local regulations (76). In countries like Denmark (77), Australia (78, 79), and New Zealand (80), the use of iodized salt in the bread-making process is mandated. Additional approaches have been explored, including sugar fortification (81), egg fortification (82), use of iodized salt in the preparation of fermented fish and fish sauce (83), and use of iodine-rich crop fertilizers (84). In addition, fortification of livestock feeds with iodine and the use of iodophors for sanitation during milking contribute to increasing iodine content in dairy products (85). Finally, annual doses of iodized vegetable oil are administered orally or intramuscularly to individuals in iodine-deficient populations who do not have access to iodized salt (4, 56).
Safety
Acute toxicity
Acute iodine poisoning is rare and usually occurs only with doses of many grams. Symptoms of acute iodine poisoning include burning of the mouth, throat, and stomach, fever, nausea, vomiting, diarrhea, a weak pulse, cyanosis, and coma (1).
Excessive iodine intakes
Risk of iodine-induced hyperthyroidism in iodine-deficient individuals
Iodine supplementation programs in iodine-deficient populations have been associated with an increased incidence of iodine-induced hyperthyroidism (IIH), especially in older people with multi-nodular goiter (86). Iodine intakes of 150-200 μg/day have been found to increase the incidence of IIH in iodine-deficient populations. Iodine deficiency increases the risk of developing autonomous thyroid nodules that are unresponsive to TSH control (see Function). These autonomous nodules may then overproduce thyroid hormones in response to sudden iodine supply. IIH symptoms include weight loss, tachycardia (high pulse rate), muscle weakness, and skin warmth. IIH can be dangerous in individuals with underlying heart disease. Yet, because the primary cause of nodular goiter and IIH is chronic iodine deficiency, the benefit of iodization programs largely outweighs the risk of IIH in iodine-deficient populations (1).
Risk of hypothyroidism in iodine-sufficient individuals
In iodine-sufficient individuals, excess iodine intake is most commonly associated with elevated blood concentrations of thyroid stimulating hormone (TSH) that inhibit thyroid hormone production, leading to hypothyroidism and goiter. A slightly elevated serum TSH concentration without a decrease in serum T4 or T3 is the earliest sign of abnormal thyroid function when iodine intake is excessive. In iodine-sufficient adults, elevated serum TSH has been found at chronic iodine intakes of ≥750 μg/day in children and ≥1,700 μg/day in adults. Because various edible seaweed species substantially contribute to traditional Asian meals, average Japanese dietary intakes are estimated to range between 1,000 and 3,000 μg of iodine/day (71). Iodine-induced goiter and hypothyroidism are not uncommon in Japan and can be reversed by restricting seaweed intake (71). Prolonged intakes of more than 18,000 μg/day (18 mg/day) increase the incidence of goiter in adults. In newborns, iodine-induced goiter and hypothyroidism can be due to either high maternal intakes or high exposure to iodized antiseptics (87). In order to minimize the risk of adverse health effects, the Food and Nutrition Board of the US Institute of Medicine set a tolerable upper intake level (UL) for iodine that is likely to be safe in almost all individuals. The UL values for iodine are listed in Table 4 by age group; the UL does not apply to individuals who are being treated with iodine under medical supervision (74).
Individuals with increased sensitivity to excess iodine intake
Individuals with iodine deficiency and those with preexisting thyroid disease, including nodular goiter, autoimmune Hashimoto thyroiditis, Graves disease, and a history of partial thyroidectomy, may be sensitive to iodine intake levels considered safe for the general population and may not be protected by the UL for iodine (9). Infants, the elderly, and pregnant and lactating women may also be more susceptible to excess iodine (see Supplements) (74).
Do elevated and/or insufficient iodine intakes increase the risk of thyroid cancer?
Over the past decades, the incidence of thyroid cancer has increased worldwide. In the US, the incidence of thyroid cancer — representing 4% of all newly diagnosed cancers — has increased from 4.9 cases per persons in 1983 to 14.7 cases per 100,000 persons in 2011, but mortality rate from thyroid cancer has remained low (about 0.5 per 100,000 persons) (88). Accounting for over 80% of all thyroid cancers, thyroid papillary cancer is less aggressive and has a better prognosis than thyroid follicular cancer or anaplastic thyroid cancer. The increasing incidence of thyroid cancer worldwide is likely due at least in part to the improved screening and diagnosis activities. However, because it has coincided with the introduction of iodine fortification programs, a possible contribution of increased iodine intakes has been hypothesized. Yet, in the US, the increasing incidence of thyroid cancers (primarily papillary cancer) over the last few decades was paralleled with a reduction in average iodine intake (89).
Ecologic studies also suggested that iodine prophylaxis in populations that were previously iodine deficient was associated with an increased incidence of the papillary rather than the follicular cancer subtype, and with a reduced incidence of the more aggressive anaplastic thyroid cancer (89). While changes in iodine intakes appear to affect the histological type of thyroid cancer, it is not yet clear whether iodine deficiency and/or iodine excess increase the risk of thyroid cancer (88).
Drug interactions
Amiodarone, a medication used to prevent abnormal heart rhythms, contains high levels of iodine and may affect thyroid function (90, 91). Antithyroid drugs used to treat hyperthyroidism, such as propylthiouracil (PTU), methimazole, and carbimazole, may increase the risk of hypothyroidism. Additionally, the long-term use of lithium to treat mood disorders may increase the risk of hypothyroidism (92). Further, the use of pharmacologic doses of potassium iodide may decrease the anticoagulant effect of warfarin (coumarin) (56).
Contaminants
Perchlorate is an oxidizing agent found in rocket propellants, airbags, fireworks, herbicides, and fertilizer. Mainly as a result of human activity, perchlorate has been found to contaminate drinking water and many foods (57). Chronic exposure to perchlorate concentrations at levels greater than 20 μg per kg body weight (bw) per day interferes with iodine uptake by the thyroid gland and may lead to hypothyroidism (93). The US Environment Protection Agency (EPA) recommends that daily oral exposure to perchlorate should not exceed 0.7 μg/kg bw to protect the most sensitive population, i.e., the fetuses of pregnant women who might be deficient in iodine and/or hypothyroid (94). Among all age groups, children aged two years have the highest estimated perchlorate intakes per day with 0.35-0.39 μg/kg bw/day. Average estimated intakes of perchlorate in US adults range between 0.08 and 0.11 μg/kg bw/day (70).
Linus Pauling Institute Recommendation
The RDA for iodine is sufficient to ensure normal thyroid function. There is presently no evidence that iodine intakes higher than the RDA are beneficial. Most people in the US consume sufficient iodine in their diets, making supplementation unnecessary.
Pregnant and breast-feeding women
Given the importance of sufficient iodine during prenatal development and infancy, pregnant and breast-feeding women should take a supplement that provides 150 μg of iodine per day (see Deficiency).
Older adults (>50 years)
Because aging has not been associated with significant changes in the requirement for iodine, the LPI recommendation for iodine intake is not different for older adults.
Authors and Reviewers
Originally written in 2001 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2003 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in July 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in March 2010 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in August 2015 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in August 2015 by:
Elizabeth N. Pearce, M.D., M.Sc.
Associate Professor of Medicine
Boston University School of Medicine
Copyright 2001-2024 Linus Pauling Institute
References
1. Zimmermann MB. Iodine and iodine deficiency disorders. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed: John Wiley & Sons; 2012:554-567.
2. Laurberg P. Iodine. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed: Lippincott Williams & Wilkins; 2014:217-224.
3. Larsen PR, Davies TF, Hay ID. The thyroid gland. In: Wilson JD, Foster DW, Kronenberg HM, Larsen PR, eds. Williams Textbook of Endocrinology. 9th ed. Philadelphia: W.B. Saunders Company; 1998:389-515.
4. WHO, UNICEF, ICCIDD. Assessment of iodine deficiency disorders and monitoring of their elimination: a guide for programme managers, 3rd ed. 2007. http://www.who.int/nutrition/publications/micronutrients/iodine_deficiency/9789241595827/en/. Accessed 8/28/15.
5. de Benoist B, McLean E, Andersson M, Rogers L. Iodine deficiency in 2007: global progress since 2003. Food Nutr Bull. 2008;29(3):195-202. (PubMed)
6. Andersson M, Karumbunathan V, Zimmermann MB. Global iodine status in 2011 and trends over the past decade. J Nutr. 2012;142(4):744-750. (PubMed)
7. Pearce EN, Andersson M, Zimmermann MB. Global iodine nutrition: Where do we stand in 2013? Thyroid. 2013;23(5):523-528. (PubMed)
8. United Nations Children's Fund. The State of the World's Children 2007, UNICEF. New York; 2006.
9. Food and Nutrition Board, Institute of Medicine. Iodine. Dietary reference intakes for vitamin A, vitamin K, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanandium, and zinc. Washington, D.C.: National Academy Press; 2001:258-289. (National Academy Press)
10. Zimmermann MB, Andersson M. Assessment of iodine nutrition in populations: past, present, and future. Nutr Rev. 2012;70(10):553-570. (PubMed)
11. Konig F, Andersson M, Hotz K, Aeberli I, Zimmermann MB. Ten repeat collections for urinary iodine from spot samples or 24-hour samples are needed to reliably estimate individual iodine status in women. J Nutr. 2011;141(11):2049-2054. (PubMed)
12. Zimmermann MB. The effects of iodine deficiency in pregnancy and infancy. Paediatr Perinat Epidemiol. 2012;26 Suppl 1:108-117. (PubMed)
13. Ma ZF, Skeaff SA. Thyroglobulin as a biomarker of iodine deficiency: a review. Thyroid. 2014;24(8):1195-1209. (PubMed)
14. Di Liegro I. Thyroid hormones and the central nervous system of mammals (Review). Mol Med Rep. 2008;1(3):279-295. (PubMed)
15. Zimmermann MB. Are weaning infants at risk of iodine deficiency even in countries with established iodized salt programs? Nestle Nutr Inst Workshop Ser. 2012;70:137-146. (PubMed)
16. Stagnaro-Green A, Abalovich M, Alexander E, et al. Guidelines of the American Thyroid Association for the diagnosis and management of thyroid disease during pregnancy and postpartum. Thyroid. 2011;21(10):1081-1125. (PubMed)
17. Andersson M, Aeberli I, Wust N, et al. The Swiss iodized salt program provides adequate iodine for school children and pregnant women, but weaning infants not receiving iodine-containing complementary foods as well as their mothers are iodine deficient. J Clin Endocrinol Metab. 2010;95(12):5217-5224. (PubMed)
18. Bouhouch RR, Bouhouch S, Cherkaoui M, et al. Direct iodine supplementation of infants versus supplementation of their breastfeeding mothers: a double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2(3):197-209. (PubMed)
19. Bath SC, Steer CD, Golding J, Emmett P, Rayman MP. Effect of inadequate iodine status in UK pregnant women on cognitive outcomes in their children: results from the Avon Longitudinal Study of Parents and Children (ALSPAC). Lancet. 2013;382(9889):331-337. (PubMed)
20. Hynes KL, Otahal P, Hay I, Burgess JR. Mild iodine deficiency during pregnancy is associated with reduced educational outcomes in the offspring: 9-year follow-up of the gestational iodine cohort. J Clin Endocrinol Metab. 2013;98(5):1954-1962. (PubMed)
21. Cobra C, Muhilal, Rusmil K, et al. Infant survival is improved by oral iodine supplementation. J Nutr. 1997;127(4):574-578. (PubMed)
22. DeLong GR, Leslie PW, Wang SH, et al. Effect on infant mortality of iodination of irrigation water in a severely iodine-deficient area of China. Lancet. 1997;350(9080):771-773. (PubMed)
23. Hetzel BS. Iodine and neuropsychological development. J Nutr. 2000;130(2S Suppl):493S-495S. (PubMed)
24. Levander OA, Whanger PD. Deliberations and evaluations of the approaches, endpoints and paradigms for selenium and iodine dietary recommendations. J Nutr. 1996;126(9 Suppl):2427S-2434S. (PubMed)
25. Bleichrodt N, Born, M.P. A meta-analysis of research on iodine and its relationship to cognitive development. In: Stanbury JB, ed. The damaged brain of iodine deficiency: cognitive, behavioral, neuromotor, educative aspects. New York: Cognizant Communication Corporation; 1994:195-200.
26. Qian M, Wang D, Watkins WE, et al. The effects of iodine on intelligence in children: a meta-analysis of studies conducted in China. Asia Pac J Clin Nutr. 2005;14(1):32-42. (PubMed)
27. Bougma K, Aboud FE, Harding KB, Marquis GS. Iodine and mental development of children 5 years old and under: a systematic review and meta-analysis. Nutrients. 2013;5(4):1384-1416. (PubMed)
28. Zimmermann MB. Iodine: it's important in patients that require parenteral nutrition. Gastroenterology. 2009;137(5 Suppl):S36-46. (PubMed)
29. Lazarus JH. Iodine status in europe in 2014. Eur Thyroid J. 2014;3(1):3-6. (PubMed)
30. Caldwell KL, Makhmudov A, Ely E, Jones RL, Wang RY. Iodine status of the U.S. population, National Health and Nutrition Examination Survey, 2005-2006 and 2007-2008. Thyroid. 2011;21(4):419-427. (PubMed)
31. Caldwell KL, Pan Y, Mortensen ME, Makhmudov A, Merrill L, Moye J. Iodine status in pregnant women in the National Children's Study and in U.S. women (15-44 years), National Health and Nutrition Examination Survey 2005-2010. Thyroid. 2013;23(8):927-937. (PubMed)
32. Gregory CO, Serdula MK, Sullivan KM. Use of supplements with and without iodine in women of childbearing age in the United States. Thyroid. 2009;19(9):1019-1020. (PubMed)
33. Kirk AB, Martinelango PK, Tian K, Dutta A, Smith EE, Dasgupta PK. Perchlorate and iodide in dairy and breast milk. Environ Sci Technol. 2005;39(7):2011-2017. (PubMed)
34. Pearce EN, Leung AM, Blount BC, et al. Breast milk iodine and perchlorate concentrations in lactating Boston-area women. J Clin Endocrinol Metab. 2007;92(5):1673-1677. (PubMed)
35. Nazeri P, Mirmiran P, Shiva N, Mehrabi Y, Mojarrad M, Azizi F. Iodine nutrition status in lactating mothers residing in countries with mandatory and voluntary iodine fortification programs: an updated systematic review. Thyroid. 2015;25(6):611-620. (PubMed)
36. Becker DV, Braverman LE, Delange F, et al. Iodine supplementation for pregnancy and lactation-United States and Canada: recommendations of the American Thyroid Association. Thyroid. 2006;16(10):949-951. (PubMed)
37. Kuriti M, Pearce EN, Braverman LE, He X, Leung AM. Iodine content of U.S. weight-loss food. Endocr Pract. 2014;20(3):232-235. (PubMed)
38. Leung AM, Lamar A, He X, Braverman LE, Pearce EN. Iodine status and thyroid function of Boston-area vegetarians and vegans. J Clin Endocrinol Metab. 2011;96(8):E1303-1307. (PubMed)
39. Brooks MJ, Post EM. Acquired hypothyroidism due to iodine deficiency in an American child. J Pediatr Endocrinol Metab. 2014;27(11-12):1233-1235. (PubMed)
40. Cheetham T, Plumb E, Callaghan J, Jackson M, Michaelis L. Dietary restriction causing iodine-deficient goitre. Arch Dis Child. 2015;100(8):784-786. (PubMed)
41. Belfort MB, Pearce EN, Braverman LE, He X, Brown RS. Low iodine content in the diets of hospitalized preterm infants. J Clin Endocrinol Metab. 2012;97(4):E632-636. (PubMed)
42. Hess SY. The impact of common micronutrient deficiencies on iodine and thyroid metabolism: the evidence from human studies. Best Pract Res Clin Endocrinol Metab. 2010;24(1):117-132. (PubMed)
43. Schneider MJ, Fiering SN, Thai B, et al. Targeted disruption of the type 1 selenodeiodinase gene (Dio1) results in marked changes in thyroid hormone economy in mice. Endocrinology. 2006;147(1):580-589. (PubMed)
44. Schomburg L. Selenium, selenoproteins and the thyroid gland: interactions in health and disease. Nat Rev Endocrinol. 2012;8(3):160-171. (PubMed)
45. Negro R, Greco G, Mangieri T, Pezzarossa A, Dazzi D, Hassan H. The influence of selenium supplementation on postpartum thyroid status in pregnant women with thyroid peroxidase autoantibodies. J Clin Endocrinol Metab. 2007;92(4):1263-1268. (PubMed)
46. Mao J, Pop VJ, Bath SC, Vader HL, Redman CW, Rayman MP. Effect of low-dose selenium on thyroid autoimmunity and thyroid function in UK pregnant women with mild-to-moderate iodine deficiency. Eur J Nutr. 2014. [Epub ahead of print] (PubMed)
47. Contempre B, Duale NL, Dumont JE, Ngo B, Diplock AT, Vanderpas J. Effect of selenium supplementation on thyroid hormone metabolism in an iodine and selenium deficient population. Clin Endocrinol (Oxf). 1992;36(6):579-583. (PubMed)
48. Contempre B, Dumont JE, Ngo B, Thilly CH, Diplock AT, Vanderpas J. Effect of selenium supplementation in hypothyroid subjects of an iodine and selenium deficient area: the possible danger of indiscriminate supplementation of iodine-deficient subjects with selenium. J Clin Endocrinol Metab. 1991;73(1):213-215. (PubMed)
49. Zimmermann MB. The influence of iron status on iodine utilization and thyroid function. Annu Rev Nutr. 2006;26:367-389. (PubMed)
50. Hess SY, Zimmermann MB, Adou P, Torresani T, Hurrell RF. Treatment of iron deficiency in goitrous children improves the efficacy of iodized salt in Cote d'Ivoire. Am J Clin Nutr. 2002;75(4):743-748. (PubMed)
51. Zimmermann MB, Wegmuller R, Zeder C, Chaouki N, Torresani T. The effects of vitamin A deficiency and vitamin A supplementation on thyroid function in goitrous children. J Clin Endocrinol Metab. 2004;89(11):5441-5447. (PubMed)
52. Zimmermann MB. Interactions of vitamin A and iodine deficiencies: effects on the pituitary-thyroid axis. Int J Vitam Nutr Res. 2007;77(3):236-240. (PubMed)
53. Zimmermann MB, Jooste PL, Mabapa NS, et al. Vitamin A supplementation in iodine-deficient African children decreases thyrotropin stimulation of the thyroid and reduces the goiter rate. Am J Clin Nutr. 2007;86(4):1040-1044. (PubMed)
54. Knudsen N, Brix TH. Genetic and non-iodine-related factors in the aetiology of nodular goitre. Best Pract Res Clin Endocrinol Metab. 2014;28(4):495-506. (PubMed)
55. Rendina D, De Palma D, De Filippo G, et al. Prevalence of simple nodular goiter and Hashimoto's thyroiditis in current, previous, and never smokers in a geographical area with mild iodine deficiency. Horm Metab Res. 2015;47(3):214-219. (PubMed)
56. Hendler SS, Rorvik DM, eds. PDR for Nutritional Supplements. 2nd ed. Montvale: Thomson Reuters; 2008.
57. Council on Environmental Health, Rogan WJ, Paulson JA, et al. Iodine deficiency, pollutant chemicals, and the thyroid: new information on an old problem. Pediatrics. 2014;133(6):1163-1166. (PubMed)
58. Leung AM, Pearce EN, Braverman LE, Stagnaro-Green A. AAP recommendations on iodine nutrition during pregnancy and lactation. Pediatrics. 2014;134(4):e1282. (PubMed)
59. Cardis E, Howe G, Ron E, et al. Cancer consequences of the Chernobyl accident: 20 years on. J Radiol Prot. 2006;26(2):127-140. (PubMed)
60. Zanzonico PB, Becker DV. Effects of time of administration and dietary iodine levels on potassium iodide (KI) blockade of thyroid irradiation by 131I from radioactive fallout. Health Phys. 2000;78(6):660-667. (PubMed)
61. Nauman J, Wolff J. Iodide prophylaxis in Poland after the Chernobyl reactor accident: benefits and risks. Am J Med. 1993;94(5):524-532. (PubMed)
62. Nuclear Regulatory Commission. Consideration of potassium iodide in emergency plans. Nuclear Regulatory Commission. Final rule. Fed Regist. 2001;66(13):5427-5440. (PubMed)
63. Guray M, Sahin AA. Benign breast diseases: classification, diagnosis, and management. Oncologist. 2006;11(5):435-449. (PubMed)
64. Anil C, Guney T, Gursoy A. The prevalence of benign breast diseases in patients with nodular goiter and Hashimoto's thyroiditis. J Endocrinol Invest. 2015;38(9):971-975. (PubMed)
65. Bhargav PR, Mishra A, Agarwal G, Agarwal A, Verma AK, Mishra SK. Prevalence of hypothyroidism in benign breast disorders and effect of thyroxine replacement on the clinical outcome. World J Surg. 2009;33(10):2087-2093. (PubMed)
66. Giustarini E, Pinchera A, Fierabracci P, et al. Thyroid autoimmunity in patients with malignant and benign breast diseases before surgery. Eur J Endocrinol. 2006;154(5):645-649. (PubMed)
67. Eskin BA, Grotkowski CE, Connolly CP, Ghent WR. Different tissue responses for iodine and iodide in rat thyroid and mammary glands. Biol Trace Elem Res. 1995;49(1):9-19. (PubMed)
68. Ghent WR, Eskin BA, Low DA, Hill LP. Iodine replacement in fibrocystic disease of the breast. Can J Surg. 1993;36(5):453-460. (PubMed)
69. Kessler JH. The effect of supraphysiologic levels of iodine on patients with cyclic mastalgia. Breast J. 2004;10(4):328-336. (PubMed)
70. Murray CW, Egan SK, Kim H, Beru N, Bolger PM. US Food and Drug Administration's Total Diet Study: dietary intake of perchlorate and iodine. J Expo Sci Environ Epidemiol. 2008;18(6):571-580. (PubMed)
71. Zava TT, Zava DT. Assessment of Japanese iodine intake based on seaweed consumption in Japan: A literature-based analysis. Thyroid Res. 2011;4:14. (PubMed)
72. Leung AM, Braverman LE, Pearce EN. History of U.S. iodine fortification and supplementation. Nutrients. 2012;4(11):1740-1746. (PubMed)
73. Pennington JAT, Schoen SA, Salmon GD, Young B, Johnson RD, Marts RW. Composition of core foods of the U.S. food supply, 1982-1991. III. Copper, manganese, selenium, iodine. J Food Comp Anal. 1995;8:171-217.
74. Leung AM, Avram AM, Brenner AV, et al. Potential risks of excess iodine ingestion and exposure: statement by the American Thyroid Association Public Health Committee. Thyroid. 2015;25(2):145-146. (PubMed)
75. Maalouf J, Barron J, Gunn JP, Yuan K, Perrine CG, Cogswell ME. Iodized salt sales in the United States. Nutrients. 2015;7(3):1691-1695. (PubMed)
76. Dasgupta PK, Liu Y, Dyke JV. Iodine nutrition: iodine content of iodized salt in the United States. Environ Sci Technol. 2008;42(4):1315-1323. (PubMed)
77. Rasmussen LB, Ovesen L, Christensen T, et al. Iodine content in bread and salt in Denmark after iodization and the influence on iodine intake. Int J Food Sci Nutr. 2007;58(3):231-239. (PubMed)
78. Charlton KE, Yeatman H, Brock E, et al. Improvement in iodine status of pregnant Australian women 3 years after introduction of a mandatory iodine fortification programme. Prev Med. 2013;57(1):26-30. (PubMed)
79. Clifton VL, Hodyl NA, Fogarty PA, et al. The impact of iodine supplementation and bread fortification on urinary iodine concentrations in a mildly iodine deficient population of pregnant women in South Australia. Nutr J. 2013;12:32. (PubMed)
80. Skeaff SA, Lonsdale-Cooper E. Mandatory fortification of bread with iodised salt modestly improves iodine status in schoolchildren. Br J Nutr. 2013;109(6):1109-1113. (PubMed)
81. Eltom M, Elnagar B, Sulieman EA, et al. The use of sugar as a vehicle for iodine fortification in endemic iodine deficiency. Int J Food Sci Nutr. 1995;46(3):281-289. (PubMed)
82. Charoensiriwatana W, Srijantr P, Teeyapant P, Wongvilairattana J. Consuming iodine enriched eggs to solve the iodine deficiency endemic for remote areas in Thailand. Nutr J. 2010;9:68. (PubMed)
83. Chanthilath B, Chavasit V, Chareonkiatkul S, Judprasong K. Iodine stability and sensory quality of fermented fish and fish sauce produced with the use of iodated salt. Food Nutr Bull. 2009;30(2):183-188. (PubMed)
84. Weng HX, Liu HP, Li DW, Ye M, Pan L, Xia TH. An innovative approach for iodine supplementation using iodine-rich phytogenic food. Environ Geochem Health. 2014;36(4):815-828. (PubMed)
85. Zimmermann MB. Symposium on 'Geographical and geological influences on nutrition': Iodine deficiency in industrialised countries. Proc Nutr Soc. 2010;69(1):133-143. (PubMed)
86. Laurberg P, Nohr SB, Pedersen KM, et al. Thyroid disorders in mild iodine deficiency. Thyroid. 2000;10(11):951-963. (PubMed)
87. Nishiyama S, Mikeda T, Okada T, Nakamura K, Kotani T, Hishinuma A. Transient hypothyroidism or persistent hyperthyrotropinemia in neonates born to mothers with excessive iodine intake. Thyroid. 2004;14(12):1077-1083. (PubMed)
88. Davies L, Morris LG, Haymart M, et al. American Association of Clinical Endocrinologists and American College of Endocrinology Disease State Clinical Review: The Increasing Incidence of Thyroid Cancer. Endocr Pract. 2015;21(6):686-696. (PubMed)
89. Zimmermann MB, Galetti V. Iodine intake as a risk factor for thyroid cancer: a comprehensive review of animal and human studies. Thyroid Res. 2015;8:8. (PubMed)
90. Ahmed S, Van Gelder IC, Wiesfeld AC, Van Veldhuisen DJ, Links TP. Determinants and outcome of amiodarone-associated thyroid dysfunction. Clin Endocrinol (Oxf). 2011;75(3):388-394. (PubMed)
91. Kurnik D, Loebstein R, Farfel Z, Ezra D, Halkin H, Olchovsky D. Complex drug-drug-disease interactions between amiodarone, warfarin, and the thyroid gland. Medicine (Baltimore). 2004;83(2):107-113. (PubMed)
92. McKnight RF, Adida M, Budge K, Stockton S, Goodwin GM, Geddes JR. Lithium toxicity profile: a systematic review and meta-analysis. Lancet. 2012;379(9817):721-728. (PubMed)
93. US NRC. Health Implications of Perchlorate Ingestion The National Academies Press. Available at: http://www.nap.edu/openbook.php?record_id=11202. Accessed 08/11/2015.
94. US EPA. Perchlorate and Perchlorate Salts. 02/18/2005. http://www.epa.gov/iris/subst/1007.htm. Accessed 08/11/2015.
Iron
Contents
Summary
- Iron is an essential component of hundreds of proteins and enzymes that support essential biological functions, such as oxygen transport, energy production, and DNA synthesis. Hemoglobin, myoglobin, cytochromes, and peroxidases require iron-containing heme as a prosthetic group for their biological activities. (More information)
- Because the body excretes very little iron, iron metabolism is tightly regulated. In particular, the iron regulatory hormone, hepcidin, blocks dietary iron absorption, promotes cellular iron sequestration, and reduces iron bioavailability when body iron stores are sufficient to meet requirements. (More information)
- Iron status can be assessed in healthy men and nonpregnant women using laboratory tests that measure serum ferritin (iron-storage protein), serum iron, total iron binding capacity, saturation of transferrin (the main iron carrier in blood), and soluble transferrin receptor. (More information)
- Iron deficiency results from an inadequate supply of iron to cells following depletion of the body’s reserves. Microcytic anemia occurs when body iron stores are so low that hemoglobin synthesis and red blood cell formation are severely impaired. (More information)
- Iron deficiency is the most common nutritional deficiency worldwide, affecting primarily children, women of childbearing age, pregnant women, frequent blood donors, and individuals with certain medical conditions. (More information)
- Much of our iron requirement is met through recycling iron from senescent red blood cells. The recommended dietary allowance (RDA) for iron is 8 mg/day for men and postmenopausal women, 18 mg/day for premenopausal women, and 27 mg/day for pregnant women. (More information)
- Iron deficiency with or without anemia in children has been associated with poor cognitive development, poor school achievement, and abnormal behavior patterns. Limited evidence suggests that iron supplementation has no effect on psychomotor development and cognitive function of anemic iron-deficient infants younger than three years but may improve attention and concentration in older children, adolescents, and women with anemia and/or iron deficiency. (More information)
- Heme iron comes from hemoglobin and myoglobin in animal food sources and represents 10%-15% of total dietary iron intake of meat eaters. Yet, because it is much better absorbed than nonheme iron found in both plant and animal food sources, heme iron contributes up to 40% of total absorbed iron. (More information)
- Toxic iron deposition in vital organs in patients affected by hereditary hemochromatosis has been associated with numerous chronic conditions, including liver cancer and type 2 diabetes mellitus. Increased heme iron intake and/or loss of iron homeostasis might also increase the risk of chronic disease in individuals free of genetic disorders. (More information)
- Iron supplementation may cause gastrointestinal irritation, nausea, vomiting, diarrhea, or constipation, and interfere with the absorption and efficacy of certain medications, including antibiotics and drugs used to treat osteoporosis, hypothyroidism, or Parkinson’s disease symptoms. (More information)
Iron is the fourth most abundant element of Earth’s crust and one of the best studied micronutrients in nutrition science (1, 2). It is a key element in the metabolism of all living organisms. Iron exists in two biologically relevant oxidation states: the ferrous form (Fe2+) and the ferric form (Fe3+). Iron is an essential component of hundreds of proteins and enzymes supporting essential biological functions, such as oxygen transport, energy production, DNA synthesis, and cell growth and replication.
Function
Heme is an iron-containing compound found in a number of biologically important molecules (Figure 1). Some, but not all, iron-dependent proteins are heme-containing proteins (also called hemoproteins). Iron-dependent proteins that carry out a broad range of biological activities may be classified as follows (1, 3):
- Globin-heme: nonenzymatic proteins involved in oxygen transport and storage (e.g., hemoglobin, myoglobin, neuroglobin)
- Heme enzymes involved in electron transfer (e.g., cytochromes a, b, f; cytochrome c oxidase) and/or with oxidase activity (e.g., sulfite oxidase, cytochrome P450 oxidases, myeloperoxidase, peroxidases, catalase, endothelial nitric oxide synthase, cyclooxygenase)
- Iron-sulfur (Fe-S) cluster proteins with oxidoreductase activities involved in energy production (e.g., succinate dehydrogenase, isocitrate dehydrogenase, NADH dehydrogenase, aconitase, xanthine oxidase, ferredoxin-1) or involved in DNA replication and repair (DNA polymerases, DNA helicases)
- Nonheme enzymes that require iron as a cofactor for their catalytic activities (e.g., phenylalanine, tyrosine, tryptophan, and lysine hydroxylases; hypoxia-inducible factor (HIF) prolyl and asparaginyl hydroxylases; ribonucleotide reductase)
- Nonheme proteins responsible for iron transport and storage (e.g., ferritin, transferrin, haptoglobin, hemopexin, lactoferrin).
Iron-containing proteins support a number of functions, some of which are listed below.
Oxygen transport and storage
Globin-hemes are heme-containing proteins that are involved in the transport and storage of oxygen and, to a lesser extent, may act as free radical scavengers (1). Hemoglobin is the primary protein found in red blood cells and represents about two-thirds of the body's iron (3). The vital role of hemoglobin in transporting oxygen from the lungs to the rest of the body is derived from its unique ability to acquire oxygen rapidly during the short time it spends in contact with the lungs and to release oxygen as needed during its circulation through the tissues. Myoglobin functions in the transport and short-term storage of oxygen in muscle cells, helping to match the supply of oxygen to the demand of working muscles (1). A third globin called neuroglobin is preferentially expressed in the central nervous system, but its function is not well understood (4).
Electron transport and energy metabolism
Cytochromes are heme-containing enzymes that have important roles in mitochondrial electron transport required for cellular energy production and thus life. Specifically, cytochromes serve as electron carriers during the synthesis of ATP, the primary energy storage compound in cells. Cytochrome P450 (CYP) is a family of enzymes involved in the metabolism of a number of important biological molecules (including organic acids; fatty acids; prostaglandins; steroids; sterols; and vitamins A, D, and K), as well as in the detoxification and metabolism of drugs and pollutants. Nonheme iron-containing enzymes in the citric acid cycle, such as NADH dehydrogenase and succinate dehydrogenase, are also critical to energy metabolism (1).
Antioxidant and beneficial pro-oxidant functions
Catalase and some peroxidases are heme-containing enzymes that protect cells against the accumulation of hydrogen peroxide, a potentially damaging reactive oxygen species (ROS), by catalyzing a reaction that converts hydrogen peroxide to water and oxygen. As part of the immune response, some white blood cells engulf bacteria and expose them to ROS in order to kill them. The synthesis of one such ROS, hypochlorous acid, by neutrophils is catalyzed by the heme-containing enzyme myeloperoxidase (1).
In addition, in the thyroid gland, heme-containing thyroid peroxidase catalyzes the iodination of thyroglobulin for the production of thyroid hormones such that thyroid metabolism can be impaired in iron deficiency and iron-deficiency anemia (see Nutrient Interactions).
Oxygen sensing
Inadequate oxygen (hypoxia), such as that experienced by those who live at high altitudes or those with chronic lung disease, induces compensatory physiologic responses, including increased red blood cell formation (erythropoiesis), increased blood vessel growth (angiogenesis), and increased production of enzymes utilized in anaerobic metabolism. Hypoxia is also observed in pathological conditions like ischemia/stroke and inflammatory disorders. Under hypoxic conditions, transcription factors known as hypoxia-inducible factors (HIF) bind to response elements in genes that encode various proteins involved in compensatory responses to hypoxia and increase their synthesis. Iron-dependent enzymes of the dioxygenase family, HIF prolyl hydroxylases and asparaginyl hydroxylase (factor inhibiting HIF-1 [FIH-1]), have been implicated in HIF regulation. When cellular oxygen tension is adequate, newly synthesized HIF-α subunits (HIF-1α, HIF-2α, HIF-3α) are modified by HIF prolyl hydroxylases in an iron/2-oxoglutarate-dependent process that targets HIF-α for rapid degradation. FIH-1-induced asparaginyl hydroxylation of HIF-α impairs the recruitment of co-activators to HIF-α transcriptional complex and therefore prevents HIF-α transcriptional activity. When cellular oxygen tension drops below a critical threshold, prolyl hydroxylase can no longer target HIF-α for degradation, allowing HIF-α to bind to HIF-1β and form a transcription complex that enters the nucleus and binds to specific hypoxia response elements (HRE) on target genes like the erythropoietin gene (EPO) (5).
DNA replication and repair
Ribonucleotide reductases (RNRs) are iron-dependent enzymes that catalyze the synthesis of deoxyribonucleotides required for DNA replication. RNRs also facilitate DNA repair in response to DNA damage. Other enzymes essential for DNA synthesis and repair, such as DNA polymerases and DNA helicases, are Fe-S cluster proteins. Although the underlying mechanisms are still unclear, depletion of intracellular iron was found to inhibit cell cycle progression, growth, and division. Inhibition of heme synthesis also induced cell cycle arrest in breast cancer cells (6).
Iron is required for a number of additional vital functions, including growth, reproduction, healing, and immune function.
Regulation
Systemic regulation of iron homeostasis
While iron is an essential mineral, it is potentially toxic because free iron inside the cell can lead to the generation of free radicals causing oxidative stress and cellular damage. Thus, it is important for the body to systemically regulate iron homeostasis. The body tightly regulates the transport of iron throughout various body compartments, such as developing red blood cells (erythroblasts), circulating macrophages, liver cells (hepatocytes) that store iron, and other tissues (7). Intracellular iron concentrations are regulated according to the body’s iron needs (see below), but extracellular signals also regulate iron homeostasis in the body through the action of hepcidin.
Hepcidin, a peptide hormone primarily synthesized by liver cells, is the key regulator of systemic iron homeostasis. Hepcidin can induce the internalization and degradation of the iron-efflux protein, ferroportin-1; ferroportin-1 regulates the release of iron from certain cells, such as enterocytes, hepatocytes, and iron-recycling macrophages, into plasma (8). When body iron concentration is low and in situations of iron-deficiency anemia, hepcidin expression is minimal, allowing for iron absorption from the diet and iron mobilization from body stores. In contrast, when there are sufficient iron stores or in the case of iron overload, hepcidin inhibits dietary iron absorption, promotes cellular iron sequestration, and reduces iron bioavailability. Hepcidin expression is up-regulated in conditions of inflammation and endoplasmic reticulum stress and down-regulated in hypoxia (9). In Type 2B hemochromatosis, deficiency in hepcidin due to mutations in the hepcidin gene, HAMP, causes abnormal iron accumulation in tissues (see Iron Overload). Of note, hepcidin is also thought to have a major antimicrobial role in the innate immune response by limiting iron availability to invading microorganisms (see Iron withholding defense during infection) (10).
Regulation of intracellular iron
Iron-responsive elements (IREs) are short sequences of nucleotides found in the messenger RNAs (mRNAs) that code for key proteins in the regulation of iron storage, transport, and utilization. Iron regulatory proteins (IRPs: IRP-1, IRP-2) can bind to IREs and control mRNA stability and translation, thereby regulating the synthesis of specific proteins, such as ferritin (iron storage protein) and transferrin receptor-1 (TfR; controls cellular iron uptake) (1, 2).
When the iron supply is low, iron is not available for storage or release into plasma. Less iron binds to IRPs, allowing the binding of IRPs to IREs. The binding of IRPs to IREs located in the 5’end of mRNAs coding for ferritin and ferroportin-1 (iron efflux protein) inhibits mRNA translation and protein synthesis. Translation of mRNA that codes for the key regulatory enzyme of heme synthesis in immature red blood cells is also reduced to conserve iron. In contrast, IRP binding to IREs in the 3’ end of mRNAs that code for TfR and divalent metal transporter-1 (DMT1) stimulates the synthesis of iron transporters, thereby increasing iron uptake into cells (1, 2).
When the iron supply is high, more iron binds to IRPs, thereby preventing the binding of IRPs to IREs on mRNAs. This allows for an increased synthesis of proteins involved in iron storage (ferritin) and efflux (ferroportin-1) and a decreased synthesis of iron transporters (TfR and DMT1) such that iron uptake is limited (2). In the brain, IRPs are also prevented from binding to the 5’end of amyloid precursor protein (APP) mRNA, allowing for APP expression. APP stimulates iron efflux from neurons through stabilizing ferroportin-1. In Parkinson’s disease (PD), APP expression is inappropriately suppressed, leading to iron accumulation in dopaminergic neurons (11, 12).
Iron withholding defense during infection
Iron is required by most infectious agents to grow and spread, as well as by the infected host in order to mount an effective immune response. Sufficient iron is critical for the differentiation and proliferation of T lymphocytes and the generation of reactive oxygen species (ROS) required for killing pathogens (13). During infection and inflammation, hepcidin synthesis is up-regulated, serum iron concentrations decrease, and concentrations of ferritin (the iron storage protein) increase, supporting the idea that sequestering iron from pathogens is an important host defense mechanism (2).
Recycling of iron
Total body content of iron in adults is estimated to be 2.3 g in women and 3.8 g in men (2). The body excretes very little iron; basal losses, menstrual blood loss, and the need of iron for the synthesis of new tissue are compensated by the daily absorption of a small proportion of dietary iron (1 to 2 mg/day). Body iron is primarily found in red blood cells, which contain 3.5 mg of iron per g of hemoglobin. Senescent red blood cells are engulfed by macrophages in the spleen, and about 20 mg of iron can be recovered daily from heme recycling. The released iron is either deposited to the ferritin of spleen macrophages or exported by ferroportin-1 (iron efflux protein) to transferrin (the main iron carrier in blood) that delivers iron to other tissues. Iron recycling is very efficient, with about 35 mg being recycled daily (1).
Assessment of iron status
Measurements of iron stores, circulating iron, and hematological parameters may be used to assess the iron status of healthy people in the absence of inflammatory disorders, parasitic infection, and obesity. Commonly used iron status biomarkers include serum ferritin (iron-storage protein), serum iron, total iron binding capacity (TIBC), and saturation of transferrin (the main iron carrier in blood; TSAT). Soluble transferrin receptor (sTfR) is also an indicator of iron status when iron stores are depleted. In iron deficiency and iron-deficiency anemia, the abundance of cell surface-bound transferrin receptors that bind diferric transferrin is increased in order to maximize the uptake of available iron. Therefore, the concentration of sTfR generated by the cleavage of cell-bound transferrin receptors is increased in iron deficiency. Hematological markers, including hemoglobin concentration, mean corpuscular hemoglobin concentration, mean corpuscular volume of red blood cells, and reticulocyte hemoglobin content can help detect abnormality if anemia is present (9, 14).
Of note, serum ferritin is an acute-phase reactant protein that is up-regulated by inflammation. Importantly, serum hepcidin concentration is also increased by inflammation to limit iron availability to pathogens. Therefore, it is important to include inflammation markers (e.g., C-reactive protein, fibrinogen) when assessing iron status to rule out inflammation (14).
Nutrient Interactions
Vitamin A
Vitamin A deficiency often coexists with iron deficiency and may exacerbate iron-deficiency anemia by altering iron metabolism (15). Vitamin A supplementation has been shown to have beneficial effects on iron-deficiency anemia and improve iron nutritional status among children and pregnant women (15, 16). The combination of vitamin A and iron seems to reduce anemia more effectively than either supplemental iron or vitamin A alone (17). Vitamin A may facilitate the mobilization of iron from storage sites to developing red blood cells for incorporation into hemoglobin (15, 16). Moreover, studies in rats have shown that iron deficiency alters plasma and liver levels of vitamin A (18, 19).
Copper
Adequate copper nutritional status is necessary for normal iron metabolism and red blood cell formation. Anemia is a clinical sign of copper deficiency, and iron has been found to accumulate in the livers of copper-deficient animals, indicating that copper (via copper-containing ceruloplasmin) is required for iron transport to the bone marrow for red blood cell formation (20). The connection between copper availability and iron metabolism has also been established in humans; copper deficiency can lead to secondary ceruloplasmin deficiency and hepatic iron overload and/or cirrhosis (21). Oral copper supplementation restored normal ceruloplasmin levels and plasma ferroxidase activity and corrected the iron metabolism disorder in a copper-deficient subject (22). Moreover, infants fed a high-iron formula absorbed less copper than infants fed a low-iron formula, suggesting that high iron intakes may interfere with copper absorption in infants (23).
Zinc
Zinc is essential to maintain adequate erythropoiesis. When zinc deficiency coexists with iron deficiency, it may exacerbate iron-deficiency anemia (24). On the other hand, high doses of iron supplements, taken together with zinc supplements on an empty stomach, may inhibit the absorption of zinc. When taken with food, supplemental iron does not appear to inhibit zinc absorption. Iron-fortified foods have not been found to impair zinc absorption (25, 26).
Calcium
The presence of calcium decreases iron absorption from both nonheme (i.e., most supplements and food sources other than meat, poultry, and seafood) and heme sources (27). However, calcium supplementation up to 12 weeks has not been found to change iron nutritional status, probably due to a compensatory increase in iron absorption (28). Individuals taking iron supplements should take them two hours apart from calcium-rich food or supplements to maximize iron absorption.
Iodine
Severe iron-deficiency anemia can impair thyroid metabolism in the following ways: (1) by altering the thyroid-stimulating hormone response of the pituitary gland; (2) by reducing the activity of thyroid peroxidase that catalyzes the iodination of thyroglobulin for the production of thyroid hormones; and (3) in the liver by limiting the conversion of T4 to T3, increasing T3 turnover, and decreasing T3 binding to nuclear receptors (29). It is estimated that goiter and iron-deficiency anemia coexist in up to 25% of school-age children in west and north Africa (30). A randomized controlled study in iron-deficient children with goiter showed a greater reduction in thyroid size following the consumption of iodized salt together with 60 mg/day of iron four times per week compared to placebo (31). Additional interventions have confirmed that correcting iron-deficiency anemia improved the efficacy of iodine supplementation to mitigate thyroid disorders (reviewed in 29, 30).
Deficiency
Levels of iron deficiency
Iron deficiency is the most common nutrient deficiency in the US and the world. Levels of iron deficiency are listed below from least to most severe.
Storage iron depletion
Iron stores are depleted, but the functional iron supply is not limited.
Early functional iron deficiency
Before the development of frank anemia, the supply of functional iron to tissues, including bone marrow, is inadequate such as to impair erythropoiesis.
Iron-deficiency anemia
By definition, anemia is present when individual hemoglobin concentrations fall below two standard deviations of the distribution mean for hemoglobin in a healthy population of the same gender and age and living at the same altitude (32). In 2013, iron-deficiency anemia was the leading cause of years lived with disability in children and adolescents in the 50 most populous countries. The countries with the highest prevalence of iron-deficiency anemia in individuals younger than 19 years were Afghanistan (41%) and Yemen (39.8%); India contributed the largest number of cases of anemia (147.9 million). The prevalence in the US was estimated to be 19.3% with nearly 16 million cases of iron-deficiency anemia in children and adolescents (33).
Iron-deficiency anemia occurs when there is inadequate iron to support normal red blood cell formation. The anemia of iron deficiency is usually characterized as microcytic and hypochromic, i.e., red blood cells are measurably smaller than normal and their hemoglobin content is decreased such that they are paler than normal. At this stage of iron deficiency, symptoms may be a result of inadequate oxygen delivery due to anemia and/or suboptimal function of iron-dependent enzymes. Changes in hematological parameters are used in the clinical diagnosis of iron-deficiency anemia (see Assessment of iron status). It is important to remember that iron deficiency is not the only cause of anemia, and that the diagnosis or treatment of iron deficiency solely on the basis of anemia may lead to misdiagnosis or inappropriate treatment of the underlying cause (34). See also the articles on Folate and Vitamin B12 for information on other nutritional causes of anemia.
Symptoms of iron deficiency
Most of the symptoms of iron deficiency are a result of the associated anemia and may include fatigue, rapid heart rate, palpitations, and rapid breathing on exertion. Iron deficiency impairs athletic performance and physical work capacity in several ways. In iron-deficiency anemia, the reduced hemoglobin content of red blood cells results in decreased oxygen delivery to active tissues. Decreased myoglobin levels in muscle cells limit the amount of oxygen that can be delivered to mitochondria for oxidative metabolism. Iron depletion also decreases the oxidative capacity of muscle by diminishing the mitochondrial content of cytochromes and other iron-dependent enzymes required for electron transport and ATP synthesis (see Function) (35).
Poor thyroid function and impaired thyroid hormone synthesis likely disrupt the ability to maintain a normal body temperature on exposure to cold in iron-deficient individuals (see Function). Iron deficiency may also impair neutrophil phagocytosis and microbicidal activity and T-lymphocyte proliferative responses to infection (1). Severe iron-deficiency anemia may result in brittle and spoon-shaped nails, sores at the corners of the mouth, taste bud atrophy, and a sore tongue. In rare cases, advanced iron-deficiency anemia may cause difficulty in swallowing due to the formation of webs of tissue in the throat and esophagus due to a degradation of the pharyngeal muscles (36). The development of esophageal webs, also known as Plummer-Vinson syndrome, may require a genetic predisposition in addition to iron deficiency. Iron deficiency and iron-deficiency anemia in early childhood have been shown to impair psychomotor development and induce short- and long-term behavioral and cognitive alterations (reviewed in 37). Further, pica, a behavioral disturbance characterized by the consumption of non-food items, may be a symptom and a cause of iron deficiency (38).
Individuals at increased risk of iron deficiency
Life stage groups with increased requirements
Neonates and infants up to six months of age: Inadequate maternal iron body stores and anemia during pregnancy may reduce the duration of gestation and birth weight; preterm and/or low body-weight newborns are at increased risk of iron-deficiency anemia (14). Pregnancy complications, including preeclampsia and gestational diabetes mellitus, may also lead to low iron stores in preterm and term infants (14).
Most of the 150 to 250 mg of iron present in a full-term healthy newborn is accumulated during the third trimester of pregnancy and is sufficient for the first four to six months of life (34). Iron stores are essential for infants less than six months of age because breast milk is relatively poor in iron (0.2 mg/L-0.4 mg/L), and intestinal absorption of iron remains low until six months of age. High iron requirements during this period of sustained and rapid growth rate can worsen the deficit in body iron in preterm infants (14). Moreover, a review of randomized controlled trials suggested that infants with an early umbilical cord clamping (≤1 min after birth) are at least twice more likely to be iron-deficient at three to six months compared to those with delayed cord clamping (39). Yet, healthy full-term infants have little need for external sources of iron before six months of age (1).
Infants and children between the ages of 6 months and 3 years: A full-term infant's iron stores are usually sufficient to last for the early months of life, but there is an increased risk of iron deficiency for infants older than six months (1). Given the sustained need of iron for increasing tissue mass, blood volume, and replenishing iron stores, the recommended dietary allowance (RDA) for iron is 11 mg/day for infants aged seven to 12 months, as established by the US Institute of Medicine (see Table 1).
The RDA for iron is 7 mg/day for toddlers aged 1 to 3 years old. Based on the US National Health and Nutrition Examination Survey (NHANES) 1999-2002 data, the prevalence of iron deficiency in toddlers aged 12 to 35 months varies from 6.6%-15.2%, and the prevalence of iron-deficiency anemia is 0.9%-4.4%, depending on ethnicity and socioeconomic status (14).
Of note, the World Health Organization (WHO) and the American Academy of Pediatrics recommend universal screening for anemia at one year of age. Yet, a recent report by the US Preventive Services Task Force (USPSTF) stated there was insufficient evidence to assess the benefits versus harms of screening (34, 40).
Adolescents: Early adolescence is period of rapid growth. Blood loss that occurs with menstruation in adolescent girls adds to the increased iron requirement of adolescence (1). The RDA of iron is 11 mg/day and 15 mg/day for adolescent boys and girls, respectively (see Table 1).
Nonpregnant women of childbearing age: Based on data from NHANES 2003-2006, the percentage of US women with two out of three markers of iron status (i.e., hemoglobin, ferritin, and % transferrin saturation) below cutoff values for deficiency was 9.8% in nonpregnant women (41).
The use of oral contraceptives decreases menstrual blood losses and is thus associated with improved iron status compared to intrauterine devices (copper coil) (1).
Breast-feeding is associated with lower dietary iron needs, allowing for the repletion of iron stores depleted during pregnancy and delivery. However, iron repletion may be incomplete in high-parity women who are therefore at increased risk of iron deficiency (41).
Pregnant women: The requirement for iron is significantly increased during pregnancy due to increased iron utilization by the developing fetus and placenta, as well as maternal blood volume expansion (42). Analysis of data from NHANES 2005-2006 found that 18.1% of pregnant women (mean age, 27.5 years) were deficient in iron, as assessed by the log ratio of soluble transferrin receptor to serum ferritin (43). The prevalence of iron deficiency was greater during the second (20.7%) and third (29.7%) trimesters compared to the first trimester (4.5%) of gestation. Further, iron deficiency in pregnancy was found to be more prevalent in Mexican (23.6%) and Black Americans (29.6%) than in non-Hispanic White Americans (13.9%) (43).
Individuals with chronic blood losses
Chronic bleeding or acute blood loss may result in iron deficiency. One milliliter (mL) of blood with a hemoglobin concentration of 150 g/L contains 0.5 mg of iron. Thus, chronic loss of very small amounts of blood may result in iron deficiency.
Parasitic infestation: A common cause of chronic blood loss and iron deficiency in developing countries is intestinal parasitic infection (44).
Frequent blood donation: Individuals who donate blood frequently, especially menstruating women, may need to increase their iron intake to prevent deficiency because each 500 mL of blood donated contains between 200 and 250 mg of iron (45, 46).
Regular intense exercise: Daily iron losses have been found to be greater in athletes involved in intense endurance training. This may be due to expanding blood cell mass and muscle mass, increased microscopic bleeding from the gastrointestinal tract (with the regular use of anti-inflammatory drugs), or increased fragility and hemolysis of red blood cells (47). The Food and Nutrition Board estimates that the average requirement for iron may be 30% higher for those who engage in regular intense exercise (25).
Individuals with decreased iron absorption
Celiac disease: Celiac disease (celiac sprue) is an autoimmune disorder estimated to occur in 1% of the population. When people with celiac disease consume food or products that contain gluten, the immune system response damages the intestinal mucosa, which may result in nutrient malabsorption and iron-deficiency anemia (48).
Atrophic gastritis: This condition is usually associated with the presence of antibodies directed towards stomach cells and has been implicated in pernicious anemia (see the article on Vitamin B12). Atrophic gastritis simultaneously impairs the absorption of both vitamin B12 and iron; yet, in menstruating women, iron deficiency may occur years before the depletion of vitamin B12 body stores (47).
Helicobacter pylori infection: H. pylori infection is associated with iron-deficiency anemia, especially in children, even in the absence of gastrointestinal bleeding. Data from NHANES 2000-2001 in individuals older than three years showed that the presence of iron deficiency (based on serum ferritin concentrations) was 40% more prevalent in those infected with H. pylori than in H. pylori-free individuals (49). Occult gastrointestinal bleeding and competition for dietary iron by bacteria may explain iron deficiency in infected individuals. Moreover, Helicobacter pylori infection may also play a role in the pathogenesis of atrophic gastritis (47).
Inflammatory bowel diseases (IBD): Iron-deficiency anemia is commonly reported among patients with IBD (e.g., ulcerative colitis, Crohn’s disease), likely due to both impaired intestinal absorption of iron and blood loss from ulcerated mucosa (50).
Gastric bypass surgery: Some types of gastric bypass (bariatric) surgery increase the risk of iron deficiency by causing malabsorption of iron, among other nutrients (51).
Obesity: An inverse association between body weight and iron status has been reported in several observational studies in children and adults (52, 53). Higher hepcidin expression in obese people may impair iron absorption despite adequate dietary intake of iron. Weight loss might lower serum hepcidin concentration and improve iron status in obese individuals (9).
Anemia of chronic disease: Acute and chronic inflammation may lead to abnormally low circulating concentrations of iron and to the development of anemia. This type of anemia of inflammation, also known as anemia of chronic disease (ACD), is commonly observed in inflammatory disorders, cancer, critical illness, trauma, chronic infection, and parasitic infestation. It is thought that anemia develops because dietary iron absorption and iron mobilization from body stores are inhibited by inflammation-induced hepcidin up-regulation (see also Systemic regulation of iron homeostasis) (9).
Other causes of iron deficiency
Vegetarian diet with inadequate sources of iron: Because iron from plants (nonheme iron) is less efficiently absorbed than that from animal sources (see Sources), the US Food and Nutrition Board (FNB) of the Institute of Medicine (IOM) estimated that the bioavailability of iron from a vegetarian diet was only 10% versus 18% from a mixed Western diet. Therefore, the recommended dietary allowance (RDA) of iron for individuals consuming a completely vegetarian diet may be 1.8 times higher than the RDA for non-vegetarians (25). Yet, a vegetarian diet does not appear to be associated with an increased risk of iron deficiency when it includes whole grains, legumes, nuts, seeds, dried fruit, iron-fortified cereal, and green leafy vegetables (see Sources) (54).
Chronic kidney disease (CKD): Iron losses in CKD patients are due to significant gastrointestinal blood loss (1.2 L blood loss/year corresponding to ~400 mg iron/year) compared to individuals with normal kidney function (0.83 mL blood loss/day corresponding to ~100 mg iron/year). Estimated blood losses are even larger in patients on hemodialysis, and iron losses may be 1,000 to 2,000 mg/year or higher. Persistent inflammation in CKD patients may also contribute to inadequate iron supply for red blood cell formation despite adequate body iron stores (55).
The Recommended Dietary Allowance (RDA)
The RDA for iron was revised in 2001 and is based on the prevention of iron deficiency and maintenance of adequate iron stores in individuals eating a mixed diet (Table 1; 25).
| Life Stage | Age | Males (mg/day) | Females (mg/day) |
|---|---|---|---|
| Infants | 0-6 months | 0.27 (AI) | 0.27 (AI) |
| Infants | 7-12 months | 11 | 11 |
| Children | 1-3 years | 7 | 7 |
| Children | 4-8 years | 10 | 10 |
| Children | 9-13 years | 8 | 8 |
| Adolescents | 14-18 years | 11 | 15 |
| Adults | 19-50 years | 8 | 18 |
| Adults | 51 years and older | 8 | 8 |
| Pregnancy | all ages | - | 27 |
| Breast-feeding | 18 years and younger | - | 10 |
| Breast-feeding | 19 years and older | - | 9 |
Disease Prevention
Prevention or alleviation of iron deficiency or iron-deficiency anemia can limit the impact of iron inadequacy and defective erythropoiesis on the following health conditions and diseases.
Impaired psychomotor, cognitive, and intellectual development in children
Iron is critical for the development of the central nervous system, and iron deficiency is thought to be especially detrimental during the prenatal and early postnatal periods. Iron-dependent enzymes are required for nerve myelination, neurotransmitter synthesis, and normal neuronal energy metabolism (56). Most observational studies have found relationships between iron deficiency — with or without anemia — in children and poor cognitive development, poor school achievement, and abnormal behavior patterns (reviewed in 37). Whether psychomotor and mental deficits may be attributed to the lack of iron, only, or to a combination effect of iron deficiency and low hemoglobin concentrations — like in iron-deficiency anemia and anemia of inflammation — in early childhood remains unclear (14).
A recent systematic review of six small placebo-controlled trials (published between 1978 and 1989) in children with iron-deficiency anemia younger than 27 months found no convincing evidence that iron therapy (for less than 11 days) had any consistent effect on measures of psychomotor and mental development within 30 days of treatment initiation (57). Only one randomized, double-blind trial in anemic, iron-deficient infants examined the impact of iron therapy for four months and found a significant benefit on indices of cognitive development that needs to be further confirmed (58). A review of five randomized controlled trials in non-anemic, iron-deficient infants (0-9 months old) suggested an improvement in psychomotor (but not mental) development throughout the first 18 months of life (59). Iron supplementation in early infancy (4 to 6 months) also failed to demonstrate any long-term effect on cognitive performance and school performance at the age of 9 years compared to placebo (60). At present, evidence supporting any benefits of iron therapy on neurodevelopment outcomes in infants with iron deficiency, with or without anemia, remains limited.
Iron therapy might be more effective at improving cognitive outcomes in older children with anemia and/or iron deficiency. A systematic review of 17 randomized controlled trials found that iron supplementation had no effect on mental development of children under the age of 27 months but modestly improved scores of mental development in children over seven years of age (61). A more recent meta-analysis of randomized controlled trials in children older than six years, adolescents, and women with iron deficiency, anemia, or iron-deficiency anemia suggested that supplemental iron could improve attention and concentration irrespective of participants’ iron status (62). A potential improvement in IQ measures with iron therapy was also reported in anemic participants regardless of their iron status. No additional benefits were observed regarding measures of memory performance, psychomotor function, and school achievements.
Alterations in brain functions due to iron deficiency are likely to be resistant to iron therapy when they occur in early childhood. Long-term consequences of early life iron deficiency may include poor socioeconomic achievements and increased risk of certain psychopathologies, including anxiety, depression, and schizophrenia (56).
Adverse pregnancy outcomes
Epidemiological studies provide strong evidence of an association between severe anemia in pregnant women and adverse pregnancy outcomes, such as low birth weight, preterm birth, and neonatal and maternal mortality (63). Although iron deficiency can be a major contributing factor to severe anemia, evidence that iron-deficiency anemia causes poor pregnancy outcomes is still lacking. In addition, iron supplementation during pregnancy was shown to improve iron status and hematological parameters in women but failed to significantly reduce adverse pregnancy outcomes, including low birth weight and/or prematurity, neonatal death, and congenital anomalies (64). Moreover, routine supplementation during pregnancy had no effect on the length of gestation or newborn Apgar scores (40). Nevertheless, most experts consider the control of maternal anemia to be an important part of prenatal health care, and the IOM recommends screening for anemia in each trimester of pregnancy (65).
The requirement for iron is greatly increased in the second and third trimesters, and the RDA for pregnant women is 27 mg/day of iron (see The Recommended Dietary Allowance) (25). The American College of Obstetricians and Gynecologists recommend screening all pregnant women for anemia and advise iron supplementation when required (66). Nonetheless, the US Preventive Services Task Force (40) and the American Academy of Family Physicians (67) consider that evidence is lacking to evaluate the harms and benefits of screening for iron-deficiency anemia and supplementing with iron during pregnancy.
In malaria-endemic regions, however, iron supplementation may improve pregnancy outcomes when provided in conjunction with measures of prevention and management of malaria. Two recent randomized, placebo-controlled trials failed to find an increased risk of malaria infection in both iron-deficient and iron-replete pregnant women supplemented with iron, supporting the use of universal iron supplementation in malaria-endemic countries that adopt malaria intermittent preventive treatment (IPT) (68, 69).
Lead toxicity
Children who are chronically exposed to lead, even in small amounts, are more likely to develop learning disabilities, behavioral problems, and have low IQs. Deficits in growth and neurologic development may occur in the infants of women exposed to lead during pregnancy and lactation. In adults, lead toxicity may result in kidney damage and high blood pressure. Although the use of lead in paint products, gasoline, and food cans has been discontinued in the US, lead toxicity continues to be a significant health problem, especially in children living in inner cities (70). In 2012, the US Centers for Disease Control and Prevention set the reference value for blood lead concentration at 5 micrograms per deciliter (μg/dL) to identify children at risk. Yet, there is no known blood lead concentration below which children are 100% safe (71).
Iron deficiency and lead poisoning share a number of the same risk factors, including low socioeconomic status, ethnic minority groups, and residence in urban areas. Iron deficiency may increase the risk of lead poisoning in children, especially by increasing the intestinal absorption of lead via the DMT1 intestinal transporter (72). However, the use of iron supplementation in lead poisoning might be reserved for children who are truly iron deficient or for iron-replete children with chronic lead exposure (e.g., living in lead-exposed housing) (72).
Disease Treatment
Restless legs syndrome
Restless legs syndrome (RLS; also called Willis-Ekbom disease) is a neurologic movement disorder of unknown etiology. People with RLS experience unpleasant sensations resulting in an irresistible urge to move their legs and transient relief with movement. These sensations are more common at rest and often interfere with sleep (73). The prevalence of RLS is higher in women than in men and increases with age (74). This syndrome appears to be inherited in about 50% of patients but has also been related to chronic kidney failure (73). Iron deficiency may be involved in RLS development, possibly by affecting the activity of tyrosine hydroxylase, a rate limiting iron-dependent enzyme in the synthesis of the neurotransmitter, dopamine (74). The management of RLS includes iron therapy and the use of drugs like dopamine agonists (73). Current clinical evidence is insufficient to evaluate whether iron therapy may help relieve some RLS symptoms (74). Yet, the Medical Advisory Board of the Willis-Ekbom Disease Syndrome Foundation suggests that iron status should be assessed in all patients with RLS, and iron therapy be attempted on a case-by-case basis in those who might benefit from it (73).
Sources
Food sources
The amount of iron in food or supplements that is absorbed and used by the body is influenced by the iron nutritional status of the individual and whether or not the iron is in the form of heme. Because it is absorbed by a different mechanism than nonheme iron, heme iron is more readily absorbed and its absorption is less affected by other dietary factors (2). In an attempt to improve body iron status, iron absorption is enhanced in individuals who are anemic or iron deficient compared to iron-replete individuals.
Heme iron
Heme iron comes mainly from hemoglobin and myoglobin in meat, poultry, and fish. Although heme iron accounts for only 10%-15% of the iron found in the diet, it may provide up to one-third of total absorbed dietary iron (54). The absorption of heme iron is less influenced by other dietary factors than that of nonheme iron (27).
Nonheme iron
Plants, dairy products, meat, and iron salts added to food and supplements are all sources of nonheme iron. The absorption of nonheme iron is strongly influenced by enhancers and inhibitors present in the same meal (27).
Enhancers of nonheme iron absorption
- Vitamin C (ascorbic acid): Vitamin C strongly enhances the absorption of nonheme iron by reducing dietary ferric iron (Fe3+) to ferrous iron (Fe2+) and forming an absorbable, iron-ascorbic acid complex (75).
- Other organic acids: Citric, malic, tartaric, and lactic acids have some enhancing effects on nonheme iron absorption (1).
- Meat, poultry, and fish: Aside from providing highly absorbable heme iron, meat, fish, and poultry also enhance nonheme iron absorption. The mechanism for this enhancement of nonheme iron absorption is not clear (1, 25).
Inhibitors of nonheme iron absorption
- Phytic acid (phytate): Phytic acid, present in legumes, whole grains, nuts, and seeds, inhibits nonheme iron absorption, probably by binding to it. Small amounts of phytic acid (5 to 10 mg) can reduce nonheme iron absorption by 50%. The absorption of iron from legumes, such as soybeans, black beans, lentils, mung beans, and split peas, has been shown to be as low as 2% (25). Food preparation, including soaking, germination, fermentation, and cooking, can help remove or degrade phytic acid (27).
- Polyphenolic compounds: Polyphenolic compounds in coffee, black tea, and herbal tea can markedly inhibit the absorption of nonheme iron (76). This effect may be reduced by the presence of vitamin C (27, 77).
- Soy protein: Soy protein, such as that found in tofu, has an inhibitory effect on iron absorption that is only partly related to its phytic acid content (27, 77).
- Calcium: Calcium appears to affect iron absorption from both heme and nonheme sources. Yet, its effect appears to be limited when one consumes a wide variety of food with varied levels of enhancers and inhibitors of iron absorption (27).
National surveys in the US have indicated that the average dietary intake of iron is 16 to 18 mg/day in men, 12 mg/day in pre- and postmenopausal women, and about 15 mg/day in pregnant women (25). Thus, the majority of premenopausal and pregnant women in the US consume less than the RDA for iron, and many men consume more than the RDA (see The Recommended Dietary Allowance). In the US, most grain products are fortified with nonheme iron. The iron content of some relatively iron-rich foods is listed in milligrams (mg) in Table 2. For more information on the nutrient content of specific foods, search USDA's FoodData Central.
Supplements
Iron supplements are indicated for the prevention and treatment of iron deficiency and iron deficiency anemia. Individuals who are not at risk of iron deficiency (e.g., adult men and postmenopausal women) should not take iron supplements without an appropriate medical evaluation. A number of iron supplements are available, and different forms provide different proportions of elemental iron. Ferrous sulfate heptahydrate is 20% elemental iron, ferrous sulfate monohydrate is 33% elemental iron, ferrous gluconate is 12% elemental iron, and ferrous fumarate is 33% elemental iron. If not stated otherwise, the iron discussed in this article is elemental iron.
Iron Overload
Deregulation of intestinal iron absorption will result in iron overload because the body cannot excrete excess iron (2). However, iron overload due to prolonged iron supplementation is very rare in healthy individuals without a genetic predisposition. Several genetic disorders may lead to pathological accumulation of iron in the body despite normal iron intake. Supplementation of individuals who are not iron deficient should be avoided due to the frequency of undetected inherited diseases and recent concerns about the more subtle effects of chronic excess iron intake (see Diseases associated with iron overload).
Inherited iron-overload diseases
Hereditary hemochromatosis
Hereditary hemochromatosis (HH) refers to late-onset, autosomal recessive disorders of iron metabolism that result in iron accumulation in the liver, heart, and other tissues. Disorders may lead to cirrhosis of the liver, diabetes mellitus, cardiomyopathy (heart muscle damage), hypogonadism, arthropathy (joint problems), and increased skin pigmentation (reviewed in 78). There are four main types of HH, which are classified according to the specific gene that is mutated. The most common type of HH, called Type 1 or HFE-related HH, results from mutations in the HFE gene (79, 80). The majority of Type 1 HH cases are homozygous for mutation C282Y G>A (rs1800560) in the HFE gene. Another mutation found in 4% of patients with Type 1 HH is H63D C>G (rs1799945) in the HFE gene. The protein encoded by the HFE gene is thought to play a role in regulating intestinal absorption of dietary iron and with sensing the body’s iron stores (81). HFE gene mutations are associated with an increased cellular uptake of iron. With a typical disease onset before age 30, juvenile hemochromatosis (HH Type 2) is much rarer than Type 1 HH and results from genetic mutations affecting either hemojuvelin (Type 2A) or hepcidin (Type 2B) function (82). HH Type 3 results from mutations in the transferrin receptor 2 gene (TFR2), and HH Type 4 (also called Ferroportin disease) results from mutations in the gene encoding ferroportin-1 (SLC40A1), a protein important in the export of iron from cells (see Regulation). Type 4 HH is the second most common inherited iron overload disorder after Type 1 HH (78).
Iron overload in HH is treated by phlebotomy, the removal of 500 mL of blood at a time, at intervals determined by the severity of the iron overload. Chelation therapy is an alternate option to deplete iron in HH patients who cannot undergo phlebotomy treatment. Individuals with HH are advised to avoid supplemental iron, but generally not told to avoid iron-rich food. High-dose vitamin C regimens may worsen iron overload in patients with HH (75). Alcohol consumption is strongly discouraged due to the increased risk of cirrhosis of the liver (83). Genetic testing, which requires a blood sample, is available for those who may be at risk for HH, for example, individuals with a family history of hemochromatosis.
Other inherited conditions
Other genetic disorders leading to iron overload include aceruloplasminemia, hypotransferrinemia, Friedreich’s ataxia, and porphyria cutanea tarda (2).
Acquired iron-overload diseases
Iron overload may develop in individuals with severe hereditary anemias that are not caused by iron deficiency. Excessive dietary absorption of iron may occur in response to the body's continued efforts to form red blood cells. Beta-thalassemia is characterized by defective hemoglobin A synthesis due to mutations in the β-globin gene. Patients affected by thalassemia intermedia do not require blood transfusion as do those affected by the most severe form of the disease (called thalassemia major), yet they develop iron overload due to increased intestinal iron absorption (84). Other anemic patients at risk of iron overload include those with sideroblastic anemia, hemolytic anemia, pyruvate kinase deficiency, and thalassemia major, especially because they are treated with numerous transfusions. Patients with hereditary spherocytosis and thalassemia minor do not usually develop iron overload unless they are misdiagnosed as having iron deficiency and treated with large doses of iron over many years. Iron overload has also been associated with hemodialysis and chronic liver diseases (metabolic, viral, and alcoholic) (2).
Diseases associated with iron overload
Toxic iron deposition in vital organs in hereditary hemochromatosis (HH) has been associated with an increased incidence of liver cancer, type 2 diabetes mellitus, and neurodegenerative disease. Iron overload might also increase the risk of chronic disease in HH-free individuals. Nevertheless, whether iron tissue accumulation in those unaffected by genetic disorders is due to high dietary iron intakes is not yet fully understood (1).
Cancer
Hereditary hemochromatosis that is characterized by abnormal hepatic iron accumulation is a risk factor for liver cancer (hepatocellular carcinoma; HCC). Iron accumulation is thought to function as a carcinogen by increasing oxidative stress that causes damage to lipids, proteins, and DNA. A meta-analysis of nine observational studies found an increased HCC risk with the C282Y mutation in the HFE gene of healthy participants and patients with chronic liver disease (see Iron Overload) (85). Other meta-analyses have reported associations between HFE gene mutations C282Y and H63D and increased risks of overall cancer (86, 87). However, studies reporting on HFE gene mutations and risk of cancer at extra-hepatic sites are rather scarce and/or inconsistent. Some, but not all, observational studies found significant associations between the C282Y mutation and risk of colorectal (88), breast (88, 89), and epithelial ovarian cancer (90). The presence of the H63D mutation in the HFE gene was linked to an increased risk of leukemia (91, 92) and gastric cancer (93).
Whether high dietary iron could increase the risk of cancer in individuals without hemochromatosis has also been investigated. The consumption of red or processed meat (but not white meat), rich in heme iron, has been linked to an increased risk of colorectal cancer (CRC) (94). Exposure to carcinogenic compounds (called heterocyclic amines) generated when meat is cooked at high temperatures and to carcinogenic N-nitroso compounds formed in the gastrointestinal tract following consumption of red and processed meat may explain such an association (95). Several meta-analyses of observational studies have also suggested a potential association of heme iron in red meat with CRC (96-98). This has been explained by an increased exposure of colonic cells to potentially damaging N-nitroso compounds and lipid peroxidation end-products derived from heme iron-catalyzed reactions (99). Further, recent results from the large European Prospective Investigation into Cancer and Nutrition (EPIC) study suggested a higher risk of esophageal adenocarcinoma with high intakes of red/processed meat and heme iron (100).
Cardiovascular disease
Experimental studies have suggested a role for iron-induced oxidative stress in vessel wall damage and the development of atherosclerosis, which underlies most forms of cardiovascular disease (101). However, epidemiological studies of iron nutritional status and cardiovascular disease in humans have yielded conflicting results. A recent systematic review and meta-analysis of 17 prospective cohort studies in 156,427 participants (9,236 cases of coronary heart disease [CHD] or myocardial infarction [MI]) did not find evidence to support the existence of strong associations between a number of different measures of iron status and CHD/MI (102). Only individuals in the highest versus lowest tertile of serum transferrin saturation exhibited an 18% lower incidence of CHD/MI (102). Another meta-analysis of 21 prospective studies found serum transferrin saturation and serum iron to be inversely associated with the risk of CHD. However, the authors noted that most studies failed to adjust for the confounding effects of inflammation (103). The review also reported an inverse association between CHD incidence and total dietary iron intake, but dietary heme iron was positively associated with CHD incidence (103). Although the relationship between iron stores and CHD/MI requires further clarification, it would be prudent for those who are not at risk of iron deficiency (e.g., adult men and postmenopausal women) to avoid excess iron intake (see the LPI Rx for Health).
Type 2 diabetes mellitus and metabolic syndrome
Individuals with hereditary hemochromatosis (HH) are known to be at a heightened risk of developing type 2 diabetes mellitus (104). Increasing evidence also suggests a role for iron excess in the pathogenesis of type 2 diabetes independent of hemochromatosis. Cross-sectional, case-control, and prospective cohort studies have reported an increased risk of type 2 diabetes (105) and metabolic syndrome (106) with high versus low ferritin concentrations (reflecting iron body stores) after adjustment for inflammation. It is currently unclear how other indices of iron status relate to the risk of type 2 diabetes (107-110). Iron overload-induced oxidative stress in patients with HH is thought to damage pancreatic β-cells and impair insulin secretion. In subjects free of HH, iron excess might damage the liver, interfering with glucose metabolism and triggering insulin resistance, rather than impair β-cell function (111, 112). Iron removal by phlebotomy has been shown to improve metabolic parameters in subjects with type 2 diabetes (113) and metabolic syndrome (114). Additional randomized controlled trials are needed to determine whether lowering body stores of iron will aid in the prevention of type 2 diabetes and metabolic syndrome.
Neurodegenerative disease
Iron is required for normal brain and nerve function through its involvement in cellular metabolism, as well as in the synthesis of neurotransmitters and myelin. Deregulation of iron homeostasis has been observed in a number of neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis (ALS; Lou Gehrig’s disease) (115-117). The abnormal accumulation of iron in the brain does not appear to be a result of increased dietary iron, but rather a disruption in the complex process of cellular iron regulation (117). Brain iron accumulation can result in increased oxidative stress, and the brain is particularly susceptible to oxidative damage. Mechanisms behind the disruption of iron homeostasis in the brain of patients affected by neurodegenerative disease are actively being investigated. For example, studies using genetically modified mouse models indicated that the suppression of amyloid precursor protein (APP) expression by upstream nitric oxide (NO) elevation (11) or loss of Tau protein (12) could impair neuronal export of iron and lead to iron accumulation in specific brain regions affected in Parkinson’s disease. A pilot, double-blind, placebo-controlled trial in patients with early-stage Parkinson’s disease demonstrated oral administration of the iron chelator, deferiprone, for 12 months reduced iron deposition in the part of the brain called substantia nigra and improved motor performance without compromising systemic iron homeostasis (118, 119).
Safety
Toxicity
Overdose
Accidental overdose of iron-containing products is the single largest cause of poisoning fatalities in children under six years of age. Although the oral lethal dose of elemental iron is approximately 180 to 250 mg/kg of body weight, considerably less has been fatal. Symptoms of acute toxicity may occur with iron doses of 20 to 60 mg/kg of body weight. Iron overdose is an emergency situation because the severity of iron toxicity is related to the amount of elemental iron absorbed. Acute iron poisoning produces symptoms in four stages: (1) Within one to six hours of ingestion, symptoms may include nausea, vomiting, abdominal pain, tarry stools, lethargy, weak and rapid pulse, low blood pressure, fever, difficulty breathing, and coma; (2) If not immediately fatal, symptoms may subside for about 24 hours; (3) Symptoms may return 12 to 48 hours after iron ingestion and may include serious signs of failure in the following organ systems: cardiovascular, kidney, liver, hematologic (blood), and central nervous system; and (4) Long-term damage to the central nervous system, liver (cirrhosis), and stomach may develop two to six weeks after ingestion (25, 120).
Adverse effects
At therapeutic levels used to treat iron deficiency, iron supplements may cause gastrointestinal irritation, nausea, vomiting, diarrhea, or constipation. Stools will often appear darker in color. Iron-containing liquids can temporarily stain teeth, but diluting the liquid helps to prevent this effect (120). Taking iron supplements with food instead of on an empty stomach may relieve gastrointestinal effects. The Food and Nutrition Board (FNB) of the Institute of Medicine based the tolerable upper intake level (UL) for iron on the prevention of gastrointestinal distress (Table 3). The UL for adolescents (14-18 years) and adults, including pregnant and breast-feeding women, is 45 mg/day. It should be noted that the UL is not meant to apply to individuals being treated with iron under close medical supervision. Individuals with hereditary hemochromatosis or other conditions of iron overload, as well as individuals with alcoholic cirrhosis and other liver diseases, may experience adverse effects at iron intake levels below the UL (25).
| Age Group | UL (mg/day) |
|---|---|
| Infants 0-12 months | 40 |
| Children 1-13 years | 40 |
| Adolescents 14-18 years | 45 |
| Adults 19 years and older | 45 |
Drug interactions
Medications that decrease stomach acidity, such as antacids, histamine (H2) receptor antagonists (e.g., cimetidine, ranitidine), and proton-pump inhibitors (e.g., omeprazole, lansoprazole), may impair iron absorption. Taking iron supplements at the same time as the following medications may result in decreased absorption and efficacy of the medication: carbidopa and levodopa (Sinemet), levothyroxine (Synthroid, Levoxyl), methyldopa (Aldomet), penicillamine (Cuprimine, Depen), quinolones, tetracyclines, and bisphosphonates (120). Therefore, it is best to take these medications two hours apart from iron supplements. Cholestyramine (Questran) and colestipol (Colestid), used to lower blood cholesterol concentrations, should also be taken at least four hours apart from iron supplements because they may interfere with iron absorption (121).
Does iron supplementation increase the risk of malaria in malaria-endemic regions?
Despite the critical functions of iron in the immune response, the nature of the relationship between iron status and susceptibility to infection, especially with respect to malaria, has been controversial. Because iron withholding is a recognized defense mechanism against pathogens (see Iron withholding defense during infection), concerns have been raised regarding the safety of iron supplementation, especially in iron-replete children living in malaria-endemic regions (122).
Iron supplementation of children residing in the tropics has been associated with increased risk of clinical malaria and other infections like pneumonia (123, 124). A randomized controlled trial in 24,076 children (ages, 1-35 months) living in a malaria-endemic region of eastern Africa (Tanzania) investigated the effects of supplemental iron and folic acid, with or without zinc, compared to the effects of zinc alone or a placebo, on all-cause mortality and hospital admissions (125). The administration of iron, folic acid, and/or zinc was found to increase the risk of serious adverse effects, hospital admission, and death, and was therefore prematurely halted. Further analyses of the trial revealed that iron-replete children were more likely than iron deficient-children (with or without anemia) to be at risk of adverse effects following iron supplementation (125). Such a potential risk of adverse effects with routine iron supplementation was not observed in preschool children in settings without malaria (southern Nepal) (126).
A recent review of 35 trials indicated that iron supplementation did not increase the risk of clinical malaria or other parasitic diseases, infections, and all-cause mortality in children living in malaria-endemic regions in which prevention and management of malaria are available (127). Moreover, a pooled analysis of three high-quality trials demonstrated that supplemental iron combined with anti-malarial treatment protected children against clinical malaria and improved hematological parameters (127). The World Health Organization (WHO) currently recommends the provision of iron supplementation in infants and children, together with measures of malaria prevention, diagnosis, and treatment in malaria-endemic areas (128).
Linus Pauling Institute Recommendation
Following the RDA for iron should provide sufficient iron to prevent deficiency without causing adverse effects in most individuals. Although sufficient iron can be obtained through a varied diet, a considerable number of people do not consume adequate iron to prevent deficiency. A multivitamin/mineral supplement containing 100% of the daily value (DV) for iron provides 18 mg of elemental iron. While this amount of iron may be beneficial for premenopausal women, it is well above the RDA for men and postmenopausal women.
Adult men and postmenopausal women
Since hereditary hemochromatosis is not uncommon and the effects of long-term dietary iron excess on chronic disease risk are not yet clear, men and postmenopausal women who are not at risk of iron deficiency should take a multivitamin/mineral supplement without iron. A number of multivitamins formulated specifically for men or those over 50 years of age do not contain iron.
Older adults (>50 years)
Moderately elevated iron stores might be much more common than iron deficiency in middle-age and older individuals (129). Thus, older adults should not generally take nutritional supplements containing iron unless they have been diagnosed with iron deficiency. Moreover, it is extremely important to determine the underlying cause of the iron deficiency, rather than simply treating it with iron supplements (see The Recommended Dietary Allowance).
Authors and Reviewers
Originally written in 2001 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in March 2003 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in January 2006 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in August 2009 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2016 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in May 2016 by:
Marianne Wessling-Resnick, Ph.D.
Professor of Nutritional Biochemistry
Department of Genetics and Complex Diseases
Harvard T.H. Chan School of Public Health
The 2016 update of this article was underwritten, in part, by a grant from Bayer Consumer Care AG, Basel, Switzerland.
Copyright 2001-2024 Linus Pauling Institute
References
1. Aggett PJ. Iron. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Ames: Wiley-Blackwell; 2012:506-520.
2. Wessling-Resnick M. Iron. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed: Lippincott Williams & Wilkins; 2014:176-188.
3. Winter WE, Bazydlo LA, Harris NS. The molecular biology of human iron metabolism. Lab Med. 2014;45(2):92-102. (PubMed)
4. Burmester T, Hankeln T. What is the function of neuroglobin? J Exp Biol. 2009;212(Pt 10):1423-1428. (PubMed)
5. Salminen A, Kauppinen A, Kaarniranta K. 2-Oxoglutarate-dependent dioxygenases are sensors of energy metabolism, oxygen availability, and iron homeostasis: potential role in the regulation of aging process. Cell Mol Life Sci. 2015;72(20):3897-3914. (PubMed)
6. Zhang C. Essential functions of iron-requiring proteins in DNA replication, repair and cell cycle control. Protein Cell. 2014;5(10):750-760. (PubMed)
7. Anderson GJ, Darshan D, Wilkins SJ, Frazer DM. Regulation of systemic iron homeostasis: how the body responds to changes in iron demand. Biometals. 2007;20(3-4):665-674. (PubMed)
8. Fleming MD. The regulation of hepcidin and its effects on systemic and cellular iron metabolism. Hematology Am Soc Hematol Educ Program. 2008:151-158. (PubMed)
9. Tussing-Humphreys L, Pusatcioglu C, Nemeth E, Braunschweig C. Rethinking iron regulation and assessment in iron deficiency, anemia of chronic disease, and obesity: introducing hepcidin. J Acad Nutr Diet. 2012;112(3):391-400. (PubMed)
10. Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090-2093. (PubMed)
11. Ayton S, Lei P, Hare DJ, et al. Parkinson's disease iron deposition caused by nitric oxide-induced loss of beta-amyloid precursor protein. J Neurosci. 2015;35(8):3591-3597. (PubMed)
12. Lei P, Ayton S, Finkelstein DI, et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med. 2012;18(2):291-295. (PubMed)
13. Bhaskaram P. Immunobiology of mild micronutrient deficiencies. Br J Nutr. 2001;85 Suppl 2:S75-80. (PubMed)
14. Baker RD, Greer FR, Committee on Nutrition American Academy of Pediatrics. Diagnosis and prevention of iron deficiency and iron-deficiency anemia in infants and young children (0-3 years of age). Pediatrics. 2010;126(5):1040-1050. (PubMed)
15. Semba RD, Bloem MW. The anemia of vitamin A deficiency: epidemiology and pathogenesis. Eur J Clin Nutr. 2002;56(4):271-281. (PubMed)
16. Allen LH. Iron supplements: scientific issues concerning efficacy and implications for research and programs. J Nutr. 2002;132(4 Suppl):813S-819S. (PubMed)
17. Suharno D, West CE, Muhilal, Karyadi D, Hautvast JG. Supplementation with vitamin A and iron for nutritional anaemia in pregnant women in West Java, Indonesia. Lancet. 1993;342(8883):1325-1328. (PubMed)
18. Jang JT, Green JB, Beard JL, Green MH. Kinetic analysis shows that iron deficiency decreases liver vitamin A mobilization in rats. J Nutr. 2000;130(5):1291-1296. (PubMed)
19. Rosales FJ, Jang JT, Pinero DJ, Erikson KM, Beard JL, Ross AC. Iron deficiency in young rats alters the distribution of vitamin A between plasma and liver and between hepatic retinol and retinyl esters. J Nutr. 1999;129(6):1223-1228. (PubMed)
20. Turnlund JR. Copper. In: Shils ME, Shike M, Ross AC, Caballero B, Cousins RJ, eds. Modern Nutrition in Health and Disease. 10th ed. Philadelphia: Lippincott Williams & Wilkins; 2006:286-299.
21. Thackeray EW, Sanderson SO, Fox JC, Kumar N. Hepatic iron overload or cirrhosis may occur in acquired copper deficiency and is likely mediated by hypoceruloplasminemia. J Clin Gastroenterol. 2011;45(2):153-158. (PubMed)
22. Videt-Gibou D, Belliard S, Bardou-Jacquet E, et al. Iron excess treatable by copper supplementation in acquired aceruloplasminemia: a new form of secondary human iron overload? Blood. 2009;114(11):2360-2361. (PubMed)
23. Food and Nutrition Board, Institute of Medicine. Copper. Dietary Reference Intakes for Vitamin A, Vitamin K, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Washington, D.C.: National Academy Press; 2001:224-257. (National Academy Press)
24. Kelkitli E, Ozturk N, Aslan NA, et al. Serum zinc levels in patients with iron deficiency anemia and its association with symptoms of iron deficiency anemia. Ann Hematol. 2016;95(5):751-756. (PubMed)
25. Food and Nutrition Board, Institute of Medicine. Iron. Dietary Reference Intakes for Vitamin A, Vitamin K, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Washington, D.C.: National Academy Press; 2001:290-393. (National Academy Press)
26. Lynch SR. Interaction of iron with other nutrients. Nutr Rev. 1997;55(4):102-110. (PubMed)
27. Hurrell R, Egli I. Iron bioavailability and dietary reference values. Am J Clin Nutr. 2010;91(5):1461S-1467S. (PubMed)
28. Weaver CM. Calcium. In: Erdman JJ, Macdonald I, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed: John Wiley & Sons, Inc.; 2012:434-446.
29. Zimmermann MB. The influence of iron status on iodine utilization and thyroid function. Annu Rev Nutr. 2006;26:367-389. (PubMed)
30. Hess SY. The impact of common micronutrient deficiencies on iodine and thyroid metabolism: the evidence from human studies. Best Pract Res Clin Endocrinol Metab. 2010;24(1):117-132. (PubMed)
31. Hess SY, Zimmermann MB, Adou P, Torresani T, Hurrell RF. Treatment of iron deficiency in goitrous children improves the efficacy of iodized salt in Cote d'Ivoire. Am J Clin Nutr. 2002;75(4):743-748. (PubMed)
32. World Health Organization, United Nations Children's Fund, United Nations University. Iron deficiency anaemia: assessment, prevention and control - A guide for programme managers 2001.
33. Global Burden of Disease Pediatrics C, Kyu HH, Pinho C, et al. Global and National Burden of Diseases and Injuries Among Children and Adolescents Between 1990 and 2013: Findings From the Global Burden of Disease 2013 Study. JAMA Pediatr. 2016;170(3):267-287. (PubMed)
34. Wang M. Iron deficiency and other types of anemia in infants and children. Am Fam Physician. 2016;93(4):270-278. (PubMed)
35. Beard JL. Iron biology in immune function, muscle metabolism and neuronal functioning. J Nutr. 2001;131(2S-2):568S-579S; discussion 580S. (PubMed)
36. Changela K, Haeri NS, Krishnaiah M, Reddy M. Plummer-Vinson syndrome with proximal esophageal web. J Gastrointest Surg. 2015;20(5):1074-1075. (PubMed)
37. Jauregui-Lobera I. Iron deficiency and cognitive functions. Neuropsychiatr Dis Treat. 2014;10:2087-2095. (PubMed)
38. Lee GR. Disorders of iron metabolism and heme synthesis. In: Lee GR, Foerster J, Paraskevas F, Greer JP, Rogers GM, eds. Wintrobe's Clinical Hematology. Baltimore: Williams and Wilkins; 1999:979-1070.
39. McDonald SJ, Middleton P, Dowswell T, Morris PS. Effect of timing of umbilical cord clamping of term infants on maternal and neonatal outcomes. Evid Based Child Health. 2014;9(2):303-397. (PubMed)
40. Siu AL, Force USPST. Screening for iron deficiency anemia in young children: USPSTF recommendation statement. Pediatrics. 2015;136(4):746-752. (PubMed)
41. Miller EM. Iron status and reproduction in US women: National Health and Nutrition Examination Survey, 1999-2006. PLoS One. 2014;9(11):e112216. (PubMed)
42. Brody T. Nutritional Biochemistry. 2nd ed. San Diego: Academic Press; 1999.
43. Mei Z, Cogswell ME, Looker AC, et al. Assessment of iron status in US pregnant women from the National Health and Nutrition Examination Survey (NHANES), 1999-2006. Am J Clin Nutr. 2011;93(6):1312-1320. (PubMed)
44. Khuroo MS, Khuroo MS, Khuroo NS. Trichuris dysentery syndrome: a common cause of chronic iron deficiency anemia in adults in an endemic area (with videos). Gastrointest Endosc. 2010;71(1):200-204. (PubMed)
45. Brittenham GM. Iron deficiency in whole blood donors. Transfusion. 2011;51(3):458-461. (PubMed)
46. Li H, Condon F, Kessler D, et al. Evidence of relative iron deficiency in platelet- and plasma-pheresis donors correlates with donation frequency. J Clin Apher. 2016; doi: 10.1002/jca.21448. [Epub ahead of print]. (PubMed)
47. Hershko C, Skikne B. Pathogenesis and management of iron deficiency anemia: emerging role of celiac disease, helicobacter pylori, and autoimmune gastritis. Semin Hematol. 2009;46(4):339-350. (PubMed)
48. Mahadov S, Green PH. Celiac disease: a challenge for all physicians. Gastroenterol Hepatol (N Y). 2011;7(8):554-556. (PubMed)
49. Cardenas VM, Mulla ZD, Ortiz M, Graham DY. Iron deficiency and Helicobacter pylori infection in the United States. Am J Epidemiol. 2006;163(2):127-134. (PubMed)
50. Dignass AU, Gasche C, Bettenworth D, et al. European consensus on the diagnosis and management of iron deficiency and anaemia in inflammatory bowel diseases. J Crohns Colitis. 2015;9(3):211-222. (PubMed)
51. Aron-Wisnewsky J, Verger EO, Bounaix C, et al. Nutritional and Protein Deficiencies in the Short Term following Both Gastric Bypass and Gastric Banding. PLoS One. 2016;11(2):e0149588. (PubMed)
52. Lecube A, Carrera A, Losada E, Hernandez C, Simo R, Mesa J. Iron deficiency in obese postmenopausal women. Obesity (Silver Spring). 2006;14(10):1724-1730. (PubMed)
53. Nead KG, Halterman JS, Kaczorowski JM, Auinger P, Weitzman M. Overweight children and adolescents: a risk group for iron deficiency. Pediatrics. 2004;114(1):104-108. (PubMed)
54. Saunders AV, Craig WJ, Baines SK, Posen JS. Iron and vegetarian diets. Med J Aust. 2013;199(4 Suppl):S11-16. (PubMed)
55. Macdougall IC, Bircher AJ, Eckardt KU, et al. Iron management in chronic kidney disease: conclusions from a "Kidney Disease: Improving Global Outcomes" (KDIGO) Controversies Conference. Kidney Int. 2016;89(1):28-39. (PubMed)
56. Doom JR, Georgieff MK. Striking while the iron is hot: Understanding the biological and neurodevelopmental effects of iron deficiency to optimize intervention in early childhood. Curr Pediatr Rep. 2014;2(4):291-298. (PubMed)
57. Wang B, Zhan S, Gong T, Lee L. Iron therapy for improving psychomotor development and cognitive function in children under the age of three with iron deficiency anaemia. Cochrane Database Syst Rev. 2013;6:CD001444. (PubMed)
58. Idjradinata P, Pollitt E. Reversal of developmental delays in iron-deficient anaemic infants treated with iron. Lancet. 1993;341(8836):1-4. (PubMed)
59. Szajewska H, Ruszczynski M, Chmielewska A. Effects of iron supplementation in nonanemic pregnant women, infants, and young children on the mental performance and psychomotor development of children: a systematic review of randomized controlled trials. Am J Clin Nutr. 2010;91(6):1684-1690. (PubMed)
60. Pongcharoen T, DiGirolamo AM, Ramakrishnan U, Winichagoon P, Flores R, Martorell R. Long-term effects of iron and zinc supplementation during infancy on cognitive function at 9 y of age in northeast Thai children: a follow-up study. Am J Clin Nutr. 2011;93(3):636-643. (PubMed)
61. Sachdev H, Gera T, Nestel P. Effect of iron supplementation on mental and motor development in children: systematic review of randomised controlled trials. Public Health Nutr. 2005;8(2):117-132. (PubMed)
62. Falkingham M, Abdelhamid A, Curtis P, Fairweather-Tait S, Dye L, Hooper L. The effects of oral iron supplementation on cognition in older children and adults: a systematic review and meta-analysis. Nutr J. 2010;9:4. (PubMed)
63. Burke RM, Leon JS, Suchdev PS. Identification, prevention and treatment of iron deficiency during the first 1000 days. Nutrients. 2014;6(10):4093-4114. (PubMed)
64. Pena-Rosas JP, De-Regil LM, Garcia-Casal MN, Dowswell T. Daily oral iron supplementation during pregnancy. Cochrane Database Syst Rev. 2015;7:CD004736. (PubMed)
65. Institute of Medicine Committee on Preventive Services for Women; Board on Population Health and Public Health Practice. Clinical prevention services for women - closing the gaps: The National Academies Press; 2011. (The National Academies Press)
66. American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 95: anemia in pregnancy. Obstet Gynecol. 2008;112(1):201-207. (PubMed)
67. American Academy of Family Physicians. Clinical preventive service recommendation: iron deficiency anemia. Available at: http://www.aafp.org/patient-care/clinical-recommendations/all/iron-deficiency-anemia.html. Accessed 4/17/16.
68. Etheredge AJ, Premji Z, Gunaratna NS, et al. Iron supplementation in iron-replete and nonanemic pregnant women in Tanzania: a randomized clinical trial. JAMA Pediatr. 2015;169(10):947-955. (PubMed)
69. Mwangi MN, Roth JM, Smit MR, et al. Effect of daily antenatal iron supplementation on plasmodium infection in Kenyan women: a randomized clinical trial. JAMA. 2015;314(10):1009-1020. (PubMed)
70. Mielke HW, Gonzales C, Powell E, Mielke PW. Evolving from reactive to proactive medicine: community lead (Pb) and clinical disparities in pre- and post-Katrina New Orleans. Int J Environ Res Public Health. 2014;11(7):7482-7491. (PubMed)
71. Centers for Disease Control and Prevention. New blood lead level information. [Web page]. Available at: http://www.cdc.gov/nceh/lead/acclpp/blood_lead_levels.htm. Accessed 6/1/16.
72. Kwong WT, Friello P, Semba RD. Interactions between iron deficiency and lead poisoning: epidemiology and pathogenesis. Sci Total Environ. 2004;330(1-3):21-37. (PubMed)
73. Silber MH, Becker PM, Earley C, Garcia-Borreguero D, Ondo WG, Medical Advisory Board of the Willis-Ekbom Disease F. Willis-Ekbom Disease Foundation revised consensus statement on the management of restless legs syndrome. Mayo Clin Proc. 2013;88(9):977-986. (PubMed)
74. Trotti LM, Bhadriraju S, Becker LA. Iron for restless legs syndrome. Cochrane Database Syst Rev. 2012;5:CD007834. (PubMed)
75. Johnston CS. Vitamin C. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Ames: Wiley-Blackwell; 2012:248-260.
76. Morck TA, Lynch SR, Cook JD. Inhibition of food iron absorption by coffee. Am J Clin Nutr. 1983;37(3):416-420. (PubMed)
77. Natural Medicines. Iron: Interactions with Herbs & Supplements [professional monograph]; 2016. Available at: https://naturalmedicines.therapeuticresearch.com. Accessed 6/1/16.
78. Liu J, Pu C, Lang L, Qiao L, Abdullahi MA, Jiang C. Molecular pathogenesis of hereditary hemochromatosis. Histol Histopathol. 2016:11762. [Epub ahead of print]. (PubMed)
79. Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13(4):399-408. (PubMed)
80. Franchini M, Veneri D. Recent advances in hereditary hemochromatosis. Ann Hematol. 2005;84(6):347-352. (PubMed)
81. Ayonrinde OT, Milward EA, Chua AC, Trinder D, Olynyk JK. Clinical perspectives on hereditary hemochromatosis. Crit Rev Clin Lab Sci. 2008;45(5):451-484. (PubMed)
82. Wallace DF, Subramaniam VN. Non-HFE haemochromatosis. World J Gastroenterol. 2007;13(35):4690-4698. (PubMed)
83. Powell LW, Seckington RC, Deugnier Y. Haemochromatosis. Lancet. 2016; pii: S0140-6736(15)01315-X. doi: 10.1016/S0140-6736(15)01315-X. [Epub ahead of print]. (PubMed)
84. Oikonomidou PR, Casu C, Rivella S. New strategies to target iron metabolism for the treatment of beta thalassemia. Ann N Y Acad Sci. 2016; 1368(1):162-168. (PubMed)
85. Jin F, Qu LS, Shen XZ. Association between C282Y and H63D mutations of the HFE gene with hepatocellular carcinoma in European populations: a meta-analysis. J Exp Clin Cancer Res. 2010;29:18. (PubMed)
86. Shen LL, Gu DY, Zhao TT, Tang CJ, Xu Y, Chen JF. Implicating the H63D polymorphism in the HFE gene in increased incidence of solid cancers: a meta-analysis. Genet Mol Res. 2015;14(4):13735-13745. (PubMed)
87. Zhang M, Xiong H, Fang L, et al. Meta-Analysis of the Association between H63D and C282Y Polymorphisms in HFE and Cancer Risk. Asian Pac J Cancer Prev. 2015;16(11):4633-4639. (PubMed)
88. Osborne NJ, Gurrin LC, Allen KJ, et al. HFE C282Y homozygotes are at increased risk of breast and colorectal cancer. Hepatology. 2010;51(4):1311-1318. (PubMed)
89. Liu X, Lv C, Luan X, Lv M. C282Y polymorphism in the HFE gene is associated with risk of breast cancer. Tumour Biol. 2013;34(5):2759-2764. (PubMed)
90. Gannon PO, Medelci S, Le Page C, et al. Impact of hemochromatosis gene (HFE) mutations on epithelial ovarian cancer risk and prognosis. Int J Cancer. 2011;128(10):2326-2334. (PubMed)
91. Kennedy AE, Kamdar KY, Lupo PJ, et al. Examination of HFE associations with childhood leukemia risk and extension to other iron regulatory genes. Leuk Res. 2014;38(9):1055-1060. (PubMed)
92. Viola A, Pagano L, Laudati D, et al. HFE gene mutations in patients with acute leukemia. Leuk Lymphoma. 2006;47(11):2331-2334. (PubMed)
93. Agudo A, Bonet C, Sala N, et al. Hemochromatosis (HFE) gene mutations and risk of gastric cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Carcinogenesis. 2013;34(6):1244-1250. (PubMed)
94. Larsson SC, Wolk A. Meat consumption and risk of colorectal cancer: a meta-analysis of prospective studies. Int J Cancer. 2006;119(11):2657-2664. (PubMed)
95. Cross AJ, Ferrucci LM, Risch A, et al. A large prospective study of meat consumption and colorectal cancer risk: an investigation of potential mechanisms underlying this association. Cancer Res. 2010;70(6):2406-2414. (PubMed)
96. Bastide NM, Pierre FH, Corpet DE. Heme iron from meat and risk of colorectal cancer: a meta-analysis and a review of the mechanisms involved. Cancer Prev Res (Phila). 2011;4(2):177-184. (PubMed)
97. Fonseca-Nunes A, Jakszyn P, Agudo A. Iron and cancer risk--a systematic review and meta-analysis of the epidemiological evidence. Cancer Epidemiol Biomarkers Prev. 2014;23(1):12-31. (PubMed)
98. Qiao L, Feng Y. Intakes of heme iron and zinc and colorectal cancer incidence: a meta-analysis of prospective studies. Cancer Causes Control. 2013;24(6):1175-1183. (PubMed)
99. Bastide NM, Chenni F, Audebert M, et al. A central role for heme iron in colon carcinogenesis associated with red meat intake. Cancer Res. 2015;75(5):870-879. (PubMed)
100. Jakszyn P, Lujan-Barroso L, Agudo A, et al. Meat and heme iron intake and esophageal adenocarcinoma in the European Prospective Investigation into Cancer and Nutrition study. Int J Cancer. 2013;133(11):2744-2750. (PubMed)
101. de Valk B, Marx JJ. Iron, atherosclerosis, and ischemic heart disease. Arch Intern Med. 1999;159(14):1542-1548. (PubMed)
102. Das De S, Krishna S, Jethwa A. Iron status and its association with coronary heart disease: systematic review and meta-analysis of prospective studies. Atherosclerosis. 2015;238(2):296-303. (PubMed)
103. Hunnicutt J, He K, Xun P. Dietary iron intake and body iron stores are associated with risk of coronary heart disease in a meta-analysis of prospective cohort studies. J Nutr. 2014;144(3):359-366. (PubMed)
104. Swaminathan S, Fonseca VA, Alam MG, Shah SV. The role of iron in diabetes and its complications. Diabetes Care. 2007;30(7):1926-1933. (PubMed)
105. Orban E, Schwab S, Thorand B, Huth C. Association of iron indices and type 2 diabetes: a meta-analysis of observational studies. Diabetes Metab Res Rev. 2014;30(5):372-394. (PubMed)
106. Abril-Ulloa V, Flores-Mateo G, Sola-Alberich R, Manuel-y-Keenoy B, Arija V. Ferritin levels and risk of metabolic syndrome: meta-analysis of observational studies. BMC Public Health. 2014;14:483. (PubMed)
107. Huth C, Beuerle S, Zierer A, et al. Biomarkers of iron metabolism are independently associated with impaired glucose metabolism and type 2 diabetes: the KORA F4 study. Eur J Endocrinol. 2015;173(5):643-653. (PubMed)
108. Montonen J, Boeing H, Steffen A, et al. Body iron stores and risk of type 2 diabetes: results from the European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam study. Diabetologia. 2012;55(10):2613-2621. (PubMed)
109. Podmore C, Meidtner K, Schulze MB, et al. The association of multiple biomarkers of iron metabolism and type 2 diabetes: the EPIC-InterAct study. Diabetes Care. 2016; 39(4):572-581. (PubMed)
110. Yeap BB, Divitini ML, Gunton JE, et al. Higher ferritin levels, but not serum iron or transferrin saturation, are associated with Type 2 diabetes mellitus in adult men and women free of genetic haemochromatosis. Clin Endocrinol (Oxf). 2015;82(4):525-532. (PubMed)
111. Fernandez-Real JM, McClain D, Manco M. Mechanisms linking glucose homeostasis and iron metabolism toward the onset and progression of type 2 diabetes. Diabetes Care. 2015;38(11):2169-2176. (PubMed)
112. Huang J, Karnchanasorn R, Ou HY, et al. Association of insulin resistance with serum ferritin and aminotransferases-iron hypothesis. World J Exp Med. 2015;5(4):232-243. (PubMed)
113. Fernandez-Real JM, Penarroja G, Castro A, Garcia-Bragado F, Hernandez-Aguado I, Ricart W. Blood letting in high-ferritin type 2 diabetes: effects on insulin sensitivity and β-cell function. Diabetes. 2002;51(4):1000-1004. (PubMed)
114. Houschyar KS, Ludtke R, Dobos GJ, et al. Effects of phlebotomy-induced reduction of body iron stores on metabolic syndrome: results from a randomized clinical trial. BMC Med. 2012;10:54. (PubMed)
115. Belaidi AA, Bush AI. Iron neurochemistry in Alzheimer's disease and Parkinson's disease: targets for therapeutics. J Neurochem. 2015; doi: 10.1111/jnc.13425. [Epub ahead of print]. (PubMed)
116. Kwan JY, Jeong SY, Van Gelderen P, et al. Iron accumulation in deep cortical layers accounts for MRI signal abnormalities in ALS: correlating 7 tesla MRI and pathology. PLoS One. 2012;7(4):e35241. (PubMed)
117. Wong BX, Duce JA. The iron regulatory capability of the major protein participants in prevalent neurodegenerative disorders. Front Pharmacol. 2014;5:81. (PubMed)
118. Devos D, Moreau C, Devedjian JC, et al. Targeting chelatable iron as a therapeutic modality in Parkinson's disease. Antioxid Redox Signal. 2014;21(2):195-210. (PubMed)
119. Grolez G, Moreau C, Sablonniere B, et al. Ceruloplasmin activity and iron chelation treatment of patients with Parkinson's disease. BMC Neurol. 2015;15:74. (PubMed)
120. Hendler SS, Rorvik DM. PDR for Nutritional Supplements. 2nd ed. Montvale: Thomson Reuters; 2008.
121. Natural Medicines. Iron: Interactions with Drugs [professional monograph]; 2016. Available at: https://naturalmedicines.therapeuticresearch.com. Accessed 6/1/16.
122. Wander K, Shell-Duncan B, McDade TW. Evaluation of iron deficiency as a nutritional adaptation to infectious disease: an evolutionary medicine perspective. Am J Hum Biol. 2009;21(2):172-179. (PubMed)
123. Oppenheimer SJ. Iron and its relation to immunity and infectious disease. J Nutr. 2001;131(2S-2):616S-633S; discussion 633S-635S. (PubMed)
124. van den Hombergh J, Dalderop E, Smit Y. Does iron therapy benefit children with severe malaria-associated anaemia? A clinical trial with 12 weeks supplementation of oral iron in young children from the Turiani Division, Tanzania. J Trop Pediatr. 1996;42(4):220-227. (PubMed)
125. Sazawal S, Black RE, Ramsan M, et al. Effects of routine prophylactic supplementation with iron and folic acid on admission to hospital and mortality in preschool children in a high malaria transmission setting: community-based, randomised, placebo-controlled trial. Lancet. 2006;367(9505):133-143. (PubMed)
126. Tielsch JM, Khatry SK, Stoltzfus RJ, et al. Effect of routine prophylactic supplementation with iron and folic acid on preschool child mortality in southern Nepal: community-based, cluster-randomised, placebo-controlled trial. Lancet. 2006;367(9505):144-152. (PubMed)
127. Neuberger A, Okebe J, Yahav D, Paul M. Oral iron supplements for children in malaria-endemic areas. Cochrane Database Syst Rev. 2016;2:CD006589. (PubMed)
128. World Health Organization. Guideline: daily iron supplementation in infants and children. Geneva: World Health Organization; 2016.
129. Fairweather-Tait SJ, Wawer AA, Gillings R, Jennings A, Myint PK. Iron status in the elderly. Mech Ageing Dev. 2014;136-137:22-28. (PubMed)
Magnesium
Contents
Summary
-
Magnesium is an essential mineral and a cofactor for hundreds of enzymes. Magnesium is involved in many physiologic pathways, including energy production, nucleic acid and protein synthesis, ion transport, cell signaling, and also has structural functions. (More information)
-
Severe magnesium deficiency can impede vitamin D and calcium homeostasis. Certain individuals are more susceptible to magnesium deficiency, especially those with gastrointestinal or renal disorders, those suffering from chronic alcoholism, and older people. (More information)
-
Inadequate dietary intakes and/or low serum concentrations of magnesium have been associated with increased risk of cardiovascular disease, osteoporosis, and metabolic disorders, including metabolic syndrome, hypertension, and type 2 diabetes mellitus. Preliminary studies have shown that magnesium improved insulin sensitivity in individuals at risk for type 2 diabetes mellitus. Randomized controlled trials have also investigated the role of magnesium supplementation in the prevention of complications following stroke or heart surgery. (More information)
-
Magnesium sulfate is used in obstetric care for the prevention of seizures in pregnant women with preeclampsia or eclampsia. Observational studies and randomized controlled trials also support a role for magnesium in preventing brain damage in premature infants. (More information)
-
The use of magnesium supplementation is currently being explored in the management of various conditions, including hypertension, cardiovascular disease, type 2 diabetes mellitus, asthma and pain. (More information)
-
About half of the US adult population may have insufficient magnesium intakes to support nutritional adequacy. Dietary sources rich in magnesium include green leafy vegetables, unrefined grains, legumes, beans, and nuts. (More information)
-
The tolerable upper intake level (UL) for supplemental magnesium is 350 mg/day. Excessive intake of supplemental magnesium can result in adverse effects, especially in individuals with impaired kidney functions. (More information)
Magnesium plays important roles in the structure and the function of the human body. The adult human body contains about 25 grams (g) of magnesium. About 50 to 60% of all the magnesium in the body is found in the skeleton and the remainder is found in soft tissue, primarily in muscle. Magnesium is the second most abundant intracellular cation after potassium. Blood contains less than 1% of total body magnesium. Only the free, ionized form of magnesium (Mg2+) is physiologically active. Protein-bound and chelated magnesium serve to buffer the pool of free, ionized magnesium (1).
Function
Magnesium is involved in more than 300 essential metabolic reactions, some of which are discussed below (2).
Energy production
The metabolism of carbohydrates and fats to produce energy requires numerous magnesium-dependent chemical reactions. Magnesium is required by the adenosine triphosphate (ATP)-synthesizing protein in mitochondria. ATP, the molecule that provides energy for almost all metabolic processes, exists primarily as a complex with magnesium (MgATP) (3).
Synthesis of essential molecules
Magnesium is required for a number of steps during synthesis of deoxyribonucleic acid (DNA), ribonucleic acid (RNA), and proteins. Several enzymes participating in the synthesis of carbohydrates and lipids require magnesium for their activity. Glutathione, an important antioxidant, requires magnesium for its synthesis (3).
Structural roles
Magnesium plays a structural role in bone, cell membranes, and chromosomes (3).
Ion transport across cell membranes
Magnesium is required for the active transport of ions like potassium and calcium across cell membranes. Through its role in ion transport systems, magnesium affects the conduction of nerve impulses, muscle contraction, and normal heart rhythm (3).
Cell signaling
Cell signaling requires MgATP for the phosphorylation of proteins and the formation of the cell-signaling molecule, cyclic adenosine monophosphate (cAMP). cAMP is involved in many processes, including the secretion of parathyroid hormone (PTH) from the parathyroid glands (see the articles on Vitamin D and Calcium for information on the role of PTH in the body) (3).
Cell migration
Calcium and magnesium concentrations in the fluid surrounding cells affect the migration of a number of different cell types. Such effects on cell migration may be important in wound healing (3).
Nutrient interactions
Zinc
High doses of zinc in supplemental form apparently interfere with the absorption of magnesium. One study reported that zinc supplements of 142 mg/day (well above the tolerable upper intake level (UL) of 40 mg/day for zinc) in healthy adult males significantly decreased magnesium absorption and disrupted magnesium balance (the difference between magnesium intake and magnesium loss) (4).
Fiber
Large increases in the intake of dietary fiber have been found to decrease magnesium utilization in experimental studies. However, the extent to which dietary fiber affects magnesium nutritional status in individuals with a varied diet outside the laboratory is not clear (2, 3).
Protein
Dietary protein intake may affect magnesium absorption. One study in adolescent boys found that magnesium absorption was directly related to protein intake, with magnesium absorption the lowest when protein intake was less than 30 g/day (5).
Vitamin D and calcium
The active form of vitamin D (calcitriol) may slightly increase intestinal absorption of magnesium (6). However, it is not clear whether magnesium absorption is calcitriol-dependent as is the absorption of calcium and phosphate. High calcium intake has not been found to affect magnesium balance in most studies. Inadequate blood magnesium concentrations are known to result in low blood calcium concentrations, resistance to parathyroid hormone (PTH) action, and resistance to some of the effects of vitamin D (2, 3).
Deficiency
Risk factors
Magnesium deficiency in healthy individuals who are consuming a balanced diet is quite rare because magnesium is abundant in both plant and animal foods and because the kidneys are able to limit urinary excretion of magnesium when intake is low. The following conditions increase the risk of magnesium deficiency (7):
- Gastrointestinal disorders: Prolonged diarrhea, Crohn's disease, malabsorption syndromes, celiac disease, surgical removal of a portion of the small intestine, and intestinal inflammation due to radiation may all lead to magnesium depletion.
- Renal disorders (magnesium wasting): Diabetes mellitus and long-term use of certain diuretics (see Drug interactions) may result in increased urinary loss of magnesium. Multiple other medications can also result in renal magnesium wasting (3).
- Endocrine and metabolic disorders: Several conditions, such as diabetes mellitus, parathyroid gland disorders, phosphate depletion, primary aldosteronism, and even excessive lactation, can lead to magnesium depletion.
Poor dietary intake, gastrointestinal problems, and increased urinary loss of magnesium may all contribute to magnesium depletion in people suffering from alcoholism. Older adults have relatively low dietary intakes of magnesium (8, 9). Intestinal magnesium absorption tends to decrease with age, and urinary magnesium excretion tends to increase with age; thus, suboptimal dietary magnesium intake may increase the risk of magnesium depletion in the elderly (2).
Signs and symptoms
Although severe magnesium deficiency is uncommon, it has been induced experimentally. When magnesium deficiency was induced in humans, the earliest sign was a decrease in serum magnesium concentration. Hypomagnesemia usually describes serum magnesium concentrations less than 0.74 millimoles/liter (mmol/L) or 1.40 milliequivalents/liter (mEq/L) or 1.70 milligrams/deciliter (mg/dL). Over time, serum calcium concentration also began to decrease (hypocalcemia) despite adequate dietary calcium. Hypocalcemia persisted despite increased secretion of parathyroid hormone (PTH), which regulates calcium homeostasis. Usually, increased PTH secretion quickly results in the mobilization of calcium from bone and normalization of blood calcium concentration. As the magnesium depletion progressed, PTH secretion diminished to low concentrations. In addition to hypomagnesemia, signs of severe magnesium deficiency included hypocalcemia, low serum potassium concentrations (hypokalemia), retention of sodium, low circulating PTH concentrations, neurological and muscular symptoms (tremor, muscle spasms, tetany), loss of appetite, nausea, vomiting, and personality changes (3).
While mild magnesium deficiency may not elicit clinical symptoms, it may be associated with an increased risk of developing chronic diseases (see Disease Prevention) (1).
Assessing magnesium status
There is currently no reliable indicator of magnesium status. The magnesium tolerance test, which basically determines magnesium retention (using 24-h urine collection) following the intravenous administration of magnesium, is considered to be the gold standard (1). If this method is a good indicator of hypomagnesemia in adults, it appears to be poorly sensitive to changes in magnesium status in healthy people. Moreover, the method is invasive and cumbersome, and thus difficult to use routinely (10). Another method to assess magnesium status is through measurements of plasma ionized magnesium, which represents the physiologically active form of magnesium. However, it is unknown whether plasma ionized magnesium reflect body stores (10).
In practice, magnesium status is usually determined through assessments of dietary magnesium intake, serum magnesium concentration, and/or urinary magnesium concentration (10). However, each of these indicators has limitations. Although predominantly used in epidemiological studies and the sole indicator available to clinicians, serum magnesium concentration has been found to poorly respond to magnesium supplementation. Regarding dietary intakes of magnesium, about 30 to 40% of ingested magnesium is absorbed, yet absorption varies with the amount of magnesium ingested and with the food matrix composition. Finally, a state of magnesium deficiency has not been associated to a clear cutoff concentration of magnesium in the urine. Urinary magnesium concentration fluctuates rapidly with dietary intakes, but measurements of 24-hour urinary magnesium can be used in addition to other indicators to assess population status. Presently, a combination of all three markers — dietary, serum, and urinary magnesium — may be used to get a valid assessment of magnesium status (reviewed in 10).
The Recommended Dietary Allowance (RDA)
In 1997, the Food and Nutrition Board of the Institute of Medicine increased the Recommended Dietary Allowance (RDA) for magnesium, based on the results of tightly controlled balance studies that utilized more accurate methods of measuring magnesium (Table 1) (2). Balance studies are useful for determining the amount of a nutrient that will prevent deficiency; however, such studies provide little information regarding the amount of a nutrient required for chronic disease prevention or optimal health.
| Life Stage | Age | Males (mg/day) | Females (mg/day) |
|---|---|---|---|
| Infants | 0-6 months | 30 (AI*) | 30 (AI) |
| Infants | 7-12 months | 75 (AI) | 75 (AI) |
| Children | 1-3 years | 80 | 80 |
| Children | 4-8 years | 130 | 130 |
| Children | 9-13 years | 240 | 240 |
| Adolescents | 14-18 years | 410 | 360 |
| Adults | 19-30 years | 400 | 310 |
| Adults | 31 years and older | 420 | 320 |
| Pregnancy | 18 years and younger | - | 400 |
| Pregnancy | 19-30 years | - | 350 |
| Pregnancy | 31 years and older | - | 360 |
| Breast-feeding | 18 years and younger | - | 360 |
| Breast-feeding | 19-30 years | - | 310 |
| Breast-feeding | 31 years and older | - | 320 |
| *Adequate Intake |
Disease Prevention
Metabolic syndrome
Metabolic syndrome refers to the concomitant presentation of several metabolic disorders in an individual, including dyslipidemia, hypertension, insulin resistance, and obesity (11). People with metabolic syndrome are at greater risk of developing type 2 diabetes mellitus, cardiovascular disease, and some types of cancer (12-14). A 2015 analysis of data from the US National Health and Nutrition Examination Survey (NHANES 2001-2010) in 9,148 adults (mean age, 50 years) found a 32% lower risk of metabolic syndrome in those in the highest versus lowest quantile of magnesium intake (≥355 mg/day versus <197 mg/day) (15). Several meta-analyses of primarily cross-sectional studies have also reported an inverse association between dietary magnesium intake and risk of metabolic syndrome (16-18). Moreover, lower serum magnesium concentrations have been reported in individuals with metabolic syndrome compared to controls (18, 19). However, circulating magnesium represents only 1% of total body stores and is tightly regulated; thus, serum magnesium concentrations do not best reflect magnesium status (1). At present, additional evidence is needed from prospectively designed studies to inform the potential relationship between dietary and circulating magnesium and the risk of metabolic syndrome.
Systemic inflammation, which contributes to the development of metabolic disorders, has been inversely correlated with magnesium intakes in a cross-sectional study of 11,686 women (≥45 years). In this study, the lowest prevalence of metabolic syndrome was found in the group of women in the highest quintile of magnesium intakes (median intake, 422 mg/day) (20). Several randomized controlled trials also reported a reduction in circulating C-reactive protein (CRP) — a marker of inflammation — following oral magnesium supplementation (21). This might constitute a potential mechanism through which magnesium could play a role in the prevention of metabolic disorders.
Cardiovascular disease
Hypertension (high blood pressure)
Large prospective cohort studies have examined the relationship between magnesium and blood pressure. However, the fact that foods high in magnesium (fruit, vegetables, whole grains) are frequently high in potassium and dietary fiber has made it difficult to evaluate the independent effect of magnesium on blood pressure. Findings from large cohorts, including the Health Professionals Follow-up Study (HPFS) (22), the Nurses’ Health Study (NHS) (23), the Atherosclerosis Risk in Communities (ARIC) study (24), and the Coronary Artery Risk Development in Young Adults (CARDIA) study (25), have been summarized in a recent meta-analysis (26). The pooled analysis of seven prospective studies showed an 8% lower risk of hypertension with higher versus lower dietary magnesium intakes (26). In one of these studies, data from 5,511 men and women followed for a median period of 7.6 years found that the highest concentrations of urinary magnesium corresponded to a 25% reduction in the risk of hypertension, whereas there was no association between plasma magnesium concentrations and the risk of hypertension (27). There was also no evidence of an association between circulating magnesium concentrations and the risk of hypertension in a meta-analysis of three prospective cohort studies (26).
The relationship between magnesium intake and risk of hypertension suggests that improving diet quality or using magnesium supplements might play a role in the prevention of hypertension in those with inadequate dietary intakes.
Vascular calcification
The buildup of plaque inside arterial walls — a process called atherosclerosis — is an early event in the development of cardiovascular disease. The calcification of atherosclerotic plaques that occurs with the progression of atherosclerosis has been associated with a three- to four-fold increase in the risk of cardiovascular events and mortality (28).
Individuals with chronic kidney disease (CKD): Abnormalities in mineral and bone metabolism are not uncommon in individuals with impaired kidney function and have been associated with an increased risk of cardiovascular disease and mortality (29, 30). In particular, elevated blood phosphorus concentration and increased deposition of calcium phosphate within the vasculature are thought to promote vascular calcification. Since magnesium can function as a calcium antagonist, it has been suggested that it could be utilized to slow down or reverse the calcification of vessels observed in patients with CKD. In a cross-sectional study in patients with pre-dialysis CKD, higher serum magnesium concentrations were associated with lower coronary artery calcification density scores in those in the higher end of normal serum phosphorus concentrations (i.e., ≥3.4 mg/dL) (31). One small randomized, placebo-controlled trial in participants with pre-dialysis CKD examined the effect of oral, slow-release magnesium hydroxide on the calcification propensity of serum by measuring the time needed for primary calciprotein particles (containing amorphous calcium phosphate) to transform into secondary calciprotein particles (containing crystalline hydroxyapatite) (32). Increased serum calcification propensity has been associated with greater risk of mortality in patients with impaired kidney function (33, 34). The trial found an increase in serum magnesium concentration and a reduction in serum calcification propensity — i.e., an increase in the time needed for 50% of the transformation to occur (T50) — with 720 mg/day of supplemental magnesium for eight weeks compared to placebo (32). Serum calcification propensity was also reduced when magnesium concentration was increased (from 1 to 2 mEq/L for 28 days) in the dialysate of patients with established kidney failure (35). A larger randomized controlled trial in patients with pre-dialysis CKD is underway to examine further the effect of oral magnesium on markers of vascular calcification, markers of mineral and bone metabolism, incidence of cardiovascular events, and deterioration of kidney function (36).
Individuals with normal kidney function: The cross-sectional analysis of data from 2,695 middle-aged participants in the Framingham Heart Study showed that the odds of having coronary artery calcification was 58% lower in those in the highest versus lowest quartile of total magnesium intakes (median values, 427 mg/day versus 259 mg/day) (37). Serum magnesium concentration was also found to be inversely associated with vascular calcification in recent population-based cross-sectional studies (38-40). No research has yet examined whether improving magnesium status of generally healthy people could play a role in atherosclerosis prevention.
Risk of cardiovascular disease
Dietary magnesium intakes: Several large prospective cohort studies, including the Health Professionals Follow-up Study (HPFS) and the Nurses’ Health Study (NHS), have examined magnesium intakes in relation to cardiovascular outcomes. In the most recent analysis of the NHS, which followed nearly 90,000 female nurses for 28 years, those in the highest quintile of magnesium intake had a 39% lower risk of fatal myocardial infarction (but not nonfatal coronary heart disease [CHD]) compared to those in the lowest quintile (>342 mg/day versus <246 mg/day) (41). A meta-analysis of nine prospective cohort studies, mostly conducted in participants without cardiovascular disease at baseline, reported a 22% lower risk of CHD per 200 mg/day incremental intake in dietary magnesium (42). A more recent meta-analysis by Fang et al. (43) included six studies and reported a 10% lower risk of CHD with higher versus lower dietary magnesium intakes.
Higher magnesium intakes were associated with an 8 to 11% reduction in stroke risk in two meta-analyses of prospective studies, each including over 240,000 participants (44, 45). The most recent pooled analysis of 14 studies found a 12% lower risk of stroke with higher versus lower magnesium intakes and estimated a 7% risk reduction of stroke associated with each 100-mg increment in daily magnesium intake (43).
Only two prospective studies have examined the risk of heart failure in relation to magnesium intakes. The pooled analysis suggested a 31% lower risk of heart failure with higher dietary magnesium intakes (43).
Finally, a meta-analysis of 13 prospective studies in over 475,000 participants reported that the risk of total cardiovascular events, including stroke, nonfatal myocardial infarction, and CHD, was 15% lower in individuals with higher intakes of magnesium (46). However, in the recent meta-analysis of eight studies by Fang et al. (43), there was no association between dietary magnesium intake and risk of total cardiovascular disease.
It is important to note that while these prospective cohort studies assessed the association between dietary magnesium and cardiovascular disease, they did not account for the use of supplemental magnesium by a significant fraction of participants.
Serum magnesium concentrations: One large prospective study (almost 14,000 men and women) associated higher serum magnesium concentrations with a lower risk of CHD in women but not in men (47). This study was included in a meta-analysis of four studies that showed no evidence of a reduced risk of CHD with increasing serum magnesium concentrations (42). In contrast, a 0.2 mmol/L increment in serum magnesium concentration was associated with a 30% lower risk of total cardiovascular disease in a pooled analysis of eight prospective cohort studies (42). In the recently published British Regional Heart Study that followed 3,523 men for a mean 15 years, there was no association between serum magnesium concentration and incidental CHD events, yet serum magnesium concentration was inversely associated with the risk of heart failure (48).
Cardiovascular mortality
A number of early studies found lower cardiovascular-related mortality in populations who routinely consume "hard" water. Hard (alkaline) water is generally high in magnesium but may also contain more calcium and fluoride than "soft" water, making the cardioprotective effects of hard water difficult to attribute to magnesium alone (49). Additionally, meta-analyses of prospective studies have found no associations between magnesium intake and cardiovascular (50) or all-cause mortality (43). In a prospective analysis of NHANES data from 14,353 participants, followed for a median period of 28.6 years, the risk of all-cause and stroke mortality was significantly increased in those with low rather than normal serum concentrations of magnesium (<0.7 mmol/L versus 0.8-0.89 mmol/L) (51). In contrast, hypermagnesemia (serum magnesium concentration >0.89 mmol/L) — rather than hypomagnesemia — in people with heart failure was associated with an increased risk of cardiovascular and all-cause mortality (52).
Aneurysmal subarachnoid hemorrhage
Occurrence of hypomagnesemia has been reported in patients who suffered from a subarachnoid hemorrhage (a type of stroke) caused by the rupture of a cerebral aneurysm (53). Poor neurologic outcomes following an aneurysmal subarachnoid hemorrhage (aSAH) have been linked to inappropriate calcium-dependent contraction of arteries (known as cerebral arterial vasospasm), leading to delayed cerebral ischemia (54). Because magnesium is a calcium antagonist and potent vasodilator, several randomized controlled trials have examined whether intravenous magnesium sulfate infusions could reduce the incidence of vasospasm after aSAH. A meta-analysis of nine randomized controlled trials found that magnesium therapy after aSAH significantly reduced vasospasm but failed to prevent neurologic deterioration or decrease the risk of death (55). Another meta-analysis of 13 trials in 2,413 aSAH sufferers concluded that the infusion of magnesium sulfate had no benefit regarding neurologic outcome and mortality, despite a reduction in the incidence of delayed cerebral ischemia (56). The post-hoc analysis of a small randomized controlled trial suggested that maintaining magnesium sulfate infusion for 10 days post-aSAH or until signs of vasospasm disappear might protect against secondary cerebral infarction when markers of vasoconstriction and reduced brain perfusion are present (57, 58). Current evidence does not support the use of magnesium supplementation in clinical practice for aSAH patients beyond magnesium status normalization.
Complications of heart surgery
Atrial arrhythmia (also called atrial fibrillation) is a condition defined as the occurrence of persistent heart rate abnormalities; such arrhythmias often complicate the recovery of patients after cardiac surgery. The use of magnesium in the prophylaxis of postoperative atrial arrhythmia after coronary artery bypass grafting has been evaluated as a sole or adjunctive agent to classical antiarrhythmic molecules (namely, β-blockers and amiodarone) in several prospective, randomized controlled trials. A meta-analysis of 21 intervention studies showed that intravenous magnesium infusions could significantly reduce postoperative atrial arrhythmia in treated compared to untreated patients (59). The results of a more recent meta-analysis of 22 placebo-controlled trials suggested that magnesium may effectively reduce atrial arrhythmia when administered post-operatively, as a bolus, and for more than 24 hours (60). However, another meta-analysis of four trials found that magnesium was no more effective than other antiarrhythmic agents (60). Moreover, the meta-analysis of five randomized controlled trials also suggested that intravenous magnesium added to β-blocker treatment did not decrease the risk of atrial arrhythmia compared to β-blocker alone and was associated with more adverse effects (bradycardia and hypotension) (61). Presently, high-quality evidence is still lacking to support the use of magnesium in the prophylaxis of post-operative atrial fibrillation and other arrhythmias in patients with contraindications to first-line antiarrhythmic agents (60).
Diabetes mellitus
Public health concerns regarding the epidemics of obesity and type 2 diabetes mellitus and the prominent role of magnesium in glucose metabolism have led scientists to investigate the relationship between magnesium intake and type 2 diabetes mellitus. A prospective cohort study that followed more than 25,000 individuals, 35 to 65 years of age, for seven years found no difference in incidence of type 2 diabetes mellitus when comparing the highest (377 mg/day) quintile of magnesium intake to the lowest quintile (268 mg/day) (62). However, inclusion of this study in a meta-analysis of eight cohort studies showed that the risk of type 2 diabetes was inversely correlated with magnesium intake (62). The most recent meta-analysis of 25 prospective cohort studies, including 637,922 individuals and 26,828 new cases of type 2 diabetes mellitus, found that higher magnesium intakes were associated with a 17% lower risk of type 2 diabetes mellitus (63). Several meta-analyses conducted to date reported an 8 to 15% decrease in the risk of developing type 2 diabetes mellitus with each 100 mg-increment in dietary magnesium intake (63-66).
Insulin resistance, characterized by alterations in both insulin secretion by the pancreas and insulin action on target tissues, has been linked to inadequate magnesium status. A cross-sectional analysis of the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium, which included 15 cohorts with a total of 52,684 diabetes-free participants, showed that magnesium intakes were inversely associated with fasting insulin concentrations after multiple adjustments, including various lifestyle factors, body mass index (BMI), caffeine intake, and fiber intake (65). It is thought that pancreatic β-cells, which secrete insulin, could become less responsive to changes in insulin sensitivity in magnesium-deficient subjects (67). A randomized, double-blind, placebo-controlled trial that enrolled 97 healthy adults with significant hypomagnesemia (serum magnesium concentration ≤0.70 mmol/L) showed that daily consumption of 638 mg of magnesium (from a solution of magnesium chloride) for three months improved the function of pancreatic β-cells, resulting in lower fasting glucose and insulin concentrations (68). In a follow-up randomized controlled trial, the administration of 382 mg/day of magnesium for four months to participants (mean age, 42 years) with both hypomagnesemia (serum magnesium concentration <0.74 mmoles/L) and impaired fasting glucose improved serum magnesium concentrations, as well as fasting and post-load glucose concentrations (69). Other metabolic markers, including serum triglycerides, HDL-cholesterol, and a measure of insulin resistance, also improved in magnesium- versus placebo-treated individuals (69). Additionally, similar metabolic improvements have been reported following the supplementation of magnesium (382 mg/day for four months) to participants who were both hypomagnesemic and lean yet metabolically obese (i.e., with metabolic disorders usually associated with obesity) (70). In another study, supplementation with 365 mg/day of magnesium (from magnesium aspartate hydrochloride) for six months reduced insulin resistance in 27 overweight individuals with normal values of serum and intracellular magnesium (71). This latter study suggests that magnesium might have additional effects on glucose tolerance and insulin sensitivity that go beyond the normalization of serum magnesium concentrations in hypomagnesemic individuals.
Osteoporosis
Although decreased bone mineral density (BMD) is the primary feature of osteoporosis, other osteoporotic changes in the collagenous matrix and mineral composition of bone may result in bones that are brittle and more susceptible to fracture (72). Around 60% of total body magnesium is stored in the skeleton and is known to influence both bone matrix and bone mineral metabolism. Magnesium at the surface of bones is also available for dynamic exchange with blood (73). As the magnesium content of bone mineral decreases, hydroxyapatite crystals of bone may become larger and more brittle. Some studies have found lower magnesium content and larger hydroxyapatite crystals in bones of women with osteoporosis compared to disease-free women (74). Inadequate serum magnesium concentrations are known to result in low serum calcium concentrations, resistance to parathyroid hormone (PTH) action, and resistance to some of the effects of vitamin D (calcitriol), all of which can lead to increased bone loss (see the articles on Vitamin D and Calcium). Lower serum magnesium concentrations may not be unusual in postmenopausal women with osteoporosis (75), and hypomagnesemia has been reported as an adverse effect of using the prescription drug teriparatide (Forteo) in the treatment of osteoporosis (76).
Higher dietary magnesium intakes have been associated with increased site-specific (77) and total-body BMD (78) in observational studies, including studies of older adults. More recently, a large cohort study conducted in almost two-thirds of the Norwegian population found the level of magnesium in drinking water to be inversely associated with the risk of hip fracture (79). In the Women’s Health Initiative study, data analysis from 4,778 participants (mean age, 63 years) followed for about seven years showed that higher magnesium intakes were associated with higher hip and whole-body BMD but not with reduced hip or total fractures (80). Moreover, the highest versus lowest quintile of total magnesium intakes was associated with a 23% increased risk of lower arm and wrist fractures (80). In a case-cohort study nested within the European Prospective Investigation into Cancer and Nutrition (EPIC)-Norfolk study, which included 5,319 individuals, total magnesium and potassium intakes were found to be inversely associated with heel bone (calcaneus) broadband ultrasound attenuation (BUA) measurements — which are predictive of the risk of incidental fracture — and with the risk of hip fractures (81).
Few studies have addressed the effect of magnesium supplementation on BMD or osteoporosis in humans. In a small group of postmenopausal women with osteoporosis, magnesium supplementation of 750 mg/day for six months followed by 250 mg/day for 18 more months resulted in increased BMD at the wrist after one year, with no further increase after two years of supplementation (82). A study in postmenopausal women who were taking estrogen replacement therapy and a multivitamin supplement found that supplementation with an additional 400 mg/day of magnesium and 600 mg/day of calcium resulted in increased BMD at the heel compared to postmenopausal women receiving estrogen replacement therapy only (83). A more recent randomized controlled study conducted in 20 postmenopausal women with osteoporosis suggested that high-dose supplementation with magnesium citrate (1,830 mg/day) for one month could reduce the rapid rate of bone loss that characterizes osteoporosis (84). Evidence is not yet sufficient to suggest that supplemental magnesium in excess of the RDA could be effective in the prevention of osteoporosis unless normalization of serum magnesium concentration is required (85).
Sarcopenia
Sarcopenia is a condition characterized by a loss of skeletal muscle mass that increases frailty and risk of falls in older adults (86). Several cross-sectional studies have reported a positive association between dietary magnesium intakes and proxy measures of skeletal muscle mass in middle-age and older adults (87-90). A 2014 randomized controlled study in 139 physically active and healthy older women (mean age, 71.5 years) found little-to-no impact of magnesium supplementation (900 mg/day of magnesium oxide) for 12 weeks on body composition and muscle strength, yet the Short Physical Performance Battery [SPPB] test score — a composite indicator of physical function — improved (91). More research is needed to examine further the effect of magnesium supplementation on body composition, muscle strength, and physical performance in older adults, whether physically active or sedentary, and with normal or inadequate magnesium status.
Disease Treatment
The use of pharmacologic doses of magnesium to treat specific disorders is discussed below. Although many of the cited studies utilized supplemental magnesium at doses considerably higher than the tolerable upper intake level (UL), which is 350 mg/day set by the Food and Nutrition Board (see Safety), it is important to note that these studies were all conducted under medical supervision. Because of the potential side effects of high doses of magnesium, especially in the presence of impaired kidney function, any disease treatment trial using oral magnesium doses higher than the UL should be conducted under medical supervision. Moreover, intravenous magnesium has been used in the management of several conditions.
Pregnancy complications
Preeclampsia and eclampsia
Preeclampsia and eclampsia are hypertensive disorders of pregnancy that may occur at any time after 20 weeks’ gestation and persist up to six weeks following birth. Preeclampsia (sometimes called toxemia of pregnancy) affects approximately 4% of pregnant women in the US (92). Preeclampsia is defined as the presence of elevated blood pressure (hypertension), protein in the urine, and severe swelling (edema) during pregnancy (93). Eclampsia occurs with the addition of seizures to the triad of preeclamptic symptoms and is a significant cause of perinatal and maternal mortality (93, 94). Although cases of preeclampsia are at high risk of developing eclampsia, one-quarter of eclamptic women do not initially exhibit preeclamptic symptoms (95).
Although lower magnesium concentrations have been reported in the blood and brain of women with preeclampsia than in healthy pregnant women, there is no evidence that magnesium imbalance may cause adverse pregnancy events. A 2014 meta-analysis of 10 randomized controlled trials found no effect of oral magnesium salt administration during normal and at-risk pregnancies on the risk of preeclampsia, perinatal mortality, and small-for-gestational age infants (96).
For many years, high-dose intravenous magnesium sulfate has been the treatment of choice for preventing eclamptic seizures that may occur in association with severe preeclampsia in pregnancy or during labor (97, 98). A systematic review of seven randomized trials in 1,396 women with eclampsia compared the effect of magnesium sulfate administration with diazepam (a known anticonvulsant) treatment on perinatal outcomes. Risks of recurrent seizures and maternal death were significantly reduced by the magnesium regimen compared to diazepam. Moreover, the use of magnesium for the care of eclamptic women resulted in newborns with higher Apgar scores; there was no significant difference in the risk of preterm birth and perinatal mortality (95). Additional research has confirmed that infusion of magnesium sulfate should always be considered in the management of severe preeclampsia and eclampsia to prevent initial and recurrent seizures (99). Moreover, the World Health Organization (WHO) recommends the use of magnesium sulfate — administered either intramuscularly or intravenously — as first-line treatment for the prevention of eclampsia in women with severe preeclampsia, in preference to other anticonvulsants (100). Further research is needed to evaluate the efficacy of magnesium salt infusion in eclampsia prophylaxis in women with mild preeclampsia (101). In addition, it is unclear whether prolonging magnesium use post-partum in women who presented with severe preeclampsia during pregnancy is necessary to lower the risk of eclampsia after delivery (102).
Perinatal neuroprotection
While intravenous magnesium sulfate is included in the medical care of preeclampsia and eclampsia, the American College of Obstetricians and Gynecologists (ACOG) and the Society for Maternal-Fetal Medicine support its use in two additional situations: specific conditions of short-term prolongation of pregnancy and neuroprotection of the fetus in anticipated premature delivery (103).
Preterm birth, which is defined by the premature delivery of an infant between the 20th and 37th weeks of estimated gestation, is associated with an increased risk of perinatal mortality and both short- and long-term morbidity. The ACOG approves the use of different classes of drugs — known as tocolytics — that are meant to delay delivery for long enough so that antenatal corticoids can be used to accelerate lung maturation in the fetus of women at imminent risk of preterm labor (104). A 2014 meta-analysis of 37 trials found that intravenous infusion of magnesium sulfate was no more efficacious than commonly used tocolytics (e.g., β-adrenergic receptor agonists, calcium channel blockers, prostaglandin inhibitors) in delaying delivery or preventing serious infant outcomes (105). Very limited evidence also suggested that high- versus low-dose magnesium infusion may reduce the length of hospital stays in neonates admitted to intensive care units (106).
The relationship between magnesium sulfate and risk of cerebral damage in premature infants has been assessed in observational studies. A meta-analysis of six case-control and five prospective cohort studies showed that the use of magnesium significantly reduced the risk of cerebral palsy, as well as mortality (107). However, the high degree of heterogeneity among the cohort studies and the fact that corticosteroid exposure (which is known to decrease antenatal mortality) was higher in the cases of children exposed to magnesium compared to controls imply a cautious interpretation of the results. Nonetheless, a meta-analysis of five randomized controlled trials, which included 5,493 women at risk of preterm birth and 6,135 babies, found that magnesium therapy given to mothers delivering before term decreased the risk of cerebral palsy by 32% without causing severe adverse maternal events, but this treatment did not reduce the risk of other neurologic impairments or mortality in early childhood (108). Another meta-analysis conducted on five randomized controlled trials found that intravenous magnesium administration to newborns who suffered from perinatal asphyxia could be beneficial in terms of short-term neurologic outcomes, although there was no effect on mortality (109). Additional trials are needed to evaluate the long-term benefits of magnesium in pediatric care.
Cardiovascular disease
Hypertension
While results from intervention studies have not been entirely consistent (2), the latest review of the data highlighted a therapeutic benefit of magnesium supplements in treating hypertension. A 2012 meta-analysis examined 22 randomized, placebo-controlled trials of magnesium supplementation conducted in 1,173 individuals with either normal blood pressure (normotensive) or hypertension (treated with medication or untreated). Oral supplementation with magnesium (mean dose of 410 mg/day; range of 120 to 973 mg/day) for a median period of 11.3 months significantly reduced systolic blood pressure by 2 to 3 mm Hg and diastolic blood pressure by 3 to 4 mm Hg (110); a greater effect was seen at higher doses (≥370 mg/day). The results of 19 of the 22 trials included in the meta-analysis were previously reviewed together with another 25 intervention studies (111). The systematic examination of these 44 trials suggested a blood pressure-lowering effect associated with supplemental magnesium in hypertensive but not in normotensive individuals.
Magnesium doses required to achieve a decrease in blood pressure appeared to depend on whether participants with high blood pressure were treated with antihypertensive medications, including diuretics. Intervention trials on treated participants showed a reduction in hypertension with magnesium doses from 243 mg/day to 486 mg/day, whereas untreated patients required doses above 486 mg/day to achieve a significant decrease in blood pressure. A more recent meta-analysis of randomized controlled studies with 2,028 participants found that supplemental magnesium at a median dose of 368 mg/day (range: 238-960 mg/day) for a median duration of three months (range: 3 weeks-6 months) increased serum magnesium concentration by 0.05 mmol/L (27 trials) and reduced systolic blood pressure by 2 mm Hg and diastolic blood pressure by 1.78 mm Hg (37 trials) (112). A 2017 meta-analysis restricted to trials in participants with underlying preclinical (insulin resistance or prediabetes) or clinical conditions (type 2 diabetes mellitus or coronary heart disease) found a 4.18 mm Hg reduction in systolic blood pressure and a 2.27 mm Hg reduction in diastolic blood pressure with supplemental doses of magnesium ranging between 365 mg/day and 450 mg/day for one to six months (113).
While oral magnesium supplementation may be helpful in hypertensive individuals who are depleted of magnesium due to chronic diuretic use and/or inadequate dietary intake (7), several dietary factors play a role in hypertension. For example, adherence to the DASH diet — a diet rich in fruit, vegetables, and low-fat dairy and low in saturated and total fats — has been linked to significant reductions in systolic and diastolic blood pressures (114). See the topic page: High Blood Pressure.
Atherosclerosis
Vascular endothelial cells line arterial walls where they are in contact with the blood that flows through the circulatory system. Normally functioning vascular endothelium promotes vasodilation when needed, for example, during exercise, and inhibits the formation of blood clots. Conversely, endothelial dysfunction results in widespread vasoconstriction and coagulation abnormalities. In cardiovascular disease, chronic inflammation is associated with the formation of atherosclerotic plaques in arteries. Atherosclerosis impairs normal endothelial function, increasing the risk of vasoconstriction and clot formation, which may lead to heart attack or stroke (reviewed in 115). A recent systematic review identified six randomized controlled trials that examined the effect of pharmacologic doses of oral magnesium on vascular endothelial function (116). Three out of six trials, which included individuals with coronary artery disease (117), diabetes mellitus (118), or hypertension (119), reported an improvement in flow-mediated dilation (FMD) with supplemental magnesium compared to control. In other words, the normal dilation response of the brachial (arm) artery to increased blood flow was improved. In contrast, there was no evidence of an effect of magnesium supplementation on FMD in three trials conducted in hemodialysis patients (120) or healthy participants with normal (121) or high body mass index (BMI) (122). A pooled analysis of the six trials in 262 participants found that supplementation with 107 to 730 mg/day of magnesium for one to six months resulted in an overall improvement of FMD, regardless of the health status or baseline magnesium concentrations of participants (116).
The measurement of the thickness of the inner layers of the carotid arteries is sometimes used as a surrogate marker of atherosclerosis (123). Higher serum magnesium concentrations have been associated with reduced carotid intima-media thickness (CIMT) in all women and in Caucasian men participating in the Atherosclerosis Risk in Communities (ARIC) study (124). A meta-analysis of four small (145 participants in total) and heterogeneous intervention studies found no effect of magnesium supplementation (98.6 to 368 mg/day for 2 to 6 months) on CIMT (116).
Survival after myocardial infarction
Results from several randomized, placebo-controlled trials have suggested that an intravenous magnesium administered early after a suspected myocardial infarction could decrease the risk of death. The most influential study was a randomized, placebo-controlled trial in 2,316 patients that found a significant reduction in mortality in the group of patients given intravenous magnesium sulfate within 24 hours of suspected myocardial infarction (7.8% all-cause mortality in the experimental group vs. 10.3% all-cause mortality in the placebo group) (125). Follow-up from one to five years after treatment revealed that the mortality from cardiovascular disease was 21% lower in the magnesium-treated group (126). However, a larger placebo-controlled trial in more than 58,000 patients found no significant reduction in five-week mortality in patients treated with intravenous magnesium sulfate within 24 hours of suspected myocardial infarction, resulting in controversy regarding the efficacy of this treatment (127). A US survey of the treatment of more than 173,000 individuals with acute myocardial infarction found that only 5% were given intravenous magnesium in the first 24 hours post-infarction and that mortality was higher in this group compared to the group of patients not treated with magnesium (128). A 2007 systematic review of 26 clinical trials, including 73,363 participants, concluded that intravenous magnesium administration does not appear to reduce post-myocardial infarction mortality and thus should not be utilized as a treatment (129). Thus, the use of intravenous magnesium sulfate in the therapy of acute myocardial infarction remains controversial.
Diabetes mellitus
Magnesium depletion has been associated with type 1 (insulin-dependent) and type 2 diabetes mellitus, as well as with gestational diabetes. Low serum concentrations of magnesium (hypomagnesemia) have been reported in 13.5 to 47.7% of individuals with type 2 diabetes mellitus (130). One cause of the depletion may be an increased urinary loss of magnesium caused by an increased urinary excretion of glucose that accompanies poorly controlled diabetes. Magnesium depletion has been shown to increase insulin resistance in a few studies and may adversely affect blood glucose control in diabetes mellitus (see also Diabetes mellitus under Disease Prevention) (131). A small study in nine individuals with type 2 diabetes mellitus reported that supplemental magnesium (300 mg/day for 30 days), in the form of a liquid, magnesium-containing salt solution, improved fasting insulin but not fasting glucose concentrations (132). One randomized, double-blind, placebo-controlled study in 63 individuals with type 2 diabetes mellitus and hypomagnesemia found that those taking an oral magnesium chloride solution (638 mg/day of elemental magnesium) for 16 weeks had improved measures of insulin sensitivity and glycemic control compared to those taking a placebo (133). The most recent meta-analysis of nine randomized, double-blind, controlled trials concluded that oral supplemental magnesium lowered fasting plasma glucose concentrations in individuals with diabetes (134). However, magnesium supplementation did not improve other markers of glucose homeostasis, such as glycated hemoglobin (HbA1c) concentration, fasting and post-glucose load insulin concentrations, and measures of insulin resistance (134). Another meta-analysis of trials that included participants either at-risk of diabetes mellitus or with diabetes mellitus suggested that evidence to support a benefit of magnesium supplementation on measures of insulin resistance was stronger in subjects who were magnesium deficient than in those with normal serum concentrations of magnesium (135). Correcting existing magnesium deficiencies may improve glucose metabolism and insulin sensitivity in subjects with diabetes, but it remains uncertain whether magnesium supplementation can have any therapeutic benefit in patients with adequate magnesium status.
Asthma
The occurrence of hypomagnesemia may be greater in patients with asthma than in individuals without asthma (136). Several clinical trials have examined the effect of intravenous magnesium infusions on acute asthmatic attacks in children or adults who did not respond to initial treatment in the emergency room. Indeed, magnesium can promote bronchodilation in subjects with asthma by interfering with mechanisms like the activation of N-methyl D-aspartate (NMDA) receptors that trigger bronchoconstriction through facilitating calcium influx in airway smooth muscle cells (137). In a meta-analysis of six (quasi) randomized controlled trials in 325 children with acute asthma treated with a short-acting β2-adrenergic receptor agonist (e.g., salbutamol) and systemic steroids, intravenous magnesium sulfate treatment improved measurements of the respiratory function and reduced the risk of hospital admission by 30% compared to control (138). Another meta-analysis of randomized controlled trials primarily conducted in adults with asthma exacerbations indicated that single infusions of 1.2 to 2 g of magnesium sulfate over 15 to 30 minutes could reduce the risk of hospital admission and improve lung function after initial treatments failed (i.e., oxygen, short-acting β2 agonist, and steroids) (139).
The use of nebulized, inhaled magnesium for treating asthma has also been investigated. A recent systematic review of 25 randomized controlled trials, including adults, children, or both, found little evidence that inhaled magnesium sulfate alone or along with a β2-adrenergic receptor agonist and/or a muscarinic anticholinergic (e.g., ipratropium) could improve pulmonary function in patients with acute asthma (140). In addition, oral magnesium supplementation is of no known value in the management of chronic asthma (141-143).
Pain management
The potential analgesic effect of magnesium is attributed in particular to its capacity to block NMDA receptors, which are located in the brain and spinal cord and are involved in pain transduction (144).
Post-operative pain
Several intervention studies have examined the role of magnesium on pain control and analgesic requirement in patients during the immediate post-surgery period.
After cesarean section: Pain management strategies after cesarean section usually involve the injection of an analgesic into either the epidural space (for epidural analgesia) or the subarachnoid space (for spinal [intrathecal] analgesia). A recent meta-analysis of nine randomized controlled trials summarized the evidence regarding the potential use of magnesium sulfate to control or relieve postoperative pain in 827 women who underwent cesarean section (145). All the trials evaluated the effect of a first-line analgesic regimen (i.e., bupivacaine or lidocaine, with or without opioids) with and without the addition of magnesium sulfate. The results suggested that the anesthesia (8 studies) and sensory blockade (6 studies) lasted longer in women who received the additional magnesium sulfate. The use of magnesium sulfate also resulted in lower pain score (3 studies) and in lower postoperative consumption of analgesics (4 studies). Additionally, there was no difference in occurrence of side effects between regimens (145). A recent randomized controlled trial in 60 healthy women undergoing elective cesarean section confirmed that the addition of magnesium sulfate to a bupivacaine/opioid regimen increased the duration of spinal anesthesia and lowered the pain level, yet did not improve the potency of bupivacaine (146). In another study in women with mild preeclampsia who received an epidural injection of ropivacaine after cesarean section, spinal infusion of magnesium sulfate increased the duration of sensory and motor blockade, as well as the time before patients requested an analgesic, compared to midazolam (147).
After a variety of other surgeries: The efficacy of intravenous magnesium has also been examined for local, regional, or systemic pain control following a range of different surgeries. A review of four small randomized controlled studies suggested that, when added to local analgesics, magnesium infusion to patients undergoing tonsillectomy might decrease pain and incidence of laryngospasm, extend the time to first post-operative analgesic requirement, and reduce the number of post-operative analgesic requests (148). Similar observations were reported in two additional meta-analyses, yet there was discrepancy regarding the ability of magnesium to alleviate pain (149, 150). Indeed, the review of eight trials by Xie et al. (150), of which only two scored pain using the same scale, showed no pain reduction with magnesium compared to control. Finally, both meta-analyses reported no reduction in risk of post-operative nausea and vomiting with intravenous magnesium administration (148, 150). A 2018 meta-analysis of four randomized controlled trials in 263 patients also suggested that magnesium sulfate infusion may help reduce pain scores at 2 and 8 hours (but not 24 hours) after laparoscopic cholecystectomy (151). Recent studies have examined the use of magnesium sulfate for pain control after other surgeries, including hysterectomy (152, 153), spinal surgery (154, 155), or during endoscopic sinus (156) or cochlear implantation (157) surgery. Despite conflicting results or reports of limited benefits of magnesium, further research is needed before any conclusions can be drawn.
Neuropathic pain
The effect of magnesium on neuropathic pain has been examined in some clinical studies. The intravenous administration of magnesium sulfate was found to partially or completely alleviate pain in patients with postherpetic neuralgia, a type of neuropathic pain caused by herpes zoster infection (shingles) (158, 159). In a more recent randomized controlled trial in 45 patients with either postherpetic neuralgia or neuropathic pain of traumatic or surgical origin, oral supplementation of magnesium failed to improve measures of pain and quality of life compared to a placebo (160). Another trial is underway to examine the impact of intravenous magnesium with ketamine on neuropathic pain (161).
Migraine headaches
Lower intracellular magnesium concentrations (in both red blood cells and white blood cells) have been reported in individuals who suffer from recurrent migraine headaches compared to migraine-free individuals (162). Additionally, the incidence of hypomagnesemia also appeared to be greater in women who experience migraines with menstruation compared to women without menstrual migraines (163).
A few intervention studies have examined whether an increase in intracellular magnesium concentration with supplemental (oral) magnesium could help decrease the frequency and severity of migraine headaches in affected individuals. Two early placebo-controlled trials demonstrated modest decreases in the frequency of migraine headaches after supplementation with 600 mg/day of magnesium (162, 164). Another placebo-controlled trial in 86 children with frequent migraine headaches found that oral magnesium oxide (9 mg/kg body weight/day) reduced headache frequency over the 16-week intervention (165). However, there was no reduction in the frequency of migraine headaches with 485 mg/day of magnesium in another placebo-controlled study conducted in 69 adults suffering migraine attacks (166). The efficiency of magnesium absorption varies with the type of oral magnesium complex, and this might explain the conflicting results. Although no serious adverse effects were noted during these migraine headache trials, 19 to 40% of individuals taking the magnesium supplements have reported diarrhea and gastric irritation.
The efficacy of magnesium infusions was also investigated in a randomized, single-blind, placebo-controlled, cross-over trial of 30 patients with migraine headaches (167). The administration of 1 gram of intravenous magnesium sulfate ended the attacks, abolished associated symptoms, and prevented recurrence within 24 hours in nearly 90% of the subjects. While this promising result was confirmed in another trial (168), two additional randomized, placebo-controlled studies found that magnesium sulfate was less effective than other molecules (e.g., metoclopramide) in treating migraines (169, 170). The most recent meta-analysis of five randomized, double-blind, controlled trials reported no beneficial effect of magnesium infusion for migraine in adults (171). Another two more recent intervention studies suggested that magnesium sulfate infusion could be more effective and faster than dexamethasone/metoclopramide (172) or caffeine citrate (173) to relieve pain in patients with acute migraine.
The efficacy of magnesium should be examined in larger studies that consider the magnesium status of migraine sufferers (174).
Critical injury or illness
Hypomagnesemia is not uncommon in patients admitted to intensive care units (ICU). Two recent meta-analyses of prospective and retrospective cohort studies reported serum magnesium concentrations ≤0.75 mmol/L in ICU patients at admission or within 24 hours following admission to be associated with a greater need for mechanical ventilation, longer ICU stay, and higher risk of hospital mortality (175, 176). A pooled analysis of three studies also suggested a higher risk of sepsis in ICU patients with hypomagnesemia (175). A recent prospective study conducted in patients admitted with severe head injury found better neurological outcomes after six months in those who presented with normal serum magnesium concentrations at admission compared to those with hypomagnesemia (serum magnesium concentrations <0.65 mmol/L) (177). However, evidence is currently unavailable to suggest that magnesium administration could improve outcomes in critically ill or severely injured patients (178).
Sources
Food sources
The analysis of US national nutrition survey data (NHANES 2003-2006) showed an average magnesium intake in adults (ages ≥19 years) of 278 mg/day when only unfortified food sources were considered (179). Considering all sources of magnesium intakes (i.e., unfortified and fortified food and supplements), the average intake in adults was estimated to be around 330 mg/day — a value close to the estimated average requirements (EAR) for magnesium — suggesting that about one-half of the adult population may be at risk of magnesium inadequacy (179). Yet, the long-term consequences of inadequate dietary intakes remain unclear (1).
Since magnesium is part of chlorophyll, the green pigment in plants, green leafy vegetables are good sources of magnesium. Unrefined grains (whole grains) and nuts also have high magnesium content. Meats and milk have an intermediate content of magnesium, while refined foods generally have the lowest. Water is a variable source of intake; harder water usually has a higher concentration of magnesium salts (2). Some foods that are relatively rich in magnesium are listed in Table 2, along with their magnesium content in milligrams (mg). For more information on the nutrient content of foods, search USDA's FoodData Central.
| Food | Serving | Magnesium (mg) |
|---|---|---|
| Brazil nuts | 1 ounce (6 kernels) | 107 |
| Cereal, oat bran | ½ cup dry | 96 |
| Brown rice, medium-grain, cooked | 1 cup | 86 |
| Cashews | 1 ounce (16 kernels) | 83 |
| Fish, mackerel, cooked | 3 ounces | 82 |
| Spinach, frozen, chopped, cooked | ½ cup | 78 |
| Almonds | 1 ounce (23 kernels) | 77 |
| Swiss chard, chopped, cooked | ½ cup | 75 |
| Lima beans, large, immature seeds, cooked | ½ cup | 63 |
| Cereal, shredded wheat | 2 biscuits | 61 |
| Avocado | 1 fruit | 58 |
| Cereal, all bran (whole wheat) | ½ cup, dry | 57 |
| Peanuts | 1 ounce (28 peanuts) | 48 |
| Molasses, blackstrap | 1 tablespoon | 48 |
| Hazelnuts | 1 ounce (21 kernels) | 46 |
| Chickpeas, mature seeds, cooked | ½ cup | 39 |
| Milk, 1% fat | 8 fluid ounces | 39 |
| Banana | 1 medium | 32 |
Supplements
Magnesium supplements are available as magnesium oxide, magnesium hydroxide, magnesium gluconate, magnesium chloride, and magnesium citrate salts, as well as a number of amino acid chelates like magnesium aspartate. Magnesium hydroxide, oxide, or trisilicate salts are used as antacids to mitigate gastric hyperacidity and symptoms of gastroesophageal reflux disease (180).
Safety
Toxicity
Adverse effects have not been identified from magnesium occurring naturally in food. However, adverse effects from excessive magnesium have been observed with intakes of various magnesium salts (i.e., supplemental magnesium) (6). The initial symptom of excess magnesium supplementation is diarrhea — a well-known side effect of magnesium that is used therapeutically as a laxative. Individuals with impaired kidney function are at higher risk for adverse effects of magnesium supplementation, and symptoms of magnesium toxicity have occurred in people with impaired kidney function taking moderate doses of magnesium-containing laxatives or antacids. Elevated serum concentrations of magnesium (hypermagnesemia) may result in a fall in blood pressure (hypotension). Some of the later effects of magnesium toxicity, such as lethargy, confusion, disturbances in normal cardiac rhythm, and deterioration of kidney function, are related to severe hypotension. As hypermagnesemia progresses, muscle weakness and difficulty breathing may occur. Severe hypermagnesemia may result in cardiac arrest (2, 3). The Food and Nutrition Board (FNB) of the US Institute of Medicine set the tolerable upper intake level (UL) for magnesium at 350 mg/day; this UL represents the highest level of daily supplemental magnesium intake likely to pose no risk of diarrhea or gastrointestinal disturbance in almost all individuals. The FNB cautions that individuals with renal impairment are at higher risk for adverse effects from excess supplemental magnesium intake. However, the FNB also notes that there are some conditions that may warrant higher doses of magnesium under medical supervision (2).
Drug interactions
Magnesium interferes with the absorption of digoxin (a heart medication), nitrofurantoin (an antibiotic), and certain anti-malarial drugs, which could potentially reduce drug efficacy. Bisphosphonates (e.g., alendronate, etidronate), which are drugs used to treat osteoporosis, and magnesium should be taken two hours apart so that the absorption of the bisphosphonates is not inhibited (181, 182). Magnesium has also been found to reduce the efficacy of chlorpromazine (a tranquilizer), penicillamine, oral anticoagulants, and the quinolone and tetracycline classes of antibiotics (181, 182). Intravenous magnesium might inhibit calcium entry into smooth muscle cells and lead to hypotension and muscular weakness if administered with calcium channel blockers (e.g., nifedipin, nicardipin) (182). Because intravenous magnesium has increased the effects of certain muscle-relaxing medications used during anesthesia, it is advisable to let medical staff know if you are taking oral magnesium supplements, laxatives, or antacids prior to surgical procedures. Moreover, long-term use (three months or longer) of proton-pump inhibitors (drugs used to reduce the amount of stomach acid) may increase the risk of hypomagnesemia (183, 184). High doses of furosemide (Lasix) and some thiazide diuretics (e.g., hydrochlorothiazide), if taken for extended periods, may interfere with magnesium reabsorption in the kidneys and result in magnesium depletion (182). Many other medications may also result in renal magnesium loss (3).
Linus Pauling Institute Recommendation
The Linus Pauling Institute supports the latest RDA for magnesium intake (400 to 420 mg/day for men and 310 to 320 mg/day for women). Despite magnesium being plentiful in foods, it is considered a shortfall nutrient (see the article on Micronutrient Inadequacies in the US Population). Following the Linus Pauling Institute recommendation to take a daily multivitamin/mineral supplement might ensure an intake of at least 100 mg/day of magnesium. Few multivitamin/mineral supplements contain more than 100 mg of magnesium due to its bulk. Eating a varied diet that provides green vegetables, whole grains, and nuts daily should provide the rest of an individual's magnesium requirement.
Older adults (>50 years)
Older adults are less likely than younger adults to consume enough magnesium to meet their needs and should therefore take care to eat magnesium-rich foods in addition to taking a multivitamin/mineral supplement daily (see the article on Micronutrient Inadequacies: Subpopulations at Risk). Since older adults are also more likely to have impaired kidney function, they should avoid taking more than 350 mg/day of supplemental magnesium without medical consultation (see Safety).
Authors and Reviewers
Originally written in 2001 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2003 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in August 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in October 2013 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in November 2018 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in February 2019 by:
Stella L. Volpe, Ph.D., RDN, ACSM-CEP, FACSM
Professor and Chair
Department of Nutrition Sciences
Drexel University
Copyright 2001-2024 Linus Pauling Institute
References
1. Volpe SL. Magnesium. In: Erdman Jr. JW, Macdonald IA, Ziegler EE, eds. Present Knowledge in Nutrition. 10th ed: ILSI Press; 2012:459-474.
2. Food and Nutrition Board, Institute of Medicine. Magnesium. Dietary Reference Intakes: Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington, D.C.: National Academy Press; 1997:190-249. (National Academy Press)
3. Rude RK, Shils ME. Magnesium. In: Shils ME, Shike M, Ross AC, Caballero B, Cousins RJ, eds. Modern Nutrition in Health and Disease. 10th ed. Baltimore: Lippincott Williams & Wilkins; 2006:223-247.
4. Spencer H, Norris C, Williams D. Inhibitory effects of zinc on magnesium balance and magnesium absorption in man. J Am Coll Nutr. 1994;13(5):479-484. (PubMed)
5. Schwartz R, Walker G, Linz MD, MacKellar I. Metabolic responses of adolescent boys to two levels of dietary magnesium and protein. I. Magnesium and nitrogen retention. Am J Clin Nutr. 1973;26(5):510-518. (PubMed)
6. Navarro-Gonzalez JF, Mora-Fernandez C, Garcia-Perez J. Clinical implications of disordered magnesium homeostasis in chronic renal failure and dialysis. Semin Dial. 2009;22(1):37-44. (PubMed)
7. Rude RK. Magnesium. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. China: Williams & Wilkins; 2014:159-175.
8. Moshfegh A, Goldman J, Ahuja J, Rhodes D, LaComb R. What We Eat in America, NHANES 2005-2006: Usual Nutrient Intakes from Food and Water Compared to 1997 Dietary Reference Intakes for Vitamin D, Calcium, Phosphorus, and Magnesium. 2009.
9. Sebastian RS, Cleveland LE, Goldman JD, Moshfegh AJ. Older adults who use vitamin/mineral supplements differ from nonusers in nutrient intake adequacy and dietary attitudes. J Am Diet Assoc. 2007;107(8):1322-1332. (PubMed)
10. Costello RB, Nielsen F. Interpreting magnesium status to enhance clinical care: key indicators. Curr Opin Clin Nutr Metab Care. 2017;20(6):504-511. (PubMed)
11. Grundy SM, Cleeman JI, Daniels SR, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112(17):2735-2752. (PubMed)
12. Esposito K, Chiodini P, Colao A, Lenzi A, Giugliano D. Metabolic syndrome and risk of cancer: a systematic review and meta-analysis. Diabetes Care. 2012;35(11):2402-2411. (PubMed)
13. Ninomiya JK, L'Italien G, Criqui MH, Whyte JL, Gamst A, Chen RS. Association of the metabolic syndrome with history of myocardial infarction and stroke in the Third National Health and Nutrition Examination Survey. Circulation. 2004;109(1):42-46. (PubMed)
14. Sung KC, Lee MY, Kim YH, et al. Obesity and incidence of diabetes: Effect of absence of metabolic syndrome, insulin resistance, inflammation and fatty liver. Atherosclerosis. 2018;275:50-57. (PubMed)
15. Moore-Schiltz L, Albert JM, Singer ME, Swain J, Nock NL. Dietary intake of calcium and magnesium and the metabolic syndrome in the National Health and Nutrition Examination (NHANES) 2001-2010 data. Br J Nutr. 2015;114(6):924-935. (PubMed)
16. Dibaba DT, Xun P, Fly AD, Yokota K, He K. Dietary magnesium intake and risk of metabolic syndrome: a meta-analysis. Diabet Med. 2014;31(11):1301-1309. (PubMed)
17. Ju SY, Choi WS, Ock SM, Kim CM, Kim DH. Dietary magnesium intake and metabolic syndrome in the adult population: dose-response meta-analysis and meta-regression. Nutrients. 2014;6(12):6005-6019. (PubMed)
18. Sarrafzadegan N, Khosravi-Boroujeni H, Lotfizadeh M, Pourmogaddas A, Salehi-Abargouei A. Magnesium status and the metabolic syndrome: A systematic review and meta-analysis. Nutrition. 2016;32(4):409-417. (PubMed)
19. La SA, Lee JY, Kim DH, Song EL, Park JH, Ju SY. Low magnesium levels in adults with metabolic syndrome: a meta-analysis. Biol Trace Elem Res. 2016;170(1):33-42. (PubMed)
20. Song Y, Ridker PM, Manson JE, Cook NR, Buring JE, Liu S. Magnesium intake, C-reactive protein, and the prevalence of metabolic syndrome in middle-aged and older U.S. women. Diabetes Care. 2005;28(6):1438-1444. (PubMed)
21. Dibaba DT, Xun P, He K. Dietary magnesium intake is inversely associated with serum C-reactive protein levels: meta-analysis and systematic review. Eur J Clin Nutr. 2014;68(4):510-516. (PubMed)
22. Ascherio A, Rimm EB, Giovannucci EL, et al. A prospective study of nutritional factors and hypertension among US men. Circulation. 1992;86(5):1475-1484. (PubMed)
23. Ascherio A, Hennekens C, Willett WC, et al. Prospective study of nutritional factors, blood pressure, and hypertension among US women. Hypertension. 1996;27(5):1065-1072. (PubMed)
24. Peacock JM, Folsom AR, Arnett DK, Eckfeldt JH, Szklo M. Relationship of serum and dietary magnesium to incident hypertension: the Atherosclerosis Risk in Communities (ARIC) Study. Ann Epidemiol. 1999;9(3):159-165. (PubMed)
25. He K, Liu K, Daviglus ML, et al. Magnesium intake and incidence of metabolic syndrome among young adults. Circulation. 2006;113(13):1675-1682. (PubMed)
26. Han H, Fang X, Wei X, et al. Dose-response relationship between dietary magnesium intake, serum magnesium concentration and risk of hypertension: a systematic review and meta-analysis of prospective cohort studies. Nutr J. 2017;16(1):26. (PubMed)
27. Joosten MM, Gansevoort RT, Mukamal KJ, et al. Urinary magnesium excretion and risk of hypertension: the prevention of renal and vascular end-stage disease study. Hypertension. 2013;61(6):1161-1167. (PubMed)
28. Rennenberg RJ, Kessels AG, Schurgers LJ, van Engelshoven JM, de Leeuw PW, Kroon AA. Vascular calcifications as a marker of increased cardiovascular risk: a meta-analysis. Vasc Health Risk Manag. 2009;5(1):185-197. (PubMed)
29. Major RW, Cheng MRI, Grant RA, et al. Cardiovascular disease risk factors in chronic kidney disease: A systematic review and meta-analysis. PLoS One. 2018;13(3):e0192895. (PubMed)
30. Palmer SC, Hayen A, Macaskill P, et al. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: a systematic review and meta-analysis. JAMA. 2011;305(11):1119-1127. (PubMed)
31. Sakaguchi Y, Hamano T, Nakano C, et al. Association between density of coronary artery calcification and serum magnesium levels among patients with chronic kidney disease. PLoS One. 2016;11(9):e0163673. (PubMed)
32. Bressendorff I, Hansen D, Schou M, et al. Oral magnesium supplementation in chronic kidney disease stages 3 and 4: efficacy, safety, and effect on serum calcification propensity-a prospective randomized double-blinded placebo-controlled clinical trial. Kidney Int Rep. 2017;2(3):380-389. (PubMed)
33. Pasch A, Block GA, Bachtler M, et al. Blood calcification propensity, cardiovascular events, and survival in patients receiving hemodialysis in the EVOLVE trial. Clin J Am Soc Nephrol. 2017;12(2):315-322. (PubMed)
34. Smith ER, Ford ML, Tomlinson LA, et al. Serum calcification propensity predicts all-cause mortality in predialysis CKD. J Am Soc Nephrol. 2014;25(2):339-348. (PubMed)
35. Bressendorff I, Hansen D, Schou M, Pasch A, Brandi L. The effect of increasing dialysate magnesium on serum calcification propensity in subjects with end stage kidney disease: a randomized, controlled clinical trial. Clin J Am Soc Nephrol. 2018;13(9):1373-1380. (PubMed)
36. Bressendorff I, Hansen D, Schou M, Kragelund C, Brandi L. The effect of magnesium supplementation on vascular calcification in chronic kidney disease-a randomised clinical trial (MAGiCAL-CKD): essential study design and rationale. BMJ Open. 2017;7(6):e016795. (PubMed)
37. Hruby A, O'Donnell CJ, Jacques PF, Meigs JB, Hoffmann U, McKeown NM. Magnesium intake is inversely associated with coronary artery calcification: the Framingham Heart Study. JACC Cardiovasc Imaging. 2014;7(1):59-69. (PubMed)
38. Hisamatsu T, Miura K, Fujiyoshi A, et al. Serum magnesium, phosphorus, and calcium levels and subclinical calcific aortic valve disease: A population-based study. Atherosclerosis. 2018;273:145-152. (PubMed)
39. Lee SY, Hyun YY, Lee KB, Kim H. Low serum magnesium is associated with coronary artery calcification in a Korean population at low risk for cardiovascular disease. Nutr Metab Cardiovasc Dis. 2015;25(11):1056-1061. (PubMed)
40. Posadas-Sanchez R, Posadas-Romero C, Cardoso-Saldana G, et al. Serum magnesium is inversely associated with coronary artery calcification in the Genetics of Atherosclerotic Disease (GEA) study. Nutr J. 2016;15:22. (PubMed)
41. Chiuve SE, Sun Q, Curhan GC, et al. Dietary and plasma magnesium and risk of coronary heart disease among women. J Am Heart Assoc. 2013;2(2):e000114. (PubMed)
42. Del Gobbo LC, Imamura F, Wu JH, de Oliveira Otto MC, Chiuve SE, Mozaffarian D. Circulating and dietary magnesium and risk of cardiovascular disease: a systematic review and meta-analysis of prospective studies. Am J Clin Nutr. 2013;98(1):160-173. (PubMed)
43. Fang X, Wang K, Han D, et al. Dietary magnesium intake and the risk of cardiovascular disease, type 2 diabetes, and all-cause mortality: a dose-response meta-analysis of prospective cohort studies. BMC Med. 2016;14(1):210. (PubMed)
44. Larsson SC, Orsini N, Wolk A. Dietary magnesium intake and risk of stroke: a meta-analysis of prospective studies. Am J Clin Nutr. 2012;95(2):362-366. (PubMed)
45. Nie ZL, Wang ZM, Zhou B, Tang ZP, Wang SK. Magnesium intake and incidence of stroke: meta-analysis of cohort studies. Nutr Metab Cardiovasc Dis. 2013;23(3):169-176. (PubMed)
46. Qu X, Jin F, Hao Y, et al. Magnesium and the risk of cardiovascular events: a meta-analysis of prospective cohort studies. PLoS One. 2013;8(3):e57720. (PubMed)
47. Liao F, Folsom AR, Brancati FL. Is low magnesium concentration a risk factor for coronary heart disease? The Atherosclerosis Risk in Communities (ARIC) Study. Am Heart J. 1998;136(3):480-490. (PubMed)
48. Wannamethee SG, Papacosta O, Lennon L, Whincup PH. Serum magnesium and risk of incident heart failure in older men: The British Regional Heart Study. Eur J Epidemiol. 2018;33(9):873-882. (PubMed)
49. Catling LA, Abubakar I, Lake IR, Swift L, Hunter PR. A systematic review of analytical observational studies investigating the association between cardiovascular disease and drinking water hardness. J Water Health. 2008;6(4):433-442. (PubMed)
50. Xu T, Sun Y, Xu T, Zhang Y. Magnesium intake and cardiovascular disease mortality: A meta-analysis of prospective cohort studies. Int J Cardiol. 2013;167(6):3044-3047. (PubMed)
51. Zhang X, Xia J, Del Gobbo LC, Hruby A, Dai Q, Song Y. Serum magnesium concentrations and all-cause, cardiovascular, and cancer mortality among U.S. adults: Results from the NHANES I Epidemiologic Follow-up Study. Clin Nutr. 2018;37(5):1541-1549. (PubMed)
52. Angkananard T, Anothaisintawee T, Eursiriwan S, et al. The association of serum magnesium and mortality outcomes in heart failure patients: A systematic review and meta-analysis. Medicine (Baltimore). 2016;95(50):e5406. (PubMed)
53. van den Bergh WM, Algra A, van der Sprenkel JW, Tulleken CA, Rinkel GJ. Hypomagnesemia after aneurysmal subarachnoid hemorrhage. Neurosurgery. 2003;52(2):276-281; discussion 281-272. (PubMed)
54. Chen T, Carter BS. Role of magnesium sulfate in aneurysmal subarachnoid hemorrhage management: A meta-analysis of controlled clinical trials. Asian J Neurosurg. 2011;6(1):26-31. (PubMed)
55. Yarad EA, Hammond NE, Winner ABNRPsbE. Intravenous magnesium therapy in adult patients with an aneurysmal subarachnoid haemorrhage: A systematic review and meta-analysis. Aust Crit Care. 2013;26(3):105-117. (PubMed)
56. Golan E, Vasquez DN, Ferguson ND, Adhikari NK, Scales DC. Prophylactic magnesium for improving neurologic outcome after aneurysmal subarachnoid hemorrhage: systematic review and meta-analysis. J Crit Care. 2013;28(2):173-181. (PubMed)
57. Kunze E, Lilla N, Stetter C, Ernestus RI, Westermaier T. Magnesium protects in episodes of critical perfusion after aneurysmal SAH. Transl Neurosci. 2018;9:99-105. (PubMed)
58. Westermaier T, Stetter C, Vince GH, et al. Prophylactic intravenous magnesium sulfate for treatment of aneurysmal subarachnoid hemorrhage: a randomized, placebo-controlled, clinical study. Crit Care Med. 2010;38(5):1284-1290. (PubMed)
59. Arsenault KA, Yusuf AM, Crystal E, et al. Interventions for preventing post-operative atrial fibrillation in patients undergoing heart surgery. Cochrane Database Syst Rev. 2013;1:CD003611. (PubMed)
60. Fairley JL, Zhang L, Glassford NJ, Bellomo R. Magnesium status and magnesium therapy in cardiac surgery: A systematic review and meta-analysis focusing on arrhythmia prevention. J Crit Care. 2017;42:69-77. (PubMed)
61. Wu X, Wang C, Zhu J, Zhang C, Zhang Y, Gao Y. Meta-analysis of randomized controlled trials on magnesium in addition to beta-blocker for prevention of postoperative atrial arrhythmias after coronary artery bypass grafting. BMC Cardiovasc Disord. 2013;13:5. (PubMed)
62. Schulze MB, Schulz M, Heidemann C, Schienkiewitz A, Hoffmann K, Boeing H. Fiber and magnesium intake and incidence of type 2 diabetes: a prospective study and meta-analysis. Arch Intern Med. 2007;167(9):956-965. (PubMed)
63. Fang X, Han H, Li M, et al. Dose-response relationship between dietary magnesium intake and risk of type 2 diabetes mellitus: a systematic review and meta-regression analysis of prospective cohort studies. Nutrients. 2016;8(11). (PubMed)
64. Dong JY, Xun P, He K, Qin LQ. Magnesium intake and risk of type 2 diabetes: meta-analysis of prospective cohort studies. Diabetes Care. 2011;34(9):2116-2122. (PubMed)
65. Hruby A, Ngwa JS, Renstrom F, et al. Higher magnesium intake is associated with lower fasting glucose and insulin, with no evidence of interaction with select genetic loci, in a meta-analysis of 15 CHARGE Consortium Studies. J Nutr. 2013;143(3):345-353. (PubMed)
66. Larsson SC, Wolk A. Magnesium intake and risk of type 2 diabetes: a meta-analysis. J Intern Med. 2007;262(2):208-214. (PubMed)
67. Simental-Mendia LE, Rodriguez-Moran M, Guerrero-Romero F. Failure of beta-cell function for compensate variation in insulin sensitivity in hypomagnesemic subjects. Magnes Res. 2009;22(3):151-156. (PubMed)
68. Guerrero-Romero F, Rodriguez-Moran M. Magnesium improves the beta-cell function to compensate variation of insulin sensitivity: double-blind, randomized clinical trial. Eur J Clin Invest. 2011;41(4):405-410. (PubMed)
69. Guerrero-Romero F, Simental-Mendia LE, Hernandez-Ronquillo G, Rodriguez-Moran M. Oral magnesium supplementation improves glycaemic status in subjects with prediabetes and hypomagnesaemia: A double-blind placebo-controlled randomized trial. Diabetes Metab. 2015;41(3):202-207. (PubMed)
70. Rodriguez-Moran M, Guerrero-Romero F. Oral magnesium supplementation improves the metabolic profile of metabolically obese, normal-weight individuals: a randomized double-blind placebo-controlled trial. Arch Med Res. 2014;45(5):388-393. (PubMed)
71. Mooren FC, Kruger K, Volker K, Golf SW, Wadepuhl M, Kraus A. Oral magnesium supplementation reduces insulin resistance in non-diabetic subjects - a double-blind, placebo-controlled, randomized trial. Diabetes Obes Metab. 2011;13(3):281-284. (PubMed)
72. Castiglioni S, Cazzaniga A, Albisetti W, Maier JA. Magnesium and osteoporosis: current state of knowledge and future research directions. Nutrients. 2013;5(8):3022-3033. (PubMed)
73. Vormann J. Magnesium: Nutrition and Homeostasis. AIMS Public Health. 2016;3(2):329-340. (PubMed)
74. Sojka JE, Weaver CM. Magnesium supplementation and osteoporosis. Nutr Rev. 1995;53(3):71-74. (PubMed)
75. Zheng J, Mao X, Ling J, He Q, Quan J, Jiang H. Association between serum level of magnesium and postmenopausal osteoporosis: a meta-analysis. Biol Trace Elem Res. 2014;159(1-3):8-14. (PubMed)
76. Begin MJ, Ste-Marie LG, Coupal L, Ethier J, Rakel A. Hypomagnesemia during teriparatide treatment in osteoporosis: incidence and determinants. J Bone Miner Res. 2018;33(8):1444-1449. (PubMed)
77. Tucker KL, Hannan MT, Chen H, Cupples LA, Wilson PW, Kiel DP. Potassium, magnesium, and fruit and vegetable intakes are associated with greater bone mineral density in elderly men and women. Am J Clin Nutr. 1999;69(4):727-736. (PubMed)
78. Ryder KM, Shorr RI, Bush AJ, et al. Magnesium intake from food and supplements is associated with bone mineral density in healthy older white subjects. J Am Geriatr Soc. 2005;53(11):1875-1880. (PubMed)
79. Dahl C, Sogaard AJ, Tell GS, et al. Nationwide data on municipal drinking water and hip fracture: Could calcium and magnesium be protective? A NOREPOS study. Bone. 2013;57(1):84-91. (PubMed)
80. Orchard TS, Larson JC, Alghothani N, et al. Magnesium intake, bone mineral density, and fractures: results from the Women's Health Initiative observational study. Am J Clin Nutr. 2014;99(4):926-933. (PubMed)
81. Hayhoe RP, Lentjes MA, Luben RN, Khaw KT, Welch AA. Dietary magnesium and potassium intakes and circulating magnesium are associated with heel bone ultrasound attenuation and osteoporotic fracture risk in the EPIC-Norfolk cohort study. Am J Clin Nutr. 2015;102(2):376-384. (PubMed)
82. Stendig-Lindberg G, Tepper R, Leichter I. Trabecular bone density in a two year controlled trial of peroral magnesium in osteoporosis. Magnes Res. 1993;6(2):155-163. (PubMed)
83. Abraham GE, Grewal H. A total dietary program emphasizing magnesium instead of calcium. Effect on the mineral density of calcaneous bone in postmenopausal women on hormonal therapy. J Reprod Med. 1990;35(5):503-507. (PubMed)
84. Aydin H, Deyneli O, Yavuz D, et al. Short-term oral magnesium supplementation suppresses bone turnover in postmenopausal osteoporotic women. Biol Trace Elem Res. 2010;133(2):136-143. (PubMed)
85. Nieves JW. Bone. Maximizing bone health--magnesium, BMD and fractures. Nat Rev Endocrinol. 2014;10(5):255-256. (PubMed)
86. Landi F, Liperoti R, Russo A, et al. Sarcopenia as a risk factor for falls in elderly individuals: results from the ilSIRENTE study. Clin Nutr. 2012;31(5):652-658. (PubMed)
87. Hayhoe RPG, Lentjes MAH, Mulligan AA, Luben RN, Khaw KT, Welch AA. Cross-sectional associations of dietary and circulating magnesium with skeletal muscle mass in the EPIC-Norfolk cohort. Clin Nutr. 2019;38(1):317-323. (PubMed)
88. Scott D, Blizzard L, Fell J, Giles G, Jones G. Associations between dietary nutrient intake and muscle mass and strength in community-dwelling older adults: the Tasmanian Older Adult Cohort Study. J Am Geriatr Soc. 2010;58(11):2129-2134. (PubMed)
89. Welch AA, Kelaiditi E, Jennings A, Steves CJ, Spector TD, MacGregor A. Dietary magnesium is positively associated with skeletal muscle power and indices of muscle mass and may attenuate the association between circulating C-reactive protein and muscle mass in women. J Bone Miner Res. 2016;31(2):317-325. (PubMed)
90. Welch AA, Skinner J, Hickson M. Dietary magnesium may be protective for aging of bone and skeletal muscle in middle and younger older age men and women: cross-sectional findings from the UK Biobank cohort. Nutrients. 2017;9(11). (PubMed)
91. Veronese N, Berton L, Carraro S, et al. Effect of oral magnesium supplementation on physical performance in healthy elderly women involved in a weekly exercise program: a randomized controlled trial. Am J Clin Nutr. 2014;100(3):974-981. (PubMed)
92. Bibbins-Domingo K, Grossman DC, Curry SJ, et al. Screening for preeclampsia: US Preventive Services Task Force recommendation statement. JAMA. 2017;317(16):1661-1667. (PubMed)
93. Jeyabalan A. Epidemiology of preeclampsia: impact of obesity. Nutr Rev. 2013;71 Suppl 1:S18-25. (PubMed)
94. Duley L. The global impact of pre-eclampsia and eclampsia. Semin Perinatol. 2009;33(3):130-137. (PubMed)
95. Duley L, Henderson-Smart DJ, Chou D. Magnesium sulphate versus phenytoin for eclampsia. Cochrane Database Syst Rev. 2010(10):CD000128. (PubMed)
96. Makrides M, Crosby DD, Bain E, Crowther CA. Magnesium supplementation in pregnancy. Cochrane Database Syst Rev. 2014(4):Cd000937. (PubMed)
97. Sibai BM. Diagnosis, prevention, and management of eclampsia. Obstet Gynecol. 2005;105(2):402-410. (PubMed)
98. Altman D, Carroli G, Duley L, et al. Do women with pre-eclampsia, and their babies, benefit from magnesium sulphate? The Magpie Trial: a randomised placebo-controlled trial. Lancet. 2002;359(9321):1877-1890. (PubMed)
99. McDonald SD, Lutsiv O, Dzaja N, Duley L. A systematic review of maternal and infant outcomes following magnesium sulfate for pre-eclampsia/eclampsia in real-world use. Int J Gynaecol Obstet. 2012;118(2):90-96. (PubMed)
100. World Health Organization. WHO recommendations for prevention and treatment of pre-eclampsia and eclampsia. Implications and actions. Available at: http://www.who.int/reproductivehealth/publications/maternal_perinatal_health/program-action-eclampsia/en/. Accessed 10/9/18.
101. Berhan Y, Berhan A. Should magnesium sulfate be administered to women with mild pre-eclampsia? A systematic review of published reports on eclampsia. J Obstet Gynaecol Res. 2015;41(6):831-842. (PubMed)
102. Vigil-DeGracia P, Ludmir J, Ng J, et al. Is there benefit to continue magnesium sulphate postpartum in women receiving magnesium sulphate before delivery? A randomised controlled study. Bjog. 2018;125(10):1304-1311. (PubMed)
103. American College of Obstetricians and Gynecologists Committee. Committee opinion no. 573: magnesium sulfate use in obstetrics. Obstet Gynecol. 2013;122(3):727-728. (PubMed)
104. Roberts D, Brown J, Medley N, Dalziel SR. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst Rev. 2017;3:Cd004454. (PubMed)
105. Crowther CA, Brown J, McKinlay CJ, Middleton P. Magnesium sulphate for preventing preterm birth in threatened preterm labour. Cochrane Database Syst Rev. 2014(8):Cd001060. (PubMed)
106. McNamara HC, Crowther CA, Brown J. Different treatment regimens of magnesium sulphate for tocolysis in women in preterm labour. Cochrane Database Syst Rev. 2015(12):Cd011200. (PubMed)
107. Wolf HT, Hegaard HK, Greisen G, Huusom L, Hedegaard M. Treatment with magnesium sulphate in pre-term birth: a systematic review and meta-analysis of observational studies. J Obstet Gynaecol. 2012;32(2):135-140. (PubMed)
108. Crowther CA, Middleton PF, Voysey M, et al. Assessing the neuroprotective benefits for babies of antenatal magnesium sulphate: An individual participant data meta-analysis. PLoS Med. 2017;14(10):e1002398. (PubMed)
109. Tagin M, Shah PS, Lee KS. Magnesium for newborns with hypoxic-ischemic encephalopathy: a systematic review and meta-analysis. J Perinatol. 2013;33(9):663-669. (PubMed)
110. Kass L, Weekes J, Carpenter L. Effect of magnesium supplementation on blood pressure: a meta-analysis. Eur J Clin Nutr. 2012;66(4):411-418. (PubMed)
111. Rosanoff A. Magnesium supplements may enhance the effect of antihypertensive medications in stage 1 hypertensive subjects. Magnes Res. 2010;23(1):27-40. (PubMed)
112. Zhang X, Li Y, Del Gobbo LC, et al. Effects of magnesium supplementation on blood pressure: a meta-analysis of randomized double-blind placebo-controlled trials. Hypertension. 2016;68(2):324-333. (PubMed)
113. Dibaba DT, Xun P, Song Y, Rosanoff A, Shechter M, He K. The effect of magnesium supplementation on blood pressure in individuals with insulin resistance, prediabetes, or noncommunicable chronic diseases: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2017;106(3):921-929. (PubMed)
114. Appel LJ, Moore TJ, Obarzanek E, et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N Engl J Med. 1997;336(16):1117-1124. (PubMed)
115. Maier JA. Endothelial cells and magnesium: implications in atherosclerosis. Clin Sci (Lond). 2012;122(9):397-407. (PubMed)
116. Darooghegi Mofrad M, Djafarian K, Mozaffari H, Shab-Bidar S. Effect of magnesium supplementation on endothelial function: A systematic review and meta-analysis of randomized controlled trials. Atherosclerosis. 2018;273:98-105. (PubMed)
117. Shechter M, Sharir M, Labrador MJ, Forrester J, Silver B, Bairey Merz CN. Oral magnesium therapy improves endothelial function in patients with coronary artery disease. Circulation. 2000;102(19):2353-2358. (PubMed)
118. Barbagallo M, Dominguez LJ, Galioto A, Pineo A, Belvedere M. Oral magnesium supplementation improves vascular function in elderly diabetic patients. Magnes Res. 2010;23(3):131-137. (PubMed)
119. Cunha AR, D'El-Rei J, Medeiros F, et al. Oral magnesium supplementation improves endothelial function and attenuates subclinical atherosclerosis in thiazide-treated hypertensive women. J Hypertens. 2017;35(1):89-97. (PubMed)
120. Mortazavi M, Moeinzadeh F, Saadatnia M, Shahidi S, McGee JC, Minagar A. Effect of magnesium supplementation on carotid intima-media thickness and flow-mediated dilatation among hemodialysis patients: a double-blind, randomized, placebo-controlled trial. Eur Neurol. 2013;69(5):309-316. (PubMed)
121. Cosaro E, Bonafini S, Montagnana M, et al. Effects of magnesium supplements on blood pressure, endothelial function and metabolic parameters in healthy young men with a family history of metabolic syndrome. Nutr Metab Cardiovasc Dis. 2014;24(11):1213-1220. (PubMed)
122. Joris PJ, Plat J, Bakker SJ, Mensink RP. Effects of long-term magnesium supplementation on endothelial function and cardiometabolic risk markers: A randomized controlled trial in overweight/obese adults. Sci Rep. 2017;7(1):106. (PubMed)
123. Mookadam F, Moustafa SE, Lester SJ, Warsame T. Subclinical atherosclerosis: evolving role of carotid intima-media thickness. Prev Cardiol. 2010;13(4):186-197. (PubMed)
124. Ma J, Folsom AR, Melnick SL, et al. Associations of serum and dietary magnesium with cardiovascular disease, hypertension, diabetes, insulin, and carotid arterial wall thickness: the ARIC study. Atherosclerosis Risk in Communities Study. J Clin Epidemiol. 1995;48(7):927-940. (PubMed)
125. Woods KL, Fletcher S, Roffe C, Haider Y. Intravenous magnesium sulphate in suspected acute myocardial infarction: results of the second Leicester Intravenous Magnesium Intervention Trial (LIMIT-2). Lancet. 1992;339(8809):1553-1558. (PubMed)
126. Woods KL, Fletcher S. Long-term outcome after intravenous magnesium sulphate in suspected acute myocardial infarction: the second Leicester Intravenous Magnesium Intervention Trial (LIMIT-2). Lancet. 1994;343(8901):816-819. (PubMed)
127. Fourth International Study of Infarct Survival (ISIS-4) Collaborative Group. A randomised factorial trial assessing early oral captopril, oral mononitrate, and intravenous magnesium sulphate in 58,050 patients with suspected acute myocardial infarction. Lancet. 1995;345(8951):669-685. (PubMed)
128. Ziegelstein RC, Hilbe JM, French WJ, Antman EM, Chandra-Strobos N. Magnesium use in the treatment of acute myocardial infarction in the United States (observations from the Second National Registry of Myocardial Infarction). Am J Cardiol. 2001;87(1):7-10. (PubMed)
129. Li J, Zhang Q, Zhang M, Egger M. Intravenous magnesium for acute myocardial infarction. Cochrane Database Syst Rev. 2007(2):CD002755. (PubMed)
130. Pham PC, Pham PM, Pham SV, Miller JM, Pham PT. Hypomagnesemia in patients with type 2 diabetes. Clin J Am Soc Nephrol. 2007;2(2):366-373. (PubMed)
131. Takaya J, Higashino H, Kobayashi Y. Intracellular magnesium and insulin resistance. Magnes Res. 2004;17(2):126-136. (PubMed)
132. Yokota K, Kato M, Lister F, et al. Clinical efficacy of magnesium supplementation in patients with type 2 diabetes. J Am Coll Nutr. 2004;23(5):506S-509S. (PubMed)
133. Rodriguez-Moran M, Guerrero-Romero F. Oral magnesium supplementation improves insulin sensitivity and metabolic control in type 2 diabetic subjects: a randomized double-blind controlled trial. Diabetes Care. 2003;26(4):1147-1152. (PubMed)
134. Veronese N, Watutantrige-Fernando S, Luchini C, et al. Effect of magnesium supplementation on glucose metabolism in people with or at risk of diabetes: a systematic review and meta-analysis of double-blind randomized controlled trials. Eur J Clin Nutr. 2016;70(12):1354-1359. (PubMed)
135. Simental-Mendia LE, Sahebkar A, Rodriguez-Moran M, Guerrero-Romero F. A systematic review and meta-analysis of randomized controlled trials on the effects of magnesium supplementation on insulin sensitivity and glucose control. Pharmacol Res. 2016;111:272-282. (PubMed)
136. Hashimoto Y, Nishimura Y, Maeda H, Yokoyama M. Assessment of magnesium status in patients with bronchial asthma. J Asthma. 2000;37(6):489-496. (PubMed)
137. Irazuzta JE, Chiriboga N. Magnesium sulfate infusion for acute asthma in the emergency department. J Pediatr (Rio J). 2017;93 Suppl 1:19-25. (PubMed)
138. Su Z, Li R, Gai Z. Intravenous and nebulized magnesium sulfate for treating acute asthma in children: a systematic review and meta-analysis. Pediatr Emerg Care. 2018;34(6):390-395. (PubMed)
139. Kew KM, Kirtchuk L, Michell CI. Intravenous magnesium sulfate for treating adults with acute asthma in the emergency department. Cochrane Database Syst Rev. 2014(5):Cd010909. (PubMed)
140. Knightly R, Milan SJ, Hughes R, et al. Inhaled magnesium sulfate in the treatment of acute asthma. Cochrane Database Syst Rev. 2017;11:Cd003898. (PubMed)
141. Monteleone CA, Sherman AR. Nutrition and asthma. Arch Intern Med. 1997;157(1):23-34. (PubMed)
142. Beasley R, Aldington S. Magnesium in the treatment of asthma. Curr Opin Allergy Clin Immunol. 2007;7(1):107-110. (PubMed)
143. Fogarty A, Lewis SA, Scrivener SL, et al. Oral magnesium and vitamin C supplements in asthma: a parallel group randomized placebo-controlled trial. Clin Exp Allergy. 2003;33(10):1355-1359. (PubMed)
144. Na HS, Ryu JH, Do SH. The role of magnesium in pain. In: Vink R, Nechifor M, eds. Magnesium in the Central Nervous System. Adelaide (AU): University of Adelaide Press; 2011. (PubMed)
145. Wang SC, Pan PT, Chiu HY, Huang CJ. Neuraxial magnesium sulfate improves postoperative analgesia in Cesarean section delivery women: A meta-analysis of randomized controlled trials. Asian J Anesthesiol. 2017;55(3):56-67. (PubMed)
146. Xiao F, Xu W, Feng Y, et al. Intrathecal magnesium sulfate does not reduce the ED50 of intrathecal hyperbaric bupivacaine for cesarean delivery in healthy parturients: a prospective, double blinded, randomized dose-response trial using the sequential allocation method. BMC Anesthesiol. 2017;17(1):8. (PubMed)
147. Paleti S, Prasad PK, Lakshmi BS. A randomized clinical trial of intrathecal magnesium sulfate versus midazolam with epidural administration of 0.75% ropivacaine for patients with preeclampsia scheduled for elective cesarean section. J Anaesthesiol Clin Pharmacol. 2018;34(1):23-28. (PubMed)
148. Vlok R, Melhuish TM, Chong C, Ryan T, White LD. Adjuncts to local anaesthetics in tonsillectomy: a systematic review and meta-analysis. J Anesth. 2017;31(4):608-616. (PubMed)
149. Cho HK, Park IJ, Yoon HY, Hwang SH. Efficacy of adjuvant magnesium for posttonsillectomy morbidity in children: a meta-analysis. Otolaryngol Head Neck Surg. 2018;158(1):27-35. (PubMed)
150. Xie M, Li XK, Peng Y. Magnesium sulfate for postoperative complications in children undergoing tonsillectomies: a systematic review and meta-analysis. J Evid Based Med. 2017;10(1):16-25. (PubMed)
151. Chen C, Tao R. The impact of magnesium sulfate on pain control after laparoscopic cholecystectomy: a meta-analysis of randomized controlled studies. Surg Laparosc Endosc Percutan Tech. 2018;28(6):349-353. (PubMed)
152. Abd-Elsalam KA, Fares KM, Mohamed MA, Mohamed MF, El-Rahman AMA, Tohamy MM. Efficacy of magnesium sulfate added to local anesthetic in a transversus abdominis plane block for analgesia following total abdominal hysterectomy: a randomized trial. Pain Physician. 2017;20(7):641-647. (PubMed)
153. Imani F, Rahimzadeh P, Faiz HR, Abdullahzadeh-Baghaei A. An evaluation of the adding magnesium sulfate to ropivacaine on ultrasound-guided transverse abdominis plane block after abdominal hysterectomy. Anesth Pain Med. 2018;8(4):e74124. (PubMed)
154. Martin DP, Samora WP, 3rd, Beebe AC, et al. Analgesic effects of methadone and magnesium following posterior spinal fusion for idiopathic scoliosis in adolescents: a randomized controlled trial. J Anesth. 2018;32(5):702-708. (PubMed)
155. Srivastava VK, Mishra A, Agrawal S, Kumar S, Sharma S, Kumar R. Comparative evaluation of dexmedetomidine and magnesium sulphate on propofol consumption, haemodynamics and postoperative recovery in spine surgery: a prospective, randomized, placebo controlled, double-blind study. Adv Pharm Bull. 2016;6(1):75-81. (PubMed)
156. Hamed MA. Comparative study between magnesium sulfate and lidocaine for controlled hypotension during functional endoscopic sinus surgery: a randomized controlled study. Anesth Essays Res. 2018;12(3):715-718. (PubMed)
157. Hassan PF, Saleh AH. Dexmedetomidine versus magnesium sulfate in anesthesia for cochlear implantation surgery in pediatric patients. Anesth Essays Res. 2017;11(4):1064-1069. (PubMed)
158. Brill S, Sedgwick PM, Hamann W, Di Vadi PP. Efficacy of intravenous magnesium in neuropathic pain. Br J Anaesth. 2002;89(5):711-714. (PubMed)
159. Tanaka M, Shimizu S, Nishimura W, et al. Relief of neuropathic pain with intravenous magnesium [Article in Japanese]. Masui. 1998;47(9):1109-1113. (PubMed)
160. Pickering G, Morel V, Simen E, et al. Oral magnesium treatment in patients with neuropathic pain: a randomized clinical trial. Magnes Res. 2011;24(2):28-35. (PubMed)
161. Delage N, Morel V, Picard P, Marcaillou F, Pereira B, Pickering G. Effect of ketamine combined with magnesium sulfate in neuropathic pain patients (KETAPAIN): study protocol for a randomized controlled trial. Trials. 2017;18(1):517. (PubMed)
162. Mauskop A, Altura BM. Role of magnesium in the pathogenesis and treatment of migraines. Clin Neurosci. 1998;5(1):24-27. (PubMed)
163. Mauskop A, Altura BT, Altura BM. Serum ionized magnesium levels and serum ionized calcium/ionized magnesium ratios in women with menstrual migraine. Headache. 2002;42(4):242-248. (PubMed)
164. Peikert A, Wilimzig C, Kohne-Volland R. Prophylaxis of migraine with oral magnesium: results from a prospective, multi-center, placebo-controlled and double-blind randomized study. Cephalalgia. 1996;16(4):257-263. (PubMed)
165. Wang F, Van Den Eeden SK, Ackerson LM, Salk SE, Reince RH, Elin RJ. Oral magnesium oxide prophylaxis of frequent migrainous headache in children: a randomized, double-blind, placebo-controlled trial. Headache. 2003;43(6):601-610. (PubMed)
166. Pfaffenrath V, Wessely P, Meyer C, et al. Magnesium in the prophylaxis of migraine--a double-blind placebo-controlled study. Cephalalgia. 1996;16(6):436-440. (PubMed)
167. Demirkaya S, Vural O, Dora B, Topcuoglu MA. Efficacy of intravenous magnesium sulfate in the treatment of acute migraine attacks. Headache. 2001;41(2):171-177. (PubMed)
168. Bigal ME, Bordini CA, Tepper SJ, Speciali JG. Intravenous magnesium sulphate in the acute treatment of migraine without aura and migraine with aura. A randomized, double-blind, placebo-controlled study. Cephalalgia. 2002;22(5):345-353. (PubMed)
169. Corbo J, Esses D, Bijur PE, Iannaccone R, Gallagher EJ. Randomized clinical trial of intravenous magnesium sulfate as an adjunctive medication for emergency department treatment of migraine headache. Ann Emerg Med. 2001;38(6):621-627. (PubMed)
170. Cete Y, Dora B, Ertan C, Ozdemir C, Oktay C. A randomized prospective placebo-controlled study of intravenous magnesium sulphate vs. metoclopramide in the management of acute migraine attacks in the Emergency Department. Cephalalgia. 2005;25(3):199-204. (PubMed)
171. Choi H, Parmar N. The use of intravenous magnesium sulphate for acute migraine: meta-analysis of randomized controlled trials. Eur J Emerg Med. 2014; 21(1):2-9. (PubMed)
172. Shahrami A, Assarzadegan F, Hatamabadi HR, Asgarzadeh M, Sarehbandi B, Asgarzadeh S. Comparison of therapeutic effects of magnesium sulfate vs. dexamethasone/metoclopramide on alleviating acute migraine headache. J Emerg Med. 2015;48(1):69-76. (PubMed)
173. Baratloo A, Mirbaha S, Delavar Kasmaei H, Payandemehr P, Elmaraezy A, Negida A. Intravenous caffeine citrate vs. magnesium sulfate for reducing pain in patients with acute migraine headache; a prospective quasi-experimental study. Korean J Pain. 2017;30(3):176-182. (PubMed)
174. Mauskop A, Varughese J. Why all migraine patients should be treated with magnesium. J Neural Transm. 2012;119(5):575-579. (PubMed)
175. Jiang P, Lv Q, Lai T, Xu F. Does hypomagnesemia impact on the outcome of patients admitted to the intensive care unit? A systematic review and meta-analysis. Shock. 2017;47(3):288-295. (PubMed)
176. Upala S, Jaruvongvanich V, Wijarnpreecha K, Sanguankeo A. Hypomagnesemia and mortality in patients admitted to intensive care unit: a systematic review and meta-analysis. Qjm. 2016;109(7):453-459. (PubMed)
177. Nayak R, Attry S, Ghosh SN. Serum magnesium as a marker of neurological outcome in severe traumatic brain injury patients. Asian J Neurosurg. 2018;13(3):685-688. (PubMed)
178. Ardehali SH, Dehghan S, Baghestani AR, Velayati A, Vahdat Shariatpanahi Z. Association of admission serum levels of vitamin D, calcium, Phosphate, magnesium and parathormone with clinical outcomes in neurosurgical ICU patients. Sci Rep. 2018;8(1):2965. (PubMed)
179. Fulgoni VL, 3rd, Keast DR, Bailey RL, Dwyer J. Foods, fortificants, and supplements: Where do Americans get their nutrients? J Nutr. 2011;141(10):1847-1854. (PubMed)
180. Natural Medicines. Magnesium. Professional handout/Effectiveness. Available at: https://naturalmedicines.therapeuticresearch.com/. Accessed 10/29/18.
181. Hendler SS, Rorvik DM. PDR for Nutritional Supplements. Montvale: Thomson Reuters; 2008.
182. Natural Medicines. Magnesium. Professional handout/Drug Interactions. Available at: https://naturalmedicines.therapeuticresearch.com/. Accessed 10/19/18.
183. Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6(4):443-451. (PubMed)
184. US Food and Drug Administration. Proton pump inhibitor drugs (PPIs): drug safety communication - low magnesium levels can be associated with long-term use. 08/04/2017 Available at: https://www.fda.gov/Drugs/DrugSafety/ucm245011.htm. Accessed 10/22/18.
Manganese
Contents
Manganese is a mineral element that is both nutritionally essential and potentially toxic. The derivation of its name from the Greek word for magic remains appropriate, because scientists are still working to understand the diverse effects of manganese deficiency and manganese toxicity in living organisms (1).
Function
Manganese (Mn) plays an important role in a number of physiologic processes as a constituent of multiple enzymes and an activator of other enzymes (2).
Antioxidant function
Manganese superoxide dismutase (MnSOD) is the principal antioxidant enzyme in the mitochondria. Because mitochondria consume over 90% of the oxygen used by cells, they are especially vulnerable to oxidative stress. The superoxide radical is one of the reactive oxygen species produced in mitochondria during ATP synthesis. MnSOD catalyzes the conversion of superoxide radicals to hydrogen peroxide, which can be reduced to water by other antioxidant enzymes (3).
Metabolism
A number of manganese-activated enzymes play important roles in the metabolism of carbohydrates, amino acids, and cholesterol (4). Pyruvate carboxylase, a manganese-containing enzyme, and phosphoenolpyruvate carboxykinase (PEPCK), a manganese-activated enzyme, are critical in gluconeogenesis — the production of glucose from non-carbohydrate precursors. Arginase, another manganese-containing enzyme, is required by the liver for the urea cycle, a process that detoxifies ammonia generated during amino acid metabolism (3). In the brain, the manganese-activated enzyme, glutamine synthetase, converts the amino acid glutamate to glutamine. Glutamate is an excitotoxic neurotransmitter and a precursor to an inhibitory neurotransmitter, γ-aminobutyric acid (GABA) (5, 6).
Bone and cartilage formation
Manganese deficiency results in abnormal skeletal development in a number of animal species. Manganese is the preferred cofactor of enzymes called glycosyltransferases; these enzymes are required for the synthesis of proteoglycans that are needed for the formation of healthy cartilage and bone (7).
Wound healing
Wound healing is a complex process that requires increased production of collagen. Manganese is required for the activation of prolidase, an enzyme that functions to provide the amino acid, proline, for collagen formation in human skin cells (8). A genetic disorder known as prolidase deficiency results in abnormal wound healing among other problems and is characterized by abnormal manganese metabolism (7). Glycosaminoglycan synthesis, which requires manganese-activated glycosyltransferases, may also play an important role in wound healing (9).
Nutrient interactions
Iron
Iron and manganese share common absorption and transport proteins, including the divalent metal transporter 1, the lactoferrin receptor, transferrin, and ferroportin (reviewed in 10). Absorption of manganese from a meal decreases as the meal's iron content increases (7). Iron supplementation (60 milligrams [mg]/day for four months) has been associated with decreased blood manganese concentrations and decreased MnSOD activity in leukocytes, indicating a reduction in manganese nutritional status (11).
Additionally, an individual's iron status can affect manganese bioavailability. Intestinal absorption of manganese is increased during iron deficiency and decreased when iron stores are elevated (i.e., high ferritin concentrations) (12). Small studies have found increased blood concentrations of manganese in iron-deficient infants (13) and children (14), and a national survey of adults residing in South Korea found men and women with low ferritin levels had higher blood concentrations of manganese compared to those with normal ferritin levels (15). In this analysis, anemia was associated with higher blood concentrations of manganese in women but not in men (15). Men generally absorb less manganese than women, which may be related to the fact that men usually have higher iron stores than women (16). Iron deficiency has also been shown to increase the risk of manganese accumulation in the brain (17).
Magnesium
Supplemental magnesium (200 mg/day) has been shown to slightly decrease manganese bioavailability in healthy adults, either by decreasing manganese absorption or by increasing its excretion (18).
Calcium
In one set of studies, supplemental calcium (500 mg/day) slightly decreased manganese bioavailability in healthy adults. As a source of calcium, milk had the least effect, while calcium carbonate and calcium phosphate had the greatest effect (18). Several other studies have found minimal effects of supplemental calcium on manganese metabolism (19).
Regulation
Although manganese is a nutritionally essential mineral, it is potentially toxic; thus, it is important for the body to tightly regulate manganese homeostasis. While the exact mechanisms that govern manganese homeostasis are not completely understood, systemic regulation is achieved through intestinal control of manganese absorption and hepatic excretion of manganese into bile (20). At the cellular level, influx of manganese into cells is regulated by several different transport proteins, including the transferrin receptor, the divalent metal transporter 1 (DMT 1), zinc-interacting proteins 8 and 14 (ZIP8 and ZIP14), as well as others (reviewed in 21). Efflux of manganese from cells is accomplished by various transporters, including SLC30A10; the sodium-calcium exchanger; and the iron transporter, ferroportin (reviewed in 22). Moreover, subcellular organelles (i.e., the nucleus, mitochondria, Golgi apparatus, lysosome, endosome) utilize various transporters for manganese trafficking within the cell, but the exact mechanisms of regulation are not fully understood (21).
Deficiency
Manganese deficiency has been observed in a number of animal species, but manganese deficiency is not a concern in humans. Signs of manganese deficiency vary among animal species and may include impaired growth, impaired reproductive function, skeletal abnormalities, impaired glucose tolerance, and altered carbohydrate and lipid metabolism. In humans, demonstration of a manganese deficiency syndrome has been less clear (2, 7). A child on long-term total parenteral nutrition (TPN) lacking manganese developed bone demineralization and impaired growth that were corrected by manganese supplementation (23). Young men who were fed a low-manganese diet developed decreased serum cholesterol concentrations and a transient skin rash (24). Blood concentrations of calcium, phosphorus, and alkaline phosphatase were also elevated, which may indicate increased bone remodeling as a consequence of insufficient dietary manganese. Young women fed a manganese-poor diet developed mildly abnormal glucose tolerance in response to an intravenous (IV) infusion of glucose (19). Overall, manganese deficiency is quite rare, and there is more concern for toxicity related to manganese overexposure (see Safety).
The Adequate Intake (AI)
Because there was insufficient information on manganese requirements to set a Recommended Dietary Allowance (RDA), the Food and Nutrition Board (FNB) of the Institute of Medicine (now the National Academy of Medicine) set an adequate intake (AI). Since overt manganese deficiency has not been documented in humans eating natural diets, the FNB based the AI on average dietary intakes of manganese determined by the Total Diet Study — an annual survey of the mineral content of representative American diets (4). AI values for manganese are listed in Table 1 in milligrams (mg)/day by age and gender. Manganese requirements are increased in pregnancy and lactation (4).
Disease Prevention
Low dietary manganese intake or low levels of manganese in blood or tissue have been associated with various chronic diseases. Although manganese insufficiency is not currently thought to cause the diseases discussed below, more research may be warranted to determine whether suboptimal manganese nutritional status contributes to certain disease processes.
Osteoporosis
Early studies found women with osteoporosis had decreased plasma or serum concentrations of manganese and an enhanced plasma response to an oral dose of manganese (25, 26), suggesting they may have lower manganese status than women without osteoporosis. However, more recent studies in postmenopausal women have reported conflicting results, with one study finding lower blood manganese concentrations among women with osteoporosis compared to those without osteoporosis (27) and another finding no differences (28). A study in healthy postmenopausal women found that a supplement containing manganese (5 mg/day), copper (2.5 mg/day), and zinc (15 mg/day) in combination with a calcium supplement (1,000 mg/day) was more effective than the calcium supplement alone in preventing spinal bone loss over a two-year period (29). However, the presence of other trace elements in the supplement makes it impossible to determine whether manganese supplementation was the beneficial agent for maintaining bone mineral density.
Diabetes mellitus
In animal models, manganese deficiency results in impaired insulin secretion and glucose intolerance similar to diabetes mellitus (30); however, results of human studies on manganese and type 2 diabetes have been somewhat conflicting. Manganese intake was inversely associated with type 2 diabetes in 71,270 French women participating in the E3N-EPIC cohort study (31). In an analysis of two prospective cohorts of Chinese adults (ages 20-74 years at baseline), followed for a mean of 4.2 and 5.3 years, higher dietary intakes of manganese were associated with a lower risk of type 2 diabetes; these associations were independent of dietary total antioxidant capacity, a measure of dietary antioxidant intake (32). Most recently, a prospective cohort study of 19,862 adults (ages 40-79 at baseline) participating in the Japan Collaborative Cohort Study, followed for five years, found higher dietary intake of manganese to be associated with a lower risk of type 2 diabetes in women but not in men (33).
While these studies of dietary manganese intake are suggestive of a protective association with type 2 diabetes, studies employing biomarkers of manganese intake – either blood concentrations of manganese or urinary manganese excretion — have reported mixed results. Most studies conducted have been small case-control studies; studies to date have reported blood manganese concentrations in patients with diabetes were higher (34-36), lower (37), or similar (38-40) compared to blood manganese concentrations in controls without diabetes. Additionally, a case-control study that included 3,228 adults in China reported a U-shaped relationship between plasma concentration of manganese and type 2 diabetes, meaning those with low or high blood concentrations had a higher odds of diabetes when compared to those with an intermediate blood concentration of manganese (41). In a cross-sectional analysis of adults participating in the Korean National Health and Nutrition Examination Survey, blood concentrations of manganese were significantly lower among those with diabetes compared to those without diabetes (42). Further, one case-control study found higher urinary manganese excretion in patients with diabetes compared to controls without diabetes (37). Results of case-control studies are more likely to be distorted by bias (i.e., the selection bias with the selection of cases and controls, as well as dietary recall bias) than results of prospective cohort studies.
Moreover, one study of functional manganese status found the activity of the antioxidant enzyme, MnSOD, to be lower in the white blood cells of patients with diabetes than in those without diabetes (43). When given at the same time as an oral glucose challenge, an acute oral dose of 15 mg or 30 mg manganese did not improve glucose tolerance in subjects with diabetes or in controls without diabetes (44).
Although manganese appears to play a role in glucose metabolism, it is not clear whether higher manganese status might improve glucose tolerance and protect against the development of type 2 diabetes mellitus.
Epilepsy (seizure disorders)
Manganese deficient rats are more susceptible to seizures than manganese sufficient rats, and rats that are genetically prone to epilepsy have lower than normal brain and blood manganese concentrations. Certain subgroups of humans with epilepsy reportedly have lower whole blood manganese concentrations than control subjects without epilepsy (45). One study found blood manganese concentrations of individuals with epilepsy of unknown origin were lower than those of individuals whose epilepsy was induced by trauma (e.g., head injury) or disease (46), suggesting a possible genetic relationship between epilepsy and abnormal manganese metabolism. While manganese deficiency does not appear to be a cause of epilepsy in humans, the relationship between manganese metabolism and epilepsy deserves further research (7, 45, 47).
Sources
Food sources
In the US, estimated average intakes of dietary manganese range from 2.1 to 2.3 mg/day for men and 1.6 to 1.8 mg/day for women (4). Surveys have found those adhering to a vegetarian diet have manganese intakes of up to 7.0 mg/day (reviewed in 48). Rich sources of manganese include whole grains, legumes, nuts, leafy vegetables, and teas (49). Foods high in phytic acid, such as beans, seeds, nuts, whole grains, and soy products, or foods high in oxalic acid, such as cabbage, spinach, and sweet potatoes, may slightly inhibit manganese absorption. Although teas are rich sources of manganese, the tannins present in tea may moderately reduce the absorption of manganese (18). Intakes of other minerals, including iron, calcium, and phosphorus, have been found to limit retention of manganese (4). The manganese content of some manganese-rich foods is listed in milligrams (mg) in Table 2. For more information on the nutrient content of foods, search USDA’s FoodData Central database (50).
Breast milk and infant formulas
Infants are exposed to varying amounts of manganese depending on their source of nutrition. Manganese concentrations in breast milk, cow-based formula, and soy-based formula range from 3 to 10 micrograms/liter (μg/L), 30 to 50 μg/L, and 200 to 300 μg/L, respectively. However, the bioavailability of manganese from breast milk is higher than from infant formulas, and manganese deficiencies in breast-fed infants or toxicities in formula-fed infants have not been reported (20).
Water
Manganese concentrations in drinking water range from 1 to 100 micrograms (μg)/L, but most sources contain less than 10 μg/L (51). The US Environmental Protection Agency (EPA) recommends 0.05 mg (50 μg)/L as the maximum allowable manganese concentration in drinking water (52).
Supplements
Several forms of manganese are found in supplements, including manganese gluconate, manganese sulfate, manganese ascorbate, and amino acid chelates of manganese. Manganese is available as a stand-alone supplement or in combination products (53). Relatively high levels of manganese ascorbate may be found in a bone/joint health product containing chondroitin sulfate and glucosamine hydrochloride (see Safety).
Parenteral nutrition
Manganese may be present in solutions of parenteral nutrition either included as a trace element or as an incidental contaminant (see Intravenous manganese in the Safety section) (54, 55).
Safety
Toxicity
Inherited manganese-overload disorders
Autosomal recessive mutations in the SLC30A10 gene, which encodes a manganese transporter expressed in the liver and brain, causes a manganese overload syndrome. Such loss-of-function mutations lead to manganese accumulation in certain brain regions and in the liver, causing hypermanganesemia, dystonia parkinsonism, and hepatic dysfunction at an early age (56, 57). Autosomal recessive mutations in the SLC39A14 gene have also been reported, leading to hypermanganesemia and similar a neurological phenotype, but liver disease is absent with this specific mutation (58). Chelation therapy is important to treat these inherited manganese-overload disorders (59).
Inhaled manganese
Manganese toxicity may result in multiple neurologic problems and is a well-recognized health hazard for people who inhale manganese dust, such as welders, miners, and smelters (1, 4). Unlike ingested manganese, inhaled manganese is transported directly to the brain before it can be metabolized in the liver (60, 61). The symptoms of manganese toxicity generally appear slowly over a period of months to years. In its worst form, manganese toxicity can result in a permanent neurological disorder with symptoms similar to those of Parkinson's disease, including tremors, difficulty walking, and facial muscle spasms. This syndrome, often called manganism, is sometimes preceded by psychiatric symptoms, such as irritability, aggressiveness, and even hallucinations (62, 63). Additionally, environmental or occupational inhalation of manganese can cause an inflammatory response in the lungs (64), with clinical symptoms including cough, acute bronchitis, and decreased lung function (65).
Methylcyclopentadienyl manganese tricarbonyl (MMT)
Methylcyclopentadienyl manganese tricarbonyl (MMT) is a manganese-containing compound used in gasoline as an anti-knock additive. Although it has been used for this purpose in Canada for more than 20 years, uncertainty about adverse health effects from inhaled exhaust emissions kept the US EPA from approving its use in unleaded gasoline. In 1995, a US court decision made MMT available for widespread use in unleaded gasoline (60). A study in Montreal, where MMT had been used for more than 10 years, found airborne manganese levels to be similar to those in areas where MMT was not used (66). Another Canadian study found higher concentrations of respirable manganese in an urban versus a rural area, but average concentrations in both areas were below the safe level set by the US EPA (67). The impact of long-term exposure to low levels of MMT combustion products, however, has not been thoroughly evaluated and will require additional study (68).
A single case of reversible neurotoxicity and seizures following unintentional MMT ingestion has been documented: a 54-year old man accidentally drank an MMT-containing anti-knock agent that he assumed was an energy drink due so similar product labeling (69).
Ingested manganese
Limited evidence suggests that high manganese intakes from drinking water may be associated with neurological symptoms similar to those of Parkinson's disease. Severe neurological symptoms were reported in 25 people who drank water contaminated with manganese — and probably other contaminants — from dry cell batteries for two to three months (70). Water manganese concentrations were found to be 14 mg/L almost two months after symptoms began and may have already been declining (1). A study of older adults in Greece found a high prevalence of neurological symptoms in those exposed to water manganese levels of 1.8 to 2.3 mg/L (71), while a study in Germany found no evidence of increased neurological symptoms in people drinking water with manganese levels ranging from 0.3 to 2.2 mg/L compared to those drinking water containing less than 0.05 mg/L of manganese (72). Manganese in drinking water may be more bioavailable than manganese in food. However, none of the studies measured dietary manganese, so total manganese intake in these cases is unknown. In the US, the EPA recommends 0.05 mg/L as the maximum allowable manganese concentration in drinking water (52), but the World Health Organization does not currently have a health-based limit for manganese in drinking water (73, 74).
Additionally, several cross-sectional studies have associated high levels of manganese in drinking water with cognitive and behavioral deficits in children (reviewed in 75). For example, a cross-sectional study of 362 children (ages 6-13 years) in Canada found children with the highest manganese concentrations in home tap water (median of 216 μg/L) had a 6.2-point lower Full Scale IQ (lower Performance IQ but not Verbal IQ) than those with lowest manganese levels in home tap water (median of 1 μg/L) (76). A cohort study that followed 287 of these children for a mean of 4.4 years found that exposure to higher concentrations of manganese in drinking water was linked to a lower Performance IQ among girls but a higher Performance IQ among boys (77). Additionally, a prospective cohort study among 1,265 children in Bangladesh did not find manganese concentration in drinking water (medians of 0.20 mg/L during pregnancy and 0.34 mg/L at 10 years) to be associated with any measure of cognitive ability (i.e., IQ, verbal comprehension, perceptual reasoning, working memory, processing speed) when assessed at age 10 (78). Yet, this study associated manganese in drinking water with higher risks of conduct problems among boys and low prosocial scores among girls (78). In a population-based cohort study in Denmark that followed 643,401 children, exposure to higher manganese concentrations in drinking water was linked to a heightened risk of one subtype of attention-deficit hyperactive disorder (79). Specifically, exposure to a manganese concentration in drinking water of at least 100 μg/L was associated with a 51% higher risk of the ADHD-Inattentive subtype in girls and a 20% higher risk in boys, in comparison to exposure of <5 μg/L — using these exposure comparisons, a 9% increased risk of ADHD-Overall was observed in girls and no difference found in boys (79).
Only a few adverse effects of manganese intake from supplements have been documented. A single case of manganese toxicity was reported in a person who took large amounts of mineral supplements for years (51), while another case was reported as a result of a person taking a Chinese herbal supplement (62). More recently, Parkinson’s disease was reported in a woman taking 100 mg/day of manganese chloride for at least two years, followed by 30 mg/day for two months (80).
Manganese toxicity resulting from food alone has not been reported in humans, even though certain vegetarian diets could provide up to 20 mg/day of manganese (4, 51).
Intravenous manganese
Manganese neurotoxicity has been observed in individuals receiving total parenteral nutrition (TPN), both as a result of excessive manganese in the solution and as an incidental contaminant (54). Neonates are especially vulnerable to manganese-related neurotoxicity (81). Infants receiving manganese-containing TPN can be exposed to manganese concentrations about 100-fold higher than breast-fed infants (20). Because of potential toxicities, some argue against including manganese in parenteral nutrition (82).
Individuals with increased susceptibility to manganese toxicity
- Chronic liver disease: Manganese is eliminated from the body mainly in bile. Thus, impaired liver function may lead to decreased manganese excretion. Manganese accumulation in individuals with cirrhosis or liver failure may contribute to neurological problems and Parkinson's disease-like symptoms (1, 53).
- Infants and children: Compared to adults, infants and children have higher intestinal absorption of manganese, as well as lower biliary excretion of manganese (83). Thus, infants and children are especially susceptible to any negative, neurotoxic effects of manganese. Indeed, several studies in school-aged children have reported deleterious cognitive and behavioral effects following excessive manganese exposure (76, 84-90). Additional studies have associated higher manganese exposures during pregnancy with cognitive and motor deficits in children under six years of age (reviewed in 75).
- Iron-deficient populations: Iron deficiency has been shown to increase the risk of manganese accumulation in the brain (17).
- Individuals with occupational exposures to airborne manganese, such as welders, miners, and smelters (reviewed in 21).
- Abusers of the illicit drug, methcathinone (ephedrone): Intravenous use of manganese-contaminated methcathinone (i.e., when the drug is synthesized with potassium permanganate as the oxidant) can cause lasting neurological damage and a parkinsonism disorder (91, 92).
Due to the severe implications of manganese neurotoxicity, the Food and Nutrition Board (FNB) of the Institute of Medicine (now the National Academy of Medicine) set very conservative tolerable upper intake levels (UL) for manganese; the ULs are listed in Table 3 according to age (4).
Drug interactions
Magnesium-containing antacids and laxatives and the antibiotic medication, tetracycline, may decrease the absorption of manganese if taken together with manganese-containing foods or supplements (53).
High levels of manganese in supplements marketed for bone/joint health
Two studies have found that supplements containing a combination of glucosamine hydrochloride, chondroitin sulfate, and manganese ascorbate are beneficial in relieving pain due to mild or moderate osteoarthritis of the knee when compared to a placebo (93, 94). The dose of elemental manganese supplied by the supplements was 30 mg/day for eight weeks in one study (94) and 40 mg/day for six months in the other study (93). No adverse effects were reported during either study, and blood manganese concentrations were not measured. Neither study compared the treatment containing manganese ascorbate to a treatment containing glucosamine hydrochloride and chondroitin sulfate without manganese ascorbate, so it is impossible to determine whether the supplement would have resulted in the same benefit without high doses of manganese.
Linus Pauling Institute Recommendation
The adequate intake (AI) for manganese (2.3 mg/day for adult men and 1.8 mg/day for adult women) appears sufficient to prevent deficiency in most individuals. The daily intake of manganese most likely to promote optimum health is not known. Following the Linus Pauling Institute’s recommendation to take a multivitamin/mineral supplement containing 100% of the daily values (DV) of most nutrients will generally provide 2.3 mg/day of manganese. Because of the potential for toxicity and the lack of information regarding benefit, manganese supplementation beyond 100% of the DV (2.3 mg/day) is not recommended. There is presently no evidence that the consumption of a manganese-rich plant-based diet results in manganese toxicity.
Older adults (>50 years)
The requirement for manganese is not known to be higher for older adults. However, liver disease is more common in older adults and may increase the risk of manganese toxicity by decreasing the elimination of manganese from the body (see Toxicity). Manganese supplementation beyond 100% of the DV (2.3 mg/day) is not recommended.
Authors and Reviewers
Originally written in 2001 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in June 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in March 2010 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2021 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in May 2021 by:
Michael Aschner, Ph.D.
Professor and Chair, Department of Molecular Pharmacology
Professor, Dominick P. Purpura Department of Neuroscience
Professor, Department of Pediatrics
Albert Einstein College of Medicine
Copyright 2001-2024 Linus Pauling Institute
References
1. Keen CL, Ensunsa JL, Watson MH, et al. Nutritional aspects of manganese from experimental studies. Neurotoxicology. 1999;20(2-3):213-223. (PubMed)
2. Nielsen FH. Ultratrace minerals. In: Shils M, Olson JA, Shike M, Ross AC, eds. Modern Nutrition in Health and Disease. 9th ed. Baltimore: Williams & Wilkins; 1999:283-303.
3. Leach RM, Harris ED. Manganese. In: O'Dell BL, Sunde RA, eds. Handbook of nutritionally essential minerals. New York: Marcel Dekker, Inc; 1997:335-355.
4. Food and Nutrition Board, Institute of Medicine. Manganese. Dietary reference intakes for vitamin A, vitamin K, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. Washington, D.C.: National Academy Press; 2001:394-419. (National Academy Press)
5. Wedler FC. Biochemical and nutritional role of manganese: an overview. In: Klimis-Tavantzis DJ (ed). Manganese in health and disease. Boca Raton: CRC Press, Inc.; 1994:1-37.
6. Albrecht J, Sonnewald U, Waagepetersen HS, Schousboe A. Glutamine in the central nervous system: function and dysfunction. Front Biosci. 2007;12:332-343. (PubMed)
7. Keen CL, Zidenberg-Cherr S. Manganese. In: Ziegler EE, Filer LJ, eds. Present Knowledge in Nutrition. 7th ed. Washington D.C.: ILSI Press; 1996:334-343.
8. Muszynska A, Palka J, Gorodkiewicz E. The mechanism of daunorubicin-induced inhibition of prolidase activity in human skin fibroblasts and its implication to impaired collagen biosynthesis. Exp Toxicol Pathol. 2000;52(2):149-155. (PubMed)
9. Shetlar MR, Shetlar CL. The role of manganese in wound healing. In: Klimis-Tavantzis DL, ed. Manganese in health and disease. Boca Raton: CRC Press, Inc.; 1994:145-157.
10. Ye Q, Park JE, Gugnani K, Betharia S, Pino-Figueroa A, Kim J. Influence of iron metabolism on manganese transport and toxicity. Metallomics. 2017;9(8):1028-1046. (PubMed)
11. Davis CD, Greger JL. Longitudinal changes of manganese-dependent superoxide dismutase and other indexes of manganese and iron status in women. Am J Clin Nutr. 1992;55(3):747-752. (PubMed)
12. Finley JW. Manganese absorption and retention by young women is associated with serum ferritin concentration. Am J Clin Nutr. 1999;70(1):37-43. (PubMed)
13. Park S, Sim CS, Lee H, Kim Y. Blood manganese concentration is elevated in infants with iron deficiency. Biol Trace Elem Res. 2013;155(2):184-189. (PubMed)
14. Rahman MA, Rahman B, Ahmed N. High blood manganese in iron-deficient children in Karachi. Public Health Nutr. 2013;16(9):1677-1683. (PubMed)
15. Kim Y, Lee BK. Iron deficiency increases blood manganese level in the Korean general population according to KNHANES 2008. Neurotoxicology. 2011;32(2):247-254. (PubMed)
16. Finley JW, Johnson PE, Johnson LK. Sex affects manganese absorption and retention by humans from a diet adequate in manganese. Am J Clin Nutr. 1994;60(6):949-955. (PubMed)
17. Aschner M, Dorman DC. Manganese: pharmacokinetics and molecular mechanisms of brain uptake. Toxicol Rev. 2006;25(3):147-154. (PubMed)
18. Kies C. Bioavailability of manganese. In: Klimis-Tavantzis D, ed. Manganese in Health and Disease. Boca Raton: CRC Press, Inc.; 1994:39-58.
19. Johnson P, Lykken G. Manganese and calcium absorption and balance in young women fed diets with varying amounts of manganese and calcium. J Trace Elem Exp Med. 1991;4:19-35.
20. Aschner JL, Aschner M. Nutritional aspects of manganese homeostasis. Mol Aspects Med. 2005;26(4-5):353-362. (PubMed)
21. Chen P, Bornhorst J, Aschner M. Manganese metabolism in humans. Front Biosci (Landmark Ed). 2018;23:1655-1679. (PubMed)
22. Horning KJ, Caito SW, Tipps KG, Bowman AB, Aschner M. Manganese is essential for neuronal health. Annu Rev Nutr. 2015;35:71-108. (PubMed)
23. Norose N, Terai M, Norose K. Manganese deficiency in a child with very short bowel syndrome receiving long-term parenteral nutrition. J Trace Elem Exp Med. 1992;5:100-101 (abstract).
24. Friedman BJ, Freeland-Graves JH, Bales CW, et al. Manganese balance and clinical observations in young men fed a manganese-deficient diet. J Nutr. 1987;117(1):133-143. (PubMed)
25. Freeland-Graves J, Llanes C. Models to study manganese deficiency. In: Klimis-Tavantzis D, ed. Manganese in Health and Disease. Boca Raton: CRC Press, Inc.; 1994:59-86.
26. Reginster JY, Strause LG, Saltman P, Franchimont P. Trace elements and postmenopausal osteoporosis: a preliminary study of decreased serum manganese. Med Sci Res. 1988;16:337-338.
27. Yakout SM, Alharbi F, Abdi S, Al-Daghri NM, Al-Amro A, Khattak MNK. Serum minerals (Ca, P, Co, Mn, Ni, Cd) and growth hormone (IGF-1 and IGF-2) levels in postmenopausal Saudi women with osteoporosis. Medicine (Baltimore). 2020;99(27):e20840. (PubMed)
28. Odabasi E, Turan M, Aydin A, Akay C, Kutlu M. Magnesium, zinc, copper, manganese, and selenium levels in postmenopausal women with osteoporosis. Can magnesium play a key role in osteoporosis? Ann Acad Med Singapore. 2008;37(7):564-567. (PubMed)
29. Strause L, Saltman P, Smith KT, Bracker M, Andon MB. Spinal bone loss in postmenopausal women supplemented with calcium and trace minerals. J Nutr. 1994;124(7):1060-1064. (PubMed)
30. Baly DL, Curry DL, Keen CL, Hurley LS. Effect of manganese deficiency on insulin secretion and carbohydrate homeostasis in rats. J Nutr. 1984;114(8):1438-1446. (PubMed)
31. Mancini FR, Dow C, Affret A, et al. Micronutrient dietary patterns associated with type 2 diabetes mellitus among women of the E3N-EPIC (Etude Epidemiologique aupres de femmes de l'Education Nationale) cohort study. J Diabetes. 2018;10(8):665-674. (PubMed)
32. Du S, Wu X, Han T, et al. Dietary manganese and type 2 diabetes mellitus: two prospective cohort studies in China. Diabetologia. 2018;61(9):1985-1995. (PubMed)
33. Eshak ES, Muraki I, Imano H, Yamagishi K, Tamakoshi A, Iso H. Manganese intake from foods and beverages is associated with a reduced risk of type 2 diabetes. Maturitas. 2021;143:127-131. (PubMed)
34. Ekin S, Mert N, Gunduz H, Meral I. Serum sialic acid levels and selected mineral status in patients with type 2 diabetes mellitus. Biol Trace Elem Res. 2003;94(3):193-201. (PubMed)
35. Li XT, Yu PF, Gao Y, et al. Association between plasma metal levels and diabetes risk: a case-control study in China. Biomed Environ Sci. 2017;30(7):482-491. (PubMed)
36. Anetor JI, Asiribo OA, Adedapo KS, Akingbola TS, Olorunnisola OS, Adeniyi FA. Increased plasma manganese, partially reduced ascorbate, 1 and absence of mitochondrial oxidative stress in type 2 diabetes mellitus: implications for the superoxide uncoupling protein 2 (UCP-2) pathway. Biol Trace Elem Res. 2007;120(1-3):19-27. (PubMed)
37. Kazi TG, Afridi HI, Kazi N, et al. Copper, chromium, manganese, iron, nickel, and zinc levels in biological samples of diabetes mellitus patients. Biol Trace Elem Res. 2008;122(1):1-18. (PubMed)
38. Walter RM, Jr., Uriu-Hare JY, Olin KL, et al. Copper, zinc, manganese, and magnesium status and complications of diabetes mellitus. Diabetes Care. 1991;14(11):1050-1056. (PubMed)
39. Simic A, Hansen AF, Asvold BO, et al. Trace element status in patients with type 2 diabetes in Norway: The HUNT3 Survey. J Trace Elem Med Biol. 2017;41:91-98. (PubMed)
40. Hansen AF, Simic A, Asvold BO, et al. Trace elements in early phase type 2 diabetes mellitus-A population-based study. The HUNT study in Norway. J Trace Elem Med Biol. 2017;40:46-53. (PubMed)
41. Shan Z, Chen S, Sun T, et al. U-shaped association between plasma manganese levels and type 2 diabetes. Environ Health Perspect. 2016;124(12):1876-1881. (PubMed)
42. Koh ES, Kim SJ, Yoon HE, et al. Association of blood manganese level with diabetes and renal dysfunction: a cross-sectional study of the Korean general population. BMC Endocr Disord. 2014;14:24. (PubMed)
43. Nath N, Chari SN, Rathi AB. Superoxide dismutase in diabetic polymorphonuclear leukocytes. Diabetes. 1984;33(6):586-589. (PubMed)
44. Walter RM, Jr., Aoki T, Keen C. Acute oral manganese does not consistently affect glucose tolerance in non diabetic and type II diabetic humans. J Trace Elem Exp Med. 1991;4:73-79.
45. Gonzalez-Reyes RE, Gutierrez-Alvarez AM, Moreno CB. Manganese and epilepsy: a systematic review of the literature. Brain Res Rev. 2007;53(2):332-336. (PubMed)
46. Carl GF, Keen CL, Gallagher BB, et al. Association of low blood manganese concentrations with epilepsy. Neurology. 1986;36(12):1584-1587. (PubMed)
47. Carl G, Gallagher B. Manganese and epilepsy. In: Klimis-Tavantzis D, ed. Manganese in Health and Disease. Boca Raton: CRC Press, Inc.; 1994:133-157.
48. Freeland-Graves JH, Mousa TY, Kim S. International variability in diet and requirements of manganese: Causes and consequences. J Trace Elem Med Biol. 2016;38:24-32. (PubMed)
49. Erikson KM, Aschner M. Manganese: its role in disease and health. Met Ions Life Sci. 2019;19. doi: 10.1515/9783110527872-016. (PubMed)
50. US Department of Agriculture, Agricultural Research Service. FoodData Central, 2019. https://fdc.nal.usda.gov/.
51. Keen CL, Zidenberg-Cherr S. Manganese toxicity in humans and experimental animals. In: Klimis-Tavantzis DJ, ed. Manganese in Health and Disease. Boca Raton: CRC Press, Inc.; 1994:193-205.
52. US Environmental Protection Agency. Secondary Drinking Water Standards: Guidance for Nuisance Chemicals. 1/7/21. Available at: https://www.epa.gov/sdwa/secondary-drinking-water-standards-guidance-nuisance-chemicals. Accessed 4/30/21.
53. In: Hendler S, Rorvik D, eds. PDR for Nutritional Supplements. Montvale: Medical Economics Company, Inc.; 2001.
54. Dobson AW, Erikson KM, Aschner M. Manganese neurotoxicity. Ann N Y Acad Sci. 2004;1012:115-128. (PubMed)
55. Livingstone C. Manganese provision in parenteral nutrition: an update. Nutr Clin Pract. 2018;33(3):404-418. (PubMed)
56. Quadri M, Federico A, Zhao T, et al. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet. 2012;90(3):467-477. (PubMed)
57. Tuschl K, Clayton PT, Gospe SM, Jr., et al. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am J Hum Genet. 2012;90(3):457-466. (PubMed)
58. Tuschl K, Meyer E, Valdivia LE, et al. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat Commun. 2016;7:11601. (PubMed)
59. Kapoor D, Garg D, Sharma S, Goyal V. Inherited manganese disorders and the brain: what neurologists need to know. Ann Indian Acad Neurol. 2021;24(1):15-21. (PubMed)
60. Davis JM. Methylcyclopentadienyl manganese tricarbonyl: health risk uncertainties and research directions. Environ Health Perspect. 1998;106 Suppl 1:191-201. (PubMed)
61. O'Neal SL, Zheng W. Manganese toxicity upon overexposure: a decade in review. Curr Environ Health Rep. 2015;2(3):315-328. (PubMed)
62. Pal PK, Samii A, Calne DB. Manganese neurotoxicity: a review of clinical features, imaging and pathology. Neurotoxicology. 1999;20(2-3):227-238. (PubMed)
63. Aschner M, Aschner JL. Manganese neurotoxicity: cellular effects and blood-brain barrier transport. Neurosci Biobehav Rev. 1991;15(3):333-340. (PubMed)
64. Han J, Lee JS, Choi D, et al. Manganese (II) induces chemical hypoxia by inhibiting HIF-prolyl hydroxylase: implication in manganese-induced pulmonary inflammation. Toxicol Appl Pharmacol. 2009;235(3):261-267. (PubMed)
65. Roels H, Lauwerys R, Buchet JP, et al. Epidemiological survey among workers exposed to manganese: effects on lung, central nervous system, and some biological indices. Am J Ind Med. 1987;11(3):307-327. (PubMed)
66. Zayed J, Thibault C, Gareau L, Kennedy G. Airborne manganese particulates and methylcyclopentadienyl manganese tricarbonyl (MMT) at selected outdoor sites in Montreal. Neurotoxicology. 1999;20(2-3):151-157. (PubMed)
67. Bolte S, Normandin L, Kennedy G, Zayed J. Human exposure to respirable manganese in outdoor and indoor air in urban and rural areas. J Toxicol Environ Health A. 2004;67(6):459-467. (PubMed)
68. Aschner M. Manganese: brain transport and emerging research needs. Environ Health Perspect. 2000;108 Suppl 3:429-432. (PubMed)
69. Nemanich A, Chen B, Valento M. Toxic boost: Acute, reversible neurotoxicity after ingestion of methylcyclopentadienyl manganese tricarbonyl (MMT) mistaken for an energy drink. Am J Emerg Med. 2021;42:261 e263-261 e265. (PubMed)
70. Kawamura R. Intoxication by manganese in well water. Kisasato Arch Exp Med. 1941;18:145-169.
71. Kondakis XG, Makris N, Leotsinidis M, Prinou M, Papapetropoulos T. Possible health effects of high manganese concentration in drinking water. Arch Environ Health. 1989;44(3):175-178. (PubMed)
72. Vieregge P, Heinzow B, Korf G, Teichert HM, Schleifenbaum P, Mosinger HU. Long term exposure to manganese in rural well water has no neurological effects. Can J Neurol Sci. 1995;22(4):286-289. (PubMed)
73. Lucchini RG, Aschner M, Landrigan PJ, Cranmer JM. Neurotoxicity of manganese: Indications for future research and public health intervention from the Manganese 2016 conference. Neurotoxicology. 2018;64:1-4. (PubMed)
74. Kullar SS, Shao K, Surette C, et al. A benchmark concentration analysis for manganese in drinking water and IQ deficits in children. Environ Int. 2019;130:104889. (PubMed)
75. Liu W, Xin Y, Li Q, et al. Biomarkers of environmental manganese exposure and associations with childhood neurodevelopment: a systematic review and meta-analysis. Environ Health. 2020;19(1):104. (PubMed)
76. Bouchard MF, Sauve S, Barbeau B, et al. Intellectual impairment in school-age children exposed to manganese from drinking water. Environ Health Perspect. 2011;119(1):138-143. (PubMed)
77. Dion LA, Saint-Amour D, Sauve S, Barbeau B, Mergler D, Bouchard MF. Changes in water manganese levels and longitudinal assessment of intellectual function in children exposed through drinking water. Neurotoxicology. 2018;64:118-125. (PubMed)
78. Rahman SM, Kippler M, Tofail F, Bolte S, Hamadani JD, Vahter M. Manganese in drinking water and cognitive abilities and behavior at 10 years of age: a prospective cohort study. Environ Health Perspect. 2017;125(5):057003. (PubMed)
79. Schullehner J, Thygesen M, Kristiansen SM, Hansen B, Pedersen CB, Dalsgaard S. Exposure to manganese in drinking water during childhood and association with attention-deficit hyperactivity disorder: a nationwide cohort study. Environ Health Perspect. 2020;128(9):97004. (PubMed)
80. Schuh MJ. Possible Parkinson's disease induced by chronic manganese supplement ingestion. Consult Pharm. 2016;31(12):698-703. (PubMed)
81. Erikson KM, Thompson K, Aschner J, Aschner M. Manganese neurotoxicity: a focus on the neonate. Pharmacol Ther. 2007;113(2):369-377. (PubMed)
82. Hardy IJ, Gillanders L, Hardy G. Is manganese an essential supplement for parenteral nutrition? Curr Opin Clin Nutr Metab Care. 2008;11(3):289-296. (PubMed)
83. Ljung K, Vahter M. Time to re-evaluate the guideline value for manganese in drinking water? Environ Health Perspect. 2007;115(11):1533-1538. (PubMed)
84. Wasserman GA, Liu X, Parvez F, et al. Water manganese exposure and children's intellectual function in Araihazar, Bangladesh. Environ Health Perspect. 2006;114(1):124-129. (PubMed)
85. Bouchard M, Laforest F, Vandelac L, Bellinger D, Mergler D. Hair manganese and hyperactive behaviors: pilot study of school-age children exposed through tap water. Environ Health Perspect. 2007;115(1):122-127. (PubMed)
86. Wright RO, Amarasiriwardena C, Woolf AD, Jim R, Bellinger DC. Neuropsychological correlates of hair arsenic, manganese, and cadmium levels in school-age children residing near a hazardous waste site. Neurotoxicology. 2006;27(2):210-216. (PubMed)
87. Menezes-Filho JA, Novaes Cde O, Moreira JC, Sarcinelli PN, Mergler D. Elevated manganese and cognitive performance in school-aged children and their mothers. Environ Res. 2011;111(1):156-163. (PubMed)
88. Carvalho CF, Menezes-Filho JA, de Matos VP, et al. Elevated airborne manganese and low executive function in school-aged children in Brazil. Neurotoxicology. 2014;45:301-308. (PubMed)
89. Oulhote Y, Mergler D, Barbeau B, et al. Neurobehavioral function in school-age children exposed to manganese in drinking water. Environ Health Perspect. 2014;122(12):1343-1350. (PubMed)
90. Khan K, Factor-Litvak P, Wasserman GA, et al. Manganese exposure from drinking water and children's classroom behavior in Bangladesh. Environ Health Perspect. 2011;119(10):1501-1506. (PubMed)
91. de Bie RM, Gladstone RM, Strafella AP, Ko JH, Lang AE. Manganese-induced Parkinsonism associated with methcathinone (Ephedrone) abuse. Arch Neurol. 2007;64(6):886-889. (PubMed)
92. Sikk K, Haldre S, Aquilonius SM, Taba P. Manganese-induced parkinsonism due to ephedrone abuse. Parkinsons Dis. 2011;2011:865319. (PubMed)
93. Das A, Jr., Hammad TA. Efficacy of a combination of FCHG49 glucosamine hydrochloride, TRH122 low molecular weight sodium chondroitin sulfate and manganese ascorbate in the management of knee osteoarthritis. Osteoarthritis Cartilage. 2000;8(5):343-350. (PubMed)
94. Leffler CT, Philippi AF, Leffler SG, Mosure JC, Kim PD. Glucosamine, chondroitin, and manganese ascorbate for degenerative joint disease of the knee or low back: a randomized, double-blind, placebo-controlled pilot study. Mil Med. 1999;164(2):85-91. (PubMed)
Molybdenum
Contents
Summary
- The molybdenum atom is part of the molybdenum cofactor in the active site of four enzymes in humans: sulfite oxidase, xanthine oxidase, aldehyde oxidase, and mitochondrial amidoxime reducing component. (More information)
- Excess molybdenum intake causes fatal copper deficiency diseases in grazing animals. Their rumen is the site of high sulfide generation, and the interaction of molybdenum with sulfur results in the formation of thiomolybdates. Tetrathiomolybdate, a thiomolybdate with four sulfur atoms, can form complexes with copper preventing its absorption and blocking the activity of copper-dependent enzymes. (More information)
- In humans, tetrathiomolybdate therapy has been developed for Wilson's disease, a genetic disease in which the accumulation of copper in tissues leads to liver and brain damage. More recently, tetrathiomolybdate use has been explored for the treatment of cancer and inflammatory diseases. (More information)
- Mutations in the molybdenum cofactor biosynthetic pathway lead to the combined deficiency of all molybdenum-dependent enzymes. Molybdenum cofactor deficiency Type A is due to mutations in the MOCS1 gene, while Type B deficiency is caused by mutations in MOCS2. Both Type A and Type B deficiencies result in the loss of sulfite oxidase activity, also observed in isolated sulfite oxidase deficiency and characterized by severe neurologic abnormalities in affected patients. (More information)
- A new treatment option for molybdenum cofactor deficiency Type A is now available in the US. Intravenous administration of a replacement drug for cyclic pyranopterin monophosphate may help correct the metabolic disorder and prevent neurologic deterioration in patients with Type A deficiency. Patients with Type B deficiency do not lack this molecule and therefore cannot benefit from this treatment. However, one study showed that pyridoxine supplementation in these patients could alleviate suffering by abolishing seizures. (More information)
- The molybdenum content of foods depends on the molybdenum content of soils, which can vary considerably. Variation in esophageal cancer incidence worldwide has been linked to the molybdenum content in soils and food. Similar observations have been made in order to identify the factors associated with a population's extended lifespan. (More information)
Molybdenum is an essential trace element for virtually all life forms. It functions as a cofactor for a number of enzymes that catalyze important chemical transformations in the global carbon, nitrogen, and sulfur cycles (1). Thus, molybdenum-dependent enzymes are not only required for human health, but also for the health of our ecosystem.
Function
The biological form of the molybdenum atom is an organic molecule known as the molybdenum cofactor (Moco) present in the active site of Moco-containing enzymes (molybdoenzymes) (2). In humans, molybdenum is known to function as a cofactor for four enzymes (3):
- Sulfite oxidase catalyzes the transformation of sulfite to sulfate, a reaction that is necessary for the metabolism of sulfur-containing amino acids (methionine and cysteine). Recent evidence also indicates a role for sulfite oxidase in the reduction of nitrite to nitric oxide (4).
- Xanthine oxidase catalyzes the breakdown of nucleotides (precursors to DNA and RNA) to form uric acid, which contributes to the plasma antioxidant capacity of the blood.
- Aldehyde oxidase and xanthine oxidase catalyze hydroxylation reactions that involve a number of different molecules with similar chemical structures. Xanthine oxidase and aldehyde oxidase also play a role in the metabolism of drugs and toxins (5).
- Mitochondrial amidoxime reducing component (mARC) was described fairly recently (6), and its precise function is still under investigation. Initial studies showed that mARC forms a three-component enzyme system with cytochrome b5 and NADH/cytochrome b5 reductase that catalyzes the detoxification of mutagenic N-hydroxylated bases (7). mARC reduces various N-hydroxylated compounds and plays an important role in prodrug metabolism (8, 9). Moreover, recent studies have found a separate function of this enzyme system: the reduction of nitrite to nitric oxide (10). Two isoforms of the mARC enzyme are known to exist in humans, mARC1 and mARC2 (11).
Of these enzymes, sulfite oxidase is known to be crucial for human health (12). Hereditary xanthinuria, characterized by a deficiency in xanthine oxidase (Type I) or by a deficiency in both xanthine oxidase and aldehyde oxidase (Type II), can be asymptomatic. However, in less than half of the cases, affected individuals exhibit a range of health issues of variable severity (13, 14).
Nutrient interactions
Copper
An early study reported that molybdenum intakes of 500 μg/day and 1,500 μg/day from sorghum increased urinary copper excretion (2). However, the results of a more recent, well-controlled study indicated that very high dietary molybdenum intakes (up to 1,500 μg/day) did not adversely affect copper nutritional status in eight, healthy young men (15).
Tetrathiomolybdate
Excess dietary molybdenum has been found to result in copper deficiency in grazing animals (ruminants). In the digestive tract of ruminants, the formation of compounds containing sulfur and molybdenum, known as thiomolybdates, prevents the absorption of copper and can cause fatal copper-dependent disorders (16, 17). Tetrathiomolybdate (TM) is a molecule that can form high-affinity complexes with copper, controlling free copper (copper that is not bound to ceruloplasmin), and inhibiting copper chaperones and copper-containing enzymes (18, 19). TM's ability to lower free copper levels is exploited in the treatment of Wilson's disease, a genetic disorder characterized by copper accumulation in tissues responsible for hepatic and neurologic disorders. Neurologic worsening has been linked with toxic levels of free copper in the serum of neurologically presenting patients. TM therapy seems able to stabilize neurologic status and prevent neurologic deterioration in these patients, as opposed to the standard initial treatment of choice (20).
Copper is also a required cofactor for enzymes involved in inflammation and angiogenesis that are known to accelerate cancer progression and metastasis. Copper depletion studies employing TM have been initiated in patients with advanced malignancies with the aim of preventing disease progression or relapse. These pilot trials showed promising results in individuals with metastatic kidney cancer (21), metastatic colorectal cancer (22), and breast cancer with high risk of relapse (23). TM was relatively well-tolerated and stabilized disease or prevented relapse in correlation with copper depletion. TM's efficacy has also been investigated in animal models of inflammatory and immune-related diseases (24, 25), but clinical studies are needed to evaluate whether copper depletion might stabilize diseases and improve survival in humans, as suggested by a trial of TM therapy with patients with biliary cirrhosis (26).
Deficiency
Dietary molybdenum deficiency has never been observed in healthy people (2).
Acquired molybdenum deficiency
The only documented case of acquired molybdenum deficiency occurred in a patient with Crohn's disease on long-term total parenteral nutrition (TPN) without molybdenum added to the TPN solution (27). The patient developed rapid heart and respiratory rates, headaches, and night blindness, and ultimately became comatose. The patient was diagnosed with defects in uric acid production and sulfur amino acid metabolism. The patient's clinical condition improved and the amino acid intolerance disappeared when the TPN solution was discontinued and instead supplemented with molybdenum in the form of ammonium molybdate (300 μg/day) (27).
Inherited molybdenum cofactor deficiency
Because molybdenum functions only in the form of the Moco in humans, any disturbance of Moco metabolism can disrupt the function of all molybdoenzymes. Current understanding of the essentiality of molybdenum in humans is based largely on the study of individuals with very rare inborn metabolic disorders caused by a deficiency in Moco. Moco is synthesized de novo by a multistep metabolic pathway involving four genes: MOCS1, MOCS2, MOCS3, and GPHN (see Figure 1 below). To date, more than 60 mutations affecting mostly MOCS1 and MOCS2 have been identified (28).
The absence of a functional Moco has a direct impact on the activity of the molybdoenzymes. Metabolic disorders specifically associated with deficiency in sulfite oxidase activity include an accumulation of sulfite, taurine, S-sulfocysteine, and thiosulfate (see Figure 2 below). This metabolic profile is identical to that observed in isolated sulfite oxidase deficiency (ISOD), an inherited condition caused by mutations in the SUOX gene that codes for sulfite oxidase (29). Compared with ISOD, Moco deficiency (MocoD) also affects the xanthine pathway and leads to an accumulation of hypoxanthine and xanthine, and low to undetectable uric acid concentrations in blood (see Figure 3 below). MocoD and ISOD have been diagnosed in more than 100 individuals worldwide. However the global prevalence of MocoD is likely to be underestimated as a result of a failure to diagnose or to report (28, 30, 31). The incidence of MocoD has been recently estimated at one in 100,000 to 200,000 live births (32).
Both MocoD and ISOD result from recessive traits, meaning that only individuals who inherit two copies of the abnormal gene (one from each parent) develop the disease. Individuals who inherit only one copy of the abnormal gene are known as carriers of the trait but do not exhibit any symptoms. ISOD and MocoD can be diagnosed relatively early in pregnancy (10-14 weeks' gestation) by enzyme activity assays using amniotic cell and chorionic villus sampling and by genetic testing (30, 33). These disorders typically occur in the first days of life, although a few cases of MocoD with late presentation have been described (34-37). The loss of sulfite oxidase activity in ISOD and MocoD leads to severe neurological dysfunction characterized by cerebral atrophy, mental retardation, intractable seizures, and dislocation of ocular lenses. At present, it is not clear whether the neurologic effects are a result of the accumulation of a toxic metabolite, such as sulfite, or inadequate sulfate production. Patients with ISOD and MocoD were also found with elevated excretion of α-amino adipic semialdehyde (α-AASA) (38). α-AASA accumulation is the metabolic signature of a deficiency in α-AASA dehydrogenase observed in patients with pyridoxine-dependent epilepsy. The enzymatic deficiency in these individuals causes an increase in α-AASA and its cyclic form piperideine-6-carboxylate (P6C). P6C can trap pyridoxal-5-phosphate (PLP), the active form of vitamin B6 (pyridoxine), leading to a deficiency in PLP, which is corrected with supplemental pyridoxine. A decrease in PLP has also been observed in the cerebrospinal fluid from ISOD and MocoD patients (39). It is not clear whether sulfite is responsible for the accumulation of α-AASA and the deficiency in PLP in ISOD and MocoD patients. Nevertheless, pyridoxine and folic acid supplementation in patients with MocoD successfully normalized the PLP level and abolished seizures in two patients with mutations in MOCS2 (MocoD Type B) (40). Although anti-seizure medications and dietary restriction of sulfur-containing amino acids may be beneficial in some cases (41), there are no treatment options for patients with mutations in the MOCS2, GPHN (MocoD Type C), or SUOX genes. Pyridoxine supplementation is an option being considered to alleviate specific clinical features in patients.
A successful treatment using cyclic pyranopterin monophosphate (cPMP) has been described for patients with mutations in the MOCS1 gene (i.e., those with MocoD Type A) (42). The MOCS1 gene controls the initial step in the Moco biosynthetic pathway, catalyzing the conversion of guanosine triphosphate into cPMP. Therefore, patients with mutations in the MOCS1 gene lack cPMP. Daily administration of cPMP to patients resolved all metabolic abnormalities associated with defective sulfite oxidase and xanthine pathways and prevented further signs of neurologic deterioration (43, 44). Early diagnosis and initiation of treatment are essential to ensure success (44). A prospective cohort study of 16 young infants, followed for five years, found that intravenous cPMP treatment was associated with clinical improvement in most infants with MocoD Type A but not in those with MocoD Type B (42). The US Food and Drug Administration recently approved the cPMP replacement drug, fosdenopterin (brand name: Nulibry), for intravenous treatment of MocoD Type A (45). Since cPMP replacement therapy can only benefit MocoD Type A, additional treatment methods are required. A mouse model for MocoD Type B has been recently developed, which may aid in the development of a therapy for those suffering from the MOSC2 mutation (46).

The Recommended Dietary Allowance (RDA)
The recommended dietary allowance (RDA) for molybdenum was most recently revised in January 2001 by the Food and Nutrition Board of the Institute of Medicine (now the National Academy of Medicine) (2). It was based on the results of nutritional balance studies conducted in eight, healthy young men under controlled laboratory conditions (47, 48). The RDA values for molybdenum are listed in Table 1 in micrograms (μg)/day by age and gender. Adequate intake (AI) levels were set for infants based on mean molybdenum intake from human milk, exclusively. The Daily Value (DV), derived from the RDA, is 45 μg/day for individuals 4 years of age or older (49).
| Life Stage | Age | Males (μg/day) | Females (μg/day) |
|---|---|---|---|
| Infants | 0-6 months | 2 (AI) | 2 (AI) |
| Infants | 7-12 months | 3 (AI) | 3 (AI) |
| Children | 1-3 years | 17 | 17 |
| Children | 4-8 years | 22 | 22 |
| Children | 9-13 years | 34 | 34 |
| Adolescents | 14-18 years | 43 | 43 |
| Adults | 19 years and older | 45 | 45 |
| Pregnancy | all ages | - | 50 |
| Breast-feeding | all ages | - | 50 |
Disease Prevention
Esophageal cancer
Linxian is a small region in northern China where the incidence of cancer of the esophagus and stomach is very high (10 times higher than the average in China and 100 times higher than the average in the US). The soil in this region is low in molybdenum and other mineral elements; therefore, dietary molybdenum intake is also low. Studies conducted in other areas of low and high incidence of esophageal cancer showed that content of molybdenum and zinc in hair and nails is significantly lower in inhabitants of high-risk regions compared to cold spots. Moreover, esophageal cancer patients display reduced content of the trace elements compared to healthy relatives (50, 51).
Increased intake of nitrosamines, which are known carcinogens, may be one of a number of dietary and environmental factors that contributes to the development of esophageal cancer in residents of high-risk regions. Adding ammonium molybdate to the soil may help decrease the risk of esophageal cancer by limiting nitrosamine exposure. It is not clear whether dietary molybdenum supplementation is beneficial in decreasing the risk of esophageal cancer. A placebo-controlled, intervention trial conducted in the Linxian area found that supplementation with molybdenum (30 μg/day) and vitamin C (120 mg/day) for 5.25 years did not decrease the incidence or mortality from esophageal cancer (52-54). The 25-year follow-up of this trial found that co-supplementation with these two micronutrients actually led to small increases in risk of death from gastric cardia cancer and cancer in general, but not death from esophageal cancer (54). However, a subanalysis of this 25-year follow-up revealed that co-supplementation with molybdenum and vitamin C slightly increased risk of mortality from esophageal cancer in those who were 55 years or older when the trial initially began (HR, 1.16; 95% CI: 1.04-1.30) (54).
Longevity
Rugao is a county in Jiangsu province (China) renowned for the longevity of its residents. Extended longevity can hardly be attributed to significant differences in traditions, lifestyles, or dietary habits among the residents, and most longevous people are not related to one another, limiting the possible influence of genetics. However, the county has a large number of different soils whose compositions could affect the distribution of trace elements in water and crops and ultimately be linked with human health and longevity. Significant correlations were found between the ratio of people over 90 years old per 100,000 inhabitants and trace elements, including molybdenum, in soils, drinking water, and rice, which constitute key elements of their natural environment (55). The percentage of long-lived people (>80 years old) in Zhongxiang (Hubei province) was also positively linked to the content of molybdenum in their staple food, rice (56). In these regions, it is likely that combinations of trace elements contribute to optimum health and longevity as opposed to the sole effect of molybdenum.
Sources
Food sources
The Total Diet Study, an annual survey of the mineral content in the typical American diet, indicates that the dietary intake of molybdenum averages 76 μg/day for women and 109 μg/day for men. Thus, usual molybdenum intakes are well above the RDA for molybdenum. Legumes, such as beans, lentils, and peas, are the richest sources of molybdenum. Grain products and nuts are considered good sources, while animal products, fruit, and many vegetables are generally low in molybdenum (2). Because the molybdenum content of plants depends on the soil molybdenum content and other environmental conditions, the molybdenum content of foods can vary considerably (51, 57).
Supplements
Molybdenum in single-nutrient and multiple nutrient supplements is of various forms, including sodium molybdate, ammonium molybdate, molybdenum citrate, molybdenum chloride, and molybdenum glycinate, among others (58).
Parenteral nutrition
Molybdenum may be present in solutions of parenteral nutrition either included as a trace element or as an incidental contaminant (59, 60).
Safety
Toxicity
The toxicity of molybdenum compounds appears to be relatively low in humans. Increased serum concentrations of uric acid and ceruloplasmin (an iron-oxidizing enzyme) have been reported in occupationally exposed workers in a molybdenite roasting plant (61). Gout-like symptoms have also been reported in an Armenian population consuming 10 to 15 milligrams (mg) of molybdenum from food daily (62). In other studies, blood and urinary uric acid concentrations were not elevated by molybdenum intakes up to 1.5 mg/day (2). There has been only one report of acute toxicity related to molybdenum from a dietary supplement: an adult male reportedly consumed a total of 13.5 mg of molybdenum over a period of 18 days (300 to 800 μg/day) and developed acute psychosis with hallucinations, seizures, and other neurologic symptoms (63). However, a controlled study in four, healthy young men found that molybdenum intakes, ranging from 22 μg/day to 1,490 μg/day (almost 1.5 mg/day), elicited no serious adverse effects when molybdenum was given for 24 days (47).
The Food and Nutrition Board (FNB) of the Institute of Medicine (now the National Academy of Medicine) found little evidence that molybdenum excess was associated with adverse health outcomes in generally healthy people. To determine the tolerable upper intake level (UL), the FNB selected adverse reproductive effects in rats as the most sensitive index of toxicity and applied a large uncertainty factor because animal data were used (2). The UL for molybdenum is listed by age group in Table 2.
Drug interactions
High doses of molybdenum have been found to inhibit the metabolism of acetaminophen in rats (64); however, it is not known whether this occurs at clinically relevant doses in humans.
Linus Pauling Institute Recommendation
The RDA for molybdenum (45 μg/day for adults) is sufficient to prevent deficiency. Although the intake of molybdenum most likely to promote optimum health is not known, there is presently no evidence that intakes higher than the RDA are beneficial. Most people in the US consume more than sufficient molybdenum in their diets, making supplementation unnecessary. Following the Linus Pauling Institute's general recommendation to take a multivitamin/mineral supplement that contains 100% of the daily values (DV) for most nutrients is likely to provide 45 μg/day of molybdenum.
Older adults (>50 years)
Because aging has not been associated with significant changes in the requirement for molybdenum (2), our recommendation for older adults is the same as that for adults 50 years and younger.
Authors and Reviewers
Originally written in 2001 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in June 2013 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in May 2021 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in June 2021 by:
Ralf R. Mendel, Ph.D.
Institute for Plant Biology
Braunschweig University of Technology
Braunschweig, Germany
Copyright 2001-2024 Linus Pauling Institute
References
1. Wuebbens MM, Liu MT, Rajagopalan K, Schindelin H. Insights into molybdenum cofactor deficiency provided by the crystal structure of the molybdenum cofactor biosynthesis protein MoaC. Structure Fold Des. 2000;8(7):709-718. (PubMed)
2. Food and Nutrition Board, Institute of Medicine. Molybdenum. In: Dietary reference intakes for vitamin A, vitamin K, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. Washington, D.C.: National Academy Press; 2001:420-441. (National Academy Press)
3. Schwarz G, Mendel RR, Ribbe MW. Molybdenum cofactors, enzymes and pathways. Nature. 2009;460(7257):839-847. (PubMed)
4. Wang J, Krizowski S, Fischer-Schrader K, et al. Sulfite oxidase catalyzes single-electron transfer at molybdenum domain to reduce nitrite to nitric oxide. Antioxid Redox Signal. 2015;23(4):283-294. (PubMed)
5. Eckhert C. Other trace elements In: Shils ME, Shike M, Ross AC, Caballero B, Cousins RJ, eds. Modern Nutrition in Health and Disease. 10th ed. Philadelphia: Lippincott, Williams & Wilkins; 2006:338-350.
6. Wahl B, Reichmann D, Niks D, et al. Biochemical and spectroscopic characterization of the human mitochondrial amidoxime reducing components hmARC-1 and hmARC-2 suggests the existence of a new molybdenum enzyme family in eukaryotes. J Biol Chem. 2010;285(48):37847-37859. (PubMed)
7. Havemeyer A, Bittner F, Wollers S, Mendel R, Kunze T, Clement B. Identification of the missing component in the mitochondrial benzamidoxime prodrug-converting system as a novel molybdenum enzyme. J Biol Chem. 2006;281(46):34796-34802. (PubMed)
8. Plitzko B, Ott G, Reichmann D, et al. The involvement of mitochondrial amidoxime reducing components 1 and 2 and mitochondrial cytochrome b5 in N-reductive metabolism in human cells. J Biol Chem. 2013;288(28):20228-20237. (PubMed)
9. Ott G, Havemeyer A, Clement B. The mammalian molybdenum enzymes of mARC. J Biol Inorg Chem. 2015;20(2):265-275. (PubMed)
10. Sparacino-Watkins CE, Tejero J, Sun B, et al. Nitrite reductase and nitric-oxide synthase activity of the mitochondrial molybdopterin enzymes mARC1 and mARC2. J Biol Chem. 2014;289(15):10345-10358. (PubMed)
11. Mayr SJ, Mendel RR, Schwarz G. Molybdenum cofactor biology, evolution and deficiency. Biochim Biophys Acta Mol Cell Res. 2021;1868(1):118883. (PubMed)
12. Beedham C. Molybdenum hydroxylases as drug-metabolizing enzymes. Drug Metab Rev. 1985;16(1-2):119-156. (PubMed)
13. Zannolli R, Micheli V, Mazzei MA, et al. Hereditary xanthinuria type II associated with mental delay, autism, cortical renal cysts, nephrocalcinosis, osteopenia, and hair and teeth defects. J Med Genet. 2003;40(11):e121. (PubMed)
14. Fujiwara Y, Kawakami Y, Shinohara Y, Ichida K. A case of hereditary xanthinuria type 1 accompanied by bilateral renal calculi. Intern Med. 2012;51(14):1879-1884. (PubMed)
15. Turnlund JR, Keyes WR. Dietary molybdenum: Effect on copper absorption, excretion, and status in young men. In: Roussel AM, ed. Trace Elements in Man and Animals. Vol 10. New York: Kluwer Academic Press.; 2000:951-953.
16. Suttle NF. Copper imbalances in ruminants and humans: unexpected common ground. Adv Nutr. 2012;3(5):666-674. (PubMed)
17. Lopez-Alonso M, Miranda M. Copper supplementation, a challenge in cattle. Animals (Basel). 2020;10(10):1890. (PubMed)
18. Helz GR, Erickson BE. Extraordinary stability of copper(I)-tetrathiomolybdate complexes: possible implications for aquatic ecosystems. Environ Toxicol Chem. 2011;30(1):97-102. (PubMed)
19. Alvarez HM, Xue Y, Robinson CD, et al. Tetrathiomolybdate inhibits copper trafficking proteins through metal cluster formation. Science. 2010;327(5963):331-334. (PubMed)
20. Brewer GJ, Askari F, Dick RB, et al. Treatment of Wilson's disease with tetrathiomolybdate: V. Control of free copper by tetrathiomolybdate and a comparison with trientine. Transl Res. 2009;154(2):70-77. (PubMed)
21. Redman BG, Esper P, Pan Q, et al. Phase II trial of tetrathiomolybdate in patients with advanced kidney cancer. Clin Cancer Res. 2003;9(5):1666-1672. (PubMed)
22. Gartner EM, Griffith KA, Pan Q, et al. A pilot trial of the anti-angiogenic copper lowering agent tetrathiomolybdate in combination with irinotecan, 5-flurouracil, and leucovorin for metastatic colorectal cancer. Invest New Drugs. 2009;27(2):159-165. (PubMed)
23. Jain S, Cohen J, Ward MM, et al. Tetrathiomolybdate-associated copper depletion decreases circulating endothelial progenitor cells in women with breast cancer at high risk of relapse. Ann Oncol. 2013;24(6):1491-1498. (PubMed)
24. Hou G, Abrams GD, Dick R, Brewer GJ. Efficacy of tetrathiomolybdate in a mouse model of multiple sclerosis. Transl Res. 2008;152(5):239-244. (PubMed)
25. Wei H, Zhang WJ, McMillen TS, Leboeuf RC, Frei B. Copper chelation by tetrathiomolybdate inhibits vascular inflammation and atherosclerotic lesion development in apolipoprotein E-deficient mice. Atherosclerosis. 2012;223(2):306-313. (PubMed)
26. Askari F, Innis D, Dick RB, et al. Treatment of primary biliary cirrhosis with tetrathiomolybdate: results of a double-blind trial. Transl Res. 2010;155(3):123-130. (PubMed)
27. Abumrad NN, Schneider AJ, Steel D, Rogers LS. Amino acid intolerance during prolonged total parenteral nutrition reversed by molybdate therapy. Am J Clin Nutr. 1981;34(11):2551-2559. (PubMed)
28. Reiss J, Hahnewald R. Molybdenum cofactor deficiency: Mutations in GPHN, MOCS1, and MOCS2. Hum Mutat. 2011;32(1):10-18. (PubMed)
29. Tan WH, Eichler FS, Hoda S, et al. Isolated sulfite oxidase deficiency: a case report with a novel mutation and review of the literature. Pediatrics. 2005;116(3):757-766. (PubMed)
30. Shalata A, Mandel H, Dorche C, et al. Prenatal diagnosis and carrier detection for molybdenum cofactor deficiency type A in northern Israel using polymorphic DNA markers. Prenat Diagn. 2000;20(1):7-11. (PubMed)
31. Kikuchi K, Hamano S, Mochizuki H, Ichida K, Ida H. Molybdenum cofactor deficiency mimics cerebral palsy: differentiating factors for diagnosis. Pediatr Neurol. 2012;47(2):147-149. (PubMed)
32. Atwal PS, Scaglia F. Molybdenum cofactor deficiency. Mol Genet Metab. 2016;117(1):1-4. (PubMed)
33. Johnson JL. Prenatal diagnosis of molybdenum cofactor deficiency and isolated sulfite oxidase deficiency. Prenat Diagn. 2003;23(1):6-8. (PubMed)
34. Hughes EF, Fairbanks L, Simmonds HA, Robinson RO. Molybdenum cofactor deficiency-phenotypic variability in a family with a late-onset variant. Dev Med Child Neurol. 1998;40(1):57-61. (PubMed)
35. Vijayakumar K, Gunny R, Grunewald S, et al. Clinical neuroimaging features and outcome in molybdenum cofactor deficiency. Pediatr Neurol. 2011;45(4):246-252. (PubMed)
36. Alkufri F, Harrower T, Rahman Y, et al. Molybdenum cofactor deficiency presenting with a parkinsonism-dystonia syndrome. Mov Disord. 2013;28(3):399-401. (PubMed)
37. Scelsa B, Gasperini S, Righini A, Iascone M, Brazzoduro VG, Veggiotti P. Mild phenotype in Molybdenum cofactor deficiency: A new patient and review of the literature. Mol Genet Genomic Med. 2019;7(6):e657. (PubMed)
38. Mills PB, Footitt EJ, Ceyhan S, et al. Urinary AASA excretion is elevated in patients with molybdenum cofactor deficiency and isolated sulphite oxidase deficiency. J Inherit Metab Dis. 2012;35(6):1031-1036. (PubMed)
39. Footitt EJ, Heales SJ, Mills PB, Allen GF, Oppenheim M, Clayton PT. Pyridoxal 5'-phosphate in cerebrospinal fluid; factors affecting concentration. J Inherit Metab Dis. 2011;34(2):529-538. (PubMed)
40. Struys EA, Nota B, Bakkali A, Al Shahwan S, Salomons GS, Tabarki B. Pyridoxine-dependent epilepsy with elevated urinary α-amino adipic semialdehyde in molybdenum cofactor deficiency. Pediatrics. 2012;130(6):e1716-1719. (PubMed)
41. Johnson JL, Duran M. Molybdenum cofactor deficiency and isolated sulfite deficiency. In: Scriver RC, ed. Metabolic and molecular bases of inherited disease. New York: Mcgraw-Hill; 2001:3163-3177.
42. Schwahn BC, Van Spronsen FJ, Belaidi AA, et al. Efficacy and safety of cyclic pyranopterin monophosphate substitution in severe molybdenum cofactor deficiency type A: a prospective cohort study. Lancet. 2015;386(10007):1955-1963. (PubMed)
43. Veldman A, Santamaria-Araujo JA, Sollazzo S, et al. Successful treatment of molybdenum cofactor deficiency type A with cPMP. Pediatrics. 2010;125(5):e1249-1254. (PubMed)
44. Hitzert MM, Bos AF, Bergman KA, et al. Favorable outcome in a newborn with molybdenum cofactor type A deficiency treated with cPMP. Pediatrics. 2012;130(4):e1005-1010. (PubMed)
45. US Food and Drug Administration. FDA Approves First Treatment for Molybdenum Cofactor Deficiency Type A. February 26, 2021. Available at: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-molybdenum-cofactor-deficiency-type. Accessed 6/25/21.
46. Jakubiczka-Smorag J, Santamaria-Araujo JA, Metz I, et al. Mouse model for molybdenum cofactor deficiency type B recapitulates the phenotype observed in molybdenum cofactor deficient patients. Hum Genet. 2016;135(7):813-826. (PubMed)
47. Turnlund JR, Keyes WR, Peiffer GL. Molybdenum absorption, excretion, and retention studied with stable isotopes in young men at five intakes of dietary molybdenum. Am J Clin Nutr. 1995;62(4):790-796. (PubMed)
48. Turnlund JR, Keyes WR, Peiffer GL, Chiang G. Molybdenum absorption, excretion, and retention studied with stable isotopes in young men during depletion and repletion. Am J Clin Nutr. 1995;61(5):1102-1109. (PubMed)
49. US Food and Drug Administration. Daily Value and Percent Daily Value: Changes on the New Nutrition and Supplement Facts Labels. March 2020. Available at: https://www.fda.gov/food/new-nutrition-facts-label/daily-value-new-nutrition-and-supplement-facts-labels#referenceguide. Accessed 5/28/21.
50. Nouri M, Chalian H, Bahman A, et al. Nail molybdenum and zinc contents in populations with low and moderate incidence of esophageal cancer. Arch Iran Med. 2008;11(4):392-396. (PubMed)
51. Ray SS, Das D, Ghosh T, Ghosh AK. The levels of zinc and molybdenum in hair and food grain in areas of high and low incidence of esophageal cancer: a comparative study. Glob J Health Sci. 2012;4(4):168-175. (PubMed)
52. Blot WJ, Li JY, Taylor PR, et al. Nutrition intervention trials in Linxian, China: supplementation with specific vitamin/mineral combinations, cancer incidence, and disease-specific mortality in the general population. J Natl Cancer Inst. 1993;85(18):1483-1492. (PubMed)
53. Qiao YL, Dawsey SM, Kamangar F, et al. Total and cancer mortality after supplementation with vitamins and minerals: follow-up of the Linxian General Population Nutrition Intervention Trial. J Natl Cancer Inst. 2009;101(7):507-518. (PubMed)
54. Wang SM, Taylor PR, Fan JH, et al. Effects of nutrition intervention on total and cancer mortality: 25-year post-trial follow-up of the 5.25-year Linxian Nutrition Intervention Trial. J Natl Cancer Inst. 2018;110(11):1229-1238. (PubMed)
55. Huang B, Zhao Y, Sun W, et al. Relationships between distributions of longevous population and trace elements in the agricultural ecosystem of Rugao County, Jiangsu, China. Environ Geochem Health. 2009;31(3):379-390. (PubMed)
56. Lv J, Wang W, Krafft T, Li Y, Zhang F, Yuan F. Effects of several environmental factors on longevity and health of the human population of Zhongxiang, Hubei, China. Biol Trace Elem Res. 2011;143(2):702-716. (PubMed)
57. Mills CF, Davis GK. Molybdenum. In: Mertz W, ed. Trace elements in human and animal nutrition. 5th ed. San Diego: Academic Press; 1987:429-463.
58. National Institutes of Health. Dietary Supplement Label Database. Version 7.0.12. August 2020. Available at: https://dsld.od.nih.gov/dsld. Accessed 5/28/21.
59. Hardy G, Menendez AM, Manzanares W. Trace element supplementation in parenteral nutrition: pharmacy, posology, and monitoring guidance. Nutrition. 2009;25(11-12):1073-1084. (PubMed)
60. Stehle P, Stoffel-Wagner B, Kuhn KS. Parenteral trace element provision: recent clinical research and practical conclusions. Eur J Clin Nutr. 2016;70(8):886-893. (PubMed)
61. Walravens PA, Moure-Eraso R, Solomons, CC, Chapell, R, Bentley G. Biochemical abnormalities in workers exposed to molybdenum dust. Arch Environ Health. 1979;34(5):302-308. (PubMed)
62. Vyskocil A, Viau C. Assessment of molybdenum toxicity in humans. J Appl Toxicol. 1999;19(3):185-192. (PubMed)
63. Momcilovic B. A case report of acute human molybdenum toxicity from a dietary molybdenum supplement--a new member of the "Lucor metallicum" family. Arh Hig Rada Toksikol. 1999;50(3):289-297. (PubMed)
64. Boles JW, Klaassen CD. Effects of molybdate and pentachlorophenol on the sulfation of acetaminophen. Toxicology. 2000;146(1):23-35. (PubMed)
Phosphorus
Contents
Summary
- Phosphorus is an essential structural component of cell membranes and nucleic acids but is also involved in several biological processes, including bone mineralization, energy production, cell signaling through phosphorylation reactions, and regulation of acid-base homeostasis. (More information)
- Dietary phosphorus deficiency is uncommon and often only observed in cases of near-total starvation or in rare inherited disorders involving renal phosphorus wasting. Symptoms include loss of appetite, muscle weakness, bone fragility, numbness in the extremities, and rickets in children. (More information)
- The Recommended Dietary Allowance (RDA), 700 mg/day of phosphorus for healthy adults, is meant to sustain serum phosphorus concentrations within the physiologic range of 2.5 to 4.5 mg/dL. (More information)
- Phosphorus is found in most food sources and is a component of many commonly used food additives. The bioavailability of phosphorus from food is usually very high with the exception of phytate phosphorus in plant sources, such as grains, legumes, and seeds, which is poorly digested. (More information)
- Estimates of dietary phosphorus intakes in the US population are likely to be inaccurate because the amounts of phosphorus-based food additives used in processed foods are not always included in the nutrient content database used to compute nutrient intakes. (More information)
- High serum phosphorus concentrations have been associated with increased rates of cardiovascular disease and mortality in subjects with or without kidney disease. Abnormal deposition of calcium phosphate in soft tissues may predispose individuals to vascular dysfunction and cardiovascular disease. (More information)
- Hyperphosphatemia, which is common in individuals with impaired kidney function, characterizes a condition in which there is an abnormally high accumulation of phosphorus in blood, because the kidneys are not able to effectively excrete it. (More information)
- The tolerable upper intake level (UL) for phosphorus is 4,000 mg/day for generally healthy adults. Yet, daily phosphorus intakes in excess of the RDA have been linked to an increased risk of all-cause mortality in healthy individuals. (More information)
- Observational studies suggest that a low calcium-to-phosphorus intake ratio may be detrimental to bone health, especially in women at increased risk for osteoporosis. (More information)
Phosphorus is an essential mineral that is required by every cell in the body for normal function (1). Bound to oxygen in all biological systems, phosphorus is found as phosphate (PO43-) in the body. Approximately 85% of the body's phosphorus is found in bones and teeth (2).
Function
Phosphorus is a major structural component of bone in the form of a calcium phosphate salt called hydroxyapatite. Phospholipids (e.g., phosphatidylcholine) are major structural components of cell membranes. All energy production and storage are dependent on phosphorylated compounds, such as adenosine triphosphate (ATP) and creatine phosphate. Nucleic acids (DNA and RNA), which are responsible for the storage and transmission of genetic information, are long chains of phosphate-containing molecules. A number of enzymes, hormones, and cell-signaling molecules depend on phosphorylation for their activation. Phosphorus also helps maintain normal acid-base balance (pH) by acting as one of the body's most important buffers. Additionally, the phosphorus-containing molecule 2,3-diphosphoglycerate (2,3-DPG) binds to hemoglobin in red blood cells and regulates oxygen delivery to the tissues of the body (1).
Regulation
Parathyroid hormone-vitamin D and FGF-23-endocrine axis
Dietary phosphorus is readily absorbed in the small intestine, and in healthy individuals, excess phosphorus is excreted by the kidneys under the regulatory action of the endocrine hormones: parathyroid hormone (PTH), vitamin D, and fibroblast growth factor-23 (FGF-23). The acute regulation of blood calcium and phosphorus concentrations is controlled through the actions of PTH and the active form of vitamin D. A slight drop in blood calcium levels (e.g., in the case of inadequate calcium intake) is sensed by the parathyroid glands, resulting in their increased secretion of PTH, which rapidly decreases urinary excretion of calcium but increases urinary excretion of phosphorus and stimulates bone resorption. This results in the release of bone mineral (calcium and phosphate) — actions that restore serum calcium concentrations. Although the action is not immediate, PTH also stimulates conversion of vitamin D to its active form (1,25-dihydroxyvitamin D; calcitriol) in the kidneys. Increased circulating 1,25-dihydroxyvitamin D in turn stimulates increased intestinal absorption of both calcium and phosphorus. A third hormone, FGF-23, plays a central role in phosphorus homeostasis. FGF-23 is secreted by bone-forming cells (osteoblasts/osteocytes) in response to increases in phosphorus intake. In a negative feedback loop, FGF-23 inhibits the production and stimulates the degradation of 1,25-dihydroxyvitamin D, as well as promotes an increase in urinary phosphorus excretion independently of PTH and 1,25-dihydroxyvitamin D (3).
Deficiency
Inadequate phosphorus intake rarely results in abnormally low serum phosphorus levels (hypophosphatemia) because renal reabsorption of phosphorus increases to compensate for decreased intake. The effects of moderate to severe hypophosphatemia may include loss of appetite, anemia, muscle weakness, bone pain, rickets (in children), osteomalacia (in adults), increased susceptibility to infection, numbness and tingling of the extremities, difficulty walking, and respiratory failure. Severe hypophosphatemia may occasionally be life threatening. Since phosphorus is so widespread in food, dietary phosphorus deficiency is usually seen only in cases of near-total starvation. Other individuals at risk of hypophosphatemia include alcoholics, diabetics recovering from an episode of diabetic ketoacidosis, patients with respiratory alkalosis, and starving or anorexic patients on refeeding regimens that are high in calories but too low in phosphorus (reviewed in 4). Hypophosphatemia caused by inherited disorders of phosphorus homeostasis (phosphorus wasting disorders) has been linked to elevated urinary excretion or impaired renal reabsorption of phosphorus in affected subjects (reviewed in 5).
The Recommended Dietary Allowance (RDA)
The Recommended Dietary Allowance (RDA) for phosphorus is based on the maintenance of normal serum phosphorus levels in adults (2.5-4.5 milligrams/deciliter [mg/dL]) and is believed to represent adequate phosphorus intakes to meet cellular and bone formation needs (6; Table 1). The RDA, which is the average daily intake that meets the requirements of 97.5% of healthy individuals in a specific life stage and gender group, is based on the Estimated Average Requirement (EAR; 580 mg/day of phosphorus for adults) — the nutrient intake that meets the requirements of 50% of healthy individuals in a particular life stage and gender group.
| Life Stage | Age |
Males (mg/day) |
Females (mg/day) |
|---|---|---|---|
| Infants | 0-6 months | 100 (AI) | 100 (AI) |
| Infants | 7-12 months | 275 (AI) | 275 (AI) |
| Children | 1-3 years | 460 | 460 |
| Children | 4-8 years | 500 | 500 |
| Children | 9-13 years | 1,250 | 1,250 |
| Adolescents | 14-18 years | 1,250 | 1,250 |
| Adults | 19 years and older | 700 | 700 |
| Pregnancy | 18 years and younger | - | 1,250 |
| Pregnancy | 19 years and older | - | 700 |
| Breast-feeding | 18 years and younger | - | 1,250 |
| Breast-feeding | 19 years and older | - | 700 |
Sources
Food sources
Phosphorus is found in most food because it is a critical constituent of all living organisms. Dairy foods, cereal products, meat, and fish are particularly rich sources of phosphorus (7). Phosphorus is also a component of many food additives that are used in food processing and is present in cola soft drinks as phosphoric acid (8). In the nationally representative NHANES survey, phosphorus intakes were well above the EAR and RDA, with average daily intakes of 1,602 mg in men and 1,128 mg in women (8). Dietary phosphorus derived from food additives is not always included in food nutrient composition databases, so the total amount of phosphorus consumed by the average person in the US can be underestimated by more than 20% (9). Segments of the US population who consume more highly processed foods and whose phosphorus intakes approach the tolerable upper intake level of 4,000 mg/day are thought by some to be at high risk of developing adverse health outcomes (see Safety) (7, 9).
Bioavailability
The phosphorus in plant seeds (beans, peas, cereals, and nuts) is present in a storage form of phosphate called phytic acid or phytate. Only about 50% of the phosphorus from phytate is available to humans because we lack enzymes (phytases) that liberate phosphorus from phytate (10). Yeasts possess phytases, so whole grains incorporated into leavened breads have more bioavailable phosphorus than whole grains incorporated into breakfast cereals or flat breads (6). Because reducing dietary phosphorus absorption may benefit individuals with impaired kidney function who are at risk of hyperphosphatemia (serum phosphorus at or above the high-normal range), protein sources of phosphorus in grain-based vegetarian diets may be preferred over meat-based diets (11). Table 2 lists a number of phosphorus-rich foods, along with their phosphorus content in milligrams (mg). For more information on the nutrient content of food, search USDA's FoodData Central.
| Food | Serving | Phosphorus (mg) |
|---|---|---|
| Salmon (chinook, cooked) | 3 ounces* | 315 |
| Yogurt (plain, nonfat) | 8 ounces | 306 |
| Milk (skim) | 8 ounces | 247 |
| Halibut (Atlantic or Pacific, cooked) | 3 ounces | 244 |
| Turkey (light meat, cooked) | 3 ounces | 217 |
| Chicken (light meat, cooked) | 3 ounces | 135-196 |
| Beef (chuck eye steak, cooked) | 3 ounces | 179 |
| Lentils# (cooked) | ½ cup | 178 |
| Almonds# | 1 ounce (23 nuts) | 136 |
| Cheese, mozzarella (part skim) | 1 ounce | 131 |
| Peanuts# | 1 ounce | 108 |
| Egg (hard-boiled) | 1 large | 86 |
| Bread, whole-wheat | 1 slice | 68 |
| Carbonated cola drink | 12 ounces | 41 |
| Bread, enriched white | 1 slice | 25 |
|
*A three-ounce serving of meat or fish is about the size of a deck of cards. #Phosphorus from nuts, seeds, and grains is about 50% less bioavailable than phosphorus from other sources (12). |
||
Supplements
Phosphorus content of multivitamin/mineral (MVM) supplements varies; a US national survey found that MVM supplements contributed an average of 108 mg to daily phosphorus intake (10). Sodium phosphate and potassium phosphate salts are used for the treatment of hypophosphatemia that occurs in hereditary disorders of phosphate wasting, and their use requires medical supervision. Calcium phosphate salts are sometimes used as calcium supplements (13). Commonly used over-the-counter and prescription drugs also contribute to phosphorus intakes at levels yet to be defined (10).
Safety
Toxicity
Several disorders characterized by serum phosphorus levels above normal (hyperphosphatemia) have been described, including those resulting from increased intestinal absorption of phosphate salts taken by mouth or by colonic absorption of the phosphate salts in enemas (1). Yet, the disruption of phosphorus homeostasis is most often associated with excretion failure in patients with chronic kidney disease (CKD) or end-stage renal disease (advanced CKD). When kidney function is only 20% of normal, even phosphorus intakes within the recommended range may lead to hyperphosphatemia. Hyperphosphatemia may also affect individuals with inappropriately low parathyroid hormone (PTH) levels (hypoparathyroidism) as they lack PTH stimulation of renal phosphate excretion and fail to stimulate synthesis of 1,25-dihydroxyvitamin D (the active form of vitamin D). These individuals cannot excrete excess phosphorus in the absence of both hormones (14). Elevated serum phosphorus concentrations have been associated with accelerated disease progression in individuals with impaired kidney function and have been linked to increased risk of adverse health outcomes in the general population (9, 15).
High serum phosphorus concentrations in the general population
High serum phosphorus within the normal range (2.5-4.5 mg/dL) has recently been associated with increased incidence of cardiovascular disease (CVD) in individuals with normal kidney function. Two studies conducted in the general population and in individuals with prior CVD have linked high-normal serum phosphorus concentrations (≥3.5 mg/dL) to a greater cardiovascular risk (16, 17). Additional observational studies found that serum phosphorus concentrations equal to or above 4 mg/dL were associated with a doubling of the risk of developing incident CKD and end-stage renal disease in individuals free of renal disease at study inception (18). In a prospective cohort study, which followed 4,005 healthy young adults for more than 15 years, higher serum phosphorus within the normal range was also associated with left ventricular hypertrophy, a condition often linked to adverse cardiovascular outcomes (19). In another study of 3,088 middle-aged healthy participants followed for over 17 years, serum phosphorus concentrations in the top quartile of the normal range were associated with a two-fold higher risk of heart failure compared to the lowest quartile (≥3.5 mg/dL vs. <2.9 mg/dL) (16). It is thought that vascular calcification, which may explain the relationship between high phosphorus and cardiovascular disease risk in CKD patients (see Hyperphosphatemia in subjects with kidney disease), contributes to this association in individuals with normal kidney function, even when their serum phosphorus is within the normal range and their intakes are below the tolerable upper intake level (UL) (20, 21).
Phosphorus homeostasis is tightly regulated by the PTH/vitamin D/FGF-23 axis in individuals with normal kidney function (see Regulation). Increased secretion of PTH and FGF-23 helps maintain phosphorus serum concentrations in the normal range (2.5-4.5 mg/dL) even in the setting of high phosphorus intake (9). This contributes to serum phosphorus being only weakly correlated to phosphorus consumption (22). Of note, sustained increases in FGF-23 and PTH are commonly observed during CKD in order to maintain normal serum phosphorus concentrations despite a reduction in urinary phosphorus excretion (23). Elevated FGF-23, rather than serum phosphorus, appears to be an early marker of disordered phosphorus homeostasis and a predictor of adverse health outcomes in patients with early-stage CKD (23, 24). Thus, it is reasonable to assume that measuring serum phosphorus in people with normal renal function cannot adequately reflect early disturbances in phosphorus metabolism due to high phosphorus consumption.
Hyperphosphatemia in subjects with kidney disease
Observational studies have reported high rates of mortality and cardiovascular events in association with high blood phosphorus levels in subjects with CKD. A meta-analysis of 13 prospective cohort studies, conducted in over 90,000 CKD patients, found an 18% increase in all-cause mortality per 1 mg/dL increase in serum phosphorus concentration above 3.5 mg/dL. A 10% increased risk in cardiovascular disease (CVD)-related death was also calculated for each 1 mg/dL higher concentration in the meta-analysis of three studies (25).
Although no causality has been established between vitamin D deficiency and CVD risk, it has been suggested that failure to produce 1,25-dihydroxyvitamin D in hyperphosphatemic individuals may modify the risk of developing cardiovascular and renal disease, as well as worsen kidney insufficiency in CKD patients (26). Another plausible mechanism for hyperphosphatemia-induced cardiovascular dysfunction is the deposition of calcium phosphate in non-skeletal tissues, especially the vasculature (27). Indeed, high phosphorus concentrations may stimulate the expression of bone specific markers in blood vessel-forming cells, resulting in a shift in their functions; this process, called osteochondrogenic differentiation, transforms vascular smooth muscle cells (VSMCs) into bone-like cells. The culture of human aortic VSMCs in hyperphosphatemic conditions was found to result in the mineralization of the extracellular media, mimicking in vivo vascular calcification (28). Vascular calcification has been associated with at least a three-fold increase in risk for cardiovascular events and mortality; the risk for cardiovascular events is twice as high (i.e., six-fold increased risk) in individuals with kidney insufficiency (29).
In CKD patients, disorders in bone remodeling may result in excess release of phosphorus and calcium into the blood, which exacerbates hyperphosphatemia and vascular calcification and accelerates the decline of kidney function. Currently, dietary phosphorus restriction is recommended to normalize serum concentrations in CKD patients, although the impact on CVD and mortality risks is not known.
The Tolerable Upper Intake Level (UL)
To avoid the adverse effects of hyperphosphatemia, the US Food and Nutrition Board set a tolerable upper intake level (UL) for oral phosphorus in generally healthy individuals (6; Table 3). The lower UL for individuals over 70 years of age, compared to younger age groups, reflects the increased likelihood of impaired kidney function in elderly individuals. The UL does not apply to individuals with significantly impaired kidney function or other health conditions known to increase the risk of hyperphosphatemia.
Adverse health outcomes have been associated with normal serum phosphorus concentrations, suggesting that in individuals with adequate kidney function, the measurement of tightly controlled serum phosphorus levels may misrepresent the detrimental effect of high dietary phosphorus intake (see High serum phosphorus concentrations in the general population). While phosphorus intakes below the UL of 4,000 mg/day should not result in hyperphosphatemia or cardiovascular risk in healthy adults ages 19-70 years, a recent study found that daily phosphorus intakes more than twice the RDA (i.e., >1,400 mg/day) were significantly associated with an increased risk of all-cause mortality (30).
Is high phosphorus intake detrimental to bone health?
Some investigators are concerned about the increasing amounts of phosphates in the diet, which they largely attribute to phosphoric acid in some soft drinks and the increasing use of phosphate additives in processed foods (31, 32). High serum phosphorus has been shown to impair synthesis of the active form of vitamin D (1,25-dihydroxyvitamin D) in the kidneys, reduce blood calcium, and lead to increased PTH release by the parathyroid glands (8). PTH stimulation then results in decreased urinary calcium excretion and increased bone resorption; both contribute to serum calcium concentrations returning to normal (8). If sustained, elevated PTH levels could have an adverse effect on bone mineral content, but this effect appears to be observed with diets that are high in phosphorus and low in calcium, underscoring the importance of a balanced dietary calcium-to-phosphorus ratio. In a small cross-sectional study, which enrolled 147 premenopausal women with adequate calcium intakes, participants with lower calcium-to-phosphorus (Ca:P) intakes (ratios ≤0.5) had significantly higher serum PTH levels and urinary calcium excretion than those with higher Ca:P ratios (ratios >0.5) (33). A controlled trial in 10 young women found no adverse effects of a phosphorus-rich diet (3,000 mg/day) on bone-related hormones and biochemical markers of bone resorption when dietary calcium intakes were maintained at almost 2,000 mg/day (Ca:P = 0.66), again demonstrating the importance of the balance between dietary calcium and phosphorus (34).
A cross-sectional study conducted in 2,344 Brazilian men and women (median age, 58 years) showed an association between higher phosphorus intakes and increased risk of fracture. Yet, intakes of other minerals and vitamins relevant to bone health, such as calcium, magnesium, and vitamin D, were below the RDA in this population, whereas phosphorus intakes were close to the RDA (35). While it appears that hormonal and calcium disorders might be prevented by an adequate calcium-to-phosphorus intake ratio, there is no convincing evidence that the dietary phosphorus levels experienced in the US adversely affect bone mineral density. Nevertheless, the substitution of phosphate-containing soft drinks and snack foods for milk and other calcium-rich food may represent a serious risk to bone health (see the article on Calcium) (36).
Drug interactions
Aluminum-containing antacids reduce the absorption of dietary phosphorus by forming aluminum phosphate, which is unabsorbable. When consumed in high doses, aluminum-containing antacids can produce abnormally low blood phosphorus levels (hypophosphatemia), as well as aggravate phosphorus deficiency due to other causes (37). The reduction of stomach acidity by proton-pump inhibitors may also limit the efficacy of phosphate-binder therapy in patients with kidney failure (38). Excessively high doses of 1,25-dihydroxyvitamin D, the active form of vitamin D, or its analogs, may result in hyperphosphatemia (6).
Potassium supplements or potassium-sparing diuretics taken together with phosphorus supplements may result in high blood levels of potassium (hyperkalemia). Hyperkalemia can be a serious problem, resulting in life-threatening heart rhythm abnormalities (arrhythmias). People taking such a combination must inform their health care provider and have their serum potassium levels checked regularly (37).
Additionally, prevention of bone demineralization by hormone replacement therapy in postmenopausal women is associated with higher urinary phosphorus excretion and lower serum phosphorus levels in treated compared to untreated women (39, 40).
Linus Pauling Institute Recommendation
The Linus Pauling Institute supports the RDA for phosphorus (700 mg/day for adults). Although some multivitamin/mineral supplements contain more than 15% of the current RDA for phosphorus, a varied diet should easily provide adequate phosphorus for most people.
Older adults (>50 years)
At present, there is no evidence that phosphorus requirements of older adults differ from that of younger adults, and a varied diet should easily provide the RDA (700 mg/day) of phosphorus for those over 50 years of age.
Authors and Reviewers
Originally written in 2001 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2003 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in August 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in June 2014 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in June 2014 by:
Mona S. Calvo, Ph.D.
Office of Applied Research and Safety Assessment
Center for Food Safety and Applied Nutrition
US Food and Drug Administration
The findings and conclusions of this reviewer do not necessarily represent the views and opinions of the US Food and Drug Administration.
Copyright 2001-2024 Linus Pauling Institute
References
1. Knochel JP. Phosphorus. In: Shils ME, Shike M, Ross AC, Caballero B, Cousins RJ, eds. Modern Nutrition in Health and Disease. 10th ed. Baltimore: Lippincott Williams & Wilkins; 2006:211-222.
2. Heaney RP. Phosphorus. In: Erdman Jr. JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Ames: Wiley-Blackwell; 2012; 447-458.
3. Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev. 2012;92(1):131-155. (PubMed)
4. Amanzadeh J, Reilly RF, Jr. Hypophosphatemia: an evidence-based approach to its clinical consequences and management. Nat Clin Pract Nephrol. 2006;2(3):136-148. (PubMed)
5. Alizadeh Naderi AS, Reilly RF. Hereditary disorders of renal phosphate wasting. Nat Rev Nephrol. 2010;6(11):657-665. (PubMed)
6. Food and Nutrition Board, Institute of Medicine. Phosphorus. Dietary Reference Intakes: Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington D.C.: National Academy Press; 1997:146-189. (National Academy Press)
7. Takeda E, Yamamoto H, Yamanaka-Okumura H, Taketani Y. Dietary phosphorus in bone health and quality of life. Nutr Rev. 2012;70(6):311-321. (PubMed)
8. Calvo MS, Moshfegh AJ, Tucker KL. Assessing the health impact of phosphorus in the food supply: issues and considerations. Adv Nutr. 2014;5(1):104-113. (PubMed)
9. Calvo MS, Uribarri J. Public health impact of dietary phosphorus excess on bone and cardiovascular health in the general population. Am J Clin Nutr. 2013;98(1):6-15. (PubMed)
10. Calvo MS, Uribarri J. Contributions to total phosphorus intake: all sources considered. Semin Dial. 2013;26(1):54-61. (PubMed)
11. Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol. 2011;6(2):257-264. (PubMed)
12. National Research Council, Food and Nutrition Board. Recommended Dietary Allowances. 10th ed. Washington, D.C.: National Academy Press; 1989:184-187.
13. Phosphorus. In: Hendler SS, Rorvik DM, eds., eds. PDR for Nutritional Supplements. 2nd ed. Montvale: Physicians' Desk Reference; 2008:494-497.
14. Al-Azem H, Khan AA. Hypoparathyroidism. Best Pract Res Clin Endocrinol Metab. 2012;26(4):517-522. (PubMed)
15. Menon MC, Ix JH. Dietary phosphorus, serum phosphorus, and cardiovascular disease. Ann N Y Acad Sci. 2013; 1301:21-26. (PubMed)
16. Dhingra R, Sullivan LM, Fox CS, et al. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med. 2007;167(9):879-885. (PubMed)
17. Tonelli M, Sacks F, Pfeffer M, et al. Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation. 2005;112(17):2627-2633. (PubMed)
18. O'Seaghdha CM, Hwang SJ, Muntner P, Melamed ML, Fox CS. Serum phosphorus predicts incident chronic kidney disease and end-stage renal disease. Nephrol Dial Transplant. 2011;26(9):2885-2890. (PubMed)
19. Foley RN, Collins AJ, Herzog CA, Ishani A, Kalra PA. Serum phosphate and left ventricular hypertrophy in young adults: the coronary artery risk development in young adults study. Kidney Blood Press Res. 2009;32(1):37-44. (PubMed)
20. Shuto E, Taketani Y, Tanaka R, et al. Dietary phosphorus acutely impairs endothelial function. J Am Soc Nephrol. 2009;20(7):1504-1512. (PubMed)
21. Tuttle KR, Short RA. Longitudinal relationships among coronary artery calcification, serum phosphorus, and kidney function. Clin J Am Soc Nephrol. 2009;4(12):1968-1973. (PubMed)
22. de Boer IH, Rue TC, Kestenbaum B. Serum phosphorus concentrations in the third National Health and Nutrition Examination Survey (NHANES III). Am J Kidney Dis. 2009;53(3):399-407. (PubMed)
23. Isakova T, Wahl P, Vargas GS, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79(12):1370-1378. (PubMed)
24. Isakova T, Xie H, Yang W, et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA. 2011;305(23):2432-2439. (PubMed)
25. Palmer SC, Hayen A, Macaskill P, et al. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: a systematic review and meta-analysis. JAMA. 2011;305(11):1119-1127. (PubMed)
26. Li YC. Vitamin D: roles in renal and cardiovascular protection. Curr Opin Nephrol Hypertens. 2012;21(1):72-79. (PubMed)
27. Hruska KA, Mathew S, Lund R, Qiu P, Pratt R. Hyperphosphatemia of chronic kidney disease. Kidney Int. 2008;74(2):148-157. (PubMed)
28. Jono S, McKee MD, Murry CE, et al. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000;87(7):E10-17. (PubMed)
29. Rennenberg RJ, Kessels AG, Schurgers LJ, van Engelshoven JM, de Leeuw PW, Kroon AA. Vascular calcifications as a marker of increased cardiovascular risk: a meta-analysis. Vasc Health Risk Manag. 2009;5(1):185-197. (PubMed)
30. Chang AR, Lazo M, Appel LJ, Gutierrez OM, Grams ME. High dietary phosphorus intake is associated with all-cause mortality: results from NHANES III. Am J Clin Nutr. 2014;99(2):320-327. (PubMed)
31. Calvo MS, Park YK. Changing phosphorus content of the US diet: potential for adverse effects on bone. J Nutr. 1996;126(4 Suppl):1168S-1180S. (PubMed)
32. Calvo MS. Dietary considerations to prevent loss of bone and renal function. Nutrition. 2000;16(7-8):564-566. (PubMed)
33. Kemi VE, Karkkainen MU, Rita HJ, Laaksonen MM, Outila TA, Lamberg-Allardt CJ. Low calcium:phosphorus ratio in habitual diets affects serum parathyroid hormone concentration and calcium metabolism in healthy women with adequate calcium intake. Br J Nutr. 2010;103(4):561-568. (PubMed)
34. Grimm M, Muller A, Hein G, Funfstuck R, Jahreis G. High phosphorus intake only slightly affects serum minerals, urinary pyridinium crosslinks and renal function in young women. Eur J Clin Nutr. 2001;55(3):153-161. (PubMed)
35. Pinheiro MM, Schuch NJ, Genaro PS, Ciconelli RM, Ferraz MB, Martini LA. Nutrient intakes related to osteoporotic fractures in men and women--the Brazilian Osteoporosis Study (BRAZOS). Nutr J. 2009;8:6. (PubMed)
36. Calvo MS, Tucker KL. Is phosphorus intake that exceeds dietary requirements a risk factor in bone health? Ann N Y Acad Sci. 2013; 1301:29-35. (PubMed)
37. Minerals. Drug Facts and Comparisons. St. Louis: Facts and Comparisons; 2000:27-51.
38. Cervelli MJ, Shaman A, Meade A, Carroll R, McDonald SP. Effect of gastric acid suppression with pantoprazole on the efficacy of calcium carbonate as a phosphate binder in haemodialysis patients. Nephrology (Carlton). 2012;17(5):458-465. (PubMed)
39. Zhang D, Maalouf NM, Adams-Huet B, Moe OW, Sakhaee K. Effects of sex and postmenopausal estrogen use on serum phosphorus levels: a cross-sectional study of the National Health and Nutrition Examination Survey (NHANES) 2003-2006. Am J Kidney Dis. 2014;63(2):198-205. (PubMed)
40. Bansal N, Katz R, de Boer IH, et al. Influence of estrogen therapy on calcium, phosphorus, and other regulatory hormones in postmenopausal women: the MESA study. J Clin Endocrinol Metab. 2013;98(12):4890-4898. (PubMed)
Potassium
Contents
Summary
- Potassium is considered to be a "nutrient of public health concern" according to the 2015-2020 Dietary Guidelines for Americans since its underconsumption in the US population is associated with adverse health effects (hypertension and cardiovascular disease). (More information)
- Normal body function depends on tight regulation of potassium concentrations both inside and outside of cells. (More information)
- Low potassium concentration in blood (hypokalemia) can result in muscular paralysis or abnormal heart rhythms and can be fatal. Hypokalemia is usually due to excessive loss of potassium as with prolonged vomiting or diarrhea, use of diuretics, or with kidney disease. (More information)
- Chronic hypertension damages the heart, blood vessels, and kidneys, thereby increasing the risk of cardiovascular disease. Increasing dietary potassium intake may help lower blood pressure in normotensive and hypertensive individuals. (More information)
- Results from observational studies reported higher dietary potassium intakes to be associated with lower risks of stroke and kidney stone formation. Evidence of a role for potassium intakes in promoting bone health remains weak. (More information)
- The adequate intake (AI) for potassium is 2,600 mg/day for women and 3,400 mg/day for men. The AI for each age/life stage group was set based on the level of intake reported in apparently healthy populations. (More information)
- Good dietary sources of potassium include fruit and vegetables, some nuts and seeds, and dairy products. (More information)
- Safety concerns with consuming potassium are limited in healthy people because the kidneys adjust urinary potassium excretion to potassium intake. Concomitant use of potassium supplements with certain drugs can increase the risk of potassium toxicity. (More information)
Potassium is an essential dietary mineral and electrolyte. The term electrolyte refers to a substance that dissociates into ions (charged particles) in solution, making it capable of conducting electricity. Normal body function depends on tight regulation of potassium concentrations both inside and outside of cells (1).
Function
Maintenance of membrane potential
Potassium (K+) is the principal positively charged ion (cation) in the fluid inside of cells, while sodium (Na+) is the principal cation in the extracellular fluid. Potassium concentrations are about 30 times higher inside than outside cells, while sodium concentrations are more than 10 times lower inside than outside cells. The concentration differences between potassium and sodium across cell membranes create an electrochemical gradient known as the membrane potential. A cell's membrane potential is maintained by ion pumps in the cell membrane, especially the Na+/K+-ATPase pumps. These pumps use ATP (energy) to pump sodium out of the cell in exchange for potassium (Figure 1). Their activity has been estimated to account for 20%-40% of the resting energy expenditure in a typical adult. The large proportion of energy dedicated to maintaining sodium/potassium concentration gradients emphasizes the importance of this function in sustaining life. Tight control of cell membrane potential is critical for nerve impulse transmission, muscle contraction, and heart function (2-4).
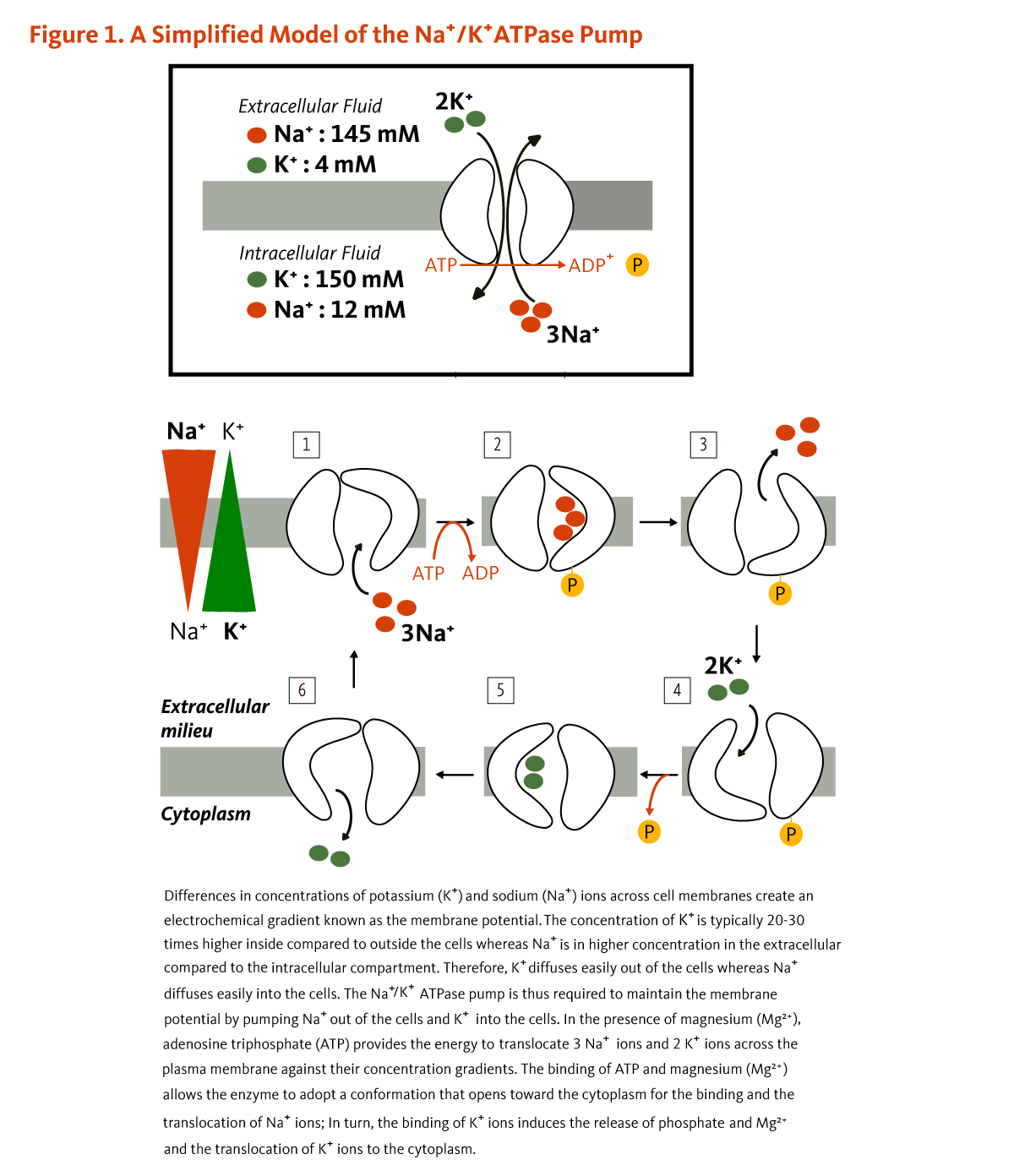
Cofactor for enzymes
A limited number of enzymes require the presence of potassium for their activity. The activation of Na+/K+-ATPase requires the presence of sodium and potassium. The presence of potassium is also required for the activity of pyruvate kinase, an important enzyme in carbohydrate metabolism (5).
Deficiency
An abnormally low plasma potassium concentration is referred to as hypokalemia. Hypokalemia is most commonly a result of excessive loss of potassium, e.g., from prolonged vomiting or diarrhea, use of some diuretics and other medications (see Drug interactions), some forms of kidney disease, or metabolic disturbances. The symptoms of hypokalemia are related to alterations in membrane potential and cellular metabolism (1). They include fatigue, muscle weakness and cramps, and intestinal paralysis, which may lead to bloating, constipation, and abdominal pain. Chronic hypokalemia is associated with hypertension and kidney stone formation (see Disease Prevention and Disease Treatment). Severe hypokalemia may result in muscular paralysis or abnormal heart rhythms (cardiac arrhythmias) that can be fatal (1, 6).
Conditions that increase the risk of hypokalemia (see also Drug interactions; 1):
- The use of potassium-wasting diuretics (e.g., thiazide diuretics or furosemide)
- Prolonged vomiting or diarrhea
- Overuse or abuse of laxatives
- Anorexia nervosa or bulimia
- Excessive sweating
- Nephropathies
- Polyuria
- Abnormally high production of aldosterone (hyperaldosteronism)
- Magnesium depletion
- Recovery from prolonged undernutrition
Low dietary potassium intake alone does not generally result in hypokalemia. However, insufficient dietary potassium in patients at risk of hypokalemia can precipitate hypokalemia (1).
In rare cases, habitual consumption of large amounts of black licorice has resulted in hypokalemia (7, 8). Licorice contains a compound (i.e., glycyrrhizic acid) with similar physiologic effects to those of aldosterone, a hormone that increases urinary excretion of potassium.
The Adequate Intake (AI)
The Dietary Reference Intakes (DRIs) for potassium have been recently revised by the Food and Nutrition Board (FNB) of the National Academy of Medicine. The FNB did not find sufficient evidence to determine an Estimated Average Requirement (EAR) and derive a Recommended Dietary Allowance (RDA); instead, they established an adequate intake (AI) based on median intakes in generally healthy people (Table 1) (9). The FNB found insufficient evidence from human studies that examined potassium intakes in relation to chronic disease and mortality (reviewed recently by the Agency for Healthcare Research and Quality; 10) to inform the DRIs for potassium (11).
Disease Prevention
The diets of people residing in Western industrialized countries are quite different from those that were consumed before the agricultural revolution and the shift towards the consumption of highly refined, processed food (12). Among other differences, the daily intake of sodium chloride (salt) in modern diets is about three times higher than the daily intake of potassium on a molar basis, whereas salt intake in primitive cultures is about seven times lower than potassium intake (13). The relative deficiency of dietary potassium in the modern diet and a higher sodium-to-potassium ratio may contribute to the development of some chronic diseases.
Stroke
Observational studies have consistently reported an increased risk of cardiovascular disease with elevated dietary sodium intakes (14, 15). Several prospective cohort studies have also found an inverse association between potassium intake and risk of stroke. A meta-analysis of nine prospective cohort studies showed that daily potassium intakes ranging between 3,510 mg and 4,680 mg were associated with a 30% reduced risk of stroke (16). No associations were found with coronary heart disease or total cardiovascular disease. In a more recent meta-analysis of 16 studies, the highest versus lowest dietary potassium intake was found to be associated with a 13% lower risk of stroke after multiple adjustments (including for blood pressure) (17). The lowest risk of stroke corresponded to daily potassium intakes around 3,500 mg. Subgroup analyses showed a reduced risk of ischemic stroke, but not hemorrhagic stroke. Finally, in a recent meta-analysis of 16 observational studies, each 1-unit increase in the dietary sodium-to-potassium ratio was found to be associated with a 22% higher risk of stroke (12).
Kidney stones
Abnormally high urinary calcium (hypercalciuria) increases the risk of developing kidney stones. In individuals with a history of developing calcium-containing kidney stones, increased dietary acid load has been significantly associated with increased urinary calcium excretion (18). Increasing dietary potassium (and alkali) intake by increasing fruit and vegetable intake or by taking potassium bicarbonate (KHCO3) supplements has been found to decrease urinary calcium excretion. Conversely, potassium deprivation has been found to increase urinary calcium excretion (19, 20).
Three large US prospective cohort studies — the Health Professionals Follow-up Study and the Nurses’ Health Studies I and II — which included 193,676 participants, have examined dietary potassium intake and animal protein-to-potassium ratio (a marker of dietary acid load) in the diet in relation to the risk of developing kidney stones (21). In all three cohorts, dietary potassium intake was derived almost entirely from potassium-rich foods, such as fruit and vegetables. Across the three cohorts, individuals in the highest quintile of potassium intake were found to be 33%-56% less likely to develop symptomatic kidney stones than those in the lowest quintile of intake. Additionally, a pooled analysis of the data from all three cohorts showed that those with the highest versus lowest animal protein-to-potassium ratio were 41% more likely to develop kidney stones (21).
Urinary alkalinization with supplemental potassium citrate is used in stone formers to reduce the risk of recurrent stone formation (reviewed in 22). However, potassium citrate therapy should only be initiated under the supervision of a medical provider.
Osteoporosis
In a 2015 case-cohort study nested within the European Prospective Investigation into Cancer and Nutrition (EPIC)-Norfolk study, which included 5,319 individuals, dietary intakes of potassium (alone or combined with intakes of magnesium) were found to be inversely associated with heel bone (calcaneus) broadband ultrasound attenuation (BUA) measurements (a predictor of the risk of incidental fracture) and risk of hip fracture in women but not in men (23). More recently, a cross-sectional study in older Korean adults reported higher total hip and femur neck bone mineral density (BMD) in those in the top versus bottom tertile of potassium intakes (24). Although these observational studies suggest a link between potassium intakes and bone health, they cannot establish whether there is a cause-and-effect relationship.
The mechanisms by which potassium might influence bone health are poorly understood. Modern (Western) diets tend to be relatively low in sources of alkali (fruit and vegetables) and high in sources of acid (fish, meat, and cheese) (25). When the quantity of bicarbonate ions is insufficient to maintain normal pH, the body is capable of mobilizing alkaline calcium salts from bone in order to neutralize acids consumed in the diet or generated by metabolism (26). Because fruit and vegetables are rich in both potassium and precursors to bicarbonate ions, increasing their consumption might help reduce the net acid content of the diet and preserve calcium in bones, which might otherwise be mobilized to maintain normal pH (see the article on Fruit and Vegetables).
Alternatively, potassium bicarbonate supplementation might decrease urinary acid and calcium excretion and influence bone turnover — a small trial in postmenopausal women found that potassium bicarbonate supplementation increases biomarkers of bone formation and a decreased biomarkers of bone resorption (27). A two-year randomized, double-blind, controlled trial in 201 older adults without osteoporosis (mean age, 69 years) found evidence of increased lumbar spine, hip, femoral neck, and total-body BMD, as well as trabecular BMD of the radius and tibia, with supplemental potassium citrate (2,340 mg/day) compared to placebo (28). Potassium citrate was also found to increase the serum concentration of N-terminal propeptide of type I procollagen (PINP) — a marker of bone formation — and reduce the urine concentration of N-telopeptide of collagen type I (NTX) — a marker of bone resorption (28). Another three-month, randomized, placebo-controlled trial in 244 adults (≥50 years) examined the effect of oral potassium bicarbonate, at either 39 mg/kg/day or 58.5 mg/kg/day, on markers of bone turnover (29). Both dosage regimens led to reductions in serum PINP concentration and urine NTX concentration, yet evidence of an effect was stronger with the lowest dose (median dose administered, 3,160 mg/day) rather than with the highest dose (median dose administered, 4,760 mg/day). In contrast, a two-year randomized controlled trial found that neither supplementation with potassium citrate (721 mg/day or 2,165 mg/day) nor an increase in fruit and vegetable intake (300 mg/day) had an impact on markers of bone turnover or increased BMD in postmenopausal women (30). A 2015 meta-analysis of intervention studies found that supplemental potassium citrate or potassium bicarbonate could reduce urinary net acid and calcium excretion, but evidence to support an effect on markers of bone turnover and bone density was weak (31). The most recent randomized, double-blind, controlled trial in 40 postmenopausal women with osteopenia found no difference in markers of bone turnover over a six-month period between those supplemented with potassium citrate and those taking a placebo (32). The authors highlighted the possibility that an effect of supplemental potassium might have an impact on bone health in a subset of subjects with low potassium intakes and/or signs of low-grade acidosis. In this study, all participants received daily supplements of calcium carbonate (500 mg/day) and vitamin D (10 µg/day).
Overall, whether consuming potassium-rich fruit and vegetables can influence bone health and help lower the risk of osteoporosis remains uncertain (see also the article on Fruit and Vegetables).
Disease Treatment
Hypertension
Forty-five percent of US adults have hypertension (blood pressure levels ≥130/80 mm Hg) (33). Chronic hypertension damages the heart, blood vessels, and kidneys, thereby increasing the risk of heart disease and stroke, as well as hypertensive kidney disease (34, 35). Modern diets, which are high in sodium and low in potassium, are recognized as largely contributing to the high prevalence of hypertension (see the article on Sodium). Unlike 24-hour dietary recalls, 24-hour urine collections provide accurate estimates of dietary intakes of sodium and potassium (36). An analysis of the 2014 US National Health and Nutrition Examination Survey (NHANES) showed an increase in systolic blood pressure with increasing sodium excretion and increasing sodium-to-potassium ratio in the urine (37). In this study, the highest versus lowest quartile of urinary potassium excretion (mid-values, 3,043 mg/day versus 1,484 mg/day) was associated with a 62% lower risk of hypertension (37).
The Dietary Approaches to Stop Hypertension (DASH) trial provided evidence of the blood pressure-lowering effect of a diet higher in potassium and calcium, modestly higher in protein, and lower in total fat, saturated fat, cholesterol, red meat, sweets, and sugar-containing beverages compared to the typical US diet (38). Indeed, compared to the control diet providing only 3.5 servings/day of fruit and vegetables and 1,700 mg/day of potassium, adherence to the DASH diet that included 8.5 servings/day of fruit and vegetables and 4,100 mg/day of potassium lowered systolic/diastolic blood pressures by an average 11.4/5.5 mm Hg in people with hypertension and 3.5/2.1 mm Hg in those without hypertension (38). A 2014 meta-analysis of 17 randomized controlled trials that examined the effect of the DASH diet compared to a control diet in a total of 2,561 adults found overall reductions in systolic and diastolic blood pressure by 6.7 mm Hg and 3.5 mm Hg, respectively (39). However effective the DASH diet is, the blood pressure-lowering effects can hardly be solely attributed to potassium intakes (40).
A 2015 meta-analysis of 15 randomized controlled trials, including 917 individuals, assessed the effects of increased potassium intake, mostly in the form of potassium chloride (KCl) supplements, on blood pressure (41). Thirteen studies included hypertensive participants who were not taking anti-hypertensive medication and two studies included normotensive or at-risk subjects. Most studies used supplemental potassium doses between 2,340 and 2,535 mg/day (60-65 mmol/L). Increased potassium intake resulted in overall reductions of systolic blood pressure by 4.7 mm Hg and diastolic blood pressure by 3.5 mm Hg. The blood pressure-lowering effect of supplemental potassium was more pronounced when the analysis was restricted to individuals with hypertension: systolic and diastolic blood pressure were found to be reduced by 6.8 mm Hg and 4.6 mm Hg, respectively (41). Two additional meta-analyses published in 2017 also confirmed a blood pressure-lowering effect of supplemental potassium. Findings suggested some evidence of a greater effect when baseline potassium intake was less than 3,510 mg/day (vs. ≥3,510 mg/day [90 mmol]) (42). Meta-analyses have also reported a dose-response relationship between the intake of potassium and the lowering of blood pressure (42, 43).
Supplemental potassium can help lower blood pressure, but potassium supplements should only be used in consultation with a medical provider (see Supplements). Increasing potassium intake to recommended levels (see Adequate Intake) by consuming a diet rich in fruit and vegetables can help lower blood pressure and may have additional benefits to health (see the article on Fruit and Vegetables). Blood pressure is a reliable cardiovascular risk marker (44). Yet, although reducing sodium consumption while increasing potassium intake helps with lowering blood pressure (45), current evidence suggests that dietary advice and support interventions may not be sufficient to deliver long-term cardiovascular benefits in individuals with hypertension (46).
Sources
Food sources
The richest sources of potassium are fruit and vegetables. Nuts, seeds, and dairy products are also good sources of potassium. A dietary survey in the US indicated that the average dietary potassium intake was 2,408 mg/day for adult women and 3,172 mg/day for adult men (47). Because many individuals in the population consume potassium in amounts that are well below the AI and because underconsumption of potassium is linked with adverse health effects, potassium has been recognized as a "nutrient of public health concern" in the 2015-2020 Dietary Guidelines for Americans. In 2016, the US Food and Drug Administration (FDA) required manufacturers to display potassium content of foods on the Nutrition Facts food label (48).
Some relatively good dietary sources of potassium are listed in the Table 2, along with their potassium content in milligrams (mg). For more information on the nutrient content of foods, search USDA's FoodData Central (49).
Supplements
Multivitamin-mineral supplements in the US do not contain more than 99 mg of potassium per serving (50). One milliequivalent (mEq) or one millimole (mmol) corresponds to about 39 mg of potassium. Higher doses of supplemental potassium are generally prescribed to prevent and treat potassium depletion and hypokalemia. The use of more potent potassium supplements in potassium deficiency requires close monitoring of serum potassium concentrations. Potassium is available in different supplemental forms, including potassium chloride, potassium citrate, potassium gluconate, potassium bicarbonate, potassium aspartate, and potassium orotate (50). Because of the potential for serious side effects, one should seek medical advice before deciding to use a potassium supplement (see Safety). The best way to increase one’s potassium intake is by increasing the consumption of potassium-rich food and beverages (50).
Finally, many salt substitutes contain potassium chloride, and acesulfame potassium (Ace-K) is an FDA-approved general purpose sweetener.
Safety
Toxicity
Abnormally elevated serum potassium concentrations are referred to as hyperkalemia. Hyperkalemia occurs when potassium intake exceeds the capacity of the kidneys to eliminate it. Acute or chronic kidney failure, the use of potassium-sparing diuretics, and insufficient aldosterone secretion (hypoaldosteronism) may result in the accumulation of potassium due to a decreased urinary potassium excretion. Oral doses of potassium >18 g taken at one time in individuals not accustomed to high intakes may lead to severe hyperkalemia, even in those with normal kidney function (6, 50). Hyperkalemia may also result from a shift of intracellular potassium into the circulation, which may occur with the rupture of red blood cells (hemolysis) or tissue damage (e.g., trauma or severe burns). Symptoms of hyperkalemia may include tingling of the hands and feet, muscular weakness, and temporary paralysis. The most serious complication of hyperkalemia is the development of an abnormal heart rhythm (cardiac arrhythmia), which can lead to cardiac arrest (51). A meta-analysis of randomized controlled studies showed that heart rate in healthy adults was unlikely to be affected by the chronic use of supplemental potassium doses of 2 to 3 g/day (52).
See the section on Drug interactions for a discussion of the medications that increase the risk of hyperkalemia.
Adverse reactions to potassium supplements
Gastrointestinal symptoms are the most common side effects of potassium supplements, including nausea, vomiting, abdominal discomfort, and diarrhea. Intestinal ulceration has been reported after the use of enteric-coated potassium chloride tablets. Taking potassium with meals or taking a microencapsulated form of potassium may reduce gastrointestinal side effects (50). Rashes may occasionally occur. The most serious adverse reaction to potassium supplementation is hyperkalemia, yet is rare in subjects with normal kidney function (see Toxicity). Individuals with abnormal kidney function and those on potassium-sparing medications (see Drug interactions) should be monitored closely to prevent hyperkalemia (50, 53).
Drug interactions
Table 3 lists the classes of medications known to increase the risk of hyperkalemia (elevated serum potassium) in patients who also use potassium supplements (50, 51, 54).
Several classes of medications are known to induce hypokalemia (low serum potassium; Table 4; 55) . In the absence of treatment, hypokalemia can have serious complications and even be fatal (see Deficiency). Various mechanisms explain how certain medications can lead to potassium depletion. For example, both loop and thiazide diuretics increase the urinary excretion of potassium. Corticoids cause sodium retention that leads to a compensatory increase in urinary potassium excretion. Penicillins formulated as sodium salts also stimulate potassium excretion. Several medications, including aminoglycosides, anti-fungal agents (amphotericin-B, fluconazole), and cisplatin, can damage the renal tubular epithelium and lead to severe potassium loss. Outdated tetracycline antibiotics have been linked to electrolyte disturbances.
Linus Pauling Institute Recommendation
There is substantial evidence suggesting that a diet high in potassium-rich food and beverages may be associated with lower risks of stroke, hypertension, kidney stones, and possibly osteoporosis. However, currently there is insufficient evidence to establish a causal relationship between potassium intakes and the risk of these chronic conditions (10). As a consequence, median potassium intakes observed in apparently healthy people were used to set adequate intakes (AI) by age/life stage in the recent revision of the Dietary Reference Intakes (DRIs) for potassium. The revised AI values are 2.6 g/day for women and 3.4 g/day for men (see The Adequate Intake).
Fruit and vegetables are among the richest sources of dietary potassium, and a large body of evidence supports the association of increased fruit and vegetable intakes with reduced risk of cardiovascular disease (see the article on Fruit and Vegetables). The Linus Pauling Institute recommends the consumption of a diet high in potassium-rich foods (see Sources), especially fruit, vegetables, nuts, and dairy products to ensure adequate potassium intakes.
Older adults (>50 years)
A diet rich in fruit and vegetables that supplies 2.6-3.4 g/day of potassium (see AI) should contribute to maintaining a low risk of chronic disease in generally healthy older adults. This recommendation does not apply to individuals who have been advised to limit potassium consumption by a health care professional (see Safety).
Authors and Reviewers
Originally written in 2001 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in February 2004 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in December 2010 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in April 2019 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in April 2019 by
Connie Weaver, Ph.D.
Distinguished Professor and Department Head
Department of Nutrition Science
Purdue University
Copyright 2001-2024 Linus Pauling Institute
References
1. Bailey JL, Sands JM, Franch HA. Water, electrolytes, and acid — Base Metabolism In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease: Lippincott Williams & Wilkins; 2014:102-132.
2. Clausen T. Quantification of Na+,K+ pumps and their transport rate in skeletal muscle: functional significance. J Gen Physiol. 2013;142(4):327-345. (PubMed)
3. Larsen BR, Stoica A, MacAulay N. Managing brain extracellular K(+) during neuronal activity: the physiological role of the Na(+)/K(+)-ATPase subunit isoforms. Front Physiol. 2016;7:141. (PubMed)
4. Shattock MJ, Ottolia M, Bers DM, et al. Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J Physiol. 2015;593(6):1361-1382. (PubMed)
5. Sheng H-W. Sodium, chloride and potassium. In: Stipanuk M, ed. Biochemical and Physiological Aspects of Human Nutrition. Philadelphia: W.B. Saunders Company; 2000:686-710.
6. Food and Nutrition Board, Institute of Medicine. Potassium. Dietary Reference Intakes for Water, Potassium, Sodium, Chloride, and Sulfate. Washington, D.C.: National Academies Press; 2005:186-268. (The National Academies Press)
7. Mumoli N, Cei M. Licorice-induced hypokalemia. Int J Cardiol. 2008;124(3):e42-44. (PubMed)
8. Walker BR, Edwards CR. Licorice-induced hypertension and syndromes of apparent mineralocorticoid excess. Endocrinol Metab Clin North Am. 1994;23(2):359-377. (PubMed)
9. Food and Nutrition Board, National Academy of Medicine. Dietary Reference Intakes for Sodium and Potassium - uncorrected proofs. The National Academies of Sciences, Engineering, and Medicine. Washington, D.C.: The National Academies Press; 2019. (The National Academies Press)
10. Newberry SJ, Chung M, Anderson CAM, et al. AHRQ Comparative Effectiveness Reviews. Sodium and potassium intake: effects on chronic disease outcomes and risks. Rockville (MD): Agency for Healthcare Research and Quality (US); 2018. (PubMed)
11. Food and Nutrition Board, National Academy of Medicine. Potassium: Dietary Reference Intakes based on chronic disease. Dietary Reference Intakes for Sodium and Potassium - uncorrected proofs. The National Academies of Sciences, Engineering, and Medicine. Washington, D.C.: National Academy Press; 2019:121-154. (The National Academies Press)
12. Weaver CM. Potassium and health. Adv Nutr. 2013;4(3):368s-377s. (PubMed)
13. Young DB, Lin H, McCabe RD. Potassium's cardiovascular protective mechanisms. Am J Physiol. 1995;268(4 Pt 2):R825-837. (PubMed)
14. Aburto NJ, Ziolkovska A, Hooper L, Elliott P, Cappuccio FP, Meerpohl JJ. Effect of lower sodium intake on health: systematic review and meta-analyses. BMJ. 2013;346:f1326. (PubMed)
15. Jayedi A, Ghomashi F, Zargar MS, Shab-Bidar S. Dietary sodium, sodium-to-potassium ratio, and risk of stroke: A systematic review and nonlinear dose-response meta-analysis. Clin Nutr. 2018; doi: 10.1016/j.clnu.2018.05.017. [Epub ahead of print]. (PubMed)
16. Aburto NJ, Hanson S, Gutierrez H, Hooper L, Elliott P, Cappuccio FP. Effect of increased potassium intake on cardiovascular risk factors and disease: systematic review and meta-analyses. BMJ. 2013;346:f1378. (PubMed)
17. Vinceti M, Filippini T, Crippa A, de Sesmaisons A, Wise LA, Orsini N. Meta-analysis of potassium intake and the risk of stroke. J Am Heart Assoc. 2016;5(10). (PubMed)
18. Trinchieri A, Zanetti G, Curro A, Lizzano R. Effect of potential renal acid load of foods on calcium metabolism of renal calcium stone formers. Eur Urol. 2001;39 Suppl 2:33-36; discussion 36-37. (PubMed)
19. Lemann J, Jr., Pleuss JA, Gray RW. Potassium causes calcium retention in healthy adults. J Nutr. 1993;123(9):1623-1626. (PubMed)
20. Morris RC, Jr., Schmidlin O, Tanaka M, Forman A, Frassetto L, Sebastian A. Differing effects of supplemental KCl and KHCO3: pathophysiological and clinical implications. Semin Nephrol. 1999;19(5):487-493. (PubMed)
21. Ferraro PM, Mandel EI, Curhan GC, Gambaro G, Taylor EN. Dietary protein and potassium, diet-dependent net acid load, and risk of incident kidney stones. Clin J Am Soc Nephrol. 2016;11(10):1834-1844. (PubMed)
22. Suarez M, Youssef RF. Potassium citrate: treatment and prevention of recurrent calcium nephrolithiasis. J Clin Nephrol Res. 2015;2(1).
23. Hayhoe RP, Lentjes MA, Luben RN, Khaw KT, Welch AA. Dietary magnesium and potassium intakes and circulating magnesium are associated with heel bone ultrasound attenuation and osteoporotic fracture risk in the EPIC-Norfolk cohort study. Am J Clin Nutr. 2015;102(2):376-384. (PubMed)
24. Kong SH, Kim JH, Hong AR, Lee JH, Kim SW, Shin CS. Dietary potassium intake is beneficial to bone health in a low calcium intake population: the Korean National Health and Nutrition Examination Survey (KNHANES) (2008-2011). Osteoporos Int. 2017;28(5):1577-1585. (PubMed)
25. Fenton TR, Eliasziw M, Lyon AW, Tough SC, Hanley DA. Meta-analysis of the quantity of calcium excretion associated with the net acid excretion of the modern diet under the acid-ash diet hypothesis. Am J Clin Nutr. 2008;88(4):1159-1166. (PubMed)
26. Morris RC, Jr., Frassetto LA, Schmidlin O, Forman A, Sabastian A. Expression of osteoporosis as determined by diet-disordered electrolyte and acid-base metabolism. In: Burckhardt P, Dawson-Hughes B, Heaney R, eds. Nutritional Aspects of Osteoporosis. San Diego: Academic Press; 2001:357-378.
27. Sebastian A, Harris ST, Ottaway JH, Todd KM, Morris RC, Jr. Improved mineral balance and skeletal metabolism in postmenopausal women treated with potassium bicarbonate. N Engl J Med. 1994;330(25):1776-1781. (PubMed)
28. Jehle S, Hulter HN, Krapf R. Effect of potassium citrate on bone density, microarchitecture, and fracture risk in healthy older adults without osteoporosis: a randomized controlled trial. J Clin Endocrinol Metab. 2013;98(1):207-217. (PubMed)
29. Dawson-Hughes B, Harris SS, Palermo NJ, et al. Potassium bicarbonate supplementation lowers bone turnover and calcium excretion in older men and women: a randomized dose-finding trial. J Bone Miner Res. 2015;30(11):2103-2111. (PubMed)
30. Macdonald HM, Black AJ, Aucott L, et al. Effect of potassium citrate supplementation or increased fruit and vegetable intake on bone metabolism in healthy postmenopausal women: a randomized controlled trial. Am J Clin Nutr. 2008;88(2):465-474. (PubMed)
31. Lambert H, Frassetto L, Moore JB, et al. The effect of supplementation with alkaline potassium salts on bone metabolism: a meta-analysis. Osteoporos Int. 2015;26(4):1311-1318. (PubMed)
32. Granchi D, Caudarella R, Ripamonti C, et al. Potassium citrate supplementation decreases the biochemical markers of bone loss in a group of osteopenic women: the results of a randomized, double-blind, placebo-controlled pilot study. Nutrients. 2018;10(9). (PubMed)
33. Centers for Disease Control and Prevention. High Blood Pressure Facts. November 2016. Available at: https://www.cdc.gov/bloodpressure/facts.htm. Accessed 11/30/18.
34. Mente A, O'Donnell M, Rangarajan S, et al. Associations of urinary sodium excretion with cardiovascular events in individuals with and without hypertension: a pooled analysis of data from four studies. Lancet. 2016;388(10043):465-475. (PubMed)
35. Sanghavi S, Vassalotti JA. Dietary sodium: a therapeutic target in the treatment of hypertension and CKD. J Ren Nutr. 2013;23(3):223-227. (PubMed)
36. Cogswell ME, Loria CM, Terry AL, et al. Estimated 24-hour urinary sodium and potassium excretion in US adults. JAMA. 2018;319(12):1209-1220. (PubMed)
37. Jackson SL, Cogswell ME, Zhao L, et al. Association between urinary sodium and potassium excretion and blood pressure among adults in the United States: National Health and Nutrition Examination Survey, 2014. Circulation. 2018;137(3):237-246. (PubMed)
38. Appel LJ, Moore TJ, Obarzanek E, et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N Engl J Med. 1997;336(16):1117-1124. (PubMed)
39. Saneei P, Salehi-Abargouei A, Esmaillzadeh A, Azadbakht L. Influence of Dietary Approaches to Stop Hypertension (DASH) diet on blood pressure: a systematic review and meta-analysis on randomized controlled trials. Nutr Metab Cardiovasc Dis. 2014;24(12):1253-1261. (PubMed)
40. Weaver CM, Stone MS, Lobene AJ, Cladis DP, Hodges JK. What is the evidence base for a potassium requirement? Nutr Today. 2018;53(5):184-195. (PubMed)
41. Binia A, Jaeger J, Hu Y, Singh A, Zimmermann D. Daily potassium intake and sodium-to-potassium ratio in the reduction of blood pressure: a meta-analysis of randomized controlled trials. J Hypertens. 2015;33(8):1509-1520. (PubMed)
42. Filippini T, Violi F, D'Amico R, Vinceti M. The effect of potassium supplementation on blood pressure in hypertensive subjects: A systematic review and meta-analysis. Int J Cardiol. 2017;230:127-135. (PubMed)
43. Poorolajal J, Zeraati F, Soltanian AR, Sheikh V, Hooshmand E, Maleki A. Oral potassium supplementation for management of essential hypertension: A meta-analysis of randomized controlled trials. PLoS One. 2017;12(4):e0174967. (PubMed)
44. Viera AJ. Screening for hypertension and lowering blood pressure for prevention of cardiovascular disease events. Med Clin North Am. 2017;101(4):701-712. (PubMed)
45. Appel LJ, Giles TD, Black HR, et al. ASH position paper: dietary approaches to lower blood pressure. J Am Soc Hypertens. 2010;4(2):79-89. (PubMed)
46. Adler AJ, Taylor F, Martin N, Gottlieb S, Taylor RS, Ebrahim S. Reduced dietary salt for the prevention of cardiovascular disease. Cochrane Database Syst Rev. 2014(12):Cd009217. (PubMed)
47. Hoy MK, Goldman JD. Potassium Intake of the US Population: What We Eat In America, NHANES 2009-2010. 2012.
48. Food and Drug Administration. Food Labeling: Revision of the Nutrition and Supplement Facts Labels. Available at: https://www.regulations.gov/document?D=FDA-2012-N-1210-0875. Accessed 12/12/18.
49. US Department of Agriculture, Agricultural Research Service. FoodData Central, 2019. fdc.nal.usda.gov.
50. Hendler SS, Rorvik DR. PDR for Nutrititional Supplements. Montvale: Thomson Reuters; 2008.
51. Mandal AK. Hypokalemia and hyperkalemia. Med Clin North Am. 1997;81(3):611-639. (PubMed)
52. Gijsbers L, Molenberg FJ, Bakker SJ, Geleijnse JM. Potassium supplementation and heart rate: A meta-analysis of randomized controlled trials. Nutr Metab Cardiovasc Dis. 2016;26(8):674-682. (PubMed)
53. Gennari FJ. Hypokalemia. N Engl J Med. 1998;339(7):451-458. (PubMed)
54. Natural Medicines. Potassium/Professional Monograph/Interactions with Drugs. June 26, 2018. Available at: https://naturalmedicines.therapeuticresearch.com. Accessed 11/19/18.
55. Natural Medicines. Potassium/Professional Monograph/Nutrient Depletion. June 26, 2018. Available at: https://naturalmedicines.therapeuticresearch.com. Accessed 11/19/18.
Selenium
Contents
Summary
- Selenium exerts various biological functions mainly as part of the amino acid, selenocysteine, which is found in at least 25 selenocysteine-containing proteins (selenoproteins) in humans. (More information)
-
Five glutathione peroxidases, three thioredoxin reductases, three iodothyronine deiodinases, and one methionine sulfoxide reductase B1 are among the best characterized selenoproteins with known functions. (More information)
-
Impaired antioxidant protection in selenium-deficient individuals may affect physiological responses to stress. Keshan cardiomyopathy and Kashin-Beck osteoarthropathy are diseases occurring specifically in areas of selenium deficiency in Asia. (More information)
-
The current recommended dietary allowance (RDA) set by the US National Academy of Medicine is 55 μg/day for adolescents and most adults. (More information)
-
Overall, early observational studies have found either null or inverse (protective) associations between selenium exposure and risk of site-specific cancers. However, most recent evidence from intervention trials in selenium-replete participants does not support a protective effect of selenium supplementation against cancer. (More information)
-
Preliminary evidence from randomized controlled clinical trials suggests that selenium supplementation may prevent viral load progression and increase immune cell count in HIV-positive patients. (More information)
-
The levels and chemical forms of selenium in plant-based foods vary according to the composition and selenium content of soil in which the plants are grown. Selenium-rich food sources include Brazil nuts (nuts from the Bertholletia excelsa tree), grains, seafood, organ meats, poultry, and dairy products. (More information)
-
The tolerable upper intake level (UL) for selenium is 400 μg/day for adolescents and adults and includes both selenium obtained from food, which averages about 100 μg/day for adults in the US, and selenium from supplements. (More information)
-
Because some evidence suggests that high serum selenium concentrations may have adverse effects on glycemic control, individuals with high selenium status and/or those at risk for type 2 diabetes mellitus should avoid taking selenium supplements. (More information)
Selenium is a trace mineral that is essential for humans in small amounts, but like all essential elements, selenium can be toxic at high levels. Unlike plants, most animals — including humans — require selenium for the appropriate functioning of a number of selenium-dependent enzymes known as selenoproteins. During protein synthesis (translation), the amino acid selenocysteine is incorporated into elongating proteins at very specific locations in the amino acid sequence in order to form functional selenoproteins. Although higher plants do not appear to require selenium for survival, they can incorporate it nonspecifically into sulfur-containing molecules when the mineral is present in the soil (1). Of note, in animals, the amino acid selenomethionine can be nonspecifically incorporated into proteins in place of methionine (2). However, only selenocysteine-containing proteins are regarded as selenoproteins (Figure 1).

Function
Selenoproteins
Twenty-five genes coding for selenoproteins have been identified in humans (3). The insertion of selenocysteine into selenoproteins during translation is directed by the presence of a selenocysteine-insertion sequence (SECIS) within selenoprotein mRNAs. Briefly, the recognition of SECIS by the translational machinery results in the recruitment of specific translational factors that decode in-frame UGA codons by inserting selenocysteine into elongating selenoproteins (4).
Research is gradually uncovering the metabolic functions of all human selenoproteins, including splicing variants (5). Some of the selenoproteins with identified functions are discussed below.
Glutathione peroxidases
Five selenium-containing glutathione peroxidases (GPx1-4 and GPx6) have been identified: GPx1 (cytosolic GPx), GPx2 (epithelial cell-specific GPx first identified in intestinal lining and lungs), GPx3 (highly expressed in thyroid gland and kidneys), GPx4 (phospholipid-hydroperoxide GPx; PHGPx), and GPx6 (expressed in the olfactory epithelium) (6). GPx isoenzymes are all antioxidant enzymes that reduce potentially damaging reactive oxygen species (ROS), such as hydrogen peroxide and lipid hydroperoxides, to harmless products like water and alcohols by coupling their reduction with the oxidation of glutathione (Figure 2a). Spermatogenesis and male fertility are highly dependent on GPx4 and selenoprotein P (SELENOP, formerly SEPP1 or SELP; see below). In the testes, GPx4 reduces phospholipid hydroperoxides, hence protecting immature spermatozoa cells against oxidative stress. GPx4 is also a major structural protein of the capsule embedding mature sperm mitochondrial helix involved in sperm motility. SELENOP is essential for selenium supply to the testes, and animal models lacking the SELENOP gene are infertile due to poor selenium tissue bioavailability, defective GPx4 synthesis, and impaired sperm maturation (7).

Thioredoxin reductases
In mammals, three selenocysteine-containing thioredoxin reductase (TXNRD) isoenzymes have been identified in the thioredoxin system: cytosolic TXNRD1, mitochondrial TXNRD3, and testes-specific thioredoxin glutathione reductase (TXNRD3, also known as TGR). TXNRDs are homodimeric enzymes, and each monomer contains FAD- and NADPH-binding domains and a selenocysteine-containing catalytic site. TXNRDs catalyze the reduction of a wide range of substrates, including thioredoxin and protein disulfide isomerase (PDI) (see Figure 2b). TXNRDs also serve as electron donors for the regeneration of small antioxidants, possibly recycling ascorbic acid (vitamin C), α-lipoic acid, α-tocopherol (vitamin E), and coenzyme Q10 from their oxidized forms (8). The maintenance of thioredoxin in a reduced form by TXNRDs is important for regulating cell growth and survival. The protein thioredoxin, together with TXNRD1 (or TXNRD3), NADPH, and FAD, constitute the thioredoxin antioxidant system involved in the reduction of antioxidant enzymes (e.g., peroxiredoxins, methionine sulfoxide reductases, and ribonucleotide reductase) and of many oxidation/reduction (redox)-sensitive signaling proteins (9). TXNRD1 is one of the most investigated selenoproteins and regarded as one of the major antioxidant enzymes and redox regulators in mammalian cells.
Iodothyronine deiodinases (thyroid hormone deiodinases)
The thyroid gland releases very small amounts of biologically active thyroid hormone (triiodothyronine or T3) and larger amounts of an inactive form of thyroid hormone (T3 precursor: thyroxine or T4) into the circulation. Most of the biologically active T3 in the circulation and inside cells is generated by the removal of one iodine atom from T4 in a reaction catalyzed by selenium-dependent iodothyronine deiodinase enzymes. Two different selenium-dependent iodothyronine deiodinases (DIOs type 1 and 2) can deiodinate T4, thus increasing circulating T3, while a third iodothyronine deiodinase (DIO type 3) can convert both T3 and T4 to inactive metabolites (Figure 3) (10, 11). Of note, inactivation of the genes encoding DIOs in rodent models has revealed a role for DIO type 1 in iodine homeostasis and the importance of DIOs type 2 and 3 in the maturation of auditory and visual systems during fetal development (10). Thus the importance of selenium in normal development, growth, and metabolism is not limited to its role in the regulation of thyroid gland function.

Selenoprotein P
Selenoprotein P (SELENOP, formerly SEPP1 or SELP) is predominantly produced by the liver, a major storage site for selenium, and secreted in the plasma. The full-length glycoprotein contains a selenium-rich domain with nine selenocysteine residues, as well as a thioredoxin-like catalytic site with one selenocysteine residue. SELENOP constitutes the major form of selenium transport to peripheral tissues (12, 13). SELENOP also functions as an antioxidant that protects cells from oxidative damage by enabling full activity of thioredoxin reductases and glutathione peroxidases through adequate supply of selenium to extrahepatic tissues (see Glutathione peroxidases). SELENOP appears to be especially critical for selenium homeostasis in the brain and testes where apolipoprotein E receptor 2 (apoER2) facilitates the uptake of SELENOP. Megalin is another SELENOP-specific lipoprotein receptor that helps limit urinary selenium loss through SEPP1 reuptake by the kidneys (3, 14). SEPP1 has been implicated in the regulation of glucose metabolism and insulin sensitivity (15). Moreover, low plasma concentrations of SELENOP have been associated with increased risk of heart failure in a recent population-based, prospective cohort study (16).
Selenoprotein W
Selenoprotein W (SELENOW, formerly SEPW or SelW) exists in different isoforms (homologues) and is highly conserved across species. In humans, SELENOW is expressed in numerous tissues, with highest levels found in skeletal muscle and heart (17). SELENOW contains a selenocysteine residue and a cysteine residue that binds to a glutathione molecule, suggesting a role in redox regulation (18). The expression of SELENOW is correlated with selenium status and appears to be sensitive to low-selenium supply (19, 20). SELENOW expression in the brain of mice has been found to confer protection against oxidative stress-induced neuronal cell death (21). SELENOW also appears to be a negative regulator for 14-3-3 proteins. Indeed, 14-3-3 inhibition by SELENOW in breast cancer cells was found to increase cell proliferation and cell survival through increasing resistance to genotoxic stress in human breast and lung cancer cells (22). In skeletal muscle cells, SELENOW was shown to reduce the binding of 14-3-3 to a transcriptional co-activator with PDZ-binding motif (TAZ) (23), allowing TAZ translocation to the nucleus and subsequent activation of muscle cell differentiation genes (24). Finally, SELENOW was found to prevent the degradation of the epidermal growth factor receptor (EGFR) in breast and prostate epithelial cells in culture. EGFR is constitutively activated in many tumors, and evidence of a role for SELENOW in EGFR activation and signaling may help shed light on the relationship between selenium status and cancer risk (25).
Selenophosphate synthetase 2
There is no free pool of the amino acid selenocysteine in cells such that selenocysteine synthesis takes place on a specialized tRNA immediately preceding the translation of selenoprotein mRNAs. The reaction is catalyzed by pyridoxal 5’-phosphate (PLP)-dependent L-seryl-tRNASec selenium transferase and uses selenophosphate (monoselenium phosphate) as the selenium donor (Figure 4) (26). Selenophosphate synthetase 2 is the selenoenzyme that catalyzes the ATP-dependent synthesis of selenophosphate from hydrogen selenide (Figure 4) (5).

Methionine-R-sulfoxide reductase B1 (formerly selenoprotein R)
The methionine sulfoxide reduction system is involved in the protection against oxidative stress and is especially critical for the regeneration of proteins damaged by ROS. Indeed, ROS can oxidize methionine residues (methionine sulfoxides) within proteins and potentially impair their activities. In humans, two stereospecific families of methionine sulfoxide reductases (MsrA and MsrB) are encoded by a single MSRA gene and three MSRB genes (MSRB1-3). MsrA catalyzes the reduction of the S-form of methionine sulfoxide; the R-form of methionine sulfoxide is reduced by MsrB1, 2, or 3. Only MsrB1 has been characterized as a selenoprotein with one selenocysteine residue in its catalytic site. MsrB1 appears to be involved in the redox regulation of certain proteins. In macrophages, reorganization of the actin cytoskeleton necessary for chemotaxis and phagocytosis requires MsrB1-dependent reduction of methionine-R-sulfoxide residues within actin (27). Studies using MSR gene inactivation in mice have also shown that methionine sulfoxide reduction is implicated in the regulation of the methionine cycle (reviewed in 28). Both the thioredoxin (Trx) and GSH-dependent glutaredoxin (Grx) antioxidant systems have been found to reduce methionine sulfoxide reductases in vitro and/or in vivo (see Figure 2) (28).
15 kDa selenoprotein
The 15 kDa selenoprotein SELENOF (formerly selenoprotein 15; SEP15) is highly expressed in several tissues, including prostate, kidney, testes, liver, and brain (29). Although its function continues to be elucidated, SELENOF was found to interact with the endoplasmic reticulum UDP-glucose:glycoprotein glucosyltransferase (UGGT), an enzyme involved in the quality control of glycoprotein folding in the endoplasmic reticulum (30, 31). Because SELENOF has a thioredoxin-like catalytic site, SELENOF is thought to either regulate UGGT activity or the redox state of UGGT substrates (32). Mice lacking a functional SELENOF were found to develop nuclear cataract (lens opacification) at a very early age suggesting that SELENOF may be critical to the quality control system of protein folding in the lens (33). SELENOF may also be implicated in tissue-specific anticancer mechanisms (reviewed in 34), including colorectal cancer, that may, in part, be the result of SELENOFs potential role in intestinal barrier integrity (35).
Selenoprotein S
The mammalian selenoprotein S (SELENOS, formerly known as SEPS1, SelS, or VCP-interacting membrane selenoprotein [VIMP]) is another endoplasmic reticulum (ER) membrane protein. SELENOS is involved in the cellular response to ER stress (ER-associated degradation; ERAD) activated by the detection of misfolded proteins. SEPS1 contributes to the removal and transfer (retrotranslocation) of misfolded proteins from the ER lumen to the cytosol where proteins are tagged with ubiquitin before being degraded. A polymorphism or variation in the sequence within an ER-response element located in the SELENOS promoter was found to result in reduced SELENOS promoter activity and gene expression (36). The polymorphism corresponding to the substitution of a guanine (G) by an adenine (A) at nucleotide -105 (-105G>A) has been associated with increased plasma concentrations of pro-inflammatory cytokines. In addition, a case-control study reported that the A allele was more prevalent in individuals affected by Hashimoto thyroiditis (HT) — a T-cell-mediated autoimmune disease resulting in the destruction of thyroid cells — than in healthy controls (37). Other associations between SELENOS polymorphism (including -105G>A) and susceptibility to various conditions, such as preeclampsia, coronary artery disease, or gastrointestinal cancers, strongly suggest a role for this selenoprotein in the regulation of inflammatory and immune responses (38-41).
Other less well-characterized selenoproteins, which are also localized in the ER lumen and/or membrane, include selenoproteins K (SELENOK), M (SELENOM), N (SELENON), and T (SELENOT) (42).
Nutrient interactions
Antioxidant nutrients
The importance of selenium to biological systems, and specifically to the cellular redox (pro-oxidant/antioxidant) balance, is thought to be derived primarily from its presence as selenocysteine in the catalytic site of selenoproteins (see Function). Other minerals that are critical components of antioxidant enzymes include copper (as superoxide dismutase), zinc (as superoxide dismutase), and iron (as catalase). Selenium acts via selenoenzymes in synergy with the antioxidant vitamins, vitamin C (ascorbic acid) and vitamin E (α-tocopherol), by regenerating them from their oxidized forms and promoting maximal antioxidant protection (43-45).
Iodine
While iodine is an essential component of thyroid hormones, the selenium-containing iodothyronine deiodinases (DIOs) are enzymes required for the conversion of thyroxine (T4) to the biologically active thyroid hormone, triiodothyronine (T3) (see Function). DIO1 activity may also be involved in regulating iodine homeostasis (46). Additionally, the glutathione peroxidases play a critical role in thyroid function because they catalyze the degradation of peroxides generated during thyroid hormone synthesis (10). The epidemiology of coexisting iodine and selenium deficiencies in central Africa, but not in China, has been linked to myxedematous cretinism, a severe form of congenital hypothyroidism accompanied by mental and physical retardation. Selenium deficiency may be only one of several undetermined factors that might exacerbate the detrimental effects of iodine deficiency (47). Interestingly, selenium deficiency in rodents was found to have little impact on DIO activities as it appears that selenium is being supplied in priority for adequate synthesis of DIOs at the expense of other selenoenzymes (10).
Deficiency
Insufficient selenium intake may negatively affect the activity of several selenium-responsive enzymes, including glutathione peroxidases (specifically, GPx1 and GPx3), iodothyronine deiodinases, selenoprotein W, and methionine-R-sulfoxide reductase B1 (MsrB1). Even when severe, isolated selenium deficiency does not usually result in obvious clinical illness. Yet, compared to subjects with adequate selenium status, selenium-deficient individuals might be more susceptible to additional physiological stresses (48). Prolonged selenium deficiency may likely contribute to Keshan and Kashin-Beck diseases (see below).
Individuals at increased risk of selenium deficiency
Selenium deficiency has been reported in chronically ill patients who received total parenteral nutrition (TPN) without added selenium for prolonged periods of time. Muscular weakness, muscle wasting, and cardiomyopathy (inflammation and damage to the heart muscle) have been observed in these patients. Today, TPN solutions are routinely supplemented with selenium.
The risk of selenium deficiency also may be increased following bariatric surgery or in severe gastrointestinal conditions, such as Crohn's disease (49, 50). Some specialized medical diets like those used to treat certain metabolic disorders, including phenylketonuria, homocystinuria, and maple syrup urine disease, need to be supplemented with selenium to ensure optimal selenium status in patients (51).
Keshan disease
Keshan disease is a fatal form of dilated cardiomyopathy that was first described in young women and children in a selenium-deficient region in China. The acute form of the disease is characterized by the sudden onset of cardiac insufficiency, while the chronic form results in moderate-to-severe heart enlargement with varying degrees of cardiac insufficiency (52). The incidence of Keshan disease is closely associated with very low dietary intakes of selenium and poor selenium nutritional status. Selenium supplementation (in the form of sodium selenite; Na2SeO3) was found to protect people from developing Keshan disease but could not reverse heart muscle damage once it had occurred (52). One case-control study reported that selenium-responsive glutathione peroxidase 1 (GPx1) activity was significantly lower in Keshan patients compared to healthy individuals. Interestingly, a specific GPX1 polymorphism resulting in a proline-to-leucine transition at position 198 (Pro198Leu) is associated with a reduction in GPx1 activity and found to be more prevalent in Keshan patients. This GPX1 polymorphism might confer a greater susceptibility to Keshan disease in carriers with low selenium nutritional status (53).
While selenium deficiency is a major etiological factor of Keshan disease (54), the seasonal and annual variation in disease occurrence suggested that other factors, especially an infectious agent, might be involved in addition to selenium deficiency (55). Coxsackie virus B3 is one virus type that has been isolated from Keshan patients, and animal studies have shown that this virus was capable of causing an inflammation of the heart (myocarditis) in selenium-deficient mice. Studies in mice also indicated that oxidative stress induced by selenium deficiency could result in changes in the viral genome, such as to convert a relatively harmless strain of coxsackie virus B3 into a myocarditis-causing strain (56). Although not documented in Keshan disease, it is possible that selenium deficiency may increase the virulence of viruses with the potential to invade and damage the heart muscle (57).
Kashin-Beck disease
Kashin-Beck disease (KBD) is an endemic condition that affects an estimated 2.5 million people in Tibet, northern and central China, North Korea, and southeastern Siberia, a region of Russia (58). KBD is characterized by the degeneration of articular cartilage between joints (osteoarthritis) that can result in joint deformities and dwarfism in the most severe forms of the disease. The disease affects children as young as two years old. As with Keshan disease, KBD is prevalent in selenium-deficient provinces and thus generally affects people with very low selenium intakes (58, 59). Studies have suggested that increased susceptibility to KBD in selenium-deficient populations might result from a reduced antioxidant protection associated with polymorphisms in GPX genes (60, 61), but polymorphisms in other genes have also been implicated (62). Yet, the etiology appears to be multifactorial, as a number of other causative factors have been suggested for KBD, including fungal toxins in grain, iodine deficiency, and contaminated drinking water (52).
Meta-analyses of a few small clinical trials and prospective cohort studies have indicated that improving selenium nutritional status in children living in endemic areas may help reduce KBD incidence (63). Also, there is limited evidence to suggest that selenium supplementation could be useful in the treatment of patients with KBD. A meta-analysis of 10 randomized controlled trials reported a significant increase in the repairing rate of bone lesions in KBD children supplemented with sodium selenite for at least one year (64). Larger trials of higher quality are needed to assess whether selenium supplementation could result in disease remission.
The Recommended Dietary Allowance (RDA)
The dietary reference intakes (DRIs) for selenium were last revised in 2000 by the Food and Nutrition Board of the US Institute of Medicine (now the National Academy of Medicine). The most recent RDA (Table 1) is based on the estimated average requirement (EAR) needed to maximize antioxidant enzyme glutathione peroxidase (GPx) activity in plasma (65).
| Life Stage | Age | Males (μg/day) | Females (μg/day) |
|---|---|---|---|
| Infants | 0-6 months | 15 (AI) | 15 (AI) |
| Infants | 7-12 months | 20 (AI) | 20 (AI) |
| Children | 1-3 years | 20 | 20 |
| Children | 4-8 years | 30 | 30 |
| Children | 9-13 years | 40 | 40 |
| Adolescents | 14-18 years | 55 | 55 |
| Adults | 19 years and older | 55 | 55 |
| Pregnancy | all ages | - | 60 |
| Breast-feeding | all ages | - | 70 |
Of note, a US national survey (NHANES III) reported that over 99% of the US participants had serum selenium concentrations consistent with selenium requirements being met (65), suggesting selenium supplementation is not needed for most Americans.
Disease Prevention
Cancer
There has been considerable research on the effect of selenium supplementation on the incidence of cancer using preclinical models. More than two-thirds of over 100 published studies in 20 different animal models of spontaneous, viral, and chemically induced cancers found that selenium supplementation (to at least adequate intake levels) significantly reduces tumor incidence, in comparison to selenium-deficient diets (66). Evidence of cancer-inhibiting effects of selenium has provided a strong rationale for investigating potential associations between selenium intake and cancer risk in humans.
Observational studies
Most of the early observational evidence from case-control and nested case-control studies suggested either null or inverse associations between selenium exposure and risk of site-specific cancers (67). Biomarkers of selenium exposure include toenail and blood selenium content, as well as plasma glutathione peroxidase (GPx) activity. However, it is not clear whether these biomarkers adequately reflect selenium exposure from dietary and supplemental sources or selenium distribution in tissues and organs that may be affected by cancer. In the Danish Prospective Diet, Cancer, and Health prospective study that followed over 57,000 men and women for 14 years, the risk of rectal cancer was found to be 42% higher in current smokers compared to nonsmokers. No difference between smokers and nonsmokers regarding supplemental and dietary intakes of antioxidant micronutrients, including selenium, was found to contribute to the association of smoking and rectal cancer (68). Yet, because studies have consistently reported lower blood selenium concentrations and GPx activities in smokers compared to nonsmokers (reviewed in 69), estimation of selenium intakes might not be a reliable marker of selenium exposure in this population. Also, the chemical forms of selenium found in food are varied (see Sources) and may have very different biological and toxicological effects (70, 71).
A Cochrane review included 55 completed observational studies — mostly with a nested case-control design — published over three decades (67). A meta-analysis of 16 of these observational studies, including over 144,000 individuals, reported that higher versus lower selenium status was associated with a 31% lower risk of cancer at any site and a 40% lower risk of cancer-related mortality. A significantly lower risk was reported for bladder cancer (5 studies) and prostate cancer (17 studies); however, higher selenium status was not inversely associated with risks of breast cancer (8 studies), lung cancer (12 studies), colorectal cancer (5 studies), and gastric cancer (5 studies) (67). Other meta-analyses of observational studies have reported inverse associations between serum selenium concentrations and breast cancer (72) and thyroid cancer (73). Sex differences in cancer susceptibility have been reported in some studies, although consistent evidence of different effects in men and women appears to be lacking.
Single nucleotide variations (polymorphisms) in the sequence of genes can modify gene expression level and the stability and activity of the synthesized proteins. For example, a proline-to-leucine transition caused by a specific polymorphism in the GPX1 gene (rs1050450 C>T) is associated with reduced GPx1 enzymatic activity. The description of several polymorphisms in genes encoding selenoproteins has led to evaluation of possible associations with selenium status and cancer incidence. Notably, certain polymorphisms in genes coding for selenoproteins have been associated with increased risks of gastric and colorectal cancers (74, 75). Additionally, a number of studies have investigated the associations among selenoprotein polymorphisms, selenium status, and prostate cancer risk. A nested case-control study within the EPIC-Heidelberg cohort combined genotyping for several selenoprotein variants with biomarkers of selenium status (76). Briefly, the study showed that a GPX1 gene polymorphism (rs1050450 C>T) affected the association between selenium concentrations and prostate cancer risk. Specifically, selenium concentrations were found to be inversely associated with prostate cancer risk only among carriers of the GPX1 T allele. Additional variants in selenoprotein genes may mitigate the effect of selenium status on the risk of prostate cancer (77, 78). In another nested case-control study within the Physicians’ Health Study (PHS), individuals in the highest versus lowest quartile of plasma selenium concentrations were found to have a reduced risk of prostate cancer-related mortality except if carrying a specific variant in 15kDa selenoprotein gene (SELENOF rs561104 G>A) (79). More research is needed to further unravel the mechanisms underlying the influence of gene-diet interactions on the risk of developing cancer.
Intervention trials
Community-based studies: An early intervention trial of selenium supplementation was undertaken in China in 1985 among a general population of 130,471 individuals living in a high-risk area for viral hepatitis B infection and liver cancer. The trial provided table salt enriched with sodium selenite to the population of one township (20,847 people) using four other townships as controls. During an eight-year follow-up period, the average incidence of liver cancer was reduced by 35% in the selenium-supplemented population, while no reduction was found in the control populations. In a clinical trial in the same region in 1987-1990, 226 individuals with evidence of chronic hepatitis B infection were supplemented daily with either 200 μg of selenium in the form of a selenium-enriched yeast tablet or a placebo yeast tablet. During the four-year follow-up period, 7 out of 113 individuals on the placebo developed primary liver cancer, while none of the 113 subjects supplemented with selenium developed liver cancer (80).
Randomized controlled trials: The double-blind, placebo-controlled Nutritional Prevention of Cancer (NPC) study in 1,312 older adults with a history of nonmelanoma skin cancer found that supplementation with 200 μg/day of selenium-enriched yeast (selenized yeast) for an average of 7.4 years resulted in a 52% decrease in prostate cancer incidence in men (81). The protective effect of selenium supplementation was greatest in men with lower baseline plasma selenium and prostate-specific antigen (PSA) levels. A reduced incidence in lung, colorectal, and total cancer was also associated with supplementation with 200 μg/day (82) but not with 400 μg/day of selenium-enriched yeast (83). Additionally, selenium supplementation increased the risk of one type of skin cancer (squamous cell carcinoma) by 25%. A larger randomized, placebo-controlled intervention trial (the SELECT study) in more than 35,000 middle-aged and selenium-replete men, randomized to receive selenium (in the form of selenomethionine, 200 μg/day) and/or vitamin E supplementation, was halted because of concerns regarding an increased risk of type 2 diabetes mellitus with selenium and increased risk of prostate cancer with vitamin E (84, 85). In addition, the supplementation of selenium, alone or together with vitamin E, did not show any benefits regarding the risk of prostate, lung, or colorectal cancers after 5.5 years of follow-up (86, 87). In a randomized, placebo-controlled trial in 1,374 participants who had colonoscopic removal of at least one colorectal adenoma (the Selenium and Celecoxib trial), selenium supplementation (200 μg/day with selenized yeast) for a median of 33 months had no effect on colorectal adenoma recurrence (88). However, in a subanalysis of patients with advanced adenoma at baseline (n=161), selenium supplementation reduced adenoma recurrence by 18% (88). Outcomes from other smaller trials (reviewed in 89) have suggested either a lack of an effect or the possibility of an increased risk of cancer. The lack of a beneficial effect of selenium supplementation was supported in a 2014 meta-analysis of randomized controlled trials (67).
Cardiovascular disease
Low activity levels of the selenoenzymes, glutathione peroxidases (GPx), have been reported in oxidative stress-related diseases, including cardiovascular disease (CVD) (90). Presumably, the maintenance of an optimal selenium status has the potential to protect against oxidative stress (including lipid peroxidation) and could possibly aid in the prevention of chronic inflammation and cardiovascular disorders. However, most of the available research to date on selenium status and risk of CVD comes from observational studies, and results are largely conflicting.
Analyses of cross-sectional data from 13,887 US adults included in the Third National Health and Nutrition Examination Survey (NHANES III, 1988-1994) failed to show any significant associations between serum selenium concentrations and mortality from CVD, coronary artery/heart disease (CAD), or stroke (91). In addition, while individuals with renal insufficiency are at higher risk of developing CAD compared to those with normal kidney function, that risk was not found to be greater with low rather than normal selenium concentrations in serum (≤98 μg/L vs. >98 μg/L) (92). Yet, analysis of data from NHANES 2003-2018, a US national cross-sectional survey of 39,438 adults, found an inverse association between selenium intake and stroke, with daily intakes of 105 μg/day linked to the lowest stroke risk (93). A meta-analysis of 12 observational studies (5 prospective cohort studies, 4 case-control studies, and 3 cross-sectional studies) also found blood concentrations of selenium to be inversely associated with risk of stroke (94).
Some observational studies have raised concern with high selenium status. A cross-sectional study based on NHANES 2003-2004 data from 2,638 participants ages 40 years and over found that the risk of high blood pressure (hypertension), a major contributing factor for CVD, was 73% higher in individuals in the upper versus lowest quintile of serum selenium concentrations (≥150 μg/L vs. <122 μg/L) (95). But a systematic review of the literature failed to find enough evidence to support any relationship between serum selenium concentrations and hypertension (96). A few observational studies have also reported associations between normal-to-high selenium status and elevated serum lipid levels in selenium-replete populations, speculating that selenium might interfere with lipid metabolism and adversely affect cardiovascular health (97, 98).
At present, randomized controlled trials have not provided consistent results regarding the effect of selenium supplementation on lipid levels nor have they demonstrated any additional cardiovascular benefits of selenium in individuals with suboptimal or optimal selenium intakes (99, 100). A recent meta-analysis that pooled randomized controlled trials reported no association of single-nutrient selenium supplementation with cardiovascular disease (4 trials), coronary heart disease (3 trials), stroke (3 trials), or cardiovascular-related mortality (5 trials) (101).
A meta-analysis that combined five observational studies and one randomized controlled trial found selenium status was not significantly associated with incidence of cardiovascular disease (RR, 0.66; 95% CI, 0.40-1.09), but the data were highly heterogenous (102). Yet, when 11 studies were included in a dose-response meta-analysis, a 15% lower risk in CVD incidence with each incremental increase of 10 μg/L in blood selenium concentration was found; no association with CVD incidence was seen in a dose-response analysis of eight studies measuring selenium status by toenail concentration (102).
Disease Treatment
Immune dysfunction
Selenium deficiency has been associated with impaired immunity and chronic inflammation (103). A considerable amount of research conducted in cell culture and animal models indicates that selenium plays essential roles in regulating the migration, proliferation, differentiation, activation, and optimal function of immune cells, thus influencing innate immunity, B-cell dependent antibody production, and T-cell immunity (reviewed in 104). A review of nine randomized controlled trials concluded that selenium supplementation may affect cell-mediated (T-cells, natural killer cells) immunity but has little effect on antibody-mediated (humoral) immunity (105).
Evidence on the role of selenium and selenoproteins in the production of lipid mediators (called eicosanoids) involved in inflammatory responses suggests that selenium supplementation might mitigate dysfunctional inflammatory responses that contribute to the pathogenesis of many chronic health conditions (106). At present, randomized controlled trials are needed to evaluate the potential benefits of selenium supplementation in inflammatory disorders, such as asthma (107) and inflammatory bowel disease (108).
Infectious diseases
HIV/AIDS
In areas of widespread malnutrition, deficiencies in micronutrients (including selenium) are common among individuals infected by the human immunodeficiency virus (HIV) that causes acquired immunodeficiency syndrome (AIDS). Before antiretroviral therapy (ART) became the standard for HIV treatment, observational studies had consistently reported associations between low serum selenium concentrations and HIV infection in well-nourished subjects (109). Poor selenium status has been linked to increased risks of dilated cardiomyopathy and mortality in HIV-infected children and adults, as well as mother-to-child HIV transmission and perinatal mortality (reviewed in 110). Early laboratory studies suggested that HIV might disrupt normal antioxidant defenses in infected T-cells by reducing the levels of selenoproteins, i.e., thioredoxine reductases and glutathione peroxidases (110). Interestingly, a cross-sectional study found that HIV-seropositive individuals receiving ART for more than two years had undetectable plasma viral loads, higher CD4 lymphocyte T-cell counts, and adequate serum selenium concentrations compared to ART-naïve subjects (111). Because the antioxidant activity of selenoproteins may interfere with viral replication in HIV-infected immune cells (112, 113), it has been suggested that selenium supplementation might serve as a potential adjunct to ART for HIV patients.
A few trials of selenium supplementation in HIV-infected individuals have been conducted. A randomized, double-blind, placebo-controlled trial in 186 HIV-positive adults initially found that selenium supplementation at 200 μg/day for two years significantly decreased the rate of hospital admissions (114). Another randomized, double-blind, placebo-controlled trial in 174 HIV-positive individuals reported that 200 μg/day of selenium supplementation (in the form of selenium-enriched yeast) for nine months increased serum selenium concentrations, improved CD4 lymphocyte T-cell count, and prevented any progression of the HIV viral load (115). In a third double-blind trial in Tanzania, 913 pregnant women between 12 and 27 weeks’ gestation were randomized to receive 200 μg/day of selenium (as selenomethionine) or a placebo until six months after birth. Selenium supplementation had no effect on maternal CD4, CD8, and CD3 T-cell counts and on HIV viral load, but it significantly decreased the risk of acute or persistent diarrhea (116, 117). In addition, the risk of death between six weeks and six months postpartum was significantly reduced in infants of mothers supplemented with selenium compared to placebo (117).
A four-armed trial in Botswana randomized 878 HIV-positive adults at an early stage of the infection to receive either a placebo treatment, multivitamins (vitamins B, C, and E), 200 μg/day of selenium, or both multivitamins and selenium for 24 months (118). Unlike selenium alone, supplementation with multivitamins (with or without selenium) reduced the risk of immune decline by significantly increasing the time before ART initiation became necessary (i.e., when CD4 T-cell count fell below 251 cells/mL) compared to placebo. In the study, a combined outcome of (1) CD4 T-cell count falling below 251 cells/mL; (2) occurrence of AIDS-defining conditions; and (3) AIDS-related death — whichever happened first — was used to evaluate disease progression in the different arms of treatment. Compared to placebo, there was a longer period of time from randomization to the date of the composite outcome in individuals supplemented with multivitamins plus selenium, but not in those who received multivitamins or selenium alone (118). Moreover, a systematic review of six randomized controlled trials found that selenium supplementation (200 μg/day for 9 to 24 months) in HIV-infected subjects did not suppress the virus but delayed the decline in CD4 T cells in most trials (119).
Sepsis
The systemic inflammatory response syndrome (SIRS) results from a systemic inflammatory response that can be due to an infection (sepsis) (120). Severe sepsis and septic shock — defined as persistent sepsis-induced low blood pressure — are associated with elevated mortality rates in critically ill patients (120, 121). Because systemic inflammatory responses involve excessive oxidative stress, it has been suggested that providing antioxidant nutrients like selenium may improve the outcome of critically ill patients in intensive care units. Two meta-analyses of randomized controlled trials found that intravenous selenium supplementation (as sodium selenite) in critically ill patients with SIRS, sepsis, or septic shock resulted in significantly reducing the risk of mortality by 17% to 27% (122, 123). However, in a more recent, large placebo-controlled trial in 538 patients with severe sepsis or septic shock, intravenous selenium selenite administration did not improve mortality at the 28-day mark (124). International guidelines do not support the use of intravenous selenium in the treatment of sepsis and septic shock (125).
Autoimmune thyroid diseases
Hashimoto thyroiditis (HT; chronic autoimmune thyroiditis) is an autoimmune disease characterized by T-cell infiltration in the thyroid gland and circulating autoantibodies (predominantly against thyroid peroxidase but also against thyroglobulin), causing prolonged inflammation, tissue damage, and hypothyroidism (10) and increased risk of papillary thyroid cancer (126). While the function of the thyroid gland of healthy individuals is usually protected from variations in selenium supply, selenium deficiency and genetic polymorphisms affecting the activity of selenoproteins might be potential contributing factors to autoimmune thyroid diseases. A Cochrane systematic review (127) identified four randomized controlled trials that evaluated the effect of selenium supplementation as an adjunct treatment to T4 replacement therapy (levothyroxine) in HT patients (128-131). While three out of four studies suggested a reduction in levels of circulating autoantibodies, none of them provided information on whether selenium may improve mood- and health-related symptoms to allow for a decreased dosage of levothyroxine. In a recent open-label trial in 90 patients newly diagnosed with HT and not taking levothyroxine, supplementation with 200 μg/day in the form of selenious yeast for six months lowered blood concentrations of autoantibodies (both thyroid peroxidase and thyroglobulin) compared to the ‘no treatment’ group (132). Placebo-controlled studies are needed to evaluate whether supplemental selenium might improve clinical symptoms or influence the dosage of or need for levothyroxine. At present, evidence from randomized controlled trials is largely lacking.
Graves’ disease is an autoimmune thyroid disease that leads to hyperthyroidism. One randomized controlled trial found that selenium supplementation improved the well-being of patients affected by this disease (133). The results of two ongoing, randomized, placebo-controlled trials — the CATALYST in HT patients and the GRASS trial in patients with Graves’ disease — may provide insight into an effect of selenium on thyroid-specific quality-of-life criteria and inform clinical decision making (134, 135).
Sources
Food sources
The richest food sources of selenium are organ meats and seafood, followed by muscle meats from farmed animals, as many are supplemented with selenium in their feed. Drinking water is not considered to be a significant source of selenium in North America. However, in areas where high levels of selenium in soil contribute to the selenium content of the water (e.g., California, the Dakotas), higher levels of selenium may be found in wells used for drinking water (136). In general, there is wide variation in the selenium content of plants and grains, especially because some plants, including garlic, Brazil nuts (nuts from the Bertholletia excelsa tree), and multiple Brassica species, tend to accumulate selenium ('selenium accumulators'), while others assimilate selenium to a lesser extent ('non-accumulators'). The assimilation of selenium by plants also depends on soil selenium content. Brazil nuts grown in areas of Brazil with selenium-rich soil may provide more than 100 μg of selenium in one nut, while those grown in selenium-poor soil provide an amount 10 times lower (137). In the US, grains are a good source of selenium, but fruit and vegetables tend to be relatively poor in selenium.
Various chemical forms (species) of selenium are found in selenium accumulators, including selenate (inorganic selenium), selenomethionine, selenocysteine, selenium-methyl-selenocysteine, and γ-glutamyl-selenium-methyl-selenocysteine. Although the two latter compounds are predominant in plants of the Allium and Brassicaceae families (which include garlic, onion, and broccoli), wheat, other grains (including Brazil nuts), and soy are rich in selenomethionine and contain smaller amounts of selenocysteine and selenate. Less is known about selenium species and distribution in dietary sources of animal origin. Animal nutrition and growth conditions certainly contribute to the selenium species formed and their relative quantities, and it is assumed that the metabolic pathway of dietary selenium in animals is similar to that in humans. Selenocysteine is predominantly formed in animals fed inorganic selenium, while selenomethionine is derived from dietary sources of plant origin (reviewed in 138).
In the US, the national survey NHANES III reported mean dietary intakes ranging between 100.5 μg/day and 158.5 μg/day in adults ages 19-50 years (65). Table 2 lists some good food sources of selenium and their average selenium content in micrograms (μg). For more information on the selenium content of specific foods, search USDA’s FoodData Central.
| Food | Serving | Selenium (μg) |
|---|---|---|
| Brazil nuts (from selenium-rich soil) | 1 ounce (6 kernels) | 544* |
| Oysters (Pacific, steamed) | 3 ounces | 131 |
| Tuna (yellowfin, cooked, dry heat) | 3 ounces | 91.8 |
| Clams (steamed) | 3 ounces | 54.4 |
| Halibut (Atlantic and Pacific, cooked, dry heat) | 3 ounces | 47.1 |
| Shrimp (steamed) | 3 ounces | 42.1 |
| Noodles (egg, cooked, enriched) | 1 cup | 38.2 |
| Crab (queen, steamed) | 3 ounces | 37.7 |
| Chicken (light-meat, roasted) | 1 cup | 36.1 |
| Pork (tenderloin, roasted) | 3 ounces | 32.5 |
| Salmon (sockeye, cooked, dry heat) | 3 ounces | 30.2 |
| Beef (plate steak, grilled) | 3 ounces | 28.9 |
| Sunflowers seeds (dried) | ¼ cup | 18.6 |
| Whole-wheat bread | 2 slices | 16.4 |
| Rice (brown, long-grain, cooked) | 1 cup | 11.7 |
| Milk (fat free or skim) | 8 fl oz. (1 cup) | 7.6 |
| *Above the tolerable upper intake level (UL) of 400 μg/day. | ||
Supplements
Selenium supplements are available in several forms and mostly unregulated by the FDA. Sodium selenite and sodium selenate are inorganic forms of selenium. Sodium selenate is almost completely absorbed, but a significant amount is excreted in the urine before it can be incorporated into proteins. Sodium selenite is only about 50% absorbed but is better retained than selenate once it is absorbed. Selenomethionine, an organic form of selenium, is about 90% absorbed (65); however, only about 34% may then actually be converted to free selenomethionine (139). Selenomethionine and selenium-enriched yeast, which mainly supply selenomethionine, are also available as supplements. The consumer should be aware that some forms of selenium-enriched yeast on the market contain mainly inorganic forms of selenium added to the yeast.
Humans can metabolize both inorganic and organic forms of selenium to selenocysteine and incorporate into selenoenzymes. In intervention trials, supplementation with selenomethionine more effectively increased blood selenium concentrations compared to supplementation with inorganic forms (i.e., sodium selenite and sodium selenate) (138). Yet, inorganic forms may increase plasma glutathione peroxidase (GPx) activity more effectively than organic forms (reviewed in 140). It has also been suggested that the incorporation of selenomethionine in place of methionine into tissue proteins may ensure that selenium is available upon protein turnover (138).
Selenium-enriched foods
Selenium-enriched foods have been of interest to scientists, especially because of the suggestion that some of the chemical forms of selenium produced by plants might be more potent modifiers of cancer risk than those currently available in supplements. Although there is currently no evidence of long-term health benefits associated with the consumption of selenium-enriched foods, results from animal studies and intervention trials suggest that protein-based sources of selenium are more effective at increasing GPx activity than selenium-enriched yeast and selenomethionine (140). Food fortification may also represent a cost-effective strategy to improve selenium nutritional status in populations at risk of inadequacy (141).
Safety
Toxicity
Although selenium is required for health, high doses of selenium — especially with long-term supplementation — can be toxic. Acute and fatal toxicities have occurred with accidental or suicidal ingestion of gram quantities of selenium. Clinically significant selenium toxicity was reported in 13 individuals after taking supplements that contained 27.3 mg (27,300 μg) per tablet due to a manufacturing error. Chronic selenium toxicity (selenosis) may occur with smaller doses of selenium over long periods of time. The most common symptoms of selenosis are hair and nail brittleness and loss (65, 142). Other symptoms may include gastrointestinal disturbances, skin rashes, a garlic breath odor, fatigue, irritability, and neurological disorders. In an area of China with a high prevalence of selenosis, toxic effects occurred with increasing frequency when blood selenium concentrations reached a level corresponding to an intake of 850 μg/day.
The Food and Nutrition Board of the US Institute of Medicine (now the National Academy of Medicine) set the tolerable upper intake level (UL) for selenium at 400 μg/day in adults based on the prevention of hair and nail brittleness and loss and early signs of chronic selenium toxicity (65). The UL includes both selenium obtained from food and selenium from supplements (Table 3).
Do selenium supplements increase the risk for type 2 diabetes mellitus?
A few studies have examined the relationship between selenium status and type 2 diabetes mellitus. In the cross-sectional analysis of NHANES III (1988-1994) data from 8,876 adult participants, the highest versus lowest quintile of serum selenium concentrations (≥137 μg/L vs. <111 μg/L) was associated with an increased risk of type 2 diabetes (143). Data analyses from 917 participants (≥40 years of age) of NHANES 2003-2004 also indicated an increased prevalence of type 2 diabetes in the highest versus lowest quartile of serum selenium concentrations (≥147 μg/L vs. <124 μg/L). Individuals in the highest versus lowest quartile of serum selenium concentrations also had higher levels of plasma glucose and glycated hemoglobin (HbA1c), suggestive of poor glycemic control (144). In NHANES 2013-2016, a cross-sectional analysis of 2,706 adults with normal glucose metabolism, the highest (≥209 μg/L) versus lowest (<181 μg/L) quartile of blood selenium concentration were associated with elevated markers of glucose metabolism, including fasting plasma glucose concentration, HbA1c, and circulating insulin (145). In a dose-response meta-analysis of 34 observational studies, circulating selenium concentration of at least 160 μg/L was associated with a near doubling of risk for type 2 diabetes when compared to a circulating concentration of 90 μg/L; a significantly higher risk was also observed at 120 μg/L or greater (RR, 1.27; 95% CI, 1.10-1.47). In this analysis, dietary selenium intakes greater than 80 μg/day were linked to a higher risk of type 2 diabetes compared to dietary selenium intake of 55 μg/day (146).
Type 2 diabetes has been evaluated in randomized controlled trials as a secondary endpoint. The randomized, double-blind, placebo-controlled study in 1,312 participants in the Nutritional Prevention of Cancer (NPC) trial found that selenium supplementation (200 μg/day; mean follow-up of 7.7 years) significantly increased the risk for type 2 diabetes in participants in the highest tertile of baseline plasma selenium concentration (147). In addition, in the Selenium and Vitamin E Cancer Prevention Trial (SELECT), more cases of type 2 diabetes were found in the selenium group (200 μg/day; median follow-up of 5.5 years) than in the placebo group, but this was only a trend and not statistically significant (87). However, other randomized controlled trials have reported 200 μg/day for ~3-4 years does not increase diabetes risk (88, 148).
At present, the mechanisms behind some of these observations are not well understood. An increase in insulin sensitivity has been reported in individuals with congenital (inborn) deficiency of most selenoproteins (149). Results from several animal studies also indicated that selenium supplementation and selenoproteins may interfere with insulin action and glucose homeostasis (reviewed in 150). On the other hand, studies have found that impaired glucose metabolism in patients with type 2 diabetes may affect SELENOP expression and selenium homeostasis (15, 151, 152). While more research is needed to fully understand the interplay between carbohydrate metabolism and selenium homeostasis, the use of high-dose selenium supplements in healthy individuals are currently discouraged in those with high selenium status and/or at increased risk for developing type 2 diabetes (150, 153, 154).
Drug interactions
At present, few interactions between selenium and medications have been reported (155). For example, the anticonvulsant drug valproic acid and the chemotherapeutic agent cisplatin may lower circulating selenium concentrations in treated subjects (156, 157). Also, supplemental sodium selenite was found to reduce the toxicity of the antibiotic nitrofurantoin and the herbicide paraquat in animal studies (158).
Antioxidant supplements and statins
A three-year randomized controlled trial in 160 patients with documented coronary artery disease (CAD) and low high-density-lipoprotein (HDL) levels found that a combination of simvastatin (Zocor) and niacin increased HDL2 subfraction levels, inhibited the progression of coronary artery stenosis (narrowing), and decreased the frequency of cardiovascular events (159). Surprisingly, when an antioxidant combination (1,000 mg of vitamin C, 800 IU of vitamin E (d-α-tocopherol), 100 μg of selenium, and 25 mg of β-carotene daily) was taken with the simvastatin-niacin combination, the protective effects were diminished. However, the individual contribution of selenium cannot be established, and other studies have reported that antioxidant vitamins alone could interfere with the action of HDL-raising drugs, including statins (160).
Linus Pauling Institute Recommendation
The average American diet is estimated to provide about 100 μg/day of selenium, an amount that is well above the current RDA (55 μg/day for adults) and appears sufficient to optimize plasma and cellular glutathione peroxidase (GPx) activity. A 10-week randomized controlled study in healthy British adults (ages, 50-64 years) estimated that about 105 μg/day of total selenium intake was required to maximize the plasma concentrations of selenoprotein P (SELENOP), another useful biomarker of selenium status (161). However, a similar trial in an American cohort with higher baseline plasma selenium concentrations found no effect of selenium supplementation on SELENOP concentrations (162). The amount of selenium in multivitamin/mineral (MVM) supplements varies considerably: most MVMs on the market in the US provide the Daily Value (DV) of 55 μg or a lower amount, but some contain up to 200 μg of selenium (163). Eating a varied diet and taking a daily MVM supplement should provide sufficient selenium for most people in the US and help improve selenium status in populations with lower selenium intakes outside the US.
Men
At present, the effect of selenium supplementation on cancer risk is not clear enough to support a general recommendation for an extra selenium supplement, especially in men with serum selenium concentrations consistent with adequate selenium intakes. SELECT found that 200 μg/day of selenium did not reduce the risk of prostate cancer (87), refuting the results of the NPC trial (see Cancer). Another multicenter, randomized, double-blind, placebo-controlled trial (the Negative Biopsy Trial) in 699 men at high risk of prostate cancer found no effect of either 200 μg/day or 400 μg/day of selenium on prostate cancer risk during a mean follow-up of 36 months (164). In addition, because current evidence suggests a U-shaped relationship between selenium status and prostate cancer risk (165), men with replete selenium status should avoid taking supplemental selenium that would exceed 200 μg/day.
Women
At present, there is no clinical evidence showing that selenium supplementation above recommended levels decreases the risk of breast cancer, although some, but not all, observational studies have found an inverse relationship between selenium status and breast cancer in women (72).
Older adults (>50 years)
The RDA of selenium for older adults is the same as for younger adults: 55 μg/day. A five-year, randomized, double-blind, placebo-controlled trial in healthy Danish older people (ages at inclusion, 60-74 years) found that selenium supplementation (100-300 μg/day) had little-to-no impact on circulating levels of antioxidant enzymes, including GPx (166).
Authors and Reviewers
Originally written in 2001 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in October 2003 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in November 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in November 2014 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in November 2024 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in February 2024 by:
Petra A. Tsuji, Ph.D., M.P.H.
Professor
Department of Biological Sciences
Towson University
Copyright 2001-2024 Linus Pauling Institute
References
1. Rayman MP. The importance of selenium to human health. Lancet. 2000;356(9225):233-241. (PubMed)
2. Mangiapane E, Pessione A, Pessione E. Selenium and selenoproteins: an overview on different biological systems. Curr Protein Pept Sci. 2014;15(6):598-607. (PubMed)
3. Burk RF, Hill KE. Regulation of selenium metabolism and transport. Annu Rev Nutr. 2015;35:109-134. (PubMed)
4. Berry MJ, Banu L, Harney JW, Larsen PR. Functional characterization of the eukaryotic SECIS elements which direct selenocysteine insertion at UGA codons. EMBO J. 1993;12(8):3315-3322. (PubMed)
5. Mariotti M, Ridge PG, Zhang Y, et al. Composition and evolution of the vertebrate and mammalian selenoproteomes. PLoS One. 2012;7(3):e33066. (PubMed)
6. Terry EN, Diamond JA. Selenium. In: Erdman Jr J, Macdonald J, Zeisel S, eds. Present Knowledge in Nutrition: John Wiley & Sons, Inc; 2012:568-585.
7. Boitani C, Puglisi R. Selenium, a key element in spermatogenesis and male fertility. Adv Exp Med Biol. 2008;636:65-73. (PubMed)
8. Arner ES. Focus on mammalian thioredoxin reductases--important selenoproteins with versatile functions. Biochim Biophys Acta. 2009;1790(6):495-526. (PubMed)
9. Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med. 2014;66:75-87. (PubMed)
10. Schomburg L. Selenium, selenoproteins and the thyroid gland: interactions in health and disease. Nat Rev Endocrinol. 2012;8(3):160-171. (PubMed)
11. Tsuji PA, Santesmasses D, Lee BJ, Gladyshev VN, Hatfield DL. Historical roles of selenium and selenoproteins in health and development: the good, the bad and the ugly. Int J Mol Sci. 2021;23(1):5. (PubMed)
12. Hill KE, Wu S, Motley AK, et al. Production of selenoprotein P (Sepp1) by hepatocytes is central to selenium homeostasis. J Biol Chem. 2012;287(48):40414-40424. (PubMed)
13. Schomburg L. Selenoprotein P - Selenium transport protein, enzyme and biomarker of selenium status. Free Radic Biol Med. 2022;191:150-163. (PubMed)
14. Olson GE, Winfrey VP, Hill KE, Burk RF. Megalin mediates selenoprotein P uptake by kidney proximal tubule epithelial cells. J Biol Chem. 2008;283(11):6854-6860. (PubMed)
15. Misu H, Takamura T, Takayama H, et al. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab. 2010;12(5):483-495. (PubMed)
16. Jujic A, Molvin J, Schomburg L, et al. Selenoprotein P deficiency is associated with higher risk of incident heart failure. Free Radic Biol Med. 2023;207:11-16. (PubMed)
17. Whanger PD. Selenoprotein expression and function-selenoprotein W. Biochim Biophys Acta. 2009;1790(11):1448-1452. (PubMed)
18. Jeong D, Kim TS, Chung YW, Lee BJ, Kim IY. Selenoprotein W is a glutathione-dependent antioxidant in vivo. FEBS Lett. 2002;517(1-3):225-228. (PubMed)
19. Reeves MA, Hoffmann PR. The human selenoproteome: recent insights into functions and regulation. Cell Mol Life Sci. 2009;66(15):2457-2478. (PubMed)
20. Reszka E, Jablonska E, Gromadzinska J, Wasowicz W. Relevance of selenoprotein transcripts for selenium status in humans. Genes Nutr. 2012;7(2):127-137. (PubMed)
21. Chung YW, Jeong D, Noh OJ, et al. Antioxidative role of selenoprotein W in oxidant-induced mouse embryonic neuronal cell death. Mol Cells. 2009;27(5):609-613. (PubMed)
22. Jeon YH, Park YH, Kwon JH, Lee JH, Kim IY. Inhibition of 14-3-3 binding to Rictor of mTORC2 for Akt phosphorylation at Ser473 is regulated by selenoprotein W. Biochim Biophys Acta. 2013;1833(10):2135-2142. (PubMed)
23. Kanai F, Marignani PA, Sarbassova D, et al. TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 2000;19(24):6778-6791. (PubMed)
24. Jeon YH, Park YH, Lee JH, Hong JH, Kim IY. Selenoprotein W enhances skeletal muscle differentiation by inhibiting TAZ binding to 14-3-3 protein. Biochim Biophys Acta. 2014;1843(7):1356-1364. (PubMed)
25. Alkan Z, Duong FL, Hawkes WC. Selenoprotein W controls epidermal growth factor receptor surface expression, activation and degradation via receptor ubiquitination. Biochim Biophys Acta. 2015;1853(5):1087-1095. (PubMed)
26. Ganichkin OM, Xu XM, Carlson BA, et al. Structure and catalytic mechanism of eukaryotic selenocysteine synthase. J Biol Chem. 2008;283(9):5849-5865. (PubMed)
27. Lee BC, Peterfi Z, Hoffmann FW, et al. MsrB1 and MICALs regulate actin assembly and macrophage function via reversible stereoselective methionine oxidation. Mol Cell. 2013;51(3):397-404. (PubMed)
28. Kim HY. The methionine sulfoxide reduction system: selenium utilization and methionine sulfoxide reductase enzymes and their functions. Antioxid Redox Signal. 2013;19(9):958-969. (PubMed)
29. Kumaraswamy E, Malykh A, Korotkov KV, et al. Structure-expression relationships of the 15-kDa selenoprotein gene. Possible role of the protein in cancer etiology. J Biol Chem. 2000;275(45):35540-35547. (PubMed)
30. Korotkov KV, Kumaraswamy E, Zhou Y, Hatfield DL, Gladyshev VN. Association between the 15-kDa selenoprotein and UDP-glucose:glycoprotein glucosyltransferase in the endoplasmic reticulum of mammalian cells. J Biol Chem. 2001;276(18):15330-15336. (PubMed)
31. Labunskyy VM, Ferguson AD, Fomenko DE, Chelliah Y, Hatfield DL, Gladyshev VN. A novel cysteine-rich domain of Sep15 mediates the interaction with UDP-glucose:glycoprotein glucosyltransferase. J Biol Chem. 2005;280(45):37839-37845. (PubMed)
32. Labunskyy VM, Hatfield DL, Gladyshev VN. The Sep15 protein family: roles in disulfide bond formation and quality control in the endoplasmic reticulum. IUBMB Life. 2007;59(1):1-5. (PubMed)
33. Kasaikina MV, Fomenko DE, Labunskyy VM, et al. Roles of the 15-kDa selenoprotein (Sep15) in redox homeostasis and cataract development revealed by the analysis of Sep 15 knockout mice. J Biol Chem. 2011;286(38):33203-33212. (PubMed)
34. Hatfield DL, Tsuji PA, Carlson BA, Gladyshev VN. Selenium and selenocysteine: roles in cancer, health, and development. Trends Biochem Sci. 2014;39(3):112-120. (PubMed)
35. Canter JA, Ernst SE, Peters KM, et al. Selenium and the 15kDa selenoprotein impact colorectal tumorigenesis by modulating intestinal barrier integrity. Int J Mol Sci. 2021;22(19):10651. (PubMed)
36. Curran JE, Jowett JB, Elliott KS, et al. Genetic variation in selenoprotein S influences inflammatory response. Nat Genet. 2005;37(11):1234-1241. (PubMed)
37. Santos LR, Duraes C, Mendes A, et al. A polymorphism in the promoter region of the selenoprotein S gene (SEPS1) contributes to Hashimoto's thyroiditis susceptibility. J Clin Endocrinol Metab. 2014;99(4):E719-723. (PubMed)
38. Alanne M, Kristiansson K, Auro K, et al. Variation in the selenoprotein S gene locus is associated with coronary heart disease and ischemic stroke in two independent Finnish cohorts. Hum Genet. 2007;122(3-4):355-365. (PubMed)
39. Cox AJ, Lehtinen AB, Xu J, et al. Polymorphisms in the selenoprotein S gene and subclinical cardiovascular disease in the Diabetes Heart Study. Acta Diabetol. 2013;50(3):391-399. (PubMed)
40. Moses EK, Johnson MP, Tommerdal L, et al. Genetic association of preeclampsia to the inflammatory response gene SEPS1. Am J Obstet Gynecol. 2008;198(3):336 e331-335. (PubMed)
41. Shibata T, Arisawa T, Tahara T, et al. Selenoprotein S (SEPS1) gene -105G>A promoter polymorphism influences the susceptibility to gastric cancer in the Japanese population. BMC Gastroenterol. 2009;9:2. (PubMed)
42. Shchedrina VA, Zhang Y, Labunskyy VM, Hatfield DL, Gladyshev VN. Structure-function relations, physiological roles, and evolution of mammalian ER-resident selenoproteins. Antioxid Redox Signal. 2010;12(7):839-849. (PubMed)
43. Li X, Hill KE, Burk RF, May JM. Selenium spares ascorbate and alpha-tocopherol in cultured liver cell lines under oxidant stress. FEBS Lett. 2001;508(3):489-492. (PubMed)
44. May JM, Mendiratta S, Hill KE, Burk RF. Reduction of dehydroascorbate to ascorbate by the selenoenzyme thioredoxin reductase. J Biol Chem. 1997;272(36):22607-22610. (PubMed)
45. Murer SB, Aeberli I, Braegger CP, et al. Antioxidant supplements reduced oxidative stress and stabilized liver function tests but did not reduce inflammation in a randomized controlled trial in obese children and adolescents. J Nutr. 2014;144(2):193-201. (PubMed)
46. Schneider MJ, Fiering SN, Thai B, et al. Targeted disruption of the type 1 selenodeiodinase gene (Dio1) results in marked changes in thyroid hormone economy in mice. Endocrinology. 2006;147(1):580-589. (PubMed)
47. Hess SY. The impact of common micronutrient deficiencies on iodine and thyroid metabolism: the evidence from human studies. Best Pract Res Clin Endocrinol Metab. 2010;24(1):117-132. (PubMed)
48. Thomson CD. Assessment of requirements for selenium and adequacy of selenium status: a review. Eur J Clin Nutr. 2004;58(3):391-402. (PubMed)
49. Shahmiri SS, Eghbali F, Ismaeil A, et al. Selenium deficiency after bariatric surgery, incidence and symptoms: a systematic review and meta-analysis. Obes Surg. 2022;32(5):1719-1725. (PubMed)
50. Yan W, Meihao W, Zihan S, et al. Correlation between Crohn's disease activity and serum selenium concentration. Clin Ther. 2022;44(5):736-743 e3. (PubMed)
51. Cooper A, Mones RL, Heird WC. Nutritional management of infants and children with specific diseases and other conditions. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore: Lippincott Williams & Wilkins; 2014:988-1005.
52. Sunde RA. Selenium. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore: Lippincott Williams & Wilkins; 2014:265-276.
53. Lei C, Niu X, Wei J, Zhu J, Zhu Y. Interaction of glutathione peroxidase-1 and selenium in endemic dilated cardiomyopathy. Clin Chim Acta. 2009;399(1-2):102-108. (PubMed)
54. Zhou H, Wang T, Li Q, Li D. Prevention of Keshan disease by selenium supplementation: a systematic review and meta-analysis. Biol Trace Elem Res. 2018;186(1):98-105. (PubMed)
55. Chen J. An original discovery: selenium deficiency and Keshan disease (an endemic heart disease). Asia Pac J Clin Nutr. 2012;21(3):320-326. (PubMed)
56. Beck MA, Kolbeck PC, Rohr LH, Shi Q, Morris VC, Levander OA. Benign human enterovirus becomes virulent in selenium-deficient mice. J Med Virol. 1994;43(2):166-170. (PubMed)
57. Harthill M. Review: micronutrient selenium deficiency influences evolution of some viral infectious diseases. Biol Trace Elem Res. 2011;143(3):1325-1336. (PubMed)
58. Stone R. Diseases. A medical mystery in middle China. Science. 2009;324(5933):1378-1381. (PubMed)
59. Wang K, Yu J, Liu H, et al. Endemic Kashin-Beck disease: A food-sourced osteoarthropathy. Semin Arthritis Rheum. 2020;50(2):366-372. (PubMed)
60. Du XH, Dai XX, Xia Song R, et al. SNP and mRNA expression for glutathione peroxidase 4 in Kashin-Beck disease. Br J Nutr. 2012;107(2):164-169. (PubMed)
61. Xiong YM, Mo XY, Zou XZ, et al. Association study between polymorphisms in selenoprotein genes and susceptibility to Kashin-Beck disease. Osteoarthritis Cartilage. 2010;18(6):817-824. (PubMed)
62. Yu FF, Sun L, Zhou GY, Ping ZG, Guo X, Ba Y. Meta-analysis of association studies of selenoprotein gene polymorphism and Kashin-Beck disease: an updated systematic review. Biol Trace Elem Res. 2022;200(2):543-550. (PubMed)
63. Zou K, Liu G, Wu T, Du L. Selenium for preventing Kashin-Beck osteoarthropathy in children: a meta-analysis. Osteoarthritis Cartilage. 2009;17(2):144-151. (PubMed)
64. Jirong Y, Huiyun P, Zhongzhe Y, et al. Sodium selenite for treatment of Kashin-Beck disease in children: a systematic review of randomised controlled trials. Osteoarthritis Cartilage. 2012;20(7):605-613. (PubMed)
65. Food and Nutrition Board, Institute of Medicine. Selenium. Dietary reference intakes for vitamin C, vitamin E, selenium, and carotenoids. Washington, D.C.: National Academy Press; 2000:284-324. (National Academy Press)
66. Combs GF, Jr., Gray WP. Chemopreventive agents: selenium. Pharmacol Ther. 1998;79(3):179-192. (PubMed)
67. Vinceti M, Dennert G, Crespi CM, et al. Selenium for preventing cancer. Cochrane Database Syst Rev. 2014;3:CD005195. (PubMed)
68. Hansen RD, Albieri V, Tjonneland A, Overvad K, Andersen KK, Raaschou-Nielsen O. Effects of smoking and antioxidant micronutrients on risk of colorectal cancer. Clin Gastroenterol Hepatol. 2013;11(4):406-15.e3. (PubMed)
69. Northrop-Clewes CA, Thurnham DI. Monitoring micronutrients in cigarette smokers. Clin Chim Acta. 2007;377(1-2):14-38. (PubMed)
70. Hazane-Puch F, Champelovier P, Arnaud J, et al. Long-term selenium supplementation in HaCaT cells: importance of chemical form for antagonist (protective versus toxic) activities. Biol Trace Elem Res. 2013;154(2):288-298. (PubMed)
71. Weekley CM, Harris HH. Which form is that? The importance of selenium speciation and metabolism in the prevention and treatment of disease. Chem Soc Rev. 2013;42(23):8870-8894. (PubMed)
72. Babaknejad N, Sayehmiri F, Sayehmiri K, et al. The relationship between selenium levels and breast cancer: a systematic review and meta-analysis. Biol Trace Elem Res. 2014;159(1-3):1-7. (PubMed)
73. Hao R, Yu P, Gui L, Wang N, Pan D, Wang S. Relationship between serum levels of selenium and thyroid cancer: a systematic review and meta-analysis. Nutr Cancer. 2023;75(1):14-23. (PubMed)
74. Meplan C, Hesketh J. The influence of selenium and selenoprotein gene variants on colorectal cancer risk. Mutagenesis. 2012;27(2):177-186. (PubMed)
75. Li J, Zhu Y, Zhou Y, et al. The SELS rs34713741 polymorphism is associated with susceptibility to colorectal cancer and gastric cancer: a meta-analysis. Genet Test Mol Biomarkers. 2020;24(12):835-844. (PubMed)
76. Steinbrecher A, Meplan C, Hesketh J, et al. Effects of selenium status and polymorphisms in selenoprotein genes on prostate cancer risk in a prospective study of European men. Cancer Epidemiol Biomarkers Prev. 2010;19(11):2958-2968. (PubMed)
77. Gerstenberger JP, Bauer SR, Van Blarigan EL, et al. Selenoprotein and antioxidant genes and the risk of high-grade prostate cancer and prostate cancer recurrence. Prostate. 2015;75(1):60-69. (PubMed)
78. Meplan C, Rohrmann S, Steinbrecher A, et al. Polymorphisms in thioredoxin reductase and selenoprotein K genes and selenium status modulate risk of prostate cancer. PLoS One. 2012;7(11):e48709. (PubMed)
79. Penney KL, Schumacher FR, Li H, et al. A large prospective study of SEP15 genetic variation, interaction with plasma selenium levels, and prostate cancer risk and survival. Cancer Prev Res (Phila). 2010;3(5):604-610. (PubMed)
80. Yu SY, Zhu YJ, Li WG. Protective role of selenium against hepatitis B virus and primary liver cancer in Qidong. Biol Trace Elem Res. 1997;56(1):117-124. (PubMed)
81. Duffield-Lillico AJ, Dalkin BL, Reid ME, et al. Selenium supplementation, baseline plasma selenium status and incidence of prostate cancer: an analysis of the complete treatment period of the Nutritional Prevention of Cancer Trial. BJU Int. 2003;91(7):608-612. (PubMed)
82. Clark LC, Combs GF, Jr., Turnbull BW, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA. 1996;276(24):1957-1963. (PubMed)
83. Reid ME, Duffield-Lillico AJ, Slate E, et al. The nutritional prevention of cancer: 400 mcg per day selenium treatment. Nutr Cancer. 2008;60(2):155-163. (PubMed)
84. National Cancer Institute. Review of Prostate Cancer Prevention Study Shows No Benefit for Use of Selenium and Vitamin E Supplements. [Web page]. http://www.cancer.gov/newscenter/pressreleases/SELECTresults2008. Accessed 10/28/08.
85. Hatfield DL, Gladyshev VN. The Outcome of Selenium and Vitamin E Cancer Prevention Trial (SELECT) reveals the need for better understanding of selenium biology. Mol Interv. 2009;9(1):18-21. (PubMed)
86. Klein EA, Thompson IM, Jr., Tangen CM, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2011;306(14):1549-1556. (PubMed)
87. Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2009;301(1):39-51. (PubMed)
88. Thompson PA, Ashbeck EL, Roe DJ, et al. Selenium supplementation for prevention of colorectal adenomas and risk of associated type 2 diabetes. J Natl Cancer Inst. 2016;108(12):djw152. (PubMed)
89. Vinceti M, Crespi CM, Malagoli C, Del Giovane C, Krogh V. Friend or foe? The current epidemiologic evidence on selenium and human cancer risk. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2013;31(4):305-341. (PubMed)
90. Flores-Mateo G, Carrillo-Santisteve P, Elosua R, et al. Antioxidant enzyme activity and coronary heart disease: meta-analyses of observational studies. Am J Epidemiol. 2009;170(2):135-147. (PubMed)
91. Bleys J, Navas-Acien A, Guallar E. Serum selenium levels and all-cause, cancer, and cardiovascular mortality among US adults. Arch Intern Med. 2008;168(4):404-410. (PubMed)
92. Eaton CB, Abdul Baki AR, Waring ME, Roberts MB, Lu B. The association of low selenium and renal insufficiency with coronary heart disease and all-cause mortality: NHANES III follow-up study. Atherosclerosis. 2010;212(2):689-694. (PubMed)
93. Shi W, Su L, Wang J, Wang F, Liu X, Dou J. Correlation between dietary selenium intake and stroke in the National Health and Nutrition Examination Survey 2003-2018. Ann Med. 2022;54(1):1395-1402. (PubMed)
94. Ding J, Zhang Y. Relationship between the circulating selenium level and stroke: a meta-analysis of observational studies. J Am Nutr Assoc. 2022;41(5):444-452. (PubMed)
95. Laclaustra M, Navas-Acien A, Stranges S, Ordovas JM, Guallar E. Serum selenium concentrations and hypertension in the US Population. Circ Cardiovasc Qual Outcomes. 2009;2(4):369-376. (PubMed)
96. Kuruppu D, Hendrie HC, Yang L, Gao S. Selenium levels and hypertension: a systematic review of the literature. Public Health Nutr. 2014;17(6):1342-1352. (PubMed)
97. Laclaustra M, Stranges S, Navas-Acien A, Ordovas JM, Guallar E. Serum selenium and serum lipids in US adults: National Health and Nutrition Examination Survey (NHANES) 2003-2004. Atherosclerosis. 2010;210(2):643-648. (PubMed)
98. Stranges S, Laclaustra M, Ji C, et al. Higher selenium status is associated with adverse blood lipid profile in British adults. J Nutr. 2010;140(1):81-87. (PubMed)
99. Rees K, Hartley L, Day C, Flowers N, Clarke A, Stranges S. Selenium supplementation for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2013;1:CD009671. (PubMed)
100. Flores-Mateo G, Navas-Acien A, Pastor-Barriuso R, Guallar E. Selenium and coronary heart disease: a meta-analysis. Am J Clin Nutr. 2006;84(4):762-773. (PubMed)
101. Jenkins DJA, Kitts D, Giovannucci EL, et al. Selenium, antioxidants, cardiovascular disease, and all-cause mortality: a systematic review and meta-analysis of randomized controlled trials. Am J Clin Nutr. 2020;112(6):1642-1652. (PubMed)
102. Kuria A, Tian H, Li M, et al. Selenium status in the body and cardiovascular disease: a systematic review and meta-analysis. Crit Rev Food Sci Nutr. 2021;61(21):3616-3625. (PubMed)
103. McKenzie RC, Beckett GJ, Arthur JR. Effects of selenium on immunity and aging. In: Hatfield DL, Berry MJ, Gladyshev VN, eds. Selenium: Its Molecular Biology and Role in Human Health. 2nd ed. New York: Springer; 2006:311-323.
104. Huang Z, Rose AH, Hoffmann PR. The role of selenium in inflammation and immunity: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2012;16(7):705-743. (PubMed)
105. Fairweather-Tait SJ, Filippini T, Vinceti M. Selenium status and immunity. Proc Nutr Soc. 2023;82(1):32-38. (PubMed)
106. Mattmiller SA, Carlson BA, Sordillo LM. Regulation of inflammation by selenium and selenoproteins: impact on eicosanoid biosynthesis. J Nutr Sci. 2013;2:e28. (PubMed)
107. Norton RL, Hoffmann PR. Selenium and asthma. Mol Aspects Med. 2012;33(1):98-106. (PubMed)
108. Speckmann B, Steinbrenner H. Selenium and selenoproteins in inflammatory bowel diseases and experimental colitis. Inflamm Bowel Dis. 2014;20(6):1110-1119. (PubMed)
109. Drain PK, Kupka R, Mugusi F, Fawzi WW. Micronutrients in HIV-positive persons receiving highly active antiretroviral therapy. Am J Clin Nutr. 2007;85(2):333-345. (PubMed)
110. Stone CA, Kawai K, Kupka R, Fawzi WW. Role of selenium in HIV infection. Nutr Rev. 2010;68(11):671-681. (PubMed)
111. de Menezes Barbosa EG, Junior FB, Machado AA, Navarro AM. A longer time of exposure to antiretroviral therapy improves selenium levels. Clin Nutr. 2015;34(2):248-51. (PubMed)
112. Baum MK, Miguez-Burbano MJ, Campa A, Shor-Posner G. Selenium and interleukins in persons infected with human immunodeficiency virus type 1. J Infect Dis. 2000;182 Suppl 1:S69-73. (PubMed)
113. Kalantari P, Narayan V, Natarajan SK, et al. Thioredoxin reductase-1 negatively regulates HIV-1 transactivating protein Tat-dependent transcription in human macrophages. J Biol Chem. 2008;283(48):33183-33190. (PubMed)
114. Burbano X, Miguez-Burbano MJ, McCollister K, et al. Impact of a selenium chemoprevention clinical trial on hospital admissions of HIV-infected participants. HIV Clin Trials. 2002;3(6):483-491. (PubMed)
115. Hurwitz BE, Klaus JR, Llabre MM, et al. Suppression of human immunodeficiency virus type 1 viral load with selenium supplementation: a randomized controlled trial. Arch Intern Med. 2007;167(2):148-154. (PubMed)
116. Kupka R, Mugusi F, Aboud S, Hertzmark E, Spiegelman D, Fawzi WW. Effect of selenium supplements on hemoglobin concentration and morbidity among HIV-1-infected Tanzanian women. Clin Infect Dis. 2009;48(10):1475-1478. (PubMed)
117. Kupka R, Mugusi F, Aboud S, et al. Randomized, double-blind, placebo-controlled trial of selenium supplements among HIV-infected pregnant women in Tanzania: effects on maternal and child outcomes. Am J Clin Nutr. 2008;87(6):1802-1808. (PubMed)
118. Baum MK, Campa A, Lai S, et al. Effect of micronutrient supplementation on disease progression in asymptomatic, antiretroviral-naive, HIV-infected adults in Botswana: a randomized clinical trial. JAMA. 2013;310(20):2154-2163. (PubMed)
119. Muzembo BA, Ngatu NR, Januka K, et al. Selenium supplementation in HIV-infected individuals: A systematic review of randomized controlled trials. Clin Nutr ESPEN. 2019;34:1-7. (PubMed)
120. Bone RC, Balk RA, Cerra FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101(6):1644-1655. (PubMed)
121. Mann EA, Baun MM, Meininger JC, Wade CE. Comparison of mortality associated with sepsis in the burn, trauma, and general intensive care unit patient: a systematic review of the literature. Shock. 2012;37(1):4-16. (PubMed)
122. Alhazzani W, Jacobi J, Sindi A, et al. The effect of selenium therapy on mortality in patients with sepsis syndrome: a systematic review and meta-analysis of randomized controlled trials. Crit Care Med. 2013;41(6):1555-1564. (PubMed)
123. Huang TS, Shyu YC, Chen HY, et al. Effect of parenteral selenium supplementation in critically ill patients: a systematic review and meta-analysis. PLoS One. 2013;8(1):e54431. (PubMed)
124. Bloos F, Trips E, Nierhaus A, et al. Effect of sodium selenite administration and procalcitonin-guided therapy on mortality in patients with severe sepsis or septic shock: a randomized clinical trial. JAMA Intern Med. 2016;176(9):1266-1276. (PubMed)
125. Rhodes A, Evans LE, Alhazzani W, et al. Surviving Sepsis Campaign: International guidelines for management of sepsis and septic shock: 2016. Crit Care Med. 2017;45(3):486-552. (PubMed)
126. Xu S, Huang H, Qian J, et al. Prevalence of Hashimoto thyroiditis in adults with papillary thyroid cancer and its association with cancer recurrence and outcomes. JAMA Netw Open. 2021;4(7):e2118526. (PubMed)
127. van Zuuren EJ, Albusta AY, Fedorowicz Z, Carter B, Pijl H. Selenium supplementation for Hashimoto's thyroiditis: summary of a Cochrane systematic review. Eur Thyroid J. 2014;3(1):25-31. (PubMed)
128. Karanikas G, Schuetz M, Kontur S, et al. No immunological benefit of selenium in consecutive patients with autoimmune thyroiditis. Thyroid. 2008;18(1):7-12. (PubMed)
129. Krysiak R, Okopien B. The effect of levothyroxine and selenomethionine on lymphocyte and monocyte cytokine release in women with Hashimoto's thyroiditis. J Clin Endocrinol Metab. 2011;96(7):2206-2215. (PubMed)
130. Negro R, Greco G, Mangieri T, Pezzarossa A, Dazzi D, Hassan H. The influence of selenium supplementation on postpartum thyroid status in pregnant women with thyroid peroxidase autoantibodies. J Clin Endocrinol Metab. 2007;92(4):1263-1268. (PubMed)
131. Turker O, Kumanlioglu K, Karapolat I, Dogan I. Selenium treatment in autoimmune thyroiditis: 9-month follow-up with variable doses. J Endocrinol. 2006;190(1):151-156. (PubMed)
132. Hu Y, Feng W, Chen H, et al. Effect of selenium on thyroid autoimmunity and regulatory T cells in patients with Hashimoto's thyroiditis: A prospective randomized-controlled trial. Clin Transl Sci. 2021;14(4):1390-1402. (PubMed)
133. Marcocci C, Kahaly GJ, Krassas GE, et al. Selenium and the course of mild Graves' orbitopathy. N Engl J Med. 2011;364(20):1920-1931. (PubMed)
134. Watt T, Cramon P, Bjorner JB, et al. Selenium supplementation for patients with Graves' hyperthyroidism (the GRASS trial): study protocol for a randomized controlled trial. Trials. 2013;14:119. (PubMed)
135. Winther KH, Watt T, Bjorner JB, et al. The chronic autoimmune thyroiditis quality of life selenium trial (CATALYST): study protocol for a randomized controlled trial. Trials. 2014;15:115. (PubMed)
136. Peplow D, Edmonds R. Health risks associated with contamination of groundwater by abandoned mines near Twisp in Okanogan County, Washington, USA. Environ Geochem Health. 2004;26(1):69-79. (PubMed)
137. Chang JC, Gutenmann WH, Reid CM, Lisk DJ. Selenium content of Brazil nuts from two geographic locations in Brazil. Chemosphere. 1995;30(4):801-802. (PubMed)
138. Rayman MP, Infante HG, Sargent M. Food-chain selenium and human health: spotlight on speciation. Br J Nutr. 2008;100(2):238-253. (PubMed)
139. Reyes LH, Encinar JR, Marchante-Gayon JM, Alonso JI, Sanz-Medel A. Selenium bioaccessibility assessment in selenized yeast after "in vitro" gastrointestinal digestion using two-dimensional chromatography and mass spectrometry. J Chromatogr A. 2006;1110(1-2):108-116. (PubMed)
140. Bermingham EN, Hesketh JE, Sinclair BR, Koolaard JP, Roy NC. Selenium-enriched foods are more effective at increasing glutathione peroxidase (GPx) activity compared with selenomethionine: a meta-analysis. Nutrients. 2014;6(10):4002-4031. (PubMed)
141. Giacosa A, Faliva MA, Perna S, Minoia C, Ronchi A, Rondanelli M. Selenium fortification of an Italian rice cultivar via foliar fertilization with sodium selenate and its effects on human serum selenium levels and on erythrocyte glutathione peroxidase activity. Nutrients. 2014;6(3):1251-1261. (PubMed)
142. Lei XG, Combs GF, Jr., Sunde RA, Caton JS, Arthington JD, Vatamaniuk MZ. Dietary selenium across species. Annu Rev Nutr. 2022;42:337-375. (PubMed)
143. Bleys J, Navas-Acien A, Guallar E. Serum selenium and diabetes in U.S. adults. Diabetes Care. 2007;30(4):829-834. (PubMed)
144. Laclaustra M, Navas-Acien A, Stranges S, Ordovas JM, Guallar E. Serum selenium concentrations and diabetes in U.S. adults: National Health and Nutrition Examination Survey (NHANES) 2003-2004. Environ Health Perspect. 2009;117(9):1409-1413. (PubMed)
145. Yang J, Chen E, Choi C, et al. Cross-sectional association of blood selenium with glycemic biomarkers among U.S. adults with normoglycemia in the National Health and Nutrition Examination Survey 2013-2016. Nutrients. 2022;14(19):3972. (PubMed)
146. Vinceti M, Filippini T, Wise LA, Rothman KJ. A systematic review and dose-response meta-analysis of exposure to environmental selenium and the risk of type 2 diabetes in nonexperimental studies. Environ Res. 2021;197:111210. (PubMed)
147. Stranges S, Marshall JR, Natarajan R, et al. Effects of long-term selenium supplementation on the incidence of type 2 diabetes: a randomized trial. Ann Intern Med. 2007;147(4):217-223. (PubMed)
148. Karp DD, Lee SJ, Keller SM, et al. Randomized, double-blind, placebo-controlled, phase III chemoprevention trial of selenium supplementation in patients with resected stage I non-small-cell lung cancer: ECOG 5597. J Clin Oncol. 2013;31(33):4179-4187. (PubMed)
149. Schoenmakers E, Agostini M, Mitchell C, et al. Mutations in the selenocysteine insertion sequence-binding protein 2 gene lead to a multisystem selenoprotein deficiency disorder in humans. J Clin Invest. 2010;120(12):4220-4235. (PubMed)
150. Steinbrenner H. Interference of selenium and selenoproteins with the insulin-regulated carbohydrate and lipid metabolism. Free Radic Biol Med. 2013;65:1538-1547. (PubMed)
151. Kaur P, Rizk NM, Ibrahim S, et al. iTRAQ-based quantitative protein expression profiling and MRM verification of markers in type 2 diabetes. J Proteome Res. 2012;11(11):5527-5539. (PubMed)
152. Yang SJ, Hwang SY, Choi HY, et al. Serum selenoprotein P levels in patients with type 2 diabetes and prediabetes: implications for insulin resistance, inflammation, and atherosclerosis. J Clin Endocrinol Metab. 2011;96(8):E1325-1329. (PubMed)
153. Steinbrenner H, Duntas LH, Rayman MP. The role of selenium in type-2 diabetes mellitus and its metabolic comorbidities. Redox Biol. 2022;50:102236. (PubMed)
154. Cardoso BR, Braat S, Graham RM. Selenium status is associated with insulin resistance markers in adults: findings from the 2013 to 2018 National Health and Nutrition Examination Survey (NHANES). Front Nutr. 2021;8:696024. (PubMed)
155. Azrak RG, Cao S, Pendyala L, et al. Efficacy of increasing the therapeutic index of irinotecan, plasma and tissue selenium concentrations is methylselenocysteine dose dependent. Biochem Pharmacol. 2007;73(9):1280-1287. (PubMed)
156. Graf WD, Oleinik OE, Glauser TA, Maertens P, Eder DN, Pippenger CE. Altered antioxidant enzyme activities in children with a serious adverse experience related to valproic acid therapy. Neuropediatrics. 1998;29(4):195-201. (PubMed)
157. Vernie LN, de Goeij JJ, Zegers C, de Vries M, Baldew GS, McVie JG. Cisplatin-induced changes of selenium levels and glutathione peroxidase activities in blood of testis tumor patients. Cancer Lett. 1988;40(1):83-91. (PubMed)
158. Flodin NW. Micronutrient supplements: toxicity and drug interactions. Prog Food Nutr Sci. 1990;14(4):277-331. (PubMed)
159. Brown BG, Zhao XQ, Chait A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345(22):1583-1592. (PubMed)
160. Tousoulis D, Antoniades C, Stefanadis C. Statins and antioxidant vitamins: should co-administration be avoided? J Am Coll Cardiol. 2006;47(6):1237; author reply 1237-1238. (PubMed)
161. Hurst R, Armah CN, Dainty JR, et al. Establishing optimal selenium status: results of a randomized, double-blind, placebo-controlled trial. Am J Clin Nutr. 2010;91(4):923-931. (PubMed)
162. Burk RF, Norsworthy BK, Hill KE, Motley AK, Byrne DW. Effects of chemical form of selenium on plasma biomarkers in a high-dose human supplementation trial. Cancer Epidemiol Biomarkers Prev. 2006;15(4):804-810. (PubMed)
163. US Department of Health and Human Services, National Institutes of Health, Office of Dietary Supplements. Dietary Supplement Label Database (DSLD). [Internet]. [cited 10/25/2023]. Available from: https://dsld.od.nih.gov.
164. Algotar AM, Stratton MS, Ahmann FR, et al. Phase 3 clinical trial investigating the effect of selenium supplementation in men at high-risk for prostate cancer. Prostate. 2013;73(3):328-335. (PubMed)
165. Chiang EC, Shen S, Kengeri SS, et al. Defining the optimal selenium dose for prostate cancer risk reduction: insights from the U-shaped relationship between selenium status, DNA damage, and apoptosis. Dose Response. 2009;8(3):285-300. (PubMed)
166. Ravn-Haren G, Krath BN, Overvad K, et al. Effect of long-term selenium yeast intervention on activity and gene expression of antioxidant and xenobiotic metabolising enzymes in healthy elderly volunteers from the Danish Prevention of Cancer by Intervention by Selenium (PRECISE) pilot study. Br J Nutr. 2008;99(6):1190-1198. (PubMed)
Sodium (Chloride)
Contents
Summary
- Sodium and chloride — major electrolytes of the fluid compartment outside of cells (i.e., extracellular) — work together to control extracellular volume and blood pressure. Disturbances in sodium concentrations in the extracellular fluid are associated with disorders of water balance. (More information)
- Various mechanisms act on the kidney to ensure that the amount of sodium lost via renal excretion compensates adequately for the amount of sodium consumed, thereby maintaining sodium homeostasis. (More information)
- Hyponatremia (abnormally low sodium concentrations in blood) is common among older adults and in individuals with hypertension, kidney disease, and heart disease. Hyponatremia also occurs in up to 30% of hospitalized patients. (More information)
- Acute severe hyponatremia may lead to brain edema with neurologic consequences and be lethal if not promptly diagnosed and treated. Mild chronic hyponatremia with long-term adverse health effects, such as attention deficits, gait instability, falls, and bone loss and fractures, has been associated with cardiovascular morbidity and mortality. (More information)
- In 2019, the National Academy of Medicine established an adequate intake (AI) for sodium of 1.5 grams (g)/day in adults, equivalent to 3.8 g/day of sodium chloride (salt). (More information)
- The National Academy of Medicine established a Chronic Disease Risk Reduction Intake (CDRR) for sodium of 2.3 g/day (5.8 g/day of salt) for adults based on evidence of potential long-term health benefits on blood pressure and risk of hypertension and cardiovascular disease associated with reducing sodium intakes below this level. (More information)
- Current sodium intakes of the US adult population far exceed the CDRR. Sodium has been identified as a nutrient of public health concern for overconsumption. (More information)
- Excess dietary sodium is a major contributor to hypertension, which is a leading preventable risk factor for cardiovascular disease. Randomized controlled studies demonstrated that dietary sodium reduction (by 1.8 to 3.2 g/day) could lower blood pressure in subjects with elevated blood pressure. Yet, current evidence fails to support a decrease in cardiovascular morbidity and mortality with moderate sodium restriction in patients with hypertension. (More information)
- Additional adverse health outcomes, including gastric cancer, osteoporosis, and kidney stones, have also been linked to sodium overconsumption. (More information)
Salt (sodium chloride) is essential for life. Total body sodium in an average 70-kg person is of about 4,200 mmol (~100 g), of which 40% is found in bone and 60% in the fluid inside and outside of cells (1). Total body chloride averages 2,310 mmol (~82 g), of which 70% is distributed in the extracellular fluid and the remaining is found in the collagen of connective tissue (1). Multiple mechanisms work in concert to tightly regulate the body's sodium and chloride concentrations. Although this review emphasizes the function and requirements of sodium, sodium and chloride ions work together to control extracellular volume and blood pressure (1).
Function
Sodium (Na+) and chloride (Cl-) are the principal ions in the extracellular compartment, which includes blood plasma, interstitial fluid (fluid between cells), and transcellular fluid (e.g., cerebrospinal fluid, joint fluid). As such, they play critical roles in a number of life-sustaining processes.
Maintenance of membrane potential
Sodium and chloride are electrolytes that contribute to the maintenance of concentration and charge differences across cell membranes. Potassium (K+) is the principal positively charged ion (cation) inside of cells, while sodium is the principal cation in extracellular fluid. Potassium concentrations are about 30 times higher inside than outside cells, while sodium concentrations are more than 10 times lower inside than outside cells. The concentration differences between potassium and sodium across cell membranes create an electrochemical gradient known as the membrane potential. A cell's membrane potential is maintained by ion pumps in the cell membrane, especially the Na+/K+ ATPase pumps. These pumps use ATP (energy) to pump sodium out of the cell in exchange for potassium (Figure 1). Their activity has been estimated to account for 20%-40% of the resting energy expenditure in a typical adult. The large proportion of energy dedicated to maintaining sodium/potassium concentration gradients emphasizes the importance of this function in sustaining life. Tight control of cell membrane potential is critical for nerve impulse transmission, muscle contraction, and cardiac function (2-4).
Nutrient absorption and transport
Absorption of sodium in the small intestine plays an important role in the absorption of chloride, amino acids, glucose, and water. Similar mechanisms are involved in the reabsorption of these nutrients after they have been filtered from the blood by the kidneys. Chloride, in the form of hydrochloric acid (HCl), is also an important component of gastric juice, which aids the digestion and absorption of many nutrients (5).
Maintenance of blood volume and blood pressure
Because sodium is the primary determinant of extracellular fluid volume, including blood volume, a number of physiological mechanisms that regulate blood volume and blood pressure work by adjusting the body's sodium content. In the circulatory system, pressure receptors (baroreceptors) sense changes in blood pressure and send excitatory or inhibitory signals to the nervous system and/or endocrine glands to affect sodium regulation by the kidneys. In general, sodium retention results in water retention, and sodium loss results in water loss (6). Below are descriptions of three mechanisms contributing to a larger multifactorial homeostatic control system that governs blood volume and blood pressure through regulation of sodium balance. These regulatory mechanisms are especially important for the control of sodium transport in various segments of the nephron (basic structural unit of the kidney), including the proximal and distal convoluted tubules, the thick ascending limb of the loop of Henle, and the collecting duct.
Renin-angiotensin-aldosterone system
In response to a significant decrease in blood volume or pressure (e.g., serious blood loss or dehydration), the kidneys release renin into the circulation. Renin is an enzyme that splits a small peptide (angiotensin I) from a larger protein (angiotensinogen) produced by the liver. Angiotensin I is split into a smaller peptide (angiotensin II) by angiotensin converting enzyme (ACE), an enzyme present on the inner surface of blood vessels and in the lungs, liver, and kidneys. Angiotensin II stimulates the constriction of small arteries, resulting in increased blood pressure. Angiotensin II is also a potent stimulator of aldosterone synthesis by the adrenal glands. Aldosterone is a steroid hormone that acts on the kidneys to increase the reabsorption of sodium and the excretion of potassium. Retention of sodium by the kidneys increases the retention of water, resulting in increased blood volume and blood pressure (7).
Anti-diuretic hormone
Secretion of anti-diuretic hormone (ADH; also known as arginine vasopressin [AVP]) by the posterior pituitary gland is stimulated by a significant decrease in blood volume or pressure. In conjunction with the renin-angiotensin-aldosterone system, ADH stimulates epithelial sodium channels (ENaC) in apical cell membranes along kidney nephron distal tubules to increase the reabsorption of sodium and water (8).
Dopaminergic system
Dopamine is produced from L-DOPA in the kidney proximal tubules and acts on dopamine receptors distributed along the proximal tubules and the thick ascending limbs of the loop of Henle to regulate sodium transport. Dopamine promotes sodium excretion (natriuresis) by inhibiting the Na+/H+ exchanger and Na+/phosphate (Pi) co-transporter in apical cell membranes and the Na+/bicarbonate (HCO3-) co-transporter and Na+/K+ ATPase in basolateral cell membranes. The inhibitory effect of dopamine on Na+/K+ ATPase is enhanced by the natriuretic hormone, atrial natriuretic peptide (ANP), which is secreted by heart muscle cells into the circulation (9).
Deficiency
Hyponatremia, defined as a serum sodium concentration ([Na+]) <136 mmol/liter (mM), may result from increased fluid retention (dilutional hyponatremia) or increased sodium loss. Inadequate sodium intakes rarely result in hyponatremia, even in those on very low-salt diets, because the kidneys increase the excretion of water in order to maintain serum osmolality (i.e., electrolyte-water balance). The results of the 1999-2004 US National Health And Nutrition Examination Survey (NHANES) indicated an overall prevalence of hyponatremia of 1.9% in a US population representative sample of 14,697 participants aged 18 and older (10). Hyponatremia was found to be more prevalent among older individuals (3.1% in those aged 65 to 84 years) and in those suffering from hypertension (2.9%), diabetes mellitus (3.3%), coronary heart disease (CHD; 2.6%), stroke (3.6%), chronic obstructive pulmonary disease (COPD; 3.9%), cancer (3.4%), and psychiatric disorders (2.9%) (10). Hyponatremia is also common in hospitalized patients, with an estimated 15%-30% having mild hyponatremia (serum [Na+]: 130-135 mM) and up to 7% having moderate-to-severe hyponatremia (serum [Na+] <130 mM) (11).
Causes of hyponatremia
Dilutional hyponatremia may be due to inappropriate anti-diuretic hormone (ADH) secretion, which is associated with disorders affecting the central nervous system, and with use of certain drugs (see Drug interactions). In some cases, excessive water intake may also lead to dilutional hyponatremia (see also Exercise-associated hyponatremia). Conditions that increase the loss of sodium and chloride include severe or prolonged vomiting or diarrhea, excessive and persistent sweating, the use of some diuretics, and some forms of kidney disease. Too severe restriction of dietary sodium intake in renal patients with hypertension and congestive heart failure might also result in harmful body depletion of sodium (1).
Exercise-associated hyponatremia
Exercise-associated hyponatremia (EAH) is dilutional hyponatremia occurring in individuals competing in endurance (up to 6 h in duration) and ultra-endurance (>6 h in duration) exercise events, such as marathons, Ironman triathlons, mountain bike races, hiker treks, and open-water ultra-distance swimming events. Of note, symptomatic EAH has been increasingly reported in shorter events, such as half-marathons and sprint triathlons. The development of hyponatremia during or up to 24 h after intense and/or sustained physical activity has been linked to fluid overload due to excessive water intakes, impaired urinary water excretion due to persistent ADH secretion, and very low or very high ambient temperature (12). Risk factors include pre-exercise hyperhydration, use of non-steroidal anti-inflammatory drugs (NSAIDs), and prolonged exercise (>4 h) (reviewed in 12).
Signs and symptoms of hyponatremia
Symptoms of hyponatremia include headache, nausea, vomiting, muscle cramps, fatigue, disorientation, and fainting. Complications of severe and rapidly developing hyponatremia may include cerebral edema (swelling of the brain), seizures, coma, and brain damage. Acute or severe hyponatremia may be fatal without prompt and appropriate medical treatment (13).
Chronic mild hyponatremia has been associated with deficits in gait and attention, falls, and bone loss and fractures, especially in women and the elderly (14, 15). A recent 11-year prospective cohort study in over 3,000 men free of cardiovascular disease (CVD) also reported significantly higher risks of stroke, coronary heart disease, total CVD events, CVD-related mortality, and all-cause mortality in participants with mild-to-severe hyponatremia (serum [Na+] <139 mM) compared to those with serum [Na+] between 139 mM and 144 mM (16). In addition, in a meta-analysis of 81 observational studies in patients with diverse medical conditions (including cardiovascular disease, pulmonary infections, and cirrhosis), the risk of mortality was found to be nearly three times greater in hyponatremic compared to normonatremic subjects (17). Improvement or normalization of serum [Na+] in hyponatremic subjects was associated with a reduced mortality rate in patients with diverse clinical conditions (18).
Drug interactions
Table 1 lists some medications that may increase the risk of hyponatremia (10, 19).
The Adequate Intake (AI) for Sodium
In 2019, the Food and Nutrition Board (FNB) of the National Academy of Medicine revised the Dietary Reference Intakes (DRIs) for sodium (20). The FNB did not find sufficient evidence to determine an Estimated Average Requirement (EAR) and derive a Recommended Dietary Allowance (RDA). Instead, they established an adequate intake (AI) for sodium (Table 2; 20). Considerations accounted for by the FNB for establishing an AI for sodium included that the available evidence was insufficient to identify adverse health effects associated with low dietary sodium intakes and that there was substantial evidence suggesting potential long-term health benefits associated with reducing habitual sodium intakes below 2,300 mg/day (see The Chronic Disease Risk Reduction Intake for sodium). The AI could not be derived from the average dietary intakes of apparently healthy people in the US since average intake levels are well above 2,300 mg/day (see Sources).
Sources
Most of the sodium and chloride in the diet comes from salt (21). Very little sodium occurs naturally in food. Instead, sodium is added to make certain foods shelf stable, and it is ubiquitously used in the US food supply such that all food groups contribute to sodium intake levels (21). It has been estimated that 75% of the salt intake in the US is derived from salt added during food processing or manufacturing, rather than from salt added at the table or during cooking (22). The lowest salt intakes are associated with diets that emphasize unprocessed foods, especially fruit, vegetables, and legumes. Combined data of the US National Health And Nutrition Examination Surveys (NHANES) 2007-2008 and 2009-2010 indicated average dietary sodium intakes of 3,100 mg/day in children (ages, 3-18 years), 3,800 mg/day in adults (ages, 19-50 years), and 3,300 mg/day in older adults (>50 years) (23). Usual intakes were estimated to be 4,400 mg/day and 3,100 mg/day in adult men and women (ages, 19-50 years), respectively. Overall, sodium intakes among males of all age groups were found to be 20%-45% higher than among females (23). These intakes are well above the sodium Chronic Disease Risk Reduction Intake (CDRR) of 2,300 mg/day (see The Chronic Disease Risk Reduction Intake for sodium).
Table 3 lists the sodium content (in milligrams [mg]) of some foods that are high in salt, and Table 4 lists some foods that are relatively low in salt. Most sodium is consumed in the form of sodium chloride (salt). The salt content of foods can be calculated by multiplying the sodium content by 2.5.
Example: 2,000 mg (2 g) of sodium x 2.5 = 5,000 mg (5 g) of salt.
For more information on the sodium content of foods, search USDA's FoodData Central.
The daily value (DV) for sodium is less than 2,400 mg. The % DV included on the Nutrition Facts label of packaged foods and beverages is meant to help consumers make informed choices and consider the foods with low (≤5% DV per serving) rather than high (≥20% DV per serving) sodium content (24).
Safety
Toxicity
Excessive intakes of sodium chloride lead to an increase in extracellular fluid volume as water is pulled from cells to maintain normal sodium concentrations outside of cells. However, as long as water needs can be met, normally functioning kidneys can excrete the excess sodium and restore the system to normal (1). Ingestion of large amounts of salt may lead to nausea, vomiting, diarrhea, and abdominal cramps (25). Hypernatremia, defined as serum sodium concentrations ([Na+]) >145 mM, is much less common than hyponatremia and rarely caused by excessive sodium intake (e.g., from the ingestion of large amounts of seawater or intravenous infusion of concentrated saline solution) (26). Hypernatremia generally develops from excess water loss (e.g., burns, respiratory infections, renal loss, osmotic diarrhea, hypothalamic disorders) or reduced water intake, frequently accompanied by an impaired thirst mechanism (6). Symptoms of hypernatremia with evidence of dehydration from excessive water loss may include dizziness or fainting, low blood pressure, and diminished urine production. Severe hypernatremia ([Na+] >158 mM) may result in altered mental status, lethargy, irritability, stupor, convulsions, and coma. Acute brain shrinkage can lead to intracranial and subarachnoid hemorrhage (26).
The Dietary Reference Intakes (DRIs) for sodium were recently revised by the Food and Nutrition Board of the National Academy of Medicine (20). Using a new expanded DRI model, the National Academy of Medicine did not find sufficient evidence of adverse toxicological effects of excessive sodium intake; therefore, they did not establish a tolerable upper intake level (UL) for sodium (20).
Health risks of excess dietary sodium
Hypertension
Normal blood pressure is defined by a systolic blood pressure below 120 mm Hg and a diastolic blood pressure below 80 mm Hg, often noted <120/80 mm Hg (27). Currently, about one-third of US adults have hypertension (blood pressure levels ≥140/90 mm Hg), and another one-third have elevated blood pressure (prehypertension, corresponding to levels ≥120/80 mm Hg and <140/90 mm Hg) that places them at risk for hypertension (28). Chronic hypertension damages the heart, blood vessels, and kidneys, thereby increasing the risk of heart disease and stroke, as well as hypertensive kidney disease. In a number of clinical studies, salt intake has been significantly correlated with left ventricular hypertrophy, an abnormal thickening of the heart muscle, which is associated with increased mortality from cardiovascular disease (29). Several lines of research, conducted over the last decades, have provided evidence of a relationship between sodium consumption and health outcomes. For instance, observational cohort studies, like the well-designed International Study of Salt and Blood Pressure (INTERSALT), have associated excess sodium intake with a progressive increase of blood pressure with age (30, 31). Further, a number of intervention studies, including the Trial of Nonpharmacologic Interventions in the Elderly (TONE), Trials Of Hypertension Prevention (TOHP), and Dietary Approaches to Stop Hypertension (DASH)-sodium, have demonstrated that dietary sodium reduction could effectively prevent or improve hypertension among population subgroups at elevated risk (see Blood pressure clinical trials).
Salt sensitivity: Blood pressure responses to short-term changes in sodium intake are heterogeneous. Indeed, some individuals have little-to-no change in blood pressure in response to sodium manipulation and are identified as "salt-resistant." In contrast, individuals who experience a greater change in blood pressure following dietary sodium manipulation are labeled "salt sensitive" (32, 33). Most of the protocols used in salt sensitivity studies involve extreme manipulations of sodium intake (sodium loading and sodium depletion) over a short timespan of a few days or up to one week. A typical controlled isocaloric intervention may include a two-phase, randomized, cross-over seven-day dietary sodium manipulation with a high-sodium diet (6.9 to 8.1 g/day) and a very-low-sodium diet (0.5 g/day). Salt resistance is often defined as a change of ≤5 mm Hg in 24-h mean arterial pressure (MAP) between a high-sodium diet and a low-sodium diet. Conversely, salt sensitivity is defined by changes of >5 mm Hg in MAP between high- and low-sodium diets (34).
About 26% of normotensive and 51% of hypertensive individuals are estimated to be salt sensitive (35). Salt sensitivity among normotensive subjects has been found to predict future hypertension (36, 37). Long-term prospective cohort studies also provided substantial evidence suggesting that sensitivity to salt may be an independent risk factor for cardiovascular disease (reviewed in 38). Salt sensitivity involves greater sodium reabsorption in the kidney proximal tubules and higher glomerular filtration rate (GFR) on a high-sodium diet than salt resistance. The rise in blood pressure is thought to compensate high sodium and fluid retention by triggering increased renal excretion. Conversely, salt resistance is associated with adequate excretion of excess sodium such that consumption of large amounts of sodium does not markedly increase blood pressure (1).
Earlier observations suggested that certain subgroups of the population, including African Americans, older individuals (>45 years), and hypertensive patients, tended to have greater average blood pressure responses to changes in sodium intake (38). Nevertheless, a recent meta-analysis of randomized controlled trials found marginal ethnic differences in blood pressure response to sodium reduction (for at least one week) compared to previously reported data (39). Research examining a genetic basis for salt sensitivity may eventually lead to better and reliable classification of individuals for salt sensitivity. Yet, at present, the analyses of common variations (known as polymorphisms) in the sequence of specific genes involved in sodium retention by the kidney failed to show consistent results (reviewed in 40). A recent meta-analysis of nine observational studies failed to show significant associations between specific gene polymorphisms in the renin-angiotensin-aldosterone system and blood pressure response to salt (41). Other polymorphisms in genes like those coding for G-protein coupled receptor kinase type 4 (GRK4), epithelial sodium channels (ENaC) and regulators, or α-adducin may favor sodium retention by the kidney, hence predisposing to blood pressure sensitivity to salt (reviewed in 40). Finally, in addition to genetic predispositions, factors like diet quality (e.g., the DASH diet) and body weight probably influence blood pressure sensitivity to salt (38).
Blood pressure clinical trials: Of particular importance are the results of long-term multicenter trials that are the most relevant to clinical and public health practice, i.e., TONE (42), TOHP (43), and DASH (Dietary Approaches to Stop Hypertension)-sodium. TONE showed that modest reduction in sodium intake by about 1.0 g/day (~2.5 g/day of salt) resulted in a better control of hypertension in older adults who initially were on blood pressure medication (42). TOHP-Phase II (the second of two hypertension prevention trials) showed that a similar level of sodium reduction significantly reduced systolic (but not diastolic) blood pressure by 1.2 mm Hg in overweight participants with prehypertension after three years and limited the onset of hypertension by 18% after four years compared to usual care (no dietary intervention) (43, 44). Adherence to the DASH diet, which emphasizes fruit, vegetables, whole grains, poultry, fish, nuts, and low-fat dairy products, was found to substantially lower systolic/diastolic blood pressures by 11.4/5.5 mm Hg in hypertensive and 3.5/2.1 mm Hg in normotensive people compared to a typical US diet (45). The DASH diet is also markedly higher in potassium and calcium, modestly higher in protein, and lower in total fat, saturated fat, cholesterol, red meat, sweets, and sugar-containing beverages than the typical US diet. The DASH-sodium trial compared the DASH diet to a typical US (control) diet at three levels of sodium intake: low (1.5 g/day, current AI), intermediate (2.3 g/day, recommended as an upper level by US dietary guidelines), and high (3.2 g/day, typical US intake) levels (46). At each level of sodium intake, systolic and diastolic blood pressures were systematically lower in (pre)hypertensive people (blood pressure >120/80 mm Hg) consuming the DASH diet compared to the control diet. Reduction of sodium intake also significantly reduced blood pressure in hypertensive consumers of either the DASH diet or the typical US diet. Yet, in prehypertensive participants, the blood pressure-lowering effect of sodium reduction was only significant in those consuming the typical US diet; sodium reduction failed to reduce blood pressure in all prehypertensive participants assigned to the DASH diet, with the exception of African Americans. Compared to the high-salt control diet, average blood pressure in (pre)hypertensive participants on the low-sodium DASH diet was decreased by 8.9/4.5 mm Hg. Results of the DASH trials support the idea that sodium reduction in the context of a healthful dietary pattern offers an effective approach to the prevention and treatment of hypertension (47).
A majority of randomized clinical trials have examined the effect of dietary sodium reduction on blood pressure in (pre)hypertensive rather than in normotensive (normal blood pressure) people. A recent meta-analysis assessed the results of modest sodium reduction from 22 trials in 990 participants with hypertension (blood pressure ≥140/90 mm Hg) and 12 trials in 2,240 participants without hypertension (blood pressure <140/90 mm Hg). A modest sodium reduction by 1.8 g/day (equivalent to 4.4 g/day of salt; based on 24-hour urinary sodium excretions) for at least four weeks decreased systolic and diastolic blood pressure by an average of 5.4/2.8 mm Hg in subjects with hypertension and 2.4/1.0 mm Hg in those without hypertension (48). Another pooled analysis of eight randomized controlled studies demonstrated a dose-response relationship between sodium reduction (by 1.8 to 3.2 g/day) and blood pressure in subjects with blood pressure over 130/80, but there was no such relationship in subjects whose blood pressure was below 130/80 (49).
The 2015-2020 Dietary Guidelines for Americans concurred with the 2013 American Heart Association (AHA)/American College of Cardiology (ACC) Guideline to recommend that individuals who would benefit from blood pressure lowering, particularly prehypertensive and hypertensive adults, should follow the DASH eating pattern to lower sodium intakes (50). Recommendations include sodium intakes of no more than 2.4 g/day, further sodium intake reduction to 1.5 g/day for an even greater blood pressure-lowering effect, or a reduction is sodium intake of at least 1 g/day if sodium intakes of 1.5 g/day or 2.4 g/day cannot be achieved (50, 51).
More information about the DASH diet is available from the National Heart, Lung, and Blood Institute (NHLBI) of the National Institutes of Health (NIH).
Endothelial dysfunction
Early studies in animals and humans reported that high-salt intake was associated with pathological alterations in the structure and function of large elastic arteries, independent of changes in blood pressure (reviewed in 52). Endothelial dysfunction is considered to be an early step in the development of atherosclerosis. Alterations in the structure and function of the vascular endothelium that lines the inner surface of all blood vessels are associated with the loss of normal nitric oxide (NO)-mediated endothelium-dependent vasodilation. Endothelial dysfunction results in widespread vasoconstriction and coagulation abnormalities. The measurement of brachial artery flow-mediated dilation (FMD) is often used as a functional marker of endothelial function; FMD values are inversely correlated with the risk of future cardiovascular events (53).
The link between sodium intake and cardiovascular disease has traditionally involved hypertension. Yet, recent investigations in salt-resistant normotensive individuals have reported that dietary sodium loading could impair endothelial function independent of changes in blood pressure (54, 55). A study using controlled dietary sodium conditions also demonstrated that a high-sodium diet (6.9 g/day for one week) reduced brachial artery FMD to the same extent in salt-sensitive and salt-resistant normotensive participants (56). On the other hand, a three-week dietary sodium intake of 3 g following a baseline intake of 2.4 g/day of sodium did not lower FMD in 36 untreated (pre)hypertensive adults with baseline FMD values significantly lower than those usually observed in healthy normotensive subjects (57). Circulating markers of endothelial function and low-grade inflammation were unchanged; only urinary sodium excretion and systolic blood pressure increased (57).
In healthy normotensive adults in whom FMD significantly decreased following a meal containing 1.5 g of sodium, potassium supplementation (1.5 g) could limit high sodium-induced FMD reduction in the postprandial state (58, 59). In a 12-week, randomized, cross-over study in 25 overweight/obese and normotensive subjects, a diet containing 2.3 g/day reduced FMD after two days and for six weeks, compared to a diet containing 3.5 g/day (60). In another randomized, cross-over, placebo-controlled trial, dietary sodium restriction (1.2 g/day vs. 3.5 g/day) for five weeks significantly lowered systolic (but not diastolic) blood pressure by 12 mm Hg and increased FMD by 68% in 17 (pre)hypertensive subjects (mean age, 62 years) (61). The improvements in both blood pressure and endothelial function suggest that sodium restriction has a strong potential for reducing CVD risk by preserving the vasculature.
Cardiovascular morbidity and mortality
A high dietary sodium intake is a risk factor for cardiovascular disease. A pooled analysis of 13 prospective cohort studies in 177,025 participants followed for 3 to 17 years found greater risks of cardiovascular disease (+17%) and stroke (+23%) with an average difference of 2 g of sodium (5 g of salt) between higher and lower daily consumption across the studies (62). Higher sodium intakes were also found to be associated with an increased risk of stroke (+24%) — but not with the risk of cardiovascular disease or coronary heart disease — in a more recent meta-analysis of 10 prospective cohort studies and randomized controlled trials (63).
A meta-analysis of randomized controlled studies examined the effect of dietary sodium reduction on cardiovascular events and mortality (64). Dietary interventions aiming at reducing sodium intake failed to demonstrate an effect on all-cause mortality in hypertensive and non-hypertensive individuals. Current evidence also fails to support a decrease in cardiovascular morbidity and mortality in patients with hypertension (64). Likewise, the Centers for Disease Control and Prevention (CDC)-sponsored 2013 report on Sodium Intake in Populations by the US Institute of Medicine (IOM; renamed the National Academy of Medicine) indicated that evidence was scarce and of poor quality to assess whether sodium intakes below 2.3 g/day may lower the risk of heart disease, stroke, and all-cause mortality in the general US population (65). In addition, the IOM committee found no evidence of benefits but some evidence of potential harm in reducing sodium intakes to 1.5 g/day-2.3 g/day in individuals with diabetes mellitus, kidney disease, or cardiovascular disease (65). However, in a recent review of the literature, the US Agency for Healthcare Research and Quality (AHRQ) identified low strength evidence to suggest that sodium reduction could decrease the risk of a composite measure of cardiovascular outcomes (seven trials) and the risk of combined cardiovascular morbidity and mortality (eight trials) (66).
Gastric cancer
In the evidence-based report, Food, Nutrition, Physical Activity, and the Prevention of Cancer (2007), the World Cancer Research Fund/American Institute for Cancer Research concluded that salt was a probable cause of stomach cancer (67). A recent meta-analysis of seven prospective cohort studies in nearly 270,000 participants showed a 68% greater risk of gastric cancer with the highest versus lowest salt intake level (68). Findings from observational studies, conducted mainly in Asian countries, also suggested an increased risk of gastric cancer with high intakes of salted foods, pickled foods, and processed meat products (68-70). Low intakes of fruit and vegetables, which are protective against gastric cancer, in populations with high intakes of salted foods might also contribute to increasing the risk of gastric cancer (69, 71).
Animal studies suggested that high concentrations of salt may damage the cells lining the stomach, potentially increasing the risk of bacterial infection by Helicobacter pylori (72, 73) and cancer-promoting genetic damage (74). The colonization by H. pylori is a recognized risk factor for gastric cancer. Case-control studies examining the potential interaction between salt intake and H. pylori infection in the risk of gastric cancer have provided mixed results (75-78). Although there is little evidence that salt itself is a carcinogen, high intakes of salted foods may increase the risk of gastric cancer in individuals infected with H. pylori or exposed to gastric carcinogens (67). Salty foods, like processed meat, cured meat, and salted fish, contain high levels of nitrosated compounds that may contribute to increasing the risk of gastric cancer (67).
Osteoporosis
Osteoporosis is a multifactorial skeletal disorder in which bone strength is compromised, resulting in an increased risk of fracture. Nutrition is one of many factors contributing to the development and progression of osteoporosis. Dietary sodium is a major determinant of urinary calcium loss (79). High-sodium intake results in increased loss of calcium in the urine, possibly due to competition between sodium and calcium for reabsorption in the kidneys or by an effect of sodium on parathyroid hormone (PTH) secretion (see the article on Calcium). Every 1-g increment in sodium (2.5 g of salt) excreted by the kidneys has been found to draw about 26.3 mg of calcium into the urine (79). A study conducted in adolescent girls reported that a high-salt diet had a greater effect on urinary sodium and calcium excretion in White compared to Black girls, suggesting differences among ethnic groups (80). In adult women, each extra gram of sodium consumed per day is projected to produce an additional rate of bone loss of 1% per year if all of the calcium loss comes from the skeleton.
A number of cross-sectional and intervention studies have suggested that high-sodium intakes are deleterious to bone health, especially in older women (81). In particular, high-sodium intake in conjunction with low-calcium intake may be especially detrimental to bone health (82-84). A two-year longitudinal study in postmenopausal women found increased urinary sodium excretion (an indicator of increased sodium intake) to be associated with decreased bone mineral density (BMD) at the hip (85). Linear regression analysis estimated that BMD could be maintained by reducing sodium intake to a level of 2.3 g/day and by increasing calcium intake to 1.2 g/day. A second longitudinal study in postmenopausal women found that habitual sodium intake of approximately 3 g/day was not detrimental to BMD over three years of follow-up (86). Notably, the average calcium intake in this study population was 1.3 to 1.5 g/day — slightly above the RDA for calcium in women over 50 years. Another study in 40 postmenopausal women found that adherence to a low-sodium diet (2 g/day) for six months was associated with significant reductions in sodium excretion, calcium excretion, and aminoterminal propeptide of type I collagen (a biomarker of bone resorption). Yet, these associations were only observed in women with elevated baseline urinary sodium excretions (87). Finally, in a randomized, placebo-controlled study in 60 postmenopausal women, potassium citrate supplementation prevented an increase in calcium excretion induced by the consumption of a high-sodium diet (≥5 g/day of sodium) for four weeks (88).
Kidney stones
Most kidney stones are composed of calcium oxalate or calcium phosphate. Subjects with an abnormally high level of calcium in the urine (hypercalciuria) are at higher risk of developing kidney stones (a process called nephrolithiasis) (89). A large prospective cohort study that followed more than 90,000 women over a 12-year period found that women with a sodium intake averaging 4.9 g/day (12.6 g/day of salt) had a 30% higher risk of developing symptomatic kidney stones than women whose sodium intake averaged 1.5 g/day (3.8 g/day of salt) (90). Because urinary calcium excretion is increased by high sodium intakes (79), dietary sodium restriction may reduce the risk of stone formation, especially in patients with a history of kidney stones, by limiting calcium excretion (91). A five-year, randomized intervention study that enrolled 120 men with idiopathic hypercalciuria (mean age, 45 years) reported that those assigned to a normal-to-high calcium (1.2 g/day) and low-sodium diet (1.2 g/day) had a 49% reduced risk of kidney stone recurrence compared to those on a low-calcium diet (~0.4 g/day) (92).
The Chronic Disease Risk Reduction Intake
In 2004, the Food and Nutrition Board (FNB) of the National Academy of Medicine (formerly, the Institute of Medicine [IOM]) established a tolerable upper intake level (UL) of 2.3 g/day sodium (5.8 g/day of salt) for adults based on the adverse effects of high sodium intakes on blood pressure, a major risk factor for cardiovascular and kidney diseases (93). The 2015-2020 Dietary Guidelines for Americans report also recognizes that excess sodium consumption poses potential health risks and emphasizes the reduction of sodium intake in the context of a healthful dietary pattern, following the UL set by the IOM panel (50).
In a 2019 update of the Dietary Reference Intakes (DRIs) for sodium, the National Academy of Medicine made use of a new expanded DRI model (20). As a consequence, only evidence of a toxicological risk associated with excessive sodium intakes was considered to establish a UL for sodium (20), whereas previously the UL for sodium was based on evidence of any type of adverse effects (93). In addition, the evidence of the relationship between sodium intake levels, blood pressure, and cardiovascular disease — considered toward establishing a UL in the previous report — has now been reviewed to establish a new DRI category, namely the Chronic Disease Risk Reduction Intake (CDRR) for sodium (20).
Using the expanded DRI model, the National Academy of Medicine did not find sufficient evidence of adverse toxicological effects to establish a UL for sodium (20).
In contrast, substantial evidence from trials showing a lowering of the risks of hypertension and cardiovascular disease with reduction of sodium intakes was used to set a CDRR for sodium in apparently healthy adults at 2,300 mg/day (20). The CDRR for healthy adults means that a lowering of usual sodium intakes to at least 2,300 mg/day from higher levels is expected to reduce the risk of chronic disease.
The CDRR values for other ages/life stages have been extrapolated from the CDRR value set for adults using estimated energy requirements; these CDRR-based recommendations for each age/life stage are presented in Table 5.
Of note, in its 2013 report on Sodium Intake in Populations, the FNB committee considered that recommendations to population subgroups, including those who are most sensitive to the blood pressure effects of sodium, like older people (≥51 years), African Americans, and individuals with hypertension, diabetes mellitus, or chronic kidney disease, should be similar to those for the general US population (65). The IOM committee found no supportive evidence to recommend that these subgroups lower their sodium intake to 1.5 g/day or below (65). In contrast, the 2013 American Heart Association (AHA)/American College of Cardiology (ACC) Guideline on Lifestyle Management to Reduce Cardiovascular Risk advises adults with (pre)hypertension to consume no more than 2.4 g/day of sodium, further reduce sodium intake to 1.5 g/day for an even greater blood pressure-lowering effect, or reduce their daily intake by at least 1 g if sodium intakes of 1.5 g/day or 2.4 g/day cannot be achieved (51).
Finally, the 2010 IOM report on Strategies to Reduce Sodium Intake in the United States suggested that the US Food and Drug Administration (FDA) revisit the Generally Recognized As Safe (GRAS) status of salt added to processed food, restaurant food, and food additives, in order to reduce the salt content in the food supply and assist in achieving sodium intakes consistent with US Dietary Guidelines and IOM recommendations (21). These recommendations are currently being reviewed by the FDA (22).
Drug interactions
Taking sodium bicarbonate orally may reduce the efficacy of the antibiotic cefpodoxime and the antidiabetic drug chlorpropamide by limiting drug absorption or increasing drug urinary excretion. Intravenous administration of sodium bicarbonate may also reduce the effects of aspirin and the nasal decongestant pseudoephedrine. Excessive intake of sodium bicarbonate may increase the risk of hypokalemia (abnormally low blood potassium concentration) in patients taking potassium-depleting drugs like diuretics (e.g., hydrochlorothiazide, furosemide, or bumetanide), the anti-gout agent colchicine, and calcium- or magnesium-containing antacids (94).
Linus Pauling Institute Recommendation
There is strong and consistent evidence that diets relatively low in sodium (2.3 g/day or less) and high in potassium are associated with decreased risk of high blood pressure and the related risks of cardiovascular and kidney diseases. Moreover, the DASH trial demonstrated that a diet emphasizing fruit, vegetables, whole grains, nuts, and low-fat dairy products substantially lowered blood pressure, an effect that was enhanced by reducing salt intake to 2.3 g/day of sodium (5.8 g/day of salt). The Linus Pauling Institute recommends a diet that is rich in fruit and vegetables (at least 9 servings/day) and limits processed foods that are high in salt.
Older adults (>50 years)
Since sensitivity to the blood pressure-raising effect of salt increases with age, sodium reduction in the context of a healthful dietary pattern may especially benefit older adults, who are at increased risk of high blood pressure, cardiovascular disease, and kidney disease.
Authors and Reviewers
Originally written in 2001 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in February 2004 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in November 2008 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in May 2016 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in December 2016 by:
Harry G. Preuss, M.D., M.A.C.N., C.N.S.
Professor of Biochemistry, Physiology, Medicine, and Pathology
Georgetown University Medical Center
Last updated 4/11/19 Copyright 2001-2024 Linus Pauling Institute
References
1. Preuss HG, Clouatre DL. Sodium, chloride, and potassium. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. John Wiley & Sons; 2012:475-492.
2. Clausen T. Quantification of Na+,K+ pumps and their transport rate in skeletal muscle: functional significance. J Gen Physiol. 2013;142(4):327-345. (PubMed)
3. Larsen BR, Stoica A, MacAulay N. Managing brain extracellular K(+) during neuronal activity: the physiological role of the Na(+)/K(+)-ATPase subunit isoforms. Front Physiol. 2016;7:141. (PubMed)
4. Shattock MJ, Ottolia M, Bers DM, et al. Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J Physiol. 2015;593(6):1361-1382. (PubMed)
5. Sullivan S, Alpers D, Klein S. Nutritional physiology of the alimentary tract. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Lippincott Williams & Wilkins; 2014:540-573.
6. Bailey JL, Sands JM, Franch HA. Water, electrolytes, and acid-base metabolism. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Lippincott Williams & Wilkins; 2014:102-132.
7. Kopple JD. Nutrition, diet, and the kidney. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Lippincott Williams & Wilkins; 2014:1330-1371.
8. Kortenoeven ML, Pedersen NB, Rosenbaek LL, Fenton RA. Vasopressin regulation of sodium transport in the distal nephron and collecting duct. Am J Physiol Renal Physiol. 2015;309(4):F280-299. (PubMed)
9. Rayner B, Ramesar R. The importance of G protein-coupled receptor kinase 4 (GRK4) in pathogenesis of salt sensitivity, salt sensitive hypertension and response to antihypertensive treatment. Int J Mol Sci. 2015;16(3):5741-5749. (PubMed)
10. Mohan S, Gu S, Parikh A, Radhakrishnan J. Prevalence of hyponatremia and association with mortality: results from NHANES. Am J Med. 2013;126(12):1127-1137. (PubMed)
11. Hoorn EJ, Lindemans J, Zietse R. Development of severe hyponatraemia in hospitalized patients: treatment-related risk factors and inadequate management. Nephrol Dial Transplant. 2006;21(1):70-76. (PubMed)
12. Urso C, Brucculeri S, Caimi G. Physiopathological, epidemiological, clinical and therapeutic aspects of exercise-associated hyponatremia. J Clin Med. 2014;3(4):1258-1275. (PubMed)
13. Giuliani C, Peri A. Effects of hyponatremia on the brain. J Clin Med. 2014;3(4):1163-1177. (PubMed)
14. Holm JP, Amar AO, Hyldstrup L, Jensen JE. Hyponatremia, a risk factor for osteoporosis and fractures in women. Osteoporos Int. 2016;27(3):989-1001. (PubMed)
15. Zaino CJ, Maheshwari AV, Goldfarb DS. Impact of mild chronic hyponatremia on falls, fractures, osteoporosis, and death. Am J Orthop (Belle Mead NJ). 2013;42(11):522-527. (PubMed)
16. Wannamethee SG, Shaper AG, Lennon L, Papacosta O, Whincup P. Mild hyponatremia, hypernatremia and incident cardiovascular disease and mortality in older men: A population-based cohort study. Nutr Metab Cardiovasc Dis. 2016;26(1):12-19. (PubMed)
17. Corona G, Giuliani C, Parenti G, et al. Moderate hyponatremia is associated with increased risk of mortality: evidence from a meta-analysis. PLoS One. 2013;8(12):e80451. (PubMed)
18. Corona G, Giuliani C, Verbalis JG, Forti G, Maggi M, Peri A. Correction: hyponatremia improvement is associated with a reduced risk of mortality: evidence from a meta-analysis. PLoS One. 2016;11(3):e0152846. (PubMed)
19. Adrogue HJ, Madias NE. Hyponatremia. N Engl J Med. 2000;342(21):1581-1589. (PubMed)
20. Food and Nutrition Board, National Academy of Medicine. Dietary Reference Intakes for Sodium and Potassium -- uncorrected proofs. Washington, D.C.: The National Academies Press; 2019. (The National Academies Press)
21. Institute of Medicine Committee on Strategies to Reduce Sodium Intake. Strategies to reduce sodium intake in the United States. Washington, D.C. 2010.
22. US Food and Drug Administration. Lowering salt in your diet. March 29, 2016. http://www.fda.gov/ForConsumers/ConsumerUpdates/ucm181577.htm. Accessed 5/6/16.
23. Agarwal S, Fulgoni VL, 3rd, Spence L, Samuel P. Sodium intake status in United States and potential reduction modeling: an NHANES 2007-2010 analysis. Food Sci Nutr. 2015;3(6):577-585. (PubMed)
24. US Food and Drug Administration. Sodium in your diet: use the Nutrition Facts label and reduce your intake. May 22, 2016. Available at: http://www.fda.gov/Food/ResourcesForYou/Consumers/ucm315393.htm. Accessed 5/23/16.
25. Minerals. Drug Facts and Comparisons. St. Louis: Facts and Comparisons; 2000:27-51.
26. Reynolds RM, Padfield PL, Seckl JR. Disorders of sodium balance. BMJ. 2006;332(7543):702-705. (PubMed)
27. American Heart Association. Understanding blood pressure readings. March 23, 2016. Available at: http://www.heart.org/HEARTORG/Conditions/HighBloodPressure/AboutHighBloodPressure/Understanding-Blood-Pressure-Readings_UCM_301764_Article.jsp#.VyzuIxJJmM8. Accessed 5/6/16.
28. Centers for Disease Control and Prevention. High blood pressure facts. February 19, 2015. Available at: http://www.cdc.gov/bloodpressure/facts.htm. Accessed 5/6/16.
29. Chrysant GS. High salt intake and cardiovascular disease: is there a connection? Nutrition. 2000;16(7-8):662-664. (PubMed)
30. Intersalt Cooperative Research Group. Intersalt: an international study of electrolyte excretion and blood pressure. Results for 24 hour urinary sodium and potassium excretion. BMJ. 1988;297(6644):319-328. (PubMed)
31. Elliott P, Stamler J, Nichols R, et al. Intersalt revisited: further analyses of 24 hour sodium excretion and blood pressure within and across populations. Intersalt Cooperative Research Group. BMJ. 1996;312(7041):1249-1253. (PubMed)
32. Luft FC, Weinberger MH. Heterogeneous responses to changes in dietary salt intake: the salt-sensitivity paradigm. Am J Clin Nutr. 1997;65(2 Suppl):612S-617S. (PubMed)
33. Weinberger MH. Salt sensitivity of blood pressure in humans. Hypertension. 1996;27(3 Pt 2):481-490. (PubMed)
34. Schmidlin O, Sebastian AF, Morris RC, Jr. What initiates the pressor effect of salt in salt-sensitive humans? Observations in normotensive blacks. Hypertension. 2007;49(5):1032-1039. (PubMed)
35. He FJ, MacGregor GA. Reducing population salt intake worldwide: from evidence to implementation. Prog Cardiovasc Dis. 2010;52(5):363-382. (PubMed)
36. Barba G, Galletti F, Cappuccio FP, et al. Incidence of hypertension in individuals with different blood pressure salt-sensitivity: results of a 15-year follow-up study. J Hypertens. 2007;25(7):1465-1471. (PubMed)
37. Mu J, Zheng S, Lian Q, Liu F, Liu Z. Evolution of blood pressure from adolescents to youth in salt sensitivies: a 18-year follow-up study in Hanzhong children cohort. Nutr J. 2012;11:70. (PubMed)
38. Franco V, Oparil S. Salt sensitivity, a determinant of blood pressure, cardiovascular disease and survival. J Am Coll Nutr. 2006;25(3 Suppl):247S-255S. (PubMed)
39. Graudal N, Jurgens G. The blood pressure sensitivity to changes in sodium intake is similar in Asians, Blacks, and Whites. An analysis of 92 randomized controlled trials. Front Physiol. 2015;6:157. (PubMed)
40. Armando I, Villar VA, Jose PA. Genomics and pharmacogenomics of salt-sensitive hypertension. Curr Hypertens Rev. 2015;11(1):49-56. (PubMed)
41. Sun J, Zhao M, Miao S, Xi B. Polymorphisms of three genes (ACE, AGT and CYP11B2) in the renin-angiotensin-aldosterone system are not associated with blood pressure salt sensitivity: A systematic meta-analysis. Blood Press. 2016;25(2):117-122. (PubMed)
42. Whelton PK, Appel LJ, Espeland MA, et al. Sodium reduction and weight loss in the treatment of hypertension in older persons: a randomized controlled trial of nonpharmacologic interventions in the elderly (TONE). TONE Collaborative Research Group. JAMA. 1998;279(11):839-846. (PubMed)
43. The Trials of Hypertension Prevention Collaborative Research Group. Effects of weight loss and sodium reduction intervention on blood pressure and hypertension incidence in overweight people with high-normal blood pressure. The Trials of Hypertension Prevention, phase II. Arch Intern Med. 1997;157(6):657-667. (PubMed)
44. Kumanyika SK, Cook NR, Cutler JA, et al. Sodium reduction for hypertension prevention in overweight adults: further results from the Trials of Hypertension Prevention Phase II. J Hum Hypertens. 2005;19(1):33-45. (PubMed)
45. Appel LJ, Moore TJ, Obarzanek E, et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N Engl J Med. 1997;336(16):1117-1124. (PubMed)
46. Sacks FM, Svetkey LP, Vollmer WM, et al. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N Engl J Med. 2001;344(1):3-10. (PubMed)
47. Greenland P. Beating high blood pressure with low-sodium DASH. N Engl J Med. 2001;344(1):53-55. (PubMed)
48. He FJ, Li J, Macgregor GA. Effect of longer term modest salt reduction on blood pressure: Cochrane systematic review and meta-analysis of randomised trials. BMJ. 2013;346:f1325. (PubMed)
49. Graudal N, Hubeck-Graudal T, Jurgens G, McCarron DA. The significance of duration and amount of sodium reduction intervention in normotensive and hypertensive individuals: a meta-analysis. Adv Nutr. 2015;6(2):169-177. (PubMed)
50. US Department of Health and Human Services and US Department of Agriculture. 2015-2020 Dietary Guidelines for Americans. Available at: http://health.gov/dietaryguidelines/2015/guidelines/.
51. Eckel RH, Jakicic JM, Ard JD, et al. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 Suppl 2):S76-99. (PubMed)
52. Safar ME, Thuilliez C, Richard V, Benetos A. Pressure-independent contribution of sodium to large artery structure and function in hypertension. Cardiovasc Res. 2000;46(2):269-276. (PubMed)
53. Ras RT, Streppel MT, Draijer R, Zock PL. Flow-mediated dilation and cardiovascular risk prediction: a systematic review with meta-analysis. Int J Cardiol. 2013;168(1):344-351. (PubMed)
54. DuPont JJ, Greaney JL, Wenner MM, et al. High dietary sodium intake impairs endothelium-dependent dilation in healthy salt-resistant humans. J Hypertens. 2013;31(3):530-536. (PubMed)
55. Greaney JL, DuPont JJ, Lennon-Edwards SL, Sanders PW, Edwards DG, Farquhar WB. Dietary sodium loading impairs microvascular function independent of blood pressure in humans: role of oxidative stress. J Physiol. 2012;590(21):5519-5528. (PubMed)
56. Matthews EL, Brian MS, Ramick MG, Lennon-Edwards S, Edwards DG, Farquhar WB. High dietary sodium reduces brachial artery flow-mediated dilation in humans with salt-sensitive and salt-resistant blood pressure. J Appl Physiol (1985). 2015;118(12):1510-1515. (PubMed)
57. Gijsbers L, Dower JI, Schalkwijk CG, et al. Effects of sodium and potassium supplementation on endothelial function: a fully controlled dietary intervention study. Br J Nutr. 2015;114(9):1419-1426. (PubMed)
58. Blanch N, Clifton PM, Petersen KS, Keogh JB. Effect of sodium and potassium supplementation on vascular and endothelial function: a randomized controlled trial. Am J Clin Nutr. 2015;101(5):939-946. (PubMed)
59. Dickinson KM, Clifton PM, Keogh JB. Endothelial function is impaired after a high-salt meal in healthy subjects. Am J Clin Nutr. 2011;93(3):500-505. (PubMed)
60. Dickinson KM, Clifton PM, Keogh JB. A reduction of 3 g/day from a usual 9 g/day salt diet improves endothelial function and decreases endothelin-1 in a randomised cross_over study in normotensive overweight and obese subjects. Atherosclerosis. 2014;233(1):32-38. (PubMed)
61. Jablonski KL, Racine ML, Geolfos CJ, et al. Dietary sodium restriction reverses vascular endothelial dysfunction in middle-aged/older adults with moderately elevated systolic blood pressure. J Am Coll Cardiol. 2013;61(3):335-343. (PubMed)
62. Strazzullo P, D'Elia L, Kandala NB, Cappuccio FP. Salt intake, stroke, and cardiovascular disease: meta-analysis of prospective studies. BMJ. 2009;339:b4567. (PubMed)
63. Aburto NJ, Ziolkovska A, Hooper L, Elliott P, Cappuccio FP, Meerpohl JJ. Effect of lower sodium intake on health: systematic review and meta-analyses. BMJ. 2013;346:f1326. (PubMed)
64. Adler AJ, Taylor F, Martin N, Gottlieb S, Taylor RS, Ebrahim S. Reduced dietary salt for the prevention of cardiovascular disease. Cochrane Database Syst Rev. 2014;12:CD009217. (PubMed)
65. US Institute of Medicine. Sodium intake in populations: Assessment of evidence. Washington, D.C.; 2013. (The National Academies Press)
66. Newberry SJ, Chung M, Anderson CAM, et al. AHRQ Comparative Effectiveness Reviews. Sodium and potassium intake: effects on chronic disease outcomes and risks. Rockville (MD): Agency for Healthcare Research and Quality (US); 2018. (PubMed)
67. World Cancer Research Fund/American Institute for Cancer Research. Food, Nutrition, Physical Activity, and the Prevention of Cancer: a Global Perspective. Washington, D.C. 2007.
68. D'Elia L, Rossi G, Ippolito R, Cappuccio FP, Strazzullo P. Habitual salt intake and risk of gastric cancer: a meta-analysis of prospective studies. Clin Nutr. 2012;31(4):489-498. (PubMed)
69. Fang X, Wei J, He X, et al. Landscape of dietary factors associated with risk of gastric cancer: A systematic review and dose-response meta-analysis of prospective cohort studies. Eur J Cancer. 2015;51(18):2820-2832. (PubMed)
70. Tsugane S. Salt, salted food intake, and risk of gastric cancer: epidemiologic evidence. Cancer Sci. 2005;96(1):1-6. (PubMed)
71. Liu C, Russell RM. Nutrition and gastric cancer risk: an update. Nutr Rev. 2008;66(5):237-249. (PubMed)
72. Bergin IL, Sheppard BJ, Fox JG. Helicobacter pylori infection and high dietary salt independently induce atrophic gastritis and intestinal metaplasia in commercially available outbred Mongolian gerbils. Dig Dis Sci. 2003;48(3):475-485. (PubMed)
73. Gaddy JA, Radin JN, Loh JT, et al. High dietary salt intake exacerbates Helicobacter pylori-induced gastric carcinogenesis. Infect Immun. 2013;81(6):2258-2267. (PubMed)
74. Kato S, Tsukamoto T, Mizoshita T, et al. High salt diets dose-dependently promote gastric chemical carcinogenesis in Helicobacter pylori-infected Mongolian gerbils associated with a shift in mucin production from glandular to surface mucous cells. Int J Cancer. 2006;119(7):1558-1566. (PubMed)
75. Fontham ET, Ruiz B, Perez A, Hunter F, Correa P. Determinants of Helicobacter pylori infection and chronic gastritis. Am J Gastroenterol. 1995;90(7):1094-1101. (PubMed)
76. Machida-Montani A, Sasazuki S, Inoue M, et al. Association of Helicobacter pylori infection and environmental factors in non-cardia gastric cancer in Japan. Gastric Cancer. 2004;7(1):46-53. (PubMed)
77. Peleteiro B, Lopes C, Figueiredo C, Lunet N. Salt intake and gastric cancer risk according to Helicobacter pylori infection, smoking, tumour site and histological type. Br J Cancer. 2011;104(1):198-207. (PubMed)
78. Zhong C, Li KN, Bi JW, Wang BC. Sodium intake, salt taste and gastric cancer risk according to Helicobacter pylori infection, smoking, histological type and tumor site in China. Asian Pac J Cancer Prev. 2012;13(6):2481-2484. (PubMed)
79. Weaver CM. Calcium. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. John Wiley & Sons; 2012:434-446.
80. Wigertz K, Palacios C, Jackman LA, et al. Racial differences in calcium retention in response to dietary salt in adolescent girls. Am J Clin Nutr. 2005;81(4):845-850. (PubMed)
81. Frassetto LA, Morris RC, Jr., Sellmeyer DE, Sebastian A. Adverse effects of sodium chloride on bone in the aging human population resulting from habitual consumption of typical American diets. J Nutr. 2008;138(2):419S-422S. (PubMed)
82. Bedford JL, Barr SI. Higher urinary sodium, a proxy for intake, is associated with increased calcium excretion and lower hip bone density in healthy young women with lower calcium intakes. Nutrients. 2011;3(11):951-961. (PubMed)
83. Heaney RP. Role of dietary sodium in osteoporosis. J Am Coll Nutr. 2006;25(3 Suppl):271S-276S. (PubMed)
84. Park SM, Jee J, Joung JY, et al. High dietary sodium intake assessed by 24-hour urine specimen increase urinary calcium excretion and bone resorption marker. J Bone Metab. 2014;21(3):189-194. (PubMed)
85. Devine A, Criddle RA, Dick IM, Kerr DA, Prince RL. A longitudinal study of the effect of sodium and calcium intakes on regional bone density in postmenopausal women. Am J Clin Nutr. 1995;62(4):740-745. (PubMed)
86. Ilich JZ, Brownbill RA, Coster DC. Higher habitual sodium intake is not detrimental for bones in older women with adequate calcium intake. Eur J Appl Physiol. 2010;109(4):745-755. (PubMed)
87. Carbone LD, Barrow KD, Bush AJ, et al. Effects of a low sodium diet on bone metabolism. J Bone Miner Metab. 2005;23(6):506-513. (PubMed)
88. Sellmeyer DE, Schloetter M, Sebastian A. Potassium citrate prevents increased urine calcium excretion and bone resorption induced by a high sodium chloride diet. J Clin Endocrinol Metab. 2002;87(5):2008-2012. (PubMed)
89. Lerolle N, Lantz B, Paillard F, et al. Risk factors for nephrolithiasis in patients with familial idiopathic hypercalciuria. Am J Med. 2002;113(2):99-103. (PubMed)
90. Curhan GC, Willett WC, Speizer FE, Spiegelman D, Stampfer MJ. Comparison of dietary calcium with supplemental calcium and other nutrients as factors affecting the risk for kidney stones in women. Ann Intern Med. 1997;126(7):497-504. (PubMed)
91. Nouvenne A, Meschi T, Prati B, et al. Effects of a low-salt diet on idiopathic hypercalciuria in calcium-oxalate stone formers: a 3-mo randomized controlled trial. Am J Clin Nutr. 2010;91(3):565-570. (PubMed)
92. Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med. 2002;346(2):77-84. (PubMed)
93. Food and Nutrition Board, Institute of Medicine. Sodium and Chloride. Dietary Reference Intakes for Water, Potassium, Sodium, Chloride, and Sulfate. Washington, D.C.: National Academy Press; 2004:247-392. (The National Academies Press)
94. Natural Medicines. Sodium bicarbonate: interactions with drugs; 2016. Available at: https://naturalmedicines.therapeuticresearch.com/.
Zinc
Contents
Summary
- Zinc is a nutritionally essential mineral needed for catalytic, structural, and regulatory functions in the body. (More information)
- Severe zinc deficiency is rare and caused by an inherited condition called acrodermatitis enteropathica. Acquired zinc deficiency is primarily due to malabsorption syndromes and chronic alcoholism. (More information)
- Dietary zinc deficiency is quite common in the developing world, affecting an estimated 2 billion people. Consumption of diets high in phytate and lacking foods from animal origin drive zinc deficiency in these populations. (More information)
- The recommended dietary allowance (RDA) for adult men and women is 11 mg/day and 8 mg/day of zinc, respectively. (More information)
- Long-term consumption of zinc in excess of the tolerable upper intake level (UL; 40 mg/day for adults) can result in copper deficiency. (More information)
- Dietary zinc deficiency has been associated with impaired growth and development in children, pregnancy complications, and immune dysfunction with increased susceptibility to infections. (More information)
- Supplementation with doses of zinc in excess of the UL is effective to reduce the duration of common cold symptoms. The use of zinc at daily doses of 50 to 180 mg for one to two weeks has not resulted in serious side effects. (More information)
- Current evidence suggests that supplemental zinc may be useful in the management of chronic conditions, such as age-related macular degeneration, diabetes mellitus, Wilson’s disease, and HIV/AIDS. (More information)
-
Zinc bioavailability is relatively high in meat, eggs, and seafood; zinc is less bioavailable from whole grains and legumes due to their high content in phytate that inhibits zinc absorption. (More information)
Zinc is an essential trace element for all forms of life. Clinical zinc deficiency in humans was first described in 1961, when the consumption of diets with low zinc bioavailability due to high phytate content (see Food sources) was associated with "adolescent nutritional dwarfism" in the Middle East (1). Since then, zinc insufficiency has been recognized by a number of experts as an important public health issue, especially in low-resource countries (2, 3).
Function
Numerous aspects of cellular metabolism are zinc-dependent. Zinc plays important roles in growth and development, immune function, neurotransmission, vision, reproduction, and intestinal ion transport (4). Using data mining approaches, it has been estimated that over 3,000 proteins in humans have functional zinc-binding sites (5). At the cellular level, the function of zinc can be divided into three categories: (1) catalytic, (2) structural, and (3) regulatory (6).
Catalytic role
Over 50 different enzymes depend on zinc for their ability to catalyze vital chemical reactions (7). Zinc-dependent enzymes can be found in all six classes of enzymes (8), as defined by the International Union of Biochemistry and Molecular Biology (9). During enzymatic reactions, zinc may have either a direct catalytic role or a structural role (i.e., stabilizing the structure of catalytic enzymes; see below).
Structural role
Zinc plays an essential role in the folding of some proteins. A finger-like structure, known as a zinc finger motif, stabilizes the structure several proteins. Examples of zinc finger proteins include the superfamily of nuclear receptors that bind and respond to steroids and other molecules, such as estrogens, thyroid hormones, vitamin D, and vitamin A (10). Zinc finger motifs in the structure of nuclear receptors allow them to bind to DNA and act as transcription factors to regulate gene expression (see Regulatory role). Zinc finger motifs are also involved in interactions of proteins with other proteins, ribonucleotides, and lipids (6). Removal of zinc from zinc-containing proteins results in protein misfolding and loss of function.
Metallothioneins are examples of proteins with a zinc-binding motif. Metallothioneins are small metal-binding cysteine-rich proteins with a high affinity for zinc. They work in concert with zinc transporters, regulating free zinc concentrations in the cytosol (11). Metallothioneins are also involved in the regulation of metal ion homeostasis, cellular defense against oxidative stress, and detoxification of heavy metals (11, 12).
The antioxidant enzyme, copper-zinc superoxide dismutase 1 (SOD 1), is made of two identical dimers, each including an active site with a catalytic copper ion and a structural zinc ion. Demetalation of SOD1 has been implicated in the formation of amyloid aggregates in some forms of inherited amyotrophic lateral sclerosis (ALS) — a motor neuron disease leading to muscle atrophy and paralysis (13).
Regulatory role
Zinc finger proteins have been found to regulate gene expression by acting as transcription factors (see above). Zinc also plays a role in cell signaling via the metal-response element (MRE)-binding transcription factor 1 (MTF1); MTF1 has a zinc finger domain that allows its binding to MRE sequences in the promoter of target genes and the subsequent expression of zinc-responsive genes (6). Zinc may also have a direct regulatory function, modulating the activity of cell-signaling enzymes and transcription factors (6). Extracellular zinc can also stimulate a zinc-sensing receptor that triggers the release of intracellular calcium, a second messenger in signaling pathways (14). Zinc has been found to influence hormone release (see Type 2 diabetes mellitus) (15) and nerve impulse transmission (16).
Nutrient interactions
Copper
Taking large quantities of zinc (50 mg/day or more) over a period of weeks can interfere with copper bioavailability. High intake of zinc induces the intestinal synthesis of a copper-binding protein called metallothionein (see the article on Copper). Metallothionein traps copper within intestinal cells and prevents its systemic absorption (see Wilson’s disease). More typical intakes of zinc do not affect copper absorption, and high copper intakes do not affect zinc absorption (17).
Iron
Iron and zinc compete for absorptive pathways (18). Supplemental (38-65 mg/day of elemental iron) but not dietary levels of iron may decrease zinc absorption (18, 19). This interaction is of concern in the management of iron supplementation during pregnancy and lactation and has led some experts to recommend zinc supplementation for pregnant and lactating women taking iron supplements (20, 21). Food fortification with iron has not been shown to negatively affect zinc absorption (22). In a placebo-controlled study, supplementation with zinc (10 mg/day) for three months in children aged eight to nine years significantly decreased serum iron concentrations, yet not to the extent of causing anemia (23). Additional randomized controlled studies have reported a worsening of nutritional iron status with chronic zinc supplementation (24, 25).
Calcium
High levels of dietary calcium impair zinc absorption in animals, but it is uncertain whether this occur in humans (17). One study showed that increasing the calcium intake of postmenopausal women by 890 mg/day in the form of milk or calcium phosphate (total calcium intake, 1,360 mg/day) reduced zinc absorption and zinc balance in postmenopausal women (26). However, another study found that increasing the calcium intake of adolescent girls by 1,000 mg/day in the form of calcium citrate malate (total calcium intake, 1,667 mg/day) did not affect zinc absorption or balance (27). Calcium in combination with phytate might affect zinc absorption, which would be particularly relevant to individuals who very frequently consume tortillas made with lime (i.e., calcium oxide). A study in 10 healthy women (age range, 21-47 years) found that high intake of dietary calcium (~1,800 mg/day) did not impair zinc absorption regardless of the phytate content of the diet (28). For more information on phytate, see Food sources.
Folate
The bioavailability of dietary folate (vitamin B9) is increased by the action of a zinc-dependent enzyme. Accordingly, some studies found low zinc intake decreased folate absorption. It was also suggested that supplementation with folic acid — the synthetic form of folate — might impair zinc utilization in individuals with marginal zinc status (17, 29). However, one study reported that supplementation with a relatively high dose of folic acid (800 µg/day) for 25 days did not alter zinc absorption or status in a group of students being fed a low-zinc diet (3.5 mg/day) (30).
Vitamin A
Zinc and vitamin A interact in several ways. Zinc is a component of retinol-binding protein, a protein necessary for transporting vitamin A in the blood. Zinc is also required for the enzyme that converts retinol (vitamin A) to retinal. This latter form of vitamin A is necessary for the synthesis of rhodopsin, a protein in the eye that absorbs light and thus is involved in dark adaptation. Zinc deficiency has been associated with a decreased release of vitamin A from the liver, which may contribute to symptoms of night blindness that are seen with zinc deficiency (31, 32).
Deficiency
Inherited zinc deficiency
Much of what is known about severe zinc deficiency was derived from the study of individuals born with acrodermatitis enteropathica, a genetic disorder resulting from the impaired uptake and transport of zinc (33). The symptoms of severe zinc deficiency include the slowing or cessation of growth and development, delayed sexual maturation, characteristic skin rashes, chronic and severe diarrhea, immune system deficiencies, impaired wound healing, diminished appetite, impaired taste sensation, night blindness, swelling and clouding of the cornea, and behavioral disturbances. Before the cause of acrodermatitis enteropathica was known, patients typically died in infancy. Oral zinc therapy results in the complete remission of symptoms, though it must be maintained indefinitely in individuals with the genetic disorder (33, 34).
Acquired zinc deficiency
It is now recognized that milder zinc deficiency contributes to a number of health problems, especially common in children who live in low-resource countries. An estimated 2 billion people worldwide are affected by dietary zinc deficiency (3). The lack of a sensitive and specific indicator of marginal zinc deficiency hinders the scientific study of its health implications (8). However, controlled trials of moderate zinc supplementation have demonstrated that marginal zinc deficiency contributes to impaired physical and neuropsychological development and increased susceptibility to life-threatening infections in young children (34). In fact, zinc deficiency has been estimated to cause more than 450,000 deaths annually in children under five years of age, comprising 4.4% of global childhood deaths (35). For a more detailed discussion of the relationship of zinc deficiency to health problems, see the section on Disease Prevention.
In industrialized countries, dietary zinc deficiency is unlikely to cause severe zinc deficiency in individuals without a genetic disorder, zinc malabsorption or conditions of increased zinc loss, such as severe burns or prolonged diarrhea. Severe zinc deficiency has also been reported in individuals undergoing total parenteral nutrition without zinc, in those who abuse alcohol, and in those who are taking certain medications like penicillamine (see Drug interactions) (36).
Individuals at risk of zinc deficiency (6, 36-38):
- Premature and low-birth-weight infants
- Older breast-fed infants and toddlers with inadequate intake of zinc-rich complementary foods
- Children and adolescents
- Pregnant and lactating (breast-feeding) women, especially adolescents
- Patients receiving total parenteral nutrition (intravenous feedings)
- Malnourished individuals, including those with protein-energy malnutrition and anorexia nervosa
- Individuals with severe or persistent diarrhea
- Individuals with malabsorption syndromes, including celiac disease and short bowel syndrome
- Individuals with inflammatory bowel disease, including Crohn's disease and ulcerative colitis
- Alcoholics and those with alcoholic liver disease who have increased urinary zinc excretion and low liver zinc levels
- Individuals with chronic renal disease
- Individuals with sickle cell anemia
- Individuals who use medications that decrease intestinal zinc absorption, increase zinc excretion, or impair zinc utilization (see Drug interactions)
- Older adults (65 years and older)
- Vegetarians: The requirement for dietary zinc may be as much as 50% greater for vegetarians whose major food staples are grains and legumes, because high levels of phytate in these foods reduce zinc absorption (see Food sources) (29).
Biomarkers of zinc status
Currently, there is not a sensitive and specific biomarker to detect zinc deficiency in humans. Low plasma or serum zinc concentrations are typically used as indicators of zinc status in populations and in intervention studies, but they have a number of limitations, including lack of sensitivity to detect marginal zinc deficiency, diurnal variations, and confounding by inflammation, stress, and hormones (38, 39).
The Recommended Dietary Allowance (RDA)
The recommended dietary allowance (RDA) for zinc is listed by gender and age group in Table 1. Infants, children, adolescents, and pregnant and lactating women are at increased risk of zinc deficiency. Since a sensitive indicator of zinc nutritional status is not readily available, the RDA for zinc is based on a number of different indicators of zinc nutritional status and represents the daily intake likely to prevent deficiency in nearly all individuals in a specific age and gender group (29).
| Life Stage | Age | Males (mg/day) | Females (mg/day) |
|---|---|---|---|
| Infants | 0-6 months | 2 (AI) | 2 (AI) |
| Infants | 7-12 months | 3 | 3 |
| Children | 1-3 years | 3 | 3 |
| Children | 4-8 years | 5 | 5 |
| Children | 9-13 years | 8 | 8 |
| Adolescents | 14-18 years | 11 | 9 |
| Adults | 19 years and older | 11 | 8 |
| Pregnancy | 18 years and younger | - | 12 |
| Pregnancy | 19 years and older | - | 11 |
| Breast-feeding | 18 years and younger | - | 13 |
| Breast-feeding | 19 years and older | - | 12 |
Prevention of Diseases or Conditions Related to Zinc Deficiency
Pregnancy complications and adverse pregnancy outcomes
Estimates based on national food supply indicate that dietary zinc intake is likely inadequate in most low- and middle-income countries, especially those in Sub-Saharan Africa and South Asia (40). Inadequate zinc status during pregnancy interferes with fetal development, and preterm neonates from zinc-deficient mothers suffer from growth retardation and dermatitis and are at risk of infections, necrotizing enterocolitis, chronic lung disease, and retinopathy of prematurity (4). Maternal zinc deficiency has also been associated with a number of pregnancy complications and poor outcomes. A recent case-control study conducted in an Iranian hospital reported higher odds of congenital malformations in newborns of mothers with low serum zinc concentrations during the last month of pregnancy (41). A 2016 review of 64 observational studies found an inverse relationship between maternal zinc status and the severity of preeclampsia, as well as between maternal zinc intake and the risk of low-birth-weight newborns (42). There were no apparent associations between maternal zinc status and the risk of gestational diabetes mellitus and preterm birth. However, the conclusions of this analysis were limited by the fact that most observational studies were conducted in women from populations not at risk for zinc deficiency (42).
To date, available evidence from maternal zinc intervention trials conducted worldwide does not support the recommendation of routine zinc supplementation during pregnancy. A 2015 systematic review and meta-analysis of 21 randomized controlled trials in over 17,000 women and their babies found a 14% reduction in premature deliveries with zinc supplementation during pregnancy, mainly in low-income women (43). This analysis, however, did not find zinc supplementation to benefit other indicators of maternal or infant health, including stillbirth or neonatal death, low birth weight, small-for-gestational age, and pregnancy-induced hypertension. There was also no effect of supplemental zinc on postpartum hemorrhage, maternal infections, congenital malformations, and child development outcomes (43). A recent review of 17 trials (of which 15 were conducted in low- and middle-income countries) found that maternal supplementation with multiple micronutrients (including, among others, zinc, iron, and folic acid) reduced the risk of low-birth-weight newborns and small-for-gestational age infants when compared to supplemental iron with or without folic acid (44). While multiple micronutrient supplementation would likely benefit pregnant women with coexisting micronutrient deficiencies in low- and middle-income countries, there is no evidence to recommend zinc supplementation in isolation in pregnant women from any settings (43, 45).
Impaired growth and development
Growth retardation
Significant delays in linear growth and weight gain, known as growth retardation or failure to thrive, are common features of mild zinc deficiency in children. In the 1970s and 1980s, several randomized, placebo-controlled studies of zinc supplementation in young children with significant growth delays were conducted in Denver, Colorado. Modest zinc supplementation (5.7 mg/day) resulted in increased growth rates compared to placebo (46). Several meta-analyses of growth data from zinc intervention trials have confirmed the widespread occurrence of growth-limiting zinc deficiency in young children, especially in low- and middle-income countries (47-49). A 2018 systematic review and meta-analysis identified 54 trials that examined the impact of zinc supplementation during infancy (on average, 7.6 mg/day for 30.9 weeks) or childhood (on average, 8.5 mg/day for 38.9 weeks) on child anthropometric measurements (50). There was evidence of a positive effect of supplemental zinc on children’s height, weight, and weight-for-age Z score (WAZ), but neither on height-for-age Z score (HAZ) or weight-for-height Z score (WHZ). In addition, zinc supplementation did not reduce the risks of underweight (WAZ<-2 standard deviation [SD]), wasting (WHZ<-2 SD), or stunting (HAZ<-2 SD) in children (50). Although the exact mechanisms for the growth-limiting effect of zinc deficiency are not known, research indicates that zinc availability affects cell-signaling systems that coordinate the response to the growth-regulating hormone, insulin-like growth factor-1 (IGF-1) (51).
Delayed mental and psychomotor development in young children
Adequate nutrition in essential for brain growth and development, especially during the first 1,000 days of life — a critical period of development for all organs and systems spanning from conception to 24 months of age (52). Animal studies have established that zinc deficiency in early life interferes with normal brain development and cognitive functions (reviewed in 53). Data on the effect of zinc supplementation during pregnancy on infants’ neurologic and psychomotor outcomes is very limited. In a randomized, placebo-controlled trial in African-American women, daily maternal supplementation with 25 mg of zinc from about 19 weeks’ gestation had no effect on neurologic development test scores in their children at five years of age (54).
Several studies have reported on the effect of postnatal zinc supplementation on mental and motor development. Two early randomized controlled trials, one conducted in India and the other in Guatemala, suggested that postnatal supplementation with 10 mg/day of zinc resulted in toddlers being more vigorous (55) and functionally active (56). In one trial conducted in Brazilian newborns from low-income families and weighing between 1,500 g and 2,499 g at birth, neither zinc supplementation for eight weeks with 1 mg/day or 5 mg/day improved mental and psychomotor development at 6 or 12 months of age compared to a placebo and assessed using the Bayley Scales of Infant Development (BSID) for Mental Development Index (MDI) and Psychomotor Development Index (PDI) (57). Additionally, a randomized, placebo-controlled, double-blind trial in Chilean newborns (birth weights >2,300 g) from low-income families reported no effect of zinc supplementation (5 mg/day) on mental and psychomotor development indices at 6 and 12 months (58). Two other trials found that supplemental zinc failed to improve MDI or PDI at 12 months of age when zinc (10 mg/day) was given to six-month-old infants for six months (59) or at the end of the intervention in toddlers aged 12-18 months when zinc (30 mg/day) was given for four months (60). A 2012 Cochrane review of eight clinical trials found no evidence that postnatal zinc supplementation improves mental or motor development of infants and children from populations with presumably inadequate zinc status (61).
Impaired immune system function
Adequate zinc intake is essential in maintaining the integrity of the immune system (62), specifically for normal development and function of cells that mediate both innate (neutrophils, macrophages, and natural killer cells) and adaptive (B-lymphocytes and T-lymphocytes) immune responses (63). Because pathogens also require zinc to thrive and invade, a well-established antimicrobial defense mechanism in the body sequesters free zinc away from microbes (64). Another opposite mechanism consists in intoxicating intracellular microbes within macrophages with excess zinc (65). Through weakening innate and adaptive immune responses, zinc deficiency diminishes the capacity of the body to combat pathogens (63, 64). As a consequence, zinc-deficient individuals experience an increased susceptibility to a variety of infectious agents (66).
Increased susceptibility to infectious disease in children
Diarrhea: Zinc promotes mucosal resistance to infections by supporting the activity of immune cells and the production of antibodies against invading pathogens (63, 64, 67). Therefore, a deficiency in zinc increases the susceptibility to intestinal infections and constitutes a major contributor to diarrheal diseases in children (66). In turn, persistent diarrhea contributes to zinc deficiency and malnutrition (66). Research indicates that zinc deficiency may also potentiate the effects of toxins produced by diarrhea-causing bacteria like E. coli (68). It is estimated that diarrheal diseases are responsible for the deaths of about 500,000 children under five years of age annually in low- and middle-income countries (69). Zinc supplementation in combination with oral rehydration therapy has been shown to significantly reduce the duration and severity of acute and persistent childhood diarrhea and to increase survival in a number of randomized controlled trials (70). A 2016 meta-analysis of randomized controlled trials found that zinc supplementation reduced the duration of acute diarrhea by one day in children aged >6 months who presented signs of malnutrition (5 trials; 419 children) (71). However, there was little evidence to suggest that zinc could be as efficacious to reduce the duration of acute diarrheal episodes in children aged <6 months and in well-nourished children aged >6 months. Zinc supplementation also reduced the duration of persistent diarrhea in children by more than half a day (5 trials; 529 children) (71).
The World Health Organization (WHO) and the United Nations Children's Fund (UNICEF) currently recommend supplementing young children with 10 to 20 mg/day of zinc as part of the treatment for acute diarrheal episodes and to prevent further episodes in the two to three months following zinc supplementation (72).
Pneumonia: Pneumonia — caused by lower respiratory tract viral or bacterial infections (LRTIs) — accounts for nearly 1 million deaths among children annually, primarily in low-and middle-income countries (69). Vaccinations against Haemophilus influenzae type B, pneumococcus, pertussis (whooping cough), and measles can help prevent pneumonia (73). According to a 2009 WHO report on disease risk factors, zinc deficiency may be responsible for 13% of all LRTI cases, primarily pneumonia and flu cases, in children younger than 5 years (74). Accordingly, a 2016 meta-analysis of six trials found that zinc supplementation in children under 5 years old reduced the risk of pneumonia by 13% (75). However, it remains unclear whether supplemental zinc, in conjunction with antibiotic therapy, is beneficial in the treatment of pneumonia. A recent randomized, placebo-controlled trial conducted in Gambian children who were not zinc deficient failed to show any benefit of zinc supplementation (10 mg/day or 20 mg/day [depending on child’s age] for 7 days) given alongside antibiotics in the treatment of severe pneumonia (76). A 2018 meta-analysis of five trials (1,822 participants) found no improvement when zinc was used as an adjunct to antibiotic treatment in children with pneumonia (77). There was, however, evidence that supplemental zinc reduced the risk of pneumonia-related mortality (3 trials; 1,318 participants) (77).
Malaria: Early studies have indicated that zinc supplementation may reduce the incidence of clinical attacks of malaria in children (78). A placebo-controlled trial in preschool-aged children in Papua New Guinea found that zinc supplementation reduced the frequency of health center attendance due to Plasmodium falciparum malaria by 38% (79). Additionally, the number of malaria episodes accompanied by high circulating parasite concentrations was reduced by 68%, suggesting that zinc supplementation may be of benefit in preventing more severe episodes of malaria. However, a six-month trial in more than 700 West African children did not find any difference in the frequency or severity of malaria episodes between children supplemented with zinc and those given a placebo (80). Another randomized controlled trial reported that zinc supplementation did not benefit preschool-aged children with acute, uncomplicated malaria (81). There is also little evidence to suggest that zinc supplementation could reduce the risk of malaria-related mortality in children (82). At present, there is not enough evidence to suggest a prophylactic and/or therapeutic role for supplemental zinc in the management of childhood malaria (48). A recent randomized, placebo-controlled trial did not provide clear-cut evidence of a protective effect of zinc (25 mg/day) administered to Tanzanian women during their first gestational trimester until delivery on the risk of placental malaria infection (83).
Age-related decline in immune function
Inadequate zinc status in elderly subjects is not uncommon and is thought to exacerbate the age-related decline in immune function (84). In one study, low serum zinc concentrations in nursing home residents were associated with higher risks of pneumonia and pneumonia-related and all-cause mortality (85). Trials examining the effects of zinc supplementation on immune function in middle-aged and elderly adults have given mixed results (reviewed in 86). Some studies showed mixed or no effects of zinc supplementation on parameters of immune function (87-89). However, zinc supplementation was found to have a positive impact on certain aspects of immune function that are affected by zinc deficiency, such as the decline in T-cell (a type of lymphocyte) function (90). For example, a randomized, placebo-controlled study in adults over 65 years of age found that zinc supplementation (25 mg/day) for three months increased blood concentrations of helper T-cells and cytotoxic T-cells (91). Additionally, a randomized, double-blind, placebo-controlled trial in 101 older adults (aged 50-70 years) with normal blood zinc concentrations showed that zinc supplementation at 15 mg/day for six months improved the helper T-cells/cytotoxic T-cells ratio, which tends to decline with age and is a predictor of survival (92). However, the study also suggested that a dose of 30 mg/day of zinc might reduce the number of B-lymphocytes, which play a central role in humoral immunity. Further, zinc supplementation had no effect on various immune parameters, including markers of inflammation, measures of granulocyte and monocyte phagocytic capacity, or cytokine production by activated monocytes (92).
A more recent trial examined the effect of daily supplementation with a multiple micronutrients, including 5 mg or 30 mg of zinc for three months, on zinc status and markers of immune function in institutionalized elderly participants (mean age, >80 years) with low serum zinc concentrations (93). Zinc status was improved with the 30 mg/day dose — but not with 5 mg/day — yet the most zinc-deficient individuals failed to achieve normal serum zinc concentrations within the intervention period. The number of circulating T-cells was also significantly increased in those who took the micronutrient supplement with the higher versus low dose of zinc (93).
More research is warranted before zinc supplementation could be recommended to older adults, especially those with no symptoms of declining immunity. Nonetheless, the high prevalence of zinc deficiency among institutionalized elderly adults should be addressed and would likely improve the performance of their immune systems (86).
Type 2 diabetes mellitus
There is a close relationship between zinc and insulin action. Specifically, in pancreatic β-cells, zinc is involved in insulin synthesis and storage in secretory vesicles. Zinc is released with the hormone when blood glucose concentrations increase (15). Zinc is also understood to stimulate glucose uptake and metabolism by insulin-sensitive tissues through triggering the intracellular insulin signaling pathway (94). Single-nucleotide polymorphisms (SNPs) in the SLC30A8 (solute carrier family 30 member 8) gene, coding for a zinc transporter that co-localizes with insulin in β-cells, have been associated with higher risks of type 1 and type 2 diabetes mellitus (95), though the risk for type 2 diabetes mellitus was found to be reduced with rare protein-truncating variants of the gene (96). The first prospective cohort study to examine the risk of type 2 diabetes in relation to zinc intakes — the Nurses’ Health Study (NHS) — followed 82,297 US registered female nurses for 24 years. The data analysis showed an 8% lower risk of type 2 diabetes with the highest versus lowest intake of dietary zinc (median values, 11.8 mg/day versus 4.9 mg/day) (97). This finding was consistent with the result of the Australian Longitudinal Study on Women’s Health (ALSWH) that enrolled 8,921 women for six years and showed a 50% lower risk of diabetes with the highest versus lowest intake of energy-adjusted dietary zinc (98). Both NHS and ALSWH studies also reported a reduced risk of diabetes with higher versus lower zinc-to-heme iron ratios in the diet (97, 98), although the significance is unclear as nonheme iron, rather than heme iron, is known to interfere with dietary zinc absorption (see Nutrient interactions). Heme iron may be an indicator of red meat consumption, which has been positively associated with the risk of type 2 diabetes (99). However, two other prospective cohort studies — the Multi-Ethnic Study of Atherosclerosis (MESA; 4,982 participants) and the National Institutes of Health-American Association of Retired Persons (NIH-AARP) Diet and Health Study (232,007 participants) — failed to find evidence for an association between zinc intake and risk of type 2 diabetes (100, 101). Another recent prospective cohort study, the Malmo Diet and Cancer Study in 26,132 middle-aged Swedish participants followed for 19 years, found an increased risk of diabetes with higher dietary zinc intakes yet a lower risk of diabetes in zinc supplement users (versus non-users) and in those with a higher zinc-to-iron intake ratio (102). The authors reported a stronger inverse association between zinc-to-iron intake ratio and risk of diabetes among obese participants carrying a specific SLC30A8 genotype (102).
The results of a few short-term intervention studies suggest that zinc supplementation may improve glucose handling in subjects with prediabetes. A 2015 systematic review identified three short trials (4 to 12 weeks) conducted in adults with prediabetes and found little evidence of an improvement in insulin resistance with zinc supplementation (103). However, a 2016 randomized, placebo-controlled trial in 55 Bangladeshi with prediabetes showed that daily supplementation with zinc sulfate (30 mg/day for 6 months) improved fasting blood glucose, as well as measures of β-cell function and insulin sensitivity (104). Similar observations were made in another recent trial in 100 Sri Lankan randomized to receive daily supplementation with zinc (20 mg of elemental zinc) or a placebo for one year (105). Supplemental zinc improved zinc status and measures of glycemic control (105). Large-scale, long-term studies are necessary to provide definite conclusions regarding the potential benefit of zinc supplementation in subjects at risk of type 2 diabetes.
Disease Treatment
Doses of supplemental zinc in many of the below-mentioned clinical trials exceeded the tolerable upper intake level (UL). Such high intake of supplemental zinc may lead to adverse health effects with prolonged use (see Safety).
Wilson's disease
The protein, ATP7B, is responsible for the excretion of hepatic copper into the biliary tract, and its impairment in Wilson's disease results in an increased concentration of 'free' copper (i.e., not bound to the copper-carrying protein, ceruloplasmin) in blood, an increased excretion of copper in the urine (hypercupriuria), the deposition of copper in part of the cornea (forming Kayser-Fleischer rings), and the accumulation of copper in the liver and brain (106). This inherited condition is progressive and fatal if untreated. The standard-of-care for symptomatic patients usually includes an initial phase (around 2-6 months) of copper chelation with agents, such as penicillamine or trientine (triethylenetetramine), followed by lifelong maintenance therapy with penicillamine and/or trientine and/or zinc salts (107). Patients presenting without symptoms can be treated with maintenance therapeutic doses of a chelating agent or with zinc (108). Zinc-induced metallothionein in the intestinal mucosa binds copper and prevents its absorption (see Nutrient interactions). There is growing evidence to suggest that zinc salts are a safer, much cheaper, and efficacious alternative to metal-chelating agents — which have been associated with a worsening of symptoms during the initial phase of treatment in some patients (109). The use of zinc is advocated as safe and efficacious in both pediatric (110, 111) and adult patients (112-114).
Common cold
Zinc lozenges
There is no proven treatment for the common cold (115). The use of zinc lozenges within 24 hours of the onset of cold symptoms, and continued intake every two to three hours while awake until symptoms resolve, have been advocated for reducing the duration of the common cold (116). Several clinical trials examining the effect of zinc have been published to date. A 2012 systematic review and meta-analysis of 13 randomized controlled trials reported that zinc supplementation in the form of lozenges or syrup shortened the duration of cold symptoms, but there was significant heterogeneity (inconsistent effects across the included studies) for the primary outcomes (117). A 2013 Cochrane review confirmed that oral zinc administrated within 24 hours of symptom onset could reduce the duration of cold symptoms (14 trials, 1,656 participants) (118). Subgroup analyses also suggested that oral zinc was effective regardless of the age of participants (children or adults) and the type of zinc formulation (gluconate/acetate lozenges or sulfate syrup). In addition, beneficial effects on cold duration were seen in trials that provided more than 75 mg/day of zinc but not in trials that used lower doses. The pooled analysis of five trials found no evidence of an effect of oral zinc on the severity of cold symptoms. The analysis of secondary trial outcomes suggested a faster resolution of specific cold symptoms (cough, nasal congestion, nasal drainage, sore throat) and a lower proportion of participants exhibiting cold symptoms after seven days of treatment in zinc- versus placebo-supplemented participants (118).
Inconsistent findings among trials have been partly attributed to different amounts of zinc released from various forms used in the lozenges (particularly zinc acetate and zinc gluconate) (119, 120). It has been argued that the unpleasant taste of zinc gluconate forming complexes with carbohydrates may have led to poor compliance, thereby explaining negative trial results (119, 121). However, when a meta-analysis was recently conducted on results from seven trials (575 participants) that employed zinc lozenges at doses >75 mg/day, there was no evidence of a difference in efficacy observed between trials that used either zinc acetate (3 trials) or zinc gluconate (4 trials) (122).
With numerous well-controlled trials and meta-analyses, the efficacy of zinc lozenges or syrup in treating common cold symptoms is no longer questionable. A meta-analysis of seven trials recently reported a 33% reduction in the duration of cold symptoms with the intake of zinc lozenges (>75 mg/day of elemental zinc) (122). However, many supplemental zinc formulations available over-the-counter have been found to release zero zinc ions (i.e., the biologically active form of zinc) or to contain additives (e.g., magnesium, certain amino acids, citric acid) that either cancel out the benefit of zinc or worsen cold symptoms (119).
Finally, although taking zinc lozenges for a cold every two to three hours while awake will result in daily zinc intakes well above the tolerable upper intake level (UL) of 40 mg/day for adults (see Safety), the use of zinc at daily doses of 50 to 180 mg for one to two weeks has not resulted in serious side effects (117). Bad taste and nausea were the most frequent adverse effects reported in therapeutic trials (117). Use of zinc lozenges for prolonged periods (e.g., 6-8 weeks) is likely to result in copper deficiency (see Nutrient interactions and Safety).
Intranasal zinc (zinc nasal gels and nasal sprays)
Intranasal zinc preparations, designed to be applied directly to the nasal epithelium (cells lining the nasal passages), are marketed as over-the-counter cold remedies. While two placebo-controlled trials found that intranasal zinc gluconate modestly shortened the duration of cold symptoms (123, 124), another one found intranasal zinc to be of no benefit (125). The pooled analysis of these three trials showed no overall benefit of intranasal zinc on the risk of still experiencing cold symptoms by day 3 (126). The existence of a mouth-nose biologically close electric circuit (BCEC) has been proposed to explain the efficacy of oral rather than intranasal zinc delivery (119). Specifically, it is suggested that the positively charged interior of the nose repels ionic zinc (Zn2+) such that ionic zinc delivered by throat lozenges and migrating from the mouth to the nose are more effective against rhinovirus infection than those directly delivered into the nose (119). Of serious concern are several case reports of individuals experiencing loss of the sense of smell (anosmia) after using intranasal zinc as a cold remedy (127). Since zinc-associated anosmia may be irreversible, intranasal zinc preparations should be avoided.
Age-related macular degeneration
Age-related macular degeneration (AMD) is a degenerative disease of the macula and a leading cause of blindness in people aged >65 years in the US (128). The macula is the portion of the retina in the back of the eye involved with central vision. Zinc is hypothesized to play a role in the development of AMD for several reasons: (1) zinc is found at high concentrations in the part of the retina affected by AMD, (2) retinal zinc content has been shown to decline with age, and (3) the activities of some zinc-dependent retinal enzymes have been shown to decline with age. To date, prospective cohort studies have shown limited evidence suggesting an association between dietary zinc intake and the incidence of AMD (129-131).
However, an early randomized controlled trial provoked interest when it found that 200 mg/day of zinc sulfate (81 mg/day of elemental zinc) over two years limited the loss of vision in patients with AMD (132). Yet a later trial using the same dose and duration found no benefit to patients with a more advanced form of AMD in one eye (133). Small trials have generally not reported a protective effect of vitamin and mineral supplementation on AMD (134, 135). However, a randomized, double-blind, placebo-controlled trial in 74 patients with AMD reported that supplementation with 50 mg/day of zinc monocysteine for six months improved measures of macular function, including visual acuity, contrast sensitivity, and photorecovery (136). A large randomized, placebo-controlled trial of daily supplementation with antioxidants (500 mg of vitamin C, 400 IU of vitamin E, and 15 mg of β-carotene) and high-dose zinc (80 mg of zinc as zinc oxide and 2 mg of copper as cupric oxide) — the Age-Related Eye Disease Study (AREDS) — found that administration of high-dose zinc alone or with the antioxidant combination to individuals with signs of moderate-to-severe macular degeneration significantly reduced the risk of developing advanced macular degeneration over a mean follow-up of 6.3 years (137). A follow-up analysis conducted four years after the cessation of the trial in 2001, including nearly 85% of the surviving participants, found that the benefit of the AREDS (combined antioxidants plus zinc) formulation had persisted (138). Indeed, the odds of developing late AMD, especially neovascular AMD, was lower in both participants with a low risk of developing AMD and those who were at risk and recommended to continue taking the AREDS formulation after the trial ended. There was, however, no effect of AREDS formulation on the risk of developing central geographic atrophy (138). Another trial, AREDS2, examined the effect of an AREDS formulation without β-carotene and/or containing 25 mg instead of 80 mg of zinc (139). The trial showed no apparent difference in the risk of developing advanced AMD with the use of AREDS formulations containing either 25 mg or 80 mg of zinc and/or β-carotene (140). A recent meta-analysis of five trials (including the original AREDS study) confirmed the protective effect of supplemental zinc against neovascular and advanced AMD (141).
In conclusion, the AREDS formulation combining antioxidants and zinc (25 mg or 80 mg) may delay the progression of the disease in patients with AMD. Patients, especially smokers and those with vascular disease, are advised to discuss with their physician the benefits versus potential harms that could be associated with the long-term use of high-dose antioxidant vitamins and carotenoids (141).
Diabetes mellitus
Type 2 diabetes mellitus
Poor glycemic control and frequent urination in patients with diabetes mellitus may be driving urinary loss of zinc and contribute to marginal zinc deficiency (142, 143). Yet, because of the role of zinc in β-cell function and insulin action (see Disease Prevention), a number of randomized controlled trials have examined whether supplementation with zinc (alone or with other minerals and vitamins) could play a role in diabetes management, especially by improving glycemic control in people with type 2 diabetes (15). Out of the 12 trials that measured participants’ zinc status at baseline, supplementation with zinc (20-240 mg/day) for 4 to 16 weeks improved fasting blood glucose in patients who presented with zinc deficiency (6 studies). Supplemental zinc also reduced the proportion of glycated hemoglobin (HbA1c) in two trials conducted in zinc-deficient participants, yet not in four studies including participants without zinc deficiency (15). Patients with type 2 diabetes should ensure that their diet provides enough zinc to cover their needs, especially if their blood glucose is poorly controlled.
Gestational diabetes mellitus
Gestational diabetes mellitus is defined as hyperglycemia that is first diagnosed during pregnancy. The condition is associated with an increased risk for adverse pregnancy outcomes (144). A group of investigators in Iran conducted two small randomized, placebo-controlled trials to examine the effect of zinc supplementation in pregnant women with gestational diabetes. Supplemental zinc (30 mg/day) for six weeks during pregnancy improved zinc status, reduced fasting blood glucose, and improved insulin sensitivity in women with gestational diabetes but had no impact on pregnancy outcomes, including the need for cesarean section, need for insulin therapy, newborn’s birth size and Apgar scores, or incidence of hyperbilirubinemia (145, 146). Similar improvements of markers of glycemic control were reported in another placebo-controlled trial that randomized pregnant women with gestational diabetes to receive zinc (4 mg) together with magnesium (100 mg), calcium (400 mg), and vitamin D (200 IU) twice a day for six weeks (147). There was also some evidence suggesting that supplemental zinc might help correct other metabolic disorders (e.g., abnormal blood lipid profile) associated with gestational diabetes (147, 148).
HIV/AIDS
Sufficient zinc is essential to maintain immune system function, and HIV-infected individuals are particularly susceptible to zinc deficiency. In HIV-infected patients, low serum zinc concentrations have been associated with disease progression and increased mortality (149, 150). In one study conducted in AIDS patients, 45 mg/day of zinc for one month resulted in a decreased incidence of opportunistic infections compared to placebo (151). A placebo-controlled trial in 231 HIV-positive adults with low zinc status found that zinc supplementation (12 mg/day for women and 15 mg/day for men) for 18 months reduced the incidence of immunological failure (defined by a CD4+ count <200 cells/mm3) by 76% and the rate of diarrhea by 60% (152). A 2011 systematic review that identified three randomized controlled trials in primarily resource-poor settings concluded that zinc supplementation was safe and efficacious in reducing opportunistic infections in HIV-positive adults (153).
Evidence of benefits of zinc supplementation in HIV-positive pregnant women and children is very limited. In a double-blind, randomized, placebo-controlled trial in Tanzania, the administration of zinc (25 mg/day) to women between 12 and 27 weeks’ gestation until six months after delivery failed to reduce maternal viral load or limit mother-to-child HIV transmission (154). A randomized placebo-controlled trial of zinc supplementation (10 mg/day for 6 months) in 96 HIV-positive children (6 months to 5 years old) in South Africa showed no effect on CD4+ count and viral load (155). There was evidence showing a reduction in the incidence of watery diarrhea in zinc-supplemented children compared to those taking a placebo, yet no differences in the incidence of pneumonia, ear infection, or upper respiratory tract infection (155). Another trial in Uganda showed that supplemental zinc in children with severe pneumonia effectively reduced case fatality regardless of children’s HIV status (156). While zinc supplementation during pregnancy and infancy is recommended in populations likely to be zinc deficient (43, 71, 75), its use in HIV infection management requires further investigation (157).
Alzheimer’s disease
Abnormal homeostasis of trace metals, in particular copper and zinc, has been reported in individuals affected by Alzheimer’s disease — the most common form of dementia. Specifically, results from case-control studies have shown higher serum copper concentrations and lower serum zinc concentrations in people with Alzheimer’s disease compared to cognitively healthy controls (158-160). Based on the utilization of zinc salts in Wilson's disease, it has been proposed that zinc supplementation could improve zinc and copper status and limit further cognitive deterioration in individuals with Alzheimer’s disease. The use of slow-release zinc acetate (150 mg/day for six months) in a randomized, placebo-controlled study of 60 patients with mild-to-moderate Alzheimer’s disease corrected low zinc status and decreased serum 'free' copper (i.e., unbound to ceruloplasmin) (161). Moreover, when a post-hoc analysis was restricted to participants over 70 years of age (N=29), it was found that zinc supplementation prevented the deterioration of cognition scores over the trial period (161). Additional evidence is needed to confirm whether zinc supplementation could play a role in stabilizing cognitive deficits in older adults with dementia.
Depression
A data analysis of the Boston Area Community Health (BACH) survey, including 3,708 participants (ages, 30-79 years), reported higher odds of depression symptoms in women (but not in men) in the lowest versus highest quartiles of total (median values, 8.7 mg/day versus 26.8 mg/day) and dietary (median values, 7.6 mg/day versus 13.1 mg/day) zinc intakes (162). The possibility that zinc could play a role in preventing or alleviating depression has been explored in two trials conducted by one research group. The data from these trials were analyzed following a per-protocol approach (i.e., restricted to the participants who completed the studies). A preliminary randomized, double-blind, placebo-controlled trial in 20 subjects (mean age, 43 years) treated for major depression showed that supplementation with 25 mg/day of zinc reduced depression symptoms at 6 and 12 weeks as assessed by the Hamilton Depression Rating Scale (HDRS) and Beck Depression Inventory (BDI) scores (163). A second placebo-controlled trial in 60 participants treated with the antidepressant imipramine (Tofranil; 100-200 mg/day) assessed the therapeutic response to supplemental zinc (25 mg/day) using HDRS, BDI, Clinical Global Impression scale (CGI), and Montgomery-Åsberg Depression Rating Scale (MADRS) scores (164). Zinc supplementation improved score-based measures of therapeutic response and remission after six weeks but only when the analysis was restricted to participants resistant to imipramine. There was, however, no evidence of an effect of zinc after 12 weeks (164).
Neonatal sepsis
Sepsis is a life-threatening condition that causes organ dysfunction as a consequence of a dysregulated host’s response to infection (165). Sepsis is accompanied by changes in zinc homeostasis characterized in particular by a decrease in serum zinc concentration and an increase in liver zinc concentration (166). These changes in zinc distribution are thought to be part of a host’s defense mechanism whereby the host can limit zinc availability to pathogens, as well as stimulate the immune system. Such a mechanism has been described for other transition metals, including iron and manganese (167). However, lower serum zinc concentrations in critically ill patients at high risk of organ failure have been associated with recurrent sepsis episodes and poorer outcomes (168, 169). A 2018 systematic review identified four trials that examined the effect of zinc supplementation in newborns with sepsis (166). Zinc supplementation was found to result in decreased inflammation (170) and better neurological development (171, 172). Three out of four trials that examined the rate of mortality showed no effect of zinc supplementation (170, 172, 173).
Sources
Food sources
Shellfish, beef, and other red meats are rich sources of zinc; nuts and legumes are relatively good plant sources of zinc. Zinc bioavailability (the fraction of zinc retained and used by the body) is relatively high in meat, eggs, and seafood because of the relative absence of compounds that inhibit zinc absorption and the presence of sulfur-containing amino acids (cysteine and methionine) that improve zinc absorption. Zinc in whole-grain products and plant proteins is less bioavailable due to their relatively high content of phytate, which inhibits zinc absorption (174). The enzymatic action of yeast reduces the level of phytate in foods; therefore, leavened whole-grain breads have more bioavailable zinc than unleavened whole-grain breads.
National dietary surveys in the US estimate that average dietary zinc intake from naturally and fortified food is about 12.3 mg/day in adults, with about 12% of the adult population being at risk for inadequate intake (175). The zinc content of some foods relatively rich in zinc is listed in Table 2 in milligrams (mg). For more information on the nutrient content of specific foods, search USDA's FoodData Central (176).
Supplements
A number of zinc supplements are commercially available, including zinc acetate, zinc gluconate, zinc picolinate, and zinc sulfate. Zinc picolinate has been promoted as a more absorbable form of zinc, but few data support this idea in humans. Limited work in animals suggests that increased intestinal absorption of zinc picolinate may be offset by increased elimination (29).
Safety
Toxicity
Acute toxicity
Isolated outbreaks of acute zinc toxicity have occurred as a result of the consumption of food or beverages contaminated with zinc released from galvanized containers. Signs of acute zinc toxicity are abdominal pain, diarrhea, nausea, and vomiting. Single doses of 225 to 450 mg of zinc usually induce vomiting. Milder gastrointestinal distress has been reported at doses of 50 to 150 mg/day of supplemental zinc. Metal fume fever has been reported after the inhalation of zinc oxide fumes. Specifically, profuse sweating, weakness, and rapid breathing may develop within eight hours of zinc oxide inhalation and persist for 12 to 24 hours after exposure is terminated (6, 29).
Adverse effects
The major consequence of long-term consumption of excessive zinc is copper deficiency. Total zinc intakes of 60 mg/day (50 mg supplemental and 10 mg dietary zinc) for up to 10 weeks have been found to result in signs of copper deficiency (29). Copper deficiency has also been reported following chronic use of excessive amounts of zinc-containing denture creams (≥2 tubes per week containing 17-34 mg/g of zinc) (177). In order to prevent copper deficiency, the US Food and Nutrition Board set the tolerable upper intake level (UL) for adults at 40 mg/day, including dietary and supplemental zinc (Table 3) (29).
Intranasal zinc
Intranasal zinc is known to cause a loss of the sense of smell (anosmia) in laboratory animals (178), and there have been several case reports of individuals who developed anosmia after using intranasal zinc gluconate (127). Since zinc-associated anosmia may be irreversible, the use of zinc nasal gels and sprays should be avoided.
Drug interactions
The use of zinc supplements decreases the absorption of certain medications, including cephalexin (Keplex) and penicillamine (Cuprimine, Depen), as well as the antiretroviral drugs atazanavir (Reyataz) and ritonavir (Norvir) (179). Concomitant administration of zinc supplements with certain medications like tetracycline and quinolone antibiotics may decrease the absorption of both zinc and the medications, potentially reducing drug efficacy. Taking zinc supplements and these medications at least two hours apart should prevent this interaction.
The therapeutic use of metal-chelating agents, such as penicillamine (used to treat copper overload in Wilson's disease) and diethylenetriamine pentaacetate (DTPA; used to treat iron overload), has resulted in severe zinc deficiency. Anticonvulsant drugs, especially sodium valproate, may also precipitate zinc deficiency. Prolonged use of diuretics may increase urinary zinc excretion, resulting in increased loss of zinc. Because supplemental zinc can lower blood glucose, those taking anti-diabetic agents are advised to use zinc supplements with caution.
Linus Pauling Institute Recommendation
The RDA for zinc (8 mg/day for adult women and 11 mg/day for adult men) appears sufficient to prevent deficiency in most individuals, but the lack of sensitive indicators of zinc nutritional status in humans makes it difficult to determine the level of zinc intake most likely to promote optimum health. Following the Linus Pauling Institute recommendation to take a multivitamin/mineral supplement will generally provide at least the RDA for zinc. Daily total (supplemental + dietary) intakes of zinc should not exceed the UL (40 mg/day for adults) in order to limit the risk of copper deficiency in particular (see Safety).
Older adults (>50 years)
Although the requirement for zinc is not known to be higher for older adults, many have inadequate dietary zinc intakes (180, 181). A reduced capacity to absorb zinc, increased likelihood of disease states that alter zinc utilization, and increased use of drugs that decrease zinc bioavailability may all contribute to an increased risk of mild zinc deficiency in older adults. Adequate dietary intake of zinc is essential for older adults because the consequences of mild zinc deficiency, such as impaired immune system function, are especially relevant to maintenance of their health.
Authors and Reviewers
Originally written in 2001 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in December 2003 by:
Jane Higdon, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in October 2007 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in June 2013 by:
Victoria J. Drake, Ph.D.
Linus Pauling Institute
Oregon State University
Updated in February 2019 by:
Barbara Delage, Ph.D.
Linus Pauling Institute
Oregon State University
Reviewed in May 2019 by:
Emily Ho, Ph.D.
Endowed Director, Moore Family Center for Whole Grain Foods,
Nutrition and Preventive Health
Professor, School of Biological and Population Health Sciences
Principal Investigator, Linus Pauling Institute
Oregon State University
Copyright 2001-2024 Linus Pauling Institute
References
1. Prasad AS, Halsted JA, Nadimi M. Syndrome of iron deficiency anemia, hepatosplenomegaly, hypogonadism, dwarfism and geophagia. Am J Med. 1961;31:532-546. (PubMed)
2. Penny ME. Zinc supplementation in public health. Ann Nutr Metab. 2013;62 Suppl 1:31-42. (PubMed)
3. Prasad AS. Impact of the discovery of human zinc deficiency on health. J Trace Elem Med Biol. 2014;28(4):357-363. (PubMed)
4. Terrin G, Berni Canani R, Di Chiara M, et al. Zinc in early life: a key element in the fetus and preterm neonate. Nutrients. 2015;7(12):10427-10446. (PubMed)
5. Andreini C, Banci L, Bertini I, Rosato A. Counting the zinc-proteins encoded in the human genome. J Proteome Res. 2006;5(1):196-201. (PubMed)
6. King JC, Cousins RJ. Zinc. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore: Lippincott Williams & Wilkins; 2014:189-205.
7. Vallee BL, Falchuk KH. The biochemical basis of zinc physiology. Physiol Rev. 1993;73(1):79-118. (PubMed)
8. King JC. Zinc: an essential but elusive nutrient. Am J Clin Nutr. 2011;94(2):679S-684S. (PubMed)
9. Cornish-Bowden A. Current IUBMB recommendations on enzyme nomenclature and kinetics. Perspectives in Science. 2014;1(1-6):74-87.
10. Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83(6):835-839. (PubMed)
11. Atrian-Blasco E, Santoro A, Pountney DL, Meloni G, Hureau C, Faller P. Chemistry of mammalian metallothioneins and their interaction with amyloidogenic peptides and proteins. Chem Soc Rev. 2017;46(24):7683-7693. (PubMed)
12. Hijova E. Metallothioneins and zinc: their functions and interactions. Bratisl Lek Listy. 2004;105(5-6):230-234. (PubMed)
13. Sirangelo I, Iannuzzi C. The role of metal binding in the amyotrophic lateral sclerosis-related aggregation of copper-zinc superoxide dismutase. Molecules. 2017;22(9). (PubMed)
14. Hershfinkel M, Moran A, Grossman N, Sekler I. A zinc-sensing receptor triggers the release of intracellular Ca2+ and regulates ion transport. Proc Natl Acad Sci U S A. 2001;98(20):11749-11754. (PubMed)
15. Ruz M, Carrasco F, Rojas P, Basfi-Fer K, Hernandez MC, Perez A. Nutritional effects of zinc on metabolic syndrome and type 2 diabetes: mechanisms and main findings in human studies. Biol Trace Elem Res. 2019; 188(1):177-188. (PubMed)
16. Takeda A, Tamano H. The impact of synaptic Zn(2+) dynamics on cognition and its decline. Int J Mol Sci. 2017;18(11). (PubMed)
17. Holt RR, Uriu-Adams JY, Keen CL. Zinc. In: Erdman Jr JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Washington D.C.: ILSI Press; 2012:521-539.
18. Sandstrom B. Micronutrient interactions: effects on absorption and bioavailability. Br J Nutr. 2001;85 Suppl 2:S181-185. (PubMed)
19. Zaman K, McArthur JO, Abboud MN, et al. Iron supplementation decreases plasma zinc but has no effect on plasma fatty acids in non-anemic women. Nutr Res. 2013;33(4):272-278. (PubMed)
20. O'Brien KO, Zavaleta N, Caulfield LE, Wen J, Abrams SA. Prenatal iron supplements impair zinc absorption in pregnant Peruvian women. J Nutr. 2000;130(9):2251-2255. (PubMed)
21. Fung EB, Ritchie LD, Woodhouse LR, Roehl R, King JC. Zinc absorption in women during pregnancy and lactation: a longitudinal study. Am J Clin Nutr. 1997;66(1):80-88. (PubMed)
22. Davidsson L, Almgren A, Sandstrom B, Hurrell RF. Zinc absorption in adult humans: the effect of iron fortification. Br J Nutr. 1995;74(3):417-425. (PubMed)
23. de Brito NJ, Rocha ED, de Araujo Silva A, et al. Oral zinc supplementation decreases the serum iron concentration in healthy schoolchildren: a pilot study. Nutrients. 2014;6(9):3460-3473. (PubMed)
24. Carter RC, Kupka R, Manji K, et al. Zinc and multivitamin supplementation have contrasting effects on infant iron status: a randomized, double-blind, placebo-controlled clinical trial. Eur J Clin Nutr. 2018;72(1):130-135. (PubMed)
25. de Oliveira Kde J, Donangelo CM, de Oliveira AV, Jr., da Silveira CL, Koury JC. Effect of zinc supplementation on the antioxidant, copper, and iron status of physically active adolescents. Cell Biochem Funct. 2009;27(3):162-166. (PubMed)
26. Wood RJ, Zheng JJ. High dietary calcium intakes reduce zinc absorption and balance in humans. Am J Clin Nutr. 1997;65(6):1803-1809. (PubMed)
27. McKenna AA, Ilich JZ, Andon MB, Wang C, Matkovic V. Zinc balance in adolescent females consuming a low- or high-calcium diet. Am J Clin Nutr. 1997;65(5):1460-1464. (PubMed)
28. Hunt JR, Beiseigel JM. Dietary calcium does not exacerbate phytate inhibition of zinc absorption by women from conventional diets. Am J Clin Nutr. 2009;89(3):839-843. (PubMed)
29. Food and Nutrition Board, Institute of Medicine. Zinc. Dietary reference intakes for vitamin A, vitamin K, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. Washington, D.C.: National Academy Press; 2001:442-501. (National Academy Press)
30. Kauwell GP, Bailey LB, Gregory JF, 3rd, Bowling DW, Cousins RJ. Zinc status is not adversely affected by folic acid supplementation and zinc intake does not impair folate utilization in human subjects. J Nutr. 1995;125(1):66-72. (PubMed)
31. Boron B, Hupert J, Barch DH, et al. Effect of zinc deficiency on hepatic enzymes regulating vitamin A status. J Nutr. 1988;118(8):995-1001. (PubMed)
32. Christian P, West KP, Jr. Interactions between zinc and vitamin A: an update. Am J Clin Nutr. 1998;68(2 Suppl):435S-441S. (PubMed)
33. Ciampo I, Sawamura R, Ciampo LAD, Fernandes MIM. Acrodematitis enteropathica: clinical manifestations and pediatric diagnosis. Rev Paul Pediatr. 2018;36(2):238-241. (PubMed)
34. Hambidge M. Human zinc deficiency. J Nutr. 2000;130(5S Suppl):1344S-1349S. (PubMed)
35. Fischer Walker CL, Ezzati M, Black RE. Global and regional child mortality and burden of disease attributable to zinc deficiency. Eur J Clin Nutr. 2009;63(5):591-597. (PubMed)
36. Prasad AS. Discovery of human zinc deficiency: 50 years later. J Trace Elem Med Biol. 2012;26(2-3):66-69. (PubMed)
37. International Zinc Nutrition Consultative Group, Brown KH, Rivera JA, et al. International Zinc Nutrition Consultative Group (IZiNCG) technical document #1. Assessment of the risk of zinc deficiency in populations and options for its control. Food Nutr Bull. 2004;25(1 Suppl 2):S99-203. (PubMed)
38. Krebs NF. Update on zinc deficiency and excess in clinical pediatric practice. Ann Nutr Metab. 2013;62 Suppl 1:19-29. (PubMed)
39. Gibson RS, Hess SY, Hotz C, Brown KH. Indicators of zinc status at the population level: a review of the evidence. Br J Nutr. 2008;99 Suppl 3:S14-23. (PubMed)
40. Wessells KR, Brown KH. Estimating the global prevalence of zinc deficiency: results based on zinc availability in national food supplies and the prevalence of stunting. PLoS One. 2012;7(11):e50568. (PubMed)
41. Moghimi M, Ashrafzadeh S, Rassi S, Naseh A. Maternal zinc deficiency and congenital anomalies in newborns. Pediatr Int. 2017;59(4):443-446. (PubMed)
42. Wilson RL, Grieger JA, Bianco-Miotto T, Roberts CT. Association between maternal zinc status, dietary zinc intake and pregnancy complications: a systematic review. Nutrients. 2016;8(10). (PubMed)
43. Ota E, Mori R, Middleton P, et al. Zinc supplementation for improving pregnancy and infant outcome. Cochrane Database Syst Rev. 2015(2):Cd000230. (PubMed)
44. Haider BA, Bhutta ZA. Multiple-micronutrient supplementation for women during pregnancy. Cochrane Database Syst Rev. 2017;4:Cd004905. (PubMed)
45. Petry N, Olofin I, Boy E, Donahue Angel M, Rohner F. The effect of low dose iron and zinc intake on child micronutrient status and development during the first 1000 days of life: a systematic review and meta-analysis. Nutrients. 2016;8(12). (PubMed)
46. Walravens PA, Hambidge KM, Koepfer DM. Zinc supplementation in infants with a nutritional pattern of failure to thrive: a double-blind, controlled study. Pediatrics. 1989;83(4):532-538. (PubMed)
47. Hambidge M, Krebs N. Zinc and growth. In: Roussell AM, ed. Trace elements in man and animals 10: Proceedings of the tenth international symposium on trace elements in man and animals. New York: Plenum Press; 2000:977-980.
48. Brown KH, Peerson JM, Baker SK, Hess SY. Preventive zinc supplementation among infants, preschoolers, and older prepubertal children. Food Nutr Bull. 2009;30(1 Suppl):S12-40. (PubMed)
49. Imdad A, Bhutta ZA. Effect of preventive zinc supplementation on linear growth in children under 5 years of age in developing countries: a meta-analysis of studies for input to the lives saved tool. BMC Public Health. 2011;11 Suppl 3:S22. (PubMed)
50. Liu E, Pimpin L, Shulkin M, et al. Effect of zinc supplementation on growth outcomes in children under 5 years of age. Nutrients. 2018;10(3). (PubMed)
51. MacDonald RS. The role of zinc in growth and cell proliferation. J Nutr. 2000;130(5S Suppl):1500S-1508S. (PubMed)
52. Thousand day global initiative. Available at: https://thousanddays.org/. Accessed 2/14/19.
53. Bhatnagar S, Taneja S. Zinc and cognitive development. Br J Nutr. 2001;85 Suppl 2:S139-145. (PubMed)
54. Tamura T, Goldenberg RL, Ramey SL, Nelson KG, Chapman VR. Effect of zinc supplementation of pregnant women on the mental and psychomotor development of their children at 5 y of age. Am J Clin Nutr. 2003;77(6):1512-1516. (PubMed)
55. Sazawal S, Bentley M, Black RE, Dhingra P, George S, Bhan MK. Effect of zinc supplementation on observed activity in low socioeconomic Indian preschool children. Pediatrics. 1996;98(6 Pt 1):1132-1137. (PubMed)
56. Bentley ME, Caulfield LE, Ram M, et al. Zinc supplementation affects the activity patterns of rural Guatemalan infants. J Nutr. 1997;127(7):1333-1338. (PubMed)
57. Ashworth A, Morris SS, Lira PI, Grantham-McGregor SM. Zinc supplementation, mental development and behaviour in low birth weight term infants in northeast Brazil. Eur J Clin Nutr. 1998;52(3):223-227. (PubMed)
58. Castillo-Duran C, Perales CG, Hertrampf ED, Marin VB, Rivera FA, Icaza G. Effect of zinc supplementation on development and growth of Chilean infants. J Pediatr. 2001;138(2):229-235. (PubMed)
59. Lind T, Lonnerdal B, Stenlund H, et al. A community-based randomized controlled trial of iron and zinc supplementation in Indonesian infants: effects on growth and development. Am J Clin Nutr. 2004;80(3):729-736. (PubMed)
60. Taneja S, Bhandari N, Bahl R, Bhan MK. Impact of zinc supplementation on mental and psychomotor scores of children aged 12 to 18 months: a randomized, double-blind trial. J Pediatr. 2005;146(4):506-511. (PubMed)
61. Gogia S, Sachdev HS. Zinc supplementation for mental and motor development in children. Cochrane Database Syst Rev. 2012;12:CD007991. (PubMed)
62. Baum MK, Shor-Posner G, Campa A. Zinc status in human immunodeficiency virus infection. J Nutr. 2000;130(5S Suppl):1421S-1423S. (PubMed)
63. Maares M, Haase H. Zinc and immunity: An essential interrelation. Arch Biochem Biophys. 2016;611:58-65. (PubMed)
64. Subramanian Vignesh K, Deepe GS, Jr. Immunological orchestration of zinc homeostasis: The battle between host mechanisms and pathogen defenses. Arch Biochem Biophys. 2016;611:66-78. (PubMed)
65. Subramanian Vignesh K, Landero Figueroa JA, Porollo A, Caruso JA, Deepe GS, Jr. Granulocyte macrophage-colony stimulating factor induced Zn sequestration enhances macrophage superoxide and limits intracellular pathogen survival. Immunity. 2013;39(4):697-710. (PubMed)
66. Fischer Walker C, Black RE. Zinc and the risk for infectious disease. Annu Rev Nutr. 2004;24:255-275. (PubMed)
67. Shankar AH, Prasad AS. Zinc and immune function: the biological basis of altered resistance to infection. Am J Clin Nutr. 1998;68(2 Suppl):447S-463S. (PubMed)
68. Wapnir RA. Zinc deficiency, malnutrition and the gastrointestinal tract. J Nutr. 2000;130(5S Suppl):1388S-1392S. (PubMed)
69. Liu L, Oza S, Hogan D, et al. Global, regional, and national causes of child mortality in 2000-13, with projections to inform post-2015 priorities: an updated systematic analysis. Lancet. 2015;385(9966):430-440. (PubMed)
70. Black RE. Progress in the use of ORS and zinc for the treatment of childhood diarrhea. J Glob Health. 2019;9(1):010101. (PubMed)
71. Lazzerini M, Wanzira H. Oral zinc for treating diarrhoea in children. Cochrane Database Syst Rev. 2016;12:Cd005436. (PubMed)
72. WHO and UNICEF. Clinical management of acute diarrhoea. Available at: https://www.who.int/publications/i/item/WHO_FCH_CAH_04.7. Accessed 3/19/24.
73. World Health Organization. Fact sheets: pneumonia. November 6, 2016. Available at: https://www.who.int/news-room/fact-sheets/detail/pneumonia. Accessed 2/11/19.
74. World Health Organization. Global health risks: mortality and burden of disease attributable to selected major risks. 2009. Available at: https://apps.who.int/iris/handle/10665/44203. Accessed 2/11/19.
75. Lassi ZS, Moin A, Bhutta ZA. Zinc supplementation for the prevention of pneumonia in children aged 2 months to 59 months. Cochrane Database Syst Rev. 2016;12:Cd005978. (PubMed)
76. Howie S, Bottomley C, Chimah O, et al. Zinc as an adjunct therapy in the management of severe pneumonia among Gambian children: randomized controlled trial. J Glob Health. 2018;8(1):010418. (PubMed)
77. Wang L, Song Y. Efficacy of zinc given as an adjunct to the treatment of severe pneumonia: A meta-analysis of randomized, double-blind and placebo-controlled trials. Clin Respir J. 2018;12(3):857-864. (PubMed)
78. Black MM. Zinc deficiency and child development. Am J Clin Nutr. 1998;68(2 Suppl):464S-469S. (PubMed)
79. Shankar AH. Nutritional modulation of malaria morbidity and mortality. J Infect Dis. 2000;182 Suppl 1:S37-53. (PubMed)
80. Müller O, Becher H, van Zweeden AB, et al. Effect of zinc supplementation on malaria and other causes of morbidity in west African children: randomised double blind placebo controlled trial. BMJ. 2001;322(7302):1567. (PubMed)
81. Zinc Against Plasmodium Study Group. Effect of zinc on the treatment of Plasmodium falciparum malaria in children: a randomized controlled trial. Am J Clin Nutr. 2002;76(4):805-812. (PubMed)
82. Sazawal S, Black RE, Ramsan M, et al. Effect of zinc supplementation on mortality in children aged 1-48 months: a community-based randomised placebo-controlled trial. Lancet. 2007;369(9565):927-934. (PubMed)
83. Darling AM, Mugusi FM, Etheredge AJ, et al. Vitamin A and zinc supplementation among pregnant women to prevent placental malaria: a randomized, double-blind, placebo-controlled trial in Tanzania. Am J Trop Med Hyg. 2017;96(4):826-834. (PubMed)
84. Mocchegiani E, Romeo J, Malavolta M, et al. Zinc: dietary intake and impact of supplementation on immune function in elderly. Age (Dordr). 2013;35(3):839-860. (PubMed)
85. Meydani SN, Barnett JB, Dallal GE, et al. Serum zinc and pneumonia in nursing home elderly. Am J Clin Nutr. 2007;86(4):1167-1173. (PubMed)
86. Haase H, Rink L. The immune system and the impact of zinc during aging. Immun Ageing. 2009;6:9. (PubMed)
87. Bogden JD, Oleske JM, Lavenhar MA, et al. Effects of one year of supplementation with zinc and other micronutrients on cellular immunity in the elderly. J Am Coll Nutr. 1990;9(3):214-225. (PubMed)
88. Bogden JD, Oleske JM, Lavenhar MA, et al. Zinc and immunocompetence in elderly people: effects of zinc supplementation for 3 months. Am J Clin Nutr. 1988;48(3):655-663. (PubMed)
89. Provinciali M, Montenovo A, Di Stefano G, et al. Effect of zinc or zinc plus arginine supplementation on antibody titre and lymphocyte subsets after influenza vaccination in elderly subjects: a randomized controlled trial. Age Ageing. 1998;27(6):715-722. (PubMed)
90. Prasad AS. Clinical, immunological, anti-inflammatory and antioxidant roles of zinc. Exp Gerontol. 2008;43(5):370-377. (PubMed)
91. Fortes C, Forastiere F, Agabiti N, et al. The effect of zinc and vitamin A supplementation on immune response in an older population. J Am Geriatr Soc. 1998;46(1):19-26. (PubMed)
92. Hodkinson CF, Kelly M, Alexander HD, et al. Effect of zinc supplementation on the immune status of healthy older individuals aged 55-70 years: the ZENITH Study. J Gerontol A Biol Sci Med Sci. 2007;62(6):598-608. (PubMed)
93. Barnett JB, Dao MC, Hamer DH, et al. Effect of zinc supplementation on serum zinc concentration and T cell proliferation in nursing home elderly: a randomized, double-blind, placebo-controlled trial. Am J Clin Nutr. 2016;103(3):942-951. (PubMed)
94. Norouzi S, Adulcikas J, Sohal SS, Myers S. Zinc stimulates glucose oxidation and glycemic control by modulating the insulin signaling pathway in human and mouse skeletal muscle cell lines. PLoS One. 2018;13(1):e0191727. (PubMed)
95. Gu HF. Genetic, epigenetic and biological effects of zinc transporter (SLC30A8) in type 1 and type 2 diabetes. Curr Diabetes Rev. 2017;13(2):132-140. (PubMed)
96. Flannick J, Thorleifsson G, Beer NL, et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet. 2014;46(4):357-363. (PubMed)
97. Sun Q, van Dam RM, Willett WC, Hu FB. Prospective study of zinc intake and risk of type 2 diabetes in women. Diabetes Care. 2009;32(4):629-634. (PubMed)
98. Vashum KP, McEvoy M, Shi Z, et al. Is dietary zinc protective for type 2 diabetes? Results from the Australian longitudinal study on women's health. BMC Endocr Disord. 2013;13:40. (PubMed)
99. Schwingshackl L, Hoffmann G, Lampousi AM, et al. Food groups and risk of type 2 diabetes mellitus: a systematic review and meta-analysis of prospective studies. Eur J Epidemiol. 2017;32(5):363-375. (PubMed)
100. de Oliveira Otto MC, Alonso A, Lee DH, et al. Dietary intakes of zinc and heme iron from red meat, but not from other sources, are associated with greater risk of metabolic syndrome and cardiovascular disease. J Nutr. 2012;142(3):526-533. (PubMed)
101. Song Y, Xu Q, Park Y, Hollenbeck A, Schatzkin A, Chen H. Multivitamins, individual vitamin and mineral supplements, and risk of diabetes among older U.S. adults. Diabetes Care. 2011;34(1):108-114. (PubMed)
102. Drake I, Hindy G, Ericson U, Orho-Melander M. A prospective study of dietary and supplemental zinc intake and risk of type 2 diabetes depending on genetic variation in SLC30A8. Genes Nutr. 2017;12:30. (PubMed)
103. El Dib R, Gameiro OL, Ogata MS, et al. Zinc supplementation for the prevention of type 2 diabetes mellitus in adults with insulin resistance. Cochrane Database Syst Rev. 2015(5):Cd005525. (PubMed)
104. Islam MR, Attia J, Ali L, et al. Zinc supplementation for improving glucose handling in pre-diabetes: A double blind randomized placebo controlled pilot study. Diabetes Res Clin Pract. 2016;115:39-46. (PubMed)
105. Ranasinghe P, Wathurapatha WS, Galappatthy P, Katulanda P, Jayawardena R, Constantine GR. Zinc supplementation in prediabetes: A randomized double-blind placebo-controlled clinical trial. J Diabetes. 2018;10(5):386-397. (PubMed)
106. Mak CM, Lam CW. Diagnosis of Wilson's disease: a comprehensive review. Crit Rev Clin Lab Sci. 2008;45(3):263-290. (PubMed)
107. Poujois A, Woimant F. Wilson's disease: A 2017 update. Clin Res Hepatol Gastroenterol. 2018;42(6):512-520. (PubMed)
108. Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47(6):2089-2111. (PubMed)
109. Avan A, de Bie RMA, Hoogenraad TU. Wilson's disease should be treated with zinc rather than trientine or penicillamine. Neuropediatrics. 2017;48(5):394-395. (PubMed)
110. Brewer GJ, Dick RD, Johnson VD, Fink JK, Kluin KJ, Daniels S. Treatment of Wilson's disease with zinc XVI: treatment during the pediatric years. J Lab Clin Med. 2001;137(3):191-198. (PubMed)
111. Eda K, Mizuochi T, Iwama I, et al. Zinc monotherapy for young children with presymptomatic Wilson disease: A multicenter study in Japan. J Gastroenterol Hepatol. 2018;33(1):264-269. (PubMed)
112. Gupta P, Choksi M, Goel A, et al. Maintenance zinc therapy after initial penicillamine chelation to treat symptomatic hepatic Wilson's disease in resource constrained setting. Indian J Gastroenterol. 2018;37(1):31-38. (PubMed)
113. Shimizu N, Fujiwara J, Ohnishi S, et al. Effects of long-term zinc treatment in Japanese patients with Wilson disease: efficacy, stability, and copper metabolism. Transl Res. 2010;156(6):350-357. (PubMed)
114. Sinha S, Taly AB. Withdrawal of penicillamine from zinc sulphate-penicillamine maintenance therapy in Wilson's disease: promising, safe and cheap. J Neurol Sci. 2008;264(1-2):129-132. (PubMed)
115. Centers for Disease Control and Prevention. Common colds: protect yourself and others. February 12, 2018. Available at: https://www.cdc.gov/features/rhinoviruses/. Accessed 2/7/19.
116. Rao G, Rowland K. PURLs: Zinc for the common cold--not if, but when. J Fam Pract. 2011;60(11):669-671. (PubMed)
117. Science M, Johnstone J, Roth DE, Guyatt G, Loeb M. Zinc for the treatment of the common cold: a systematic review and meta-analysis of randomized controlled trials. CMAJ. 2012;184(10):E551-561. (PubMed)
118. Singh M, Das RR. Zinc for the common cold. Cochrane Database Syst Rev. 2013(6):Cd001364. (PubMed)
119. Eby GA, 3rd. Zinc lozenges as cure for the common cold--a review and hypothesis. Med Hypotheses. 2010;74(3):482-492. (PubMed)
120. Hemila H. Zinc lozenges may shorten the duration of colds: a systematic review. Open Respir Med J. 2011;5:51-58. (PubMed)
121. Jackson JL, Lesho E, Peterson C. Zinc and the common cold: a meta-analysis revisited. J Nutr. 2000;130(5S Suppl):1512S-1515S. (PubMed)
122. Hemila H. Zinc lozenges and the common cold: a meta-analysis comparing zinc acetate and zinc gluconate, and the role of zinc dosage. JRSM Open. 2017;8(5):2054270417694291. (PubMed)
123. Mossad SB. Effect of zincum gluconicum nasal gel on the duration and symptom severity of the common cold in otherwise healthy adults. QJM. 2003;96(1):35-43. (PubMed)
124. Hirt M, Nobel S, Barron E. Zinc nasal gel for the treatment of common cold symptoms: a double-blind, placebo-controlled trial. Ear Nose Throat J. 2000;79(10):778-780, 782. (PubMed)
125. Belongia EA, Berg R, Liu K. A randomized trial of zinc nasal spray for the treatment of upper respiratory illness in adults. Am J Med. 2001;111(2):103-108. (PubMed)
126. D'Cruze H, Arroll B, Kenealy T. Is intranasal zinc effective and safe for the common cold? A systematic review and meta-analysis. J Prim Health Care. 2009;1(2):134-139. (PubMed)
127. DeCook CA, Hirsch AR. Anosmia due to inhalational zinc: a case report. Chem Senses. 2000;25(5):659.
128. Centers for Disease Control and Prevention. Learn about age-related macular degeneration. July 18, 2018. Available at: https://www.cdc.gov/visionhealth/resources/features/macular-degeneration.html. Accessed 3/19/24.
129. Cho E, Stampfer MJ, Seddon JM, et al. Prospective study of zinc intake and the risk of age-related macular degeneration. Ann Epidemiol. 2001;11(5):328-336. (PubMed)
130. van Leeuwen R, Boekhoorn S, Vingerling JR, et al. Dietary intake of antioxidants and risk of age-related macular degeneration. JAMA. 2005;294(24):3101-3107. (PubMed)
131. VandenLangenberg GM, Mares-Perlman JA, Klein R, Klein BE, Brady WE, Palta M. Associations between antioxidant and zinc intake and the 5-year incidence of early age-related maculopathy in the Beaver Dam Eye Study. Am J Epidemiol. 1998;148(2):204-214. (PubMed)
132. Newsome DA, Swartz M, Leone NC, Elston RC, Miller E. Oral zinc in macular degeneration. Arch Ophthalmol. 1988;106(2):192-198. (PubMed)
133. Stur M, Tittl M, Reitner A, Meisinger V. Oral zinc and the second eye in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1996;37(7):1225-1235. (PubMed)
134. Evans JR. Antioxidant vitamin and mineral supplements for slowing the progression of age-related macular degeneration. Cochrane Database Syst Rev. 2006(2):CD000254. (PubMed)
135. Evans J. Antioxidant supplements to prevent or slow down the progression of AMD: a systematic review and meta-analysis. Eye (Lond). 2008;22(6):751-760. (PubMed)
136. Newsome DA. A randomized, prospective, placebo-controlled clinical trial of a novel zinc-monocysteine compound in age-related macular degeneration. Curr Eye Res. 2008;33(7):591-598. (PubMed)
137. Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119(10):1417-1436. (PubMed)
138. Chew EY, Clemons TE, Agron E, et al. Long-term effects of vitamins C and E, beta-carotene, and zinc on age-related macular degeneration: AREDS report no. 35. Ophthalmology. 2013;120(8):1604-1611.e1604. (PubMed)
139. Age-Related Eye Disease Study 2 Research Group. Lutein + zeaxanthin and omega-3 fatty acids for age-related macular degeneration: the Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial. JAMA. 2013;309(19):2005-2015. (PubMed)
140. Aronow ME, Chew EY. Age-related Eye Disease Study 2: perspectives, recommendations, and unanswered questions. Curr Opin Ophthalmol. 2014;25(3):186-190. (PubMed)
141. Evans JR, Lawrenson JG. Antioxidant vitamin and mineral supplements for slowing the progression of age-related macular degeneration. Cochrane Database Syst Rev. 2017;7:Cd000254. (PubMed)
142. Blostein-Fujii A, DiSilvestro RA, Frid D, Katz C, Malarkey W. Short-term zinc supplementation in women with non-insulin-dependent diabetes mellitus: effects on plasma 5'-nucleotidase activities, insulin-like growth factor I concentrations, and lipoprotein oxidation rates in vitro. Am J Clin Nutr. 1997;66(3):639-642. (PubMed)
143. Perez A, Rojas P, Carrasco F, et al. Association between zinc nutritional status and glycemic control in individuals with well-controlled type-2 diabetes. J Trace Elem Med Biol. 2018;50:560-565. (PubMed)
144. Billionnet C, Mitanchez D, Weill A, et al. Gestational diabetes and adverse perinatal outcomes from 716,152 births in France in 2012. Diabetologia. 2017;60(4):636-644. (PubMed)
145. Karamali M, Heidarzadeh Z, Seifati SM, et al. Zinc supplementation and the effects on metabolic status in gestational diabetes: A randomized, double-blind, placebo-controlled trial. J Diabetes Complications. 2015;29(8):1314-1319. (PubMed)
146. Karamali M, Heidarzadeh Z, Seifati SM, et al. Zinc supplementation and the effects on pregnancy outcomes in gestational diabetes: a randomized, double-blind, placebo-controlled trial. Exp Clin Endocrinol Diabetes. 2016;124(1):28-33. (PubMed)
147. Karamali M, Bahramimoghadam S, Sharifzadeh F, Asemi Z. Magnesium-zinc-calcium-vitamin D co-supplementation improves glycemic control and markers of cardiometabolic risk in gestational diabetes: a randomized, double-blind, placebo-controlled trial. Appl Physiol Nutr Metab. 2018;43(6):565-570. (PubMed)
148. Ostadmohammadi V, Samimi M, Mobini M, et al. The effect of zinc and vitamin E cosupplementation on metabolic status and its related gene expression in patients with gestational diabetes. J Matern Fetal Neonatal Med. 2018:1-8. (PubMed)
149. Lai H, Lai S, Shor-Posner G, Ma F, Trapido E, Baum MK. Plasma zinc, copper, copper:zinc ratio, and survival in a cohort of HIV-1-infected homosexual men. J Acquir Immune Defic Syndr. 2001;27(1):56-62. (PubMed)
150. Wellinghausen N, Kern WV, Jochle W, Kern P. Zinc serum level in human immunodeficiency virus-infected patients in relation to immunological status. Biol Trace Elem Res. 2000;73(2):139-149. (PubMed)
151. Mocchegiani E, Muzzioli M. Therapeutic application of zinc in human immunodeficiency virus against opportunistic infections. J Nutr. 2000;130(5S Suppl):1424S-1431S. (PubMed)
152. Baum MK, Lai S, Sales S, Page JB, Campa A. Randomized, controlled clinical trial of zinc supplementation to prevent immunological failure in HIV-infected adults. Clin Infect Dis. 2010;50(12):1653-1660. (PubMed)
153. Zeng L, Zhang L. Efficacy and safety of zinc supplementation for adults, children and pregnant women with HIV infection: systematic review. Trop Med Int Health. 2011;16(12):1474-1482. (PubMed)
154. Villamor E, Aboud S, Koulinska IN, et al. Zinc supplementation to HIV-1-infected pregnant women: effects on maternal anthropometry, viral load, and early mother-to-child transmission. Eur J Clin Nutr. 2006;60(7):862-869. (PubMed)
155. Bobat R, Coovadia H, Stephen C, et al. Safety and efficacy of zinc supplementation for children with HIV-1 infection in South Africa: a randomised double-blind placebo-controlled trial. Lancet. 2005;366(9500):1862-1867. (PubMed)
156. Srinivasan MG, Ndeezi G, Mboijana CK, et al. Zinc adjunct therapy reduces case fatality in severe childhood pneumonia: a randomized double blind placebo-controlled trial. BMC Med. 2012;10:14. (PubMed)
157. McHenry MS, Dixit A, Vreeman RC. A systematic review of nutritional supplementation in HIV-infected children in resource-limited settings. J Int Assoc Provid AIDS Care. 2015;14(4):313-323. (PubMed)
158. Li DD, Zhang W, Wang ZY, Zhao P. Serum copper, zinc, and iron levels in patients with Alzheimer's disease: a meta-analysis of case-control studies. Front Aging Neurosci. 2017;9:300. (PubMed)
159. Ventriglia M, Brewer GJ, Simonelli I, et al. Zinc in Alzheimer's disease: a meta-analysis of serum, plasma, and cerebrospinal fluid studies. J Alzheimers Dis. 2015;46(1):75-87. (PubMed)
160. Ventriglia M, Bucossi S, Panetta V, Squitti R. Copper in Alzheimer's disease: a meta-analysis of serum, plasma, and cerebrospinal fluid studies. J Alzheimers Dis. 2012;30(4):981-984. (PubMed)
161. Brewer GJ. Copper excess, zinc deficiency, and cognition loss in Alzheimer's disease. Biofactors. 2012;38(2):107-113. (PubMed)
162. Maserejian NN, Hall SA, McKinlay JB. Low dietary or supplemental zinc is associated with depression symptoms among women, but not men, in a population-based epidemiological survey. J Affect Disord. 2012;136(3):781-788. (PubMed)
163. Nowak G, Siwek M, Dudek D, Zieba A, Pilc A. Effect of zinc supplementation on antidepressant therapy in unipolar depression: a preliminary placebo-controlled study. Pol J Pharmacol. 2003;55(6):1143-1147. (PubMed)
164. Siwek M, Dudek D, Paul IA, et al. Zinc supplementation augments efficacy of imipramine in treatment resistant patients: a double blind, placebo-controlled study. J Affect Disord. 2009;118(1-3):187-195. (PubMed)
165. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus definitions for sepsis and septic shock (sepsis-3). JAMA. 2016;315(8):801-810. (PubMed)
166. Alker W, Haase H. Zinc and Sepsis. Nutrients. 2018;10(8). (PubMed)
167. Hood MI, Skaar EP. Nutritional immunity: transition metals at the pathogen-host interface. Nat Rev Microbiol. 2012;10(8):525-537. (PubMed)
168. Hoeger J, Simon TP, Beeker T, Marx G, Haase H, Schuerholz T. Persistent low serum zinc is associated with recurrent sepsis in critically ill patients - A pilot study. PLoS One. 2017;12(5):e0176069. (PubMed)
169. Saleh NY, Abo El Fotoh WMM. Low serum zinc level: The relationship with severe pneumonia and survival in critically ill children. Int J Clin Pract. 2018;72(6):e13211. (PubMed)
170. Banupriya N, Vishnu Bhat B, Benet BD, Sridhar MG, Parija SC. Efficacy of zinc supplementation on serum calprotectin, inflammatory cytokines and outcome in neonatal sepsis - a randomized controlled trial. J Matern Fetal Neonatal Med. 2017;30(13):1627-1631. (PubMed)
171. Banupriya N, Bhat BV, Benet BD, Catherine C, Sridhar MG, Parija SC. Short term oral zinc supplementation among babies with neonatal sepsis for reducing mortality and improving outcome - a double-blind randomized controlled trial. Indian J Pediatr. 2018;85(1):5-9. (PubMed)
172. Newton B, Bhat BV, Dhas BB, Mondal N, Gopalakrishna SM. Effect of zinc supplementation on early outcome of neonatal sepsis--a randomized controlled trial. Indian J Pediatr. 2016;83(4):289-293. (PubMed)
173. Mehta K, Bhatta NK, Majhi S, Shrivastava MK, Singh RR. Oral zinc supplementation for reducing mortality in probable neonatal sepsis: a double blind randomized placebo controlled trial. Indian Pediatr. 2013;50(4):390-393. (PubMed)
174. Gupta RK, Gangoliya SS, Singh NK. Reduction of phytic acid and enhancement of bioavailable micronutrients in food grains. J Food Sci Technol. 2015;52(2):676-684. (PubMed)
175. Fulgoni VL, 3rd, Keast DR, Bailey RL, Dwyer J. Foods, fortificants, and supplements: Where do Americans get their nutrients? J Nutr. 2011;141(10):1847-1854. (PubMed)
176. US Department of Agriculture, Agricultural Research Service. FoodData Central, 2019. fdc.nal.usda.gov.
177. Nations SP, Boyer PJ, Love LA, et al. Denture cream: an unusual source of excess zinc, leading to hypocupremia and neurologic disease. Neurology. 2008;71(9):639-643. (PubMed)
178. McBride K, Slotnick B, Margolis FL. Does intranasal application of zinc sulfate produce anosmia in the mouse? An olfactometric and anatomical study. Chem Senses. 2003;28(8):659-670. (PubMed)
179. Natural Medicines. Zinc: professional handout/drug interactions. Available at: https://naturalmedicines.therapeuticresearch.com. Accessed 1/28/19.
180. Ervin RB, Kennedy-Stephenson J. Mineral intakes of elderly adult supplement and non-supplement users in the third national health and nutrition examination survey. J Nutr. 2002;132(11):3422-3427. (PubMed)
181. Kvamme JM, Gronli O, Jacobsen BK, Florholmen J. Risk of malnutrition and zinc deficiency in community-living elderly men and women: the Tromso Study. Public Health Nutr. 2015;18(11):1907-1913. (PubMed)